

Abstract

Dual-specificity tyrosine-regulated kinase 1A (DYRK1A) regulates the proliferation and differentiation of neuronal progenitor cells during brain development. Consequently, DYRK1A has attracted interest as a target for the treatment of neurodegenerative diseases, including Alzheimer’s disease (AD) and Down’s syndrome. Recently, the inhibition of DYRK1A has been investigated as a potential treatment for diabetes, while DYRK1A’s role as a mediator in the cell cycle has garnered interest in oncologic indications. Structure–activity relationship (SAR) analysis in combination with high-resolution X-ray crystallography leads to a series of pyrazolo[1,5-b]pyridazine inhibitors with excellent ligand efficiencies, good physicochemical properties, and a high degree of selectivity over the kinome. Compound 11 exhibited good permeability and cellular activity without P-glycoprotein liability, extending the utility of 11 in an in vivo setting. These pyrazolo[1,5-b]pyridazines are a viable lead series in the discovery of new therapies for the treatment of diseases linked to DYRK1A function.
Introduction
Therapeutic applications of antagonists of the serine/threonine kinase, dual-specificity tyrosine-regulated kinase 1A (DYRK1A), have been under investigation for two decades. Diseases associated with the abnormal expression of DYRK1A (Alzheimer’s disease (AD), Down’s syndrome, cancer, and diabetes) represent significant unmet medical needs. Targeting the underlying mechanisms that cause these diseases is desirable in the search for novel approaches to treatment. DYRK1A plays a crucial role in neurogenesis and neuronal differentiation during embryonic development.1 DYRK1A is located on the Down’s syndrome critical region (DSCR) of chromosome 21; hence, individuals with Trisomy 21 (Down’s syndrome) express elevated levels of DYRK1A.2 As a result, overexpression of DYRK1A is thought to contribute to cognitive impairments associated with Down’s syndrome.2 AD has been associated with the accumulation of Aβ amyloid plaques and neurofibrillary tangles (NFTs) in the brain.3 Increased DYRK1A activity promotes both NFT formation and aggregation,4 in addition to accelerating the cleavage of Aβ peptides that comprise amyloid plaques.5 The ability of DYRK1A to effect the phosphorylation of nuclear factors of activated T cells (NFAT) and promote the proliferation of β-cells has piqued interest in DYRK1A inhibitors as a potential treatment for diabetes.6
Several DYRK1A inhibitors have previously been developed.7 Despite these efforts, few have progressed to in vivo models of disease, largely as a result of limited pharmacokinetics or poor selectivity for DYRK1A. Leucettine L41,8 SM07883 (structure not disclosed),9 GNF2133,10 and compound 2-2c11 represent DYRK1A antagonists that have been investigated in vivo (Figure 1). However, Leucettine 41, SM07883, and GNF2133 potently inhibit other CMGC kinases, so they are more accurately described as dual DYRK1/CLK inhibitors.
Figure 1.

Advanced DYRK1/CLK inhibitors reported in the literature (Leucettine 41,8 GNF2133,10 and compound 2–2c11) and previously characterized DYRK1A inhibitors 6b and 8b with DYRK1A IC50 values of 186 and 76 nM, respectively. 6b (shown in yellow) bound to DYRK1A such that the N-methyl group was directed toward a small pocket on the kinase hinge region (PDB 6S1I). Key hydrogen-bonding interactions between 6b, a consensus water, and DYRK1A (Leu241, Glu239, and Lys188) are depicted by black dotted lines. Compound 8b had a selectivity score (S(40)) against DYRK1A of 0.01.12
Previously, we reported the cheminformatics-based design of a series of selective DYRK1A inhibitors, exemplified by 8b and 6b (Figure 1), which had DYRK1A IC50 values of 76 and 186 nM, respectively.12 Here, we report the structure-guided development of this pyrazolo[1,5-b]pyridazine series, resulting in potent and selective inhibitors of DYRK1A with physical properties compatible with treatment of diseases associated with DYRK1A function. In agreement with our previous conclusions,12 the favorable selectivity profile of the series was achieved by maintaining hydrogen-bonding interactions between the ligand, a consensus water, and amino acid residues Glu239, Leu241, and Lys188, while directing a methyl substituent, or similar moiety, into a shallow lipophilic pocket, exclusive to the hinge of the DYRK and CLK kinases.
Results and Discussion
Three regions of 8b were selected for structure–activity relationship (SAR) analysis: (a) the N-Me-aryl substituent, (b) the pyrazolo[1,5-b]pyridazine heterocycle, and (c) the pyrimidine hinge-binding moiety. To maintain and improve upon the selectivity of the series, it was important to preserve the unusual binding mode of the series in the ATP-binding site of DYRK1A. Therefore, a synthetic strategy was devised to access a range of N-alkyl analogues (9–55). The synthesis of key pyrazolo[1,5-b]pyridazine intermediates 6 and 8 followed literature procedures (Scheme 1).12,13 N-Amination of pyridazine was completed with hydroxylamine-O-sulfonic acid. The pyrazolo[1,5-b]pyridazine heterocycle was constructed by reacting 1,3-dipole 2 with 3-butyne-2-one in a Huisgen cycloaddition. Dimethylformamide–dimethylacetamide (DMF–DMA) condensation of ketone 3 gave intermediate 4 in a high yield. Performing a urea melt on 4, followed by dehydration and chlorination with phosphorus(V)oxychloride afforded key intermediate 6. Alternatively, thioether 7 was accessed by treating 4 with thiourea and sodium methoxide, followed by iodomethane. Upon oxidation of 7 with m-CPBA, the key intermediate 8 was isolated.
Scheme 1. Synthesis of Key Intermediates 6 and 8.

Reagents and conditions: (i) hydroxylamine-O-sulfonic acid, KHCO3, KI, H2O, 70 °C, 1.5 h, 94%; (ii) 3-butyne-2-one, KOH, CH2Cl2, rt, 16 h, 60%; (iii) DMF–DMA, 100 °C, 16 h, 91%; (iv) urea, sodium hydride, 140 °C, 10 min, 91%; (v) phosphorus(V)oxychloride, 110 °C, 3 h, 92%; (vi) thiourea, sodium methoxide, tert-butanol, 90 °C, 16 h, then iodomethane, 30 °C, 4 h, 27%; (vii) m-CPBA, CH2Cl2, rt, 16 h, 67%.
Compounds 6 and 8 were used as key intermediates for nucleophilic aromatic substitution with assorted primary and secondary amines to afford 9–55 (Scheme 2). N-Alkyl analogues not prepared from commercially available amines were prepared in two steps: initial reaction of 6 or 8 with the primary amine was followed by alkylation with sodium hydride and appropriate haloalkane (Scheme 2).
Scheme 2. Synthesis of Compounds 9–55.

Reagents and conditions: (i) amine, 2-propanol, 110–140 °C, 2–16 h; (ii) sodium hydride, haloalkane, DMF, rt, 5 h; (iii) secondary amine, 2-propanol, 140 °C, 16–96 h or microwave 25 min.
Aminopyrimidine 9 exhibited marginally weaker binding affinity for DYRK1A than 6b but with superior LE (0.55 vs 0.41) and LLE (5.59 vs 3.55). Compound 10 bound with equal affinity to DYRK1A as 6b, also conferring superior LE (0.56) and LLE (5.75). Compound 11 bound with stronger affinity than 6b, while maintaining favorable lipophilicity (clog P = 1.62), LE (0.55) and LLE (5.50), representing a good starting point for further elaboration. Compound 12 showed similar binding affinity to 11. Overall, maintaining one amino substituent as methyl while increasing the steric bulk of the other amino substituent was tolerated; however, two larger substituents (13–15) led to a reduction in DYRK1A binding affinity, a 10-fold reduction in the case of the matched-molecular pairs 12 and 13, highlighting the importance of an N-methyl substituent for maintaining the binding mode seen in the co-crystal structure of DYRK1A:6b (Figure 1). The longer 4-carbon-chain amino substituent of 18 led to increased binding affinity compared to 17 (Table 1).
Table 1. DYRK1A Binding Affinity of Pyrazolo[1,5-b]pyridazines.


pIC50 in TR-FRET-based ligand-binding displacement assay measured with two technical replicates (n = 1 biological replicate). LE = 1.4(pIC50)/nonhydrogen atoms. LLE = pIC50 – clog P.
Co-crystal structures of DYRK1A bound to 10, 11, and 12 were determined at a high resolution (Figure 2, Table 2). The N-methyl and N-H matched pair, 11 and 10, adopt the same binding mode in DYRK1A, respectively. The selectivity profiles of N-methyl and N-H matched pairs were previously shown to differ significantly, with N-methyl analogues being more selective for DYRK1A.12 The N-methyl groups in 6b, 11, and 12 all occupy the same pocket on the kinase hinge region. The retention of binding mode gave confidence that the selectivity profile of 6b, and by inference, 8b, would be maintained in the chemical series.12
Figure 2.

Co-crystal structures of pyrazolo[1,5-b]pyridazines (compounds 6b, 10, 11, 12, and 30) and compound 78 (yellow) in complex with DYRK1A. The backbones of the kinase N-lobe, hinge, and C-lobe are colored in gray, pink, and purple, respectively, and polar contacts are indicated by black dotted lines. The conserved water molecule at the back of the pocket is indicated by a red sphere. The electron density of each compound (2Fo – Fc map) is shown contoured at 1.0σ (blue mesh). For clarity, DYRK1A strands β1 and β2 are hidden. (A) Previously identified compound 6b (PDB 6S1I),12 (B) compound 10 (PDB 6S1B), (C) compound 11 (PDB 6S17), (D) compound 12 (PDB 6S14), (E) compound 30 (PDB 6S1J), and (F) compound 78 (PDB 6S1H).
Table 2. Data Collection and Refinement Statistics for DYRK1A Co-crystal Structures.
| Compound | 10 | 11 | 12 | 30 | 78 |
|---|---|---|---|---|---|
| PDB ID | 6S1B | 6S17 | 6S14 | 6S1J | 6S1H |
| space group | C2 | C2 | C2 | C2 | C2 |
| unit cell dimensions a, b, c (Å), α, β, γ (deg) | 101.3, 70.4, 68.7, 90.0, 118.0, 90.0 | 100.9, 70.6, 68.3, 90.0, 117.8, 90.0 | 101.5, 70.3, 68.5, 90.0, 118.0, 90.0 | 99.2, 69.7, 67.4, 90.0, 117.5, 90.0 | 99.6, 69.7, 67.7, 90.0, 117.8, 90.0 |
| Data collection | |||||
| resolution range (Å)a | 30.60–1.30 (1.33–1.30) | 30.46–1.10 (1.13–1.10) | 30.39–1.05 (1.08–1.05) | 59.80–1.41 (1.48–1.41) | 30.11–1.05 (1.08–1.05) |
| unique observationsa | 103 325 (7306) | 137 242 (3030) | 149 683 (1105) | 75 809 (8676) | 174 657 (6548) |
| average multiplicitya | 2.7 (2.0) | 4.0 (2.0) | 2.9 (1.1) | 12.2 (8.8) | 3.0 (1.7) |
| completeness (%)a | 98.9 (95.0) | 80.2 (24.3) | 75.9 (7.6) | 96.4 (76.0) | 91.9 (47.0) |
| Rmergea | 0.04 (0.70) | 0.05 (0.06) | 0.04 (0.61) | 0.20 (2.68) | 0.05 (0.63) |
| mean ((I)/σ(I))a | 10.1 (1.1) | 11.7 (1.3) | 15.4 (1.2) | 9.5 (1.8) | 11.1 (1.0) |
| mean CC(1/2) | 0.999 (0.549) | 0.998 (0.552) | 0.999 (0.323) | 0.996 (0.400) | 0.997 (0.509) |
| Refinement | |||||
| resolution range (Å) | 30.60–1.30 | 30.46–1.10 | 30.39–1.05 | 59.80–1.41 | 30.11–1.05 |
| R-value, Rfree | 0.16, 0.18 | 0.15, 0.17 | 0.15, 0.16 | 0.18, 0.18 | 0.14, 0.15 |
| r.m.s. deviation from ideal bond length (Å) | 0.012 | 0.022 | 0.013 | 0.013 | 0.006 |
| r.m.s. deviation from ideal bond angle (deg) | 1.15 | 1.62 | 1.22 | 1.28 | 1.03 |
| Ramachandran Outliersb Most favored | 0.0% 96.7% | 0.0% 96.1% | 0.0% 96.1% | 0.0% 95.8% | 0.0% 95.8% |
Values within parentheses refer to the highest-resolution shell.
Values from Molprobity.
With multiple crystal structures and SAR analysis supporting the existence of a shallow pocket on DYRK1A’s hinge motif that could be occupied by small substituents such as methyl, in silico modeling was used to identify possible replacements. The modeling suggested that a constrained methylene unit would be an appropriate substitution for the methyl group. Therefore, a range of cyclic amine analogues were synthesized to probe the size of the lipophilic pocket (Table 3).
Table 3. DYRK1A Binding Affinity of Pyrazolo[1,5-b]pyridazines.

