

Abstract
Background
In cardiovascular diseases, atherosclerotic disorder are the most frequent and important with respect to morbidity and mortality. Inflammation mediated by immune cells is central in all parts of the atherosclerotic progress, and further understanding of the underlying mechanisms is needed. Growing evidence suggests that deamination of adenosine‐to‐inosine in RNA is crucial for a correct immune response; nevertheless, the role of adenosine‐to‐inosine RNA editing in atherogenesis has barely been studied. Several proteins have affinity for inosines in RNA, one being ENDOV (endonuclease V), which binds and cleaves RNA at inosines. Data on ENDOV in atherosclerosis are lacking.
Methods and Results
Quantitative polymerase chain reaction on ENDOV mRNA showed an increased level in human carotid atherosclerotic plaques compared with control veins. Inosine‐ribonuclease activity as measured by an enzyme activity assay is detected in immune cells relevant for the atherosclerotic process. Abolishing EndoV in atherogenic apolipoprotein E‐deficient (ApoE−/− ) mice reduces the atherosclerotic plaque burden, both in size and lipid content. In addition, in a brain stroke model, mice without ENDOV suffer less damage than control mice. Finally, lack of EndoV reduces the recruitment of monocytes to atherosclerotic lesions in atherogenic ApoE−/− mice.
Conclusions
ENDOV is upregulated in human atherosclerotic lesions, and data from mice suggest that ENDOV promotes atherogenesis by enhancing the monocyte recruitment into the atherosclerotic lesion, potentially by increasing the effect of CCL2 activation on these cells.
Keywords: atherosclerosis, A‐to‐I editing, CCL2, endonuclease V, monocyte recruitment, stroke
Subject Categories: Basic Science Research, Vascular Biology, Cell Signalling/Signal Transduction
Nonstandard Abbreviations and Acronyms
- ADAR1
adenosine deaminase acting on RNA 1
- BMDM
bone marrow–derived macrophage
- ENDOV
endonuclease V
- PBMC
peripheral blood mononuclear cell
- VSMC
vascular smooth muscle cell
Clinical Perspective
What Is New?
RNA‐editing enzymes and edited ribonucleotide inosine are dysregulated in human carotid atherosclerosis.
Abolishing endonuclease V, a central regulator of these processes, reduces plaque size and ischemic stroke severity in preclinical models.
What Are the Clinical Implications?
Endonuclease V may represent a novel therapeutic target in patients with atherosclerotic disorders.
Despite decreased prevalence of cardiovascular disease during the past 20 years, cardiovascular disease is still the leading cause of death in both developing and developed countries and accounts for 17 million deaths each year according to the World Health Organization.1 In cardiovascular diseases, atherosclerotic disorders are the most frequent and important with respect to morbidity and mortality.2 Atherosclerosis is a progressive disease with a bidirectional interaction between lipids and inflammation as a major pathogenic hallmark. Although great progress has been achieved in the understanding of atherogenesis during the past decades, the identification of all the signaling pathways that are involved in this complex process has not been fulfilled.
Upon activation, immune cells respond rapidly through a balanced secretion of cytokines, chemokines, interferons, and growth factors to evoke a properly tuned innate immune response. For example, hundreds of interferon‐stimulated genes are upregulated within 2 to 3 hours after macrophage activation.3 One of these genes is ADAR1 (adenosine deaminase acting on RNA 1),4, 5 which is responsible for deamination of adenosine to inosine in RNA.6 Growing evidence suggests that ADAR1‐mediated adenosine‐to‐inosine editing is crucial for a correct immune response, in particular the ability of recognition of endogenous (self) double‐stranded RNA that can be released during tissue damage.7
Mammalian cells have enzymes that recognize inosines in RNA. One of these is endonuclease V (ENDOV), which binds and cleaves RNA at inosines.8, 9 In humans, there seems to be a complex alternative splicing of ENDOV mRNA and a specific regulation of the various isoforms in different macrophage phenotypes.10 Although the in vitro activity of mammalian ENDOV is well studied, the in vivo function is still unknown.
The role of adenosine‐to‐inosine RNA editing in atherogenesis has barely been studied and data on ENDOV are lacking. One study showed that ADAR1 was essential for vascular remodeling and vascular smooth muscle cell (VSMC) phenotypic modulation in vivo by the inactivation of splice sites in VSMC contractile genes.11 Recently, Stellos et al showed that adenosine‐to‐inosine editing controls the mRNA stability of the extracellular matrix degradation enzyme cathepsin S and that increased expression of ADAR1 in atherosclerotic inflammatory diseases results in increased levels of cathepsin S.12
We hypothesized that the interplay between ADAR1, adenosine‐to‐inosine editing, and ENDOV could play a pathogenic role in the development and progression of atherosclerosis. This hypothesis was examined by different experimental approaches including clinical and experimental studies particularly focusing on leukocyte influx into the lesion and the role of the prototypical proatherogenic chemokine/chemokine receptors CCL2/CCR2 as well as VSMC remodeling.
Methods
Because of ethical restrictions from the Regional Committee for Medical and Research Ethics in South‐East Norway, the data from the individual patients will unfortunately not be made available to other researchers for purposes of reproducing the results or replicating the procedure.
Ethics
The protocols for human samples were approved by the Regional Committee for Medical and Research Ethics in South‐East Norway, reference numbers S‐0923a 2009/6065 and 2017/2202. All participants gave signed informed consent, and the study protocols were in agreement with the principles of the Declaration of Helsinki.
The animal experiments have been approved by the Norwegian National Animal Research Authority with FOTS project license numbers 7927, 8395, 5336, and 21681. All animal experiments were performed in accordance with the European Directive 2010/63/EU.
Patient Material
Patients who were referred to carotid endarterectomy were consecutively enrolled in the Oslo Cohort Study between 2005 and 2014 at the Department of Neurology, Oslo University Hospital Rikshospitalet. The indication for carotid endarterectomy was based on current guidelines.13 Characteristics of the 163 patients (mean age, 68.7 years; 67.1% men) who donated carotid atherosclerotic plaques retrieved during carotid endarterectomy are presented in detail in the Table. The patients with no relevant symptoms had carotid stenosis that was coincidentally detected during clinical examination of patients with coronary or peripheral artery disease. For comparison of plaque analyses, healthy arterial tissue samples were obtained from nonatherosclerotic common iliac arteries of organ donors with no history of cardiovascular disease, deceased from sudden death and approved for organ transplantation. Characteristics on patients donating blood for peripheral blood mononuclear cells (PBMCs) analysis are listed in Table S1. For comparison, PBMCs were also isolated from healthy controls (n=33) based on disease history (mean age, 66.6 years; 66.7% men).
Table 1.
Baseline Variables in Patients (Carotid Plaques; n=163)
| Variable | Percentage (No.) or Mean±SD |
|---|---|
| Age, y | 68.7±8.1 |
| Male sex | 67.1 (110) |
| BMI, kg/m2 | 26.2±3.8 |
| Current smoking | 44.5 (73) |
| Hypertension | 64.6 (106) |
| Diabetes mellitus | 22.0 (36) |
| Aspirin treatment | 88.4 (145) |
| Statin treatment | 90.9 (149) |
| CRP, mg/L | 8.3±34.8 |
| Leucocyte count, 109/L | 7.8±2.1 |
| Platelets, 109/L | 277.7±76.6 |
| Total cholesterol, mmol/L | 4.3±1.2 |
| LDL cholesterol, mmol/L | 2.5±0.9 |
| HDL cholesterol, mmol/L | 1.3±0.5 |
| Triglycerides, mmol/L | 1.5±1.0 |
| HbA1c, % | 6.2±1.5 |
BMI indicates body mass index; CRP, C‐reactive protein; HbA1c, hemoglobin A1c; HDL, high‐density lipoprotein; and LDL, low‐density lipoprotein.
