

Abstract
Leigh syndrome is a severe mitochondrial neurodegenerative disease with no effective treatment. In the Ndufs4−/− mouse model of LS, continuously breathing 11% O2 (hypoxia) prevents neurodegeneration and leads to a dramatic extension (~5-fold) in lifespan. We investigated the effect of hypoxia on the brain metabolism of Ndufs4−/− mice by studying blood gas tensions and metabolite levels in simultaneously sampled arterial and cerebral internal jugular venous (IJV) blood. Relatively healthy Ndufs4−/− and wildtype (WT) mice breathing air until postnatal age ~38 d were compared to Ndufs4−/− and WT mice breathing air until ~38 days old followed by 4-weeks of breathing 11% O2. Compared to WT control mice, Ndufs4−/− mice breathing air have reduced brain O2 consumption as evidenced by an elevated partial pressure of O2 in IJV blood (PijvO2) despite a normal PO2 in arterial blood, and higher lactate/pyruvate (L/P) ratios in IJV plasma revealed by metabolic profiling. In Ndufs4−/− mice, hypoxia treatment normalized the PijvO2 and L/P ratios, and decreased levels of nicotinate in IJV plasma. Brain concentrations of nicotinamide adenine dinucleotide (NAD+) were lower in Ndufs4−/− mice breathing air than in WT mice, but preserved at WT levels with hypoxia treatment. Although mild hypoxia (17% O2) has been shown to be an ineffective therapy for Ndufs4−/− mice, we find that when combined with nicotinic acid supplementation it provides a modest improvement in neurodegeneration and lifespan. Therapies targeting both brain hyperoxia and NAD+ deficiency may hold promise for treating Leigh syndrome.
Keywords: Leigh syndrome, Ndufs4, Hypoxia, Nicotinamide adenine dinucleotide, NAD, Nicotinic acid, Niacin, Arteriovenous difference, Arterial-venous difference, A-V difference
1. Introduction
Inherited defects of the mitochondrial respiratory chain can produce severe neurological disease in children and adults. Leigh syndrome is the most common pediatric clinical manifestation of mitochondrial disease and is caused by variants in more than 75 different genes [1, 2]. Children with Leigh syndrome appear healthy at birth, but soon thereafter develop progressive neurodegenerative brain lesions, and their median length of survival is 2 years [3]. There is currently no proven therapy to prevent or delay neurodegeneration in patients with Leigh syndrome.
One of the more severe forms of Leigh syndrome is caused by inactivation of the NDUFS4 gene, which encodes the NADH:ubiquinone oxidoreductase subunit S4 of the mitochondrial complex I [4]. Complex I oxidizes the reduced form of nicotinamide adenine dinucleotide (NADH) to generate NAD+ and electrons which form the reducing power used to fuel oxidative phosphorylation to produce ATP. Patients with defects in the electron transport chain exhibit an increase in the cytosolic NADH/NAD+ ratio, which manifests in plasma as an elevated lactate/pyruvate (L/P) ratio.
Mice deficient in Ndufs4 have decreased activity of mitochondrial complex I and serve as a model of Leigh syndrome by recapitulating many of the neuropathological and clinical features [5]. Although born healthy, Ndufs4−/− mice breathing air (21% O2) die at a median postnatal age of 55 days [5]. Compared to Ndufs4−/− mice breathing air, continuously breathing normobaric 11% O2 prevents and reverses neurological disease and increases lifespan ~5-fold [6, 7]. In addition to continuously breathing 11% O2, chronic inhalation of 600 ppm carbon monoxide and chronic anemia also prevent neurological disease and increase survival duration of Ndufs4−/− mice [8]. Furthermore, each of these therapeutic strategies is associated with a reduction in elevated brain tissue partial pressure of O2 (PbO2) measured in anesthetized (0.5–1% isoflurane) and mechanically ventilated Ndufs4−/− mice using an optical fluorescence PO2 probe [8]. The most parsimonious explanation for these observations is that brain hyperoxia may be pathogenic in Leigh syndrome.
Sampling blood simultaneously from an artery and a cerebral vein, such as the internal jugular vein (IJV), enables determination of the cerebral arterial-internal jugular venous (A-IJV) difference in blood gas tensions and metabolite levels. Measuring cerebral A-IJV differences of blood gas tensions and plasma metabolite levels has been employed to study the effect of hypoxia on brain metabolism in mammals, such as humans at high-altitude and in simulated-diving Weddell seals [9–12]. However, to our knowledge, this technique has not previously been used in a mouse model of mitochondrial disease.
The objective of this study was to investigate the effect of continuously breathing normobaric 11% O2 on the brain metabolism of Ndufs4-deficient mice by measuring blood gas tensions and the levels of metabolites in simultaneously sampled arterial and cerebral IJV blood. Cerebral A-IJV blood sampling in anesthetized (ketamine 120 mg/kg and fentanyl 9 µg/kg) and mechanically ventilated Ndufs4−/− mice breathing air, allowed the detection of reduced cerebral O2 consumption that produces elevated cerebral venous O2 levels, which is consistent with the higher PbO2 reported previously in these mice [8]. In addition, blood sampling revealed higher brain NADH/NAD+ ratio, that was accompanied by lower NAD+ concentrations in the brain. Importantly, breathing 11% O2 normalized both O2 and NAD+ levels in the brain of Ndufs4−/− mice. Milder hypoxia (17% O2) is an ineffective therapy by itself in this model, however we observe a modest improvement in neurodegeneration and survival duration when 17% O2 is combined with NAD+ precursor supplementation.
2. Results
2.1. Breathing 11% O2 for four weeks decreases the elevated PO2 in internal jugular venous blood of anesthetized and ventilated Ndufs4-deficient mice
To investigate the mechanisms by which chronic hypoxic breathing increases lifespan and improves neurological disease in Ndufs4−/− mice, we studied brain oxygenation and metabolism. We measured the partial pressure of O2 (PO2) and CO2 (PCO2) in simultaneously sampled arterial and IJV blood collected from anesthetized (ketamine 120 mg/kg and fentanyl 9 µg/kg) and ventilated Ndufs4−/− and WT mice exposed to either normoxia (breathing air until a postnatal age of ~38 d) or hypoxia (breathing air for the same length of time followed by breathing 11% O2 for four weeks to a postnatal age of ~66 d).
The partial pressure of O2 in arterial blood (PaO2) supplying the brain was not different between Ndufs4−/− and WT mice breathing air (98.8 ± 4.0 vs. 103.0 ± 4.5 mm Hg; P=0.57; Figure 1 A). In mice breathing air, the partial pressure of O2 in IJV blood (PijvO2) leaving the brain, however, was higher in Ndufs4−/− mice compared to WT mice (56.0 ± 1.4 vs. 46.5 ± 3.0 mm Hg; P=0.006; Figure 1 B). The difference between the PaO2 in arterial blood supplying the brain and the PijvO2 in IJV blood leaving the brain is a proxy for cerebral O2 consumption if cerebral blood flow (CBF) is constant. In mice breathing air, the arterial-IJV PO2 difference (Pa-ijvO2) was smaller in Ndufs4−/− mice as compared to WT mice (42.8 ± 16.1 vs. 56.7 ± 19.0 mm Hg; P=0.03; Figure 1 C). These data suggest that Ndufs4−/− mice breathing air have an elevated PO2 in cerebral IJV blood, which is likely due to impaired O2 utilization by the brain assuming blood flow is constant.
Figure 1. Partial pressure and blood O2 content in simultaneously sampled arterial and internal jugular venous blood collected from anesthetized and ventilated Ndufs4−/− and WT mice breathing air or after 4-weeks of 11% O2.
