

Abstract
Aims
Type 1 diabetes (T1D) has a strong genetic predisposition and requires an environmental trigger to initiate the beta‐cell autoimmune destruction. The rate of childhood obesity has risen in parallel to the proportion of T1D, suggesting high‐fat diet (HFD)/obesity as potential environmental triggers for autoimmune diabetes. To explore this, non‐obese diabetic (NOD) mice were subjected to HFD and monitored for the development of diabetes, insulitis and beta‐cell stress.
Materials and Methods
Four‐week‐old female NOD mice were placed on HFD (HFD‐NOD) or standard chow‐diet. Blood glucose was monitored weekly up to 40 weeks of age, and glucose‐ and insulin‐tolerance tests performed at 4, 10 and 15 weeks. Pancreata and islets were analysed for insulin secretion, beta‐cell mass, inflammation, insulitis and endoplasmic reticulum stress markers. Immune cell levels were measured in islets and spleens. Stool microbiome was analysed at age 4, 8 and 25 weeks.
Results
At early ages, HFD‐NOD mice showed a significant increase in body weight, glucose intolerance and insulin resistance; but paradoxically, they were protected from developing diabetes. This was accompanied by increased insulin secretion and beta‐cell mass, decreased insulitis, increased splenic T‐regulatory cells and altered stool microbiome.
Conclusions
This study shows that HFD protects NOD mice from autoimmune diabetes and preserves beta‐cell mass and function through alterations in gut microbiome, increased T‐regulatory cells and decreased insulitis. Further studies into the exact mechanism of HFD‐mediated prevention of diabetes in NOD mice could potentially lead to interventions to prevent or delay T1D development in humans.
Keywords: Bacteroidetes, high‐fat diet, insulin resistance, microbiome, NOD mice, obesity, T‐regulatory cells, Type 1 diabetes, Verrucomicrobia
1. INTRODUCTION
Type 1 diabetes (T1D) is caused by the autoimmune destruction of insulin producing beta‐cells. 1 Multiple genes are known to modulate susceptibility to the development of T1D. 2 , 3 , 4 , 5 , 6 However, clinical studies of T1D have shown a relatively low concordance rate in identical twins, suggesting a strong environmental component in the development of the disease. 7 , 8 While the exact triggers of T1D are not well established, many environmental factors such as infections, diet, gut microbiome and vitamin D deficiency have been suggested to be involved. 9 Interestingly, the incidence of T1D has been increasing in parallel with the childhood obesity epidemic, 10 , 11 , 12 suggesting that a high calorie diet or obesity may be environmental triggers for the increasing incidence of T1D.
The rate of T1D and other autoimmune diseases has been steadily increasing in westernized societies. 12 , 13 , 14 , 15 The high‐fat Western diet has been proposed to play a role in the development of many autoimmune diseases. 16 High‐fat diet (HFD) has been previously associated with worsening autoimmune disease pathogenesis in animal models of lupus and multiple sclerosis. 17 , 18 In cell and animal models, HFD is known to induce insulin resistance, beta‐cell endoplasmic reticulum (ER) stress, mitochondrial damage, oxidative stress, dysfunctional insulin production and beta‐cell death. 19 , 20 , 21 , 22 In addition, stimulation of beta‐cell stress pathways has been shown to lead to enhanced production of neo‐antigens and activation of diabetogenic T cells. 23 , 24 , 25 , 26 These findings support the hypothesis that increasing levels of beta‐cell oxidative and ER stress via HFD feeding may contribute to both the initiation and acceleration of T1D.
The highest incidence of T1D in humans occurs during puberty, which is a period of elevated insulin resistance. 27 , 28 Puberty represents a time when beta‐cells are under stress because of an insulin resistance‐mediated increase in insulin demand, supporting the concept that insulin resistance induced beta‐cell stress may trigger or accelerate the onset of T1D. 29 Indeed, several clinical studies have shown a correlation between overweight or obese individuals and development of T1D, as well as an association between elevated weight gain in early life and higher risk of T1D development. 30 , 31 , 32 Clinical studies have also shown a faster decline in residual C‐peptide production in newly diagnosed T1D adolescents with a higher body mass index (BMI) at diagnosis, compared with non‐overweight adolescents. 33 These clinical findings suggest that HFD‐induced obesity and insulin resistance may function as environmental triggers or accelerators in the development of T1D.
While many studies suggest a detrimental effect of HFD on beta‐cell health, others have found that HFD has protective effects against the development of diabetes. For instance, HFD is well known to stimulate beta‐cell replication and increase beta‐cell mass. 34 In addition, HFD feeding has been shown to improve glucose levels in the KATP channel gain of function mouse model of monogenic neonatal diabetes. 35 In addition, while some clinical studies suggest an association between elevated BMI and development of T1D, other clinical studies have indicated that elevated BMI does not alter T1D progression. 36 These data reveal the importance of investigating the effect of HFD upon the development of T1D as seemingly detrimental to beta‐cell function, while HFD has been also shown to have beneficial effects in some models of diabetes.
To determine the effect of HFD‐induced obesity on the development of T1D in genetically predisposed individuals, we utilized non‐obese diabetic (NOD) mice. NOD mice develop spontaneous autoimmune diabetes with similar pathophysiology to humans with T1D. 37 , 38 To determine the effect of HFD on the development of T1D, 4‐week‐old NOD mice were placed on standard chow diet (SCD) or HFD to simulate Western diet feeding and induce obesity. Then, animals from both groups were monitored for the development of diabetes, insulin production, beta‐cell mass, islet inflammation and islet stress markers.
2. MATERIALS AND METHODS
NOD/ShiLtJ (RRID:IMSR_Jax:001976) mice were purchased from the Jackson Laboratory (Bar Harbor, Maine). Animal protocols were approved by the Institutional Animal Care and Use Committee (IACUC). All mice were kept in a pathogen‐free facility. Four‐week‐old NOD female mice were randomly separated into two groups and fed with (a) SCD (PicoLab mouse diet 20‐21.559% calories from fat, 55.231% calories from carbohydrates, 23.210% calories from protein), or (b) HFD (Envigo TD.008811/irradiated, 44.6% calories from fat, 40.7% from carbohydrate, 14.7% from protein). Body weight and blood glucose (via tail‐vein using a Bayer Contour meter; Ascensia Diabetes Care, Basel, Switzerland) were monitored weekly. In a subset of mice, 24‐h food consumption was monitored once a week from weeks 5 to 10. Mice were considered diabetic once they had two random (12.00‐16.00 h) blood glucose values >250 mg/dL on two separate days. Once ascertained as diabetic, the animals were killed. A subset of mice was treated with 200 mg/kg of cyclophosphamide (EMD Millipore 239 785, Burlington, MA) via IP injection at 25 weeks to reduce T‐regulatory cells.
