

Summary
Objective:
Fibroblast growth factor homologous factors (FHFs) are brain and cardiac sodium channel binding proteins that modulate channel density and inactivation gating. A recurrent de novo gain-of-function missense mutation in the FHF1(FGF12) gene (p.Arg52His) is associated with early infantile epileptic encephalopathy (EIEE47, OMIM 617166). To determine whether the FHF1 missense mutation is sufficient to cause EIEE and to establish an animal model for EIEE47, we sought to engineer this mutation into mice.
Methods:
The Arg52His mutation was introduced into fertilized eggs by CRISPR editing to generate Fhf1R52H/+ mice. Spontaneous epileptiform events in Fhf1R52H/+ mice were assessed by cortical electroencephalography (EEG) and video monitoring. Basal heart rhythm and seizure-induced arrhythmia in were recorded by electrocardiogram (ECG). Modulation of cardiac sodium channel inactivation by FHF1BR52H protein was assayed by voltage clamp recordings of FHF-deficient mouse cardiomyocytes infected with adenoviruses expressing wild-type FHF1B or FHF1BR52H protein.
Results:
All Fhf1R52H/+ mice experienced seizure or seizure-like episodes with lethal ending between 12-26 days of age. EEG recordings in 19-20 day-old mice confirmed SUDEP as severe tonic seizures immediately preceding loss of brain activity and death. Within 2-53 seconds after lethal seizure onset, heart rate abruptly declined from 572 +/− 16 bpm to 108 +/− 15 bpm, suggesting a parasympathetic surge accompanying seizures that may have contributed to SUDEP. Although ectopic overexpression of FHF1BR52H in cardiomyocytes induced a 15 mV depolarizing shift in voltage of steady-state sodium channel inactivation and slowed the rate of channel inactivation, heart rhythm was normal in Fhf1R52H/+ mice prior to seizure.
Significance:
The EIEE47 FHF1 missense mutation p.Arg52His induces epileptic encephalopathy with full penetrance in mice. Both FHF1 (p.Arg52His) and SCN8A (p.Asn1768Asp) missense mutations enhance sodium channel Nav1.6 currents and induce SUDEP with bradycardia in mice, suggesting an FHF1/Nav1.6 functional axis underlying altered brain sodium channel gating in epileptic encephalopathy.
Keywords: EIEE47, FHF1(FGF12), CRISPR, SUDEP, bradycardia
1. INTRODUCTION
Sudden unexpected death in epilepsy (SUDEP) accounts for up to 40% of all fatalities among individuals with therapeutically refractory epilepsy 1, 2. Risk for SUDEP is enhanced by early developmental onset of generalized seizures, in its severest form termed early infantile epileptic encephalopathy (EIEE). Causes underlying SUDEP are not easily revealed by post-mortem examination, thereby restricting determination of mechanism to individuals undergoing polymodal physiological monitoring at time of death and to studies employing animal models. SUDEP may result from seizure-induced respiratory or cardiac arrest due to ictal or postictal firing anomalies in forebrain or brainstem regions controlling respiration and heart rhythm 3-8. Alternatively, SUDEP may occur, in principle, independent of seizures due to genetic and developmental factors that may affect in parallel both seizure propensity and respiratory or cardiac dysfunction.
Causes of EIEE and SUDEP include mutations in genes encoding pore-forming subunits of voltage-gated sodium channels (Nav1.x). Hemizygous loss-of-function mutations in SCN1A encoding Nav1.1 (EIEE6) are the most common cause of human Dravet syndrome, a form of EIEE associated with myoclonic seizures occurring spontaneously or triggered by fever 9,10. SUDEP in an Scn1a+/− mouse model of Dravet syndrome appears to be due to seizure-induced cardiac arrest triggered by parasympathetic cardiac suppression 3,4,8, and breathing disorders in Scn1a-deficient mice have also been noted 11. An Scn1a conditional loss-of-function mouse has been used to show that Scn1a deficiency in inhibitory neurons leads to epilepsy by increasing excitatory drive of neural networks 12.
In contrast, de novo heterozygous gain-of-function missense mutations of the SCN8A gene encoding Nav1.6, (EIEE13) have been identified in over 100 children with refractory infantile-onset generalized seizures 13-15, and some EIEE13 individuals succumb to SUDEP 13. The mutant isoforms of Nav1.6 protein have altered gating properties that are expected to increase sodium currents under physiological conditions and thereby enhance neuronal excitability 16. Some of these mutations to Nav1.6 channels, including Nav1.6(N1768D), result in slower inactivation, a depolarizing shift in voltage dependence of steady-state inactivation and elevated persistent sodium current 13, 17, while another EIEE13-associated mutation causes a hyperpolarizing shift in voltage-dependent activation 18. In the EIEE13 mouse models Scn8aN1768D/+ and Scn8aN1768D/N1768D, animals undergo SUDEP brought on by tonic seizures 19 and have bradycardia due to enhanced tonic parasympathetic drive that worsens in the days preceding SUDEP 20.
EIEE is also associated with mutations in genes encoding sodium channel-binding proteins. EIEE52, characterized by febrile and myoclonic seizures, is associated with homozygous deletion of the SCN1B gene encoding the beta-1 subunit of sodium channels 21. EIEE47 involves mainly tonic and tonic-clonic seizures and is associated with a recurrent dominant missense mutation in the FHF1(FGF12) gene 22-26. FHF1 (Fibroblast growth factor Homologous Factor 1) encodes cytosolic Nav-binding proteins that are broadly expressed in neurons throughout the central nervous system, including the cerebral cortex, hippocampus, and brainstem 27-33. FHF1A and FHF1B protein isoforms have different N-terminal moieties extending from a common β-trefoil core that includes the Nav binding interface 28, 34-37. The EIEE47-associated arginine-to-histidine substitution within the FHF1 Nav-binding domain (FHF1AR114H, FHF1BR52H) is gain-of-function with respect to Nav1.6 modulation, such that the mutant isoforms induce greater depolarizing shifts in the voltage of steady-state inactivation than the wild-type protein counterparts 22. Two children carrying the FHF1 EIEE47 mutation experienced sudden death, while others have survived to date despite recurrent seizures. These findings show that epilepsy-inducing mutations in different genes, SCN8AN1768D and FHF1R52H(R114H), have similar effects on delaying Nav1.6 inactivation.
Here we report the generation and phenotypic characterization of mice heterozygous for the EIEE47 FHF1 missense mutation (abbreviated hereafter as Fhf1R52H). All Fhf1R52H/+ mice experience spontaneous seizures (epilepsy) and die of SUDEP between 12-26 days of age immediately following a terminal seizure episode. The terminal seizure onset is accompanied by sudden bradycardia, likely as the result of an autonomic surge. The comparison of our findings to prior studies on EIEE13 Scn8aN1768D/+ mice 16,20 shows that dominant gain-of-function substitutions in Nav1.6 or its binding partner FHF1 induce similar progression for epileptic encephalopathy, bradycardia, and SUDEP.
