

Abstract
The skeleton is affected by numerous primary and metastatic solid and hematopoietic malignant tumors, which can cause localized sites of osteolysis or osteosclerosis that can weaken bones and increase the risk of fractures in affected patients. Chemotherapeutic drugs can eliminate some tumors in bones or reduce their volume and skeletal-related events, but adverse effects on non-target organs can significantly limit the amount of drug that can be administered to patients. In these circumstances, it may be impossible to deliver therapeutic drug concentrations to tumor sites in bones. One attractive mechanism to approach this challenge is to conjugate drugs to bisphosphonates, which can target them to bone where they can be released at diseased sites. Multiple attempts have been made to do this since the 1990s with limited degrees of success. Here, we review the results of pre-clinical and clinical studies made to target FDA-approved drugs and other antineoplastic small molecules to bone to treat diseases affecting the skeleton, including osteoporosis, metastatic bone disease, multiple myeloma and osteosarcoma. Results to date are encouraging and indicate that drug efficacy can be increased and side effects reduced using these approaches. Despite these successes, challenges remain: no drugs have gone beyond small phase 2 clinical trials, and major pharmaceutical companies have shown little interest in the approach to repurpose any of their drugs or to embrace the technology. Nevertheless, interest shown by smaller biotechnology companies in the technology suggests that bone targeting of drugs with bisphosphonates has a viable future.
Keywords: Bone metastasis, bone-targeting, bisphosphonate, bortezomib, drug conjugate, chloroquine
1. Introduction
Bone is one of the most common and preferred sites to which cancer cells metastasize [1, 2]. Prostate and breast cancer are among the most common cancer types that metastasize to bone [1, 2] and they are responsible for up to 70% of bone metastasis cases [3]; other tumor types spread to bone less frequently, including lung, kidney, thyroid, melanoma, lymphoma, gynecologic, and gastrointestinal cancers [4]. Up to 75% of patients dying from prostate or breast cancers have evidence of bone metastasis [3]. A large recent retrospective study of 382,733 cancer patients with a variety of solid tumors reported that the overall incidence of bone metastasis ranged from 2.9% at 30 days from the original diagnosis of the primary tumor to 8.4% at 10 years from diagnosis [4]. However, the incidence of bone metastasis in this review was highly variable depending on the primary tumor type, with a range at 10 years from 2.9% for gynecologic to 29.2% for prostatic cancer. Patients with prostate cancer had the highest risk of developing bone metastases, followed by those with lung, renal or breast cancer [4]. Bone metastases currently cannot be cured and affected patients have a poor prognosis [1], with population-based studies reporting that cancer patients with bone metastases have a significantly shorter survival after the diagnosis of bone metastases [5–7]. Thus, there is a great unmet need to develop drugs that will eliminate cancers that have spread to bone.
The most common primary bone malignancies are osteosarcoma (35%), chondrosarcoma (25%), and Ewing’s sarcoma (16%) [8]. Osteosarcoma is the most common malignant bone tumor, particularly among children and adolescents [9]. It represents 56% of malignant bone tumors in children [10], but it also occurs in older adults when it can complicate Paget’s disease of bone [11], prior radiation exposure [12] and fibrous dysplasia [13]. The 5-year relative survival rate of localized osteosarcoma is 77% [14, 15], but it drops to 27% when the disease has spread to distant organs [14] (https://www.cancer.org/cancer/osteosarcoma/detection-diagnosis-staging/survival-rates.html).
Multiple myeloma (MM) is a plasma cell neoplasm and the second most common adult hematologic malignancy in the US [16]. It arises in the bone marrow (BM) where it causes multiple foci of localized bone destruction with associated bone pain and increased risk of fractures [17]. MM accounts for 1% of all types of cancer and 2% of cancer deaths [16, 18]. It is an incurable disease of the elderly with a median age at diagnosis of 70 years, with 35–40% of patients being older than 75 years [19–21]. Myeloma cells originate from plasma cells in the BM where they interact with BM cells to form vicious cycles in which factors released by mesenchymal cells promote myeloma cell growth and myeloma cells in turn promote release of factors from mesenchymal cells [22]. Thus, alternation in cancer cells or/and the bone microenvironment contribute to MM progression [23]. Development of drugs that could target both myeloma cells and bone cells would represent a new therapy for MM.
Delivery of chemotherapeutic drugs preferentially to bone using bone targeting strategies, such as bisphosphonate (BP) drug conjugates [24, 25] offers potential improvements in efficacy and side-effect profiles. A targeted delivery design provides an effective concentration of a chemotherapeutic agent in bone, in particular at sites of higher bone turnover in the skeleton, while maintaining low systemic levels. It may also overcome drug resistance by increasing concentrations of drugs in the local bone microenvironment to well above those achievable with approved doses of non-targeted drugs, which are limited because of adverse effects [25]. Another benefit is that BP-linked conjugates could function as a drug releasing depot to deliver anti-cancer therapies locally to the bone and BM. In addition to treatment of metastatic bone disease, therapeutic indications for bone-targeting of anti-cancer drugs include treatment of patients with primary BM malignancies and osteosarcomas that have metastasized to extra-skeletal sites. This paper will review previous and current attempts to target FDA-approved anti-neoplastic drugs and other small molecules to bone, the strategies used, and their successes and failures.
2. Development of bone-targeted drugs using bisphosphonates (BPs)
BPs have been studied extensively since the 1960s and this led to development of numerous BP-based drugs becoming commercially available for the treatment of a variety of bone diseases, including osteoporosis, Paget’s disease, metastatic bone disease, and multiple myeloma [26]. Mechanistically, BPs have varying degrees of affinity for bone by chelating calcium ions present in bone hydroxyapatite [27]. The involvement of phosphonate groups in BPs in their mechanism of binding to hydroxyapatite was further exemplified in an investigation of the comparative binding ability of BP esters [28]. As a result of their high affinity for bone, BPs have been used for drug targeting mainly in pre-clinical studies for over 30 years [25, 29–31].
One of the earliest advanced studies with this approach to target anticancer therapies to bone was carried out by scientists at Ariad Pharmaceuticals and the University of Rochester Medical Center that involved development of Src tyrosine kinase small molecule inhibitors, in which they used stable linkages to BP analogs to target the inhibitors to bone [32–34]. Previous studies had shown that Src tyrosine kinase was expressed by cancer cells and enhanced their proliferation, migration and metastatic potential [35]. Later studies reported, unexpectedly, that Src tyrosine kinase expression was required for osteoclast ruffled border formation in mice [36]. Thus, the goal of these studies was to develop novel drugs that would inhibit not only osteoclastic bone resorption, but also the growth of cancer cells that had metastasized to bone. Their studies indicated that the lead bone-targeted compound, AP23451 (Fig. 1A), dose-dependently inhibited PTH-induced hypercalcemia in mice, and that this effect required the attached bisphosphonate because the inhibitor alone was ineffective [33]. AP23451 also dose-dependently inhibited ovariectomy-induced bone loss in mice as effectively as alendronate, albeit with a high dose, (Fig. 1) and inhibited osteolysis induced by metastatic MDA-MB-231 breast cancer cells in nude mice as effectively as zoledronate (Figs. 1A, 2A–C). However, unlike zoledronate, AP23541 also significantly reduced tumor cell volume inside and outside bones at sites of metastasis (Fig. 2). It more effectively reduced osteoclast numbers (Fig. 3), but it also induced significantly less apoptosis of osteoclasts than zoledronate, a well-recognized anti-resorptive action of BPs [37]. Despite these encouraging in-vivo data, AP23451 was not developed as a new drug. This was because serious adverse CNS effects were observed in a subsequent investigational new drug-enabling high-dose toxicology study in non-human primates and presumably occurred because enough of the Src inhibitor crossed into the brain where Src is highly expressed, but does not have an essential function [38]. Of note, are the higher in vivo doses administered in these comparisons with alendronate and zoledronate. While different biochemical targets within the osteoclast are invoked by the respective classes of drugs, similar uptake through endocytotic mechanisms likely occurs. The greatest difference in biodistribution is likely at the level of bone uptake. While high uptake on bone has been demonstrated for AP23451, its bone targeting moiety, a phosphinylmethylphosphonate, is known to exhibit significantly lower mineral affinity than hydroxyalkylbisphosphonates, such as alendronate and zoledronate [39, 40]. This raises the possibility that this Src inhibitor could be conjugated with a bisphosphonate with much higher mineral affinity with the result that more of it would be targeted to bone and away from other organs.
Figure 1. Chemical structures of AP23451, alendronate and zoledronate and effects of Src inhibitor, AP23451, and alendronate on ovariectomy-induced bone loss.