PBMC Isolation
PBMCs were isolated from heparinized blood from healthy controls and patients with stroke by Isopaque‐Ficoll (Lymphoprep, Fresenius Kabi Norge AS) gradient centrifugation within 1 hour after blood collection and stored at −80°C as cell pellets before RNA isolation.
Animal Studies
The apolipoprotein E‐/‐ (ApoE −/−) EndoV −/− mouse strain was generated by crossing EndoV−/− mice described previously14 and ApoE −/− mice (C57BL/6N, Taconic Biosciences; Figure S1). Wild‐type controls in the EndoV−/− experiments were C57BL/6N mice. All mice were housed under a 12‐hour light–dark cycle with ad libitum access to food and water unless otherwise stated.
For assessment of aortic root atherosclerosis, female ApoE−/− and ApoE−/− EndoV−/− mice were fed an atherogenic diet (Research Diets Inc., Diet No. D12451 enriched with 0.25% cholesterol) for 10 weeks before termination by cardiac puncture. Blood was collected using a 1‐mL syringe coated with 0.5 mol/L of EDTA (Fluka, Sigma‐Aldrich). EDTA blood was immediately placed on ice and centrifuged within 30 minutes at 2000g (4°C) for 20 minutes to obtain platelet‐poor plasma. Cranial halves of the hearts were placed in Tissue‐Tek optimal cutting temperature (OCT) compound (Tissue‐Tek, Sakura Finetek) and frozen at −80°C until analysis.
In the monocyte lesion recruitment experiment, male ApoE−/− and ApoE−/− EndoV−/− mice received an injection of Fluoresbrite Polychromatic Red 1.0 µm beads (2.5% solids‐latex, Polysciences, Inc., No. 18660). Beads were diluted in sterile PBS (1:4 dilution) and 250 µL was administered through caudal vein injection. The mice were then fed an atherogenic diet (Diet No. D12451) for 5 weeks before termination by cardiac puncture. Blood and tissue samples were collected as described previously.
Model of Hypoxia–Ischemic Stroke
Wild‐type and EndoV −/− mice underwent cerebral ischemic stroke at postnatal day 10 by permanent occlusion of the common carotid artery followed by hypoxia (Levine15and Sheldon et al16). Briefly, the mice were administered an analgesic with buprenorphine (0.1 mg/kg) before surgery and anesthetized with isoflurane (4% induction, 2.5% maintenance in a 2:1 mixture of ambient air and oxygen). After a small skin incision, the left common carotid artery was isolated and ligated by cauterization. All surgical procedures were performed within 5 minutes. The pups were then returned to the dam for 1 hour to recover before 60 minutes of hypoxia with 10% O2 at 36°C in a hypoxia chamber (Invivo2, Thermo Fisher Scientific). After the procedure, the pups were returned to the dam until harvesting at day 42.
Histological Analysis of the Aortic Root
Frozen hearts embedded in Tissue‐Tek OCT compound were sectioned from the caudal to the cranial direction at 10‐µm intervals on a cryostat. The sections were collected, starting at a 90‐µm distance after the appearance of the aortic cusps and stopping at 880 µm. Great caution was taken to prevent oblique sections. Sections were air dried and fixated with either 4% paraformaldehyde or cold acetone. Plaque size and lipid content were investigated in paraformaldehyde‐fixed sections collected at 100‐µm intervals, stained with Oil‐Red‐O (Sigma‐Aldrich) and counterstained using hematoxylin (H‐3404, Vector Laboratories). Smooth muscle cell content was determined using fluorescence staining with rabbit anti–α‐smooth muscle actin (ab124964, Abcam) and Alexa‐488 goat anti‐rabbit secondary antibody (A‐11008, Thermo Fisher Scientific) on acetone‐fixed aortic root slides. Sirius Red (Histolab, Västra Frölunda) staining was done on paraformaldehyde‐fixated slides. Stained sections were scanned (Axio Scan.Z1, Zeiss) and imported to an in‐house, web‐based digital slide storage‐ and analysis system (z9.nird.sigma2.no) system for evaluation and quantitative examination. Here, atherosclerotic plaques in the aortic root were digitally analyzed, and areal marker prevalence was quantified using colorimetric detection of the Picrosirius red/fluorescence/oil‐red‐O staining within the hue, saturation, value/red, green, blue color space.
Bone Marrow–Derived Macrophages
Bone marrow was isolated by flushing femurs and tibiae and culturing cells in Roswell Park Memorial Institute 1640 medium (Invitrogen) containing 10% heat‐inactivated FCS, 100 U/mL penicillin, 100 mg/mL streptomycin, 2 mmol/L l‐glutamine, and 10 mmol/L Hepes and supplemented with 15% L929‐conditioned medium to generate bone marrow–derived macrophages (BMDMs). Medium was replaced every 3 days, and differentiated BMDMs were used for in vitro assays 7 days after isolation.
For monitoring oxidized low‐density lipoprotein accumulation, oxidized low‐density lipoprotein (Kalen Biomedical, LLC) was stained with pHrodo Red, succinimidyl ester (Invitrogen, catalog number P36600) as described by the manufacturer. Briefly, 100 µL of 1 mg/mL oxidized low‐density lipoprotein was incubated with 1 µL of 10.2 mmol/L pHrodo for 18 hours at 4°C in the dark before use. PBS with pHrodo alone was used for negative controls. BMDMs were seeded in 96‐well plates (0.02 million cells/well) and placed in the Incucyte S3 live cell system. Pictures were taken with 15‐minute time intervals and analyzed with the Incucyte S3 2018C software. A fluorescent signal was related to confluent area in the wells.
For assessing random migration of BMDMs in vitro, cells were seeded on 12‐well plates (MatTek Life Science) coated with 0.02% gelatin (American Type Culture Collection) for 15 minutes. The cells were left overnight to attach and stimulated with the regular cell culture media described previously or with media containing 0.2% FCS and 100 ng/mL CCL2 (R&D Biotechne). Live cell imaging of migrating cells was acquired using a Zeiss Axio Observer Z1 wide‐field microscope controlled by the Micro‐Manager software 1 (version 2.0b). The microscope was equipped with a 20×0.8 Numerical Aperture air objective, a Complementary Metal‐Oxide Semiconductor camera (ORCA‐Flash4.0, Hamamatsu) and a high‐precision stage. During imaging, cells were maintained at 37°C and 5% CO2. A grid of 4×4 adjacent images was acquired once every 4 minutes using transmitted light and a Differential Interference Contrast (II=2) condenser setting. Subsequently, image panels were stitched together into mosaic images. Image stacks were processed in a 3‐step manner. First, background correction was performed with ImageJ (ImageJ2 and Fiji) by subtracting the mean intensity of each stack across the time points from each slice in that stack. Second, a random subset of the images at various time points were imported into Ilastik (imagej.net), and cell nuclei were annotated using a pixel‐wise classification setup. Once the predictions from Ilastik were comparable with the ability of the operator to discern individual nuclei, the probability maps of the nuclei detections were exported for the full range of images. Lastly, in ImageJ, a workflow was used that automatically applied TrackMate particle detection for the probability maps of the nuclei. The workflow further used the results from the detected particles to calculate absolute and Euclidean means by analyzing the movement of each individually detected cell and also overlaying all cells in each well onto a migration map, where the position of every cell was normalized to its position at the first detected time point.
