

ABSTRACT
The highly contagious whooping cough agent Bordetella pertussis has evolved as a human-restricted pathogen from a progenitor which also gave rise to Bordetella parapertussis and Bordetella bronchiseptica. While the latter colonizes a broad range of mammals and is able to survive in the environment, B. pertussis has lost its ability to survive outside its host through massive genome decay. Instead, it has become a highly successful human pathogen by the acquisition of tightly regulated virulence factors and evolutionary adaptation of its metabolism to its particular niche. By the deployment of an arsenal of highly sophisticated virulence factors it overcomes many of the innate immune defenses. It also interferes with vaccine-induced adaptive immunity by various mechanisms. Here, we review data from in vitro, human and animal models to illustrate the mechanisms of adaptation to the human respiratory tract and provide evidence of ongoing evolutionary adaptation as a highly successful human pathogen.
KEYWORDS: Pertussis, Bordetella, metabolism, virulence factors, evolution, adaptive immunity, innate immunity
Graphical Abstract
Introduction
Bordetella pertussis is the main causative agent of pertussis or whooping cough, an acute respiratory disease that severely affects young children [1], but also constitutes a significant disease burden in adolescents and adults. Despite a very high global vaccination coverage close to 85% [2], the most recent models put estimates at 24 million cases annually causing more than 160,000 deaths in children less than 5 years of age. Nevertheless, many cases remain undiagnosed. This disease overwhelmingly affects low-income countries. However, in spite of strong immunization policies since the 1950s, even industrialized countries have failed to eradicate the disease. In fact, over the past 10 years, a reemergence of this pathogen has been reported in several countries, including more than 15,000 cases diagnosed in 2019 in the United States. For children that become infected under 6 months of age, the hospitalization rate is over 40%. Several factors might explain the upward trend of pertussis, including improvements of diagnostic techniques [3], vaccine-driven antigenic drift of circulating strains [4], the short duration of protection induced by acellular pertussis vaccines (aPV) [5], and the failure of the current vaccines to prevent B. pertussis infection and transmission [6].
Whooping cough is highly contagious and transmitted by aerosol droplets [7]. In the pre-vaccination era, the reproduction number R0 was estimated at 12 to 17, similar to that of measles (R0 = 12–18 in unvaccinated populations), but much larger than that of smallpox (R0 3.5–6) [8–10]. This makes B. pertussis one of the most contagious pathogens. Furthermore, whereas the R0 of measles dropped to almost zero in vaccinated populations, that of pertussis remained between 5 and 6 in areas with high vaccination rates, such as in Europe [11], implying that B. pertussis persists and circulates even in populations with high vaccination coverage.
The evolutionary history of B. pertussis
The genus Bordetella contains many species, most of which have been isolated from severely immunocompromised individuals. Only a few of them, including Bordetella petrii, have so far only been isolated from natural milieus [12–14]. This contrasts with the closely related genus Achromobacter, which comprises mostly environmental species and only a few opportunistic pathogens. Among the 16 known Bordetella species, 3 closely related species that form the “Bordetella bronchiseptica complex”, B. bronchiseptica, B. pertussis and Bordetella parapertussis have attracted most of the attention, as they are bona fide pathogens for humans and/or other mammals.
Given the paucity of information on most environmental Bordetella, studies on the evolutionary trajectory in this genus have mainly focused on the B. bronchiseptica complex, even though they are not the only professional pathogens of the genus [15]. In the B. bronchiseptica complex, the two-component regulatory system BvgAS enables the bacteria to switch from a virulent Bvg+ phase in which virulence-associated genes (vags) are expressed, to an avirulent Bvg− phase, in which they are silenced [16]. The avirulent phase is also characterized by the expression of a set of virulence-repressed genes (vrgs), including those coding for a flagellum and for metabolic pathways allowing growth in nutrient-limited media [15,17,18].
With its complex life cycle alternating between the soil and mammalian hosts, B. bronchiseptica is a good model to understand the transition from one environment to the other and how Bvg regulation is instrumental to this adaptation [15]. The finding that B. bronchiseptica survives and multiplies within amoebae, such as Dictyostelium discoideum, was key to understanding the selective pressure that has led to the acquisition of its virulence regulon [19]. Amoebae are protozoa that predate on bacteria as a source of nutrients, and their digestion mechanisms are very similar to the bactericidal mechanisms of mammalian phagocytic cells [20].
Similar evolutionary trajectories have been described for other pathogens, including Pseudomonas, Legionella, Listeria, Francisella, Burkholderia, Mycobacteria and others [21–25]. With their reduced genomes compared to that of the broad host range pathogen B. bronchiseptica, human-restricted B. pertussis and B. parapertussis are no longer able to survive in the environment [26]. B. pertussis also lost the ability to survive within amoeba and is considered primarily an extracellular pathogen, although some reports have described its admittedly limited capacity to survive within respiratory epithelial and phagocytic cells, which may be an evolutionary remnant [27–29]. Instead, B. pertussis and B. parapertussis have adopted specific infection strategies adapted to their cognate hosts.
B. bronchiseptica infects various mammals, and serological investigations of domestic animals have indicated that it is an almost ubiquitous pathogen [30–32]. Paradoxically, humans are rarely infected with this bacterium [33], whereas they are readily colonized by B. pertussis and B. parapertussis [34]. Unlike B. bronchiseptica, which is not very virulent or contagious in animal populations but instead causes chronic infections, B. pertussis and, to a lesser extent B. parapertussis, rely on high levels of virulence and transmissibility [35]. They cause acute, relatively short-lived infections, except in individuals unable to control the infection. It is likely that B. pertussis and B. parapertussis emerged and adopted this life style thanks to the evolution of the social organization of humans [35,36], whose population density has steadily increased throughout history. Host abundance and proximity enable easily-transmitted bacteria to persist in the population. Conversely, lower-density populations select pathogens able to persist in their host for longer periods, as they cannot quickly colonize new hosts.
It has been proposed that B. pertussis and B. parapertussis emerged independently from a common ancestor similar to current-day B. bronchiseptica and adopted similar modes of infection of their human host [37]. While a set of adaptive mutations enabled the former two species to cause acute infections in humans, the wide range of host species available to B. bronchiseptica limited the evolutionary pressure on the latter to adapt to human hosts. Its poor ability to infect humans may be due to the principle that two populations cannot survive within the same ecological niche if their requirements are similar [38]. Therefore, indirect rather than direct competition between bacteria may be mediated by the host. Since the three species are genetically extremely close, infection with any of them may trigger the development of adaptive immune defenses targeting virulence factors common to all three. In this context, B. pertussis and B. parapertussis appear to be favored by their infection strategy, as acute disease and rapid spread allow these bacteria to persist in the population. Since a large part of the human population has developed immune defenses against the virulence factors of the B. bronchiseptica complex, B. bronchiseptica is disadvantaged by its lower level of transmission.
The initial emergence of B. pertussis and the disappearance of B. bronchiseptica in humans are thought to have been favored by modifications of the immunogenic profile of B. pertussis to partially circumvent preexisting immunity against B. bronchiseptica. As such, B. pertussis has replaced lipopolysaccharide (LPS) with lipooligosaccharide (LOS) and lost its flagellum, two highly immunogenic components (see below). Furthermore, B. pertussis produces large amounts of pertussis toxin (PTX) to limit the recruitment and the activation of phagocytes, as it lost most genes required to survive phagocytosis. Thereby, it has taken advantage of the limited anti-PTX immune response elicited by B. bronchiseptica infection, whose lifestyle appears to require at most very low levels of PTX. These differences may represent evolutionary steps that have allowed B. pertussis to partially escape the immunity induced by the competing species
An arsenal of virulence factors
Regulatory network in B. pertussis
Evolution has endowed B. pertussis with many virulence factors that make it an extremely contagious pathogen. The virulence regulon of B. pertussis contains more than 200 genes, which are controlled in a coordinated manner by the two-component system BvgAS [18,39,40] (Figure 1). In the Bvg+ phase the system is active by default. The sensor-kinase BvgS requires no activating ligand to induce its auto-phosphorylation and the subsequent transfer of the phosphate group to the transcriptional activator BvgA. Phosphorylated BvgA binds to the promoter regions of the vags and transactivates their expression. These genes notably encode toxins and adhesins and their secretion machineries. They also include bvgR, encoding a c-di-GMP phosphodiesterase, which indirectly down-regulates the expression of the vrgs. In laboratory conditions, BvgS responds to chemical stimuli, such as nicotinate and sulfate ions, by turning off its auto-phosphorylation, which shifts the bacteria to the Bvg− phase [41]. Low temperatures (<25°C) also shift B. bronchiseptica to the Bvg− phase, which involves remodeling of membrane lipid composition, whereas B. pertussis responds only partially to low temperatures [42]. An intermediate Bvgi phase also exists at intermediate concentrations of modulators, and possibly at intermediate temperatures corresponding to those of the nasal cavity. In the Bvgi phase, certain adhesins are produced, but not the toxins [43,44]. The signals to which BvgS might respond in vivo are not known [45].
Figure 1.
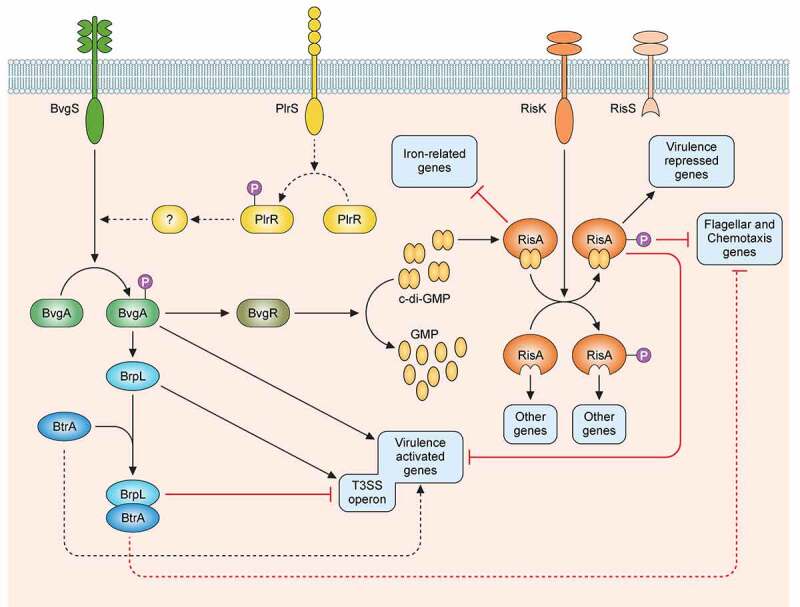
Regulatory network of the virulence factors in B. pertussis. BvgS is a sensor-kinase that is active by default at a temperature of 37°C. Its autophosphorylation is followed by the phosphorylation of its cognate response regulator BvgA. Phosphorylated BvgA triggers the expression of vir-activated genes (vags). BvgS responds to chemical stimuli such as sulfate or nicotinate ions by switching off the phosphorylation cascade, which increases the expression of vir-repressed genes (vrgs). BvgR is the product of a vag and hydrolyzes c-di-GMP to GMP. c-di-GMP affects the activity of the RisAK two-component system by binding to the response regulator RisA. The sensor-kinase RisS is truncated and nonfunctional in B. pertussis, and the partner of RisA is the sensor-kinase RisK. The targets of RisA depend on its phosphorylation and on the concentration of c-di-GMP. Non-phosphorylated RisA bound to c-di-GMP represses the expression of iron-related genes. With both c-di-GMP and phosphorylation RisA induces the expression of the vrgs and represses that of the vags and of the mobility genes, whereas in the absence of c-di-GMP, RisA induces the expression of other sets of genes depending on its phosphorylation. The sensor-kinase PlrS responds to CO2 by phosphorylating the response regulator PlrR. The regulon of PlrSR is unknown, but one of its target(s) interact(s) directly or indirectly with the BvgAS system in an uncharacterized manner. BrpL is a vag coding for a sigma factor that triggers the expression of the T3SS. BtrA antagonizes the activity of BrpL by titrating it in a BtrA/BrpL complex, which results in the repression of the T3SS and the mobility genes and the overexpression of certain vags. The dotted arrows represent interactions identified in B. bronchiseptica and suspected in B. pertussis.
Recent studies have revealed a more complex picture of virulence regulation in B. pertussis, involving several additional systems [46] (Figure 1). The two-component RisA/S system is an ortholog of EnvZ-OmpR, known to regulate responses to osmolarity, motility and in some cases virulence in many Gram-negative bacteria [47]. B. pertussis has a truncated nonfunctional RisS sensor-kinase, but another sensor-kinase, RisK, phosphorylates the response regulator RisA [48]. Phospho-RisA triggers the differential expression of many genes, including the vrgs, depending on the intracellular concentration of c-di-GMP, which is hydrolyzed by BvgR when the bacterium is in the Bvg+ phase [49].
An additional two-component system, PlrRS, is thought to be involved in the colonization of the lower respiratory tract [50,51]. This system reacts to the concentration of CO2 and modulates the expression of certain vags, which might be useful in minimizing the inflammatory response of the host [46].
In addition to those two-component systems, sigma/anti-sigma factors regulate specific aspects of virulence. One such system is the BtrA/BrpL pair involved in the regulation of the type III secretion system (T3SS) [52]. However, it also intersects with the regulation of several other virulence factors, such as filamentous hemagglutinin (FHA), adenylate cyclase toxin (ACT) and pertactin (Prn). BtrA/BrpL is sensitive to in vitro stresses, such as glutamate limitation and the oxidation state of the culture medium [53,54]. The BrpL sigma factor is one of the vags.
Major virulence factors of B. pertussis
Many of the B. pertussis virulence factors have immunomodulatory properties that play important roles in host-pathogen interactions, including adhesins and toxins (Table 1). Several virulence factors, such as FHA, PTX, Prn and fimbriae, are included in the current aPV.
Table 1.
Major virulence factors of B. pertussis.
| Major virulence factors | ||||
|---|---|---|---|---|
| Regulators | Type | Stimuli | Regulon | |
| BvgAS | TCS* | In lab: nicotinate, MgSO4 | [40,48] | |
| RisAK | TCS | Unknown | [49,107] | |
| BtrA | Sigma factor | In lab: oxidative conditions, iron starvation, glutamate limitation … | [52] | |
| PlrRS** | TCS | CO2 | Unknown [51] | |
| RseA/RpoE | Sigma factor | Envelope stress | [55] | |
| Adhesins | Name | Structure | Secretion | |
| FHA | Filamentous hemagglutinin | β helix [56] | Two-Partner Secretion [67] | |
| Prn | Pertactin | β helix [57] | Autotransporter [72] | |
| Fim | Fimbriae | Polymer of pilin subunits [58] | Chaperone-usher pathway [59] | |
| Toxins | Name | Structure | Secretion mechanism | Activity |
| PTX | Pertussis toxin | AB5-type exotoxin [78] | Type IV secretion system Ptl [79] | ADP ribosylation of G proteins [80] |
| ACT | Adenylate cyclase | RTX toxin [83] | Type I secretion system CyaBDE [83] | Pore forming and calmodulin-dependent adenylate cyclase [83] |
| TCT | Tracheal cytotoxin | Disaccharide-tetrapeptide [60] | Peptidoglycan fragment [85,86] triggering an inflammatory response | |
| DNT | Dermonecrotic toxin | Homologous to Pasteurella multocida toxin [61] | Not secreted [62] | Transglutaminase activating Rho GTPases [63] |
| BteA/BopC | T3SS effector | [195] | Type III secretion system [64] | Cytotoxicity, unknown mechanism |
| BopN | T3SS effector | Homologous to Yersinia effector [65] | Type III secretion system [64] | Cytotoxicity, unknown mechanism |
* Abbreviations: TCS, two-component systems; RTX, repeats in toxin; T3SS, type three secretion system
Numbers in brackets refer to references.
FHA is a major adhesin of the genus Bordetella [66]. It forms a long beta helix and results from the proteolytic maturation of the 370-kDa precursor FhaB. It is secreted by a two-partner secretion system (TPS) [67] and mediates adherence to ciliated cells in the respiratory tract [68]. FHA also mediates bacterial aggregation [69].
The Bordetella fimbriae are type I pili. Two fimbrial serotypes are produced by B. pertussis, encoded by the fim2 and fim3 genes, respectively [70]. The pili are assembled via a chaperone-usher pathway, with the FimD subunit found at the distal end of the pili made up of Fim2 or Fim3 protein monomers. The fimbriae are involved in the adherence to the host’s ciliated cells [71].
