

Abstract
ATG9A, the only multi‐pass transmembrane protein among core ATG proteins, is an essential regulator of autophagy, yet its regulatory mechanisms and network of interactions are poorly understood. Through quantitative BioID proteomics, we identify a network of ATG9A interactions that includes members of the ULK1 complex and regulators of membrane fusion and vesicle trafficking, including the TRAPP, EARP, GARP, exocyst, AP‐1, and AP‐4 complexes. These interactions mark pathways of ATG9A trafficking through ER, Golgi, and endosomal systems. In exploring these data, we find that ATG9A interacts with components of the ULK1 complex, particularly ATG13 and ATG101. Using knockout/reconstitution and split‐mVenus approaches to capture the ATG13‐ATG101 dimer, we find that ATG9A interacts with ATG13‐ATG101 independently of ULK1. Deletion of ATG13 or ATG101 causes a shift in ATG9A distribution, resulting in an aberrant accumulation of ATG9A at stalled clusters of p62/SQSTM1 and ubiquitin, which can be rescued by an ULK1 binding‐deficient mutant of ATG13. Together, these data reveal ATG9A interactions in vesicle‐trafficking and autophagy pathways, including a role for an ULK1‐independent ATG13 complex in regulating ATG9A.
Keywords: ATG13, ATG9A, autophagy, BioID, p62
Subject Categories: Autophagy & Cell Death, Membranes & Trafficking
This study presents the ATG9A proximity interactome, revealing interactions with vesicle trafficking complexes, core autophagy regulators, and an ULK1‐independent ATG13‐ATG101 complex that promotes the basal turnover of ubiquitin‐rich p62/SQSTM1 clusters.

Introduction
The recycling of misfolded proteins, dysfunctional organelles, and other molecules through macroautophagy (referred to here as autophagy) is critical for maintaining cellular homeostasis and promoting cell survival during stress. Deregulated autophagy underlies the pathophysiology of many human diseases, including a variety of degenerative disorders, cancer, autoimmunity, and infectious disease. The central event in autophagy is the formation of the autophagosome, which begins as a double‐membrane cisterna that expands and captures portions of the cytosol/cell and ultimately closes to form a sealed vesicle. The autophagosome then fuses with the lysosome for degradation and recycling of the autophagosome contents. The flux of autophagy substrates through this degradative pathway increases in breadth and rate under nutrient deprivation. In contrast, under nutrient replete conditions, a more selective, lower level of autophagy (referred to here as “basal autophagy”) maintains organelle and protein homeostasis (Komatsu et al, 2005; Hara et al, 2006; Komatsu et al, 2006; Antonucci et al, 2015). Defects in basal autophagy can lead to the accumulation of defective mitochondria and toxic protein aggregates that underlie a variety of degenerative diseases (Hara et al, 2006; Komatsu et al, 2006; Dikic & Elazar, 2018).
Our understanding of the upstream signaling that controls autophagy mainly derives from studies on nutrient deprivation, in which the inhibition of mTORC1 results in the activation of the ULK1 kinase complex that includes FIP200, ATG101, and ATG13 (Hosokawa et al, 2009a; Lee et al, 2010; Egan et al, 2011; Kim et al, 2011; Shang et al, 2011). Active ULK1 complex then coordinates a variety of autophagy events, such as recruitment of VPS34 lipid kinase complex, that stimulates formation of the membrane precursor to the autophagosome, referred to as the isolation membrane (IM) (Zachari & Ganley, 2017). The location of this emergent autophagosome is also called the phagophore assembly site (PAS). Additional autophagy regulatory proteins are recruited to the IM/PAS, including ATG5‐ATG12‐ATG16L1 conjugation systems that attach the ubiquitin‐like protein LC3 to autophagosomes.