For RNA analysis, BMDMs were seeded in 24‐well plates (Thermo Fisher Scientific; 0.5 million cells/well) with normal cell culture media. After overnight attachment, the cells were incubated for 4 hours in normal cell culture media with 100 ng/mL recombinant CCL2 (R&D Biotechne) before being washed with PBS and lysed with RLT buffer from the RNeasy RNA isolation kit (Qiagen).
mRNA Analysis
Total RNA was isolated under RNase‐free conditions using RNeasy spin columns, as described by the company (Qiagen). Isolated RNA was treated with DNase (Qiagen) and stored at −80°C until further analysis. RNA concentrations and purity based on the 260/280 and the 260/230 ratios were assessed by spectrophotometer absorbance (NanoDrop ND‐1000, Thermo Fisher Scientific). Equal amounts of total RNA were used for synthesis of cDNA using the q‐Script cDNA Synthesis kit (Quanta Bioscience). Reverse transcriptase–quantitative polymerase chain reaction was performed using Perfecta SYBR Green Fastmix ROX (Quanta Bioscience) and the 7900HT Fast Real‐Time PCR System (Applied Biosystems) with the accompanying software SDS 2.4. Briefly, polymerase chain reaction conditions were 95°C for 3 minutes, followed by 40 cycles of 95°C for 15 seconds and 60°C for 60 seconds. Primers are listed in Table S2. Target transcript levels were quantified by the comparative Ct method using the nonregulated reference gene b‐actin as endogenous control.
Paired‐end RNA sequencing of total RNA from BMDMs from EndoV−/− and WT mice was performed on Novaseq 6000 system from Illumina. Raw files were trimmed with Fastp (version 0.20.0)17 in paired‐end mode to remove contaminated adapters and reads with Phred quality scores <30. Filtered reads were mapped to mouse cDNA (GRCm38.p6, Ensembl release 98)18 by Kallisto (version 0.46.0) with 200 bootstrap iterations.19 The output was then processed by Sleuth (version 0.30.0) to obtain the significant differentially expressed transcripts with the Wald test.20 The b value is analogous to the fold change, and it is on the natural‐log scale, taking into account the inferential variance estimated from bootstraps.
Quantification of Inosine
RNA was hydrolysed to ribonucleosides with 20 U benzonase (Santa Cruz Biotechnology) and 0.2 U nuclease P1 (Sigma‐Aldrich) in 10 mmol/L ammonium acetate pH 6.0 and 1 mmol/L MgCl2 at 40°C for 45 minutes and then ammonium bicarbonate to 50 mmol/L and 0.002 U phosphodiesterase I and 0.1 U alkaline phosphatase (Sigma‐Aldrich) were added and incubated further at 37°C for 45 minutes. The hydrolysates were added to 3 volumes of acetonitrile and centrifuged (16 000g, 30 minutes, 4°C). The supernatants were dried and dissolved in 50 μL of water for the liquid chromatography with tandem mass spectrometry analysis of modified and unmodified ribonucleosides. Chromatographic separation was performed using an Agilent 1290 Infinity II ultra high performance liquid chromatography system with a ZORBAX RRHD Eclipse Plus C18 150×2.1 mm ID (1.8 µm) column protected with a ZORBAX RRHD Eclipse Plus C18 5×2.1 mm ID (1.8 µm) guard column (Agilent). The mobile phase consisted of water and methanol (both added 0.1% formic acid) run at 0.23 mL/min, for modifications starting with 5% methanol for 30 seconds, followed by a 3‐minute gradient of 5% to 15% methanol, a 3‐minute gradient of 15% to 90% methanol, and a 4‐minute re‐equilibration with 5% methanol. A portion of each sample was diluted for the analysis of unmodified ribonucleosides, which was chromatographed isocratically with 20% methanol. Mass spectrometric detection of inosines was performed using an Agilent 6495 Triple Quadrupole system operating in positive electrospray ionization mode, monitoring the mass transitions with parameters as follows: molecular weight, 268.1 Da; Q1 molecular ion, 267.1 m/z, atomic mass unit (amu); Q3 product ion, 135.1 m/z, amu; and negative ionization polarity.
ENDOV Activity Assay
THP‐1 cells and Jurkat cells (American Type Culture Collection, LGC standards) were harvested by centrifugation at 500g for 5 minutes, and T‐rex cells overexpressing ENDOV (positive control21) were harvested by trypsinization. All cells were washed once in PBS and then homogenized in radio immuno precipitation assay buffer (50 mmol/L Tris HCl pH 7.5, 150 mmol/L NaCl, 0.1% SDS, 0.5% Na‐Deoxycholate, 0.5% Triton X100) and freshly added protease inhibitor cocktail (PIC 1:100, Sigma‐Aldrich), 1 mmol/L dithithreitol (DTT), 100 U/mL RNase OUT (Invitrogen), and 400 µmol/L vanadyl ribonucleoside complex (VRC) (BioNordika/New England Biolabs). Extracts were cleared by centrifugation at 21 000g for 20 minutes, and protein concentration was measured with the Pierce BCA Protein assay (Thermo Fisher Scientific). Endogenous ENDOV was immunoprecipitated from protein extracts (1.5 mg) by incubating with 2 µg ENDOV antibody (ab69400, Abcam) in 1 mL of buffer containing 100 mmol/L KCl, 5 mmol/L MgCl2, 10 mmol/L HEPES pH 7.0, 0.5% IGEPAL CA‐630 (Sigma‐Aldrich), 1 mmol/L DTT, 100 U/mL RNase OUT, 400 µmol/L VRC and PIC (1:100). The mixtures were incubated for 2 hours with slow rotation at 4°C. Protein A/G Plus agarose beads (Santa Cruz) were added, and incubation continued overnight. The beads were collected by centrifugation at 1000g for 5 minutes and washed 3 times with 1 mL NT2 buffer (50 mmol/L Tris HCl pH 7.4, 150 mmol/L NaCl, 1 mmol/L MgCl2, and 0.5% IGEPAL CA 630). After the last wash, the liquid was removed, and ENDOV activity was measured by adding 2 µL 32P‐end‐labeled single‐stranded RNA substrate (5‐ACUGGACA[rI][rI]U[rI]CUCCGAGG‐3), 4 µL reaction buffer (final 10 mmol/L Tris HCl pH 7.5, 0.5 mmol/L MnCl2, 50 mmol/L KCl, 1 mmol/L DTT, and 5% glycerol) and 8 µL water as described in Berges et al.10 Reactions proceeded at 37°C for 10 minutes and were stopped by adding 10 µL formamide loading dye (80% formamide, 10 mmol/L EDTA, 0.1% xylene cyanol, and 0.1% bromophenol blue). The samples were heated at 50°C for 5 minutes, and the reaction products separated on 20% polyacrylamide/urea gels at 200 V for 1 hour in 1× taurine buffer. The 32P‐labeled fragments were visualized by phosphorimaging (Typhoon 9410 Variable Mode Imager).
Cholesterol and Triglyceride Measurements
Plasma cholesterol and triglycerides were analyzed using a colorimetric method according to manufacturer's guidelines (Wako Chemicals GmbH).
Cytokine Measurements
Plasma interferon γ, interleukin (IL)‐1β, IL‐4, IL‐5, keratinocytes‐derived chemokine, tumor necrosis factor, and IL‐10 levels were analyzed by using U‐plex Biomarker Group1 (mouse) kit (Meso Scale Diagnostics) using a QuickPlex SQ120 instrument.