Prn is an auto-transporter [72]. Its biological role is poorly understood. Some in vitro studies have provided evidence that it mediates adherence to ciliated cells of the epithelium [73], but its deletion does not affect mouse lung colonization by B. pertussis [74], and many recent clinical isolates no longer produce it, most likely because of vaccine-induced selective immune pressure [4] (see below). Additional autotransporter proteins found in B. pertussis include adhesins, such as the tracheal colonization factor Tcf [75] and factors involved in resistance to complement, such as BrkA [76] and Vag8 [77].
PTX is an AB5-type holotoxin of 105 kDa [78] secreted by the Ptl type IV secretion system [79]. The A subunit expresses an ADP-ribosyltransferase activity, and the B moiety is a pentamer responsible for binding of the toxin to the target cells [80]. The targets of the ADP-ribosyltransferase activity are the α subunits of many Giα and Goα subunit isoforms of trimeric G proteins, leading to dysregulation of several signaling pathways [81]. Notably, ADP-ribosylation of the G protein that transduces the adrenergic β-receptor-triggered negative regulation of the membrane-bound adenylate cyclase reverses this inhibition, resulting in unregulated cAMP production [82].
ACT is a member of the repeats in toxin (RTX) family. It is secreted via a Type I secretion system. The C-terminal part of ACT contains the RTX domain as well as 4 hydrophobic segments, which mediate attachment to target cells and form a pore in the plasma membrane [83,84]. Its N-terminal domain expresses a calmodulin-dependent adenylate cyclase activity, which converts ATP into cAMP. This toxin therefore causes ionic imbalances in the target cells through pore formation, as well as deregulation of their signaling pathways due to accumulation of cyclic AMP and depletion of ATP [83,84]. ACT specifically targets complement receptor (CR)3-expressing sentinel cells of the myeloid lineage [83].
Another major B. pertussis toxin is tracheal cytotoxin (TCT), a disaccharide-tetrapeptide produced during the remodeling of the bacterial peptidoglycan. Most Gram-negative bacteria recycle this fragment, but B. pertussis does not re-import it into the cytoplasm and, instead, releases it in large quantities to induce a pro-inflammatory response [85] and overexpression of the inducible nitric oxide synthase [86], triggering the destruction of ciliated cells in the upper respiratory tract [87].
Dermonecrotic toxin permanently activates the Rho GTPase of target cells by polyamination and deamidation of glutamines [88,89]. However, its biological role in pertussis pathogenesis is poorly understood because it is not secreted but remains within the bacterial cytoplasm. Currently, no study has been able to demonstrate how this toxin is delivered to target cells. However, recently, its cellular receptor was identified as the T-type voltage-gated Ca2+ channel CaV3.1, a protein particularly present in the nervous system [90].
Bordetellae also possess a BvgAS-regulated T3SS. The effector protein BteA (also known as BopC) was shown to cause necrosis of certain cell types in vitro [91]. A more recent study carried out with B. bronchiseptica has demonstrated the action of BteA on signaling mechanisms of the actin cytoskeleton, inducing necrosis of certain cell types but also the inhibition of phagocytosis by macrophages [92]. In B. pertussis, a specific amino acid substitution in the BteA protein [93] appears to decrease its cytotoxicity. BopN, another effector, was reported to be necessary for the cytotoxicity of BteA in certain epithelial cell lines [94]. BopN harbors nuclear targeting signals, suggesting its involvement in gene regulation of target cells [95].
Metabolic adaptation and pathogenicity
The basic metabolism of B. pertussis
The metabolism of B. pertussis has been mostly studied in vitro, essentially due to the need to identify optimal B. pertussis growth requirements for vaccine production. B. pertussis grows slowly, even under optimal conditions. It is not able to utilize sugars as carbon source [96], as three genes encoding enzymes involved in the glycolytic pathway (glucokinase, phosphofructokinase and fructose-1,6-bisphosphate) are lacking in its genome [97]. The molecules most readily metabolized by B. pertussis are amino acids, the most efficient of which is glutamate [96]. B. pertussis can be grown with glutamate as the sole carbon source and cysteine as a source of sulfur, demonstrating that all building blocks of the cell can be synthesized from just a few sources [98]. Glutamate enters the TCA cycle via glutamate dehydrogenase, from which it can feed into different branches, such as gluconeogenesis for synthesizing sugars of LOS and the capsule, or anabolism of other amino acids. Although it was initially assumed that B. pertussis had an incomplete TCA cycle, since it could not grow on citrate, all genes required for a fully functional TCA cycle are present in its genome [97] and are expressed. Furthermore, their products are functionally active [99].
Compared to B. pertussis, B. bronchiseptica is able to utilize a wider range of substrates, including sugars, which is compatible with its environmental niche [97]. This difference in metabolism between the two species may also be related to the difference in host specificity. Since B. pertussis is highly host restricted, it likely has adapted its metabolism to take advantage of what is readily available in its specific niche of the human nasopharynx and may have lost the ability to metabolize various substrates, including sugars. Instead, it has retained a broad metabolic potential and is able to synthesize most of what is necessary for growth, with few exceptions, such as nicotinic acid [100] and glutathione.
Early difficulties in growing B. pertussis in the laboratory were due to inhibitory molecules present either in media or produced during growth, in particular fatty acids. Therefore, substances such as starch, blood or heptakis ((2,6-O-dimethyl) β-cyclodextrin) [101], are required in growth media to prevent growth inhibition by the presence of fatty acids [102,103]. Palmitic acids and other fatty acids were found in cell extracts of B. pertussis in concentrations likely to be inhibitory, suggesting that they are byproducts of growth. Interestingly, sensitivity to fatty acids appears to have been specifically selected for in B. pertussis and is not shared by B. bronchiseptica. The lack of B. pertussis growth in the presence of fatty acids can be overcome by restoring the functionality of an operon coding for an efflux pump whose gene is inactivated in the B. pertussis genome due to a frame-shift insertion [104]. The production of autoinhibitory free fatty acids during growth seems counterintuitive. It may be that growth within the infected host does not result in the production of these fatty acids in enough quantity to affect growth, or that fatty acids are washed away or neutralized during in vivo growth. This is suggested by the high bacterial densities (up to 108 CFU/ml) that can be recovered from nasal washes of infected baboons [105], which do not include bacteria attached to the tissue and forming biofilms. Thus, the bacteria are not inhibited from reaching high densities in vivo. The in vivo relevance of the production of and sensitivity to fatty acids is not known and has not been explored.
Metabolism with host perspective: Linking metabolism and virulence
The link between metabolism and B. pertussis virulence is not yet fully understood. However, it has been shown that B. pertussis virulence may depend on the medium used to grow the bacteria, since proteins are differentially secreted depending on the growth medium [106]. Whereas the most abundant proteins secreted in glutamate-rich as well as in glutamate-poor media are well-known virulence factors, a number of less abundant proteins, including some thought to be virulence associated were found uniquely to be secreted in glutamate-poor media [53]. Other proteins were found to be secreted only in glutamate-rich medium. Glutamate-sensitive protein secretion might reflect metabolic adaptation to physiological concentrations of glutamate during infection.
In addition to bona fide virulence genes, both B. bronchiseptica and B. pertussis regulate many metabolic genes via the BvgAS two-component system. In B. bronchiseptica, which can switch between mammalian hosts and environmental niches, functional vrgs are much more abundant than in B. pertussis. For the former, it may be helpful to be able to switch between profoundly different metabolic states, using BvgAS-dependent gene regulation to rapidly alternate the Bvg+ and Bvg− phases. For B. pertussis, which has no environmental phase, the purpose of the Bvg− phase is not known. It is possible that the Bvg− phase of B. pertussis is an evolutionary remnant, since it shares a common ancestor with B. bronchiseptica. Alternatively, the avirulent state of B. pertussis may still play a role in the infection cycle, which is yet to be discovered. The first hypothesis is consistent with the observation that virulence factors expressed in the Bvg+ phase are sufficient for infection in animal models. However, since the Bvg− phase has been conserved in B. pertussis, at least in part, it may be responsible for more than turning off virulence factor production, despite the absence of an environmental phase of growth. Many genes involved in multiple metabolic pathways are negatively regulated by the BvgAS system and are thus upregulated in the Bvg− phase [107]. They include genes involved in lipid metabolism, the TCA cycle, amino acid transport systems, peptidoglycan synthesis and the glycolate oxidation pathway. Thus, B. pertussis undergoes multiple metabolic changes when shifting between Bvg phases. It may be that metabolic genes are up-regulated in the Bvg intermediate phase (Bvgi) in conditions such as those encountered in the nasal cavity. The resulting metabolic changes might favor maximal bacterial proliferation during the catarrhal phase to optimize the probability of transmission via aerosol droplets. However, the Bvgi phase is poorly understood and more work is required to link specific gene expression changes in this phase to distinct steps in the infection cycle.
B. pertussis genes involved in cellular respiration, cell division, cell wall synthesis and sulfur metabolism are expressed at lower levels in mice than in vitro [108]. This observation suggests that the bacteria grow more slowly in vivo than in vitro or/and that the metabolism is altered in vivo. Although this study highlighted the difference between in vitro and in vivo metabolism, many aspects of the B. pertussis metabolism during infection, including its carbon source in vivo, still remain unknown.
In addition to transcriptomic studies, essential gene studies can help elucidate basic physiology of an organism, both in vitro and in vivo. Transposon-directed insertion sequencing (TraDIS) is a powerful tool that has been used to identify genes involved in viability in different growth conditions. As an example, genes can be essential depending on whether the bacteria were grown in the Bvg+ or Bvg− phase [109]. Interestingly, that study showed some of the genes involved in cell wall synthesis are essential only in the Bvg− phase. This is the case for the entire mre locus and genes involved in energy metabolism, including all four genes involved in synthesizing the succinate dehydrogenase complex. Such studies have shown that conditionally essential genes of B. pertussis depend on the Bvg phase, and entire processes, such as cell wall biosynthesis, can proceed differently depending on the Bvg phase. These observations provide further evidence that BvgAS not only regulates virulence but is a key regulator in wider growth and metabolism, implying coordination between virulence and metabolism for B. pertussis. It should be noted that the Bvg− phase described in [109] was obtained using 50 mM MgSO4, which might by itself have some impact on gene expression.
Another technology, termed Tn-seq, has been used to discover B. pertussis genes that are essential for growth in vivo [110], during infection in mice. At day 3 post-infection, more genes were found to be conditionally essential than at day 1 after infection. Interestingly, only few classical virulence factors were identified as essential in these conditions, probably due to redundancy in their function. Of the essential BvgAS-regulated genes, the majority were associated with metabolism, illustrating the importance of a coordination of metabolism and virulence by BvgAS in vivo. In fact, most genes required for infection are associated with metabolism, regardless of regulation by BvgAS, and include genes involved in transport and biosynthesis of secondary metabolites, many of unknown function. An example of genes which were found to be essential on day 3 but not day 1 post-infection is the bhu operon, encoding a heme iron acquisition system, which shows that the process of acquiring iron from heme at this stage in infection is critical [111]. Furthermore, the cyoABCD operon encoding a quinol oxidase is also essential only in vivo, which may be due to adaptation to low oxygen concentrations in these conditions.
Through functional genomic studies it has thus become clear that metabolism is coordinated during infection by B. pertussis, at least partly in tandem with virulence through the BvgAS system. These studies have provided valuable insight into an organism which has adapted its metabolism to its host environment, but which has still retained the ability to change its metabolism and adapt to the changing parameters during infection, potentially to optimize growth and enhance transmission.
Interaction with host metabolism: Hyperinsulinemia and metal acquisition
Some virulence factors of B. pertussis may have effects on host metabolism. Very early studies [112] reported hypoglycemia in infants with pertussis. This was confirmed in more recent studies showing that insulin levels are higher in the patients with pertussis than in controls [113]. Mice infected with B. pertussis also develop hyperinsulinemia which led to a state of hypoglycemia [114]. PTX is likely to be a key virulence factor responsible for these observations, as it induces insulin secretion by pancreatic β cells and has been referred to as islet-activating protein [115–117]. Injection of PTX into diabetic rats leads to normalization of glucose tolerance [116]. In humans, insulin levels were enhanced following a single injection of PTX, but glucose concentrations were not modified, indicating that hyperinsulinemia in humans is not necessarily accompanied by hypoglycemia. Nevertheless, these studies show a systemic effect of PTX during infection in humans and suggest that this toxin is able to play a role in changing the host metabolism. Hypoglycemia in B. pertussis-infected patients may potentially diminish the capacity of their immune response to effectively combat the infection, since activated immune cells require enormous amounts of glucose for their effector functions [118–120]. Effective inflammatory responses often lead to insulin resistance, in order to augment glycemia to fuel their energy requirements. PTX may thus counterbalance this mechanism through its hyper-insulinemic effect.
Another metabolic adaptation of B. pertussis to its host is its ability to acquire essential, host-sequestered oligo-elements, such as iron and copper. B. pertussis gains iron from host proteins through the production of siderophores [121,122], such as alcaligin, whose synthesis is negatively regulated by the presence of iron. Mutants defective in siderophore synthesis are affected for growth and survival in mouse models of infection [123]. B. pertussis also acquires iron from heme [111], which occurs through the Bhu system. The expression of the bhu gene is iron-regulated and specifically heme-inducible [124]. Bhu-deficient mutants have a fitness defect in prolonged mouse colonization, providing evidence for a crucial role in iron acquisition from heme especially during later stages of infection [125]. A study on the regulation of siderophore production during infection in mice confirmed that alcaligin is important early in infection and that the Bhu heme system is important later in infection, probably reflecting the changing availability of iron during infection, perhaps as the specific niche in the respiratory tract changes with time [126]. An in vitro transcriptomic study carried out in iron-starved conditions revealed a previously undescribed ferric iron transport system, FbpABC, which is iron-repressed [127]. Subsequently, a ferrous iron transport system named FtrABCD was identified and shown to be active especially in acidic conditions in vitro, suggesting adaptation to acidic microenvironments during infection [128], when siderophores may become ineffective. Thus, the expression of genes which B. pertussis uses to gain iron is tightly controlled and appears to change in a temporal fashion during infection.
Transcriptomic and proteomic studies have revealed the interplay between iron metabolism and virulence [127,129]. Upregulation of genes under iron starvation causes greater attachment to epithelial respiratory cells in vitro, as a result of increased mucin binding [129]. Furthermore, the genes coding for the T3SS are up-regulated during iron starvation [127]. In contrast, several other virulence factors, including FHA, fimbriae, Prn and ACT are less abundant under iron-starved conditions, which was speculated to decrease the recognition of the bacteria by immune cells in conditions in which the bacteria may be stressed [129]. The gain of iron appears thus to be linked to the production of virulence factors, providing further evidence for the coordination of metabolism and virulence. During iron starvation, B. pertussis is metabolically stressed and, in addition to upregulating bhu expression and siderophore production, it increases the production of poly-hydroxybutyrate, a storage compound found previously to build up when growing in an excess of carbon source [130].
Like iron, copper is an essential element in most organisms because of its role as a cofactor in various proteins. However, the toxicity of this metal is also used by protozoa and phagocytic cells to poison ingested bacteria [20,131]. Copper homeostasis systems found in bacteria are correlated with their lifestyles [132]. B. pertussis harbors genes of copper homeostasis classically found in free living bacteria, but most of them are inactivated by insertion sequences, unlike in B. bronchiseptica. The only system active in B. pertussis is a copper-induced tolerance system composed of a metallochaperone for copper passivation and two peroxide detoxification enzymes [133]. This system is probably an evolutionary remnant of mechanisms developed to survive amoebal predation that nowadays help the bacteria to survive immune phagocytosis in mammals [134]. Interestingly, B. pertussis and other Bordetella species possess an original copper-regulated operon for uptake of this metal, suggesting that the bacteria might actually be starved of copper in certain host microenvironments (A.R.-M. and F. J.-D., unpublished data).
