

Abstract
Cyclic AMP has been implicated as second messenger in a wide range of cellular processes. In the protozoan parasite Trypanosoma cruzi, cAMP is involved in the development of the parasite’s life cycle. While cAMP effectors have been widely studied in other eukaryotic cells, little is known about cAMP’s mechanism of action in T. cruzi. To date, only a cAMP-dependent protein kinase A (PKA) has been cloned and characterised in this parasite; however experimental evidence indicates the existence of cAMP-dependent, PKA-independent events. In order to identify new cAMP binding proteins as potential cAMP effectors, we carried out in silico studies using the predicted T. cruzi proteome. Using a combination of search methods 27 proteins with putative cNMP binding domains (CBDs) were identified. Phylogenetic analysis of the CBDs presented a homogeneous distribution, with sequences segregated into two main branches: one containing kinases-like proteins and the other gathering hypothetical proteins with different function or no other known. Comparative modelling of the strongest candidates provides support for the hypothesis that these proteins may give rise to structurally viable cyclic nucleotide binding domains. Pull-down and nucleotide displacement assays strongly suggest that TcCLB.508523.80 could bind cAMP and eventually be a new putative PKA-independent cAMP effector in T. cruzi.
Keywords: Trypanosoma cruzi, cAMP signalling, cAMP binding proteins, cAMP novel effectors
1. Introduction
Chagas disease is a potentially life-threatening disease caused by the protozoan parasite, Trypanosoma cruzi. Endemic to the Americas, it represents a serious health threat among people living in poor rural populations in Latin America. In addition to an estimated 10 million infected people and an alarming 25,000deaths per year, at least 55,600 individuals from endemic areas annually require etiological treatment against T. cruzi infection [1]. Moreover, this regional issue is now becoming global due to migration of infected people to developed countries [2,3]. There is currently no vaccine, and treatment is limited to two old antiparasitic drugs (nifurtimox and benznidazol) that have important limitations that include variable efficacy, long treatment courses and toxicity [4]. For this reason, there is an urgent need for new therapies, which requires the identification of new potential targets for the design of novel drugs for anti-trypanosomal therapy.
T. cruzi has a complex life cycle involving four developmental stages that alternate between the insect blood-sucking vector and the mammalian host. In the insect gut, a proliferative non-infective form of the parasite (epimastigote) differentiates into a non-replicative infective form (metacyclic trypomastigote). Metacyclic trypomastigotes gain access to the mammalian host through faeces contamination at the insect bite wound and infect a wide range of cells. Upon invasion of the host cell, trypomastigotes rapidly differentiate into amastigotes, an intracellular form that first replicates and then differentiates back into trypomastigotes. Trypomastigotes are released into the bloodstream, spread and infect other tissues, and can be picked up by the bug vector when the insect feeds from the host.The molecular mechanisms involved in the stage specific transformations occurring in the T. cruzi life cycle are not completely understood [5]. Several studies suggested the involvement of cAMP in proliferation/differentiation [6]. Early reports demonstrated a negative role for cAMP in epimastigote proliferation [7,8]; high cAMP levels blocked DNA synthesis [9] and consistent with this observation, known parasite mitogens were able to decrease cAMP levels [10]. However, inhibition of protein kinase A (PKA, the only known T. cruzi cAMP effector todate) by genetic (PKI expression) as well as pharmacologic (H89) approaches led to epimastigote death, instead of the expected rescue [11]. Early evidence also indicated a potential role for cAMP in T. cruzi differentiation: the passage from epimastigote to metacyclic stage was accompanied by an increase in cAMP levels, an effect mimicked by cAMP analogues, inhibition of PDE, and a peptide from hindguts of the insect vector that activates adenylyl cyclase activity in T. cruzi [8,12,13].
Similarly, Trypanosoma brucei, the etiological agent of the Human African Trypanosomiasis or sleeping sickness, has a multi-stage life cycle that involves morphological changes as it is transmitted among the mammalian host and the insect vector, usually the tsetse fly. In vitro differentiation of T. brucei from replicating long slender bloodstream forms to non-dividing short stumpy forms was induced by a soluble factor, SIF (stumpy inducing factor), which could stimulate an immediate twofold to threefold elevation of intracellular cAMP. Additionally, SIF effects could be mimic by membrane-permeable cAMP analogues or the PDE inhibitor etazolate [14]. More recently, the relevance of cAMP-mediated signalling in T. brucei was demonstrated by the pharmacological validation of trypanosomal PDEs as drug targets [15], where inhibition of PDE by CpdA caused a dramatic increase of intracellular cAMP, immediately stalling cell proliferation and triggering cell death within 3 days. Furthermore, in searching for resistance to the T. brucei PDE inhibitor Gould et al. [16] identified a group of kinetoplastid unique proteins (cAMP response proteins (CARPs)) as mediators of the cAMP signalling involved in cell death. These results clearly show the significance of the cAMP cascade as a novel target for antiparasitic drug development.
Although much attention is focused on the putative machinery involved in synthesis (i.e. cyclase) and degradation (i.e. phosphodiesterase) of cAMP [17], the role of cAMP effector pathways is currently unknown.
In mammalian cells, cAMP-dependent pathways are transduced by two ubiquitously expressed intracellular effectors, the classic PKA and the more recently discovered Exchange protein directly activated by cAMP (Epac), as well as cAMP gated-ion channels in specific tissues [18]. However, in the case of Trypanosomatids, while reported evidence indicates the presence of PKA-independent pathways, the genome of the parasite does not appear to have Epac or cAMP gated-ion channels, suggesting a highly unusual cAMP signalling mechanism [19]. This prompted us to search for proteins containing cyclic nucleotide binding domains (CBDs) that could potentially be new cAMP effectors in the parasite biology. In silico studies using the predicted T. cruzi proteome lead to the identification of 27 proteins with CBDs. The strongest in silico candidates were in vitro evaluated as cAMP-binding proteins. Our results indicated thatTcCLB.508523.80 could bind cAMP and potentially play a role as cAMP sensor in a PKA-independent pathway.
