

SUMMARY
Programmed −1 ribosomal frameshifting (−1PRF) is a widely used translation recoding mechanism. HIV-1 expresses Gag-Pol protein from the Gag-coding mRNA through −1PRF, and the ratio of Gag to Gag-Pol is strictly maintained for efficient viral replication. Here, we report that the interferon-stimulated gene product C19orf66 (herein named Shiftless) is a host factor that inhibits the −1PRF of HIV-1. Shiftless (SFL) also inhibited the −1PRF of a variety of mRNAs from both viruses and cellular genes. SFL interacted with the −1PRF signal of target mRNA and translating ribosomes and caused premature translation termination at the frameshifting site. Downregulation of translation release factor eRF3 or eRF1 reduced SFL-mediated premature translation termination. We propose that SFL binding to target mRNA and the translating ribosome interferes with the frameshifting process. These findings identify SFL as a broad-spectrum inhibitor of −1PRF and help to further elucidate the mechanisms of −1PRF.
In Brief
A host HIV restriction factor inhibits programmed ribosomal frameshifting via direct interaction with ribosomes and frameshifting RNA to cause premature translation termination.
Graphical Abstract
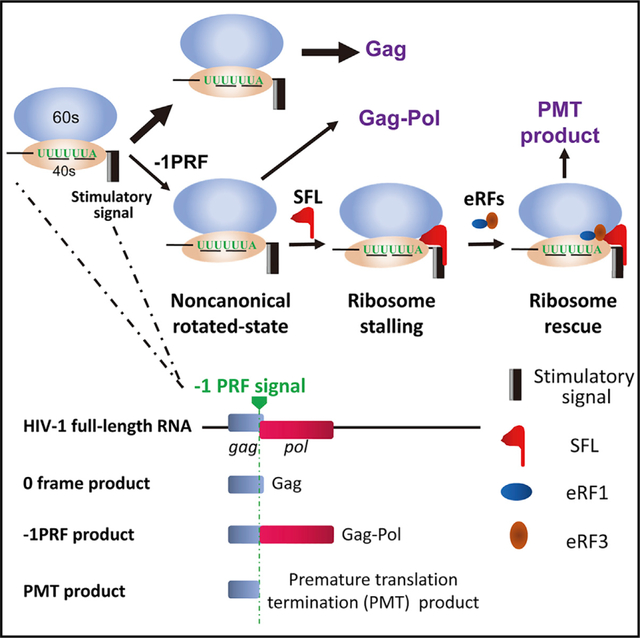
INTRODUCTION
Viral infection induces the production of type I interferons, which subsequently upregulate the expression of interferon-stimulated genes (ISGs). Multiple ISGs have been reported to inhibit the replication of HIV-1 at different steps of the viral life cycle by a variety of mechanisms (Schneider et al., 2014). HIV-1 uses the programmed −1 ribosomal frameshifting (−1PRF) mechanism to produce Gag-Pol polyprotein from the Gag-coding mRNA (Jacks et al., 1988; Wilson et al., 1988). How the −1PRF of HIV-1 is regulated by host factors remains to be elucidated.
−1PRF is a translation-recoding mechanism commonly used by viruses, wherein a proportion of translating ribosomes slip back one nucleotide such that the translation continues in a new reading frame (Atkins et al., 2016). HIV-1 Gag is translated from the unspliced viral RNA, which harbors a −1PRF signal near the end of the open reading frame of Gag. A fraction of the translation shifts the reading frame to −1, resulting in the expression of Gag-Pol. The ratio of Gag to Gag-Pol is strictly maintained for efficient virus assembly, genome packaging, and maturation (Brakier-Gingras et al., 2012; Dulude et al., 2006; Hung et al., 1998; Karacostas et al., 1993; Shehu-Xhilaga et al., 2001). −1PRF also exists in organisms from bacteria to higher eukaryotes (Belew et al., 2008; Caliskan et al., 2015). In mammalian cells, −1PRF of PEG10 (Clark et al., 2007) and Ma3 (Wills et al., 2006) mRNAs leads to the production of extended proteins, and −1PRF of CCR5 results in a premature stop codon and thereby mRNA degradation by nonsense-mediated mRNA decay (NMD) (Belew et al., 2014).
The −1PRF signal comprises two basic elements. One is the heptameric slippery sequence X XXY YYZ, wherein X is any nucleotide; Y is A or U; and Z is A, U, or C. In the original frame (0 frame), the codons are XXY and YYZ, and in the −1 frame, the codons are XXX and YYY. The slippery sequence is the place where −1PRF takes place. The other element is the stimulatory signal, an mRNA secondary structure at a particular distance downstream of the slippery sequence (Belew and Dinman, 2015; Caliskan et al., 2015).
The efficiency of −1PRF is regulated by both the cis-acting signal and trans-acting factors. Mutations in the stimulatory signal can significantly affect the efficiency of HIV-1 −1PRF (Dulude et al., 2006). Encephalomyocarditis virus (EMCV) protein 2A binding to the stimulatory signal dramatically increases the −1PRF efficiency (Napthine et al., 2017). MicroRNAs have been reported to increase −1PRF efficiency through binding and stabilizing the pseudoknot of CCR5 mRNA (Belew et al., 2014).
Here, we report that the host interferon-stimulated gene product C19orf66 (herein named Shiftless) is a trans-acting −1PRF inhibitor. We provide evidence suggesting that Shiftless inhibits −1PRF through binding to −1PRF RNA and the translating ribosome and thereby interfering with the −1PRF process.
RESULTS
SFL Inhibits the −1PRF of HIV-1
We first explored whether type I interferon (IFN) inhibits the expression of HIV-1 Gag-Pol. MT4 cells were infected with the replication-competent HIV-1 virus NL4–3 and then treated with IFN. Considering that IFN treatment may affect multiple aspects of the virus, we specifically focused on the expression of Gag-Pol versus Gag. IFN treatment slightly inhibited the expression of the viral proteins. Nonetheless, IFN treatment reproducibly reduced the ratio of Gag-Pol to Gag (Figure 1A, compare lanes 1 and 4), suggesting that IFN inhibited the −1PRF of HIV-1. We speculated that IFN inhibited −1PRF through ISGs and thus screened a subset of ISGs using a dual-luciferase reporter, pDual-HIV(−1). In this reporter, the −1PRF signal sequence was inserted between the coding sequences of Renilla luciferase (Rluc) and firefly luciferase (Fluc), with Fluc in −1 frame (Figure S1A). A 0 frame reporter pDual-HIV(0) was used as a control, in which the slippery sequence was mutated such that Fluc and Rluc were in the same reading frame (Figure S1A). Out of the 99 ISGs we screened (Table S1), C19orf66 displayed the highest inhibitory activity against the −1PRF reporter (Figure S1B) but had little effect on the 0 frame reporter (Figure S1C). Based on its function to inhibit −1PRF, C19orf66 was named Shiftless (SFL) hereafter.
Figure 1. SFL Inhibits HIV-1 Gag-Pol Expression.
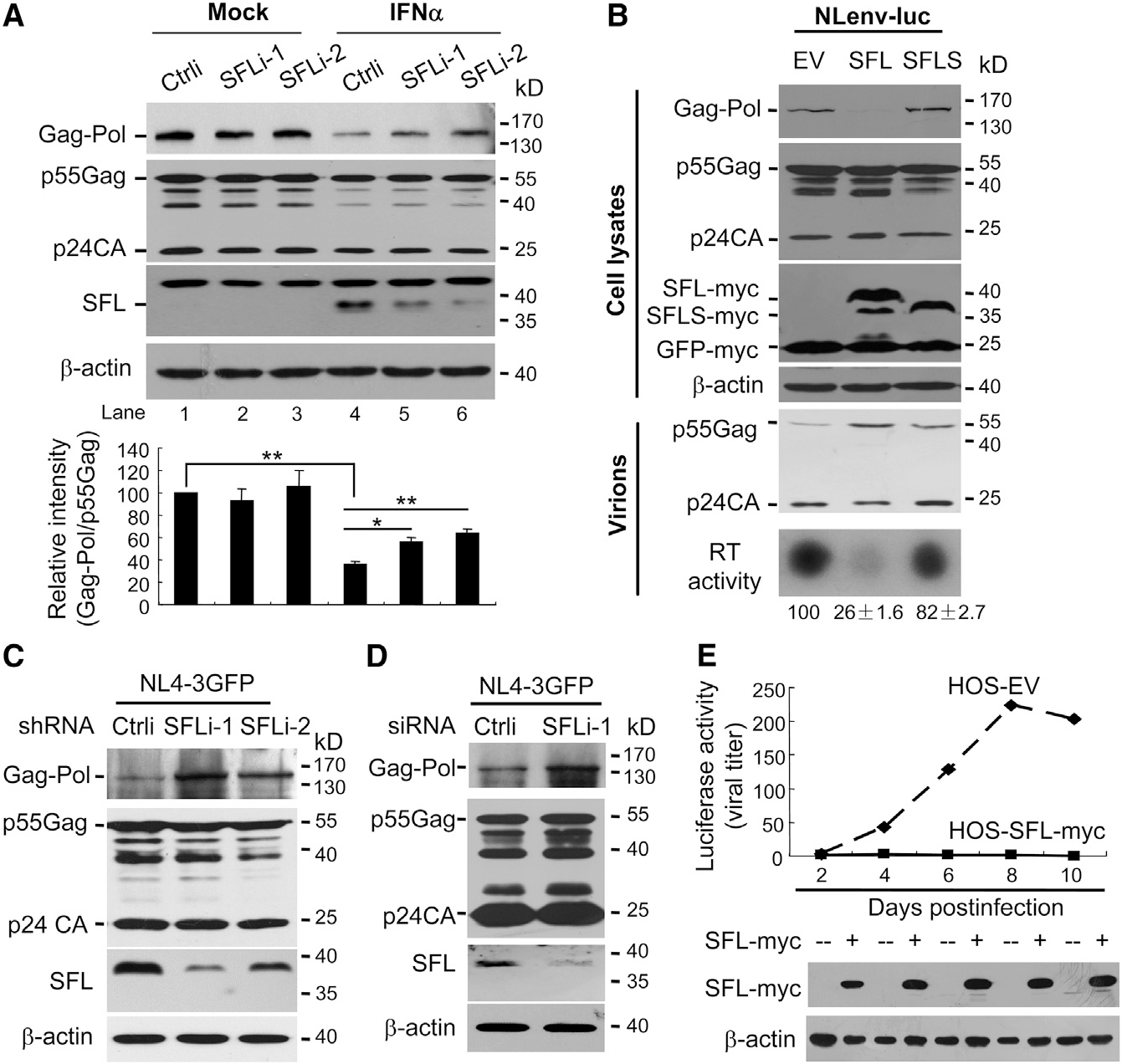
(A) MT4 cells stably expressing a control shRNA(Ctrli) or shRNAs targeting SFL (SFLi) were infected with NL4–3 virus, followed by treatment with IFN. Cell lysates were analyzed by western blotting. The band intensities were measured using ImageJ software. The intensity of Gag-Pol was normalized with that of p55Gag. The relative intensity in the control cells was set as 100. Data presented are means ± SD of three independent experiments. *p < 0.05; **p < 0.01.
(B) NLenv-luc was expressed in 293T cells together with SFL-Myc or SFLS-Myc. GFP-Myc was included to serve as a control. Cells were assayed for protein expressions. Virions in the supernatants were purified and analyzed for protein levels and RT activity. The relative RT activities are indicated. Data presented are means ± SD of three independent experiments. EV, empty vector.
(C) THP-1 cells stably expressing the shRNAs were transduced with VSV-G-pseudotyped NL4–3 GFP.
(D) Human monocyte-derived-macrophage (MDM) cells were transfected with a control siRNA or a siRNA targeting SFL and infected with VSV-G-pseudotyped NL4–3-GFP.
(E) HOS-CD4-CCR5 cells were infected with HIV-1 and then treated with doxycycline to induce SFL expression. At time points indicated, supernatants were tittered on TZM-bl cells. EV, cells stably transfected with an empty vector; HOS-SFL-Myc, cells expressing SFL-Myc.
See also Figures S1 and S2 and Table S1.
To probe the role of SFL in IFN inhibition of HIV-1 −1PRF, SFL expression was downregulated in MT4 cells using two short hairpin RNAs (shRNAs). The cells were infected with NL4–3 virus and then treated with IFN. Without IFN treatment, the expression of SFL in MT4 cells was below detection (Figure 1A, lanes 1–3). IFN treatment significantly upregulated SFL expression (Figure 1A, compare lanes 1 and 4), and the shRNAs reduced the expression levels (Figure 1A, lanes 4–6). Downregulation of SFL significantly increased Gag-Pol levels without obviously affecting Gag levels (Figure 1A, lanes 4–6). As noted, downregulation of SFL did not fully restore Gag-Pol expression (Figure 1A). One plausible explanation is that SFL was still expressed at low levels. Alternatively, SFL might not be the only player in IFN inhibition of HIV-1 −1PRF. Nonetheless, these results indicate that the endogenous SFL inhibits Gag-Pol expression and plays an important role in IFN inhibition of HIV-1 −1PRF.
There are two isoforms of C19orf66 arising from alternative splicing. The long form (SFL) comprises 291 amino acids. In the short form (SFLS), 36 amino acids (aas) (164–199) are missing. To test their functions, the two proteins were transiently expressed in 293T cells, together with NLenv-luc, an HIV-1 vector in which the coding sequence of firefly luciferase was inserted into that of Nef in NL4–3 (Dang et al., 2006). SFL significantly reduced the ratio of Gag-Pol to Gag (Figure 1B, upper panels). In contrast, SFLS had little effect (Figure 1B, upper panels). Gag-Pol is processed into multiple structural and enzymatic proteins, including the protease and reverse transcriptase. In agreement with the reduced Gag-Pol expression, SFL overexpression led to reduced RT activity and processing of p55Gag into p24CA in the virions (Figure 1B, lower panels). Similar results were observed with NL4–3 (Figure S2).