Creation of a privileged niche in naïve hosts
Interactions with the respiratory epithelium
B. pertussis is a strictly mucosal pathogen. It efficiently colonizes the upper respiratory tract of the human host. Infection of the lower respiratory tract can accidentally occur during severe pertussis [135]. In the first steps of infection by B. pertussis adhesins, principally FHA and fimbriae, mediate bacterial attachment to the respiratory epithelium. The N-terminal part of FHA contains a heparin-binding domain that interacts with sulfated glycolipids and proteoglycans, key components on the surface of epithelial cells and the extracellular matrix [136]. FHA contains an RGD (arginine-glycine-aspartate) motif, which has initially been proposed to allow B. pertussis to attach to integrin CR3 on the surface of monocytes [137]. However, direct binding studies with purified CR3 have shown that, while FHA is able to bind to CR3, this binding is not directly mediated by the RGD sequence [138]. Instead, the RGD sequence of FHA directly interacts with the leukocyte-response integrin and integrin-associated protein (LRI/AIP), which upregulates CR3 binding activity [139]. A carbohydrate-recognition-domain mediates adherence to ciliated epithelial cells [68,140]. Finally, the C-terminal domain beyond the β-helical region is involved in adherence to certain cell types such as rat lung epithelial (L2) cells and J774A.1 macrophages [141].
The minor B. pertussis fimbrial subunit FimD has been shown to bind to very late antigen-5 (VLA-5) on the surface of monocytes, and the interaction of FimD with VLA-5 activates CR3 through protein tyrosine kinase signaling, which may enhance FHA-mediated B. pertussis binding [142]. Studies in mice have shown that FimD is important for the colonization especially of the trachea and the lungs [143].
The role of Prn in colonization of the respiratory tract is unclear, as there is little convincing evidence of its importance for adherence to epithelial cells [144]. Experiments with B. bronchiseptica have shown that the loss of pertactin leads to a defect in colonization only in SCID mice [145], which rather suggests interactions with immune cells. The tracheal colonization factor Tcf is a poorly characterized virulence factor exclusive to B. pertussis. It also contains integrin-binding RGD motifs and seems specific for colonization of the trachea [75].
Adhesins are not the only mediators of interactions with epithelial cells. Other virulence factors such as ACT, TCT and the T3SS also indirectly interact with the epithelium to allow the bacteria to persist in its niche. TCT plays a major role in the disruption of the epithelium. By inducing lysis of ciliated epithelial cells and overproduction of mucus, it enables the bacteria to avoid mucociliary clearance [87]. The enzymatic activity of ACT targets regulatory pathways of cells and also disorganizes the epithelium by altering the tight junctions [146]. Moreover, ACT physically associates with FHA, which inhibits biofilm formation in vitro [147]. The T3SS also induces cytotoxicity in the respiratory epithelium [91,92], although B. pertussis BteA is less potent than that of B. bronchiseptica [93].
Strategies to escape recognition by naive hosts: A question of balance
In addition to specific interactions with the respiratory epithelium, B. pertussis has also evolved mechanisms to escape recognition by the first line of immunity, in particular by innate immune cells, without completely silencing the host immune response. To achieve this delicate balance, B. pertussis has modified surface molecules to decrease their immunogenicity, such as LPS that composes the outer leaflet of the outer membrane of Gram-negative bacteria. LPS is made of a lipid A membrane anchor, an oligosaccharide core and a polysaccharidic O antigen, and is a strong triggering molecule of the innate inflammatory response. The B. pertussis LPS-like molecule, named LOS, is devoid of O antigen, and therefore is less inflammatory and immunogenic than LPS. The presence of an unrepeated tri-saccharide composed of α-N-acetylglucosamine, β-2-acetamido-3-acetamido-2,3-dideoxy-mannuronic acid and β-l-2-acetamido-4-methylamino-fucose protects the bacteria from the bactericidal effect of the pulmonary surfactant proteins SP-A and SP-D [148,149]. In addition, the reactogenicity of the lipid A moiety of B. pertussis LOS is further decreased thanks to the inactivation of pagP, the gene coding for a transferase that adds a palmitic acid residue to lipid A. The absence of the palmitate reduces LOS recognition by the innate immune Toll-like receptor 4 (TLR4) [150]. Furthermore, in the Bvg+ phase the phosphate of lipid A is substituted by glucosamine [151], which intriguingly slightly increases the endotoxic activity of B. pertussis LOS and results in the induction of the pro-inflammatory cytokines IL-6, IL-1β and TNF-α [152,153]. However, at the same time, this modification enhances the resistance of the outer membrane to cationic antimicrobial peptides [154].
Together with surface modifications, B. pertussis also forms biofilms providing protection against the immune system. The architecture of this biofilm is complex. Major components include FHA, the surface-associated polysaccharide poly-β-1,6-N-acetyl-d-glucosamine produced by enzymes encoded by the BpsABCD genes [155] and extracellular DNA [156]. In a mouse model of infection, bacteria were found in biofilms up to 19 days post-inoculation in the nasal cavity, suggesting a role of biofilms for bacterial persistence [156].
Subversion of anti-B. pertussis Immunity
Subversion of innate immune responses
In addition to surface modifications and biofilm formation to decrease its visibility by the immune system, B. pertussis has evolved strategies to subvert the innate immune defense mechanisms and re-shape both innate and adaptive immune responses. Resident cells, infiltrating monocytes and immature DCs are the first immune cells to sense B. pertussis molecules via recognition by TLR and NOD and NOD-like receptors [157,158]. Activated immune cells and infected epithelial cells release cytokines and chemokines that mediate recruitment and activation of polymorphonuclear neutrophils (PMN), macrophages, natural killer (NK) cells, and certain types of T lymphocytes [159,160]. Activated innate cells ingest and kill bacteria and therefore help to clear the infection, while antigen-presenting cells (APCs) provide a link between innate and adaptive immunity important for long-term protection against (re)infection. The strategies deployed by B. pertussis to overcome these lines of defense comprise 1° the inhibition of cell trafficking to the respiratory tract, 2° the suppression of phagocyte activation, 3° triggering cell death and 4° blocking complement-mediated killing.
Among phagocytes, PMNs are the most numerous and have the greatest bactericidal capacity. Despite activation of neutrophil recruitment pathways in mice within 6 hours after B. pertussis infection, neutrophil influx in the lungs is delayed up to 3 days [159,161]. This blockage of neutrophil migration is mainly due to PTX, which prevents early neutrophil chemotaxis by inhibiting the production of attracting chemokines released in response to TLR-4 activation of alveolar macrophages (AM) and probably epithelial cells [162]. Decreased chemokine release may also contribute to the reduced early infiltration of monocytes and lymphocytes in the airways [163,164]. Likewise, TCT and ACT may inhibit trafficking of professional phagocytic cells to the respiratory tract [165–167].
Monocytes that arrive at the site of infection differentiate into functional effector macrophages to replenish the resident sentinel cells. However, exposure of primary human monocytes to ACT inhibits their differentiation [168], which makes them poorly phagocytic and devoid of pseudopodia. In vitro, ACT also causes de-differentiation of primary human alveolar macrophages into monocyte-like cells with upregulated CD14 levels [168]. Reprogramming of infiltrating monocytes and alveolar macrophages to less bactericidal and short-lived monocyte-like cell types might be particularly relevant to infection of infants and the elderly, whose immune defense relies strongly on innate immunity, with monocytes and macrophages playing significant protective roles.
Several circulating and membrane proteins of innate immune cells collaborate with TLRs in sensing microbial ligands and promoting inflammatory responses [169]. Phagocytic cells that encounter B. pertussis recognize the bacterium via surface-exposed molecules or surface-bound opsonins, such as immunoglobulins and complement proteins. Upon phagocytosis, the route of intracellular trafficking determines the fate of internalized B. pertussis. In the absence of opsonizing serum, B. pertussis might be able to shift the balance toward the non-killing CR3-mediated internalization, thereby escaping clearance by the more efficient Fc receptors (FcR)-mediated pathway [137,170].
Non-opsonic recognition of B. pertussis by CR3 depends on the fibronectin receptor VLA-5 that binds to FimD, and the CR3-VLA-5 complex increases CR3 binding via FHA [142,171]. Interaction of FHA via its RGD motif to the LRI/IAP, mediating is cross-linking, also results in upregulation of CR3 and enhances CR3 recognition by an unidentified FHA domain [139,141]. It is likely that FHA adhesion to the cells promote cell intoxication with ACT that notably blocks the oxidative burst capacity of neutrophils [172].
When bacteria are internalized through the non-opsonic route, a significant fraction of macrophage-phagocytosed B. pertussis can prevent phagosome-lysosome fusion and eventually replicate in compartments with early endosomal characteristics. This escape mechanism involves microtubule assembly, lipid raft integrity, and the activation of a tyrosine-kinase-mediated signaling [27–29]. Deletion of the mgtC gene leads to a higher proportion of bacteria trafficked to lysosomes, suggesting that MgtC may enable the bacterium to prevent phagosome-lysosome fusion [173]. The enzymatic activity of ACT also appears to extend the intracellular survival of non-opsonized bacteria internalized by human and murine macrophages [174,175], while inhibiting opsonophagocytis in primary neutrophils [176]. This indicates that cAMP activates signaling pathways that result in the blockade of phagocytic killing of bacteria [175–177]. ACT was further reported to block phagocytosis by hijacking RhoA activity and by blocking Syk signaling and membrane micropinocytosis [178–180].
PTX inhibits the antimicrobial activity of resident airway macrophages [181] and can also suppress the phagocytic activity of monocytes [182]. It may also intoxicate other resident cells in the lung tissues and inhibit their secretion of neutrophil-attracting chemokines, thereby significantly reducing neutrophil recruitment [162]. Furthermore, the administration of PTX to mice strongly reduced the amplification of the inflammatory response of TLR-stimulated epithelial cells and macrophages by serum or serum-borne lipids, such as lysophosphatidylcholine, confirming an anti-inflammatory activity of PTX [169].
Among key strategies to subvert host immune defenses, B. pertussis also induces phagocyte apoptosis via ACT-mediated activation of the macrophage effector caspases 3 and 7 [183,184]. The pro-apoptotic signaling of ACT depends on its enzymatic activity [185,186]. ACT‐produced cAMP also triggers macrophage apoptosis by the mitochondrial route [185]. Programmed cell death is further promoted by the ability of ACT to form cation-selective pores in the cell membranes [187]. B. pertussis FHA might also affect apoptosis. Purified FHA was shown to induce dose-dependent apoptosis in human phagocytic and epithelial cells, possibly through the inhibition of the NF-κB pathway [188,189]. However, this effect may be nonspecific as purified FHA is generally contaminated with LOS, a potent TLR-2 ligand that triggers apoptosis via TNF-α secretion.
B. pertussis has also evolved defense mechanisms against the complement system. By interacting with the C1 complex, the autotransporter BrkA prevents the deposition of the C3, C4 and C9 proteins on the bacterial surface thereby inhibiting both the classical and the alternative complement pathways [190]. Moreover, B. pertussis hijacks two proteins, C4BP and C1INH, that block the complement cascade. C4BP is recruited to the bacteria via FHA [191], and C1INH is subverted by the autotransporter Vag8 [77,192,193]. ACT also plays a role in complement resistance, by preventing complement-mediated opsonophagocytosis. It invades cells carrying CR3 and diverts the phosphatase–dependent dephosphorylation pathway, thereby inhibiting the complement-dependent oxidative burst and inducing non-apoptotic cell death [175,178,194].
Thus, B. pertussis deploys an arsenal of effectors that subvert innate immune responses. On the other hand, it has evolved specific adaptations that reduce its capacity to suppress the host inflammatory response via modifications in effector molecules that are transported by the T3SS. In B. bronchiseptica, the T3SS BteA effector is necessary and sufficient for cytotoxicity in vitro [195,196], inducing necrosis through an actin cytoskeleton signaling pathway and inhibiting phagocytosis by macrophages [92]. On the other hand, BteA was reported to interfere with NF-κB to modulate host immune responses [197]. In B. pertussis BteA has very low specific cytotoxic activity in vitro due to the insertion of one Ala residue in its sequence [93], and furthermore the T3SS expression is tightly regulated by the anti-σ factor BtrA. Deletion of btrA in B. pertussis derepresses the T3SS genes and confers readily detectable BteA-dependent cytotoxicity in vitro [52]. It is thus likely that the reduced cytotoxic activity of the B. pertussis T3SS results from a combination of the inhibition of its expression by BtrA activity and of the reduced specific activity of BteA. In line with this, the deletion of the additional Ala residue increased lethality of B. pertussis in a mouse model of intranasal infection and decreased inflammation in the mouse lungs at sublethal doses of infection, most likely due to its immunosuppressive activity [93]. The latter observation suggests that the reduced activity of B. pertussis BteA might contribute to the acute course of the pulmonary form of human infant pertussis.
Subversion of innate control of adaptive immunity
As the infection progresses the bacteria are challenged by the adaptive immune response. The orientation of the adaptive immune response depends on the primary responses induced by epithelial cells and innate immune cells, in particular DCs as powerful APCs. Both humoral and cellular immune responses contribute to protection against pertussis.
After recognition by bacterial TLR ligands immature intra-epithelial DCs mature and migrate to lymphoid tissues, where they present pathogen-derived antigens to naïve CD4+ and CD8+ T cells. This immunological synapse initiates the adaptive T cell response by priming the expansion and differentiation of naïve CD4+ T cells into different T-helper cell subsets, among which the Th1 and Th17 cells are the most important subsets for protection against B. pertussis [198,199]. There is strong evidence that Th1 cells are responsible for effective clearance of B. pertussis from the lungs of infected mice, while Th17 cells – and in particular resident memory IL-17-producing CD4+ T cells – mediate clearance from the nasal cavity [199–202]. They do so by mobilizing neutrophils probably through granulopoeisis and chemotactic cytokine and chemokine induction, as well as by increasing neutrophil survival [203]. IFN-γ produced by Th1 cells mediates activation of neutrophils and macrophages, triggers the formation of opsonizing antibodies and enhances FcR and CR expression on myeloid cells. It also plays a role in the induction of HLA class II expression on the cell surface. Antigen processing and presentation by class II HLA activates CD4+ helper-T cells, that promote the maturation and differentiation of antigen-specific B cells and the production of antibodies that are critical for preventing reinfection.
B. pertussis is apt at perturbing the deployment of the adaptive immune response by significantly delaying Th17 responses in the blood and in the lungs of mice [204], which is caused in part by the T3SS [205]. Moreover, internalization of B. pertussis by myeloid cells triggers the release of IFN-α, which directly suppresses IL-17 responses [204,206]. In addition, FHA-triggered IFN-α responses by peripheral blood mononuclear cells also compromise Th17 cell differentiation and potentially promote suppressive T cells [204]. In parallel, PTX prevents the migration of IFN-α-expressing DCs from the lungs to the lymphoid tissues [207], and thereby enriches the local IFN-α environment able to reduce the Th17 responses, which facilitates bacterial colonization.
At the peak of infection, PTX stimulates pro-inflammatory responses, including Th17 responses in the airways, and these responses are long-lived [208]. In addition to exacerbating airway inflammatory responses at the peak of infection in mice, PTX also inhibits the host mechanisms to resolve inflammation [209]. The local Th17 responses trigger the recruitment of neutrophils, which has been associated with cough, bronchial hyperactivity and hypersecretion in lung diseases [210]. This neutrophil influx may prolong paroxysmal cough typical for B. pertussis infection in humans. Eventually, however, Th17 and associated cytokines, such as IL-22, may potentially synergize with IL-17 to control infection [211]. Th17 cells play a crucial role in the resolution of infection and in memory immune responses.
To avoid protective Th1 responses, several B. pertussis virulence factors alter the priming of T cells. Through its enzymatic activity ACT perturbs the T-cell activation cascade [212,213] by disassembling the immune synapse [213,214]. In addition, ACT can intoxicate DCs to alter the Th1/Th17 balance and limit Th1 expansion in mice [215–217]. ACT activity also strongly interferes with human monocyte-derived DC functions [218], decreasing their capacity to present antigens to CD4+ T cells [219] and to induce CD8+ T cell proliferation, while enhancing IL-10 and IL-17-production [220]. Both ACT and PTX promote innate IL-1β production by myeloid cells through the activation of the NALP3 inflammasome and caspase-1 [221,222]. Interestingly, ACT-mediated activation of caspase-1, which polarizes the T cell response toward the Th17 subtype and promotes clearance of the bacteria, did not depend on the enzyme activity of this toxin but was due to its pore-forming activity [221]. In parallel, increased levels of cAMP can enhance the capacity of DC to promote regulatory T cells [220], which may alter protective immune responses against B. pertussis.
In addition, intoxication of epithelial cells by ACT during the course of infection impacts their cross-talk with resident immune cells [223]. As mucosal epithelial cells respond to IFN-γ, the presence of IFN-γ in the airways likely influences how they regulate downstream immune responses, including the recruitment of NK cells and T cells [224,225]. ACT exposure enhances IL-6 secretion by broncho-tracheal and nasal epithelial cells [223], a pleiotropic cytokine that promotes Th2 differentiation and simultaneously inhibits Th1 polarization [226]. IL-6 also plays an important role in regulating the balance between Th17 cells and regulatory T cells [223,227].
