

Abstract
The initial interactions between incoming, pre-replicated virion RNA and host protein factors are important in infection and immunity. Yet currently there are no methods to study these crucial events. We established VIR-CLASP (VIRal Cross-Linking And Solid-phase Purification) to identify the primary viral RNA-host protein interactions. First, host cells are infected with 4-thiouridine (4SU)-labeled RNA viruses and irradiated with 365 nm light to crosslink 4SU-labeled viral genomes and interacting proteins from host or virus. The cross-linked RNA binding proteins (RBPs) are purified by solid-phase reversible immobilization (SPRI) beads with protein denaturing buffers, and then identified by proteomics. With VIR-CLASP, only the incoming virion RNAs are labeled with 4SU, so cross-linking events specifically occur between proteins and pre-replicated virion RNA. Since solid-phase purification under protein-denaturing conditions, rather than sequence-specific nucleic acid purification, is used to pull-down total RNA and cross-linked RBPs, this method facilitates investigation of potentially all RNA viruses, regardless of RNA sequence. Preparation of 4SU-labeled virus takes ~7 days and VIR-CLASP takes 1 day.
Introduction
We developed a method to capture interactions between incoming virion RNA and cellular proteins, as these events are crucial to both viral infection and host innate immunity. VIR-CLASP innovates on previous techniques such as RAP-MS1, TUX-MS2, cRIC3, and vPAR-CL4 by specifically isolating interactions with pre-replicated virion RNA and by using sequence-independent methods to isolate RBP-RNA complexes5,6. Since VIR-CLASP does not use sequence-dependent isolation of RBP-RNA complexes, this technique does not suffer from biases associated with changing probe hybridization efficiencies due to dynamic ribonucleoprotein assembly over time, experimental condition, or cellular context.
Development of the protocol
VIR-CLASP identifies early viral-host interactions by using a three-step strategy: first, viruses are propagated in the presence of 4SU; second, isolated 4SU-labeled viruses are infected into unlabeled host cells followed by ultraviolet (UV) irradiation with 365 nm light; and third, crosslinked viral RNA-protein complexes are purified by a modified solid-phase reversible immobilization method (CLASP, Figure 1). As we outline in detail below, VIR-CLASP is ideally suited for the identification of RNA binding proteins interacting with infecting viral genomic RNA.
Fig. 1: Outline of VIR-CLASP.

VIR-CLASP is a method for identifying the primary interactions between viral RNA and host proteins. First, viruses are propagated with or without 4SU to label their RNA genome (Steps 1–18). Second, unlabeled cells are infected with 4SU-labeled or unlabeled viruses (Steps 19–28). Finally, the RNP complexes are purified with SPRI beads under denaturing conditions, RNA is digested with benzonase, and the resulting proteins are analyzed by Silver staining or LC–MS/MS (Steps 29-55). 4SU: 4-thiouridine, CHIKV: Chikungunya Virus, 4SU-CHIKV: 4SU labeled Chikungunya Virus, CLASP: Cross-Linking And Solid-phase Purification.
To label the viral genome with 4SU, RNA viruses are propagated in the presence of 4-thiouridine (4SU). This ribonucleoside is readily taken up by cells and incorporated into RNA in vivo12. 4SU allows for efficient crosslinking between RNA and proteins upon exposure to 365nm UV light. The virus is then purified by ultracentrifugation with a sucrose cushion. For VIR-CLASP to specifically identify proteins bound to pre-replicated virion RNA, 4SU should only be present in the viral genome. To ensure this, we wash the pelleted virus to remove any remaining free 4SU in the viral supernatant. Then, cells are infected with un-labeled or 4SU-labeled viruses and any uninfected virus is washed away. The infected cells are irradiated with 365 nm UV light to activate 4SU, driving efficient RNA protein crosslinking7. Cells are lysed under denaturing buffer conditions and boiled in order to minimize non-specific interactions between proteins or between protein and RNA. To implement a method that could be applied to all RNA viruses in an unbiased way, we used solid-phase reversible immobilization (SPRI) beads for the purification of protein-RNA interactions. SPRI beads are used in established methods for purification and size-selection of DNA or RNA but was not optimized for purification of protein cross-linked to RNA8. To optimize for specific purification of covalently crosslinked RNA-protein complexes, we adapted the buffer conditions for protein denaturation. In protein denaturing conditions (SPRI beads with sodium dodecyl sulfate (SDS)-containing buffer), total nucleic acids are purified but the only proteins recovered are those that are covalently crosslinked to the RNA viral genomes containing 4SU5. The SPRI beads and complexes are washed under the denaturing conditions used for the initial binding. The crosslinked RNA-protein complexes are eluted, the RNA is digested by nuclease (Benzonase), and then the proteins are identified by LC-MS/MS.
Applications of the method
Our experience with VIR-CLASP indicates that it can be used for a wide variety of RNA viruses and is quick to implement because it uses a SPRI paramagnetic bead-based technology that does not require sequence-specific optimization to purify the viral RNA. This facilitates comparisons across time points, different cell lines, and different strains for the same virus. Owing to its reliance on photocrosslinks between RNA and protein, we anticipate that VIR-CLASP would be most effective for investigating the host protein interactome with the genomes of single-stranded RNA viruses. Double-stranded RNA viruses have the added complexity of intra-strand crosslinks, thus reducing the probability of RNA-protein photoadducts, as has also been observed for methods which rely on crosslinking to stabilize RNA-protein complexes, e.g. crosslinking-immunoprecipitation (CLIP) techniques such as HITs-CLIP9, eCLIP10, iCLIP11, and PAR-CLIP12,13. We have performed initial VIR-CLASP experiments on viruses from seven distinct families, including plus- and minus-stranded RNA viruses: CHIKV (Chikungunya virus, Togaviridae), EMCV(Encephalomyocarditis virus, Picornaviridae), MHV (Mouse hepatitis virus, Coronaviridae), ZIKV (Zika virus, Flaviviridae), RVFV (Rift Valley Fever Virus, Phenuiviridae), IAV (Influenza A virus subtype H3N2, Orthomyxoviridae), and VSV (Vesicular stomatitis virus, Rhabdoviridae) 5,6. We have observed an array of distinct interactions between cellular proteins and 4SU-labeled viral genomes. We have used VIR-CLASP to discover the earliest host protein-viral RNA interactions between human cells (U2OS or A549) and Chikungunya virus (CHIKV) or Influenza A (IAV) virus. CHIKV has a positive-sense RNA genome and IAV has a negative sense RNA genome, thus these viruses likely form distinct interactions upon infection into host cells. Using VIR-CLASP followed by mass spectrometry, we identified hundreds of proteins that directly bind CHIKV and IAV. We found proteins that distinctly interact with CHIKV or IAV, and proteins that interact with both5. VIR-CLASP will enhance our understanding of the early events in viral replication, opening new avenues for therapeutic innovation and prophylactic treatment of CHIKV and other RNA viruses. VIR-CLASP can also be used to broadly identify the RNA binding proteins interacting with different RNA species (e.g., viral RNA within virion or cellular RNA). To identify RBPs in the virion, VIR-CLASP can be performed on just purified virus, without infection into host cells. We observed that viral proteins E1, E2, and Capsid interact with the CHIKV genome5. Recently, Zhou & Routh have characterized viral RNA-protein interactions similarly by isolating 4SU-labeled RNA viruses4. The capsid binding sites in genomic viral RNA were identified using vPAR-CL. vPAR-CL differs from VIR-CLASP in that it identifies protein binding sites on RNA within the virion via high-throughput sequencing, while VIR-CLASP identifies proteins bound to RNA virus genomes after release into the host cytoplasm via LC-MS/MS. With VIR-CLASP and vPAR-CL, viral RNA-protein interactions in virions can be analyzed from both perspectives, identifying the RBPs and the RBP binding sites on virion RNA. Finally, a subsection of the VIR-CLASP protocol is also amenable for the broad identification of cellular RNA binding proteins that directly interact with bulk cellular RNA; labeling of cultured cells with 4SU allows for the isolation of RNA binding proteins with the CLASP method, in a sequence-independent way5. This application of CLASP adds to the growing list of interactome-wide RBP identification methods11-15.