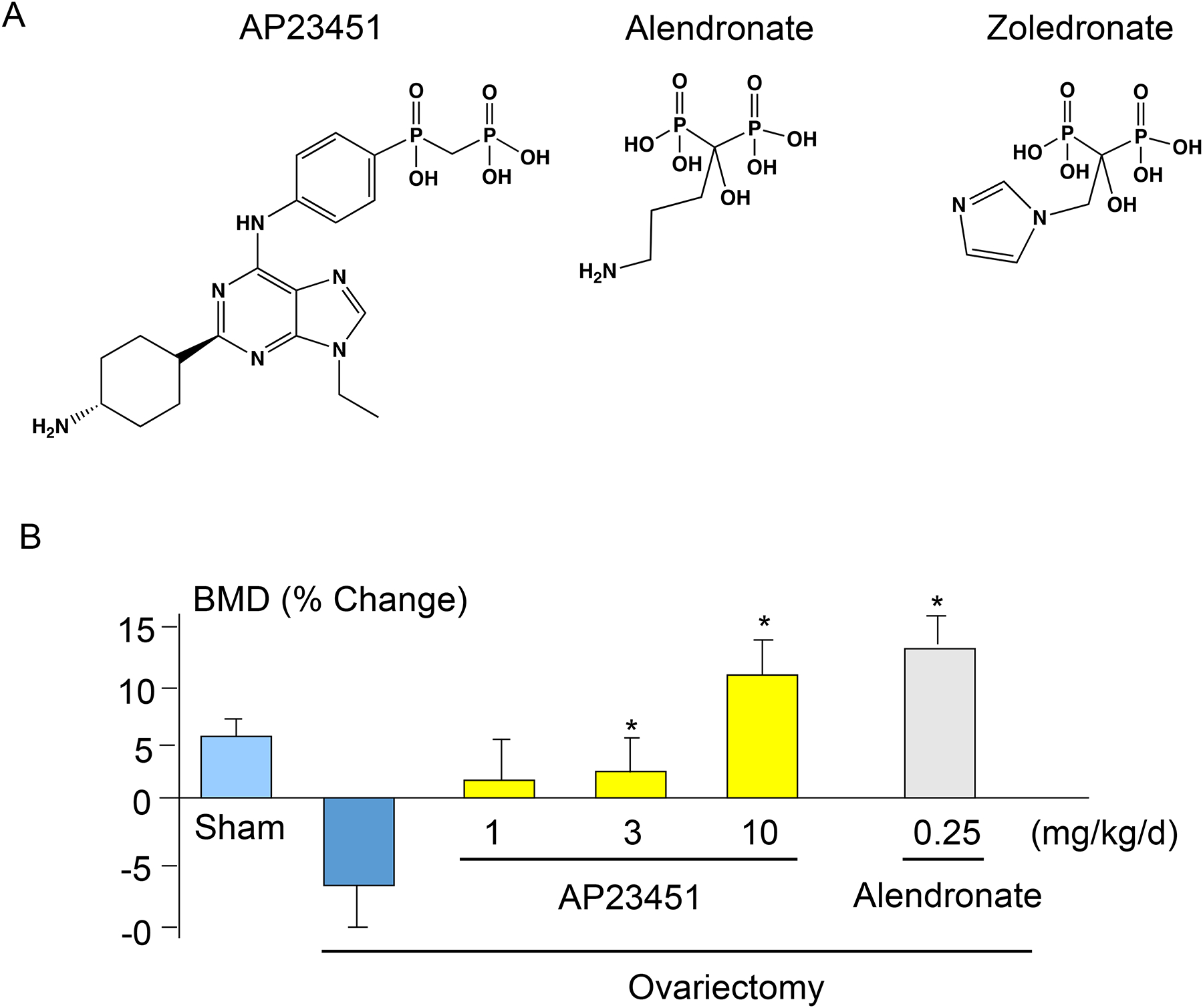
(A). Chemical structures of AP23451, alendronate and zoledronate. (B). 3-m-old female mice were sham operated or ovariectomized and treated with vehicle, AP23451 or alendronate at the indicated doses once per day for 35 days. After sacrifice, bone mineral density (BMD) was measured in L2–4 lumber vertebrae using dual X-ray absorptiometry. Data are means +/− SD. One-way ANOVA with Turkey’s test. *: p<0.05 vs ovariectomy.
Figure 2. AP23451 prevents MDA-MB-231 breast cancer-induced osteolysis and reduces, but does not eliminate tumor cell growth in long bones.
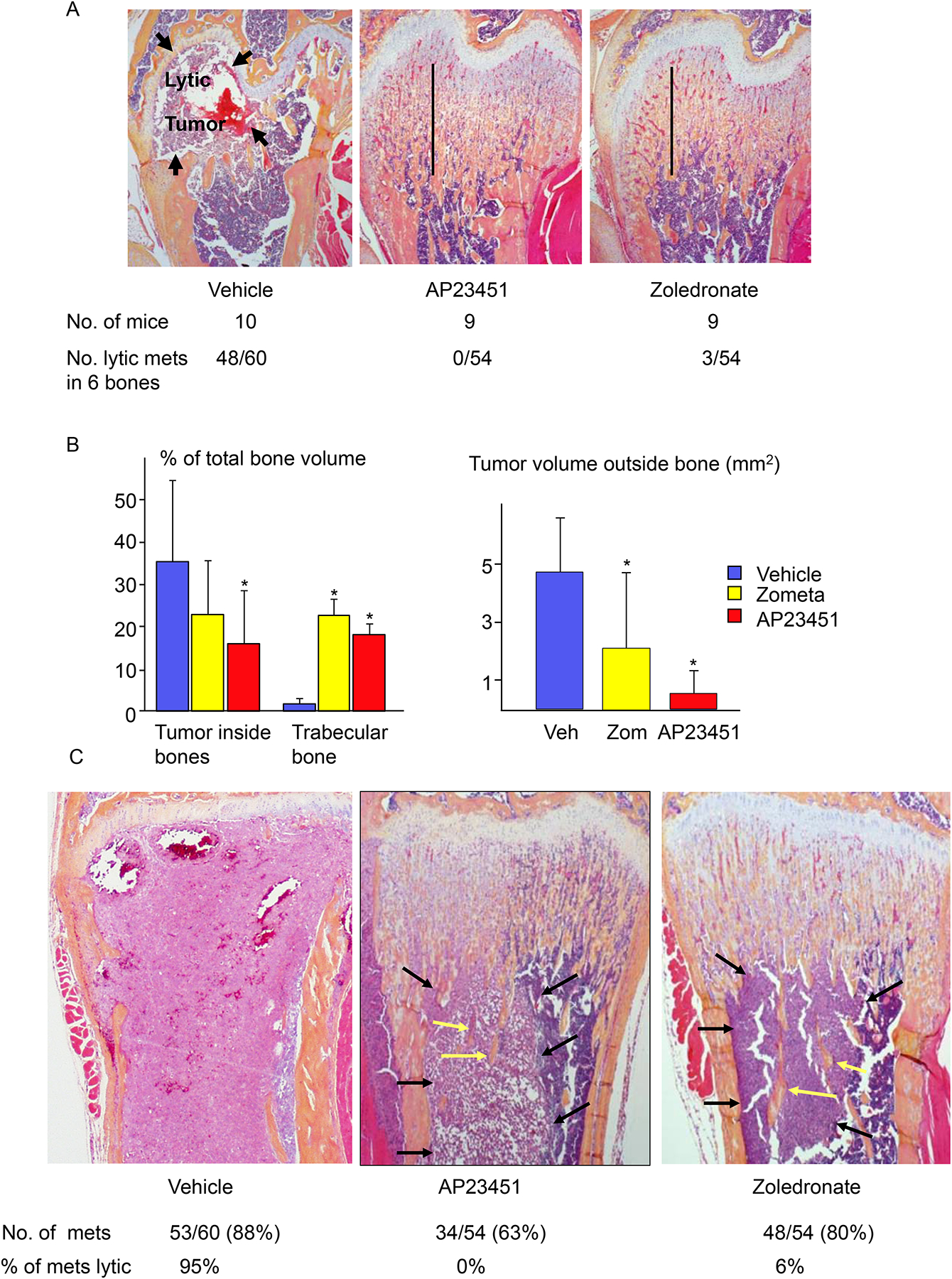
MDA-MB-231 cells (104–106) were inoculated into the left cardiac ventricle of 3-m-old female nude mice, which were given daily s.c. injections of AP23451 (10 mg/kg) or zoledronate (0.25mg/kg) for 28 days. After sacrifice, all femora, tibiae and humeri were removed and fixed in 10% phosphate-buffered formalin, decalcified in 14% EDTA and embedded in paraffin. (A). H&E-stained sections of distal femora showing an osteolytic lesion in the metaphysis of a vehicle-treated mouse (arrows), but not in AP23451- or zoledronate-treated mice, which developed osteopetrosis (vertical lines, where unresorbed bone largely fills the BM cavity) during the 28 days of treatment as a consequence of the inhibitory effects of these agents on bone resorption. The numbers of osteolytic metastases were counted in these 6 bones from each mouse. (B). Volume of tumor inside bones and trabecular bone volume (left panel) and volumes of tumor deposits outside bones (right panel). C. H&E-stained sections of distal femora showing replacement of metaphyseal bone and diaphyseal bone marrow by tumor cells in a vehicle-treated mouse. Osteopetrosis in the metaphysis of AP23451- and zoledronate-treated mice as well as tumor deposits (black arrows) in the BM where there are surviving unresorbed bone trabeculae (yellow arrows), and the numbers of metastases (mets) and % of metastases with osteolysis. Data are means +/− SD. One-way ANOVA with Bonferroni test. *: p<0.05 vs vehicle-treated mice.
Figure 3. Differing effects of AP23451 and zoledronate on osteoclasts in metaphyses of mice inoculated with MDA-MB-231 breast cancer cells.
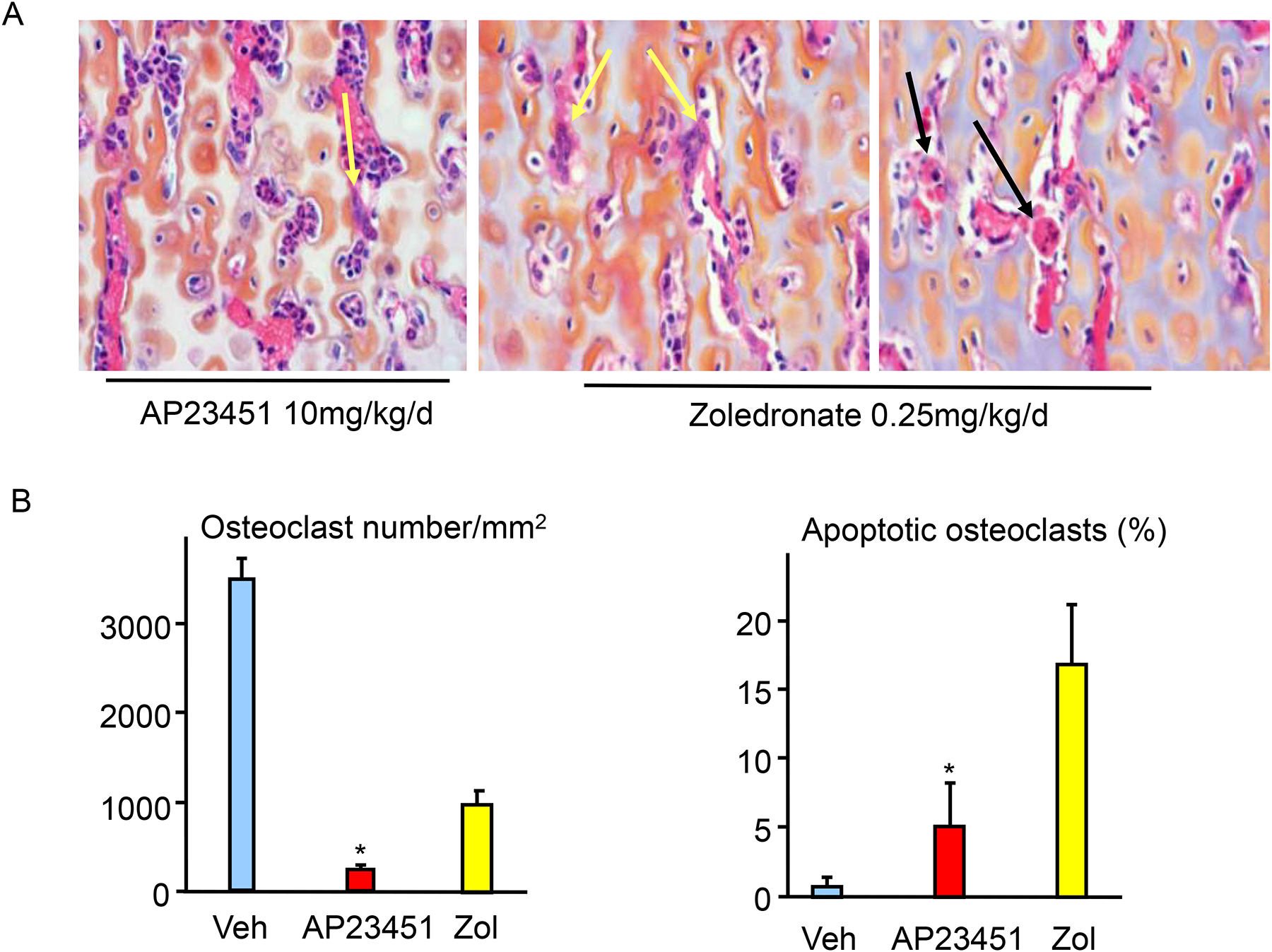
(A). H&E-stained sections of distal femora of mice showing occasional surviving (yellow arrows) osteoclasts in AP23451-treated mice and surviving and apoptotic (black arrows) osteoclasts in zoledronate-treated mice. (B). Total numbers of osteoclasts in femoral metaphyses (left panel) and % of osteoclasts with the typical features of apoptosis (cytoplasmic contraction and nuclear condensation and fragmentation). Data are means +/− SD. One-way ANOVA with Bonferroni test. * p < 0.05 vs zoledronate.