Other B. pertussis virulence factors suppress IL-12 production and skew the response toward anti-inflammatory cytokines. McGuirk and Mills have provided evidence that FHA inhibits the secretion of IL-12 by LPS-stimulated macrophages through upregulation of IL-6 and IL-10, thereby avoiding protective Th1 immunity and favoring the generation of tolerogenic regulatory T cells [228]. However, it has also been reported that this effect might be due to the presence of contaminating LOS [229]. Other studies have shown that wild type FHA can induce the secretion of IL-10 by human monocyte-derived dendritic cells, even in the presence of LOS-neutralizing polymyxin B, while a truncated version of FHA did not, even in the presence of LOS [230]. These studies suggest a role of FHA in the induction of regulatory cytokines by innate immune cells, perhaps in synergy with LOS. Fedele et al. have indeed shown that B. pertussis LOS can modulate human dendritic cell function and influences T cell polarization [231,232]. Prn also has an immunomodulatory function and dampens the production of pro-inflammatory cytokines by DCs [233]. Finally, the T3SS effector BopN may facilitate survival of B. pertussis by inhibiting local innate inflammatory responses and the subsequent induction of antigen-specific Th1, Th17, and antibody responses [205]. In DCs BopN can also induce the expression of IL-10 through the inhibition of both extracellular signal-regulated kinase [95] and NF-κB pathways.
Overall, the B. pertussis anti-immune strategies summarized in Figure 2 allow the bacteria to very efficiently colonize the respiratory tract of susceptible hosts, which may explain its high contagiousness. It is possible that factors other than those described here may also modulate adaptive immune responses to B. pertussis, but they require further investigation. There is also a significant gap in our knowledge of how preexisting immunity may affect immune responses upon re-infection. An effort in identifying adaptive signatures of B. pertussis in vaccinated populations is a first step toward understanding escape from preexisting B. pertussis-specific immune responses.
Figure 2.
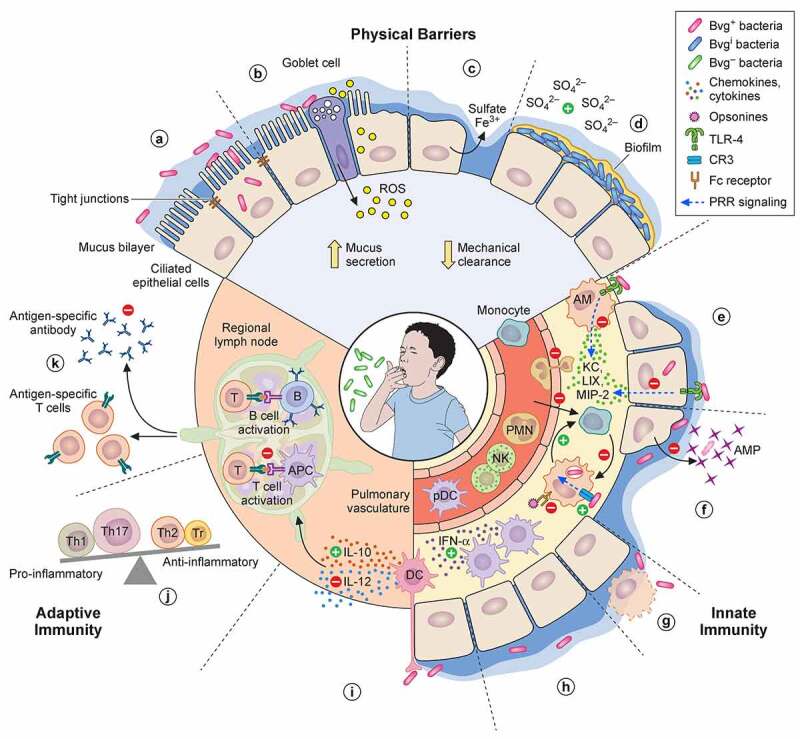
Anti-immune strategies of B. pertussis. (a) Upon infection, several virulence factors (FHA, Prn, Tcf, Fim, BteA, ACT, PTX) regulated by the BvgA/S two-component system (Bvg+ bacteria) enable B. pertussis adherence to epithelial cells of the nasal mucosa. The role for ACT in adherence is likely indirect. Some bacteria may invade epithelial cells. (b) TCT is internalized by epithelial cells and, along with LOS, induces the release of NO by the intoxicated cells, which is a major destructor of epithelial cells. ACT and BteA may also contribute to the disruption of the epithelial barrier. (c) Once the epithelial barrier is broken and submucosal glands have fired, iron may be delivered onto the surface of epithelial cells by the plasma exudate [234]. In response to the infection, the respiratory epithelium may produce highly sulfated mucins. Sulfate might be released from mucins by glycosyltransferases of B. pertussis and commensal bacteria [235,236], which might induce the Bvgi phase and hence trigger biofilm formation (d), thereby allowing long-term colonization. Up-regulation of the pendrin anion exchanger of epithelial cell drives the production of mucus, which along with ciliary damage, reduces mechanical clearance of the bacteria. (e) B. pertussis toxins, in particular PTX, suppress the early recruitment of immune cells. PTX prevents neutrophil chemotaxis indirectly by inhibiting the production of neutrophil-attracting chemokines KC, LIX and MIP-2 by alveolar macrophages (AM) and epithelial cells following disruption of TLR-4 signaling. (f) ACT may elicit reprogramming of infiltrating monocytes or alveolar macrophages to less bactericidal and short-lived monocyte-like cell types. Alternatively or additionally, FHA may enhance non-opsonic CR3-phagocytosis to escape effective clearance mediated by FcR recognition and suppresses – along with BrkA, LOS and ACT, and most of all Vag8 – complement-mediated killing (not represented). Expression of MgtC may enable the bacterium to escape opsonophagocytic killing by preventing phagosome-lysosome fusion, while ACT, PTX, FHA and Prn dampen the antimicrobial activity of the phagocytes, including oxidative stress and release of antimicrobial peptides (AMP). (g) To further avoid clearance by professional phagocytes, B. pertussis virulence factors such as ACT could trigger phagocyte apoptosis. (h) Several virulence factors may contribute to the accumulation of IFN-α-expressing DCs in the lungs that mediate early suppression of Th17 cell differentiation. (i) Intraepithelial DCs (and other myeloid cells) are reprogrammed to up-regulate IL-10 and down-regulate IL-12 production in order to favor the differentiation of regulatory T cells (Tr), Th2 and Th17 cells over Th1 cells. (j) ACT-intoxicated DCs are impaired in their capacity to stimulate T cells. The arrival of potentially tolerogenic DCs into the draining lymph nodes may alter cellular protective immune responses against B. pertussis. (k) Additionally, PTX is able to suppress serum antibody responses
B. pertussis adaptations to escape preexisting immunity
Pertussis is rare among recently vaccinated children, but its incidence quickly increases with age [237]. Protection following immunization with aPV wanes faster than that afforded by whole cell pertussis vaccines [238]. aPV used in most industrialized countries are composed of one to 5 different antigens, all combined with tetanus and diphtheria toxoids. All aPV contain detoxified PTX, most contain FHA and Prn in addition, and some further contain serotype 2 and serotype 3 fimbriae. Vaccines with one or more antigens, in addition to PTX, are more effective than vaccines containing PTX only [239]. However, similar to other bacteria for which acellular vaccines are widely used, B. pertussis evolves to escape vaccine-induced immunity by modifying the structure or production levels of the antigens present in the aPV.
PTX variations
PTX increases the severity of B. pertussis infections because closely related B. parapertussis, which does not produce PTX, generally causes less severe infections [240]. PTX causes leukocytosis in humans, which correlates with increased rates of hospitalizations and deaths [241]. Since the introduction of aPV, several variations in the ptx gene and its promoter region have been identified, some of which have been shown to increase ptx expression. In vaccinated hosts, increased PTX production might be beneficial for the pathogen because higher levels of antibodies are required for its neutralization. Seventeen ptxP alleles, with mutations in the ptx promoter, have been reported in the B. pertussis population worldwide [242,243], suggesting positive diversifying selection [244]. In contrast, there is minimal sequence diversity in the ptx coding sequences [244–248]. Only five protein variants of the enzymatic subunit PtxA have been identified, of which PtxA1 and PtxA2 predominate [242]. Very few Ptx-deficient isolates have been reported [249–251] so far, and the disease caused by these variants is much less severe than disease caused by PTX-producing strains, confirming the important role of PTX in pertussis pathogenesis [250].
With the progressive disappearance of the pre-vaccination ptxP2 variant, the ptxP1 and ptxP3 variants predominate globally [244,252]. Variants with ptxP3 promoters have been described to produce increased levels of PTX and were found to be associated with the resurgence of pertussis in the Netherlands [244,252]. They spread quickly and caused enhanced disease in vaccinated and unvaccinated populations. However, the spread and persistence of ptxP3 variants in vaccinated populations may not necessarily be due to the escape of anti-PTX antibodies but may also be the result of PTX-mediated immune suppression described above.
Compared to the pre-vaccination variants, ptxP3 strains also differentially express other virulence genes. The expression of the prn and tcfA genes is enhanced in the predominant ptxP3 strains in Australia, which may result in improved adhesion, while the downregulation of the T3SS and other immunogenic proteins, such as Vag8 and BipA may reduce immune recognition, which overall may contribute to the increased fitness of these strains [253]. Additionally, mutations in bscI – a T3SS gene – found in those strains may be advantageous in reducing immune recognition [253]. It is therefore not possible to ascribe the success of ptxP3 strains in vaccinated populations solely to enhanced production of PTX due to vaccine pressure.
Antigenic variations of Prn, FHA and fim
In contrast to PTX, a substantial sequence diversification has been detected for Prn and FHA, suggesting that B. pertussis benefits from changing these proteins in response to vaccine pressure [254]. Thirteen Prn variants have been identified, of which Prn1-Prn3 predominate in B. pertussis populations. Prn represents the only antigen of aPV that inducse bactericidal antibodies [255,256], while natural infection with B. pertussis induces bactericidal antibodies against a broad range of antigens [256]. Most variations in Prn occur in two regions comprised of five- and three-amino acid repeats [252]. Expansion or contraction of the tandem repeats may affect the binding avidity of anti-Prn antibodies [257].
Strains completely lacking Prn are currently isolated twice as frequently from vaccinated children than from unvaccinated children, and these strains outcompete Prn-producing strains in vaccinated, but not in unvaccinated mice [4,258,259]. The expansion of Prn− strains has reached a prevalence of 78–85% in some regions [260–262]. These findings suggest that the loss of Prn is favorable to B. pertussis when a vaccine-induced anti-Prn immune response is present, and that these strains have emerged and expanded because of aPV pressure.
FHA also exhibits diversification in highly antigenic sites but conservation within its functional domains. If diversified sites allow for anti-FHA antibody escape without impacting cytoadherence, this might enhance B. pertussis fitness. However, FHA variants have not exhibited the same widespread level of diversity in protein size and presence as Prn [254], most likely because of its importance in pertussis pathogenesis and the fact that current aPV do not induce bactericidal antibodies against FHA. Nevertheless, specific selection of fhaB frameshift mutations was found to be most prominent in the lungs of aPV-immunized mice, while they were rarely detected in the noses, suggesting that FHA is more important for colonization of the nose than of the lungs [263].
Like anti-PTX and anti-Prn antibodies, anti-fimbriae antibodies positively correlate with clinical protection [264]. Two variants of fim2 and 3 variants of fim3 have been found in circulating strains [252]. The relation between fimbrial types and pathogenicity remains elusive [245,265]. Genomic rearrangements leading to expression of either fim2 or fim3 have been observed in some ptxP3 strains [266], but differences in immune recognition remain untested.
Mutations in ptxP, prn and fim3 are associated with clonal sweep, i.e., these mutations are becoming dominant in the B. pertussis population through positive selection, suggesting that they increase strain fitness or are associated with additional mutations that do so [252]. Strain variation was shown to affect vaccine efficacy in mouse models [267–269]. The antigenic divergence observed between vaccine strains and circulating strains may act synergistically with the ptxP3 polymorphism by enhancing transmission from vaccinated hosts. The recent emergence of strains lacking Prn that also contain the ptxP3 allele is alarming. However, infected patients who had been vaccinated still experienced less severe forms of the disease than those who had not been vaccinated [248]. Nevertheless, adaptive changes of B. pertussis to aPV pressure may increase waning immunity. Of potential concern, an increase in fitness of vaccine-escape strains may lead to a rapid rise in the proportion of cases due to these strains and an overall increase in pertussis case numbers over a 20-year period [270].
Conclusion: Ongoing evolution of bacterial-host adaptation
The evolution of B. pertussis from the B. bronchiseptica complex was primarily associated with genome decay, mainly due to recombination between IS element repeat regions. Considering the scale of the gene loss in B. pertussis, which results in severe fitness cost in the environment, it is likely that it is a result of sophisticated adaptation to its specific niche, which is the human upper respiratory tract [271]. This high level of adaptation is reflected in the relatively monomorphic nature of B. pertussis strains, although adaptation and evolution continue to take place. Thus, recent isolates have undergone further loss of genetic material through insertion elements, the frequency of which is increasing with time [272]. Comparative genomic studies provide evidence of B. pertussis changes since the introduction of vaccine programs, in addition to those mentioned above. A study on 343 strains from 19 countries found two distinct branches, although one of them represented only 1.7% of strains, none of which was isolated in recent years [247]. Among the 12 alleles of the prn gene, the most diverse virulence factor gene described, three predominate in this collection, and among the eight ptxA alleles three are dominant, with ptxA1 representing 78% of strains tested. However, the ptxP3 allele frequency has increased overtime, suggesting an increasing advantage of this allele [247].
As described above, the other most striking example of recent continued adaptation by B. pertussis to its host is the emergence of strains lacking prn as a result of vaccine pressure. The prevalence of these strains varies from 27% of recent isolates from Japan [273] to more than 85% of strains recently isolated in the US [262], and prn loss was caused by 10 different types of mutations, including nucleotide substitutions resulting in a stop codon, or insertions resulting in frameshifts. However, the most common mutation was the insertion of IS481 in approximately 90% of prn mutations.
Thus, it appears that B. pertussis continues to adapt to its niche, and that the process of restriction to the human respiratory tract by genome reduction is still ongoing. Not unexpectedly, B. pertussis continues to adapt to changing human behavior, such as the shift from whole-cell vaccines to aPV, containing only a few antigens. Intuitively, one might expect that this continued adaptation would lead to decreased virulence, especially because of the loss of some of the classical virulence factors. However, this may be counterbalanced by compensating adaptations, such as the emergence of the ptxP3 allele. Most of such compensating adaptations are still to be discovered.
That the 2012 pertussis outbreak in the United Kingdom was caused by a number of closely related strains rather than the emergence of one hypervirulent clone, argues against specific large-scale genetic changes being responsible for the resurgence in pertussis [274]. In the isolates collected during this outbreak genes encoding antigens included in the aPV evolved faster than other genes encoding cell surface proteins. Although this was already seen before the introduction of the aPV, the rate of evolution of genes encoding aPV antigens has increased since the introduction of aPV.
Evolution of B. pertussis is not limited to genome decay but occurs also via genome rearrangement. A recent comparison of the genomes of 469 US strains showed 107 unique chromosome structures [275], with little diversity at the sequence level, indicating that structural rearrangement is also an important source of variability between strains. Chromosomal rearrangements can affect gene expression and may influence the phenotype, as shown in a recent study [276]. Adaptation of B. pertussis by genome rearrangement is only just beginning to be understood and its true significance will have to be evaluated through in vitro and in vivo experiments.
While evolution has enabled B. pertussis to adapt very specifically to its host, the host also undergoes evolutionary pressures to resist infection. This evolutionary principle is summarized in the ‘Red Queen” hypothesis [277,278] referring to the work of Lewis Carrol “Through the Looking-glass” where the two protagonists always remain at an equal distance during their chase. In an analogy for coevolution, this theory stipulates that a specific organism in a specific environment must constantly evolve in order to survive, because other living organisms also evolve in the same environment and therefore exert selective pressure on the first one. This hypothesis was extended to the relationship between parasites, more broadly including pathogens and symbionts, and their hosts [279].
