

Cellular senescence is considered a crucial process for tumour suppression, which can be facilitated by immune surveillance. However, when senescent cells persist in tissues, they can also trigger a plethora of tumour‐promoting effects. Here, we discuss the main hallmarks, mechanisms and roles of senescence in cancer and provide a comprehensive revision of the available tools for its detection.

Keywords: cancer, cellular senescence, detection, senoprobes, tumour microenvironment
Abstract
Senescence refers to a cellular state featuring a stable cell‐cycle arrest triggered in response to stress. This response also involves other distinct morphological and intracellular changes including alterations in gene expression and epigenetic modifications, elevated macromolecular damage, metabolism deregulation and a complex pro‐inflammatory secretory phenotype. The initial demonstration of oncogene‐induced senescence in vitro established senescence as an important tumour‐suppressive mechanism, in addition to apoptosis. Senescence not only halts the proliferation of premalignant cells but also facilitates the clearance of affected cells through immunosurveillance. Failure to clear senescent cells owing to deficient immunosurveillance may, however, lead to a state of chronic inflammation that nurtures a pro‐tumorigenic microenvironment favouring cancer initiation, migration and metastasis. In addition, senescence is a response to post‐therapy genotoxic stress. Therefore, tracking the emergence of senescent cells becomes pivotal to detect potential pro‐tumorigenic events. Current protocols for the in vivo detection of senescence require the analysis of fixed or deep‐frozen tissues, despite a significant clinical need for real‐time bioimaging methods. Accuracy and efficiency of senescence detection are further hampered by a lack of universal and more specific senescence biomarkers. Recently, in an attempt to overcome these hurdles, an assortment of detection tools has been developed. These strategies all have significant potential for clinical utilisation and include flow cytometry combined with histo‐ or cytochemical approaches, nanoparticle‐based targeted delivery of imaging contrast agents, OFF‐ON fluorescent senoprobes, positron emission tomography senoprobes and analysis of circulating SASP factors, extracellular vesicles and cell‐free nucleic acids isolated from plasma. Here, we highlight the occurrence of senescence in neoplasia and advanced tumours, assess the impact of senescence on tumorigenesis and discuss how the ongoing development of senescence detection tools might improve early detection of multiple cancers and response to therapy in the near future.
Abbreviations
- 5‐FU
5‐fluorouracil
- AAH
atypical adenomatous hyperplasia
AIS adenocarcinoma in situ
- ATM
ataxia‐telangiectasia mutated
- ATR
ATM‐ and Rad3‐related
- B2M
β2‐microglobulin
- BAX
BCL2‐associated protein X
- BCL‐2
B‐cell lymphoma 2
- BrdU
5‐bromo‐2′‐deoxyuridine
- C/EBPβ
CCAAT/enhancer‐binding protein beta
- CCF
cytoplasmic chromatin fragment
- CDK
cyclin‐dependent kinase
- cfDNA
cell‐free DNA
- cGAS‐STING
cyclic GMP‐AMP synthase linked to stimulator of interferon genes
- CHK
checkpoint kinase
- CIS
carcinoma innonbreakingspacesitu
- CKI
CDK inhibitors
- CM
conditioned medium
- CSLC
cancer stem‐like cell
- CT
computed tomography
- CXCL
C‐X‐C‐motif ligand
- DDR
DNA damage response
- DSB
double‐strand breaks
- EdU
5‐ethynyl‐2′‐deoxyuridine
- EMT
epithelial–mesenchymal transition
- ER
endoplasmic reticulum
- EV
extracellular vesicle
- FFPE
formalin‐fixed and paraffin‐embedded
- FOXO
forkhead box O
- H3K20me3
trimethylation of lysine 20 on histone 3
- H3K27ac
acetylation of lysine 27 on histone 3
- H3K9me3
trimethylation of lysine 9 on histone 3
- HER
human epidermal growth factor receptor
- HMGB1
high mobility group box‐1
- IL
interleukin
- LMNB1
lamin B1
- MAPK
mitogen‐activated protein kinase
- MDM2
mouse double minute 2
- MEK
MAPK/ERK kinase
- MHC
major histocompatibility complex
- MIA
minimally invasive adenocarcinoma
- MMP
matrix metalloproteinase
- MRI
magnetic resonance imaging
- mTOR
mammalian target of rapamycin
- nanoMIP
molecularly imprinted nanoparticle
- NB
Nile blue
- NF‐κB
nuclear factor kappa light‐chain enhancer of activated B cells
- NIR
near‐infrared
- NK
natural killer
- NP
nanoparticle
- NSCLC
non‐smallcell lung cancer
- OIS
oncogene‐induced senescence
- PDH
pyruvate dehydrogenase
- PDK1
PDH‐inhibitory enzyme pyruvate dehydrogenase kinase 1
- PDP2
PDH‐activating enzyme pyruvate dehydrogenase phosphatase 2
- PDTX
patient‐derived tumour xenograft
- PET
positron emission tomography
- PI3K
phosphatidylinositol 3‐kinase
- PTBP1
polypyrimidine tract binding protein 1
- PTEN
phosphatase and tensin homolog
- RB
retinoblastoma protein
- ROS
reactive oxygen species
- RPS14
ribosomal protein S14
- SAHF
senescence‐associated heterochromatin foci
- SASP
senescence‐associated secretory phenotype
- SAβG
senescence‐associated β‐galactosidase
- SBB
Sudan Black B
- TGFβ
transforming growth factor β
- TIF
telomere dysfunction‐induced foci
- TIS
therapy‐induced senescence
- TKI
tyrosine kinase inhibitor
- TS
tumour suppressor
- UPS
unfolded protein response
- VEGF
vascular endothelial growth factor
- γH2AX
phosphorylation of the histone H2AX
1. Cellular senescence: Introduction
The term senescence derives etymologically from senex, the Latin word for old. Based on the in vitro observation of the finite proliferation capacity of human fibroblasts upon serial cultivation, at the beginning of the 1960s Leonard Hayflick and Paul Moorhead introduced the concept of cellular senescence [1]. Such limited proliferative capacity was attributed to the gradual attrition, through multiple cell divisions, of the telomeres located at both ends of the chromosome, consisting of the repetitive TTAGGG DNA sequence as protective structures [2, 3]. This is referred to as replicative senescence [4]. Cellular senescence is a stable state of cell‐cycle arrest that is triggered in proliferative cells by multiple types of damage, including replicative stress, and is characterised by the implementation of a complex pro‐inflammatory secretory phenotype associated with altered metabolism (the so‐called ‘senescence‐associated secretory phenotype’, or SASP).
Cellular senescence is a very heterogeneous programme that varies depending on the different stimuli and cellular contexts to which it responds. Senescence is involved in several physiological and pathological processes, ageing and cancer being probably the most notorious. Cellular senescence has been characterised for over a half‐century, and a plethora of studies have proposed several types of senescence based on diverse stimuli [5]. While DNA damage‐induced senescence refers to the response to irreparable DNA damage caused by either endogenous sources [e.g., telomere shortening or double‐strand breaks (DSBs) occurring during proliferation], or exogenous sources (such as ionising radiation and DNA‐damaging agents), oncogene‐induced senescence (OIS) springs to action upon either the activation of oncogenes or the inactivation of tumour suppressors [6, 7]. In the context of cancer cells, therapy‐induced senescence (TIS) can emerge in response to a therapeutic regimen (e.g., cytotoxic chemotherapy or radiation) [8]. Furthermore, mitochondrial dysfunctionality and oxidative stress can also induce cellular senescence [9, 10]. Despite the fact that a diversity of stimuli can trigger it, cellular senescence exhibits certain distinct hallmarks that are not observed in other cellular states, providing promise for universal methods of detection and therapeutic targeting.
1.1. Hallmarks of senescence
Senescent cells usually acquire some structural changes as a result of stress‐induced signalling cascades, including flattened, enlarged and aberrant morphologies with modified cytoplasmic compositions. However, some characteristics of senescent cells (e.g., stable withdrawal from the cell‐cycle and specific morphological changes) are shared with other cellular states such as quiescence and terminal differentiation. The International Cell Senescence Association has proposed a consensus set of the defining hallmarks of senescence phenotypes based on the following four features: (a) cell‐cycle withdrawal; (b) macromolecular damage; (c) secretory phenotype, and (d) deregulated metabolism [11] (Fig. 1). Of note, while these are distinct cellular features, they have complex interplays and interdependence.
Fig. 1.
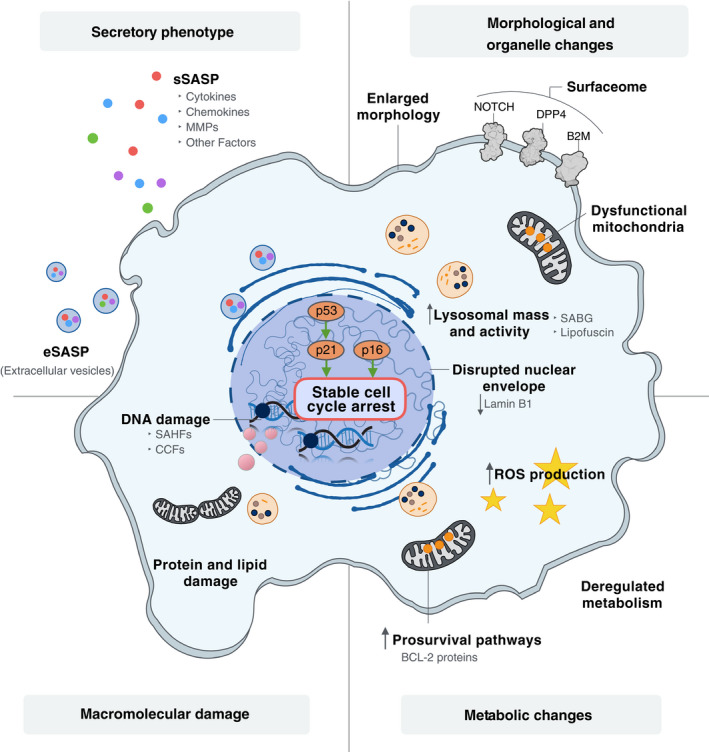
Hallmarks of cellular senescence. Senescence is triggered in response to a variety of stimuli, with senescent cells acquiring phenotypes derived from changes in morphology, the nucleus and the cytoplasm. B2M, β2 microglobulin; BCL‐2, B‐cell lymphoma 2; CCF, cytoplasmic chromatin fragment; DPP4, dipeptidyl‐peptidase 4; MMPs, matrix metalloproteinases; ROS, reactive oxygen species; SAβG, senescence‐associated β‐galactosidase; SAHF, senescence‐associated heterochromatin foci; SASP, senescence‐associated secretory phenotype.
1.1.1. Cell‐cycle withdrawal
Mammalian cell‐cycle progression is driven by the dynamics of cyclins and cyclin‐dependent kinases (CDKs), which ensure timely phase transition. Cyclin D/CDK4‐6 complexes promote cell‐cycle progression into G1, followed by cyclin E/CDK2 complexes eliciting G1/S phase transition; the progression into S phase and subsequent transition to S/G2 phase require competent cyclin A/CDK2 complexes; and cyclin B/CDK1 facilitates G2/M phase transition [12, 13]. Careful modulation of the activity of cyclin/CDKs by various CDK inhibitors (CKIs), as occurs in cellular quiescence and senescence, ensures efficient context‐dependent control of the cell cycle [14]. Increased levels of p27KIP1 mediate cell‐cycle arrest in G0 in quiescent cells [15, 16] while a decrease in p27KIP1 levels upon mitogenic stimulation reverses this arrest and leads to cell‐cycle re‐entry [17]. By contrast, the upregulation of p21WAF1/CIP1 (CDKN1A) and p16INK4A (CDKN2A) commonly halts cell proliferation leading to cellular senescence. Accumulation of p21WAF1/CIP1 and p16INK4A first leads to the hypo‐phosphorylation of retinoblastoma protein (RB) and then inhibits the transactivation of the E2F genes involved in nucleotide metabolism and DNA synthesis [18, 19], resulting in a stable cell‐cycle arrest [20].
Alongside the upregulation of CKIs, cellular senescence is also associated with some fundamental epigenetic changes [21, 22]. Distinct histone modifications, including elevated trimethylation of lysine 9 or lysine 20 on histone H3 (H3K9me3 or H3K20me3) may facilitate cell‐cycle arrest [21, 23], while acetylation of lysine 27 on histone H3 (H3K27ac) has a role in promoting the SASP [24]. Senescence‐associated heterochromatin foci (SAHF), which form to various extents in senescent cells depending on the stimuli used [23], are a gene repressing mechanism exerted through the induction of focal hypermethylation of DNA via cooperation between heterochromatin protein 1 (HP1) and DNA methyltransferase [25, 26]. The location of SAHF in the vicinity of E2F target genes implicated in cell‐cycle progression thereby imposes stable cell‐cycle withdrawal [23, 27, 28]. Additionally, loss of the nuclear structural protein lamin B1 (LMNB1) compromises the integrity of the nuclear envelope, leading to the production of cytoplasmic chromatin fragments (CCFs), which are involved in the regulation of the secretory phenotype of senescent cells [29]. Loss of LMNB1 and resultant CCFs are viewed as hallmarks of senescence [30, 31].
1.1.2. Macromolecular damage
The substantial accumulation of macromolecular damage, such as DNA and protein damage, is another discriminating feature that distinguishes cellular senescence from cell differentiation. The progressive telomere shortening owing to successive cell divisions is eventually recognised and processed as DNA damage by the DNA damage response (DDR) machinery. When left unresolved, this induces cellular senescence [32]. In the case of OIS, oncogene‐driven hyperproliferation leads to the collision and subsequent collapse of replication forks, ultimately generating DSBs [33, 34], leading to induction of senescence. Senescent cells are metabolically active, and the accumulation of reactive oxygen species (ROS) contribute to oxidative DNA damage at telomeric G‐rich repeats, forming so‐called telomere dysfunction‐induced foci (TIFs) that are part of the DDR response [35, 37]. DDR processes lead to the phosphorylation of histones H2AX (γH2AX) and H3K9me3 which facilitate the binding and assembly of DNA repair machineries or proteins involved in the DDR signalling cascade. The detection of these modifications is widely used to identify DNA damage‐induced senescence [38, 39].
Furthermore, ROS oxidation of cysteine residues in protein tyrosine phosphatases and their subsequent removal by the proteasome‐dependent protein degradation system leads to hyperactivation of ERK signalling which in turn triggers cellular senescence similarly to when driven by oncogenic stress [35, 40, 41]. The accumulation of damaged proteins also increases endoplasmic reticulum (ER) stress, setting off the unfolded protein response (UPS), which then triggers reduced protein synthesis, ER expansion and accelerated protein export [42].
In addition to the macromolecular damage already described, senescent cells also manifest an upregulation of pro‐survival (anti‐apoptotic) pathways and mechanisms aimed at alleviating the impact of DNA and protein damage in the cells [43]. Senescent human fibroblasts are resistant to apoptosis due to persistently elevated levels of anti‐apoptotic BCL2 proteins and the reduced levels of pro‐apoptotic BCL2‐associated protein X (BAX), which may be attributed to senescence‐associated histone modifications [44, 45, 46]. BCL‐W and BCL‐XL, members of the anti‐apoptotic BCL2 family of proteins, are also upregulated in senescent fibroblasts irrespective of the senescence trigger [46]. Moreover, senescence can interfere with apoptosis implementation via downregulation of the main effector caspase‐3 [47]. This senescence‐mediated intrinsic resistance to apoptosis is abrogated upon genetic or pharmacological perturbation of p21WAF1/CIP1 or BCL‐2 family members, thus implying the significance of senescence in the promotion of cell survival [46, 48].
1.1.3. Secretory phenotype
The SASP plays an important role in the reinforcement and propagation of senescence phenotypes [49] and contributes to tissue homeostasis [50]. However, it can also have detrimental effects depending on the nature of the triggers, the specific cell types involved and whether senescent cells persist in tissues [51]. The SASP is composed of diverse pro‐inflammatory cytokines, chemokines, growth factors, and matrix metalloproteinases and can function in cell‐autonomous (autocrine) or non‐cell‐autonomous (paracrine) fashions, exerting particular physiological or pathological effects depending on the context [49, 52, 53, 54]. Regulation of the SASP is a complex process that involves different drivers, including nuclear factor kappa light‐chain‐enhancer of activated B cells (NF‐κB) [55], CCAAT/enhancer‐binding protein beta (C/EBPβ) [56], mammalian target of rapamycin (mTOR) [57, 58] and NOTCH1 [59]. CCFs (discussed previously) also activate the cytosolic DNA‐sensing GMP‐AMP synthase stimulator of interferon genes (cGAS‐STING) pathway, an important contributor to innate immunity that participates in driving pro‐inflammatory responses and SASP regulation [29, 60]. The senescence‐associated Alarmin high mobility group box 1 (HMGB1) protein is also implicated in SASP regulation. HMGB1 is exported from the nucleus to the extracellular milieu of senescent cells in a p53‐dependent manner. Depletion of HMGB1 attenuates the secretion of the canonical SASP factor interleukin‐6 (IL‐6) [61]. Intriguingly, the expression levels of HMGB1 were recently proposed to be the key determinant of the fate between senescence and apoptosis in various types of cancer cells following genotoxic stress [62].
1.1.4. Deregulated metabolism
Production of the senescent secretome is highly energy‐demanding, and senescent cells rely heavily on augmented mitochondrial metabolism and glycolysis to meet their ATP needs [63]. Oncogene BRAFV600E ‐induced senescent cells undergo a metabolic reprogramming that is dependent on the mitochondrial gatekeeper pyruvate dehydrogenase (PDH). This process is accompanied by the simultaneous suppression of PDH‐inhibitory enzyme pyruvate dehydrogenase kinase 1 (PDK1) and an increase in the production of the PDH‐activating enzyme pyruvate dehydrogenase phosphatase 2 (PDP2) to promote the use of pyruvate in the tricarboxylic acid cycle, helping senescent cells meet their higher energy requirements [64]. Given the state of perturbed mitochondrial metabolism and its accompanying proteotoxic stress, it is crucial that senescent cells maintain balance between anabolism and catabolism. They do so by a process that couples mTOR to autolysosomes in a distinctive cellular compartment that is known as the TOR‐autophagy spatial coupling compartment, located at the trans side of the Golgi apparatus [65]. Currently, an elevated number of lysosomes showing enhanced lysosomal β‐galactosidase activity are the most widely and intensively used marker for the detection of senescence. This can be detected using the senescence‐associated β‐galactosidase (SAβG) assay, where β‐galactosidase activity can be assessed (both in vitro and in vivo) using a chromogenic reagent at a restrictive pH (pH 6) [66]. Lysosomes in senescent cells also have higher levels of lipofuscin, composed of insoluble lipid‐containing aggregates of lysosomal digestion. Using dyes, these aggregates can also be visualised as an indicator of senescence [67].
Taken together, these hallmarks of senescence discriminate senescent from quiescent or differentiated cells, resulting in their potential utility as biomarkers for senescence detection. Nevertheless, it must be borne in mind that senescence is a highly heterogeneous phenomenon and some of the features discussed above may vary according to different cellular contexts and senescence‐inducing stimuli. Taking into account the complexity and heterogeneity of human tissues in health and disease, the detection of senescent cells in clinical settings requires much more comprehensive studies.
1.2. Mechanistic insights into senescence induction in cancer
While a variety of stimuli may trigger cellular senescence (Fig. 2), DDR induction remains one of the most intensively studied mechanisms [32]. Once sensor protein complexes recognise DNA damage, either apical kinase ataxia‐telangiectasia mutated (ATM) or ATM‐ and Rad3‐related (ATR) is recruited and activated, resulting in the phosphorylation and activation of downstream checkpoint kinases, either CHK1 or CHK2 [68]. Regardless of the specific trigger, DDR signalling cascades ultimately converge on activating the effector protein p53 that subsequently transactivates the CKI p21WAF1/CIP1, arresting the cell in G1 or G2/M by blocking activity of CDK2 or CDK1, respectively [69, 70]. In addition to DDR, ageing and epigenetic de‐repression of the CDKN2A gene, which encodes the tumour suppressor ARF, also promotes p53‐p21WAF1/CIP1‐mediated cellular senescence through the inhibition of MDM2‐mediated p53 degradation [71, 72]. Contrastingly, p16INK4A, the other product of the CDKN2A gene, interferes directly with cyclin/CDK complexes to impose cell‐cycle arrest [20]. The response to the accumulation of ROS owing to metabolic perturbation includes activation not only of DDR but also mitogen‐activated protein kinase (MAPK)/p38 pathways to promote cellular senescence [73, 74].
Fig. 2.
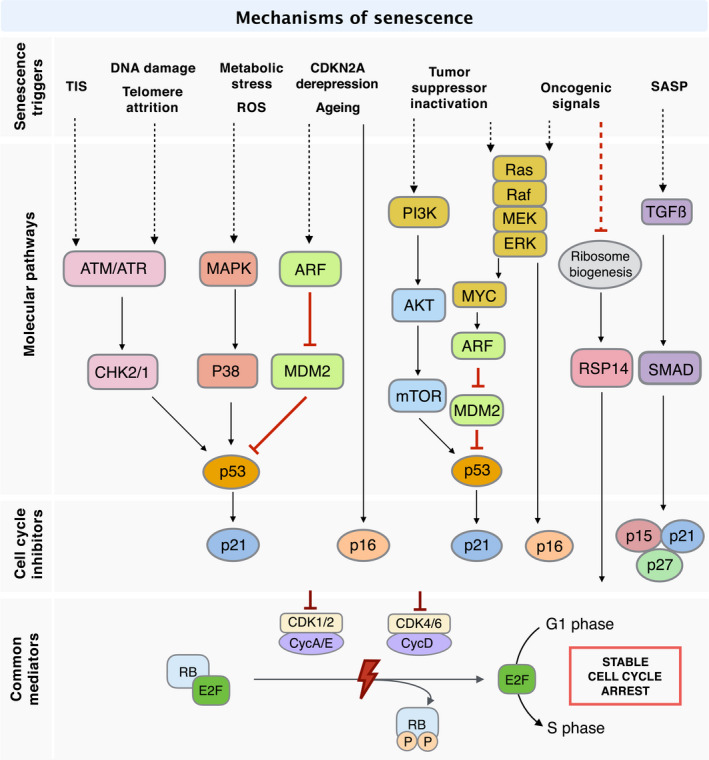
Signalling pathways of senescence induction in cancer. DNA damage and telomere shortening activate a DNA damage response that imposes cell‐cycle arrest through the p53‐p21 axis while ARF and p16 upregulation due to ageing and CDKN2A de‐repression block cell‐cycle progression via both the p53‐p21 and p16 axis. ROS and metabolic alterations implement senescence through MAPK/p38 signalling whereas SASP reinforces senescence by means of TGFβ signalling. Inactivation of tumour suppressors not only induces the Ras/Raf/MEK signalling pathway as oncogenic signals, but also modulates the p53‐p21 axis via the PI3K/AKT/mTOR pathway. In addition to the conventional CKI‐dependent pathway, oncogenic signals trigger cell‐cycle withdrawal by downregulating ribosome biogenesis, thereby increasing RPS14 for direct inhibition of CDK/cyclin‐mediated RB phosphorylation. ATM, ataxia‐telangiectasia mutated; ATR, ATM‐ and Rad3‐related; CDK, cyclin‐dependent kinase; CHK, checkpoint kinase; MAPK, mitogen‐activated protein kinase; MEK, MAPK/ERK kinase; mTOR, mammalian target of rapamycin; PI3K, phosphatidylinositol 3‐kinase; RB, retinoblastoma protein; RPS14, ribosomal protein S14; TGFβ, transforming growth factor β.