To further demonstrate that SFL inhibits −1PRF at the endogenous level, the two shRNAs targeting SFL were expressed in THP-1 cells, which express endogenous SFL at a detectable level (Figure 1C). The cells were transduced with vesicular stomatitis virus G protein (VSV-G) pseudotyped NL4–3-GFP and analyzed for Gag and Gag-Pol expressions. As expected, downregulation of SFL significantly increased Gag-Pol expression levels (Figure 1C). To validate the function of endogenous SFL in primary human cells, monocyte-derived macrophages from a healthy human donor were transfected with a small interfering RNA (siRNA) to downregulate SFL expression. The cells were incubated with Vpx-loaded simian immunodeficiency virus (SIV)-like particles (VLPs) to suppress the antiviral activity of endogenous SAMHD1 to promote subsequent transduction of the cells with VSV-G-pseudotyped NL4–3-GFP. Downregulation of SFL increased Gag-Pol levels without obviously affecting Gag levels (Figure 1D), indicating that the endogenous SFL inhibits Gag-Pol expression in these primary cells. We noticed that the p24CA levels in the cell lysates were relatively high compared to the p55Gag levels. One likely explanation is that the p24CA protein was from the infecting virions.
It has been well documented that adequate −1PRF efficiency and thus correct ratio of Gag-Pol to Gag is critical for HIV-1 replication (Brakier-Gingras et al., 2012; Dulude et al., 2006; Hung et al., 1998; Karacostas et al., 1993). Based on the above results, one would expect that SFL expression should alter the ratio and thereby inhibit HIV-1 replication. To test this idea, Myc-tagged SFL (SFL-Myc) was expressed in a doxycycline-inducible manner in HOS-CD4-CCR5 cells, a human osteosarcoma cell line engineered to express HIV-1 receptors and commonly used as HIV-1 reporter cells (Deng et al., 1996). The cells were infected with HIV-1 virus strain JRCSF. To avoid possible cytotoxicity of long-term SFL overexpression, the cells were used to grow the virus for only two days. A fraction of the culture supernatant was used to infect freshly seeded cells, and a fraction was used to infect TZM-bl cells to monitor the propagation of the virus. Indeed, propagation of the virus was significantly restricted in the SFL-expressing cells (Figure 1E). These results demonstrate that SFL inhibits HIV-1 replication.
SFL Is a Broad-Spectrum Inhibitor of −1PRF
We next explored whether SFL inhibits the −1PRF of other mRNAs. The −1PRF sequences from Rous sarcoma virus (RSV) (Marczinke et al., 1998), human T cell leukemia virus type II (HTLV-2) (Kim et al., 2001), mouse mammary tumor virus (MMTV) (Chamorro et al., 1992), HIV-2 (Le et al., 1991), simian immunodeficiency virus (SIVmac) (Marcheschi et al., 2007), Sindibis virus (SINV) (Snyder et al., 2013), and human cellular genes PEG10 (Clark et al., 2007) and CCR5 (Belew et al., 2014) were cloned into the dual-luciferase reporter and tested for response to SFL expression. SFL significantly reduced the −1PRF efficiencies of all the reporters (Figure 2A; Table S2), suggesting that SFL is a broad-spectrum inhibitor of −1PRF. To substantiate this notion, we analyzed the effect of SFL on the production or replication of the viruses or expression of the −1PRF products of the cellular genes. Like HIV-1, HIV-2 and SIVmac use the −1PRF mechanism to express Gag-Pol. VSV-G pseudotyped vectors NL4–3-GFP, HIV-2-GFP, and SIVmac-GFP were produced in 293T cells with or without SFL-Myc. The virion particles were used to infect recipient HeLa cells, with the number of GFP-positive recipient cells indicating the production of infectious virion particles. Overexpression of SFL significantly reduced the production of both HIV-2 and SIVmac (Figure 2B). SINV encodes a small protein 6K. During the translation of 6K, −1PRF leads to the expression of protein TF, which shares the same N-terminal domain with but differs at the C-terminal domain from 6K. TF deletion was reported to result in decreased viral production (Firth et al., 2008; Snyder et al., 2013). SFL inhibition of SINV −1PRF is expected to inhibit the expression of TF and thus the replication of the virus. SFL was expressed in a doxycycline-inducible manner in 293TREx cells. The cells were infected with SINV-nLuc, a replication-competent SINV carrying a nano luciferase reporter. The luciferase activity is expected to reflect the levels of viral replication. In line with the above results, SFL significantly inhibited the replication of the virus (Figure 2C).
Figure 2. SFL Is a Broad-Spectrum Inhibitor of −1PRF.
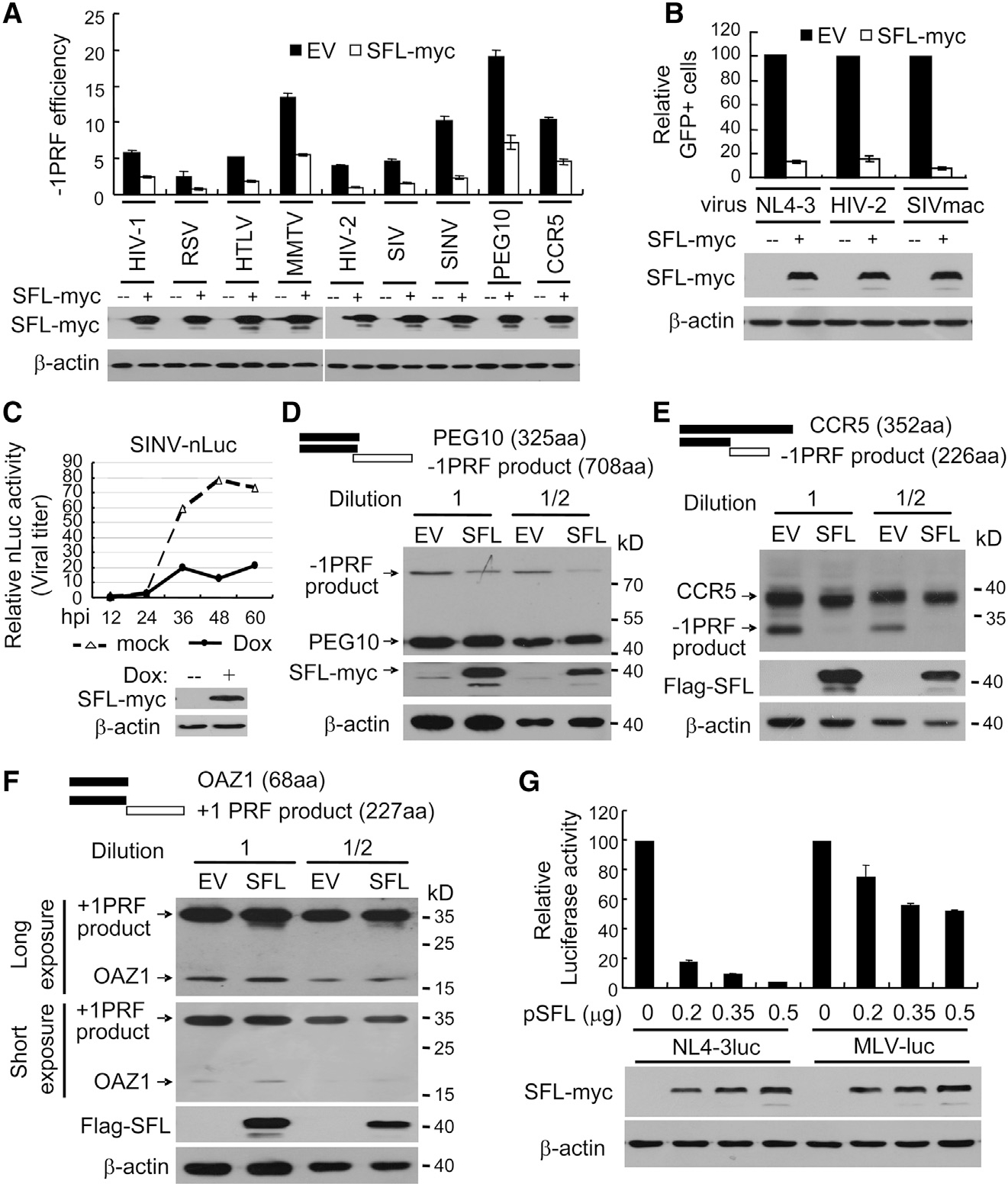
(A) Dual-luciferase reporters were expressed in 293T cells with or without SFL-Myc. The −1PRF efficiency of each reporter was calculated as the Fluc/Rluc ratio divided by that of the HIV(0) reporter.
(B) VSV-G-pseudotyped GFP-expressing lentivectors were produced in 293T cells with or without SFL-Myc and used to infect HeLa cells. The relative number of GFP-positive HeLa cells without SFL was set as 100.
(C) 293TREx-SFL-Myc cells were infected with SVNI-nLuc virus and mock treated or treated with doxycycline to induce SFL-Myc expression. At time points indicated, viral titers in culture supernatants were measured. hpi, hours post infection.
(D–F) N-terminally Myc-tagged PEG-10 (D), CCR5 (E), or OAZ1 (F) was transiently expressed in 293T cells, with or without SFL. Cell lysates were analyzed by western blotting.
(G) Plasmids producing VSV-G-pseudotyped NL4–3luc or MLV-luc were transfected into 293T cells with increasing amounts of a plasmid expressing SFL-Myc. The viruses were collected to infect 293T cells followed by luciferase activity measurement. The relative luciferase activity without SFL was set as 100. Data presented are means ± SD of three independent measurements, representative of three independent experiments.
See also Table S2.
Cellular human gene PEG10 encodes a protein of 325 aas from 0 frame and a −1PRF product of 708 aas (Clark et al., 2007; Figure 2D). The coding sequence of PEG10 was cloned into a mammalian expression vector with a Myc-tag at the N terminus and expressed in 293T cells with or without SFL-Myc. SFL reduced the ratio of the −1PRF product to the 0 frame product (Figure 2D). The −1PRF of endogenous CCR5 mRNA modulates the stability of the RNA (Belew et al., 2014). But −1PRF in the cDNA expression leads to the production of two proteins, one of 352 aas from 0 frame and one −1PRF product of 226 aas (Figure 2E). SFL considerably reduced the ratio of the −1PRF product to the full-length protein (Figure 2E). Collectively, these results indicate that SFL inhibits the −1PRF of a variety of mRNAs from both viruses and host.
To probe whether SFL affects other translation-recoding processes, we analyzed the effects of SFL on the expression of OAZ1 and the production of murine leukemia virus (MLV). OAZ1 mRNA expresses two proteins using the +1PRF mechanism (Matsufuji et al., 1995; Figure 2F). SFL had little effect on the expression of the +1PRF product (Figure 2F). MLV expresses Gag-Pol using a readthrough mechanism (Yoshinaka et al., 1985). Reduced Gag-Pol expression would result in reduced production of infectious virion particles. We thus used the production of the MLV vector carrying a luciferase reporter (MLV-luc) as an indicator of Gag-Pol expression. SFL overexpression had only marginal inhibitory effect on the production of infectious MLV-luc, although, under the same condition, SFL dramatically inhibited the production of HIV-1 vector NL4–3luc (Figure 2G). These results implicated that SFL did not significantly inhibit MLV Gag-Pol expression. The marginal inhibitory effect could be accounted for by the slight toxicity of SFL overexpression to the cells. Collectively, these results indicate that SFL specifically inhibits −1PRF.
SFL Interacts with the Ribosome
The process of −1PRF involves the translating ribosome and the −1PRF signal RNA. To understand how SFL inhibits this process, we first analyzed whether SFL interacts with ribosomes. SFL-Myc, SFLS-Myc, or GFP-Myc was expressed in 293T cells. Ribosomes in the cell lysates were pelleted through sucrose cushion, and associated proteins were detected by western blotting. SFL-Myc was easily detected in the pellet (Figure 3A), suggesting that SFL associates with ribosomes. In contrast, SFLS-Myc was not detected in the pellet, nor was GFP-Myc (Figure 3A). To test whether the endogenous SFL is associated with ribosomes, SFL was downregulated using two siRNAs in HeLa cells. The endogenous SFL was easily detected in the ribosome pellet (Figure 3B). When SFL was downregulated, little SFL was detected in the pellet (Figure 3B), confirming that the protein in the pellet from the control cells was indeed the endogenous SFL. To test whether SFL is associated with actively translating ribosomes, SFL-Myc or SFLS-Myc was co-expressed with GFP-Myc in 293T cells. The cell lysates were subjected to the polysome profiling assay. SFL-Myc was detected in most of the fractions (Figure 3C, left panel). In contrast, GFP-Myc was not detected in the ribosome-containing fractions (Figure 3C, left panel). In line with the above results, SFLS was not detected in the ribosome-containing fractions (Figure 3C, right panel). Collectively, these results indicate that SFL, but not SFLS, is associated with actively translating ribosomes.
Figure 3. SFL Interacts with Ribosomes.