In contrast to starvation‐induced autophagy, basal autophagy is primarily driven by a variety of autophagy adaptors, including p62/SQSTM1, Optineurin, and TAX1BP1, that selectively deliver cargo to the autophagosome. For example, p62/SQSTM1 interacts with poly‐ubiquitinated cargo via its ubiquitin association domain and then tethers these cargo to the LC3‐decorated autophagosomes via its LC3‐interacting region (Seibenhener et al, 2004; Pankiv et al, 2007). Transition of these p62/SQSTM1‐poly‐ubiquitinated protein complexes into phase‐separated droplets appears to be a precursor to cargo degradation (Cloer et al, 2018; Sun et al, 2018; Jakobi et al, 2020). However, given that basal autophagy occurs under conditions in which ULK1 activity is low (and MTORC1 activity is high), the hierarchy of signaling that governs basal autophagy, including how core autophagy machinery (e.g., ATG9A) is engaged and regulated, is not yet clear.
ATG9A is essential for the formation of autophagosomes (Kuma et al, 2004; Saitoh et al, 2009; Yamamoto et al, 2012), but is one of the least understood of the core ATG proteins. Studies from yeast and mammalian cells suggest that ATG9A (referred to as Atg9 in yeast) traffics on small membrane vesicles and accumulates at several sites within vesicular trafficking pathways, including the Golgi, endosomes, and ER where it colocalizes with IM/PAS markers (Young et al, 2006; Mari et al, 2010; Orsi et al, 2012; Imai et al, 2016; Takahashi et al, 2016; Kakuta et al, 2017; Nishimura et al, 2017). A few proteins have been identified as regulators of ATG9A trafficking, including the coat adaptors AP‐1, AP‐2, and AP‐4, components of the ULK1 complex, BIF‐1, and p38IP (Young et al, 2006; Takahashi et al, 2011; Tang et al, 2011; Guo et al, 2012; Orsi et al, 2012; Popovic & Dikic, 2014; Ktistakis & Tooze, 2016; Mattera et al, 2017; Davies et al, 2018). The trafficking/mobilization of ATG9A to the IM/PAS is considered an apical step in autophagy (Itakura et al, 2012; Kishi‐Itakura et al, 2014; Karanasios et al, 2016). While at the IM/PAS, ATG9A is thought to supply membrane to growing autophagosomes, although the mechanism by which this may occur is still unclear (Yamamoto et al, 2012; Judith et al, 2019).
Several recent studies indicate that, in addition to the role of ATG9A in starvation‐induced autophagy, ATG9A is essential for basal autophagy—potentially in ways that do not easily fit within current autophagy paradigms. Although ATG9A KO MEFs still display LC3B puncta (suggesting that autophagosomes still form in the absence of ATG9A) (Saitoh et al, 2009), studies focused on the basal lysosomal turnover of autophagy adaptors demonstrate a strong requirement for ATG9A. For example, degradative flux of the autophagy adaptor NBR1 is largely independent of ULK1 and ATG factors required for LC3 lipidation, but is entirely dependent on ATG9A. Similarly, ATG9A emerged as a top hit in a genome‐wide CRISPR/Cas9 screen for proteins required for basal lysosomal degradation of p62/SQSTM1, while a variety of core ATG proteins were notably not essential (Goodwin et al, 2017). In addition, the tyrosine kinase Src phosphorylates ATG9A at Tyr8 to maintain active ATG9A trafficking under basal conditions (Zhou et al, 2017). Furthermore, defective ATG9A trafficking (or genetic loss of ATG9A) is associated with impaired clearance of protein aggregates (Winslow et al, 2010; De Pace et al, 2018; Yamaguchi et al, 2018). Together, these data support a central role for ATG9A in basal autophagy. However, the general mechanisms that control basal autophagy are poorly understood, including how ATG9A may interact with autophagy machinery to promote the constitutive turnover of basal autophagy cargo/adaptors.
Here, we take advantage of BioID and quantitative LC‐MS/MS to identify a network of proximity‐based ATG9A interactions that include a variety of vesicular trafficking complexes and autophagy regulators. In exploring these interactions further, we discover that ATG9A interacts with an ULK1‐independent ATG13 “subcomplex” that is essential for proper ATG9A trafficking and basal turnover of p62/SQSTM1. Together, our data elucidate a diverse array of novel ATG9A interactions and reveal, to our knowledge, the first ULK1‐independent role for ATG13 in regulating ATG9A function.