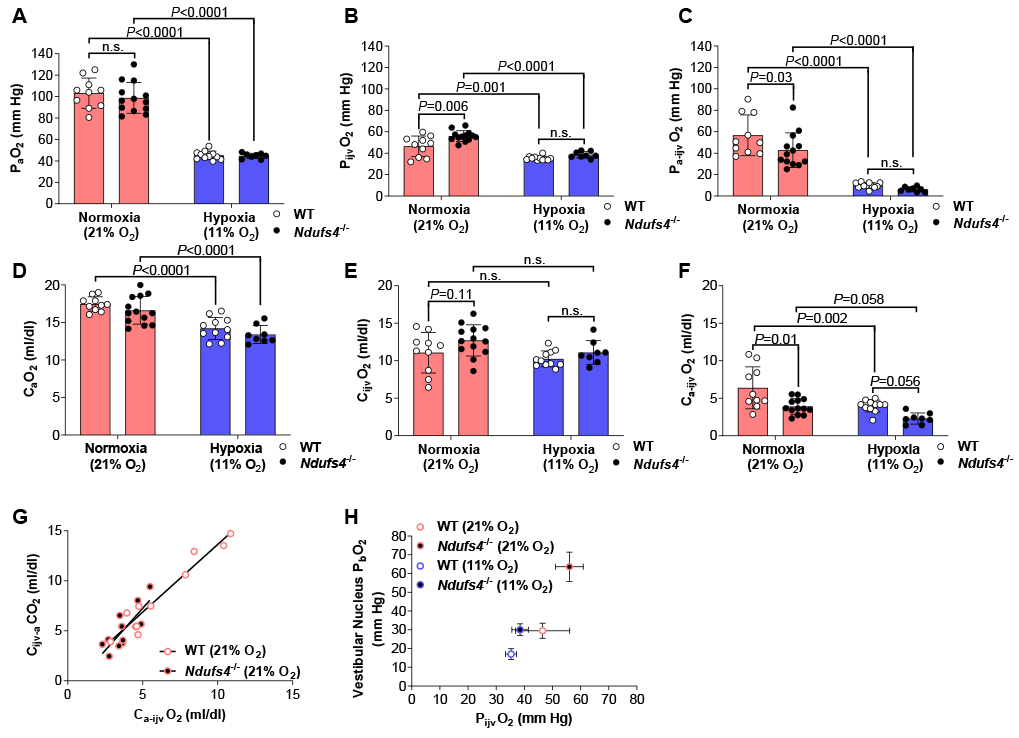
Partial pressure of O2 was measured in (A) arterial (PaO2) and (B) internal jugular venous (PijvO2) blood. (C) Arterial-internal jugular venous PO2 difference (Pa-ijvO2). Blood O2 content was measured in (D) arterial (CaO2) and (E) internal jugular venous (CijvO2) blood. (F) Arterial-internal jugular venous O2 content difference (Ca-ijvO2). (G) Pearson correlation between the arterial-internal jugular venous O2 content difference (Ca-ijvO2) and the internal jugular venous-arterial CO2 content difference (Cijv-aCO2) in Ndufs4−/− mice (r =0.80, P<0.002) and WT (r =0.97, P<0.0001) mice breathing air. (H) PijvO2 plotted against values previously reported in the vestibular nucleus brain tissue PO2 (PbO2) of Ndufs4−/− and WT mice breathing either air or 11% O2 [8]. Statistical significance was determined using two-way ANOVA with Sidak’s multiple comparisons test. Data are mean ± SD.
To quantify O2 consumption by the brain, we calculated the O2 content in arterial blood (CaO2), the O2 content in IJV blood (CijvO2), and arterial-IJV O2 content difference (Ca-ijvO2). O2 content of blood is determined by PO2, the O2 saturation of hemoglobin (SO2) and hemoglobin concentration. The SO2 paralleled the PO2 trends measured in arterial and IJV blood (Figure S1 A–C). The concentration of hemoglobin in both arterial and IJV blood was similar between Ndufs4−/− and WT mice breathing air (Figure S1 D & E). As a result, calculated CaO2 was similar between Ndufs4−/− and WT mice breathing air (17.5 ± 0.3 vs. 16.6 ± 0.5 ml/dl; Figure 1 D). The CijvO2 trended higher in Ndufs4−/− mice compared to WT mice (12.7 ± 0.6 vs. 11.1 ± 0.9; P=0.11; Figure 1 E). Therefore, the Ca-ijvO2 was smaller in Ndufs4−/− mice compared to WT mice breathing air (3.9 ± 0.3 vs. 6.4 ± 0.9 ml/dl; P=0.01; Figure 1 F). These data indicate reduced O2 consumption in the brain of Ndufs4−/− mice breathing air. Interestingly, compared to WT mice, impaired O2 utilization in the brain of Ndufs4−/− mice is visibly evident as the IJV appeared a lighter shade of red, indicating a higher venous SO2, thus PO2 (Figure S2).
If cerebral O2 consumption is reduced in Ndufs4−/− mice than in WT mice breathing air, a decrease in cerebral CO2 production would also be expected in Ndufs4−/− mice. The total CO2 carrying capacity of blood (CO2 content), comprised of the partial pressure of CO2 (PCO2) and bicarbonate concentration, was measured in the identical blood samples used to calculate O2 content; the results mirrored the trends of O2 content (Figure S3 A–H). Thus, in Ndufs4−/− mice breathing air, the smaller Ca-ijvO2 correlated with a smaller IJV-arterial CO2 content difference (Cijv-aCO2) compared to WT mice breathing air (Figure 1 G). These results support the concept of impaired O2 utilization in the brain of Ndufs4−/− mice breathing air.
Since the PijvO2 likely reflects global brain tissue PO2 (PbO2), a higher PijvO2 level in Ndufs4−/− mice breathing air compared to WT mice may reflect an elevated PbO2 [13]. We previously reported that vestibular nucleus PbO2 measured with an optical fluorescence O2 sensor was higher in anesthetized (0.5–1% isoflurane) and ventilated Ndufs4−/− mice breathing air compared to WT mice [8]. Therefore, the increased PijvO2 in Ndufs4−/− mice measured by cerebral A-IJV blood sampling in the present study was associated with the elevated vestibular nucleus PbO2 levels previously reported (Figure 1 H).
In mice breathing 11% O2, the PaO2 was similar between Ndufs4−/− and WT mice (45.7 ± 0.9 vs. 45.2 ± 1.2 mm Hg; Figure 1 A) but was reduced compared to Ndufs4−/− and WT mice breathing air. The PijvO2 was also similar between Ndufs4−/− and WT mice breathing 11% O2 (34.5 ± 1.0 vs. 35.3 ± 0.6 mm Hg; Figure 1 B), and was lower compared to Ndufs4−/− and WT mice breathing air. The Pa-ijvO2 did not differ between Ndufs4−/− and WT mice breathing 11% O2 (6.2 ± 2.3 vs. 9.9 ± 2.6 mm Hg; Figure 1 C) and reduced compared to Ndufs4−/− and WT mice breathing air. The results indicate when Ndufs4−/− and WT mice breathe 11% O2 for four weeks the PO2 of arterial and IJV blood is reduced to a comparable level in both genotypes.
In Ndufs4−/− and WT mice breathing 11% O2, the concentration of hemoglobin in both arterial and IJV blood was greater than in Ndufs4−/− and WT mice breathing air, but did not differ between the genotypes (Figure S1 F&G). Despite a compensatory increase in hemoglobin concentration, the CaO2 was lower in Ndufs4−/− and WT mice breathing 11% O2 (13.4 ± 0.4 vs. 14.2 ± 0.4 ml/dl; Figure 1 D) as compared to Ndufs4−/− and WT mice breathing air. The CijvO2 in Ndufs4−/− and WT mice breathing 11% O2, was similar between Ndufs4−/− and WT mice and did not differ from mice breathing air (Figure 1 E). Thus, the Ca-ijvO2 decreased similarly in Ndufs4−/− and WT mice breathing 11% O2 as compared to Ndufs4−/− and WT mice breathing air (Figure 1 F). The results indicate that arterial O2 content, and likely cerebral O2 delivery, is reduced when Ndufs4−/− and WT mice breathe 11% O2 for four weeks, and this would be expected to lower the PO2 in IJV blood and brain tissue.
Blood gas tensions and metabolite levels in arterial and IJV blood are influenced by CBF, and CBF is affected by the depth of anesthesia. In our study, anesthesia depth may have differed between the Ndufs4−/− and WT mice because Ndufs4−/− mice are known to have a slight insensitivity to ketamine [14]. Although we were not able to quantify CBF in Ndufs4−/− mice, we instead measured systemic hemodynamics, including heart rate and mean arterial blood pressure, in Ndufs4−/− and WT mice undergoing blood sampling. There was no difference in basal heart rate or mean arterial blood pressure between Ndufs4−/− and WT mice breathing either air or 11% O2 (Table S1). The results suggest that differences in arterial and IJV blood gas tensions or metabolite levels from Ndufs4−/− and WT mice breathing either air or 11% O2 would not have been driven by changes in systemic hemodynamics.
2.2. Breathing 11% O2 for four weeks normalizes brain NAD+ metabolism in Ndufs4-deficient mice
To more broadly explore the metabolic differences between arterial and cerebral venous blood, we performed metabolic profiling of paired arterial and IJV plasma samples collected from anesthetized (ketamine 120 mg/kg and fentanyl 9 µg/kg) and ventilated Ndufs4−/− and WT mice exposed to normoxia or hypoxia (described above). Mitochondrial disorders exhibit an elevated L/P ratio in the circulation, which reflects an elevated cytosolic NADH/NAD+ ratio [15]. Previous reports have shown that circulating lactate is elevated in Ndufs4−/− mice breathing air [6, 7, 16], in addition we have previously shown that circulating lactate is normalized by continuously breathing normobaric 11% O2 [6, 7]. Using liquid chromatography–mass spectrometry (LC-MS), we measured levels of lactate and pyruvate, and calculated the L/P ratio, in paired arterial and IJV plasma samples. We observed a positive linear relationship in L/P ratios between arterial and IJV plasma (Figure 2 A & B). Compared to WT mice breathing air, in Ndufs4−/− mice breathing air the L/P ratios in IJV plasma were higher for a given L/P ratio in arterial plasma (Figure 2 A). In contrast, the correlation of L/P ratios between arterial and IJV plasma were similar between Ndufs4−/− and WT mice breathing 11% O2 for four weeks (Figure 2 B). These results likely reflect an increased cytosolic NADH/NAD+ ratio in the brain of Ndufs4−/− mice breathing air, and that breathing 4-weeks of 11% O2 preserves the NADH/NAD+ ratio.
Figure 2. Lactate/pyruvate ratios and nicotinate levels measured in simultaneously sampled arterial and internal jugular venous plasma, brain tissue concentration of NAD+ and Naprt mRNA expression, collected from Ndufs4−/− and WT mice breathing air or after 4-weeks of 11% O2.