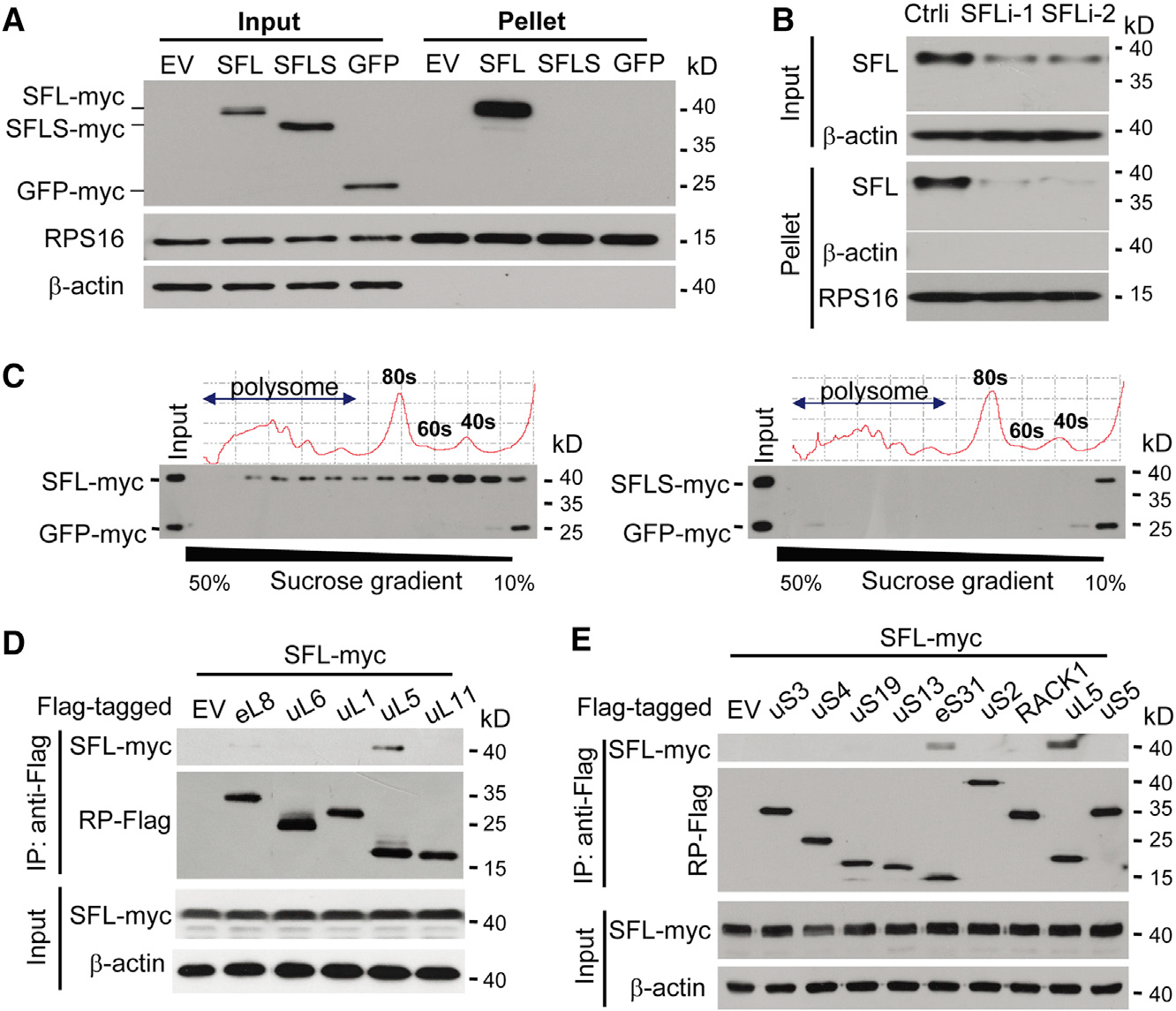
(A) Ribosomes in the lysates of 293T cells transiently expressing SFL-Myc, SFLS-Myc, or GFP-Myc were pelleted and analyzed by western blotting. RPS16 is an endogenous ribosomal protein.
(B) HeLa cells were transfected with a control siRNA or siRNAs targeting SFL. Ribosomes were pelleted and analyzed by western blotting.
(C) SFL-Myc (left) or SFLS-Myc (right) was transiently expressed in 293T cells with GFP-Myc. The cell lysates were subjected to sucrose gradient centrifugation, followed by polysome profiling analysis. Protein levels in each fraction were analyzed by western blotting.
(D and E) Interactions of SFL-Myc with Flag-tagged ribosome large-subunit (D) and small-subunit (E) proteins in 293T cells.
See also Figure S3.
We next analyzed which ribosomal proteins SFL interacts with. SFL-Myc was co-expressed with a subset of Flag-tagged ribosomal proteins in 293T cells and analyzed for their interactions by co-immunoprecipitation assays. To prevent possible nonspecific RNA and DNA tethering, cell lysates were treated with RNase A and DNase I. Immunoprecipitation of the large-subunit component uL5 (Figure 3D) and the small-subunit component eS31 (Figure 3E) coprecipitated SFL-Myc. In a reciprocal experiment, immunoprecipitation of SFL-Myc coprecipitated eS31 and uL5 (Figure S3A). To test whether the interactions of SFL with uL5 and eS31 were direct, bacterially expressed SFL fused with glutathione S-transferase (GST) was analyzed for interactions with bacterially expressed eS31 and uL5 fused with maltose binding protein (MBP) in the presence of RNase A. GST-SFL interacted with MBP-tagged eS31 and uL5, but not uL6 (Figures S3B and S3C). These results indicate that SFL interacts with the ribosomal proteins eS31 and uL5, though the interactions seemed to be weak. Surprisingly, SFLS also interacted with eS31 and uL5 in the co-immunoprecipitation assay (Figure S3D), suggesting that interactions with eS31 and uL5 alone were not enough to support sustained association of SFL with the ribosome (see below for discussion).
SFL Interacts with RNAs Harboring the −1PRF Signal
We next analyzed whether SFL interacts with the −1PRF RNA. The coding sequence of HIV-1 Gag-Pol/Gag was cloned into a mammalian expression vector. As a control, a mutant reporter was constructed in which the stimulatory sequence was mutated to disrupt the secondary structure. The reporters were expressed in 293T cells together with SFL-Myc or SFLS-Myc. The SFL proteins were immunoprecipitated, and the amounts of associated RNA were measured. SFL precipitation specifically enriched the wild-type Gag-Pol mRNA, but not the mutant reporter or the endogenous GAPDH mRNA (Figure 4A, upper panel), although comparable levels of the reporter mRNAs were expressed in the cells (Figure 4A, lower panel). SFLS failed to enrich any of the reporters (Figure 4A). These results indicate that SFL interacts with target mRNA.
Figure 4. SFL Interacts with −1PRF Signal RNA.
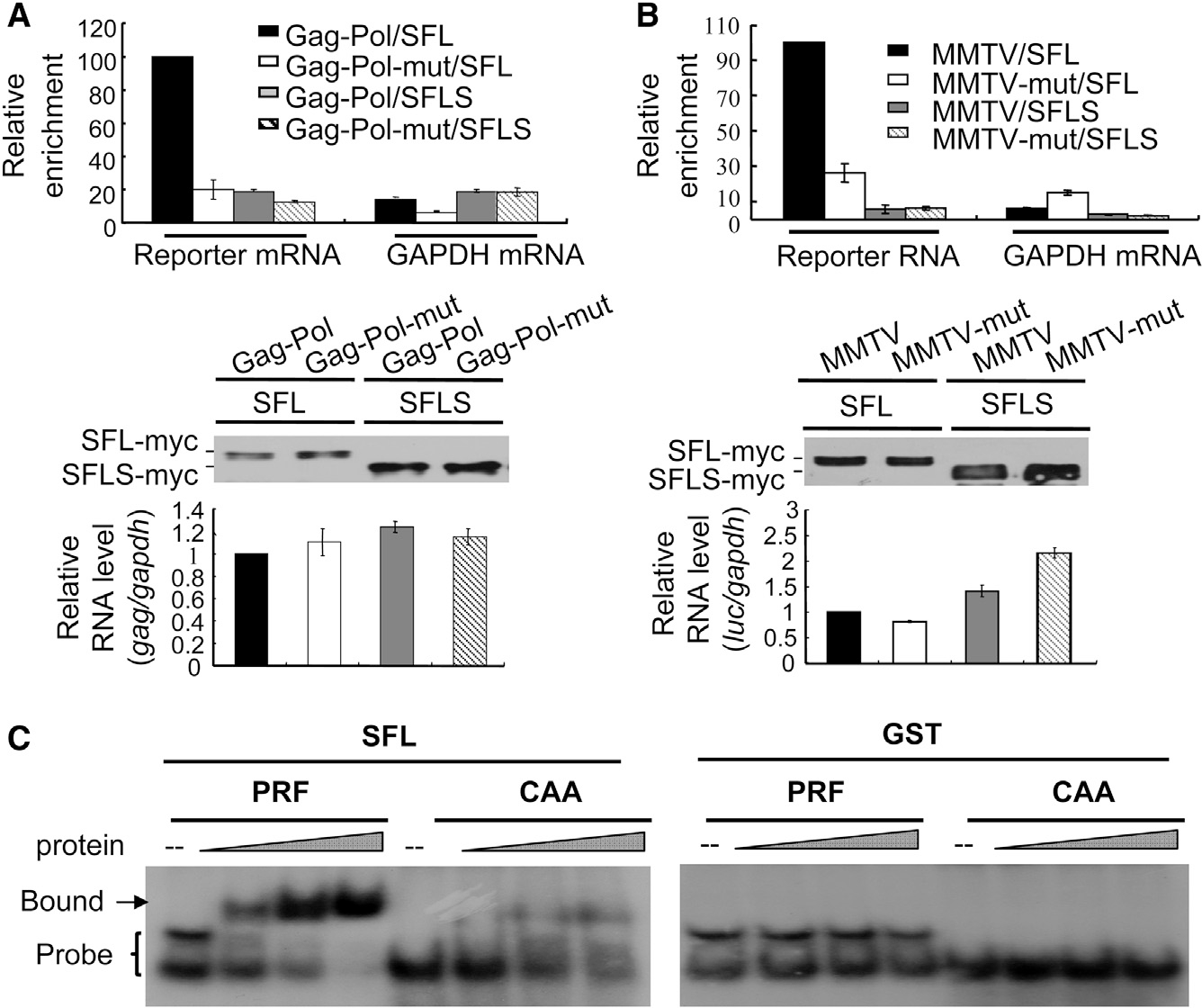
(A and B) A reporter expressing HIV-1 Gag-Pol (A) or the MMTV −1PRF signal RNA (B) was transiently transfected into 293T cells, with or without a plasmid expressing SFL-Myc or SFLS-Myc. Cell lysates were immunoprecipitated with anti-Myc antibody. Association of the RNA with SFL or SFLS was indicated by relative enrichment, which was calculated as the amount of RNA in the precipitates divided by that in the cell lysates (upper). The relative enrichment in the cells expressing the wild-type reporter and SFL was set as 100. Data presented are means ± SD of two independent measurements, representative of three independent experiments. That comparable amounts of proteins were immunoprecipitated was confirmed by western blotting (middle). The reporter RNA expression levels in the cells were measured (lower).
(C) EMSA of radio-labeled RNA with increasing amounts of SFL (left) or GST (right) protein. CAA, RNA probe of repeated CAA; PRF, RNA probe containing the −1PRF signal of HIV-1.
Because the translating mRNA is associated with ribosomes and SFL interacts with ribosomes, the association of SFL with Gag-Pol mRNA could be mediated by ribosomes even though the interaction was analyzed in the presence of EDTA. To further exclude this possibility, we analyzed the interaction of SFL with a −1PRF signal RNA that cannot be translated. The −1PRF sequence of MMTV in fusion with a fragment of luciferase sequence was cloned into a vector under the transcription control of H1 promoter. The RNA transcribed from this vector is not expected to have the 5′ Cap and 3′ polyA tail. The fragment of luciferase sequence allowed us to measure the RNA levels by reverse transcription-qPCR. In the control reporter (MMTV-mut), the −1PRF sequence was deleted. The reporters were expressed in 293T cells with SFL-Myc or SFLS-Myc. SFL proteins were immunoprecipitated, and the amounts of associated RNA were measured. SFL significantly enriched the wild-type reporter (Figure 4B, upper panel). In comparison, SFL enrichment of the mutant reporter was much lower (Figure 4B, upper panel), although comparable levels of reporter mRNAs were expressed in the cells (Figure 4B, lower panel). In line with the above results, SFLS failed to enrich any of the reporters (Figure 4B).
To test whether the interaction of SFL with target RNA is direct, we employed the electrophoretic mobility shift assay (EMSA). The −1PRF sequence of HIV-1 was generated by in vitro transcription and radio labeled. The RNA probe was incubated with increasing amounts of purified SFL protein and analyzed by native polyacrylamide electrophoresis. Two bands of the probe were observed (Figure 4C), which could likely be two structured forms of the RNA. SFL associated with the RNA in a dose-dependent manner (Figure 4C). To test whether the interaction of SFL with target RNA was specific, a control RNA containing repeated CAA, which is not expected to form any secondary structure, was analyzed for its interaction with SFL. Compared with the −1PRF signal RNA, the interaction of the control RNA with SFL was much weaker, though detectable (Figure 4C). As noted, SFLS would be an ideal control in this assay. However, SFLS proved to be unstable in vitro (data not shown). As a surrogate, we used purified GST as a control. GST did not bind to either of the RNA probes (Figure 4C). These results suggest that SFL is an RNA-binding protein and preferentially binds to −1PRF signal RNA.
SFL Inhibition of −1PRF Causes Premature Translation Termination
Previous studies indicated that the stimulatory signal causes the translating ribosome pausing at the −1PRF site to allow frameshifting to take place (Caliskan et al., 2014; Chen et al., 2014; Lopinski et al., 2000; Somogyi et al., 1993). We reasoned that SFL could either prolong the pausing to translation termination or help the ribosome to pass the blocking secondary structure to translate in the original frame. In the first scenario, a premature translation termination (PMT) product might appear. In the second scenario, no PMT product would be generated. To test this idea, we constructed a new reporter, in which the HIV-1 −1PRF signal was inserted between GST and Fluc with a tandem Flag-tag fused at the N terminus of GST (Figure 5A). This reporter is expected to express two proteins, one of 38 kDa from the 0 frame and a −1PRF product of 99 kDa. Premature translation termination around the slippery sequence would result in the third product of about 30 kDa (Figure 5A). Two mutant reporters were constructed to serve as controls (Figure 5A). In the 0 frame reporter, one nucleotide was inserted into the slippery sequence such that GST and Fluc were in the same reading frame. In the −1PRF-mut reporter, the slippery sequence was mutated such that −1PRF would be prevented and Fluc was still in the −1 frame. The reporters were expressed in 293T cells with or without SFL, and the products were analyzed by western blotting. For the −1PRF reporter, without SFL, only the 99-kDa and 38-kDa products were detected. However, in the presence of SFL, a product of 30 kDa was detected (Figure 5B). SFL had little effect on the expressions of the two control reporters (Figure 5B), strongly arguing against the possibility that the 30-kDa protein was a degradation product from either the 99-kDa or the 38-kDa protein. The absence of PMT product from the mutant reporter further suggested that PMT was caused by SFL inhibition of frameshifting. To probe whether SFL inhibition of HIV-1 Gag-Pol expression also causes premature translation termination, the protease coding sequence was deleted from NL4–3luc to minimize the background Gag proteins. A truncated Gag that ends at the first codon of the slippery sequence was used as a marker. SFL expression indeed led to the appearance of a specific band of the same size as the marker (Figure S4). Collectively, these results support the hypothesis that SFL inhibition of −1PRF causes premature translation termination.