pIC50 in TR-FRET-based ligand-binding displacement assay measured with two technical replicates (n = 1 biological replicate). LE = 1.4(pIC50)/nonhydrogen atoms. LLE = pIC50 – clog P.
Several cyclic amine analogues exhibited a similar level of binding affinity for DYRK1A as former lead compound 8b but with superior LE.12 All analogues exhibited lower LE than 11, but a small number exhibited comparable levels of binding affinity (22, 24, 28, 30, 31, 33 and 34) and superior LLE (22, 33 and 34). Analogues bearing substituents with smaller ring sizes (4 or 5 carbon atoms) showed, in general, greater DYRK1A binding affinity than analogues with larger rings (>5 carbon atoms), reinforcing the hypothesis that steric bulk is permitted, but limited, close to the hinge of the kinase. The X-ray co-crystal structure of 30 bound to DYRK1A (Figure 2 and Table 2) showed a retention of the binding mode, with a methylene group of the cyclopentyl ring occupying the same pocket as the methyl groups of 6b, 11, and 12, validating the prediction made by molecular modeling. Compound 22 and single R-enantiomers 31 and 34 were the only compounds with binding affinity for DYRK1A (IC50) of less than 100 nM.
At this stage, as 11 possessed favorable LE (0.55) and had been shown to maintain the same binding mode as 6b, it was used as the reference compound/matched-molecular pair for further SAR analysis. The data so far had not identified which of the atoms within the pyrazolo[1,5-b]pyridazine core contributed to selectivity and binding affinity, and were therefore, essential pharmacophoric features to maintain. The pyrazolo[1,5-b]pyridazine core, although not prevalent in the literature, is not novel and was being explored by others as antibiotic adjuvants14 and as treatments against neglected tropical diseases.15 Therefore changes to the pyrazolo[1,5-b]pyridazine were investigated for their effects on DYRK1A affinity and selectivity as well as on compound solubility and metabolic stability. Close inspection of the DYRK1A:11 co-crystal structure revealed the presence of a water molecule located toward the back of the DYRK1A binding site, with hydrogen bonds to the inhibitor and to the backbone amide nitrogen of Asp307 and the carboxylic acid of Glu203 (Figure 2C). We hypothesized that changing the pyrazolo[1,5-b]pyridazine head for a bioisostere or other heteroaromatic group might affect binding affinity and selectivity through replacement of the water, or perturbation or enhancement of the H-bonding network between the water, Glu203 and the catalytic lysine, Lys188. Iterative addition of substituents around the pyrazolo[1,5-b]pyridazine heterocycle was also hypothesized as a method to achieve selectivity over closely related kinases.
Synthesis of heterocyclic analogues of the pyrazolo[1,5-b]pyridazine motif required bespoke routes; the 2-methyl analogue 60 was synthesized following the literature procedure,13 until the 1,3-dipolar cycloaddition step (Scheme 3A). For the first time with 1,3-dipole 2, we report the organic base-catalyzed cycloaddition to furnish 58. The main advantage of this procedure is that it avoids the formation of a KOH/CH2Cl2 emulsion that occurs under the usual biphasic reaction conditions. The DBU catalyzed cycloaddition was found to be rather versatile. The 4-, 5- and 6- substituted pyrazolo[1,5-b]pyridazines were all accessed using the appropriately substituted pyridazine, following closely the literature procedure described for similar analogues.13 It was discovered that isolation and purification of intermediates were not a prerequisite for the synthesis of compounds 62–66. In this instance purification was performed on the final products only. 5,6-Nitrogeneous heterocyclic analogues were synthesized using one of two major routes; the 5,6-heterocyclic ketone was constructed from commercially available building blocks, then treated, in a similar manner to the pyrazolo[1,5-b]pyridazines (cf.Scheme 1), with DMF–DMA condensation, followed by cyclization with suitable guanidine (Scheme 3B,C).
Scheme 3. Synthesis of 5,6-Heterocyclic Replacements of the Pyrazolo[1,5-b]pyridazine.

Reagents and conditions: (A) (i) Bis(triphenylphosphine)palladium(II) dichloride, copper iodide, TEA, propyne (ca. 5% in THF, ca. 1 mol/L), THF, rt, overnight, 87%; (ii) pyridazin-1-ium-1-amine iodide, DBU, acetonitrile, 50 °C, 16 h, 73%; (iii) m-CPBA, CH2Cl2, rt, overnight, 80%; (iv) 2 M dimethylamine in THF, 110 °C, 2 h, 74%. (B) (i) Hydroxylamine-O-sulfonic acid, KHCO3, KI, H2O, 70 °C, 2 h; (ii) 3-butyne-2-one, KOH, CH2Cl2, rt, overnight; (iii) DMF–DMA, 75 °C, 16 h; (iv) 2-methoxyethanol, potassium carbonate, 1,1-dimethylguanidine sulfate salt, 125 °C, overnight. (C) (i) DMF–DMA, toluene, 90 °C, 2 h—overnight then chloroacetone, EtOH, reflux, overnight; (ii) DMF–DMA, 75 °C, 16 h; (iii) 2-methoxyethanol, potassium carbonate, 1,1-dimethylguanidine sulfate salt, 125 °C, overnight.
Alternatively, 4-chloro-N,N-dimethyl-pyrimidin-2-amine (compound 74) was reacted with appropriate boronic heterocycle, either purchased commercially or produced following literature procedures, under Suzuki conditions (Scheme 4).
Scheme 4. Synthesis of Heterocyclic Analogues.

Reagents and conditions: (i) Sodium hydride, iodomethane, THF, rt, 16 h, 89%; (ii) bis[2-(di-tert-butylphosphanyl)cyclopenta-2,4-dien-1-yl]iron; dichloropalladium, boronic acid or ester, 1,4-dioxane, 70 °C, 16 h, 24–83%; (iii) 2-methyl-2-propanyl 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrrolo[2,3-b]pyridine-1-carboxylate, tris(dibenzylideneacetone)dipalladium(0), potassium fluoride, tri-tert-butylphosphonium tetrafluoroborate, 120 °C, 20 min, microwave, then 4 N HCl in 1,4-dioxane, rt, 2 h, 91%; (iv) sodium hydride, haloalkane, DMF, rt, 1 h; (v) tetrabutylammonium hydrogensulfate, 33% sodium hydroxide, p-toluenesulfonyl chloride, toluene, rt, overnight, 92%; (vi) N-bromosuccinimide, DMF, rt, overnight, 14%; (vii) bis(pinacolato)diboron, bis(triphenylphosphine)palladium(II) dichloride, potassium acetate, 1,4-dioxane, 100 °C, 2 h, 85%; (viii) compound 85, potassium fluoride, tris(dibenzylideneacetone)dipalladium(0), tri-tert-butylphosphonium tetrafluoroborate, acetonitrile/water (4:1), 120 °C, 5 h, then 1 M sodium hydroxide, 100 °C, 1 h, microwave, 12%; (ix) sodium hydride, iodomethane, DMF, rt, 30 min, 15%.
Compound 60 showed no binding affinity for DYRK1A (Table 4), probably due to unfavorable clashes with the phenylalanine (Phe238) gatekeeper residue of DYRK1A. Substitution in the 6-position resulted in more than a 10-fold drop in DYRK1A binding affinity for 62 and 65. This is likely due to unfavorable clashes between the molecules and the DYRK1A protein surface close to Lys188. Compound 64, bearing a methyl substituent at the 4-position, also showed an approximate 10-fold loss in DYRK1A binding affinity, while the 4-methoxy analogue 66 showed approximately a 2-fold reduction in affinity, despite the potential change in orientation. A methyl substituent in the 5-position (63) was well tolerated.
Table 4. DYRK1A Binding Affinity of Pyrazolo[1,5-b]pyridazine Analogues.


pIC50 in TR-FRET-based ligand-binding displacement assay measured with two technical replicates (n = 1 biological replicate). LE = 1.4(pIC50)/nonhydrogen atoms. LLE = pIC50 – clog P.
All bioisosteric replacements of the pyrazolo[1,5-b]pyridazine motif, and compounds 70–87, designed to interact with, or to displace the structural water molecule, exhibited diminished DYRK1A binding affinity. Potentially, the loss of hydrogen bonding from the water to the inhibitor and to the protein was not compensated for by any other interaction, leading to a reduction in binding affinity. Another possibility is that the phenylalanine gatekeeper obstructs access to the back-pocket of DYRK1A, preventing the ligand from being able to displace the consensus water. Compound 70 showed strong binding affinity for DYRK1A, comparable to 63 and 66 but with better LE (0.53) and LLE (6.18).
The binding affinity and LE of 78 were favorable, despite the change in binding orientation of compound 78 relative to the pyrazolo[1,5-b]pyridazine series (Figure 2F). Compound 78 could be considered as a distinct series and would potentially serve as an alternative scaffold for DYRK1A inhibition.
The final point of diversification to investigate was the pyrimidine motif while retaining the pyrazolo[1,5-b]pyridazine moiety and the N,N-dimethylamine appendage. Several synthetic strategies were employed to furnish 5-substituted pyrimidine analogues. Compounds 95–100 were accessed via Suzuki reaction between pyrazolo[1,5-b]pyridazine boronic ester and appropriate 5-substituted pyrimidine. To construct the pyrazolo[1,5-b]pyridazine boronic ester, 1-aminopyridinium iodide 2 underwent 1,3-dipolar cycloaddition with methyl propiolate. Ester hydrolysis with lithium hydroxide furnished carboxylic acid 90 (Scheme 5A). Facile bromodecarboxylation under mild conditions gave the 3-bromo intermediate 91. Miyaura borylation of 91 furnished the desired boronic ester 92, which was used crude in subsequent Suzuki reactions.
Scheme 5. Synthesis of Key Boronic Ester and Pyrazolo[1,5-b]pyridazines.

Reagents and conditions: (A) (i) Intermediate 2, potassium carbonate, CH2Cl2, rt, 16 h, 36%; (ii) lithium hydroxide, MeOH, rt, 16 h then 1 M HCl, rt, 30 min, 74%; (iii) N-bromosuccinimide, DMF, rt, 3 h, 78%; (iv) bis(pinacolato)diboron, [1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium(II), complex with CH2Cl2, potassium acetate, 1,4-dioxane, 90 °C, 16 h. (B) (i) Sodium hydride, iodomethane, DMF, rt, 16 h; (ii) 92, bis[2-(di-tert-butylphosphanyl)cyclopenta-2,4-dien-1-yl]iron; dichloropalladium, sodium carbonate, acetonitrile/water (10:1), 140 °C overnight. (C) (i) 92, bis[2-(di-tert-butylphosphanyl)cyclopenta-2,4-dien-1-yl]iron; dichloropalladium, potassium carbonate, DMF, 60 °C, 1.5 h; (ii) 2 M dimethylamine in THF, 100 °C, 2 h. (D) (i) NXS, acetonitrile, rt, 2 h; (ii) sodium hydride, iodomethane, THF, rt, overnight.
The 5-substituted pyrimidine was prepared from either the commercial aminopyrimidine or 2,4-dichloropyrimidine (Scheme 5B,C). Alkylation with sodium hydride and iodomethane gave the required N,N-dimethylamine substituent (Scheme 5B). Alternatively, starting from the 2,4-dichloropyrimidine, the Suzuki reaction was performed first to install the pyrazolo[1,5-b]pyridazine moiety, followed by displacement of the chloride with dimethylamine, in a two-step one-pot reaction (Scheme 5C). After some investigation, it was found that 5-halo analogues 105–107 could be accessed reliably via electrophilic halogenation of compound 9, followed by alkylation of the free amino group (Scheme 5D).
Triazine 109 and pyridine 111 were prepared from 2,4-dichlorotriazine and 2,6-dibromopyridine, respectively (Scheme 6); nucleophilic aromatic substitution with dimethylamine followed by installation of the pyrazolo[1,5-b]pyridazine motif via Suzuki protocol gave 109 and 111. Dimethylation of building block 112 under the usual conditions followed by Suzuki protocol to install the pyrazolo[1,5-b]pyridazine motif gave compound 113.
Scheme 6. Synthesis of Pyrimidine Isosteres.

Reagents and conditions: (i) dimethylamine hydrochloride, N,N-diisopropylethylamine, THF, rt, 3 h; (ii) 92, bis[2-(di-tert-butylphosphanyl)cyclopenta-2,4-dien-1-yl]iron; dichloropalladium, 2 M potassium carbonate, 1,4-dioxane, 80 °C, 16 h; (iii) iodomethane, sodium hydride, DMF, rt, overnight.
Of the pyrimidine motif alterations, only the 5-fluoro analogue, 95, exhibited stronger DYRK1A binding affinity than 11 (Table 5). Increasing the size of the halogen (F → Cl → Br → I) was detrimental to DYRK1A binding affinity for the series. This suggests that when the pyrazolo[1,5-b]pyridazine series binds to DYRK1A, the space around the phenylalanine gatekeeper is restricted, limiting the size of substituent that can be placed favorably in the 5-position. The increased binding affinity observed for compound 95 may be the result of a combination of factors. A favorable interaction may have been established between the lipophilic fluorine atom and the phenylalanine gatekeeper (F−π interaction).16 A second possibility for the increased binding affinity observed for compound 95 could be the result of a conformational effect. The fluorine at the 5-position of pyrimidine may be acting as a weak hydrogen-bond acceptor with the proton of the 2-position of the pyrazolo[1,5-b]pyridazine, limiting rotation around the Ar-Ar bond, and preorganizing the molecule into an energetically favorable binding conformation. Substituents added to the 6-position of the pyrimidine were not well tolerated, probably as a result of unfavorable clashes between the ligand and the amino acid residues on the hinge region of DYRK1A.
Table 5. DYRK1A Binding Affinity of Pyrazolo[1,5-b]pyridazines.