2.1. Glucose and insulin tolerance tests
Glucose tolerance tests were performed in 10‐ and 15‐week‐old animals after overnight (16 h) fasting, and in 4‐week‐old animals after 6‐h fasting. Mice were intraperitoneally injected with 2 g/kg dextrose and blood glucose monitored at 0, 15, 30, 60 and 120 min. Plasma insulin was measured before (0 min), and 30 and 60 min after glucose load using a Mouse Insulin Elisa kit (Crystal Chem, Elk Grove Village, IL). Insulin tolerance tests were performed after a 4‐h fast. Mice were intraperitoneally injected with 0.3 u/kg (4‐week‐old mice) or 0.4 u/kg (10‐ and 15‐week‐old mice). Blood glucose was measured via the tail vein at 0, 15, 30, 45, 60, 90 and 120 min after insulin injection. Of note, NOD mice that developed blood glucose lower than 50 mg/dL after 4‐h fasting were excluded from this test.
2.2. Islet isolation
Mice were killed with isoflurane followed by cervical dislocation. The pancreas was perfused with collagenase (Sigma, St. Louis, MO) in Hanks' balanced salt solution via bile‐duct cannulation. The pancreas was digested in collagenase solution, and washed with Hanks' balanced salt solution and RPMI media. Islets were handpicked using a dissecting microscope into RPMI + 10% fetal bovine serum + 100 μg/mL penicillin/streptomycin and used immediately for experiments. Total insulin content was measured using a Mouse Insulin Elisa kit (Crystal Chem).
2.3. Immunohistochemistry and insulitis scoring
At 10 and 40 weeks of age, mice were killed and pancreata immediately fixed in formalin, then embedded in paraffin. For immunohistochemical staining, slides were deparaffinized, rehydrated and underwent antigen retrieval with sodium citrate. Sections were blocked in 3% bovine serum albumin and incubated overnight in anti‐insulin (CST 1:100, Danvers, MA) and anti‐glucagon (Abcam 1:200, Cambridge, MA) primary antibodies, followed by 2‐h secondary antibodies (goat antirabbit AF‐488 1:500 and goat antimouse AF‐594 1:500; Abcam) incubation, and mounted using prolong anti‐fade media with DAPI. Images were obtained using an EXC‐500 fluorescence microscope (Visual Dynamix, Chesterfield, MO, USA). For beta‐cell mass, an insulin‐positive area was measured in ImageJ (ImageJ, RRID:SCR 003070) from five slides separated by 100 μM and calculated by taking the total insulin positive area divided by the total section area and multiplying by the weight of the pancreas (g). To grade insulitis, five sections per pancreas were stained with haematoxylin and eosin and all islets (>50 islets for each animal analysed) in the sections scored using the following grading system; 1, no islet associated mononuclear cell infiltrates; 2, peri‐insulitis involving <50% of islet circumference; 3, peri‐insulitis involving >50% of the islet circumference; and 4, invasion of the interior of the islet by immune cells.
2.4. Beta‐cell replication and apoptosis analysis
Paraffin‐embedded pancreatic sections from 10‐week‐old SCD‐NOD mice and HFD‐NOD mice were stained with DAPI, insulin antibody and either Ki67 or TUNEL assay (Roche, Basel, Switzerland). More than 50 islets per animal were analysed via blinded counting by two individuals. Insulin‐positive cells per islet were counted via the assistance of automation via cell profiler software. Ki67+insulin+ or TUNEL+insulin+cells were counted by the investigators. Data were expressed as the percentage of insulin+KI67+ or insulin+TUNEL+ cells over the total number of insulin+ cells/mouse.
2.5. Quantitative polymerase chain reaction analysis
Total RNA was extracted from 80 to 100 islets per mouse using the Qiagen RNAeasy kit (Qiagen, Germantown, MD), followed by reverse transcriptase reaction using a high‐capacity cDNA reverse‐transcription‐kit (Applied Biosystems, Waltham, MA). Gene expression was calculated using the delta‐delta C t method, using mL32 as a housekeeping gene. The following primers were used in this study: mL32 forward 5′‐TTCCTGGTCCAC‐AATGTCAA‐3′, reverse 5′‐GGCTTTTCGGTTCTTAGAGGA‐3′; msXBP1 forward 5′‐GTCCATGGGAAGATGTTCTGG‐3′, reverse 5′‐CTGAGTCCGAATCAGGTG‐CAG‐3′; mChop forward 5′‐CCACCACACCTGAAAGCAGAA‐3′, reverse 5′‐AGG‐TGAAAGGCAGGGACTCA‐3′. The following primer pairs for interleukin (IL)‐1β (MP206724), transforming growth factor‐β (MP217184) and IL‐6 (MP206798) were purchased from Origene (Rockville, MD).
2.6. Flow cytometry
Mouse spleens were isolated and strained through a 70‐μM strainer to disperse cells. Red blood cells were lysed using RBC lysis buffer (Invitrogen, Waltham, MD). Non‐specific antibody binding sites were blocked by incubation with the CD16/32 Fc‐receptor blocking antibody (BioLegend, San Diego, CA; cat. no. 101201, RRID:AB‐312800). For analyses of cell surface markers, cells were labelled with fluorescent‐conjugated antibodies in phosphate‐buffered saline containing 2% bovine serum albumin. For FOXP3 staining, cells were labelled with cell surface markers, then fixed using the FoxP3 fix and perm kit (eBioscience, Waltham, MA), and then incubated with APC‐Foxp3 antibody (ThermoFisher Scientific, Waltham, MA; cat. no. 17‐4776‐41, RRID:AB_1603281). Flow cytometry measurements were performed with the Canto II instrument (BD Scientific, San Jose, CA; Canto II Flow Cytometer, RRID:SCR_018056) using the flow cytometry core (https://pathology.wustl.edu/research/core‐facilities/flow‐cytometry‐fluorescence‐activated‐cell‐sorting/). FACS data were analysed with FlowJo software (FlowJo, Ashland, OR; RRID:SCR_008520).
2.7. Stool microbiome studies
Fresh stool samples were collected from SCD‐NOD and HFD‐NOD mice serially at age 4, 8 and 25 weeks and frozen at −80 before DNA extraction. DNA was prepared from faecal samples by bead beating, followed by isolation using the DNA‐Kit (Qiagen). The V4 region of the 16S rRNA gene was polymerase chain reaction‐amplified using the barcoded primer described previously (https://stm.sciencemag.org/content/7/276/276ra24?ijkey=f600e2dc0f70c723aa047539d95962a6ca6dccd1&keytype2=tf_ipsecsha) and sequenced using the Illumina MiSeq Platform (2 × 250‐bp/paired‐end‐reads). Data analysis was performed using the Kraken tool via Partek Flow Software.