2. MATERIALS AND METHODS
2.1. DNA oligonucleotides
F1-R52H-124b (for CRISPR-guided introduction of R52H and silent mutations): 5’-TTTTCTTTTCTTTTTTCCTTCTAGCCCTCTTCAATCTAATTCCTGTAGGACTGCACGTCGTCGCGATTCAGGGGGTTAAAGCCAGCCTTTATGTGGCCATGAATGGAGAAGGCTATCTCTACAG-3’
F1-R52H-For (for PCR): 5’-GTAGGACTGCACGTCGTCGCGATT-3’
F1-IVS3-Rev (for PCR): 5’-GACTCACTCCATAGGGCATGA-3’
F1-IVS2-For (for PCR): 5’-GCAGTGTGCCTCTCTTTGTGGTT-3’
F1-Ex3-AS2 (for sequencing): 5’-CACACGACTGAACTTACTGAG-3’
F1-R52H-S (mutagenesis): 5’-GAACCCCAGCTCAAAGGGTAATAGACAAGGTTATTCAGCCAGC-3’
F1-R52H-AS (mutagenesis): 5’- GCTGGCTGAATAACCTTGTCTATTACCCTTTGAGCTGGGGTTC-3’
F1-Delta-S (mutagenesis): 5’-CTAATTCCCGTGGGCCTGCATGTAGTGGCCATCCAAGG-3’
F1-Delta-AS (mutagenesis): 5’-CCTTGGATGGCCACTACATGCAGGCCCACGGGAATTAG-3’
2.2. CRISPR derivation of Fhf1R52H/+ mice
All mice in these studies were generated, bred, and experimentally utilized under protocols approved by Institutional Animal Care and Use Committees at Icahn School of Medicine at Mount Sinai, New York Medical College, New York University Langone Medical Center, and Hunter College of City University New York.
The Fhf1R52H mutation was generated in mice by CRISPR editing via reagent pronuclear microinjection into F2 generation C57Bl/6 x DBA/2 fertilized eggs. Two guide RNAs (MML001056257 and MMR0001056262) and Cas9D10A nickase messenger RNA (Sigma Aldrich) directed single-strand nicks at PAM sites on opposite strands within Fhf1 exon 3 flanking the Arg52 codon (Figure 1A), and the mutagenic DNA oligonucleotide F1-R52H-124b mediated the introduction of the R52H missense mutation together with six silent mutations into the Fhf1 gene by homologous DNA strand invasion and repair (Figure 1A), as originally described 38,39. Reimplantation of injected eggs into foster females yielded ninety-eight progeny that were screened by polymerase chain reaction (PCR) and DNA sequencing for successful mutagenesis.
Figure 1. Generation and survival of Fhf1R52H/+ mice.

A) Strategy for embryonic editing of the Fhf1 gene. Reagents for gene editing were microinjected into pronuclei of fertilized eggs that were reimplanted into foster mothers to generate candidate founder mice. Two CRISPR guide RNAs included sequences (shaded gray) near the Arg52 codon (shaded green) directing single strand nicking by Cas9D10A at the adjacent PAM sites (shaded blue) on opposite DNA strands. Introduction of the R52H missense mutation (shaded pink) and six linked silent mutations (shaded yellow) were directed by mutagenic DNA oligonucleotide F1-R52H-124b. The sites corresponding to PCR and sequencing oligonucleotides are indicated with arrows above the gene schematic. B) PCR screening for pups bearing an Fhf1R52H allele. Genomic DNA was amplified with exon 3-flanking oligos F1-IVS2-For and F1-IVS3-Rev, and 500 bp products were used as template for PCR with F1-IVS3-Rev and F1-R52H-For, which only anneals without mismatch to the mutant allele sequence. Pups #5 and #86 yielded mutant diagnostic 270 bp products. C) DNA sequencing of Fhf1 alleles in first- and second-generation mice. The 500 bp amplicons spanning exon 3 from potential founder mice and progeny of mosaic founder #5 were sequenced with primer F1-ex3-AS. Fhf1R52H/+ templates #86 and #5-22 showed heterozygosity at the seven edited residue sites, while founder #5 is mosaic Fhf1R52H/+: Fhf1+/+, showing very weak representation of the mutant residues in chromatograms. The complementary sequence to the R52 and H52 codons are shaded green. Fhf1R52H/+ heterozygotes from #5 and #86 founder lineages died as juveniles. D) Survival of Fhf1R52H/+ versus wild-type mice. All 91 Fhf1R52H/+ mice, including 44 males and 47 females, died between postnatal days 12 and 26, while all Fhf1+/+ mice (n = 200) were viable. E) Phenytoin delays death of Fhf1R52H/+ mice. Residual survival time plot of Fhf1R52H/+ mice administered phenytoin daily commencing postnatal day 12 (n = 23) versus untreated Fhf1R52H/+ mice (n = 68). Phenytoin marginally, but significantly, extended survival time (treated 18.7±0.4 days vs. untreated 16.1±0.3 days, P<0.00005).
2.3. Genotyping and estimation of Fhf1R52H/+: Fhf1+/+ mosaicism in founder mouse #5.
All thermal cycling employed Phire Hot Start DNA Polymerase and buffers (Thermo Fisher). Tail DNAs served as templates for unbiased amplification of a 500 base pair (bp) fragment spanning Fhf1 exon 3 using PCR primers F1-IVS2-For and F1-IVS3-Rev (Figure 1A). Purified 500 bp amplicons were tested for the presence of the mutant allele by reamplification with nested mutant primer F1-R52H-For, which yielded a positive diagnostic 270 bp product for two of the ninety-eight progeny (Figure 1A,B). The presence of the mutant allele in these pups and in the progeny of mosaic founder #5 was confirmed by sequencing of the 500 bp amplicon with primer F1-Ex3-AS2 (Figure 1A). Male founder #5 was mated with 129/SvPas females to derive Fhf1R52H/+ and wild-type experimental animals. At age 16 months, mosaic founder #5 was sacrificed and sperm was cryopreserved for further propagation of the mutant allele by in vitro fertilization and reimplantation into foster mothers. The percentage of Fhf1R52H/+ germ cell precursors in mouse #5 was calculated as twice the percentage of Fhf1R52H/+ offspring. Mosaicism in somatic tissues was approximated by comparing sequence chromatograms of 500 bp amplicons derived from these tissues to those of Fhf1R52H/+ and wild-type offspring. The ratio of peak amplitudes for adjacent identical residues in wild-type DNA versus that for these residues in Fhf1R52H/+ DNA, where one of these peaks was reduced by an introduced silent mutation, provided a consistent metric against which the ratio of these peak amplitudes in a #5 mosaic tissue could be compared to assess level of mosaicism. The percentage of mosaicism was taken from the analysis of three nucleotide doublets affected by silent mutations.