pIC50 in TR-FRET-based ligand-binding displacement assay measured with two technical replicates (n = 1 biological replicate). LE = 1.4(pIC50)/nonhydrogen atoms. LLE = pIC50 – clog P.
Scaffold hopping from the pyrimidine to all other nitrogenous heterocycles resulted in reduced binding affinity for DYRK1A (Table 6). Removal of the nitrogen atom that serves as a hydrogen-bond acceptor to the hinge region of DYRK1A in compound 111 led to an approximately 10-fold reduction in binding affinity. Likewise, removal of the nitrogen atom from the 3-position of the pyrimidine and replacement with C–H (compound 113) also resulted in a 10-fold loss in binding affinity. The diminished binding affinity observed for compound 113 is likely the result of a conformational effect. Co-crystal structures of the pyrazolo[1,5-b]pyridazine series (compounds 10, 11, 12, and 30) show retention in the relative orientation of the pyrimidine and pyrazolo[1,5-b]pyridazine rings (Figure 2, SI Figure 2). It is possible that the N3 of the pyrimidine and the C–H at the 4-position of the pyrazolo[1,5-b]pyridazine form an attractive interaction, preorganizing some compounds into an energetically favorable conformation for binding. Removal of N3 (compound 113) may prevent the prearrangement of the energetically favorable binding conformation. Although triazine 109 exhibits >2-fold weaker affinity than 11, the LE is still high (0.52). If potency could be improved by other alterations, then the triazine may lead to a molecular probe for DYRK1A with superior properties to the pyrimidine.
Table 6. DYRK1A Binding Affinity of Pyrazolo[1,5-b]pyridazinesa.

| compound | A | B | C | DYRK1A pIC50 | clog P | LE | LLE |
|---|---|---|---|---|---|---|---|
| 109 | N | N | N | 6.74 | 1.88 | 0.52 | 4.86 |
| 111 | C | N | C | 6.04 | 2.25 | 0.47 | 3.79 |
| 113 | N | C | C | 6.17 | 1.86 | 0.48 | 4.31 |
pIC50 in TR-FRET-based ligand-binding displacement assay measured with two technical replicates (n = 1 biological replicate). LE = 1.4(pIC50)/nonhydrogen atoms. LLE = pIC50 – clog P.
Surface Plasmon Resonance Measurement of Lead Compounds
A set of the most potent compounds were assessed for DYRK1A binding by surface plasmon resonance (SPR) to measure the binding constants and association and dissociation rates (Table 7). The measured KD values were in good agreement with the data from the TR-FRET-based binding displacement assay. All compounds showed fast binding kinetics. Compound 95 had the slowest off-rate (longest residence time) of 0.06 s–1.
Table 7. SPR Measurement of Lead Compounds.
| compound | ka (M–1 s–1) | kd (s–1) | KD (nM) |
|---|---|---|---|
| 9 | 7.3 (±0.1) × 106 | 0.97 (±0.02) | 134.1 (±0.5) |
| 10 | 6.0 (±0.1) × 106 | 0.33 (±0.1) | 55.0 (±0.1) |
| 11 | 2.8 (±0.1) × 106 | 0.24 (±0.01) | 85 (±1) |
| 12 | 6.3 (±0.2) × 106 | 0.57 (±0.01) | 90.4 (±0.0.3) |
| 22 | 3.9 (±0.4) × 106 | 0.25 (±0.03) | 64 (±2) |
| 33 | 2.2 (±0.1) × 106 | 0.16 (±0.01) | 72.8 (±0.3) |
| 34 | 1.8 (±0.1) × 106 | 0.12 (±0.02) | 65.5 (±0.4) |
| 95 | 4.7 (±0.1) ×106 | 0.06 (±0.01) | 12.0 (±0.1) |
| 105 | 1.6 (±0.1) × 106 | 1.58 (±0.02) | 963 (±2) |
| 109 | 2.9 (±0.1) × 106 | 0.44 (±0.01) | 153.2 (±0.5) |
Metabolic Stability and Solubility Profiles of Lead Compounds
Several analogues had emerged with superior LE, LLE and binding affinity for DYRK1A compared to the original starting point 6b (LE = 0.41, LLE = 3.55 and pIC50 = 6.73). These analogues also possessed a lower cLog P, a reduced number of aromatic rings, and were analyzed to determine whether the improvements in physicochemical properties would translate to improvements in metabolic stability and solubility (Table 8), aiming to identify the compounds most suitable for development into in vivo chemical probes.
Table 8. Metabolic Stability and Solubility.
| compound | DYRK1A pIC50a | HLM (μL/(min mg)) | RLM (μL/(min mg)) | solubility (mg/mL) |
|---|---|---|---|---|
| 6b | 6.73 | 449.8b | 340.3b | 0.05 |
| 8b | 7.10 | 75 ± 5 | 783.7 ± 31 | 0.01 |
| 11 | 7.12 | 242 ± 32 | 231 ± 31 | 0.10 |
| 12 | 6.94 | 71 ± 2 | 434 ± 58 | 0.06 |
| 22 | 7.10 | 126 ± 9 | 119 ± 6 | 0.06 |
| 24 | 6.96 | 328 ± 25 | 929 ± 116 | 0.01 |
| 28 | 6.94 | 378 ± 66 | 448 ± 20 | 0.02 |
| 29 | 6.83 | 389 ± 23 | 184 ± 18 | 0.05 |
| 31 | 7.18 | 296 ± 14 | 130 ± 17 | 0.04 |
| 33 | 6.87 | 199b | 48 ± 23 | 1.92 |
| 34 | 7.14 | 40b | 66 ± 24 | 4.37 |
| 63 | 6.94 | 160 ± 66 | 164b | 0.01 |
| 66 | 6.86 | 89 ± 11 | 77 ± 17 | 0.07 |
| 70 | 6.80 | 154 ± 6 | 96 ± 25 | 0.47 |
| 78 | 6.87 | 153.9 ± 39 | 234.2 ± 17 | 0.00 |
| 95 | 7.24 | 697 ± 50 | 983 ± 39 | 0.03 |
| 109 | 6.74 | 93.6 ± 1 | 105.3 ± 7.0 | 0.05 |
pIC50 in TR-FRET-based ligand-binding displacement assay measured with two technical replicates (n = 1 biological replicate). HLM and RLM determinations mean of n = 2 ± standard deviation (unless otherwise stated).
Data derived from single experiment. HLM = human liver microsomes, RLM = rat liver microsomes; thermodynamic solubility data was derived from a single experiment at pH 6.8.
In general, a reduction in lipophilicity and aromatic ring count led to modest improvements in in vitro metabolic stability and solubility. Larger reductions in lipophilicity, observed for compounds 11, 22, 33, 34, and 70, showed marked improvements in solubility and metabolic stability. It is likely that a number of other factors are affecting the solubility of the series, including π-stacking due to the planarity of the series. In terms of metabolic stability, N-dealkylation is likely to be a major issue, especially for N,N-dimethylamine analogues, and to some extent cyclic analogues 33 and 34 circumvent this issue. Notably, an increase in polarity led to better solubility and metabolic stability (22, 33, and 34). The addition of substituents onto the pyrazolo[1,5-b]pyridazine motif 66 and substitution of the pyrazolo[1,5-b]pyridazine heterocycle for 70 also appears to improve solubility and metabolic stability.
Selectivity Profiles of Lead Compounds
During inhibitor development selected compounds were analyzed for selectivity against an in-house panel of 48 representative kinases by measuring the change in thermal stabilization of the kinase in the presence and absence of inhibitor (Supporting Information). This data revealed that the series showed generally good selectivity for DYRK1A, DYRK2, CLK1, and Haspin (GSG2). Compound 78 was a notable exception which cross-reacted with several additional kinases including the NAK kinases AAK1 and BMP2K, indicating the importance of the binding mode of the pyrazolo[1,5-b]pyridazine core for selectivity (Figure 2). In contrast to the pyrazolo[1,5-b]pyridazine series, compound 78 appears unable to establish a hydrogen-bonding interaction with the consensus water at the back-pocket of DYRK1A and lacks a substituent, such as a methyl, to occupy the shallow cleft on the hinge of DYRK1A (Figure 2).
The kinome-wide selectivity of key pyrazolo[1,5-b]pyridazines was further assessed at a 1 μM inhibitor concentration, using the KINOMEscan panel (DiscoverX) of >400 wild-type human kinases (summary Table 9, whole datasets in the Supporting Information). The analysis revealed good selectivity scores (S35) for key compounds with common off-targets of the series being the CDC-like kinases (CLKs) and other DYRKs. Crucially, all lead exemplars were selective against the majority of the human kinome and maintained selectivity for DYRK1A and CLKs over CMGC kinases CDK2 and GSK3β, which were known targets of the pyrazolo[1,5-b]pyridazine chemotype and are involved in some of the same mechanistic pathways as DYRK1A.12,13
Table 9. Selectivity Profiles of Lead Compoundsa.
| compound | DYRK1A | DYRK1B | DYRK2 | CLK1 | CLK2 | CLK3 | CDK2 | GSK3β | S score (35) |
|---|---|---|---|---|---|---|---|---|---|
| 11 | 0.4 | 2.7 | 19 | 7.1 | 9.4 | 54 | 100 | 94 | 0.027 |
| 12 | 34 | 43 | 100 | 79 | 95 | 96 | 100 | 91 | 0.013 |
| 22 | 0.7 | 8.3 | 11 | 33 | 13 | 54 | 100 | 75 | 0.042 |
| 24 | 33 | 81 | 66 | 71 | 61 | 100 | 100 | 92 | 0.004 |
| 28 | 2.4 | 7.2 | 15 | 17 | 16 | 88 | 100 | 77 | 0.050 |
| 29 | 1.5 | 9.1 | 25 | 16 | 15 | 73 | 95 | 100 | 0.035 |
| 31 | 20 | 34 | 39 | 48 | 62 | 96 | 95 | 98 | 0.011 |
| 33 | 1.7 | 18 | 29 | 36 | 72 | 95 | 100 | 100 | 0.027 |
| 34 | 1.3 | 10 | 28 | 47 | 51 | 89 | 86 | 100 | 0.032 |
| 63 | 1.1 | 1.6 | 25 | 15 | 13 | 76 | 89 | 100 | 0.027 |
| 66 | 0.2 | 0.7 | 24 | 7.8 | 12 | 83 | 100 | 100 | 0.022 |
| 70 | 0.7 | 6.2 | 35 | 20 | 31 | 56 | 86 | 77 | 0.022 |
| 95 | 18 | 71 | 55 | 53 | 54 | 84 | 100 | 93 | 0.002 |
% Remaining activity as determined by DiscoverX KINOMEscan experiment. Inhibitor concentration = 1 μM.
The selectivity profile of 11 was further investigated by acquiring KD values for kinase off-targets identified in the KINOMEscan analysis (Table 10). Compound 11 showed approximately 20-fold selectivity for DYRK1A over DYRK1B, 100-fold selectivity over DYRK2, more than 20-fold selectivity over CLK1 and CLK4 and approximately 70-fold selectivity over CLK2, making 11 one of the most target-specific DYRK1A inhibitors reported. Other potential off-targets, such as JAK3, which had been identified in the KINOMEscan analysis were determined to be false positives following dose–response KD measurement (Table 10). The only kinase apart from DYRK1A with strong affinity for 11 was the mitotic kinase Haspin. The lead compounds in Table 9 have comparable selectivity profiles to 11 with good physical properties and would serve as advanced starting points for future studies. Our data shows that modifications could be made to the series that result in improved kinome selectivity and more desirable physicochemical properties.
Table 10. Selectivity Profile of Compound 11.
| kinase | KDa |
|---|---|
| DYRK1A | 3 |
| Haspin | 15 |
| DYRK1B | 56 |
| CLK4 | 61 |
| CLK1 | 70 |
| CLK2 | 220 |
| DYRK2 | 360 |
| HIPK2 | 1200 |
| MKNK2 | 1800 |
| CAMK1B | 10 000 |
| JAK3 (JH1domain-catalytic) | 1800 |
| STK39 | 10 000 |
| WNK2 | 10 000 |
KD (nM) determined in competition binding assay (DiscoverX, n = 2).
Inhibition of DYRK1A Phosphorylation
To demonstrate the DYRK1A inhibitory activity of the series, compound 11 was profiled in a radiometric kinase assay (33PanQinase Activity Assay) provided by Proqinase GMBH. The assay measured the kinase activity of DYRK1A in the presence and absence of an inhibitor at 1 μM concentration. Follow-up IC50 profiling confirmed that the phosphorylation activity of DYRK1A was inhibited by compound 11 with an IC50 of 220 nM (Table 11).
Table 11. DYRK1A Phosphorylation Inhibition by Compound 11.
% inhibition at 1 μM inhibitor concentration determined in 33PanQinase Activity Assay (Proqinase, n = 1).
10-point IC50 (nM) determined in 33PanQinase Activity Assay (Proqinase, n = 1).
Cellular Profiling of Lead Compound 11
Compound 11 was tested for direct binding to DYRK1A in live cells using a nanoluciferase (nanoLuc) bioluminescence resonance energy transfer (BRET) (NanoBRET, Promega) target engagement assay (Figure 3). The NanoBRET assay measured the ability of compound 11 to displace a fluorescent tracer molecule from the ATP-binding site of DYRK1A protein N-terminally fused with NanoLuc luciferase (Nluc) in HEK293 cells. In the absence of compound 11, the fusion protein and tracer molecule were in proximity and able to generate a detectable BRET signal. Compound 11 demonstrated acceptable cellular activity, with IC50 of 434 nM compared to the enzymatic IC50 of 75 nM (Table 1).
Figure 3.

