

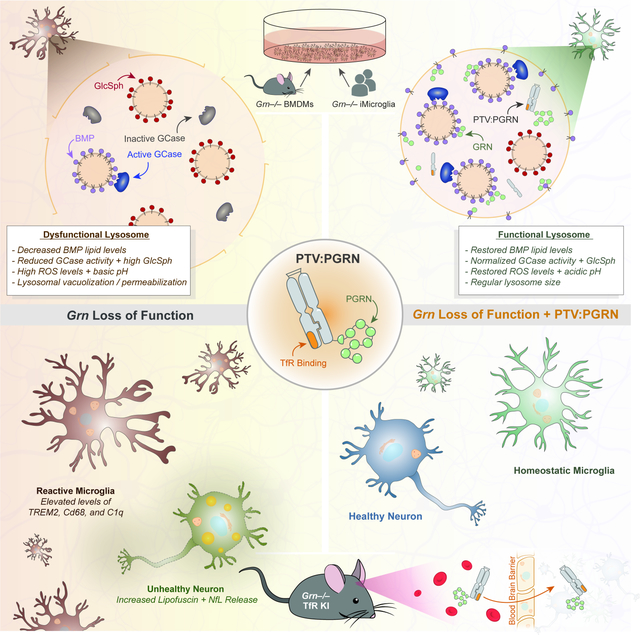
SUMMARY
GRN mutations cause frontotemporal dementia (GRN-FTD) due to deficiency in progranulin (PGRN), a lysosomal and secreted protein with unclear function. Here, we found that Grn−/− mice exhibit a global deficiency in bis(monoacylglycero)phosphate (BMP), an endolysosomal phospholipid we identified as a pH-dependent PGRN interactor as well as a redox-sensitive enhancer of lysosomal proteolysis and lipolysis. Grn−/− brains also showed an age-dependent, secondary storage of glucocerebrosidase substrate glucosylsphingosine. We investigated a protein replacement strategy by engineering protein transport vehicle (PTV):PGRN – a recombinant protein linking PGRN to a modified Fc domain that binds human transferrin receptor for enhanced CNS biodistribution. PTV:PGRN rescued various Grn−/− phenotypes in primary murine macrophages and human iPSC-derived microglia, including oxidative stress, lysosomal dysfunction and endomembrane damage. Peripherally delivered PTV:PGRN corrected levels of BMP, glucosylsphingosine and disease pathology in Grn−/− CNS, including microgliosis, lipofuscinosis and neuronal damage. PTV:PGRN thus represents a potential biotherapeutic for GRN-FTD.
Keywords: Neurodegenerative disease, disease associated microglia, lysosome, lipids, lysobisphosphatidic acid, LBPA, galectin-3, GBA, GCase, lipofuscin, lipidomics, metabolomics
ETOC:
Peripherally delivered progranulin biologic with enhanced CNS biodistribution corrects CNS disease pathology of the GRN mouse model of frontotemporal dementia.
Graphical Abstract
INTRODUCTION
GRN-FTD represents 10–15% of FTD cases and is characterized by lipofuscinosis, microgliosis, TAR DNA-binding protein 43 (TDP-43) pathology and cortical neuronal loss (Kao et al., 2017; Wauters et al., 2017). Most heterozygous GRN-FTD mutations result in haploinsufficiency of PGRN, while various missense or nonsense mutations in a subset of cases cause PGRN loss-of-function (LoF) (Baker et al., 2006; Cruts et al., 2006; Wauters et al., 2017). Rare homozygous GRN mutations lead to a lysosomal storage disorder termed neuronal ceroid lipofuscinosis (NCL) (Smith et al., 2012). Lipofuscin, a lysosomal pathology caused by oxidative stress, is also found in the brain and retina of GRN-FTD patients, suggesting common pathogenic mechanisms with NCL (Ward et al., 2017). Along with data showing a sortilin-dependent lysosomal localization of PGRN (Hu et al., 2010), this genetic link points to a key function of PGRN in lysosomes. PGRN has also been implicated in the regulation of cathepsins, primarily CatD (Beel et al., 2017; Butler et al., 2019; Gotzl et al., 2018; Gotzl et al., 2014; Tanaka et al., 2017; Valdez et al., 2017; Wils et al., 2012; Zhou et al., 2017a), lysosomal pH (Tanaka et al., 2017), and glucocerebrosidase (GCase) activity, a lipase involved in Gaucher disease and GBA-linked Parkinson’s disease (Arrant et al., 2019; Jian et al., 2016; Valdez et al., 2020; Zhou et al., 2019). PGRN was shown to regulate GCase activity via direct binding (Jian et al., 2016), recruitment of co-chaperone Hsp70 (Jian et al., 2016), and lysosomal delivery and processing of prosaposin into saposin C, which activates GCase (Arrant et al., 2019; Paushter et al., 2018; Valdez et al., 2020; Zhou et al., 2019; Zhou et al., 2017c).
PGRN is processed intracellularly by CatB and CatL into seven ~6 kDa granulin (GRN) peptides that may mediate its lysosomal functions (Holler et al., 2017; Lee et al., 2017; Zhou et al., 2017b). Given that key lysosomal PGRN interactors – CatD, prosaposin, GCase – are genetically linked to neurodegeneration, defects in lysosomal functions of PGRN or GRN peptides likely underlie pathogenic mechanisms in GRN-FTD and NCL. This is corroborated by findings that a modifier of GRN-FTD age of onset is TMEM106B (Cruchaga et al., 2011; Finch et al., 2011), a gene encoding a transmembrane lysosomal protein whose ablation worsens lysosomal defects and FTD-like pathology of Grn−/− mice (Feng et al., 2020; Werner et al., 2020; Zhou et al., 2020).
In the CNS, Grn LoF profoundly impacts microglial state and function (Gotzl et al., 2019; Gotzl et al., 2018; Kao et al., 2017; Krabbe et al., 2017; Lui et al., 2016; Martens et al., 2012; Zhang et al., 2020). Microglia express the highest CNS PGRN levels and show higher expression in reactive states, as a result of injuries (Byrnes et al., 2011; Moisse et al., 2009; Tanaka et al., 2013), aging (Zhang et al., 2020) or disease pathology (Chen-Plotkin et al., 2010). Grn LoF in mice causes inflammation and microgliosis, a hallmark of disease, along with enhanced neurotoxicity due in part to secretion of complement factors (Gotzl et al., 2019; Krabbe et al., 2017; Lui et al., 2016; Martens et al., 2012; Zhang et al., 2020). Along with data showing Grn LoF microglia induce aberrant TDP-43 in cultured neurons (Zhang et al., 2020), these studies suggest microglial dysfunction may be pathogenic in patients. Since Grn−/− microglia share common features with other LSD models, including expression profiles typical of reactive microglia (Colombo et al., 2021; Evers et al., 2017; Gotzl et al., 2019; Ullman et al., 2020), lysosomal dysfunction may contribute to driving the hyperreactive state of Grn LoF microglia. Because lipofuscinosis is also a feature of PGRN-deficient neurons (Kao et al., 2017; Paushter et al., 2018), we hypothesize that correcting lysosomal dysfunction in both microglia and neurons in the CNS of GRN-FTD patients may slow or arrest disease progression.
Since GRN-FTD results from PGRN deficiency, a potentially attractive therapeutic strategy is a CNS-penetrant PGRN replacement therapy. Here, we capitalized on recently-engineered transport vehicles (TVs) that enable receptor-mediated transcytosis of large molecules across endothelial cells of the blood brain barrier (BBB) by binding to the human transferrin receptor apical domain (huTfR) (Kariolis et al., 2020; Ullman et al., 2020). We fused a TV to recombinant human PGRN, termed PTV:PGRN, to assess efficacy in correcting Grn LoF-associated phenotypes in vitro and in vivo. Our results uncover a relationship between PGRN and lysosomal lipid metabolism by defining the PGRN deficiency model as an atypical LSD associated with a deficiency of the lysosomal lipid bis(monoacylglycero)phosphate (BMP), leading to a secondary storage of GCase substrates. Importantly, our data show PTV:PGRN rescues a range of lysosomal defects, including BMP deficiency, GCase substrate accumulation, lysosomal vacuolization and endomembrane damage, as well as inflammation and neuronal damage. Our data provide a pre-clinical proof of concept supporting the use of PTV:PGRN for treatment of GRN-FTD.
RESULTS
Grn−/− brains show lysosomal lipid alterations
Previous liquid chromatography-mass spectrometry (LCMS) studies have shown lipid metabolism is altered in various Grn−/− models (Evers et al., 2017; Marschallinger et al., 2020). However, lipid panels previously assessed were limited in coverage of glycosphingolipids and other lipids often dysregulated in LSDs, such as BMP (also known as lysobisphosphatidic acid). To determine if Grn LoF is associated with profiles suggestive of LSDs, we performed targeted lipidomics and metabolomics on young (2 month) and old (13 month) Grn+/+ and Grn−/− mouse brain with LCMS. We selected Grn−/− rather than Grn+/− models as they best replicate the GRN-FTD pathologies (Kao et al., 2017). We found a decrease in various BMP species including BMP(22:6/22:6) and BMP(18:1/18:1) as the largest genotype-dependent changes (Fig. 1A–D). This anionic phospholipid is enriched in late endosomes/lysosomes, where it is believed to stimulate lysosomal lipases and cholesterol egress into the cytosol (Breiden and Sandhoff, 2020; Gruenberg, 2020; McCauliff et al., 2019). BMP showed an age effect, but no age-genotype interaction (Fig. 1C and 1D). Conversely, there was a genotype- and age-dependent increase in brain levels of glucosylsphingosine (GlcSph), a GCase substrate (Dai et al., 2016; Taguchi et al., 2017) (Fig. 1A,1B and 1E). Consistent with GCase substrate accumulation, Grn−/− brain showed a decrease in GCase activity, but only at 13 months (Fig. 1F). Grn−/− plasma, liver and urine also revealed a decrease in one or more BMP species, while Grn−/− plasma and liver exhibited a GlcSph increase (Fig. S1A–C). Overall, these data establish BMP deficiency and GlcSph accumulation as defining molecular features of Grn−/− mice and point to lysosomal dysfunction.
Figure 1. Grn−/− brains show lysosomal lipid dysregulation.

(A) Volcano plot of lipid and metabolites elevated (red) or reduced (blue) in frontal cortex from Grn−/− mice relative to age-matched Grn+/+ mice (2 and 13mo mice, log2 transformed, n=4/group). Plotted p-values adjusted for age and genotype interaction. Adjusted analyte values with an FDR < 5% and a fold change of >20% are labeled with filled squares (linear regression model with Benjamini-Hochberg multiple comparisons correction). Analytes with significant genotype differences prior to adjustment indicated with open squares. (B) Heatmap of all analytes with unadjusted p-values of <0.1. Columns represent individual mice. Plotted values are log2 fold transformed and normalized to mean of 2mo. Grn+/+ mice. (C-E) Characterization of genotype and age effects on frontal cortex levels of BMP(22:6/22:6) (C), BMP(18:1/18:1) (D) and glucosylsphingosine (GlcSph) (E). LC/MS area ratios normalized to mean 2mo. Grn+/+ area ratios (left, grey circles) for each analyte. (F) GCase activity in 2mo. vs. 13mo. Grn−/− mouse brain. For (C-E), Two-way ANOVA, Sidak’s multiple comparison was used (n=4/group). *p<0.05, ***p<0.001, ****p<0.0001 (unadjusted). Data represented as geometric mean±SEM. See also Fig. S1.
Next, we analyzed plasma and cerebrospinal fluid (CSF) from healthy controls, non-GRN FTD and GRN-FTD patients (Table S1). PGRN deficiency was validated in the biofluids of GRN mutation carriers (Fig. S1D–E). Neurofilament light chain (Nf-L) increase was also confirmed in those diagnosed with FTD (Fig. S1F–G), indicative of neurodegeneration (Ghidoni et al., 2008; Meeter et al., 2016). We found a subtle deficiency in CSF BMP(18:1/18:1) (Fig. S1H) and a decreasing trend of BMP(22:6/22:6) in GRN-FTD CSF (Fig. S1I), but not in plasma (Fig. S1J–K). Non-GRN FTD patients’ CSF also exhibited a mild reduction of BMP(18:1/18:1) (Fig. S1H), suggesting potential converging lysosomal defects in FTD patients. GlcSph was significantly increased in GRN-FTD plasma, but not in non-GRN FTD plasma (Fig. S1L), replicating the Grn−/− mouse plasma data. GlcSph was below the assay detection limit in human CSF. Despite the small effect size of BMP and GlcSph alterations, likely reflecting the fact GRN-FTD patients carry one functional copy of the GRN gene, these lysosomal lipids represent candidate clinical biomarkers in patients’ biofluids, though more patient samples need to be analyzed to confirm their utility.
PGRN regulates lysosomal function via BMP
To investigate a potential functional relationship between PGRN and BMP, murine bone marrow-derived macrophages (BMDMs) were stained for PGRN, LAMP-2 and BMP. PGRN was primarily found in lysosomes based on colocalization with LAMP-2 in Grn+/+ BMDMs (Fig. 2A). Super-resolution microscopy showed PGRN both on the limiting membrane and within the lumen of lysosomes, a subset of which also contained BMP. Like PGRN, BMP localized to the limiting membrane and lumen of lysosomes, as well as additional compartments in proximity to the plasma membrane (Fig. 2A and 2B, Fig. S2A–C). PGRN was virtually absent and the anti-BMP immunoreactivity was decreased by ~50% in Grn−/− BMDMs (Fig. 2C and 2D). LCMS showed a decrease in BMP species containing poly-unsaturated fatty acids (PUFAs), such as BMP(20:4/20:4) and BMP(22:6/22:6), replicating Grn−/− mouse tissue data (Fig. 2E). However, BMP(18:1/18:1) was uniquely upregulated by ~2.5-fold in mutant cells (p < 0.05; n = 3 biological replicates), in contrast with in vivo findings, suggesting Grn−/− cells are not defective in BMP synthesis.
Figure 2. PGRN regulates lysosomal function via control of BMP levels.

(A) Epifluorescence microscopy analysis of PGRN, BMP, and LAMP-2 in BMDMs showing no PGRN signal and reduced BMP signal in Grn−/− cells. Scale bar: 50 μm. (B) Super-resolution microscopy analysis of markers in (A) highlighting lysosomal localization of PGRN and BMP in Grn+/+ cells. Scale bar: 2 μm. (C,D) Levels of PGRN (C) and BMP (D) from immunofluorescence in (A) showing PGRN loss and reduced BMP in Grn−/− BMDMs. (n=3 independent experiments; Student’s t-test). (E) LCMS analysis of BMP species in BMDMs showing a decrease in PUFA-BMP levels in Grn−/− cells. (n=3 independent experiments; Student’s t-test). (F) Cartoon of liposome sedimentation assay with recombinant huPGRN and PC vs. BMP liposomes after incubation at acidic or neutral pH. (G) Coomassie Blue stain of PGRN after sedimentation and SDS-PAGE. (H) PGRN levels in pellet (bound) and supernatant (free) fractions from (G). (n = 4 independent experiments; one-way ANOVA, Dunnett’s multiple comparison). (I) Cartoon of liposome supplementation paradigm for the DQ-BSA assay in BMDMs. (J) Fluorescence microscopy of lysosomal proteolysis using the DQ-BSA assay after artificial coloring of the fluorescence intensity. Scale bar: 50 μm. (K) Quantification of the DQ-BSA fluorescence from (J) showing rescue of lysosomal proteolysis in Grn−/− BMDMs treated with BMP and PG liposomes. (n = 5 independent experiments; one-way ANOVA, Dunnett’s multiple comparison). (L) Cartoon of liposome supplementation paradigm to assess GCase activity in brain homogenate. (M) Quantification of GCase activity in detergent-free 6–7 mo. brain extracts showing rescue of GCase activity in Grn−/− extracts treated with BMP and PG liposomes. (n = 4 mice, representative of 2 independent experiments; one-way ANOVA, Dunnett’s multiple comparison). #p<0.1, *p<0.05, **p<0.01, ****p<0.0001. Data shown as geometric mean±SEM. See also Fig. S2.
Given the partial PGRN/BMP colocalization in endolysosomes, we tested if PGRN physically binds to BMP with a liposome sedimentation assay. Liposomes containing phosphatidylcholine (PC) or PC supplemented with BMP(22:6/22:6) were incubated with recombinant human PGRN at neutral (7) or acidic (4.5) pH, prior to sedimentation (Fig. 2F). PGRN bound selectively to BMP liposomes, but only at acidic pH (Fig. 2G and 2H), suggesting it undergoes a conformational change or protonation under conditions mimicking the acidic environment of lysosomes, allowing for an interaction with anionic lipid BMP.
Next, we investigated the functional consequence of Grn LoF-associated BMP dysregulation in BMDMs. Grn−/− BMDMs showed a ~50% decrease in DQ-BSA puncta fluorescence relative to controls, indicating defects in lysosomal proteolysis (p < 0.01; n = 3 biological replicates). To assess if alteration of BMP levels underlie these defects in Grn−/− BMDMs, serum-deprived BMDMs were treated with liposomes containing PC with various test lipids, including BMP(22:6/22:6), phosphatidylglycerol (PG) (i.e., the precursor of BMP), or phosphatidylserine (PS) (i.e., a control anionic phospholipid) using pure PC liposomes as a control (Fig. 2I). LCMS showed BMP(22:6/22:6) liposomes increase cellular levels of the corresponding BMP species, while control lipids PC and PS did not (Fig. S2D). However, BMP(22:6/22:6) liposomes surprisingly decreased BMP(20:4/20:4) levels (Fig. S2E). BMP and PG liposomes significantly increased DQ-BSA fluorescence in Grn−/− BMDMs, indicating lysosomal proteolysis rescue (Fig. 2J and 2K). BMP liposomes increased levels of mature CatD, but not CatD precursor, in Grn−/− KO BMDMs, where levels were slightly elevated at baseline (Fig. S2F–2H). Our data thus suggest BMP is a positive regulator of lysosomal proteolysis.
BMP has been shown to stimulate GCase activity via saposin C in cell-free assays by enhancing electrostatic interactions between the anionic lipid and protonated saposin C and GCase proteins (Abdul-Hammed et al., 2017). We thus tested if BMP liposomes regulate GCase activity in a more physiological context, providing a potential mechanistic link between BMP deficiency and accumulation of GCase substrate, GlcSph. Total GCase activity measurements in detergent-free mouse brain extracts incubated with liposomes of different composition showed reduced GCase activity, but no change in GCase protein levels, in Grn−/− extracts relative to controls and a stimulatory effect of PC-only liposomes on GCase activity in both genotypes (Fig. 2L, 2M, S2I and 2J). BMP liposomes rescued GCase activity, while PG liposomes only showed a trend and PS liposomes had no effect, compared to PC liposomes (Fig. 2M). BMP- and PG-containing liposomes also stimulated GCase activity in Grn−/− liver extracts, where GCase protein levels were also normal (Fig. S2K and S2L). BMP liposomes were ineffective on GCase activity in the presence of detergent, indicating that intact bilayers are required for this effect (data not shown). This highlights BMP is a positive regulator of GCase activity and suggests BMP deficiency underlies GCase activity defects and thus accumulation of substrate, GlcSph, in Grn−/− mice.
Generation and characterization of the candidate biotherapeutic PTV:PGRN
We recently developed a BBB transport vehicle (TV) consisting of an engineered Fc domain that binds to huTfR and enhances CNS penetrance and pharmacodynamic responses of large molecules, including a lysosomal enzyme, in mice expressing huTfR (Kariolis et al., 2020; Ullman et al., 2020). Here, we developed an analogous platform, referred to as protein transport vehicle:PGRN (PTV:PGRN), to enhance brain uptake of recombinant huPGRN, using huPGRN fused to a normal Fc as a control (Fc:PGRN) (Fig. 3A). PGRN was fused to the TV C-terminus, leaving its own C-terminus free to interact with sortilin, a key receptor for uptake and lysosomal delivery of PGRN (Hu et al., 2010). After purifying PTV:PGRN and Fc:PGRN to homogeneity (Fig. S3A), PTV:PGRN affinity to huTfR was measured to be ~130 nM. Both PTV:PGRN and Fc:PGRN retained the ability to bind to human sortilin and another key ligand, prosaposin, with affinities of ~2 nM and ~5 nM, respectively (Fig. S3B and S3C).
Figure 3. PTV:PGRN rescues lysosomal and inflammatory phenotypes in Grn−/− BMDMs.