Pearson correlation between arterial and internal jugular venous plasma lactate/pyruvate ratios in (A) Ndufs4−/− (r=0.78, P=0.012) and WT (r=0.77, P=0.016) mice breathing air or in Ndufs4−/− (r=0.98, P<0.0001) and WT (r=0.97, P=0.005) mice breathing 4-weeks of 11% O2. (C) Volcano plot of the arterial-internal jugular venous (A-IJV) metabolite level difference depicting on the X-axis log2fold-changes (log2FC) and on the Y-axis statistical significance in Condition (Hypoxia vs. Normoxia) as -log10 P-values in both Ndufs4−/− and WT mice breathing 4-weeks of 11% O2 (Hypoxia) compared to both Ndufs4−/− and WT mice breathing air (Normoxia). Tissue concentration of NAD+ measured in the (D) brainstem, (E) cerebellum and (F) cerebrum. Tissue mRNA expression of Naprt measured in the (G) brainstem, (H) cerebellum and (I) cerebrum. Statistical significance was determined using two-way ANOVA with Sidak’s multiple comparisons test. All P-values generated from metabolic profiling were corrected for multiple hypothesis testing using the Benjamini-Hochberg procedure and false-discovery rate (FDR) <0.05 considered statistically significant. For Pearson correlation the ROUT method (Q = 0.5%) was used to identify and exclude 1 outlier [40]. n.s.= non-significant. Data are mean ± SD.
Using LC-MS we measured the levels of 126 metabolites (including lactate and pyruvate) in arterial and IJV plasma. To determine the metabolites that are altered by the brain we calculated the arterial-IJV (A-IJV) metabolite level difference for all 126 metabolites (Table S2). The A-IJV metabolite level difference was compared by Condition (Hypoxia vs. Normoxia), Genotype (Ndufs4−/− mice vs. WT mice), and an Interaction term (Genotype:Condition; Table S3–5). We found that the A-IJV difference of nicotinate (vitamin B3/niacin), a precursor for the biosynthesis of NAD+, increased in both Ndufs4−/− and WT mice breathing air compared to Ndufs4−/− and WT mice breathing 11% O2 (Figure 2 C). This difference was driven by a decrease in the nicotinate level in IJV plasma as compared to Ndufs4−/− and WT mice breathing air (Figure S4 A–C). These data suggest that nicotinate is released from the brain into the venous circulation, and that the amount that is released decreases when mice of either genotype breathe 11% O2 for four weeks.
Since nicotinate is a metabolic precursor for NAD+, and neurodegenerative lesions in Ndufs4−/− mice are predominatly localised to the brainstem and cerebellum, we sought to determine whether NAD+ levels are altered in a region specific way. Notably, Lee and colleagues previously reported a decline in whole brain NAD+ levels in 65 to 75 day old Ndufs4−/− mice breathing air [17]. We measured regional brain tissue concentrations of reduced (NADH) and oxidized (NAD+) NAD forms in ~38 day old Ndufs4−/− and WT mice breathing air or after 4-weeks of 11% O2. In Ndufs4−/− mice breathing air, the brainstem concentration of NAD+ was less than in WT mice (1.31 ± 0.43 vs. 2.36 ± 0.55 nmol/mg protein; P=0.01; Figure 2 D), but in mice breathing 11% O2 brainstem NAD+ concentrations did not differ between Ndufs4−/− and WT mice. Similarly, in the cerebellum of mice breathing air, the concentration of NAD+ trended lower in Ndufs4−/− mice as compared to WT mice (5.28 ± 1.10 vs. 7.56 ± 2.64 nmol/mg protein; P=0.050; Figure 2 E), and were similar between Ndufs4−/− and WT mice breathing 11% O2. In the cerebrum, the concentration of NAD+ was also decreased in Ndufs4−/− mice compared to WT mice breathing air (5.19 ± 0.57 vs. 7.61 ± 2.02 nmol/mg protein; P=0.03; Figure 2 F), and cerebrum NAD+ concentrations increased in Ndufs4−/− mice breathing 11% O2 (5.19 ± 0.57 vs. 8.94 ± 2.04 nmol/mg protein; P=0.0009) to similar levels found in WT mice (Figure 2 F). NADH concentrations in the brainstem and cerebrum did not differ between Ndufs4−/− and WT mice breathing either air or 11% O2 (Figure S4 D & F). The NADH concentration in the cerebellum of Ndufs4−/− mice breathing air was greater than in WT mice, but similar in mice breathing 11% O2 (2.89 ± 0.18 vs. 2.47 ± 0.51 nmol/mg protein; P=0.03; Figure S4 E). The results indicate that in Ndufs4−/− mice breathing air the NAD+ level is decreased in the brainstem, cerebellum and cerebrum, which is preserved by breathing 4-weeks of 11% O2.
We measured mRNA expression of nicotinate phosphoribosyltransferase (Naprt), the rate-limiting enzyme in the biosynthesis of NAD+ from nicotinic acid (Preiss-Handler pathway [18, 19]) in the brain of Ndufs4−/− and WT mice breathing either air or 11% O2 for four weeks [20]. In Ndufs4−/− mice breathing air, the mRNA expression of Naprt, was higher in the brainstem (1.38 ± 0.21 vs. 1.00 ± 0.17; P=0.008), cerebellum (1.44 ± 0.20 vs. 1.00 ± 0.18; P=0.002) and cerebrum (1.58 ± 0.47 vs. 1.00 ± 0.27; P=0.007) compared to WT mice breathing air (Figure 2 G–I). Moreover, Naprt levels did not differ between Ndufs4−/− and WT mice breathing 4-weeks of 11% O2 (Figure 2 G–I). The results indicate increased gene expression of the Preiss-Handler pathway, that generates NAD+ from nicotinic acid, in all brain regions of Ndufs4−/− mice breathing air, which is normalised when Ndufs4−/− mice breathe 11% O2 for four weeks.
2. 3. Combining nicotinic acid supplementation with breathing 17% O2 increases lifespan and delays neurological disease in Ndufs4-deficient mice
Since in Ndufs4−/− mice breathing 4-weeks of 11% O2 normalizes both the PijvO2 and brain NAD+ concentrations, and alters brain nicotinic acid metabolism, we hypothesized that pharmacologically replenishing brain NAD+ levels would prevent neurodegeneration in Ndufs4−/− mice. To test this hypothesis, we supplemented Ndufs4−/− and WT mice breathing air (21% O2) with the NAD+ biosynthetic precursor, nicotinic acid (240 mg/kg twice daily by i.p. injection), but observed no benefit to neurological symptoms or lifespan (Figure S5 E–H). We next tested nicotinic acid supplementation in the setting of reduced PijvO2 by treating Ndufs4−/− and WT mice breathing 17% O2. Of note, Ferrari and colleagues have previously shown that chronic exposure to mild hypoxia (17% O2) alone does not improve either survival duration or the manifestations of neurologic disease in Ndufs4−/− mice.
Ndufs4−/− mice that continuously breathe 17% O2 from 30 days of age will survive for at least three weeks, therefore we measured the PijvO2 in anesthetized (ketamine 120 mg/kg and fentanyl 9 µg/kg) and ventilated Ndufs4−/− mice breathing 17% O2 for 3-weeks. The PijvO2 was higher in Ndufs4−/− mice breathing air compared to Ndufs4−/− mice breathing 3-weeks of 17% O2 (56.0 ± 4.9 vs. 43.8 ± 2.9 mm Hg; Figure 3 A). In Ndufs4−/− mice breathing 17% O2, the PijvO2 was similar to the PijvO2 in WT mice breathing air (43.8 ± 2.9 vs. 46.5 ± 9.6 mm Hg; Figure 3 A). Thus breathing 17% O2 for 3-weeks lowers the O2 tension in IJV blood of Ndufs4−/− mice to a comparable PO2 to WT mice breathing air.
Figure 3. Nicotinic acid treatment provides a modest increase in lifespan and delay in the time of onset of neurological symptoms and neurodegenerative brain lesions of Ndufs4−/− mice breathing 17% O2.