Statistical Analysis
Statistical analyses were performed using the Student t test. Correlation analysis was calculated using the Spearman rank correlation coefficient. Statistical analyses were conducted using Prism version 6.0 (GraphPad software). P<0.05 was considered statistically significant. Data are presented as mean±SD.
Results
Higher Levels of ENDOV, ADAR1, and Inosines in Human Carotid Plaque mRNA
ENDOV has affinity for and cleaves inosine‐containing RNA.8, 9 As RNA processing recently has been linked to the atherosclerotic process,12 we examined mRNA levels of ENDOV in nonatherosclerotic control arteries (n=13) and in carotid plaque from patients with atherosclerotic disease (n=163). Patients with carotid atherosclerosis had 2 times higher ENDOV mRNA levels compared with control arteries (Figure 1A). When dividing the patients according to the presence of preoperative symptoms of ischemic stroke or transient ischemic attack ipsilateral to the stenotic carotid artery ≤1 month (n=40), >1 and ≤2 months (n=25), >2 months and ≤6 month (n=24), and those with symptoms for >6 months or without any symptoms (n=74), we found no differences in the levels of ENDOV mRNA in relation to symptom duration (Table S3). Moreover, ENDOV mRNA levels were positively correlated with RNA expression of the pan‐leukocyte marker CD45, linking ENDOV to leukocyte infiltration within the lesion (n=183; Figure 1B).
Figure 1. ENDOV, ADAR1, and inosine levels are increased in mRNA from human atherosclerotic plaques.
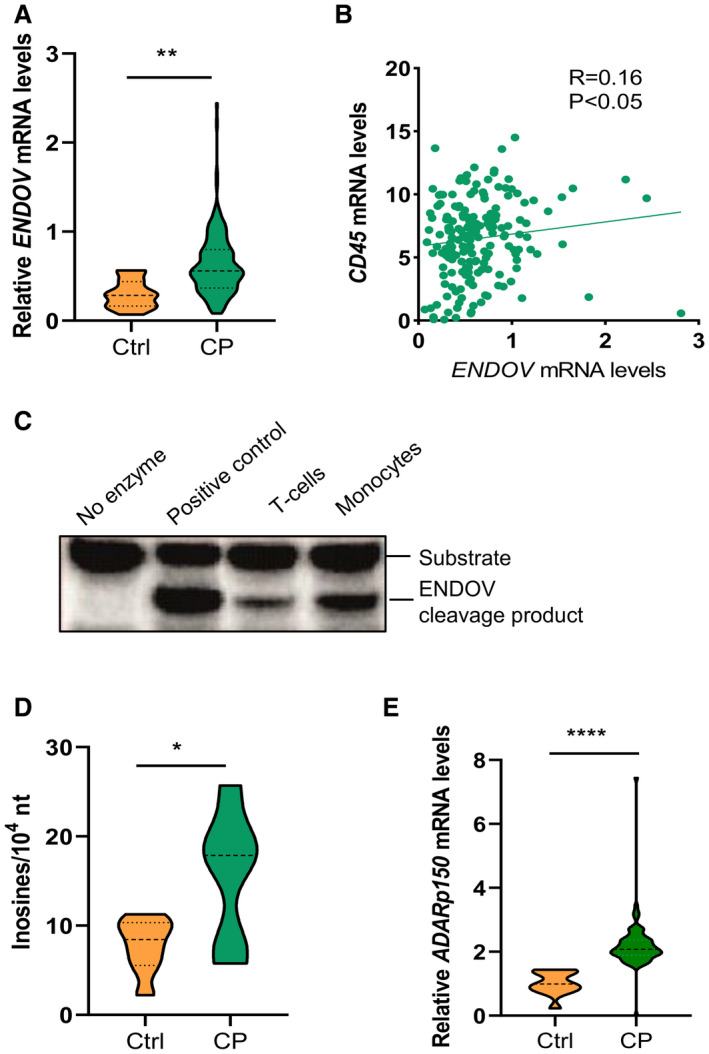
A, Reverse transcriptase–quantitative polymerase chain reaction analysis of ENDOV gene expression in human carotid plaques (n=163) compared with control arteries (n=13). B, Correlation of ENDOV gene expression to the gene expression of the pan leucocyte marker CD45. C, Enzyme activity assay of endogenous ENDOV immunoprecipitated from T cells (Jurkat) and monocytes (THP‐1), shows cleavage of adenosine‐to‐inosine modified RNA. D, Mass‐spectrometric detection of inosines in mRNA in the carotid plaque compared (n=6) to control arteries (n=8). E, Reverse transcriptase–quantitative polymerase chain reaction analysis of the main adenosine‐to‐inosine editing enzyme ADAR1 in carotid plaques (n=76) compared with control arteries (n=13). Data are presented as means, and statistical analyses were performed using the Student t test or Spearman correlation. ADAR1 indicates adenosine deaminase acting on RNA 1; CP, carotid plaque; Ctrl, controls; ENDOV, endonuclease V; and nt, nucleotides. *P<0.05, **P<0.005 and ****P<0.0001.
We next examined ENDOV enzyme activity in 2 human leukocyte cell lines, Jurkat (T cells) and THP‐1 (monocytes) to verify the presence of functional ENDOV protein in immune cell types relevant for atherogenesis. When endogenous ENDOV isolated from these 2 cell lines was incubated with inosine‐containing RNA, we detected a clear ENDOV cleavage product, showing enzyme activity of ENDOV in both T cells and monocytes (Figure 1C).
Finally, inosine levels in mRNA were significantly upregulated (≈2‐fold) in carotid atherosclerotic lesions (n=6) compared with nonatherosclerotic arteries (n=8; Figure 1D). In line with this, mRNA levels of ADAR1 p150, the interferon‐stimulated isoform and main adenosine‐to‐inosine editing enzyme in human cells6 were also increased (≈2‐fold) in carotid atherosclerotic samples (n=76) compared with nonatherosclerotic samples (n=13; Figure 1E).
EndoV Deficiency Attenuates Atherosclerosis in ApoE−/− Mice
To elucidate the potential role of ENDOV in the atherosclerotic process, we next examined the effect of EndoV abolishment in experimental atherosclerosis by crossing EndoV−/− and atherogenic ApoE−/− mice (ApoE−/− EndoV−/− ; Figure S1). The ApoE−/− EndoV−/− and ApoE−/− mice were fed an atherogenic diet for 10 weeks before termination. Quantification of the aortic root displayed significantly decreased plaque areas in ApoE−/− EndoV−/− mice compared with ApoE−/− mice (Figure 2A and 2B). In addition, the ApoE−/− EndoV−/− mice had a decreased lipid accumulation in the plaques compared with the ApoE−/− mice as measured by Oil‐Red‐O staining (Figure 2C). In contrast, there were no differences in body weight, cholesterol levels (Figure 2D), or circulating inflammatory markers (Table S4) between the 2 genotypes.
Figure 2. Reduced plaque area and lipid accumulation in the aortic root harvested from ApoE−/− EndoV−/− mice.