2. Materials and methods
2.1. Sequence search and phylogenetic analysis
In order to identify T. cruzi proteins containing cNMP domains, the parasite’s complete predicted proteome was run against the Simple Modular Architecture Research Tool (SMART, http://smart.embl-heidelberg.de/) and the Prosite database (http://prosite.expasy.org/). Potential cAMP-binding proteins were identified using the following cNMP binding motifs: Pfam PF00027, Smart SM00100, Prosite PS00888, PS00889 and PSPS50042. As an additional method, a selection of cNMP-containing sequences from T. cruzi, Escherichia coli, and Homo sapiens were used in BLAST searches against trypanosomatid databases (TriTrypDB v5.1). BLAST hits having log E-values less than 1E–9 were used in further analysis against Hidden Markov Model profiles (HMMER 3.0). The HMMER searches were continued until no potential proteins with cNMPs could be found. The final list of putative CBS proteins was generated from hits that emerged in searches using at least two different methods. Multiple sequence alignments were performed using DNAStar package with the default alignment parameters. For the calculation of protein distances, the neighbour-joining method was used. Percent divergences between all pairs of sequence from the multiple alignments were calculated, and the neighbour-joining (NJ) method was applied to the distance matrix. The unrooted tree obtained was plotted using Treeview software (taxonomy.zoology.gla.ac.uk/rod/treeview.html).
The output list of cNMP-domain containing sequences obtained for T. cruzi was used as a query in searches against the T. brucei and Leishmania major genomes in order to identify orthologs (Table 1). To this end we used the TriTrypDB (www.tritrypdb.org) ortholog transform tool based on OrthoMCL.
Table 1.
Trypanosomatid genes containing cNMP domains. A list of T. cruzi (CL Brener strain), T. brucei (TREU927 strain) and L. major (Friedlin strain) orthologs are shown. Most of the genes are ortholog across the three species. Organisms are indicated above each column and systematic gene name are displayed in rows. –, no ortholog found.
| Trypanosoma cruzi | Trypanosoma brucei | Leishmania major |
|---|---|---|
| TcCLB.506605.220 | Tb11.v5.0149 | LmjF.14.0050 |
| TcCLB.506227.150 | Tb11.v5.0587 | LmjF.13.0160 |
| TcCLB.510297.110 | Tb11.v5.0233 | LmjF.36.0830 |
| TcCLB.509891.20 | – | LmjF.34.4620 |
| TcCLB.510879.50 | – | LmjF.34.2820 |
| TcCLB.507035.110 | Tb927.11.16210 | LmjF.32.3970 |
| TcCLB.507041.10 | Tb927.11.15730 | LmjF.32.2530 |
| TcCLB.503643.20 | Tb927.3.5020 | LmjF.29.1040 |
| TcCLB.504153.20 | Tb927.11.2380 | – |
| TcCLB.507993.210 | – | LmjF.18.0130 |
| TcCLB.505977.13 | – | LmjF.15.1200 |
| TcCLB.504449.30 | – | LmjF.15.1190 |
| TcCLB.508995.10 | Tb11.v5.0534 | LmjF.20.0770 |
| TcCLB.508859.70 | Tb11.v5.0149 | LmjF.14.0050 |
| TcCLB.511809.80 | Tb927.7.2320 | – |
| TcCLB.506009.109 | – | – |
| TcCLB.510691.30 | – | LmjF.34.4620 |
| TcCLB.504013.60 | – | LmjF.34.2820 |
| TcCLB.508523.80 | Tb927.11.16210 | LmjF.32.3970 |
| TcCLB.508273.30 | Tb927.11.15730 | LmjF.32.2530 |
| TcCLB.506485.109 | Tb927.3.5020 | LmjF.29.1040 |
| TcCLB.508355.140 | Tb927.11.2140 | LmjF.27.1480 |
| TcCLB.511279.20 | – | LmjF.18.0130 |
| TcCLB.506447.19 | – | LmjF.15.1190 |
| TcCLB.470521.10 | – | LmjF.15.1200 |
| TcCLB.506477.60 | Tb11.v5.0534 | LmjF.20.0770 |
| TcCLB.418221.20 | Tb927.8.2130 | LmjF.23.0160 |
2.2. Molecular modelling ofT. cruzi CBDs
Human homologues with known 3D structure comprising the putative CBD of each sequence of the T. cruzi’s strongest putative cAMP binding proteins were identified using the HHpred 2.0 programme [20]. The closest human homologue ofTcCLB.508523.80/TcCLB.507035.110 (different size alleles) and TcCLB.510297.110 was the regulatory domain of PKAIα, which has been solved by X-ray crystallography and annotated with the PDB code 3PNA [21]. Both sequences match the template with E-values of 5E–23 and 5.3E–23, respectively. Homology models based on this template were constructed using Modeller v9.11 [22]. The coordinates of the bound cAMP were taken from the original X-ray structure after a structural alignment performed with STAMP [23]. To optimize the orientation of the side chains around the ligand, the cAMP bound models underwent energy minimisation and 10 ns of molecular dynamics simulation using the hybrid solvation approach of the SIRAH force field (www.sirahff.com), as reported in [24]. The final minimised models contained only two and one residue outside allowed regions of the Ramachandran plots for sequences TcCLB.508523.80 and TcCLB.510297.110, respectively (see Supplementary Figs. 2 and 3). PDB files of both models are provided as supplementary material).