Subsequent attempts to target drugs to bone with BPs have had limited success for a number of reasons. These include insufficient financial support to sustain programs and the use of non-cleavable linkages between the BP and drug that can limit the activity of the drug. For example, Agyin et al. synthesized a BP-linked bortezomib (Btz) conjugate to treat multiple myeloma [41]. The BP-Btz conjugate inhibited myeloma cell growth in vitro, but its effects were not tested in vivo due to lack of funding (personal communication). They linked Btz to a BP using a chemically and biochemically stable linkage, aiming to ensure the success of these bone-targeted drug conjugates [41], but this would likely reduce cellular transport in vivo. The investigators also planned to develop releasable proteasome inhibitors in the tumor microenvironment by linking them to BPs containing more labile linkers, although they have never reported any results with these. These investigators incorporated alendronate, an FDA-improved bone resorption inhibitor, as the bone-targeting agent, but this could make it difficult to determine the precise source of any osteoclast inhibitory activity since Btz also inhibits osteoclast formation [41]. As with all dual action drugs, even with drug releasing conjugates, consideration would need to be given to the relative concentration of each component of the drug combination that releases locally as the concentration of each required to produce the desired pharmacologic effect would likely differ for each biochemical target.
Recent attempts to target estrogen analogs [42], prostaglandins [42] [43], and antibiotics [42, 43] to bone through a carbamate linker have led to promising results in preclinical models. This chemical linkage was designed to allow the targeting and subsequent release of an active agent at the bone surface. This approach improved the efficacy of estradiol to inhibit bone resorption in ovariectomized mice, while limiting side-effects, such as endometrial hyperplasia [42]. In a related effort, this BP-drug conjugation technology facilitated adequate delivery to bone and the slow release of prostaglandin to stimulate bone formation in an animal model of osteoporosis and a radio-labelled, pharmacokinetics study to estimate concentrations of delivered drug [44, 45]. Some carbamate linkers appear to be cleaved by enzymatic or hydrolytic means in the acidic microenvironment under the OC ruffled border in bone resorption lacunae, thus releasing the active drug. However, this release mechanism may be substrate-specific because carbamate-linked glycopeptide antibiotics, such as vancomycin, dalbavancin, ortavancin and telavancin, failed to produce in vivo activity in mouse models of osteomyelitis [46].
Bisphosphonates are highly effective anti-resorptive drugs in diseases associated with increased bone resorption [47]. However, adverse effects associated with some of the potent recent generation BP therapies, including osteonecrosis of the jaw in some patients with MM or metastatic bone disease, and atypical femoral fractures in a small percentage of patients with osteoporosis, have reduced both patients’ willingness to take and physicians’ comfort to prescribe BPs [48]. The exquisite bone-targeting property of BPs, however, is an attribute that has not been fully exploited in new drug design [49]. BPs with high bone-binding activity and minimal anti-resorptive activity should therefore be ideal carriers to deliver desired drugs to bone with minimal risk of the adverse effects of aminobisphosphonates [50].
3. Clinical and preclinical studies of BP- linked anti-cancer drugs
Chemotherapeutic drugs are used widely to kill rapidly multiplying malignant cells by a variety of mechanisms, but they can also kill healthy dividing cells, including those in the gastrointestinal tract [51], central nerve system [52] and hair follicles [24]. In an attempt to reduce adverse effects and more specifically target cancer cells and bone, several chemotherapy drugs have been linked to BPs and tested in mainly preclinical and some human studies outlined below.
3.1. OsteoDex (ODX)
OsteoDex (ODX) is a BP-dextran-guanidine complex developed by DexTech Medical AB, to treat metastatic bone disease. ODX contains a carbohydrate backbone (dextran) that enables the incorporation of multiple alendronate molecules (average 8 alendronate per dextran) and guanidine. In vitro studies demonstrated that ODX had similar potency to inhibit bone resorption as zoledronate, but it also killed PC3 cells, a cell line established in 1979 from a bone metastasis of a 62-year-old Caucasian male with grade IV prostate cancer, with 10-fold higher potency than zoledronate (IC50 2 μm vs. 25 μm) [53]. DexTech completed a Phase 1 open-label, multiple ascending dose, multicenter clinical trial of ODX (ClinicalTrials.gov NCT02825628) and reported the results in 2016 [54]. Twenty-eight patients with castration-resistant prostate cancer and confirmed bone metastases were given 7 intravenous infusions of ODX every third week. In total, 206 adverse events were recorded. Of these, 13.6% were classified as treatment-related, mostly acute-phase reactions, and none was considered to be serious or severe. No cumulative toxicity and no renal toxicity were recorded [54].
DexTech initiated a phase IIb randomized, double-blind, placebo-controlled multicenter study evaluating efficacy and tolerability of Osteodex in patients with metastatic castration-resistant prostate cancer (CRPC) in 2015, but it was terminated in 2016 because of poor patient recruitment. They initiated a second phase II randomized, double-blind, dose-finding, repeat dose Phase II multicenter study ODX in 2016 of 55 patients with castration-resistant prostate cancer with skeletal metastases (ClinicalTrials.gov NCT02825628). The primary objective was to evaluate relative changes from baseline in serum alkaline phosphatase and P1NP at 12 weeks of three different doses of ODX (3.0, 6.0 and 9.0 mg/kg ODX). Secondary objectives are progression-free and overall survival. The company has reported encouraging results without serious side effects on their website, and suggested that ODX treatment slowed down the course of the skeletal disease in the majority of patients who completed the 5 months of treatment (https://news.cision.com/dextech/r/promising-follow-up-results-from-dextech-s-phase-iib-study-for-osteodex,c2932053), but results from the last recruited patients will not be reported until June 2020.
3.2. Cytarabine-etidronate conjugate (MBC-11)
MBC Pharma, Inc, Aurora, CO, USA conjugated cytarabine to etidronate, the first bisphosphonate approved to treat osteoporosis and Paget’s disease [55], to generate MBC-11. Cytarabine, also known as cytosine arabinoside, is used to treat acute myeloid leukemia, acute lymphocytic leukemia, chronic myelogenous leukemia, and non-Hodgkin’s lymphoma. Cytosine arabinoside interferes with DNA synthesis through its rapid conversion to cytosine arabinoside triphosphate, which damages DNA during the cell cycle S phase (synthesis of DNA). MBC-11 was licensed to Osteros Biomedica Ltd., part of the Russian holding company, Maxwell Biotech, for clinical development. Osteros Biomedica Ltd. sponsored the first-in-human trial of BP-linked anti-cancer drugs, which was conducted in St. Petersburg, Russia. The phase I open-label, nonrandomized, dose escalation study, which included 15 patients with advanced breast, cervical or prostate cancers and bone metastases who were given MBC-11 daily for 5 days intravenously, every 4 weeks for 4 cycles (ClinicalTrials.gov NCT02673060), was completed in 2019. Zinnen et al. reported a maximal tolerated dose of 5 mg/kg, based on 2 of 3 patients given 10 mg/kg experiencing dose-limiting grade 4 neutropenia and thrombocytopenia. This lower dose had similar myelosuppressive adverse effects as effective doses of the parent drug, cytosine arabinoside, which are typically 2–20-fold higher, suggesting that the bone-targeting of lower amounts of cytosine arabinoside delivered sufficient effective concentrations of the drug to the bone microenvironment. Evidence for this is that treatment significantly reduced cancer cell activity in 111 of 206 bone lesions, as assessed by fluorodeoxyglucose positron emission tomography/computed tomography imaging compared to levels detected at baseline [56]. Although 3 patients had a partial metabolic response and 3 others had stable metabolic disease, as assessed by PET scanning, 8 had progressive metabolic disease, despite often displaying significant reduction or stabilization of bone lesions. On the basis of these phase 1 findings, the company is planning a phase II study to investigate the effects of MBC-11 in patients with bone metastases as the dominant site of disease due to castration-resistant prostate or breast cancer. MBC-11 also was tested in a pilot phase-II trial in 5 dogs with spontaneous osteosarcomas. An initial report indicated that an effective dose was identified, and 4 of the 5 dogs had a positive response to MBC-11, as indicated by reduced lameness in the affected limb via force platform gait analysis. Owner-reported ‘quality of life and pain scores’ also improved and FDG-PET/CT imaging indicated reduced tumor-specific metabolic activity in 3 dogs [57].
3.3. Gemcitabine-ibandronate conjugate (GEM-IB)
GEM-IB is a 2nd generation bone-targeted conjugate from MBC Pharma, Inc. consisting of the nucleoside metabolic inhibitor, gemcitabine (GEM), chemically linked to ibandronate (IB). GEM is used in combination with other chemotherapeutic drugs for advanced or metastatic cancers, such as lung and breast cancers, and as a first-line treatment alone for pancreatic cancer. GEM-IB was tested in vivo in a mouse model of prostate cancer-induced bone disease and significantly reduced metastases with a trend for improved survival. It was also used in combination with the mitotic inhibitor drug, docetaxel, and higher efficacy with regard to tumor reduction and survival benefit was reported in a murine model of osteosarcoma (https://patents.justia.com/patent/20190350958).