NanoBRET analysis of the direct binding of 11 to DYRK1A in HEK293 cells. Two biological replicates were made at different tracer concentrations, plotted separately on the graph. The IC50 values were 576 nM in the presence of a 2 μM tracer and 434 nM in the presence of a 1 μM tracer.
Compounds 11 and 95 were profiled in the ClariCELL DYRK1A/Tau cellular phosphorylation assay to demonstrate a functional effect in vitro. The tau phosphorylation activity of DYRK1A was measured in the absence or presence of various concentrations of compound in human embryonic kidney cells (HEK293) transiently transfected with DYRK1A and tau. The activity of 11 was compared to the activity of harmine, a known DYRK1A inhibitor. Table 12 shows that compounds 11 and 95 inhibited tau phosphorylation with IC50’s of 0.59 and 0.25 μM, respectively, and are comparable to harmine (IC50 0.20 μM).
Table 12. Inhibition of Tau Phosphorylation by DYRK1A by Selected Compoundsa.
| compound | IC50 for tau phosphorylation (μM) |
|---|---|
| 11 | 0.59 |
| 95 | 0.25 |
| Harmine | 0.20 |
IC50 determined in ClariCELL DYRK1A/Tau cellular phosphorylation assay (Cell Assay Innovations, n = 2).
Compound 11 was also progressed to a Madin–Darby canine kidney cells transfected with the human MDR1 gene (MDCK-MDR1) assay to determine whether there would be any issues with P-gp liability or permeability (Table 13). The high central nervous system multiparameter optimization (CNS MPO) score calculated for compound 11 correlated with good levels of permeability and low P-gp efflux, suggesting that 11 is a good starting point for development into a CNS-penetrant DYRK1A chemical probe.
Table 13. DYRK1A CNS MPO and MDCK-MDR1.
| direction = A2B | direction = B2A | |||
|---|---|---|---|---|
| compound | CNS MPOa score | mean Papp (10–6 cms–1)b | efflux ratio (mean Papp B2A/mean Papp A2B)b | |
| 11 | 6 | 71.4 ± 0.65 | 58.7 ± 1.45 | 0.822 |
CNS MPO score calculated using CNS MPO KNIME workflow with ChemAxon nodes.
Data generated by Cyprotex in MDCK-MDR1 assay. Permeability coefficient (Papp) calculated across cells in direction: A2B (Apical to Basolateral) and B2A (Basolateral to Apical). Determinations ± standard deviation (mean of n = 2 unless otherwise stated).
In Vivo Pharmacokinetic Profiling of Lead Compound 11
Compound 11 had good physicochemical properties and had demonstrated high levels of enzymatic potency, favorable aqueous solubility, exquisite kinome selectivity, and promising on-target in vitro inhibition. Although compound 11 was not optimized for in vitro clearance, we sought to benchmark 11 as a DYRK1A tool for future optimization.
Compound 11 was dosed intravenously at 1 mg/kg in Sprague–Dawley rats (n = 3) in the fed state. Plasma and brain samples were collected. Consistent with the in vitro findings, high clearance was observed for compound 11in vivo. As a consequence of the high clearance and low volume of distribution, the half-life of compound 11 is not optimal (Table 14). There were no adverse effects reported in the study and the brain:plasma ratio indicated good brain penetration for compound 11 (total br/pl > 0.35), consistent with what was predicted from the MDCK-MDR1 assay.
Table 14. In Vivo Pharmacokinetic Study of Compound 11a.
| PK parameter | compound 11 |
|---|---|
| half-life (h) | 0.2 |
| Tmax (h) | 0.03 |
| Cmax (ng/mL) | 1082.8 |
| AUClast (h·ng/mL) | 275.3 |
| AUCinf (h·ng/mL) | 276.4 |
| Cl (mL/(min kg)) | 61.6 |
| Vd | 0.8 |
| MRT (h) | 0.2 |
| C0 (ng/mL) | 1331.5 |
Compound 11 was dosed as a suspension of the amorphous solid at 1 mg/kg i.v.
Conclusions
Starting from DYRK1A inhibitors previously published by our group (6b and 8b),12 we were able to design compound 11, and derivatives thereof, that maintained and improved upon the selectivity profiles of 6b and 8b. The binding mode appeared to be the same for compounds 11 and 6b, providing further evidence that this unusual binding mode, and in particular, possession of a motif that occupies a shallow lipophilic pocket close to the hinge of DYRK1A, further stabilized by a hydrogen-bonding interaction between the ligand and consensus water, appears to afford isoform and kinome-selective DYRK1A inhibitors. Iterative crystallography and structure-based design using the elucidated binding mode of 6b, 11, and other pyrazolo[1,5-b]pyridazine analogues allowed rapid expansion to selective and high-affinity binders of DYRK1A with improved physicochemical properties.
Through focused design to reduce the number of aromatic rings and lipophilicity, compounds such as 11 showed improvements in metabolic stability and solubility relative to 6b. The permeability and on-target cellular activity of 11 were demonstrated. Compound 11 was not a P-gp substrate, giving confidence that 11, and analogues thereof, can be used with confidence as tool compounds to determine the role of DYRK1A. Compound 11 represents a small, highly selective, and ligand-efficient starting point for future drug discovery efforts on DYRK1A in any area of medicine. Further optimization of the in vivo clearance of the series may lead to clinical DYRK1A inhibitors.
Experimental Section
The syntheses of lead compounds (Table 9) are described below. The synthetic procedures for all other compounds are found in the Supporting Information. The purity of all target compounds was confirmed to be >95% by HPLC or 1H NMR.
General Methods
All commercial reagents were purchased from Sigma-Aldrich, Alfa Aesar, Apollo Scientific, Fluorochem, or Tokyo Chemical Industry and were of the highest available purity. Unless otherwise stated, chemicals were used as supplied without further purification. Anhydrous solvents were purchased from Acros (AcroSeal) or Sigma-Aldrich (SureSeal) and were stored under nitrogen. Anhydrous solvents and reagents were used as purchased. Thin-layer chromatography (TLC) was carried out using glass plates precoated with Merck silica gel 60 F254. Melting point measurements are recorded on an MPA100 OptiMelt apparatus and are uncorrected. Proton nuclear magnetic resonance spectra were recorded at 500 MHz on a Varian VNMRS 500 MHz spectrometer or at 500 MHz on a Bruker Biospin GmbH 500 MHz spectrometer or at 600 MHz on a Varian VNMRS 600 MHz spectrometer, using residual isotopic solvent (CHCl3, δH = 7.27 ppm, DMSO δH = 2.50 ppm, MeOH δH = 3.31 ppm, DMF δH = 2.92) as an internal reference. Chemical shifts are quoted in parts per million (ppm). Coupling constants (J) are recorded in hertz (Hz). Carbon nuclear magnetic resonance spectra were recorded at 125 MHz on a Varian 500 MHz spectrometer or 151 MHz on a Varian 600 MHz spectrometer, using residual isotopic solvent (CHCl3, δC = 77.00 ppm, DMSO δC = 39.52 ppm, MeOH δC = 49.00 ppm) as an internal reference. Proton and carbon spectra assignments are supported by DEPT editing. Chemical shifts (δC) are quoted in ppm. High-resolution mass spectrometry data (ESI) were recorded on Bruker Daltonics, Apex III, ESI source: Apollo ESI with methanol as spray solvent. Only molecular ions, fractions from molecular ions, and other major peaks are reported as mass/charge (m/z) ratios. LCMS-LCQ data was recorded on a Waters 2695 HPLC using a Waters 2487 UV detector and a Thermo LCQ ESI-MS. Samples were eluted through a Phenomenex Lunar 3 μm C18 50 mm × 4.6 mm column, using water and acetonitrile acidified by 0.1% formic acid at 1 mL/min and detected at 254 nm. The gradient employed was a 5 min method 10–90% MeCN over a 4 min gradient, held at 90% MeCN for 1 min, then reequilibrated over 1 min. LCMS-MDAP analytical data was recorded on a Shimadzu Prominence Series coupled to an LCMS-2020 ESi and APCI mass spectrometer. Samples were eluted through a Phenomenex Gemini 5μ C18 110A 250 mm × 4.6 mm column, using water and acetonitrile acidified by 0.1% formic acid at 1 mL/min and detected at 254 nm. The gradient employed was a 30 min method either (A) 5–95% MeCN over a 20 min gradient, held at 95% MeCN for 4 min, then reequilibrated to 30% MeCN over 5 min or (B) 30–95% MeCN over 20 min gradient, held at 95% MeCN for 4 min, then reequilibrated to 30% MeCN over 5 min. Specific Optical Rotations were measured using Schmidt and Haensch UniPol L polarimeter, in cells with a path length of 5 cm. The light source was maintained at 589 nm. The concentration (c) is expressed in g/100 mL (equivalent to g/0.1 dm3). Specific rotations are denoted [α]DT and are given in implied units of 10–1 deg cm2 g–1 (T = ambient temperature in °C).
Pyridazin-1-ium-1-amine Iodide (2)
Aminosulfuric acid (13.10 g, 115.84 mmol) was dissolved in water (25 mL), and the reaction mixture was cooled to 10 °C in an ice bath. Potassium bicarbonate solution (48 mL, 2.4 M, 115.2 mmol) was added to the reaction mixture until it was at pH 5. Pyridazine (5.62 mL, 77.4 mmol) was added in one portion, and the reaction mixture was heated to 70 °C for 1.5 h. The pH was adjusted to pH 7 by the addition of a 2.4 M potassium bicarbonate solution (10.0 mL, 24 mmol). The reaction mixture was cooled to 40 °C and was stirred for 1 h. Potassium iodide (12.85 g, 77.41 mmol) in water (25 mL) was added to the reaction mixture, and the reaction was stirred for a further 1 h. The solvent was removed under reduced pressure. A solution of 5% MeOH in EtOH (100 mL) was then added to the residue. The resulting solids were collected by filtration and dried under reduced pressure to furnish the title compound, pyridazin-1-ium-1-amine iodide, 2, as a yellow solid (17 g, 72.3 mmol, 94% yield). 1H NMR (600 MHz, DMSO-d6) δH 9.82 (s, 2H), 9.29–9.22 (m, 1H), 9.11 (d, J = 6.2 Hz, 1H), 8.52–8.43 (m, 1H), 8.12 (dd, J = 8.2, 5.2 Hz, 1H).
1-Pyrazolo[1,5-b]pyridazin-3-ylethanone (3)
To a slurry of pyridazin-1-ium-1-amine iodide (16.0 g, 71.74 mmol) in CH2Cl2 (200 mL) was added 3-butyn-2-one (2.76 mL, 35.26 mmol). The reaction flask was cooled in an ice bath before adding a solution of potassium hydroxide (5.0 g, 89.11 mmol) in water (100 mL) in one portion. The reaction mixture was stirred at rt for 16 h. The organic layer was separated and the aqueous layer was extracted with CH2Cl2 (2 × 200 mL). The combined organic components were dried over MgSO4, filtered, and concentrated under reduced pressure to afford the title compound, 1-pyrazolo[1,5-b]pyridazin-3-ylethanone, 3, as a red-black solid (3.43 g, 21.28 mmol, 60% yield). 1H NMR (500 MHz, DMSO-d6) δH 8.78 (s, 1H), 8.71–8.63 (m, 2H), 7.57 (dd, J = 8.9, 4.6 Hz, 1H), 2.56 (s, 3H). LC-MS (ESI): calculated for C8H8N3O [M + H]+ is 162.1, found 162.1 [M + H]+.