(A) Architecture of PTV:PGRN fusion protein, showing the huTfR binding site in the Fc domain (orange) linked to huPGRN. (B-C) Epifluorescence (B) and super-resolution microscopy (C) representative images of PGRN, LAMP-2, and BMP in untreated Grn+/+ and Grn−/− BMDMs or Grn−/− BMDMs treated with 50nM PTV:PGRN for 72h. PTV:PGRN restores PGRN and BMP immunoreactivity in mutant cells. Scale bar: 25 μm (B), 2 μm (C). (D-E) Levels of PGRN (D) and BMP (E) from immunofluorescence in (B) showing rescue in Grn−/− BMDMs after treatment with PTV:PGRN, but not IgG control. (n = 3 independent experiments; one-way ANOVA, Dunnett’s multiple comparison). (F) Fluorescence microscopy of lysosomal proteolysis using the DQ-BSA assay. PTV:PGRN, but not IgG control, rescued the decrease in DQ-BSA fluorescence, and thus lysosomal proteolysis, in Grn−/− BMDMs. Artificial coloring of fluorescence intensity was used. Scale bar: 50 μm. (G) Quantification of the DQ-BSA fluorescence from (F) showing rescue of lysosomal proteolysis in the Grn−/− BMDMs with PTV:PGRN, but not IgG control. (n = 4 independent experiments; one-way ANOVA, Dunnett’s multiple comparison). (H) Lysosomal pH measurements showing rescue of alkalinized pH in the Grn−/− BMDMs treated with PTV:PGRN, but not IgG control. (n = 3 independent experiments; one-way ANOVA, Dunnett’s multiple comparison). (I) Fluorescence microscopy of reactive oxygen species (ROS) using the DCFDA assay. PTV:PGRN, but not IgG control, rescued the increase in DCFDA fluorescence, and thus ROS production, in Grn−/− BMDMs. Scale bar: 25 μm. (J) Quantification of the DCFDA fluorescence from (I) showing rescue of ROS levels in the Grn−/− BMDMs treated with PTV:PGRN, but not IgG control. (n = 3 independent experiments; one-way ANOVA, Dunnett’s multiple comparison). (K) Quantification of BMP (20:4/20:4) levels showing a decrease in both Grn+/+ and Grn−/− after treatment with oxLDL, but not vehicle control. (n = 3 independent experiments; two-way ANOVA, Tukey’s multiple comparison). (L) Quantification of soluble TREM2 levels in conditioned media from BMDM analyzed in (J) showing an increase in Grn−/− BMDMs and a rescue after PTV:PGRN treatment, but not control IgG. (n = 3 independent experiments; one-way ANOVA, Dunnett’s multiple comparison). *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Data shown as geometric mean±SEM. See also Fig. S3.
PTV:PGRN rescues lysosomal and inflammatory phenotypes in Grn−/− BMDMs
To test if PTV:PGRN can restore PGRN levels in Grn−/− cells, BMDMs expressing a chimeric human/mouse TfR that harbors the human TfR apical domain (TfRmu/hu mice) were used. Immunostainings showed PTV:PGRN is taken up in Grn−/− BMDMs, based on PGRN immunoreactivity in LAMP-2 positive compartments with standard (Fig. 3B) and super-resolution (Fig. 3C) microscopy, with a similar pattern to endogenous protein in Grn+/+ BMDMs (Fig. 2B). Consistent with correct subcellular targeting, PTV:PGRN, but not IgG control, rescued PGRN protein levels and BMP deficiency (Fig. 3B–E). Further, PTV:PGRN corrected defects in lysosomal proteolysis and lysosomal acidification (Fig. 3F–3H), indicating that PTV:PGRN can rescue key aspects of lysosomal function.
Grn−/− BMDMs also showed higher levels of reactive oxygen species (ROS), which were rescued with PTV:PGRN (Fig. 3I–J). Next, we hypothesized the reduction in PUFA-BMP species in Grn−/− cells may reflect increased lysosomal oxidative stress, consistent with ROS increase and lipofuscin pathology observed in Grn LoF mice and patients (Kao et al., 2017). Treatment of BMDMs with oxidized LDL (oxLDL), which increases oxidative stress upon internalization based on increased oxidized PC immunoreactivity detected in oxLDL-treated BMDMs (Fig. S3D), decreased PUFA-BMP in Grn+/+ BMDMs and exacerbated the PUFA-BMP depletion phenotype in Grn−/− BMDMs (Fig. 3K and S3E). These data indicate PGRN may stabilize PUFA-BMP levels in lysosomes by preventing BMP oxidation, thus acting as an antioxidant.
Finally, Grn−/− myeloid cells have been shown to have higher levels of the immune receptor TREM2 (Gotzl et al., 2019). Similarly, Grn−/− BMDMs showed higher cellular and secreted TREM2 levels, both of which were rescued with PTV:PGRN (Fig. 3L and S3F). PTV:PGRN can thus rescue lysosomal dysfunction and a key innate immune phenotype in Grn−/− BMDMs.
PTV:PGRN rescues lysosomal membrane damage in human GRN−/− iPSC-microglia
Severe microgliosis and CNS inflammation may be pathogenic drivers in GRN-FTD patients (Gotzl et al., 2019; Kao et al., 2017; Lui et al., 2016; Zhang et al., 2020). We thus tested PTV:PGRN in human GRN−/− iPSC-derived microglia (iMG), a disease-relevant cellular model. Treatment of two independent GRN−/− iMG clones with PTV:PGRN restored cellular levels of PGRN, comparable to endogenous PGRN in GRN+/+ iMG (Fig. 4A and 4B). Based on super-resolution microscopy, GRN−/− iMG exhibited more vacuolated LAMP-1 positive endolysosomal compartments, which was rescued with PTV:PGRN (Fig. 4C). DQ-BSA filled these enlarged vacuoles in GRN−/− iMG and PTV:PGRN rescued this phenotype in both mutant clones (Fig. 4D and 4E). Time-lapse DQ-BSA imaging revealed more homotypic vacuolar fusion events as well as abrupt loss of DQ-BSA fluorescence from these vacuoles (Fig. S4A), reflecting either a fusion of these vacuoles with the plasma membrane or a physical rupture. To assess the latter, we stained for galectin-3 (Gal-3), a marker of endolysosomal membrane damage (Papadopoulos et al., 2020; Yim and Mizushima, 2020). Quantification of Gal-3-positive cells and fluorescence intensity showed a 4-fold and 2-fold increase in the two GRN−/− iMG clones, respectively (Fig. 4F, 4G and S4B). Super-resolution microscopy showed Gal-3 fluorescence is increased in close proximity to the LAMP-1 compartment in a greater proportion of GRN−/− iMG, indicative of endomembrane damage (Fig. 4H and 4I). Gal-3-positive GRN+/+ iMG showed similar Gal-3/LAMP-1 colocalization pattern, but the frequency of Gal-3 positive cells was much lower than in GRN−/− iMG (Fig. 4G; not shown). PTV:PGRN rescued the Gal-3 phenotype in mutant iMG, indicating it can preserve the physical integrity of endolysosomes (Fig. 4F, 4G and S4B). Thus, PTV:PGRN can rescue lysosomal phenotypes in a CNS and disease-relevant human cell type.
Figure 4. PTV:PGRN rescues endolysosomal vacuolization and endomembrane damage in human GRN−/− iMG.

(A) Epifluorescence microscopy of PGRN in GRN+/+ and GRN−/− human iPSC-derived microglia (iMG) treated with 100 nM control IgG or PTV:PGRN each day for 3 days. PTV:PGRN restores PGRN in mutant cells. Scale bar: 25 μm. (B) Quantification of PGRN levels in (A) showing rescue of the protein in two independent GRN−/− iMG clones after treatment with PTV:PGRN. NT, no treatment. (n = 3 independent experiments; one-way ANOVA, Dunnett’s multiple comparison). (C) Super-resolution microscopy of PGRN and LAMP-1 in GRN−/− iMG showing PTV:PGRN restores PGRN localization in the lysosomes and corrects the vacuolization phenotype. Insets: higher magnification of lysosomes with single channels and merge. Scale bar: 10 μm. (D) Fluorescence microscopy of DQ-BSA fluorescence in GRN+/+ and GRN−/− iMG showing PTV:PGRN rescues the vacuolization of GRN−/− iMG. Images show increase in fraction of cells containing one or more large vacuoles filled with the fluorescent dye GRN−/− iMG. Scale bar: 25 μm. (E) Quantification of % cells with DQ-BSA-filled vacuoles GRN+/+ and GRN−/− iMG with no treatment (NT) or after treatment with IgG control or PTV:PGRN. (n = 3 independent experiments; one-way ANOVA, Dunnett’s multiple comparison). (F) Epifluorescence microscopy of galectin-3 (Gal-3) in iMG showing an increase in endolysosomal membrane damage, as denoted by the increased Gal-3 fluorescence, in GRN−/− iMG and a rescue with PTV:PGRN. Scale bar: 25 μm. (G) Quantification of Gal-3 positive GRN+/+ and GRN−/− iMG with no treatment (NT) or after a treatment with IgG control or PTV:PGRN. (n = 4 independent experiments; one-way ANOVA, Dunnett’s multiple comparison). (H) Super-resolution microscopy of Gal-3 and LAMP-1 in GRN−/− iMG. Insets: higher magnification of lysosomes with single channels and merge. Scale bar: 10 μm. (I) Quantification of colocalization of Gal-3 and LAMP-1 from (H) using Mander’s overlap coefficients. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Data shown as geometric mean±SEM. See also Fig. S4.
PTV:PGRN exhibits enhanced brain biodistribution in Grn−/−; TfRmu/hu mice
We next tested if peripherally-administered PTV:PGRN exhibits more efficient brain uptake than non-TfR binding recombinant Fc:PGRN. Our previous studies described the TfRmu/hu KI mice (Kariolis et al., 2020; Ullman et al., 2020), which enable assessment of our huTfR binding biologics. TfRmu/hu KI mice crossed to Grn−/− mice (Grn−/− x TfRmu/hu KI) were dosed intravenously (IV) with 5 or 50 mg/kg of PTV:PGRN or Fc:PGRN (Fig. 5A). Drug concentrations were determined using a dual ELISA to detect the Fc portion of the fusion protein (Fc/Fc) (Fig. 5B–D), or the intact molecule (Fc/PGRN) (Fig. S5A–D). Until 4h post-dose, plasma concentrations were similar between PTV:PGRN and Fc:PGRN (Fig. 5B and S5A) with PTV:PGRN showing lower concentration in plasma over time due to huTfR mediated clearance. In contrast, PTV:PGRN was ~3–10 fold greater in the brain (Fig. 5C and S5B) and ~1.25–1.5 greater in CSF at both 4 and 24h post-dose (Fig. 5D). As expected, liver uptake was comparable for both PTV:PGRN and Fc:PGRN (Fig. S5D).
Figure 5: PTV:PGRN increases brain uptake and lysosomal lipid rescue in Grn−/−; TfRmu/hu mice relative to Fc:PGRN.

(A) Schematic of 24h biodistribution study in 3mo Grn−/−; TfRmu/hu mice. (B-D) Concentration of Fc domain in plasma (B), brain (C) and CSF (D) as determined by Fc sandwich ELISA (n=4/group, Two-way ANOVA with Sidak’s multiple comparison test). (B) PTV:PGRN (dark orange = 50 mg/kg, light orange = 5mg/kg) is cleared more rapidly from plasma than Fc:PGRN (dark blue = 50mg/kg, light blue = 5mg/kg) at both dose levels (50mg/kg: padj<0.0001, 5mg/kg: padj<0.0001). (C) Brain PTV:PGRN concentrations are higher at either dose than 50mg/kg Fc:PGRN (50mg/kg PTV:PGRN: padj<0.0001, 5mg/kg PTV:PGRN: padj<0.001). (D) CSF Fc concentrations are greater for PTV:PGRN than Fc:PGRN at 50mg/kg dose (50mg/kg: padj<0.01, 5mg/kg: padj=0.3946). (E) Capillary depletion allows for separation of parenchymal and vascular brain fractions. (F) The parenchymal:vascular ratio of drug levels is increased by PTV:PGRN at 5 and 50mg/kg doses (n=4/group, Two-way ANOVA with Sidak’s multiple comparison test, 50mg/kg: padj<0.0001, 5mg/kg: padj<0.0001). (G) Representative fluorescence image 3D projections of thalamic huIgG (magenta) distribution 24h after a 50mg/kg IV dose of Fc:PGRN (left) or PTV:PGRN (right). Neuronal (NEUN), microglial (IBA1) and vascular (podocalyxin/CD31) compartments also labeled (scale bar: 15 μm). Inset: single cells with intracellular huIgG uptake. Scale bar: 5μm. (H) Relative brain abundance of BMP(22:6/22:6) 24h after 5mg/kg dose. PTV:PGRN increases huIgG concentrations vs. Fc:PGRN (n=4/group; one way ANOVA, Dunnett’s multiple comparison). (I) CSF BMP (22:6/22:6) in Grn−/−; TfRmu/hu mice treated with PTV:PGRN vs. Fc:PGRN. Only PTV:PGRN significantly increases BMP levels (n=4/group; one way ANOVA, Dunnett’s multiple comparison). (J) Correlation of brain and CSF BMP(22:6/22:6) levels in PTV:PGRN-treated mice (n=8, linear regression). (K) Schematic of 6-week repeat dosing study in 7mo Grn−/−; TfRmu/hu mice. (L) Rescue of brain BMP(22:6/22:6) by Fc:PGRN and PTV:PGRN. (n=7–10/group; one way ANOVA, Dunnett’s multiple comparison). (M) Rescue of brain GlcSph by Fc:PGRN and PTV:PGRN. (n=7–10/group; Kruskal-Wallis with Dunn’s post-hoc multiple comparisons). (N-P) Volcano plots of lipid and metabolite changes in Grn−/− vs. Grn+/+ mice (N), Fc:PGRN-treated (O), and PTV:PGRN-treated Grn−/− mice (P). **p<0.01, ***p<0.001, ****p<0.0001. Data shown as mean±SEM. See also Fig. S5.
To confirm PTV:PGRN reaches the brain parenchyma, we used a capillary-depletion method to separate vascular and parenchymal brain fractions (Fig. 5E) (Kariolis et al., 2020). The parenchymal:vascular ratio of PTV:PGRN rose over 24h unlike Fc:PGRN that largely saturated at ~5% by 4h post-dose, regardless of dose level (Fig. 5F). Parenchymal PTV:PGRN concentrations were higher, suggesting the increased parenchymal:vascular ratio reflects rapid drug clearance from the vascular fraction (Fig S5C). We then assessed delivery of PTV:PGRN to CNS cells using super-resolution microscopy. Anti-huIgG immunoreactivity was found in the vasculature as well as brain parenchyma, where it localized to the soma of NeuN- and IBA1-positive cells, highlighting PTV:PGRN delivery to neurons and microglia, respectively. In contrast, Fc:PGRN staining was less abundant in vascular, neuronal and microglial compartments (Fig. 5G). We conclude PTV:PGRN is delivered more efficiently to the brain parenchyma than Fc:PGRN.
PTV:PGRN rescues lysosomal lipids in the CNS of Grn−/−; TfRmu/hu mice
We next tested if PTV:PGRN shows greater rescue of lysosomal lipids than Fc:PGRN in young (3 month-old) Grn−/−; TfRmu/hu brains. At 4h post IV dosing, no rescue of brain or CSF BMP levels was observed by PTV:PGRN or Fc:PGRN. By 24h, 5mg/kg PTV:PGRN restored BMP(22:6/22:6) to ~80% of WT levels, while Fc:PGRN had negligible effects (Fig. 5H). In contrast, BMP(22:6/22:6) levels were corrected by both Fc:PGRN and PTV:PGRN as early as 4h post-dose in mutant liver and plasma (Fig. S5E and S5F). Correction of CSF BMP(22:6/22:6) levels by PTV:PGRN vs. Fc:PGRN at 24h followed the same pattern to that observed in the brain (Fig. 5I), further supported by linear regression analysis (Fig. 5J). GlcSph levels were not corrected by 24h with either dose level of PTV:PGRN or Fc:PGRN in the brain and liver (Fig. S6G and S6H), consistent with the notion GlcSph storage is secondary to BMP deficiency, likely requiring longer dosing for correction. However, there was partial correction of plasma GlcSph with both fusion proteins at 24h (Fig. S6I).
To test if PTV:PGRN preferentially rescues brain BMP and GlcSph with a multi-dose treatment paradigm, young (4 month-old) Grn−/−; TfRmu/hu mice were dosed intraperitoneally (IP) at 5mg/kg PTV:PGRN or Fc:PGRN, weekly for 6 weeks (Fig. 5K). Plasma drug concentrations measured 1 week after the final dose showed lower PTV:PGRN levels than Fc:PGRN, consistent with hTfR-mediated clearance (Fig. S5J). Chronic treatment with PTV:PGRN, but not Fc:PGRN, significantly rescued brain BMP(22:6/22:6) and GlcSph (Fig. 5L and 5M). This was corroborated by volcano plots showing multiple species of BMP downregulated in Grn−/− brain (Fig. 5N), virtually unchanged by Fc:PGRN (Fig. 5O), but significantly corrected by PTV:PGRN (Fig. 5P). In contrast, both Fc:PGRN and PTV:PGRN fully rescued BMP(22:6/22:6) and GlcSph in the liver (Fig. S5K–L). PTV:PGRN elevated BMP(22:6/22:6) in plasma (Fig. S5M). Both fusion proteins rescued GlcSph in plasma (Fig. S5N). Our data indicate multi-dose treatment with PTV:PGRN corrects lysosomal lipid anomalies in Grn−/−; TfRmu/hu CNS.
PTV:PGRNv2 chronic treatment rescues lipid anomalies in key CNS cell types
PTV:PGRN was stably expressed in CHO cells to generate sufficient material for larger scale in vivo studies. However, CHO expression resulted in C-terminal truncation and loss of sortilin binding (data not shown). To increase PTV:PGRN stability, we replaced the C-terminal amino acids of human PGRN (QLL) with corresponding amino acids from orthologous PGRN sequences and analyzed the stability of each new variant (Table S2). The engineered variant with the most stable C-terminus was termed PTV:PGRNv2, and had comparable stability and binding affinity to sortilin and PSAP relative to PTV:PGRN expressed in Expi293 cells (Table S2 and S3, Fig. S3G and S3H). Importantly, PTV:PGRNv2 showed similar rescue of brain BMP(22:6/22:6) levels 2 weeks following IV dosing, relative to PTV:PGRN (Fig S6A). To determine the minimum PTV:PGRNv2 dose required to rescue BMP and GlcSph and the effect duration, we treated Grn−/− x TfRmu/hu KI mice with a single IV dose of drug at 1, 2.5, or 5 mg/kg for 2, 3 and 6 weeks (Fig. 6A). At 5 mg/kg, partial rescue of BMP(22:6/22:6) was observed at 6 weeks. At 2 weeks, 1 mg/kg rescued BMP back to WT levels (Fig. 6B). Remarkably, 1mg/kg of PTV:PGRNv2 was sufficient to partially rescue GlcSph at the 6 week timepoint (Fig 6C). GCase activity was also restored by 5mg/kg PTV:PGRNv2 two weeks post dose (Fig. 6D). PTV:PGRNv2 treatment thus generates both a rapid and sustained rescue of lysosomal lipids in Grn−/−; TfRmu/hu CNS.
Figure 6: PTV:PGRNv2 rescues lipid anomalies across CNS cell types.

(A) Schematic of single dose, 6-week IV dose titration study in 4mo. mice. (B-C) Time course of PTV:PGRNv2 driven rescue of BMP(22:6/22:6) (1mg/kg: padj<0.0001) (B) or GlcSph (1mg/kg: padj<0.0001) (C) in Grn−/−; TfRmu/hu at 5mg/kg (dark green), 2.5mg/kg (medium green) or 1mg/kg (light green) dose (n=6/group, Two-way ANOVA with Sidak’s multiple comparison test). (D) Rescue of GCase activity 2 weeks following IV dosing of 5mg/kg PTV:PGRNv2 (n=6/group; one way ANOVA, Dunnett’s multiple comparison). (E) Schematic of 8 week repeat dosing study with weekly or every other week (EOW) PTV:PGRNv2 starting at 7mo. in Grn−/−; TfRmu/hu mice (n=7–10/group). (F-G) Rescue of BMP(22:6/22:6) in brain (F) and CSF (G) (Kruskal-Wallis with Dunn’s post-hoc multiple comparison (F), and one way ANOVA, Dunnett’s multiple comparison, respectively). (H) Rescue of brain GlcSph (one way ANOVA, Dunnett’s multiple comparison). (I) Restoration of brain GCase activity (one way ANOVA, Dunnett’s multiple comparison). (J) Heatmap of the top differentially regulated lipids and metabolites in cell-type enriched populations. Analyte abundance normalized to Grn+/+; TfRmu/hu (left). Columns represent biological replicates (n=7–10/group). (K-M) Bar plots of BMP(18:1/18:1) (K), BMP(22:6/22:6) (L), and GlcCer(18:1/18:0) (M) from all three cell types, demonstrating rescue across CNS cell types with both weekly (Wkly) and EOW PTV:PGRNv2 treatment (Kruskal-Wallis with Dunn’s post-hoc multiple comparison (K, M (microglia)), or one way ANOVA with Dunnett’s multiple comparison, (M, L)). #p<0.1, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Data shown as mean±SEM. See also Fig. S6.
To test if PTV:PGRNv2 can alter disease-associated endpoints, we treated older (7 month-old) Grn−/−; TfRmu/hu mice, which exhibit more robust phenotypes (Zhang et al., 2020) with weekly or every other week (EOW) 5mg/kg IP administered doses for 8 weeks (Fig. 6E). We confirmed IP dosing does not alter efficacy by demonstrating equivalent BMP(22:6/22:6) rescue at 1 and 2 weeks post-dosing with 5mg/kg PTV:PGRNv2 (Fig. S6B). PTV:PGRNv2 is human sequence-derived and may trigger an immune response in mice. We thus co-treated mice EOW with an IP 0.5mg dose of anti-mouse CD4 antibody, which causes targeted immunosuppression and prevents anti-drug antibodies from interfering with drug exposure or efficacy (Weber et al., 2018). To confirm that a CD4 co-administration does not interfere with phenotypes of interest, we dosed a separate cohort of Grn−/− x TfRmu/hu KI mice with an anti-CD4 antibody EOW for 8 weeks and found no modification of lipids or gliosis typical of Grn−/− mice (Fig. S6C–H). For these studies, we also developed a quantitative LCMS assay (“tier 2”) to measure absolute BMP concentrations. Both weekly and EOW treatments with PTV:PGRNv2 fully rescued brain and CSF BMP(22:6/22:6) levels, brain GlcSph levels, and brain GCase activity after 8 weeks (Fig. 6F–6I).