Although no study has specifically examined coevolution of humans and B. pertussis, some observations prompt us to speculate on the adaptation of the human host to B. pertussis infection. Host cells use TLR2 and TLR4 to detect bacterial LPS, but the B. pertussis LOS triggers this signaling pathway only poorly [280]. This may be counterbalanced by PTX, which causes activation of TLR2/4 [281]. Likewise, the addition of a glucosamine residue onto the phosphate group of lipid A allows resistance to antimicrobial peptides [151,154], but it also causes increase in IL-6, IL-1β and TNF-α production [152,153]. Furthermore, in synergy with TCT B. pertussis LOS triggers the induction of IL-1α and inducible NO synthase (iNOS) in hamster tracheal organ cultures, while either molecule alone does not [282]. TCT is directly recognized by cytosolic peptidoglycan-recognition proteins from insects to mammals, including NOD1, and triggers NF-κB signaling pathways. However, while in NOD1-transfected HEK293 cells, TCT was well recognized by mouse NOD1, it was poorly recognized by human NOD1 [283]. More recent studies have shown that human NOD1 is a sensor for TCT, but TCT must be transported to the cytosol by the peptidoglycan-specific transporter SLC46A2 to activate NOD1 [284], which suggests that humans have adapted their TCT-mediated signaling through a specific mechanism.
The hemolysin RTX domain of ACT forms cation-selective pores by oligomerisation, resulting in efflux of potassium ions from target cells. Perturbation of potassium homeostasis activates dendritic cells and triggers a cell-mediated immune response in the host [177,285,286], thereby turning this property of ACT to humans’ advantage [221].
Funding Statement
The author(s) reported there is no funding associated with the work featured in this article.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data Availability statement:
I do not have a DOI yet, and since this is an invited signature review, I don’t think this applies here
References
- [1].Yeung KHT, Duclos P, Nelson EAS, et al. An update of the global burden of pertussis in children younger than 5 years: a modelling study. Lancet Infect Dis. 2017;17(9):974–980. [DOI] [PubMed] [Google Scholar]
- [2].WHO . https://www.who.int/data/gho/data/themes/immunization.
- [3].Guiso N, Wirsing von Knig CH, Forsyth K, et al. The global pertussis initiative: report from a round table meeting to discuss the epidemiology and detection of pertussis, Paris, France, 11–12 January 2010. Vaccine. 2011;29(6):1115–1121. [DOI] [PubMed] [Google Scholar]
- [4].Hegerle N, Dore G, Guiso N.. Pertactin deficient Bordetella pertussis present a better fitness in mice immunized with an acellular pertussis vaccine. Vaccine. 2014;32(49):6597–6600. [DOI] [PubMed] [Google Scholar]
- [5].Wilkinson K, Righolt CH, Elliott LJ, et al. Pertussis vaccine effectiveness and duration of protection - A systematic review and meta-analysis. Vaccine. 2021;39(23):3120–3130. [DOI] [PubMed] [Google Scholar]
- [6].Althouse BM, Scarpino SV. Asymptomatic transmission and the resurgence of Bordetella pertussis. BMC Med. 2015;13(1):146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Warfel JM, Beren J, Merkel TJ. Airborne transmission of Bordetella pertussis. J Infect Dis. 2012;206(6):902–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Anderson RM, May RM. Directly transmitted infections diseases: control by vaccination. Science. 1982;215(4536):1053–1060. [DOI] [PubMed] [Google Scholar]
- [9].Guerra FM, Bolotin S, Lim G, et al. The basic reproduction number (R0) of measles: a systematic review. Lancet Infect Dis. 2017;17(12):e420–e428. e420-e428. [DOI] [PubMed] [Google Scholar]
- [10].Gani R, Leach S. Transmission potential of smallpox in contemporary populations. Nature. 2001;414(6865):748–751. [DOI] [PubMed] [Google Scholar]
- [11].Kretzschmar M, Teunis PFM, Pebody RG. Incidence and reproduction numbers of pertussis: estimates from serological and social contact data in five European Countries. PLoS Med. 2010;7(6):e1000291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].von Wintzingerode F, Schattke A, Siddiqui RA, et al. Bordetella petrii sp. nov., isolated from an anaerobic bioreactor, and emended description of the genus Bordetella. Int J System Evol Microbiol. 2001;51(4):1257–1265. [DOI] [PubMed] [Google Scholar]
- [13].Wang F, Grundmann S, Schmid M, et al. Isolation and characterization of 1,2,4-trichlorobenzene mineralizing Bordetella sp. and its bioremediation potential in soil. Chemosphere. 2007;67(5):896–902. [DOI] [PubMed] [Google Scholar]
- [14].Bachate SP, Khapare RM, Kodam KM. Oxidation of arsenite by two beta-proteobacteria isolated from soil. Appl Microbiol Biotechnol. 2012;93(5):2135–2145. [DOI] [PubMed] [Google Scholar]
- [15].Hamidou Soumana I, Linz B, Harvill ET. Environmental origin of the genus Bordetella. Front Microbiol. 2017;8:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Melvin JA, Scheller EV, Miller JF, et al. Bordetella pertussis pathogenesis: current and future challenges. Nature Rev Microbiol. 2014;12(4):274–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hoffman CL, Gronyar LA, Zacca F, et al. Bordetella pertussis can be motile and express flagellum-like structures. mBio. 2019;10(3):e00787–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Cummings CA, Bootsma HJ, Relman DA, et al. Species- and strain-specific control of a complex, flexible regulon by Bordetella BvgAS. J Bacteriol. 2006;188(5):1775–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Taylor-Mulneix DL,Bendor L, Linz B, et al. Bordetella bronchiseptica exploits the complex life cycle of Dictyostelium discoideum as an amplifying transmission vector. PLoS Biol. 2017;15(4):e2000420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hao X, Lthje F, Ronn R, et al. A role for copper in protozoan grazing – two billion years selecting for bacterial copper resistance. Mol Microbiol. 2016;102(4):628–641. [DOI] [PubMed] [Google Scholar]
- [21].José Maschio V, Corçao G, Rott MB. Identification of Pseudomonas spp. as amoeba-resistant microorganisms in isolates of acanthamoeba. Rev Inst Med Trop São Paulo. 2015;57(1):81–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Greub G, Raoult D. Microorganisms resistant to free-living amoebae. Clin Microbiol Rev. 2004;17(2):413–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hägele S, Khler R, Merkert H, et al. Dictyostelium discoideum: a new host model system for intracellular pathogens of the genus Legionella. Cell Microbiol. 2000;2(2):165–171. [DOI] [PubMed] [Google Scholar]
- [24].Cirillo JD, Falkow S, Tompkins L, et al. Interaction of Mycobacterium avium with environmental amoebae enhances virulence. Infect Immun. 1997;65(9):3759–3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].DiSalvo S, Haselkorn TS, Bashir U, et al. Burkholderia bacteria infectiously induce the proto-farming symbiosis of Dictyostelium amoebae and food bacteria. Proc Natl Acad Sci USA. 2015;112(36):E5029–E5037. E5029-E5037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Rivera I, Linz B, Harvill ET. Evolution and conservation of Bordetella intracellular survival in eukaryotic host cells. Front Microbiol. 2020;11:557819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lamberti Y, Gorgojo J, Massillo C, et al. Bordetella pertussis entry into respiratory epithelial cells and intracellular survival. Pathog Dis. 2013;69(3):194–204. [DOI] [PubMed] [Google Scholar]
- [28].Lamberti Y,Perez Vidakovics ML, van der Pol LW, et al. Cholesterol-rich domains are involved in Bordetella pertussis phagocytosis and intracellular survival in neutrophils. Microb Pathog. 2008;44(6):501–511. [DOI] [PubMed] [Google Scholar]
- [29].Lamberti YA, Hayes JA, Perez Vidakovics ML, et al. Intracellular trafficking of Bordetella pertussis in human macrophages. Infect Immun. 2010;78(3):907–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Englund L, Jacobs AAC, Klingeborn B, et al. Seroepidemiological survey of Bordetella bronchiseptica and canine parainfluenza-2 virus in dogs in Sweden. Vet Rec. 2003;152(9):251–254. [DOI] [PubMed] [Google Scholar]
- [31].Hoskins JD,Williams J, Roy AF, et al. Isolation and characterization of Bordetella bronchiseptica from cats in southern Louisiana. Vet Immunol Immunopathol. 1998;65(2):173–176. [DOI] [PubMed] [Google Scholar]
- [32].By S, Nr U, Te S. Serologic survey for Bordetella bronchiseptica in Nebraska specific-pathogen-free pigs. Am J Vet Res. 1983;44(6):1123–1125. [PubMed] [Google Scholar]
- [33].Gupta S, Goyal P, Mattana J. Bordetella bronchiseptica pneumonia a thread in the diagnosis of human immunodeficiency virus infection. IDCases. 2019;15:e00509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].He Q, Viljanen MK, Arvilommi H, et al. Whooping cough caused by Bordetella pertussis and Bordetella parapertussis in an immunized population. JAMA. 1998;280(7):635–637. [DOI] [PubMed] [Google Scholar]
- [35].Bjørnstad ON, Harvill ET. Evolution and emergence of Bordetella in humans. Trends Microbiol. 2005;13(8):355–359. [DOI] [PubMed] [Google Scholar]
- [36].Novembre J, Han E. Human population structure and the adaptive response to pathogen-induced selection pressures. Philos Trans R Soc Lond, B, Biol Sci. 2012;367(1590):878–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Diavatopoulos DA, Cummings CA, Schouls LM, et al. Bordetella pertussis, the causative agent of whooping cough, evolved from a distinct, human-associated lineage of B. bronchiseptica. PLoS Pathog. 2005;1(4):e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Hardin G. The competitive exclusion principle. Science. 1960;131(3409):1292–1297. [DOI] [PubMed] [Google Scholar]
- [39].Chen Q, Stibitz S. The BvgASR virulence regulon of Bordetella pertussis. Curr Opin Microbiol. 2019;47:74–81. [DOI] [PubMed] [Google Scholar]
- [40].Coutte L, Antoine R, Slupek S, et al. Combined RNAseq and ChIPseq analyses of the BvgA virulence regulator of Bordetella pertussis. mSystems. 2020;5(3):e00208–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Melton AR, Weiss AA. Environmental regulation of expression of virulence determinants in Bordetella pertussis. J Bacteriol. 1989;171(11):6206–6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Seydlova G, Beranova G, Bibova I, et al. The extent of the temperature-induced membrane remodeling in two closely related Bordetella species reflects their adaptation to diverse environmental niches. J Biol Chem. 2017;292(19):8048–8058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Cotter PA, Miller JF. A mutation in the Bordetella bronchiseptica bvgS gene results in reduced virulence and increased resistance to starvation, and identifies a new class of Bvg-regulated antigens. Mol Microbiol. 1997;24(4):671–685. [DOI] [PubMed] [Google Scholar]
- [44].Deora R, Bootsma HJ, Miller JF, et al. Diversity in the Bordetella virulence regulon: transcriptional control of a Bvg-intermediate phase gene. Mol Microbiol. 2001;40(3):669–683. [DOI] [PubMed] [Google Scholar]
- [45].Melton AR, Weiss AA. Characterization of environmental regulators of Bordetella pertussis. Infect Immun. 1993;61(3):807–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Gestal MC, Whitesides LT, Harvill ET. Integrated signaling pathways mediate Bordetella immunomodulation, persistence, and transmission. Trends Microbiol. 2019;27(2):118–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Tipton KA, Rather PN. An OmpR-EnvZ two-component system ortholog regulates phase variation, osmotic tolerance, motility, and virulence in Acinetobacter baumannii strain AB5075. J Bacteriol. 2017;199(3):e00705–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Chen Q, Ng V, Warfel JM, et al. Activation of Bvg-repressed genes in Bordetella pertussis by RisA requires cross talk from noncooperonic histidine kinase RisK. J Bacteriol. 2017;199(22):e00475-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Coutte L, Huot L, Antoine R, et al. The multifaceted RisA regulon of Bordetella pertussis. Sci Rep. 2016;6(1):32774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Kaut CS, Duncan MD, Yei Kim JI, et al. A novel sensor kinase is required for Bordetella bronchiseptica to colonize the lower respiratory tract. Infect Immun. 2011;79(8):3216–3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Bone MA, Wilk AJ, Perault AI, et al. Bordetella PlrSR regulatory system controls BvgAS activity and virulence in the lower respiratory tract. Proc Natl Acad Sci USA. 2017;114(8):E1519–E1527. E1519-E1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Ahuja U, Shokeen B, Cheng N, et al. Differential regulation of type III secretion and virulence genes in Bordetella pertussis and Bordetella bronchiseptica by a secreted anti-σ factor. Proc Natl Acad Sci USA. 2016;113(9):2341–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Hanawa T, Kamachi K, Yonezawa H, et al. Glutamate limitation, BvgAS activation, and (p)ppGpp regulate the expression of the Bordetella pertussis type 3 secretion system. J Bacteriol. 2016;198(2):343–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Goto M, HanawaT, Abe A, et al. transcriptional downregulation of a type III secretion system under reducing conditions in Bordetella pertussis. J Bacteriol. 2020;202(21):e00400–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Barbier M, Boehm DT, Sen-Kilic E, et al. Modulation of pertussis and adenylate cyclase toxins by sigma factor RpoE in Bordetella pertussis. Infect Immun. 2017;85(1):e00565–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Clantin B, Hodak H, Willery C, et al. The crystal structure of filamentous hemagglutinin secretion domain and its implications for the two-partner secretion pathway. Proc Natl Acad Sci USA. 2004;101(16):6194–6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Emsley P, Charles IG, Fairweather NF, et al. Structure of Bordetella pertussis virulence factor P.69 pertactin. Nature. 1996;381(6577):90–92. [DOI] [PubMed] [Google Scholar]
- [58].Heck DV, Trus BL, Steven AC. Three-dimensional structure of Bordetella pertussis fimbriae. J Struct Biol. 1996;116(2):264–269. [DOI] [PubMed] [Google Scholar]
- [59].Scheller EV, Cotter PA. Bordetella filamentous hemagglutinin and fimbriae: critical adhesins with unrealized vaccine potential. Pathog Dis. 2015;73(8):ftv079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Chang C-I, Chelliah Y, Borek D, et al. Structure of tracheal cytotoxin in complex with a heterodimeric pattern-recognition receptor. Science. 2006;311(5768):1761–1764. [DOI] [PubMed] [Google Scholar]
- [61].Kitadokoro K, Kamitani S, Miyazawa M, et al. Crystal structures reveal a thiol protease-like catalytic triad in the C-terminal region of Pasteurella multocida toxin. Proc Natl Acad Sci USA. 2007;104(12):5139–5144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Cowell JL, Hewlett EL, Manclark CR. Intracellular localization of the dermonecrotic toxin of Bordetella pertussis. Infect Immun. 1979;25(3):896–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Fukui A, Horiguchi Y. Bordetella dermonecrotic toxin exerting toxicity through activation of the small GTPase Rho. J Biochem. 2004;136(4):415–419. [DOI] [PubMed] [Google Scholar]
- [64].Kamanova J. Bordetella type III secretion injectosome and effector proteins. Front Cell Infect Microbiol. 2020;10:466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Yahalom A, Davidov G, Kolusheva S, et al. Structure and membrane-targeting of a Bordetella pertussis effector N-terminal domain. Biochim Biophys Acta Biomembr. 2019;1861(12):183054. [DOI] [PubMed] [Google Scholar]
- [66].Tuomanen E, Weiss A. Characterization of two adhesins of Bordetella pertussis for human ciliated respiratory-epithelial cells. J Infect Dis. 1985;152(1):118–125. [DOI] [PubMed] [Google Scholar]
- [67].Guerin J, Bigot S, Schneider R, et al. Two-partner secretion: combining efficiency and simplicity in the secretion of large proteins for bacteria-host and bacteria-bacteria interactions. Front Cell Infect Microbiol. 2017;7:148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Tuomanen E, Towbin H, Rosenfelder G, et al. Receptor analogs and monoclonal antibodies that inhibit adherence of Bordetella pertussis to human ciliated respiratory epithelial cells. J Exp Med. 1988;168(1):267–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Menozzi FD, Boucher PE, Riveau G, et al. Surface-associated filamentous hemagglutinin induces autoagglutination of Bordetella pertussis. Infect Immun. 1994;62(10):4261–4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Ashworth LA, Robinson A, Funnell S, et al. Agglutinogens and fimbriae of Bordetella pertussis. Tokai J Exp Clin Med. 1988;13(Suppl: p):203–210. [PubMed] [Google Scholar]