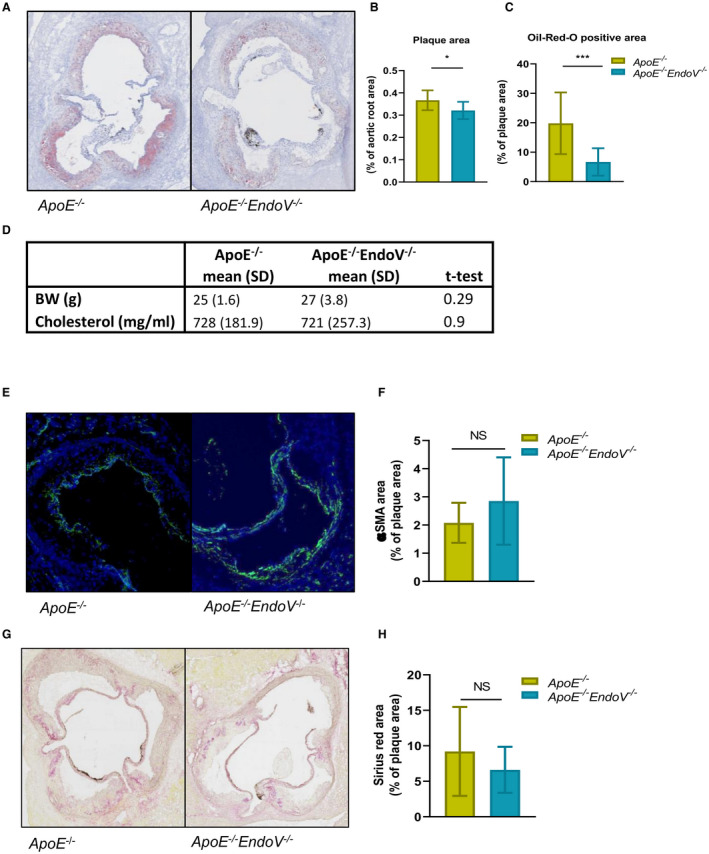
A, Lesion area and Oil‐Red‐O staining of the aortic root of ApoE−/− EndoV−/− and ApoE−/− mice after 10 weeks on an atherogenic diet. B, Calculated plaque area, presented as percentage of aortic root. C, Lipid accumulation measured as percentage Oil‐Red‐O–positive area of the total plaque area. D, BW and cholesterol levels of ApoE−/− EndoV−/− and ApoE−/− mice. E, Staining of smooth muscle cells in the lesion with α‐SMA antibody and (F) calculated α‐SMA–positive area presented as percentage of total plaque area. G, Collagen staining with sirius red of the lesion area and (H) calculated sirius red–positive area presented as percentage of total plaque area. Data are presented as mean (standard deviation). α‐SMA indicates α–smooth muscle actin; BW, body weight; EndoV, endonucleaseV; ApoE, apolipoprotein E; and NS, not significant. *P<0.05, ***P<0.0001.
VSMCs play a key role in the development and the stability of the atherosclerotic plaque,22 and notably, ADAR1 has been suggested to influence VSMC phenotype.11 Herein, however, there were no significant differences in VSMC content in the plaque area between the 2 genotypes (Figure 2E and 2F). In accordance with this, we found no difference in collagen level as measured by Sirius red staining (Figure 2G and 2H).
ENDOV Deficiency Attenuates Atherosclerosis in ApoE−/− Mice by Impairing CCL2‐Mediated Monocyte Infiltration Into the Lesion
Recruitment and activation of monocytes to the atherosclerotic plaque leading to foam‐cell formation within the lesion is a hallmark of atherogenesis.23 We therefore examined the lipid‐accumulating capacity of BMDMs of EndoV−/− and wild‐type mice. Using real‐time monitoring of pHrodo‐stained oxidized low‐density lipoprotein accumulation by macrophages, we found a time‐dependent and concentration‐dependent increase of lipid engulfment by the macrophages, but no difference between the two genotypes was observed (24‐hour point presented in Figure 3A and 3B). Similar findings were seen (no differences between the two genotypes) when examining the migration capacity of BMDM by real‐time monitoring of macrophages plated with cell culture media containing 10% FCS (Figure 3C and 3D [left panels]). However, when BMDMs from EndoV−/− mice were stimulated with CCL2, one of the main chemoattractants for recruiting monocytes to the atherosclerotic plaque,24 the cells had a drastic reduction in migration capacity compared with wild‐type BMDMs (Figure 3C and 3D [right panels]). Finally, to study the in vivo migration and recruitment capacity of EndoV−/− monocytes, we labeled circulating monocytes by injecting ApoE−/− EndoV−/− and ApoE−/− mice with fluorescent latex beads, shown to be engulfed by circulating monocytes. After 5 weeks of feeding an atherogenic diet, quantification of monocytes within the plaque in the aortic root revealed a significantly reduced number of infiltrated monocytes into the atherosclerotic plaque in ApoE−/− EndoV−/− mice compared with ApoE−/− mice (Figure 3E and 3F).
Figure 3. No change in oxLDL clearance, but reduced migration capacity of macrophages abolished of EndoV.
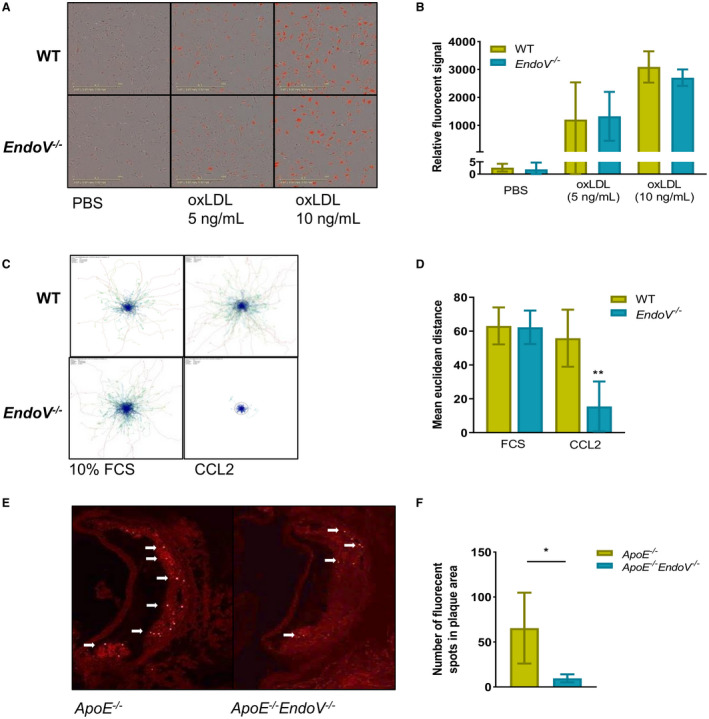
Engulfment capacity of macrophages derived from bone marrow of EndoV−/− mice measured with oxLDL stained with pHrodo (Invitrogen) and followed over time using the Incucyte system. A, Representative pictures of the 24‐hour time point and (B) calculated fluorescent signal at the 24‐hour time point. In vivo tracking of macrophages derived from bone marrow of EndoV−/− mice stimulated with 10% FCS or CCL2 (C) track plot and (D) graph of mean Euclidean distance. (E and F) Representative pictures of in vivo tracking of monocytes stained with fluorescent‐labeled beads to the aortic root of the ApoE−/− EndoV−/− mice compared with ApoE−/− mice and graphical presentations of the numbers of fluorescent signals in the plaque area. Data are presented as mean±SD. oxLDL indicates oxidized low‐density lipoprotein; EndoV, endonucleaseV; and WT, wild type. *P<0.05, **P<0.01.
Altered Response to CCL2 in BMDM From ENDOV‐Deficient Mice
To further examine how ENDOV affect the CCL2 signaling, we stimulated BMDMs from both EndoV−/− and wild‐type mice with recombinant CCL2 and analyzed the transcriptome using mRNA sequencing analysis (Figure 4A). In total, 567 transcripts were significantly and differently regulated (q<0.05) between the two genotypes after CCL2 stimuli; 337 was higher and 230 was lower in the CCL2‐stimulated EndoV−/− BMDMs compared with the CCL2‐stimulated wild‐type BMDMs (Figure 4B).