To determine if PTV:PGRNv2 corrects lipid anomalies across CNS cell types, we performed lipidomics on sorted microglia, astrocytes and neurons, after confirming enrichment for the respective cell types by qPCR (Fig. S6I). LCMS data showed Grn−/− microglia lipids were more profoundly affected than astrocytic and neuronal lipids (Fig. 6J), confirming results from others suggesting microglia are more severely impacted by Grn LoF (Zhang et al., 2020). Consistent with a global depletion of BMP in Grn−/− mice, one or more BMP species were downregulated in all three CNS cell types (Fig. 6J–6L). Sorted Grn−/− microglia also showed a robust upregulation of glucosylceramide (GlcCer) species, another GCase substrate, consistent with a reduction in lipase activity (Fig. 6J and 6M). A mild increase in GCase substrate was also found in astrocytes, but no changes were observed in neurons (Fig. 6J and 6M). Unfortunately, GlcSph was below the lower limit of detection in sorted cells with our current methodology. Lipid changes were significantly rescued with weekly and EOW PTV:PGRNv2 treatments (Fig. 6I–6M), indicating PTV:PGRN rescues lipid dysregulation across different CNS cell types.
To test if lysosomal lipid anomalies in the Grn−/− mice are seen in another Grn LoF model and are treatable with PTV:PGRNv2, we crossed a mouse harboring homozygous mutations in the Grn gene (R504X), homologous to the common R493X GRN-FTD disease mutation (Nguyen et al., 2018), onto the TfRmu/hu background. Brain BMP and GlcSph phenotypes were comparable to those found in Grn−/− mice and IV dosing of 5mg/kg PTV:PGRNv2 in Grn R493X; TfRmu/hu mice restored normal lipid levels after 7 days (Fig. S6K–6M). Total TREM2 levels were also evaluated in the Grn R493X; TfRmu/hu mouse brain, and showed a significant, but milder TREM2 elevation than in the Grn−/− mouse model. Treatment with 5mg/kg PTV:PGRNv2 did not rescue TREM2 7 days after a single dose (Fig. S6N). Together, these data support the treatment potential of PTV:PGRN, but that drug concentration, dose frequency and treatment duration may impact efficacy in various Grn LoF models.
PTV:PGRNv2 rescues disease pathology in Grn−/−; TfRmu/hu mice
To understand PTV:PGRNv2 impact on CNS inflammation and gliosis, brain sections were stained for IBA1 (total microglia), CD68 and C1q (reactive microglia), and GFAP (reactive astrocytes) (Fig. 7A–7F). Each glial marker was quantified in the thalamus, a vulnerable brain region in Grn−/− mice (Lui et al., 2016; Zhang et al., 2020). PTV:PGRNv2 – dosed weekly or EOW – significantly rescued IBA1, CD68, C1q and GFAP elevation. Super resolution imaging revealed a vesicular expression pattern of CD68 in the Grn−/− mouse thalamus, consistent with lysosomal localization, that is corrected by PTV:PGRNv2 (Fig. S7A). We also found an increase in total brain TREM2 levels by MSD immunoassay in mutant mice, which was normalized by PTV:PGRNv2 (Fig. 7F). Similarly, TREM2 levels were corrected in the liver and plasma of Grn−/−; TfRmu/hu mice, consistent with a rescue of peripheral inflammation (Fig. S7B and S7C). These findings highlight PTV:PGRNv2 ability to rescue Grn LoF-associated CNS gliosis, which is a key disease manifestation in GRN-FTD patients (Lui et al., 2016).
Figure 7: PTV:PGRNv2 rescues gliosis, lipofuscin and neurodegeneration.

(A-F) Representative images and quantification of markers of microgliosis (IBA1), astrogliosis (GFAP) and reactive microglia (CD68, and C1Q), in thalamus of 9mo. Grn−/−; TfRmu/hu mice after weekly or EOW dosing with PTV:PGRNv2 for 8 weeks, as shown in Fig. 6E (n=6–10/group; one way ANOVA, Dunnett’s multiple comparison). (A) Low (left) and high (right) magnification images of thalamic gliosis markers after weekly PTV:PGRNv2 treatment. Left scale bar: 500 μm, right scale bar: 20μm. (B) Low (left) and high (right) magnification images of C1Q expression in thalamus and rescue with weekly PTV:PGRNv2 treatment. Left scale bar: 500 μm, right scale bar: 10μm. Increased total gliosis as measured by IBA1 (C), astrogliosis marker GFAP (D), and reactive microgliosis markers CD68 (E) and C1Q (F) are rescued by PTV:PGRNv2. (G) Reduction of total brain TREM2 levels measured by MSD in Grn−/−; TfRmu/hu by PTV:PGRNv2. (H-I) Representative images (H) and quantification (I) of thalamic lipofuscin in Grn−/−; TfRmu/hu and rescue by PTV:PGRNv2 (n=7–10/group; one way ANOVA, Dunnett’s multiple comparison). (J) Reduced CSF Nf-L levels by PTV:PGRNv2 in Grn−/−; TfRmu/hu mice (n=6–9/group; one way ANOVA, Dunnett’s multiple comparison). (K) Model for PGRN deficiency driven lysosomal dysfunction. Under normal conditions, PGRN is delivered to lysosomes via interaction with sortilin (or indirectly via prosaposin, not shown). There, it is proteolytically cleaved into granulin peptides. PGRN and/or GRN peptides stabilize BMP via both direct physical interaction and prevention of ROS mediated oxidation. This preserves the stimulatory function of BMP toward GCase activity, possibly via saposin C peptides (not shown). In the context of Grn LoF, lysosomal PGRN and GRN peptide deficits result in destabilization of BMP, and vulnerability of lysosomal limiting membrane to oxidative damage, ultimately leading to membrane permeabilization and proton leak. Additionally, depletion of BMP disrupts the interaction of GCase enzyme with the surface of internal vesicles. Impairing lipase activity and driving accumulation of the Gcase substrate GlcSph. #p<0.1, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Data shown as mean±SEM. See also Fig. S7.
Lipofuscinosis and neuronal damage/loss are also salient pathological features in cortices of GRN-FTD patients and aged Grn−/− mouse brain (Lui et al., 2016; Ward et al., 2017; Zhang et al., 2020). To assess lipofuscinosis, we quantified autofluorescence in the thalamus and found weekly and EOW treatments with PTV:PGRNv2 significantly reduced this lysosomal pathology (Fig. 7H and 7I). Finally, treatments also reduced Nf-L levels in Grn−/− CSF (Fig. 7J), suggesting that PTV:PGRNv2 is neuroprotective. In conclusion, our candidate biotherapeutic can rescue lysosomal dysfunction and disease-relevant pathology in the brain of Grn−/−; TfRmu/hu mice upon chronic dosing.
DISCUSSION
Our data provide an in vivo proof of concept that a brain-penetrant PGRN biologic administered peripherally is efficacious in pre-clinical models of Grn LoF, identifying a promising therapeutic avenue for GRN-FTD. Our study also uncovered a key role of PGRN in lysosomal lipid metabolism, including BMP homeostasis, regulation of GCase activity, and GCase substrate levels (i.e., GlcSph), as well as preservation of lysosomal membrane integrity. Mechanistic studies revealed PGRN physically binds to and likely stabilizes BMP. This establishes Grn LoF models as atypical lysosomal storage disorders, where the primary defect is a lipid deficiency (i.e., BMP) rather than an accumulation and the secondary defect is a storage of GCase substrate. Finally, we identified candidate clinical biomarkers, CSF BMP and plasma GlcSph, which may help to evaluate the therapeutic efficacy of PTV:PGRN and other therapeutics in GRN-FTD patients.
Human genetics established GRN-FTD results from reduced levels or function of PGRN (Baker et al., 2006; Cruts et al., 2006). However, the pathogenic mechanisms underlying neurodegeneration are largely unclear. Observations that intracellular PGRN localizes to lysosomes in a sortilin- or PSAP-dependent fashion and that homozygous GRN mutations cause NCL pointed to a PGRN role in neuronal lysosomes in the CNS. Functional interactions with other lysosomal proteins such as CatD, prosaposin, GCase, and TMEM106B, all which are genetically linked to neurodegenerative disorders, provided additional clues that Grn LoF alters lysosomal function (Kao et al., 2017; Paushter et al., 2018). Our LCMS data identifying key lysosomal lipid phenotypes strengthened the rationale for drawing analogies between LSDs and GRN-FTD, and as such, for devising a therapeutic strategy akin to enzyme replacement therapy (ERT). However, a major limitation of ERT for CNS disorders has been the restriction of biotherapeutics’ access to the CNS by the BBB.
In this study, we employed a large molecule transport vehicle to enhance brain uptake of biologics via TfR-mediated trancytosis across brain endothelial cells at the BBB (Kariolis et al., 2020; Ullman et al., 2020). We showed that PTV:PGRN, a recombinant protein with engineered huTfR binding Fc fused to huPGRN, rescues various cellular dysfunctions and disease endpoints in Grn−/−; TfRms/hu mouse brain, including gliosis, lipofuscinosis and neurodegeneration. Our unbiased LCMS analyses of Grn−/− mice identified a ubiquitous and largely age-independent deficiency of anionic phospholipid BMP, which is enriched in the limiting membrane and internal vesicles of late endosomes/lysosomes (Fig. 7I). BMP has been proposed to facilitate LDL-derived cholesterol egress from endolysosomes, the biogenesis of intraluminal vesicles and sphingolipid catabolism in these organelles (Breiden and Sandhoff, 2020; Gruenberg, 2020; McCauliff et al., 2019). BMP is also secreted in exosomes (Miranda et al., 2018), potentially explaining its presence in plasma, CSF and urine. BMP elevation is often seen in tissues and biofluids from LSD preclinical models or patients, where it is considered a biomarker of lysosomal dysfunction (Showalter et al., 2020). BMP upregulation is thought to be a beneficial, though not always effective, homeostatic response counteracting lysosomal lipid accumulation. In contrast, Grn−/− mice uniquely exhibit a profound decrease in BMP across all tested tissues, key CNS cell types, and biofluids. We also found a subtle BMP deficiency in the CSF of GRN-FTD patients (who have one functional copy of the GRN allele), suggesting BMP may also be affected by partial Grn LoF. Interestingly, non-GRN FTD patients’ CSF exhibited a mild decrease in BMP levels, though the underlying mechanism is unclear. These findings suggest CSF BMP may represent a clinically translatable, lysosomal function-related biomarker for FTD, though the small effect size may limit its utility for this disease indication.
Grn−/− mice displayed an elevation of GlcSph, which, unlike BMP, showed a CNS age-dependent effect. Additionally, GlcCer accumulates in Grn−/− microglia. We thus hypothesized this increase may represent a lysosomal storage secondary to BMP deficiency relating to a defect in GCase activity, as GlcSph and GlcCer are known GCase substrates (Dai et al., 2016; Taguchi et al., 2017). Accordingly, we noted a decrease in GCase activity in Grn−/− liver and brain, consistent with other studies (Zhou et al., 2019). However, our work defines a distinct molecular basis for this GCase activity deficit, as BMP liposomes rescue this defect in Grn−/− mouse brain extract, suggesting BMP deficiency at least partly underlies the GCase activity defect. This is in line with previous cell-free studies using recombinant saposin C and GCase showing BMP liposomes stimulate GCase activity (Abdul-Hammed et al., 2017). Indeed, BMP preserves its negative charge in the acidic pH of lysosomes and may facilitate electrostatic interactions with positively charged saposin peptides and GCase protein at the surface of lysosomal internal vesicles (Breiden and Sandhoff, 2020). We show PGRN physically interacts with BMP at acidic pH, suggesting this binding may take place in endolysosomes, where both PGRN and BMP are concentrated. This lends support to the notion PGRN (or cleaved GRN peptides) and BMP, in combination with saposin C, physiologically regulate GCase activity. PGRN deficiency impairs this regulation by reducing BMP levels, likely by enhancing its turnover (Fig.7I).
In addition to rescuing lysosomal lipids acutely, PTV:PGRN treatment reduces lipofuscinosis chronically in Grn−/− brain. Whether lipofuscin deposition stems from chronic deficiency of BMP, GlcSph accumulation or other disease mechanisms remains to be determined. The fact that PUFA-BMP species, such as BMP(22:6/22:6), are decreased in Grn−/− brain may result from lipid peroxidation, which drives lipofuscin deposition in lysosomes. This notion, which is also supported by ROS accumulation in Grn−/− cells, suggests PGRN may act physiologically as a lysosomal antioxidant (Fig. 7I).
While our mechanistic work largely focused on lysosomal biology, it also showed myeloid cells, such as macrophages and microglia, are profoundly affected by PGRN deficiency, consistent with others (Gotzl et al., 2019; Krabbe et al., 2017; Lui et al., 2016; Martens et al., 2012; Zhang et al., 2020). Growing evidence indicates Grn LoF in microglia is critical for disease progression in pre-clinical models, perhaps best exemplified by studies showing Grn−/− microglia conditioned media induces cell death and aberrant TDP-43 in primary neurons via complement activation (Zhang et al., 2020). Along with data from GRN-FTD patients’ cortices showing severe microgliosis, these studies strongly suggest that the hyperreactivity of Grn LoF microglia significantly contributes to disease pathogenesis.
A key question is which cellular pathways contribute to the hyperreactive and neurotoxic features of Grn LoF microglia. While loss of PGRN extracellular signaling functions may be involved, multiple data points suggest microglial lysosomal dysfunction from loss of the lysosomal PGRN pool may be a major inflammatory contributor. First, lysosomal dysfunction in other myeloid models of LSDs is typically associated with a reactive transcriptional state, where many immune genes including TREM2, complement factors and galectin-3 are upregulated (Ullman et al., 2020). Second, transcription factors (e.g., TFEB, TFE3) that activate lysosomal gene expression in LSDs also control genes mediating inflammatory responses in myeloid cells (Brady et al., 2018). Third, our work shows GRN−/− iMG undergo lysosomal membrane permeabilization (LMP), based on the recruitment of galectin-3 on lysosomes, suggesting the lysosomal PGRN pool may prevent LMP and an associated proton leak. Because LMP is known to cause pro-inflammatory responses such as NLRP3 inflammasome (Lewcock et al., 2020), PGRN’s anti-inflammatory properties may be partly related to its ability to minimize LMP via direct effects on lysosomes, perhaps via BMP or GlcSph regulation.
In conclusion, PTV:PGRN represents a promising biotherapeutic for the treatment of GRN-FTD and perhaps other neurological conditions associated with decreased PGRN, BMP or GCase activity. The potential of PTV:PGRN outside GRN-FTD is yet to be investigated via additional pre-clinical and biomarker studies. Recent genome-wide association studies showed common GRN polymorphisms resulting in lower PGRN levels are associated with higher risks of developing idiopathic Parkinson’s and Alzheimer’s disease (Jansen et al., 2019; Nalls et al., 2019). Other therapeutic approaches are currently being tested in the clinic, including AAV-based gene therapy and an anti-sortilin antibody, which raises extracellular intact PGRN levels by preventing uptake and/or lysosomal delivery by sortilin. In contrast, our data suggest PTV:PGRN is a pro-drug that acquires activity upon processing in endolysosomes, following cellular uptake by TfR-based internalization, likely in conjunction with sortilin- and prosaposin-based mechanisms (Paushter et al., 2018). Specifically, the pharmacodynamic effect observed in Grn−/− brain several weeks after the intact drug has been cleared suggests it is the granulin peptides produced in lysosomes rather than intact PGRN per se that drive prolonged efficacy. If this hypothesis is correct, assessing extracellular levels of intact PGRN in pre-clinical models or clinical settings may not be sufficient to predict efficacy. In summary, our study demonstrates PTV:PGRN is a molecule worthy of further pre-clinical and clinical studies for treatment of GRN-FTD.
Limitations of the study
It is unclear if BMP deficiency stems from increased turnover and drives a specific Grn Lof phenotype (i.e., decreased GCase activity) vs. a broader range of in vivo phenotypes, including storage of other glycosphingolipids (e.g., gangliosides), lipofuscinosis, gliosis, TDP-43 pathology and neuronal loss. Our patient data was obtained from a single cohort, and will require validation in additional cohorts as well as optimization of CSF GlcSph detection to enable quantification in future studies. Finally, method development is needed to accurately identify and quantify individual granulin peptides to verify the hypothesis that the extended pharmacodynamic effect achieved by PTV:PGRN reflects processing of the pro-drug into stable granulin peptides mediating drug activity.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Gil Di Paolo (dipaolo@dnli.com)
Material Availability
All unique reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
Raw LCMS area ratio data presented in heatmaps and volcano plots in the figures is provided at: DeVos, Sarah (2021), “Logan, Simon et al-2021-LC/MS Dataset”, Mendeley Data, V1, doi: 10.17632/s4y5hgm7xt.1
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Grn−/− and Grn−/−; TfRmu/hu mouse model generation
Previously generated and characterized Grn−/− on a C57Bl6/J background were obtained from Jackson Laboratories (JAX strain 013175) (Yin et al., 2010). TfRmu/hu KI mice (also on a C57Bl6/J background) expressing a chimeric TfR receptor (human TfR apical domain knocked into the mouse receptor) were developed by generating a knock-in of the human apical TfR mouse line using CRISPR, as described previously (Kariolis et al., 2020). Homozygous TfRmu/hu male mice were bred to female Grn+/− mice to generate Grn−/− x TfRmu/hu mice. To generate Grn R493X x TfRmu/hu mice, homozygous TfRmu/hu male mice were bred to female Grn R493X mice (Jax strain 029919) (Nguyen et al., 2018). Both female and male mice were used for all studies in equal percentages, when possible (see below).
Age and sex of mice used throughout manuscript.
All mouse procedures adhered to regulations and protocols approved by Denali Therapeutics Institutional Animal Care and Use Committee. Mice were housed under a 12-hour light/dark cycle and group housed when possible. Animal ages/sexes for each experiment are indicated in figure legends and in the study detail summary table. Within studies, age/sex variability was minimized as much as possible, and if variability existed, ages/sexes were evenly distributed across groups, where possible. Animal weights were collected at the start of each study and at each dose for multi-dose studies. Animals were monitored weekly for health/immune issues before and during studies and were excluded from study inclusion if serious concerns were observed. Mice were housed with enrichment and bedding was changed weekly for group housed animals and monthly for single house animals. For study group assignment, littermate animals and when possible – cage-mate animals were assigned to unique experimental groups. Each animal was also assigned a unique, blinded study ID that then served to blind and randomize samples for downstream analyses. For each analyte of interest, a post-hoc analysis was performed for normality and sex-dependent effects (see quantification and statistical analysis section).
Human biofluid sample collection
De-identified plasma and CSF samples from FTD patients (n=42, among them n=16 with GRN mutation and n=1 with MAPT mutation) and clinically normal controls (n=22, among them n=2 with GRN mutation) were obtained through the NCRAD Biofluid Repository and UCSF collected from LEFFTDS and ARTFL studies. Samples were allocated based on genotype and disease status. To limit the risk of loss of confidentiality, NCRAD did not provide the gender information, nor the exact age, but an age range for the subjects. Since this was an exploratory analysis in the absence of a treatment group, no sample size estimation was performed. Biofluid samples were blinded, randomized and subjected to metabolite/lipid extraction by methanol and analyzed on a quantitative LC/MS/MS platform. Sample demographic data can be found in Table S1.
Cell line and primary culture conditions
Primary BMDMs were harvested from 3–6 month old male and female mice. Cells were cultured in 10mL RPMI-1640 (ThermoFisher) + 10% Hyclone FBS (GE Healthcare) + Penicillin-Streptomycin (ThermoFisher) at 37°C and 5% CO2. Expi293F cells (ThermoFisher) were maintained according to the manufacturer’s specifications in Expi293 Expression medium (ThermoFisher #A1435101). Human induced-pluripotent stem cell lines (hiPSCs) were generated from a female clone from ThermoFisher (#A18945) and routinely passaged as clumps onto Geltrex (Thermo #A1413302)-coated plates with mTeSR1 media (StemCell Technologies #85850) according to manufacturer’s instructions. All the experiments using GRN+/+ and GRN−/− iMG were performed in IMDM (Gibco) media supplemented with 10% defined FBS (Gibco), 1% Penicillin/Streptomycin (Gibco), 20 ng/mL of hIL3 (Peprotech), 20 ng/mL of hGM-CSF (Peprotech) and 20 ng/mL of hM-CSF (Peprotech) (referred to as “C+++ Media”) and maintained at 37°C and 5% CO2. Full characterization of human iPSC-derived microglia and additional details on the differentiation protocol has been published elsewhere (Andreone et al., 2020).
METHOD DETAILS
Mouse handling and tissue collection
Mice received therapeutic treatment via intravenous (IV) tail vein injection (~200 μl total injection volume) or intraperitoneal (IP) injection (~200 μl total injection volume). For all tissue collection, animals were anesthetized with tribromoethanol. Whole blood was collected via cardiac puncture collected into EDTA coated tubes spun down at 12,700rpm for 7min at 4°C before collecting the top plasma layer for analysis. CSF, if collected, was then extracted via cisterna magna puncture, spun down at 12,700rpm for 7min at 4°C and supernatant was transferred to a new tube for analysis. Following plasma and CSF collection, mice were transcardiac perfused with ice-cold PBS. Tissues were then collected, weighed, frozen on dry ice and stored at −80 °C for subsequent analysis.