2.3. Fusion protein expression and purification
TcCLB.508523.80 full-length coding sequence was amplified by PCR from 100 ng of total genomic DNA of T. cruzi CL-Brener using the following primers:
8523 fw (5′-CACCATGTACGGGACCTTTTTTAAAGGG-3′);
8523 rev (5′-TCAAATTGCATCACCCTTTACATGGCATTTCC-3′).
The sequence was cloned into pGEM-T Easy vector (Promega) and subcloned into the EcoRI site of pGEX-3X vector (Amersham Biosciences), generating a glutathione S-transferase (GST) fusion protein and transformed into E. coli BL21. Cultures were induced with 0.1 mM isopropyl β-D-thiogalactopyranoside for 16 h at 18 °C. GST fusion proteins were purified using a glutathione–agarose column (Sigma). pGEX-4T3-PKA-R1β was kindly provided by Dr. Daniel Hochbaum.
2.4. cAMP binding assay
For the binding assays 500 ng of the recombinant protein in binding buffer [Tris–HCl/NaCl 50 mM pH: 7.4; β-mercaptoethanol 10 mM (Sigma); N-lauroylsarcosine sodium salt 0.6% (Sigma), 2% of Triton X-100] was incubated with cAMP-conjugated agarose resin (A0144, Sigma) for 2h at 4 °C with rocking. The resin was extensively washed with washing buffer (Tris–HCl/NaCl 50 mM, pH 7.4) and eluted with SDS-cracking buffer at 60 °C for 10 min. For the nucleotide displacement experiments, 1 or 10 mM of the indicated free nucleotide (Sigma) was added to the binding reaction.
2.5. SDS-PAGE and immunoblot
The eluted protein was separated on a 10% SDS-polyacrylamide gel. For the input a percentage of the protein used for the binding assay was load: 10–20% for PKA and 5% for TcCLB.508523.80 and GST. The gel was transferred onto Hybond C nitrocellulose membrane (Amersham Pharmacia Biosciences). Membranes were incubated in blocking buffer (TBS containing 5% milk and 0.5% Tween 20) for 1 h at room temperature (RT). An anti–GST antibody (Sigma, 1/2500 dilution) was used as primary antibody (4°C ON). Horseradish peroxidase-conjugated goat anti-mouse IgG was used as secondary antibody (2 h at RT). Supersignal Western blotting detection reagents (Pierce Thermo Scientific) were used for visualisation of the reactive bands.
3. Results
3.1. Identification ofputative cNMP binding proteins in T. cruzi’s genome
In order to identify potential cAMP binding proteins that could ultimately have an in vivo function as cAMP effector in T. cruzi, we performed an in silico search for putative proteins harbouring cyclic nucleotide binding domains using the whole parasite predicted proteome. A combination of searching methods allowed us to identify several proteins with putative CBDs: 70 hits were obtained from the SMART server, 24 hits from Prosite, 25 from HMMER profiles searches and 31 using BLAST algorithm (see Section 2). In total, 80 different putative cNMP binding proteins were obtained by at least one of the mentioned methods (Table S1). From these sequences, 27 hits were found by at least two approaches, 24 by three different strategies and 18 proteins were found by all search methods (Table S1 and Fig. S1). Our final list of candidates included putative cNMP binding proteins that were hits in at least two different approaches (Fig. 1).
Fig. 1.

Identification of putative cNMP binding proteins in T. cruzi. 27 proteins were identified in the parasite genome using a combination of search methods: HMMER and BLAST for local similarity and Smart and Prosite as databases for protein domains. Proteins were identified by at least two of the previously mentioned techniques and listed according to the Pfam E-values, in descending order. In blackand grey the cNMP binding domains detected by Pfam database (http://pfam.sanger.ac.uk) are shown. Inyellow the cNMP binding domains according to SmartDB (http://smart.embl-heidelberg.de/help/smart_about.shtml) are shown. Bracketsonthe left indicate pairs of paralogs; due to the hybrid genotype of the T. cruzi CL Brener clone. H, haplotype; E, Esmeraldo-like; N-E, Non-Esmeraldo-like; N/A, not available. Green IDs, protein kinases. Red and orange IDs: potential cAMP-binding proteins analysed inthis study.
The presence of the identified cNMP proteins in ortholog candidates from T. brucei and L. major was investigated. To this end, we obtained trypanosomatid protein containing cNMP domains that were orthologs of the T. cruzi proteins using the TriTrypDB web server (www.tritrypdb.org). Interestingly, when these sequences were classified (see Table 1), ortholog sequences were found for all cNMP proteins, exception made for TcCLB.506009.109, one gene that was unique to T. cruzi. Two genes were present only in T. cruzi and T. brucei and 10 genes were conserved in T. cruzi and L. major but not in the African trypanosome. Notice that in most cases, the T. cruzi list contains two paralogs, presumably because the hybrid genotype of the CL Brener clone [19]. Overall all three organisms have similar sets of cNMP-containing protein orthologs, suggesting common signal transduction pathways in kinetoplastids.