(E)-3-(Dimethylamino)-1-pyrazolo[1,5-b]pyridazin-3-yl-prop-2-en-1-one (4)
To a flask containing 1-pyrazolo[1,5-b]pyridazin-3-ylethanone (0.68 g, 4.22 mmol) was added DMF–DMA (3.78 mL, 28.44 mmol). The reaction mixture was heated to 100 °C for 16 h. The reaction mixture was concentrated under reduced pressure and triturated with diethyl ether to afford the title compound, (E)-3-(dimethylamino)-1-pyrazolo[1,5-b]pyridazin-3-yl-prop-2-en-1-one, 4, as a black solid (0.87 g, 3.82 mmol, 91% yield). 1H NMR (600 MHz, DMSO-d6) δH 8.73 (dd, J = 9.0, 2.0 Hz, 1H), 8.70 (s, 1H), 8.57 (dd, J = 4.5, 2.0 Hz, 1H), 7.68 (d, J = 12.3 Hz, 1H), 7.41 (dd, J = 9.0, 4.4 Hz, 1H), 5.83 (d, J = 12.3 Hz, 1H), 3.20–2.80 (m, 6H). LC-MS (ESI): calculated for C11H13N4O [M + H]+ is 217.1, found 217.0 [M + H]+.
4-Pyrazolo[1,5-b]pyridazin-3-yl-pyrimidin-2-ol (5)
To (E)-3-(dimethylamino)-1-pyrazolo[1,5-b]pyridazin-3-yl-prop-2-en-1-one (2.5 g, 11.56 mmol) was added urea (15 g, 250 mmol) followed by sodium hydride (1.25 g, 31.25 mmol), and the reaction mixture was heated to 140 °C under nitrogen until melted fully. The melt was then maintained at this temperature for a further 5 min. Once the melt had cooled to rt, water (10 mL) was added slowly while stirring. The solution was made to pH 3 with 1 M HCl solution and stirred overnight at rt. The solid formed was filtered and triturated with ethanol to afford the title compound, 4-pyrazolo[1,5-b]pyridazin-3-yl-pyrimidin-2-ol, 5, as a brown solid (2.3 g, 9.71 mmol, 84% yield). Rf 0.32 (MeOH/CH2Cl2 1:9); 1H NMR (500 MHz, DMSO-d6) δH 11.60 (s, 1H), 8.94 (dd, J = 9.0, 1.9 Hz, 1H), 8.89 (s, 1H), 8.61 (dd, J = 4.5, 2.0 Hz, 1H), 7.90 (d, J = 6.5 Hz, 1H), 7.50 (dd, J = 9.1, 4.5 Hz, 1H), 6.93 (d, J = 6.6 Hz, 1H); HRMS m/z (ESI+) calculated for C10H8N5O [M + H]+ is 214.0723, found: 214.0721 [M + H]+; LC-MS (ESI): calculated for C10H8N5O [M + H]+ is 214.1, found 214.3 [M + H]+
3-(2-Chloropyrimidin-4-yl)pyrazolo[1,5-b]pyridazine (6)
To a pressure vial were added 6-pyrazolo[1,5-b]pyridazin-3-yl-1H-pyrimidin-2-one (500 mg, 2.35 mmol) and phosphorus(V) oxychloride (2 mL, 21.46 mmol). The vial was sealed and heated to 110 °C for 6 h. The crude reaction mixture was added slowly to a water/ice mix and stirred at rt for 1 h. The brown suspension that formed was filtered and washed with water to afford the title compound, 3-(2-chloropyrimidin-4-yl)pyrazolo[1,5-b]pyridazine, 6, as a brown solid (540 mg, 2.17 mmol, 92% yield). Rf 0.83 (MeOH/CH2Cl2 1:9); mp 293–295 °C; 1H NMR (500 MHz, DMSO-d6) δH 9.00 (s, 1H), 8.88 (dd, J = 9.1, 1.9 Hz, 1H), 8.70 (d, J = 5.3 Hz, 1H), 8.66 (dd, J = 4.5, 1.9 Hz, 1H), 8.04 (d, J = 5.4 Hz, 1H), 7.56 (dd, J = 9.1, 4.5 Hz, 1H); LC-MS (ESI): calculated for C10H6N535Cl [M]+ is 231.0 and C10H6N537Cl [M + 2]+ is 233.0, found 231.9 [M]+ and 233.9 [M + 2]+.
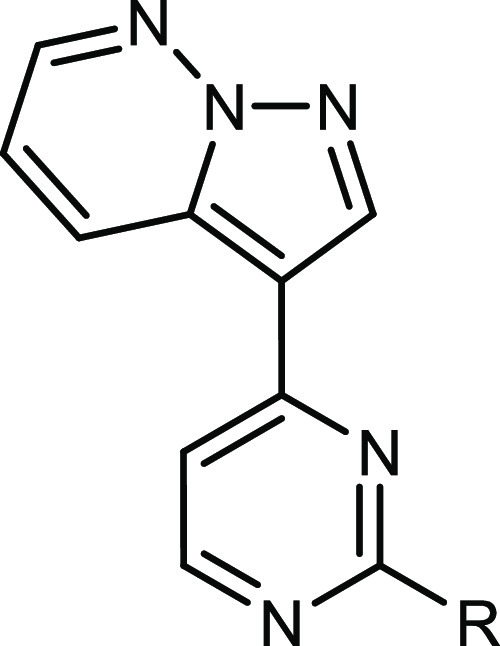
N,N-Dimethyl-4-pyrazolo[1,5-b]pyridazin-3-yl-pyrimidin-2-amine (11)
To a microwave vial containing 3-(2-chloropyrimidin-4-yl)pyrazolo[1,5-b]pyridazine (80 mg, 0.35 mmol) was added dimethylamine (0.03 mL, 0.69 mmol). The reaction mixture was heated at 110 °C for 2 h. The reaction mixture was concentrated under reduced pressure. The residue was purified using flash silica column chromatography on an Isco Combiflash system (12 g silica, elution with 0–50% 20% MeOH in CH2Cl2/CH2Cl2 gradient) to afford the title compound, N,N-dimethyl-4-pyrazolo[1,5-b]pyridazin-3-yl-pyrimidin-2-amine, 11, as an off-white solid (70 mg, 0.26 mmol, 77% yield). Rf 0.68 (MeOH/ CH2Cl2 1:9); mp 241–242 °C; 1H NMR (500 MHz DMSO-d6) δH 8.96 (dd, J = 9.0, 1.9 Hz, 1H), 8.84 (s, 1H), 8.61–8.58 (m, 1H), 8.35 (d, J = 5.2 Hz, 1H), 7.46 (dd, J = 9.0, 4.4 Hz, 1H), 7.14 (d, J = 5.2 Hz, 1H), 3.22 (s, 6H);13C NMR (125 MHz, DMSO-d6) δC 161.9, 159.9, 157.9, 143.9, 139.7, 132.3, 129.1, 118.9, 110.5, 104.6, 37.0; HRMS m/z (ESI+) calculated for C12H12N6 [M + H]+ is 241.1196, found: 241.1197 [M + H]+; LCMS (MDAP): Rt = 3.6 min, >95% (method B); LC-MS (ESI): calculated for C12H12N6 [M + H]+ is 241.1, found 241.0 [M + H]+.
N-Cyclopropyl-N-methyl-4-pyrazolo[1,5-b]pyridazin-3-yl-pyrimidin-2-amine (12)
To a microwave vial containing 3-(2-chloropyrimidin-4-yl)pyrazolo[1,5-b]pyridazine (80 mg, 0.35 mmol) in 2-propanol (2 mL) was added N-cyclopropyl-methylamine hydrochloride (74 mg, 0.69 mmol). The vial was sealed and heated at 150 °C for 30 min under microwave irradiation. The reaction was concentrated under reduced pressure. The crude was purified using flash silica column chromatography on an Isco Combiflash system (12 g silica, elution with 0–50% 20% MeOH in CH2Cl2/CH2Cl2 gradient) to afford the title compound, N-cyclopropyl-N-methyl-4-pyrazolo[1,5-b]pyridazin-3-yl-pyrimidin-2-amine, 12, as an off-white solid (10 mg, 0.04 mmol, 10% yield). Mp 162–164 °C; 1H NMR (500 MHz, DMSO-d6) δH 9.13–9.09 (m, 1H), 8.86 (s, 1H), 8.60–8.56 (m, 1H), 8.38 (d, J = 5.1 Hz, 1H), 7.46 (dd, J = 9.0, 4.4 Hz, 1H), 7.23 (d, J = 5.1 Hz, 1H), 3.15 (s, 3H), 2.90–2.80 (m, 1H), 0.95–0.89 (m, 2H), 0.70–0.64 (m, 2H); 13C NMR (125 MHz, DMSO-d6) δC 163.2, 158.8, 157.8, 144.0, 139.7, 132.5, 129.4, 118.9, 110.4, 105.7, 35.7, 31.6, 8.3; HRMS m/z (ESI+) calculated for C14H15N6 [M + H]+ is 267.1353, found: 267.1348 [M + H]+; LCMS (MDAP): Rt = 9.93 min, >95% (method A); LC-MS (ESI): calculated for C14H15N6 [M + H]+ is 267.1, found 267.1 [M + H]+.
1-(4-Pyrazolo[1,5-b]pyridazin-3-yl-pyrimidin-2-yl)-6-oxa-1-azaspiro[3.3]heptane (22)
To a pressure vial containing 3-(2-chloropyrimidin-4-yl)pyrazolo[1,5-b]pyridazine (60 mg, 0.26 mmol) in 2-propanol (2 mL) was added 6-oxa-1-azaspiro[3.3]heptane (51 mg, 0.52 mmol). The vial was sealed and heated to 140 °C for 16 h. The reaction mixture was concentrated under reduced pressure. The residue was adsorbed onto solid load material. The crude was purified using flash silica column chromatography on a Biotage system (12 g silica, elution with 0–100% EtOAc/petroleum ether gradient). Desired fractions were combined and concentrated under reduced pressure. The crude was purified using reversed-phase column chromatography on a Biotage system (12 g C-18, elution with 10–100% MeOH/water gradient) to afford the title compound, 1-(4-pyrazolo[1,5-b]pyridazin-3-yl-pyrimidin-2-yl)-6-oxa-1-azaspiro[3.3]heptane, 22, as a colorless solid (17 mg, 0.05 mmol, 21% yield). Rf 0.29 (EtOAc); decomposed > 246 °C; 1H NMR (600 MHz, MeOD) δH 9.50 (br.s, 1H), 8.74 (s, 1H), 8.52–8.49 (m, 1H), 8.34 (d, J = 5.2 Hz, 1H), 7.35 (dd, J = 8.8, 4.4 Hz, 1H), 7.24 (d, J = 5.2 Hz, 1H), 5.64 (d, J = 7.1 Hz, 2H), 4.95–4.76 (m, 2H), 4.02 (t, J = 7.1 Hz, 2H), 2.71 (t, J = 7.2 Hz, 2H); 13C NMR (150 MHz, CDCl3) δC 160.9, 159.8, 158.3, 143.2, 139.6, 133.5, 130.5, 118.4, 110.5, 106.2, 81.5, 68.3, 45.6, 29.1; HRMS m/z (ESI+) calculated for C15H15N6O [M + H]+ is 295.1302, found: 295.1301 [M + H]+; LCMS (MDAP): Rt = 11.9 min, >95% (method A); LC-MS (ESI): calculated for C15H15N6O [M + H]+ is 295.1, found 295.0 [M + H]+
3-[2-(2-Methylpyrrolidin-1-yl)pyrimidin-4-yl]pyrazolo[1,5-b]pyridazine (24)
To a microwave vial containing 3-(2-methylsulfonylpyrimidin-4-yl)pyrazolo[1,5-b]pyridazine (100 mg, 0.33 mmol) in 2-propanol (1 mL) was added 2-methylpyrrolidine (0.03 mL, 0.33 mmol). The vial was sealed and heated to 140 °C overnight. The reaction mixture was concentrated under reduced pressure. The crude was purified using flash silica column chromatography on an Isco Combiflash system (12 g silica, elution with 0–100% EtOAc/petroleum ether gradient) to afford the title compound 3-[2-(2-methylpyrrolidin-1-yl)pyrimidin-4-yl]pyrazolo[1,5-b]pyridazine, 24, as a white solid (96 mg, 0.33 mmol, 99% yield). Rf 0.38 (EtOAc/petroleum ether 1:1); mp 205–207 °C; 1H NMR (600 MHz, DMSO-d6) δH 8.98 (d, J = 9.1 Hz, 1H), 8.84 (s, 1H), 8.61–8.57 (m, 1H), 8.36–8.32 (m, 1H), 7.46 (dd, J = 9.1, 4.4 Hz, 1H), 7.13 (d, J = 5.0 Hz, 1H), 4.42–4.21 (m, 1H), 3.79–3.22 (m, 2H), 2.15–2.02 (m, 2H), 2.02–1.89 (m, 1H), 1.77–1.67 (m, 1H), 1.26 (s, 3H); 13C NMR (150 MHz, DMSO-d6) δC 159.8, 159.0, 158.0, 143.9, 139.6, 132.4, 129.2, 118.9, 110.4, 104.5, 52.6, 46.8, 32.3, 22.8, 19.3; HRMS m/z (ESI+) calculated for C15H17N6 [M + H]+ is 281.1509, found: 281.1519 [M + H]+; LCMS (MDAP): Rt = 11.2 min, >95% (method A); LC-MS (ESI): calculated for C15H17N6 [M + H]+ is 281.2, found 281.0 [M + H]+.
3-[2-[(3S)-3-Methylpyrrolidin-1-yl]pyrimidin-4-yl]pyrazolo[1,5-b]pyridazine (28)
To a microwave vial containing a suspension of 3-(2-chloropyrimidin-4-yl)pyrazolo[1,5-b]pyridazine (60 mg, 0.26 mmol) in 2-propanol (2 mL) was added (S)-3-methyl-pyrrolidine hydrochloride (63 mg, 0.52 mmol). The vial was sealed, and the reaction mixture was heated to 140 °C for 16 h. The reaction mixture was concentrated under reduced pressure. The crude was purified using flash silica column chromatography on an Isco Combiflash system (12 g silica, elution with 0–100% EtOAc/petroleum ether gradient). Desired fractions were combined and concentrated under reduced pressure. The crude was purified using flash reversed-phase column chromatography on a Biotage system (12 g silica, elution with 10–100% MeOH/water gradient) to afford the title compound, 3-[2-[(3S)-3-methylpyrrolidin-1-yl]pyrimidin-4-yl]pyrazolo[1,5-b]pyridazine, 28, as a colorless solid (21 mg, 0.07 mmol, 27% yield). Rf 0.34 (EtOAc/petroleum ether 1:1); [α]D25 = −60.1 (c = 3.3 × 10–4 g/mL, CHCl3); mp 213–215 °C; 1H NMR (600 MHz, DMSO-d6) δH 8.99 (app. d, J = 9.0 Hz, 1H), 8.84 (s, 1H), 8.61–8.57 (m, 1H), 8.33 (d, J = 5.1 Hz, 1H), 7.46 (dd, J = 9.2, 4.4 Hz, 1H), 7.13 (d, J = 5.1 Hz, 1H), 3.96–3.40 (m, 3H), 3.25–2.97 (m, 1H), 2.46–2.31 (m, 1H), 2.23–2.04 (m, 1H), 1.71–1.53 (m, 1H), 1.12 (s, 3H); 13C NMR (150 MHz, DMSO-d6) δC 160.0, 159.0, 158.0, 143.9, 139.6, 132.4, 129.4, 118.9, 110.3, 104.4, 53.4, 46.1, 32.9, 32.6, 17.9; HRMS m/z (ESI+) calculated for C15H17N6 [M + H]+ is 281.1509, found: 281.1508 [M + H]+; LCMS (MDAP): Rt = 11.6 min, >95% (method A); LC-MS (ESI): calculated for C15H17N6 [M + H]+ is 281.2, found 281.0 [M + H]+. Chiral HPLC, chiralcel OJ column (20:80 heptane: isopropanol (0.2% v/v NH3), 1.0 mL/min). Retention time = 8.04 min > 98% e.e.
3-[2-[(3R)-3-Methylpyrrolidin-1-yl]pyrimidin-4-yl]pyrazolo[1,5-b]pyridazine (29)