Comparison with other methods
VIR-CLASP differs from RNA-antisense purification mass-spectrometry (RAP-MS1), thiouracil cross-linking mass-spectrometry (TUX-MS2) and comparative RNA-interactome capture (cRIC3) in two fundamental ways: first, VIR-CLASP only identifies interactions with the pre-replicated virion RNA; second, VIR-CLASP uses a sequence-independent method to purify total RNA under protein-denaturing conditions. With VIR-CLASP we can identify host proteins that interact with the pre-replicated genomes of various RNA viruses.
Recently, other non-sequence-based methods of crosslinking RBP complexes and purification have been developed. Acid guanidinium thiocyanate-phenol-chloroform extraction (TRIZOL)14 and Phenol Toluol extraction (PTex) methods are based on chemically driven liquid phase separation. Silica-based solid-phase extraction15,16 uses silica matrices to purify RBP-RNA complexes. Unlike these methods, the CLASP method uses paramagnetic beads to isolate crosslinked RBP complexes. Compared to other methods for nucleic acid purification, crosslinked RBPs are more easily and specifically separated from the samples when using magnetic beads17.
Experimental design
Propagation and host cells
Propagation and host cells can be chosen based upon the researcher’s interests. Because the yield of cross-linked protein relies on the amount of 4SU-labeled viral RNA that is released as infectious virus, a high 4SU incorporation rate in propagation cells and high infectivity in host cells will yield the best signal. To incorporate 4SU into viral RNA during propagation, 4SU needs to be imported into propagation cells by at least one member of equilibrative nucleoside transporters (ENT1 and SLC29A1)13. Modulating transporter expression in propagation cells can be considered to increase 4SU uptake18. Also, overexpression of viral entry receptors in host cells can be an option to increase cell entry of the virus19.
Control (Non-labeled virus)
VIR-CLASP should be performed with non-labeled virus and 4SU-labeled virus simultaneously. The non-labeled virus acts as a blank to assess the extent of non-specific purification of proteins with CLASP; we used the LC-MS/MS-identified proteins from the sample with non-labeled virus to define enrichment of the candidate set of proteins in the sample with 4SU-labeled virus.
4SU incorporation into viral genomes
Because VIR-CLASP requires 4SU labeled RNA, the 4SU incorporation rate in viral genomes is important. Depending upon the cell lines and virus, 4SU concentration and treatment time should be considered. If the viral propagation cycle is fast (e.g. picornavirus: ~ 8 hours20), pre-treatment of cells with 4SU is helpful because it allows cells to take up and phosphorylate 4SU before viral RNA replication starts. If the propagation cell line has not been used for a 4SU-labeling experiment previously, we normally treat the cells with 100 μM 4SU overnight (16 h) and analyze the 4SU incorporation rate of cellular RNA via HPLC to verify that this cell line can uptake 4SU. Although a higher 4SU concentration would result in more 4SU incorporation into viral genomes, it could also impact the yield of virus propagation. We observed that compared to unlabeled virus, titers of viral supernatant were decreased 2~3 fold with CHIKV and ~10 fold with IAV after treating with 1 mM 4SU in the propagation medium. We typically use 100 μM 4SU for 16 h to pretreat and 1 mM 4SU for 2 days during propagation. The average amount of 4SU incorporation in the purified CHIKV particles was measured to be 2.5% (of total uridine content) using HPLC5.
Test the timing of pre-replication with titer assay and qPCR
To define the timepoints when interactions between pre-replicated virion RNA and host proteins are most important for viral replication and virion production, a viral titer assay of UV-irradiated 4SU-labeled virus at timepoints before and during infection can be used. Covalent crosslinks between viral RNA and proteins inhibit maturation of new virions only when the incoming RNA genomes are necessary for critical steps including translation and replication. With 4SU-labeled CHIKV, we observed that the initial decrease in titer when irradiating infected cells at 0 hpi was from ~109 to 107; therefore, we estimate that ~99% of viral particles contain a biologically meaningful amount of 4SU substitution5. The UV irradiated CHIKV titers recovered to approximately control levels at 3 hpi. RT-qPCR analysis also revealed that viral RNA increases between 2 and 3 hours post infection (hpi), confirming that viral transcription and replication had begun5. Therefore, for CHIKV, we chose to examine pre-replicated virion RNA and host protein interactions between 0 hpi and 3 hpi. We observed that IAV replication is fully dependent upon the pre-replicated virion RNA at ~ 1 hpi and does not become independent until ~ 4 hpi5. Thus, we chose to assay interactions with IAV at 1 hpi.
Determine the bead buffer ratio
The SPRI method uses carboxyl-coated magnetic beads that can reversibly bind RNA in SPRI bead buffer (20% (wt/vol) polyethylene glycol (PEG) and 2.5M NaCl)8. By increasing the ratio of SPRI bead buffer volume to sample volume, the efficiency of binding smaller RNA will be increased21. In the VIR-CLASP method, we employed protein denaturing conditions and SPRI beads to purify crosslinked RNA-protein complexes. As in the SPRI method, the CLASP bead buffer to sample ratio can affect the efficiency of binding smaller RNA. Although increasing the bead buffer to sample buffer ratio can increase the efficiency of purifying smaller RNA, this modification may be not suitable for the CLASP method since the denaturing agents in the sample buffer may result in solubility issues due to PEG in the bead buffer. Based on the length of the RNA viral genome, the CLASP bead to sample buffer ratio should be optimized. VIR-CLASP can use up to 1:1 (v/v) ratio of CLASP bead buffer to sample buffer (e.g. 50 μl bead buffer and 50 μl sample buffer), which can recover RNA longer than 300 nt (Supplementary Fig. 1). In VIR-CLASP with CHIKV, we used a 2:3 ratio sample buffer to bead buffer to recover CHIKV viral RNA and crosslinked protein.