Figure 5. SFL Inhibition of −1PRF Causes Premature Translation Termination.
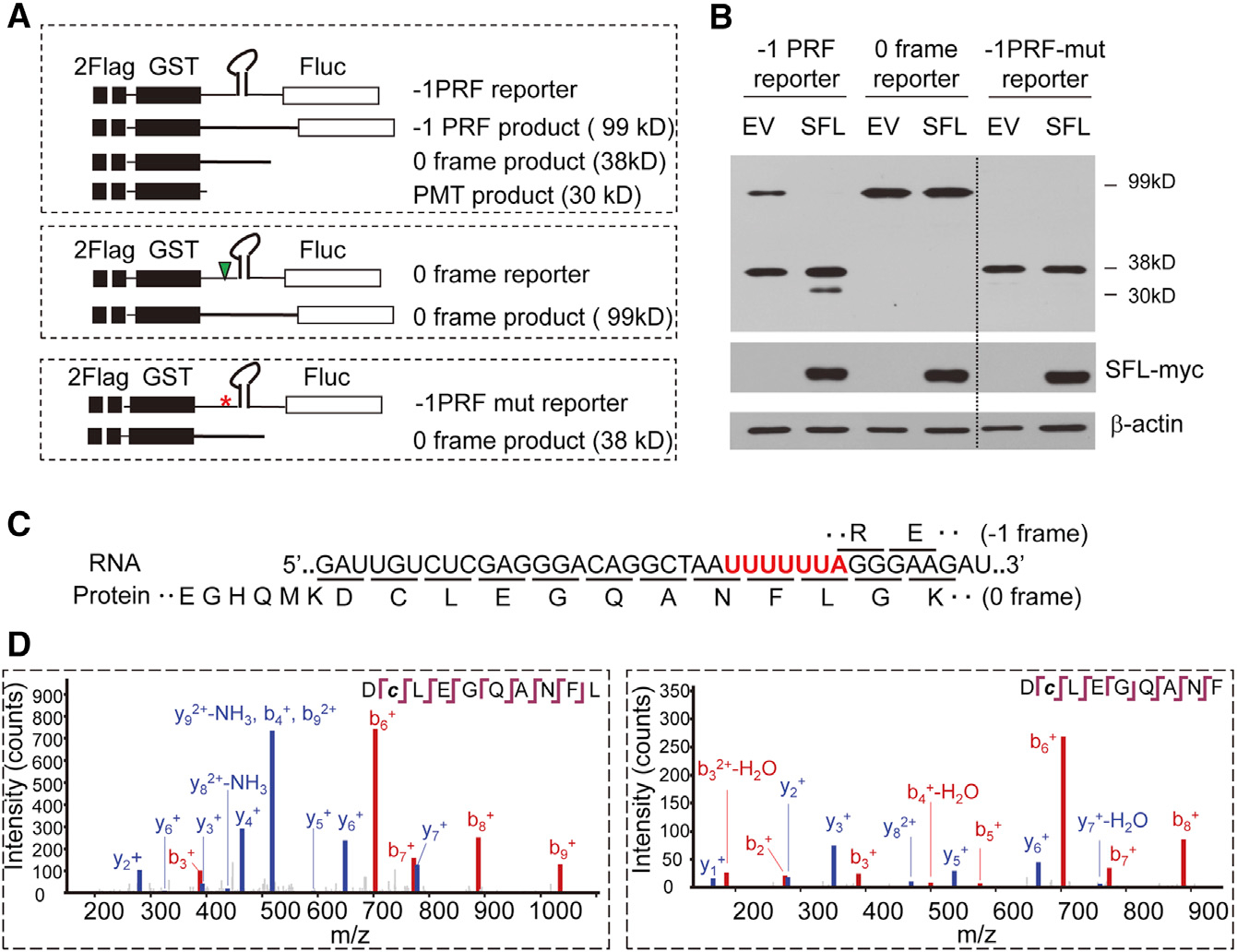
(A) Schematic representation of reporters and predicted protein products. The asterisk denotes mutations in the slippery sequence. The triangle denotes insertion of a nucleotide. PMT, premature translation termination.
(B) Reporters were transiently expressed in 293T cells with or without SFL-Myc and analyzed for protein expression. The dotted line indicates that irrelevant lanes were removed for easier comparison.
(C) Predicted amino acid sequences translated from 0 and −1 frames. The slippery sequence is in red with frames underlined.
(D) Tandem mass spectra for two peptides identified from the PMT products.
See also Figure S4.
To confirm the identity of the PMT product, mass spectrometric (MS) analysis was employed. According to the previously proposed model (Caliskan et al., 2014), at the slippery sequence, the ribosome harboring the nascent peptide-Asn-tRNAPhe would pause at the P site. After tRNALeu entering the A site and being incorporated into the peptide, frameshifting may take place. In 0 frame translation, the peptide sequence would be ANFLGK, and in −1 frame, the sequence would be ANFLRE (Figure 5C). The PMT product would end with either ANF or ANFL. The −1PRF reporter was transiently expressed in 293T cells together with SFL-Myc. The proteins were immunoprecipitated with anti-Flag antibody, resolved on SDS-PAGE, trypsinized, and subjected to MS analysis. In addition to the trypsinized peptides that typically end with amino acid K or R, two peptides with sequences of DCLEGQANF and DCLEGQANFL were detected (Figure 5D). These results are highly consistent with that premature translation termination occurred at the slippery sequence. These results also implicate that SFL inhibition of −1PRF can cause translation termination both before and after the incorporation of the leucine amino acid.
Downregulation of eRF1 or eRF3 Reduces SFL-Mediated Premature Translation Termination
The foregoing results showed that SFL interacted with the −1PRF signal of target mRNA and translating ribosome and caused premature translation termination. We reasoned that SFL could interfere with the rotation of the translating ribosome, causing ribosome stalling. In this case, a mechanism would be needed to rescue the ribosome. Two complexes, Pelo-HBS1L and eRF3-eRF1, have been reported in ribosome rescue in eukaryotic cells (Buskirk and Green, 2017; Chiabudini et al., 2014). To explore their roles in SFL-mediated premature translation termination, each factor was downregulated by a siRNA in 293T cells and analyzed for the effect on reporter expression. Downregulation of eRF3 significantly reduced the PMT product level (Figure 6A). Considering that the concentration of eRF1 has been reported to be much higher than that of eRF3 in human cells (Janzen and Geballe, 2004), we used two concentrations of targeting siRNA to downregulate the expression of endogenous eRF1. Under the same condition as eRF3 was downregulated, the effect of eRF1 downregulation was modest (Figure 6B). However, when eRF1 was further downregulated using a higher concentration of the siRNA, the level of the PMT product was significantly reduced (Figure 6B). In contrast, downregulation of Pelo or HBS1L (Figure S5) had little effect (Figure 6A). Noticeably, downregulation of eRF1 or eRF3 did not restore the levels of the −1PRF product (Figures 6A and 6B; see below for discussion). These results suggest that the eRF3-eRF1 complex is required for SFL-mediated premature translation termination.
Figure 6. Downregulation of eRF1 and eRF3 Reduces SFL-Mediated PMT.
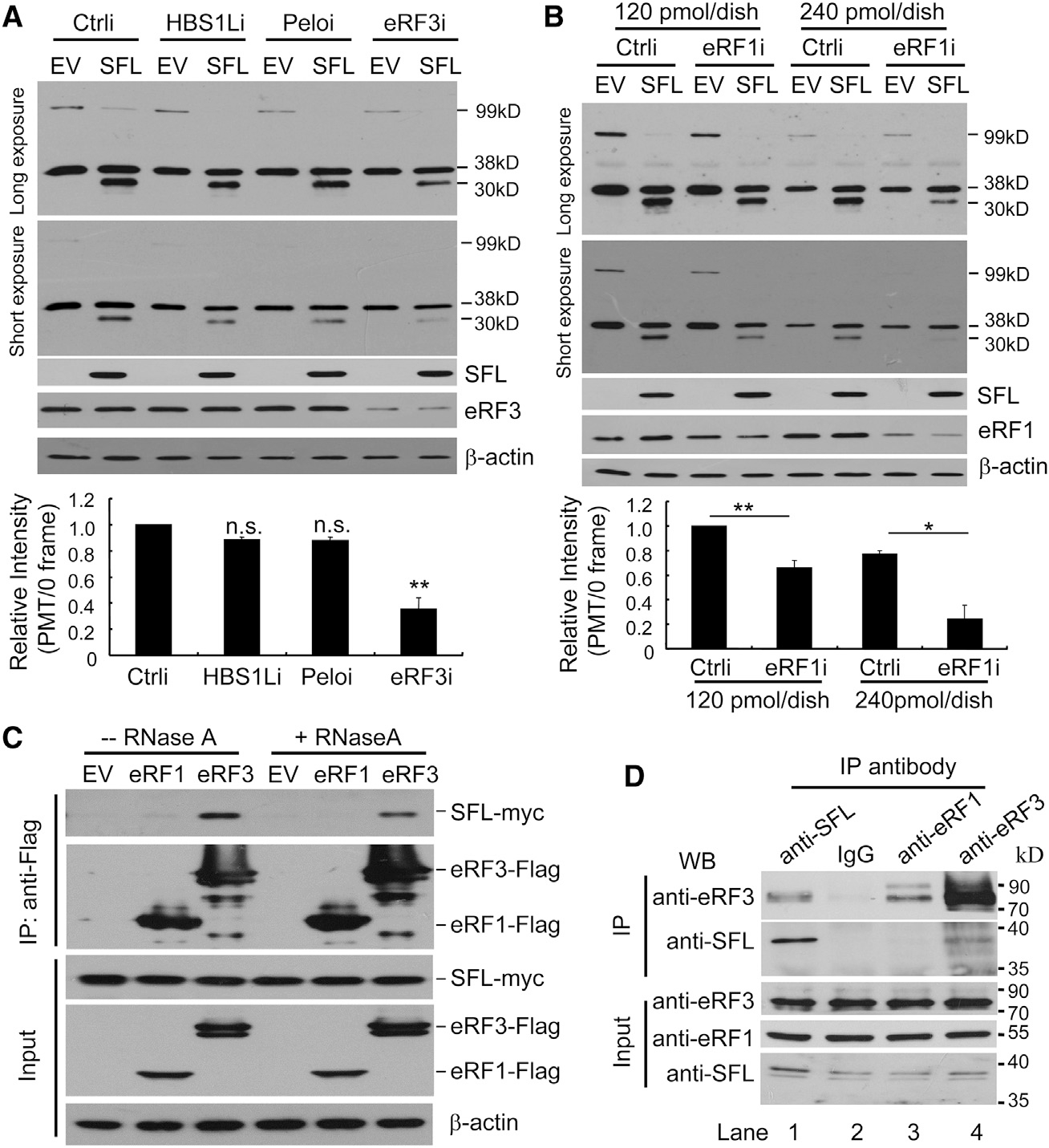
(A and B) The −1PRF reporter was expressed in 293T cells with or without SFL-Myc, followed by transfection of a siRNA targeting HBS1L, Pelo or eRF3 (A) or eRF1 (B). Cell lysates were analyzed for protein expressions. The band intensities were measured using ImageJ software. The intensity of the 30-kDa PMT product was normalized with that of the 38-kDa protein. The relative intensity in the control cells (Ctrli) with SFL-Myc was set as 1. Data presented are means ± SD of three experiments. n.s., p > 0.05; *p < 0.05; **p < 0.01. The amounts of the siRNA used in the transfection are indicated in (B).
(C) Interactions of SFL-Myc with Flag-tagged eRF1 and eRF3 in 293T cells.
(D) THP-1 cells were treated with IFN to enhance the expression of endogenous SFL. Cell lysates were immunoprecipitated with the antibodies indicated in the presence of RNase A and analyzed by western blotting.
See also Figures S5 and S6.
We next tested whether SFL interacts with eRF1 and eRF3. Flag-tagged eRF1 or eFR3 was co-expressed with SFL-Myc in 293T cells, and the interactions were analyzed by co-immunoprecipitation assays. SFL specifically interacted with eRF3, but not eRF1, with or without RNase A treatment (Figure 6C). To analyze the interaction between the endogenous eRF3 and SFL, THP-1 cells were treated with IFN to upregulate SFL expression. The cell lysates were immunoprecipitated with an anti-SFL, anti-eRF1, or anti-eRF3 antibody. Immunoprecipitation of SFL coprecipitated eRF3 (Figure 6D, lane 1). Consistently, immunoprecipitation of eRF3 coprecipitated SFL (Figure 6D, lane 4). In contrast, immunoprecipitation of eRF1 failed to coprecipitate SFL (Figure 6D, lane 3), although it coprecipitated eRF3 (Figure 6D, lane 3). These results indicate that the endogenous eRF3 and SFL interact. To test whether the interaction between eRF3 and SFL is direct, we analyzed the interactions between bacterially expressed MBP-eRF3 and GST-SFL. Indeed, they interacted (Figure S6). Collectively, these results indicate that SFL interacts with eRF3.