To a microwave vial containing a suspension of 3-(2-chloropyrimidin-4-yl)pyrazolo[1,5-b]pyridazine (60 mg, 0.26 mmol) in 2-propanol (2 mL) was added (R)-3-methyl-pyrrolidine hydrochloride (63 mg, 0.52 mmol). The vial was sealed, and the reaction mixture was heated to 140 °C for 16 h. The reaction mixture was concentrated under reduced pressure. The crude was purified using flash silica column chromatography on an Isco Combiflash system (12 g silica, elution with 0–100% EtOAc/petroleum ether gradient). Desired fractions were combined and concentrated under reduced pressure. The crude was purified using flash reversed-phase column chromatography on a Biotage system (12 g silica, elution with 10–100% MeOH/water gradient) to afford the title compound, 3-[2-[(3R)-3-methylpyrrolidin-1-yl]pyrimidin-4-yl]pyrazolo[1,5-b]pyridazine, 29, as a pale yellow solid (13 mg, 0.04 mmol, 17% yield). Rf 0.34 (EtOAc/petroleum ether 1:1); [α]D25 = +60.1 (c = 3.3 × 10–4 g/mL, CHCl3); mp 213–215 °C; 1H NMR (600 MHz, DMSO-d6) δH 9.00 (app. d, J = 9.0 Hz, 1H), 8.84 (s, 1H), 8.61–8.58 (m, 1H), 8.34 (d, J = 5.1 Hz, 1H), 7.47 (dd, J = 9.2, 4.4 Hz, 1H), 7.13 (d, J = 5.1 Hz, 1H), 3.96–3.43 (m, 3H), 3.24–2.94 (m, 1H), 2.45–2.32 (m, 1H), 2.21–2.05 (m, 1H), 1.70–1.54 (m, 1H), 1.13 (s, 3H); 13C NMR (150 MHz, DMSO-d6) δC 160.0, 159.0, 158.0, 143.9, 139.6, 132.4, 129.4, 118.9, 110.4, 104.4, 53.4, 46.1, 32.9, 32.6, 17.9; HRMS m/z (ESI+) calculated for C15H17N6 [M + H]+ is 281.1509, found: 281.1508 [M + H]+; LCMS (MDAP): Rt = 11.6 min, >95% (method A); LC-MS (ESI): calculated for C15H17N6 [M + H]+ is 281.2, found 281.0 [M + H]+. Chiral HPLC, chiralcel OJ column (20:80 heptane: isopropanol (0.2% v/v NH3), 1.0 mL/min). Retention time = 7.07 min > 96% e.e.
3-[2-[(3R)-3-Fluoropyrrolidin-1-yl]pyrimidin-4-yl]pyrazolo[1,5-b]pyridazine (31)
To a microwave vial containing 3-(2-chloropyrimidin-4-yl)pyrazolo[1,5-b]pyridazine (67 mg, 0.29 mmol) in 2-propanol (1 mL) was added R-(−)-3-fluoropyrrolidine hydrochloride (36 mg, 0.29 mmol). The vial was sealed and heated to 140 °C for 6 h. The reaction mixture was concentrated under reduced pressure. The crude was purified using flash silica column chromatography on an Isco Combiflash system (12 g silica, elution with 0–100% EtOAc/petroleum ether gradient) to afford the title compound 3-[2-[(3R)-3-fluoropyrrolidin-1-yl]pyrimidin-4-yl]pyrazolo[1,5-b]pyridazine, 31, as a cream solid (70 mg, 0.23 mmol, 80% yield). Rf 0.24 (EtOAc/petroleum ether 1:1); [α]D25 = −60.1 (c = 3.67 × 10–3 g/mL, CHCl3); mp 228–230 °C; 1H NMR (600 MHz, DMSO-d6) δH 8.97 (app. d, J = 9.0 Hz, 1H), 8.82 (s, 1H), 8.59–8.55 (m, 1H), 8.34 (d, J = 5.1 Hz, 1H), 7.43 (dd, J = 9.2, 4.4 Hz, 1H), 7.16 (d, J = 5.1 Hz, 1H), 5.47 (d, J = 53.3 Hz, 1H), 4.05–3.46 (m, 4H), 2.37–2.12 (m, 2H); 13C NMR (150 MHz, DMSO-d6) δC 160.0, 159.1, 158.1, 143.9, 139.7, 132.5, 129.4, 119.0, 110.2, 105.1, 93.2 (d, J = 170.5 Hz), 53.2 (d, J = 22.5 Hz), 44.2, 31.6 (d, J = 22.0 Hz); HRMS m/z (ESI+) calculated for C14H14FN6 [M + H]+ is 285.1258, found: 285.1267 [M + H]+; LCMS (MDAP): Rt = 11.6 min, >95% (method A); LC-MS (ESI): calculated for C14H14FN6 [M + H]+ is 285.1, found 285.0 [M + H]+; Chiral SFC, Lux A2 column (50:50 isopropanol:CO2 (0.2% v/v NH3), 4.0 mL/min). Retention time = 2.04 min > 99% e.e.
(3S)-1-(4-pyrazolo[1,5-b]pyridazin-3-yl-pyrimidin-2-yl)pyrrolidin-3-amine (33)
To a microwave vial containing a suspension of 3-(2-chloropyrimidin-4-yl)pyrazolo[1,5-b]pyridazine (60 mg, 0.26 mmol) in 2-propanol (2 mL) was added (3S)-(−)-3-aminopyrrolidine (22 mg, 0.26 mmol). The vial was sealed, and the reaction mixture was heated to 140 °C for 16 h. The reaction mixture was concentrated under reduced pressure. The crude was purified using flash silica column chromatography on an Isco Combiflash system (12 g silica, elution with 0–50% 20% MeOH in CH2Cl2/CH2Cl2 gradient) to afford the title compound. Desired fractions were combined and concentrated under reduced pressure. The crude was purified using flash reversed-phase column chromatography on a Biotage system (12 g silica, elution with 10–100% MeOH/water gradient) to afford the title compound, (3S)-1-(4-pyrazolo[1,5-b]pyridazin-3-yl-pyrimidin-2-yl)pyrrolidin-3-amine, 33, as a pale yellow solid (32 mg, 0.11 mmol, 42% yield). Rf 0.09 (MeOH/ CH2Cl2 1:9); [α]D25 = +89.6 (c = 6.7 × 10–4 g/mL, CHCl3); decomposed > 210 °C; 1H NMR (600 MHz, DMSO-d6) δH 8.99 (app. d, J = 9.1 Hz, 1H), 8.82 (s, 1H), 8.60–8.56 (m, 1H), 8.32 (d, J = 5.1 Hz, 1H), 7.45 (dd, J = 9.1, 4.4 Hz, 1H), 7.11 (d, J = 5.1 Hz, 1H), 3.92–2.90 (m, 5H), 2.30–1.52 (m, 4H); 13C NMR (150 MHz, DMSO-d6) δC 160.1, 158.9, 158.0, 143.9, 139.6, 132.4, 129.3, 118.8, 110.4, 104.4, 54.9, 50.7, 44.9, 34.1; HRMS m/z (ESI+) calculated for C14H16N7 [M + H]+ is 282.1462, found: 282.1455 [M + H]+; LCMS (MDAP): Rt = 8.1 min, >95% (method A); LC-MS (ESI): calculated for C14H16N7 [M + H]+ is 282.1, found 282.0 [M + H]+. Chiral HPLC, Lux C1 column (50:50 heptane/EtOH (0.2% v/v NH3), 1.0 mL/min). Retention time = 12.50 min > 99% e.e.
(3R)-1-(4-pyrazolo[1,5-b]pyridazin-3-yl-pyrimidin-2-yl)pyrrolidin-3-amine (34)
To a microwave vial containing a suspension of 3-(2-chloropyrimidin-4-yl)pyrazolo[1,5-b]pyridazine (60 mg, 0.26 mmol) in 2-propanol (2 mL) was added (R)-pyrrolidin-3-amine (22 mg, 0.26 mmol). The vial was sealed, and the reaction mixture was heated to 140 °C for 16 h. The reaction mixture was concentrated under reduced pressure. The crude was purified using flash silica column chromatography on an Isco Combiflash system (12 g silica, elution with 0–50% 20% MeOH in CH2Cl2/CH2Cl2 gradient) to afford the title compound. Desired fractions were combined and concentrated under reduced pressure. The crude was purified using flash reversed-phase column chromatography on a Biotage system (12 g silica, elution with 10–100% MeOH/water gradient) to afford the title compound, (3R)-1-(4-pyrazolo[1,5-b]pyridazin-3-yl-pyrimidin-2-yl)pyrrolidin-3-amine, 34, as a pale yellow solid (36 mg, 0.12 mmol, 44% yield). Rf 0.09 (MeOH/ CH2Cl2 1:9); [α]D25 = −59.7 (c = 6.7 × 10–4 g/mL, CHCl3); decomposed > 210 °C; 1H NMR (600 MHz, DMSO-d6) δH 8.99 (app. d, J = 9.1 Hz, 1H), 8.82 (s, 1H), 8.60–8.56 (m, 1H), 8.32 (d, J = 5.1 Hz, 1H), 7.45 (dd, J = 9.1, 4.4 Hz, 1H), 7.11 (d, J = 5.1 Hz, 1H), 3.96–2.78 (m, 5H), 2.36–1.68 (m, 4H); 13C NMR (150 MHz, DMSO-d6) δC 160.1, 158.9, 158.0, 143.9, 139.6, 132.4, 129.3, 118.8, 110.4, 104.4, 54.7, 50.7, 44.9, 34.0; HRMS m/z (ESI+) calculated for C14H16N7 [M + H]+ is 282.1462, found: 282.1456 [M + H]+; LCMS (MDAP): Rt = 7.9 min, >95% (method A); LC-MS (ESI): calculated for C14H16N7 [M + H]+ is 282.1, found 282.0 [M + H]+. Chiral HPLC, Lux C1 column (50:50 heptane/EtOH (0.2% v/v NH3), 1.0 mL/min). Retention time = 9.55 min > 99% e.e.
N,N-Dimethyl-4-(5-methylpyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-amine (63)
To a solution of 4-methylpyridazine (256 mg, 2.72 mmol) in pH 5 acetate buffer (1.5 mL) was added aminosulfuric acid (462 mg, 1.36 mmol) in water (0.1 mL). The reaction mixture was neutralized to pH 7.5 by slow addition of 2.4 M potassium bicarbonate (1.98 mL, 4.75 mmol). The reaction mixture was heated to 70 °C and was stirred for 2 h. The reaction mixture was cooled to rt, and CH2Cl2 (4.5 mL) was added. The reaction mixture was cooled in an ice bath, and 3-butyn-2-one (0.09 mL, 1.15 mmol) was added in one portion followed by dropwise addition of potassium hydroxide (171 mg, 3.05 mmol) in water (0.45 mL). The reaction mixture was allowed to warm to rt and stirred overnight. The aqueous layer was extracted with CH2Cl2 (2 × 20 mL). The combined organic layers were washed with water (10 mL), dried over MgSO4, filtered, and concentrated under reduced pressure. To the residue was added N,N-dimethylformamide dimethyl acetal (3.37 mL, 25.38 mmol). The reaction mixture was heated to 75 °C for 16 h. The reaction mixture was concentrated under reduced pressure. To the crude were added 1,1-dimethylguanidine sulfate salt (600 mg, 2.2 mmol), 2-methoxyethanol (30 mL), and potassium carbonate (400 mg, 2.89 mmol). The reaction mixture was heated to 125 °C overnight. The reaction mixture was concentrated under reduced pressure. The residue was partitioned between EtOAc (50 mL) and water (50 mL), and the organic layer was separated. The aqueous layer was extracted with further EtOAc (2 × 50 mL). The combined organics were dried over MgSO4, filtered, and concentrated to dryness under reduced pressure. The crude was purified using flash silica column chromatography on an Isco Combiflash system (12 g silica, elution with 0–100% EtOAc/petroleum ether gradient), trituration with petroleum ether followed by recrystallization in EtOAc furnished the title compound, N,N-dimethyl-4-(5-methylpyrazolo[1,5-b]pyridazin-3-yl)pyrimidin-2-amine, 63, as a salmon pink solid (15 mg, 0.05 mmol, 9% yield). Rf 0.46 (EtOAc/ petroleum ether 2:3); mp 236–238 °C; 1H NMR (600 MHz, DMSO-d6) δH 8.77 (s, 1H), 8.70–8.66 (m, 1H), 8.48 (d, J = 2.4 Hz, 1H), 8.33 (d, J = 5.1 Hz, 1H), 7.10 (d, J = 5.1 Hz, 1H), 3.22 (s, 6H), 2.45 (s, 3H); 13C NMR (150 MHz, DMSO-d6) δC 161.9, 159.0, 157.8, 145.8, 139.9, 132.1, 129.3, 126.7, 109.2, 104.4, 36.9, 18.3; HRMS m/z (ESI+) calculated for C13H15N6 [M + H]+ is 255.1353, found: 255.1350 [M + H]+; LCMS (MDAP): 11.6 min, >95% (method A); LC-MS (ESI): calculated for C13H15N6 [M + H]+ is 255.1, found 255.9 [M + H]+.
4-(4-Methoxypyrazolo[1,5-b]pyridazin-3-yl)-N,N-dimethyl-pyrimidin-2-amine (66)