Expertise needed to implement the protocol
A competent graduate student or postdoc can perform all procedures for VIR-CLASP. It is essential for investigators working with infectious viral pathogens to obtain the appropriate level of biological safety training required by their respective institutions, and to operate the protocol within approved and certified biosafety cabinets and facilities. Core facilities for proteomics and operation of the LC–MS/MS instrument are also required.
Limitations
In the VIR-CLASP method, the yield of cross-linked protein is dependent on 4SU-labeled RNA that is released from infected virus. To yield enough signal, we typically infect host cells with a viral MOI of 200 to 5000. If viral propagation is not efficient, total virus quantity might also be an issue. As with all proteome-wide screens, it is recommended that identified hits are validated. For viral nucleic acid-host protein interactions, it is recommended that validation experiments are performed at lower MOI (e.g. 0.01 to 1). We also highly recommend to follow-up with a secondary screen to assay the biological relevance of the VIR-CLASP hits, also performed at lower MOI. The specific design of this validation will be dependent upon the biological function(s) of interest. It is possible that depending on the virus, small amounts of cellular RNA can be packaged within the virion, and thus contain 4SU. This is reported to occur for some retroviruses and therefore extra precaution should be exercised22. For example, it may be necessary to assess the extent of cellular RNA that can carry-over using reverse-transcription followed by PCR or sequencing methods. For IAV and CHIKV, we found that they contained either no or very little cellular RNA within the virion (IAV: < 3%23, CHIKV: < 1.3%5,24). It is also possible that proteins from the propagation cell line are packaged within the viral particle. If this is a concern, we recommend performing mass-spec analysis on viral particles after purification to determine the extent of cellular protein carryover during propagation. Lastly, the concentration and timing of 4SU incorporation during viral propagation may need to be optimized for the RNA virus of interest to achieve optimal labeling efficiency.
Materials
Biological materials
CAUTION: The cell lines used in your research should be regularly checked to ensure that they are authentic and that they are not infected with mycoplasma. Cell lines for viral propagation and infection should be chosen based on the RNA virus used.
Cell line for Chikungunya virus propagation: BHK-21 (ATCC, cat. no. CCL-10; https://scicrunch.org/resolver/CVCL_1915)
Host cell line for Chikungunya viral infection: U2OS (ATCC, cat. no. HTB-96; https://scicrunch.org/resolver/CVCL_0042)
Virus: CHIKV strain 181/25 (provided by Terence S. Dermody, University of Pittsburgh School of Medicine)
CAUTION: Virus must be handled according to approved biosafety protocols and regulations set by each individual lab and institution. Improper handling of samples may lead to exposure to infectious pathogens.
REAGENTS
DMEM (Gibco, cat. no. 11965-118)
L-Glutamine (Gibco, cat. no. A2916801)
HEPES (Gibco, cat. no. 15630080)
Sodium Pyruvate (Gibco, cat. no. 11360070)
MEM Non-Essential Amino Acids Solution (Gibco, cat. no. 11140050)
2-Mercaptoethanol (Gibco, cat. no. 21985023)
McCoy's 5A (Modified) Medium (Gibco, cat. no. 16600082)
4-Thiouridine (Sigma, cat. no. T4509)
DMSO (sigma, cat. no. D2650)
PBS (National Diagnostics, cat. no. CL-253)
FBS (Peak Serum, cat. no. PS-FB2)
Tris-base (Sigma, cat. no. 93362)
HCl (Sigma, cat. no. H1758)
NaCl (Sigma, cat. no. S3014)
EDTA (Sigma, cat. no. E6758)
NaOH (Sigma, cat. no. S8045)
Sucrose (Sigma, cat. no. 84097)
SDS (Sigma, cat. no. 74255)
NP-40 (Sigma, cat. no. I8896)
Glycerol (Sigma, cat. no. G5516)
Sera-mag SpeedBeads (Fisher, cat. no. 09-981-123)
Tween 20 (Sigma, cat. no. P9416)
PEG-8000 (Sigma, cat. no. 89510)
Benzonase (Novagen, cat. no. 70746)
MgCl2 (Sigma, cat. no. M8266)
DTT (Sigma, cat. no. D9779)
Methanol (Sigma, cat. no. 34860)
Chloroform (Sigma, cat. no. C2432)
NuPAGE LDS Sample Buffer (4X) (Invitrogen, cat. no. NP0007)
NuPAGE Sample Reducing Agent (10X) (Invitrogen, cat. no. NP0004)
NuPAGE 10%, Bis-Tris, 1.0 mm, Mini Protein Gel, 10-well (Invitrogen, cat. no. NP0301BOX)
NuPAGE MOPS SDS Running Buffer (20X) (Invitrogen, cat. no. NP0001)
SimplyBlue SafeStain (Invitrogen, cat. no. LC6065)
Trifluoroacetic Acid (Sigma-Aldrich, cat. no. 302031)
Ammonium bicarbonate (NH4HCO3; Sigma-Aldrich, cat. no. A6141)
Trypsin Gold (Promega, cat. no. V5280)
Iodoacetamide (Sigma-Aldrich, cat. no. I1149)
Acetonitrile (Sigma-Aldrich, cat. no. 34851)
Formic acid (Pierce, cat. no. 28905)
EQUIPMENT
Floor Low Speed Centrifuge (Thermo Fisher, cat. no. Sorvall RC-3B Plus)
H-6000A 6 x 1000mL Swinging-Bucket Rotor (Thermo Fisher, cat. no. 11250)
H-6000A bucket adapter for 500-ml tubes (Thermo Fisher, cat. no. 00-444)
Ultracentrifuge (Beckman coulter, cat. no. Optima LE-80k)
SW 32 Ti Swinging-Bucket Rotor (Beckman coulter, cat. no. 369650)
Spectrolinker XL-1500 (Spectronics, cat. no. XL-1500A)
DynaMag-2 Magnet (Thermo Fisher, cat. no. 12321D)
Microcentrifuges (Eppendorf, cat. no. 5404000138)
Vortex-Genie 2 (Scientific Industries, cat. no. SI-0236)
Digital Dry Bath (Fisher Scientific, cat. no. 11-715-125D)
ThermoMixer (Eppendorf, cat. no. 5382000023)
Q Exactive Plus mass spectrometer (Thermo Scientific)
Plasticware
150 mm TC-treated Cell Culture Dish (Corning, cat. no. 353025)
500 mL PP Centrifuge Tubes (Corning, cat. no. 431123)
Cell Scraper (VWR, cat. no. 101093-452)
1.5ml-tubes (Denville, cat. no. C2170)
Thinwall Polypropylene Tube, 25x89mm (Beckman coulter, cat. no. 326823)
0.22 μm filter (Millipore, cat. no. S2GPU05RE)
0.45 μm filter (Millipore, cat. no. SCHVU05RE)
REAGENT SETUP
Virus diluent
Virus diluent is 10 mM HEPES and 1% (vol/vol) FBS in DMEM medium. Store at 4 °C for up to 3 months.