DISCUSSION
−1PRF is an important translation-recoding mechanism utilized by all domains of life to enhance the information content of the genome (Caliskan et al., 2015; Dinman, 2012). In addition, −1PRF may result in a premature stop codon, which could lead to the degradation of the mRNA (Belew et al., 2011, 2014; Plant et al., 2004). Computational analysis predicted that ~10% of cellular mRNAs might contain −1PRF signals in higher eukaryotes (Belew et al., 2008), though only a few of them have been confirmed. Here, we identified SFL as a broad-spectrum inhibitor of −1PRF (Figure 2). Further studies of SFL should be helpful in the validation of cellular −1PRF mRNAs.
SFL was associated with actively translating ribosomes (Figures 3A–3C). In contrast, SFLS, which did not inhibit HIV-1 −1PRF (Figure 1B), was not (Figures 3A and 3C). These results suggest that association with the translating ribosome is required for SFL inhibition of −1PRF. How SFL interacts with the translating ribosome is not clear. However, results from previous studies and results reported here provide some clues. SFL interacted with the ribosomal proteins uL5 and eS31 in the co-immunoprecipitation and pull-down assays (Figures 3 and S3). The interactions seemed to be weak, and we did not detect the interactions of endogenous SFL with uL5 or eS31 (data not shown). SFLS also interacted with uL5 and eS31 in the co-immunoprecipitation assay (Figure S3D), suggesting that interactions with uL5 and eS31 are not enough for SFL to stably associate with ribosomes. A difference between SFL and SFLS is in their ability to bind to −1PRF signal RNA (Figure 4), although other possible differences cannot be excluded. These results suggest that binding to the −1PRF signal RNA may be important for SFL association with ribosomes and inhibition of −1PRF.
Structural analyses revealed that uL5 is located in the large subunit close to the subunit interface and eS31 is located in the small subunit close to the interface (Svidritskiy et al., 2014; Figure 7A). The −1PRF signal RNA is supposed to be located in proximity to the mRNA entry tunnel. During the process of −1PRF, the ribosome undergoes conformational rearrangements of the ribosomal proteins, intersubunit rotations, and prolonged pausing at a noncanonical rotated state (Caliskan et al., 2014; Chen et al., 2014; Kim et al., 2014). The clockwise rotation of the small subunit relative to the large one brings uL5 closer to eS31 and the mRNA entry tunnel (Figure 7A). We speculate that the conformational rearrangements and intersubunit rotations might create a new binding surface for SFL to bind stably to the ribosome and the stimulatory RNA. The strengthened interactions of SFL with the translating ribosome might render the ribosome “stuck” in a non-productive, semi-rotated conformation. Noticeably, SFL protein molecules interacted with each other (Figure S7), which would allow SFL to act as a dimer to extend its ability to simultaneously interact with proteins and RNAs that are located far away from each other in space.
Figure 7. Working Model of SFL.
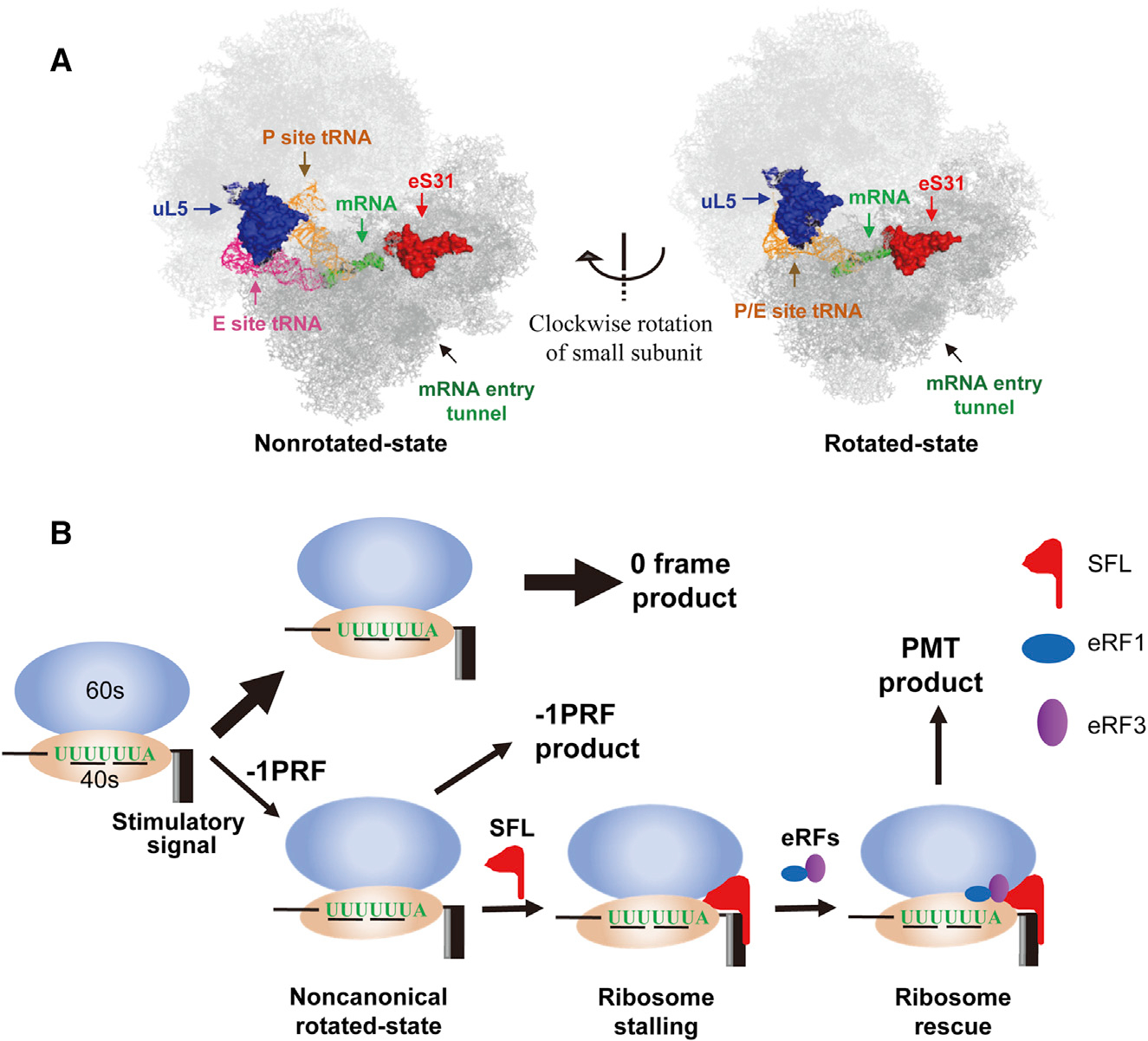
(A) Positions of uL5, eS31, and mRNA entry tunnel in the nonrotated state (left) and rotated state (right) of the ribosome. The structures are adapted from a previously published paper (Svidritskiy et al., 2014).
(B) A working model for SFL inhibition of −1PRF. When the translating ribosome is in a noncanonical rotated state during the process of −1PRF, SFL stably binds to the ribosome, causing ribosome stalling. The eRF1-eRF3 complex is recruited to rescue the stalled ribosome, leading to the production of PMT product.
See also Figure S7.
SFL did not obviously inhibit the expression of the 0 frame product from the −1PRF reporters (Figures 1, 2, 5, and 6), indicating that it does not interfere with the ribosomal rotations in 0 frame translation. The ribosomes translating in 0 and −1 frames differ in at least two aspects. First, the pausing time in the rotated state in −1 frame translation is much longer (Chen et al., 2014). Second, in 0 frame translation, the ribosome is in a canonical rotated state, and in −1 frame translation, the ribosome is in a noncanonical rotated state (Chen et al., 2014). It is conceivable that the binding affinities of SFL for the ribosomes in the two rotated states may be different. Prolonged pausing at the noncanonical rotated state would further facilitate the interactions of SFL with the ribosome.
SFL inhibition of −1PRF caused premature translation termination at the slippery site (Figure 5), and downregulation of eRF1 or eRF3 reduced the PMT product (Figure 6). It has been reported that, in bacterial −1PRF, enhanced pseudoknot structures acted as road blocks to cause ribosome stalling, leading to the production of a series of polypeptides (Tholstrup et al., 2012). In yeast, translation of the polyA tail of a no-stop mRNA caused ribosome stalling and the production of a PMT product and reduction in eRF3 expression level prevented the accumulation of the PMT product (Chiabudini et al., 2014). By analogy, SFL may cause ribosome stalling and lead to the production of premature polypeptides.
In the conventional translation termination, eRF1 in complex with eRF3 recognizes a stop codon and releases the peptide (Jackson et al., 2012). It has also been well documented that eRF3 can mediate translation termination in a manner independent of a stop codon, though the underlying mechanisms are not fully understood (Chiabudini et al., 2014; Doronina et al., 2008; Yan et al., 2010). In bacterial −1PRF, multiple premature translation products around the slippery sequence were detected (Yan et al., 2015). It was proposed that, during the frameshifting process, multiple ribosome translocation attempts led to branching of frameshifting pathways and that, in some pathways, translation was aborted by the retrospective fidelity check mechanism executed by the translation release factors (Yan et al., 2015; Zaher and Green, 2011). In SFL inhibition of −1PRF, the stalled ribosomes might be rescued by a similar mechanism. SFL interaction with eRF3 (Figures 6C and 6D) would be expected to facilitate the rescue. Without the translation release factors, SFL could still interfere with the frameshifting process, but the ribosome might eventually be able to “squeeze” across the secondary structure to continue the translation in 0 frame. Alternatively, the mRNA harboring stalled ribosomes could be degraded by the quality control mechanism. This would explain the observation that downregulation of eRF1 or eRF3 significantly reduced the PMT product but did not restore the expression level of the −1PRF product (Figures 6A and 6B).
Taking into account the results in this report and the above discussions, we propose a working model for SFL to inhibit −1PRF (Figure 7B). During the process of −1PRF, conformational rearrangements and intersubunit rotations of the ribosomes create a new binding surface for SFL, possibly acting as a dimer, to bind simultaneously to the translating ribosome and the stimulatory RNA, rendering the ribosome stuck in a non-productive, semi-rotated conformation. The eRF1-eRF3 complex is recruited to rescue the stalled ribosome, resulting in premature translation termination. SFL might also inhibit −1PRF by other mechanisms. For example, SFL could recruit RNA helicases to help unwind the secondary structure to facilitate the translation in 0 frame, thereby reducing the chance of −1PRF. These possible mechanisms do not seem to exclude each other. Further investigation is needed for in-depth understanding of the mechanisms for SFL inhibition of −1PRF.
SFL (C19orf66) was previously reported to inhibit the replication of a variety of viruses, including Dengue virus, hepatitis C virus (HCV), Kunjin virus (WNVkun), and Chikungunya virus (CHIKV) (Balinsky et al., 2017; Suzuki et al., 2016). It was proposed that C19orf66 inhibited the viral replication through suppressing the translation of the viral RNAs. To be noted, HCV (Choi et al., 2003), WNVkun (Firth and Atkins, 2009; Melian et al., 2010), and CHIKV (Ramsey and Mukhopadhyay, 2017) contain −1PRF signals. Whether Shiftless inhibits the replication of these viruses through inhibiting the −1PRF awaits further investigation. It is also possible that SFL may be a multifunctional protein that uses different mechanisms against different viruses.
In summary, here, we identified SFL as a broad-spectrum inhibitor of −1PRF. We propose that SFL causes ribosome stalling during the process of −1PRF and that the stalled ribosome is rescued by the eRF1-eRF3 complex. Further studies of SFL may help to better understand the mechanisms of −1PRF.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Guangxia Gao (gaogx@moon.ibp.ac.cn).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
293T (ATCC CRL-11268), HeLa (ATCC CCL-2), TZM-bl (NIH AIDS Reagent Program 8129), HOS-CD4-CCR5 (NIH AIDS Reagent Program 3318), BHK-21 (ATCC CCL-10) and 293TREx (Thermo Fisher R710–07) cells were maintained in DMEM supplemented with 10% FBS at 37°C, 5% CO2. MT4 (NIH AIDS Reagent Program 120), THP-1 (ATCC TIB-202) and MDM (this study) cells were maintained in RPMI-1640 supplemented with 10% heat-inactivated FBS at 37°C, 5% CO2.
The sex of HEK293T, HeLa, TZM-bl, HOS-CD4-CCR5 and 293TREx cells are female, while THP-1, MT4, BHK-21 and MDM cells are male.
The 293TREx cells expressing SFL-myc in a doxycycline-inducible manner were generated by transfecting 293TREx cells with pcDNA4-SFL-myc, followed by selection in zeocin-containing medium. Single colonies were picked, expanded and tested for doxycycline-inducible expression of the protein.
To generate HOS-CD4-CCR5 cells expressing SFL-myc, pEasiLV-MCS or pEasiLV-SFL-myc were packaged into VSV_G pseudotypted lentivectors in 293T cells to transduce HOS-CD4-CCR5 cells. After treatment with doxycycline for 48 h, E2-Crimson positive cells were collected by FACS. Doxycycline-inducible expression of SFL-myc was detected by western blotting. VSV_G pseudotyped pSuper-retro-GFP vectors expressing the control shRNA or shRNAs targeting SFL were generated in 293T cells to transduce MT4 or THP-1 cells, followed by FACS sorting for GFP-positive cells.
METHOD DETAILS
Plasmid construction
pLPCX-SFL and pLPCX-SFLS express SFL-myc and SFLS-myc, respectively. The coding sequences of C19orf66 isoform 1 (SFL; Genbank: NM_018381.3) and isoform 2 (SFLS; Genbank: NM_001308277.1) were PCR-amplified from a cDNA library from THP-1 cells, and cloned into the expression vector pLPCX (Clontech). A myc-tag was fused to the C terminus. SFL was also cloned into pcDNA4/TO/myc-HisB (Invitrogen) with a myc-tag at the C terminus and pCMV-HF with a Flag-tag at the N terminus. The SFL coding sequence was cloned into pGEX-5X-3 (GE Healthcare) to express GST-SFL in Ecoli. BL21. The SFL-myc coding sequence was cloned into the lentivector pEasiLV-MCS to express SFL-myc in a doxycycline-inducible manner.