3. RESULTS
3.1. High‐fat diet feeding induces impaired glucose tolerance and reduced insulin sensitivity in non‐obese diabetic mice
At 4 weeks of age, female NOD mice were divided into two groups and provided ad‐lib access to HFD (HFD‐NOD) or SCD (SCD‐NOD). Female NOD mice were studied as they exhibit an increased incidence of diabetes compared with male mice. 39 Beta‐cell function, mass and islet morphology were evaluated at 10 weeks of age. This time point was chosen as it is before a significant loss of beta‐cell mass or development of glucose intolerance in NOD. 40 As expected, HFD‐NOD mice gained significantly more weight than SCD‐NOD mice (Figure 1A). Weekly follow‐up also revealed an elevation of non‐fasted blood glucose in HFD‐NOD mice compared with SCD‐NOD mice at age 6‐10 weeks, with SCD‐NOD beginning to have higher blood sugar at 14 weeks (Figure 1B). Food consumption was largely similar in HFD‐NOD and SCD‐NOD between 5 and 10 weeks of age, except for 8‐9 weeks where HFD‐NOD mice ate significantly less food (Figure 1C). While fasting blood glucose was not significantly different between groups (Figure 2D‐F), HFD‐NOD mice did exhibit significantly impaired glucose tolerance compared with SCD‐NOD at 10 weeks (Figure 2G‐I). To determine if impaired glucose tolerance was mediated by insulin resistance, we performed insulin tolerance tests. Ten‐week‐old HFD‐NOD mice showed a significant impairment in insulin sensitivity compared with SCD‐NOD mice (Figure 2K), an expected outcome considering the significantly higher weight gain in the HFD group (Figure 1A).
FIGURE 1.
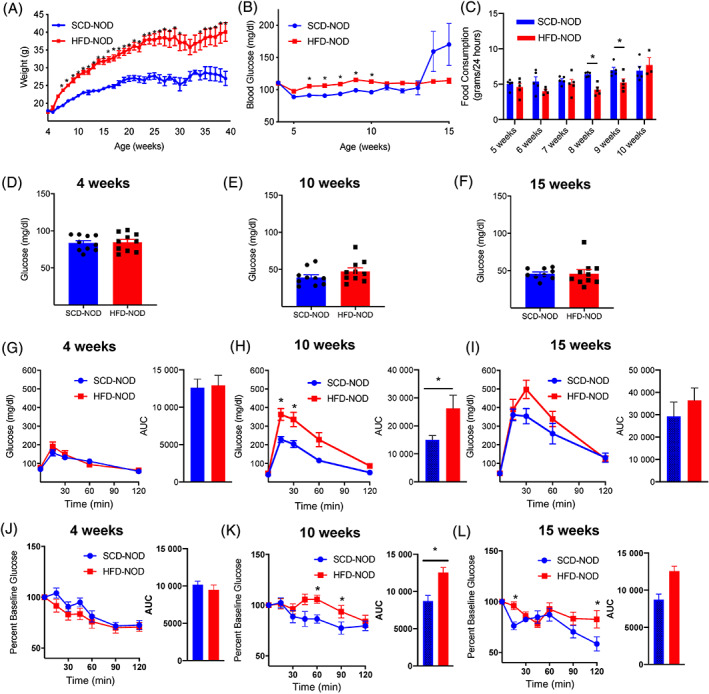
High‐fat diet (HFD) non‐obese diabetic (NOD) mice develop increased weight, impaired glucose tolerance and insulin resistance by 10 weeks of age. (A) Weekly weight measurements in HFD‐NOD (red) and standard chow diet (SCD)‐NOD (blue). (B) Weekly random blood glucose for HFD‐NOD and SCD‐NOD mice from 4 to 15 weeks of age. (C) Food consumption for HFD‐NOD and SCD‐NOD mice from 5 to 10 weeks. Fasting blood glucose for HFD‐NOD and SCD‐NOD mice at (D) 4, (E) 10 and (F) 15 weeks of age. Intraperitoneal glucose tolerance testing at age (G) 4, (H) 10 and (I) 15 weeks with area under the curve (AUC). Insulin tolerance testing at (J) 4, (K) 10 and (L) 15 weeks of age with AUC. N = 5 mice/group for (C) and n = 10 mice/group for all other panels, data represent mean ± SEM. *p < .05 by two‐way ANOVA with Sidak‐correction for multiple comparisons for (A, B, H, I, K, L) and (C) by non‐parametric T‐test for, and AUC
FIGURE 2.
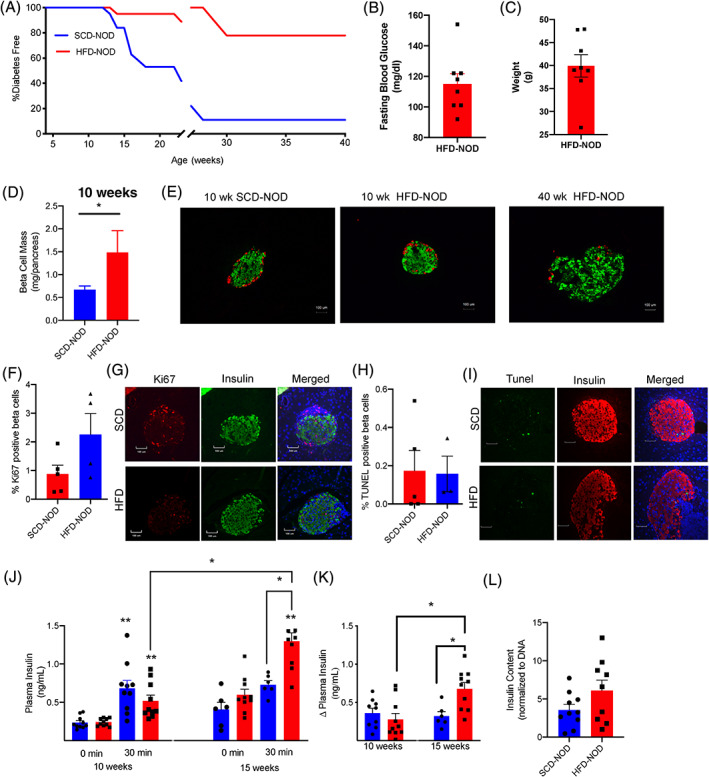
High‐fat diet (HFD) non‐obese diabetic (NOD) mice are protected from the development of diabetes and have increased beta‐cell mass and plasma insulin levels. (A) Incidence of diabetes in HFD‐NOD (red) compared with standard chow diet (SCD)‐NOD (blue) until age 40 weeks (19 mice/group were followed until 25 weeks, 9 mice/group were followed to 40 weeks as indicated by broken x‐axis). (B) Fasting blood glucose and (C) weight in 40‐week‐old HFD‐NOD mice. (D) Quantification of beta‐cell mass in 10‐week‐old SCD‐NOD and HFD‐NOD mice. (E) Representative images of islet sections stained for insulin (green) and glucagon (red). Quantification of percentage beta‐cells positive for (F) Ki67 and (H) TUNEL and (G,I) representative images. (J) Plasma insulin levels during glucose tolerance testing at 10 and 15 weeks of age. (K) Delta plasma insulin levels after glucose tolerance testing. (L) Islet insulin content measured in islets from 15‐week‐old HFD‐NOD and SCD‐NOD. N = 8 mice/group for (B,C), and 3‐5 mice/group for (D), 6‐10 mice/group for (F,G) and 9‐10 mice/group for (H). Data represent mean ± SE. *p < .05 by non‐parametric T‐test for (D). **p < .05 compared with same diet and age at time 0, or *p < .05 compared with group indicated by brackets by two‐way ANOVA with Tukey's and Sidak's multiple comparison testing for (J,K)
3.2. High‐fat diet non‐obese diabetic mice are protected from the development of diabetes
Given the increase in insulin resistance and the impaired glucose tolerance in HFD‐NOD, we expected an accelerated onset of diabetes. Surprisingly, our data showed that the HFD‐NOD mice were protected from diabetes. Animals were monitored for diabetes development up to 40 weeks of age (Figure 2A). While as expected, only 11% of SCD‐NOD mice remained diabetes free at 40 weeks, a significantly greater fraction (78%) of the HFD‐NOD mice were protected from diabetes (Figure 2A). Four‐week‐old HFD‐NOD mice had an average fasting blood glucose of 117 mg/dL and continued to be overweight (Figure 2B,C).