Complete DMEM
Complete DMEM is 10mM HEPES, 2 mM L-Glutamine, 1 mM Sodium Pyruvate, 1x MEM Non-Essential Amino Acids, 50 μM 2-Mercaptoethanol and 10% (vol/vol) FBS in DMEM medium. Store at 4 °C for up to 3 months.
4SU stock solution
Prepare 4SU stock solution at a concentration of 1 M in DMSO and then make aliquots in sterilized 1.5 ml Eppendorf tubes. Store at −20 °C in the dark up to 1 year.
CRITICAL: 4SU is light sensitive.
20% (wt/vol) sucrose in TNE buffer
20% (wt/vol) sucrose in TNE buffer is 50 mM Tris–HCl [pH 7.4], 100 mM NaCl, 0.1 mM EDTA and 20% (wt/vol) sucrose in water. Filter the solution with a 0.22 μm bottle top filter in a Biological Safety Cabinet. Store at 4 °C for up to 6 months.
3x denaturation buffer
3x denaturation buffer is 150 mM Tris–HCl [pH 6.8], 30% (vol/vol) Glycerol, 7.5% (wt/vol) SDS and 2% (vol/vol) NP-40 in water. Store the buffer at room temperature (25 °C) for up to 3 months.
SPRI bead suspension
Sera-mag SpeedBeads are provided as a concentration of 50 mg/ml. After washing with TE (10 mM Tris–HCl [pH 8.0] and 1 mM EDTA) buffer, SPRI beads are reconstituted as a concentration of 1 mg/ml in 10 mM Tris–HCl [pH 8.0], 1 mM EDTA, 18% (wt/vol) PEG-8000, 1 M NaCl and 0.055% (vol/vol) Tween-20 in water. Store at 4 °C for up to 1 month.
Beads buffer
Beads buffer is 2.5 M NaCl and 20% (wt/vol) PEG-8000 in water. Store the buffer at room temperature (25 °C) for up to 1 months.
WASH buffer
WASH buffer is prepared by mixing 1x denaturation buffer and Beads buffer in a 2:3 ratio by volume (e.g. mix 2 ml 1x denaturation buffer and 3 ml Beads buffer) . WASH buffer should be freshly prepared before use.
4x Benzonase reaction buffer
4x Benzonase reaction buffer is 80 mM Tris–HCl [pH 7.5], 20 mM MgCl2, 600 mM NaCl, 40% (wt/vol) Glycerol and 4 mM DTT in water. Store the buffer at room temperature (25 °C) for up to 1 year.
CRITICAL: DTT should be added freshly before use.
Procedure
Propagation of 4SU-labeled virus; Timing ~3 d
CRITICAL
As described above in Experimental design section about Control (Non-labeled virus), the virus should be propagated with/without 4SU to prepare control (Non-labeled virus) and 4SU-labeled virus (Figure 1).
1. Seed 5x106 BHK-21 cells in a 150 mm Cell Culture Dish in 20 ml of complete DMEM with 100 μM 4SU. In parallel, seed an equal number of cells in complete DMEM without 4SU. We typically use 12 plates per condition.
2. Incubate cells at 37 °C overnight.
3. Remove medium from the dish and incubate both the control and 4SU cells with 2.5 ml of diluted inoculum of CHIKV at MOI 0.124, at 37 °C for 1 h in a cell culture incubator with rocking every 10-15 min to prevent cells from drying.
4. After 1 h, remove inoculum and wash cells twice with 1X with PBS.
CRITICAL STEP: Ensure complete washing of extracellular CHIKV that is not labeled with 4SU, as this could affect the measured 4SU incorporation rate.
5. Add 17 ml of complete DMEM with or without 1 mM 4SU to the cell culture dish, and incubate cells until cytopathic effects are observed (typically 48 h post-infection) at which point most of the cells will have lifted or rounded.
?TROUBLESHOOTING
6. After cells have rounded and lifted, harvest and transfer the viral supernatants to a 500 ml conical tube.
7. Clear cell debris by centrifugation at 1,000 x g for 10 min at 4 °C and filter through a 0.45 μm filter.
8. Transfer and aliquot in 50 ml Falcon tubes.
CAUTION: Each aliquot is only to be thawed once to maintain an accurate titer.
PAUSE POINT: The virus can be frozen at −80 °C until use.
Concentration of 4SU labeled CHIKV; Timing ~1 d
9. Add ml of sterile 20% (wt/vol) sucrose in TNE buffer to each ultracentrifuge tube.
10. Slowly layer up to 30 ml of viral supernatant above the 20 % (w/v) sucrose.
CRITICAL STEP: The concentration of the sucrose cushion should be adjusted based upon the density of the virion which is determined by density gradient centrifugation25. The density of CHIKV is 1.22 g/cm3 in sucrose26.
11. Ultracentrifuge with an SW32Ti swinging bucket rotor at 121,000 × g for 3 h at 4 °C.
12. Remove and discard the supernatant. The pellet may or may not be visible, but the virus will be stuck to bottom of tube.
13. To wash away free 4SU, add 5 ml of sterile 20% (wt/vol) sucrose in TNE buffer on top of the pellet, and overlay with 30 ml of cold PBS.
14. Ultracentrifuge with an SW32Ti swinging bucket rotor at 121,000 × g for 30 min at 4 °C and remove and discard supernatant.
15. Repeat Steps 13-14 two more times to completely rinse the free 4SU out of the virus pellet.
16. Add 1 ml (or less) of cold virus diluent to the pellet (do not pipette up and down yet).
17. Seal the tubes with parafilm and allow the virus to “soak” at 4 °C for 4-6 hours.
PAUSE POINT: The virus pellet can be “soaked” for up to 12 hours (overnight).
18. Mix the soaked viral pellet well by pipetting up and down many times (>20).
CRITICAL STEP: Ensure complete suspension of the viral pellet, otherwise titer estimation may be inaccurate. To infect with a specific MOI, the concentrated viral titer should be determined.
Infection of unlabeled cells with 4SU-CHIKV; Timing ~1 d (~2 h hands-on)
19. For each condition, Seed 3x106 U2OS cells in a 150 mm cell culture dish in 20 ml of complete DMEM.
CRITICAL STEP: To ensure that the cells are ready at the same time as the concentrated virus, perform step 19 at the same time as virus concentration (steps 9-17). Therefore steps 18 and 22 will occur on the same day.
20. Incubate cells at 37 °C overnight.
21. Remove medium from dish and wash twice with 5ml cold PBS.
22. Remove PBS from dish and incubate cells with 2.5 ml of diluted inoculum of CHIKV at MOI 1000 (from Step 18), at 4 °C for 1 h with rocking every 10-15 min to prevent cells from drying.