Figure 4. Altered CCL2 gene regulation in macrophages from EndoV−/− mice as compared with WT mice.
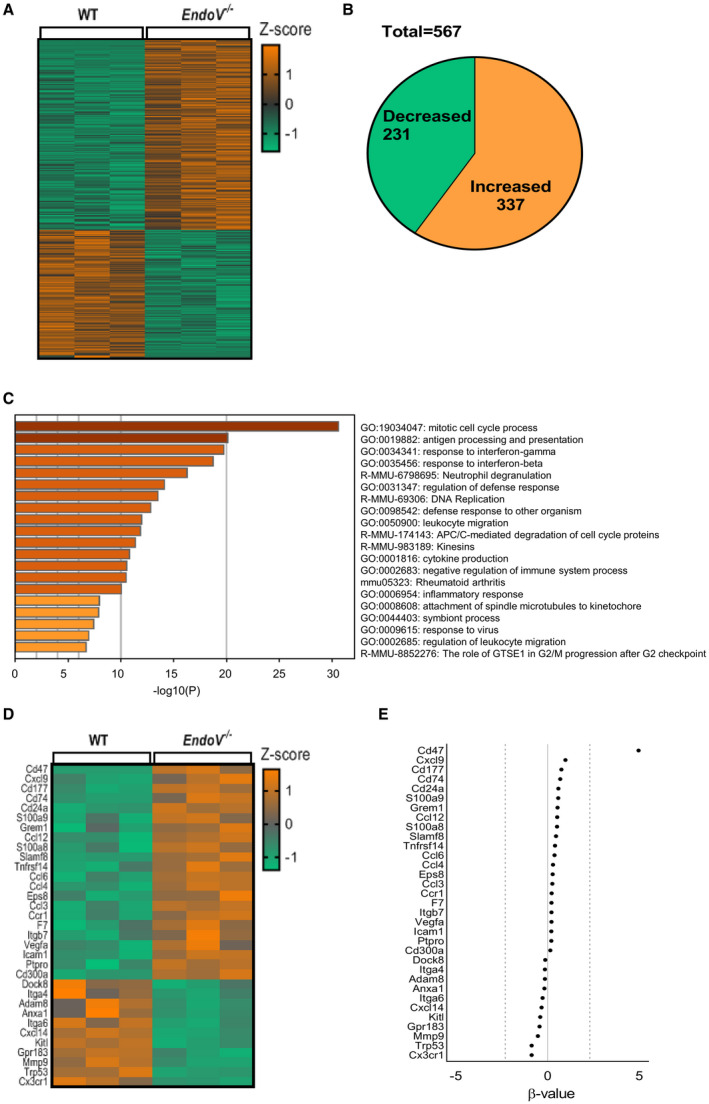
RNA sequencing analysis of BMDMs from EndoV −/− mice compared with BMDMs from WT mice stimulated with CCL2: (A) Heat map of differentially expressed transcripts with a q<0.05, (B) pie chart visualizing the alteration of gene expression in EndoV−/− macrophages compared with WT macrophages, and (C) pathway analysis. D, Heat map presentation of the genes annotated to migration in the pathway analysis. E, Natural‐log transformed b value as an estimator of the fold change of genes in D. BMDMs indicates bone marrow derived macrophages; WT, wild type; and EndoV, EndonucleaseV.
Gene ontology pathway analysis using Metascape25 revealed regulation of several pathways involved in atherosclerotic plaque development (Figure 4C). Of these, there were 2 migration‐associated gene ontology terms, GO:0050900, “leukocyte migration,” and GO:0002685, “regulation of leukocyte migration” (Figure 4C). These gene ontology terms identified 33 uniquely regulated genes, where 22 were upregulated and 11 downregulated (Figure 4D). As shown in Figure 4E, the most upregulated gene in BMDMs from EndoV−/− was CD47 with a β value of 4.9.
Dysregulated Interaction Between CD47 and ENDOV in Human Atherosclerosis?
CD47 is a “don't eat me signal” on the cell surface and is involved in a range of cellular processes, including apoptosis, proliferation, adhesion, and migration.26 EndoV is shown to cleave RNA at inosines, and the high expression of CD47 in the EndoV−/− mice could be attributed to a lack of transcript degradation in the absence of ENDOV. However, inspection of adenosine‐to‐inosine editing (observed as Adenosine to Guanine mutations) in the RNA sequencing data when compared with the mouse genomic sequence (GRCm38.p6, Ensembl release 98)18 in the CD47 transcript showed no adenosine‐to‐inosine editing in either wild‐type or EndoV−/− mRNA (data not shown).
As CD47 mRNA could be a putative target for ENDOV, we next measured the CD47 mRNA expression in human carotid plaques (n=75) and nonatherosclerotic arteries (n=13). Like ENDOV, CD47 was markedly upregulated in the carotid plaque compared with nonatherosclerotic arteries (Figure 5A). Moreover, although ENDOV mRNA levels were negatively correlated with mRNA levels of CD47 in PBMCs from healthy individuals (n=35; R=−0.35; P<0.05; Figure 5B), this correlation was not found in PBMCs from patients with carotid atherosclerosis (n=24; R=−0.09; P=0.66; Figure 5B), potentially indicating a systemic dysregulated interaction between CD47 and ENDOV during clinical atherosclerosis.
Figure 5. Gene expression of CD47 in human carotid atherosclerotic plaque and correlation between CD47 and ENDOV gene expression in human PBMCs.
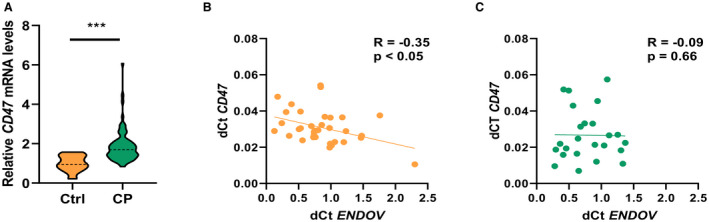
A, Reverse transcriptase–quantitative polymerase chain reaction analysis of CD47 gene expression in human carotid plaque compared with control arteries. B, Correlation analysis of ENDOV and CD47 gene expression in peripheral blood mononuclear cells isolated from healthy controls and (C) between ENDOV and CD47 gene expression in peripheral blood mononuclear cells isolated from patients with ischemic stroke. Data are presented as means, and statistical analyses were performed using the Student t test or Spearman correlation. CP indicates carotid plaque; Ctrl, controls; and ENDOV, endonuclease V. ***P<0.001.
ENDOV Deficiency Protects Against Hypoxia–Ischemic Stroke
The most important consequence of carotid atherosclerosis is an ischemic stroke. To examine the role of EndoV during stress‐induced ischemic stroke, we applied the widely used Levine method,15 modified for use in perinatal mice16 on the EndoV−/− and wild‐type mice. A combination of hypoxia and cerebral ischemia produces injury confined to the brain hemisphere ipsilateral to the occluded common carotid artery. The mice were left for 42 days after injury to allow for regeneration. When inspecting the brains, we found that the damaged brain structures were partially restored in both genotypes, but with reduced volume and extensive scarring in the ipsilateral hemisphere. Serial cross‐sectioning of the entire forebrain was conducted, and neuronal tissue loss was significantly decreased in several slides of the EndoV−/− brains compared with wild‐type brains (Figure 6A and 6B). In addition, tissue loss was significantly lower in the EndoV−/− animals when measuring total area loss as area under the curve (Figure 6C), suggesting that ENDOV also may impact acute stroke severity.