3.3. High‐fat diet non‐obese diabetic mice have increased beta‐cell mass and increased glucose‐stimulated plasma insulin levels
To determine the mechanism underlying the protective effects of HFD, we analysed beta‐cell mass and islet morphology, beta‐cell replication and death. At 10 weeks, HFD‐NOD mice showed a significantly higher beta‐cell mass and normal islet morphology compared with SCD‐NOD (Figure 2D,E). Staining and percentage of beta‐cells positive for Ki67 (replication) or TUNEL (death) did not reveal any significant differences between SCD‐NOD and HFD‐NOD animals (Figure 2G‐I). Next, to determine if islets from HFD‐NOD mice showed improved function we performed glucose‐stimulated insulin‐secretion studies in vivo by measuring plasma insulin levels during a glucose tolerance test. While SCD‐NOD mice showed loss of insulin‐secretory capacity from 10 to 15 weeks, HFD‐NOD mice did not (Figure 2J‐K). In addition, at 15 weeks HFD‐NOD mice were able to secrete significantly more insulin during glucose challenge than SCD‐NOD mice did (Figure 2J‐K) and showed no changes in whole islet insulin content with respect to SCD‐MOD mice (Figure 2L). These data showed that despite having early glucose intolerance and insulin resistance, HFD‐NOD mice had increased beta‐cell mass and improved insulin secretion compared with SCD‐NOD mice.
3.4. High‐fat diet non‐obese diabetic mice exhibit reduced islet immune cell infiltration
Insulitis scoring of pancreatic sections revealed that 10‐ and 40‐week old HFD‐NOD mice trended towards having less islet infiltration than 10‐week‐old SCD‐NOD mice (Figure 3A‐C), although it was probably not statistically significant because of islet heterogeneity and the small sample size. We also examined potential differences in the types of immune cells entering the islets by flow cytometry analysis and found that HFD‐NOD mice trended towards lower total numbers of CD45+ immune cells per islet (Figure SS1A). We found no significant differences in the populations of major immune cells including F480+ macrophages, B220+ B cells, CD3+ T cells, CD4+ T cells and CD8+ T cells between islets of HFD‐NOD and SCD‐NOD mice (Figure SS1B‐P).
FIGURE 3.
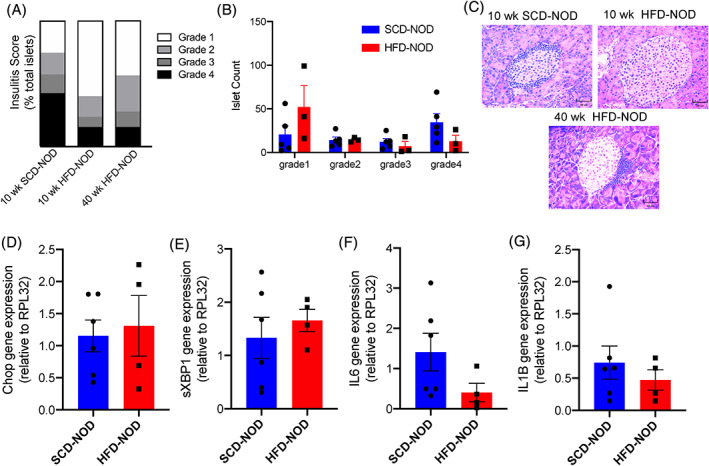
High‐fat diet (HFD) non‐obese diabetic (NOD) islets have decreased insulitis and no significant difference in endoplasmic reticulum stress markers and inflammatory markers at 10 weeks of age. (A) Insulitis scoring for islets at 10 and 40 weeks of age, and (B) mean islet count by grade for individual mice. (C) Representative islets stained with haematoxylin and eosin at 10 weeks of age and 40 weeks of age. (D‐G) Quantitative polymerase chain reaction analysis of gene expression from whole islet samples compared with the housekeeping gene (RPL32). N = 3‐5 mice/group for (A,B), and 4‐6 mice/group for (D‐G)
3.5. Cell stress and inflammatory markers were not increased in high‐fat diet non‐obese diabetic mice
Islets from young NOD mice have been previously reported to have steadily increasing levels of ER stress and inflammatory markers with age. 41 To investigate the effect of HFD on islet ER stress, islets from 4‐ and 10‐week‐old SCD‐NOD and HFD‐NOD mice were analysed for gene expression. As expected, transcripts of the ER stress marker chop and the inflammatory cytokines IL1‐β and IL6 significantly increased from 4 to 10 weeks in both SCD‐NOD and HFD NOD mice (Figure S2A‐C). However, there were no significant differences in ER stress markers between SCD‐NOD and HFD‐NOD mice at 10 weeks (Figure 3D,E). We then examined levels of the inflammatory cytokines IL6 and IL1‐β within the islets. Although the results did not reach statistical significance because of high inter‐animal variability, HFD‐NOD mice trended towards lower levels of IL6 and IL1‐β (Figure 3F,G). This decrease in inflammatory cytokines correlates with decreased levels of insulitis in HFD‐NOD mice (Figure 3A‐C).
3.6. T‐regulatory cells were increased in the spleen of high‐fat diet non‐obese diabetic mice
We next investigated T‐regulatory cell levels in HFD‐NOD mice. T‐regulatory cells are important repressors of autoimmunity in multiple human diseases including T1D. 42 , 43 In support of this association, NOD mice showed lower levels of T‐regulatory cells compared with other mouse strains. 44 To determine if HFD feeding resulted in increased immune regulation by increasing T‐regulatory cells, we performed FACs analysis on splenocytes from 10‐week‐old HFD‐NOD and SCD‐NOD mice. Spleens, but not the pancreatic lymph nodes of HFD‐NOD mice showed increased levels of CD25+FoxP3+CD4+ T‐regulatory cells compared with SCD‐NOD mice (Figure 4A‐C). To examine the contribution of T‐regulatory cells in the protection of HFD‐NOD mice from development of diabetes, we reduced the T‐regulatory cells in HFD‐NOD animals by treatment with cyclophosphamide. Cyclophosphamide treatment has been previously shown to reduce T‐regulatory cells efficiently in animal models, 45 and it was confirmed in our mice (Figure S3A,B). Depletion of T‐regulatory cells in HFD‐NOD mice significantly increased the incidence of diabetes to 60%, compared with 10.5% in age‐matched untreated HFD‐NOD mice (Figure 4D), suggesting that T‐regulatory cells are necessary for HFD‐mediated protection from diabetes.
FIGURE 4.