The hyperproliferative nature of premalignant cells is associated with the progressive shortening of telomeres [75]. The recognition of shortened telomeres as sites of DNA damage induces replicative senescence via the activation of the DDR signalling cascade (Fig. 2; DNA damage and telomere attrition). It is noteworthy that certain cancer treatments in widespread use effectively implement TIS [76]. Many cancer therapeutics, despite significant mechanistic diversity, induce senescence through the generation of DNA damage, thus eliciting DDR signalling (Fig. 2; TIS).
Senescence can also be triggered in the absence of DNA damage. Since the observation of the first senescence phenotype, in cells ectopically expressing HRasG12V, several studies have unravelled the molecular mechanisms underlying OIS and the concomitant accumulation of p53 and p16INK4A [77]. The constitutively active MEK/MAPK cascade upregulates p53 and p16INK4A upon HRas‐induced premature senescence in human fibroblasts [78]. Similarly, Zhu et al. [79] proposed that the MEK/MAPK signalling pathway also participates in eliciting senescence in human fibroblasts under conditional activation of Raf, the downstream kinase signal transmitter of Ras. However, they showed that Raf‐induced senescence requires p16INK4A, but not p53 and p21WAF1/CIP1, thus implying a more nuanced Ras/Raf/MEK/MAPK regulation of OIS [79]. Oncogenic Myc‐induced senescence in B lymphocytes requires both the DDR mediator ATM and p53, highlighting the plausible existence of crosstalk between the DDR and Ras/Raf/MEK/MAPK pathways in OIS regulation [80, 81, 82]. Myc‐induced senescence correlates with delayed lymphoma onset, a process that is suppressed by Cdk2 contributing to lymphomagenesis [83]. Further, a more recent study revealed that Myc promotes OIS through the transactivation of the ubiquitin‐specific protease USP10, which stabilises ARF and thus maintains downstream p53‐mediated senescence [84]. Epigenetic modifications (e.g., H3K9me3 methylation catalysed by the histone methyltransferase Suv39h1), one of the hallmarks of cellular senescence, are involved in Ras‐ or Myc‐driven OIS by suppressing the E2F‐mediated transactivation of proliferative genes [82, 85]. The phosphatidylinositol 3‐kinase (PI3K)/AKT pathway constitutes an additional route to the establishment of OIS since it promotes mTOR‐regulated translation and stabilisation of p53 [86]. In addition to OIS, there are other (indirect) ways to achieve p53‐dependent senescence both in vivo and in vitro, including the inactivation of other tumour suppressors (TS), such as the phosphatase and tensin homolog (PTEN), an antagonist of the PI3K/AKT pathway [87].
Although the upregulation of CKIs is at the heart of most of the strategies leading to the induction of cellular senescence, a recent study reported a novel mechanism for the induction of senescence that is independent of canonical CKIs. In this study, the accumulation of ribosomal protein S14 (RPS14) arising from reduced ribosome biogenesis triggers senescence through the direct binding of RPS14 to CDK4, thereby inhibiting CDK4 itself as well as RB phosphorylation [88]. Particularly, when we consider that many cancers harbour dysfunctional DDR signalling, this study provides an important insight into the manipulation, and potential redundancy, of senescence pathways in such complex disease settings.
1.3. Role of senescence: a double‐edged sword
Although originally viewed as a cellular response to stress and related to ageing and cancer, cellular senescence is also implicated in a plethora of physiological and pathological processes. The ultimate impact of senescence depends on the triggers, stimuli, signalling pathways involved and, crucially, whether senescent cells are efficiently cleared or persist in the tissues [5]. In this regard, transient senescence may play an important role in certain developmental or physiological conditions. More specifically, senescence‐initiated repair processes include SASP‐driven signalling to nearby cells and recruitment of immune cells, clearance of senescent cells by phagocytic cells and activation of nearby stem or progenitor cells to promote repopulation the damaged tissue. These sequential events are brought together in the model of senescence‐clearance‐regeneration [7], where cellular senescence results in a transient (resolved) process that facilitates wound healing [50, 89], limits fibrotic scarring [90] and promotes regeneration [91, 92, 93]. Even during embryonic development, senescence plays a fundamental role in tissue remodelling and organogenesis and facilitates the programmed elimination of transitory embryonic structures and the maintenance of cell balance [94, 95].
However, upon persistent damage or stress, or during ageing, the process of clearance can be compromised by a number of factors, resulting in the accumulation of senescent cells and a chronic inflammatory microenvironment in tissues. The persistence of senescent cells in tissues with age [96, 97] may be also attributed to the decline or exhaustion of immune function, gradually resulting in perturbed tissue homeostasis [7]. As a consequence, senescence is associated with multiple age‐related disorders that include lung fibrosis [98, 99], cardiovascular diseases and atherosclerosis [100, 101], type 1 and 2 diabetes mellitus [102, 103], liver steatosis [104], obesity‐induced metabolic syndrome [105], osteoarthritis [106], sarcopenia [107, 108] and neurological disorders [109, 110, 111]. Accumulation of senescent cells in aged tissues and increased expression of p16INK4A account for the attenuated regenerative functions of stem or progenitor cells [108, 112, 113, 114]. Importantly, in progeroid and naturally aged mice, the selective elimination of senescent cells delays the onset of ageing‐related disorders, resulting in extended median murine life span and health span [107, 115, 116].
Notably, the functions of cellular senescence in cancer development are stage‐ and context‐dependent. Senescence may prevent propagation of premalignant cells by inducing durable cell‐cycle arrest. Conversely, senescence may also promote a tumour‐prone microenvironment and stemness of tumour cells. In Section 2, we will discuss these tumour‐preventing and tumour‐promoting aspects of senescence in more detail.
2. Occurrence of senescence in cancer and impact on tumorigenesis
Multicellular eukaryotes have developed mechanisms to counteract the deleterious effects of potentially tumorigenic events – these are the induction of cell death (apoptosis) and a mechanism of permanent cell‐cycle arrest (senescence). Despite these processes sharing an activating mechanism via the DDR‐p53 axis, it is the context that ultimately determines whether cells implement pro‐senescent or pro‐apoptotic programmes [43, 117, 118]. Once activated in premalignant cells, p53 initially transactivates CKI p21WAF1/CIP1 inducing cell‐cycle arrest while enhancing the DNA repair response. However, persistent DDR, due to irreparable damage, can result in p53‐mediated transactivation of several pro‐apoptotic genes, including p53 upregulated modulator of apoptosis (PUMA), BAX and BCL2 antagonist/killer (BAK), leading to mitochondria‐mediated apoptotic cell death [119]. However, an alternative mechanism to the elimination of precancerous cells via apoptosis exists, through the p53– p21WAF1/CIP1 axis leading to persistent halting of the cell cycle and senescence. This axis also modulates the microenvironment through the SASP, which not only reinforces senescence in situ, thus potentially preventing the expansion of precancerous cells, but also promotes immune surveillance to enhance clearance of precancerous cells [51]. Despite both processes appearing vital in tumour suppression, apoptosis and senescence programmes antagonise each other. In apoptotic cancer cells, the p53‐targeted DNA methyltransferase 3a (DNMT3a) represses the senescence programme [120] whereas the levels of anti‐apoptotic BCL‐2 proteins are increased in senescent fibroblasts [44, 45, 46]. We focus here on the induction of cellular senescence in neoplastic growths, both endogenously and as a result of anticancer treatments, and on their respective influences on tumorigenic processes.
2.1. Oncogene‐induced senescence in neoplasia
Oncogene‐induced senescence was first observed in cells ectopically expressing HRASG12V [77]. This was then followed up by studies revealing the molecular mechanisms underlying OIS implementation, which involve Raf/MEK/MAPK and the downstream effectors p53 and p16INK4A [78, 79]. These results, obtained in vitro, laid the initial foundations for the concept of cellular senescence acting as a barrier against oncogene‐driven tumorigenesis before further in vivo validation was obtained (Table 1, Mouse models of OIS). The first in vivo validation was reported in 20 05 by five independent groups employing different mouse models of OIS [85, 87, 121, 122, 123]. Normal human skin melanocytes bearing the BRAFV600E mutation acquired a short‐term enhancement in proliferation owing to persistent activation of the Raf/MAPK mitogenic signalling [121]. However, in the longer term, BRAFV600E ‐expressing melanocytes exhibited cell‐cycle arrest with elevated levels of p16INK4A and SAβG activity. Although p16INK4A expression was observed to be heterogeneous among the melanocyte population, the absence of the proliferation marker Ki67 indicated a prevailing growth arrest in human nevi, strongly suggestive of OIS occurrence [121]. Likewise, overexpression of the cell‐cycle related oncogene E2F3 initially promoted cell proliferation resulting in pituitary hyperplasia in mice. Sustained activity of E2F3, however, renders melanotrophs permanently refractory to mitogenic stimulation and induces irreversible cell‐cycle arrest with increased levels of SAHFs and other senescent biomarkers, for example p16INK4A and ARF [123]. Mice bearing a constitutively active NRasG12D oncogene develop invasive T‐cell lymphomas, usually within one year [85]. The progression of this disease accelerates after selective inactivation of histone methyltransferase Suv39h1, which is required for senescence‐associated H3K9me3 and SAβG activation in response to NRasG12D . Consistent with the suppression of apoptosis by senescence, the loss of Suv39h1 and therefore of OIS competence renders NRas‐driven lymphomas responsive to apoptosis induction [85]. In the case of KRasG12V ‐driven neoplasia in the lung, premalignant lung adenomas exhibited weak proliferation with elevated expression of the senescence biomarkers p16INK4A, p15INK4B, Dec1, and DcR2 as well as SAβG activity and formation of SAHFs, whereas senescence biomarkers were hardly observed in lung adenocarcinomas, instead staining positive for the proliferative marker Ki67, which implies the presence of OIS in premalignant lesions but not in the established, malignant disease [122]. In a mouse model, the loss of the tumour suppressor PTEN alone promotes development of invasive prostate cancer, whereas the loss of p53 does not [87]. Strikingly, the combined loss of p53, and thereby the competence to induce senescence, and PTEN results in much earlier onset of invasive and highly aggressive prostate cancer. Immunohistochemical analysis confirmed the existence of OIS in Pten‐null prostates by the detection of elevated levels of SAβG activity and of expression of ARF, p53 and p21WAF1/CIP1, whereas OIS was shown to be absent in double‐null prostates [87]. While preneoplastic cells bearing Ras mutations are more prone to senescence induction, Myc‐driven premalignant cells preferentially favour apoptosis induction [124], suggesting oncogene‐specific effects. However, this is also context‐dependent, as seen in the case of Myc‐driven lymphomagenesis, where DDR mediators and SASP factor TGF‐β1 are both required for senescence induction, suggesting that stroma‐derived pro‐senescence signals play a role in tumorigenesis in this setting [82].
Table 1.
Incidence of senescence in cancer and premalignancy. 5‐FU, 5‐fluorouracil; γH2AX, phosphorylation of the Ser‐139 residue of the histone variant H2AX; CCL2, C–C‐motif chemokine ligand 2; CDK1, cyclin‐dependent kinase 1; EGFR, epidermal growth factor receptor; ELISA, enzyme‐linked immunosorbent assay; GRO‐alpha/CXCL1, chemokine (C‐X‐C‐motif) ligand 1; HER, human epidermal growth factor receptor; IGFBP‐2, insulin‐like growth factor binding protein 2; IF, immunofluorescence; IHC, immunohistochemistry; IL, interleukin; IR, ionising radiation; NF1, neurofibromin; NSCLC, non‐smallcell lung carcinoma; p53BP1, p53‐binding protein 1; PAI‐1, plasminogen activator inhibitor‐1; PDX, patient‐derived xenografts; Pten, phosphatase and tensin homolog; Rb1, retinoblastoma protein 1; Rheb, Ras homolog enriched in brain; Stat3, signal transducer and activator of transcription 3; TKI, tyrosine kinase inhibitor; VEFGA, vascular endothelial growth factor A.
| Gene | Tumour or premalignancy | Mechanism | Reference |
|---|---|---|---|
| Mouse models of oncogene‐induced senescence | |||
| HrasG12V | Breast tumour, bladder tumour, skin papilloma and angiosarcoma | Oncogene activation | [258, 259, 260, 261, 262] |
| KrasG12V | Lung adenoma and pancreatic intraductal neoplasia | Oncogene activation | [122] |
| KrasG12D | Lung adenoma | Oncogene activation | [263] |
| NrasG12D | Lymphoproliferative disorder | Oncogene activation | [85] |
| BrafV600E | Nevi, lung adenoma and melanoma | Oncogene activation | [121, 126, 264, 265] |
| Rheb | Prostate intraepithelial neoplasia | Oncogene activation | [266] |
| E2f3 | Pituitary hyperplasia | Oncogene activation | [123] |
| Akt1 | Prostate intraepithelial neoplasia | Oncogene activation | [267] |
| Myc | Lymphoma, osteosarcoma, liver and lung carcinoma | Oncogene inactivation | [142, 268] |
| Trp53 | Sarcoma and liver carcinoma | Tumour suppressor activation | [141, 269] |
| Pten | Prostate intraepithelial neoplasia | Tumour suppressor inactivation | [87, 270] |
| Rb1 | Thyroid C cell adenoma | Tumour suppressor inactivation | [271] |
| Stat3 | Breast tumour | Tumour suppressor inactivation | [272] |
| Human tissues with oncogene‐induced senescence | |||
| BRAFV600E | Papillary thyroid carcinomas and nevi | Oncogene activation | [125, 126] |
| NF1 | Dermal neurofibromas | Tumour suppressor inactivation | [127] |
| Therapeutic agent | Tumour or malignancy | Mechanism | Reference |
|---|---|---|---|
| Therapy‐induced senescence with irradiation (IR) or drugs | |||
| 5‐aza‐2′‐deoxycytidine (Dacogen) | Colorectal tumour, renal cell carcinoma, hepatoma and NSCLC | Inhibition of DNA methyltransferase | [273] |
| 5‐aza‐cytidine (Decitabine) | Malignant pleural mesothelioma | Inhibition of DNA methyltransferase | [274] |
| Axitinib (Inlyta®) | Glioblastoma | TKI | [275] |
| BRD4770 | Pancreatic adenocarcinoma | Inhibition of histone methyltransferase | [276] |
| Doxorubicin | MMTV‐Wnt1 mice with mammary tumour | DNA damage (DNA intercalator) | [140] |
| Erlotinib and IR |
NSCLC Xenografts of A549 NSCLC |
DNA damage and TKI | [129] |
|
Imidazoacridinone C‐1311 (Symadex™) |
Colorectal tumour, NSCLC and oesophageal carcinoma | DNA damage (inhibition of topoisomerase II) | [277] |
| IR |
Lung adenoma, breast tumour, colorectal tumour, glioblastoma and neuroblastoma Xenografts of H460 lung carcinoma |
DNA damage | [130, 278, 279, 280] |
| Lapatinib | Breast tumour | HER2‐targeted TKI | [135] |
| LBH589 (Panobinostat) |
Osteosarcoma Xenografts of osteosarcoma |
Inhibition of histone deacetylase | [281] |
| MLN4924 |
Colorectal tumour, NSCLC, glioblastoma, lymphoma, gastric tumour and osteosarcoma Xenografts of SJSA‐1 osteosarcoma |
DNA damage (inhibition of NEDD8 activating enzyme) | [282, 283, 284, 285] |
| MLN8054 |
Colorectal tumour and NSCLC Xenografts of HCT116 colorectal tumour |
Inhibition of Aurora A kinase | [286] |
| Neratinib and afatinib | Breast tumour | panHER TKI | [135] |
| Palbociclib |
Patient‐derived sarcoma cells PDX of sarcoma |
Inhibition of CDK4/6 | [287] |
| Palbociclib |
Melanoma Xenografts of 983B/983BR melanoma |
Inhibition of CDK4/6 | [288] |
| Palbociclib and chloroquine or hydroxychloroquine |
Breast tumour Xenografts of MCF7‐T breast tumour |
Inhibition of CDK4/6 and autophagy | [289] |
| Ribociclib |
Neuroblastoma Xenografts of BE2C, 1643 and EBC1 neuroblastoma |
Inhibition of CDK4/6 | [290] |
| Sunitinib (SU11248) |
Renal cell carcinoma Xenografts of OS‐RC‐2 renal cell carcinoma |
TKI | [137] |
| Vemurafenib (PLX4032) |
Melanoma Xenografts of SK‐MEL‐28 melanoma |
Inhibition of BRAF | [291] |
| VO‐OHpic |
MEF and prostate cancer Xenografts of MDA PCa‐2b prostate cancer |
Inhibition of PTEN | [292] |
| WM‐1119 |
MEF Xenografts of EMRK1184 lymphoma |
Inhibition of histone acetyltransferases | [136] |
| WM‐8014 |
MEF Zebrafish model of hepatocarcinoma |
Inhibition of histone acetyltransferases | [136] |
| Therapeutic regimens | Malignancy | Evidence of senescence | Reference |
|---|---|---|---|
| Human tissues with therapy‐induced senescence | |||
| Neoadjuvant chemotherapy containing cyclophosphamide, doxorubicin, and 5‐FU | Breast cancer | SAβG and IHC of p53 and p21WAF1/CIP1 | [293] |
| Neoadjuvant chemotherapy containing carboplatin and docetaxel | NSCLC | SAβG and IHC of CDK1 | [185] |
| Neoadjuvant chemotherapy containing cisplatin and gemcitabine or pemetrexed | Malignant pleural mesothelioma | SAβG and IHC of PAI‐1 and p21WAF1/CIP1 | [294] |
| Neoadjuvant chemotherapy containing mitoxantrone | Prostate cancer | qPCR of p16INK4a , p21WAF1/CIP1 and SASP factors including IL‐6, IL‐8, GRO‐alpha/CXCL1, IGFBP‐2, and IL‐1beta | [167] |
| Neoadjuvant chemotherapy containing sunitinib (SU11248) | Renal cell carcinoma | SAβG and IHC of p53, Dec1, and Ki67 | [137] |
| Neoadjuvant or adjuvant chemotherapy containing anthracycline | Breast cancer | qPCR of p16INK4a and ARF; ELISA of SASP factor VEGFA and CCL2 (assessed using peripheral blood T lymphocytes and patient sera) | [139] |
| Radiotherapy | Head and neck cancer | IF of γH2AX and p53BP1; IHC of p21WAF1/CIP1 (assessed using patient salivary gland) | [177] |
| Neoadjuvant chemoradiotherapy containing 5‐FU | Rectal cancer | qPCR of p21WAF1/CIP1 , p16INK4a and IL‐8 | [138] |
Evidence of OIS is also found in human tissues although literature reports are sparse, likely due to the scarcity of available specimens and a paucity of dedicated research. Relevant studies are highlighted in Table 1 (Human tissues with OIS). For example, the expression of the senescence markers SAβG and p16INK4A has been shown to decrease with the increasing malignancy of pigmented lesions. SAβG and p16INK4A were readily detected in benign nevi, but p16INK4A expression was predominantly low in half of dysplastic nevi samples and negative in most areas of invasive melanoma samples [125]. In snap‐frozen tissue sections taken from cases of BRAFV600E‐expressing papillary thyroid carcinoma, p16INK4A‐positive cells correlated with SAβG‐positive cells, and stable cell‐cycle arrest was further validated by the absence of Ki67 staining [126]. As with senescence induced by inactivation of tumour suppressors, samples from patients with dermal neurofibromas bearing mutated neurofibromin (NF1), which antagonises Ras/MAPK signalling by being a Ras GTPase‐activating protein, tested positive for expression of p16INK4A and for SAβG activity [127].
In summary, the above studies provide the first in vivo evidence of OIS and support the notion of cellular senescence being an important tumour suppression mechanism both in mice and in humans [128]. It is crucial that we now enhance our knowledge of OIS in clinical settings, and develop optimised tools for the efficient detection of senescence to facilitate this, including senescence probes (senoprobes) to be used for translational studies in archived and fresh human tissues.
2.2. Therapy‐induced senescence in cancer
Cellular senescence takes place not only in the early stages of tumorigenesis but also in more advanced tumours, including in response to DNA damage induced by therapeutic regimens [76]. TIS is in fact a positive outcome of treatment as the proliferation of cancer cells is hampered and immunosurveillance of senescent cells may facilitate clearance of cancer cells, alongside therapy‐induced apoptosis [117]. Radiotherapy, a critically important cancer treatment, is an efficient way of inducing senescence in various p53‐proficient cancer cell types. For instance, 10 Gray radiation doses induce senescence in A549 lung cancer cells [129] and MCF‐7 breast cancer cells, but not in MDA‐MB231 breast cancer cells with hypomorphic p53 [130]. Cell fate decision between radiation‐induced senescence and apoptosis depends in part on the presence of tumour suppressors, as the radiation of PTEN‐deficient human glioma cells induces senescence whereas in PTEN‐proficient cells it triggers apoptosis [131].