- [71].Edwards JA, Groathouse NA, Boitano S. Bordetella bronchiseptica adherence to cilia is mediated by multiple adhesin factors and blocked by surfactant protein A. Infect Immun. 2005;73(6):3618–3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Leo JC, Grin I, Linke D. Type V secretion: mechanism(s) of autotransport through the bacterial outer membrane. Philos Trans R Soc Lond, B, Biol Sci. 2012;367(1592):1088–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Everest P, Li J, Douce G, et al. Role of the Bordetella pertussis P.69/pertactin protein and the P.69/pertactin RGD motif in the adherence to and invasion of mammalian cells. Microbiol-SGM. 1996;142(Pt 11):3261–3268. [DOI] [PubMed] [Google Scholar]
- [74].Khelef N, Bachelet CM, Vargaftig BB, et al. Characterization of murine lung inflammation after infection with parental Bordetella pertussis and mutants deficient in adhesins or toxins. Infect Immun. 1994;62(7):2893–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Finn TM, Stevens LA. Tracheal colonization factor: a Bordetella pertussis secreted virulence determinant. Mol Microbiol. 1995;16(4):625–634. [DOI] [PubMed] [Google Scholar]
- [76].Fernandez RC, Weiss AA. Cloning and sequencing of a Bordetella pertussis serum resistance locus. Infect Immun. 1994;62(11):4727–4738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Marr N, Shah NR, Lee R, et al. Bordetella pertussis autotransporter Vag8 binds human C1 esterase inhibitor and confers serum resistance. PLoS ONE. 2011;6(6):e20585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Stein PE, Boodhoo A, Armstrong, GD, et al. The crystal structure of pertussis toxin. Structure. 1994;2(1):45–57. [DOI] [PubMed] [Google Scholar]
- [79].Weiss AA, Johnson FD, Burns DL. Molecular characterization of an operon required for pertussis toxin secretion. Proc Natl Acad Sci USA. 1993;90(7):1569–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Locht C, Coutte L, Mielcarek N. The ins and outs of pertussis toxin. FEBS J. 2011;278(23):4668–4682. [DOI] [PubMed] [Google Scholar]
- [81].Kikuchi A, Kozawa Q, Kaibuchi T, et al. Direct evidence for involvement of a guanine nucleotide-binding protein in chemotactic peptide-stimulated formation of inositol bisphosphate and trisphosphate in differentiated human leukemic (HL-60) cells. reconstitution with Gi or Go of the plasma membranes ADP-ribosylated by pertussis toxin. J Biol Chem. 1986;261(25):11558–11562. [PubMed] [Google Scholar]
- [82].Katada T, Ui M. Direct modification of the membrane adenylate cyclase system by islet-activating protein due to ADP-ribosylation of a membrane protein. Proc Natl Acad Sci USA. 1982;79(10):3129–3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Novak J, Cherry O, Osickova A, et al. Structure-function relationships underlying the capacity of Bordetella adenylate cyclase toxin to disarm host phagocytes. Toxins (Basel). 2017;9(10):300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Guiso N. Bordetella adenylate cyclase-hemolysin toxins. Toxins (Basel). 2017;9(9):277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Heiss LN, Moser SA, Unanue ER, et al. Interleukin-1 is linked to the respiratory epithelial cytopathology of pertussis. Infect Immun. 1993;61(8):3123–3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Flak TA, Goldman WE. Signalling and cellular specificity of airway nitric oxide production in pertussis. Cell Microbiol. 1999;1(1):51–60. [DOI] [PubMed] [Google Scholar]
- [87].Kessie DK, Lodes N, Oberwinkler H, et al. Activity of tracheal cytotoxin of Bordetella pertussis in a human tracheobronchial 3D tissue model. Front Cell Infect Microbiol. 2021;10:614994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Horiguchi Y, Nakai T, Kume K. Purification and characterization of Bordetella bronchiseptica dermonecrotic toxin. Microb Pathog. 1989;6(5):361–368. [DOI] [PubMed] [Google Scholar]
- [89].Schmidt G, Goehring UM, Schirmer J, et al. Identification of the C-terminal part of Bordetella dermonecrotic toxin as a transglutaminase for Rho GTPases. J Biol Chem. 1999;274(45):31875–31881. [DOI] [PubMed] [Google Scholar]
- [90].Teruya S, Hiramatzu Y, Nakamura K, et al. Bordetella dermonecrotic toxin is a neurotropic virulence factor that uses CaV3.1 as the cell surface receptor. mBio. 2020;11(2):e03146–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Kuwae A, Matsuzawa T, Ishikawa N, et al. BopC is a novel type III effector secreted by Bordetella bronchiseptica and has a critical role in type III-dependent necrotic cell death. J Biol Chem. 2006;281(10):6589–6600. [DOI] [PubMed] [Google Scholar]
- [92].Kuwae A, Momose F, Nagamatsu K, et al. BteA secreted from the Bordetella bronchiseptica type III secretion system induces necrosis through an actin cytoskeleton signaling pathway and inhibits phagocytosis by macrophages. PLoS ONE. 2016;11(2):e0148387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Bayram J, Malcova I, Sinkovec L, et al. Cytotoxicity of the effector protein BteA was attenuated in Bordetella pertussis by insertion of an alanine residue. PLoS Pathog. 2020;16(8):e1008512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Abe A, Nishimura R, Kuwae A. Bordetella effector BopN is translocated into host cells via its N-terminal residues. Microbiol Immunol. 2017;61(6):206–214. [DOI] [PubMed] [Google Scholar]
- [95].Nagamatsu K, Kuwae A, Konaka T, et al. Bordetella evades the host immune system by inducing IL-10 through a type III effector, BopN. J Exp Med. 2009;206(13):3073–3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Jebb WHH, Tomlinson AHY. The catabolic activity of washed suspensions of Haemophilus pertussis. Microbiology. 1951;5(5):951–965. [DOI] [PubMed] [Google Scholar]
- [97].Parkhill J, Sebaihia M, Preston A, et al. Comparative analysis of the genome sequences of Bordetella pertussis, Bordetella parapertussis and Bordetella bronchiseptica. Nat Genet. 2003;35(1):32–40. [DOI] [PubMed] [Google Scholar]
- [98].Stainer DW, Scholte MJY. A simple chemically defined medium for the production of phase I Bordetella pertussis. J Gen Microbiol. 1970;63(2):211–220. [DOI] [PubMed] [Google Scholar]
- [99].Izac M, Garnier D, Speck D, et al. A functional tricarboxylic acid cycle operates during growth of Bordetella pertussis on amino acid mixtures as sole carbon substrates. PLoS ONE. 2015;10(12):e0145251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Brickman TJ, Suhadolc RJ, McKelvey PJ, et al. Essential role of Bordetella NadC in a quinolinate salvage pathway for NAD biosynthesis. Mol Microbiol. 2017;103(3):423–438. [DOI] [PubMed] [Google Scholar]
- [101].Fröhlich BT, d'Alarcao M, Feldberg RS, et al. Formation and cell-medium partitioning of autoinhibitory free fatty acids and cyclodextrin’s effect in the cultivation of Bordetella pertussis. J Biotechnol. 1996;45(2):137–148. [DOI] [PubMed] [Google Scholar]
- [102].Pollock MR. The growth of H. pertussis on media without blood. Br J Exp Pathol. 1947;28(4):295–307. [PMC free article] [PubMed] [Google Scholar]
- [103].Field LH, Parker CD. Effects of fatty acids on growth of Bordetella pertussis in defined medium. J Clin Microbiol. 1979;9(6):651–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].MacArthur I, Belcher T, King JD, et al. The evolution of Bordetella pertussis has selected for mutations of acr that lead to sensitivity to hydrophobic molecules and fatty acids. Emerg Microb Infect. 2019;8(1):603–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Eby JC, Gray MC, Warfel JM, et al. Quantification of the adenylate cyclase toxin of Bordetella pertussis in vitro and during respiratory infection. Infect Immun. 2013;81(5):1390–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Thalen M, Venema M, Dekker A, et al. Fed-batch cultivation of Bordetella pertussis: metabolism and pertussis toxin production. Biologicals. 2006;34(4):289–297. [DOI] [PubMed] [Google Scholar]
- [107].Moon K, Bonocora RP, Kim DD, et al. The BvgAS regulon of Bordetella pertussis. mBio. 2017;8(5):e01526–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Wong TY, Hall JM, Nowak ES, et al. Analysis of the in vivo transcriptome of Bordetella pertussis during infection of mice. mSphere. 2019;4(2):e00154–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Belcher T, MacArthur I, King JD, et al. Fundamental differences in physiology of Bordetella pertussis dependent on the two-component system Bvg revealed by gene essentiality studies. Microb Genom. 2020;6(12):e000496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Gonyar LA, Gelbach PE, McDuffie DG, et al. In vivo gene essentiality and metabolism in Bordetella pertussis. mSphere. 2019;4(3):e00694–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Vanderpool CK, Armstrong SK. The Bordetella bhu locus is required for heme iron utilization. J Bacteriol. 2001;183(14):4278–4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Regan JC, Tolstoouhov A. Relations of acid base equilibrium to the pathogenesis and treatment of whooping cough. NY State J. Med. 1936;36:1075–1087. [Google Scholar]
- [113].Furman BL, Walker E, Sidey FM, et al. Slight hyperinsulinaemia but no hypoglycaemia in pertussis patients. J Med Microbiol. 1988;25(3):183–186. [DOI] [PubMed] [Google Scholar]
- [114].Furman BL, Sidey FM, Wardlaw AC. Role of insulin in the hypoglycaemic effect of sublethal Bordetella pertussis infection in mice. Br J Exp Pathol. 1986;67(2):305–312. [PMC free article] [PubMed] [Google Scholar]
- [115].Katada T, Ui M. Slow interaction of islet-activating protein with pancreatic islets during primary culture to cause reversal of alpha-adrenergic inhibition of insulin secretion. J Biol Chem. 1980;255(20):9580–9588. [PubMed] [Google Scholar]
- [116].Toyota T, Kai Y, Kakizaki M, et al. Effects of islet-activating protein (IAP) on blood glucose and plasma insulin in healthy volunteers (phase 1 studies). Tohoku J Exp Med. 1980;130(2):105–116. [DOI] [PubMed] [Google Scholar]
- [117].Elahi S, Brownlie R, Korzeniowski J, et al. Infection of newborn piglets with Bordetella pertussis: a new model for pertussis. Infect Immun. 2005;73(6):3636–3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Straub RH. Interaction of the endocrine system with inflammation: a function of energy and volume regulation. Arthritis Res Ther. 2014;16(1):203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Macintyre AN, Gerriets VA, Nichols AG, et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 2014;20(1):61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Palmer CS, Ostrowski M, Balderson B, et al. Glucose metabolism regulates T cell activation, differentiation, and functions. Front Immunol. 2015;6:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Gorringe AR, Woods G, Robinson A. Growth and siderophore production by Bordetella pertussis under iron-restricted conditions. FEMS Microbiol Lett. 1990;66(1–3):101–105. [DOI] [PubMed] [Google Scholar]
- [122].Pradel E, Guiso N, Locht C. Identification of AlcR, an AraC-type regulator of alcaligin siderophore synthesis in Bordetella bronchiseptica and Bordetella pertussis. J Bacteriol. 1998;180(4):871–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Brickman TJ, Armstrong SK. Impact of alcaligin siderophore utilization on in vivo growth of Bordetella pertussis. Infect Immun. 2007;75(11):5305–5312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Vanderpool CK, Armstrong SK. Integration of environmental signals controls expression of Bordetella heme utilization genes. J Bacteriol. 2004;186(4):938–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Brickman TJ, Vanderpool CK, Armstrong SK. Heme transport contributes to in vivo fitness of Bordetella pertussis during primary infection in mice. Infect Immun. 2006;74(3):1741–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Brickman TJ, Hanawa T, Anderson MT, et al. Differential expression of Bordetella pertussis iron transport system genes during infection. Mol Microbiol. 2008;70(1):3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Brickman TJ, Cummings CA, Liew SY, et al. Transcriptional profiling of the iron starvation response in Bordetella pertussis provides new insights into siderophore utilization and virulence gene expression. J Bacteriol. 2011;193(18):4798–4812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Brickman TJ, Armstrong SK. Iron and pH-responsive FtrABCD ferrous iron utilization system of Bordetella species. Mol Microbiol. 2012;86(3):580–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Hayes JA, Lamberti Y, Surmann K, et al. Shotgun proteome analysis of Bordetella pertussis reveals a distinct influence of iron availability on the bacterial metabolism, virulence, and defense response. Proteomics. 2015;15(13):2258–2266. [DOI] [PubMed] [Google Scholar]
- [130].Thalen M, van den Ijssel J, Jiskoot W, et al. Rational medium design for Bordetella pertussis: basic metabolism. J Biotechnol. 1999;75(2):147–159. [DOI] [PubMed] [Google Scholar]
- [131].White C, Lee J, Kambe T, et al. A role for the ATP7A copper-transporting ATPase in macrophage bactericidal activity. J Biol Chem. 2009;284(49):33949–33956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Antoine R, Rivera-Millot A, Roy G, et al. Relationships between copper-related proteomes and lifestyles in β proteobacteria. Frontiers Microbiol. 2019;10:2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Rivera-Millot A, Slupek S, Chatagnon J, et al. Streamlined copper defenses make Bordetella pertussis reliant on custom-made operon. Commun Biol. 2021;4(1):46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Taylor-Mulneix DL, Hamidou Soumana I, Linz B, et al. Evolution of Bordetellae from environmental microbes to human respiratory pathogens: amoebae as a missing link. Front Cell Infect Microbiol. 2017;7:510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Greenberg D, Bamberger E, Ben-Shimol S, et al. Pertussis is under diagnosed in infants hospitalized with lower respiratory tract infection in the pediatric intensive care unit. Med Sci Monit. 2007;13(11):CR475–480. [PubMed] [Google Scholar]
- [136].Menozzi FD, Mutombo R, Renauld G, et al. Heparin-inhibitable lectin activity of the filamentous hemagglutinin adhesin of Bordetella pertussis. Infect Immun. 1994;62(3):769–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Relman D, Tuomanen E, Falkow S, et al. Recognition of a bacterial adhesin by an integrin: macrophage CR3 (αMβ2, CD11bCD18) binds filamentous hemagglutinin of Bordetella pertussis. Cell. 1990;61(7):1375–1382. [DOI] [PubMed] [Google Scholar]
- [138].Van Strijp JA, Russell DG, Tuomanen E, et al. Ligand specificity of purified complement receptor type three (CD11b/CD18, alpha m beta 2, Mac-1). Indirect effects of an arg-gly-asp (RGD) sequence. J Immunol. 1993;151(6):3324–3336. [PubMed] [Google Scholar]
- [139].Ishibashi Y, Claus S, Relman DA. Bordetella pertussis filamentous hemagglutinin interacts with a leukocyte signal transduction complex and stimulates bacterial adherence to monocyte CR3 (CD11b/CD18). J Exp Med. 1994;180(4):1225–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Prasad SM, Yi Y, Rodzinski E, et al. Identification of a carbohydrate recognition domain in filamentous hemagglutinin from Bordetella pertussis. Infect Immun. 1993;61(7):2780–2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Julio SM, Inatsuka CS, Mazar J, et al. Natural-host animal models indicate functional interchangeability between the filamentous haemagglutinins of Bordetella pertussis and Bordetella bronchiseptica and reveal a role for the mature C-terminal domain, but not the RGD motif, during infection. Mol Microbiol. 2009;71(6):1574–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Hazenbos WL, van den Berg BM, Geuijen CW, et al. Binding of FimD on Bordetella pertussis to very late antigen-5 on monocytes activates complement receptor type 3 via protein tyrosine kinases. J Immunol. 1995;155(8):3972–3978. [PubMed] [Google Scholar]
- [143].Geuijen CA, Willems RJ, Bongaerts M, et al. Role of the Bordetella pertussis minor fimbrial subunit, fimd, in colonization of the mouse respiratory tract. Infect Immun. 1997;65(10):4222–4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Roberts M, Fairweather N, Leininger E, et al. Construction and characterization of Bordetella pertussis mutants lacking the vir-regulated P.69 outer membrane protein. Mol Microbiol. 1991;5(6):1393-1404. [DOI] [PubMed] [Google Scholar]