23. After 1 h, remove and discard inoculum and wash twice with PBS.
24. Add 17 ml complete DMEM and incubate cells in a cell culture incubator at 37 °C until the crosslinking time point.
25. Remove medium from dish and discard and wash twice with 5ml cold PBS.
26. Remove PBS from dish and add 2.5 ml PBS.
27. Irradiate uncovered dish twice with 0.6 J/cm2 of 365 nm UV light in a Spectrolinker XL-1500 or similar device.
28. Scrape cells from the plate with a cell scraper, transfer and divide 600 μl each to four 1.5-ml tubes and add 300 μl 3x denaturation buffer per tube. Mix well by pipetting up and down.
PAUSE POINT: Unless you want to continue directly with cell lysis, collect the cells by centrifugation at 500 x g for 5 min at 4 °C and discard the supernatant. Freeze the cell pellet in liquid nitrogen and store at −80 °C. When ready to proceed with CLASP, you can then add 1x denaturation buffer directly into cell pellets (~900 μl/106 cells) and mix well by pipetting up and down.
CLASP
Preparation of whole cell lysate and protein denaturation; Timing ~30 min
29. Incubate cells from Step 28 at 95°C for 10 min. Save two 100 μl aliquots in 1.5 ml Eppendorf tubes (after boiling) for “input” samples as in Figure 2. These are would be processed with VIR-CLASP samples on step 42 for RNA and on step 43 for protein.
Fig. 2: Anticipated results for VIR-CLASP samples.
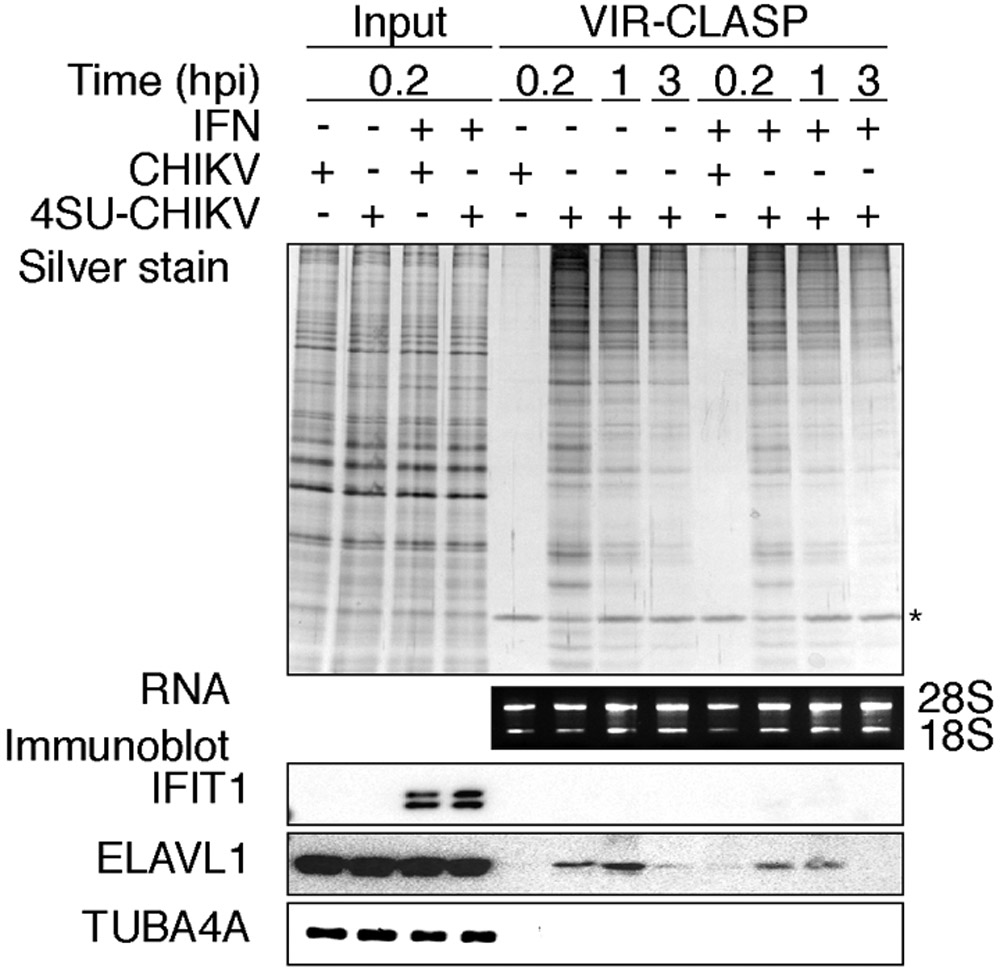
SDS-PAGE and silver stain (top), agarose gel (middle), or immunoblot (bottom) of proteins purified using VIR-CLASP from host (U2OS) cells that were infected with unlabeled or 4SU-labeled viruses (CHIKV). Condition (IFN): pre-treatment with recombinant interferon-β for 16 h before infection. Data represent three biologically independent repeats. The asterisk indicates benzonase. Adapted with permission from5.
PAUSE POINT: Cell lysate can be stored at −20 °C and the protocol restarted on Step 29 (i.e., after thawing, repeat the incubation at 95°C for 10 min before proceeding to step 30).
30. Incubate sample at RT for 10 min.
Solid-phase purification; Timing ~3 hr
31. Add 600 μl SPRI bead suspension (see ‘Reagent setup’) to 900 μl denatured-lysates to result in a 2:3 bead buffer to sample ratio.
CRITICAL STEP: Before adding the SPRI bead suspension to samples, warm up the beads to room temperature (RT).
?TROUBLESHOOTING
32. Mix by pipetting 10 times and incubate samples at 25 °C for 10 min; then place tubes on magnetic stand. Allow the SPRI beads to pellet to the magnet. Settling times will vary, and may be as long as 3 minutes.
33. Remove and discard the supernatant. Add 1 ml WASH buffer.
34. Mix by vortexing (max. setting) and allow the SPRI beads to re-pellet by magnet. Repeat Steps 33 and 34 four times for a total of five washes.
?TROUBLESHOOTING
35. Remove the last supernatant. Add 600 μl 1x denaturation buffer, and mix by vortexing – then incubate at RT for 5 minutes.
36. Add 400 μl beads buffer and mix the total reaction volume by pipetting 10 times and incubate sample at 25 °C for 10 min, then place tubes on magnetic stand.
37. Remove and discard the supernatant. Add 1 ml WASH buffer.
38. Mix by vortexing (max. setting) and allow the SPRI beads to re-pellet by magnet. Repeat Steps 37 and 38 four times for a total of five washes.
39. Remove final WASH supernatant then add 200 μl 1x denaturation buffer. Mix the total reaction volume by vortexing and incubate at RT for 5 minutes.
40. Collect beads by placing on magnetic stand.
?TROUBLESHOOTING
41. Transfer supernatant to a new tube. Save a 20 μl aliquot in 1.5 ml Eppendorf tube for step 42.
42. (optional) If you want to check the recovery of RNA by the CLASP method, treat the saved aliquot from step 41 with 1 mg/ml proteinase K for 2 h at 55 °C and purify the RNA with Trizol according to the manufacturer’s instructions.