Multiple sequence alignment of T. cruzi CBD using bacterial CBD CAP.ED as reference, showed conserved sequence motifs within the core of the domain (Fig. S1). Phylogenetic analysis showed that these proteins segregated into two main branches, one containing hypothetical protein kinases (Fig. 2A, green boxes) and the other branch including hypothetical proteins with and without other associated domain. On the other hand, phylogenetic analysis of the CBDs presented a more homogeneous distribution, consistent with an evolutionary conserved nucleotide-binding domain (Fig. 2B). Similar to other organisms, putative effectors share a conserved CBD that would allow the recognition of the cyclic nucleotide by proteins with different functional domains; such as a PAS fold and phosphoglycerate kinase domain, among others (Fig. 1).
Fig. 2.

Phylogenetic analysis. (A) Phylogenetic analysis of putative cNMP binding proteins. Alignments of the 27 full-length sequences were performed with the ClustalW algorithm using the DNAStar package. The unrooted tree was plotted with TreeView software (see Section 2). Green box: protein kinases. Red, orange and blue boxes: potential cNMP-binding proteins analysed in this study. (B) Phylogenetic analysis of CBDs. The key is as for (A) but CBDs were used instead of full-length sequences. Green box, domains present in protein kinases. Red, orange and blue boxes: potential CBDs analysed in this study.
3.2. cNMP binding proteins in T. cruzi
To achieve our ultimate goal of finding proteins that could bind cAMP and eventually play a role as cAMP effectors of the PKA-independent pathway in T. cruzi, we analysed the strongest cNMP-binding proteins from the in silico list of candidates. The proteins presenting the CBD with the lowest E-value, according to the Pfam database (http://pfam.sanger.ac.uk), were: TcCLB.510297.110 (E-value: 2.3E–13), TcCLB.508523.80 (E-value: 8E–13) and TcCLB.507035.110 (E-value: 1.5E–13).
TcCLB.510297.110 codes for a hypothetical 1691 amino acids long protein with a molecular weight of 190 KDa, that presented high RNA expression in metacyclic trypomastigote stages (http://tritrypdb.org) [25].TcCLB.507035.110 andTcCLB.508523.80 are alleles that share an overall 70% identity, with a 100% identity within the CBD. TcCLB.507035.110 encodes a 708 amino-acids long protein, with high levels of mRNA expression in trypomastigotes and metacyclic trypomastigotes; while TcCLB.508523.80 codes for a 218 amino-acids shorter protein, with a 99% overall identity within the shared sequence. TcCLB.507035.110 and TcCLB.508523.80 contain two CBDs within the shared sequence (residues: 233–345/372–489 and residues: 5–117/144–261, respectively). No other known function or functional domain could be identified in these proteins (Fig. 1). Noteworthy, in searching for resistance to the T. brucei PDE inhibitor CpdA using a genome-wide RNA interference library screening approach, Gould et al. [16] identified a group of kinetoplastid unique proteins (CARPs), as cAMP sensors. Although cAMP binding was not experimentally verified, the prediction of the presence of CBDs in CARP1 clearly suggested an important role for this protein in cAMP dependent signalling in T. brucei. Significantly, the sequence analysis of TcCLB.507035.110 and TcCLB.508523.80 shown that these proteins are orthologs of the gene that codes for the T. brucei CARP1 (Tb427tmp.01.7890) (Table 1).
3.3. CBDs features in T. Cruzi
In general, the CBD is a small conserved region of around 120aa that includes both β strands and helical elements. The structure of the cAMP-binding site is organised as an eight-stranded β barrel that function as a pocket for the nucleotide. One of the important features of the domain is the phosphate-binding cassette (PBC) constituted by the β strand 6, a short turn of helix, and the β strand 7. Within the PBC sequence there are few conserved residues such as an arginine that binds to the exocyclic phosphate of cAMP and a pair glycine–glutamate that binds the ribose. Moreover, it has been shown that the arginine has co-evolved with a set of glycines located in different loops across the domain, of particular importance, a glycine in a distal β2-β3 loop that allosterically couples cAMP binding to distal regulatory sites [26,27], features that are all conserved in the studied candidates. A multiple sequence comparison between the CBDs identified in T. cruzi and other known cAMP effectors showed the presence of the key conserved motifs (Fig. 3).
Fig. 3.

Analysis of the CBDs sequences. Múltiple sequence alignment between CBDs identified in T. cruzi and other known cAMP effectors. An orange arrow indicates the Serine H-bonding the phosphate group in TcCLB.508523.80. In red the PBC (phosphate-binding cassette) is shown.
3.4. Molecular modelling ofT. cruzi’s CBDs
Aimed at gaining structural insights on the topology of T. cruzi’s CBDs, we generated comparative models using mammalian cAMP-bound PKA-RIα as template. A well-conserved overall organisation of the cAMP-binding pocket was observed. The alignment ofTcCLB.508523.80 against the PKA-RIα template (Fig. 3) presented an insertion comprised between amino acids 57 and 67. This generates a more elongated loop between β strands 4 and 5 (Fig. 4B), which is the region of highest sequence and length variability in the CBD family [28]. In fact, the homologue PKA-RIIβ presents a similarly long loop, which is not resolved in the electronic density of the PDB structure 1CX4 [29]. In contrast, TcCLB.510297.110 aligned without gaps in the modelled region (Fig. 3), resulting in a higher similarity between the structural model and template (compare Fig. 4A and C and Supplementary Movie). Structural alignment of both models against the target structure (Supplementary Fig. 5) resulted in values as low as 0.9 Å for TcCLB.508523.80 and 0.75 Å for TcCLB.510297.110.
Fig. 4.