The coding sequences of human PEG10 (Genbank: NM_015068.3), CCR5 (Genbank: NM_000579.3) and rat OAZ1 (Genbank: NM_139081.2) were cloned into pCMV-myc, with a myc-tag at the N terminus. The coding sequences of eRF1 (Genbank: NM_004730.3) and eRF3 (Genbank: NM_002094.3) were cloned into pcDNA4/TO/myc-HisB, with a tandem Flag-tag at the C terminus. The coding sequences of human ribosomal proteins uS3 (Genbank: NM_001005), uS4 (Genbank: NM_001013.3) uS19 (Genbank: NM_001018.4), uS13 (Genbank: NM_022551.2), eS31 (Genbank: NM_002954.5), uS2 (Genbank: NM_002295.5), RACK1 (Genbank: NM_006098.4), uS5 (Genbank: NM_002952.3), uL5 (Genbank: NM_000975.5), eL8 (Genbank: NM_000972.2), uL2 (Genbank: NM_000973.4), uL6 (Genbank: NM_000661.4), uL1 (Genbank: NM_007104.4) and uL11 (Genbank: NM_000976.3) were cloned into pcDNA4/TO/myc-HisB, with a tandem Flag-tag at the C terminus to express the ribosomal proteins. The coding sequences of eRF3, uL6, uL5 and eS31 were cloned into pMAL-C2X (New England Biolabs) to express MBP fusion proteins in Ecoli. BL21.
In pDual-HIV(−1), the −1PRF sequence from HIV-1 was inserted between the coding sequences of renilla luciferase (Rluc) and firefly luciferase (Fluc). For the control reporter pDual-HIV(0), an additional adenine was inserted immediately after the mutant slippery sequence such that Fluc and Rluc were in the same reading frame. The −1PRF signals from SIV (Marcheschi et al., 2007), HIV-2 (Le et al., 1991), SINV (Snyder et al., 2013), HTLV-2 (Kim et al., 2001), MMTV (Chamorro et al., 1992), RSV (Marczinke et al., 1998) and cellular genes PEG10 (Clark et al., 2007) and CCR5 (Belew et al., 2014) were synthesized or PCR-amplified to replace the −1PRF signal sequence of pDual-HIV(−1) to generate the dual luciferase reporters. The plasmids pSIVmac-GFP, pHIV-2rod-GFP, pSVNI-nluc, pCMV-myc-PEG10 and pCMV-myc-CCR5 were used as PCR templates. The −1PRF sequences of HTLV-2 (Genbank: NC_001488.1), MMTV (Genbank: NC_001503.1, the −1PRF between the coding sequences of Gag and Pro) and RSV (Genbank: AF033808.1) were synthesized (Sangon Biotech).
pEF1α-Gag-Pol expresses HIV-1 Gag-Pol under the transcription control of the EF1α promoter. To construct pEF1α-Gag-Pol, the CMV promoter and renilla luciferase coding sequence of phRL-CMV (Promega) were replaced with EF1α promoter and Gag-Pol coding sequence, respectively. To generate a control mutant, silent mutations were introduced into the stimulatory sequence (5′-TTTTTTAGGGAAGATCTGGCCTTCCTACAAGGGAAGGCCA mutated to 5′-TTTTTTAGGCAAGATATGGCCATCATACAAGGGAAGACCT).
pSuper-retro-MMTV(−1) expresses the MMTV −1PRF signal RNA under the transcription control of the H1 promoter. The MMTV −1PRF signal and part of Fluc sequence were PCR-amplified from pDual-MMTV(−1) and cloned into pSuper-Retro-GFP (OligoEngine). To generate a control mutant, the MMTV −1PRF signal was deleted.
pGST-HIV(−1)-Fluc is a −1PRF reporter modified from pDual–HIV(−1) for detecting reporter protein expression by western blotting. The coding sequence of GST in fusion with a tandem Flag-tag at the N terminus was used to replace the Rluc coding sequence of pDual-HIV-(1). To generate the mutant reporter, the slippery sequence (TTTTTTA) was mutated to CTTCCTC. To generate the 0 frame reporter pGST-HIV(0)-Fluc, the −1PRF signal was replaced with the mutant −1PRF signal from pDual-HIV(0). The reporter coding cassette was cloned into pEF1α-Gag-Pol to replace the Gag-Pol coding sequence.
To generate pNL4–3luc-DelPro, which expresses HIV-1 Gag-Pol with protease deleted, the SpeI-SbfI fragment covering part of the Gag-Pol coding sequence was cloned into an intermediate vector to delete the protease coding sequence. The fragment was then cloned back into pNL4–3luc. pCMV-PMT expresses truncated HIV-1 Gag corresponding to the predicted PMT product. To construct pCMV-PMT, the Gag-coding sequence with a stop codon immediately downstream of the slippery sequence was PCR-amplified from pNL4–3luc and cloned into pcDNA4-to-myc/hisB. The primers are listed in Table S3.
The plasmids expressing the shRNAs were generated by annealing pairs of oligonucleotides and cloning into pSuper-retro-GFP (OligoEngine) using BglII and HindIII sites following the manufacture’s instruction.
To generate −1PRF RNA in vitro, the sequence of 100 nt was PCR-amplified from pEF1α-Gag-Pol and cloned into pMD19-T (TaKaRa). The T7 promoter sequence was built in the upstream primer (PRF-SP) and an EcoRI site was built in the downstream primer (PRF-RP) (Table S3) for linearization of the plasmid for in vitro transcription. To generate an unstructured control RNA, 60 nt of repeated CAA with T7 promoter sequence at the 5′ terminus was synthesized and cloned into pMD19-T.
Virus preparation and infection
VSV_G pseudotyped NL4–3luc was produced by transfection of pVSV_G and pNL4–3luc into 293T cells. VSV_G pseudotyped MLV-luc was produced by transfection of pVSV_G, pHIT60 and pMLV-luc into 293T cells. Replication-competent HIV-1 viruses NL4–3 and JRCSF were produced by transfecting pNL4–3 or pJRCSF into 293T cells. A plasmid expressing renilla luciferase was included to serve as a control for transfection efficiency. For NL4–3luc, culture supernatants were used to infect recipient HeLa cells. For replication-competent HIV-1 viruses, culture supernatants were used to infect recipient TZM-bl cells. Firefly luciferase activity in the recipient cells was normalized with the renilla luciferase activity in the producer cells.
For western blotting analysis of virion particles, culture supernatants were loaded on a 20%–45% sucrose cushion in TNE buffer (10 mM Tris, pH 7.4; 0.2 M NaCl; 1 mM EDTA), centrifuged at 25,000 rpm for 2 h at 4°C. The virions were further pelleted through a 20% sucrose cushion and pellets were resuspended in SDS-PAGE loading buffer.
The production and titration of replication-competent Sindbis virus SINV-nluc have been previously described (Wang et al., 2016b). To prepare the virus, the infectious clone SINV-nsP3-nluc was linearized with XhoI and in vitro transcribed into RNA with SP6 RNA polymerase (Promega) in the presence of a Cap analog (Promega). The RNA was transfected into BHK-21 cells using Lipofectamine 2000 (Thermo Fisher) following the manufacturer’s instruction. At 24 h posttransfection, virus in the culture supernatant was harvested and stored at −80°C. Virus samples were titrated in duplicate by infection of BHK-21 cells at serial dilutions in DMEM supplemented with 1% FBS. At 1 h postinfection, cells were covered with DMEM overlay containing 1.2% agarose and 2% FBS. Plaques were enumerated at 1–2 d postinfection.
For the replication assay, 293TREx-SFL cells were infected with SINV-nluc (MOI = 0.1) for 1 h. Cells were then mock treated or treated with doxycycline to induce SFL-myc expression. At different time points postinfection, supernatants were collected to infect BHK-21 cells for 5 h, followed by luciferase assays.
VSV_G pseudotypted Vpx-loaded SIV VLP was produced as described previously (St Gelais et al., 2012). Human monocytes were isolated from PBMC with CD14 Microbeads (Miltenyi Biotec, MACS) following the manufacturer’s instruction. Monocyte-derived-macrophages (MDMs) were generated from purified monocytes by treatment with 20 ng/ml M-CSF for 7 d. MDMs were transfected with 40 pmol siRNA. At 4 h posttransfection, cells were incubated with Vpx-loaded SIV VLP for 2 h, followed by infection with VSV_G pseudotyped NL4–3-GFP. At 48 h postinfection with NL4–3-GFP, protein expression was analyzed by western blotting.
siRNA and siRNA transfection
The control siRNA and gene specific siRNAs were obtained from GenePharma. siRNA oligos were transfected into cells using lipofectamine 2000 (Thermo Fisher) following the manufacturer’s instruction.
RT assay
The assay was modified from a procedure described previously (Hoffman et al., 1985). Briefly, VLPs were resuspended in 60 μL reaction buffer [50 mM Tris–HCl (pH 8.0), 150 mM KCl, 5 mM DTT, 5 mM MgCl2, 0.1% Triton X-100, and 0.5 mM EGTA, 5 μg/ml oligo dT, 10 μg/ml poly(rA), 3 μCi of 32P-TTP] and incubated at room temperature for 1 h. Five μl of the reaction was dropped on DE81 paper (Whatman). The paper was washed twice with 2 X SSC buffer, followed by autoradiography or Phophoimager.
Electrophoretic Mobility Shift Assay (EMSA)
32P-CTP labeled RNA probes were prepared by in vitro transcription with T7 RNA polymerase (Promega). Insect cell-expressed SFL and bacterially expressed GST proteins were purified to near homogeneity. Probes and proteins were incubated at room temperature for 15 min in a binding buffer (25 mM HEPES pH 7.5, 100 mM NaCl, 1 mM MgCl2 and 4% glycerol) and then resolved on 5% native PAGE gels, followed by autoradiography.
Polysome profiling analysis
A plasmid expressing SFL-myc or SFLS-myc was transfecetd into 293T cells. A plasmid expressing GFP-myc was included to serve as a control. At 24 h posttransfection, cycloheximide was added to the medium at a final concentration of 50 μg/ml to stop translation. Cells were lysed with Lysis Buffer [100mM KCl, 5 mM MgCl2, 30 mM HEPES (pH 7.4), 0.5% NP-40 and 100 μg/ml cycloheximide] and the lysate was clarified by centrifugation for 10 min at 4°C. The lysate was then applied to a 10%–50% sucrose continuous gradient and centrifuged at 36 000 rpm (Hitachi, P40ST) for 3.3 h at 4°C. Thirteen fractions, 1 mL each, were collected on ice. The absorbance at 254nm was monitored and recorded to indicate the positions of polysomes and ribosome subunits. The protein in each fraction was pelleted with trichloroacetic acid, washed with acetone and subjected to western blotting.
Ribosome pelleting assay
The method has been described previously (Mu et al., 2015). Briefly, HeLa cells were transfected with either a plasmid expressing SFL-myc or SFLS-myc, or the siRNAs targeting SFL. At 24 h posttransfection, cells were lysed with RNC buffer (50 mM HEPES, pH 7.4; 100 mM KAc; 5 mM MgCl2) supplemented with Triton X-100 at a final concentration of 0.1% (v/v). The lysates were incubated on ice for 10 min before loading onto a 0.5 M sucrose cushion in RNC buffer. The samples were centrifuged at 78,000 rpm for 90 min at 4°C. The pellet was suspended in RNC buffer and subjected to protein detections.
Co-immunoprecipitation assay
293T cells in 60 mm dishes were transfected with 2 μg total plasmids. At 48 h posttransfection, cells were lysed in 500 μL Co-IP buffer (30 mM HEPES, pH 7.5; 150 mM NaCl; 0.5% NP-40; 30 mM EDTA) and the protease inhibitor cocktail, with or without RNase A (100 μg/ml) and DNase I (100 μg/ml). The lysate was clarified and mixed with the antibody and protein G beads (Amersham Pharmacia) or Anti-Flag affinity gel (Sigma-Aldrich) at 4°C for 2 h. The beads were washed with PBS three times and the bound proteins were resolved on SDS-PAGE electrophoresis, transferred to PVDF membrane and detected by western blotting.
Pull-down assay
MBP-uL6, MBP-uL5, MBP-eS31 and MBP-eRF3 proteins were expressed in E. coli BL21 and purified with pMAL Protein Fusion and Purification System (New England Biolabs) following manufacturer’s instruction. Bacterially expressed GST-SFL was immobilized on Glutathione Sepharose 4B and then incubated with MBP fusion proteins in 500 μL buffer (30 mM HEPES, pH 7.5; 100 mM NaCl; 0.5% NP-40; 30 mM EDTA) supplemented with RNase A (100 μg/ml) and DNase I (100 μg/ml) for 2 h at 4°C. The resin was washed three times with PBS, and then analyzed by western blotting. In a reciprocal experiment, the procedures were similar except that MBP fusion proteins were immobilized on Amylose resin and incubated with GST-SFL.
RNA detection
The method for detecting protein-associated RNA has been described previously (Xuan et al., 2013). Briefly, 293T cells were transfected with a plasmid expressing SLF-myc or SFLS-myc, together with the reporters. At 48 h posttransfection, cells were lysed with RNase-free passive lysis buffer (Promega) supplemented with 30 mM EDTA. The lysate was immunoprecipitated with anti-myc antibody. RNA was extracted with TRIzol and treated with RNase-free DNase followed by heat inactivation of the enzyme.
For RNA quantification, the RNA was reverse transcribed and detected by SYBR green real-time PCR in a Rotor-gene 6000 (Corbett Life Science) using the following program: (i) 95°C for 5 min, 1 cycle; (ii) 95°C for 15 s, 60°C for 30 s, and 72°C for 30 s, 40 cycles; and (iii) 72°C for 10 min, 1 cycle. The amplification specificity was confirmed by melting point analysis and sequencing of the PCR products.