Benzonase treatment; Timing ~2 hr
43. Add 100 μl 4x Benzonase reaction buffer and 200 μl water with 2 μl Benzonase to the 200 μl supernatant from step 41.
44. Incubate samples at 37 °C for 2 h.
Methanol and Chloroform Precipitation; Timing ~30 min
45. Split 800 μl benzonase-treated samples into two tubes with equal volume (400 μl)
46. Add an equal volume of 100% MeOH to each sample (50% (vol/vol) MeOH final). Vortex (max. setting) for 3 sec.
47. Add 100 μl Chloroform. Vortex (max. setting) for 3 sec.
48. Spin for 2 min at 15,000 x g at RT.
49. Remove the water/MeOH mix on top of the interface, being careful not to disturb the interface. Often the precipitated proteins do not make a visibly white interface, and care should be taken not to disturb the interface.
?TROUBLESHOOTING
50. Add 400 μl of MeOH. Vortex (max. setting) for 10 sec.
51. Spin for 3 min at 15,000 x g at RT; the protein precipitate should now pellet to the bottom of the tube.
52. Pipette as much MeOH as possible from the tube without disturbing the pellet and briefly dry the pellets in a vacuum centrifuge for 5 min at RT.
?TROUBLESHOOTING
53. Re-suspend the pellets in 20-50 μl of 1x NuPAGE LDS Sample Buffer (diluted with H2O), supplemented with 1 x NuPAGE Sample Reducing Agent.
CRITICAL STEP: The amount of 1x NuPAGE LDS Sample Buffer used to re-suspend pellets can vary across virus/cell types based on 4SU incorporation, crosslink efficiency, and protein precipitation. Based on our experience, 20-50 μl is a typical range.
54. Incubate at 95°C for 5 min.
55. Proceed to silver staining to visualize precipitated proteins27 (Figure 2) or LC-MS/MS analysis
?TROUBLESHOOTING
Mass Spectrometric Analysis
Sample preparation; Timing ~2 - 3 d
56. Run the samples from step 54 on a NuPAGE™ 10% Bis-Tris protein gel in NuPAGE™ MOPS SDS buffer according to the manufacturer protocol. Run the proteins to a distance of about 1cm into the gel. Stain with SimplyBlue™ SafeStain according to the manufacturer protocol to visualize protein bands.
57. Carefully remove the gel lanes and dice into 1 mm3 cubes.
58. Treat the gel pieces with 45mM DTT (add enough solution to cover the gel pieces) at 56°C for 30 minutes. Spin down the gel pieces and remove solution.
59. Add 100mM iodoacetamide in 25mM ammonium bicarbonate (to cover gel pieces) to carbamidomethylate available Cys residues. Incubate for 45 minutes at RT in the dark.
60. Spin down gel pieces to remove buffer, then add 50% MeCN in 25mM ammonium bicarbonate (to cover gel pieces) to further destain the gel. Repeat this step until gel is destained. If following with in-gel trypsin digestion (see protocol28 for more detail), do a final wash with 100% MeCN.
CRITICAL STEP: It is important that the gel pieces are fully dehydrated if in-gel trypsin digestion is to be performed.
Trypsin digestion; Timing ~2 - 3 d
61. Digest the proteins with 10ng/uL trypsin in 25mM ammonium bicarbonate (to cover gel pieces) at 37°C overnight (see protocol28 for more detail).
62. Extract the peptides in 60% MeCN, 0.1% TFA, and dry by speed-vac centrifugation.
63. Reconstitute the peptides in 0.1% formic acid.
LC-MS/MS Analysis; Timing ~1 d
64. Inject up to 1ug of protein into LC column. Use a gradient of HPLC solvent B that is best suited to the complexity of the protein sample (see previous publication5 for example HPLC conditions).
65. MS parameters will be specific to the instrument used (see example5), but will not typically require alterations to a standard protein identification or quantification protocol for MS.
CRITICAL STEP: It is important to use the same MS parameters for each sample (including the −4SU controls).
Proteomics Data Analysis
Peptide-spectrum matching; Timing variable
66. Construct a fasta database containing protein sequences from the host species and the virus of interest.
67. Search raw data files from the LC-MS/MS instrument using preferred peptide-spectrum matching software with appropriate search parameters (i.e., full tryptic specificity, two missed cleavages, fixed carbamidomethyl modification, variable acetylation modification), and FDR cut-off.
CRITICAL STEP: search parameters should be discussed and optimized with input from a proteomics/mass-spec analysis expert.
Quantitative analysis; Timing ~1 hr
68. Using the ion intensity values from the peptide-spectrum matching, calculate the ratio of peptide intensities for each experimental condition compared to the unlabeled (no 4SU) sample. For each protein, calculate the average peptide intensity ratio for each biological replicate.
69. Test the hypothesis that the log2-transformed ratios are different from 0 using a moderated t test, such as the one implemented in the Bioconductor package limma29.
70. Correct the p-values for multiple testing and apply a significance cut-off.
Semi-quantitative analysis; Timing ~1 hr
71. For proteins with peptides not identified in the unlabeled samples, an intensity ratio cannot be calculated. For these proteins, apply a semi-quantitative approach by tabulating the number of replicates in which each peptide was identified 3,30 (Figure 3).
Fig. 3: Proteomic Analysis of VIR-CLASP.

Flow-chart illustrating an example computational analysis of VIR-CLASP performed with CHIKV. Semiquantitative analysis shows the matrix used to calculate FDRs for proteins which cannot be tested quantitatively due to the low complexity of the −4SU samples. Peptides were sorted into boxes based on the number of biological replicates in which they were identified. Green boxes contain peptides with FDR < 0.01. Quantitative analysis shows the representative scatterplots of log2-intensity ratios of quantifiable proteins in +4SU over −4SU samples of VIR-CLASP with CHIKV (pairwise comparisons between three biological replicates). Red dots indicate significantly enriched proteins between all three biological replicates. Functional analysis shows an UpSet diagram34 summarizing the shared proteins that were identified in different timepoints and conditions of VIR-CLASP for CHIKV. The vertical bar chart shows the number of protein identifications common to the conditions marked by the colored and connected dots below. The horizontal bar chart shows the total count of proteins in each condition. Blue or red coloring highlights protein identifications unique to either (−) or (+) IFN pre-treatment. Adapted with permission from5.
72. Estimate an FDR by calculating the ratios resulting from dividing the transposed matrix.
73. Apply an FDR cut-off, and include proteins comprising peptides in cells with estimated FDRs below the cut-off.
Functional Analysis; Timing variable
74. Perform functional analyses depending on the researcher’s specific experimental goals. The UpSetR package31 can be used to construct an UpSet diagram (an alternative to a Venn diagram) as in Figure 3. Other functional analyses that can be performed include gene ontology or disease ontology, analysis of protein domains, or comparisons to other screens for host factors that promote or inhibit viral replication.