Expression and purification of fusion proteins
Generation of the transport vehicle (TV) from the effector-less Fc portion of a human IgG1 was previously described (Ullman et al., 2020). Briefly, the TV was fused N-terminally to the complete native sequence of mature human PGRN, allowing for PGRN to interact with sortilin via its C-terminal region. PTV:PGRN, PTV:PGRNv2 and Fc:PGRN were expressed as knob-in-hole heterodimeric proteins. For PTV:PGRN and FcPGRN, Expi293 cells (Thermo Fisher Scientific, Waltham, MA USA) were transfected with plasmid DNA encoding the TV Fc polypeptide and PGRN fused to an Fc polypeptide in PEIMax (MW 40,000, Linear, Polysciences). Cultures were harvested 5 days post-transfection. Expression of recombinant Fc:PGRN was carried out as described above with the exception of wild-type Fc being used in place of the TV Fc polypeptide. PTV:PGRN and Fc:PGRN were subsequently purified using a series of chromatography steps commonly used for the purification of monoclonal antibodies and Fc-fusion proteins including affinity chromatography and size exclusion chromatography. Fractions from the size exclusion columns were eluted with an arginine-succinate buffer (pH 5) and assessed for purity by Labchip Caliper and analytical SEC-HPLC. A modified version of PTV:PGRN termed PTV:PGRNv2 was expressed via transient transfection of a CHO-K1 cell line using PEIMax (MW 40,000, Linear, Polysciences). Cultures were harvested 7 days post transfection and material was purified using a series of chromatography steps commonly used for the purification of monoclonal antibodies and Fc-fusion proteins including affinity chromatography and ion exchange chromatography. The purified protein was exchanged into acetate and sucrose buffer (pH 5) and analyzed by analytical size exclusion chromatography and SDS-PAGE. The purity was more than 95% for both analytic methods. The monoclonal antibody to the F protein in the A antigenic site of the anti-respiratory syncytial virus (with effector-less IgG1) was expressed in Expi293 cells, purified as described above and used as a control IgG1 for in vitro experiments.
Engineering PTV:PGRN variants with improved stability
The UniProtKB/TrEMBL sequence database and the NCBI reference protein and non-redundant protein sequences databases were searched for PGRN sequences that differ at the last three C-terminal amino acids from the human sequence (QLL). Eight PGRN sequences from different species were selected. PTV:PGRN variants with the last three C-terminal amino acids of human PGRN replaced with the corresponding amino acids from the selected orthologous sequences were expressed in CHO-K1 cells and purified as described above (Table S2). The percent of each purified PTV:PGRN variant with intact C-terminus was determined by top-down mass spectrometry or by peptide mapping (Table S3). For top-down mass spectrometry, protein was separated by liquid chromatography on an Ultimate 3000 (Thermo Scientific, CA,USA) coupled to an Exactive Plus EMR Orbitrap electrospray ionization mass spectrometer (Thermo Scientific, CA,USA). For each analysis, 10 μL of sample (10–15 μg of protein) was injected on a Thermo MabPac RP 4μm 2.1 × 50 mm (P/N 88648) using a flow rate of 0.30 mL/min at 65°C. Mobile phase A consisted of water with 0.1% trifluoroacetic acid (TFA); mobile phase B consisted of 100% acetonitrile. The gradient was programmed as follows: 0.0–1.0 min 20% B, 1–11 min, 20% to 70% B, 11–12 min 70% B, 12–14 min 70% to 20% B, 20% B for 1 min. Total run time was 15min. Data was collected using EMR-AIF acquisition under positive mode scanning from 350–1800 m/z. The In-source CID was set to 90 eV, AIF using 5 microscans, resolution at 35000, with AGC target 1e6, maximum IT 800 ms, and CE at 200. ESI source conditions were set as following. Sheath gas flow rate: 25 psi. Aux gas flow rate 4 psi. Spray voltage 3.8 kV, Capillary temp: 250 C, S-Lens RF level: 100, and Aux gas Temp: 250C. AUC values were extracted using Xcalibur software (Thermo Scientific, San Jose, CA) with 20 ppm window. For peptide mapping, PTV-PGRN variants were enzymatically digested using endoproteinase AspN (New England Biolabs, Ipswich, MA). Briefly, AspN enzyme was prepared in Mili-Q water to 0.5 μg/μL. Fifteen to twenty μg of PTV-PGRN proteins were diluted to 20 μL final volume in water and mixed with 60 μL of digest buffer supplied with the AspN kit. Solutions were incubated in a ThermoMixer C heater and shaker (Eppendorf) at 37C for 30 min, shaking at 700 rpm. Reactions were quenched with 1% formic acid and transferred to LCMS vials for analysis. Peptide mapping analyses were performed by liquid chromatography on UHPLC Vanquish (Thermo Scientific, CA,USA) coupled Q Exactive Orbitrap electrospray ionization mass spectrometer (Thermo Scientific, CA,USA), with an LC draw speed of 5 ul/min and autosampler temp set to 10C. For each analysis, 20 μL of sample was injected on a CSH C18 1.7 μm, 2.1×150 mm column (Waters) using a flow rate of 0.20 mL/min at 40°C under positive ionization mode. Mobile phase A consisted of water with 0.1% formic acid; mobile phase B consisted of acetonitrile with 0.1% formic acid. The gradient was programmed as follows: 0.0–3.0 min 1% B, 3–12 min, 1% to 10% B, 12–40 min 10%−40% B, 40–45 min 40 to 70% B, keep 70% B for 3 min, 48–50 min from 70% to 1% B. Hold at 1% B for 10 min. Total run time was 60 min. Data was collected using Full MS-ddMS2 acquisition under positive mode. The default charge was 2+, for Full MS, resolution was set to 70,000 with AGC target 1E6 and Maximal IT (injection time) 125 ms, scan range 300–1800 m/z. For dd-MS2 settings, resolution was set to 17,500 with AGC target 5E4 and 150 ms Maximal injection time (IT), with Loop count at 5 (top 5) and isolation window 2 amu and NCE 25 unit. The minimal AGC for dd-MS2 was set to 5E3, equal to intensity Threshold of 3.3E5. Charge stage 1 and isotrope were excluded. Thermo Xcalibur software was used to extract the peak area or area under curve (AUC) of major target C-terminal peptides (2+) and the truncated peptides as shown in Table S2 (QLL (wt) and an exemplary variant). The % listed for each peptide is the AUC of the peptide divided by the sum of the AUC values of all four peptides.
HuTfR binding affinity determination by SPR
Affinities of PTV:PGRN, PTV:PGRNv2 and Fc:PGRN for hTfRapical were determined by surface plasmon resonance (SPR) using a BIAcore 8K instrument. PGRN fusion proteins were immobilized on a BiacoreTM Series S sensor chip protein A (GE, # 29127555), then 3-fold dilutions of recombinant hTfRapical protein were injected at a flow rate of 30 μL/min in HBS-EP+ running buffer (GE, #BR100669). Binding response was corrected by subtracting the RU from a blank flow cell. After each injection, the chip was regenerated using 10 mM glycine-HCl (pH 1.5). A 1:1 Langmuir model of simultaneous fitting of kon and koff was used for kinetics analysis using Biacore™ Insight Evaluation Software.
HuPSAP and huSortilin binding affinity by ELISA
His-tagged recombinant human sortilin (R&D Systems, Catalog #3153-ST) or human prosaposin (PSAP) (Abcam, Catalog #ab167924) proteins were diluted in PBS and incubated with Ni-coated plates (Thermo Scientific, Catalog # 15442) for 2 hours at RT while shaking at 600 rpm. Plates were washed 3 times with PBST and blocked with 3% BSA in PBS. Post-PBST wash, plates were incubated with PTV:PGRN or Fc:PGRN proteins with serial dilution from 1000 nM to 0.03 nM in half-log steps and incubated for 2 hours at RT while shaking to attain the dose-response curve. Anti-human IgG Fc gamma HRP (Jackson Immuno Research, Catalog # 109–035-098) was used as a detection antibody and was developed with TMB substrate. 450 nm absorbance was read on the Biotek plate reader. EC50 were calculated with Graph Pad Prism 8 and curves were fitted with a 4-parameter sigmoidal curve.
FACS isolation of microglia, astrocytes and neurons
CNS cell types were isolated by cell sorting using previously established protocols (Nugent et al., 2020; Ullman et al., 2020). Briefly, to prepare a single cell suspension, brain tissue was dissected and processed into a single cell suspension according to the manufacturers’ protocol using the adult brain dissociation kit (Miltenyi Biotec 130–107-677). Cells were Fc blocked (Biolegend #101320, 1:100) and stained for flow cytometric analysis with Fixable Viability Stain BV510 (BD Biosciences #564406, 1:100) to exclude dead cells, CD11b-BV421 (BD Biosciences 562605, 1:100), ACSA2-APC (Miltenyi #130–117-386, 1:100), and Thy1-PE (R&D #FAB7335P, 1:100). Cells were washed with PBS/1% BSA and strained through a 100μm filter before sorting CD11b+ microglia, ACSA2+ astrocytes, and Thy1+ neurons on a FACS Aria III (BD Biosciences) with a 100μm nozzle. Sorted cells were collected directly into MS grade methanol with added internal standards for lipidomic and metabolomic analysis or Qiagen RLT-plus + β-mercaptoethanol for RNA analysis.
mRNA analysis by qPCR
To evaluate cell type enrichment of FACS sorted cell populations, a minimum of 10,000 cells were sorted directly into Qiagen RLT-plus + β-mercaptoethanol, then RNA was extracted using the RNeasy Plus Micro Kit (Qiagen, 74034). cDNA libraries were generated by reverse transcription (ThermoFisher VILO Superscript IV #11756050). Cell type expression was evaluated with 10 cell type Taqman probes (ThermoFisher, Syn1: Mm00449772_m1, Tubb3: Mm00727586_s1, Mbp: Mm01266402_m1, Mog: Mm01266402_m1, Aqp4: Mm00802131_m1, Gfap: Mm01253033_m1, Aldh1l1: Mm03048957_m1, Itgam: Mm00434455_m1 and Cldn5: Mm00727012_s1). For each sample, expression of cell type markers was normalized to the housekeeping gene Gapdh (ThermoFisher Mm99999915_g1). qPCR was performed using a QuantStudio 6 flex system (Applied Biosystems) and average CT values were collected for each probe between duplicate reads. DDCT values were calculated relative to the average CT of all samples for a given probe. Log2 transformation of the fold change values were used for visualization and heatmaps were generated using the ComplexHeatmap R package.
Brain immunofluorescence/lipofuscin imaging
Following PBS transcardiac perfusion and overnight post-fixation in 4% PFA, mouse hemibrains were coronally sectioned at Neuroscience Associates using Multibrain sectioning service. Briefly, up to 40 brains were trimmed and mounted in a single gelatin block, then coronally sectioned at a thickness of 40mm. Gelatin sheets with embedded brain sections were then stored in antigen preservation solution (50% PBS:50% ethlyene glycol + 1% PVP) until staining. Sections were incubated at room temperature, rocking, for 4h in blocking buffer (PBS + 1% BSA + 0.1% fish gelatin + 0.5% triton X-100), then transferred to antibody dilution buffer (1% BSA in PBS + 0.3% Triton x-100 + 0.1% sodium azide) with added primary antibodies of interest (Fig 5: NeuN: Millipore MAB377 (1:250), huIgG: Jackson Immuno Research 109–605-003 (1:250), Iba1: Abcam ab178847 (1:250), CD31: R&D systems AF3628 (1:250) podocalyxin: R&D Systems AF1556 (1:250), Fig 7:CD68: BioRad MCA1957 (1:500), IBA1: Novus NB100–1028 (1:500), GFAP: Novus NBP1–05198 (1:1000), C1Q: Abcam ab182451 (1:500)), and stored, rocking, at 4°C overnight. Following 3 washes in PBS, samples were then transferred to antibody dilution buffer with secondary antibodies (see key resources list, 1:500 dilution for all) and incubated, rocking, at room temperature for 4h. Samples were washed with PBS + DAPI (Invitrogen D1306 1:10,000) for 20min, then washed twice more with PBS before mounting on 2” x 3” slides with Prolong Glass hardset mounting media (Life tech P36984) and allowed to dry overnight at room temperature.
For quantification, full brain hemispheres were imaged at 20X using a Zeiss Axio Scan.Z1 digital slide scanner. Image analysis was completed using Zeiss Zen Blue 3.2 software. Thalamus ROIs were drawn, and a rolling ball thresholding approach was used to determine the area of each gliosis marker relative to total thalamus area. 1–3 sections were analyzed per brain and average percent coverage values were calculated across images. Super resolution representative images were acquired on a Leica SP8 microscope using a 63x/1.4 n.A. oil immersion objective with a pixel size of 36nm (Fig 5G) or 39nm (Fig. 7A) in lightning acquisition mode and processed using the adaptive algorithm. 3D stacks were visualized in Imaris.
For imaging of lipofuscin, full brain hemispheres of DAPI-only treated brain sections were imaged at 20X using a Zeiss Axio Scan.Z1 digital slide scanner. Autofluorescent signal was quantified in the 594 channel in Zeiss Zen Blue 3.2 software and quantified as described above. Representative images of autofluorescence were acquired at 550nm excitation/600–650nm emission using a 63x/1.4N.A. oil immersion objective with a pixel size of 35nm, imaging in the lightning acquisition mode with the Adaptive algorithm.
Capillary depletion
The capillary depletion was performed as previously described (Kariolis et al., 2020). Freshly PBS-perfused brains were collected into HBSS buffer. The meninges and choroid plexus were dissected off, and the brains were homogenized by 10 strokes with a Dounce homogenizer using a smaller diameter pestle. The homogenate was centrifuged at 1,000 g for 10 min at 4°C and the supernatant was removed. The remaining pellet was resuspended in 2 mL of 17% dextran (MW 60,000; Sigma 31397) then centrifuged at 4,122 g for 15min at 4°C. The pellet was collected as the vasculature fraction. The supernatant containing parenchymal cells and myelin was transferred to a new tube, diluted with HBSS, then centrifuged at 4,122 g for 15min at 4°C. After removing the supernatant, the resulting pellet contained vascular depleted parenchymal cells. Both the vascular pellet and parenchymal pellet were resuspended in cold 1% NP40 in PBS with protease and phosphatase inhibitors (Roche 04693159001 and 04906837001), vortexed for 20 sec, and incubated at 4°C for 20 min. Samples were centrifuged once more at 12,700 g for 10 min at 4°C and the lysate total protein concentration was measured using by BCA assay (Thermo 23225).
Fc/Fc and Fc/PGRN ELISA for in vivo studies
To simultaneously determine Fc concentrations and intact Fc:PGRN concentrations in tissues, a dual sandwich ELISA was developed. The day before the assay, 384-well clear microplates (ThermoFisher #464718) were coated with a donkey polyclonal antibody against human Fc (Jackson ImmunoResearch #709–006-098) at a concentration of 1μg/mL, diluted in sodium bicarbonate buffer and incubated overnight at 4°C. The next day plates were washed 3X with PBS-T buffer, then blocked for 2+ hours (5% BSA in PBS-T) at room temperature. Following another 3X wash with PBS-T, samples (diluted in 1% BSA in PBS-T were then added to the plate along with a standard curve generated with dosing material ranging from 2nM to 0nM and incubated for 2h at room temperature. Samples were again washed, then either HRP conjugated detection antibody against human FC was added (Jackson ImmunoResearch #109–036-098; 20ng/mL) or a biotinylated goat polyclonal antibody against huPGRN was added (R&D #BAF2420; 0.5μg/mL), followed by a streptavidin-HRP conjugate (Jackson ImmunoResearch #016–030-084, 1:50,000). Samples were incubated for 1h at room temperature, washed 3X with PBS-T, followed by the addition of TMB stop solution (ThermoFisher #34029. The color reaction was allowed to develop for ~10 min. Immediately prior to reading, stop solution (Surmodics #LSTP-1000–01) was added. Absorbance was then measured with a BioTek plate reader. To determine concentrations, background was subtracted, and concentrations were determined by fitting of a four-parameter logistic model curve to standard samples using Graphpad Prism. Samples were run in duplicate for each assay.
Liquid chromatography-Mass spectrometry (LCMS)
Sample preparation for LCMS
For tissue LC-MS preparation, during tissue collection, roughly 20mg (± 2 mg) of prefrontal cortex was dissected, weighed and flash frozen. 400μl of methanol spiked with internal standards (see LC/MS methods below) were added to each sample and homogenized with a 3mm tungsten carbide bead (shaken at 25Hz for 30 seconds). The methanol fraction was then isolated via centrifugation (20 min at 4°C, 14,000 x g, followed by transfer of supernatant to a 96 well plate, 1 h incubation at −20°C followed by an additional 20 min centrifugation (4,000g at 4°C) and transferred to glass vials for LC-MS analysis. For analysis of a GlcCer/GalCer panel, an aliquot of the methanol fraction was dried under N2 gas and then resuspended in 100μl of 92.5/5/2.5 CAN/IPA/H2) (MS grade) with 5mM ammonium formate (MS grade) and 0.5% Formic Acid (MS grade). For plasma and urine, samples were diluted 1:100 in methanol + internal standards. CSF was diluted 1:500. Samples were vortexed, then centrifuged (as described above) and transferred to glass vials for LC-MS or prepped for the GlcCer/GalCer/GlcSph/GalSph analysis (as described above). For sorted cells preparation, 40,000 cells were sorted directly into MS grade methanol + internal standards. Sample volumes were adjusted up to 800μL with MS grade H2O, then 800 uL of tert-butyl methyl ether (MTBE) was added, vortexed, then centrifuged at 14,000 g for 10 min at 4C. The two phases generated by centrifugation were separated. The top phase was transferred to glass vials, dried with N2 gas, then resuspended in MS grade methanol for non-polar lipid analysis. A fraction of this was further dried and resuspended as described above for GlcCer/GalCer/GlcSph/GalSph panel analysis. For polar metabolites, the bottom fraction was dried overnight using a Genevac EZ3, resuspended in 90% methanol:water mixture, then transferred to glass vials for LCMS.
Unless otherwise noted, relative quantification of lipids and metabolites were performed using the Shimadzu Nexera X2 LC system (Shimadzu Scientific Instrument, Columbia, MD, USA) coupled to Sciex QTRAP 6500+ mass spectrometer (Sciex, Framingham, MA, USA).
Lipidomic analysis
For each analysis, 5 μL of sample was injected on a BEH C18 1.7 μm, 2.1×100 mm column (Waters Corporation, Milford, Massachusetts, USA) using a flow rate of 0.25 mL/min at 55°C. Mobile phase A consisted of 60:40 acetonitrile/water (v/v); mobile phase B consisted of 90:10 isopropyl alcohol/acetonitrile (v/v). These buffers were fortified with 10 mM ammonium formate with 0.1% formic acid (positive ionization) or with 10 mM ammonium acetate (negative ionization). The gradient was programmed as follows: 0.0–8.0 min from 45% B to 99% B, 8.0–9.0 min at 99% B, 9.0–9.1 min to 45% B, and 9.1–10.0 min at 45% B. Source settings were as follows: curtain gas at 30 psi; collision gas was set at medium; ion spray voltage at 5500 V (positive mode) or 4500 V (negative mode); temperature at 250°C (positive mode) or 600°C (negative mode); ion source Gas 1 at 55 psi; ion source Gas 2 at 60 psi. Data acquisition was performed using Analyst 1.6.3 (Sciex) in multiple reaction monitoring mode (MRM). Area ratios of endogenous metabolites and surrogate internal standards were quantified using MultiQuant 3.02 (Sciex) (see Table S4 and S5 for acquisition parameters).
Analysis of bis(monoacylglycero)phosphate (BMP)
For each analysis, 5 μL of sample was injected on a BEH C18 1.7 μm, 2.1×100 mm column (Waters Corporation, Milford, Massachusetts, USA) using a flow rate of 0.35 mL/min at 62°C. Mobile phase A consisted of 60:40 acetonitrile/water (v/v) with 10 mM ammonium acetate Mobile phase B consisted of 90:10 isopropyl alcohol /acetonitrile (v/v) with 10 mM ammonium acetate. The gradient was programmed as follows: 0.01–3.0 min from 50% B to 90% B, 3.0–3.01 min to 50% B, and 3.01–3.50 min at 50% B. Electrospray ionization was performed in negative ion mode applying the following settings: curtain gas at 30 psi; collision gas was set at medium; ion spray voltage at −4500 V; temperature at 600°C; ion source Gas 1 at 50 psi; ion source Gas 2 at 60 psi. LC-MS data acquisition, parameters of which can be found below, was performed using Analyst 1.6.3 (Sciex) in multiple reaction monitoring mode (MRM). BMP species were quantified using surrogate internal standard, BMP(14:0/14:0). Quantification was performed using MultiQuant 3.02 (Sciex).
LCMS acquisition parameters for the BMP assay.
| Lipid | Internal Standard | Q1 (m/z) | Q3 (m/z) | CE (V) |
|---|---|---|---|---|
| BMP(14:0/14:0) | Internal Standard | 665.3 | 227.2 | −50 |
| BMP(20:4/20:4) | BMP(14:0/14:0) | 817.5 | 303.3 | −50 |
| BMP(22:6/22:6) | BMP(14:0/14:0) | 865.5 | 327.3 | −50 |
| BMP(18:1/18:1) | BMP(14:0/14:0) | 773.5 | 281.3 | −50 |
| BMP(16:1/16:1) | BMP(14:0/14:0) | 717.5 | 253.1 | −50 |
| BMP(18:1_22:6) | BMP(14:0/14:0) | 819.5 | 281.3 | −50 |
| BMP(16:0_22:6) | BMP(14:0/14:0) | 795.5 | 255.3 | −50 |
| BMP(18:0_18:1) | BMP(14:0/14:0) | 775.5 | 283.3 | −50 |
| BMP(18:0_22:6) | BMP(14:0/14:0) | 823.5 | 283.3 | −50 |
Absolute quantification of BMP(22:6/22:6) and (18:1/18:1)
To quantify absolute levels of BMP(22:6/22:6) and BMP(18:1/18:1) in biological matrices (e.g., tissue lysate, CSF, serum, plasma), aliquots of calibration standards (STDs), quality control samples (QCs), and study samples were transferred to a clean 96-well plate and extracted with methanol containing BMP(14:0/14:0) as an internal standard. After mixing and centrifugation, the supernatant was further diluted if needed and injected into LCMS for analysis. LCMS analyses were performed on an ExionLC AD UHPLC system coupled with a Sciex API 6500+ Triple Quad mass spectrometer (AB Sciex, Redwood City, CA). HPLC chromatography was established on an Acquity BEH C8 column (1.7 μm, 50 × 2.1 mm) (Waters Co., Milford, MA) and the column was kept at 55 °C during the run. For the LC separation of interested BMP species from the matrix interferences, the two mobile phases used were 0.01% Ammonium Hydroxide, 1 mM Ammonium Acetate in Water (Mobile Phase A) and 0.01% Ammonium Hydroxide in ACN (Mobile Phase B). The combined flow rate was kept at 0.4 mL/min. The reference standards and internal standard were purchased from Avanti Polar Lipids (Alabaster, Al). To prepare calibration curve and QCs, the standard stock solutions were spiked in surrogate matrices to avoid the MS signal contributions from endogenous BMP species in authentic biological matrices. During each run, a Matrix QC prepared from pooled authentic biological matrices was also used to monitor the assay performance.