- [145].Inatsuka CS, Xu Q, Vujkovic-Cvijin I, et al. Pertactin is required for Bordetella species to resist neutrophil-mediated clearance. Infect Immun. 2010;78(7):2901–2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Hasan S, Kulkarni NN, Asbjarnarson A, et al. Bordetella pertussis adenylate cyclase toxin disrupts functional integrity of bronchial epithelial layers. Infect Immun. 2018;86(3):e00445–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Hoffman C, Eby J, Gray M, et al. Bordetella adenylate cyclase toxin interacts with filamentous haemagglutinin to inhibit biofilm formation in vitro. Mol Microbiol. 2017;103(2):214–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Schaeffer LM, McCormack FX, Wu H, et al. Interactions of pulmonary collectins with Bordetella bronchiseptica and Bordetella pertussis lipopolysaccharide elucidate the structural basis of their antimicrobial activities. Infect Immun. 2004;72(12):7124–7130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Schaeffer LM, McCormack FX, Wu H, et al. Bordetella pertussis lipopolysaccharide resists the bactericidal effects of pulmonary surfactant protein A. J Immunol. 2004;173(3):1959–1965. [DOI] [PubMed] [Google Scholar]
- [150].Geurtsen J, Steeghs L, Hamstra HJ, et al. Expression of the lipopolysaccharide-modifying enzymes PagP and PagL modulates the endotoxic activity of Bordetella pertussis. Infect Immun. 2006;74(10):5574–5585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Marr N, Tirsoaga A, Blanot D, et al. Glucosamine found as a substituent of both phosphate groups in Bordetella lipid A backbones: role of a BvgAS-activated arnt ortholog. J Bacteriol. 2008;190(12):4281–4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Geurtsen J, Dzieciatkowska M, Steeghs L, et al. Identification of a novel lipopolysaccharide core biosynthesis gene cluster in Bordetella pertussis, and influence of core structure and lipid A glucosamine substitution on endotoxic activity. Infect Immun. 2009;77(7):2602–2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Marr N, Hajjar AM, Shah NR, et al. Substitution of the Bordetella pertussis lipid A phosphate groups with glucosamine is required for robust NF-κb activation and release of proinflammatory cytokines in cells expressing human but not murine toll-like receptor 4-MD-2-CD14. Infect Immun. 2010;78(5):2060–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Shah NR, Hancock REW, Fernandez RC. Bordetella pertussis lipid A glucosamine modification confers resistance to cationic antimicrobial peptides and increases resistance to outer membrane perturbation. Antimicrob Agents Chemother. 2014;58(8):4931–4934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Parise G, Mishra M, Itoh Y, et al. Role of a putative polysaccharide locus in Bordetella biofilm development. J Bacteriol. 2007;189(3):750–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Conover MS, Mishra M, Deora R. Extracellular DNA is essential for maintaining Bordetella biofilm integrity on abiotic surfaces and in the upper respiratory tract of mice. PLoS ONE. 2011;6(2):e16861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124(4):783–801. [DOI] [PubMed] [Google Scholar]
- [158].Hovingh ES, van Gent M, Hamstra HJ, et al. Emerging Bordetella pertussis strains induce enhanced signaling of human pattern recognition receptors TLR2, NOD2 and secretion of IL-10 by dendritic cells. PLoS ONE. 2017;12(1):e0170027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].McGuirk P, Mahon BP, Griffin F, et al. Compartmentalization of T cell responses following respiratory infection with Bordetella pertussis: hyporesponsiveness of lung T cells is associated with modulated expression of the co-stimulatory molecule CD28. Eur J Immunol. 1998;28(1):153–163. [DOI] [PubMed] [Google Scholar]
- [160].Rollins BJ. Chemokines. Blood. 1997;90(3):909–928. [PubMed] [Google Scholar]
- [161].Kirimanjeswara GS, Agosto LM, Kennett MJ, et al. Pertussis toxin inhibits neutrophil recruitment to delay antibody-mediated clearance of Bordetella pertussis. J Clin Invest. 2005;115(12):3594–3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Andreasen C, Carbonetti NH. Pertussis toxin inhibits early chemokine production to delay neutrophil recruitment in response to Bordetella pertussis respiratory tract infection in mice. Infect Immun. 2008;76(11):5139–5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Meade BD, Kind PD, Manclark CR. Altered mononuclear phagocyte function in mice treated with the lymphocytosis promoting factor of Bordetella pertussis. Dev Biol Stand. 1985;61:63–74. [PubMed] [Google Scholar]
- [164].Spangrude GJ, Sacchi F, Hill HR, et al. Inhibition of lymphocyte and neutrophil chemotaxis by pertussis toxin. J Immunol. 1985;135(6):4135–4143. [PubMed] [Google Scholar]
- [165].Friedman RL, Fiederlein RL, Glasser L, et al. Bordetella pertussis adenylate cyclase: effects of affinity-purified adenylate cyclase on human polymorphonuclear leukocyte functions. Infect Immun. 1987;55(1):135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Cundell DR, Kanthakumar K, Taylor GW, et al. Effect of tracheal cytotoxin from Bordetella pertussis on human neutrophil function in vitro. Infect Immun. 1994;62(2):639–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Guermonprez P, Khelef N, Blouin E, et al. The adenylate cyclase toxin of Bordetella pertussis binds to target cells via the αmβ2 integrin (Cd11b/Cd18). J Exp Med. 2001;193(9):1035–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Ahmad JN, Holubova J, Benada O, et al. Bordetella adenylate cyclase toxin inhibits monocyte-to-macrophage transition and dedifferentiates human alveolar macrophages into monocyte-like cells. mBio. 2019;10(5):e01743–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Sharma N, Akhade AS, Ismaeel S, et al. Serum-borne lipids amplify TLR-activated inflammatory responses. J Leukocyte Biol. 2021;109(4):821–831. [DOI] [PubMed] [Google Scholar]
- [170].Hellwig SM, van Oirschot HF, Hazenbos WL, et al. Targeting the Fcgamma receptors, but not CR3 (CD11b/CD18), increases clearance of Bordetella pertussis. J Infect Dis. 2001;183(6):871–879. [DOI] [PubMed] [Google Scholar]
- [171].Hazenbos WL, Berg BMVD, Furth RV. Very late antigen-5 and complement receptor type 3 cooperatively mediate the interaction between Bordetella pertussis and human monocytes. J Immunol. 1993;151(11):6274–6282. [PubMed] [Google Scholar]
- [172].Mobberley-Schuman PS, Weiss AA. Influence of CR3 (CD11b/CD18) expression on phagocytosis of Bordetella pertussis by human neutrophils. Infect Immun. 2005;73(11):7317–7323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Cafiero JH, Lamberti YA, Surmann K, et al. A Bordetella pertussis MgtC homolog plays a role in the intracellular survival. PLoS ONE. 2018;13(8):e0203204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Masure HR. The adenylate cyclase toxin contributes to the survival of Bordetella pertussis within human macrophages. Microb Pathog. 1993;14(4):253–260. [DOI] [PubMed] [Google Scholar]
- [175].Cerny O, Kamanova J, Masin J, et al. Bordetella pertussis adenylate cyclase toxin blocks induction of bactericidal nitric oxide in macrophages through camp-dependent activation of the SHP-1 phosphatase. J Immunol. 2015;194(10):4901–4913. [DOI] [PubMed] [Google Scholar]
- [176].Cerny O, Anderson KE, Stephens LR, et al. cAMP signaling of adenylate cyclase toxin blocks the oxidative burst of neutrophils through Epac-mediated inhibition of phospholipase C activity. J Immunol. 2017;198(3):1285–1296. [DOI] [PubMed] [Google Scholar]
- [177].Pearson RD, Symes P, Conboy M, et al. Inhibition of monocyte oxidative responses by Bordetella pertussis adenylate cyclase toxin. J Immunol. 1987;139(8):2749–2754. [PubMed] [Google Scholar]
- [178].Kamanova J, Kofronova O, Masin J, et al. Adenylate cyclase toxin subverts phagocyte function by RhoA inhibition and unproductive ruffling. J Immunol. 2008;181(8):5587–5597. [DOI] [PubMed] [Google Scholar]
- [179].Fiser R, Masin J, Bumba L, et al. Calcium influx rescues adenylate cyclase-hemolysin from rapid cell membrane removal and enables phagocyte permeabilization by toxin pores. PLoS Pathog. 2012;8(4):e1002580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Osicka R, Osickova A, Hasan S, et al. Bordetella adenylate cyclase toxin is a unique ligand of the integrin complement receptor 3. Elife. 2015;4:e10766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Carbonetti NH, Artamonova GV, Van Rooijen N, et al. Pertussis toxin targets airway macrophages to promote Bordetella pertussis infection of the respiratory tract. Infect Immun. 2007;75(4):1713–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Schaeffer LM, Weiss AA. Pertussis toxin and lipopolysaccharide influence phagocytosis of Bordetella pertussis by human monocytes. Infect Immun. 2001;69(12):7635–7641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Cheung GY, Kelly SM, Jess TJ, et al. Functional and structural studies on different forms of the adenylate cyclase toxin of Bordetella pertussis. Microb Pathog. 2009;46(1):36–42. [DOI] [PubMed] [Google Scholar]
- [184].Gueirard P, Druilhe A, Pretolani M, et al. Role of adenylate cyclase-hemolysin in alveolar macrophage apoptosis during Bordetella pertussis infection in vivo. Infect Immun. 1998;66(4):1718–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Ahmad JN, Cerny O, Linhartova I, et al. cAMP signalling of Bordetella adenylate cyclase toxin through the SHP-1 phosphatase activates the bimel-bax pro-apoptotic cascade in phagocytes. Cell Microbiol. 2016;18(3):384–398. [DOI] [PubMed] [Google Scholar]
- [186].Basler M, Masin J, Osicka R, et al. Pore-forming and enzymatic activities of Bordetella pertussis adenylate cyclase toxin synergize in promoting lysis of monocytes. Infect Immun. 2006;74(4):2207–2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Hewlett EL, Donato GM, Gray MC. Macrophage cytotoxicity produced by adenylate cyclase toxin from Bordetella pertussis: more than just making cyclic AMP! Mol Microbiol. 2006;59(2):447–459. [DOI] [PubMed] [Google Scholar]
- [188].Abramson T, Kedem H, Relman DA. Proinflammatory and proapoptotic activities associated with Bordetella pertussis filamentous hemagglutinin. Infect Immun. 2001;69(4):2650–2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Abramson T, Kedem H, Relman DA. Modulation of the NF-κB pathway by Bordetella pertussis filamentous hemagglutinin. PLoS ONE. 2008;3(11):e3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Barnes MG, Weiss AA. BrkA protein of Bordetella pertussis inhibits the classical pathway of complement after C1 deposition. Infect Immun. 2001;69(5):3067–3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Berggard K, Johnsson E, Mooi FR, et al. Bordetella pertussis binds the human complement regulator C4BP: role of filamentous hemagglutinin. Infect Immun. 1997;65(9):3638–3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Brookes C, Freire-Martin I, Cavelle B, et al. Bordetella pertussis isolates vary in their interactions with human complement components. Emerg Microbes Infect. 2018;7(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Hovingh ES, van den Broek B, Kuipers B, et al. Acquisition of C1 inhibitor by Bordetella pertussis virulence associated gene 8 results in C2 and C4 consumption away from the bacterial surface. PLoS Pathog. 2017;13(7):e1006531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Confer DL, Eaton JW. Phagocyte impotence caused by an invasive bacterial adenylate cyclase. Science. 1982;217(4563):948–950. [DOI] [PubMed] [Google Scholar]
- [195].Panina EM, Mattoo S, Griffith N, et al. A genome-wide screen identifies a Bordetella type III secretion effector and candidate effectors in other species. Mol Microbiol. 2005;58(1):267–279. [DOI] [PubMed] [Google Scholar]
- [196].French CT, Panina EM, Yeh SH, et al. The Bordetella type III secretion system effector btea contains a conserved N-terminal motif that guides bacterial virulence factors to lipid rafts. Cell Microbiol. 2009;11(12):1735–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Yuk MH, Harvill ET, Cotter PA, et al. Modulation of host immune responses, induction of apoptosis and inhibition of NF-kappaB activation by the Bordetella type III secretion system. Mol Microbiol. 2000;35(5):991–1004. [DOI] [PubMed] [Google Scholar]
- [198].Ryan M, Murphy G, Ryan E, et al. Distinct T-cell subtypes induced with whole cell and acellular pertussis vaccines in children. Immunology. 1998;93(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Warfel JM, Merkel TJ. Bordetella pertussis infection induces a mucosal IL-17 response and long-lived Th17 and Th1 immune memory cells in nonhuman primates. Mucosal Immunol. 2013;6(4):787–796. [DOI] [PubMed] [Google Scholar]
- [200].Ross PJ, Sutton CE, Higgins S, et al. Relative contribution of Th1 and Th17 cells in adaptive immunity to Bordetella pertussis: towards the rational design of an improved acellular pertussis vaccine. PLoS Pathog. 2013;9(4):e1003264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Dubois V, Chatagnon J, Thiriard A, et al. Suppression of mucosal Th17 memory responses by acellular pertussis vaccines enhances nasal Bordetella pertussis carriage. Npj Vaccines. 2021;6(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Borkner L, Curham LM, Wilk MM, et al. IL-17 mediates protective immunity against nasal infection with Bordetella pertussis by mobilizing neutrophils, especially siglec-F(+) neutrophils. Mucosal Immunol. 2021;14(5):1183-1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Kolls JK, Lindén A. Interleukin-17 family members and inflammation. Immunity. 2004;21(4):467–476. [DOI] [PubMed] [Google Scholar]
- [204].Wu V, Smith AA, You H, et al. Plasmacytoid dendritic cell-derived IFNα modulates Th17 differentiation during early Bordetella pertussis infection in mice. Mucosal Immunol. 2016;9(3):777–786. [DOI] [PubMed] [Google Scholar]
- [205].Fennelly NK, Sisti F, Higgins SC, et al. Bordetella pertussis expresses a functional type III secretion system that subverts protective innate and adaptive immune responses. Infect Immun. 2008;76(3):1257–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Dieterich C, Relman DA. Modulation of the host interferon response and ISGylation pathway by B. pertussis filamentous hemagglutinin. PLoS ONE. 2011;6(11):e27535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Nguyen TM, Ravindra D, Kwong B, et al. Differential expression of alpha 4 integrins on effector memory T helper cells during Bordetella infections. delayed responses in Bordetella pertussis. PLoS ONE. 2012;7(12):e52903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Andreasen C, Powell DA, Carbonetti NH. Pertussis toxin stimulates IL-17 production in response to Bordetella pertussis infection in mice. PLoS ONE. 2009;4(9):e7079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Connelly CE, Sun Y, Carbonetti NH. Pertussis toxin exacerbates and prolongs airway inflammatory responses during Bordetella pertussis infection. Infect Immun. 2012;80(12):4317–4332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Lindén A, Laan M, Anderson GP. Neutrophils, interleukin-17A and lung disease. Eur Resp J. 2005;25(1):159–172. [DOI] [PubMed] [Google Scholar]
- [211].Aujla SJ, Dubin PJ, Kolls JK. Interleukin-17 in pulmonary host defense. Exp Lung Res. 2007;33(10):507–518. [DOI] [PubMed] [Google Scholar]
- [212].Arumugham VB, Ulivieri C, Onnis A, et al. Compartmentalized cyclic AMP production by the Bordetella pertussis and Bacillus anthracis adenylate cyclase toxins differentially affects the immune synapse in T lymphocytes. Front Immunol. 2018;9:919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Paccani SR, Finetti F, Davi M, et al. The Bordetella pertussis adenylate cyclase toxin binds to T cells via LFA-1 and induces its disengagement from the immune synapse. J Exp Med. 2011;208(6):1317–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Rossi Paccani S, Benagiano M, Capitani N, et al. The adenylate cyclase toxins of Bacillus anthracis and Bordetella pertussis promote Th2 cell development by shaping T cell antigen receptor signaling. PLoS Pathog. 2009;5(3):e1000325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Bagley KC, Abdelwahab SF, Tuskan RG, et al. Pertussis toxin and the adenylate cyclase toxin from Bordetella pertussis activate human monocyte-derived dendritic cells and dominantly inhibit cytokine production through a cAMP-dependent pathway. J Leukocyte Biol. 2002;72(5):962–969. [PubMed] [Google Scholar]