To a solution of 4-methoxypyridazine (300 mg, 2.72 mmol) in pH 5 acetate buffer (1.5 mL) was added aminosulfuric acid (462 mg, 1.36 mmol) in water (0.1 mL). The reaction mixture was neutralized to pH 7.5 by slow addition of 2.4 M potassium bicarbonate (1.98 mL, 4.75 mmol). The reaction mixture was heated to 70 °C and was stirred for 2 h. The reaction mixture was cooled to rt, and CH2Cl2 (4.5 mL) was added. The reaction mixture was cooled in an ice bath, and 3-butyn-2-one (0.09 mL, 1.15 mmol) was added in one portion followed by dropwise addition of potassium hydroxide (171 mg, 3.05 mmol) in water (0.45 mL). The reaction mixture was allowed to warm to rt and stirred overnight. The aqueous layer was extracted with CH2Cl2 (2 × 20 mL). The combined organic layers were washed with water (10 mL), dried over MgSO4, filtered, and concentrated under reduced pressure. To the residue was added N,N-dimethylformamide dimethyl acetal (3.37 mL, 25.38 mmol). The reaction mixture was heated to 75 °C for 16 h. The reaction mixture was concentrated under reduced pressure. To the crude were added 1,1-dimethylguanidine sulfate salt (600 mg, 2.2 mmol), 2-methoxyethanol (30 mL), and potassium carbonate (400 mg, 2.89 mmol). The reaction mixture was heated to 125 °C overnight. The reaction mixture was concentrated under reduced pressure. The residue was partitioned between EtOAc (50 mL) and water (50 mL), and the organic layer was separated. The aqueous layer was extracted with further EtOAc (2 × 50 mL). The combined organics were dried over MgSO4, filtered, and concentrated to dryness under reduced pressure. The crude was purified using flash silica column chromatography on an Isco Combiflash system (12 g silica, elution with 0–100% EtOAc/petroleum ether gradient), trituration with petroleum ether followed by recrystallization in EtOAc furnished the title compound, 4-(4-methoxypyrazolo[1,5-b]pyridazin-3-yl)-N,N-dimethyl-pyrimidin-2-amine, 66, as a white solid (12 mg, 0.04 mmol, 8% yield). Rf 0.41 (EtOAc); mp 180–182 °C; 1H NMR (600 MHz, DMSO-d6) δH 8.54 (s, 1H), 8.42 (d, J = 5.6 Hz, 1H), 8.34 (d, J = 5.1 Hz, 1H), 7.13 (d, J = 5.1 Hz, 1H), 6.88 (d, J = 5.6 Hz, 1H), 4.08 (s, 3H), 3.18 (s, 6H); 13C NMR (150 MHz, DMSO-d6) δC 161.8, 158.3, 157.9, 157.7, 144.0, 139.8, 126.5, 112.6, 108.2, 96.9, 56.8, 36.6; HRMS m/z (ESI+) calculated for C13H15N6O [M + H]+ is 271.1302, found: 271.1298 [M + H]+; LCMS (MDAP): 11.5 min, >95% (method A); LC-MS (ESI): calculated for C13H15N6O [M + H]+ is 271.1, found 271.9 [M + H]+.
1-Imidazo[1,2-a]pyrimidin-3-ylethanone (68a)
Pyrimidin-2-amine (1 g, 10.52 mmol) was dissolved in toluene (7 mL), and N,N-dimethylformamide dimethyl acetal (3 mL, 22.58 mmol) was added. The reaction mixture was heated to 90 °C for 2 h. The reaction mixture was concentrated under reduced pressure. The crude was dissolved in EtOH (10 mL), and chloroacetone (1.6 mL, 20.1 mmol) was added to the mixture. The reaction mixture was heated to reflux and stirred overnight. The reaction mixture was concentrated under reduced pressure. The crude was purified using flash silica column chromatography on an Isco Combiflash system (24 g silica, elution with 0–50% 20% MeOH in CH2Cl2/CH2Cl2 gradient) to afford 1-imidazo[1,2-a]pyrimidin-3-ylethanone, 68a, as a gold solid (800 mg, 4.59 mmol, 44% yield). Rf 0.10 (EtOAc); 1H NMR (600 MHz, DMSO-d6) δH 9.79 (dd, J = 6.8, 2.1 Hz, 1H), 8.83 (dd, J = 4.2, 2.1 Hz, 1H), 8.78 (s, 1H), 7.40 (dd, J = 6.8, 4.2 Hz, 1H), 2.58 (s, 3H); LC-MS (ESI): calculated for C8H8N3O [M + H]+ is 162.07, found 162.13 [M + H]+.
(E)-3-(Dimethylamino)-1-imidazo[1,2-a]pyrimidin-3-yl-prop-2-en-1-one (69a)
To 1-imidazo[1,2-a]pyrimidin-3-ylethanone (100 mg, 0.62 mmol) was added N,N-dimethylformamide dimethyl acetal (4 mL, 30.11 mmol). The reaction mixture was heated to 75 °C for 16 h. The reaction mixture was concentrated under reduced pressure to afford the title compound, (E)-3-(dimethylamino)-1-imidazo[1,2-a]pyrimidin-3-yl-prop-2-en-1-one, 69a, as a red solid (150 mg, 0.62 mmol, 99% yield) plus impurities. Rf 0.23 (MeOH/CH2Cl2 5:95); 1H NMR (600 MHz, CDCl3) δH 10.05 (dd, J = 6.9, 2.1 Hz, 1H), 8.66 (dd, J = 4.1, 2.1 Hz, 1H), 8.35 (s, 1H), 7.79 (d, J = 12.3 Hz, 1H), 7.03 (dd, J = 6.9, 4.1 Hz, 1H), 5.66 (d, J = 12.3 Hz, 1H), 3.28–2.76 (m, 6H); LC-MS (ESI): calculated for C11H13N4O [M + H]+ is 217.1, found 217.2 [M + H]+.
4-Imidazo[1,2-a]pyrimidin-3-yl-N,N-dimethyl-pyrimidin-2-amine (70)
To a solution of (E)-3-(dimethylamino)-1-imidazo[1,2-a]pyrimidin-3-yl-prop-2-en-1-one (100 mg, 0.46 mmol) in 2-methoxyethanol (35 mg, 0.46 mmol) were added 1,1-dimethylguanidine sulfate salt (190 mg, 0.70 mmol) and potassium carbonate (133 mg, 0.96 mmol). The reaction mixture was heated to 125 °C overnight. The reaction mixture was concentrated under reduced pressure. The residue was partitioned between EtOAc (50 mL) and water (50 mL), and the organic layer was separated. The aqueous layer was extracted with further EtOAc (2 × 50 mL). The combined organics were dried over MgSO4, filtered, and concentrated to dryness under reduced pressure. The crude was purified using flash silica column chromatography on an Isco Combiflash system (12 g silica, elution with 0–50% 20% MeOH in CH2Cl2/CH2Cl2 gradient) to afford the title compound, 4-imidazo[1,2-a]pyrimidin-3-yl-N,N-dimethyl-pyrimidin-2-amine, 70, as a pale yellow solid (21 mg, 0.08 mmol, 18% yield). Rf 0.63 (EtOAc); mp 237–239 °C; 1H NMR (600 MHz, DMSO-d6) δH 10.12 (dd, J = 7.0, 2.0 Hz, 1H), 8.70–8.65 (m, 2H), 8.35 (d, J = 5.2 Hz, 1H), 7.32 (dd, J = 7.0, 4.1 Hz, 1H), 7.17 (d, J = 5.2 Hz, 1H), 3.19 (s, 6H); 13C NMR (150 MHz, DMSO-d6) δC 161.5, 157.9, 156.3, 151.4, 150.2, 138.9, 136.6, 120.2, 110.3, 104.0, 37.2; HRMS m/z (ESI+) calculated for C12H12N6Na [M + Na]+ is 263.1016, found: 263.1012 [M + Na]+; LCMS (MDAP): 10.1 min, >95% (method A); LC-MS (ESI): calculated for C12H13N6 [M + H]+ is 241.1, found 241.7 [M + H]+.
Methyl pyrazolo[1,5-b]pyridazine-3-carboxylate (89)
To a slurry of pyridazin-1-ium-1-amine iodide (3.13 g, 14.05 mmol) in CH2Cl2 (100 mL) was added methyl propiolate (0.5 mL, 5.62 mmol). The reaction flask was cooled in an ice bath before slowly adding a solution of potassium carbonate (2.33 g, 16.86 mmol) in water (100 mL) in one portion. The reaction mixture was stirred at rt for 16 h. The organic layer was separated. The aqueous layer was washed with CH2Cl2 (3 × 100 mL). The combined organic layers were washed with brine solution (200 mL), and dried over MgSO4, filtered, and concentrated under reduced. The crude was purified using flash silica column chromatography on an Isco Combiflash system (12 g silica, elution with 0–100% EtOAc/petroleum ether gradient) to afford the title compound, methyl pyrazolo[1,5-b]pyridazine-3-carboxylate, 89, as a pale yellow solid (380 mg, 2.04 mmol, 36% yield). Rf 0.38 (CH2Cl2); mp 119–121 °C; 1H NMR (500 MHz, DMSO-d6) δH 8.70–8.65 (m, 1H), 8.58–8.53 (m, 2H), 7.55 (dd, J = 9.1, 4.5 Hz, 1H), 3.86 (s, 3H); LC-MS (ESI): calculated for C8H8N3O2 [M + H]+ is 178.1, found 178.1 [M + H]+.
Pyrazolo[1,5-b]pyridazine-3-carboxylic Acid (90)
To a solution of methyl pyrazolo[1,5-b]pyridazine-3-carboxylate (350 mg, 1.98 mmol) in MeOH (2.5 mL) was added lithium hydroxide (135 mg, 3.22 mmol) in water (2.5 mL). The reaction mixture was stirred at rt for 16 h. The reaction mixture was concentrated under reduced pressure. Water (10 mL) was added to the residue, and the resulting solution was cooled in an ice bath. HCl (10 mL, 1 M) was added, and the mixture was stirred at rt for 30 min. A suspension immediately formed and was filtered and washed with water. The resulting solid was dried under reduced pressure to afford the title compound pyrazolo[1,5-b]pyridazine-3-carboxylic acid, 90, as a white solid (250 mg, 1.46 mmol, 74% yield). Rf 0 (EtOAc/petroleum ether 1:1); decomposed > 295 °C; 1H NMR (500 MHz, DMSO-d6) δH 12.83 (s, 1H), 8.63 (dd, J = 4.4, 2.1 Hz, 1H), 8.55–8.44 (m, 2H), 7.50 (dd, J = 9.1, 4.4 Hz, 1H); LC-MS (ESI): calculated for C7H6N3O2 [M + H]+ is 164.0, found 164.1 [M + H]+.
3-Bromopyrazolo[1,5-b]pyridazine (91)
To a solution of pyrazolo[1,5-b]pyridazine-3-carboxylic acid (360 mg, 2.21 mmol) in DMF (4 mL) was added N-bromosuccinimide (790 mg, 4.41 mmol), and the reaction was stirred at rt for 3 h. The reaction mixture was concentrated under reduced pressure. The crude was purified using flash silica column chromatography on an Isco Combiflash system (12 g silica, elution with 0–60% EtOAc/petroleum ether gradient) to afford 3-bromopyrazolo[1,5-b]pyridazine, 91, as a white crystalline solid (360 mg, 1.73 mmol, 78% yield). Rf 0.74 (EtOAc/petroleum ether 1:1); mp 102–104 °C; 1H NMR (500 MHz, DMSO-d6) δH 8.52 (dd, J = 4.4, 1.8 Hz, 1H), 8.28 (s, 1H), 8.20 (dd, J = 9.0, 1.8 Hz, 1H), 7.31 (dd, J = 9.0, 4.4 Hz, 1H); LC-MS (ESI): calculated for C6H5BrN3 [M]+ is 198.0 and [M + 2]+ is 200.0, found 198.0 [M]+ and 200.0 [M + 2]+.
3-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)pyrazolo[1,5-b]pyridazine (92)
To a solution of 3-bromopyrazolo[1,5-b]pyridazine (100 mg, 0.50 mmol) in 1,4-dioxane (1 mL) were added bis(pinacolato)diboron (141 mg, 0.56 mmol), [1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium(II), complex with CH2Cl2 (21 mg, 0.03 mmol), and potassium acetate (99 mg, 1.01 mmol). The reaction mixture was heated to 90 °C and stirred for 16 h. The reaction mixture was filtered through celite and concentrated under reduced pressure. The crude was purified using flash silica column chromatography on an Isco Combiflash system (12 g silica, elution with 0–50% EtOAc/petroleum ether gradient). Desired fractions were combined and concentrated under reduced pressure. The crude was purified using flash silica column chromatography on an Isco Combiflash system (12 g silica, elution with 0–50% 20% MeOH in CH2Cl2/CH2Cl2 gradient) to afford the title compound, 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrazolo[1,5-b]pyridazine, 92, as a white crystalline solid (30 mg, 0.12 mmol, 23% yield). Rf 0.38 (EtOAc/petroleum ether 1:1); mp 160–162 °C; 1H NMR (500 MHz, DMSO-d6) δH 8.53 (dd, J = 4.4, 1.9 Hz, 1H), 8.27 (dd, J = 9.0, 1.9 Hz, 1H), 8.23 (s, 1H), 7.34 (dd, J = 9.0, 4.4 Hz, 1H), 1.30 (s, 12H); LC-MS (ESI): calculated for C12H17BN3O2 [M + H]+ is 246.1, found 246.1 [M + H]+.
5-Fluoro-N,N-dimethyl-4-pyrazolo[1,5-b]pyridazin-3-yl-pyrimidin-2-amine (95)
To a solution of 4-chloro-5-fluoropyrimidin-2-amine (100 mg, 0.68 mmol) in DMF (2 mL) was added sodium hydride (81 mg, 3.39 mmol) at 0 °C. After warming to rt and stirring for 5 min, iodomethane (0.08 mL, 1.36 mmol) was added dropwise, and the reaction was stirred at rt for 16 h. The reaction mixture was concentrated under reduced pressure. The crude was dissolved in water (100 mL) and extracted into EtOAc (3 × 100 mL). The combined EtOAc phases were dried over MgSO4, filtered, and concentrated under reduced pressure to afford the intermediate, 4-chloro-5-fluoro-N,N-dimethyl-pyrimidin-2-amine, as a crude orange solid (130 mg). LC-MS (ESI+): calculated for intermediate C6H8ClFN3 [M + H]+ is 176.1, found 176.1 [M + H]+.