Analysis of GlcCer, GalCer, GlcSph and GalSph
For each analysis,8μL of sample was injected on a HALO HILIC 2.0 μm, 3.0 × 150 mm column (Advanced Materials Technology, PN 91813–701) using a flow rate of 0.45 mL/min at 45°C. Mobile phase A consisted of 92.5/5/2.5 ACN/IPA/H2O with 5 mM ammonium formate and 0.5% formic Acid. Mobile phase B consisted of 92.5/5/2.5 H2O/IPA/ACN with 5 mM ammonium formate and 0.5% formic Acid. The gradient was programmed as follows: 0.0–2 min at 100% B, 2.1 min at 95% B, 4.5 min at 85% B, hold to 6.0 min at 85% B, drop to 0% B at 6.1min and hold to 8.5 min. Electrospray ionization was performed in the positive-ion mode applying the following settings: curtain gas at 35 psi; collision gas was set at medium; ion spray voltage at 5500 V; temperature at 400°C; ion source Gas 1 at 55 psi; ion source Gas 2 at 60 psi. Data acquisition was performed using Analyst 1.6 (Sciex) in multiple reaction monitoring mode (MRM) Lipids were quantified using a mixture of stable isotopically labeled internal standards. Quantification was performed using MultiQuant 3.02 (Sciex) (see Table S6 for acquisition parameters).
LCMS assay for Gangliosides
Ganglioside analyses were performed by liquid chromatography (ACQUITY I-Class Plus UPLC FL) coupled to electrospray mass spectrometry (XEVO TQ-S Micro). For each analysis, 4 μL of sample was injected on a BEH C18 1.7 μm, 2.1×100 mm column (Waters) using a flow rate of 0.25 mL/min at 55°C. Mobile phase A consisted of 60:40 acetonitrile/water (v/v) with 10 mM ammonium acetate Mobile phase B consisted of 90:10 isopropyl alcohol /acetonitrile (v/v) with 10 mM ammonium acetate. The gradient was programmed as follows: 0.0–8.0 min from 45% B to 99% B, 8.0–9.0 min at 99% B, 9.0–9.1 min to 45% B, and 9.1–10.0 min at 45% B. For the XEVO TQ-S Micro we applied the following settings: capillary voltage 1.9kV; source temperature at 150°C; desolvation temperature at 600°C; desolvation gas flow at 1000 L/h; cone gas flow at 50 L/h; cone voltage at 40 V; nebulizer gas at 7 bar. LC-MS data acquisition, parameters of which can be found below, was performed using MassLynx v4.2 in multiple reaction monitoring mode (MRM). Quantification was performed using Skyline (v19.1; University of Washington).
LC-MS acquisition parameters for the ganglioside assay.
| Lipid | Internal Standard | Q1 (m/z) | Q3 (m/z) | CE (V) |
|---|---|---|---|---|
| GM3(d18:1/18:0(d5)) | Internal Standard | 1184.8 | 290.1 | 55 |
| GM3(d34:1) | GM3(d18:1/18:0(d5)) | 1151.7 | 290.1 | 55 |
| GM3(d36:1) | GM3(d18:1/18:0(d5)) | 1179.8 | 290.1 | 55 |
| GM3(d38:1) | GM3(d18:1/18:0(d5)) | 1207.8 | 290.1 | 55 |
| GM3(d40:1) | GM3(d18:1/18:0(d5)) | 1235.8 | 290.1 | 55 |
Metabolomics analysis
For each analysis, 5 μL of sample was injected on a BEH amide 1.7 μm, 2.1×150 mm column (Waters Corporation, Milford, Massachusetts, USA) using a flow rate of 0.40 mL/min at 40°C. Mobile phase A consisted of water with 10 mM ammonium formate + 0.1% formic acid. Mobile phase B consisted of acetonitrile with 0.1% formic acid. The gradient was programmed as follows: 0.0–1.0 min at 95% B; 1.0–7.0 min to 50% B; 7.0–7.1 min to 95% B; and 7.1–10.0 min at 95% B. Electrospray ionization was performed in positive ion mode using the following settings: curtain gas at 30 psi; collision gas was set at medium; ion spray voltage at 5500 V; temperature at 600°C; ion source Gas 1 at 50 psi; ion source Gas 2 at 60 psi. Data acquisition was performed using Analyst 1.6 (Sciex) in multiple reaction monitoring mode (MRM) (see Table S7 for acquisition parameters).
iPSC maintenance and genome editing
iPSCs were obtained from ThermoFisher (#A18945) and routinely passaged as clumps onto Geltrex (Thermo #A1413302)-coated plates with mTeSR1 media (StemCell Technologies #85850) according to manufacturer’s instructions. Knockout lines were generated using a nucleofection-based RNP approach to introduce Cas9 (NEB # M0646M) and sgRNAs against GRN (crRNAs ordered from IDT) were introduced into iPSCs via nucleofection (Lonza P3 kit # V4XP-3032) as described before (Andreone et al., 2020). sgRNAs were designed using the Broad Institute design tool based on a previous study (Doench et al., 2016) and had the following sequence: GCTGGGTGGCCTTAACAGCA. Clones were screened by T7 endonuclease and positive clones were further screened by TOPO cloning (Thermo cat #450030) to identify precise mutations. Clones with indels introduced into both alleles that result in a null mutation were grown up and used as GRN knockout clones.
Differentiation of iPSCs to microglia
iPSCs were first differentiated into hematopoietic progenitor cells following manufacturer’s instructions using a commercially available kit (StemCell Technologies #05310) as described before (Andreone et al., 2020). HPCs positive for identity markers CD34, CD43, and CD45 were transferred to a plate containing primary human astrocytes and co-cultured using media C adapted from a previous study (Pandya et al., 2017). Once floating cells in co-culture are predominantly (>80%) mature microglia, cells were plated for 3–4 days prior to experiments. Full characterization of human iPSC-derived microglia and additional details on the differentiation protocol has been published elsewhere (Andreone et al., 2020). All the experiments using GRN+/+ and GRN−/− iMG were performed in IMDM (Gibco) media supplemented with 10% defined FBS (Gibco), 1% Penicillin/Streptomycin (Gibco), 20 ng/mL of hIL3 (Peprotech), 20 ng/mL of hGM-CSF (Peprotech) and 20 ng/mL of hM-CSF (Peprotech) (referred to as “C+++ Media”). Immunostainings of PGRN were conducted using the anti-progranulin goat polyclonal antiserum (R&D Systems # AF2420, 1:250) to verify the absence of immunoreactivity in knockout iMG.
Primary bone marrow-derived macrophage cultures
Primary bone marrow-derived macrophages (BMDMs) were prepared as described previously (Nugent et al. 2020) from Grn+/+ and Grn−/− mice in a normal background or in a hTfR mu/hu homozygous mutant background. Mouse femur and tibia bones were dissected and briefly sterilized with 70% ethanol. The bones were washed twice with HBSS, then cracked in 10mL HBSS by mortar and pestle. The cell suspension was filtered through a 70μm cell strainer, spun at 300xg for 5 min, and supernatant was discarded. The cell pellet was resuspended in ACK Lysing Buffer (ThermoFisher A1049201) for 4 min at room temperature. 10mL RPMI-1640 (ThermoFisher) + 10% Hyclone FBS (GE Healthcare) + Penicillin-Streptomycin (ThermoFisher) was added to stop ACK lysis, then spun 300xg 5 min, and supernatant was discarded. Cells were resuspended in RPMI-1640 + 10% Hyclone FBS + Penicillin-Streptomycin with 50ng/mL murine M-CSF (Life Technologies, PMC2044), counted and diluted to 1×106 cells/mL, then plated on non-tissue culture treated petri dishes. Three days after seeding, fresh murine M-CSF (50ng/mL) was added. Five days after seeding, cell culture media was aspirated and cells were washed once with PBS. Cells were resuspended in RPMI/FBS/Pen-Strep and harvested with a cell scraper. Cells were spun at 300xg for 5 min, supernatant was discarded, and cells were either diluted 1×106 cells/mL for direct culture on tissue-culture treated plates, or frozen in RPMI/FBS/Pen-Strep + 10% DMSO for later use.
Lysosomal proteolysis assay
For assessment of lysosomal proteolysis, BMDMs were plated in 96-well format in Cell Carrier Ultra plates (Perkin Elmer) at 75,000 cells/well and cultured for 48–72h. DQ Red BSA (Thermo Fisher Scientific, D12051) was resuspended in sterile H2O at a concentration of 1mg/mL and reconstituted with 10 × 1s pulses at 50% amplitude with a probe sonicator (Fisher Scientific). DQ Red BSA was then diluted to 10mg/mL final concentration in complete BMDM media and incubated with cells for 6h. Cells were washed 3x in warmed PBS and then immediately fixed in 4% Paraformaldehyde for 15 min. After fixation, cells were washed 3x with PBS and then stained with DAPI (Thermo Scientific, 62248: 1:1,000 from 1mg/mL stock) for 10 min. DQ Red BSA fluorescence (561nm excitation, 570–630nm emission filter) was visualized on an Opera Phenix (Perkin Elmer) high content microscope at 40X. Fluorescent DQ Red BSA puncta were segmented and quantified automatically within Harmony HCS software (Perkin Elmer), and lysosomal proteolysis was calculated as the total DQ Red BSA spot area per well, normalized to cell number. For the liposome supplementation experiment, BMDMs were treated for 16h with serum-free RPMI 1640 media supplemented with liposomes to a final concentration of 50μg/mL. Cells were washed in complete media with serum and either immediately incubated in DQ Red BSA reagent or harvested for LCMS analysis.
Measurement of endolysosomal pH
For measurement of endo-lysosomal pH in BMDMs using LysoSensor Yellow/Blue DND-160 (Invitrogen), cells were seeded on black 96-well plates at a density of 100,000 cells per well and cultured for 72h in either untreated complete media or media supplemented with 50nM RSV or PTV:PGRN. Prior to labeling cells, pH calibration solutions were made fresh. pH calibration solutions were generated in 20mM MES (4- Morpholineethanesulfonic acid), 110 mM KCl, and 20mM NaCl, supplemented with 30μM nigericin and 15 μM monensin, sterile filtered, and titrated to pH values of 4.0, 4.5, 5, 5.5, and 6. Cells were labeled with 2uM LysoSensor Yellow/Blue DND-160 in isotonic solution (Hanks Buffered Salt Solution (GIBCO, Life Technologies) supplemented with 60mM mannitol). After 20 min, cells were rinsed 2x with isotonic solution and incubated for 10 min with isotonic solution (test wells) or with pH calibration solutions at 37C/5%CO2. The ratio of light excited at 330nm over 380 nm, detected at a 527 nm emission, was measured at 37 °C using a Spectramax M5 plate reader (Molecular Devices), collected every 2min for 3 cycles and averaged. Absolute pH values were then calculated using a standard curve generated from the pH calibrations, plated in adjacent wells and measured simultaneously.
Reactive oxygen species assay
For measurements of cellular reactive oxygen species (ROS), the DCFDA Cellular ROS Assay Kit was used according to the manufacturer’s instructions (Abcam # ab113851), with some modifications. Briefly, BMDMs were plated at 100,000 cells/well for 72h and washed once with the wash buffer. Cells were then incubated with the diluted DCFDA solution (1:1,000 dilution of a 20mM stock) and NucBlue™ Live ReadyProbes™ Reagent (Hoechst 33342) for 45 min at 37°C in the dark. The DCFDA solution was then removed and 100ml of 1X buffer was added. Cells were live imaged at Ex/Em of 485/535 nm.
Assessment of lysosomal morphology
Assessment of endolysosomal compartments in iMG was conducted with an anti-progranulin goat polyclonal antiserum (R&D Systems # AF2420, 1:250) and an anti-LAMP-1 mouse monoclonal antibody (mAb) (Cell Signaling Technology #9091, 1:250). In murine BMDMs, an anti-LAMP-2 mouse mAb (Abcam Inc #13524, 1:250) and an anti-LBPA (BMP) mouse mAb (Echelon Inc #Z-PLBPA, 1:100) were used. For immunostainings in a 96 well-plate format, iMG or BMDMs were fixed for 5 minutes with 3.7% formaldehyde in PBS and rinsed 2X with a PBS solution containing 0.05% saponin and 5% BSA. Cells were then permeabilized and blocked with the same solution for 1h at room temperature. Primary antibodies were diluted in the same solution and incubated overnight at 4°C. Samples were then rinsed twice with PBS containing 0.05% Saponin and 5% BSA and incubated with secondary antibodies for 1.5 h at room temperature in the dark. Finally, samples were rinsed 2X with 1X PBS prior to imaging with an Opera Phenix (Perkin Elmer) high content microscope at 40X or 63X magnification (water immersion) or on a Leica SP8 microscope using a 63x/1.4 N.A. oil immersion objective with a pixel size of ~30nm at 63X magnification in lightning acquisition mode. For super-resolution images, lightning processing was performed in adaptive mode and images were then visualized and processed in LAS X and ImageJ.
Endolysosomal membrane damage was assessed in iMG by immunofluorescence analysis using an anti-galectin-3 mouse mAb (BD Biosciences #556904, 1:250). Galectin-3 fluorescence intensity was quantified automatically with the Harmony HSC software (Perkin Elmer). Assessment of galectin-3 positive cells was conducted with ImageJ. Briefly, galectin-3 positive cells were marked and counted using the “Analyze particle” function. DAPI fluorescence was used to count the total number of cells and to represent the proportion of galectin-3-positive cells. Vacuolization of iMGs was assessed using the DQ Red BSA reagent (Thermo Fisher Scientific, D12051). DQ Red BSA was incubated with iMGs at a final concentration of 10ug/mL in complete media and the cells were imaged live. Time lapse images were acquired every hour for 16h. Images from 5h post-DQ Red BSA treatment were used to score vacuolization. Briefly, vacuoles were counted using ImageJ and cells exhibiting one or more DQ-BSA-filled vacuole(s) larger than 5 pixels were scored as positive for the presence of DQ-BSA vacuoles. DAPI fluorescence was used to count the total number of cells and calculate the proportion of vacuolated cells.
Liposome preparation
For liposome preparation, 70 molar % DOPC (1,2-dioleoyl-sn-glycero-3-phosphocholine, Avanti Polar Lipids) and 30 molar % of the test lipid were combined in chloroform in a glass vial and dried under a stream of N2 gas for 1–2h, or until completely dry. Test lipids included BMP(22:6/22:6), PG(22:6/22:6) and POPS (1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine) (all from Avanti). The lipid mixture was re-suspended in HBSS (1mg/mL final lipid concentration) and vortexed for 2–3 min. Then liposomes were extruded 12 times using an Avanti mini-extruder constructed with one 100nm pore size membrane to form small unilamellar vesicles.
Liposome-PGRN binding assay
Liposomes were prepared in HBSS (1mg/ml final lipid concentration) as described above. 200μL of these liposome preparations were spun for 30 min at 100,000 x g at 22°C and supernatants were carefully removed. Liposome pellets were resuspended in 75μL of either MES buffer pH 4.5 (30mM MES, 110mM KCl, 20mM NaCl) or HEPES buffer pH 7.0 (30mM HEPES, 110mM KCl, 20mM NaCl). Liposomes were incubated for 20 min at room temperature with 4μg of recombinant human PGRN protein (Cat # 2420-PG, R&D systems, USA) on a horizontal shaker. Samples were then centrifuged at 100,000 x g for 30 min at 22°C and 75μL of supernatants were harvested. Liposome pellets were resuspended in 100μL of 1X LDS buffer (containing 4X LDS, Cat # NP007, Invitrogen; 10X Reducing agent, Cat # NP0009, Invitrogen; Protease Inhibitors, Roche and PBS) and supernatants were resuspended in 25μL of 4X LDS buffer (containing 4X LDS, Cat # NP007, Invitrogen; 10X Reducing agent, Cat # NP0009, Invitrogen; Protease Inhibitors, Roche). Samples were boiled for 5 min at 95°C and run on an SDS-PAGE, prior to staining of the gel with Coomassie Blue (InstantBlue® Coomassie, Abcam # 119211, USA). Image acquisition was performed with the 700nm channel on a LI-COR Odyssey (LI-Cor Biosciences) and protein signals were quantified using Image J.
Tissue sample homogenization for protein assays
Weighed tissue samples (brain or liver) were processed for protein assays by addition of 10X volume NP-40 buffer with added cOmplete Protease Inhibitor (Roche #04693132001) and PhosStop (Roche 04906837001) phosphatase inhibitors. Samples were then homogenized using a 3mm tungsten carbide bead, shaken using the Qiagen TissueLyzer II (Cat No./ID: 85300) (2×3 minutes at 30 Hz). Samples were centrifuged at 14,000g for 20 min at 4°C, then supernatant was aliquoted for analysis. For assays normalized for total protein (e.g., GCase activity), BCA was used to quantify protein levels.
GCase activity assay in brain and tissue extracts
Protein estimation of the lysate was done by BCA assay and then the lysate was normalized to a concentration of 2 mg/mL before GCase activity assay was performed. Normalized tissue lysate was diluted 50-fold, and 4-Methylumbelliferyl β-D-glucopyranoside stock (30mM; Sigma-Aldrich, M3633–1G) was diluted 3-fold respectively in the GCase activity buffer (100 mM Phosphate Citrate Buffer (pH 5.2), 0.5% Sodium Taurocholate, 0.25% Triton-X 100). 90 μL of the diluted tissue lysate and 10 μL of diluted 4-Methylumbelliferyl β-D-glucopyranoside was added to a 96-well round bottom black Polypropylene plate (Corning #3356). Plate was sealed and vortexed for 5 minutes at 700 rpm before placing the plate in a non-CO2 37°C incubator for 3 hours. 100 μL/well of STOP solution (500 mM Glycine, 300 mM NaOH pH 9.8) was added to stop the reaction, and the signal intensity was read using a BioTek plate reader (Read Ex 365/Em 445).
For the GCase assay in the presence of liposomes, tissue lysates and buffers were prepared as described above with the following modifications. Tissue from 6–7 months old mice was homogenized in PBS with protease and phosphatase inhibitors and sonicated at 75% amplitude for 20×1sec pulses before spinning. Lysates were normalized to 1 mg/mL following a BCA assay. GCase assay activity buffer was prepared without 0.2% Triton-X 100 to preserve liposome integrity. BCA normalized lysates (1 mg/mL) were incubated with liposomes at various lysate:liposome ratios for 30 minutes at room temperature with shaking (700 rpm). Lysate with liposome was then diluted 50-fold in GCase activity buffer and incubated with 4-Methylumbelliferyl β-D-glucopyranoside as described above. GCase assay for brain:liposome condition was done overnight to increase the overall signal, whereas liver:liposome GCase assay was kept for 5h before adding STOP solution.
Western blot analysis of GCase and Cathepsin D
Brain and liver homogenates were generated as detailed above. BMDMs cultured in 96-well format were washed 2x with PBS and then collected in 25uL 1x CST Buffer (Cell Signaling, #9803S) supplemented with PMSF. 1mg/mL tissue/cell lysates were prepared for SDS-PAGE by diluting with 4X NuPage Sample Buffer (Invitrogen #NP007), 10X NuPage Reducing Buffer (Invitrogen #NP009), and RIPA Buffer (Teknova #R379). Samples were boiled for 5 min, and then run on a 4–12% Bis-Tris gel (Invitrogen #NP0322) in MES buffer (Invitrogen #NP0002), 120V for 1.5 hours. Gel was then transferred on the Bio-Rad semi-dry transfer apparatus mixed molecule weight program onto nitrocellulose. Transferred blot was blocked with Intercept protein-free blocking buffer (Licor, #927–80001). Blot was then probed for GCase (Sigma #G4171, 1:1000) and GAPDH (Sigma #G8795, 1:2500) for liver and brain tissues or Cathepsin D (Abcam #ab78552, 1:1000) and beta-Actin (Sigma #A2228, 1:3000) for BMDM cell lysates and detected via fluorescent secondaries (Licor) while incubating in LI-COR Intercept Blocking Buffer. After washing, blot was imaged and quantified on the Odyssey scanner, and analyzed in Prism by one-way ANOVA and Dunnett’s multiple comparison test.