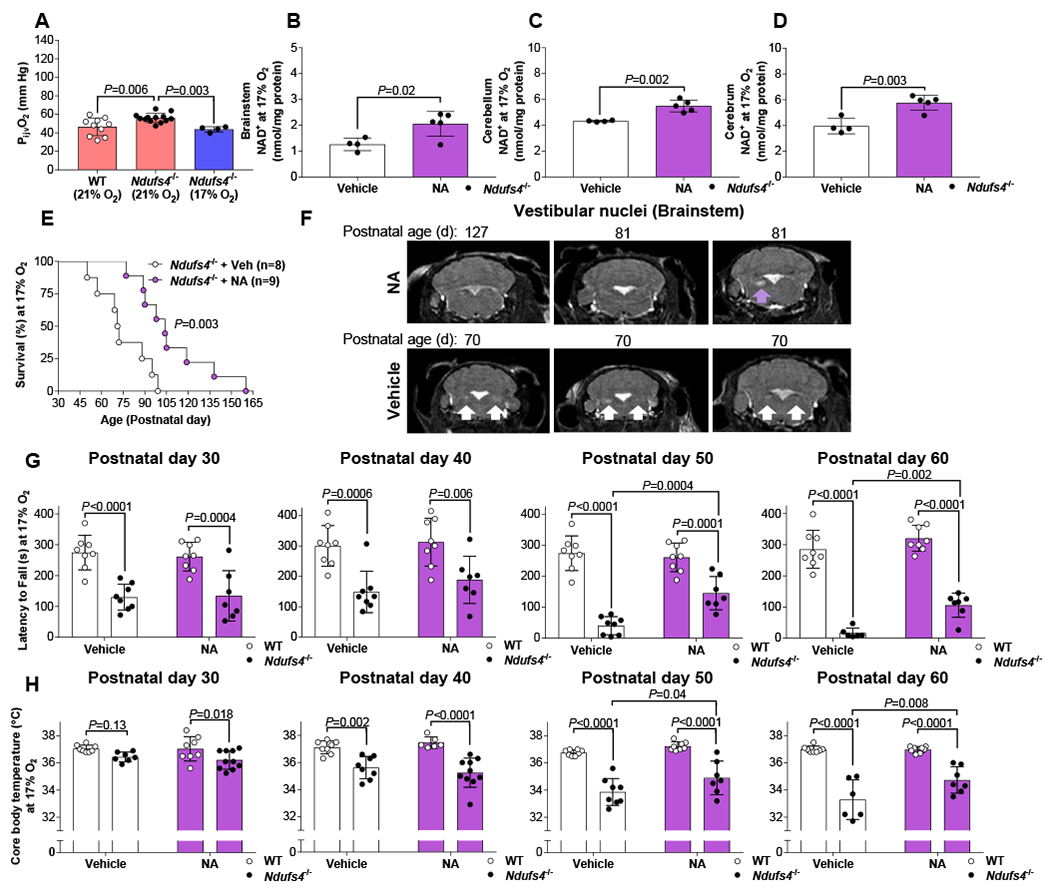
(A) The partial pressure of internal jugular venous blood (PijvO2) in Ndufs4−/− mice breathing 17% O2 for 3-weeks compared to the same PijvO2 values for Ndufs4−/−and WT mice breathing air (21% O2) taken from Figure 1. Tissue concentration of NAD+ measured in the (B) brainstem, (C) cerebellum and (D) cerebrum of Ndufs4−/− mice breathing 17% O2 for 3-weeks and treated with either vehicle or nicotinic acid (NA; 240 mg/kg twice daily by intraperitoneal injection). Brain tissue was harvested 12 h after the previous dose. (E) Survival duration, (F) MRI images of the vestibular nuclei (arrow used to indicate a lesion), (G) latency to fall from an accelerating rotarod, and (H) core body temperature of Ndufs4−/− and WT mice breathing 17% O2 and treated with either vehicle or NA. For a single comparison an unpaired, two-tailed, Students T-test was used. For multiple comparisons statistical significance was determined using two-way ANOVA with Sidak’s multiple comparisons test. Log-rank (Mantel-Cox) test was used to compare Kaplan-Meier survival curves. Data are mean ± SD.
We measured regional brain tissue concentrations of NAD+ and NADH of Ndufs4−/− mice breathing 17% O2 who were treated with either nicotinic acid or vehicle for three weeks. Brain tissue was harvested 12 h after the previous dose to obtain a trough measurement. In Ndufs4−/− mice breathing 17% O2, nicotinic acid treatment increased trough NAD+ concentrations in the brainstem (2.06 ± 0.43 vs. 1.26 ± 0.21 nmol/mg protein; P=0.02), cerebellum (5.50 ± 0.41 vs. 4.33 ± 0.05 nmol/mg protein; P=0.002) and cerebrum (5.77 ± 0.52 vs. 3.96 ± 0.52 nmol/mg protein; P=0.003; Figure 3 B–D) compared to vehicle treated Ndufs4−/− mice breathing 17% O2. In contrast, NADH concentrations in the brainstem, cerebellum and cerebrum did not differ between nicotinic acid and vehicle treated Ndufs4−/− mice breathing 17% O2 (Figure S5 B–D). These data demonstrate that treating Ndufs4−/− mice breathing 17% O2 with nicotinic acid for three weeks modestly increases NAD+ concentrations in all three brain regions (brainstem, cerebellum and cerebrum).
Survival duration was modestly increased in nicotinic acid-treated Ndufs4−/− mice breathing 17% O2 as compared to vehicle-treated mice (median age of survival of 104 vs. 72 d; P=0.003; Figure 3 E). Bodyweight was not improved and remained similar between nicotinic acid and vehicle treated Ndufs4−/− mice breathing 17% O2 (Figure S5 A). In mice breathing 17% O2, bilateral neurodegenerative lesions in the vestibular nuclei detected by MRI were observed in 70 day old Ndufs4−/− mice treated with vehicle, whereas in older (81–127 day old) Ndufs4−/− mice treated with nicotinic acid a single unilateral neurodegenerative lesion in the vestibular nucleus was detected (Figure 3 F). From postnatal age 30 to 60 days, in Ndufs4−/− mice breathing 17% O2 and treated with vehicle, both the core body temperature and the latency of the mice to fall off an accelerating rotarod progressively declined, similar to that previously reported in Ndufs4−/− mice breathing air (Figure 3 G & H)[6, 16, 21]. However, compared to vehicle treated-mice, decrease in core body temperature and the latency to fall from an accelerating rotarod was significantly blunted in nicotinic acid-treated Ndufs4−/− mice breathing 17% O2 at 50 and 60 days of age (Figure 3 G & H). The results indicate that the combination of nicotinic acid treatment and breathing 17% O2 – interventions that individually are not beneficial – modestly increase lifespan and delays neurodegeneration in Ndufs4−/− mice.
3. Discussion
We sought to evaluate the effect of continuously breathing normobaric 11% O2 for four weeks on the brain metabolism of Ndufs4−/− mice. As a proxy for brain metabolism, we measured blood gas tensions and the levels of metabolites in arterial and cerebral IJV blood obtained from anesthetized (ketamine 120 mg/kg and fentanyl 9 µg/kg) and mechanically ventilated Ndufs4−/− and WT mice breathing either air or after 4-weeks of 11% O2. Compared to WT mice breathing air, in Ndufs4−/− mice breathing air we detected reduced cerebral O2 consumption (smaller Ca-ijvO2) that produced an elevated PijvO2 and PbO2, indicating impaired brain O2 utilization in Ndufs4−/− mice – this result is consistent with our previous report that the brain PO2 of Ndufs4−/− mice is higher [8]. Ndufs4−/− mice breathing air had higher L/P ratios in IJV plasma relative to arterial L/P ratios and lower brain tissue NAD+ concentrations. In both Ndufs4−/− and WT mice breathing 4-weeks of 11% O2, both PijvO2 and CaO2 were reduced indicating chronic hypoxic breathing lowers brain O2 levels by decreasing O2 delivery to the brain. In addition, when Ndufs4−/− mice breathed 11% O2 for four weeks, the L/P ratios in IJV plasma and brain tissue NAD+ concentrations were normalized to WT levels. In both Ndufs4−/− and WT mice breathing 4-weeks of 11% O2, metabolic profiling revealed increased A-IJV difference of the NAD+ precursor nicotinate, suggesting chronic hypoxic breathing altered nicotinic acid metabolism in the brain. Treating Ndufs4−/− mice with a combination of mild hypoxia (17% O2) and nicotinic acid supplementation, each of which was ineffective, delayed the onset of neurological symptoms and neurodegeneration and increased lifespan, though not as dramatically as breathing 11% O2.
Impaired peripheral O2 extraction by skeletal muscle during exercise has been documented in patients with mitochondrial disorders such as mitochondrial encephalopathy, lactic acidosis and stroke-like episodes (MELAS) [22, 23]. Furthermore, decreased brain O2 consumption has been reported in patients with Leigh syndrome or MELAS measured as a decrease in the cerebral metabolic rate of oxygen (CMRO2) using either PET or MRI [24–27]. We previously reported reduced whole-body O2 consumption in conscious Ndufs4−/− mice breathing air and elevated PbO2 measured in the vestibular nucleus of anesthetized (0.5–1% isoflurane) and ventilated Ndufs4−/− mice breathing air suggesting impaired brain O2 utilization [8]. Using cerebral A-IJV blood sampling we now observe decreased cerebral O2 consumption and reduced cerebral CO2 production in Ndufs4−/− mice breathing air, which is consistent with and may explain the elevated vestibular nucleus PbO2. Breathing 11% O2 for four weeks decreased the PijvO2 in Ndufs4−/− mice due to a reduction in cerebral O2 delivery, suggested by a lower arterial O2 content with unchanged systemic hemodynamics.
Metabolite profiling revealed two important results. First, the L/P ratio of IJV plasma is higher for a given arterial L/P ratio in Ndufs4−/− mice breathing air, providing metabolic evidence of altered brain metabolism with increased aerobic glycolysis and indicating a higher brain NADH/NAD+ ratio, that is normalized when Ndufs4−/− mice breathe 11% O2 for four weeks. Second, we searched metabolome wide and observed 4-weeks of breathing 11% O2 consistently increased the A-IJV nicotinate level difference independent of genotype. In follow-up studies we find decreased levels of NAD+ and higher Naprt expression in the brainstem, cerebellum and cerebrum of Ndufs4−/− mice breathing air, which appeared to be preserved to WT levels Ndufs4−/− mice breathed 11% O2 for four weeks.