A wide number of anticancer drugs with distinct mechanisms of action (many resulting in DNA damage) have been used in in vitro and in vivo studies of TIS, including bleomycin, cisplatin, cyclophosphamide, docetaxel, doxorubicin, etoposide and palbociclib [8, 132], some of which are also known to induce senescence in clinical settings [Table 1; TIS with irradiation (IR) or drugs]. As it is also the case with therapeutic radiation, the balance between induction of senescence and apoptosis relies heavily on the dose administered. For example, 250 nM of doxorubicin triggers apoptotic cell death of prostate cancer cells [133] while 25 nM of the same drug induces senescence‐like cell‐growth arrest [134]. In addition to TIS caused by DNA‐damaging agents, novel drugs that interfere with oncogenic signalling [e.g., tyrosine kinase inhibitors (TKI)], or epigenetic modifications (e.g., inhibitors of DNA or histone acetyl‐methyltransferase), also contribute to TIS both in vitro and in vivo. The TKI lapatinib, which targets human epidermal growth factor receptor (HER) family members EGFR and HER2, triggers senescence in breast cancer cells, which showed elevated SAβG activity and expression of the CKIs p15 and p27KIP1 following treatment [135]. Also, administration of the small molecule WM‐1119 in lymphoma cells inhibits histone acetyltransferases and induces TIS with upregulated p16INK4A and ARF, leading to lower tumour burden in mouse xenograft experiments [136].
Importantly, increasing numbers of retrospective studies using archived specimens provided by cancer patients who received neoadjuvant or palliative anticancer drugs are being reported (Table 1, Human tissues with TIS). Administration of neoadjuvant sunitinib (a multitargeted TKI) to patients with renal cell carcinoma increases tumoral SAβG activity and senescent biomarkers p53 and DEC1, while downregulating Ki67 [137]. Neoadjuvant treatment of rectal cancer with chemoradiation [including 5‐fluorouracil (5‐FU)] induces senescence with upregulated expression of CKI p21WAF1/CIP1 and p16INK4A and of canonical SASP factor IL‐8 at the transcriptional level [138]. Intriguingly, some senescence biomarkers including p16INK4A, ARF and SASP components were detected in mRNA extracted from peripheral blood T lymphocytes and patient sera obtained from breast cancer patients treated with chemotherapy. While this likely reflects senescence induction in nontumoral cells, this observation opens up the possibility of assessing TIS using noninvasive methods [139].
In summary, the results described above provide solid evidence for TIS in preclinical and clinical settings and pave the way for studies to fully understand the role of senescent cells in treatment response and ultimately cancer progression. Tools to allow the detection of TIS are crucial for unleashing the clinical utility of TIS determination in prognostic and therapeutic scenarios, including in the detection of cancer recurrence.
2.3. Senescence as a tumour‐suppressive response
As described previously, persistent cell‐cycle withdrawal imposed by cellular senescence acts as the first barrier against tumour initiation (Fig. 3, Transient senescence) [85, 87, 121, 122, 123]. Human skin melanocytes bearing the BRAFV600E mutation exhibit enhanced proliferation in the short term, but in the long term show cell‐cycle arrest as a result of OIS, preventing progression of benign nevi into melanomas [121]. Implementation of OIS as a result of prolonged activity of E2F3 prevents progression of hyperplasia to pituitary tumours, also by inducing irreversible cell‐cycle arrest [123]. The importance of senescence‐mediated cell‐cycle withdrawal in tumour suppression was further highlighted in an NRas‐driven lymphoma model, where deficiency of p53 or histone methyltransferase Suv39h1 compromises OIS implementation, leading to lymphomagenesis [85]. The presence of senescence markers in KRas‐driven premalignant lung adenomas and their absence in invasive lung adenocarcinomas reflects the importance of OIS in suppressing tumorigenesis [122]. Similarly, the additional loss of p53 in Pten‐null mice, and the consequential compromising of OIS, accelerates the progression of premalignant intraepithelial neoplasias [87].
Fig. 3.
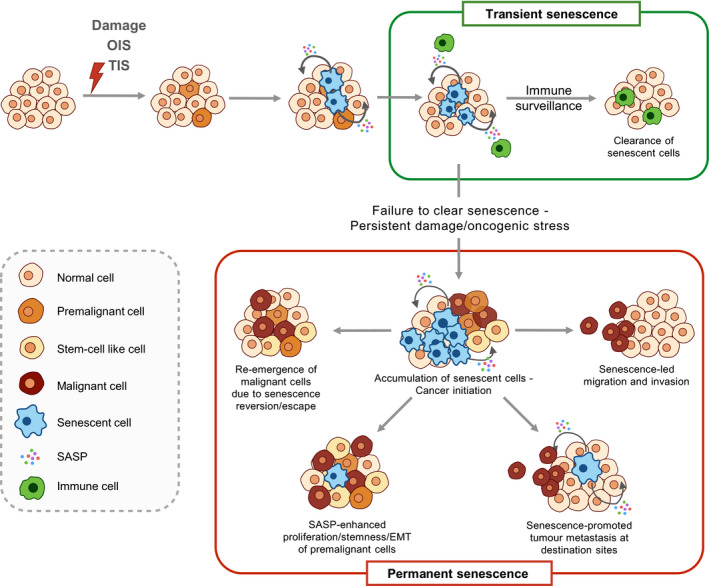
Dual role of senescence in tumorigenesis. Senescence triggered by OIS or TIS initially halts proliferation of premalignant cells and elicits immunosurveillance of senescent cells via SASP secretion, which in turn mediates clearance of premalignant cells, conferring tumour suppression. In contrast, failure to clear senescent cells leads to chronic inflammation by SASP, which cultivates a pro‐tumorigenic microenvironment that promotes proliferation, EMT and stemness of premalignant/malignant cells. Senescence reversion or escape may result in the re‐emergence of malignant cells that may have higher aggressiveness. SASP also contributes to paracrine senescence and induces chemotaxis of malignant cells, resulting in tumour migration, immune evasion and metastasis in distant organs. EMT, epithelial–mesenchymal transition; OIS, oncogene‐induced senescence; SASP, senescence‐associated secretory phenotype; TIS, therapy‐induced senescence.
In addition to providing cell‐cycle arrest as a tumour‐suppressive mechanism, the implementation of cellular senescence also facilitates tumour reversion. Re‐establishment of p53 induces senescence in mouse models of breast cancer and liver carcinoma, resulting in tumour arrest and tumour regression, respectively [140, 141]. In murine models of lymphoma, hepatocellular carcinoma and osteosarcoma, it was observed that cellular senescence programmes remain latently functional in established tumours, and suppression of c‐Myc oncogene‐induced signalling reactivates cellular senescence and promotes tumour regression [142]. Restoration of p53 in Ras‐driven non‐smallcell lung carcinomas decreases the proportion of high‐grade tumours despite failing to induce tumour regression [143]. Interestingly, high‐grade tumours responsive to p53‐mediated tumour arrest show a higher Ras signal as well as ARF expression compared to nonresponsive low‐grade tumours, possibly implying that OIS driven by Ras activation is a prerequisite for efficient tumour arrest by p53 restoration [143].
The SASP, a classic hallmark of senescence, reinforces senescence through secretomes that not only promote the execution of senescence in a cell‐autonomous manner [54, 144], but also contribute to paracrine senescence via IL‐1 signalling and NF‐κB, thereby strengthening the tumour‐suppressive effect [49]. It is worth noting, however, that the senescence‐associated inflammatory response following persistent DNA damage may also result in a form of para‐inflammation that contributes to tumour progression, accelerating growth and invasiveness [145].
Senescence also promotes immunosurveillance for precancerous cells through SASP secretion. Oncogene‐induced senescent hepatocytes promote CD4‐positive T‐cell infiltration and elimination of senescent cells, preventing further advancement of premalignant disease [146]. The association between senescence‐mediated immunosurveillance and tumour suppression was further validated in a model where senescence induction was inhibited by p53 knockdown, resulting in invasive hepatocarcinoma [141]. The restoration of p53 function, and therefore of senescence induction, elicited the infiltration of innate immune cells required for tumour clearance and led to tumour regression [141]. Further evidence in support of a role of SASP in senescence‐mediated tumour suppression was provided using a NRas‐driven hepatoma model where blockade of cGAS‐STING‐mediated production of pro‐inflammatory SASP impaired the immunosurveillance‐mediated clearance of NRasG12V‐expressing hepatocytes, resulting in intrahepatic tumorigenesis [29]. It is plausible that senescence modulates the tumour‐suppressive immune response via SASP, since senescent human melanocytes upregulate the expression of the major histocompatibility complex (MHC) class II antigen presentation apparatus in response to secreted IL‐1β, thus enhancing T‐cell proliferation in vivo, which correlates with better prognosis in melanoma patients [147]. Of note, OIS is accompanied by a dynamic fluctuation of NOTCH1 activity in senescent cells, which can digitally dictate which of two functionally distinct SASP secretomes become predominant [59]. One secretome (first wave) is enriched for TGFβ, contributes to the ‘lateral induction of senescence’ through a juxtacrine NOTCH‐JAG1 pathway and suppresses the senescence‐associated pro‐inflammatory secretome through inhibition of C/EBPβ. The second secretome (second wave) is associated with reduced NOTCH1 activity and involves the upregulation of pro‐inflammatory cytokines, promoting lymphocyte recruitment and senescence surveillance. Interestingly, ligands of the stimulatory natural killer (NK) cell receptor NKG2D were shown to be upregulated in senescent cells irrespective of the trigger, thus increasing NK‐cell‐mediated cytotoxicity towards premalignant cells [148].
In conclusion, cellular senescence is a barrier against tumorigenesis, through halting the proliferation of precancerous/cancer cells, leading to tumour arrest or even regression. SASP‐facilitated paracrine senescence can amplify these tumour‐suppressive effects. The SASP engages in the modulation of senescent cell immunosurveillance, ensuring the clearance of potentially malignant cells. Nevertheless, pro‐inflammatory SASP may be deleterious when the immune system is exhausted, or when senescence is bypassed or compromised (e.g., with the selective inactivation of components required for the implementation of senescence, such as p53, during tumorigenesis), ultimately resulting in cellular escape from senescence‐mediated repression and acquisition of more malignant phenotypes.
2.4. Cancer promotion by senescence
Cancer is broadly regarded as a disease of ageing, resulting from the progressive accumulation of damage and stress. Intriguingly, most age‐related stressors, for example DNA damage and replicative exhaustion, induce senescence. In addition to its association with age, cellular senescence can modulate tumorigenesis via the SASP that can nurture chronic inflammation within the tumour microenvironment, which in turn can promote specific aspects of tumour development [149, 150]. Alongside the tumour‐suppressive aspects of senescence described in the preceding section, mounting evidence implicates cellular senescence in tumour progression, including involvement in cancer initiation, promotion, and invasion to metastasis [5, 51]. The impact of cellular senescence on tumour progression can be dissected into proximal effects and distant effects; these will be discussed in the following sections (Fig. 3, Permanent senescence).
2.4.1. Proximal and distant effects of cellular senescence
Since the publication of the first study demonstrating that human fibroblasts undergoing replicative senescence were able to promote the growth of co‐cultured preneoplastic or neoplastic epithelial cells [151], it has been postulated that senescence can manipulate the pro‐tumorigenic microenvironment by means of SASP secretion [51, 52, 152]. Promotion of growth by senescence‐associated secretion can be invoked by multiple senescence triggers, and the stimulating effect on growth observed in preneoplastic epithelial cells can be recapitulated in co‐cultures with senescent cells induced by oxidative stress, oncogenic Ras, or ARF overexpression [151]. Gene expression profiles of senescent human prostate fibroblasts revealed an enrichment of transcripts encoding proteins that promote epithelial proliferation, including the transmembrane glycoprotein amphiregulin, the inhibition of which attenuates the proliferation of epithelial cells in response to conditioned medium (CM) of senescent fibroblasts [153]. With a well‐recognised role in cancer initiation and progression [154], matrix metalloproteinases (MMPs) secreted during senescence facilitate tumour growth in mouse xenografts of breast tumour, which can be abrogated by the broad‐spectrum MMP inhibitor GM6001 [155]. Osteopontin, another SASP factor, stimulates the proliferation of preneoplastic keratinocytes in vitro and in vivo [156], potentially via the activation of the MAPK signalling pathway [157]. Senescence also appears to account, at least in part, for obesity‐associated cancer development. Dietary/genetic‐induced obesity alters the composition and therefore the metabolites of gut microbiota, one of which is the DNA‐damaging deoxycholic acid [158]. Hepatic stellate cells induce senescence and SASP with abundant pro‐inflammatory factors in response to stimulation with deoxycholic acid. The secreted IL‐1β reinforces the SASP inflammasome, thus promoting the development of hepatocellular carcinoma [158]. Mechanistic studies have further revealed that pro‐tumorigenic SASP is mediated at both the transcriptional and translational level by mTOR, which promotes an IL‐1α/NF‐κB feedback loop for SASP production that led to tumour growth in mouse xenografts of prostate tumour cells [58]. A similar paracrine effect of the SASP was also seen in melanoma, pancreatic cancer and oral squamous cell carcinoma; invasive ability of cancer cells was augmented by CM from senescent fibroblasts, further implicating senescent stromal cells in tumour promotion [159, 160, 161]. Other SASP regulators are beginning to be discovered, for example polypyrimidine tract binding protein 1 (PTBP1) which regulates the pro‐inflammatory SASP by alternative splicing of genes involved in intracellular trafficking [162]. Knockdown of PTBP1 attenuates secretion of pro‐inflammatory factors including IL‐6, IL‐8 and IL‐1α without affecting NRasG12V‐driven OIS, thereby impeding tumour growth in mouse livers and in squamous cell carcinoma xenografts. This study identifies SASP inhibition as a promising potential therapeutic strategy against SASP inflammation‐driven cancer [162].
Interestingly, the pro‐tumorigenic effects of the senescent secretome refer not only to increased tumour progression but also to cancer initiation. A recent study demonstrated the potential of the SASP in promoting transformation and tumour initiation of non‐tumorigenic cells [163]. Pituitary embryonic precursor cells can be transformed by expressing mutant oncogenic β‐catenin, resulting in tumours resembling human adamantinomatous craniopharyngioma with clusters of nondividing cells. The presence of markers related to DDR (e.g., γH2AX and phospho‐ATM) and senescence (e.g., p53, p16INK4A and p21WAF1/CIP1) within clusters of cells confirmed the implementation of OIS, and the corresponding SASP modulated the pro‐tumorigenic microenvironment. Consistent with the requirement of senescence and SASP in tumour development, precursor cells deficient in the tumour suppressor Apc led to reduced OIS and SASP production, as well as to smaller senescent β‐catenin cluster formation, and a mitigated tumorigenic effect [163].
Senescence drives epithelial‐to‐mesenchymal transition (EMT) [164], a cellular transition that helps tumour cells acquire enhanced migratory and invasive abilities [165, 166]. Nonaggressive human breast cancer cells treated with SASP produced by senescent fibroblasts showed reduced cell surface levels of β‐catenin and E‐cadherin, reduced cytokeratin 8/18 expression and increased vimentin expression, all hallmarks of EMT [167]. Further investigation into SASP components revealed that IL‐6 and IL‐8 are major drivers of SASP‐mediated EMT and invasiveness of premalignant or malignant cancer cells, given that their inhibition attenuates the migration into the basement membrane by cells stimulated by senescent CM [167]. Additionally, malignant pleural mesothelioma cells treated with pemetrexed – a standard chemotherapeutic agent used clinically for mesothelioma treatment – undergo accelerated senescence and the addition of senescent CM drives EMT of mesothelioma cells [168]. Pretreatment of mesothelioma cells with senescent CM results in a higher rate of tumour development and earlier onset in mouse xenografts, suggestive of a role for SASP‐mediated EMT in tumour promotion [168]. Similar results were also observed in human colorectal cancer cells upon treatment with senescent CM [138]. Interestingly, in patients receiving neoadjuvant chemoradiotherapy, EMT‐related proteins are upregulated in rectal tumour niches enriched in senescent cells, but not in nearby tumour niches that lack senescent cells, providing in vivo evidence for senescence‐associated EMT programming [138].
The increased tumour vascularisation observed when tumorigenic epithelial cells were subcutaneously co‐injected with senescent fibroblasts into mice suggests that senescence may promote tumorigenesis through the stimulation of angiogenesis [169]. A similar angiogenic effect was also reported in hypoxia‐induced senescence in mouse retina cells, where the senescent secretome contributed to pathological retinopathic angiogenesis [170]. SASP factors secreted by senescent (owing to aneuploidy‐related chromosomal instability) retinal pigment epithelial cells confer angiogenic capabilities, as the CM collected from cultured senescent cells promotes the vascular sprouting of mouse choroid explants [171]. Moreover, highly aneuploid and senescent cells are located at the invasive edge of the tumour in samples from patients with invasive ductal breast carcinoma [171]. Interestingly, vascular endothelial growth factor (VEGF) is only partly responsible for promoting angiogenesis, as the senescent CM pretreated with VEGF‐neutralising antibody was unable to completely block the invasion of endothelial cells into the basement membrane, implying the presence of other angiogenic factors in the SASP [169].
In contrast to senescence‐mediated immunosurveillance of precancerous cells, an age‐related accumulation of p16INK4A‐positive senescent T cells, which is implicated in negative modulation of the adaptive immune response, may be responsible for pro‐tumorigenic effects through reduced immune clearance of premalignant cells [97, 172]. Indeed, lineage‐specific deletion of p16INK4A can rescue severe age‐related functional decline of T cells, thus facilitating homeostatic proliferation and antigen‐specific immune responses [173]. Although senescent stroma can induce infiltration of immune cells via a pro‐inflammatory SASP, the increase in myeloid cells and the concurrent decrease in lymphocytes within the infiltrating population are indicative of an immunosuppressive microenvironment that may correlate with potentially limited immunosurveillance [174]. Accordingly, cancer cells co‐injected with senescent fibroblasts into immune‐competent mice induced larger tumours volumes than those co‐injected with nonsenescent fibroblasts, whereas co‐injection in immunocompromised nude mice led to equivalent tumour growth irrespective of fibroblast status [174]. In addition, immunosuppression and therefore evasion of senescent cells can be accomplished through the pro‐inflammatory SASP factor IL‐6, which induces the expression of MHC molecule HLA‐E [175]. Once expressed on the surface of senescent cells, HLA‐E interacts with inhibitory receptors on NK cells and CD8‐positive T cells, resulting in the evasion of senescent cells from immune surveillance [175]. As the tumour‐suppressive effects of senescence appear to rely heavily on immune surveillance, the decline of immune cytotoxicity of senescent cells leads to increased senescent cell accumulation and chronic inflammation (Fig. 3, Permanent senescence), which not only correlates with poor health span and shorter life span, but also fosters a pro‐tumorigenic microenvironment [176].
The systemic impact of cancer therapies is evidenced by the increase in the levels of senescence markers and functional decline in noncancerous tissues after radiation or chemotherapy. For instance, the blood of patients after chemotherapy shows higher amounts of CD3‐positive lymphocytes expressing p16INK4a [139], and patients with head and neck cancer who received radiotherapy are predisposed in an ‘in field’ and senescence‐driven loss of salivary gland function [177]. Cellular senescence may also therefore be able to adapt the distant microenvironment to affect tumour invasion and metastasis, in addition to its capacity to modulate the proximal microenvironment in tumorigenesis. In line with the systemic effects of senescence observed in cancer patients, SASP factors including IL‐11 and angiopoietin‐like 4 were detected in the plasma of mice engrafted with HER2‐driven senescent tumour cells, contributing to larger metastases from proliferating tumour cells [178]. Consistent with the known increase of the senescence burden with age, the abundance of SASP factor Chemerin is higher in skin dermal fibroblasts from older donors than from young ones [179]. In vitro studies with Chemerin revealed that it increases the migratory ability of cutaneous squamous cell carcinoma cell lines. Such augmented migration promoted by the senescent stroma can be attributed not only to SASP‐mediated chemotaxis [179] but also to SASP‐induced reorganisation of actin and microtubule cytoskeleton networks, leading to reduced focal adhesion and traction forces, thus endowing cancer cells with more aggressive migratory behaviours [180]. In addition to the influence exerted by the senescent stroma, senescent cells within tumour clusters can also function as navigators for tumour invasion, being strongly implicated in the collective invasion and metastasis in tissues from patients with BRAFV600E ‐expressing papillary thyroid carcinoma [126]. Moreover, senescent tumour cells were found as aggregating centres for formation of three‐dimensional tumour clusters in densely plated (monolayer) MDA‐MB‐231 breast cancer cells [181]. Despite comprising only a small portion of the total isolated primary tumour cells, senescent tumour cells exhibited higher migration ability than nonsenescent tumour cells [126]. Further in vivo investigation unravelled that senescent tumour cells generated a gradient of C‐X‐C‐motif ligand (CXCL)12, a SASP factor, in the invasive region of the tumours, orchestrating the collective invasion so that senescent cells led the primary invasion followed by nonsenescent cells. Genetic perturbation of the CXCL12 gradient not only markedly attenuated tumour invasion, but also abrogated anoikis resistance, highlighting the significance of SASP in mediating tumour invasion and potential metastasis [126].
Considering that metastatic bone lesions are prevalent in patients with advanced breast cancer, it was proposed that senescent osteoblasts (which may be induced by treatment) increase local osteoclastogenesis via SASP factor IL‐6 and nourish a pro‐metastatic niche for subsequent seeding and outgrowth of breast cancer cells [182]. Management of the primary breast tumour by systemic doxorubicin treatment is overcome by tumour resistance within weeks, with subsequent metastases in the liver and lung. However, elimination of p16INK4a‐positive senescent nontumour cells in hosts resulted in significantly fewer metastases [183]. In an attempt to simulate a standard paradigm of breast cancer treatment, researchers surgically removed breast tumours prior to administering doxorubicin treatment. After a short latency, primary tumours recurred in all mice, although mice in which the senescent cells had been eliminated showed smaller recurring tumours (in size) and fewer metastases. This dramatic improvement suggests that chemotherapy can promote tumour growth and metastasis by inducing senescence in nontumour cells [183].
In summary, senescent cells modulate neighbouring preneoplastic cells via SASP, which promotes cancer initiation, proliferation and progression. Senescence induces EMT of malignant cells that enables enhanced migratory and invasive abilities. To support tumour growth, SASP factors including VEGF also stimulate angiogenesis. Importantly, senescent cells attenuate immunosurveillance for premalignant cells either through cultivating an immunosuppressive microenvironment or through reducing immune cytotoxicity of senescent cells, facilitating tumour progression. The above studies also provide evidence for the role of senescence in facilitating tumour metastasis, which is accomplished either by promoting the migratory ability of tumours in situ or by preparing the microenvironment in the distant organ for tumour seeding. While these factors complicate our understanding of TIS, they are crucial for understanding systemic contributions of senescence to cancer, with important clinical implications for both local and distant tumour control.