TREM2 analysis in brain tissue and BMDMs
For the analysis of total cellular and soluble TREM2 in BMDMs, cell supernatants were removed and cells were lysed in a solution containing Cell lysis buffer (Cell Signaling # 9803) supplemented with the protease inhibitor phenylmethanesulfonyl fluoride (PMSF). Cell lysates and supernatants were diluted 1:10 and 1:20, respectively, and an aliquot of 50 μl was used for analyses. For tissue analysis, samples were homogenized as described above. For the TREM2 Mesoscale Discovery (MSD) measurements, MSD small spot streptavidin plates were coated with biotinylated anti-mTREM2 polyclonal antibody (R&D Systems BAF1729) overnight at 40°C. The next day, plates were blocked with 3% BSA/TBST for at least 1h at room temperature, shaking at 650 rpm. After washing 3X with TBST, samples were diluted in 3% BSA/TBST and added to the assay plate after blocking. A mouse TREM2-His protein diluted in 3% BSA/TBST was used as a standard for quantification. Following 2h incubation at room temperature (650 rpm), plates were washed again 3X with TBST. A sulfo-tagged detection antibody (sheep anti-mouse TREM2, AF1729 R&D Systems), diluted in 3% BSA/TBST was added and incubated for 1h at room temperature (650 rpm). After washing with TBST, MSD plates were developed using 2x MSD read buffer T, followed by detection using an MSD Meso Sector S600 reader. MSD values were converted to absolute quantities of TREM2 by interpolating from a 4-parameter logistic curve fit to the mouse TREM2-His protein standard using Graphpad Prism software. For the same samples BCA protein analysis was performed and TREM2 levels were normalized using the protein concentrations.
CSF neurofilament light chain analysis
Cerebrospinal fluid (CSF) neurofilament light chain (Nf-L) levels were analyzed as described previously (Ullman et al., 2020) using the Quanterix Simoa Neurofilament Light Advantage (NFL) kit. Briefly, CSF was diluted 100x in sample diluent (Quanterix 102252) then Simoa detector reagent and bead reagent (Quanterix 103159, 102246) were added and samples were incubated for 30 mins, at 30°C, shaking at 800rpm. Following this, the sample plate was washed with Simoa Wash Buffer A (Quanterix 103078) using a Simoa Microplate Washer according to the Quanterix two step protocol. SBG reagent (Quanterix 102250) was added, and samples were again incubated at 30°C, 800rpm for an additional 10 min. The two-step washer protocol was continued, with the sample beads being twice resuspended in Simoa Wash Buffer B (Quanterix 103079) before final aspiration of buffer. After drying for 10 min at RT, sample Nf-L concentrations were measured using the Nf-L analysis protocol on the Quanterix SR-X instrument and interpolated against a calibration curve provided with the Quanterix assay kit.
Biomarker analyses in human biofluid samples
Analyte identities were confirmed with authentic compounds and each analyte’s signal was normalized to a corresponding internal standard. PGRN levels were measured using an ELISA assay (#DPGRN0, R&D systems) and Nf-L levels were measured using the Quanterix Simoa assay (#103400), as described above.
QUANTIFICATION AND STATISTICAL ANALYSIS
For the LCMS analysis of young and old Grn mutant mice in Figure 1, measured values were transformed using a log base 2 transformation. Differences between Grn+/+ and Grn−/− levels in select lipids and metabolites are estimated using a robust linear model adjusting for covariates of gender, age and the interaction between age and genotype in the model. Due to the exploratory nature of this analysis, p-values were adjusted for multiple comparison using the Benjamin-Hochberg methodology. However, for the BMP and GlcSph analyses in Figure S1, unadjusted p values were reported because multiple LCMS analyses prior to this analysis had established the deficiency of BMP species and the increase in GlcSph and therefore this component of the analysis was not exploratory. All the in vitro experiments were conducted with 3 or more biological replicates and, unless otherwise indicated, analyzed with one-way ANOVA with post-hoc Dunnett’s multiple comparison test. Normality was assessed in all preclinical in vivo datasets using Graphpad Prism. Normal distribution was tested for using the Shapiro-Wilk test. For datasets with low inter-group variability, the residuals of the entire dataset were evaluated. For datasets with high inter-group variability (e.g., Fig. 5B), the normality of individual groups was assessed. For large datasets (e.g., Fig. S1D–L), normality was also assessed with the Kolmogorov-Smirnov test. If either test was passed, linear regression analysis was completed as described above. In instances in which a normal distribution was not observed, non-parametric analysis (Kruskal-Wallis with Dunn’s post-hoc multiple comparisons) was used instead. Parametric in vivo experiments were analyzed with one-way ANOVA with Dunnett’s post-hoc multiple comparison test or with two-way ANOVA with Sidak’s post-hoc multiple comparison test. For correlation analysis in preclinical studies, a simple linear regression analysis was performed. All studies and samples analyses were performed blinded and in randomized order. All analytes were run in technical replicates of 2–3 (when feasible, e.g. – not for CSF Nf-L samples due to sample limitations). Studies with multiple assays were designed to be powered for the most variable analyte based on previous data. Exclusion of mice only occurred if confounding health concerns were identified during the study. Data was only excluded in circumstances when extreme outliers were identified (ROUT, Q=0.5%). Post-hoc analysis of the impact of sex on phenotypes or treatment response was performed with results from the 8-week chronic dosing study by unpaired t-test. As no differences were detected here, no further characterization of sex was evaluated for additional studies. Statistical comparisons for sorted cell lipidomic heatmaps were performed using MetaboAnalyst 4.0 (www.metaboanalyst.ca). All heatmaps were generated in R using the Complex Heatmap package. Data represented as mean ± SEM for in vitro and in vivo graphs. N is defined as the number of independent experiments or biological replicates in an in vitro study and as the number of mice in a in vivo study.
For statistical analysis of FTD patient data, differentiations were made by diagnosis (FTD vs Clinically Normal) and/or mutation status (GRN+ vs Non-GRN). Note that the latter information is missing for many of the FTD patients. In instances where mutation status is unknown, the subjects are grouped in the non-GRN category. Estimates and p-values are based on a model fit adjusted for data source (UCSF vs NCRAD). Given that key demographic information (Sex, Age and Race) is either homogenous or missing from a number of subjects, these are omitted from the model until we can obtain any additional information. Note that p-values are reported as unadjusted p-values as there is ambiguity with the level of multiple adjustment that would be required. Figures show geometric mean of measured analytes for each subject.
Supplementary Material
Table S4. LCMS acquisition parameters for lipidomics in negative ionization mode. Related to STAR Methods section “Lipidomic analysis”.
Table S5. LCMS acquisition parameters for lipidomics in positive ionization mode. Related to STAR Methods section “Lipidomic analysis”.
Table S7. LCMS acquisition parameters for metabolomics. Related to STAR Methods section “Metabolomics analysis”.
Figure S1: GRN-FTD patients’ biofluids show reduced PGRN, increased Nf-L, and aberrant lysosomal lipids, related toFigure 1. (A-C) Volcano plot visualization of lipids and metabolites elevated (orange) or reduced (blue) in tissues and fluids from 6mo. Grn−/− mice relative to age-matched Grn+/+ mice, both on the TfRmu/hu knock-in background (log2 transformed). Plotted p-values were adjusted for covariates of sex. Adjusted analyte values with an FDR < 5% and a fold change of >20% are labeled with filled squares (linear regression model with Benjamini-Hochberg multiple comparisons correction). (A) Volcano plot depiction of lipids and metabolites in plasma (log2 transformed, n=9–10/ genotype). (B) Volcano plot depiction of lipids and metabolites in liver tissue (log2 transformed, n=9–10/ genotype). (C) Volcano plot depiction of lipids and metabolites in urine (log2 transformed, n=9/genotype). (D-L) Abundance of lipid and protein biomarkers in plasma of FTD patients with (Y) or without (N) GRN mutations relative to healthy controls (FTD diagnosis: −) Healthy controls with GRN mutations indicated in blue. (D-E) Human PGRN levels are reduced in the CSF (D, Linear regression, p < 0.0001) and plasma (E, Linear regression, p < 0.0001) of GRN-FTD patients relative to healthy controls. (F-G) CSF and plasma Nf-L levels are elevated in FTD patients with or without GRN mutations (CSF: Linear regression, GRN(Y):FTD: p < 0.0001, GRN(N):FTD: p < 0.0001, Plasma: GRN(Y):FTD: p < 0.0001, GRN(N):FTD: p < 0.001). (H) CSF BMP(18:1/18:1) is reduced in FTD patients regardless of GRN mutation status (Linear regression, GRN(Y):FTD: p< 0.05, GRN(N):FTD: p< 0.05). (I) CSF BMP(22:6/22:6) shows a trend towards reduction in FTD patients (Linear regression, GRN(Y):FTD: p= 0.37, GRN(N):FTD: p= 0.41) (J) Plasma BMP(18:1/18:1) levels are unchanged in FTD patients (Linear regression, GRN(Y):FTD: p= 0.46, GRN(N):FTD: p= 0.97) regardless of GRN mutation status. (K) Plasma BMP(22:6/22:6) levels are unchanged in FTD patients (Linear regression, GRN(Y):FTD: p= 0.76, GRN(N):FTD: p= 0.73). (G) (L) Plasma GlcSph levels are increased selectively in the GRN-FTD patients relative to the healthy controls (Linear regression, p < 0.001). *p < 0.05, ***p < 0.001, ****p < 0.0001. Data represented as geometric mean ± SEM.
Figure S2. PGRN regulates lysosomal function via control of BMP levels, related toFigure 2. (A) Representative super resolution microscopic images of BMDMs showing partial colocalization of PGRN, BMP and LAMP-2 within the endolysosomal compartment. Scale bar: 2 μm. (B) Enlarged super resolution image of a Grn+/+ BMDM with selected lysosomal structures (i-iv) illustrating PGRN residing either within the lysosomal lumen, on the lysosomal limiting membrane, or both. (C) Representative line scans of selected lysosomes from (B) demonstrating colocalization of PGRN and LAMP-2 on the limiting membrane and of PGRN localized within the lysosomal lumen. (D) LCMS analysis of BMP(22:6/22:6) levels in Grn+/+ and Grn−/− BMDMs treated with the various liposome preparations for 16h. Data show an increase in BMP(22:6/22:6) levels in mutant cells treated with BMP(22:6/22:6) and PG(22:6/22:6) liposomes, but not with the other liposomes. (n = 3 independent experiments; ****, p < 0.0001). (E) LCMS analysis of BMP(20:4/20:4) levels in Grn+/+ and Grn−/− BMDMs treated with the various liposome preparations for 16h. Data show an decrease in BMP(20:4/20:4) levels in mutant cells treated with BMP(22:6/22:6) and PG(22:6/22:6) liposomes, but not with the other liposomes. (n = 3 independent experiments; ****, p < 0.0001). (F) Representative western blot of pro- and mature forms of Cathepsin D (CatD) in Grn+/+ and Grn−/− BMDM lysates (G) Quantification of pro-CatD levels showing no difference between Grn+/+ and Grn−/− BMDMs, or in response to various liposome treatments. (H) Quantification of mature CatD levels in Grn+/+ and Grn−/− BMDMs showing increase in mature cathepsin D levels in either PC-treated or BMP-treated Grn−/− BMDMs relative to PC-treated Grn+/+ BMDMs. (n = 3 independent experiments; * p < 0.05, ** p < 0.01 in one-way ANOVA and Dunnett’s multiple comparison). (I) Representative western blot of GCase levels in brain lysates from TfR KI or Grn−/−; TfR KI mice treated with either saline, Fc:PGRN or PTV:PGRN. (J, K) Quantification of GCase protein levels in mouse brain lysates showing no difference in GCase protein levels in Grn mutant brain (J) or liver (K) at baseline or in response to either Fc:PGRN or PTV:PGRN treatment. (n = 6 mice, ns by one-way ANOVA and Dunnett’s multiple comparison). (L) Quantification of the GCase activity in Grn+/+ and Grn−/− detergent-free liver extracts showing increase of GCase activity in the Grn−/− liver extracts treated with BMP and PG liposomes. (n = 4 mice, representative of 2 independent experiments; **** p < 0.0001 in one-way ANOVA and Dunnett’s multiple comparison). Data represented as geometric mean ± SEM.
Figure S3. Generation and characterization of the PTV:PGRN biologic, related toFigure 3. (A) SDS-PAGE of PTV:PGRN and Fc:PGRN constructs. 3 μg PTV:PGRN and Fc:PGRN fusion proteins (predicted MW ~115kDa) were run on SDS-PAGE under either non-reducing (- DTT) or reducing (+ DTT) conditions and stained with InstantBlue Coomassie. (B) Binding of PTV:PGRN and Fc:PGRN to human sortilin by ELISA. Recombinant human sortilin was plate-immobilized and then incubated with serial dilutions of either PTV:PGRN or Fc:PGRN fusion proteins or IgG control. PTV:PGRN and Fc:PGRN bound to sortilin with apparent affinities of 2.2nM and 1.8nM respectively. Curve values denote means ± SEM (n = 3 independent experiments). (C) Binding of PTV:PGRN and Fc:PGRN to human prosaposin by ELISA. Recombinant human prosaposin was plate-immobilized and then incubated with serial dilutions of either PTV:PGRN or Fc:PGRN fusion proteins or IgG control. PTV:PGRN and Fc:PGRN bound to prosaposin with apparent affinities of 5.1nM and 3.9nM respectively (n = 3 independent experiments). (D) Representative confocal microscopic images showing an increase in oxidized PC (oxPC) staining in Grn+/+ and Grn−/− BMDMs after treatment with oxidized LDL (oxLDL) particles. (E) Quantification of BMP (22:6/22:6) levels showing a decrease in both Grn+/+ and Grn−/− after a 72h treatment with 50ug/mL oxLDL, but not vehicle control. (n = 3 independent experiments; *, p < 0.05, ** p < 0.01 in two-way ANOVA with Tukey’s test). (F) Quantification of total TREM2 levels in lysates from BMDM analyzed in Fig. 3K showing an increase in Grn−/− BMDMs and a rescue after a 72h treatment with 50 nM PTV:PGRN, but not control IgG. Total TREM2 levels were measured by immunoassay. (n = 3 independent experiments; n.s. p > 0.1, **, p < 0.05 in one-way ANOVA with Dunnett’s multiple comparison test). (G) Binding of PTV:PGRNv2 to human sortilin by ELISA. Recombinant human sortilin was plate-immobilized and then incubated with serial dilutions of either PTV:PGRN, PTV:PGRNv2 or Fc:PGRN fusion proteins. PTV:PGRNv2 bound to sortilin with comparable apparent affinity (0.54nM) relative to PTV:PGRN and Fc:PGRN (0.47nM and 0.31nM respectively) (n = 2 independent experiments). (H) Binding of PTV:PGRNv2 to human prosaposin by ELISA. Recombinant human prosaposin was plate-immobilized and then incubated with serial dilutions of either PTV:PGRN, PTV:PGRNv2 or Fc:PGRN fusion proteins. PTV:PGRNv2 bound to prosaposin with comparable apparent affinity (1.28nM) relative to PTV:PGRN and Fc:PGRN (1.74nM and 1.14nM respectively) (n = 2 independent experiments). Data represented as geometric mean ± SEM.
Figure S4. PTV:PGRN rescues the endolysosomal vacuolization and membrane damage in GRN−/−iMG, related toFigure 4. (A) Time-lapse images showing DQ-BSA fluorescence in GRN−/− iMG. Images were acquired every hour using fluorescence microscopy. Images show fusion of vacuoles (thin arrow) and rupture of vacuoles (thick arrow) over the time-course. Scale bar: 25 μm. (B) Quantification of galectin-3 fluorescence intensity in GRN+/+ and GRN−/− iMG with no treatment (NT) or after a treatment with IgG control or PTV:PGRN each day for 3 days. PTV:PGRN rescued the endolysosomal membrane damage in two independent GRN−/− iMG clones. (n = 4 independent experiments; n.s., p > 0.1, *, p < 0.05, **, p < 0.01, ***, p < 0.001, ****, p < 0.0001 in one-way ANOVA with Dunnett’s multiple comparison test). Data represented as geometric mean ± SEM.
Figure S5. Increased brain penetrance of PTV:PGRN vs. Fc:PGRN resulting in greater lysosomal rescue in both acute and chronic dosing paradigms, related toFigure 5. (A-B) Concentration of intact PTV:PGRN in plasma (A) and brain (B) as determined by an Fc/PGRN sandwich ELISA in 3mo. Grn−/−; TfRmu/hu mice (n=4/group). (A) PTV:PGRN (dark orange = 50 mg/kg, light orange = 5mg/kg) and Fc:PGRN (dark blue = 50mg/kg, light blue = 5mg/kg) exhibit decline of full length drug concentration in plasma in addition to clearance observed for PTV:PGRN. (B) Brain concentrations of PTV:PGRN are higher than Fc:PGRN at either dose(n=4/group, Two-way ANOVA with Sidak’s multiple comparison test, 50mg/kg PTV:PGRN: padj<0.0001, 5mg/kg PTV:PGRN: padj<0.001). (C) Isolation of brain parenchyma reveals higher concentrations of PTV:PGRN at either dose, beyond the vascular barrier as measured by Fc/Fc ELISA (n=4/group, Two-way ANOVA with Sidak’s multiple comparison test, 50mg/kg PTV:PGRN: padj<0.0001, 5mg/kg PTV:PGRN: padj<0.001). (D) At 4h post-dose, Fc:PGRN shows equal or greater liver concentrations of Fc than PTV:PGRN at an equivalent dose (n=4/group, Two-way ANOVA with Sidak’s multiple comparison test, 50mg/kg PTV:PGRN: padj<0.0001, 5mg/kg PTV:PGRN: padj=0.9993). (E-F) Peripheral rescue of BMP(22:6/22:6) by PTV:PGRN or Fc:PGRN in liver (E) or plasma (F) following IV dosing. At 24h, rescue in these tissues/fluids is achieved completely (liver, One way ANOVA with Dunnett’s multiple comparison) or partially (plasma) with either PGRN construct (n=4/group, Two-way ANOVA with Sidak’s multiple comparison test). (G-I) Rescue of GlcSph in brain (G) liver (H) or plasma (I) by Fc:PGRN (blue) or PTV:PGRN (orange) (n=4/group, One way ANOVA with Dunnett’s multiple comparison (G,H), or Two-way ANOVA with Sidak’s multiple comparison test (I)). (G) A trend towards brain GlcSph rescue is observed at 24h following PTV:PGRN treatment (orange), but no correction is detected at 4h. (H) Modest modulation of liver GlcSph by PTV:PGRN 24h following IV delivery. (I) Plasma GlcSph is partially corrected by any PGRN fusion variant at 24h, but no differences are detected at earlier timepoints (n=4/group). (J) Plasma concentration of Fc as detected by ELISA 1 week following the final dose in a 6 week study with either 5mg/kg Fc:PGRN or PTV:PGRN. Fc only above the detection threshold in plasma with Fc:PGRN treatment, not with PTV:PGRN. (K-N) Pharmacokinetic and pharmacodynamic responses to 6 weeks of weekly 5mg/kg doses of either Fc:PGRN (blue) or PTV:PGRN (orange) in Grn−/− mice (n=7–10/group, One way ANOVA with Dunnett’s multiple comparison). (K) Equivalent rescue of liver BMP (22:6/22:6) by weekly administration of both Fc:PGRN and PTV:PGRN. (L) Rescue of GlcSph in the liver of Grn−/− is driven by both Fc:PGRN and PTV:PGRN. (M) PTV:PGRN significantly increases plasma BMP(22:6/22:6). (N) GlcSph in the plasma is rescued by both Fc:PGRN & PTV:PGRN. #p<0.1, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Data represented as mean ± SEM.
Figure S7: Peripheral TREM2 elevation in Grn−/−; TfRmu/humice is rescued by PTV:PGRNv2, related toFigure 7. (A) Representative images of microgliosis immunofluorescence markers (IBA1, GFAP, & CD68) in thalamus and rescue with PTV:PGRNv2 (scale bars = 10μm). (B-C) Total TREM2 levels in peripheral tissues (B, liver) and fluids (C, plasma) following 8 weeks of weekly or every other week (EOW) PTV:PGRNv2 treatment in 9mo Grn−/−; TfRmu/hu mice. (n=6/group, One-way ANOVA with Dunnett’s multiple comparison test). (A) Liver TREM2 levels are significantly upregulated in Grn−/−; TfRmu/hu mice and rescued by either weekly or EOW PTV:PGRNv2. (B) Plasma soluble TREM2 levels are significantly upregulated in Grn−/−; TfRmu/hu mice and reduced by both treatment intervals of PTV:PGRNv2. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Data represented as mean ± SEM.
Figure S6: Validation of methods involved with 8-week chronic dosing study and additional outcomes, related toFigure 6. (A) Dose titration indicates that BMP(22:6/22:6) correction by PTV:PGRN or PTV:PGRNv2 can be achieved at doses as low as 1.5 mg/kg, 7 days following IV delivery in 3mo. Grn−/−; TfRmu/hu mice (n=6–8/group, no statistical analysis). (B) Timecourse of brain BMP(22:6/22:6) rescue out to two weeks with IV or IP dosed PTV:PGRNv2 at 5mg/kg in 3mo. Grn−/−; TfRmu/hu mice (n=6/group, Two-way ANOVA with Sidak’s multiple comparison test). (C-H) Confirmation that EOW anti-CD4 treatment for 8 weeks does not impact primary lysosomal and gliosis phenotypes in 3mo. Grn−/−; TfRmu/hu mice (n=6/group, One way ANOVA with Dunnett’s multiple comparison). (C) Brain BMP(22:6/22:6) does not change with 8 weeks of anti-CD4 dosing (D) Representative coronal hemibrain images of gliosis markers with 8 weeks of anti-CD4 dosing (DAPI = blue, GFAP = green, CD68 = red, IBA1 = magenta, n=6/group, scale bars = 1mm). (E) Brain TREM2 levels in Grn−/−; TfRmu/hu are not impacted by 8 weeks of anti-CD4 dosing. (F) Quantification of total brain GFAP immunofluorescence area does not change with 8 weeks of anti-CD4 dosing. (G) Total brain CD68 does not change with 8 weeks of anti-CD4 dosing. (H) Brain IBA1 signal does not change with 8 weeks of anti-CD4 dosing. (I) Heatmap representation of cell type-marker transcript expression in sorted cell populations from 9mo. Grn−/−; TfRmu/hu mice, as quantified by qPCR. Columns represent biological replicates from each cell population, rows represent transcripts. Values are log2 transformation of fold change values (n=6/group). (J) Bar plots of BMP (20:4/20:4) from microglia, astrocytes and neurons, demonstrating increased levels across CNS cell types with weekly and EOW PTV:PGRNv2 treatment (9mo. mice). (K-M) Characterization of lipid changes in 4mo. Grn R493X; TfRmu/hu mouse brain and responsiveness to PTV:PGRNv2 (n=6/group, One way ANOVA with Dunnett’s multiple comparison). (K) BMP(22:6/22:6) is reduced in 4mo. Grn R493X mouse brain and is rescued by PTV:PGRNv2. (L) Brain BMP(18:1/18:1) is reduced in Grn R493X mice and is restored by PTV:PGRNv2. (M) GlcSph is elevated in brain of Grn R493X mice and reduced by PTV:PGRNv2. (N) TREM2 is elevated in the brain of Grn R493X mice but is not corrected by PTV:PGRNv2. #p<0.1, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Data represented as mean ± SEM.