2.4. Long-term video monitoring of nursing pups
Litters containing mother with Fhf1R52H/+ and wild-type pups were placed in the circular recording cage with regular nesting material, food ad libitum and tap water source. One or two infrared cameras were positioned above and/or on the side of the cage for continuous recordings under standard twelve-hour light/twelve-hour dark cycling (lights on at 7:00 am). Mice were recorded continuously until all mutants had died. Some pups were administered the anti-seizure drug phenytoin (50 mg/kg intraperitoneal daily) between postnatal days 11 -18 in an attempt to delay lethality.
2.5. Surgeries for electroencephalogram (EEG) and simultaneous EEG/electrocardiogram (ECG)
All surgeries were performed at postnatal day 19. Epidural silver ball EEG electrodes attached to a cranial tower (Figure 2A) were implanted as previously described 40. Briefly, each pup was subjected to deep isoflurane anesthesia continuously monitored as lack of response to tail and paw pinch (induction 5% in oxygen; maintenance 3% in oxygen), and placed in a stereotaxic apparatus (Heinrich Kopf) with anesthesia mask attached. After cleaning the skull, four small openings were made bilaterally over the frontal and occipital cortices using a 19-gauge needle, and silver ball electrodes were placed into these openings onto the cortex and attached to a 6-pin dual-in-line connector with 0.125 mm silver wire. Jeweler screws with silver wire attached were secured in the nasal bone and above the cerebellum to provide reference (nasal) and ground (cerebellum) electrodes. These screws also served as anchors for the dental acrylic cap. After recovery, the mouse was placed in a recording chamber with food and water supply, attached to the video and EEG recording systems, and recorded continuously under infrared (IR) illumination. The procedure for implantation of both EEG and ECG electrodes was similar, only differing in the use of an 8-pin dual in-line connector allowing for two pins dedicated to ECG electrodes. These ECG electrodes (either insulated stranded stainless steel wire or Teflon-insulated silver wire) were inserted under the skin on the right pectoral muscle and on the left anterior chest surface at the fourth intercostal space, and secured with a single silk stitch as described elsewhere 41. The ECG signal was recorded between the uninsulated tips of these leads.
Figure 2. EEG recordings during non-lethal seizures, terminal seizures, and interictal periods in Fhf1R52H/+ mice.
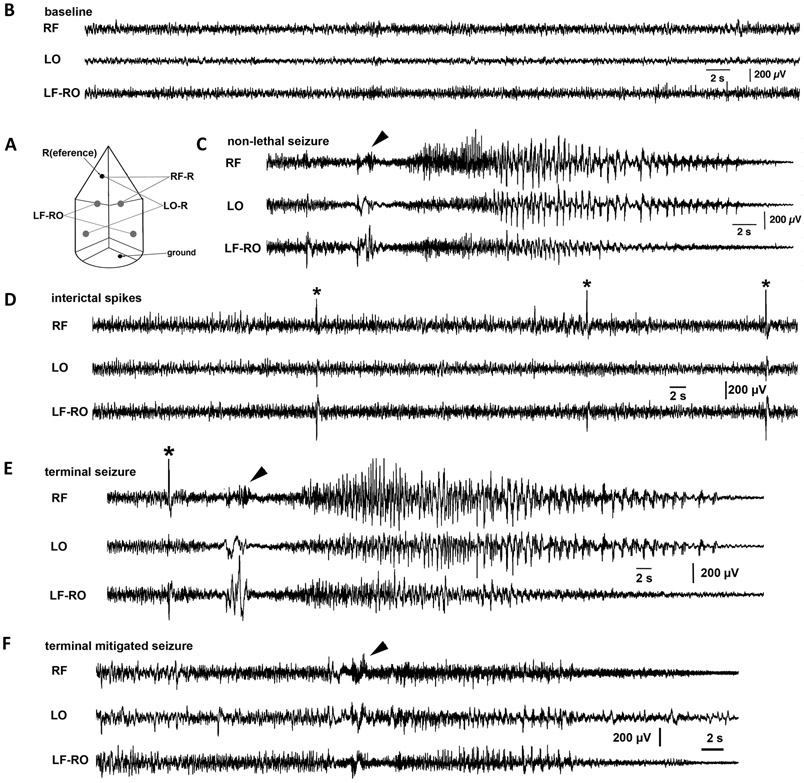
A) Scheme for all three-channel EEG recordings. RF (right frontal, sensorimotor) and LO (left occipital, visual) were unipolar mounts versus a reference electrode (R; an anchoring jeweler’s screw) in the nasal bone. The third channel is LF-RO (left frontal vs. right occipital, a bipolar mount). Common ground was placed in the skull above the cerebellum (anchoring jeweler’s screw). B) Baseline recording during awake period. Low amplitude fast activity beta waves were abundant in all channels. C) A non-lethal seizure. This mutant mouse had four seizures between P19-P21. One of the non-lethal seizures initiated coincident with a body twitch (arrowhead) followed by a tonic-clonic seizure with typical EEG signature consisting of fast spikes followed by spike-and-wave activity with slowing and decrease in amplitude and a period of post-ictal suppression (not shown). D) Interictal spikes. The same animal as in (C) had frequent interictal spikes (asterisks) without behavioral correlates. E) Terminal seizure. In the same animal as in (C) and (D), a final interictal spike (asterisk) preceded a body twitch (arrowhead) and tonic-clonic seizure with fast spike and spike-and-wave phases indistinguishable from a preceding non-lethal seizure (C). F) Terminal seizure with muted EEG signature. This mutant experienced near-identical behavioral signature preceding death as in (E) commencing with body twitch (arrowhead) and a similar late spike-and-wave phase, but fast spikes were not readily detected.
2.6. Simultaneous EEG, ECG, and video recordings
We used tethered Pinnacle Technology (Lawrence, KS) system with Sirenia 1.7.10-2.0.8 software. The EEG recording arrangement is shown in Fig 2A. Signals from the right frontal (RF) and left occipital (LO) electrodes were recorded against the reference electrode (R). Signal from left frontal electrode was recorded in bipolar arrangement against the signal from the right occipital electrode (LF-RO). For EEG-only recordings, signals were sampled at 800 Hz and subjected to 100 Hz low-pass and 1 Hz high-pass filtering. For simultaneous EEG/ECG recordings, signals were sampled at 5 kHz, with the same filtering as above for EEG traces and with 2 kHz low-pass and 1 Hz high-pass filtering for ECG traces. Mice were individually placed into circular recording cages with overhead cameras with IR illumination for continuous recordings.
2.7. EEG and ECG evaluation
For off-line data evaluation, we first scanned video-recordings to identify the terminal seizure event. Then, EEGs were visually inspected between the recording onset and the terminal point determined by the video-recording. Clear EEG ictal activity was replayed with the video and correlated to behaviors. ECG data collected together with EEG were evaluated off-line. Baseline heart rate in beats per minute (bpm) was sampled and averaged over a six-hour period subsequent to recovery from surgery anesthesia-induced bradycardia. P-R interval was measured as the time between P and R peaks and averaged over ten beats. Instantaneous heart rate in bpm at time t(n) seconds for beat n is calculated as bpm = 60 ÷ {t(n) - t(n-1)}. Surgical anesthesia suppressed heart rate in some mice for between 1-3 hours following awakening and initiation of recording. Therefore, EEG and ECG data were not analyzed until heart rhythm had stabilized.