2.5. Tumour progression facilitated by senescence reversion
The well‐established concept of irreversible senescence in normal cells [184] has been challenged by studies showing that therapy‐induced senescent cancer cells can escape the imposed cell‐cycle arrest and resume cell‐cycle progression. In a pioneering work using human non‐smallcell lung cancer (NSCLC) H1299 cell lines deficient in p53 and p16INK4A but with a competent RB pathway, senescence induction in response to camptothecin was proposed to be reversed in minority cells after extended observation. This was rare, occurring in ~ 1 in 106 cells, and was dependent on the increased expression of CDC2/CDK1 protein in senescence‐escaping cells [185]. A recent study performed using a B‐cell lymphoma mouse model further uncovered the potential pro‐tumorigenic threat posed by these previously senescent cells. Adriamycin (doxorubicin)‐induced senescent cancer cells acquired gene expression patterns similar to those of adult tissue stem cells, which then re‐entered the cell cycle and exhibited a higher tumour‐promoting capacity [186]. However, this process was shown using an experimentally induced exit from TIS, through genetic manipulation to reverse senescence‐associated histone methylation (and its related epigenetic changes). Lineage tracing of senescent H460 human lung cancer cells after etoposide treatment confirmed that a few cells with senescence phenotypes regained their ability to divide while the majority of senescent cells remained under persistent growth arrest [187]. The re‐emergent dividing cells also acquired stem cell‐like self‐renewal capacity and led to tumour formation when injected into immunodeficient mice. Similar observations were obtained when doxorubicin‐induced senescent 4T1 breast cancer cells were implanted into the mammary fat pad of both immunocompetent and immunodeficient mice, indicating the in vivo tumorigenic capability of senescent cells [187]. The mechanism underlying senescence reversion remains to be elucidated, although a recent study proposed that the SASP factor thrombospondin‐1 and its receptor CD47 may play key roles in preventing senescence escape of cancer cells upon TIS [188]. This raises the possibility that ‘senescence reversion’ observed for therapy‐induced senescent cancer cells might instead reflect ‘senescence escape’, a property that might be acquired during the previous and sequential steps required for the full implementation of the senescence programme – that is, these cells never become fully senescent. Therefore, such (pre)senescent cells might be more likely to re‐enter the cell cycle than bona fide (fully) senescent cells, which require the successful completion of a number of genetic and epigenetic alterations [4]. Whether senescence reversion and/or escape occur in other biological contexts and clinical settings still requires formal demonstration.
Regardless of the mechanisms by which such cells regain the capacity to divide, previously senescent cancer cells may acquire their aggressiveness through the process of senescence entry and exit, after which they then exhibit enhanced migratory and invasive behaviour [189]. Previous studies have demonstrated that damage‐ or ageing‐induced senescence may precondition the microenvironment for cell reprogramming, in part through secretion IL‐6 [190, 191], whereas OIS promotes the regenerative competence of primary mouse keratinocytes via upregulating transcripts associated with somatic and cancer stem cells [192]. In multiple myeloma and human kidney premalignant cells, the SASP arising upon TIS or OIS might drive the emergence of cancer stem‐like cells (CSLCs), thus promoting tumorigenesis and cancer progression [193, 194]. Senescence induction of p53‐competent nonstem leukaemia cells by adriamycin treatment leads to increased expression of leukaemia stem cell surface markers and upregulation of stem cell‐related transcripts, which was minimal in their p53‐deficient nonsenescent counterparts [186]. After genetic reversion of senescence by p53 inactivation, these previously senescent cells prompted leukaemia initiation while cells that had never experienced senescence rarely induced leukaemia in the recipient mice, implying the pro‐tumorigenic potential of senescence‐associated cell reprogramming [186]. Nevertheless, more research using refined lineage tracing of senescent cells (in diverse cancer types) is required to verify the association of senescence‐mediated reprogramming with acquired stemness of cancer cells.
In summary, although cellular senescence is implemented as a barrier to early tumorigenesis and is also induced as positive outcome in the initial response to cancer therapy, the inefficient clearance of senescent cells by immunosurveillance can result in persistent senescence. SASP‐mediated chronic inflammation within the tumour microenvironment favours tumour initiation, progression, angiogenesis, invasion and migration. Senescence and accompanying SASP also engage in modulating niche locations in distal organs to promote tumour metastasis. Furthermore, emerging evidence suggests that a small number of cancer cells can be made senescent by cytotoxic therapies but then revert to active proliferation. While this phenomenon of senescence reversion or escape remains the subject of intense debate, we must urgently and intensively study this possibility given the stem cell‐like and aggressive features that have been shown already and the associated implications for tumour progression/recurrence and metastatic spread. Better tools for detecting senescent cells in vivo will be crucial to elucidate the translational importance of each of these aspects.
3. Approaches for the detection of senescence in cancer
Given that no single universal senescence marker has been identified to date [11, 24, 195] detection of senescent cells in tissues is conventionally attained by using a battery of immunohistochemical approaches [196] to probe for the presence of molecular biomarkers involved in signalling pathways specific for tumour suppression (e.g., p53 and RB) or cell‐cycle arrest (e.g., p16INKA and p21WAF1/CIP1), senescence‐associated epigenetic changes (e.g., SAHFs and H3K9me3), lack of proliferative capacity assessed by monitoring the incorporation of nucleoside analogues [e.g., 5‐bromo‐2′‐deoxyuridine (BrdU) or 5‐ethynyl‐2′‐deoxyuridine (EdU)] into newly synthesised DNA, together with the detection of elevated SAβG activity [197]. In addition to their intrinsic specificity issues, the conventional methods also require fresh or deep‐frozen tissues, which further restrict their use in in vivo settings for real‐time senescence detection.
Bearing in mind that even a low senescence burden may contribute to tumour suppression or, when pathological, exacerbate tumour progression and facilitate relapse, it is important that we develop tools to accurately, and sensitively, identify and track senescent cells in vivo. Tracking senescent cells in vivo may help to identify the presence of premalignant lesions attributed to OIS as well as to establish the distribution of senescent cells within these lesions, which can indicate of the potential of tumour progression (prognosis) and be used as an additional tool to facilitate patient stratification and early (preventative) intervention. Additionally, in vivo detection of senescent cells can be applied to assess patient response to radiotherapy and chemotherapy interventions, where TIS occurrence may be considered for devising specific therapeutic strategies and proactive follow‐up. Last but not least, tracking senescent cells will be pivotal for post‐treatment assessment since the presence of TIS cells may pose a potential risk of recurrence. Although TIS‐focused cancer therapies have been widely proposed [198, 199], their potential efficacy in vivo will require acute assessment of the response coupled to the precise tracking of senescent cells. We discuss here some recent inventions designed to provide precise and real‐time tracking of senescent cells in preclinical models with potential clinical applications (see Fig. 4 and Table 2).
Fig. 4.
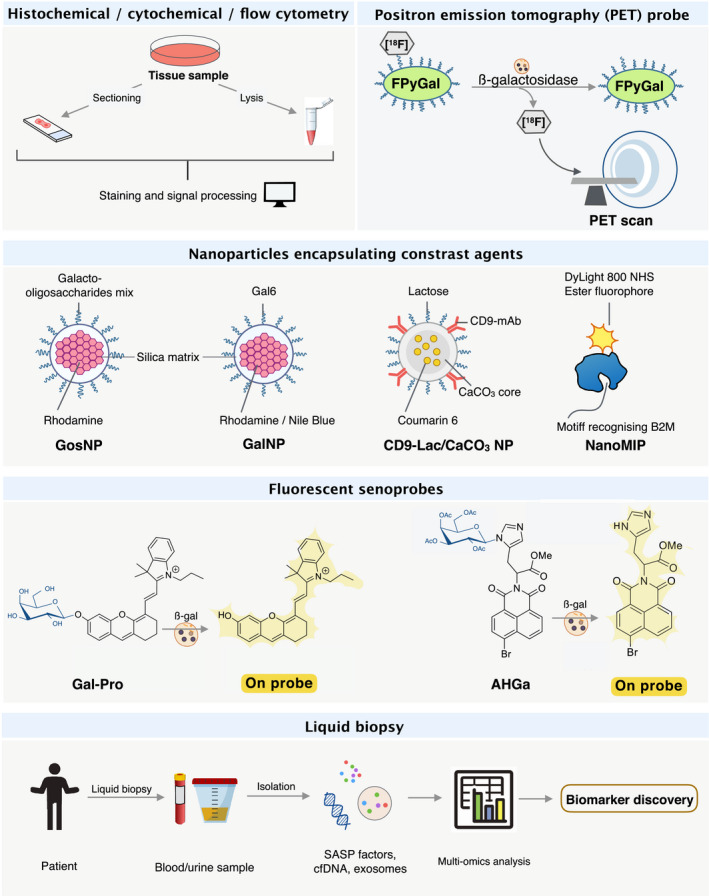
Novel approaches for in vivo senescence detection. In addition to conventional detection methods relying on IHC detection of multiple senescence biomarkers in deep‐frozen or fixed tissues, recent development of approaches combining histochemical, cytochemical and flow cytometry offer higher efficiency for senescence detection in fresh tissues. The fine tuning of nanoparticles for recognising senescent cells strengthens further the targeted delivery of cargoes, that is, image contrasting agents, into senescent tumour cells. Avoiding potential cytotoxicity, OFF‐ON Senoprobes facilitate the real‐time detection and tracking of living senescent cells with elevated SAβG activity. In the human setting, the senescent‐specific PET probe FPyGal may be used to assess senescence burden within tumours pre‐ and post‐treatment, which would provide valuable information in the design of therapeutic strategies and inpatient response. The emerging field of cell‐free DNA (cfDNA) analysis in liquid biopsy provides the least invasive senescence detection tool that is also usable in large‐scale and longitudinal patient screening and monitoring. B2M, β2 microglobulin; nanoMIP, molecular imprinted nanoparticle; NP, nanoparticle; SAβG, senescence‐associated β‐galactosidase.
Table 2.
Summary of novel & potential inventions for senescence detection in cancer. γH2AX, phosphorylation of the Ser‐139 residue of the histone variant H2AX; B2M, β2 microglobulin; HGMB1, high mobility group box‐1; nanoMIP, molecularly imprinted nanoparticles; NB, Nile blue; NIR, near‐infrared; NP, nanoparticle; PET, positron emission tomography; SAβG, senescence‐associated β‐galactosidase; SBB, Sudan Black B.
| Type | Markers | Method of detection | Application |
|---|---|---|---|
| Histochemical, cytochemical and flow cytometry |
SAβG activity Ki67, HGMB1, γH2AX |
ImageStreamX (flow cytometry and image analysis) | In vitro & in vivo [201] |
| Lipofuscin accumulation |
SBB with biotin conjugation
|
In vitro & in vivo [67] | |
| Senoprobe | SAβG activity |
NIR fluorescent probes
|
In vitro [220] In vivo [221] |
| SAβG activity |
Two‐photon fluorescence probe
|
In vitro [223] In vitro & in vivo [224] |
|
| Nanoparticle | SAβG activity | GosNPs | In vitro [210] |
| SAβG activity | GalNPs | In vitro & in vivo [99] | |
| SAβG activity | S3 (GalNPs with NIR dye NB) | In vitro & in vivo [211] | |
|
SAβG activity CD9 receptor |
CD9‐Lac/CaCO3 NPs | In vitro [215] | |
| B2M epitope | B2M nanoMIPs | In vitro & in vivo [216] | |
| PET probe | SAβG activity | PET tracer – [18F]FPyGal |
In vitro & in vivo First‐in‐man [229] |
| Liquid biopsy | DNA methylation | Genome‐wide DNA methylation analysis |
In vitro & in vivo [235] In vitro & in vivo (human samples) [236] |
3.1. Senescence detection: histochemical, cytochemical and flow cytometry
In addition to SAβG activity, used widely in senescence‐detecting assays in tissues [197], the pigment granule lipofuscin is another potential candidate for assay development. Lipofuscin accumulates upon senescence induction and consists of lipid‐containing residua of lysosomal digestion. The granules can be readily detected by staining with Sudan Black B (SBB) dye [200]. The heterogeneity in granule size and background noise (partially owing to impurities during commercial manufacturing) compromises the diagnostic power of SBB. This led to the development of a de novo synthesised compound structurally simulating SBB and employing biotin conjugation – GL13 – which can be used for signal amplification by an immunohistochemical–enzymatic reaction. Following a two‐step staining procedure, it was determined that the biotinylated compound stains senescent cells with an improved sensitivity and increased signal‐to‐noise ratio across a range of biological materials from cultured cells, fresh/frozen tissues, to formalin‐fixed and paraffin‐embedded (FFPE) tissues, the latter importantly broadening its use to archived clinical samples [67].
Conventional identification of senescent cells requires immunohistochemical staining of multiple senescence biomarkers to prevent false positives seen when senescence is assumed on the basis of a single positive marker. Current protocols for the assessment of SAβG activity, however, do not usually support the simultaneous detection of additional senescence biomarkers or any other co‐stains to reliably identify cell types. Precise quantification of senescent cells within tissues would facilitate a better understanding of how senescence is involved in cell and tissue biology in different contexts, but it is not possible with currently available approaches. In an attempt to circumvent these issues, the identification of senescent cells on a single‐cell basis was pursued using flow cytometry techniques combined with high‐resolution image analysis where SAβG assays are performed in parallel to the determination of cell type‐specific markers or other senescence‐related markers such as HGMB1, γH2AX and absence of Ki67. This method allows a more accurate annotation of cellular states in certain cell types and detects senescent cells in murine tumours and fibrotic tissues. Also, this approach is quantitative, allowing larger‐sized senescent cells and normal cells to be distinguished in vivo [201].
The combination of flow cytometry techniques with histochemical or cytochemical approaches strengthens the accurate detection and quantification of senescent cells in vivo. However, this requires physical tissue collection, which may limit the application of this combination approach. Tools that allow in situ detection of senescent cells would allow broader research and clinical utility.
3.2. Nanoparticles targeting senescence
In the pursuit of precision cancer diagnostics and therapy, nanoparticles have emerged as versatile candidates with incredibly broad potentials for clinical application [202, 203, 204]. Nanoparticles, defined as any material in which at least one of its dimensions ranges between 1 and 100 nm, possess distinct properties from their bulk counterparts that allow for widespread application across diverse fields, including the medical application of smaller, inorganic‐based nanoparticles which have had significant recent interest [205, 206, 207]. With a core‐shell (or multiple shell) structure, inorganic nanoparticles function as a degradable gated system to protect their encapsulated cargoes and to release them deliberately when appropriate stimuli are detected [208]. Given that such controllable release can be achieved using biodegradable nanoparticles [209], the characteristically elevated levels of SAβG activity in senescent cells have become an exciting feature to be exploited for targeted delivery of drugs and detection probes. Of relevance to this field, the first senescence‐targeted mesoporous silica‐based nanoparticles were manufactured endowed with a galacto‐oligosaccharide‐coated shell (GosNPs), which is a substrate of SAβG. Once these nanoparticles are endocytosed by the cells and fused with lysosomes, SAβG‐mediated hydrolysis of galacto‐oligosaccharides enables the release of an encapsulated fluorescent dye (rhodamine) in human senescent fibroblasts but not in nonsenescent cells [210]. The delivery system was improved further by capping silica‐based nanoparticles homogeneously with 6‐mer galacto‐oligosaccharides (GalNPs) since this enhances the specificity of SAβG‐mediated release of cargoes. This approach was validated in mouse xenografts of SK‐MEL‐103 melanoma and NCI‐H226 non‐small‐cell lung cancer (NSCLC) cells, where GalNPs released the encapsulated fluorophore in palbociclib‐treated senescent tumour xenografts but not in untreated tumour xenografts, providing the first in vivo proof of principle of senescence detection and therapeutic targeting using nanoparticles [99]. The detection power of GalNPs was recently further improved with the combined use of a near‐infrared (NIR) dye Nile blue (NB), termed S3 [211]. Mesoporous silica GalNPs have a high loading capacity so that NB can be entrapped at high concentration for optimal emission quenching. As a result, S3 administration to mice bearing breast tumours elicited only negligible fluorescence signals while its administration to breast tumour xenografts subjected to TIS by palbociclib treatment contributed to sharp fluorescence signals, showing efficient quenching and specific release of NB in senescent cells that had increased SAβG activity [211].
Despite the preferential release of cargoes in senescent cells compared with nonsenescent cells, the uptake of GosNPs and GalNPs is ubiquitous, which poses a threat of (cargo and coat‐dependent) cytotoxicity in normal cells, important when considering potential clinical applications. While nanoparticles may have some passive accumulation in tumour tissues owing to the enhanced permeability and retention (EPR) effect from often dysfunctional vascular and lymphatic systems [212, 213], in compilation studies derived from 232 datasets, despite some preferential retention of nanoparticles in tumours via EPR, only 0.7% (median) of administered nanoparticles reached tumours [214]. This effect remains highly controversial in humans. Consequently, the additional advantage of bioengineered and functionalised nanoparticles can be exploited by targeting cell surface receptors overexpressed on specific or particular cell types, in order to enhance preferential uptake by certain cells. Accordingly, and given that CD9 is overexpressed in senescent cells, researchers conjugated calcium carbonate nanoparticles with CD9 monoclonal antibody, followed by wrapping the nanoparticles with a lactose‐polyethylene glycol conjugate for optimised stabilisation in the blood and prevention of opsonisation (CD9‐Lac/CaCO3 NPs). CD9‐Lac/CaCO3 NPs were specifically taken up by senescent cells via interaction with CD9 receptors, before SAβG‐mediated release of cargoes occurred intracellularly, as evidenced by the presence of encapsulated fluorescent probe Coumarin 6 in senescent primary human dermal fibroblasts but not in their nonsenescent counterparts [215]. A recent study using molecularly imprinted nanoparticles (nanoMIPs) to target an extracellular component of MHC I molecules – β2 microglobulin (B2M) – allowed selective detection of senescent cells induced by overexpressing p16INK4A [216]. When intravenously injected, B2M nanoMIPs tagged with DyLight 800 NHS Ester showed fluorescent signals in elder but not in young mice, at least partially reflecting the higher number of senescent cells present in aged tissues. However, the preferential accumulation of B2M nanoMIPs in the gastrointestinal tract rather than in other organs might hamper its application in whole‐body senescence detection [216].
The development of senescent cell‐targeted nanoparticles capable of encapsulating various types of contents extends their potential use in the clinical setting, and may allow both drug and imaging contrast delivery. The low delivery rate of administered nanoparticles to tumours and potential toxicities might, however, reduce their therapeutic windows [204].
3.3. Fluorescent senoprobes
The development of OFF‐ON fluorescent senoprobes has drawn considerable attention in recent years. As the presence of elevated numbers of lysosomes is an important hallmark of senescence [11], the accompanying high‐level SAβG activity has garnered significant attention in the development of detection assays based on this lysosomal enzymatic activity. Building on traditional SAβG assays relying on chromogenic changes upon hydrolysis of its substrates, a wide number of chromogenic or fluorogenic probes are now available for the detection of senescent cells [217]. However, drawbacks such as low tissue penetrance and autofluorescence from specimens may hamper the in vivo use of commercially available fluorescent senoprobes such as SPiDER‐βGal [218]. Although some NIR probes have been designed to circumvent the issue of penetrance, they were validated using cells expressing ectopic β‐galactosidase via LacZ gene transfection instead of endogenous SAβG [219]. The recent design of OFF‐ON NIR fluorescent probes – Gal‐Pro [220] and NIR‐BG [221] – remarkably strengthen our ability to detect (in real time) senescent cells, based on the endogenous (lysosomal) SAβG activity in in vitro and in vivo settings, respectively. The feature of Gal‐Pro anchoring to intracellular proteins to promote its accumulation makes it possible to achieve single‐cell resolution when using these probes in living cells [220] while the longer emission wavelengths (708 nm) of NIR‐BG further overcome limits for deep‐tissue imaging, making the first NIR imaging of genuine SAβG activity in xenografts of human tumours in mice possible [221]. S3 (discussed above), which is composed of GalNPs and the NIR dye Nile blue, quenches autofluorescence by packing NB at high concentration in GalNPs, ensuring signals are detected in senescent cells only and with negligible background [211].
The development of two‐photon microscopy has further circumvented issues with the light‐scattering nature of most biological tissues, allowing high‐resolution deep imaging in organs of animals [222]. The combined use of two‐photon microscopy with the ratiometric two‐photon fluorescence probe SG1, which produces intensified emission upon reaction with SAβG, provides a more precise and quantitative detection of senescent cells in rat skin tissues [223]. Taking advantage of the deeper tissue penetrance offered by the two‐photon fluorescence technology, the OFF‐ON probe AHGa accomplished the first in vivo detection of senescent cells in a model of palbociclib‐treated tumour xenografts, which can be transformed into AH by SAβG in senescent cells, resulting in intense fluorescence signals for visualisation [224].
The OFF‐ON NIR and two‐photon fluorescent probes enable the precision detection and tracking of senescent cells within whole animals without raising significant cytotoxicity concerns. When it comes to human use, however, the utility for whole‐body screening using these senoprobes remains questionable. In this regard, the design of probes based on currently available whole‐body imaging technologies would therefore undoubtedly be a great advance.
3.4. Positron emission tomography senoprobes
Nuclear medicine allows to couple noninvasive imaging techniques to small amounts of radioactive materials (or radiopharmaceuticals) that can target tumours or even the tumour microenvironment [225]. Positron emission tomography (PET) imaging is a potent tool for collecting information about physiological, anatomic and biochemical properties of cancer cells [226]. In contrast to the aforementioned detection approaches, PET has already been widely used in clinical settings for cancer detection and staging and for assessing therapeutic response [227, 228]. Radioactive tracers are required for PET imaging – these can be designed for visualising biochemical changes, including SAβG activity within senescent cells. A novel β‐galactosidase specific PET tracer – [18F]FPyGal – and the first‐in‐man study was reported [229]. The specific uptake of FPyGal by senescent cells was first validated in in vitro models and subsequently applied to mouse tumour models subjected to senescence induction. In this study, consistent results were observed showing correlation of lysosomal β‐galactosidase activity and FPyGal uptake in tumours. A case study in a cancer patient treated with the anticancer agent alisertib further revealed a high uptake of FPyGal in a liver metastasis [229]. In addition to enzymatic changes, to enable more precise detection the radioactive tracers can be designed to target specific proteins/pathways related to cellular senescence (e.g., p16INKA or p53 in DDR). As far as we are aware, such probes targeting senescent biomarkers have not yet been developed, although some proof‐of‐concept studies tracking immune cells in vivo using anti‐CD8 PET probes are available [230, 231].