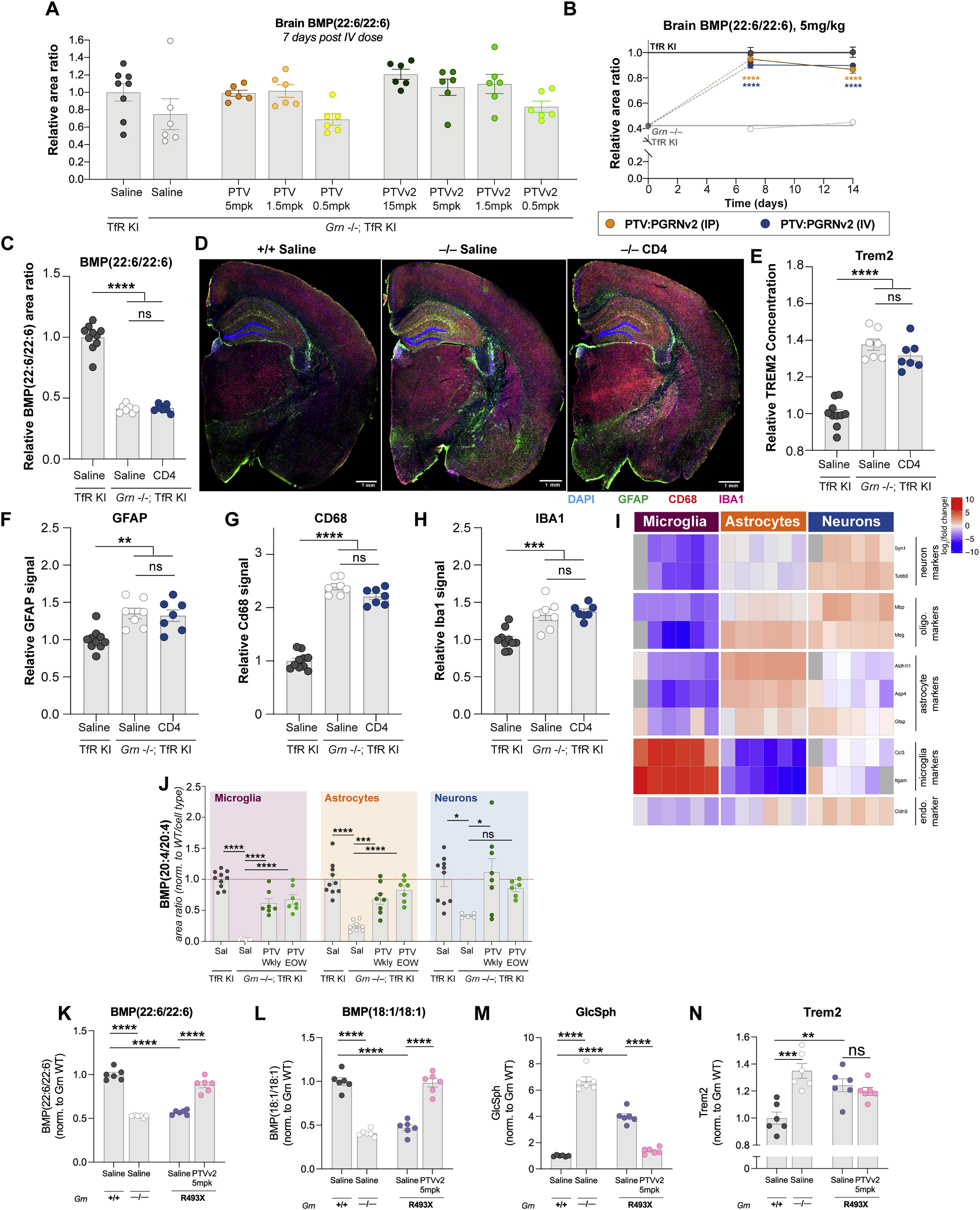{kind=link}
Highlights.
Grn−/− mice exhibit robust BMP lipid deficiency with concurrent GlcSph accumulation
BMP stimulates glucocerebrosidase activity and lysosome function in Grn−/− models
PGRN was fused to a human transferrin receptor binding Fc to increase CNS exposure
PTV:PGRN biologic rescues Grn−/− CNS lysosomal, microglial and neuronal dysfunction
ACKNOWLEDGEMENTS
We thank Adam Catherman, Victoria Assimon, Hai Tran, and Yuda Zhu for their work at the project early stage; Mike Maloney, Nick Propson and John Lin for help with various assays; and Heather Hogg, Mark Jimenez-Canet, Tony Delucchi, Zach Sweeney, Karen Lai and Robert Thorne for helpful discussions. CSF and plasma samples were obtained from Samples from the National Centralized Repository for Alzheimer’s Disease and Related Dementias (NCRAD) under a cooperative agreement grant (U24 AG021886) awarded by NIA. Biofluid samples and clinic data presented here were from the LEFFTDS & ARTFL Consortia (now combined into ALLFTD: U19AG063911): LEFFTDS: U01 AG045390 funded by NIA and NINDS; ARTFL: U54 NS092089, funded by NINDS and NCATS. The authors acknowledge ALLFTD investigators, contributions of participants in ARTFL & LEFFTDS and assistance of the support staffs at participating sites.
DECLARATION OF INTERESTS
All authors, except ALB, HH, HJR and BFB, are full time employees and/or shareholders of Denali Therapeutics. ALB reports consultancy for AGTC, Alector, Arkuda, Arvinas, Asceneuron, AZTherapies, Bioage, GSK, Humana, Lundbeck, Ono, Roche, Samumed, Sangamo, Stealth Therapeutics, Third Rock, Transposon, UCB and Wave and research support from the Association for Frontotemporal Degeneration, Biogen, Bluefield Project to Cure Frontotemporal Dementia, Eli Lilly, Eisai, National Institutes of Health (grant numbers U19AG063911, U54NS092089, R01AG031278), and the Rainwater Charitable Foundation. BFB reports research support from the NIH (U19AG063911). HJR reports funding from the NIH and consultancy with Alector, Ionis, Biogen, Wave, Takeda, and AFTD. All other authors declare no competing interests. This work has been in part described in one or more pending patent applications.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Abdul-Hammed M, Breiden B, Schwarzmann G, and Sandhoff K (2017). Lipids regulate the hydrolysis of membrane bound glucosylceramide by lysosomal beta-glucocerebrosidase. J Lipid Res 58, 563–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreone BJ, Przybyla L, Llapashtica C, Rana A, Davis SS, van Lengerich B, Lin K, Shi J, Mei Y, Astarita G, et al. (2020). Alzheimer’s-associated PLCgamma2 is a signaling node required for both TREM2 function and the inflammatory response in human microglia. Nat Neurosci 23, 927–938. [DOI] [PubMed] [Google Scholar]
- Arrant AE, Roth JR, Boyle NR, Kashyap SN, Hoffmann MQ, Murchison CF, Ramos EM, Nana AL, Spina S, Grinberg LT, et al. (2019). Impaired beta-glucocerebrosidase activity and processing in frontotemporal dementia due to progranulin mutations. Acta Neuropathol Commun 7, 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, et al. (2006). Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442, 916–919. [DOI] [PubMed] [Google Scholar]
- Beel S, Moisse M, Damme M, De Muynck L, Robberecht W, Van Den Bosch L, Saftig P, and Van Damme P (2017). Progranulin functions as a cathepsin D chaperone to stimulate axonal outgrowth in vivo. Hum Mol Genet 26, 2850–2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady OA, Martina JA, and Puertollano R (2018). Emerging roles for TFEB in the immune response and inflammation. Autophagy 14, 181–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breiden B, and Sandhoff K (2020). Mechanism of Secondary Ganglioside and Lipid Accumulation in Lysosomal Disease. Int J Mol Sci 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler VJ, Cortopassi WA, Gururaj S, Wang AL, Pierce OM, Jacobson MP, and Kao AW (2019). Multi-Granulin Domain Peptides Bind to Pro-Cathepsin D and Stimulate Its Enzymatic Activity More Effectively Than Progranulin in Vitro. Biochemistry 58, 2670–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrnes KR, Washington PM, Knoblach SM, Hoffman E, and Faden AI (2011). Delayed inflammatory mRNA and protein expression after spinal cord injury. J Neuroinflammation 8, 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calafate S, Flavin W, Verstreken P, and Moechars D (2016). Loss of Bin1 Promotes the Propagation of Tau Pathology. Cell Rep 17, 931–940. [DOI] [PubMed] [Google Scholar]
- Chen-Plotkin AS, Xiao J, Geser F, Martinez-Lage M, Grossman M, Unger T, Wood EM, Van Deerlin VM, Trojanowski JQ, and Lee VM (2010). Brain progranulin expression in GRN-associated frontotemporal lobar degeneration. Acta Neuropathol 119, 111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruchaga C, Graff C, Chiang HH, Wang J, Hinrichs AL, Spiegel N, Bertelsen S, Mayo K, Norton JB, Morris JC, et al. (2011). Association of TMEM106B gene polymorphism with age at onset in granulin mutation carriers and plasma granulin protein levels. Arch Neurol 68, 581–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, Rademakers R, Vandenberghe R, Dermaut B, Martin JJ, et al. (2006). Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 442, 920–924. [DOI] [PubMed] [Google Scholar]
- Dai M, Liou B, Swope B, Wang X, Zhang W, Inskeep V, Grabowski GA, Sun Y, and Pan D (2016). Progression of Behavioral and CNS Deficits in a Viable Murine Model of Chronic Neuronopathic Gaucher Disease. PLoS One 11, e0162367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deleon J, and Miller BL (2018). Frontotemporal dementia. Handb Clin Neurol 148, 409–430. [DOI] [PubMed] [Google Scholar]
- Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, et al. (2016). Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol 34, 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evers BM, Rodriguez-Navas C, Tesla RJ, Prange-Kiel J, Wasser CR, Yoo KS, McDonald J, Cenik B, Ravenscroft TA, Plattner F, et al. (2017). Lipidomic and Transcriptomic Basis of Lysosomal Dysfunction in Progranulin Deficiency. Cell Rep 20, 2565–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng T, Mai S, Roscoe JM, Sheng RR, Ullah M, Zhang J, Katz II, Yu H, Xiong W, and Hu F (2020). Loss of TMEM106B and PGRN leads to severe lysosomal abnormalities and neurodegeneration in mice. EMBO Rep, e50219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finch N, Carrasquillo MM, Baker M, Rutherford NJ, Coppola G, Dejesus-Hernandez M, Crook R, Hunter T, Ghidoni R, Benussi L, et al. (2011). TMEM106B regulates progranulin levels and the penetrance of FTLD in GRN mutation carriers. Neurology 76, 467–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghidoni R, Benussi L, Glionna M, Franzoni M, and Binetti G (2008). Low plasma progranulin levels predict progranulin mutations in frontotemporal lobar degeneration. Neurology 71, 1235–1239. [DOI] [PubMed] [Google Scholar]
- Glaser M, Wanaski S, Buser CA, Boguslavsky V, Rashidzada W, Morris A, Rebecchi M, Scarlata SF, Runnels LW, Prestwich GD, et al. (1996). Myristoylated alanine-rich C kinase substrate (MARCKS) produces reversible inhibition of phospholipase C by sequestering phosphatidylinositol 4,5-bisphosphate in lateral domains. J Biol Chem 271, 26187–26193. [DOI] [PubMed] [Google Scholar]
- Gotzl JK, Brendel M, Werner G, Parhizkar S, Sebastian Monasor L, Kleinberger G, Colombo AV, Deussing M, Wagner M, Winkelmann J, et al. (2019). Opposite microglial activation stages upon loss of PGRN or TREM2 result in reduced cerebral glucose metabolism. EMBO Mol Med 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotzl JK, Colombo AV, Fellerer K, Reifschneider A, Werner G, Tahirovic S, Haass C, and Capell A (2018). Early lysosomal maturation deficits in microglia triggers enhanced lysosomal activity in other brain cells of progranulin knockout mice. Mol Neurodegener 13, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotzl JK, Mori K, Damme M, Fellerer K, Tahirovic S, Kleinberger G, Janssens J, van der Zee J, Lang CM, Kremmer E, et al. (2014). Common pathobiochemical hallmarks of progranulin-associated frontotemporal lobar degeneration and neuronal ceroid lipofuscinosis. Acta Neuropathol 127, 845–860. [DOI] [PubMed] [Google Scholar]
- Gruenberg J (2020). Life in the lumen: The multivesicular endosome. Traffic 21, 76–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, McManus RM, and Latz E (2018). Inflammasome signalling in brain function and neurodegenerative disease. Nat Rev Neurosci 19, 610–621. [DOI] [PubMed] [Google Scholar]
- Holler CJ, Taylor G, Deng Q, and Kukar T (2017). Intracellular Proteolysis of Progranulin Generates Stable, Lysosomal Granulins that Are Haploinsufficient in Patients with Frontotemporal Dementia Caused by GRN Mutations. eNeuro 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu F, Padukkavidana T, Vaegter CB, Brady OA, Zheng Y, Mackenzie IR, Feldman HH, Nykjaer A, and Strittmatter SM (2010). Sortilin-mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron 68, 654–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S, Sealock J, Karlsson IK, Hagg S, Athanasiu L, et al. (2019). Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet 51, 404–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jian J, Tian QY, Hettinghouse A, Zhao S, Liu H, Wei J, Grunig G, Zhang W, Setchell KDR, Sun Y, et al. (2016). Progranulin Recruits HSP70 to beta-Glucocerebrosidase and Is Therapeutic Against Gaucher Disease. EBioMedicine 13, 212–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao AW, McKay A, Singh PP, Brunet A, and Huang EJ (2017). Progranulin, lysosomal regulation and neurodegenerative disease. Nat Rev Neurosci 18, 325–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kariolis MS, Wells RC, Getz JA, Kwan W, Mahon CS, Tong R, Kim DJ, Srivastava A, Bedard C, Henne KR, et al. (2020). Brain delivery of therapeutic proteins using an Fc fragment blood-brain barrier transport vehicle in mice and monkeys. Sci Transl Med 12. [DOI] [PubMed] [Google Scholar]
- Krabbe G, Minami SS, Etchegaray JI, Taneja P, Djukic B, Davalos D, Le D, Lo I, Zhan L, Reichert MC, et al. (2017). Microglial NFkappaB-TNFalpha hyperactivation induces obsessive-compulsive behavior in mouse models of progranulin-deficient frontotemporal dementia. Proc Natl Acad Sci U S A 114, 5029–5034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CW, Stankowski JN, Chew J, Cook CN, Lam YW, Almeida S, Carlomagno Y, Lau KF, Prudencio M, Gao FB, et al. (2017). The lysosomal protein cathepsin L is a progranulin protease. Mol Neurodegener 12, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewcock JW, Schlepckow K, Di Paolo G, Tahirovic S, Monroe KM, and Haass C (2020). Emerging Microglia Biology Defines Novel Therapeutic Approaches for Alzheimer’s Disease. Neuron. [DOI] [PubMed] [Google Scholar]
- Lui H, Zhang J, Makinson SR, Cahill MK, Kelley KW, Huang HY, Shang Y, Oldham MC, Martens LH, Gao F, et al. (2016). Progranulin Deficiency Promotes Circuit-Specific Synaptic Pruning by Microglia via Complement Activation. Cell 165, 921–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marschallinger J, Iram T, Zardeneta M, Lee SE, Lehallier B, Haney MS, Pluvinage JV, Mathur V, Hahn O, Morgens DW, et al. (2020). Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat Neurosci 23, 194–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens LH, Zhang J, Barmada SJ, Zhou P, Kamiya S, Sun B, Min SW, Gan L, Finkbeiner S, Huang EJ, et al. (2012). Progranulin deficiency promotes neuroinflammation and neuron loss following toxin-induced injury. J Clin Invest 122, 3955–3959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCauliff LA, Langan A, Li R, Ilnytska O, Bose D, Waghalter M, Lai K, Kahn PC, and Storch J (2019). Intracellular cholesterol trafficking is dependent upon NPC2 interaction with lysobisphosphatidic acid. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeter LH, Dopper EG, Jiskoot LC, Sanchez-Valle R, Graff C, Benussi L, Ghidoni R, Pijnenburg YA, Borroni B, Galimberti D, et al. (2016). Neurofilament light chain: a biomarker for genetic frontotemporal dementia. Ann Clin Transl Neurol 3, 623–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda AM, Lasiecka ZM, Xu Y, Neufeld J, Shahriar S, Simoes S, Chan RB, Oliveira TG, Small SA, and Di Paolo G (2018). Neuronal lysosomal dysfunction releases exosomes harboring APP C-terminal fragments and unique lipid signatures. Nat Commun 9, 291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moisse K, Volkening K, Leystra-Lantz C, Welch I, Hill T, and Strong MJ (2009). Divergent patterns of cytosolic TDP-43 and neuronal progranulin expression following axotomy: implications for TDP-43 in the physiological response to neuronal injury. Brain Res 1249, 202–211. [DOI] [PubMed] [Google Scholar]
- Nalls MA, Blauwendraat C, Vallerga CL, Heilbron K, Bandres-Ciga S, Chang D, Tan M, Kia DA, Noyce AJ, Xue A, et al. (2019). Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol 18, 1091–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen AD, Nguyen TA, Zhang J, Devireddy S, Zhou P, Karydas AM, Xu X, Miller BL, Rigo F, Ferguson SM, et al. (2018). Murine knockin model for progranulin-deficient frontotemporal dementia with nonsense-mediated mRNA decay. Proc Natl Acad Sci U S A 115, E2849–E2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent AA, Lin K, van Lengerich B, Lianoglou S, Przybyla L, Davis SS, Llapashtica C, Wang J, Kim DJ, Xia D, et al. (2020). TREM2 Regulates Microglial Cholesterol Metabolism upon Chronic Phagocytic Challenge. Neuron 105, 837–854 e839. [DOI] [PubMed] [Google Scholar]
- Pandya H, Shen MJ, Ichikawa DM, Sedlock AB, Choi Y, Johnson KR, Kim G, Brown MA, Elkahloun AG, Maric D, et al. (2017). Differentiation of human and murine induced pluripotent stem cells to microglia-like cells. Nat Neurosci 20, 753–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos C, Kravic B, and Meyer H (2020). Repair or Lysophagy: Dealing with Damaged Lysosomes. J Mol Biol 432, 231–239. [DOI] [PubMed] [Google Scholar]
- Paushter DH, Du H, Feng T, and Hu F (2018). The lysosomal function of progranulin, a guardian against neurodegeneration. Acta Neuropathol 136, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan E, Seehra G, Sharma P, and Sidransky E (2019). GBA1-associated parkinsonism: new insights and therapeutic opportunities. Curr Opin Neurol 32, 589–596. [DOI] [PubMed] [Google Scholar]
- Schueler UH, Kolter T, Kaneski CR, Blusztajn JK, Herkenham M, Sandhoff K, and Brady RO (2003). Toxicity of glucosylsphingosine (glucopsychosine) to cultured neuronal cells: a model system for assessing neuronal damage in Gaucher disease type 2 and 3. Neurobiol Dis 14, 595–601. [DOI] [PubMed] [Google Scholar]
- Showalter MR, Berg AL, Nagourney A, Heil H, Carraway KL 3rd, and Fiehn O (2020). The Emerging and Diverse Roles of Bis(monoacylglycero) Phosphate Lipids in Cellular Physiology and Disease. Int J Mol Sci 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skibinski G, Parkinson NJ, Brown JM, Chakrabarti L, Lloyd SL, Hummerich H, Nielsen JE, Hodges JR, Spillantini MG, Thusgaard T, et al. (2005). Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet 37, 806–808. [DOI] [PubMed] [Google Scholar]
- Skowyra ML, Schlesinger PH, Naismith TV, and Hanson PI (2018). Triggered recruitment of ESCRT machinery promotes endolysosomal repair. Science 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KR, Damiano J, Franceschetti S, Carpenter S, Canafoglia L, Morbin M, Rossi G, Pareyson D, Mole SE, Staropoli JF, et al. (2012). Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage. Am J Hum Genet 90, 1102–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi YV, Liu J, Ruan J, Pacheco J, Zhang X, Abbasi J, Keutzer J, Mistry PK, and Chandra SS (2017). Glucosylsphingosine Promotes alpha-Synuclein Pathology in Mutant GBA-Associated Parkinson’s Disease. J Neurosci 37, 9617–9631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Matsuwaki T, Yamanouchi K, and Nishihara M (2013). Exacerbated inflammatory responses related to activated microglia after traumatic brain injury in progranulin-deficient mice. Neuroscience 231, 49–60. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Suzuki G, Matsuwaki T, Hosokawa M, Serrano G, Beach TG, Yamanouchi K, Hasegawa M, and Nishihara M (2017). Progranulin regulates lysosomal function and biogenesis through acidification of lysosomes. Hum Mol Genet 26, 969–988. [DOI] [PubMed] [Google Scholar]
- Ullman JC, Arguello A, Getz JA, Bhalla A, Mahon CS, Wang J, Giese T, Bedard C, Kim DJ, Blumenfeld JR, et al. (2020). Brain delivery and activity of a lysosomal enzyme using a blood-brain barrier transport vehicle in mice. Sci Transl Med 12. [DOI] [PubMed] [Google Scholar]
- Valdez C, Wong YC, Schwake M, Bu G, Wszolek ZK, and Krainc D (2017). Progranulin-mediated deficiency of cathepsin D results in FTD and NCL-like phenotypes in neurons derived from FTD patients. Hum Mol Genet 26, 4861–4872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdez C, Ysselstein D, Young TJ, Zheng J, and Krainc D (2020). Progranulin mutations result in impaired processing of prosaposin and reduced glucocerebrosidase activity. Hum Mol Genet 29, 716–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward ME, Chen R, Huang HY, Ludwig C, Telpoukhovskaia M, Taubes A, Boudin H, Minami SS, Reichert M, Albrecht P, et al. (2017). Individuals with progranulin haploinsufficiency exhibit features of neuronal ceroid lipofuscinosis. Sci Transl Med 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wauters E, Van Mossevelde S, Van der Zee J, Cruts M, and Van Broeckhoven C (2017). Modifiers of GRN-Associated Frontotemporal Lobar Degeneration. Trends Mol Med 23, 962–979. [DOI] [PubMed] [Google Scholar]
- Weber F, Bohrmann B, Niewoehner J, Fischer JAA, Rueger P, Tiefenthaler G, Moelleken J, Bujotzek A, Brady K, Singer T, et al. (2018). Brain Shuttle Antibody for Alzheimer’s Disease with Attenuated Peripheral Effector Function due to an Inverted Binding Mode. Cell Rep 22, 149–162. [DOI] [PubMed] [Google Scholar]
- Werner G, Damme M, Schludi M, Gnorich J, Wind K, Fellerer K, Wefers B, Wurst W, Edbauer D, Brendel M, et al. (2020). Loss of TMEM106B potentiates lysosomal and FTLD-like pathology in progranulin-deficient mice. EMBO Rep, e50241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wils H, Kleinberger G, Pereson S, Janssens J, Capell A, Van Dam D, Cuijt I, Joris G, De Deyn PP, Haass C, et al. (2012). Cellular ageing, increased mortality and FTLD-TDP-associated neuropathology in progranulin knockout mice. J Pathol 228, 67–76. [DOI] [PubMed] [Google Scholar]
- Yim WW, and Mizushima N (2020). Lysosome biology in autophagy. Cell Discov 6, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin F, Banerjee R, Thomas B, Zhou P, Qian L, Jia T, Ma X, Ma Y, Iadecola C, Beal MF, et al. (2010). Exaggerated inflammation, impaired host defense, and neuropathology in progranulin-deficient mice. J Exp Med 207, 117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Velmeshev D, Hashimoto K, Huang YH, Hofmann JW, Shi X, Chen J, Leidal AM, Dishart JG, Cahill MK, et al. (2020). Neurotoxic microglia promote TDP-43 proteinopathy in progranulin deficiency. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Brooks M, Jiang P, Koga S, Zuberi AR, Baker MC, Parsons TM, Castanedes-Casey M, Phillips V, Librero AL, et al. (2020). Loss of Tmem106b exacerbates FTLD pathologies and causes motor deficits in progranulin-deficient mice. EMBO Rep, e50197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Paushter DH, Feng T, Pardon CM, Mendoza CS, and Hu F (2017a). Regulation of cathepsin D activity by the FTLD protein progranulin. Acta Neuropathol 134, 151–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Paushter DH, Feng T, Sun L, Reinheckel T, and Hu F (2017b). Lysosomal processing of progranulin. Mol Neurodegener 12, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Paushter DH, Pagan MD, Kim D, Nunez Santos M, Lieberman RL, Overkleeft HS, Sun Y, Smolka MB, and Hu F (2019). Progranulin deficiency leads to reduced glucocerebrosidase activity. PLoS One 14, e0212382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Sun L, Bracko O, Choi JW, Jia Y, Nana AL, Brady OA, Hernandez JCC, Nishimura N, Seeley WW, et al. (2017c). Impaired prosaposin lysosomal trafficking in frontotemporal lobar degeneration due to progranulin mutations. Nat Commun 8, 15277. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S4. LCMS acquisition parameters for lipidomics in negative ionization mode. Related to STAR Methods section “Lipidomic analysis”.