Results
BioID reveals proximity‐based interactions between ATG9A and a network of trafficking proteins and complexes
With the ultimate goal of elucidating the interactome of ATG9A, we first assessed potential protein–protein docking regions along the putative ATG9A structure. The long C terminus of ATG9A bears some hallmarks of a signaling hub, including a high degree of predicted intrinsic disorder and a concentration of phosphorylation sites that are repeatedly identified in global PTM mass spectrometry studies (Fig EV1A). These include several phosphorylations with over 20 independent mass spectrometry identifications (S735, S738, S741, S828) and an AMPK‐mediated phosphorylation at S761 that we identified as a 14‐3‐3ζ docking site (Weerasekara et al, 2014). In addition, there is evidence from structural and molecular studies that ATG9A self‐associates via its C termini, which might further expand its ability to act as a protein docking site or signaling hub (He et al, 2008; Staudt et al, 2016; Lai et al, 2020). In support of this idea, we found that ATG9A fused to split‐mVenus molecules at its C termini produced robust BiFC signal in a perinuclear pattern (Fig EV1B), consistent with known localization patterns of ATG9A (Young et al, 2006; Orsi et al, 2012). In addition, we found that a C‐terminally truncated ATG9A was unable to fully rescue defective LC3 processing in an ATG9A KO line (Fig EV1C). These data suggest that the ATG9A C terminus is critical for ATG9A function and likely a hub of multiple protein–protein interactions.
Figure EV1. The C terminus of ATG9A harbors hallmarks of a signaling hub and is involved in self‐association, and ATG9A trafficking.
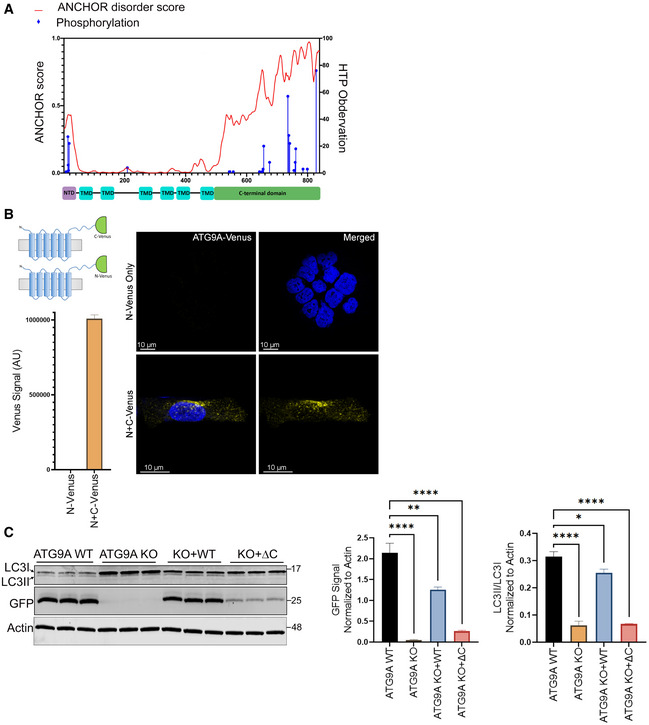
- Graph showing disorder tendency of ATG9A across its amino acid sequence (predicted by ANCHOR web server at http://anchor.elte.hu/) and PTM distribution across the protein quantified by High Throughput Paper (HTP) observations (generated by PhosphoSitePlus® at https://www.phosphosite.org/psrSearchAction) (top). Schematic of ATG9A domain structure (bottom).
- Schematic representation showing N‐ or C‐terminal halves of mVenus fused to C terminus of ATG9A (top left). HCT‐116 cells expressing ATG9A C‐terminally labeled N mVenus or N + C mVenus constructs were grown in full DMEM media, fixed, and imaged (Scale bar = 10 µm) (right). Fluorescence intensity of cells expressing ATG9A C‐terminally labeled mVenus constructs was measured via flow cytometry. Mean ± SEM, n = 1(bottom left).