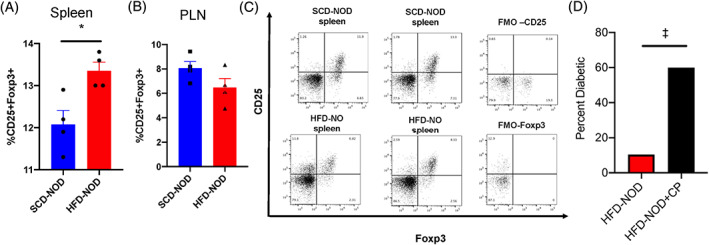
High‐fat diet (HFD) non‐obese diabetic (NOD) mice have increased levels of splenic T‐regulatory cells. Quantification of the percentage of CD45+ immune cells, which were CD45+CD4+CD25+FoxP3+ T‐regulatory cells in (A) the spleen and (B) pancreatic lymph node (PLN). (C) Representative dot plots from FACs analysis of standard chow diet (SCD)‐NOD and HFD‐NOD CD45+CD4+CD25+FoxP3 splenic and PLN immune cells with fluorescence minus one (FMO) control for CD25 and FoxP3. (D) 25‐week‐old non‐diabetic HFD‐NOD mice were treated with cyclophosphamide (CP) and monitored for the development of diabetes over the following 4 weeks. N = 4‐5 mice/group, *p < .05 non‐parametric t‐test, ‡p < .05, Fisher's exact test
3.7. Stool microbiome is significantly altered in high‐fat diet non‐obese diabetic mice
To determine the role of gut microbiome in HFD‐mediated prevention of diabetes we analysed the stool microbiome in SCD‐NOD and HFD‐NOD mice at 4, 8 and 25 weeks. Stool samples were taken from the same animals over time to determine changes in microbiome during diabetes development. As expected, no differences in beta diversity of the stool microbiome were found in SCD‐NOD mice at 4 weeks (Figure 5A,B). However, at 8 and 25 weeks of age SCD‐NOD and HFD‐NOD mice showed significantly different microbiota as shown by beta diversity plots (Figure 5C‐F), with increased abundance of Bacteroidetes in SCD‐NOD mice, and increased abundance of Verrucomicrobia in HFD‐NOD mice (Figure 5D,F).
FIGURE 5.
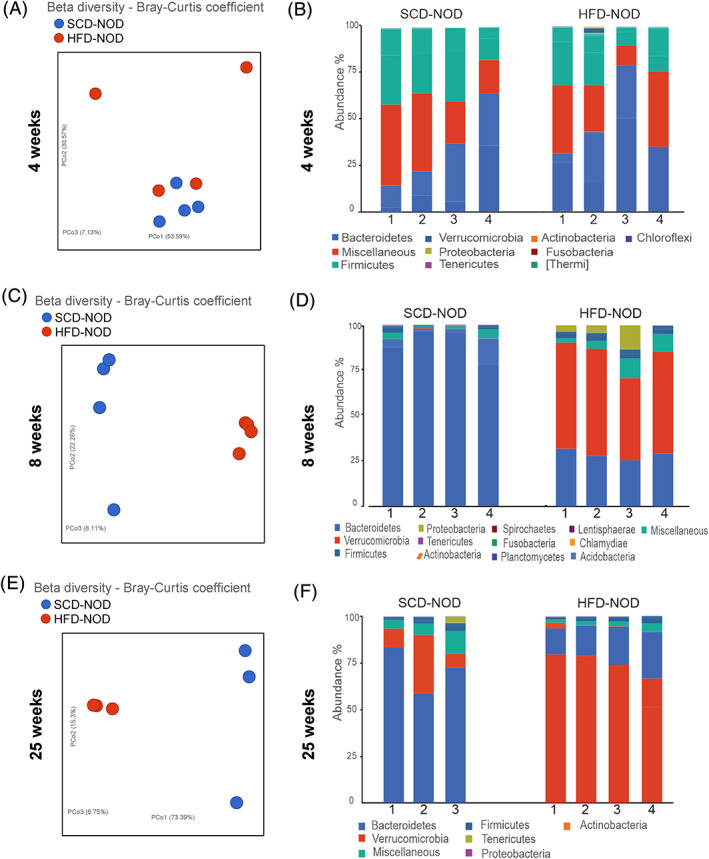
High‐fat diet (HFD) non‐obese diabetic (NOD) mice have altered gut microbiome compared with standard chow diet (SCD)‐NOD mice. Beta diversity of stool microbiome was analysed for SCD‐NOD (blue) or HFD‐NOD (red) mice at (A) 4, (C) 8 and (E) 25 weeks of age. Relative abundance data are depicted for SCD‐NOD and HFD‐NOD mice at (B) 4, (D) 8 and (F) 25 weeks of age. N = 3‐4 mice/group
4. DISCUSSION
To determine if HFD and obesity functions as a trigger or accelerator for T1D, we used an NOD mouse model of T1D with HFD. To our surprise, HFD‐NOD mice were protected from the development of diabetes. This protective phenotype was associated with increased beta‐cell mass and glucose‐stimulated insulin secretion, alterations in gut microbiome, increased splenic T‐regulatory cells and reduced islet immune cell infiltration.
T‐regulatory cells are important repressors of autoimmunity in multiple human diseases including T1D. 42 , 43 Deficiencies in T‐regulatory cells are known to initiate autoimmune diabetes in patients with immunodysregulation polyendocrinopathy enteropathy X‐linked (IPEX) syndrome. Individuals with IPEX are deficient in foxp3 and have dysfunctional T‐regulatory cells, resulting in the development of T1D in 80% of these individuals before they are 2 years old. 46 In support of the role of T‐regulatory cells in the development of T1D, studies in NOD mice showed that a reduction of T‐regulatory cells accelerates diabetes onset, and treatment with T‐regulatory cells mitigates autoimmune diabetes onset. 45 , 47 These data strongly correlate with our findings on the role of T‐regulatory cells in protection from diabetes in HFD‐NOD mice, and with the rapid development of diabetes in HFD‐NOD mice with a reduction in T‐regulatory cells. The increase in T‐regulatory cells and diminished islet infiltration in the HFD‐NOD mice suggest that HFD alters beta‐cell immune response. Interestingly, our data on HFD‐NOD mice correlate with findings in NOD‐liver insulin receptor knockout (NOD‐LIRKO) mice, which developed early insulin resistance, had increased levels of T‐regulatory cells, decreased insulitis, increased beta‐cell mass, and were protected from diabetes. NOD‐LIRKO mice also showed lower levels of diabetogenic autoantigens, decreased major histocompatibility class I proteins, and that many islet cells produced both glucagon and insulin. 48 As the HFD model results in insulin resistance‐mediated increased beta mass, further investigation should be carried out to determine if altered beta‐cell identity might be responsible for the immunoregulation in HFD‐NOD mice.