We report decreased brainstem and cerebellar concentrations of NAD+ in young (~38 day old) Ndufs4−/− mice breathing air. Lee and colleagues have previously reported lower NAD+ concentrations in whole brain tissue collected from older (66–75 day old) Ndufs4−/− mice breathing air that would be expected to have severe neurodegenerative lesions [17, 28]. Our findings suggest that NAD+ depletion may precede the development of overt neurodegenerative lesions in the brainstem and cerebellum. Therefore, decrement in NAD+ may contribute to neurodegeneration.
NAD+ serves as both an electron carrier and as a substrate for NAD+-consuming enzymes such as sirtuins and poly(ADP-ribose) polymerases (PARPs)[20], the activity of which influence total NAD+ levels in cells and are altered by mitochondrial complex I deficiency [29, 30]. Excessive activation of PARP-1, stimulated by DNA damage, has been reported in Ndufs4−/− mice breathing air, and pharmacological inhibition of PARP-1 provides a small but significant delay in the time of onset of neurological symptoms [31]. Although it is unclear exactly how mitochondrial complex I deficiency in Ndufs4−/− mice contributes to neurodegeneration, our results show that chronic hypoxic (11% O2) breathing normalizes PbO2 and NAD+ levels.
Other groups have previously tested NAD+ supplementation in Ndufs4−/− mice breathing air. For example, while our work was in progress, Lee and colleagues showed that, supplementation of the NAD+ precursor nicotinamide mononucleotide (NMN; 500 mg/kg once every three days by i.p. injection) in Ndufs4−/− mice breathing air improved survival, but not neurological disease [17]. In the current study, we administered to Ndufs4−/− mice breathing air a different NAD+ precursor, nicotinic acid, which also failed to improve neurological symptoms, but also did not increase lifespan. The results of these two studies differ slightly possibly because Lee et al., commenced treatment in younger (21 day old) Ndufs4−/− mice where pathophysiology would likely be less progressed or due to differences in the pharmacological properties between the two NAD+ precursors, however these factors remain to be confirmed. We combined chronic mild hypoxia (17% O2), an intervention that on its own is not effective in the Ndufs4−/− mouse [7], with nicotinic acid to increase brain NAD+ levels. This combination provided a small, modest improvement in neurological disease and lifespan. Further investigation using alternative NAD+ precursors with superior pharmacodynamic and pharmacokinetic properties is warranted.
A limitation of this study is that, for technical reasons, we sampled cerebral venous blood from the extracranial portion of the murine IJV and not intracranially, from the IJV bulb, which may have contaminated cerebral venous blood with venous blood from extra-cerebral sources. A second limitation of our study is that we have not quantified CBF in Ndufs4−/− and WT mice, which could in principle impact the observed O2 and metabolite extractions. This is relevant given Ndufs4−/− mice have a slight insensitivity to ketamine [14], however we would have expected the Ndufs4−/− and WT mice to be fully anesthetized at the dose (120 mg/kg) ketamine we administered. Moreover, we saw no differences in systemic hemodynamics between genotypes and conditions suggesting anesthetic depth was comparable. In addition, we have previously reported that lower thoracic aortic blood flow, a surrogate measure of cardiac output, did not differ between anesthetized (ketamine 120 mg/kg and fentanyl 9 µg/kg) and mechanically ventilated Ndufs4−/− and WT mice breathing air and was unchanged after breathing 11% O2 for three weeks [32].
In conclusion, we showed that Ndufs4−/− mice breathing air have decreased brain O2 consumption, elevated cerebral venous PO2 and depleted brain NAD+ levels, which together may contribute to neurodegeneration. Furthermore, breathing 4-weeks of 11% O2 normalized cerebral venous PO2 and brain NAD+ levels in Ndufs4−/− mice. However, at present the precise molecular mechanisms and temporal sequence of events linking mitochondrial defects, O2, and NAD+ levels are not known. Our results suggest that hypoxia inspired therapies for mitochondrial disease may benefit from concomitant NAD+ precursor supplementation.
4. Material and Methods
4.1. Animals
Animal experiments were approved by the Institutional Animal Care and Use Committee of the Massachusetts General Hospital (MGH). Ndufs4 heterozygous (Ndufs4+/-) mice on a C57BL6/J background were a kind gift from the Palmiter laboratory, University of Washington. Ndufs4+/- mice were bred to generate WT (Ndufs4+/+) and Ndufs4−/− mice. A mix of male and female Ndufs4−/− and WT mice were studied. Unless otherwise stated, WT mice breathing air (21% O2) were a median (interquartile range) postnatal age of 38 d (36–40 d) with a bodyweight of 15 ± 1.7 g (mean ± SD). Ndufs4−/− mice breathing air were a postnatal day of 38 d (35–40 d) with a bodyweight of 13 ±1.9 g. Ndufs4−/− and WT mice breathing air for ~38 d were placed into hypoxia chambers and continuously breathed 11% O2 for four weeks. WT mice at the end of breathing 11% O2 were 70 d (67–79 d) old and weighed 21 ± 3.8 g. Ndufs4−/− mice at the end of breathing 11% O2 were 69 d (68–73 d) old and weighed 16 ± 1.3 g.
4.2. Chronic breathing of air, 17% O2 or 11% O2
Mice breathed an inspired O2 fraction (FiO2) of either 21%, 17% or 11%. For A-IJV difference studies, ~38 day old Ndufs4−/− mice were used because this age corresponded with peak bodyweight (~15 g) improving successful vessel cannulation [5, 7]. Ndufs4−/− mice older than ~38 d breathing air were not studied because from this age Ndufs4−/− mice develop progressive neurodegenerative lesions and ultimately die at a median age of ~55 d [5–7, 16, 28]. Breathing 11% O2 for four weeks was studied because this has previously been shown to prevent, and reverse established, neurodegenerative lesions in Ndufs4−/− mice [6, 7].
Mice breathing air were housed in MGH’s animal resource facility (Center for Comparative Medicine at MGH). Mice that chronically breathed 11% O2 were housed in cages kept inside 80 l transparent acrylic boxes used as hypoxia chambers. A FiO2 of either 11% or 17% was obtained using a N2 generator (MAG-20; Higher Peak, Winchester, MA, USA) that concentrated atmospheric N2 to create a hypoxic gas mixture. O2 levels were monitored daily using an O2 sensor (MiniOx 1; Ohio Medical, Gurnee, IL, USA) placed at the outlet port of the hypoxia chamber and was calibrated weekly using an 5.55 % O2 reference tank (Airgas; Radnor, PA, USA). CO2 concentrations inside the chamber were kept below 0.4% (monitored using an Extech CO200 Monitor; Extech Instruments, Nashua, NH, USA) using trays containing the CO2 scavenger soda lime (Sodasorb; Smiths Medical, Minneapolis, MN, USA). Temperature (24–26°C), humidity (30–70%), light cycle (12 h light:dark). Mice housed inside the chambers were provided food and water ab libitum, and additional food pellets were inserted into hydrated gel (Napa Nectar; ScottPharma, Inc., Marlborough, MA, USA) placed on the bedding.
4.3. Femoral arterial and internal jugular venous blood sampling in mice
In young (~38 d old) Ndufs4−/− and WT mice, weighing ~13 g, we catheterized the IJV (~200 µm diameter) rather than the larger external jugular vein (~2 mm diameter) because the IJV provides cerebral venous drainage, limiting the contamination of venous blood from extra-cerebral sources [33]. To collect arterial blood the femoral artery was catheterized, rather than the larger common carotid artery, because C57BL6/J mice have an incomplete Circle of Willis and occlusion of a carotid artery might decrease cerebral perfusion [34]. Two catheters implanted retrograde into either the femoral artery or the internal jugular vein (IJV), were constructed from 4 cm long polyurethane BTPU-010 tubing (250 µm outer-diameter; Instech Laboratories, Inc. Plymouth Meeting, PA, USA) inserted 1 cm into the end of polyethylene PE10 tubing 30 cm in length (610 µm outer-diameter; Intradermic; Becton Dickinson, Franklin Lakes, NJ, USA), silicone adhesive was applied to the joint creating an air-tight seal. The opposite end of the PE10 tubing was connected, using a 27-guage needle, to either a fluid-filled pressure transducer or a 1 ml pre-heparinized syringe used for blood sample collection. Before implantation, the catheters were primed with a 10 USP/ml heparin (NDC 0409–2720-02; Hospira, Inc. Lake Forest, IL, USA) saline solution, to remove any air-bubbles and to maintain patency following implantation.