Table S5. LCMS acquisition parameters for lipidomics in positive ionization mode. Related to STAR Methods section “Lipidomic analysis”.
Table S7. LCMS acquisition parameters for metabolomics. Related to STAR Methods section “Metabolomics analysis”.
Figure S1: GRN-FTD patients’ biofluids show reduced PGRN, increased Nf-L, and aberrant lysosomal lipids, related toFigure 1. (A-C) Volcano plot visualization of lipids and metabolites elevated (orange) or reduced (blue) in tissues and fluids from 6mo. Grn−/− mice relative to age-matched Grn+/+ mice, both on the TfRmu/hu knock-in background (log2 transformed). Plotted p-values were adjusted for covariates of sex. Adjusted analyte values with an FDR < 5% and a fold change of >20% are labeled with filled squares (linear regression model with Benjamini-Hochberg multiple comparisons correction). (A) Volcano plot depiction of lipids and metabolites in plasma (log2 transformed, n=9–10/ genotype). (B) Volcano plot depiction of lipids and metabolites in liver tissue (log2 transformed, n=9–10/ genotype). (C) Volcano plot depiction of lipids and metabolites in urine (log2 transformed, n=9/genotype). (D-L) Abundance of lipid and protein biomarkers in plasma of FTD patients with (Y) or without (N) GRN mutations relative to healthy controls (FTD diagnosis: −) Healthy controls with GRN mutations indicated in blue. (D-E) Human PGRN levels are reduced in the CSF (D, Linear regression, p < 0.0001) and plasma (E, Linear regression, p < 0.0001) of GRN-FTD patients relative to healthy controls. (F-G) CSF and plasma Nf-L levels are elevated in FTD patients with or without GRN mutations (CSF: Linear regression, GRN(Y):FTD: p < 0.0001, GRN(N):FTD: p < 0.0001, Plasma: GRN(Y):FTD: p < 0.0001, GRN(N):FTD: p < 0.001). (H) CSF BMP(18:1/18:1) is reduced in FTD patients regardless of GRN mutation status (Linear regression, GRN(Y):FTD: p< 0.05, GRN(N):FTD: p< 0.05). (I) CSF BMP(22:6/22:6) shows a trend towards reduction in FTD patients (Linear regression, GRN(Y):FTD: p= 0.37, GRN(N):FTD: p= 0.41) (J) Plasma BMP(18:1/18:1) levels are unchanged in FTD patients (Linear regression, GRN(Y):FTD: p= 0.46, GRN(N):FTD: p= 0.97) regardless of GRN mutation status. (K) Plasma BMP(22:6/22:6) levels are unchanged in FTD patients (Linear regression, GRN(Y):FTD: p= 0.76, GRN(N):FTD: p= 0.73). (G) (L) Plasma GlcSph levels are increased selectively in the GRN-FTD patients relative to the healthy controls (Linear regression, p < 0.001). *p < 0.05, ***p < 0.001, ****p < 0.0001. Data represented as geometric mean ± SEM.
Figure S2. PGRN regulates lysosomal function via control of BMP levels, related toFigure 2. (A) Representative super resolution microscopic images of BMDMs showing partial colocalization of PGRN, BMP and LAMP-2 within the endolysosomal compartment. Scale bar: 2 μm. (B) Enlarged super resolution image of a Grn+/+ BMDM with selected lysosomal structures (i-iv) illustrating PGRN residing either within the lysosomal lumen, on the lysosomal limiting membrane, or both. (C) Representative line scans of selected lysosomes from (B) demonstrating colocalization of PGRN and LAMP-2 on the limiting membrane and of PGRN localized within the lysosomal lumen. (D) LCMS analysis of BMP(22:6/22:6) levels in Grn+/+ and Grn−/− BMDMs treated with the various liposome preparations for 16h. Data show an increase in BMP(22:6/22:6) levels in mutant cells treated with BMP(22:6/22:6) and PG(22:6/22:6) liposomes, but not with the other liposomes. (n = 3 independent experiments; ****, p < 0.0001). (E) LCMS analysis of BMP(20:4/20:4) levels in Grn+/+ and Grn−/− BMDMs treated with the various liposome preparations for 16h. Data show an decrease in BMP(20:4/20:4) levels in mutant cells treated with BMP(22:6/22:6) and PG(22:6/22:6) liposomes, but not with the other liposomes. (n = 3 independent experiments; ****, p < 0.0001). (F) Representative western blot of pro- and mature forms of Cathepsin D (CatD) in Grn+/+ and Grn−/− BMDM lysates (G) Quantification of pro-CatD levels showing no difference between Grn+/+ and Grn−/− BMDMs, or in response to various liposome treatments. (H) Quantification of mature CatD levels in Grn+/+ and Grn−/− BMDMs showing increase in mature cathepsin D levels in either PC-treated or BMP-treated Grn−/− BMDMs relative to PC-treated Grn+/+ BMDMs. (n = 3 independent experiments; * p < 0.05, ** p < 0.01 in one-way ANOVA and Dunnett’s multiple comparison). (I) Representative western blot of GCase levels in brain lysates from TfR KI or Grn−/−; TfR KI mice treated with either saline, Fc:PGRN or PTV:PGRN. (J, K) Quantification of GCase protein levels in mouse brain lysates showing no difference in GCase protein levels in Grn mutant brain (J) or liver (K) at baseline or in response to either Fc:PGRN or PTV:PGRN treatment. (n = 6 mice, ns by one-way ANOVA and Dunnett’s multiple comparison). (L) Quantification of the GCase activity in Grn+/+ and Grn−/− detergent-free liver extracts showing increase of GCase activity in the Grn−/− liver extracts treated with BMP and PG liposomes. (n = 4 mice, representative of 2 independent experiments; **** p < 0.0001 in one-way ANOVA and Dunnett’s multiple comparison). Data represented as geometric mean ± SEM.
Figure S3. Generation and characterization of the PTV:PGRN biologic, related toFigure 3. (A) SDS-PAGE of PTV:PGRN and Fc:PGRN constructs. 3 μg PTV:PGRN and Fc:PGRN fusion proteins (predicted MW ~115kDa) were run on SDS-PAGE under either non-reducing (- DTT) or reducing (+ DTT) conditions and stained with InstantBlue Coomassie. (B) Binding of PTV:PGRN and Fc:PGRN to human sortilin by ELISA. Recombinant human sortilin was plate-immobilized and then incubated with serial dilutions of either PTV:PGRN or Fc:PGRN fusion proteins or IgG control. PTV:PGRN and Fc:PGRN bound to sortilin with apparent affinities of 2.2nM and 1.8nM respectively. Curve values denote means ± SEM (n = 3 independent experiments). (C) Binding of PTV:PGRN and Fc:PGRN to human prosaposin by ELISA. Recombinant human prosaposin was plate-immobilized and then incubated with serial dilutions of either PTV:PGRN or Fc:PGRN fusion proteins or IgG control. PTV:PGRN and Fc:PGRN bound to prosaposin with apparent affinities of 5.1nM and 3.9nM respectively (n = 3 independent experiments). (D) Representative confocal microscopic images showing an increase in oxidized PC (oxPC) staining in Grn+/+ and Grn−/− BMDMs after treatment with oxidized LDL (oxLDL) particles. (E) Quantification of BMP (22:6/22:6) levels showing a decrease in both Grn+/+ and Grn−/− after a 72h treatment with 50ug/mL oxLDL, but not vehicle control. (n = 3 independent experiments; *, p < 0.05, ** p < 0.01 in two-way ANOVA with Tukey’s test). (F) Quantification of total TREM2 levels in lysates from BMDM analyzed in Fig. 3K showing an increase in Grn−/− BMDMs and a rescue after a 72h treatment with 50 nM PTV:PGRN, but not control IgG. Total TREM2 levels were measured by immunoassay. (n = 3 independent experiments; n.s. p > 0.1, **, p < 0.05 in one-way ANOVA with Dunnett’s multiple comparison test). (G) Binding of PTV:PGRNv2 to human sortilin by ELISA. Recombinant human sortilin was plate-immobilized and then incubated with serial dilutions of either PTV:PGRN, PTV:PGRNv2 or Fc:PGRN fusion proteins. PTV:PGRNv2 bound to sortilin with comparable apparent affinity (0.54nM) relative to PTV:PGRN and Fc:PGRN (0.47nM and 0.31nM respectively) (n = 2 independent experiments). (H) Binding of PTV:PGRNv2 to human prosaposin by ELISA. Recombinant human prosaposin was plate-immobilized and then incubated with serial dilutions of either PTV:PGRN, PTV:PGRNv2 or Fc:PGRN fusion proteins. PTV:PGRNv2 bound to prosaposin with comparable apparent affinity (1.28nM) relative to PTV:PGRN and Fc:PGRN (1.74nM and 1.14nM respectively) (n = 2 independent experiments). Data represented as geometric mean ± SEM.
Figure S4. PTV:PGRN rescues the endolysosomal vacuolization and membrane damage in GRN−/−iMG, related toFigure 4. (A) Time-lapse images showing DQ-BSA fluorescence in GRN−/− iMG. Images were acquired every hour using fluorescence microscopy. Images show fusion of vacuoles (thin arrow) and rupture of vacuoles (thick arrow) over the time-course. Scale bar: 25 μm. (B) Quantification of galectin-3 fluorescence intensity in GRN+/+ and GRN−/− iMG with no treatment (NT) or after a treatment with IgG control or PTV:PGRN each day for 3 days. PTV:PGRN rescued the endolysosomal membrane damage in two independent GRN−/− iMG clones. (n = 4 independent experiments; n.s., p > 0.1, *, p < 0.05, **, p < 0.01, ***, p < 0.001, ****, p < 0.0001 in one-way ANOVA with Dunnett’s multiple comparison test). Data represented as geometric mean ± SEM.
Figure S5. Increased brain penetrance of PTV:PGRN vs. Fc:PGRN resulting in greater lysosomal rescue in both acute and chronic dosing paradigms, related toFigure 5. (A-B) Concentration of intact PTV:PGRN in plasma (A) and brain (B) as determined by an Fc/PGRN sandwich ELISA in 3mo. Grn−/−; TfRmu/hu mice (n=4/group). (A) PTV:PGRN (dark orange = 50 mg/kg, light orange = 5mg/kg) and Fc:PGRN (dark blue = 50mg/kg, light blue = 5mg/kg) exhibit decline of full length drug concentration in plasma in addition to clearance observed for PTV:PGRN. (B) Brain concentrations of PTV:PGRN are higher than Fc:PGRN at either dose(n=4/group, Two-way ANOVA with Sidak’s multiple comparison test, 50mg/kg PTV:PGRN: padj<0.0001, 5mg/kg PTV:PGRN: padj<0.001). (C) Isolation of brain parenchyma reveals higher concentrations of PTV:PGRN at either dose, beyond the vascular barrier as measured by Fc/Fc ELISA (n=4/group, Two-way ANOVA with Sidak’s multiple comparison test, 50mg/kg PTV:PGRN: padj<0.0001, 5mg/kg PTV:PGRN: padj<0.001). (D) At 4h post-dose, Fc:PGRN shows equal or greater liver concentrations of Fc than PTV:PGRN at an equivalent dose (n=4/group, Two-way ANOVA with Sidak’s multiple comparison test, 50mg/kg PTV:PGRN: padj<0.0001, 5mg/kg PTV:PGRN: padj=0.9993). (E-F) Peripheral rescue of BMP(22:6/22:6) by PTV:PGRN or Fc:PGRN in liver (E) or plasma (F) following IV dosing. At 24h, rescue in these tissues/fluids is achieved completely (liver, One way ANOVA with Dunnett’s multiple comparison) or partially (plasma) with either PGRN construct (n=4/group, Two-way ANOVA with Sidak’s multiple comparison test). (G-I) Rescue of GlcSph in brain (G) liver (H) or plasma (I) by Fc:PGRN (blue) or PTV:PGRN (orange) (n=4/group, One way ANOVA with Dunnett’s multiple comparison (G,H), or Two-way ANOVA with Sidak’s multiple comparison test (I)). (G) A trend towards brain GlcSph rescue is observed at 24h following PTV:PGRN treatment (orange), but no correction is detected at 4h. (H) Modest modulation of liver GlcSph by PTV:PGRN 24h following IV delivery. (I) Plasma GlcSph is partially corrected by any PGRN fusion variant at 24h, but no differences are detected at earlier timepoints (n=4/group). (J) Plasma concentration of Fc as detected by ELISA 1 week following the final dose in a 6 week study with either 5mg/kg Fc:PGRN or PTV:PGRN. Fc only above the detection threshold in plasma with Fc:PGRN treatment, not with PTV:PGRN. (K-N) Pharmacokinetic and pharmacodynamic responses to 6 weeks of weekly 5mg/kg doses of either Fc:PGRN (blue) or PTV:PGRN (orange) in Grn−/− mice (n=7–10/group, One way ANOVA with Dunnett’s multiple comparison). (K) Equivalent rescue of liver BMP (22:6/22:6) by weekly administration of both Fc:PGRN and PTV:PGRN. (L) Rescue of GlcSph in the liver of Grn−/− is driven by both Fc:PGRN and PTV:PGRN. (M) PTV:PGRN significantly increases plasma BMP(22:6/22:6). (N) GlcSph in the plasma is rescued by both Fc:PGRN & PTV:PGRN. #p<0.1, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Data represented as mean ± SEM.
Figure S7: Peripheral TREM2 elevation in Grn−/−; TfRmu/humice is rescued by PTV:PGRNv2, related toFigure 7. (A) Representative images of microgliosis immunofluorescence markers (IBA1, GFAP, & CD68) in thalamus and rescue with PTV:PGRNv2 (scale bars = 10μm). (B-C) Total TREM2 levels in peripheral tissues (B, liver) and fluids (C, plasma) following 8 weeks of weekly or every other week (EOW) PTV:PGRNv2 treatment in 9mo Grn−/−; TfRmu/hu mice. (n=6/group, One-way ANOVA with Dunnett’s multiple comparison test). (A) Liver TREM2 levels are significantly upregulated in Grn−/−; TfRmu/hu mice and rescued by either weekly or EOW PTV:PGRNv2. (B) Plasma soluble TREM2 levels are significantly upregulated in Grn−/−; TfRmu/hu mice and reduced by both treatment intervals of PTV:PGRNv2. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Data represented as mean ± SEM.
Figure S6: Validation of methods involved with 8-week chronic dosing study and additional outcomes, related toFigure 6. (A) Dose titration indicates that BMP(22:6/22:6) correction by PTV:PGRN or PTV:PGRNv2 can be achieved at doses as low as 1.5 mg/kg, 7 days following IV delivery in 3mo. Grn−/−; TfRmu/hu mice (n=6–8/group, no statistical analysis). (B) Timecourse of brain BMP(22:6/22:6) rescue out to two weeks with IV or IP dosed PTV:PGRNv2 at 5mg/kg in 3mo. Grn−/−; TfRmu/hu mice (n=6/group, Two-way ANOVA with Sidak’s multiple comparison test). (C-H) Confirmation that EOW anti-CD4 treatment for 8 weeks does not impact primary lysosomal and gliosis phenotypes in 3mo. Grn−/−; TfRmu/hu mice (n=6/group, One way ANOVA with Dunnett’s multiple comparison). (C) Brain BMP(22:6/22:6) does not change with 8 weeks of anti-CD4 dosing (D) Representative coronal hemibrain images of gliosis markers with 8 weeks of anti-CD4 dosing (DAPI = blue, GFAP = green, CD68 = red, IBA1 = magenta, n=6/group, scale bars = 1mm). (E) Brain TREM2 levels in Grn−/−; TfRmu/hu are not impacted by 8 weeks of anti-CD4 dosing. (F) Quantification of total brain GFAP immunofluorescence area does not change with 8 weeks of anti-CD4 dosing. (G) Total brain CD68 does not change with 8 weeks of anti-CD4 dosing. (H) Brain IBA1 signal does not change with 8 weeks of anti-CD4 dosing. (I) Heatmap representation of cell type-marker transcript expression in sorted cell populations from 9mo. Grn−/−; TfRmu/hu mice, as quantified by qPCR. Columns represent biological replicates from each cell population, rows represent transcripts. Values are log2 transformation of fold change values (n=6/group). (J) Bar plots of BMP (20:4/20:4) from microglia, astrocytes and neurons, demonstrating increased levels across CNS cell types with weekly and EOW PTV:PGRNv2 treatment (9mo. mice). (K-M) Characterization of lipid changes in 4mo. Grn R493X; TfRmu/hu mouse brain and responsiveness to PTV:PGRNv2 (n=6/group, One way ANOVA with Dunnett’s multiple comparison). (K) BMP(22:6/22:6) is reduced in 4mo. Grn R493X mouse brain and is rescued by PTV:PGRNv2. (L) Brain BMP(18:1/18:1) is reduced in Grn R493X mice and is restored by PTV:PGRNv2. (M) GlcSph is elevated in brain of Grn R493X mice and reduced by PTV:PGRNv2. (N) TREM2 is elevated in the brain of Grn R493X mice but is not corrected by PTV:PGRNv2. #p<0.1, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Data represented as mean ± SEM.
Data Availability Statement
Raw LCMS area ratio data presented in heatmaps and volcano plots in the figures is provided at: DeVos, Sarah (2021), “Logan, Simon et al-2021-LC/MS Dataset”, Mendeley Data, V1, doi: 10.17632/s4y5hgm7xt.1
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request