Figure 6. ENDOV deficiency reduces tissue loss after hypoxia‐ischemia.
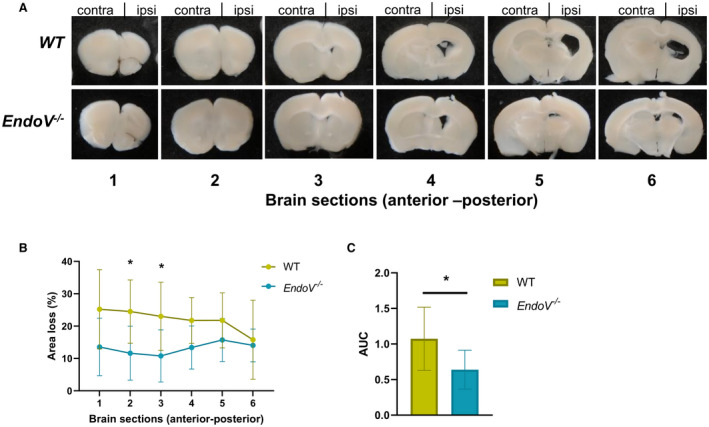
A, Pictures of representative brain sections from WT and EndoV−/− mice (n=6) 42 days after infarction. B, Infarction size was calculated as total volume loss of the ipsilateral hemispheres relative to the contralateral hemisphere in all sections extending from brain levels 1 to 6 (anterior‐posterior). C, Total area loss calculated as AUC. Data are presented as mean and SD. AUC indicates area under the curve; contra, contralateral; EndoV, endonuclease V; ipsi, ipsilateral; and WT, wild type. *P<0.01 (scale bars=1 mm).
Discussion
ENDOV homologs have been identified in all domains of life, including bacteria, archaea, and eukaryotes. In contrast to EndoV from bacteria acting on inosines in DNA, mammalian ENDOVs cleave at inosines in single‐stranded and double‐stranded RNA. Although the in vitro role of ENDOV has been studied, the in vivo role is still unclear. In this report, we show for the first time that ENDOV is expressed in human arteries, with enhanced expression in human carotid plaque as compared with a nonatherosclerotic vessel. Within the lesion, ENDOV was positively correlated with the leukocyte marker CD45. Furthermore, we showed that abolishing EndoV in an atherogenic mouse model reduced the plaque area and lipid content alongside with a decrease in recruitment of monocytes into the lesion. Moreover, the effects of EndoV deficiency in this mouse model seemed to involved in attenuated CCL2 signaling response. Finally, EndoV deficiency attenuated the severity of hypoxia–ischemic stroke.
To the best of our knowledge, this is the first report to study the role of ENDOV in human atherosclerosis. There are a few reports on the ENDOV substrate, inosine, and the main enzyme introducing inosine in RNA, ADAR1, in atherosclerotic disease. Stellos et al found increased inosine levels and ADAR1 expression in atherosclerotic lesions,12 which is confirmed by this current study. However, Fei et al showed that ADAR1‐deficient mice had an altered VSMC organization,11 an observation not seen in the EndoV−/− mice. In our study, there was no difference in the VSMC organization between ApoE −/− EndoV−/− and ApoE−/− mice, suggesting that the ADAR1/inosine effect on VSMC is independent of the function of ENDOV.
One of the main chemoattractants in the atherosclerotic lesion is CCL2.24 Herein we found that when macrophages from EndoV−/− mice were stimulated with CCL2, the cells had a marked reduction of random migration when compared with macrophages from wild‐type mice. In line with this, we found that ApoE−/− EndoV−/− mice had a reduced number of macrophages that had migrated to the plaque area when compared with the ApoE−/− mice. When stimulating BDMDs from EndoV−/− and wild‐type mice with CCL2, several genes were differently regulated between the genotypes, including genes that are engaged in cell migration and recruitment of monocytes. Mechanisms for how ENDOV impacts gene expression of the migration‐related genes is at present unclear. ENDOV has been shown to cleave RNA at inosines in vitro, suggesting that an upregulation of certain mRNA in EndoV−/− mice could be attributed to impaired cleaving and degradation in the absence of ENDOV. However, when examining CD47 mRNA, one of the genes that were markedly upregulated by CCL2 stimuli in EndoV−/− cells, we found no adenosine‐to‐inosine editing in the CD47 transcript. We have previously shown that there is no difference in the general inosine levels in different RNA subpopulations isolated from EndoV−/− mice compared with wild type, suggesting that ENDOV has other regulatory functions than cleaving at inosines in mRNA.14 In fact, we suggested that the function of ENDOV could rather be binding and protection of the RNA from an attack by other RNAses, leading to increased RNA levels.14 Our RNA sequencing data support this idea, as only 41% of the genes were lower in the EndoV−/− mice and 59% were higher. However, further and more direct studies are needed to understand details of the gene‐regulatory functions of ENDOV.
The most upregulated migration gene in the CCL2‐stimulated macrophages from EndoV−/− mice was CD47. This gene has recently gained much attention in regulating inflammation. Recently, Engelbertsen et al found that abolishing this protein led to increased atherosclerotic plaque burden in an atherosclerotic mouse model.27 It is possible that some of the beneficial effects of ENDOV deficiency on atherogenesis in our model could be related to the upregulation of CD47. We found that, whereas CD47 and ENDOV were inversely correlated in mononuclear cells from human healthy controls, this correlation was not seen in PBMCs from patients with carotid atherosclerosis. It is tempting to hypothesize that this ENDOV‐mediated regulation is abolished in atherosclerosis and could therefore represent an interesting target for the treatment of this disorder.
Our findings herein may also have relevance to stroke prevention and management. In fact, we present data from a preclinical model showing that EndoV deficiency decreases the stroke area after hypoxia–ischemia stroke, suggesting that this enzyme may be protecting against stroke severity. Our data also suggest that CD47 could be an interesting molecular target in the prevention of carotid atherosclerosis and stroke development. Finally, although much focus has been directed against epigenetic modifications, our findings may suggest that epitranscriptomic modifications could represent a new field in the prevention of carotid atherosclerosis and ischemic stroke.
The present study has some limitations. Correlations do not necessarily mean a causal relationship. As the cell composition is different in atherosclerotic lesions compared with nonatherosclerotic arteries, and as ENDOV is ubiquitously expressed, it is hard to assess if the regulation of ENDOV is attributed to an increase in the number of immune cells or represents a difference in gene expression between atherosclerotic carotid plaques and common iliac arteries. However ethical consideration makes it difficult to make more direct studies in human carotid arteries from healthy individuals. Furthermore, we also lack a direct pathway on how ENDOV influences gene expression, and further studies are needed. Moreover, the regulation of atherogenesis may be different between sexes, and the present study lacks a systemic comparison between female and male mice. Finally, our stroke model may not necessarily reflect the situation in carotid plaque–derived stroke, and ideally this model should have been used on ApoE −/− EndoV−/− and ApoE−/− mice.
In conclusion, ENDOV is upregulated in human atherosclerotic lesions, and our mice studies suggest that EndoV could promote atherogenesis by enhancing the monocyte recruitment into the atherosclerotic lesion, potentially by increasing the effect of CCL2 activation on these cells.
Sources of Funding
This work was supported by The South‐Eastern Norway Regional Health Authority (Nos. 201864 and 2018084) and the Norwegian Research Council (No. 144139).
Disclosures
None.