3.4. Bisphosphonate-doxorubicin conjugate (12b80)
12b80 is a conjugate of doxorubicin and a cleavable hydroxyl-bisphosphonate linked through imine bonds developed by Atlanthera Drug Discovery Company, France. Doxorubicin is a first-line drug used to treat some sarcomas, including osteosarcoma, but it has a limited therapeutic index due to severe cardiac toxicity. 12b80 has high bone affinity, with specific release of doxorubicin in acidic conditions, with tumor cell uptake of the prodrug and lower cytotoxicity in vitro than doxorubicin. 12b80 displayed rapid and sustained targeting of bone tissue and tumor-associated heterotopic bone in in-vivo mouse models and delivered higher doxorubicin concentrations in the tumor-bone environment compared to a non-vectorized doxorubicin and was more potent than a doxorubicin/zoledronate combination. Consequently, 12b80 showed much lower toxicity than doxorubicin and promoted strong antitumor effects in a rodent orthotopic osteosarcoma model [58].
4. Bortezomib-bisphosphonate conjugates.
Bortezomib (Btz) is a proteasome inhibitor used as a first-line treatment for multiple myeloma (MM) and mantle cell lymphoma [59]. It induces myeloma cell apoptosis by promoting excessive intracellular accumulation of ubiquitinated proteins [60] via reversible occupation of the active proteolytic site of the 26S proteasome [61, 62]. Btz also promotes osteoblast differentiation [63], reduces osteoclast formation [64], and increases bone volume in mice [65, 66]. However, the therapeutic efficacy of Btz is limited by the development of drug resistance [67] and systemic adverse effects on non-bone tissues, mainly peripheral neuropathy and thrombocytopenia [68]. Thus, there is a clinical need to develop methods to deliver Btz or other anti-MM drugs to bone, the major site where myeloma cells reside and induce debilitating osteolysis, at effective concentrations with a much lower risk of systemic side effects.
4.1. Bortezomib-alendronate conjugates.
Several BP-linked Btz conjugates have been developed and tested in in-vitro cell cultures or in preclinical mouse models. In 2013, Agyin et al. linked PS-341 (=Btz) as well as MG-262, an experimentally used proteasome inhibitor, to Alendronate (Aln) using a non-cleavable linkage. These conjugates inhibited myeloma cell growth in-vitro [41]. In 2014, Swami et al. engineered bone-homing polymeric nanoparticles by linking them to Aln and then developed a library of Btz-loaded Aln-nanoparticles. The Aln-nanoparticle-Btz increased survival and decreased tumor burden in a MM mouse model in a prevention protocol (mice were pretreated for 3 weeks before myeloma cell inoculation) compared to Btz alone [69]. In 2018, Wang et al. synthesized an Aln-nanoparticle-Btz complex using a pH-sensitive linkage, which increased release of Btz from 20–30% at pH 7.4 to 60–70% at pH 5. The conjugate was given to mice 3 weeks after intra-tibia injection of MDA-231 breast cancer cells. The Aln-nanoparticle-Btz complex reduced tumor burden and bone destruction more effectively than Btz alone [70]. The effects of these Btz complexes on Btz resistance and neurotoxicity, critical limitations of Btz therapy, were not investigated in these studies.
4.2. Drug-releasing BP conjugates of bortezomib.
Our group has recently synthesized and studied novel bone-targeted forms of Btz by linking it with various drug-releasing linkers to a BP, which has high affinity for bone, but lacks anti-resorptive activity. The linker was designed to be sensitive to the acidic environment of the bone surface where bone turnover occurs (Fig. 4A). We found that the lead bisphosphonate-bortezomib conjugate (BP-Btz), but not Btz, bound to bone slices and inhibited the growth of MM cells in-vitro [71]. Importantly, BP-Btz reduced tumor burden and prevented bone loss in a mouse model of MM more effectively than Btz with less systemic side effects on platelets and dorsal root ganglia [71]. It is notable that this approach has also been effective in reducing myeloma cell formation away from the bone surface, deeper into the bone marrow. More studies are required to fully understand the mechanism of this off-bone effect. Based on these findings, there is a strong rationale for developing BP-Btz as a novel therapy to treat patients with MM. However, despite these encouraging results, major pharmaceutical companies, as of yet, have shown little interest in targeting Btz or other proteasome inhibitors to bone (personal experiences). This may in part be due to the early stage or pre-clinical research status of many of these programs. Although BP-Btz more effectively increases bone mass than Btz [71], it is unlikely that it will be attractive as a novel bone anabolic agent for non-neoplastic bone diseases, such as osteoporosis, because Btz targets proteasomes and inhibits proteasomal degradation non-specifically, and there are other bone anabolic drugs with more specific molecular mechanisms to increase bone volume [72, 73].
Figure 4.
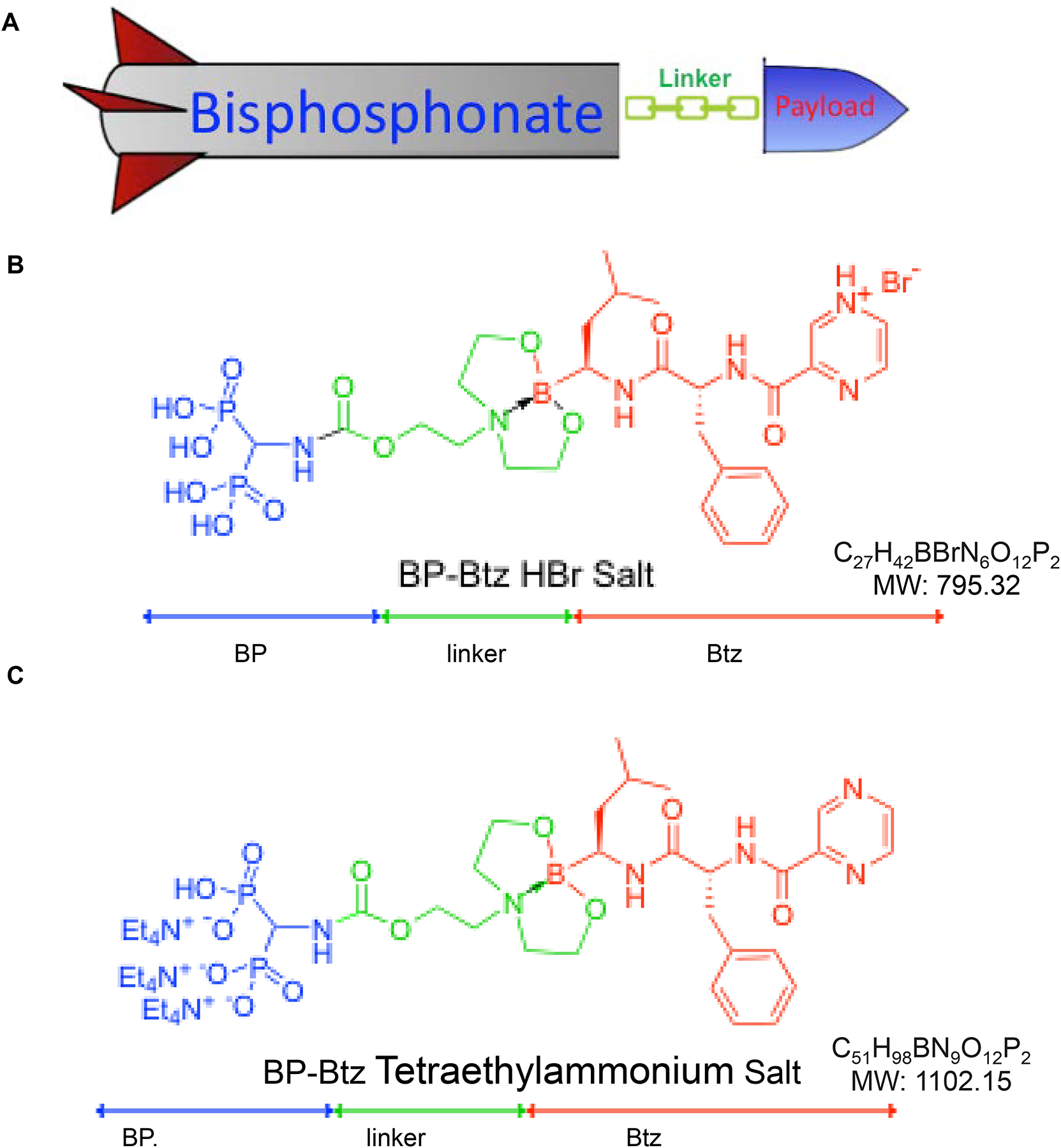
Cartoon of the bone-targeting delivery concept (A) and structure and formulation of BP-Btz (A) and UR70 (B).
4.3. Targeting bortezomib-resistant MM with a combination of bortezomib and chloroquine.
Another clinical challenge with the use of Btz is drug resistance. Bortezomib resistance (BR) develops in 50–77% of MM patients [67] via multiple mechanisms, including activation of chemo-resistance pathways, immunoproteasomes, mutations in proteasome subunits, and activation of autophagy [67]. This has led to development of 2nd and 3rd generation proteasome inhibitors [74]. These new drugs have been successful in the setting of BR, but they generally have a severe adverse effect profile [75], which has limited their widespread use. Thus, there remains an unmet need to overcome BR with novel approaches.
We hypothesized that by bone-targeting Btz, higher and sustained concentrations of Btz could be achieved locally and that these might be able to overcome Btz resistance. We measured Btz levels in plasma and bones from mice that we treated with equimolar doses of Btz or BP-Btz to determine if bone-targeting resulted in higher concentrations of Btz in bone in vivo. We detected nearly identical levels of Btz in plasma 10 minutes and 12 hours after mice were given either Btz or BP-Btz, and plasma levels had decreased to undetectable levels by 36 hours post-injection (Fig. 5). However, at 36 hours post injection, Btz levels were 4-fold higher in bone samples from mice treated with BP-Btz than from Btz-treated mice (Fig. 5).
Figure 5. BP-Btz has a longer half-life in bone than Btz.