2.8. FHF1BR52H modulation of Nav gating in cardiomyocytes
The human FHF1B coding sequence was cloned into the bicistronic adenovirus shuttle vector pAdTrack-CMV 42 to create pAdTrack-FHF1B/GFP. Quickchange mutagenesis was used with mutagenic primers F1-R52H-S and F1-R52H-AS to generate pAdTrack-FHF1BR52H/GFP or with primers F1-Delta-S and F1-Delta-AS to generate pAdTrack-FHF1BDELTA/GFP, the latter of which has nonsense stop codons Ile11stop/Val12stop, thereby serving as negative control. In vivo recombination of shuttle vectors with pAdEasy-1 was performed in E. coli, and resultant full-length adenoviral plasmids were linearized with PacI by standard procedure 42 and transfected via Lipofectamine 2000 (Life Sciences) into HEK293 cells to produce virus particles, which were purified and concentrated using Adeno-X Maxi kits (Takara Bio USA). FHF-negative dissociated ventricular cardiomyocytes from Fhf2−/Y mice were prepared as described previously 43, plated onto poly-L-lysine/laminin-coated coverslips as described elsewhere 44, and infected with the adenoviruses dually expressing FHF1B and GFP. Recordings were conducted 2-3 days post-infection from a Nikon electrophysiology workstation using MultiClamp 700 amplifier, Digidata analog/digital converter, and pClamp10 command and data acquisition software (Molecular Devices Inc). Patch pipettes with resistance of 1-2 MΩ were filled with 135 mM CsF – 5 mM NaCl – 10 mM EGTA – 5 mM Mg-ATP – 15 mM HEPES pH 7.2 and used to attain voltage-clamped whole-cell patch access into GFP-fluorescent cardiomyocytes bathed in carbogen-bubbled 24 mM NaCl – 26 mM NaHCO3 – 50 mM choline-Cl – 25 mM tetraethylamine-Cl – 3 mM KCl – 3 mM CsCl – 12.5 mM 4-aminopyridine – 2 mM MgCl2 – 1 mM CaCl2 – 1 mM BaCl2 – 0.2 mM CdCl2 – 10 mM HEPES pH7.2 – 11 mM D-glucose – 2 mM Na-pyruvate – 3 mM myoinositol. Cell capacitive and leak currents were subtracted through amplifier compensation and further during data acquisition by the pClamp P/N method. Voltage-dependence of Nav steady-state inactivation was assayed using 50% series resistance compensation through a series of n=21 consecutive command sweeps each consisting of 60 msec at −150 mV, steady-state conditioning for 100 msec at −150 + 5 x (n-1) mV, and reporting for 20 msec at −30 mV. Peak inward sodium current for each sweep was generated at the reporting voltage by channels not inactivated by the conditioning voltage step. The V1/2 voltage at which 50% of sodium channels underwent steady-state inactivation was determined by fitting data to a peak current vs. voltage Boltzmann distribution. Rate of Nav open-state inactivation was assayed from sodium current decay during voltage command steps from −150 mV to test voltages ranging from −35 mV to −10 mV. Current decays at each test voltage were fitted to a single-term exponential decay to determine time constant τ.
Cells infected with each of the three adenoviruses were lysed 48 hrs post-infection, and denatured proteins were electrophoresed through 4-20% polyacrylamide gels (NuSep), transferred to PVDF membranes, probed with antibodies specific for GFP (Abcam) or to FHF1 (Proteintech) proteins, and visualized by enhanced chemiluminscence.
2.10. Statistics
Statistical significance of differences for ECG parameters in wild-type versus Fhf1R52H/+ mice was analyzed by two-tailed Student’s t-test. Similarly, differences in cardiomyocyte Nav V1/2 steady-state inactivation and open-state inactivation rate in cardiomyocytes infected with FHF1B vs. FHF1BR52H adenovirus were analyzed by the t-test.
To reject the hypothesis that an off-site CRISPR-induced mutation unlinked to the Fhf1R52H mutation was responsible upon inheritance for juvenile lethality, we employed Poisson distribution to calculate the probability P = e−0.5n that none of n Fhf1R52H/+ mice are viable.
3. RESULTS
3.1. Generation of Fhf1R52H/+ Mice
In order to confirm that the de novo missense mutation of the FHF1 gene detected in multiple cases of human EIEE47 is sufficient to cause early-onset epilepsy, the Fhf1 mutation was recapitulated in mice by CRISPR editing 38,39. Fertilized eggs received a mixture of Cas9D10A nickase mRNA, two guide RNAs targeting complementary DNA strands within Fhf1 exon 3, and a mutagenic single-strand DNA oligonucleotide specifying the desired arginine → histidine substitution (FHF1AR114H, FHF1BR52H) along with six wobble silent mutations in adjacent codons to facilitate PCR genotyping (Figure 1A). Reimplantation of injected eggs into pseudopregnant females led to the birth of ninety-eight pups, and PCR screening with a primer specific for the mutant Fhf1 allele generated a product of predicted size in DNA from two founder mice, #5 and #86 (Figure 1B). The presence of the mutant allele in the founders was confirmed by unbiased PCR amplification spanning the targeting site in exon 3 followed by DNA sequencing. Fhf1 exon 3 amplified from founder #86 had a sequence read consistent with heterozygosity of wild-type and R52H mutant alleles, with two overlapping peaks at the seven targeted residues (Figure 1C). In contrast, the sequence of Fhf1 exon 3 amplified from founder #5 suggested mosaicism of heterozygous and wild-type homozygous cells, with the seven targeted residues showing very weak representation of the mutant allele in tail DNA (Figure 1C). After later sacrifice of this animal, heart, brain, and skeletal muscle DNA also showed weak representation of the mutant allele. The sequence chromatograms allowed estimation of 12-18% Fhf1R52H/+ cells in somatic tissues in mosaic founder #5 (see Methods for explanation).
The Fhf1R52H/+ founder #86 died suddenly and unexpectedly on postnatal day 17 in an unrecorded event suggesting SUDEP, making propagation of the mutant allele dependent upon breeding from the putative mosaic male founder #5. Breeding of founder #5 indeed gave rise to the heterozygous Fhf1R52H/+ genotype in 91 out of 398 offspring (22.8%) as confirmed by DNA sequencing of amplified Fhf1 exon 3 (examples in Figure 1C). Hence, approximately 45% of germ cell precursors are Fhf1R52H/+, constituting a much higher proportion of mutant cells compared with somatic tissues.