All mice were fasted for between two to six hours before anesthesia induction. Anesthesia was induced via i.p. injection of ketamine (120 mg/kg) and fentanyl (9 µg/kg) in saline. Once anesthetized, mice were secured supine on a heating plate to maintain core temperature at 37 ± 0.5°C via feedback from a rectal temperature probe connected to a temperature controller (TCAT-2; Physitemp Instruments, Clifton, NJ, USA). The cranium was not in contact with the heating plate to avoid directly heating the murine brain. A tracheostomy was performed followed by tracheal intubation for volume-controlled ventilation (Mini Vent 845; Harvard Apparatus, Holliston, MA) at a respiratory rate of 110 breaths/min, a tidal volume of 10 ml/kg of bodyweight, and a positive end-expiratory pressure (PEEP) of 1 cm H2O. Next, 2 mg/kg of the paralytic rocuronium (NDC 39822–4200-2; X-Gen Pharmaceuticals Inc. Big Flats, NY, USA) was injected i.p. to induce muscle relaxation. Anesthesia and paralysis were maintained by administering a bolus of one quarter of the induction dose (ketamine 30 mg/kg; fentanyl 2 µg/kg; rocuronium 0.5 mg/kg) i.p. every 10 min. To prevent arterial desaturation during surgical placement of the catheters, thus premature hypoxemia, a FiO2 of 30% was used for mice that had previously been breathing air and a FiO2 of 21% (air) used for mice previously been breathing either 11% O2 or 17% O2.
The right femoral artery was surgically isolated and catheterized retrograde using a catheter (described above) connected to a fluid-filled pressure transducer, Bridge Amp, Powerlab, and LabChart 7 (ADInstruments, Colorado Springs, CO, USA) was used to measure mean arterial blood pressure and heart rate. The murine right internal jugular vein (~200 µm diameter), found parallel and anterior to the right common carotid artery , was surgically isolated and catheterized distal to the sternocleidomastoid muscle. Once inserted the internal jugular venous catheter was fed retrograde until the tip passed distal to the branching point of the right facial vein.
Before arterial and venous blood sampling commenced, Ndufs4-/- and WT mice were transfused, via the arterial catheter, with whole blood to prevent a decrease in blood volume, thus organ perfusion, when blood was sampled. All mice used as blood donors were fasted, sex-matched, Ndufs4+/- mice kept under identical conditions as the recipient mouse. Ndufs4+/- mice were used as blood donors, rather than WT or Ndufs4−/− mice because a supply of Ndufs4+/- were more readily available. Immediately before arterial and venous blood sampling, whole-blood was collected, via cardiac puncture, into a heparinized syringe from anesthetized (described above) donor mice spontaneously breathing air. The donor blood was stored on ice until needed, then warmed to 37.0°C using a water bath. Once the artery and vein were catheterized (described above), the mice were transfused with 200 µl of pre-warmed donor mouse blood. 200 µl of blood was transfused because a minimum sampling volume of 200 µl was required for metabolic profiling or measuring blood gas tensions in both arterial and venous blood samples.
After both catheters were implanted and the mice were transfused, the FiO2 was reduced to the experimental FiO2 of either 21% O2 (air), 17% O2 or 11% O2. After allowing time to equilibrate to the experimental FiO2, and when mean arterial blood pressure and heart rate were stable, femoral arterial and internal jugular venous blood were drawn simultaneously into 1 ml pre-heparinized syringes (4041–2; Smiths Medical ASD, Inc. Keene, NH, USA). The dead-volume of the PE10 tubing was discarded to limit blood dilution. Upon collection, arterial and venous blood were transferred either to a blood gas analyzer (ABL800 FLEX analyzer; Radiometer America Inc., Westlake, OH, USA), to measure blood gas tensions (PO2 & PCO2) and the concentration of hemoglobin, or into EDTA coated tubes (07 6011; RAM Scientific, Inc. Nashville, TN, USA) followed by centrifugation (2000 g for 10 min at 4°C) to isolate plasma, used for metabolic profiling, that was snap-frozen in liquid N2 and stored at -80°C.
4.4. Measuring Oxygen dissociation curve (ODC)
Collected blood (20 µl) was diluted with 3 ml of HEMOX solution (HS-500; TCS Scientific Corporation, New Hope, PA, USA) and 6 µl of an anti-foaming agent (AFA-25; TCS Scientific Corporation, New Hope, PA, USA). HEMOX solution contains N-[Tris(hydroxymethyl)methyl]-2-aminoethanesulfonic acid (TES, 30 mM), sodium chloride (135 mM), and potassium chloride (5 mM) in Milli-Q water (pH 7.4). The O2 dissociation curve (ODC) of the diluted blood sample was measured using a HEMOX analyzer with the sample maintained at 37°C. The partial pressure of O2 at which 50% of hemoglobin is oxygenated was determined as P50 from the ODC. The O2 saturation of hemoglobin in arterial and IJV blood was determined using the average Hill slope generated from Ndufs4−/− and WT mice breathing either air (21% O2) or after breathing 4-weeks of 11% O2 (Figure S1 F).
4.5. Calculating the O2 total carrying capacity of blood (O2 content)
The O2 content of arterial and IJV blood (CaO2 and CijvO2) was calculated using the formula: O2 Content = (1.34 × Hb × SO2) + (0.0031 × PO2). CaO2 - CijvO2 difference was calculated to determine A-IJV O2 difference. The CO2 content of arterial and internal jugular venous blood (CaCO2 and CijvCO2) was calculated using the formula: CO2 Content = HCO3− + (0.0308 × PCO2). CijvCO2 - CaO2 was calculated to determine Cijv-aCO2.
4.6. Metabolic profiling using liquid chromatography–mass spectrometry
Metabolic profiling using liquid chromatography–mass spectrometry (LC-MS) was performed on a total of 62 plasma samples (31 arterial and 31 internal jugular venous). In brief, 15 µl of plasma sample was mixed with 137 µl of ice-cold acetonitrile containing internal standards (13C6 -glucose, D3-lactate, 13C3-pyruvate, D3-α-hydroxybutyrate,13C2 -β-hydroxybutyrate, 13C3 -alanine & 13C3-serine) for metabolite extraction. Samples were vortexed and incubated on ice for 30 min. After centrifugation for 20 min, at 4°C and 21,000 g, 75 µl of sample was transferred to autosampler glass vial for LC-MS analysis and 10 µl of sample was injected on Waters XBridge amide column (2.1 x 100 mm, 2.5 µm; Part # 186006091, Milford, MA, USA). The pooled Quality Control (QC) sample was prepared by mixing ~equal volume of each sample and injected every 12 samples to evaluate the analytical performance. Samples were injected in a randomized order to avoid any run order effect. The column oven temperature was 27°C and the autosampler was 4°C. Mobile phase A was 5:95/acetonitrile:water, 20 mM ammonium acetate, pH 9 (adjusted with ammonium hydroxide) and mobile phase B was acetonitrile. LC gradient conditions at flow rate of 0.220 ml/min were: 0 min 85%B, 0.5 min 85%B, 9 min 35%B, 11 min 2% B, 12 min 2% B, 13.5 min 85%B, 14.6 min 85%B ,15 min 85%B with 0.420 ml/min to 18 min. Dionex Ultimate 3000 UHPLC system was coupled to Q-Exactive Plus Orbitrap mass spectrometer (ThermoFisher Scientific, Waltham, MA, USA) with HESI probe operating in switch polarity mode. MS parameters were: sheath gas flow 50, aux gas flow 10, sweep gas flow 2, spray voltage 2.50 kV in negative & 3.8kV in positive, Capillary temperature 310°C, S-lens RF level -50 and aux gas heater temperature 370°C. Data acquisition was done using Xcalibur (ThermoFisher Scientific, Waltham, MA, USA) in range of: 70–1000 m/z, resolution 70,000, automatic gain control target 3E6 and maximum injection time of 80 ms. Data analysis was done using the Tracefinder™ 4.1. Metabolite annotation was performed based on accurate mass (± 5 ppm) and matching retention time (±0.5 min) as well as MS/MS fragmentation pattern from the pooled QC sample against in-house retention time and MS/MS library of reference chemical standards. The quality of integration for each chromatographic peak was reviewed. Metabolites reported have CV <30% in pooled quality control samples.
4.7. Brainstem, cerebellum and cerebrum tissue collection
Tissue collection was performed between the hours of 10 AM and 6 PM. Unless otherwise stated, a separate cohort of Ndufs4−/− and WT mice were used for brain tissue collection to minimize the effect of long-duration anesthesia and surgery on tissue NAD+ concentrations. A mix of male and female Ndufs4−/− and WT mice breathing air from birth until a postnatal age of ~38 d was compared to an older ~66 d old mix of male and female Ndufs4−/− and WT mice, breathing air for the same length of time followed by breathing 11% O2 for four weeks. Anesthesia was induced using ketamine and fentanyl (described above), a thoracotomy was performed, and the mouse euthanized by exsanguination. An incision was made in to the left and right atria and a needle inserted into the left ventricle, 20 ml of pre-chilled, ice-cold Dulbecco’s phosphate buffered saline (DPBS; 14190–144; Life Technologies Limited, Paisley, UK) was perfused throughout the body. Once the expelled fluid ran clear the brainstem, right and left hemisphere of the cerebellum and cerebrum, were rapidly removed on ice, snap-frozen in liquid N2 and stored at -80°C.
4.8. NADH and NAD+ tissue quantification
The total concentration of NAD (NADH + NAD+) and the NADH concentration were measured in the murine right hemisphere of the cerebrum and cerebellum, and in the entire brainstem, previously collected and stored at -80°C. Tissues were homogenized in extraction buffer then centrifuged (4000 g for 5 min) to collect the supernatant. The total NAD concentration and the NADH concentration were measured in the supernatant using a NADH/NAD+ quantitation colorimetric kit (K337–100; BioVision, Inc., Milpitas, CA, USA) in accordance with manufacturer’s protocol . The NAD+ concentration was calculated by subtracting the total concentration of NADH from the total NAD concentration.