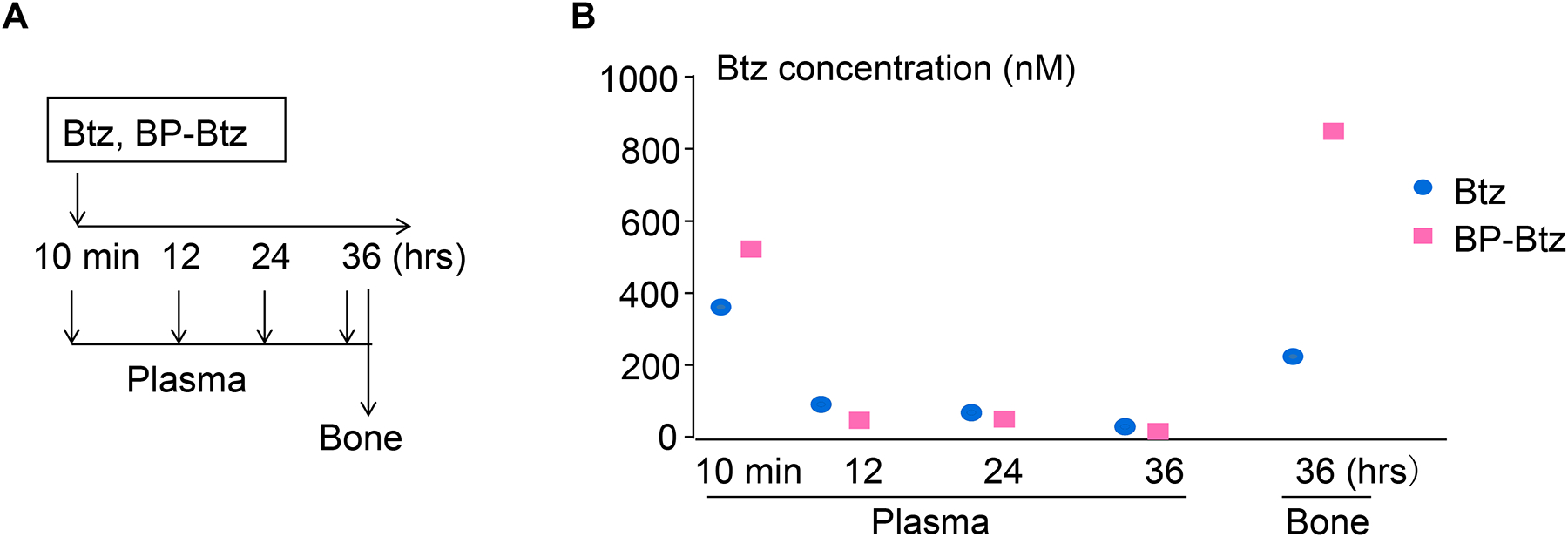
3 (A) 3.5-m-old female C57BL/6 mice were given single retro-orbital intravenous injections of equimolar doses of Btz (0.6mg/kg) or BP-Btz (1.243 mg/kg). Blood was drawn at 10 min, 12, 24, and 36 hr after injection. Leg bones were harvested at sacrifice at 36 hrs. (B) Btz levels in plasma and leg bones were measured in each mouse by liquid chromatography with liquid chromatography-mass spectrometry.
Because BP-Btz is extremely hygroscopic, which makes handling and storage challenging, we converted BP-Btz to its corresponding tris-tetraethyl ammonium salt (UR-70, Fig. 4B) to suppress its hygroscopicity and enhance shelf stability. Unlike BP-Btz, the salt remained as a free flowing solid, despite storage at room temperature for days. To determine if this new salt form of BP-Btz conjugate can affect Btz-resistant myeloma cells in vivo, we treated mice bearing 5TGM1-GFP-labeled Btz-resistant myeloma cells with UR70 or Btz and examined tumor burden using IVIS fluorescence imaging. We found that UR70 reduced tumor burden in mice bearing Btz-resistant myeloma cells, more effectively than Btz (Fig. 6). These findings suggest that our strategy to target Btz to bone could be efficacious in the clinical setting of BR.
Figure 6. Bone-targeted UR70 reduces tumor burden in mice bearing Btz-resistant myeloma cells.
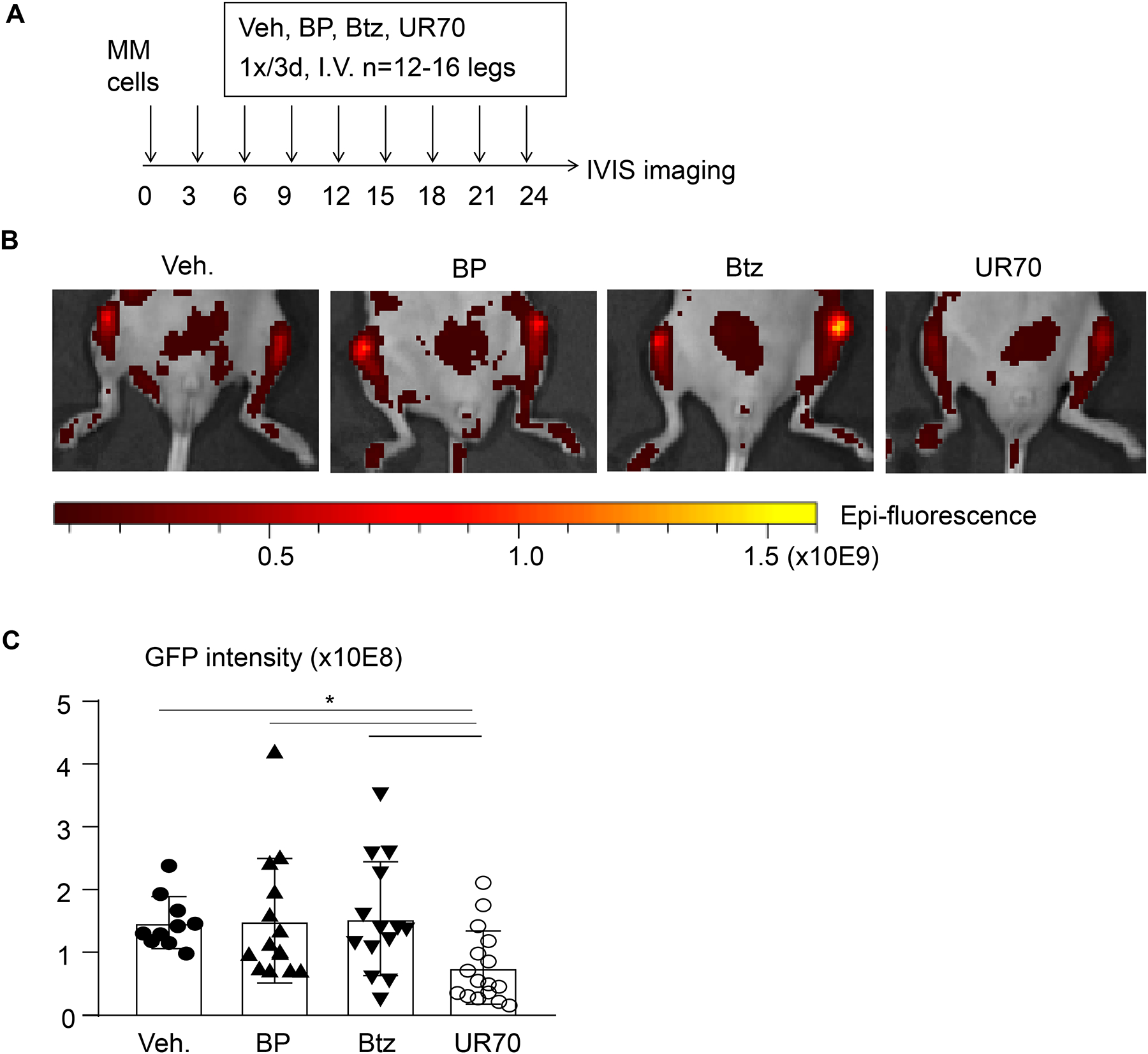
(A) Treatment protocol for 7-wk-old male NSG mice given injections of 0.5 × 106 Btz-resistant 5TGM1-GFP mouse myeloma cells into each tibia. 3 days after tumor cell inoculation, mice were given retro-orbital intravenous injections of equimolar concentrations of the BP (0.699mg/kg), Btz (0.6mg/kg) or UR70 (1.722mg/kg) every 3 days for 8 cycles. (B) Representative images of legs from mice from each group 3 days after the last injection, visualized with an IVIS Spectrum in vivo imaging system and showing GFP-positive tumor in the injected tibiae. (C) Tumor burden assessed by GFP intensity. Unit: Average Radiant Efficiency [p/s/cm2/sr] / [μW/cm2]. Data are mean ± SD. One-way ANOVA with Turkey’s test. *: p<0.05 vs vehicle.
Chloroquine (CQ) is FDA-approved to treat malaria, and was used as an anti-inflammatory drug to treat rheumatoid arthritis and systemic lupus erythematosus until the 1980s when it was replaced by hydroxychloroquine (HCQ), which has a more favorable toxicity profile, including a lower incidence of cardiomyopathy and retinopathy [76]. As autophagy inhibitors, CQ and HCQ have also been studied extensively as anti-cancer agents administered alone or in combination with more than 40 chemotherapeutic drugs and/or radiation in animal models of various cancers, including breast, lung, melanoma, colon, liver and glioblastoma [76]. Autophagy is a homeostatic cellular recycling system responsible for degrading damaged or unnecessary cellular organelles and proteins [77]. Cancer cells can use autophagy as an energy source, and thus they can survive in an environment, such as hypoxic and acidic conditions, which is unfavorable for normal cells. CQ and HCQ are able to sensitize tumor cells to chemotherapeutic drugs and/or radiation and thus enhance their therapeutic efficacy. In most of these preclinical studies CQ and HCQ have had favorable effects on preventing the growth of most of these tumors. The first clinical trial with CQ was started in 2005 in combination with standard chemotherapy for glioblastoma multiforme. Since then, CQ and HCQ have been or are being studied in more than 30 clinical trials to evaluate their efficacy in combination with various standard treatments for several types of cancers. In most of these studies a positive or partial response was reported [76]. CQ and HCQ inhibit autophagy and promote apoptosis of cancer cells, but they also work by affecting CXCR4-CXCL12 signaling, Toll-like receptor 9, and p53 in cancer cells, and tumor-associated fibroblasts and vessels as well immune responses in the tumor microenvironment retinopathy [76].
We reported that CQ, as a lysosome inhibitor, suppresses OC formation by inhibiting the degradation of TNF receptor-associated factor 3 (TRAF3), an adaptor protein in TNF receptor family signaling, which negatively regulates NF-κB signaling [78]. By this inhibitory mechanism, CQ prevented ovariectomy-induced bone loss and PTH-induced osteoclastogenesis and marrow fibrous in mice [78]. CQ also inhibited TRAF3 degradation in OB precursors and inhibition of OB differentiation induced by TGFβ1 [79]. Thus, CQ could also have beneficial effects in patients with bone metastases by inhibiting tumor-induced osteolysis and possibly enhancing bone formation. We are therefore studying the effects of Btz in combination with CQ and HCQ in MM mouse models. We also have generated BP-linked CQ and HCQ conjugates and are testing their effects in mouse models of MM and menopause- and age-related osteoporosis.