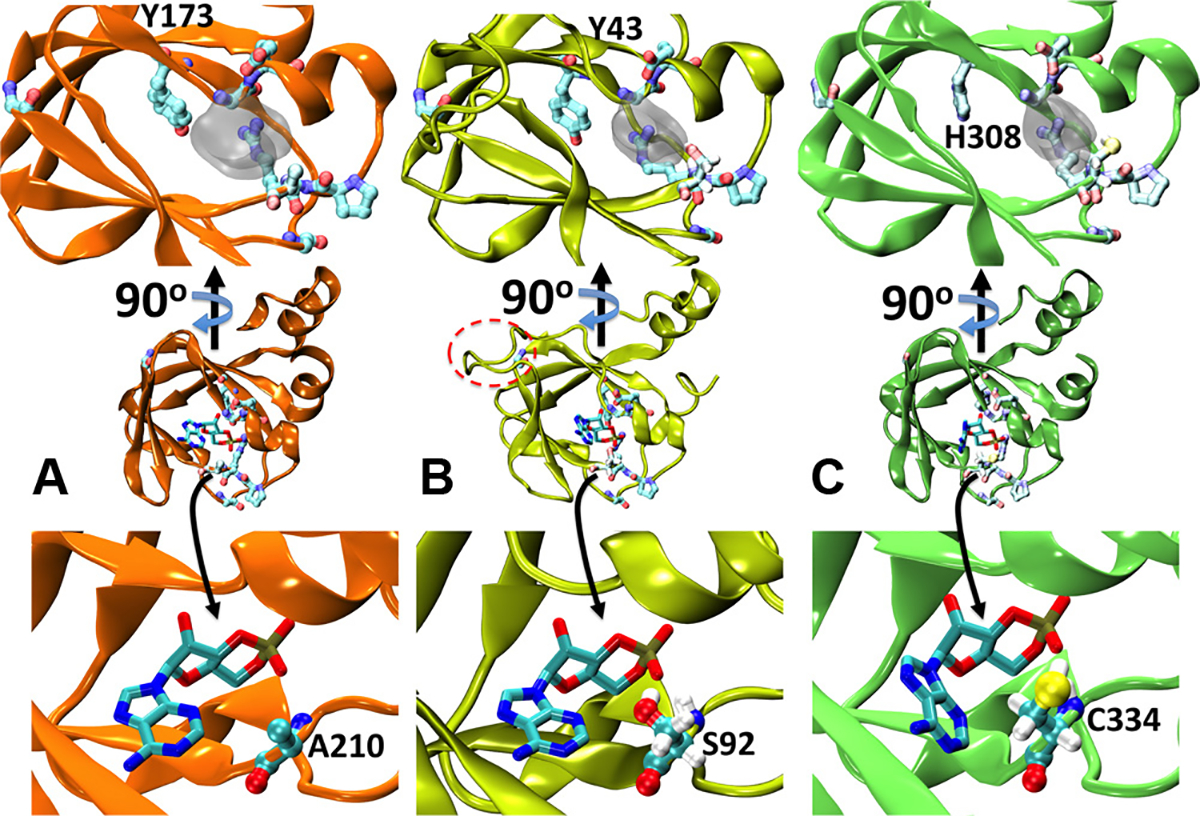
Structural modelling of T. cruzi’s cNMP binding proteins. (A) Cartoon representation of the X-ray structure of PKA-RIα (PDB id:3PNA). cAMP is shown as sticks, while the most conserved residues in Fig. 3 are presented as balls and sticks in pale colours. The inset on the top shows a different perspective on the binding site. For the sake of visual clarity, only the excluded volume of the ligand is indicated as semitransparent grey surface. (B) and (C) Homology models for TcCLB.508523.80 and TcCLB.510297.110, respectively. The label in the top insets indicates the position the tyrosine substituted by histidine in TcCLB.510297.110. The insets in the bottom depict the specific interaction between cAMP and alanine 210 in PKA-RIα (A), serine 92 in TcCLB.508523.80 (B) and cysteine 334 in TcCLB.510297.110 (C). In B, the extra loop between β strands 4 and 5 is highlighted with a dashed red oval. In B and C the hydrogen atoms of S92 and C334 are explicitly included to show the formation of a hydrogen bond with the phosphate moiety.
Placing cAMP within the binding sites of both protein models (see Section 2) suggests that the binding of the second messenger might be stabilised by both polar and non-polar interactions alike those found in the known crystallographic structures. In particular, the most highly conserved residues discussed in the previous paragraph are found in good position to interact with the ligand (Fig. 4 and Supplementary Movie). Only Tyrosine 173 in PKA-RIα is conservatively substituted by a histidine within the β barrel that constitutes the cAMP binding site of TcCLB.510297.110 (Fig. 4A and C).
The main difference encountered within the binding site was observed upon optimisation of the H-bonding pattern in the presence of cAMP and was not obvious from the sequence analysis. We found that Serine 92 and Cysteine 334 in TcCLB.508523.80 and TcCLB.510297.110, respectively, replaced Alanine at position 210 in PKA-RIα (see insets in Fig. 4). In both structural models, these Serine/Cysteine establish an H-bond with the oxygen in the cyclic phosphate group, putatively increasing the affinity for the negatively charged moiety.
3.5. Proof of principie: predicted CBD can bind cAMP
As stated before, searching for resistance to the T. brucei PDE inhibitor CpdA using a genome-wide RNA interference library screening approach, Gould et al. [16] identified a group of kinetoplastid unique proteins, cAMP Responsive Proteins (CARP 1–4). In particular, the prediction of the presence of potential CBDs in CARP1 clearly suggested an important role for this protein in cAMP signalling in T. brucei, although cAMP was not experimentally verified. Taking this into account, and considering that two of our strongest candidates, TcCLB.508523.80 and TcCLB.507035.110, are orthologs of the gene that encodes for CARP1 (Tb927.11.16210), we decided to biochemically corroborate the prediction. To examine whetherTcCLB.508523.80 could bind cAMP in vitro, the full-length version of the protein was expressed as a GST-tag fusion (E. coli expression vector pGEX) and purified from BL21 E. coli strain. Cyclic AMP binding was tested by affinity chromatography using a cAMP-conjugated agarose resin (Sigma–Aldrich). As shown in Fig. 5 binding of recombinant GST-TcCLB.508523.80 to cAMP-conjugated resin verified the prediction. In parallel, GST-PKA-R1β and GST were used as positive and negative controls respectively (Fig. 5A).