4.9. Quantitative real-time polymerase chain reaction (qPCR)
qPCR was performed similar to that described previously [35]. Total RNA was extracted using TRIzol reagent (15596018; ThermoFisher Scientific, Waltham, MA, USA) from the murine right hemisphere of the cerebrum and cerebellum, and the entire brainstem, previously collected and stored at -80°C. Brain tissue was homogenized (Power Gen 125; Fisher Scientific, Waltham, MA, USA) in TRIzol. After adding chloroform, the sample was vortexed, incubated at room temperature, then centrifuged at 12,000 g at 4°C for 10 min. The top layer was carefully transferred to a new tube. After adding isopropanol, RNA was collected by centrifugation and washed with 75% ethanol. RNA quality and quantity were measured using NanoDrop Lite (ThermoFisher Scientific, Waltham, MA, USA). To synthesize cDNA, High-Capacity cDNA Reverse Transcriptase Kit (4368813; Applied Biosciences, Foster City, CA, USA) was used according to the manufacturer’s instructions. TaqMan Fast Advance Master Mix (Applied Biosystems, Foster City, CA, USA) was used to perform qPCR. TaqMan assay (ThermoFisher Scientific, Waltham, MA, USA) for Naprt (Mm00553802_m1). Amplification and real-time quantification of transcripts was performed using a Mastercycler Realplex (Eppendorf, Hamburg, Germany). All reactions were run in duplicate. Expression data in each sample were normalized by the relative cycle threshold (∆CT) method to the expression of 18s ribosomal RNA amplified with TaqMan probe (Hs99999901_s1; ThermoFisher Scientific, Waltham, MA, USA). Relative mRNA expression data were normalized to the WT mice breathing air control group.
4.10. Nicotinic acid administration in Ndufs4−/− and WT mice breathing either air or 17% O2
30 day old Ndufs4−/− and WT mice were housed in chambers kept continuously at either 21% or 17% O2 (described above) and dosed with either vehicle control (phosphate buffered saline; PBS; 14190144; ThermoFisher Scientific, Waltham, MA, USA) or 240 mg/kg nicotinic acid (N4126; Sigma-Aldrich, St. Louis, MO, USA). 240 mg/kg nicotinic acid was used because it is the molar equivalent of a NAD+ precursor dose, 500 mg/kg nicotinamide riboside, previously reported to increase tissue NAD+ levels in mice [36]. A solution of nicotinic acid in PBS was prepared under sterile conditions and corrected to pH 7.4 using NaOH, then passed through a 0.022 µM filter. After sterilizing the injection site with an alcohol swab, mice were dosed via i.p. injection at a volume of 10 µl per gram of bodyweight twice a day (AM/PM). Nicotinic acid was administered twice per day due to its relatively short half-life in rodents [37]. Vehicle control mice were injected with PBS.
4.11. Magnetic resonance imaging (MRI)
Mice were continuously anesthetized with 0.5–1.0% isoflurane in room air and images were generated using a DICOM reader (OsiriX; University of Geneva, Switzerland). MRI scans of the brain were performed as previously described , using respiratory gated T2-weighted RARE (rapid acquisition of refocused echoes) MRI images acquired on a 4.7-T small animal scanner (Pharmascan; Bruker, Billerica, MA, USA) with the following parameters: RARE factor: 10, echo time: 60 ms, repetition time: 6000 ms, Averages: 8, 192 x 192 x 24 image matrix with a voxel size of 0.130 x 0.130 x 0.7 mm).
4.12. Latency to fall from accelerating rotarod measurements
Latency for mice to stay on an accelerating rotarod (Ugo Basile; Stoelting Co., Wood Dale, IL, USA) was performed as previously described . The parameters of the machine were: an acceleration time of 5 rpm/min, maximum speed of 40 rpm and maximum time of 420 s. 3 sets of measurements were performed on mice while breathing air, with the median time reported.
4.13. Statistics
For all data, except data generated by metabolic profiling, statistical analyses were performed using Prism 7 software (GraphPad Software; La Jolla, CA, USA). All data are expressed as mean ± standard deviation (SD). P-values <0.05 were considered statistically significant. For a single comparison an unpaired, two-tailed, Students T-test was used. For multiple comparisons statistical significance was determined using two-way ANOVA with Sidak’s multiple comparisons test. Log-rank (Mantel-Cox) test was used to compare Kaplan-Meier survival curves. For the sake of clarity all pairwise comparisons may not be indicated on every graph. The level of 126 metabolites measured in arterial and internal jugular venous plasma using LC-MS were submitted to statistical analysis. Missing values were imputed at half the minimum of the dataset. All the data were log10 transformed. After imputation and transformation, the arterial-internal jugular venous difference was found. Using the R package limma, an interaction model for the arterial-internal jugular venous difference in metabolite levels was fit using Genotype (Ndufs4−/− vs. WT) and Condition (Hypoxia vs. Normoxia) as main effects and the interaction term Genotype:Condition [38, 39]. All P-values were then corrected for multiple hypothesis testing using the Benjamini-Hochberg procedure as implemented in limma. False Discovery Rate (FDR) of <0.05 was considered statistically significant.
Supplementary Material
Aknowledgements
This work was supported by a gift from the Marriott Family Foundation (V.K.M.) and funds from the Department of Anesthesia, Critical Care and Pain Medicine at Massachusetts General Hospital (W.M.Z.). Conflicts of Interest: V.K.M. and W.M.Z. are co-inventors on a patent application submitted by Massachusetts General Hospital on the use of hypoxia as a therapy. V.K.M. owns equity stake in Raze Therapeutics and is a paid advisor for Janssen Pharmaceuticals and 5AM Ventures.
Abbreviations
- A-IJV difference
Arterial-internal jugular venous difference
- PbO2
Brain tissue PO2
- IJV
Internal Jugular Vein
- L/P ratio
Lactate/pyruvate ratio
- LS
Leigh syndrome
- LC-MS
Liquid chromatography–mass spectrometry
- Ndufs4
NADH:ubiquinone oxidoreductase subunit S4
- NAD+
Oxidized nicotinamide adenine dinucleotide
- NADH
Reduced nicotinamide adenine dinucleotide
- NA
Nicotinic acid
- CaO2
O2 content in arterial blood
- CijvO2
O2 content in IJV blood
- SaO2
O2 saturation of hemoglobin in arterial blood
- SijvO2
O2 saturation of hemoglobin in IJV blood
- PO2
Partial pressure of oxygen
- PaO2
Partial pressure of oxygen in arterial blood
- PijvO2
Partial pressure of oxygen in IJV blood
- Veh
Vehicle
References
- 1.Lake NJ, Compton AG, Rahman S, and Thorburn DR, Leigh syndrome: One disorder, more than 75 monogenic causes. Ann Neurol, 2016. 79(2): p. 190–203. [DOI] [PubMed] [Google Scholar]
- 2.Rahman S TD Nuclear Gene-Encoded Leigh Syndrome Overview. 2015. [cited 1993–2020 Jan 27 2020]; Available from: https://www.ncbi.nlm.nih.gov/books/NBK320989/.