Myeloma cells produce large amounts of immunoglobulins as well as misfolded proteins [80], accumulation of which can cause cell death [81]. Misfolded proteins are typically degraded by proteasomes and lysosomes, and a major mechanism of action of Btz is to prevent proteasomal degradation of these misfolded proteins to induce death of MM cells. A phase II clinical trial of Btz combined with CQ and cyclophosphamide carried out with the goal of testing the hypothesis that in the setting of relapsed and refractory MM simultaneous inhibition of proteasome and lysosome activity would be more effective than proteasome inhibition alone [82].
A phase II clinical trial was performed between October 2011 and April 2013 to determine if addition of chloroquine to a combination Btz and cyclophosphamide might have a synergistic therapeutic effect in 11 patients with refractory MM after 2 cycles of therapy. One patient withdrew early in cycle 1 and 2 patients progressed before completing cycle 1. Of the 8 evaluable patients who completed at least 2 cycles, 3 had a partial response, 1 had stable disease and 4 progressed, resulting in a clinical benefit rate of 4/10 (40%) with what was considered to be an acceptable toxicity profile [82].
4.4. Unsolved issues and future work for BP-Btz.
Although significant progress has been made targeting BP-Btz to bone in animal models of MM, there still are several unsolved issues, which need further investigation: 1) How does BP-Btz kill myeloma cells in cell cultures in-vitro? Is the whole conjugate taken up intact by MM cells to function as a conjugate or is Btz released intracellularly? If so, what is the mechanism, or does Btz need to be released first by a cellular mechanism on the cell membrane? 2) How far can Btz molecules released from bone surfaces travel within the bone marrow to reach and kill myeloma cells that can be hundreds of μm away from a bone surface? 3) Does BP-Btz regulate the functions of bone and myeloma cells differently from Btz because it remains in the bone microenvironment longer than Btz and at higher concentrations for longer periods? This could provide the added benefit of building new bone at sites of osteolysis. 4) Could BP-Btz be used as an anabolic drug to inhibit bone resorption and stimulate new bone formation to treat osteoporosis or enhance fracture repair? 5) Is Btz the best MM drug to target to bone to treat patients with MM, particularly those who develop Btz resistance? Would bone-targeted new generation proteasome inhibitors, such as carfilzomib, be more effective than BP-Btz? 6) Would combinations of BP-Btz with other anti-MM drugs be effective against Btz resistance than these other drugs alone? 7) Which other anti-cancer drugs should be BP-linked to treat cancers that have spread to bone, such as breast and prostate cancer?
5. Conclusions
The technology to target drugs to bone to treat diseases affecting the skeleton has been discussed for over 30 years. Since then, several small biotechnology companies and academics groups have successfully conjugated FDA-approved drugs or other small molecule drug development candidates to bisphosphonates to target them to bone. These approaches have attempted to more effectively treat common bone diseases and to reduce side effects of the drugs that limit the amounts that can be given to patients. The most promising include bisphosphonates releasably-linked to estrogen, prostaglandins, fluoroquinolones, cytarabine, gemcitabine, doxorubicin and bortezomib. Pre-clinical studies have demonstrated significant efficacy of several antineoplastic conjugates in metastatic bone disease and multiple myeloma, and in one case encouraging success with these conjugates has been reported in phase 1 and early phase 2 clinical trials. Despite these successes, no drugs have been developed or studied in phase 3 clinical trials, and no major pharmaceutical companies have shown sufficient interest in this technology to use it to target any of their drugs to bone. Some reasons for this could be the costs involved in developing and testing these conjugates, availability of 2nd and 3rd generation proteasome inhibitors, and reluctance to draw attention to side effects of drugs that they currently have on the market. Nevertheless, interest shown by smaller biotechnology companies in the technology suggests that bone targeting of drugs with bisphosphonates has a viable future.
Acknowledgements:
This work was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under Award Number R01AR43510 (to BFB), R21AR070984 (to BFB and RKB), R01AR063650 (to LX), R21AR069789 (to LX and RKB) and UR Ventures—Technology Development Fund (to BFB and RKB), and by the National Institute on Aging under award number R01AG049994 (to BFB and ZY). Tomi K. Sawyer, William C. Shakespeare and David Dalgarno worked at Ariad Pharmaceuticals where they directed Src kinase inhibitor discovery. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- 1.Roodman GD, Mechanisms of bone metastasis. N Engl J Med, 2004. 350(16): p. 1655–64. DOI: 10.1056/NEJMra030831. [DOI] [PubMed] [Google Scholar]
- 2.Mundy GR, Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer, 2002. 2(8): p. 584–93. DOI: 10.1038/nrc867. [DOI] [PubMed] [Google Scholar]
- 3.Chin H and Kim J, Bone Metastasis: Concise Overview. Fed Pract, 2015. 32(2): p. 24–30. [PMC free article] [PubMed] [Google Scholar]
- 4.Hernandez RK, Wade SW, Reich A, Pirolli M, Liede A, and Lyman GH, Incidence of bone metastases in patients with solid tumors: analysis of oncology electronic medical records in the United States. BMC Cancer, 2018. 18(1): p. 44. DOI: 10.1186/s12885-017-3922-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cetin K, Christiansen CF, Svaerke C, Jacobsen JB, and Sorensen HT, Survival in patients with breast cancer with bone metastasis: a Danish population-based cohort study on the prognostic impact of initial stage of disease at breast cancer diagnosis and length of the bone metastasis-free interval. BMJ Open, 2015. 5(4): p. e007702. DOI: 10.1136/bmjopen-2015-007702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sathiakumar N, Delzell E, Morrisey MA, Falkson C, Yong M, Chia V, Blackburn J, Arora T, and Kilgore ML, Mortality following bone metastasis and skeletal-related events among men with prostate cancer: a population-based analysis of US Medicare beneficiaries, 1999–2006. Prostate Cancer Prostatic Dis, 2011. 14(2): p. 177–83. DOI: 10.1038/pcan.2011.7. [DOI] [PubMed] [Google Scholar]
- 7.Sathiakumar N, Delzell E, Morrisey MA, Falkson C, Yong M, Chia V, Blackburn J, Arora T, Brill I, and Kilgore ML, Mortality following bone metastasis and skeletal-related events among women with breast cancer: a population-based analysis of U.S. Medicare beneficiaries, 1999–2006. Breast Cancer Res Treat, 2012. 131(1): p. 231–8. DOI: 10.1007/s10549-011-1721-x. [DOI] [PubMed] [Google Scholar]
- 8.von Eisenhart-Rothe R, Toepfer A, Salzmann M, Schauwecker J, Gollwitzer H, and Rechl H, [Primary malignant bone tumors]. Orthopade, 2011. 40(12): p. 1121–42. DOI: 10.1007/s00132-011-1866-7. [DOI] [PubMed] [Google Scholar]
- 9.Damron TA, Ward WG, and Stewart A, Osteosarcoma, chondrosarcoma, and Ewing’s sarcoma: National Cancer Data Base Report. Clin Orthop Relat Res, 2007. 459: p. 40–7. DOI: 10.1097/BLO.0b013e318059b8c9. [DOI] [PubMed] [Google Scholar]
- 10.Linabery AM and Ross JA, Childhood and adolescent cancer survival in the US by race and ethnicity for the diagnostic period 1975–1999. Cancer, 2008. 113(9): p. 2575–96. DOI: 10.1002/cncr.23866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Geller DS and Gorlick R, Osteosarcoma: a review of diagnosis, management, and treatment strategies. Clin Adv Hematol Oncol, 2010. 8(10): p. 705–18. [PubMed] [Google Scholar]
- 12.Ottaviani G and Jaffe N, The etiology of osteosarcoma. Cancer Treat Res, 2009. 152: p. 15–32. DOI: 10.1007/978-1-4419-0284-9_2. [DOI] [PubMed] [Google Scholar]
- 13.Sasikumar A, Joy A, Pillai MRA, Alex TM, and Narayanan G, 68Ga-PSMA PET/CT in Osteosarcoma in Fibrous Dysplasia. Clin Nucl Med, 2017. 42(6): p. 446–447. DOI: 10.1097/RLU.0000000000001646. [DOI] [PubMed] [Google Scholar]
- 14.Moreno F, Cacciavillano W, Cipolla M, Coirini M, Streitenberger P, Lopez Marti J, Palladino M, Morici M, Onoratelli M, Drago G, Schifino A, Cores M, Rose A, Jotomliansky J, Varel M, and Garcia Lombardi M, Childhood osteosarcoma: Incidence and survival in Argentina. Report from the National Pediatric Cancer Registry, ROHA Network 2000–2013. Pediatr Blood Cancer, 2017. 64(10). DOI: 10.1002/pbc.26533. [DOI] [PubMed] [Google Scholar]
- 15.Mirabello L, Troisi RJ, and Savage SA, Osteosarcoma incidence and survival rates from 1973 to 2004: data from the Surveillance, Epidemiology, and End Results Program. Cancer, 2009. 115(7): p. 1531–43. DOI: 10.1002/cncr.24121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kazandjian D, Multiple myeloma epidemiology and survival: A unique malignancy. Semin Oncol, 2016. 43(6): p. 676–681. DOI: 10.1053/j.seminoncol.2016.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thorsteinsdottir S, Gislason G, Aspelund T, Sverrisdottir I, Landgren O, Turesson I, Bjorkholm M, and Kristinsson SY, Fractures and survival in multiple myeloma: results from a population-based study. Haematologica, 2019. DOI: 10.3324/haematol.2019.230011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Teras LR, DeSantis CE, Cerhan JR, Morton LM, Jemal A, and Flowers CR, 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J Clin, 2016. 66(6): p. 443–459. DOI: 10.3322/caac.21357. [DOI] [PubMed] [Google Scholar]