Timing:
Step 1-18, Propagation of 4SU-labeled virus: 4 d (~4 h hands-on)
Step 19-28, Infection of unlabeled cells with 4SU-CHIKV: 1 d (~2 h hands-on)
Step 29-30, Preparation of whole cell lysate and protein denaturation: 30 min
Step 31-42, Solid-phase purification: 3 h
Step 43-44, Benzonase treatment: 2 h
Step 45-54 Methanol and Chloroform Precipitation: 30 min
Step 55-65 Silver staining or peptide identification and quantification by LC–MS/MS: 3-5 d depending on number of samples
Step 66 – 73 Preliminary data analysis: 1-2 d depending on number of samples
Step 74 Functional data analysis: 5-10 d depending on experimental goals
Troubleshooting
Troubleshooting advice can be found in Table 1.
Table 1.
Troubleshooting table
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 5 | virus titer of 4SU labeled virus was too low compared to unlabeled virus | 4SU concentration was too high | Perform a test for optimal concentration of 4SU |
| 4SU incorporation rate in 4SU labeled virus was too low. | 4SU concentration was too low or the uptake of 4SU was not efficient in the propagation cell line | Perform a test for optimal concentration of 4SU as described in “Experimental Design” | |
| 31 | beads aggregate | The concentration of nucleic acid is too high | Use fewer cells or increase the lysis volume and bead quantity |
| 33-34 | wash buffer precipitation | Temperature is lower than 25 °C or the wash buffer is not prepared freshly | Warm up the buffer |
| 40 | beads remain clumped | Nucleic acid is not eluted well | Incubate another 5 min at 37 °C |
| 52 | The size of precipitated pellets are not different between unlabeled and 4SU conditions. | Background proteins bound to the beads | Avoid beads aggregation and wash buffer precipitation |
| Low yield of precipitated protein in 4SU condition | Low 4SU incorporation rate in viral RNA | perform a test for optimal concentration of 4SU | |
| Low amount of 4SU labeled RNA | Optimize infection conditions (MOI, time) | ||
| Inefficient cross-linking | Optimize cross-linking conditions | ||
| 53 | pellet is not solublized | overdried pellets | Take more care in drying pellets |
Anticipated results
Analysis by SDS-PAGE and silver staining of VIR-CLASP samples is expected to show enrichment of proteins with unique banding patterns across conditions/timepoints of cells infected with 4SU-labeled CHIKV (Figure 2). In the −4SU samples, which represent cells infected with unlabeled CHIKV, crosslinked, and processed using VIR-CLASP, the purification is anticipated to yield little protein (Figure 2, lanes 5 and 9). Only one protein, Benzonase, is added to digest the RNAs after solid-phase purification, and this protein should show similar intensity across the samples on the silver stain (Figure 2). By VIR-CLASP, the amount of enriched and intact ribosomal RNA should be unaltered between conditions, and independent of whether a sample contained crosslinked complexes (Figure 2). VIR-CLASP samples can be validated by immunoblot with previously known interacting and non-interacting RBPs. ELAVL1 (HuR) is an RBP known to interact with the CHIKV 3’ UTR32 and the antiviral RBP IFIT1 binds only to RNA viruses with a 5’ cap-0 structure that CHIKV lacks (Figure 2)33.
The input to VIR-CLASP required in order to detect proteins with LC-MS/MS depends on the infectivity of the given virus in the given host cell. This is because only the pre-replicated virion RNA contains 4SU, so cellular protein content cannot be used as an input measure. For example, we observed that the strain of CHIKV used was much more efficient in deploying its genomic RNA into host cells than the strain of IAV used. This is reflected in the amount of viral and cellular input needed to get enough material to perform LC-MS/MS. For VIR-CLASP with CHIKV, infecting one 15 cm plate of U2OS cells (5 *106) with MOI 1000 yielded enough purified protein to perform LC-MS/MS analysis at 0.2 hpi (Figure 2), but for IAV, we infected six 15 cm plates of A549 cells (6 * 107 cells total) with MOI 10005. For other viruses, optimization will be needed to yield sufficient protein which can be dependent upon the mass spectrometry instruments used for the proteomics analysis.
We recommend performing a minimum of three biological replicates of VIR-CLASP for a given condition and virus, on different days. For VIR-CLASP, we define biological replicates as infection of distinct stocks of cells with 4SU-labeled or unlabeled virus from distinct purification and concentration, ideally performed on different days. We also recommend that VIR-CLASP recovered eluates be subjected to LC-MS/MS analysis on the same day especially if label-free quantitation will be used. To analyze the LC-MS/MS data, researchers can use their preferred software for peptide-spectrum matching with guidance from their local proteomics expert. We advise including protein sequences for viral proteins as well as host proteins. For example, for CHIKV we identified the viral proteins Capsid, E1, and E2; we identified Nucleoprotein for IAV. To define candidate “VIR-CLASP RBPs” from the LC-MS/MS data peptide intensity ratios for +4SU over −4SU samples can be calculated as described in the protocol, or alternative methods may be used with expert guidance depending on the experimental design. We found that applying the criteria of 0.01% FDR and fold change > 5 yielded ~400 proteins significantly enriched in each VIR-CLASP condition for CHIKV. After determining the candidate “VIR-CLASP RBPs”, comparisons can be made between the conditions, timepoints, viruses, or cell lines tested. For example, overlaps between conditions for CHIKV showed that ~255 candidate proteins interact with pre-replicated CHIKV RNA in all conditions tested and ~340 proteins were unique to certain subsets of conditions (Figure 3). The choice of further functional analyses to perform will be dependent upon the particular experimental goals of the researchers.
Supplementary Material
CLASP with RNA marker input. 1% Agarose gel of RNAs pulled down with CLASP performed with the RNA marker (lane 1) as input. Lanes 2-5 demonstrate that increasing the bead buffer ratio (bead buffer : sample buffer) results in purification of lower molecular weight RNAs.
Box 1. A viral titer assay of UV-irradiated 4SU-labeled virus at timepoints5.
1. Seed 2.0x105 U2OS (host) cells per well in 6 well plates in a total volume of 2 ml per well. Incubate plates 1 day at 37 °C to allow cell attachment.
2. Make 10 fold dilutions of virus samples.
3. Add sample dilutions to U2OS cell monolayers in 6-well plates.
4. Incubate for 1 h at 4 °C. Rock plates every 15 minutes to distribute inoculum and prevent dry-out of cells.
5. Remove inoculum and add the culture medium.
6. At selected timepoints for UV irradiation, remove the culture medium and irradiate uncovered plate twice with 0.6 J/cm2 of 365 nm UV light in a Spectrolinker XL-1500 or similar device. Add 2ml culture medium.
6. Incubate for 5 h (or last timepoint of the experiment) at 37 °C (The incubation time can be changed based upon virus/cell types).
7. Remove the culture medium and add plaque assay overlay (DMEM with 0.6% SeaPlaque™ Agarose (Lonza, cat# 50100) containing 5% FBS).