- 3.Sofou K, De Coo IF, Isohanni P, Ostergaard E, Naess K, De Meirleir L, Tzoulis C, Uusimaa J, De Angst IB, Lonnqvist T, Pihko H, Mankinen K, Bindoff LA, Tulinius M, and Darin N, A multicenter study on Leigh syndrome: disease course and predictors of survival. Orphanet J Rare Dis, 2014. 9: p. 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lake NJ, Bird MJ, Isohanni P, and Paetau A, Leigh syndrome: neuropathology and pathogenesis. J Neuropathol Exp Neurol, 2015. 74(6): p. 482–92. [DOI] [PubMed] [Google Scholar]
- 5.Kruse SE, Watt WC, Marcinek DJ, Kapur RP, Schenkman KA, and Palmiter RD, Mice with mitochondrial complex I deficiency develop a fatal encephalomyopathy. Cell Metab, 2008. 7(4): p. 312–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jain IH, Zazzeron L, Goli R, Alexa K, Schatzman-Bone S, Dhillon H, Goldberger O, Peng J, Shalem O, Sanjana NE, Zhang F, Goessling W, Zapol WM, and Mootha VK, Hypoxia as a therapy for mitochondrial disease. Science, 2016. 352(6281): p. 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferrari M, Jain IH, Goldberger O, Rezoagli E, Thoonen R, Cheng KH, Sosnovik DE, Scherrer-Crosbie M, Mootha VK, and Zapol WM, Hypoxia treatment reverses neurodegenerative disease in a mouse model of Leigh syndrome. Proc Natl Acad Sci U S A, 2017. 114(21): p. E4241–E4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jain IH, Zazzeron L, Goldberger O, Marutani E, Wojtkiewicz GR, Ast T, Wang H, Schleifer G, Stepanova A, Brepoels K, Schoonjans L, Carmeliet P, Galkin A, Ichinose F, Zapol WM, and Mootha VK, Leigh Syndrome Mouse Model Can Be Rescued by Interventions that Normalize Brain Hyperoxia, but Not HIF Activation. Cell Metab, 2019. 30(4): p. 824–832 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith KJ, MacLeod D, Willie CK, Lewis NC, Hoiland RL, Ikeda K, Tymko MM, Donnelly J, Day TA, MacLeod N, Lucas SJ, and Ainslie PN, Influence of high altitude on cerebral blood flow and fuel utilization during exercise and recovery. J Physiol, 2014. 592(24): p. 5507–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Murphy B, Zapol WM, and Hochachka PW, Metabolic activities of heart, lung, and brain during diving and recovery in the Weddell seal. J Appl Physiol Respir Environ Exerc Physiol, 1980. 48(4): p. 596–605. [DOI] [PubMed] [Google Scholar]
- 11.Overgaard M, Rasmussen P, Bohm AM, Seifert T, Brassard P, Zaar M, Homann P, Evans KA, Nielsen HB, and Secher NH, Hypoxia and exercise provoke both lactate release and lactate oxidation by the human brain. FASEB J, 2012. 26(7): p. 3012–20. [DOI] [PubMed] [Google Scholar]
- 12.Milledge JS and Sorensen SC, Cerebral arteriovenous oxygen difference in man native to high altitude. J Appl Physiol, 1972. 32(5): p. 687–9. [DOI] [PubMed] [Google Scholar]
- 13.Gupta AK, Hutchinson PJ, Al-Rawi P, Gupta S, Swart M, Kirkpatrick PJ, Menon DK, and Datta AK, Measuring brain tissue oxygenation compared with jugular venous oxygen saturation for monitoring cerebral oxygenation after traumatic brain injury. Anesth Analg, 1999. 88(3): p. 549–53. [DOI] [PubMed] [Google Scholar]
- 14.Quintana A, Morgan PG, Kruse SE, Palmiter RD, and Sedensky MM, Altered anesthetic sensitivity of mice lacking Ndufs4, a subunit of mitochondrial complex I. PLoS One, 2012. 7(8): p. e42904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Patgiri A, Skinner OS, Miyazaki Y, Schleifer G, Marutani E, Shah H, Sharma R, Goodman RP, To TL, Robert Bao X, Ichinose F, Zapol WM, and Mootha VK, An engineered enzyme that targets circulating lactate to alleviate intracellular NADH:NAD(+) imbalance. Nat Biotechnol, 2020. 38(3): p. 309–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johnson SC, Yanos ME, Kayser EB, Quintana A, Sangesland M, Castanza A, Uhde L, Hui J, Wall VZ, Gagnidze A, Oh K, Wasko BM, Ramos FJ, Palmiter RD, Rabinovitch PS, Morgan PG, Sedensky MM, and Kaeberlein M, mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science, 2013. 342(6165): p. 1524–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee CF, Caudal A, Abell L, Nagana Gowda GA, and Tian R, Targeting NAD(+) Metabolism as Interventions for Mitochondrial Disease. Sci Rep, 2019. 9(1): p. 3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Preiss J and Handler P, Biosynthesis of diphosphopyridine nucleotide. II. Enzymatic aspects. J Biol Chem, 1958. 233(2): p. 493–500. [PubMed] [Google Scholar]
- 19.Preiss J and Handler P, Biosynthesis of diphosphopyridine nucleotide. I. Identification of intermediates. J Biol Chem, 1958. 233(2): p. 488–92. [PubMed] [Google Scholar]
- 20.Lautrup S, Sinclair DA, Mattson MP, and Fang EF, NAD(+) in Brain Aging and Neurodegenerative Disorders. Cell Metab, 2019. 30(4): p. 630–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quintana A, Kruse SE, Kapur RP, Sanz E, and Palmiter RD, Complex I deficiency due to loss of Ndufs4 in the brain results in progressive encephalopathy resembling Leigh syndrome. Proc Natl Acad Sci U S A, 2010. 107(24): p. 10996–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Steele HE, Horvath R, Lyon JJ, and Chinnery PF, Monitoring clinical progression with mitochondrial disease biomarkers. Brain, 2017. 140(10): p. 2530–2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taivassalo T, Abbott A, Wyrick P, and Haller RG, Venous oxygen levels during aerobic forearm exercise: An index of impaired oxidative metabolism in mitochondrial myopathy. Ann Neurol, 2002. 51(1): p. 38–44. [DOI] [PubMed] [Google Scholar]
- 24.Frackowiak RS, Herold S, Petty RK, and Morgan-Hughes JA, The cerebral metabolism of glucose and oxygen measured with positron tomography in patients with mitochondrial diseases. Brain, 1988. 111 ( Pt 5): p. 1009–24. [DOI] [PubMed] [Google Scholar]
- 25.Lindroos MM, Borra RJ, Parkkola R, Virtanen SM, Lepomaki V, Bucci M, Virta JR, Rinne JO, Nuutila P, and Majamaa K, Cerebral oxygen and glucose metabolism in patients with mitochondrial m.3243A>G mutation. Brain, 2009. 132(Pt 12): p. 3274–84. [DOI] [PubMed] [Google Scholar]
- 26.Nariai T, Ohno K, Ohta Y, Hirakawa K, Ishii K, and Senda M, Discordance between cerebral oxygen and glucose metabolism, and hemodynamics in a mitochondrial encephalomyopathy, lactic acidosis, and strokelike episode patient. J Neuroimaging, 2001. 11(3): p. 325–9. [DOI] [PubMed] [Google Scholar]
- 27.Yu L, Xie S, Xiao J, Wang Z, and Zhang X, Quantitative measurement of cerebral oxygen extraction fraction using MRI in patients with MELAS. PLoS One, 2013. 8(11): p. e79859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quintana A, Zanella S, Koch H, Kruse SE, Lee D, Ramirez JM, and Palmiter RD, Fatal breathing dysfunction in a mouse model of Leigh syndrome. J Clin Invest, 2012. 122(7): p. 2359–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alano CC, Tran A, Tao R, Ying W, Karliner JS, and Swanson RA, Differences among cell types in NAD(+) compartmentalization: a comparison of neurons, astrocytes, and cardiac myocytes. J Neurosci Res, 2007. 85(15): p. 3378–85. [DOI] [PubMed] [Google Scholar]
- 30.Karamanlidis G, Lee CF, Garcia-Menendez L, Kolwicz SC Jr., Suthammarak W, Gong G, Sedensky MM, Morgan PG, Wang W, and Tian R, Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell Metab, 2013. 18(2): p. 239–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Felici R, Cavone L, Lapucci A, Guasti D, Bani D, and Chiarugi A, PARP inhibition delays progression of mitochondrial encephalopathy in mice. Neurotherapeutics, 2014. 11(3): p. 651–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schleifer G, Marutani E, Ferrari M, Sharma R, Skinner O, Goldberger O, Grange RMH, Peneyra K, Malhotra R, Wepler M, Ichinose F, Bloch DB, Mootha VK, and Zapol WM, Impaired hypoxic pulmonary vasoconstriction in a mouse model of Leigh syndrome. Am J Physiol Lung Cell Mol Physiol, 2019. 316(2): p. L391–L399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mancini M, Greco A, Tedeschi E, Palma G, Ragucci M, Bruzzone MG, Coda AR, Torino E, Scotti A, Zucca I, and Salvatore M, Head and Neck Veins of the Mouse. A Magnetic Resonance, Micro Computed Tomography and High Frequency Color Doppler Ultrasound Study. PLoS One, 2015. 10(6): p. e0129912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beckmann N, High resolution magnetic resonance angiography non-invasively reveals mouse strain differences in the cerebrovascular anatomy in vivo. Magn Reson Med, 2000. 44(2): p. 252–8. [DOI] [PubMed] [Google Scholar]
- 35.Hindle AG, Allen KN, Batten AJ, Huckstadt LA, Turner-Maier J, Schulberg SA, Johnson J, Karlsson E, Lindblad-Toh K, Costa DP, Bloch DB, Zapol WM, and Buys ES, Low guanylyl cyclase activity in Weddell seals: implications for peripheral vasoconstriction and perfusion of the brain during diving. Am J Physiol Regul Integr Comp Physiol, 2019. 316(6): p. R704–R715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Giroud-Gerbetant J, Joffraud M, Giner MP, Cercillieux A, Bartova S, Makarov MV, Zapata-Perez R, Sanchez-Garcia JL, Houtkooper RH, Migaud ME, Moco S, and Canto C, A reduced form of nicotinamide riboside defines a new path for NAD(+) biosynthesis and acts as an orally bioavailable NAD(+) precursor. Mol Metab, 2019. 30: p. 192–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Petrack B, Greengard P, and Kalinsky H, On the relative efficacy of nicotinamide and nicotinic acid as precursors of nicotinamide adenine dinucleotide. J Biol Chem, 1966. 241(10): p. 2367–72. [PubMed] [Google Scholar]
- 38.R-Core-Team, R: a language and environment for statistical computing 2016, R Foundation for Statistical Computing: Vienna, Austria. [Google Scholar]
- 39.Ritchie DB and Woodside MT, Probing the structural dynamics of proteins and nucleic acids with optical tweezers. Curr Opin Struct Biol, 2015. 34: p. 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Motulsky HJ and Brown RE, Detecting outliers when fitting data with nonlinear regression - a new method based on robust nonlinear regression and the false discovery rate. BMC Bioinformatics, 2006. 7: p. 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.