- 19.Pozzi S, Marcheselli L, Bari A, Liardo EV, Marcheselli R, Luminari S, Quaresima M, Cirilli C, Ferri P, Federico M, and Sacchi S, Survival of multiple myeloma patients in the era of novel therapies confirms the improvement in patients younger than 75 years: a population-based analysis. Br J Haematol, 2013. 163(1): p. 40–6. DOI: 10.1111/bjh.12465. [DOI] [PubMed] [Google Scholar]
- 20.Palumbo A, Bringhen S, Ludwig H, Dimopoulos MA, Blade J, Mateos MV, Rosinol L, Boccadoro M, Cavo M, Lokhorst H, Zweegman S, Terpos E, Davies F, Driessen C, Gimsing P, Gramatzki M, Hajek R, Johnsen HE, Leal Da Costa F, Sezer O, Spencer A, Beksac M, Morgan G, Einsele H, San Miguel JF, and Sonneveld P, Personalized therapy in multiple myeloma according to patient age and vulnerability: a report of the European Myeloma Network (EMN). Blood, 2011. 118(17): p. 4519–29. DOI: 10.1182/blood-2011-06-358812. [DOI] [PubMed] [Google Scholar]
- 21.Palumbo A and Anderson K, Multiple myeloma. N Engl J Med, 2011. 364(11): p. 1046–60. DOI: 10.1056/NEJMra1011442. [DOI] [PubMed] [Google Scholar]
- 22.Abe M, Hiura K, Ozaki S, Kido S, and Matsumoto T, Vicious cycle between myeloma cell binding to bone marrow stromal cells via VLA-4-VCAM-1 adhesion and macrophage inflammatory protein-1alpha and MIP-1beta production. J Bone Miner Metab, 2009. 27(1): p. 16–23. DOI: 10.1007/s00774-008-0012-z. [DOI] [PubMed] [Google Scholar]
- 23.Marino S and Roodman GD, Multiple Myeloma and Bone: The Fatal Interaction. Cold Spring Harb Perspect Med, 2018. 8(8). DOI: 10.1101/cshperspect.a031286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bodo E, Tobin DJ, Kamenisch Y, Biro T, Berneburg M, Funk W, and Paus R, Dissecting the impact of chemotherapy on the human hair follicle: a pragmatic in vitro assay for studying the pathogenesis and potential management of hair follicle dystrophy. Am J Pathol, 2007. 171(4): p. 1153–67. DOI: 10.2353/ajpath.2007.061164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Farrell KB, Karpeisky A, Thamm DH, and Zinnen S, Bisphosphonate conjugation for bone specific drug targeting. Bone Rep, 2018. 9: p. 47–60. DOI: 10.1016/j.bonr.2018.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Russell RG, Bisphosphonates: the first 40 years. Bone, 2011. 49(1): p. 2–19. DOI: 10.1016/j.bone.2011.04.022. [DOI] [PubMed] [Google Scholar]
- 27.Roelofs AJ, Stewart CA, Sun S, Blazewska KM, Kashemirov BA, McKenna CE, Russell RG, Rogers MJ, Lundy MW, Ebetino FH, and Coxon FP, Influence of bone affinity on the skeletal distribution of fluorescently labeled bisphosphonates in vivo. J Bone Miner Res, 2012. 27(4): p. 835–47. DOI: 10.1002/jbmr.1543. [DOI] [PubMed] [Google Scholar]
- 28.Puljula E, Turhanen P, Vepsalainen J, Monteil M, Lecouvey M, and Weisell J, Structural requirements for bisphosphonate binding on hydroxyapatite: NMR study of bisphosphonate partial esters. ACS Med Chem Lett, 2015. 6(4): p. 397–401. DOI: 10.1021/ml5004603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Young RN and Grynpas MD, Targeting therapeutics to bone by conjugation with bisphosphonates. Curr Opin Pharmacol, 2018. 40: p. 87–94. DOI: 10.1016/j.coph.2018.03.010. [DOI] [PubMed] [Google Scholar]
- 30.Cole LE, Vargo-Gogola T, and Roeder RK, Targeted delivery to bone and mineral deposits using bisphosphonate ligands. Adv Drug Deliv Rev, 2016. 99(Pt A): p. 12–27. DOI: 10.1016/j.addr.2015.10.005. [DOI] [PubMed] [Google Scholar]
- 31.Zhang S, Gangal G, and Uludag H, ‘Magic bullets’ for bone diseases: progress in rational design of bone-seeking medicinal agents. Chem Soc Rev, 2007. 36(3): p. 507–31. DOI: 10.1039/b512310k. [DOI] [PubMed] [Google Scholar]
- 32.Shakespeare WC, Metcalf CA 3rd, Wang Y, Sundaramoorthi R, Keenan T, Weigele M, Bohacek RS, Dalgarno DC, and Sawyer TK, Novel bone-targeted Src tyrosine kinase inhibitor drug discovery. Curr Opin Drug Discov Devel, 2003. 6(5): p. 729–41. [PubMed] [Google Scholar]
- 33.Shakespeare WC, Wang Y, Bohacek R, Keenan T, Sundaramoorthi R, Metcalf C 3rd, Dilauro A, Roeloffzen S, Liu S, Saltmarsh J, Paramanathan G, Dalgarno D, Narula S, Pradeepan S, van Schravendijk MR, Keats J, Ram M, Liou S, Adams S, Wardwell S, Bogus J, Iuliucci J, Weigele M, Xing L, Boyce B, and Sawyer TK, SAR of carbon-linked, 2-substituted purines: synthesis and characterization of AP23451 as a novel bone-targeted inhibitor of Src tyrosine kinase with in vivo anti-resorptive activity. Chem Biol Drug Des, 2008. 71(2): p. 97–105. DOI: 10.1111/j.1747-0285.2007.00615.x. [DOI] [PubMed] [Google Scholar]
- 34.Boyce BF, Xing L, Yao Z, Yamashita T, Shakespeare WC, Wang Y, Metcalf CA 3rd, Sundaramoorthi R, Dalgarno DC, Iuliucci JD, and Sawyer TK, SRC inhibitors in metastatic bone disease. Clin Cancer Res, 2006. 12(20 Pt 2): p. 6291s–6295s. DOI: 10.1158/1078-0432.CCR-06-0991. [DOI] [PubMed] [Google Scholar]
- 35.Irby RB and Yeatman TJ, Role of Src expression and activation in human cancer. Oncogene, 2000. 19(49): p. 5636–42. DOI: 10.1038/sj.onc.1203912. [DOI] [PubMed] [Google Scholar]
- 36.Boyce BF, Yoneda T, Lowe C, Soriano P, and Mundy GR, Requirement of pp60c-src expression for osteoclasts to form ruffled borders and resorb bone in mice. J Clin Invest, 1992. 90(4): p. 1622–7. DOI: 10.1172/JCI116032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hughes DE, Wright KR, Uy HL, Sasaki A, Yoneda T, Roodman GD, Mundy GR, and Boyce BF, Bisphosphonates promote apoptosis in murine osteoclasts in vitro and in vivo. J Bone Miner Res, 1995. 10(10): p. 1478–87. DOI: 10.1002/jbmr.5650101008. [DOI] [PubMed] [Google Scholar]
- 38.Soriano P, Montgomery C, Geske R, and Bradley A, Targeted disruption of the c-src proto-oncogene leads to osteopetrosis in mice. Cell, 1991. 64(4): p. 693–702. DOI: 10.1016/0092-8674(91)90499-o. [DOI] [PubMed] [Google Scholar]
- 39.Ebrahimpour A, Francis MD, Bisphosphonate therapy in acute and chronic bone loss: Physical chemical considerations in bisphosphonate-related therapies. Bisphosphonate on Bones, 1995. Chapter 8: p. 125–137. [Google Scholar]
- 40.Sunberg RJE, F. H, Mosher CT, Roof C, Designing drugs for stronger bones. Chemtech, 1991. 21: p. 304–309. [Google Scholar]
- 41.Agyin JK, Santhamma B, and Roy SS, Design, synthesis, and biological evaluation of bone-targeted proteasome inhibitors for multiple myeloma. Bioorg Med Chem Lett, 2013. 23(23): p. 6455–8. DOI: 10.1016/j.bmcl.2013.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morioka M, Kamizono A, Takikawa H, Mori A, Ueno H, Kadowaki S, Nakao Y, Kato K, and Umezawa K, Design, synthesis, and biological evaluation of novel estradiol-bisphosphonate conjugates as bone-specific estrogens. Bioorg Med Chem, 2010. 18(3): p. 1143–8. DOI: 10.1016/j.bmc.2009.12.041. [DOI] [PubMed] [Google Scholar]
- 43.Sedghizadeh PP, Sun S, Junka AF, Richard E, Sadrerafi K, Mahabady S, Bakhshalian N, Tjokro N, Bartoszewicz M, Oleksy M, Szymczyk P, Lundy MW, Neighbors JD, Russell RG, McKenna CE, and Ebetino FH, Design, Synthesis, and Antimicrobial Evaluation of a Novel Bone-Targeting Bisphosphonate-Ciprofloxacin Conjugate for the Treatment of Osteomyelitis Biofilms. J Med Chem, 2017. 60(6): p. 2326–2343. DOI: 10.1021/acs.jmedchem.6b01615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arns S, Gibe R, Moreau A, Monzur Morshed M, and Young RN, Design and synthesis of novel bone-targeting dual-action pro-drugs for the treatment and reversal of osteoporosis. Bioorg Med Chem, 2012. 20(6): p. 2131–40. DOI: 10.1016/j.bmc.2012.01.024. [DOI] [PubMed] [Google Scholar]
- 45.Liu CC, Hu S, Chen G, Georgiou J, Arns S, Kumar NS, Young RN, and Grynpas MD, Novel EP4 receptor agonist-bisphosphonate conjugate drug (C1) promotes bone formation and improves vertebral mechanical properties in the ovariectomized rat model of postmenopausal bone loss. J Bone Miner Res, 2015. 30(4): p. 670–80. DOI: 10.1002/jbmr.2382. [DOI] [PubMed] [Google Scholar]
- 46.Tanaka KS, Dietrich E, Ciblat S, Metayer C, Arhin FF, Sarmiento I, Moeck G, Parr TR Jr., and Far AR, Synthesis and in vitro evaluation of bisphosphonated glycopeptide prodrugs for the treatment of osteomyelitis. Bioorg Med Chem Lett, 2010. 20(4): p. 1355–9. DOI: 10.1016/j.bmcl.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 47.Drake MT, Clarke BL, and Khosla S, Bisphosphonates: mechanism of action and role in clinical practice. Mayo Clin Proc, 2008. 83(9): p. 1032–45. DOI: 10.4065/83.9.1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Adler RA, Duration of anti-resorptive therapy for osteoporosis. Endocrine, 2016. 51(2): p. 222–4. DOI: 10.1007/s12020-015-0748-x. [DOI] [PubMed] [Google Scholar]
- 49.Ebetino FH, Hogan AM, Sun S, Tsoumpra MK, Duan X, Triffitt JT, Kwaasi AA, Dunford JE, Barnett BL, Oppermann U, Lundy MW, Boyde A, Kashemirov BA, McKenna CE, and Russell RG, The relationship between the chemistry and biological activity of the bisphosphonates. Bone, 2011. 49(1): p. 20–33. DOI: 10.1016/j.bone.2011.03.774. [DOI] [PubMed] [Google Scholar]