All 91 Fhf1R52H/+ progeny died as juveniles between 12 to 26 days of age (average age of death 16.7±0.3 days; Figure 1D). The 100% lethality of mutant mice offers proof (P < 10−10, see Methods) that lethality is not due to an off-site CRISPR-induced mutation in mosaic founder #5. Except for seizure or seizure-like episodes (see below), mutant mice had normal body weight and showed no overt signs of morbidity. By contrast, no wild-type littermates died as juveniles (Figure 1D). Daily administration of the anti-seizure drug phenytoin (daily 50 mg/kg) to Fhf1R52H/+ mice minimally, but significantly, prolonged survival (treated 18.7±0.4 days n=23 vs. untreated 16.1±0.3 days n = 68, P<0.00005)(Figure 1E).
3.2. EIEE and SUDEP in Fhf1R52H/+ Mice.
Litters containing 12 to 19-day old Fhf1R52H/+ pups were continuously monitored by infrared video recording under standard 12-hour light/12-hour dark cycling. Twenty-five out of 40 video-monitored Fhf1R52H/+ pups that died prior to weaning underwent seizure-like episodes immediately preceding death. As shown in Supplemental Movie S1, mutant pups engaged in a brief running fit followed by a tonic phase from which they did not recover. The other mutant pups died while nesting under their mothers, obscuring their exact time and manner of death.
EEG recording in conjunction with video monitoring was performed in Fhf1R52H/+ mice surviving through P19 (n=10) 40. In some cases (n=6), ECG was also recorded. EEG and video monitoring of Fhf1R52H/+ mice all captured behavioral seizures associated with EEG ictal activity immediately preceding death (Figure 2). The EEG signatures were different among these animals. Eight Fhf1R52H/+ mice exhibited a rapid succession of events: 1) sharp electrical spike, 2) a brief running fit coincident with a preictal waveform, 3) a tonic seizure with large spike and wave components, and 4) loss of brain activity within 60 seconds signaling death (Figure 2E and Supplemental Movie S2). The other two Fhf1R52H/+ mice exhibited similar time course of observable seizure, but with low-amplitude electrographic signatures (Figure 2F), suggesting that these seizures were possibly emanating from deeper brain structures. These observations allow categorization of Fhf1R52H/+ mortality as SUDEP coincident with seizure. Three of the mutant animals experienced additional recorded seizures and interictal spikes prior to terminal seizure (examples in Figures 2C, 2D).
3.3. Sudden Bradycardia During Terminal Seizure in Fhf1R52H/+ Mice
In order to distinguish among potential causes of seizure-induced death, the six Fhf1R52H/+ mice implanted at 19 days of age with EEG and ECG electrodes were monitored continually. All six mice suffered terminal seizure within 36 hrs of surgery and recording initiation. Eight wild-type mice with similar implants were viably maintained for at least 72 hrs before terminating recording. Fhf1R52H/+ instantaneous heart rates five seconds prior to seizure onset were 597±14 bpm (Figure 3A). In all Fhf1R52H/+ mice, instantaneous heart rate abruptly plunged to <160 bpm at times ranging from 3 to 53 seconds (median 9 sec) following EEG ictal activity onset (Figure 3A). Tonic extension commenced during all fatal seizures within 4 seconds before or after bradycardia onset (Figure 3B), suggesting that both events resulted from the arrival of depolarization to the brainstem 8. Cessation of detectable cortical activity occurred between 30 to 100 seconds after onset of bradycardia and tonic extension, while slowed and irregular heart rhythm persisted for 3 to 9 minutes before cardiac arrest (data not shown).
Figure 3. Sudden bradycardia at onset of terminal seizures in Fhf1R52H/+ mice.

A) Instantaneous heart rates upon seizures. Heart rates for each of six Fhf1R52H/+ mice are marked with different color circles before and after the onset of electrogenic seizure (time = 0). All lethal seizures were accompanied by abrupt decline in heart rate to ≤160 bpm, which is marked by a larger diameter circle occuring in these mice at 3, 4, 7, 11, 15 or 53 seconds post-onset of seizure. B) Coincidence of tonic extension and bradycardia onset. For each of the six Fhf1R52H/+ mice described in (A), the time of bradycardia onset (closed circle) and tonic extension (open circle) are plotted and linked. Bradycardia and tonic extension always commenced within four seconds of one another, despite the wide temporal variability of these events relative to electrogenic seizure onset. C) Abrupt suppression of cardiac rhythm by terminal seizure in a Fhf1R52H/+ mouse. Top: EEG and ECG traces from twenty seconds prior to forty seconds after seizure onset. The EEG channel designations are the same as described in Figure 2. The tonic seizure superimposes electromyographic noise on the ECG signal. Middle: Graph of instantaneous heart rate over the same time period as above shows abrupt drop in heart rate within three seconds of seizure onset. Bottom: One-second time-scale enlargements of the ECG trace 4 seconds before and 4 or 14 seconds after seizure onset. The ECG waveform after bradycardia onset lacks detectable P waves, suggesting ventricular beats.
Figure 3C shows the EEG and ECG data for one of these lethal seizures in greater detail. Seizure onset (arrow) marked by enhanced brain activity in all EEG channels was followed by abrupt decline in instantaneous heart rate to 73 bpm within three seconds. The slower heart rhythm over the first 20 seconds of seizure may represent ventricular beats, based upon absence of detectable P waves. These data show that seizures can induce sudden ictal bradycardia that may contribute to SUDEP in Fhf1R52H/+ mice.
3.4. Normal Baseline Cardiac Rhythm in Fhf1R52H/+ Pups Despite Modulation of Cardiac Sodium Currents by FHF1BR52H
The FHF1B isoform encoded by the Fhf1 gene is strongly expressed in mouse atrial cardiomyocytes as well as in brain 28,45 (and Shekhar and Fishman, unpublished data). Since FHF1B can bind to and modulate cardiac sodium channel Nav1.5 46, we investigated how R52H substitution in FHF1B alters Nav gating in cardiomyocytes.. Cultured FHF-deficient cardiomyocytes (see Methods) were infected with adenoviruses expressing GFP together with wild-type FHF1B, FHF1BR52H, or no FHF, and sodium currents were assayed under voltage clamp protocols in fluorescent cells 2-3 days post-infection. FHF1BR52H induced a 15 mV right-shift in Nav steady-state inactivation (Figure 4A) and slowed the rate of Nav inactivation by more than 2-fold across a range of membrane potentials (Figures 4B and S1). In contrast, wild-type FHF1B expression at levels comparable to that of FHF1BR52H (Figure S1) did not significantly alter inactivation gating of cardiac sodium channels (Figures 4A, 4B, S1). Hence, FHF1BR52H is a gain-of-function modulator of both neuronal 22 and cardiac sodium channels.
Figure 4. Effects of FHF1R52H on Nav inactivation and basal heart function.