Previous studies showed that high‐protein (55% protein) and HFD/high‐protein diets (43% fat/38% protein/19% carbohydrate) accelerated the diabetes onset in NOD mice, and that HFD alone (39% fat/17% protein/43% carbohydrate) did not induce any effect on diabetes development. 49 , 50 However, our study showed protection of diabetes in NOD mice by HFD. It is difficult to compare these studies directly as the different results could arise from subtle differences in sources of fat or other components of the diets used, or lower protein content in our HFD may play a role in protection from diabetes in HFD‐NOD mice. Thus, further studies are needed to examine the effect of low‐protein diets on the development of diabetes. In addition, our study shows clear differences in the microbiota in HFD‐NOD mice, but the microbiome was not analysed in the other study. Many studies have shown that changes in gut microbiome can either potentiate or suppress the development of diabetes in NOD mice 51 , 52 , 53 , 54 , 55 Interestingly, in germ‐free mice, lack of microbial signals from the gut results in impaired immune tolerance via T‐regulatory cell dysfunction contributing to the development of autoimmunity. 56 This poses an interesting possibility that in our model HFD‐induced alteration of the gut microbiome may have altered T‐regulatory cell functionality, resulting in suppression of the islet autoimmune attack. This may explain why small differences in T cells resulted in T‐regulatory cell‐dependent protection from diabetes.
Gut microbiome differences have been shown in individuals before developing autoimmunity and after developing diabetes, with respect to individuals without diabetes. In agreement with our finding in NOD mice, alterations in the gut microbiome, specifically overabundance of Bacteroidetes, have been shown in humans at risk for and developing T1D compared with controls. 57 , 58 , 59 In our study HFD‐NOD mice showed increased abundance of Verrucomicrobia compared with SCD‐NOD. Interestingly, a previous study showed an increase in Akkermansia muciniphila, a member of the Verrucomicrobia phylum, in NOD‐resistant mice, and that transfer of A. muciniphila to NOD mice resulted in decreased blood endotoxin levels, increased islet T‐regulatory cells and delayed diabetes onset. 54 Other studies have shown that changing the gut microbiome by supplementing with probiotics in human high‐risk infants can alter the development of islet autoimmunity. 60 Our data and previous studies in NOD mice support further investigation of supplementation of the gut microbiome with members of the Verrucomicrobia phylum, which might support islet immune tolerance and prevent T1D.
This study provides important insights into the role of HFD in the development of T1D in NOD mice. We show here that NOD mice are protected from autoimmune diabetes. In addition, these results strengthen a body of research suggesting that interventions resulting in early expansion of beta‐cell mass induce decreased islet infiltration and protect NOD mice from the development of diabetes. Our data also indicated that dietary alteration of the gut microbiota might play a significant role in preventing T1D in at risk individuals. Further studies determining if lowering levels of Bacteroidetes or increaseing levels of Verrucomicrobia in the gut microbiome are responsible for HFD immune‐mediated protection against development of diabetes in NOD mice are needed. Therefore, exploration of the role of the gut microbiome and mechanisms by which HFD prevents the development of diabetes in NOD animals may lead to the development of novel treatments for individuals at high risk for T1D.
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
AUTHOR CONTRIBUTIONS
Amy L. Clark and Maria S. Remedi designed the study. Amy L. Clark wrote the manuscript. Amy L. Clark, Zihan Yan, Sophia X. Chen, Victoria Shi, Devesha H. Kulkarni and Maria S. Remedi performed experiments and data analysis. Maria S. Remedi, Devesha H. Kulkarni and Abhinav Diwan edited the manuscript. All authors read and approved the final version of the manuscript.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1111/dom.14486.
Supporting information
Figure S1. Major immune cell population frequencies were unchanged in HFD islets. FACS analysis of islet cells at 10 weeks of age from SCD‐NOD (blue) and HFD‐NOD (red) mice. Total CD45+ immune cells per pooled islets per mouse (A) with representative dot plots (B, C). Percent macrophages (D‐F), B cells (G‐I), CD3+T cells (J‐L), CD4+T cells (M‐O), CD8+T cells (P‐R) and the representative contour plots. N = 3‐6 mice/group. Data represent mean ± SEM.
Figure S2. ER stress and inflammatory marker gene expression all increase by 10 weeks of age in NOD mice. qPCR analysis of Chop, IL1‐β, and IL‐6 in whole islets from 4‐ and 10‐week‐old SCD‐NOD mice and 10‐week‐old HFD‐NOD mice. Gene expression was normalized to expression of the ML32 housekeeping‐gene and expressed as fold over expression in 4‐week‐old SCD‐NOD mice. Data represent mean ± SEM *p < .05 vs control (4‐week NOD) by non‐parametric T‐test.
Figure S3. Cyclophosphamide treatment depletes T‐regulatory cells in the spleen and pancreatic lymph node. 10‐week‐old female NOD mice were treated with cyclophosphamide and then splenic and pancreatic lymph node CD45+CD4+CD25+FoxP3+ T‐regulatory cells were counted by FACs analysis. Representative dot plots with appropriate FMO controls are depicted in panel A and quantification of CD45+CD4+CD25+FoxP3+ T‐regulatory cells in spleen and PLN is presented in panel B. N = 4 mice/group, *p < .05 by non‐parametric t‐test.
ACKNOWLEDGMENTS
We would like to acknowledge the following funding sources: Washington University Diabetes Research Center Grant No P30 DK020579 to A.L.C.; NIH‐R56DK098584 and NIH‐R01DK123163 to M.S.R. A.D was supported by grants from the Department of Veterans Affairs (I01BX004235) and NIH‐HL143431, NIH‐HL107594 and NIH‐NS094692. We acknowledge the Metabolic Tissue Function Core, Diabetes Research Center, Washington University in St Louis (supported by NIH‐P30 DK020579).