- 50.Cole LE, Vargo-Gogola T, and Roeder RK, Targeted delivery to bone and mineral deposits using bisphosphonate ligands. Adv Drug Deliv Rev, 2015. DOI: 10.1016/j.addr.2015.10.005. [DOI] [PubMed] [Google Scholar]
- 51.Escalante J, McQuade RM, Stojanovska V, and Nurgali K, Impact of chemotherapy on gastrointestinal functions and the enteric nervous system. Maturitas, 2017. 105: p. 23–29. DOI: 10.1016/j.maturitas.2017.04.021. [DOI] [PubMed] [Google Scholar]
- 52.Meyers CA, How chemotherapy damages the central nervous system. J Biol, 2008. 7(4): p. 11. DOI: 10.1186/jbiol73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Holmberg AR, Lerner UH, Alayia AA, Al-Mohanna M, Adra C, Marquez M, Meurling L, and Nilsson S, Development of a novel poly bisphosphonate conjugate for treatment of skeletal metastasis and osteoporosis. Int J Oncol, 2010. 37(3): p. 563–7. DOI: 10.3892/ijo_00000705. [DOI] [PubMed] [Google Scholar]
- 54.Thellenberg-Karlsson C, Nyman C, Nilsson S, Blom R, Marquez M, Castellanos E, and Holmberg AR, Bone-targeted Novel Cytotoxic Polybisphosphonate Conjugate in Castration-resistant Prostate Cancer: A Multicenter Phase 1 Study. Anticancer Res, 2016. 36(12): p. 6499–6504. DOI: 10.21873/anticanres.11249. [DOI] [PubMed] [Google Scholar]
- 55.Ioachimescu A and Licata A, Etidronate: what is its place in treatment of primary osteoporosis and other demineralizing diseases today? Curr Osteoporos Rep, 2007. 5(4): p. 165–9. DOI: 10.1007/s11914-007-0012-2. [DOI] [PubMed] [Google Scholar]
- 56.Zinnen SP, Karpeisky A, Von Hoff DD, Plekhova L, and Alexandrov A, First-in-Human Phase I Study of MBC-11, a Novel Bone-Targeted Cytarabine-Etidronate Conjugate in Patients with Cancer-Induced Bone Disease. Oncologist, 2019. 24(3): p. 303–e102. DOI: 10.1634/theoncologist.2018-0707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zinnen SP, Evaluation of a novel bone targeted aracytidine therapy in dogs with spontaneous osteosarcoma. Bone, 2011. 48: p. S51. [Google Scholar]
- 58.David E, Cagnol S, Goujon JY, Egorov M, Taurelle J, Benesteau C, Morandeau L, Moal C, Sicard M, Pairel S, Heymann D, Redini F, Gouin F, and Le Bot R, 12b80 - Hydroxybisphosphonate Linked Doxorubicin: Bone Targeted Strategy for Treatment of Osteosarcoma. Bioconjug Chem, 2019. 30(6): p. 1665–1676. DOI: 10.1021/acs.bioconjchem.9b00210. [DOI] [PubMed] [Google Scholar]
- 59.Robak P and Robak T, Bortezomib for the Treatment of Hematologic Malignancies: 15 Years Later. Drugs R D, 2019. 19(2): p. 73–92. DOI: 10.1007/s40268-019-0269-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hideshima T, Richardson PG, and Anderson KC, Mechanism of action of proteasome inhibitors and deacetylase inhibitors and the biological basis of synergy in multiple myeloma. Mol Cancer Ther, 2011. 10(11): p. 2034–42. DOI: 10.1158/1535-7163.MCT-11-043310/11/2034 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bonvini P, Zorzi E, Basso G, and Rosolen A, Bortezomib-mediated 26S proteasome inhibition causes cell-cycle arrest and induces apoptosis in CD-30+ anaplastic large cell lymphoma. Leukemia, 2007. 21(4): p. 838–42. DOI: 10.1038/sj.leu.2404528. [DOI] [PubMed] [Google Scholar]
- 62.Obeng EA, Carlson LM, Gutman DM, Harrington WJ Jr., Lee KP, and Boise LH, Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood, 2006. 107(12): p. 4907–16. DOI: 10.1182/blood-2005-08-3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Giuliani N, Morandi F, Tagliaferri S, Lazzaretti M, Bonomini S, Crugnola M, Mancini C, Martella E, Ferrari L, Tabilio A, and Rizzoli V, The proteasome inhibitor bortezomib affects osteoblast differentiation in vitro and in vivo in multiple myeloma patients. Blood, 2007. 110(1): p. 334–8. DOI: 10.1182/blood-2006-11-059188. [DOI] [PubMed] [Google Scholar]
- 64.Uy GL, Trivedi R, Peles S, Fisher NM, Zhang QJ, Tomasson MH, DiPersio JF, and Vij R, Bortezomib inhibits osteoclast activity in patients with multiple myeloma. Clin Lymphoma Myeloma, 2007. 7(9): p. 587–9. [DOI] [PubMed] [Google Scholar]
- 65.Khedgikar V, Kushwaha P, Gautam J, Verma A, Changkija B, Kumar A, Sharma S, Nagar GK, Singh D, Trivedi PK, Sangwan NS, Mishra PR, and Trivedi R, Withaferin A: a proteasomal inhibitor promotes healing after injury and exerts anabolic effect on osteoporotic bone. Cell Death Dis, 2013. 4: p. e778. DOI: 10.1038/cddis.2013.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mukherjee S, Raje N, Schoonmaker JA, Liu JC, Hideshima T, Wein MN, Jones DC, Vallet S, Bouxsein ML, Pozzi S, Chhetri S, Seo YD, Aronson JP, Patel C, Fulciniti M, Purton LE, Glimcher LH, Lian JB, Stein G, Anderson KC, and Scadden DT, Pharmacologic targeting of a stem/progenitor population in vivo is associated with enhanced bone regeneration in mice. J Clin Invest, 2008. 118(2): p. 491–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Niewerth D, Jansen G, Assaraf YG, Zweegman S, Kaspers GJ, and Cloos J, Molecular basis of resistance to proteasome inhibitors in hematological malignancies. Drug Resist Updat, 2015. 18: p. 18–35. DOI: 10.1016/j.drup.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 68.Mohty B, El-Cheikh J, Yakoub-Agha I, Moreau P, Harousseau JL, and Mohty M, Peripheral neuropathy and new treatments for multiple myeloma: background and practical recommendations. Haematologica, 2010. 95(2): p. 311–9. DOI: 10.3324/haematol.2009.012674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Swami A, Reagan MR, Basto P, Mishima Y, Kamaly N, Glavey S, Zhang S, Moschetta M, Seevaratnam D, Zhang Y, Liu J, Memarzadeh M, Wu J, Manier S, Shi J, Bertrand N, Lu ZN, Nagano K, Baron R, Sacco A, Roccaro AM, Farokhzad OC, and Ghobrial IM, Engineered nanomedicine for myeloma and bone microenvironment targeting. Proc Natl Acad Sci U S A, 2014. 111(28): p. 10287–92. DOI: 10.1073/pnas.1401337111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhu J, Huo Q, Xu M, Yang F, Li Y, Shi H, Niu Y, and Liu Y, Bortezomib-catechol conjugated prodrug micelles: combining bone targeting and aryl boronate-based pH-responsive drug release for cancer bone-metastasis therapy. Nanoscale, 2018. 10(38): p. 18387–18397. DOI: 10.1039/c8nr03899f. [DOI] [PubMed] [Google Scholar]
- 71.Wang H, Xiao L, Tao J, Srinivasan V, Boyce BF, Ebetino FH, Oyajobi BO, Boeckman RK Jr., and Xing L, Synthesis of a Bone-Targeted Bortezomib with In Vivo Anti-Myeloma Effects in Mice. Pharmaceutics, 2018. 10(3). DOI: 10.3390/pharmaceutics10030154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Russow G, Jahn D, Appelt J, Mardian S, Tsitsilonis S, and Keller J, Anabolic Therapies in Osteoporosis and Bone Regeneration. Int J Mol Sci, 2018. 20(1). DOI: 10.3390/ijms20010083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Seeman E and Martin TJ, Antiresorptive and anabolic agents in the prevention and reversal of bone fragility. Nat Rev Rheumatol, 2019. 15(4): p. 225–236. DOI: 10.1038/s41584-019-0172-3. [DOI] [PubMed] [Google Scholar]
- 74.Dou QP and Zonder JA, Overview of proteasome inhibitor-based anti-cancer therapies: perspective on bortezomib and second generation proteasome inhibitors versus future generation inhibitors of ubiquitin-proteasome system. Curr Cancer Drug Targets, 2014. 14(6): p. 517–36. DOI: 10.2174/1568009614666140804154511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McBride A, Klaus JO, and Stockerl-Goldstein K, Carfilzomib: a second-generation proteasome inhibitor for the treatment of multiple myeloma. Am J Health Syst Pharm, 2015. 72(5): p. 353–60. DOI: 10.2146/ajhp130281. [DOI] [PubMed] [Google Scholar]
- 76.Verbaanderd C, Maes H, Schaaf MB, Sukhatme VP, Pantziarka P, Sukhatme V, Agostinis P, and Bouche G, Repurposing Drugs in Oncology (ReDO)-chloroquine and hydroxychloroquine as anti-cancer agents. Ecancermedicalscience, 2017. 11: p. 781. DOI: 10.3332/ecancer.2017.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mizushima N and Komatsu M, Autophagy: renovation of cells and tissues. Cell, 2011. 147(4): p. 728–41. DOI: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 78.Xiu Y, Xu H, Zhao C, Li J, Morita Y, Yao Z, Xing L, and Boyce BF, Chloroquine reduces osteoclastogenesis in murine osteoporosis by preventing TRAF3 degradation. J Clin Invest, 2014. 124(1): p. 297–310. DOI: 10.1172/JCI66947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li J, Ayoub A, Xiu Y, Yin X, Sanders JO, Mesfin A, Xing L, Yao Z, and Boyce BF, TGFbeta-induced degradation of TRAF3 in mesenchymal progenitor cells causes age-related osteoporosis. Nat Commun, 2019. 10(1): p. 2795. DOI: 10.1038/s41467-019-10677-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Aronson LI and Davies FE, DangER: protein ovERload. Targeting protein degradation to treat myeloma. Haematologica, 2012. 97(8): p. 1119–30. DOI: 10.3324/haematol.2012.064923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rodriguez-Gonzalez A, Lin T, Ikeda AK, Simms-Waldrip T, Fu C, and Sakamoto KM, Role of the aggresome pathway in cancer: targeting histone deacetylase 6-dependent protein degradation. Cancer Res, 2008. 68(8): p. 2557–60. DOI: 10.1158/0008-5472.CAN-07-5989. [DOI] [PubMed] [Google Scholar]
- 82.Montanari F, Lu M, Marcus S, Saran A, Malankar A, and Mazumder A, A Phase II Trial of Chloroquine in Combination with Bortezomib and Cyclophosphamide in Patients with Relapsed and Refractory Multiple Myeloma. Blood, 2014. 124: p. 5775. [Google Scholar]