- [216].Fedele G, Spensieri F, Palazzo R, et al. Bordetella pertussis commits human dendritic cells to promote a Th1/Th17 response through the activity of adenylate cyclase toxin and MAPK-pathways. PLoS ONE. 2010;5(1):e8734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Ross PJ, Lavelle EC, Mills KHG, et al. Adenylate cyclase toxin from Bordetella pertussis synergizes with lipopolysaccharide to promote innate interleukin-10 production and enhances the induction of Th2 and regulatory T cells. Infect Immun. 2004;72(3):1568–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Spensieri F, Fedele G, Fazio C, et al. Bordetella pertussis inhibition of interleukin-12 (IL-12) p70 in human monocyte-derived dendritic cells blocks IL-12 p35 through adenylate cyclase toxin-dependent cyclic AMP induction. Infect Immun. 2006;74(5):2831–2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Henderson MW, Inatsuka CS, Sheets AJ, et al. Contribution of Bordetella filamentous hemagglutinin and adenylate cyclase toxin to suppression and evasion of interleukin-17-mediated inflammation. Infect Immun. 2012;80(6):2061–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Adkins I, Kamanova J, Kocourkova A, et al. Bordetella adenylate cyclase toxin differentially modulates toll-like receptor-stimulated activation, migration and T cell stimulatory capacity of dendritic cells. PLoS ONE. 2014;9(8):e104064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Dunne A, Ross PJ, Pospisilova E, et al. Inflammasome activation by adenylate cyclase toxin directs Th17 responses and protection against Bordetella pertussis. J Immunol. 2010;185(3):1711–1719. [DOI] [PubMed] [Google Scholar]
- [222].Ronchi F, Basso C, Preite S, et al. Experimental priming of encephalitogenic Th1/Th17 cells requires pertussis toxin-driven IL-1β production by myeloid cells. Nat Commun. 2016;7(1):11541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Bassinet L, Fitting C, Housset B, et al. Bordetella pertussis adenylate cyclase-hemolysin induces interleukin-6 secretion by human tracheal epithelial cells. Infect Immun. 2004;72(9):5530–5533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].den Hartog G, Schijf MA, Berbers GAM, et al. Bordetella pertussis induces interferon gamma production by natural killer cells, resulting in chemoattraction by respiratory epithelial cells. J Infect Dis. 2020;(jiaa140). 10.1093/infdis/jiaa140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Kampf C, Relova AJ, Sandler S, et al. Effects of TNF-alpha, IFN-gamma and IL-beta on normal human bronchial epithelial cells. Eur Resp J. 1999;14(1):84–91. [DOI] [PubMed] [Google Scholar]
- [226].Diehl S, Rincón M. The two faces of IL-6 on Th1/Th2 differentiation. Mol Immunol. 2002;39(9):531–536. [DOI] [PubMed] [Google Scholar]
- [227].Bianchi M, Sivarajan R, Walles T, et al. Susceptibility of primary human airway epithelial cells to Bordetella pertussis adenylate cyclase toxin in two- and three-dimensional culture conditions. Innate Immun. 2021;27(1):89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].McGuirk P, Mills KHG. Direct anti-inflammatory effect of a bacterial virulence factor: IL-10-dependent suppression of IL-12 production by filamentous hemagglutinin from Bordetella pertussis. Eur J Immunol. 2000;30(2):415–422. [DOI] [PubMed] [Google Scholar]
- [229].Villarino Romero R, Hasan S, Fae K, et al. Bordetella pertussis filamentous hemagglutinin itself does not trigger anti-inflammatory interleukin-10 production by human dendritic cells. Int J Med Microbiol. 2016;306(1):38–47. [DOI] [PubMed] [Google Scholar]
- [230].Dirix V, Mielcarek N, Debrie AS, et al. Human dendritic cell maturation and cytokine secretion upon stimulation with Bordetella pertussis filamentous haemagglutinin. Microbes Infect. 2014;16(7):562–570. [DOI] [PubMed] [Google Scholar]
- [231].Fedele G, Celestino I, Spensieri F, et al. Lipooligosaccharide from Bordetella pertussis induces mature human monocyte-derived dendritic cells and drives a Th2 biased response. Microb Infect. 2007;9(7):855–863. [DOI] [PubMed] [Google Scholar]
- [232].Fedele G, Nasso M, Spensieri F, et al. Lipopolysaccharides from Bordetella pertussis and Bordetella parapertussis differently modulate human dendritic cell functions resulting in divergent prevalence of Th17-polarized responses. J Immunol. 2008;181(1):208–216. [DOI] [PubMed] [Google Scholar]
- [233].Hovingh ES, Mariman R, Solans L, et al. Bordetella pertussis pertactin knock-out strains reveal immunomodulatory properties of this virulence factor. Emerg Microbes Infect. 2018;7(1):39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Rivera-Chavez F, Mekalanos JJ. Cholera toxin promotes pathogen acquisition of host-derived nutrients. Nature. 2019;572(7768):244–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].Robertson AM, Wright DP. Bacterial glycosulphatases and sulphomucin degradation. Can J Gastroenterol. 1997;11(4):361–366. [DOI] [PubMed] [Google Scholar]
- [236].Nieuw Amerongen AV, Bolscher JG, Bloemena E, et al. Sulfomucins in the human body. Biol Chem. 1998;379(1):1–18. [DOI] [PubMed] [Google Scholar]
- [237].Schwartz KL, Kwong JC, Deeks SL, et al. Effectiveness of pertussis vaccination and duration of immunity. CMAJ. 2016;188(16):E399–E406. E399-E406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [238].Zhang L, Prietsch SO, Axelsson I, et al. Acellular vaccines for preventing whooping cough in children. Cochrane Database Syst Rev. 2014;9:CD001478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [239].Olin P, Rasmussen F, Gustafsson L, et al. Randomised controlled trial of two-component, three-component, and five-component acellular pertussis vaccines compared with whole-cell pertussis vaccine. ad hoc group for the study of pertussis vaccines. Lancet. 1997;350(9091):1569–1577. [DOI] [PubMed] [Google Scholar]
- [240].Watanabe M, Nagai M. Whooping cough due to Bordetella parapertussis: an unresolved problem. Expert Rev Anti Infect Ther. 2004;2(3):447–454. [DOI] [PubMed] [Google Scholar]
- [241].Pierce C, Klein N, Peters M. Is leukocytosis a predictor of mortality in severe pertussis infection? Intensive Care Med. 2000;26(10):1512–1514. [DOI] [PubMed] [Google Scholar]
- [242].Petersen RF, Dalby T, Dragsted MD, et al. Temporal trends in Bordetella pertussis populations, Denmark, 1949–2010. Emerg Infect Dis. 2012;18(5):767–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [243].Schmidtke AJ, Boney KO, Martin SW, et al. Population diversity among Bordetella pertussis isolates, United States, 1935–2009. Emerg Infect Dis. 2012;18(8):1248–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [244].Mooi FR, van Loo IH, van Gent M, et al. Bordetella pertussis strains with increased toxin production associated with pertussis resurgence. Emerg Infect Dis. 2009;15(8):1206–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [245].Advani A, Gustafsson L, Carlsson AM, et al. Clinical outcome of pertussis in Sweden: association with pulsed-field gel electrophoresis profiles and serotype. APMIS. 2007;115(6):736–742. [DOI] [PubMed] [Google Scholar]
- [246].Octavia S, Sintchenko V, Gilbert GL, et al. Newly emerging clones of Bordetella pertussis carrying prn2 and ptxP3 alleles implicated in Australian pertussis epidemic in 2008–2010. J Infect Dis. 2012;205(8):1220–1224. [DOI] [PubMed] [Google Scholar]
- [247].Bart MJ, Harris SR, Advani A, et al. Global population structure and evolution of Bordetella pertussis and their relationship with vaccination. mBio. 2014;5(2):e01074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [248].Clarke M, McIntyre PB, Blyth CC, et al. The relationship between Bordetella pertussis genotype and clinical severity in Australian children with pertussis. J Infect. 2016;72(2):171–178. [DOI] [PubMed] [Google Scholar]
- [249].Barkoff A-M, Mertsola J, Pierard D, et al. Pertactin-deficient Bordetella pertussis isolates: evidence of increased circulation in Europe, 1998 to 2015. Eurosurveillance. 2019;24(7):1700832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [250].Bouchez V, Brun D, Cantinelli T, et al. First report and detailed characterization of B. pertussis isolates not expressing pertussis toxin or pertactin. Vaccine. 2009;27(43):6034–6041. [DOI] [PubMed] [Google Scholar]
- [251].Williams MM, Sen K, Weigand MR, et al. Bordetella pertussis strain lacking pertactin and pertussis toxin. Emerg Infect Dis. 2016;22(2):319–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [252].Mooi FR, Van Der Maas NA, De Melker HE. Pertussis resurgence: waning immunity and pathogen adaptation – two sides of the same coin. Epidemiol Infect. 2014;142(4):685–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [253].Luu LDW, Octavia S, Zhong L, et al. Comparison of the whole cell proteome and secretome of epidemic Bordetella pertussis strains from the 2008–2012 Australian epidemic under sulfate-modulating conditions. Front Microbiol. 2018;9:2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [254].Etskovitz H, Anastasio N, Green E, et al. Role of evolutionary selection acting on vaccine antigens in the re-emergence of Bordetella pertussis. Diseases. 2019;7(2):35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [255].Hellwig SM, Rodriguez ME, Berbers GA, et al. Crucial role of antibodies to pertactin in Bordetella pertussis immunity. J Infect Dis. 2003;188(5):738–742. [DOI] [PubMed] [Google Scholar]
- [256].Lesne E, Cavell BE, Freire-Martin I, et al. Acellular pertussis vaccines induce anti-pertactin bactericidal antibodies which drives the emergence of pertactin-negative strains. Front Microbiol. 2020;11:2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [257].Moot FR, van Oirschot H, Heuvelman K, et al. Polymorphism in the Bordetella pertussis virulence factors p.69/pertactin and pertussis toxin in the Netherlands: temporal trends and evidence for vaccine-driven evolution. Infect Immun. 1998;66(2):670–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [258].Mastrantonio P, Spigaglia P, Oirschot HV, et al. Antigenic variants in Bordetella pertussis strains isolated from vaccinated and unvaccinated children. Microbiol-SGM. 1999;145(8):2069–2075. [DOI] [PubMed] [Google Scholar]
- [259].Safarchi A, Octavia S, Luu LD, et al. Pertactin negative Bordetella pertussis demonstrates higher fitness under vaccine selection pressure in a mixed infection model. Vaccine. 2015;33(46):6277–6281. [DOI] [PubMed] [Google Scholar]
- [260].Zeddeman A, van Gent M, Heuvelman CJ, et al. Investigations into the emergence of pertactin-deficient Bordetella pertussis isolates in six European countries, 1996 to 2012. Eurosurveillance. 2014;19(33):20881. [DOI] [PubMed] [Google Scholar]
- [261].Lam C, Octvaia S, Ricafort L, et al. Rapid increase in pertactin-deficient Bordetella pertussis isolates, Australia. Emerg Infect Dis. 2014;20(4):626–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [262].Martin SW, Pawloski L, Williams M, et al. Pertactin-negative Bordetella pertussis strains: evidence for a possible selective advantage. Clin Infect Dis. 2015;60(2):223–227. [DOI] [PubMed] [Google Scholar]
- [263].Zeddeman A, van Schuppen E, Kok KE, et al. Effect of FHA and Prn on Bordetella pertussis colonization of mice is dependent on vaccine type and anatomical site. PLoS ONE. 2020;15(8):e0237394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [264].Storsaeter J, Hallander HO, Gutsafsson L, et al. Levels of anti-pertussis antibodies related to protection after household exposure to Bordetella pertussis. Vaccine. 1998;16(20):1907–1916. [DOI] [PubMed] [Google Scholar]
- [265].Van Buynder PG, Owen D, Vurdien JE, et al. Bordetella pertussis surveillance in England and Wales: 1995–7. Epidemiol Infect. 1999;123(3):403–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [266].Weigand MR, Peng Y, Loparev V, et al. The history of Bordetella pertussis genome evolution includes structural rearrangement. J Bacteriol. 2017;199(8):e00806–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [267].King AJ, Berbers G, van Oirschot HF, et al. Role of the polymorphic region 1 of the Bordetella pertussis protein pertactin in immunity. Microbiol-SGM. 2001;147(11):2885–2895. [DOI] [PubMed] [Google Scholar]
- [268].Watanabe M, Nagai M. Effect of acellular pertussis vaccine against various strains of Bordetella pertussis in a murine model of respiratory infection. J Health Science. 2002;48(6):560–564 [Google Scholar]
- [269].Gzyl A, Augustynowicz E, Gniadek G, et al. Sequence variation in pertussis S1 subunit toxin and pertussis genes in Bordetella pertussis strains used for the whole-cell pertussis vaccine produced in Poland since 1960: efficiency of the DTwP vaccine-induced immunity against currently circulating B. pertussis isolates. Vaccine. 2004;22(17):2122–2128. [DOI] [PubMed] [Google Scholar]
- [270].Jayasundara D, Lee E, Octavia S, et al. Emergence of pertactin-deficient pertussis strains in Australia can be explained by models of vaccine escape. Epidemics. 2020;31:100388. [DOI] [PubMed] [Google Scholar]
- [271].Preston A, Parkhill J, Maskell DJ. The Bordetellae: lessons from genomics. Nat Rev Microbiol. 2004;2(5):379–390. [DOI] [PubMed] [Google Scholar]
- [272].Bouchez V, Caro V, Levillain E, et al. Genomic content of Bordetella pertussis clinical isolates circulating in areas of intensive children vaccination. PLoS ONE. 2008;3(6):e2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [273].Otsuka N, Han HJ, Toyoizumi-Ajisaka H, et al. Prevalence and genetic characterization of pertactin-deficient Bordetella pertussis in Japan. PLoS ONE. 2012;7(2):e31985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [274].Sealey KL, Harris SR, Fry NK, et al. Genomic analysis of isolates from the united kingdom 2012 pertussis outbreak reveals that vaccine antigen genes are unusually fast evolving. J Infect Dis. 2015;212(2):294–301. [DOI] [PubMed] [Google Scholar]
- [275].Weigand MR, Peng Y, Batra D, et al. Conserved patterns of symmetric inversion in the genome evolution of Bordetella respiratory pathogens. mSystems. 2019;4(6):e00702–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [276].Dienstbier A, Amman F, Petrackova D, et al. Comparative omics analysis of historic and recent isolates of Bordetella pertussis and effects of genome rearrangements on evolution. Emerg Infect Dis. 2021;27(1):57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [277].Liow LH, Van Valen L, Stenseth NC. Red Queen: from populations to taxa and communities. Trends Ecol Evol. 2011;26(7):349–358. [DOI] [PubMed] [Google Scholar]
- [278].Van Valen L. The Red Queen. Am Nat. 1977;111(980):809–810. [Google Scholar]
- [279].Decaestecker E, Gaba S, Raeymaekers JA, et al. Host–parasite ‘red queen’ dynamics archived in pond sediment. Nature. 2007;450(7171):870–873. [DOI] [PubMed] [Google Scholar]
- [280].Marr N, Novikov A, Hajjar AM, et al. Variability in the lipooligosaccharide structure and endotoxicity among Bordetella pertussis strains. J Infect Dis. 2010;202(12):1897–1906. [DOI] [PubMed] [Google Scholar]
- [281].Nasso M, Fedele G, Spensieri F, et al. Genetically detoxified pertussis toxin induces Th1/Th17 immune response through MAPKs and IL-10-dependent mechanisms. J Immunol. 2009;183(3):1892–1899. [DOI] [PubMed] [Google Scholar]
- [282].Flak TA, Heiss LN, Engle JT, et al. Synergistic epithelial responses to endotoxin and a naturally occurring muramyl peptide. Infect Immun. 2000;68(3):1235–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [283].Magalhaes JG, Philpott DJ, Nahori MA., et al. Murine NOD1 but not its human orthologue mediates innate immune detection of tracheal cytotoxin. EMBO Rep. 2005;6(12):1201–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [284].Paik D, Monahan A, Caffrey DR, et al. SLC46 family transporters facilitate cytosolic innate immune recognition of monomeric peptidoglycans. J Immunol. 2017;199(1):263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [285].Fedele G, Schiavoni I, Adkins I, et al. Invasion of dendritic dells, macrophages and neutrophils by the Bordetella adenylate cyclase toxin: a subversive move to fool host immunity. Toxins (Basel). 2017;9(10):293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [286].Svedova M, Masin J, Fiser R, et al. Pore-formation by adenylate cyclase toxoid activates dendritic cells to prime CD8+ and CD4+ T cells. Immunol Cell Biol. 2016;94(4):322–333. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
I do not have a DOI yet, and since this is an invited signature review, I don’t think this applies here