Clark AL, Yan Z, Chen SX, et al. High‐fat diet prevents the development of autoimmune diabetes in NOD mice. Diabetes Obes Metab. 2021;23(11):2455-2465. 10.1111/dom.14486
Funding information National Institute of Health in the United states, Grant/Award Numbers: HL 107594, HL 143431, NS094692, R01DK123163, R56DK098584; U.S. Department of Veterans Affairs, Grant/Award Number: I01BX004235; Washington University Diabetes Research Center, Grant/Award Number: P30 DK020579
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Mathis D, Vence L, Benoist C. Beta‐cell death during progression to diabetes. Nature. 2001;414(6865):792‐798. [DOI] [PubMed] [Google Scholar]
- 2. Redondo MJ, Steck AK, Pugliese A. Genetics of type 1 diabetes. Pediatr Diabetes. 2018;19(3):346‐353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Noble JA, Erlich HA. Genetics of type 1 diabetes. Cold Spring Harb Perspect Med. 2012;2(1):a007732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bartsocas CS, Gerasimidi‐Vazeou A. Genetics of type 1 diabetes mellitus. Pediatr Endocrinol Rev. 2006;3(Suppl 3):508‐513. [PubMed] [Google Scholar]
- 5. Noble JA, Valdes AM, Varney MD, et al. HLA class I and genetic susceptibility to type 1 diabetes: results from the type 1 diabetes genetics consortium. Diabetes. 2010;59(11):2972‐2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Alizadeh BZ, Koeleman BP. Genetic polymorphisms in susceptibility to type 1 diabetes. Clin Chim Acta. 2008;387(1–2):9‐17. [DOI] [PubMed] [Google Scholar]
- 7. Hemminki K, Li X, Sundquist J, Sundquist K. Familial association between type 1 diabetes and other autoimmune and related diseases. Diabetologia. 2009;52(9):1820‐1828. [DOI] [PubMed] [Google Scholar]
- 8. Bogdanos DP, Smyk DS, Rigopoulou EI, et al. Twin studies in autoimmune disease: genetics, gender and environment. J Autoimmun. 2012;38(2–3):J156‐J169. [DOI] [PubMed] [Google Scholar]
- 9. Rewers M, Ludvigsson J. Environmental risk factors for type 1 diabetes. Lancet. 2016;387(10035):2340‐2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Skinner AC, Ravanbakht SN, Skelton JA, Perrin EM, Armstrong SC. Prevalence of obesity and severe obesity in US children, 1999‐2016. Pediatrics. 2018;141(3):e20173459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pettitt DJ, Talton J, Dabelea D, et al. Prevalence of diabetes in U.S. youth in 2009: the SEARCH for diabetes in youth study. Diabetes Care. 2014;37(2):402‐408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dabelea D, Mayer‐Davis EJ, Saydah S, et al. Prevalence of type 1 and type 2 diabetes among children and adolescents from 2001 to 2009. JAMA. 2014;311(17):1778‐1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang Z, Xie Z, Lu Q, Chang C, Zhou Z. Beyond genetics: what causes type 1 diabetes. Clin Rev Allergy Immunol. 2017;52(2):273‐286. [DOI] [PubMed] [Google Scholar]
- 14. Okada H, Kuhn C, Feillet H, Bach JF. The 'hygiene hypothesis' for autoimmune and allergic diseases: an update. Clin Exp Immunol. 2010;160(1):1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bach JF. The effect of infections on susceptibility to autoimmune and allergic diseases. N Engl J Med. 2002;347(12):911‐920. [DOI] [PubMed] [Google Scholar]
- 16. Manzel A, Muller DN, Hafler DA, Erdman SE, Linker RA, Kleinewietfeld M. Role of "Western diet" in inflammatory autoimmune diseases. Curr Allergy Asthma Rep. 2014;14(1):404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hanna Kazazian N, Wang Y, Roussel‐Queval A, et al. Lupus autoimmunity and metabolic parameters are exacerbated upon high fat diet‐induced obesity due to TLR7 signaling. Front Immunol. 2019;10:2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Timmermans S, Bogie JF, Vanmierlo T, et al. High fat diet exacerbates neuroinflammation in an animal model of multiple sclerosis by activation of the renin angiotensin system. J Neuroimmune Pharmacol. 2014;9(2):209‐217. [DOI] [PubMed] [Google Scholar]
- 19. Mathijs I, Da Cunha DA, Himpe E, et al. Phenylpropenoic acid glucoside augments pancreatic beta‐cell mass in high‐fat diet‐fed mice and protects beta‐cells from ER stress‐induced apoptosis. Mol Nutr Food Res. 2014;58(10):1980‐1990. [DOI] [PubMed] [Google Scholar]
- 20. Matveyenko AV, Gurlo T, Daval M, Butler AE, Butler PC. Successful versus failed adaptation to high‐fat diet‐induced insulin resistance: the role of IAPP‐induced beta‐cell endoplasmic reticulum stress. Diabetes. 2009;58(4):906‐916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu H, Javaheri A, Godar RJ, et al. Intermittent fasting preserves beta‐cell mass in obesity‐induced diabetes via the autophagy‐lysosome pathway. Autophagy. 2017;13(11):1952‐1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Remedi MS, Koster JC, Markova K, et al. Diet‐induced glucose intolerance in mice with decreased beta‐cell ATP‐sensitive K+ channels. Diabetes. 2004;53(12):3159‐3167. [DOI] [PubMed] [Google Scholar]
- 23. Marre ML, Piganelli JD. Environmental factors contribute to beta‐cell endoplasmic reticulum stress and neo‐antigen formation in type 1 diabetes. Front Endocrinol. 2017;8:262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Buitinga M, Callebaut A, Marques Camara Sodre F, et al. Inflammation‐induced Citrullinated glucose‐regulated protein 78 elicits immune responses in human type 1 diabetes. Diabetes. 2018;67(11):2337‐2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Marre ML, McGinty JW, Chow IT, et al. Modifying enzymes are elicited by ER stress, generating epitopes that are selectively recognized by CD4(+) T cells in patients with type 1 diabetes. Diabetes. 2018;67(7):1356‐1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vomund AN, Zinselmeyer BH, Hughes J, et al. Beta‐cells transfer vesicles containing insulin to phagocytes for presentation to T cells. Proc Natl Acad Sci U S A. 2015;112(40):E5496‐E5502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xia Y, Xie Z, Huang G, Zhou Z. Incidence and trend of type 1 diabetes and the underlying environmental determinants. Diabetes Metab Res Rev. 2019;35(1):e3075. [DOI] [PubMed] [Google Scholar]
- 28. Amiel SA, Sherwin RS, Simonson DC, Lauritano AA, Tamborlane WV. Impaired insulin action in puberty. A contributing factor to poor glycemic control in adolescents with diabetes. N Engl J Med. 1986;315(4):215‐219. [DOI] [PubMed] [Google Scholar]
- 29. Rogers MAM, Kim C, Banerjee T, Lee JM. Fluctuations in the incidence of type 1 diabetes in the United States from 2001 to 2015: a longitudinal study. BMC Med. 2017;15(1):199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Johansson C, Samuelsson U, Ludvigsson J. A high weight gain early in life is associated with an increased risk of type 1 (insulin‐dependent) diabetes mellitus. Diabetologia. 1994;37(1):91‐94. [DOI] [PubMed] [Google Scholar]
- 31. Yassouridis C, Leisch F, Winkler C, Ziegler AG, Beyerlein A. Associations of growth patterns and islet autoimmunity in children with increased risk for type 1 diabetes: a functional analysis approach. Pediatr Diabetes. 2017;18(2):103‐110. [DOI] [PubMed] [Google Scholar]
- 32. Hypponen E, Kenward MG, Virtanen SM, et al. Infant feeding, early weight gain, and risk of type 1 diabetes. Childhood diabetes in Finland (DiMe) study group. Diabetes Care. 1999;22(12):1961‐1965. [DOI] [PubMed] [Google Scholar]
- 33. Lauria A, Barker A, Schloot N, et al. BMI is an important driver of beta‐cell loss in type 1 diabetes upon diagnosis in 10 to 18‐year‐old children. Eur J Endocrinol. 2015;172(2):107‐113. [DOI] [PubMed] [Google Scholar]
- 34. Golson ML, Misfeldt AA, Kopsombut UG, Petersen CP, Gannon M. High fat diet regulation of beta‐cell proliferation and beta‐cell mass. Open Endocrinol J. 2010;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yan Z, Shyr ZA, Fortunato M, et al. High‐fat‐diet‐induced remission of diabetes in a subset of KATP ‐GOF insulin‐secretory‐deficient mice. Diabetes Obes Metab. 2018;20(11):2574‐2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Meah FA, DiMeglio LA, Greenbaum CJ, et al. The relationship between BMI and insulin resistance and progression from single to multiple autoantibody positivity and type 1 diabetes among TrialNet pathway to prevention participants. Diabetologia. 2016;59(6):1186‐1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Anderson MS, Bluestone JA. The NOD mouse: a model of immune dysregulation. Annu Rev Immunol. 2005;23:447‐485. [DOI] [PubMed] [Google Scholar]
- 38. Kikutani H, Makino S. The murine autoimmune diabetes model: NOD and related strains. Adv Immunol. 1992;51:285‐322. [DOI] [PubMed] [Google Scholar]
- 39. Jackson Laboratory . DIABETES ONSET IN NOD/SHILTJ. https://wwwjaxorg/jax-mice-and-services/strain-data-sheet-pages/diabetes-chart-001976.