Fig. 5.

cAMP-binding assay. Binding of recombinant TcCLB.508523.80 and PKA proteins to cAMP-coupled to agarose beads. + control: protein bound to cAMP beads in the absence of free nucleotide. Beads: beads alone. Western blots were probed with anti-GST antibody. (A) cAMP-binding for PKA and TcCLB.508523.80 is shown. Beads alone and GST binding were also assayed as negative controls. (B) cAMP displacement for both, PKA and TcCLB.508523.80 is shown. Free cAMP at 1 and 10mM was assayed. (C) Nucleotide displacement for PKA and TcCLB.508523.80 is shown. Bound PKA was displaced from cAMP-agarose in the presence of competing cAMP, but not with cGMP or AMP. TcCLB.508523.80 was displaced by cAMP and cGMP, but not with AMP.
Nucleotide-binding specificity for full length TcCLB.508523.80 was analysed by displacement assays (Fig. 5B and C). Displacement for GST-PKA-R1β positive control is also shown. As expected, free cAMP was able to displace cAMP-agarose bound GST-TcCLB.508523.80 and GST-PKA-R1β (Fig. 5B). However, while GST-PKA-R1β has shown to be only displaced by cAMP GST-TcCLB.508523.80 was also displaced by cGMP (Fig. 5C). Thus, these in vitro results showed cNMP specificity in the assayed conditions.
4. Discussion
Cyclic AMP is one of the most widely studied signalling molecules and has been implicated in a wide range of biological functions [30]. For many years, PKA was the only known cAMP effector. Accordingly, it was believed that all cAMP stimuli were mediated by this kinase. It was not until the identification of Epac as a new cAMP effector [31], that cAMP-mediated effects would be classified into two categories: PKA-dependent and PKA-independent pathways, acting independently, converging synergistically or presenting opposite effects depending on specific cellular environments [18]. Similarly, in T. cruzi the only characterised cAMP effector is PKA [11,32–34]. In addition, the absence of sequences for Epac in the T. cruzi genome strongly contributed to the idea of an exclusive PKA-mediated pathway for cAMP signalling in this parasite. However, supporting experimental evidence suggesting the existence of other biologically active cAMP effectors, prompted us to search for cNMP binding proteins as putative cAMP effectors in the parasite genome.
As a result of the in silico search, several hypothetical cNMP binding proteins were identified. We decided to characterize cNMP binding proteins harbouring the CBDs that showed the lowest E-value, representing the strongest putative cNMP-binding proteins. With this in mind, we evaluated in silico TcCLB.508523.80, TcCLB.507035.110 and TcCLB.510297.110 as potential new cAMP effectors. Sequence analysis and homology models revealed that the CBDs present in these proteins could fold in a similar manner than cNMP domains described in other effectors, creating a stable pocket that would accommodate the cyclic nucleotide.
A genome-wide RNAi screening in T. brucei recently identified four cAMP Responsive Proteins (CARP 1–4) [16]. Of particular interest to this work is CARP1, an ortholog to TcCLB.508523.80 and TcCLB.507035.110, which is specific to kinetoplastid parasites (http://tritrypdb.org/tritrypdb/) and was suggested to be a primary cAMP sensor in cAMP signalling in the parasite [16]. In particular, steady-state transcriptome of the four life stages of T. cruzi using microarrays [25] demonstrated an up-regulation in the expression of TcCLB.507035.110 transcripts in infective nonproliferative trypomastigotes. Preliminary RNAseq data confirm that TcCLB.507035.110 transcripts are elevated in trypomastigotes as compared to intracellular amastigotes at 24 h post-infection, showing higher expression in trypomastigotes over epimastigotes as well (Burleigh and El-Sayed, personal communication). Although RNA expression does not always correlate with protein abundance, these results encouraged us to hypothesize that TcCLB.507035.110 might be involved in signalling during invasion.
In this work we presented experimental in vitro evidence showing for the first time that full-length TcCLB.508523.80 was capable of binding cAMP, providing a biochemical validation to the previous observation that its ortholog in T. brucei may play a role as cAMP sensor in the parasite. However, in vitro displacement assays for TcCLB.508523.80 has shown not only affinity for cAMP but a dual cyclic nucleotide affinity, since cGMP was also able to disrupt the interaction of the protein with the cAMP-conjugated resin. In this regard, it could be important to consider the fact that PKA had shown a stronger affinity for conjugated-cNMP resin than for free cAMP in displacement assays using cAMP/cGMP-conjugated agarose [35]. A higher affinity for cAMP-agarose than for the free cyclic-nucleotide might be masking the affinity of TcCLB.508523.80 for the nucleotide also in this case. Nevertheless, when placing cGMP within the binding site of the molecular model for TcCLB.508523.80 (Fig. S4), H-bonding pattern clearly showed that the hydroxyl in Serine 92 could establish a hydrogen bond with the 2-amino group of the cGMP. In a similar manner, the affinity for cGMP was greatly enhanced by a single replacement Serine/Threonine in the PRAAT motif of the regulatory subunit of PKA [36]. Moreover, the PRSA sequence of the PBC of TcCLB.508523.80 resembles the ERSA of E. coli’s CAP PBC, which has been described to equally bind cAMP and cGMP but with only cAMP being able of stimulating a biological response [37], involving an allosteric control by the nucleotide [38].