A) Ectopic FHF1BR52H in cardiomyocytes depolarizes the voltage dependence of Nav inactivation. Dissociated adult ventricular cardiomyocytes from Fhf2−/Y mice were infected with adenoviruses expressing GFP and either wild-type FHF1B, FHF1BR52H, or no FHF. Sodium currents measured in fluorescent infected cells following equilibration at test membrane voltages were used to assess voltage dependence of steady state Nav inactivation. Wild-type FHF1B (n = 10 cells) did not significantly shift V1/2 inactivation compared to cells without FHF (n = 10), while FHF1BR52H (n = 16) induced a 15 mV depolarizing shift in V1/2 (P < 0.000005). B) Ectopic FHF1BR52H in cardiomyocytes slows rate of Nav inactivation. For all cells described in (A), sodium current decline at different membrane voltages was used to calculate the exponential τ (msec) for inactivation rate. At all test voltages, for cells expressing FHF1BR52H was >2-fold that for cells expressing either wild-type FHF1B or no FHF (P < 0.00005). C) Representative cardiac waveforms in a wild-type mouse. D) Representative cardiac waveforms in a Fhf1R52H/+ mouse prior to seizure. No significant difference compared to wild-type (C). E) Basal heart rates in wild-type and Fhf1R52H/+ mice. No significant difference. F) P-R intervals in wild-type and Fhf1R52H/+ mice. No significant difference.
The effect of FHF1BR52H substitution on gating of cardiac and neuronal sodium channels raised the possibility that heart rhythm in Fhf1R52H/+ mice could be altered by cardiac-intrinsic or autonomic mechanisms. Basal heart rhythm was assessed in 19-day old wild-type (n = 8) and Fhf1R52H/+ (n = 6) mice implanted with an ECG electrode along with EEG electrodes. The ECG waveform in mutant mice prior to seizure was indistinguishable from that for wild-type (Figure 4C,D). Furthermore, average basal heart rates recorded over a 6-hour period were not significantly different in mutant mice (Figure 4E), nor was the time delay between atrial and ventricular depolarization (P-R interval, Figure 4F). The absence of a measureable intrinsic cardiac phenotype in Fhf1R52H/+ mice likely reflects the restriction of endogenous FHF1 expression to atrial cardiomyocytes and the inability of single unipolar chest electrode recording to detect potentially subtle alterations in the P-wave.
4. DISCUSSION
All Fhf1R52H/+ mice suffered seizures with a tonic component during juvenile period, thereby providing the most compelling evidence to date that this recurrent missense mutation is sufficient to cause human EIEE disorder. Surprisingly, all Fhf1R52H/+ mice suffered a terminal seizure between 2-4 weeks of age. This lethal phenotype was observed in Fhf1R52H/+ founder #86 and in all 91 Fhf1R52H/+ progeny derived from mosaic founder #5, thereby ensuring that early death was the consequence of the Fhf1R52H mutation, and not due to a hypothetical off-target mutation during CRISPR editing.
In humans, two siblings first described with EIEE and FHF1R52H/+ genotype (EIEE47) died at three and seven years of age 22, potentially as a reflection of SUDEP, while nine other patients with the same de novo mutation also suffer from EIEE but have so far survived to between three and 32 years of age 23-26. These clinical data stand in contrast to the fully penetrant EIEE and SUDEP phenotype of the Fhf1R52H mutation in inbred mice. The heterogeneity of clinical course among individuals with the EIEE47 mutation may reflect a combination of variable genetic backgrounds and therapeutic treatment regimens employed at different institutions.
A fundamental question regarding SUDEP is whether seizure or postictal suppression compromises respiration, cardiac rhythm, or both 1-8,11,20. In the Fhf1R52H/+ mouse, heart rhythm is abruptly slowed within seconds of seizure onset. Bradycardia is likely triggered by a seizure-induced autonomic imbalance that causes or contributes to death, which in other mouse models is secondary to a wave of brain stem spreading depolarization following seizure onset 8. As this study did not include measurement of respiration, it is possible that SUDEP in Fhf1R52H/+ mice stems from a combination of cardiac and respiratory suppression during seizures. Indeed, the tonic extension displayed during all observed Fhf1R52H/+ seizures (Figure 3B, Movie S2) may constitute a sustained mechanical impediment to breathing 47 coincident with bradycardia. Alternatively, analogous to other animal models 48, the brainstem depolarization wave may also suppress activity in the pre-Botzinger complex breathing center, causing apnea that contributes to death.
The Fhf1 gene is expressed in mouse atrium as well as the brain 28,45, and we show here that ectopically expressed FHF1BR52H substitution is gain-of-function with respect the modulation of cardiac sodium channel inactivation, paralleling prior findings regarding brain Nav1.6 modulation 22. However, heart rhythm in Fhf1R52H/+ mice prior to seizure is apparently normal, at least for the readily measurable parameters of heart rate and P-R interval. Atrial FHF1BR52H expression level in Fhf1R52H/+ mice may not be sufficient to substantially alter Nav gating and conduction due to heterozygosity, endogenous coexpression of FHF2 28, and a potential less than 1:1 stoichiometry of FHF:Nav. Furthermore, potentially small effects on the P wave in Fhf1R52H/+ mice may have escaped our detection with single chest electrode ECG.
The Fhf1R52H/+ and Scn8aN1768D/+ mouse phenotypes are remarkably similar, reflecting the related effects of these mutations on Nav1.6 gating and the broadly distributed expression of both genes in the brain, including principle excitatory cortical neurons. Both mutants undergo SUDEP between 2-4 weeks of age 16,19 (and this study) accompanied by apparent autonomic-mediated bradycardia 19 (and this study), and both mutations induce large depolarizing shifts in the voltage dependence of Nav1.6 inactivation 13,22. These similarities warrant consideration of SCN8A EIEE13 and FHF1 EIEE47 as mechanistically related gain-of-function epilepsies.
Supplementary Material
Terminal seizure-like episode in P15 Fhf1R52H/+ mouse.
This nursing litter containing Fhf1+/+ and Fhf1R52H/+ pups was continuously video monitored starting at P12. Shown is a twenty-second video clip spanning the terminal seizure-like episode which commenced at the ten-second mark with a running fit followed by tonic episode and death.
Video accompanying terminal EEG-confirmed seizure in P21 Fhf1R52H/+ mouse.
This mutant mouse at P19 was implanted with cranial tower and subjected to continuous EEG and video monitoring. The onset of terminal seizure two days later commenced at ten seconds into this video clip with a quick jump followed by tonic seizure.
Adenoviral ectopic FHF1B and GFP expression and effects on sodium currents in cardiomyocytes. A-C) Representative sodium current traces from cardiomyocytes infected with adenoviruses expressing no FHF (A), wild-type FHF1B (B), or FHF1BR52H (C). Superimposed current traces from a series of voltage commands ranging from −80 mV to −20 mV in 5 mV increments are shown. Note the slower current decay in the cell expressing FHF1BR52H (C). D) Immunoblot detection of FHF1 and GFP in cells expressing wild-type FHF1B, FHF1BR52H, or no FHF (FHF1B-Δ). All viral vectors drive GFP expression, and FHF levels are similar in cells expressing wild-type FHF1B or FHF1BR52H.
Key Points.