Supporting information
Tables S1–S4
Figure S1
Acknowledgments
The inosine quantification was performed at the Proteomics and Modomics Experimental Core Facility, Norwegian University of Science and Technology. The Proteomics and Modomics Experimental Core Facility is funded by the Faculty of Medicine and Health Sciences at the Norwegian University of Science and Technology and the Central Norway Regional Health Authority. Cell imaging of random migration was performed by the advanced light microscopy core facility at Oslo University Hospital, Norway.
(J Am Heart Assoc. 2021;10:e020656. DOI: 10.1161/JAHA.120.020656.)
Supplementary Material for this article is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.120.020656
For Sources of Funding and Disclosures, see page 13.
References
- 1.Tabas I, Glass CK. Anti‐inflammatory therapy in chronic disease: challenges and opportunities. Science. 2013;339:166–172. DOI: 10.1126/science.1230720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Glass CK, Witztum JL. Atherosclerosis. The road ahead. Cell. 2001;104:503–516. DOI: 10.1016/s0092-8674(01)00238-0. [DOI] [PubMed] [Google Scholar]
- 3.Eichelbaum K, Krijgsveld J. Rapid temporal dynamics of transcription, protein synthesis, and secretion during macrophage activation. Mol Cell Proteomics. 2014;13:792–810. DOI: 10.1074/mcp.M113.030916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schoggins JW, Rice CM. Interferon‐stimulated genes and their antiviral effector functions. Curr Opin Virol. 2011;1:519–525. DOI: 10.1016/j.coviro.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Samuel CE. Adenosine deaminases acting on RNA (ADARs) are both antiviral and proviral. Virology. 2011;411:180–193. DOI: 10.1016/j.virol.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.George CX, Samuel CE. Human RNA‐specific adenosine deaminase ADAR1 transcripts possess alternative exon 1 structures that initiate from different promoters, one constitutively active and the other interferon inducible. Proc Natl Acad Sci USA. 1999;96:4621–4626. DOI: 10.1073/pnas.96.8.4621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nakahama T, Kawahara Y. Adenosine‐to‐inosine RNA editing in the immune system: friend or foe? Cell Mol Life Sci. 2020;77:2931–2948. DOI: 10.1007/s00018-020-03466-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vik ES, Nawaz MS, Strøm Andersen P, Fladeby C, Bjørås M, Dalhus B, Alseth I. Endonuclease V cleaves at inosines in RNA. Nat Commun. 2013;4:2271. DOI: 10.1038/ncomms3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morita Y, Shibutani T, Nakanishi N, Nishikura K, Iwai S, Kuraoka I. Human endonuclease V is a ribonuclease specific for inosine‐containing RNA. Nat Commun. 2013;4:2273. DOI: 10.1038/ncomms3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berges N, Nawaz MS, Borresdatter Dahl T, Hagen L, Bjoras M, Laerdahl JK, Alseth I. Complex alternative splicing of human endonuclease V mRNA, but evidence for only a single protein isoform. PLoS One. 2019;14:e0225081. DOI: 10.1371/journal.pone.0225081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fei J, Cui XB, Wang JN, Dong K, Chen SY. ADAR1‐mediated rna editing, a novel mechanism controlling phenotypic modulation of vascular smooth muscle cells. Circ Res. 2016;119:463–469. DOI: 10.1161/CIRCRESAHA.116.309003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stellos K, Gatsiou A, Stamatelopoulos K, Perisic Matic L, John D, Lunella FF, Jaé N, Rossbach O, Amrhein C, Sigala F, et al. Adenosine‐to‐inosine RNA editing controls cathepsin S expression in atherosclerosis by enabling HuR‐mediated post‐transcriptional regulation. Nat Med. 2016;22:1140–1150. DOI: 10.1038/nm.4172. [DOI] [PubMed] [Google Scholar]
- 13.Kernan WN, Ovbiagele B, Black HR, Bravata DM, Chimowitz MI, Ezekowitz MD, Fang MC, Fisher M, Furie KL, Heck DV, et al. Guidelines for the prevention of stroke in patients with stroke and transient ischemic attack: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2014;45:2160–2236. DOI: 10.1161/STR.0000000000000024. [DOI] [PubMed] [Google Scholar]
- 14.Kong XY, Vik ES, Nawaz MS, Berges N, Dahl T, Vågbø C, Suganthan R, Segers F, Holm S, Quiles‐Jiménez A, et al. Deletion of endonuclease v suppresses chemically induced hepatocellular carcinoma. Nucleic Acids Res. 2020;48:4463–4479. DOI: 10.1093/nar/gkaa115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen S, Zhou Y, Chen Y, Gu J. fastp: an ultra‐fast all‐in‐one FASTQ preprocessor. Bioinformatics. 2018;34:i884–i890. DOI: 10.1093/bioinformatics/bty560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cunningham F, Achuthan P, Akanni W, Allen J, Amode M, Armean IM, Bennett R, Bhai J, Billis K, Boddu S, et al. Ensembl 2019. Nucleic Acids Res. 2019;47:D745–D751. DOI: 10.1093/nar/gky1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bray NL, Pimentel H, Melsted P, Pachter L. Near‐optimal probabilistic RNA‐seq quantification. Nat Biotechnol. 2016;34:525–527. DOI: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 18.Pimentel H, Bray NL, Puente S, Melsted P, Pachter L. Differential analysis of RNA‐seq incorporating quantification uncertainty. Nat Methods. 2017;14:687–690. DOI: 10.1038/nmeth.4324. [DOI] [PubMed] [Google Scholar]
- 19.Nawaz MS, Vik ES, Berges N, Fladeby C, Bjørås M, Dalhus B, Alseth I. Regulation of human endonuclease V activity and relocalization to cytoplasmic stress granules. J Biol Chem. 2016;291:21786–21801. DOI: 10.1074/jbc.M116.730911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Insull W Jr. The pathology of atherosclerosis: plaque development and plaque responses to medical treatment. Am J Med. 2009;122:S3–S14. DOI: 10.1016/j.amjmed.2008.10.013. [DOI] [PubMed] [Google Scholar]
- 21.Yuan Y, Li P, Ye J. Lipid homeostasis and the formation of macrophage‐derived foam cells in atherosclerosis. Protein Cell. 2012;3:173–181. DOI: 10.1007/s13238-012-2025-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin J, Kakkar V, Lu X. Impact of MCP‐1 in atherosclerosis. Curr Pharm Des. 2014;20:4580–4588. DOI: 10.2174/1381612820666140522115801. [DOI] [PubMed] [Google Scholar]
- 23.Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, Chanda SK. Metascape provides a biologist‐oriented resource for the analysis of systems‐level datasets. Nat Commun. 2019;10:1523. DOI: 10.1038/s41467-019-09234-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dou M, Chen Y, Hu J, Ma D, Xing Y. Recent advancements in CD47 signal transduction pathways involved in vascular diseases. Biomed Res Int. 2020;2020:4749135. DOI: 10.1155/2020/4749135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Levine S. Anoxic‐ischemic encephalopathy in rats. Am J Pathol. 1960;36:1–17. [PMC free article] [PubMed] [Google Scholar]
- 26.Sheldon RA, Sedik C, Ferriero DM. Strain‐related brain injury in neonatal mice subjected to hypoxia‐ischemia. Brain Res. 1998;810:114–122. DOI: 10.1016/S0006-8993(98)00892-0. [DOI] [PubMed] [Google Scholar]
- 27.Engelbertsen D, Autio A, Verwilligen RAF, Depuydt MAC, Newton G, Rattik S, Levinsohn E, Saggu G, Jarolim P, Wang H, et al. Increased lymphocyte activation and atherosclerosis in CD47‐deficient mice. Sci Rep. 2019;9:10608. DOI: 10.1038/s41598-019-46942-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S4
Figure S1