Ongoing in vitro and in vivo characterisation of the newly described putative effector are expected to uncover new aspects of the cAMP biology in T. cruzi and provide new insights into the mechanisms of pathogenesis involved in Chagas disease. Additionally, due to the fact that cAMP pathways in T. cruzi would present some unique features [39] through divergent effectors comparing to its human counterparts, we also expect to identify a new set of potential targets for drug design.
Supplementary Material
{kind=link}
Acknowledgements
The authors would like to thank Barbara Burleigh for her valuable comments on the manuscript and, together with Najib El-Sayed, for sharing unpublished data. We wish to thank Soledad N. Gonzalez for participating in the first steps of the project. Research reported in this publication was supported by the FIC-NIH award number R03TW009001 (to DLA/MME) UBACyT-GEF 20020110200024 (to MME), the Agencia Nacional de Promoción Científica y Tecnológica (PICT-2012–0034 to AVJ and PICT-2011–1572 toJGDG), the Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET) PIP-IU number 11420100100333 and Fundación Bunge y Born (toJGDG). AVJ, JGDG and MME are members of the Research Career of CONICET, and JGM is a CONICET Research Fellow. SP was partially funded by FOCEM (MERCOSUR Structural Convergence Fund), COF 03/11.
Footnotes
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016Zj.molbiopara.2015.02.002.
References
- [1].WHO. Reporte del grupo de trabajo científico sobre la enfermedad de Chagas. World Health Organization; 2007. [TDR/GTC/09]. [Google Scholar]
- [2].Schmunis GA, Yadon ZE. Chagas disease: a Latin American health problem becoming a world health problem. Acta Trop 2010;115(1–2):14–21. [DOI] [PubMed] [Google Scholar]
- [3].Klein N, Hurwitz I, Durvasula R. Globalization of Chagas disease: a growing concern in nonendemic countries. Epidemiol Res Int 2012:13. [Google Scholar]
- [4].Bahia MT, Diniz LD, Mosqueira VC. Therapeutical approaches under investigaron for treatment of Chagas disease. Expert Opin Invest Drugs 2014:1–13. [DOI] [PubMed] [Google Scholar]
- [5].Clayton J Chagas disease 101. Nature 2010;465(7301):S4–5. [DOI] [PubMed] [Google Scholar]
- [6].Seebeck T, Schaub R, Johner A. cAMP signallin in the kinetoplastid protozoa. Curr Mol Med 2004;4(6):585–99. [DOI] [PubMed] [Google Scholar]
- [7].Rangel-Aldao R, Allende O, Triana F, Piras R, Henriquez D, Piras M. Possible role of cAMP in the differentiation of Trypanosoma cruzi. Mol Biochem Parasitol 1987;22(1):39–43. [DOI] [PubMed] [Google Scholar]
- [8].Rangel-Aldao R, Triana F, Fernandez V, Comach G, Abate T, Montoreano R. Cyclic AMP as an inducer of the cell differentiation of Trypanosoma cruzi. Biochem Int 1988;17(2):337–44. [PubMed] [Google Scholar]
- [9].Oliveira MM, Antunes A, De Mello FG. Growth of Trypanosoma cruzi epimastigotes controlled by shifts in cyclic AMP mediated by adrenergic ligands. Mol Biochem Parasitol 1984;11:283–92. [DOI] [PubMed] [Google Scholar]
- [10].Oliveira MM, Rocha ED, Rondinelli E, Arnholdt AV, Scharfstein J. Signal transduction in Trypanosoma cruzi: opposite effects of adenylcyclase and phospholipase C systems in growth control. Mol Cell Biochem 1993;124(2): 91–9. [DOI] [PubMed] [Google Scholar]
- [11].Bao Y, Weiss LM, Braunstein VL, Huang H. Role of protein kinase A in Trypanosoma cruzi. Infect Immun 2008;76(10):4757–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gonzales-Perdomo M, Romero P, Goldenberg S. Cyclic AMP and adenylate cyclase activators stimulate Trypanosoma cruzi differentiation. Exp Parasitol 1988;66(2):205–12. [DOI] [PubMed] [Google Scholar]
- [13].Fraidenraich D, Pena C, Isola EL, Lammel EM, Coso O, Anel AD, et al. Stimulation of Trypanosoma cruzi adenylyl cyclase by an alpha D-globin fragment from Triatoma hindgut: effect on differentiation of epimastigote to trypomastigote forms. Proc Natl Acad Sci U S A 1993;90(21):10140–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Vassella E, Reuner B, Yutzy B, Boshart M. Differentiation of African trypanosomes is controlled by a density sensing mechanism which signals cell cycle arrest via the cAMP pathway.J Cell Sci 1997;110(Pt 21):2661–71. [DOI] [PubMed] [Google Scholar]
- [15].de Koning HP, Gould MK, Sterk GJ, Tenor H, Kunz S, Luginbuehl E, et al. Pharmacological validation of Trypanosoma brucei phosphodiesterases as novel drug targets. J Infect Dis 2012;206(2):229–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Gould MK, Bachmaier S, Ali JA, Alsford S, Tagoe DN, Munday JC, et al. Cyclic AMP effectors in African trypanosomes revealed by genome-scale RNA interference library screening for resistance to the phosphodiesterase inhibitor CpdA. Antimicrob Agents Chemother 2013;57(10):4882–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gould MK, de Koning HP. Cyclic-nucleotide signalling in protozoa. FEMS Microbiol Rev 2011;35(3):515–41. [DOI] [PubMed] [Google Scholar]