Detection of senescent cells using PET probes would provide a noninvasive approach for the detection of OIS and TIS, alongside conventional tumour cross‐sectional imaging. There may be some concerns about the use of radioactive materials required in PET imaging, but these risks are generally low.
3.5. Senescence detection in liquid biopsies
Liquid biopsies approaches have emerged in recent years, allowing longitudinal assessment of tumour burden and subclones in a minimally invasive manner [232]. The analysis of cell‐free DNA (cfDNA), which may derive from either cancer cells or cells within the tumour microenvironment, holds promise for unravelling cancer‐associated genetic or epigenetic alterations that can in turn be used as biomarkers for early cancer detection, prognosis and monitoring [233]. In the context of early cancer detection, cfDNA analysis is a technique possessing sufficient specificity and sensitivity to discriminate signs of genomic alterations between cancer patients and healthy individuals [234]. Considering that cellular senescence is commonly a response to DNA damage and stress, which implies fundamental epigenetic changes [21, 22], it is tempting to speculate that DNA methylation signatures determined via cfDNA analysis could indicate the presence of senescence phenotypes and therefore potentially suitable for longitudinal monitoring of senescent cell burdens [235, 236]. Senescent cells generally exhibit hypermethylation of promoters associated with metabolic regulators, while transformed cells exhibit hypermethylation of promoters involved in survival and developmental genes [235]. HRas‐driven OIS, however, is associated with minimal changes in DNA methylation compared with replicative cellular senescence or early transformed cells [235]. DNAmSen is a predictor of cellular senescence that is related to three distinct senescence inducers (replicative stress, OIS and IR), developed through analysis of DNA methylation patterns that distinguish between early passage cells and senescent cells [236]. DNAmSen values correlate not only with age, reflecting a senescence burden that is higher in elder donors than in young ones, but also with the severity of age‐related diseases such as lung tumours [236]. In combination with a human tissue‐specific or cell type‐specific methylation atlas, cfDNA analysis may provide information to map senescence to a specific organ or cell type [237, 238], although there is recent evidence that cellular senescence may also block irradiation‐induced cfDNA release [239].
In addition to the direct capture of cfDNA in peripheral blood, secreted extracellular vesicles (EVs) present in liquid biopsies are another potential liquid biopsy target worth exploring [232]. EVs contain numerous molecules including proteins, lipids, RNA and DNA and may represent a source of senescence and cell type‐specific biomarkers [240]. Whole‐genome sequencing of DNA in EV isolated from plasma of patients with prostate cancer revealed genetic perturbations of senescence‐associated oncogenic signalling pathways, for example Myc, AKT and PTEN [241], implying the potential of interrogating EV contents for senescence detection. In addition to EVs, circulating SASP factors may also potentially be used to indirectly assess cellular senescence in tissues [242, 243]. Diligent assessment of circulating SASP factors showed that soluble and EV‐carried SASP proteomes correlate with human plasma ageing markers [242]. Also, the plasma concentrations of seven circulating SASP factors are highly correlated with biological age and adverse clinical outcomes [243]. Considering the abundance and heterogeneity of SASP cocktails secreted by senescent cells, advanced technologies capturing alterations of the senescent secretomes may allow detection of senescence phenotypes in liquid biopsy with increased precision.
Although the detection of senescent cells via liquid biopsy remains in its infancy, it is an exciting prospect that is worthy of further exploration in translational clinical studies in cancer patients.
4. Future perspectives and challenges
Cellular senescence is a response to stress and oncogene activation that promotes repair, contributes to tissues homeostasis and protects us from cancer by inducing a stable cell‐cycle arrest and imposing a complex secretory phenotype that affects the nearby tissue. The senescence programme is therefore presumably implemented to avoid the aberrant proliferation of oncogene‐induced cells and orchestrate tissue repair in damaged areas. More than a decade ago, a collection of landmark articles clearly demonstrated that senescent cells are abundant in premalignant lesions in multiple tissues [128], laying the foundations for the concept of OIS in vivo. Such findings included the tumour‐prone phenotype of mice lacking crucial regulators of senescence (e.g., p53 and p16), being later expanded upon by other research groups using numerous genetically engineered mouse models of different cancer types. Senescence, like cancer, is a remarkably heterogeneous process depending on the trigger(s), cellular types, tissue context and other factors, and senescence heterogeneity appears to be particularly high in human tissue samples. This feature, alongside a research focus on precancerous lesions, has led to a very incomplete understanding of cellular senescence in patient samples when compared to preclinical or mouse models. Although senescence does appear to be abundant in patients' samples of preneoplastic tissue (e.g., preneoplastic prostate gland), more detailed studies in humans are required to dissect the extent, contexts and roles of cellular senescence. We must now tackle crucially important questions about senescence in human pathology, including understanding: (a) the role of secretion of a pro‐senescent SASP by senescent cells, whether and how this expands the arrest phenotype to nearby proliferative (oncogene‐activated) cells in specific neoplastic processes, and how this may go awry during tumour progression; (b) the contexts in which senescence contributes to tumour‐promoting effects, either by non‐cell‐autonomous (paracrine) and cell‐autonomous (senescence escape or reversion) activities; (c) which biomarkers can be used as signatures of these seemingly antagonistic processes. Critically, through assessment of the senescent burden, the identification of reliable biomarkers of senescence and of its associated effects at precancerous stages may provide valuable clinical information for prognosis and/or cancer early detection. As one example, precancerous lesions in human NSCLC include a spectrum of histopathological phenotypes that are thought to be progressive, atypical adenomatous hyperplasia (AAH), adenocarcinoma in situ (AIS) and minimally invasive adenocarcinoma (MIA) [244]. These are difficult to specifically define radiologically with conventional CT cross‐sectional imaging, and patients often have multifocal lesions in the lungs (often with non‐neoplastic lesions with similar imaging characteristics) where it is unclear which, if any, will lead to a frank cancer phenotype. Given the importance of OIS, it is now crucial that we fully characterise this process in these lesions, inform prognosis and apply our senescence detection tools (see Section 3) and novel senescence‐associated biomarkers to prognostically stratify patients and select lesions for local surgical/ablative therapy.
In addition to understanding the important interplay between senescence and (pre)neoplasia, systemic anticancer drugs and radiotherapy are in routine use in the management of both localised and advanced tumours. Both therapeutic types can induce, in addition to apoptosis, cancer cell senescence; however, as we have seen from Section 2, this is a double‐edged sword, potentially contributing both to tumour growth restriction and tumour progression. This particular area is poorly explored in human cancer, despite compelling evidence from preclinical models that suggests it will be an important contributor to treatment response and resistance. Availability of human tissue under appropriate contexts (essentially before and after senescence‐inducing treatment) is undoubtedly a limiting factor here, but we must now design protocols that allow this to be comprehensively and definitely assessed. In parallel, we must develop tools (for clinical use) to detect and monitor the senescent burden so that findings from such studies can be efficiently applied.
Of particular clinical interest are the senescent cancer cells that persist after cancer treatment (either micrometastatic cells or those comprising a subset of cells in a tumour mass), which present a risk for tumour recurrence and relapse [183] due to the pro‐inflammatory, pro‐tumorigenic, pro‐proliferative and immunosuppressive activities of the SASP [51, 150, 152]. Such detrimental consequences can also derive from therapy‐induced senescent stromal cells, which can bring about a variety of unwanted effects through altering the tumour (or even metastatic niche) microenvironment [174, 182, 183]. The intriguing possibility that therapy‐induced senescent cancer cells could re‐enter the cell cycle with an aggressive proliferative potential and cancer stem‐like properties has been recently reported [186], and may even explain clinical findings regarding progressive treatment resistance and tumour growth kinetics after successive lines of cytotoxic chemotherapy. These aspects require careful and urgent investigation in human cancer. Tools to track and monitor senescent cells would help provide baseline and longitudinal assessment of the response to different cancer treatment modalities, and evaluation of the associated risks of senescent phenotypes. Furthermore, they would help define populations of cells for targeting with senescence‐targeted drugs aimed at eliminating senescent cells and monitor their success. A number of studies using preclinical in vivo models have shown the efficiency of concomitant treatment combining senescence‐inducing therapeutics and senotherapies [198, 245, 246, 247].
A crucial hurdle in the development of novel diagnostic tools for senescence is the absence of a universal marker – this may reflect incomplete understanding (there is one/more and it/they have not yet been discovered) or intrinsic heterogeneity to the process (there is none, but certain biomarkers may define specific senescence phenotypes, perhaps being cell type‐, mutational context‐ or senescence trigger‐specific). The development of senolytics will be challenged by the same uncertainty, as well as their on‐target and off‐target effects which has hampered their translation [198]. A deeper understanding of the triggers, underlying molecular mechanisms and signalling pathways, as well as how they behave in different cell types and tissues will facilitate the identification and prioritisation of diagnostic and targetable biomarkers and the development of novel tools for detection and monitoring of senescent cells. Although a collection of senescence hallmarks has been compiled and some incredibly useful markers exist, we must now prioritise research into addressing knowledge gaps for the most immediate clinical utility. This includes the identification of specific, or differentially overexpressed, targetable surface markers at the level of the cell membrane (the so‐called senescent ‘surfaceome’), which could be used to design next‐generation senoprobes and/or more precise nanocarriers encapsulating tracers/contrasting agents/drugs for preferential delivery to senescent cells. So far, only a few surface proteins are observed to be overexpressed in senescent cells, including ICAM‐1 [248], NOTCH1 [59] NOTCH3 [249], DEP1 and B2MG [122, 250], and DPP4 [251]. These proteins are likely to be more enriched in particular senescent subtypes rather than being widespread. A recent study has identified the urokinase‐type plasminogen activator receptor (uPAR) as a more widely senescence‐induced cell surface protein [252]. Promisingly, in the same study, an immunotherapy approach based on uPAR‐specific CAR T cells efficiently ablated senescent cells in vitro and in vivo.
Longitudinal monitoring of senescent areas in clinical imaging remains a formidable challenge. To date, only a small number of senoprobes are capable of tracing senescent lesions in mouse models, with almost all being activated in cells exhibiting high SAβG activity, a broad, albeit imperfect, marker of senescent cells [217]. Most are based on modified and activatable fluorophores that are suitable only when low tissue penetrance is sufficient, markedly restricting their application to some murine models and to the human disease setting. The clinical use of senoprobes in deep‐tissue penetration bioimaging techniques will require specific adaptations, for instance by producing PET‐applicable senoprobes [229]. Significant limitations in cell specificity and penetration also apply to the current nanotechnologies targeting senescent cells – improved delivery strategies could encapsulate contrasting agents (e.g., gadolinium, for MRI detection) or radionuclides (e.g., F18, for PET detection). Senescence‐activatable nanocarriers could be used as theranostic devices, simultaneously detecting and eliminating premalignant lesions or the senescent cell burden after senescence‐inducing therapies. Biosafety concerns regarding the use of senoprobes and nanocarriers to monitor senescence should be addressed in parallel to ensure that this is not the bottleneck in translation. Preclinical studies in animal models must address optimal dosing; the mechanisms of cellular uptake and the intracellular trafficking; biocompatibility and biodistribution; the routes and kinetics of clearance. These will be necessary to move promising approaches into clinical studies.
Finally, the potential use of liquid biopsies (e.g., blood or even urine) to detect and assess the senescent burden would be a less invasive alternative to bioimaging techniques, although spatial resolution to pinpoint senescent populations would not be achievable using these alone. Senescent cancer and stromal cells are characterised by distinctive genomic (e.g., mutations) and/or epigenetic (e.g., DNA and histone methylation) modifications. Genomic modifications can be detected using cfDNA and, in combination with circulating SASP factors, used as biomarkers for early detection, prognosis and monitoring of cancer. This technology can be augmented by examining senescence methylation signatures, to give a fuller (systemic) view of the longitudinal tumour microenvironments, and early promising work in this field should now be built upon to refine this approach. Since senescent cells also secrete, as part of the SASP, a complex array of EVs and exosomes containing proteins, RNA, DNA and lipids that can be sampled using liquid biopsies, we must now determine their utility as a source of clinically relevant biomarkers. Small EVs secreted by senescent cells have been proposed to act as potent pro‐tumorigenic mediators [253, 254, 255] and may not therefore simply be by‐products of senescent cells, but provide more central (mechanistic) senescence biomarkers, perhaps with greater putative clinical relevance. In addition to these promising approaches, other techniques are emerging as promising modalities to assess senescence. Raman spectroscopy, a technique that allows determination of vibrational modes of molecules in cancer cells, has been used to study physical effects of biochemical deviations occurring in cancer [256]. Raman fingerprints have emerged as promising markers of cellular senescence, with ability to visualise changes occurring in the context of ageing of the skin and other tissues [257].
Given the important implications of senescent cell populations in cancer, as well as other age‐related pathologies, and the successful validation of multiple senotherapies in preclinical models in vivo, we anticipate a rapid expansion in this field and the development of novel and more specific tools and techniques to detect, monitor and target cellular senescence.
5. Concluding remarks
Cellular senescence is a defining feature of multiple types of precancerous lesions and responses to cancer therapies. Despite recent advances in the understanding of senescence biology in cancer and the development of tools to track senescent cells, numerous challenges must still be overcome to determine if cellular senescence can be exploited as a preventative, predictive, prognostic and/or diagnostic biomarker in cancer, and how this might be efficiently achieved. These include the need for (a) more specific, detectable and targetable molecular biomarkers of cellular senescence using preclinical model systems; (b) detailed studies of senescence using myriad human cancer tissues from different diagnostic and treatment contexts, determining the context‐specific biomarkers that should be prioritised for translational investigation; (c) a more detailed knowledge of, with deeper mechanistic insights into, how cellular senescence relates to different stages of precancerous lesions (e.g., metaplasia, dysplasia, hyperplasia, carcinoma in situ, minimally invasive carcinoma, etc.) and cancer (including mutational) contexts in diverse cancer types, and how senescence changes (biochemically, and at the levels of cell and tissue biology) in response to therapy (systemic agents and radiotherapy); (d) the development of more efficient tools to track senescent cells ex vivo in tissue and liquid biopsies, and in situ using bioimaging techniques; (e) the optimisation of diagnostic probes, sensors and therapies capable of targeting specifically senescent cells, including the preclinical validation of potential toxicities, biodistribution and timelines/routes for body elimination/excretion in animal models, and other important aspects currently limiting their translatability to human settings; and (f) the most promising diagnostic and therapeutic tools to enter well‐designed clinical trials to provide clinical proof of concept. The accelerating rate of progress in this field has been astounding, and we must now ensure that we focus on the strategies of highest clinical relevance and prioritise those with the greatest probability of successful translation to human cancer patients.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
H‐LO and DM‐E conceptualized and wrote the manuscript. RH, GJD, and JEK reviewed the manuscript and provided expert opinion and critical insights. CG‐L contributed to the design of the figures and provided critical insights. All the authors commented on the text.
Acknowledgements
We thank Dr Luis Enrique Donate for critical reading, proofreading and revision of the manuscript. The Muñoz‐Espín's laboratory is supported by the Cancer Research UK (CRUK) Cambridge Centre Early Detection Programme (RG86786), by a CRUK Programme Foundation Award (C62187/A29760), by a CRUK Early Detection OHSU Project Award (C62187/A26989), by a Medical Research Council (MRC) New Investigator Research Grant (NIRG) (MR/R000530/1) and by a Royal Society Research Grant (RG160806). H‐LO is funded by a CRUK Early Detection OHSU Project Award (C62187/A26989). JEK and RH's work on this project was supported in part by Cancer Research UK and the Cancer Early Detection Advanced Research Center (CEDAR) OHSU Knight Cancer Institute, Project Award funding (grant 2018‐CRUK‐OHSU‐003).
[Correction added on 21 December 2020, after first online publication: Peer review history is not available for this article, so the peer review history statement has been removed.]