8. Incubate plates for approximately 3 days at 37°C.
9. Fix with 4% paraformaldehyde and stain with 0.1% crystal violet in 20% ethanol.
Acknowledgements
We would like to thank Dr. Kristie L. Rose and Dr. W. Hayes McDonald at the Vanderbilt Mass Spectrometry Research Center for processing of the MS samples; Matthew Albertolle for technical help in the MS analysis; Dr. Thomas Voss and Dr. James E. Crowe Jr. (Vanderbilt University Medical Center) for IAV; and Dr. Terence S. Dermody (University of Pittsburgh School of Medicine) for CHIKV. Finally, we would like to thank members of the Ascano laboratory for their support, collegiality, and critical review of the manuscript. This work was supported by the National Institutes of Health 1R35GM119569-01 (M.A.), CTSA award No.UL1TR000445 from the National Center for Advancing Translational Sciences (B.K. and M.A.), Vanderbilt University Dept. Biochemistry start-up funds (M.A.), the Chemical Biology of Infectious Disease training grant 5T32AI11254-02 (S.A.), and the Chemistry-Biology Interface training grant 5T32GM065086-14.
Footnotes
Code Availability
All code used for proteomic analysis and figure generation can be accessed under the GNU General Public License v3.0 at https://github.com/Ascano-Lab.
Competing interests
The authors have no competing financial or non-financial interests.
Data Availability
Raw and semi-processed proteomics data used to generate Figure 3 can be accessed from the PRIDE repository with the dataset identifier PXD015863 and was originally published in5
References
- 1.Phillips SL, Soderblom EJ, Bradrick SS & Garcia-Blanco MA Identification of Proteins Bound to Dengue Viral RNA In Vivo Reveals New Host Proteins Important for Virus Replication. MBio 7, e01865–15 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lenarcic EM, Landry DM, Greco TM, Cristea IM & Thompson SR Thiouracil cross-linking mass spectrometry: a cell-based method to identify host factors involved in viral amplification. J. Virol. 87, 8697–8712 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garcia-Moreno M et al. System-wide Profiling of RNA-Binding Proteins Uncovers Key Regulators of Virus Infection. Molecular Cell 74, 196–211.e11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou Y & Routh A Mapping RNA-capsid interactions and RNA secondary structure within virus particles using next-generation sequencing. Nucleic Acids Research 48, e12–e12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim B et al. Discovery of Widespread Host Protein Interactions with the Pre-replicated Genome of CHIKV Using VIR-CLASP. Molecular Cell 78, 624–640.e7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barrows NJ et al. Dual roles for the ER membrane protein complex in flavivirus infection: viral entry and protein biogenesis. Scientific Reports 9, 9711 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Favre A et al. 4-Thiouridine photosensitized RNA-protein crosslinking in mammalian cells. Biochem. Biophys. Res. Commun 141, 847–854 (1986). [DOI] [PubMed] [Google Scholar]
- 8.DeAngelis MM, Wang DG & Hawkins TL Solid-phase reversible immobilization for the isolation of PCR products. Nucleic Acids Research 23, 4742–4743 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Licatalosi DD et al. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature 456, 464–469 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Van Nostrand EL et al. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat Meth 13, 508–514 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.König J et al. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat. Struct. Mol. Biol 17, 909–915 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hafner M et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 141, 129–141 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ascano M, Hafner M, Cekan P, Gerstberger S & Tuschl T Identification of RNA-protein interaction networks using PAR-CLIP. Wiley Interdiscip Rev RNA 3, 159–177 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Trendel J et al. The Human RNA-Binding Proteome and Its Dynamics during Translational Arrest. Cell 176, 391–403.e19 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Asencio C, Chatterjee A & Hentze MW Silica-based solid-phase extraction of cross-linked nucleic acid-bound proteins. Life Sci Alliance 1, e201800088 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shchepachev V et al. Defining the RNA interactome by total RNA-associated protein purification. Mol. Syst. Biol 15, e8689 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berensmeier S Magnetic particles for the separation and purification of nucleic acids. Appl. Microbiol. Biotechnol 73, 495–504 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miller C et al. Dynamic transcriptome analysis measures rates of mRNA synthesis and decay in yeast. Mol. Syst. Biol 7, 458 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoffmann M et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 181, 271–280.e8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang P, Liu Y, Ma H-C, Paul AV & Wimmer E Picornavirus morphogenesis. Microbiol. Mol. Biol. Rev 78, 418–437 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quail MA, Swerdlow H & Turner DJ Improved protocols for the illumina genome analyzer sequencing system. Curr Protoc Hum Genet Chapter 18, Unit 18.2 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Telesnitsky A & Wolin SL The Host RNAs in Retroviral Particles. Viruses 8, 235 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Noda T et al. Importance of the 1+7 configuration of ribonucleoprotein complexes for influenza A virus genome packaging. Nat Commun 9, 54 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaur P, Lee RCH & Chu JJH Infectious Viral Quantification of Chikungunya Virus-Virus Plaque Assay. Methods Mol. Biol 1426, 93–103 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Pedersen IR Density gradient centrifugation studies on lymphocytic choriomeningitis virus and on viral ribonucleic acid. J. Virol 6, 414–420 (1970). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lim C-K Virus Isolation and Preparation of Sucrose-Banded Chikungunya Virus Samples for Transmission Electron Microscopy. Methods Mol. Biol 1426, 153–162 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Mortz E, Krogh TN, Vorum H & Görg A Improved silver staining protocols for high sensitivity protein identification using matrix-assisted laser desorption/ionization-time of flight analysis. Proteomics 1, 1359–1363 (2001). [DOI] [PubMed] [Google Scholar]
- 28.Link AJ & Labaer J In-gel trypsin digest of gel-fractionated proteins. Cold Spring Harb Protoc 2009, pdb.prot5110–pdb.prot5110 (2009). [DOI] [PubMed] [Google Scholar]
- 29.Smyth GK Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3, Article3 (2004). [DOI] [PubMed] [Google Scholar]
- 30.Sysoev VO et al. Global changes of the RNA-bound proteome during the maternal-to-zygotic transition in Drosophila. Nat Commun 7, 12128–11 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Conway JR, Lex A & Gehlenborg N UpSetR: an R package for the visualization of intersecting sets and their properties. Bioinformatics 33, 2938–2940 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dickson AM et al. Dephosphorylation of HuR protein during alphavirus infection is associated with HuR relocalization to the cytoplasm. J. Biol. Chem. 287, 36229–36238 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Daffis S et al. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature Publishing Group 468, 452–456 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lex A, Gehlenborg N, Strobelt H, Vuillemot R & Pfister H UpSet: Visualization of Intersecting Sets. IEEE Trans Vis Comput Graph 20, 1983–1992 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
CLASP with RNA marker input. 1% Agarose gel of RNAs pulled down with CLASP performed with the RNA marker (lane 1) as input. Lanes 2-5 demonstrate that increasing the bead buffer ratio (bead buffer : sample buffer) results in purification of lower molecular weight RNAs.
Data Availability Statement
Raw and semi-processed proteomics data used to generate Figure 3 can be accessed from the PRIDE repository with the dataset identifier PXD015863 and was originally published in5