Mass spectrometry analysis
The pGST-HIV(−1)-Fluc reporter was cotransfected with pLPCX-SFL-myc into 293T cells. Flag-tagged proteins were immunoprecipitated with Anti-Flag affinity gel (Sigma-Aldrich) for 3 h. The beads were washed 3 times with Washing Buffer I (50 mM Tris-HCl, pH 8.0; 100 mM NaCl; 1 mM EDTA; 0.5% NP-40), once with Washing Buffer II (50 mM Tris-HCl, pH 8.0; 100 mM NaCl; 1 mM EDTA) and twice with Milli-Q purified water. The bound proteins were eluted with 0.1% acetic acid and the elutes were precipitated by TCA- acetone, resolved on SDS-PAGE. The PMT band was excised from Coomassie stained gel and trypsinized for LC-MS/MS analysis.
LC-MS/MS analysis was performed using a nanoLC-LTQ-Orbitrap XL mass spectrometer (Thermo Fisher) in line with an easy-nLC 1200 HPLC system. Tryptic peptides generated above were loaded onto a self-packed trap column (ReproSil-Pur C18-AQ, 150 μm i.d. X 2mm, 5 μm particle) (Dr. Maisch GmbH, Ammerbuch) which was connected to the self-packed analytical column (ReproSil-Pur C18-AQ, 75 μm i.d X 200mm, 3 μm particle). The peptides were then eluted over a gradient (0%–36% B in 78 m, 36%–80% B in 12 m, where B = 100% Acetonitrile, 0.5% formic acid) at a flow rate of 300 nL/min and introduced online into the linear ion trap mass spectrometer using nano electrospray ionization (ESI).
MS data were analyzed using Proteome Discoverer (version 1.4.0.288, Thermo Fisher). MS2 spectra were searched with SEQUEST engine against a database including the 0 frame and −1 frame products encoded by the pGST-HIV(−1)-Fluc reporter and all kinds of contamination proteins. Peptides with and above +2 charge states were accepted if they were fully enzymatic. The following residue modifications were allowed in the search: carbamidomethylation on cysteine and oxidation on methionine. Peptide spectral matches (PSM) were validated by a targeted decoy database search at 1% false discovery rate (FDR). Peptide identifications were grouped into proteins according to the law of parsimony.
QUANTIFICATION AND STATISTICAL ANALYSIS
The Excel software (Microsoft) was used to determine average values and standard deviations. Unless otherwise indicated, the arithmetic mean values ± SD were calculated from three independent experiments. p values were calculated using the two-tailed paired Student’s t test. For each figure, number of experimental replicates and other information relevant for assessing the accuracy and precision of the analysis are presented in the accompanying legend.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal Flag-specific antibody M2 | Sigma-Aldrich | Cat# F3165; RRID: AB_259529 |
| Rabbit polyclonal DYKDDDDK Tag antibody | Cell Signaling Technology | Cat# 2368; RRID: AB_2217020 |
| Mouse monoclonal myc-specific antibody 9E10 | Santa Cruz Biotechnology | Cat# SC-40; RRID: AB_627268 |
| Rabbit monoclonal Myc-Tag antibody | Cell Signaling Technology | Cat# 2278; RRID: AB_490778 |
| Mouse monoclonal β-actin-specific antibody | Sigma-Aldrich | Cat# a5316; RRID: AB_476743 |
| Rabbit polyclonal C19ORF66-specific antibody | Abcam | Cat# ab122765; RRID: AB_11129894 |
| Rabbit polyclonal eRF1-specific antibody | Abcam | Cat# ab31799; RRID: AB_732264 |
| Rabbit polyclonal eRF3-specific antibody | Abcam | Cat# ab49878; RRID: AB_2115507 |
| Rabbit polyclonal RPS16-specific antibody (T-19) | Santa Cruz Biotechnology | Cat# SC-102087; RRID: AB_2269633 |
| mouse monoclonal p24-specific antibody P5F1 | Liu et al., 2007 | N/A |
| Mouse Anti-GST mAb | ZSGB-Bio | Cat# TA-03; RRID: AB_2756893 |
| Anti-MBP Monoclonal Antibody | New England Biolabs | Cat#E8032; RRID: AB_1559730 |
| Anti-Mouse IgG (H+L), HRP Conjugate | Promega | Cat# w4021; RRID: AB_430834 |
| Peroxidase-Conjugated Goat anti-Rabbit IgG(H+L) | ZSGB-Bio | Cat# ZB-2301; RRID: AB_2747412 |
| Bacterial and Virus Strains | ||
| Trans10 Chemically Competent Cell | Transgen Biotech | Cat# CD101-01 |
| BL21(DE3) Chemically Competent Cell | Transgen Biotech | Cat# CD601-01 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Cycloheximide | Sigma-Aldrich | Cat# C7698 |
| Zeocin | Thermo Fisher Scientific | Cat#:R25001 |
| Doxycycline | Sigma-Aldrich | Cat# D9891-1G |
| RNase A | Sigma-Aldrich | Cat# R6513 |
| DNase I | Sigma-Aldrich | Cat# D5025 |
| Ribo m7G Cap analog | Promega | Cat# P1711 |
| Recombinant Human Macrophage Colony Stimulating Factor | Sangon Biotech | Cat# C600148-0002 |
| Lipofectamine 2000 Transfection Reagent | Thermo Fisher Scientific | Cat#11668019 |
| TRIzol Reagent | Thermo Fisher Scientific | Cat#15596018 |
| 32P-dTTP | Perkin Elmer | NEG005H250UC |
| 32p-ctp | Perkin Elmer | BLU508H250UC |
| TrueFectTM Transfection Reagent | United Biosystems Inc | Cat#NF0866-3 |
| Critical Commercial Assays | ||
| Luciferase Assay System | Promega | Cat# E1501 |
| Passive Lysis 5X Buffer | Promega | Cat# E1941 |
| Dual-Luciferase® Reporter Assay System | Promega | Cat# E1910 |
| T7 Riboprobe® Systems | Promega | Cat# P1440 |
| Sp6 Riboprobe® Systems | Promega | Cat# P1420 |
| SuperReal PreMix Plus (SYBR Green) | Tiangen | Cat# Fp205-02 |
| CD14 Microbeads | MACS Miltenyi Biotec | 130-050-201 |
| Glutathione Sepharose 4B | GE Healthcare | 27-4574-01 |
| Amylose Resin | New England Biolabs | E8021S |
| Protein G SepharoseTM 4 Fast Flow | GE Healthcare | 17-0618-05 |
| Experimental Models: Cell Lines | ||
| HEK293T cell | ATCC | CRL-11268 |
| THP-1 cell | ATCC | TIB-202 |
| MT4 cell | NIH AIDS Reagent Program | 120 |
| TZM-bl cell | NIH AIDS Reagent Program | 8129 |
| HeLa cell | ATCC | CCL-2 |
| HOS-CD4+CCR5+ cell | NIH AIDS Reagent Program | 3318 |
| 293TREx Cell | Thermo Fisher | R710-07 |
| BHK-21(C13) | ATCC | CCL-10 |
| MT4-Ctrli | This study | N/A |
| MT4-SFLi1 | This study | N/A |
| MT4-SFLi2 | This study | N/A |
| THP-1-Ctrli | This study | N/A |
| THP-1-SFLi1 | This study | N/A |
| THP-1-SFLi2 | This study | N/A |
| T-REx-293-SFL-myc | This study | N/A |
| Human MDM | This study | N/A |
| HOS-CD4+CCR5+ -EasiLV-Ev | This study | N/A |
| HOS-CD4+CCR5+ - EasiLV-SFL-myc | This study | N/A |
| Oligonucleotides | ||
| Ctrli: 5′-UUCUCCGAACGUGUCACGU-3′ (Control siRNA sequence) | This study | N/A |
| SFLi-1: 5′-GAACUAAGUAACGAUCUGGAU-3′(siRNA targeting sequence) | This study | N/A |
| SFLi-2: 5′-CCAAGAACUAAGUAACGAUCU-3′ (siRNA targeting sequence) | This study | N/A |
| eRF3i: 5′- GAAUGUAAGGAGAAACUAG-3′ (siRNA targeting sequence) | This study | N/A |
| eRF1i: 5′-GCAAUGGCACCAGCAUGAUA-3′ (siRNA targeting sequence) | This study | N/A |
| Peloi: 5′-GGUCCAAAUUUCUUCAGGUUU-3′ (siRNA targeting sequence) | This study | N/A |
| HBS1Li: 5′-CCAGUAGAUUCCCAGACAUUU-3′ (siRNA targeting sequence) | This study | N/A |
| shCtrli: 5′-GCAAGCTGACCCTGAAG-3′ (control shRNA sequence) | This study | N/A |
| shSFLi-1: 5′-GAACTAAGTAACGATCTGGAT-3′ (shRNA targeting sequence) | This study | N/A |
| shSFLi-2: 5′-CCAAGAACTAAGTAACGATCT-3′ (shRNA targeting sequence) | This study | N/A |
| Primer sequences | See Table S3 | N/A |
| Recombinant DNA | ||
| pJRCSF | NIH AIDS Reagent Program | Cat# 394 |
| pNL4-3 | NIH AIDS Reagent Program | Cat# 114 |
| pLPCX-SFL | This study | N/A |
| pLPCX-SFLS | This study | N/A |
| pcDNA4-SFL | This study | N/A |
| pCMV-HF | Mu et al., 2015 | N/A |
| pGEX-5X-3-SFL | This study | N/A |
| pEasiLV-MCS | Goujon et al., 2013 | N/A |
| pMLV-luc | Gao et al., 2002 | N/A |
| pHIT60 | Gao et al., 2002 | N/A |
| pEasiLV-SFL-myc | This study | N/A |
| pCMV-myc-OAZ1 | This study | N/A |
| pCMV-myc-CCR5 | This study | N/A |
| pCMV-myc-PEG10 | This study | N/A |
| pcDNA4-eRF1-2*flag | This study | N/A |
| pcDNA4-eRF3-2*flag | This study | N/A |
| pcDNA4-uS3-2*flag | This study | N/A |
| pcDNA4-uS4-2*flag | This study | N/A |
| pcDNA4-uS19-2*flag | This study | N/A |
| pcDNA4-uS13-2*flag | This study | N/A |
| pcDNA4-eS31-2*flag | This study | N/A |
| pcDNA4-uS2-2*flag | This study | N/A |
| pcDNA4-RACK1-2*flag | This study | N/A |
| pcDNA4-uL5-2*flag | This study | N/A |
| pcDNA4-eL8-2*flag | This study | N/A |
| pcDNA4-uL2-2*flag | This study | N/A |
| pcDNA4-uL6-2*flag | This study | N/A |
| pcDNA4-uL1-2*flag | This study | N/A |
| pcDNA4-uL11-2*flag | This study | N/A |
| pMAL-C2X- eRF3 | This study | N/A |
| pMAL-C2X- uL6 | This study | N/A |
| pMAL-C2X- uL5 | This study | N/A |
| pMAL-C2X- eS31 | This study | N/A |
| pHIV-2Rod-GFP | Kane et al., 2013 | N/A |
| pSIVmac-GFP | Kane et al., 2013 | N/A |
| pNL4-3luc | Connor et al., 1995 | N/A |
| pNLenv-luc | Dang et al., 2006 | N/A |
| pNL4-3GFP | Wang et al., 2016a | N/A |
| pSIV(Vpx-) | St Gelais et al., 2012 | N/A |
| pCMV-Vpx | St Gelais et al., 2012 | N/A |
| pDual-HIV(−1) | Dulude et al., 2006 | N/A |
| pDual-HIV(0) | Dulude et al., 2006 | N/A |
| pDual-SIV(−1) | This study | N/A |
| pDual-HIV2(−1) | This study | N/A |
| pDual-SINV(−1) | This study | N/A |
| pDual-HTLV-2(−1) | This study | N/A |
| pDual-MMTV(−1) | This study | N/A |
| pDual-RSV(−1) | This study | N/A |
| pDual-PEG10(−1) | This study | N/A |
| pDual-CCR5(−1) | This study | N/A |
| pSVNI-nluc | Wang et al., 2016b | N/A |
| pEF1α-Gag-Pol | This study | N/A |
| pEF1α-Gag-Pol-mut | This study | N/A |
| pSuper-retro-MMTV(−1) | This study | N/A |
| pSuper-retro-MMTV(−1)-mut | This study | N/A |
| pSuper-Retro-Ctrli | This study | N/A |
| pSuper-Retro-SFLi1 | This study | N/A |
| pSuper-Retro-SFLi2 | This study | N/A |
| pGST-HIV(−1)-Fluc | This study | N/A |
| pGST-HIV(0)-Fluc | This study | N/A |
| pGST-HIV(−1)-Fluc-mut | This study | N/A |
| pNL4-3luc-DelPro | This study | N/A |
| pCMV-PMT | This study | N/A |
| pMD19-T7-PRF | This study | N/A |
| pMD19-T7-CAA | This study | N/A |
| Software and Algorithms | ||
| GraphPad Prism 5 | GraphPad software | N/A |
| ImageJ | National Institutes of Health | https://imagej.nih.gov/ij/ |
Highlights.
Shiftless is a broad-spectrum inhibitor of programmed −1 ribosomal frameshifting
Shiftless interacts with the frameshifting signal RNA and translating ribosomes
Shiftless causes premature translation termination at the frameshifting site
eRF1 and eRF3 are required for Shiftless-mediated translation termination
ACKNOWLEDGMENTS
We thank Professor Paul D. Bieniasz for kindly providing lentivectors pHIV-2-GFP and pSIVmac-GFP, Professor Michael H. Malim for pEasiLV-MCS, Professor Léa Brakier-Gingras for reporters pDual-HIV(−1) and pDual-HIV(0), and Professor Li Wu and Professor Jianhua Wang for pSIV(Vpx-) and pCMV-Vpx. We thank Jifeng Wang of Laboratory of Proteomics and Hongjie Zhang of the Core Facility of Protein Sciences, Institute of Biophysics for technical assistance. This work was supported by grants to G.G. from Chinese Academy of Sciences (XDB29010203 and KFZD-SW-209), National Health Commission (2017ZX10201101-001-005), Ministry of Science and Technology (2018YFA0507202), and National Natural Science Foundation (81530066) of China.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes three tables and seven figures and can be found with this article online at https://doi.org/10.1016/j.cell.2018.12.030.