References
- 1.Hayflick L & Moorhead PS (1961) The serial cultivation of human diploid cell strains. Exp Cell Res 25, 585–621. [DOI] [PubMed] [Google Scholar]
- 2.Blackburn EH & Gall JG (1978) A tandemly repeated sequence at the termini of the extrachromosomal ribosomal RNA genes in Tetrahymena . J Mol Biol 120, 33–53. [DOI] [PubMed] [Google Scholar]
- 3.Greider CW & Blackburn EH (1985) Identification of a specific telomere terminal transferase activity in tetrahymena extracts. Cell 43, 405–413. [DOI] [PubMed] [Google Scholar]
- 4.van Deursen JM (2014) The role of senescent cells in ageing. Nature 509, 439–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Calcinotto A, Kohli J, Zagato E, Pellegrini L, Demaria M & Alimonti A (2019) Cellular senescence: aging, cancer, and injury. Physiol Rev 99, 1047–1078. [DOI] [PubMed] [Google Scholar]
- 6.Sharpless NE & Sherr CJ (2015) Forging a signature of in vivo senescence. Nat Rev Cancer 15, 397–408. [DOI] [PubMed] [Google Scholar]
- 7.Muñoz‐Espín D & Serrano M (2014) Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol 15, 482–496. [DOI] [PubMed] [Google Scholar]
- 8.Petrova NV, Velichko AK, Razin SV & Kantidze OL (2016) Small molecule compounds that induce cellular senescence. Aging Cell 15, 999–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wiley CD, Velarde MC, Lecot P, Liu S, Sarnoski EA, Freund A, Shirakawa K, Lim HW, Davis SS, Ramanathan Aet al. (2016) Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab 23, 303–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hernandez‐Segura A, de Jong TV, Melov S, Guryev V, Campisi J & Demaria M (2017) Unmasking transcriptional heterogeneity in senescent cells. Curr Biol 27, 2652–2660.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, Campisi J, Collado M, Evangelou K, Ferbeyre Get al. (2019) Cellular senescence: defining a path forward. Cell 179, 813–827. [DOI] [PubMed] [Google Scholar]
- 12.Lim S & Kaldis P (2013) Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development 140, 3079–3093. [DOI] [PubMed] [Google Scholar]
- 13.Vermeulen K, van Bockstaele DR & Berneman ZN (2003) The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif 36, 131–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sherr CJ & Roberts JM (1999) CDK inhibitors: positive and negative regulators of G1‐phase progression. Genes Dev 13, 1501–1512. [DOI] [PubMed] [Google Scholar]
- 15.Toyoshima H & Hunter T (1994) p27, a novel inhibitor of G1 cyclin‐Cdk protein kinase activity, is related to p21. Cell 78, 67–74. [DOI] [PubMed] [Google Scholar]
- 16.Polyak K, Lee MH, Erdjument‐Bromage H, Koff A, Roberts JM, Tempst P & Massagué J (1994) Cloning of p27Kip1, a cyclin‐dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell 78, 59–66. [DOI] [PubMed] [Google Scholar]
- 17.Coats S, Flanagan WM, Nourse J & Roberts JM (1996) Requirement of p27Kip1 for restriction point control of the fibroblast cell cycle. Science 272, 877–880. [DOI] [PubMed] [Google Scholar]
- 18.Dyson N (1998) The regulation of E2F by pRB‐family proteins. Genes Dev 12, 2245–2262. [DOI] [PubMed] [Google Scholar]
- 19.Nevins JR (1998) Toward an understanding of the functional complexity of the E2F and retinoblastoma families. Cell Growth Differ 9, 585–593. [PubMed] [Google Scholar]
- 20.Salama R, Sadaie M, Hoare M & Narita M (2014) Cellular senescence and its effector programs. Genes Dev 28, 99–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng LQ, Zhang ZQ, Chen HZ & Liu DP (2017) Epigenetic regulation in cell senescence. J Mol Med 95, 1257–1268. [DOI] [PubMed] [Google Scholar]
- 22.Paluvai H, di Giorgio E & Brancolini C (2020) The histone code of senescence. Cells 9, 466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.di Micco R, Sulli G, Dobreva M, Liontos M, Botrugno OA, Gargiulo G, Dal Zuffo R, Matti V, D'Ario G, Montani Eet al. (2011) Interplay between oncogene‐induced DNA damage response and heterochromatin in senescence and cancer. Nat Cell Biol 13, 292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hernandez‐Segura A, Nehme J & Demaria M (2018) Hallmarks of cellular senescence. Trends Cell Biol 28, 436–453. [DOI] [PubMed] [Google Scholar]
- 25.Chandra T, Kirschner K, Thuret JY, Pope BD, Ryba T, Newman S, Ahmed K, Samarajiwa SA, Salama R, Carroll Tet al. (2012) Independence of repressive histone marks and chromatin compaction during senescent heterochromatic layer formation. Mol Cell 47, 203–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smallwood A, Estève PO, Pradhan S & Carey M (2007) Functional cooperation between HP1 and DNMT1 mediates gene silencing. Genes Dev 21, 1169–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beauséjour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P & Campisi J (2003) Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J 22, 4212–4222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Narita M, Nũnez S, Heard E, Narita M, Lin AW, Hearn SA, Spector DL, Hannon GJ & Lowe SW (2003) Rb‐mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 113, 703–716. [DOI] [PubMed] [Google Scholar]
- 29.Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, Simithy J, Lan Y, Lin Y, Zhou Zet al. (2017) Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 550, 402–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sadaie M, Salama R, Carroll T, Tomimatsu K, Chandra T, Young ARJ, Narita M, Pérez‐Mancera PA, Bennett DC, Chong Het al. (2013) Redistribution of the Lamin B1 genomic binding profile affects rearrangement of heterochromatic domains and SAHF formation during senescence. Genes Dev 27, 1800–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adams PD, Ivanov A, Pawlikowski J, Manoharan I, van Tuyn J, Nelson DM, Singh Rai T, Shah PP, Hewitt G, Korolchuk VIet al. (2013) Lysosome‐mediated processing of chromatin in senescence. J Cell Biol 202, 129–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.D'Adda Di Fagagna F (2008) Living on a break: cellular senescence as a DNA‐damage response. Nat Rev Cancer 8, 512–522. [DOI] [PubMed] [Google Scholar]
- 33.Gorgoulis VG & Halazonetis TD (2010) Oncogene‐induced senescence: the bright and dark side of the response. Curr Opin Cell Biol 22, 816–827. [DOI] [PubMed] [Google Scholar]
- 34.Halazonetis TD, Gorgoulis VG & Bartek J (2008) An oncogene‐induced DNA damage model for cancer development. Science 319, 1352–1355. [DOI] [PubMed] [Google Scholar]
- 35.Passos JF, Saretzki G, Ahmed S, Nelson G, Richter T, Peters H, Wappler I, Birket MJ, Harold G, Schaeuble Ket al. (2007) Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere‐dependent senescence. PLoS Biol 5, 1138–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.de Lange T (2018) Shelterin‐mediated telomere protection. Annu Rev Genet 52, 223–247. [DOI] [PubMed] [Google Scholar]
- 37.Shay JW & Wright WE (2019) Telomeres and telomerase: three decades of progress. Nat Rev Genet 20, 299–309. [DOI] [PubMed] [Google Scholar]
- 38.Celeste A, Petersen S, Romanienko PJ, Fernandez‐Capetillo O, Chen HT, Sedelnikova OA, Reina‐San‐Martin B, Coppola V, Meffre E, Difilippantonio MJet al. (2002) Genomic instability in mice lacking histone H2AX. Science 296, 922–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ayrapetov MK, Gursoy‐Yuzugullu O, Xu C, Xu Y & Price BD (2014) DNA double‐strand breaks promote methylation of histone H3 on lysine 9 and transient formation of repressive chromatin. Proc Natl Acad Sci USA 111, 9169–9174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sahin E & Depinho RA (2010) Linking functional decline of telomeres, mitochondria and stem cells during ageing. Nature 464, 520–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Deschênes‐Simard X, Gaumont‐Leclerc MF, Bourdeau V, Lessard F, Moiseeva O, Forest V, Igelmann S, Mallette FA, Saba‐El‐Leil MK, Meloche Set al. (2013) Tumor suppressor activity of the ERK/MAPK pathway by promoting selective protein degradation. Genes Dev 27, 900–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pluquet O, Pourtier A & Abbadie C (2015) The unfolded protein response and cellular senescence. A review in the theme: cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. Am J Physiol Cell Physiol 308, 415–425. [DOI] [PubMed] [Google Scholar]
- 43.Soto‐Gamez A, Quax WJ & Demaria M (2019) Regulation of survival networks in senescent cells: from mechanisms to interventions. J Mol Biol 431, 2629–2643. [DOI] [PubMed] [Google Scholar]
- 44.Sanders YY, Liu H, Zhang X, Hecker L, Bernard K, Desai L, Liu G & Thannickal VJ (2013) Histone modifications in senescence‐associated resistance to apoptosis by oxidative stress. Redox Biol 1, 8–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang E (1995) Senescent human fibroblasts resist programmed cell death, and failure to suppress bcl2 is involved. Cancer Res 5585, 2284–2292. [PubMed] [Google Scholar]
- 46.Yosef R, Pilpel N, Tokarsky‐Amiel R, Biran A, Ovadya Y, Cohen S, Vadai E, Dassa L, Shahar E, Condiotti Ret al. (2016) Directed elimination of senescent cells by inhibition of BCL‐W and BCL‐XL. Nat Commun 7, 11190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marcotte R, Lacelle C & Wang E (2004) Senescent fibroblasts resist apoptosis by downregulating caspase‐3. Mech Ageing Dev 125, 777–783. [DOI] [PubMed] [Google Scholar]
- 48.Yosef R, Pilpel N, Papismadov N, Gal H, Ovadya Y, Vadai E, Miller S, Porat Z, Ben‐Dor S & Krizhanovsky V (2017) p21 maintains senescent cell viability under persistent DNA damage response by restraining JNK and caspase signaling. EMBO J 36, 2280–2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, Athineos D, Kang TW, Lasitschka F, Andrulis Met al. (2013) A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol 15, 978–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, Laberge RM, Vijg J, VanSteeg H, Dollé METet al. (2014) An essential role for senescent cells in optimal wound healing through secretion of PDGF‐AA. Dev Cell 31, 722–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Faget DV, Ren Q & Stewart SA (2019) Unmasking senescence: context‐dependent effects of SASP in cancer. Nat Rev Cancer 19, 439–453. [DOI] [PubMed] [Google Scholar]
- 52.Coppé J‐P, Desprez P‐Y, Krtolica A & Campisi J (2010) The senescence‐associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol Mech Dis 5, 99–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kuilman T & Peeper DS (2009) Senescence‐messaging secretome: SMS‐ing cellular stress. Nat Rev Cancer 9, 81–94. [DOI] [PubMed] [Google Scholar]
- 54.Acosta JC, O'Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, Fumagalli M, da Costa M, Brown C, Popov Net al. (2008) Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 133, 1006–1018. [DOI] [PubMed] [Google Scholar]
- 55.Chien Y, Scuoppo C, Wang X, Fang X, Balgley B, Bolden JE, Premsrirut P, Luo W, Chicas A, Lee CSet al. (2011) Control of the senescence‐associated secretory phenotype by NF‐κB promotes senescence and enhances chemosensitivity. Genes Dev 25, 2125–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sebastian T, Malik R, Thomas S, Sage J & Johnson PF (2005) C/EBPβ cooperates with RB:E2F to implement RasV12‐induced cellular senescence. EMBO J 24, 3301–3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Garbers C, Kuck F, Aparicio‐Siegmund S, Konzak K, Kessenbrock M, Sommerfeld A, Häussinger D, Lang PA, Brenner D, Mak TWet al. (2013) Cellular senescence or EGFR signaling induces interleukin 6 (IL‐6) receptor expression controlled by mammalian target of rapamycin (mTOR). Cell Cycle 12, 3421–3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Laberge RM, Sun Y, Orjalo AV, Patil CK, Freund A, Zhou L, Curran SC, Davalos AR, Wilson‐Edell KA, Liu Set al. (2015) MTOR regulates the pro‐tumorigenic senescence‐associated secretory phenotype by promoting IL1A translation. Nat Cell Biol 17, 1049–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hoare M, Ito Y, Kang TW, Weekes MP, Matheson NJ, Patten DA, Shetty S, Parry AJ, Menon S, Salama Ret al. (2016) NOTCH1 mediates a switch between two distinct secretomes during senescence. Nat Cell Biol 18, 979–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Glück S, Guey B, Gulen MF, Wolter K, Kang TW, Schmacke NA, Bridgeman A, Rehwinkel J, Zender L & Ablasser A (2017) Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol 19, 1061–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Davalos AR, Kawahara M, Malhotra GK, Schaum N, Huang J, Ved U, Beausejour CM, Coppé JP, Rodoer F & Campisi J (2013) p53‐dependent release of Alarmin HMGB1 is a central mediator of senescent phenotypes. J Cell Biol 201, 613–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee JJ, Park IH, Rhee WJ, Kim HS & Shin JS (2019) HMGB1 modulates the balance between senescence and apoptosis in response to genotoxic stress. FASEB J 33, 10942–10953. [DOI] [PubMed] [Google Scholar]
- 63.Dörr JR, Yu Y, Milanovic M, Beuster G, Zasada C, Däbritz JHM, Lisec J, Lenze D, Gerhardt A, Schleicher Ket al. (2013) Synthetic lethal metabolic targeting of cellular senescence in cancer therapy. Nature 501, 421–425. [DOI] [PubMed] [Google Scholar]
- 64.Kaplon J, Zheng L, Meissl K, Chaneton B, Selivanov VA, MacKay G, van der Burg SH, Verdegaal EME, Cascante M, Shlomi Tet al. (2013) A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene‐induced senescence. Nature 498, 109–112. [DOI] [PubMed] [Google Scholar]
- 65.Narita M, Young ARJ, Arakawa S, Samarajiwa SA, Nakashima T, Yoshida S, Hong S, Berry LS, Reichelt S, Ferreira Met al. (2011) Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science 332, 966–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira‐Smith Oet al. (1995) A biomarker that identifies senescent human cells in culture and in aging skin in vivo . Proc Natl Acad Sci USA 92, 9363–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Evangelou K, Lougiakis N, Rizou SV, Kotsinas A, Kletsas D, Muñoz‐Espín D, Kastrinakis NG, Pouli N, Marakos P, Townsend Pet al. (2017) Robust, universal biomarker assay to detect senescent cells in biological specimens. Aging Cell 16, 192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shiloh Y (2003) ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer 3, 155–168. [DOI] [PubMed] [Google Scholar]
- 69.Smits VAJ, Klompmaker R, Vallenius T, Rijksen G, Mäkelä TP & Medema RH (2000) p21 inhibits Thr161 phosphorylation of Cdc2 to enforce the G2 DNA damage checkpoint. J Biol Chem 275, 30638–30643. [DOI] [PubMed] [Google Scholar]
- 70.Weinberg WC & Denning MF (2002) P21WAF1 control of epithelial cell cycle and cell fate. Crit Rev Oral Biol Med 13, 453–464. [DOI] [PubMed] [Google Scholar]
- 71.Schmitt CA, Fridman JS, Yang M, Lee S, Baranov E, Hoffman RM & Lowe SW (2002) A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell 109, 335–346. [DOI] [PubMed] [Google Scholar]
- 72.Zhang Y, Xiong Y & Yarbrough WG (1998) ARF promotes MDM2 degradation and stabilizes p53: ARF‐INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell 92, 725–734. [DOI] [PubMed] [Google Scholar]
- 73.Borodkina AV, Shatrova AN, Nikolsky NN & Burova EB (2016) The role of p38 MAP‐kinase in stress‐induced senescence of human endometrium‐derived mesenchymal stem cells. Cell Tissue Biol 10, 365–371. [PubMed] [Google Scholar]
- 74.Borodkina A, Shatrova A, Abushik P, Nikolsky N & Burova E (2014) Interaction between ROS dependent DNA damage, mitochondria and p38 MAPK underlies senescence of human adult stem cells. Aging 6, 481–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Okamoto K & Seimiya H (2019) Revisiting telomere shortening in cancer. Cells 8, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mikuła‐Pietrasik J, Niklas A, Uruski P, Tykarski A & Książek K (2020) Mechanisms and significance of therapy‐induced and spontaneous senescence of cancer cells. Cell Mol Life Sci 77, 213–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Serrano M, Lin AW, McCurrach ME, Beach D & Lowe SW (1997) Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16(INK4a). Cell 88, 593–602. [DOI] [PubMed] [Google Scholar]
- 78.Lin AW, Barradas M, Stone JC, van Aelst L, Serrano M & Lowe SW (1998) Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev 12, 3008–3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhu J, Woods D, McMahon M & Bishop JM (1998) Senescence of human fibroblasts induced by oncogenic Raf. Genes Dev 12, 2997–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LVF, Kolettas E, Niforou K, Zoumpourlis VCet al. (2006) Oncogene‐induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 444, 633–637. [DOI] [PubMed] [Google Scholar]
- 81.di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garré M, Giovanni Nuciforo P, Bensimon Aet al. (2006) Oncogene‐induced senescence is a DNA damage response triggered by DNA hyper‐replication. Nature 444, 638–642. [DOI] [PubMed] [Google Scholar]
- 82.Reimann M, Lee S, Loddenkemper C, Dörr JR, Tabor V, Aichele P, Stein H, Dörken B, Jenuwein T & Schmitt CA (2010) Tumor stroma‐derived TGF‐β limits Myc‐driven lymphomagenesis via Suv39h1‐dependent senescence. Cancer Cell 17, 262–272. [DOI] [PubMed] [Google Scholar]
- 83.Campaner S, Doni M, Hydbring P, Verrecchia A, Bianchi L, Sardella D, Schleker T, Perna D, Tronnersjö S, Murga Met al. (2010) Cdk2 suppresses cellular senescence induced by the c‐Myc oncogene. Nat Cell Biol 12, 54–59. [DOI] [PubMed] [Google Scholar]
- 84.Ko A, Han SY, Choi CH, Cho H, Lee MS, Kim SY, Song JS, Hong KM, Lee HW, Hewitt SMet al. (2018) Oncogene‐induced senescence mediated by c‐Myc requires USP10 dependent deubiquitination and stabilization of p14ARF. Cell Death Differ 25, 1050–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AHFM, Schlegelberger B, Stein H, Dörken B, Jenuwein T & Schmitt CA (2005) Oncogene‐induced senescence as an initial barrier in lymphoma development. Nature 436, 660–665. [DOI] [PubMed] [Google Scholar]
- 86.Astle MV, Hannan KM, Ng PY, Lee RS, George AJ, Hsu AK, Haupt Y, Hannan RD & Pearson RB (2012) AKT induces senescence in human cells via mTORC1 and p53 in the absence of DNA damage: implications for targeting mTOR during malignancy. Oncogene 31, 1949–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald Wet al. (2005) Crucial role of p53‐dependent cellular senescence in suppression of Pten‐deficient tumorigenesis. Nature 436, 725–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lessard F, Igelmann S, Trahan C, Huot G, Saint‐Germain E, Mignacca L, del Toro N, Lopes‐Paciencia S, le Calvé B, Montero Met al. (2018) Senescence‐associated ribosome biogenesis defects contributes to cell cycle arrest through the Rb pathway. Nat Cell Biol 20, 789–799. [DOI] [PubMed] [Google Scholar]
- 89.Jun J & Lau LF (2010) The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat Cell Biol 12, 676–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, Yee H, Zender L & Lowe SW (2008) Senescence of activated stellate cells limits liver fibrosis. Cell 134, 657–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Davaapil H, Brockes JP & Yun MH (2017) Conserved and novel functions of programmed cellular senescence during vertebrate development. Development 144, 106–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Godwin JW, Pinto AR & Rosenthal NA (2013) Macrophages are required for adult salamander limb regeneration. Proc Natl Acad Sci USA 110, 9415–9420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yun MH, Davaapil H & Brockes JP (2015) Recurrent turnover of senescent cells during regeneration of a complex structure. eLife 4, e05505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Muñoz‐Espín D, Cañamero M, Maraver A, Gómez‐López G, Contreras J, Murillo‐Cuesta S, Rodríguez‐Baeza A, Varela‐Nieto I, Ruberte J, Collado Met al. (2013) Programmed cell senescence during mammalian embryonic development. Cell 155, 1104–1118. [DOI] [PubMed] [Google Scholar]
- 95.Storer M, Mas A, Robert‐Moreno A, Pecoraro M, Ortells MC, di Giacomo V, Yosef R, Pilpel N, Krizhanovsky V, Sharpe Jet al. (2013) Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 155, 1119–1130. [DOI] [PubMed] [Google Scholar]
- 96.Herbig U, Ferreira M, Condel L, Carey D & Sedivy JM (2006) Cellular senescence in aging primates. Science 311, 1257. [DOI] [PubMed] [Google Scholar]
- 97.Liu Y, Sanoff HK, Cho H, Burd CE, Torrice C, Ibrahim JG, Thomas NE & Sharpless NE (2009) Expression of p16INK4a in peripheral blood T‐cells is a biomarker of human aging. Aging Cell 8, 439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, Oberg AL, Birch J, Salmonowicz H, Zhu Yet al. (2017) Cellular senescence mediates fibrotic pulmonary disease. Nat Commun 8, 14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Muñoz‐Espín D, Rovira M, Galiana I, Giménez C, Lozano‐Torres B, Paez‐Ribes M, Llanos S, Chaib S, Muñoz‐Martín M, Ucero ACet al. (2018) A versatile drug delivery system targeting senescent cells. EMBO Mol Med 10, e9355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J & van Deursen JM (2016) Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 354, 472–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang J, Uryga AK, Reinhold J, Figg N, Baker L, Finigan A, Gray K, Kumar S, Clarke M & Bennett M (2015) Vascular smooth muscle cell senescence promotes atherosclerosis and features of plaque vulnerability. Circulation 132, 1909–1919. [DOI] [PubMed] [Google Scholar]
- 102.Aguayo‐Mazzucato C & Midha A (2019) Β‐cell senescence in type 2 diabetes. Aging 11, 9967–9968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Thompson PJ, Shah A, Ntranos V, van Gool F, Atkinson M & Bhushan A (2019) Targeted elimination of senescent beta cells prevents type 1 diabetes. Cell Metab 29, 1045–1060. [DOI] [PubMed] [Google Scholar]
- 104.Ogrodnik M, Miwa S, Tchkonia T, Tiniakos D, Wilson CL, Lahat A, Day CP, Burt A, Palmer A, Anstee QMet al. (2017) Cellular senescence drives age‐dependent hepatic steatosis. Nat Commun 8, 15691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Palmer AK, Xu M, Zhu Y, Pirtskhalava T, Weivoda MM, Hachfeld CM, Prata LG, van Dijk TH, Verkade E, Casaclang‐Verzosa Get al. (2019) Targeting senescent cells alleviates obesity‐induced metabolic dysfunction. Aging Cell 18, e12950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jeon OH, Kim C, Laberge RM, Demaria M, Rathod S, Vasserot AP, Chung JW, Kim DH, Poon Y, David Net al. (2017) Local clearance of senescent cells attenuates the development of post‐traumatic osteoarthritis and creates a pro‐regenerative environment. Nat Med 23, 775–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Baker DJ, Wijshake T, Tchkonia T, Lebrasseur NK, Childs BG, van de Sluis B, Kirkland JL & van Deursen JM (2011) Clearance of p16 Ink4a‐positive senescent cells delays ageing‐associated disorders. Nature 479, 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sousa‐Victor P, Gutarra S, García‐Prat L, Rodriguez‐Ubreva J, Ortet L, Ruiz‐Bonilla V, Jardí M, Ballestar E, González S, Serrano ALet al. (2014) Geriatric muscle stem cells switch reversible quiescence into senescence. Nature 506, 316–321. [DOI] [PubMed] [Google Scholar]