- 40. Sreenan S, Pick AJ, Levisetti M, Baldwin AC, Pugh W, Polonsky KS. Increased beta‐cell proliferation and reduced mass before diabetes onset in the nonobese diabetic mouse. Diabetes. 1999;48(5):989‐996. [DOI] [PubMed] [Google Scholar]
- 41. Tersey SA, Nishiki Y, Templin AT, et al. Islet beta‐cell endoplasmic reticulum stress precedes the onset of type 1 diabetes in the nonobese diabetic mouse model. Diabetes. 2012;61(4):818‐827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang Y, TFrTcajoat, master of regulation , Bandala‐Sanchez E, Harrison LC. Revisiting regulatory T cells in type 1 diabetes. Curr Opin Endocrinol Diabetes Obes. 2012;19(4):271‐278. [DOI] [PubMed] [Google Scholar]
- 43. Tang Q, Bluestone JA. The Foxp3+ regulatory T cell: a jack of all trades, master of regulation. Nat Immunol. 2008;9(3):239‐244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Godoy GJ, Paira DA, Olivera C, et al. Differences in T regulatory cells between mouse strains frequently used in immunological research: Treg cell quantities and subpopulations in NOD, B6 and BALB/c mice. Immunol Lett. 2020;223:17‐25. [DOI] [PubMed] [Google Scholar]
- 45. Brode S, Raine T, Zaccone P, Cooke A. Cyclophosphamide‐induced type‐1 diabetes in the NOD mouse is associated with a reduction of CD4+CD25+Foxp3+ regulatory T cells. J Immunol. 2006;177(10):6603‐6612. [DOI] [PubMed] [Google Scholar]
- 46. Wildin RS, Ramsdell F, Peake J, et al. X‐linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet. 2001;27(1):18‐20. [DOI] [PubMed] [Google Scholar]
- 47. Harada M, Makino S. Promotion of spontaneous diabetes in non‐obese diabetes‐prone mice by cyclophosphamide. Diabetologia. 1984;27(6):604‐606. [DOI] [PubMed] [Google Scholar]
- 48. Dirice E, Kahraman S, De Jesus DF, et al. Increased beta‐cell proliferation before immune cell invasion prevents progression of type 1 diabetes. Nat Metab. 2019;1(5):509‐518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Linn T, Strate C, Schneider K. Diet promotes beta‐cell loss by apoptosis in prediabetic nonobese diabetic mice. Endocrinology. 1999;140(8):3767‐3773. [DOI] [PubMed] [Google Scholar]
- 50. Schneider K, Laube H, Linn T. A diet enriched in protein accelerates diabetes manifestation in NOD mice. Acta Diabetol. 1996;33(3):236‐240. [DOI] [PubMed] [Google Scholar]
- 51. Zheng P, Li Z, Zhou Z. Gut microbiome in type 1 diabetes: a comprehensive review. Diabetes Metab Res Rev. 2018;34(7):e3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Livanos AE, Greiner TU, Vangay P, et al. Antibiotic‐mediated gut microbiome perturbation accelerates development of type 1 diabetes in mice. Nat Microbiol. 2016;1(11):16140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Simon MC, Reinbeck AL, Wessel C, et al. Distinct alterations of gut morphology and microbiota characterize accelerated diabetes onset in nonobese diabetic mice. J Biol Chem. 2020;295(4):969‐980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hanninen A, Toivonen R, Poysti S, et al. Akkermansia muciniphila induces gut microbiota remodelling and controls islet autoimmunity in NOD mice. Gut. 2018;67(8):1445‐1453. [DOI] [PubMed] [Google Scholar]
- 55. Alam C, Bittoun E, Bhagwat D, et al. Effects of a germ‐free environment on gut immune regulation and diabetes progression in non‐obese diabetic (NOD) mice. Diabetologia. 2011;54(6):1398‐1406. [DOI] [PubMed] [Google Scholar]
- 56. Ostman S, Rask C, Wold AE, Hultkrantz S, Telemo E. Impaired regulatory T cell function in germ‐free mice. Eur J Immunol. 2006;36(9):2336‐2346. [DOI] [PubMed] [Google Scholar]
- 57. Vatanen T, Franzosa EA, Schwager R, et al. The human gut microbiome in early‐onset type 1 diabetes from the TEDDY study. Nature. 2018;562(7728):589‐594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. de Goffau MC, Luopajarvi K, Knip M, et al. Fecal microbiota composition differs between children with beta‐cell autoimmunity and those without. Diabetes. 2013;62(4):1238‐1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Vatanen T, Kostic AD, d'Hennezel E, et al. Variation in microbiome LPS immunogenicity contributes to autoimmunity in humans. Cell. 2016;165(6):1551. [DOI] [PubMed] [Google Scholar]
- 60. Uusitalo U, Liu X, Yang J, et al. Association of early exposure of probiotics and islet autoimmunity in the TEDDY study. JAMA Pediatr. 2016;170(1):20‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Major immune cell population frequencies were unchanged in HFD islets. FACS analysis of islet cells at 10 weeks of age from SCD‐NOD (blue) and HFD‐NOD (red) mice. Total CD45+ immune cells per pooled islets per mouse (A) with representative dot plots (B, C). Percent macrophages (D‐F), B cells (G‐I), CD3+T cells (J‐L), CD4+T cells (M‐O), CD8+T cells (P‐R) and the representative contour plots. N = 3‐6 mice/group. Data represent mean ± SEM.
Figure S2. ER stress and inflammatory marker gene expression all increase by 10 weeks of age in NOD mice. qPCR analysis of Chop, IL1‐β, and IL‐6 in whole islets from 4‐ and 10‐week‐old SCD‐NOD mice and 10‐week‐old HFD‐NOD mice. Gene expression was normalized to expression of the ML32 housekeeping‐gene and expressed as fold over expression in 4‐week‐old SCD‐NOD mice. Data represent mean ± SEM *p < .05 vs control (4‐week NOD) by non‐parametric T‐test.
Figure S3. Cyclophosphamide treatment depletes T‐regulatory cells in the spleen and pancreatic lymph node. 10‐week‐old female NOD mice were treated with cyclophosphamide and then splenic and pancreatic lymph node CD45+CD4+CD25+FoxP3+ T‐regulatory cells were counted by FACs analysis. Representative dot plots with appropriate FMO controls are depicted in panel A and quantification of CD45+CD4+CD25+FoxP3+ T‐regulatory cells in spleen and PLN is presented in panel B. N = 4 mice/group, *p < .05 by non‐parametric t‐test.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.