The heterozygous FHF1 missense mutation Arg52His associated with EIEE47 causes seizures and SUDEP in juvenile mice
Abrupt-onset bradycardia in Fhf1R52H/+ mice begins within seconds after the onset of terminal seizures
SUDEP with bradycardia in Fhf1R52H/+ mice bears similarity to lethality in Scn8aN1768D/+ mice, paralleling similar effects of these mutations on inactivation gating of sodium channel Nav1.6
ACKNOWLEDGMENTS
Mice used in this study were produced by the Mouse Genetics and Gene Targeting CoRE at the Icahn School of Medicine at Mount Sinai, which is supported by the Tisch Cancer Institute at Mount Sinai (P30 CA196521 - Cancer Center Grant Support). In particular, the authors would like to acknowledge the contributions of Kasper Bonderup in the IVF generation of offspring for analysis.
This research was supported in part by NIH R01NS092786 to J.V., NIH F31HL132438 to A.S., NIH R01HL105983, NIH R01HL142498 and a grant from the Fondation LeDucq to G.I.F., and CUNY awards 608100048, 802330016, and 945850101 to M.G.
Footnotes
DISCLOSURE
L.V. received support from Anavex Life Sciences, Amzell Biopharma and Aequus Biopharma for projects unrelated to this study. LV is on Scientific Advisory Board of Mallinckrodt Pharmaceuticals. Other authors have no conflict of interest to disclose. We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
REFERENCES
- 1.Jehi L, Najm IM. Sudden unexpected death in epilepsy: impact, mechanisms, and prevention. Cleve. Clin. J. Med 2008;75(Suppl 2):S66–70. [DOI] [PubMed] [Google Scholar]
- 2.Tolstykh GP, Cavazos JE. Potential mechanisms of sudden unexpected death in epilepsy. Epilepsy Behav. 2013;26:410–14. [DOI] [PubMed] [Google Scholar]
- 3.Kalume F, Westenbroek RE, Cheah CS, Yu FH, Oakley JC, Scheuer T, et al. Sudden unexplained death in a mouse model of Dravet syndrome. J. Clin. Invest 2013;123:1798–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Auerbach DS, Jones J, Clawson BC, Offord J, Lenk GM, Ogiwara I, et al. Altered cardiac electrophysiology and SUDEP in a model of Dravet syndrome. PLoS One 2013;8:e77843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim Y, Bravo E, Thirnbeck CK, Smith-Mellecker LA, Kim SH, Gehlbach BK, et al. Severe peri-ictal respiratory dysfunction is common in Dravet syndrome. J. Clin. Invest 2018;128:1141–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dlouhy BJ, Gehlbach BK, Richerson GB. Sudden unexplained death in epilepsy: basic mechanisms and clinical implications for prevention. J. Neurol. Neurosurg. Psych 2016;87:402–13. [DOI] [PubMed] [Google Scholar]
- 7.Shoemaker JK, Goswami R. Forebrain neurocircuitry associated with human reflex cardiovascular control. Front. Physiol 2015;6:240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aiba I, Noebels JL. Spreading depolarization in the brainstem mediates sudden cardiorespiratory arrest in mouse SUDEP models. Sci. Trans. Med 2015;7:282ra46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Escayg A, MacDonald BT, Meisler MH, Baulac S, Huberfeld G, An-Gourfinkel I, et al. Mutations of SCNA1, encoding a neuronal sodium channel, in two families with GEFS+2. Nature Genet. 2000;24:343–5. [DOI] [PubMed] [Google Scholar]
- 10.Marini C, Scheffer IE, Nabbout R, Suls A, De Jonghe P, Zara F, et al. The genetics of Dravet syndrome. Epilepsia. 2011;52(Suppl 2):24–9. [DOI] [PubMed] [Google Scholar]
- 11.Kuo F-S, Cleary CM, LoTurco JJ, Chen X, Mulkey DK. Disordered breathing in a mouse model of Dravet syndrome. eLife. 2019;8:e43387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rubinstein M, Han S, Tai C, Westenbroek RE, Hunker A, Scheuer T, et al. Dissecting the phenoyptes of Dravet syndrome by gene deletion. Brain. 2015;138:2219–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Veeramah KR, O’Brien JE, Meisler MH, Cheng X, Dib-Hajj SD, Waxman SG, et al. De novo pathogenic SCN8A mutation identified by whole genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. Am. J. Human Genet 2012;90:502–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ohba C, Kato M, Takahashi S, Lerman-Sagie T, Lev D, Terashima H, et al. Early onset epileptic encephalopathy caused by de novo SCN8A mutations. Epilepsia. 2014;55:994–1000. [DOI] [PubMed] [Google Scholar]
- 15.Xiao Y, Jiong J, Mao M, Liu L, Xingfang JL, Luo H, et al. Early-onset epileptic encephalopathy with de novo SCN8A mutation. Epilepsy Res. 2018;139:9–13. [DOI] [PubMed] [Google Scholar]
- 16.Lopez-Santiago LF, Yuan Y, Wagnon JL, Hull JM, Frasier CR, O’Malley HA, et al. Neuronal hyperexcitability in a mouse model of SCN8A epileptic encephalopathy. Proc. Natl. Acad. Sci. USA 2017;114:2383–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wagnon JL, Barker BS, Hounshell JA, Haaxma CA, Shealy A, Moss T, et al. Pathogenic mechanism of recurrent mutations of SCN8A in epileptic encephalopathy. Ann. Clin. Trans. Neurol 2016;3:114–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Estacion M, O’Brien JE, Conravey A, Hammer MF, Waxman SG, Dib-Hajj SD, et al. A novel de novo mutation of SCN8A (Nav1.6) with enhanced channel activation in a child with epileptic encephalopathy. Neurobiol. Dis 2014;69:117–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wagnon JL, Korn MJ, Parent R, Tarpey TA, Jones JM, Hammer MF, et al. Convulsive seizures and SUDEP in a mouse model of SCN8A epileptic encephalopathy. Hum. Mol. Genet 2014;24:506–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frasier CR, Wagnon JL, Bao YO, McVeigh LG, Lopez-Santiago LF, Meisler MH, et al. Cardiac arrhythmia in a mouse model of sodium channel SCN8A epileptic encephalopathy. Proc. Natl. Acad. Sci. USA 2016;113:12838–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patino GA, Claes LRF, Lopez-Santiago LF, Slat EA, Dondeti RSR, Chen C, et al. A functional null mutation of SCN1B in a patient with Dravet syndrome. J. Neurosci 2009;29:10764–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Siekierska A, Isrie M, Liu Y, Scheldeman C, Vanthillo N, Lagae L, et al. Gain-of-function FHF1 mutation causes early-onset epileptic encephalopathy with cerebellar atrophy. Neurology. 2016;86:2162–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Al-Mehmadi S, Splitt M, Ramesh V, DeBrosse S, Dessoffy K, Xia F, et al. FHF1 (FGF12) epileptic encephalopathy. Neurol. Genet 2016;2:e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guella I, Huh L, McKenzie MB, Toyota EB, Bebin EM, Thompson ML, et al. De novo FGF12 mutation in 2 patients with neonatal-onset epilepsy. Neurol. Genet 2016;2:e120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Villeneuve N, Abidi A, Cacciagli P, Mignon-Ravix C, Chabrol B, Villard L, et al. Heterogeneity of FHF1 related phenotype: novel case with early attacks of apnea, partial mitochondrial respiratory chain complex II deficiency, neonatal onset seizures without degeneration. Eur. J. Paediatr. Neurol 2017;21:783–6. [DOI] [PubMed] [Google Scholar]