- 109.Bussian TJ, Aziz A, Meyer CF, Swenson BL, van Deursen JM & Baker DJ (2018) Clearance of senescent glial cells prevents tau‐dependent pathology and cognitive decline. Nature 562, 578–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chinta SJ, Woods G, Demaria M, Rane A, Zou Y, McQuade A, Rajagopalan S, Limbad C, Madden DT, Campisi Jet al. (2018) Cellular senescence is induced by the environmental neurotoxin paraquat and contributes to neuropathology linked to Parkinson's disease. Cell Rep 22, 930–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhang P, Kishimoto Y, Grammatikakis I, Gottimukkala K, Cutler RG, Zhang S, Abdelmohsen K, Bohr VA, Misra Sen J, Gorospe Met al. (2019) Senolytic therapy alleviates Aβ‐associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer's disease model. Nat Neurosci 22, 719–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Janzen V, Forkert R, Fleming HE, Saito Y, Waring MT, Dombkowski DM, Cheng T, DePinho RA, Sharpless NE & Scadden DT (2006) Stem‐cell ageing modified by the cyclin‐dependent kinase inhibitor p16 INK4a. Nature 443, 421–426. [DOI] [PubMed] [Google Scholar]
- 113.Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A, Bonner‐Weir S & Sharpless NE (2006) p16INK4a induces an age‐dependent decline in islet regenerative potential. Nature 443, 453–457. [DOI] [PubMed] [Google Scholar]
- 114.Molofsky AV, Slutsky SG, Joseph NM, He S, Pardal R, Krishnamurthy J, Sharpless NE & Morrison SJ (2006) Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature 443, 448–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, Saltness RA, Jeganathan KB, Verzosa GC, Pezeshki Aet al. (2016) Naturally occurring p16 Ink4a‐positive cells shorten healthy lifespan. Nature 530, 184–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Xu M, Pirtskhalava T, Farr JN, Weigand BM, Palmer AK, Weivoda MM, Inman CL, Ogrodnik MB, Hachfeld CM, Fraser DGet al. (2018) Senolytics improve physical function and increase lifespan in old age. Nat Med 24, 1246–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Childs BG, Baker DJ, Kirkland JL, Campisi J & Deursen JM (2014) Senescence and apoptosis: dueling or complementary cell fates? EMBO Rep 15, 1139–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Rezatabar S, Karimian A, Rameshknia V, Parsian H, Majidinia M, Kopi TA, Bishayee A, Sadeghinia A, Yousefi M, Monirialamdari Met al. (2019) RAS/MAPK signaling functions in oxidative stress, DNA damage response and cancer progression. J Cell Physiol 234, 14951–14965. [DOI] [PubMed] [Google Scholar]
- 119.Aubrey BJ, Kelly GL, Janic A, Herold MJ & Strasser A (2018) How does p53 induce apoptosis and how does this relate to p53‐mediated tumour suppression? Cell Death Differ 25, 104–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhang Y, Gao Y, Zhang G, Huang S, Dong Z, Kong C, Su D, Du J, Zhu S, Liang Qet al. (2011) DNMT3a plays a role in switches between doxorubicin‐induced senescence and apoptosis of colorectal cancer cells. Int J Cancer 128, 551–561. [DOI] [PubMed] [Google Scholar]
- 121.Michaloglou C, Vredeveld LCW, Soengas MS, Denoyelle C, Kuilman T, van der Horst CMAM, Majoor DM, Shay JW, Mooi WJ & Peeper DS (2005) BRAFE600‐associated senescence‐like cell cycle arrest of human naevi. Nature 436, 720–724. [DOI] [PubMed] [Google Scholar]
- 122.Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, Barradas M, Benguría A, Zaballos A, Flores JM, Barbacid Met al. (2005) Tumour biology: senescence in premalignant tumours. Nature 436, 642. [DOI] [PubMed] [Google Scholar]
- 123.Denchi EL, Attwooll C, Pasini D & Helin K (2005) Deregulated E2F activity induces hyperplasia and senescence‐like features in the mouse pituitary gland. Mol Cell Biol 25, 2660–2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Shortt J & Johnstone RW (2012) Oncogenes in cell survival and cell death. Cold Spring Harbor Perspect Biol 4, a009829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Gray‐Schopfer VC, Cheong SC, Chong H, Chow J, Moss T, Abdel‐Malek ZA, Marais R, Wynford‐Thomas D & Bennett DC (2006) Cellular senescence in naevi and immortalisation in melanoma: a role for p16? Br J Cancer 95, 496–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kim YH, Choi YW, Lee J, Soh EY, Kim JH & Park TJ (2017) Senescent tumor cells lead the collective invasion in thyroid cancer. Nat Commun 8, 15208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Courtois‐Cox S, Genther Williams SM, Reczek EE, Johnson BW, McGillicuddy LT, Johannessen CM, Hollstein PE, MacCollin M & Cichowski K (2006) A negative feedback signaling network underlies oncogene‐induced senescence. Cancer Cell 10, 459–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Collado M & Serrano M (2010) Senescence in tumours: evidence from mice and humans. Nat Rev Cancer 10, 51–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wang M, Morsbach F, Sander D, Gheorghiu L, Nanda A, Benes C, Kriegs M, Krause M, Dikomey E, Baumann Met al. (2011) EGF receptor inhibition radiosensitizes NSCLC cells by inducing senescence in cells sustaining DNA double‐strand breaks. Cancer Res 71, 6261–6269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Jones KR, Elmore LW, Jackson‐Cook C, Demasters G, Povirk LF, Holt SE & Gewirtz DA (2005) p53‐dependent accelerated senescence induced by ionizing radiation in breast tumour cells. Int J Radiat Biol 81, 445–458. [DOI] [PubMed] [Google Scholar]
- 131.Lee JJ, Kim BC, Park MJ, Lee YS, Kim YN, Lee BL & Lee JS (2011) PTEN status switches cell fate between premature senescence and apoptosis in glioma exposed to ionizing radiation. Cell Death Differ 18, 666–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ewald JA, Desotelle JA, Wilding G & Jarrard DF (2010) Therapy‐induced senescence in cancer. J Natl Cancer Inst 102, 1536–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ewald JA, Peters N, Desotelle JA, Hoffmann FM & Jarrard DF (2009) A high‐throughput method to identify novel senescence‐inducing compounds. J Biomol Screen 14, 853–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Schwarze SR, Fu VX, Desotelle JA, Kenowski ML & Jarrard DF (2005) The identification of senescence‐specific genes during the induction of senescence in prostate cancer cells. Neoplasia 7, 816–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.McDermott MSJ, Conlon N, Browne BC, Szabo A, Synnott NC, O'Brien NA, Duffy MJ, Crown J & O'Donovan N(2019) HER2‐targeted tyrosine kinase inhibitors cause therapy‐induced‐senescence in breast cancer cells. Cancers 11, 197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Baell JB, Leaver DJ, Hermans SJ, Kelly GL, Brennan MS, Downer NL, Nguyen N, Wichmann J, McRae HM, Yang Yet al. (2018) Inhibitors of histone acetyltransferases KAT6A/B induce senescence and arrest tumour growth. Nature 560, 253–257. [DOI] [PubMed] [Google Scholar]
- 137.Zhu Y, Xu L, Zhang J, Hu X, Liu Y, Yin H, Lv T, Zhang H, Liu L, An Het al. (2013) Sunitinib induces cellular senescence via p53/Dec1 activation in renal cell carcinoma cells. Cancer Sci 104, 1052–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Tato‐Costa J, Casimiro S, Pacheco T, Pires R, Fernandes A, Alho I, Pereira P, Costa P, Castelo HB, Ferreira Jet al. (2016) Therapy‐induced cellular senescence induces epithelial‐to‐mesenchymal transition and increases invasiveness in rectal cancer. Clin Colorectal Cancer 15, 170–178.e3. [DOI] [PubMed] [Google Scholar]
- 139.Sanoff HK, Deal AM, Krishnamurthy J, Torrice C, Dillon P, Sorrentino J, Ibrahim JG, Jolly TA, Williams G, Carey LAet al. (2014) Effect of cytotoxic chemotherapy on markers of molecular age in patients with breast cancer. J Natl Cancer Inst 106, dju057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Jackson JG, Pant V, Li Q, Chang LL, Quintás‐Cardama A, Garza D, Tavana O, Yang P, Manshouri T, Li Yet al. (2012) p53‐mediated senescence impairs the apoptotic response to chemotherapy and clinical outcome in breast cancer. Cancer Cell 21, 793–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon‐Cardo C & Lowe SW (2007) Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 445, 656–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wu CH, van Riggelen J, Yetil A, Fan AC, Bachireddy P & Felsher DW (2007) Cellular senescence is an important mechanism of tumor regression upon c‐Myc inactivation. Proc Natl Acad Sci USA 104, 13028–13033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Junttila MR, Karnezis AN, Garcia D, Madriles F, Kortlever RM, Rostker F, Brown Swigart L, Pham DM, Seo Y, Evan GIet al. (2010) Selective activation of p53‐mediated tumour suppression in high‐grade tumours. Nature 468, 567–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kuilman T, Michaloglou C, Vredeveld LCW, Douma S, van Doorn R, Desmet CJ, Aarden LA, Mooi WJ & Peeper DS (2008) Oncogene‐induced senescence relayed by an interleukin‐dependent inflammatory network. Cell 133, 1019–1031. [DOI] [PubMed] [Google Scholar]
- 145.Pribluda A, Elyada E, Wiener Z, Hamza H, Goldstein RE, Biton M, Burstain I, Morgenstern Y, Brachya G, Billauer Het al. (2013) A senescence‐inflammatory switch from cancer‐inhibitory to cancer‐promoting mechanism. Cancer Cell 24, 242–256. [DOI] [PubMed] [Google Scholar]
- 146.Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, Hohmeyer A, Gereke M, Rudalska R, Potapova Aet al. (2011) Senescence surveillance of pre‐malignant hepatocytes limits liver cancer development. Nature 479, 547–551. [DOI] [PubMed] [Google Scholar]
- 147.van Tuyn J, Jaber‐Hijazi F, MacKenzie D, Cole JJ, Mann E, Pawlikowski JS, Rai TS, Nelson DM, McBryan T, Ivanov Aet al. (2017) Oncogene‐expressing senescent melanocytes up‐regulate MHC class II, a candidate melanoma suppressor function. J Invest Dermatol 137, 2197–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Sagiv A, Burton DGA, Moshayev Z, Vadai E, Wensveen F, Ben‐Dor S, Golani O, Polic B & Krizhanovsky V (2016) NKG2D ligands mediate immunosurveillance of senescent cells. Aging 8, 328–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Greten FR & Grivennikov SI (2019) Inflammation and cancer: triggers, mechanisms, and consequences. Immunity 51, 27–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Fane M & Weeraratna AT (2020) How the ageing microenvironment influences tumour progression. Nat Rev Cancer 20, 89–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Krtolica A, Parrinello S, Lockett S, Desprez PY & Campisi J (2001) Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci USA 98, 12072–12077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Gonzalez‐Meljem JM, Apps JR, Fraser HC & Martinez‐Barbera JP (2018) Paracrine roles of cellular senescence in promoting tumourigenesis. Br J Cancer 118, 1283–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Bavik C, Coleman I, Dean JP, Knudsen B, Plymate S & Nelson PS (2006) The gene expression program of prostate fibroblast senescence modulates neoplastic epithelial cell proliferation through paracrine mechanisms. Cancer Res 66, 794–802. [DOI] [PubMed] [Google Scholar]
- 154.Maurya SK, Poddar N, Tandon P & Yadav AK (2017) Matrix metalloproteinases (MMPs) in cancer initiation and progression. In Pathophysiological Aspects of Proteases (Chakraborti S, Dhalla N, eds), pp. 207–236. Springer, Singapore. [Google Scholar]
- 155.Liu D & Hornsby PJ (2007) Senescent human fibroblasts increase the early growth of xenograft tumors via matrix metalloproteinase secretion. Cancer Res 67, 3117–3126. [DOI] [PubMed] [Google Scholar]
- 156.Pazolli E, Luo X, Brehm S, Carbery K, Chung JJ, Prior JL, Doherty J, Demehri S, Salavaggione L, Piwnica‐Worms Det al. (2009) Senescent stromal‐derived osteopontin promotes preneoplastic cell growth. Cancer Res 69, 1230–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Luo X, Ruhland MK, Pazolli E, Lind AC & Stewart SA (2011) Osteopontin stimulates preneoplastic cellular proliferation through activation of the MAPK pathway. Mol Cancer Res 9, 1018–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori Met al. (2013) Obesity‐induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499, 97–101. [DOI] [PubMed] [Google Scholar]
- 159.Guan X, LaPak KM, Hennessey RC, Yu CY, Shakya R, Zhang J & Burd CE (2017) Stromal senescence by prolonged CDK4/6 inhibition potentiates tumor growth. Mol Cancer Res 15, 237–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Hassona Y, Cirillo N, Heesom K, Parkinson EK & Prime SS (2014) Senescent cancer‐associated fibroblasts secrete active MMP‐2 that promotes keratinocyte dis‐cohesion and invasion. Br J Cancer 111, 1230–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ohuchida K, Mizumoto K, Murakami M, Qian LW, Sato N, Nagai E, Matsumoto K, Nakamura T & Tanaka M (2004) Radiation to stromal fibroblasts increases invasiveness of pancreatic cancer cells through tumor‐stromal interactions. Cancer Res 64, 3215–3222. [DOI] [PubMed] [Google Scholar]
- 162.Georgilis A, Klotz S, Hanley CJ, Herranz N, Weirich B, Morancho B, Leote AC, D'Artista L, Gallage S, Seehawer Met al. (2018) PTBP1‐mediated alternative splicing regulates the inflammatory secretome and the pro‐tumorigenic effects of senescent cells. Cancer Cell 34, 85–102.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Gonzalez‐Meljem JM, Haston S, Carreno G, Apps JR, Pozzi S, Stache C, Kaushal G, Virasami A, Panousopoulos L, Neda Mousavy‐Gharavy Set al. (2017) Stem cell senescence drives age‐attenuated induction of pituitary tumours in mouse models of paediatric craniopharyngioma. Nat Commun 8, 1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Laberge RM, Awad P, Campisi J & Desprez PY (2012) Epithelial‐mesenchymal transition induced by senescent fibroblasts. Cancer Microenviron 5, 39–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Brabletz T, Kalluri R, Nieto MA & Weinberg RA (2018) EMT in cancer. Nat Rev Cancer 18, 128–134. [DOI] [PubMed] [Google Scholar]
- 166.Thiery JP, Acloque H, Huang RYJ & Nieto MA (2009) Epithelial‐mesenchymal transitions in development and disease. Cell 139, 871–890. [DOI] [PubMed] [Google Scholar]
- 167.Coppé JP, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, Nelson PS, Desprez PY & Campisi J (2008) Senescence‐associated secretory phenotypes reveal cell‐nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 6, 2853–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Canino C, Mori F, Cambria A, Diamantini A, Germoni S, Alessandrini G, Borsellino G, Galati RZ, Battistini L, Blandino Ret al. (2012) SASP mediates chemoresistance and tumor‐initiating‐activity of mesothelioma cells. Oncogene 31, 3148–3163. [DOI] [PubMed] [Google Scholar]
- 169.Coppé JP, Kauser K, Campisi J & Beauséjour CM (2006) Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence. J Biol Chem 281, 29568–29574. [DOI] [PubMed] [Google Scholar]
- 170.Oubaha M, Miloudi K, Dejda A, Guber V, Mawambo G, Germain MA, Bourdel G, Popovic N, Rezende FA, Kaufman RJet al. (2016) Senescence‐associated secretory phenotype contributes to pathological angiogenesis in retinopathy. Sci Transl Med 8, 362ra144. [DOI] [PubMed] [Google Scholar]
- 171.He Q, Au B, Kulkarni M, Shen Y, Lim KJ, Maimaiti J, Wong CK, Luijten MNH, Chong HC, Lim EHet al. (2018) Chromosomal instability‐induced senescence potentiates cell non‐autonomous tumourigenic effects. Oncogenesis 7, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Lemster BH, Michel JJ, Montag DT, Paat JJ, Studenski SA, Newman AB & Vallejo AN (2008) Induction of CD56 and TCR‐independent activation of T cells with aging. J Immunol 180, 1979–1990. [DOI] [PubMed] [Google Scholar]
- 173.Liu Y, Johnson SM, Fedoriw Y, Rogers AB, Yuan H, Krishnamurthy J & Sharpless NE (2011) Expression of p16INK4a prevents cancer and promotes aging in lymphocytes. Blood 117, 3257–3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Ruhland MK, Loza AJ, Capietto AH, Luo X, Knolhoff BL, Flanagan KC, Belt BA, Alspach E, Leahy K, Luo Jet al. (2016) Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat Commun 7, 11762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Pereira BI, Devine OP, Vukmanovic‐Stejic M, Chambers ES, Subramanian P, Patel N, Virasami A, Sebire NJ, Kinsler V, Valdovinos Aet al. (2019) Senescent cells evade immune clearance via HLA‐E‐mediated NK and CD8+ T cell inhibition. Nat Commun 10, 2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Ovadya Y, Landsberger T, Leins H, Vadai E, Gal H, Biran A, Yosef R, Sagiv A, Agrawal A, Shapira Aet al. (2018) Impaired immune surveillance accelerates accumulation of senescent cells and aging. Nat Commun 9, 5435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Marmary Y, Adar R, Gaska S, Wygoda A, Maly A, Cohen J, Eliashar R, Mizrachi L, Orfaig‐Geva C, Baum BJet al. (2016) Radiation‐induced loss of salivary gland function is driven by cellular senescence and prevented by IL6 modulation. Cancer Res 76, 1170–1180. [DOI] [PubMed] [Google Scholar]
- 178.Angelini PD, Zacarias Fluck MF, Pedersen K, Parra‐Palau JL, Guiu M, Morales CB, Vicario R, Luque‐Garciá A, Navalpotro NP, Giralt Jet al. (2013) Constitutive HER2 signaling promotes breast cancer metastasis through cellular senescence. Cancer Res 73, 450–458. [DOI] [PubMed] [Google Scholar]
- 179.Farsam V, Basu A, Gatzka M, Treiber N, Schneider LA, Mulaw MA, Lucas T, Kochanek S, Dummer R, Levesque MPet al. (2016) Senescent fibroblast‐derived Chemerin promotes squamous cell carcinoma migration. Oncotarget 7, 83554–83569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Aifuwa I, Giri A, Longe N, Lee SH, An SS & Wirtz D (2015) Senescent stromal cells induce cancer cell migration via inhibition of RhoA/ROCK/myosin‐based cell contractility. Oncotarget 6, 30516–30531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Lee HG, Kim JH, Sun W, Chi SG, Choi W & Lee KJ (2018) Senescent tumor cells building three‐dimensional tumor clusters. Sci Rep 8, 10503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Luo X, Fu Y, Loza AJ, Murali B, Leahy KM, Ruhland MK, Gang M, Su X, Zamani A, Shi Yet al. (2016) Stromal‐initiated changes in the bone promote metastatic niche development. Cell Rep 14, 82–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Demaria M, O'Leary MN, Chang J, Shao L, Liu S, Alimirah F, Koenig K, Le C, Mitin N, Deal AMet al. (2017) Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov 7, 165–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Takahashi A, Ohtani N & Hara E (2007) Irreversibility of cellular senescence: dual roles of p16INK4a/Rb‐pathway in cell cycle control. Cell Div 2, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Roberson RS, Kussick SJ, Vallieres E, Chen S‐YJ & Wu DY (2005) Escape from therapy‐induced accelerated cellular senescence in p53‐null lung cancer cells and in human lung cancers. Cancer Res 65, 2795–2803. [DOI] [PubMed] [Google Scholar]
- 186.Milanovic M, Fan DNY, Belenki D, Däbritz JHM, Zhao Z, Yu Y, Dörr JR, Dimitrova L, Lenze D, Monteiro Barbosa IAet al. (2018) Senescence‐associated reprogramming promotes cancer stemness. Nature 553, 96–100. [DOI] [PubMed] [Google Scholar]
- 187.Saleh T, Tyutyunyk‐Massey L, Murray GF, Alotaibi MR, Kawale AS, Elsayed Z, Henderson SC, Yakovlev V, Elmore LW, Toor Aet al. (2019) Tumor cell escape from therapy‐induced senescence. Biochem Pharmacol 162, 202–212. [DOI] [PubMed] [Google Scholar]
- 188.Guillon J, Petit C, Moreau M, Toutain B, Henry C, Roché H, Bonichon‐Lamichhane N, Salmon JP, Lemonnier J, Campone Met al. (2019) Regulation of senescence escape by TSP1 and CD47 following chemotherapy treatment. Cell Death Dis 10, 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Yang L, Fang J & Chen J (2017) Tumor cell senescence response produces aggressive variants. Cell Death Discov 3, 17049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Chiche A, le Roux I, von Joest M, Sakai H, Aguín SB, Cazin C, Salam R, Fiette L, Alegria O, Flamant Pet al. (2017) Injury‐induced senescence enables in vivo reprogramming in skeletal muscle. Cell Stem Cell 20, 407–414.e4. [DOI] [PubMed] [Google Scholar]
- 191.Mosteiro L, Pantoja C, Alcazar N, Marión RM, Chondronasiou D, Rovira M, Fernandez‐Marcos PJ, Muñoz‐Martin M, Blanco‐Aparicio C, Pastor Jet al. (2016) Tissue damage and senescence provide critical signals for cellular reprogramming in vivo . Science 354, aaf4445. [DOI] [PubMed] [Google Scholar]
- 192.Ritschka B, Storer M, Mas A, Heinzmann F, Ortells MC, Morton JP, Sansom OJ, Zender L & Keyes WM (2017) The senescence‐associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev 31, 172–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Cahu J, Bustany S & Sola B (2012) Senescence‐associated secretory phenotype favors the emergence of cancer stem‐like cells. Cell Death Dis 3, e446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Castro‐Vega LJ, Jouravleva K, Ortiz‐Montero P, Liu WY, Galeano JL, Romero M, Popova T, Bacchetti S, Vernot JP & Londoño‐Vallejo A (2015) The senescent microenvironment promotes the emergence of heterogeneous cancer stem‐like cells. Carcinogenesis 36, 1180–1192. [DOI] [PubMed] [Google Scholar]
- 195.González‐Gualda E, Baker AG, Fruk L and Muñoz‐Espín D (2020) A guide to assessing cellular senescence in vitro and in vivo. The FEBS Journal, In press. [DOI] [PubMed] [Google Scholar]
- 196.Giatromanolaki A, Kouroupi M, Balaska K & Koukourakis MI (2020) Immunohistochemical detection of senescence markers in human sarcomas. Pathol Res Pract 216, 152800. [DOI] [PubMed] [Google Scholar]
- 197.Debacq‐Chainiaux F, Erusalimsky JD, Campisi J & Toussaint O (2009) Protocols to detect senescence‐associated beta‐galactosidase (SA‐βgal) activity, a biomarker of senescent cells in culture and in vivo . Nat Protoc 4, 1798–1806. [DOI] [PubMed] [Google Scholar]
- 198.Paez‐Ribes M, González‐Gualda E, Doherty GJ & Muñoz‐Espín D (2019) Targeting senescent cells in translational medicine. EMBO Mol Med 11, e10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Sieben CJ, Sturmlechner I, van de Sluis B & van Deursen JM (2018) Two‐step senescence‐focused cancer therapies. Trends Cell Biol 28, 723–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Georgakopoulou EA, Tsimaratou K, Evangelou K, Fernandez‐Marcos PJ, Zoumpourlis V, Trougakos IP, Kletsas D, Bartek J, Serrano M & Gorgoulis VG (2013) Specific lipofuscin staining as a novel biomarker to detect replicative and stress‐induced senescence. A method applicable in cryo‐preserved and archival tissues. Aging 5, 37–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Biran A, Zada L, Abou Karam P, Vadai E, Roitman L, Ovadya Y, Porat Z & Krizhanovsky V (2017) Quantitative identification of senescent cells in aging and disease. Aging Cell 16, 661–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Irvine DJ & Dane EL (2020) Enhancing cancer immunotherapy with nanomedicine. Nat Rev Immunol 20, 321–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Nam J, Son S, Park KS, Zou W, Shea LD & Moon JJ (2019) Cancer nanomedicine for combination cancer immunotherapy. Nat Rev Mater 4, 398–414. [Google Scholar]