- [18].Cheng X, Ji Z, Tsalkov T, Mei F. Epac and PKA: a tale of two intracelular cAMP receptors. Acta Biochim Biophys Sin (Shanghai) 2008;40(7):651–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].El-Sayed NM, Myler PJ, Bartholomeu DC, Nilsson D, Aggarwal G, Tran AN, et al. The genome sequence of Trypanosoma cruzi, etiologic agent of Chagas disease. Science 2005;309(5733):409–15. [DOI] [PubMed] [Google Scholar]
- [20].Soding J, Biegert A, Lupas AN.The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res 2005;33(Web Server issue):W244–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Badireddy S, Yunfeng G, Ritchie M, Akamine P, Wu J, Kim CW, et al. Cyclic AMP analog blocks kinase activation by stabilizing inactive conformation: conformational selection highlights a new concept in allosteric inhibitor design. Mol Cell Proteomics 2011;10(3). M110 004390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen MY, et al. Comparative protein structure modeling using MODELLER. Curr Protoc Protein Sci 2007. [Chapter 2: p Unit 29]. [DOI] [PubMed] [Google Scholar]
- [23].Russell RB, Barton GJ. Multiple protein sequence alignment from tertiary structure comparison: assignmen of global and residue confidence levels. Proteins 1992;14(2):309–23. [DOI] [PubMed] [Google Scholar]
- [24].Gonzalez HC, Darre L, Pantano S. Transferable mixing of atomistic and coarse–grained water models. J Phys Chem B 2013. [DOI] [PubMed] [Google Scholar]
- [25].Minning TA, Weatherly DB, Atwood J 3rd, Orlando R, Tarleton RL. The steady-state transcriptome of the four major life-cycle stages of Trypanosoma cruzi. BMC Genomics 2009;10:370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Berman HM, Ten Eyck LF, Goodsell DS, Haste NM, Kornev A, Taylor SS. The cAMP binding domain: an ancient signaling module. Proc Natl Acad Sci U S A 2005;102(1):45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kannan N, Wu J, Anand GS, Yooseph S, Neuwald AF, Venter JC, et al. Evolution of allostery in the cyclic nucleotide binding module. Genome Biol 2007;8(12):R264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Canaves JM, Taylor SS. Classification and phylogenetic analysis of the cAMP-dependent protein kinase regulatory subunit family. J Mol Evol 2002;54(1):17–29. [DOI] [PubMed] [Google Scholar]
- [29].Diller TC, Madhusudan NH, Xuong SS. Taylor Molecular basis for regulatory subunit diversity in cAMP-dependent protein kinase: crystal structure of the type II beta regulatory subunit. Structure 2001;9(1):73–82. [DOI] [PubMed] [Google Scholar]
- [30].Sutherland EW. Studies on the mechanism of hormone action. Science 1972;177(47):401–8. [DOI] [PubMed] [Google Scholar]
- [31].Kawasaki H, Springett GM, Mochizuki N, Toki S, Nakaya M, Matsuda M, et al. A family of cAMP-binding proteins that directly activate Rap1. Science 1998;282(5397):2275–9. [DOI] [PubMed] [Google Scholar]
- [32].Bao Y, Weiss LM, Hashimoto M, Nara T, Huang H. Protein kinase A regulatory subunit interacts with P-Type ATPases in Trypanosoma cruzi. Am J Trop Med Hyg 2009;80(6):941–3. [PubMed] [Google Scholar]
- [33].Bao Y, Weiss LM, Ma YF, Kahn S, Huang H. Protein kinase A catalytic subunit interacts and phosphorylates member of trans-sialidase super-family in Trypanosoma cruzi. Microbes Infect 2010;12(10):716–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Huang H, Weiss LM, Nagajyothi F, Tanowitz HB, Wittner M, Orr GA, et al. Molecular cloning and characterization of the protein kinase A regulatory subunit of Trypanosoma cruzi. Mol Biochem Parasitol 2006;149(2):242–5. [DOI] [PubMed] [Google Scholar]
- [35].Scholten A, Poh MK, van Veen TA, van Breukelen B, Vos MA, Heck AJ. Analysis of the cGMP/cAMP interactome using a chemical proteomics approach in mammalian heart tissue validates sphingosine kinase type 1-interacting protein as a genuine and highly abundant AKAP. J Proteome Res 2006;5(6):1435–47. [DOI] [PubMed] [Google Scholar]
- [36].Shabb JB, Buzzeo BD, Ng L, Corbin JD. Mutating protein kinase cAMP-binding sites into cGMP-binding sites mechanism of cGMP selectivity. J Biol Chem 1991;266(36):24320–6. [PubMed] [Google Scholar]
- [37].Ebright RH, Le Grice SF, Miller JP, Krakow JS. Analogs of cyclic AMP that elicit the biochemically defined conformational change in catabolite gene activator protein (CAP) but do not stimulate bindingto DNA.J Mol Biol 1985;182(1):91–107. [DOI] [PubMed] [Google Scholar]
- [38].Popovych N, Tzeng SR, Tonelli M, Ebright RH, Kalodimos CG. Structural basis for cAMP-mediated allosteric control of the catabolite activator protein. Proc Natl Acad Sci U S A 2009;106(17):6927–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Laxman S, Beavo JA. Cyclic nucleotide signaling mechanisms in trypanosomes:possible targets for therapeutic agents. Mol Interv 2007;7(4):203–15. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.