- Hela ATG9A WT, ATG9A KO, or ATG9A KO cells reconstituted with ATG9A WT and ATG9A ΔC mutant stably expressing GFP‐LC3 were grown in full media and whole‐cell lysates followed by immunoblotting with indicated antibodies (left). Quantification of normalized GFP and normalized LCII/LC3I ratio. Mean ± SEM, n = 3 (biological replicates). Significance measured using RM one‐way ANOVA followed by Fisher’s LSD tests. ns P > 0.05, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
Our efforts to probe ATG9A protein–protein interactions by co‐IP proteomics had limited success (unpublished). The multi‐pass transmembrane nature of ATG9A presents challenges to co‐IP proteomics, most notably the difficulty of extracting ATG9A from intracellular membranes while maintaining protein–protein interactions. On the contrary, these same qualities make ATG9A a good candidate for BioID (Roux et al, 2012), in which promiscuous interactions are relatively limited by ATG9A being fixed in membrane. Thus, we fused the modified bacterial biotin ligase BirA (R118G—denoted with an asterisk) to the C terminus of hemagglutinin (HA)‐tagged ATG9A (HA‐ATG9A‐BirA*) (Roux et al, 2012; Rees et al, 2015). We verified that fusion of BirA* to the ATG9A C terminus did not impair the function of ATG9A, as the HA‐ATG9A‐BirA* construct was able to fully rescue the defects in p62/SQSTM1 degradation and LC3B lipidation in ATG9A KO cells and showed the same cellular localization patterns as endogenous ATG9A (Figs 1A and EV2A–C). We also verified that biotin signal overlaps with HA‐ATG9A‐BirA* (Fig EV2D).
Figure 1. BioID reveals a network of ATG9A interactors, including multiple trafficking regulators and members of the ULK1 complex.
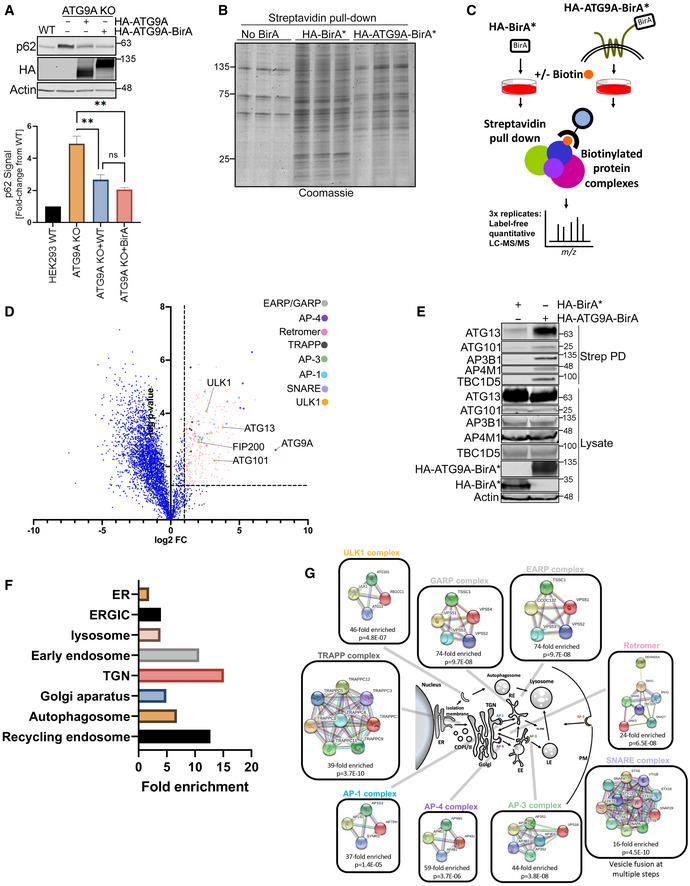
- HEK293T ATG9A WT, ATG9A KO, or ATG9A KO cells reconstituted with overexpressed HA‐ATG9A and HA‐ATG9A‐BirA* were grown in full DMEM media. Endogenous p62/SQSTM1 level was measured by immunoblot (top). Graph below shows quantification of p62 infrared signal normalized to Actin. Mean ± SEM, n = 3 (biological replicates). Significance measured using RM one‐way ANOVA followed by Fisher’s LSD tests. ns P > 0.05, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
- HEK293T parental cells or HEK293T cells stably expressing HA‐BirA* or HA‐ATG9A‐BirA* (stably integrated with lentivirus) were grown in full DMEM media, treated with 50 µM biotin for 12 h, followed by detergent lysis and incubation with streptavidin resin. Streptavidin pulldown samples were resolved in a 4–15% gradient gel and Coomassie stained.