REFERENCES
- Atkins JF, Loughran G, Bhatt PR, Firth AE, and Baranov PV (2016). Ribosomal frameshifting and transcriptional slippage: From genetic steganography and cryptography to adventitious use. Nucleic Acids Res. 44, 7007–7078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balinsky CA, Schmeisser H, Wells AI, Ganesan S, Jin T, Singh K, and Zoon KC (2017). IRAV (FLJ11286), an interferon-stimulated gene with antiviral activity against dengue virus, interacts with MOV10. J. Virol. 91, e01606–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belew AT, and Dinman JD (2015). Cell cycle control (and more) by programmed −1 ribosomal frameshifting: implications for disease and therapeutics. Cell Cycle 14, 172–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belew AT, Hepler NL, Jacobs JL, and Dinman JD (2008). PRFdb: a database of computationally predicted eukaryotic programmed −1 ribosomal frameshift signals. BMC Genomics 9, 339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belew AT, Advani VM, and Dinman JD (2011). Endogenous ribosomal frameshift signals operate as mRNA destabilizing elements through at least two molecular pathways in yeast. Nucleic Acids Res. 39, 2799–2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belew AT, Meskauskas A, Musalgaonkar S, Advani VM, Sulima SO, Kasprzak WK, Shapiro BA, and Dinman JD (2014). Ribosomal frameshifting in the CCR5 mRNA is regulated by miRNAs and the NMD pathway. Nature 512, 265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brakier-Gingras L, Charbonneau J, and Butcher SE (2012). Targeting frameshifting in the human immunodeficiency virus. Expert Opin. Ther. Targets 16, 249–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buskirk AR, and Green R (2017). Ribosome pausing, arrest and rescue in bacteria and eukaryotes. Philos. Trans. R. Soc. Lond. B Biol. Sci. 372, 20160183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caliskan N, Katunin VI, Belardinelli R, Peske F, and Rodnina MV (2014). Programmed −1 frameshifting by kinetic partitioning during impeded translocation. Cell 157, 1619–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caliskan N, Peske F, and Rodnina MV (2015). Changed in translation: mRNA recoding by −1 programmed ribosomal frameshifting. Trends Biochem. Sci. 40, 265–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamorro M, Parkin N, and Varmus HE (1992). An RNA pseudoknot and an optimal heptameric shift site are required for highly efficient ribosomal frameshifting on a retroviral messenger RNA. Proc. Natl. Acad. Sci. USA 89, 713–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Petrov A, Johansson M, Tsai A, O’Leary SE, and Puglisi JD (2014). Dynamic pathways of −1 translational frameshifting. Nature 512, 328–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiabudini M, Tais A, Zhang Y, Hayashi S, Wölfle T, Fitzke E, and Rospert S (2014). Release factor eRF3 mediates premature translation termination on polylysine-stalled ribosomes in Saccharomyces cerevisiae. Mol. Cell. Biol. 34, 4062–4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J, Xu Z, and Ou JH (2003). Triple decoding of hepatitis C virus RNA by programmed translational frameshifting. Mol. Cell. Biol. 23, 1489–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark MB, Jänicke M, Gottesbühren U, Kleffmann T, Legge M, Poole ES, and Tate WP (2007). Mammalian gene PEG10 expresses two reading frames by high efficiency −1 frameshifting in embryonic-associated tissues. J. Biol. Chem. 282, 37359–37369. [DOI] [PubMed] [Google Scholar]
- Connor RI, Chen BK, Choe S, and Landau NR (1995). Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology 206, 935–944. [DOI] [PubMed] [Google Scholar]
- Dang Y, Wang X, Esselman WJ, and Zheng YH (2006). Identification of APOBEC3DE as another antiretroviral factor from the human APOBEC family. J. Virol. 80, 10522–10533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Di Marzio P, Marmon S, Sutton RE, Hill CM, et al. (1996). Identification of a major co-receptor for primary isolates of HIV-1. Nature 381, 661–666. [DOI] [PubMed] [Google Scholar]
- Dinman JD (2012). Mechanisms and implications of programmed translational frameshifting. Wiley Interdiscip. Rev. RNA 3, 661–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doronina VA, Wu C, de Felipe P, Sachs MS, Ryan MD, and Brown JD (2008). Site-specific release of nascent chains from ribosomes at a sense codon. Mol. Cell. Biol. 28, 4227–4239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulude D, Berchiche YA, Gendron K, Brakier-Gingras L, and Heveker N (2006). Decreasing the frameshift efficiency translates into an equivalent reduction of the replication of the human immunodeficiency virus type 1. Virology 345, 127–136. [DOI] [PubMed] [Google Scholar]
- Firth AE, and Atkins JF (2009). A conserved predicted pseudoknot in the NS2A-encoding sequence of West Nile and Japanese encephalitis flaviviruses suggests NS1 ‘ may derive from ribosomal frameshifting. Virol. J. 6, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firth AE, Chung BY, Fleeton MN, and Atkins JF (2008). Discovery of frameshifting in Alphavirus 6K resolves a 20-year enigma. Virol. J. 5, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao G, Guo X, and Goff SP (2002). Inhibition of retroviral RNA production by ZAP, a CCCH-type zinc finger protein. Science 297, 1703–1706. [DOI] [PubMed] [Google Scholar]
- Goujon C, Moncorgé O, Bauby H, Doyle T, Ward CC, Schaller T, Hué S, Barclay WS, Schulz R, and Malim MH (2013). Human MX2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nature 502, 559–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman AD, Banapour B, and Levy JA (1985). Characterization of the AIDS-Associated Retrovirus Reverse-Transcriptase and Optimal Conditions for Its Detection in Virions. Virology 147, 326–335. [DOI] [PubMed] [Google Scholar]
- Hung M, Patel P, Davis S, and Green SR (1998). Importance of ribosomal frameshifting for human immunodeficiency virus type 1 particle assembly and replication. J. Virol. 72, 4819–4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacks T, Power MD, Masiarz FR, Luciw PA, Barr PJ, and Varmus HE (1988). Characterization of ribosomal frameshifting in HIV-1 gag-pol expression. Nature 331, 280–283. [DOI] [PubMed] [Google Scholar]
- Jackson RJ, Hellen CUT, and Pestova TV (2012). Termination and post-termination events in eukaryotic translation. Adv. Protein Chem. Struct. Biol. 86, 45–93. [DOI] [PubMed] [Google Scholar]
- Janzen DM, and Geballe AP (2004). The effect of eukaryotic release factor depletion on translation termination in human cell lines. Nucleic Acids Res. 32, 4491–4502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane M, Yadav SS, Bitzegeio J, Kutluay SB, Zang T, Wilson SJ, Schoggins JW, Rice CM, Yamashita M, Hatziioannou T, and Bieniasz PD (2013). MX2 is an interferon-induced inhibitor of HIV-1 infection. Nature 502, 563–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karacostas V, Wolffe EJ, Nagashima K, Gonda MA, and Moss B (1993). Overexpression of the HIV-1 gag-pol polyprotein results in intracellular activation of HIV-1 protease and inhibition of assembly and budding of virus-like particles. Virology 193, 661–671. [DOI] [PubMed] [Google Scholar]
- Kim YG, Maas S, and Rich A (2001). Comparative mutational analysis of cis-acting RNA signals for translational frameshifting in HIV-1 and HTLV-2. Nucleic Acids Res. 29, 1125–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HK, Liu F, Fei J, Bustamante C, Gonzalez RL Jr., and Tinoco I Jr. (2014). A frameshifting stimulatory stem loop destabilizes the hybrid state and impedes ribosomal translocation. Proc. Natl. Acad. Sci. USA 111, 5538–5543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le SY, Shapiro BA, Chen JH, Nussinov R, and Maizel JV (1991). RNA pseudoknots downstream of the frameshift sites of retroviruses. Genet. Anal. Tech. Appl. 8, 191–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu GJ, Wang JP, Xiao JC, Zhao ZW, and Zheng YT (2007). Preparation and characterization of three monoclonal antibodies against HIV-1 p24 capsid protein. Cell. Mol. Immunol. 4, 203–208. [PubMed] [Google Scholar]
- Lopinski JD, Dinman JD, and Bruenn JA (2000). Kinetics of ribosomal pausing during programmed −1 translational frameshifting. Mol. Cell. Biol. 20, 1095–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcheschi RJ, Staple DW, and Butcher SE (2007). Programmed ribosomal frameshifting in SIV is induced by a highly structured RNA stem-loop. J. Mol. Biol. 373, 652–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marczinke B, Fisher R, Vidakovic M, Bloys AJ, and Brierley I (1998). Secondary structure and mutational analysis of the ribosomal frameshift signal of rous sarcoma virus. J. Mol. Biol. 284, 205–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsufuji S, Matsufuji T, Miyazaki Y, Murakami Y, Atkins JF, Gesteland RF, and Hayashi S (1995). Autoregulatory frameshifting in decoding mammalian ornithine decarboxylase antizyme. Cell 80, 51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melian EB, Hinzman E, Nagasaki T, Firth AE, Wills NM, Nouwens AS, Blitvich BJ, Leung J, Funk A, Atkins JF, et al. (2010). NS1′ of flaviviruses in the Japanese encephalitis virus serogroup is a product of ribosomal frameshifting and plays a role in viral neuroinvasiveness. J. Virol. 84, 1641–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu X, Fu Y, Zhu Y, Wang X, Xuan Y, Shang H, Goff SP, and Gao G (2015). HIV-1 exploits the host factor RuvB-like 2 to balance viral protein expression. Cell Host Microbe 18, 233–242. [DOI] [PubMed] [Google Scholar]
- Napthine S, Ling R, Finch LK, Jones JD, Bell S, Brierley I, and Firth AE (2017). Protein-directed ribosomal frameshifting temporally regulates gene expression. Nat. Commun. 8, 15582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plant EP, Wang P, Jacobs JL, and Dinman JD (2004). A programmed −1 ribosomal frameshift signal can function as a cis-acting mRNA destabilizing element. Nucleic Acids Res. 32, 784–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey J, and Mukhopadhyay S (2017). Disentangling the frames, the state of research on the alphavirus 6K and TF proteins. Viruses 9, 228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider WM, Chevillotte MD, and Rice CM (2014). Interferon-stimulated genes: a complex web of host defenses. Annu. Rev. Immunol. 32, 513–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shehu-Xhilaga M, Crowe SM, and Mak J (2001). Maintenance of the Gag/Gag-Pol ratio is important for human immunodeficiency virus type 1 RNA dimerization and viral infectivity. J. Virol. 75, 1834–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder JE, Kulcsar KA, Schultz KL, Riley CP, Neary JT, Marr S, Jose J, Griffin DE, and Kuhn RJ (2013). Functional characterization of the alphavirus TF protein. J. Virol. 87, 8511–8523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somogyi P, Jenner AJ, Brierley I, and Inglis SC (1993). Ribosomal pausing during translation of an RNA pseudoknot. Mol. Cell. Biol. 13, 6931–6940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Gelais C, de Silva S, Amie SM, Coleman CM, Hoy H, Hollenbaugh JA, Kim B, and Wu L (2012). SAMHD1 restricts HIV-1 infection in dendritic cells (DCs) by dNTP depletion, but its expression in DCs and primary CD4+ T-lymphocytes cannot be upregulated by interferons. Retrovirology 9, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y, Chin WX, Han Q, Ichiyama K, Lee CH, Eyo ZW, Ebina H, Takahashi H, Takahashi C, Tan BH, et al. (2016). Characterization of RyDEN (C19orf66) as an interferon-stimulated cellular inhibitor against dengue virus replication. PLoS Pathog. 12, e1005357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svidritskiy E, Brilot AF, Koh CS, Grigorieff N, and Korostelev AA (2014). Structures of yeast 80S ribosome-tRNA complexes in the rotated and nonrotated conformations. Structure 22, 1210–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tholstrup J, Oddershede LB, and Sørensen MA (2012). mRNA pseudoknot structures can act as ribosomal roadblocks. Nucleic Acids Res. 40, 303–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Zhang X, Han Y, Wang X, and Gao G (2016a). M2BP inhibits HIV-1 virion production in a vimentin filaments-dependent manner. Sci. Rep. 6, 32736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Li MM, Zhao J, Li S, MacDonald MR, Rice CM, Gao X, and Gao G (2016b). Sindbis virus can exploit a host antiviral protein to evade immune surveillance. J. Virol. 90, 10247–10258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wills NM, Moore B, Hammer A, Gesteland RF, and Atkins JF (2006). A functional −1 ribosomal frameshift signal in the human paraneoplastic Ma3 gene. J. Biol. Chem. 281, 7082–7088. [DOI] [PubMed] [Google Scholar]
- Wilson W, Braddock M, Adams SE, Rathjen PD, Kingsman SM, and Kingsman AJ (1988). HIV expression strategies: ribosomal frameshifting is directed by a short sequence in both mammalian and yeast systems. Cell 55, 1159–1169. [DOI] [PubMed] [Google Scholar]
- Xuan Y, Gong D, Qi J, Han C, Deng H, and Gao G (2013). ZAP inhibits murine gammaherpesvirus 68 ORF64 expression and is antagonized by RTA. J. Virol. 87, 2735–2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan F, Doronina VA, Sharma P, and Brown JD (2010). Orchestrating ribosomal activity from inside: effects of the nascent chain on the peptidyltrans-ferase centre. Biochem. Soc. Trans. 38, 1576–1580. [DOI] [PubMed] [Google Scholar]
- Yan S, Wen JD, Bustamante C, and Tinoco I Jr. (2015). Ribosome excursions during mRNA translocation mediate broad branching of frameshift pathways. Cell 160, 870–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshinaka Y, Katoh I, Copeland TD, and Oroszlan S (1985). Murine leukemia virus protease is encoded by the gag-pol gene and is synthesized through suppression of an amber termination codon. Proc. Natl. Acad. Sci. USA 82, 1618–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaher HS, and Green R (2011). A primary role for release factor 3 in quality control during translation elongation in Escherichia coli. Cell 147, 396–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.