- 204.Shi J, Kantoff PW, Wooster R & Farokhzad OC (2017) Cancer nanomedicine: progress, challenges and opportunities. Nat Rev Cancer 17, 20–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Aminabad NS, Farshbaf M & Akbarzadeh A (2019) Recent advances of gold nanoparticles in biomedical applications: state of the art. Cell Biochem Biophys 77, 123–137. [DOI] [PubMed] [Google Scholar]
- 206.Hoang Thi TT, du Cao V, Nguyen TNQ, Hoang DT, Ngo VC & Nguyen DH (2019) Functionalized mesoporous silica nanoparticles and biomedical applications. Mater Sci Eng C Mater Biol Appl 99, 631–656. [DOI] [PubMed] [Google Scholar]
- 207.Wu W, Jiang CZ & Roy VAL (2016) Designed synthesis and surface engineering strategies of magnetic iron oxide nanoparticles for biomedical applications. Nanoscale 8, 19421–19474. [DOI] [PubMed] [Google Scholar]
- 208.Karlsson J, Vaughan HJ & Green JJ (2018) Biodegradable polymeric nanoparticles for therapeutic cancer treatments. Annu Rev Chem Biomol Eng 9, 105–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Aznar E, Oroval M, Pascual L, Murguía JR, Martínez‐Mánez R & Sancenón F (2016) Gated materials for on‐command release of guest molecules. Chem Rev 116, 561–718. [DOI] [PubMed] [Google Scholar]
- 210.Agostini A, Mondragõn L, Bernardos A, Martínez‐Máñez R, Dolores Marcos M, Sancenõn F, Soto J, Costero A, Manguan‐García C, Perona Ret al. (2012) Targeted cargo delivery in senescent cells using capped mesoporous silica nanoparticles. Angew Chem Int Ed Engl 51, 10556–10560. [DOI] [PubMed] [Google Scholar]
- 211.Lozano‐Torres B, Blandez JF, Galiana I, Garcia‐Fernandez A, Alfonso M, Marcos MD, Orzaez M, Sancenon F & Martínez‐Máñez R (2020) Real time in vivo detection of cellular senescence through the controlled release of the NIR fluorescent dye Nile Blue. Angew Chem Int Ed Engl 59, 15152–15156. [DOI] [PubMed] [Google Scholar]
- 212.Golombek SK, May JN, Theek B, Appold L, Drude N, Kiessling F & Lammers T (2018) Tumor targeting via EPR: Strategies to enhance patient responses. Adv Drug Deliv Rev 130, 17–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Maeda H, Nakamura H & Fang J (2013) The EPR effect for macromolecular drug delivery to solid tumors: improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo . Adv Drug Deliv Rev 65, 71–79. [DOI] [PubMed] [Google Scholar]
- 214.Wilhelm S, Tavares AJ, Dai Q, Ohta S, Audet J, Dvorak HF & Chan WCW (2016) Analysis of nanoparticle delivery to tumours. Nat Rev Mater 1, 1–12. [Google Scholar]
- 215.Thapa RK, Nguyen HT, Jeong JH, Kim JR, Choi HG, Yong CS & Kim JO (2017) Progressive slowdown/prevention of cellular senescence by CD9‐targeted delivery of rapamycin using lactose‐wrapped calcium carbonate nanoparticles. Sci Rep 7, 43299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Ekpenyong‐Akiba AE, Canfarotta F, Abd B, Poblocka M, Casulleras M, Castilla‐Vallmanya L, Kocsis‐Fodor G, Kelly ME, Janus J, Althubiti Met al. (2019) Detecting and targeting senescent cells using molecularly imprinted nanoparticles. Nanoscale Horiz 4, 757–768. [Google Scholar]
- 217.Lozano‐Torres B, Estepa‐Fernández A, Rovira M, Orzáez M, Serrano M, Martínez‐Máñez R & Sancenón F (2019) The chemistry of senescence. Nat Rev Chem 3, 426–441. [Google Scholar]
- 218.Doura T, Kamiya M, Obata F, Yamaguchi Y, Hiyama TY, Matsuda T, Fukamizu A, Noda M, Miura M & Urano Y (2016) Detection of LacZ‐positive cells in living tissue with single‐cell resolution. Angew Chem Int Ed Engl 55, 9620–9624. [DOI] [PubMed] [Google Scholar]
- 219.Gu K, Xu Y, Li H, Guo Z, Zhu S, Zhu S, Shi P, James TD, Tian H & Zhu WH (2016) Real‐time tracking and in vivo visualization of β‐galactosidase activity in colorectal tumor with a ratiometric near‐infrared fluorescent probe. J Am Chem Soc 138, 5334–5340. [DOI] [PubMed] [Google Scholar]
- 220.Zhang J, Li C, Dutta C, Fang M, Zhang S, Tiwari A, Werner T, Luo FT & Liu H (2017) A novel near‐infrared fluorescent probe for sensitive detection of β‐galactosidase in living cells. Anal Chim Acta 968, 97–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Wang Y, Liu J, Ma X, Cui C, Deenik PR, Henderson PKP, Sigler AL & Cui L (2019) Real‐time imaging of senescence in tumors with DNA damage. Sci Rep 9, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Helmchen F & Denk W (2005) Deep tissue two‐photon microscopy. Nat Methods 2, 932–940. [DOI] [PubMed] [Google Scholar]
- 223.Lee HW, Heo CH, Sen D, Byun HO, Kwak IH, Yoon G & Kim HM (2014) Ratiometric two‐photon fluorescent probe for quantitative detection of β‐galactosidase activity in senescent cells. Anal Chem 86, 10001–10005. [DOI] [PubMed] [Google Scholar]
- 224.Lozano‐Torres B, Galiana I, Rovira M, Garrido E, Chaib S, Bernardos A, Muñoz‐Espín D, Serrano M, Martínez‐Máñez R & Sancenón F (2017) An OFF‐ON two‐photon fluorescent probe for tracking cell senescence in vivo . J Am Chem Soc 139, 8808–8811. [DOI] [PubMed] [Google Scholar]
- 225.Chernov V, Zeltchan R, Medvedeva A, Sinilkin I & Bragina O (2017) Nuclear medicine in cancer diagnosis and therapy. In AIP Conference Proceedings. American Institute of Physics Inc. [Google Scholar]
- 226.Mankoff DA & Katz SI (2018) PET imaging for assessing tumor response to therapy. J Surg Oncol 118, 362–373. [DOI] [PubMed] [Google Scholar]
- 227.Grootjans W, de Geus‐Oei LF, Troost EGC, Visser EP, Oyen WJG & Bussink J (2015) PET in the management of locally advanced and metastatic NSCLC. Nat Rev Clin Oncol 12, 395–407. [DOI] [PubMed] [Google Scholar]
- 228.Langen KJ, Galldiks N, Hattingen E & Shah NJ (2017) Advances in neuro‐oncology imaging. Nat Rev Neurol 13, 279–289. [DOI] [PubMed] [Google Scholar]
- 229.Krueger MA, Cotton JM, Zhou B, Wolter K, Schwenck J, Kuehn A, Fuchs K, Maurer A, la Fougere C, Zender Let al. (2019) Abstract 1146: [18 F]FPyGal: a novel ß‐galactosidase specific PET tracer for in vivo imaging of tumor senescence. In American Association for Cancer Research Annual Meeting. American Association for Cancer Research Inc. [Google Scholar]
- 230.Tavaré R, McCracken MN, Zettlitz KA, Knowles SM, Salazar FB, Olafsen T, Witte ON & Wu AM (2014) Engineered antibody fragments for immuno‐PET imaging of endogenous CD8 + T cells in vivo . Proc Natl Acad Sci USA 111, 1108–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Tavaré R, McCracken MN, Zettlitz KA, Salazar FB, Olafsen T, Witte ON & Wu AM (2015) Immuno‐PET of murine T cell reconstitution postadoptive stem cell transplantation using anti‐CD4 and anti‐CD8 cys‐diabodies. J Nucl Med 56, 1258–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Kustanovich A, Schwartz R, Peretz T & Grinshpun A (2019) Life and death of circulating cell‐free DNA. Cancer Biol Ther 20, 1057–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Bronkhorst AJ, Ungerer V & Holdenrieder S (2019) The emerging role of cell‐free DNA as a molecular marker for cancer management. Biomol Detect Quantif 17, 100087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Alborelli I, Generali D, Jermann P, Cappelletti MR, Ferrero G, Scaggiante B, Bortul M, Zanconati F, Nicolet S, Haegele Jet al. (2019) Cell‐free DNA analysis in healthy individuals by next‐generation sequencing: a proof of concept and technical validation study. Cell Death Dis 10, 534–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Xie W, Kagiampakis I, Pan L, Zhang YW, Murphy L, Tao Y, Kong X, Kang B, Xia L, Carvalho FLFet al. (2018) DNA methylation patterns separate senescence from transformation potential and indicate cancer risk. Cancer Cell 33, 309–321.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Levine ME, Leung D, Minteer C & Gonzalez J (2019) A DNA methylation fingerprint of cellular senescence. bioRxiv 674580. 10.1101/674580 [DOI] [Google Scholar]
- 237.Moss J, Magenheim J, Neiman D, Zemmour H, Loyfer N, Korach A, Samet Y, Maoz M, Druid H, Arner Pet al. (2018) Comprehensive human cell‐type methylation atlas reveals origins of circulating cell‐free DNA in health and disease. Nat Commun 9, 5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Sun K, Jiang P, Chan KCA, Wong J, Cheng YKY, Liang RHS, Chan WK, Ma ESK, Chan SL, Cheng SHet al. (2015) Plasma DNA tissue mapping by genome‐wide methylation sequencing for noninvasive prenatal, cancer, and transplantation assessments. Proc Natl Acad Sci USA 112, E5503–E5512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Rostami A, Lambie M, Yu CW, Stambolic V, Waldron JN & Bratman SV (2020) Senescence, necrosis, and apoptosis govern circulating cell‐free DNA release kinetics. Cell Rep 31, 107830. [DOI] [PubMed] [Google Scholar]
- 240.Lázaro‐Ibáñez E, Sanz‐Garcia A, Visakorpi T, Escobedo‐Lucea C, Siljander P, Ayuso‐Sacido Á & Yliperttula M (2014) Different gDNA content in the subpopulations of prostate cancer extracellular vesicles: apoptotic bodies, microvesicles, and exosomes. Prostate 74, 1379–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Vagner T, Spinelli C, Minciacchi VR, Balaj L, Zandian M, Conley A, Zijlstra A, Freeman MR, Demichelis F, De Set al. (2018) Large extracellular vesicles carry most of the tumour DNA circulating in prostate cancer patient plasma. J Extracell Vesicles 7, 1505403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Basisty N, Kale A, Jeon OH, Kuehnemann C, Payne T, Rao C, Holtz A, Shah S, Sharma V, Ferrucci Let al. (2020) A proteomic atlas of senescence‐associated secretomes for aging biomarker development. PLoS Biol 18, e3000599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Schafer MJ, Zhang X, Kumar A, Atkinson EJ, Zhu Y, Jachim S, Mazula L, Brown AK, Berning M, Aversa Zet al. (2020) The senescence‐associated secretome as an indicator of age and medical risk. JCI Insight 5, 133668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Travis WD, Brambilla E, Nicholson AG, Yatabe Y, Austin JHM, Beasley MB, Chirieac LR, Dacic S, Duhig E, Flieder DBet al. (2015) The 2015 World Health Organization classification of lung tumors: impact of genetic, clinical and radiologic advances since the 2004 classification. J Thorac Oncol 10, 1243–1260. [DOI] [PubMed] [Google Scholar]
- 245.González‐Gualda E, Pàez‐Ribes M, Lozano‐Torres B, Macias D, Wilson JR, González‐López C, Ou HL, Mirón‐Barroso S, Zhang Z, Lérida‐Viso Aet al. (2020) Galacto‐conjugation of Navitoclax as an efficient strategy to increase senolytic specificity and reduce platelet toxicity. Aging Cell 19, e13142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Triana‐Martínez F, Picallos‐Rabina P, da Silva‐Álvarez S, Pietrocola F, Llanos S, Rodilla V, Soprano E, Pedrosa P, Ferreirós A, Barradas Met al. (2019) Identification and characterization of cardiac glycosides as senolytic compounds. Nat Commun 10, 4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Guerrero A, Herranz N, Sun B, Wagner V, Gallage S, Guiho R, Wolter K, Pombo J, Irvine EE, Innes AJet al. (2019) Cardiac glycosides are broad‐spectrum senolytics. Nat Metab 1, 1074–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Gorgoulis VG, Pratsinis H, Zacharatos P, Demoliou C, Sigala F, Asimacopoulos PJ, Papavassiliou AG & Kletsas D (2005) p53‐Dependent ICAM‐1 overexpression in senescent human cells identified in atherosclerotic lesions. Lab Invest 85, 502–511. [DOI] [PubMed] [Google Scholar]
- 249.Cui H, Kong Y, Xu M & Zhang H (2013) Notch3 functions as a tumor suppressor by controlling cellular senescence. Cancer Res 73, 3451–3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Althubiti M, Lezina L, Carrera S, Jukes‐Jones R, Giblett SM, Antonov A, Barlev N, Saldanha GS, Pritchard CA, Cain Ket al. (2014) Characterization of novel markers of senescence and their prognostic potential in cancer. Cell Death Dis 5, e1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Kim KM, Noh JH, Bodogai M, Martindale JL, Yang X, Indig FE, Basu SK, Ohnuma K, Morimoto C, Johnson PFet al. (2017) Identification of senescent cell surface targetable protein DPP4. Genes Dev 31, 1529–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Amor C, Feucht J, Leibold J, Ho Y‐J, Zhu C, Alonso‐Curbelo D, Mansilla‐Soto J, Boyer JA, Li X, Giavridis Tet al. (2020) Senolytic CAR T cells reverse senescence‐associated pathologies. Nature 583, 127–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Takasugi M, Okada R, Takahashi A, Virya Chen D, Watanabe S & Hara E (2017) Small extracellular vesicles secreted from senescent cells promote cancer cell proliferation through EphA2. Nat Commun 8, 15729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Takasugi M (2018) Emerging roles of extracellular vesicles in cellular senescence and aging. Aging Cell 17, e12734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Jakhar R & Crasta K (2019) Exosomes as emerging pro‐tumorigenic mediators of the senescence‐associated secretory phenotype. Int J Mol Sci 20, 2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Auner GW, Koya SK, Huang C, Broadbent B, Trexler M, Auner Z, Elias A, Mehne KC & Brusatori MA (2018) Applications of Raman spectroscopy in cancer diagnosis. Cancer Metastasis Rev 37, 691–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Liendl L, Grillari J & Schosserer M (2020) Raman fingerprints as promising markers of cellular senescence and aging. GeroScience 42, 377–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Chen X, Mitsutake N, LaPerle K, Akeno N, Zanzonico P, Longo VA, Mitsutake S, Kimura ET, Geiger H, Santos Eet al. (2009) Endogenous expression of HrasG12V induces developmental defects and neoplasms with copy number imbalances of the oncogene. Proc Natl Acad Sci USA 106, 7979–7984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Mo L, Zheng X, Huang HY, Shapiro E, Lepor H, Cordon‐Cardo C, Sun TT & Wu XR (2007) Hyperactivation of Ha‐ras oncogene, but not Ink4a/Arf deficiency, triggers bladder tumorigenesis. J Clin Invest 117, 314–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Sarkisian CJ, Keister BA, Stairs DB, Boxer RB, Moody SE & Chodosh LA (2007) Dose‐dependent oncogene‐induced senescence in vivo and its evasion during mammary tumorigenesis. Nat Cell Biol 9, 493–505. [DOI] [PubMed] [Google Scholar]
- 261.Sun P, Yoshizuka N, New L, Moser BA, Li Y, Liao R, Xie C, Chen J, Deng Q, Yamout Met al. (2007) PRAK is essential for ras‐induced senescence and tumor suppression. Cell 128, 295–308. [DOI] [PubMed] [Google Scholar]
- 262.Yamakoshi K, Takahashi A, Hirota F, Nakayama R, Ishimaru N, Kubo Y, Mann DJ, Ohmura M, Hirao A, Saya Het al. (2009) Real‐time in vivo imaging of p16Ink4a reveals cross talk with p53. J Cell Biol 186, 393–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Volonte D, Vyas AR, Chen C, Dacic S, Stabile LP, Kurland BF, Abberbock SR, Burns TF, Herman JG, Di YPet al. (2018) Caveolin‐1 promotes the tumor suppressor properties of oncogene‐induced cellular senescence. J Biol Chem 293, 1794–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Dhomen N, Reis‐Filho JS, da Rocha Dias S, Hayward R, Savage K, Delmas V, Larue L, Pritchard C & Marais R (2009) Oncogenic Braf induces melanocyte senescence and melanoma in mice. Cancer Cell 15, 294–303. [DOI] [PubMed] [Google Scholar]
- 265.Dankort D, Filenova E, Collado M, Serrano M, Jones K & McMahon M (2007) A new mouse model to explore the initiation, progression, and therapy of BRAFV600E‐induced lung tumors. Genes Dev 21, 379–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Nardella C, Chen Z, Salmena L, Carracedo A, Alimonti A, Egia A, Carver B, Gerald W, Cordon‐Cardo C & Pandolfi PP (2008) Aberrant Rheb‐mediated mTORC1 activation and Pten haploinsufficiency are cooperative oncogenic events. Genes Dev 22, 2172–2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Majumder PK, Grisanzio C, O'Connell F, Barry M, Brito JM, Xu Q, Guney I, Berger R, Herman P, Bikoff Ret al. (2008) A prostatic intraepithelial neoplasia‐dependent p27Kip1 checkpoint induces senescence and inhibits cell proliferation and cancer progression. Cancer Cell 14, 146–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Soucek L, Whitfield J, Martins CP, Finch AJ, Murphy DJ, Sodir NM, Karnezis AN, Swigart LB, Nasi S & Evan GI (2008) Modelling Myc inhibition as a cancer therapy. Nature 455, 679–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, Newman J, Reczek EE, Weissleder R & Jacks T (2007) Restoration of p53 function leads to tumour regression in vivo . Nature 445, 661–665. [DOI] [PubMed] [Google Scholar]
- 270.Toso A, Revandkar A, DiMitri D, Guccini I, Proietti M, Sarti M, Pinton S, Zhang J, Kalathur M, Civenni Get al. (2014) Enhancing chemotherapy efficacy in pten‐deficient prostate tumors by activating the senescence‐associated antitumor immunity. Cell Rep 9, 75–89. [DOI] [PubMed] [Google Scholar]
- 271.Shamma A, Takegami Y, Miki T, Kitajima S, Noda M, Obara T, Okamoto T & Takahashi C (2009) Rb regulates DNA damage response and cellular senescence through E2F‐dependent suppression of N‐Ras isoprenylation. Cancer Cell 15, 255–269. [DOI] [PubMed] [Google Scholar]
- 272.Tkach M, Coria L, Rosemblit C, Rivas MA, Proietti CJ, Díaz Flaqué MC, Beguelin W, Frahm I, Charreau EH, Cassataro Jet al. (2012) Targeting Stat3 induces senescence in tumor cells and elicits prophylactic and therapeutic immune responses against breast cancer growth mediated by NK cells and CD4 + T cells. J Immunol 189, 1162–1172. [DOI] [PubMed] [Google Scholar]
- 273.Venturelli S, Berger A, Weiland T, Essmann F, Waibel M, Nuebling T, Häcker S, Schenk M, Schulze‐Osthoff K, Salih HRet al. (2013) Differential induction of apoptosis and senescence by the DNA methyltransferase inhibitors 5‐azacytidine and 5‐aza‐20‐deoxycytidine in solid tumor cells. Mol Cancer Ther 12, 2226–2236. [DOI] [PubMed] [Google Scholar]
- 274.Amatori S, Bagaloni I, Viti D & Fanelli M (2011) Premature senescence induced by DNA demethylating agent (Decitabine) as therapeutic option for malignant pleural mesothelioma. Lung Cancer 71, 113–115. [DOI] [PubMed] [Google Scholar]
- 275.Morelli MB, Amantini C, Nabissi M, Cardinali C, Santoni M, Bernardini G, Santoni A & Santoni G (2017) Axitinib induces senescence‐associated cell death and necrosis in glioma cell lines: the proteasome inhibitor, bortezomib, potentiates axitinib‐induced cytotoxicity in a p21(Waf/Cip1) dependent manner. Oncotarget 8, 3380–3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Yuan Y, Wang Q, Paulk J, Kubicek S, Kemp MM, Adams DJ, Shamji AF, Wagner BK & Schreiber SL (2012) A small‐molecule probe of the histone methyltransferase G9a induces cellular senescence in pancreatic adenocarcinoma. ACS Chem Biol 7, 1152–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Skwarska A, Ramachandran S, Dobrynin G, Leszczynska KB & Hammond EM (2017) The imidazoacridinone C‐1311 induces p53‐dependent senescence or p53‐independent apoptosis and sensitizes cancer cells to radiation. Oncotarget 8, 31187–31198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Mirzayans R, Scott A, Cameron M & Murray D (2005) Induction of accelerated senescence by γ radiation in human solid tumor‐derived cell lines expressing wild‐type TP53. Radiat Res 163, 53–62. [DOI] [PubMed] [Google Scholar]
- 279.He X, Yang A, McDonald DG, Riemer EC, Vanek KN, Schulte BA & Wang GY (2017) MiR‐34a modulates ionizing radiation‐induced senescence in lung cancer cells. Oncotarget 8, 69797–69807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Kim BC, Yoo HJ, Lee HC, Kang KA, Jung SH, Lee HJ, Lee M, Park S, Ji YH, Lee YSet al. (2014) Evaluation of premature senescence and senescence biomarkers in carcinoma cells and xenograft mice exposed to single or fractionated irradiation. Oncol Rep 31, 2229–2235. [DOI] [PubMed] [Google Scholar]
- 281.Cain JE, McCaw A, Jayasekara WSN, Rossello FJ, Marini KD, Irving AT, Kansara M, Thomas DM, Ashley DM & Watkins DN (2013) Sustained low‐dose treatment with the Histone deacetylase inhibitor LBH589 induces terminal differentiation of osteosarcoma cells. Sarcoma 2013, 608964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Jia L, Li H & Sun Y (2011) Induction of p21‐dependent senescence by an NAE inhibitor, MLN4924, as a mechanism of growth suppression. Neoplasia 13, 561–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Lan H, Tang Z, Jin H & Sun Y (2016) Neddylation inhibitor MLN4924 suppresses growth and migration of human gastric cancer cells. Sci Rep 6, 24218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Wang Y, Luo Z, Pan Y, Wang W, Zhou X, Jeong LS, Chu Y, Liu J & Jia L (2015) Targeting protein neddylation with an NEDD8‐activating enzyme inhibitor MLN4924 induced apoptosis or senescence in human lymphoma cells. Cancer Biol Ther 16, 420–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Zhang Y, Shi CC, Zhang HP, Li GQ & Li SS (2016) MLN4924 suppresses neddylation and induces cell cycle arrest, senescence, and apoptosis in human osteosarcoma. Oncotarget 7, 45263–45274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Huck JJ, Zhang M, McDonald A, Bowman D, Hoar KM, Stringer B, Ecsedy J, Manfredi MG & Hyer ML (2010) MLN8054, an inhibitor of Aurora A kinase, induces senescence in human tumor cells both in vitro and in vivo . Mol Cancer Res 8, 373–384. [DOI] [PubMed] [Google Scholar]
- 287.Perez M, Muñoz‐Galván S, Jiménez‐García MP, Marín JJ & Carnero A (2015) Efficacy of CDK4 inhibition against sarcomas depends on their levels of CDK4 and p16ink4 mRNA. Oncotarget 6, 40557–40574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Yoshida A, Lee EK & Diehl JA (2016) Induction of therapeutic senescence in vemurafenib‐resistant melanoma by extended inhibition of CDK4/6. Cancer Res 76, 2990–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Vijayaraghavan S, Karakas C, Doostan I, Chen X, Bui T, Yi M, Raghavendra AS, Zhao Y, Bashour SI, Ibrahim NKet al. (2017) CDK4/6 and autophagy inhibitors synergistically induce senescence in Rb positive cytoplasmic cyclin e negative cancers. Nat Commun 8, 15916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Rader J, Russell MR, Hart LS, Nakazawa MS, Belcastro LT, Martinez D, Li Y, Carpenter EL, Attiyeh EF, Diskin SJet al. (2013) Dual CDK4/CDK6 inhibition induces cell‐cycle arrest and senescence in neuroblastoma. Clin Cancer Res 19, 6173–6182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Haferkamp S, Borst A, Adam C, Becker TM, Motschenbacher S, Windhövel S, Hufnagel AL, Houben R & Meierjohann S (2013) Vemurafenib induces senescence features in melanoma cells. J Invest Dermatol 133, 1601–1609. [DOI] [PubMed] [Google Scholar]
- 292.Alimonti A, Nardella C, Chen Z, Clohessy JG, Carracedo A, Trotman LC, Cheng K, Varmeh S, Kozma SC, Thomas Get al. (2010) A novel type of cellular senescence that can be enhanced in mouse models and human tumor xenografts to suppress prostate tumorigenesis. J Clin Invest 120, 681–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.te Poele RH, Okorokov AL, Jardine L, Cummings J & Joel SP (2002) DNA damage is able to induce senescence in tumor cells in vitro and in vivo . Cancer Res 62, 1876–1883. [PubMed] [Google Scholar]
- 294.Sidi R, Pasello G, Opitz I, Soltermann A, Tutic M, Rehrauer H, Weder W, Stahel RA & Felley‐Bosco E (2011) Induction of senescence markers after neo‐adjuvant chemotherapy of malignant pleural mesothelioma and association with clinical outcome: an exploratory analysis. Eur J Cancer 47, 326–332. [DOI] [PubMed] [Google Scholar]