- 26.Zhang L, Gao J, Liu H, Tian Y, Zhang X, Lei W, et al. Pathogenic variants identified by whole exome sequencing in 43 patients with epilepsy. Hum. Genomics 2020;14:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smallwood PM, Munoz-Sanjuan I, Tong P, Macke JP, Hendry SHC, Gilbert DJ, et al. Fibroblast growth factor (FGF) homologous factors: new members of the FGF family implicated in nervous system development. Proc. Natl. Acad. Sci. USA 1996;93:9850–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hartung H, Feldman B, Lovec H, Coulier F, Birnbaum D, Goldfarb M. Murine FGF-12 and FGF-13: expression in embryonic nervous system, connective tissue, and heart. Mech. Dev 1997;64:31–9. [DOI] [PubMed] [Google Scholar]
- 29.Wang Q, McEwen DG, Ornitz DM. Subcellular and developmental expression of alternatively spliced forms of fibroblast growth factor 14. Mech. Dev 2000;90:283–7. [DOI] [PubMed] [Google Scholar]
- 30.Goldfarb M, Schoorlemmer J, Williams A, Diwakar S, Wang Q, Huang X, et al. Fibroblast growth factor homologous factors control neuronal excitability through modulation of voltage-gated sodium channels. Neuron 2007;55:449–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Venkatesan K, Liu Y, Goldfarb M. Fast-onset long-term open-state block of sodium channels by A-type FHFs mediates classical spike accommodation in hippocampal pyramidal neurons. J. Neurosci 2014;34:16126–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature 2007;445:168–76. [DOI] [PubMed] [Google Scholar]
- 33.Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Tissue-based map of the human proteome. Science 2015;347:1260419. [DOI] [PubMed] [Google Scholar]
- 34.Munoz-Sanjuan I, Smallwood PM, Nathans J. Isoform diversity among fibroblast growth factor homologous factors is generated by alternative promoter usage and splicing. J. Biol. Chem 2000;275:2589–97. [DOI] [PubMed] [Google Scholar]
- 35.Olsen S, Garbi M, Zampieri N, Eliseenkova AV, Ornitz DM, Goldfarb M, et al. Fibroblast growth factor (FGF) homologous factors share structural but not functional homology with FGFs. J. Biol. Chem 2003;278:34226–36. [DOI] [PubMed] [Google Scholar]
- 36.Goetz R, Dover K, Laezza F, Shtraizent N, Huang X, Tchetchik D, et al. Crystal structure of a fibroblast growth factor homologous factor (FHF) defines a conserved surface on FHFs for binding and modulation of voltage-gated sodium channels. J. Biol. Chem 2009;284:17883–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang C, Chung BC, Yan H, Lee S-Y, Pitt GS. Crystal structure of the ternary complex of a NaV C-terminal domain, a fibroblast growth factor homologous factor, and calmodulin. Structure. 2012;20:1167–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, Jaenisch R. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell 2013;154:1370–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ran FA, Hsu PD, Lin C-Y, Gootenberg JS, Konermann S, Trevino AE, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 2013;154:1380–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Velíšek L, Shang E, Velíšková J, Chachua T, Macchiarulo S, Maglakelidze G, et al. GABAergic neuron deficit as an idiopathic generalized epilepsy mechanism: the role of BRD2 haploinsufficiency in juvenile myoclonic epilepsy. PloS One. 2011;6:e23656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mishra V, Gautier NM, Glasscock E. Simultaneous video-EEG-ECG monitoring to identify neurocardiac dysfunction in mouse models of epilepsy. J. Vis. Exp 2018;131:e57300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc. Natl. Acad. Sci. USA 1998;95:2509–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Park DS, Shekhar A, Marra C, Lin X, Vasquez C, Solinas S, et al. Fhf2 gene deletion causes temperature-sensitive cardiac conduction failure. Nature Commun. 2016;7:12966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang C, Hennessey JA, Kirkton RD, Wang C, Graham V, Puranam RS, et al. Fibroblast growth factor homologous factor 13 regulates Na+ channels and conduction velocity in murine hearts. Circ. Res 2011;109:775–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wiencierz AM, Kernbach M, Ecklebe J, Monnerat G, Tomiuk S, Raulf A, et al. Differential expression levels of integrin α6 enable the selective identification and isolation of atrial and ventricular cardiomyocytes. PloS One. 2015;10:e0143538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu C, Dib-Hajj SD, Reganathan M, Cummins TR, Waxman SG. Modulation of the cardiac sodium channel Nav1.5 by fibroblast growth factor homologous factor 1B. J. Biol. Chem 2003;278:1029–36. [DOI] [PubMed] [Google Scholar]
- 47.Nashef L, Walker F, Allen P, Sander JWAS, Shorvon SD, Fish DR. Apnoea and bradycardia during epileptic seziures: relationship to sudden death in epilepsy. J. Neurol. Neurosurg. Psych 1996;60:297–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jefferys JGR, Arafat AR, Irazoqui PP, Lovick TA. Brainstem activity, apnea, and death during seizures induced by intrahippocampal kainic acid in anaesthetized rats. Epilepsia 2019;60:2346–58. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Terminal seizure-like episode in P15 Fhf1R52H/+ mouse.
This nursing litter containing Fhf1+/+ and Fhf1R52H/+ pups was continuously video monitored starting at P12. Shown is a twenty-second video clip spanning the terminal seizure-like episode which commenced at the ten-second mark with a running fit followed by tonic episode and death.
Video accompanying terminal EEG-confirmed seizure in P21 Fhf1R52H/+ mouse.
This mutant mouse at P19 was implanted with cranial tower and subjected to continuous EEG and video monitoring. The onset of terminal seizure two days later commenced at ten seconds into this video clip with a quick jump followed by tonic seizure.
Adenoviral ectopic FHF1B and GFP expression and effects on sodium currents in cardiomyocytes. A-C) Representative sodium current traces from cardiomyocytes infected with adenoviruses expressing no FHF (A), wild-type FHF1B (B), or FHF1BR52H (C). Superimposed current traces from a series of voltage commands ranging from −80 mV to −20 mV in 5 mV increments are shown. Note the slower current decay in the cell expressing FHF1BR52H (C). D) Immunoblot detection of FHF1 and GFP in cells expressing wild-type FHF1B, FHF1BR52H, or no FHF (FHF1B-Δ). All viral vectors drive GFP expression, and FHF levels are similar in cells expressing wild-type FHF1B or FHF1BR52H.