To a solution of 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrazolo[1,5-b]pyridazine (80 mg, 0.3 mmol) in acetonitrile (4.5 mL) and water (0.45 mL) were added sodium carbonate (70 mg, 0.65 mmol), bis[2-(di-tert-butylphosphanyl)cyclopenta-2,4-dien-1-yl]iron (dichloropalladium) (11 mg, 0.02 mmol), and the crude orange solid [4-chloro-5-fluoro-N,N-dimethyl-pyrimidin-2-amine] (80 mg, 0.21 mmol). The reaction mixture was heated to 140 °C overnight. The reaction mixture was concentrated under reduced pressure. The crude was purified using flash silica column chromatography on an Isco Combiflash system (12 g silica, elution with 0–60% EtOAc/petroleum ether gradient). Desired fractions were concentrated under reduced pressure. The crude was purified using flash reversed-phase column chromatography on a Biotage system (12 g silica, elution with 10–100% MeOH/water gradient) to afford the title compound, 5-fluoro-N,N-dimethyl-4-pyrazolo[1,5-b]pyridazin-3-yl-pyrimidin-2-amine, 95, as an off-white solid (50 mg, 0.18 mmol, 57% yield). Rf 0.57 (EtOAc/petroleum ether 1:1); mp 181–183 °C; 1H NMR (600 MHz, CDCl3) δH 9.00 (dd, J = 9.0, 2.0 Hz, 1H), 8.66 (d, J = 3.1 Hz, 1H), 8.41 (dd, J = 4.4, 2.0 Hz, 1H), 8.23 (d, J = 3.2 Hz, 1H), 7.19 (dd, J = 9.0, 4.4 Hz, 1H), 3.25 (s, 6H); 13C NMR (150 MHz, CDCl3) δC 159.2, 148.6 (d, J = 249.1 Hz), 147.1 (d, J = 11.6 Hz), 145.4 (d, J = 24.3 Hz), 143.2, 141.8 (d, J = 16.2 Hz), 133.4, 129.7, 118.0, 107.1, 38.2; HRMS m/z (ESI+) calculated for C12H11FN6Na [M + Na]+ is 281.0921, found: 281.0918 [M + Na]+; LCMS (MDAP): 19.6 min, >95% (method A); LC-MS (ESI): calculated for C12H12FN6 [M + H]+ is 259.1, found 259.9 [M + H]+.
Acknowledgments
The authors thank Kamal R. Abdul Azeez for help with protein purification. S.H.H. acknowledges industrial sponsorship from Evotec (U.K.) Ltd. and is grateful for the use of laboratory facilities to undertake part of this work. An academic license for ChemAxon (https://www.chemaxon.com) and a license for ChemAxon Infocom KNIME nodes are gratefully acknowledged. They also acknowledge the platform KNIME (https://www.knime.com).
Glossary
Abbreviations Used
- BBB
blood–brain barrier
- CDK
cyclin-dependent kinase
- CLK
CDC-like kinases
- CMGC
including cyclin-dependent kinases (CDKs), mitogen-activated protein kinases (MAP kinases), glycogen synthase kinases (GSK) and CDK-like kinases
- CNS MPO
central nervous system multiparameter optimization
- DYRK
dual-specificity tyrosine phosphorylation-regulated kinase
- GSK
glycogen synthase kinases
- HBA
hydrogen-bond acceptor
- HLM
human liver microsomes
- LE
ligand efficiency
- LLE
lipophilic ligand efficiency
- MDCK-MDR1
Madin–Darby canine kidney cells transfected with the human MDR1 gene
- MMP
matched-molecular pair
- PDB
Protein Databank
- P-gp
permeability glycoprotein 1
- PK
pharmacokinetics
- RLM
rat liver microsomes
- SAR
structure–activity relationship
- SBDD
structure-based drug design
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c01115.
Author Present Address
◆ BenevolentAI, Minerva Building, Babraham Hall, Babraham, Cambridge, CB22 3AT, UK. (S.H.H.)
Author Contributions
S.H.H. was involved in the conception and design, acquisition of data, analysis and interpretation of data, and drafting and revising of the article. F.S. was involved in the conception and design, acquisition of data, analysis and interpretation of data, and drafting and revising of the article. J.B., O.F., P.H.G., and R.R.d.S. were involved in the acquisition, analysis, and interpretation of data. M.T.H was involved in the acquisition of data. S.R. and A.A.-K. were involved in the acquisition of data. I.H.N was involved in the acquisition of data and revising of the article. D.S.W. was involved in the analysis and interpretation of data. J.M.E. was involved in the conception of the project, analysis and interpretation of data, and drafting and revising of the article. S.E.W. was involved in the conception of the project, analysis and interpretation of data, and revising of the article. All authors approved the final version to be published.
S.H.H. is funded by the BBSRC (BB/L017105/1). S.E.W. is funded by the Wellcome Trust, MRC, BBSRC and the European Structural Funding via Sêr Cymru scheme. R.R.d.S. is funded by São Paulo Research Foundation-FAPESP 2016/17469-0 and 13/50724-5. The SGC is a registered charity (number 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada, Innovative Medicines Initiative (EU/EFPIA) [ULTRA-DD grant no. 115766], Janssen, Merck KGaA Darmstadt Germany, MSD, Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, São Paulo Research Foundation-FAPESP, Takeda, and Wellcome [106169/ZZ14/Z].
The authors declare no competing financial interest.
Notes
PDB X-ray structures: PDB 6S1B (compound 10); PDB 6S17 (compound 11); PDB 6S14 (compound 12); PDB 6S1J (compound 30); PDB 6S1H (compound 78). The authors released the atomic coordinates on June 26, 2019.
Supplementary Material
References
- Tejedor F. J.; Hammerle B. MNB/DYRK1A as a Multiple Regulator of Neuronal Development. FEBS J. 2011, 278, 223–235. 10.1111/j.1742-4658.2010.07954.x. [DOI] [PubMed] [Google Scholar]
- Wegiel J.; Kaczmarski W.; Barua M.; Kuchna I.; Nowicki K.; Wang K. C.; Yang S. M.; Frackowiak J.; Mazur-Kolecka B.; Silverman W. P.; Reisberg B.; Monteiro I.; de Leon M.; Wisniewski T.; Dalton A.; Lai F.; Hwang Y. W.; Adayev T.; Liu F.; Iqbal K.; Iqbal I. G.; Gong C. X.; et al. Link Between DYRK1A Overexpression and Several-fold Enhancement of Neurofibrillary Degeneration with 3-Repeat Tau Protein in Down Syndrome. J. Neuropathol. Exp. Neurol. 2011, 70, 36–50. 10.1097/NEN.0b013e318202bfa1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J. Alzheimer’s Disease: the Amyloid Cascade Hypothesis: an Update and Reappraisal. J. Alzheimer’s Dis. 2006, 9, 151–153. 10.3233/JAD-2006-9S317. [DOI] [PubMed] [Google Scholar]
- Smith B.; Medda F.; Gokhale V.; Dunckley T.; Hulme C. Recent Advances in the Design, Synthesis, and Biological Evaluation of Selective DYRK1A Inhibitors: a New Avenue for a Disease Modifying Treatment of Alzheimer’s?. ACS Chem. Neurosci. 2012, 3, 857–872. 10.1021/cn300094k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu Y. S.; Park S. Y.; Jung M. S.; Yoon S. H.; Kwen M. Y.; Lee S. Y.; Choi S. H.; Radnabaazar C.; Kim M. K.; Kim H.; Kim K.; Song W. J.; Chung S. H. Dyrk1A-Mediated Phosphorylation of Presenilin 1: a Functional Link Between Down Syndrome and Alzheimer’s Disease. J. Neurochem. 2010, 115, 574–584. 10.1111/j.1471-4159.2010.06769.x. [DOI] [PubMed] [Google Scholar]
- Kumar K.; Wang P.; Sanchez R.; Swartz E. A.; Stewart A. F.; DeVita R. J. Development of Kinase-Selective, Harmine-Based DYRK1A Inhibitors that Induce Pancreatic Human β-Cell Proliferation. J. Med. Chem. 2018, 61, 7687–7699. 10.1021/acs.jmedchem.8b00658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak A.; Rohilla A.; Gupta T.; Akhtar M. J.; Haider M. R.; Sharma K.; Haider K.; Yar M. S. DYRK1A Kinase Inhibition with Emphasis on Neurodegeneration: A Comprehensive Evolution Story-cum-perspective. Eur. J. Med. Chem. 2018, 158, 559–592. 10.1016/j.ejmech.2018.08.093. [DOI] [PubMed] [Google Scholar]
- Naert G.; Ferré V.; Meunier J.; Keller E.; Malmström S.; Givalois L.; Carreaux F.; Bazureau J. P.; Maurice T. Leucettine L41, a DYRK1A-preferential DYRKs/CLKs Inhibitor, Prevents Memory Impairments and Neurotoxicity Induced by Oligomeric Aβ25-35 Peptide Administration in Mice. Eur. Neuropsychopharmacol. 2015, 2170–2182. 10.1016/j.euroneuro.2015.03.018. [DOI] [PubMed] [Google Scholar]
- Melchior B.; Mittapalli G. K.; Lai C.; Duong-Polk K.; Stewart J.; Güner B.; Hofilena B.; Tjitro A.; Anderson S. D.; Herman D. S.; Dellamary L.; Swearingen C. J.; Sunil K. C.; Yazici Y. Tau Pathology Reduction with SM07883, a Novel, Potent, and Selective Oral DYRK1A Inhibitor: A Potential Therapeutic for Alzheimer’s Disease. Aging Cell. 2019, 18, e13000 10.1111/acel.13000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y. A.; Jin Q.; Zou Y.; Ding Q.; Yan S.; Wang Z.; Hao X.; Nguyen B.; Zhang X.; Pan J.; Mo T.; Jacobsen K.; Lam T.; Wu T. Y.; Petrassi H. M.; Bursulaya B.; DiDonato M.; Gordon W. P.; Liu B.; Baaten J.; Hill R.; Nguyen-Tran V.; Qiu M.; Zhang Y. Q.; Kamireddy A.; Espinola S.; Deaton L.; Ha S.; Harb G.; Jia Y.; Li J.; Shen W.; Schumacher A. M.; Colman K.; Glynne R.; Pan S.; McNamara P.; Laffitte B.; Meeusen S.; Molteni V.; Loren J. Selective DYRK1A Inhibitor for the Treatment of Type 1 Diabetes: Discovery of 6-Azaindole Derivative GNF2133. J. Med. Chem. 2020, 63, 2958–2973. 10.1021/acs.jmedchem.9b01624. [DOI] [PubMed] [Google Scholar]
- Kumar K.; Wang P.; Wilson J.; Zlatanic V.; Berrouet C.; Khamrui S.; Secor C.; Swartz E. A.; Lazarus M.; Sanchez R.; Stewart A. F.; Garcia-Ocana A.; DeVita R. J. Synthesis and Biological Validation of a Harmine-Based, Central Nervous System (CNS)-Avoidant, Selective, Human β-Cell Regenerative Dual-Specificity Tyrosine Phosphorylation-Regulated Kinase A (DYRK1A) Inhibitor. J. Med. Chem. 2020, 63, 2986–3003. 10.1021/acs.jmedchem.9b01379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson S. H.; Sorrell F.; Bennett J.; Hanley M. T.; Robinson S.; Hopkins Navratilova I.; Elkins J. M.; Ward S. E. Mining Public Domain Data to Develop Selective DYRK1A Inhibitors. ACS Med. Chem. Lett. 2020, 11, 1620–1626. 10.1021/acsmedchemlett.0c00279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavares F. X.; Boucheron J. A.; Dickerson S. H.; Griffin R. J.; Preugschat F.; Thomson S. A.; Wang T. Y.; Zhou H. Q. N-Phenyl-4-pyrazolo[1,5-b]pyridazin-3-ylpyrimidin-2-amines as Potent and Selective Inhibitors of Glycogen Synthase Kinase 3 with Good Cellular Efficacy. J. Med. Chem. 2004, 47, 4716–4730. 10.1021/jm040063i. [DOI] [PubMed] [Google Scholar]
- Schaenzer A. J.; Wlodarchak N.; Drewry D. H.; Zuercher W. J.; Rose W. E.; Ferrer C. A.; Sauer J-D.; Striker R. GW779439X and its Pyrazolopyridazine Derivatives Inhibit the Serine/Threonine Kinase Stk1 and Act as Antibiotic Adjuvants Against β-Lactam-Resistant Staphylococcus Aureus. ACS Infect Dis. 2018, 4, 1508–1518. 10.1021/acsinfecdis.8b00136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tear W. F.; Bag S.; Diaz-Gonzalez R.; Ceballos-Pérez G.; Rojas-Barros D. I.; Cordon-Obras C.; Pérez-Moreno G.; García-Hernández R.; Martinez-Martinez M. S.; Ruiz-Perez L. M.; Gamarro F.; Gonzalez Pacanowska D.; Caffrey C. R.; Ferrins L.; Manzano P.; Navarro M.; Pollastri M. P. Selectivity and Physicochemical Optimization of Repurposed Pyrazolo[1,5-b]pyridazines for the Treatment of Human African Trypanosomiasis. J. Med. Chem. 2020, 63, 756–783. 10.1021/acs.jmedchem.9b01741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heroven C.; Georgi V.; Ganotra G. K.; Brennan P.; Wolfreys F.; Wade R. C.; Fernández-Montalván A. E.; Chaikuad A.; Knapp S. Halogen-Aromatic π Interactions Modulate Inhibitor Residence Times. Angew. Chem., Int. Ed. 2018, 57, 7220–7224. 10.1002/anie.201801666. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.