- An experimental schematic of Bio‐ID workflow.
- Quantitative proteomics data from three independent experiments were analyzed by volcano plot. Significant interactors were selected based on a cutoff of P ≤ 0.05 (two‐tailed heteroscedastic t‐test) and ≥ 2‐fold increase in interaction comparing the AUC signal for each peptide from HA‐ATG9A‐BirA* versus HA‐BirA* samples. A log 2‐fold change = 1 and ‐log P ‐value 0.05 = 1.3 were marked by dash lines on the volcano plot. Significant interactors were colored in salmon, and interactors of particular interest were color coded (See heat map S2B).
- A subset of interactors were validated by immunoblotting with indicated antibodies after the streptavidin pulldown step in panel C.
- The subcellular localization of significantly scored interactors from three independent proteomics experiments was assigned by using panther GO enrichment analysis (http://www.geneontology.org/page/go‐enrichment‐analysis).
- A schematic representation of proteins identified by proteomics analysis grouped into protein complexes and associated trafficking pathways. Protein complexes were assembled from the GO enrichment and analyzed using STRING (https://string‐db.org/).
Figure EV2. Functional validation of HA‐ATG9A‐BirA* constructs.
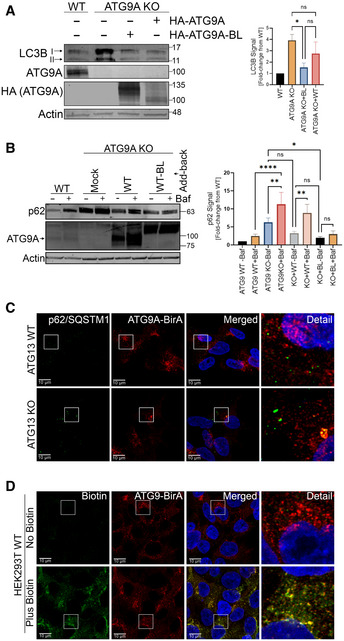
- HEK293T WT, ATG9A KO, and ATG9A KO cells reconstituted with HA‐ATG9A and HA‐ATG9A‐BirA* were grown in full DMEM media, lysed, and immunoblotted with indicated proteins (left). Quantification of normalized LC3B infrared signal. Mean ± SEM, n = 3 (biological replicates). Significance measured using RM one‐way ANOVA followed by Fisher’s LSD tests (right).
- HEK293T WT, ATG9A KO, and ATG9A KO cells reconstituted with HA‐ATG9A and HA‐ATG9A‐BirA* were grown in full DMEM media with or without 100nM Bafilomycin for 24 h, lysed, and immunoblotted with indicated proteins (left). Quantification of normalized p62 infrared signal. Mean ± SEM, n = 3 (biological replicates). Significance measured using RM one‐way ANOVA followed by Fisher’s LSD tests (right).
- Confocal images of p62/SQSTM1 colocalization with HA‐ATG9A‐BirA* in HEK293T ATG13 WT and ATG13 KO cells stably expressing HA‐ATG9A‐BirA*. Cells were grown in full DMEM media, fixed, and labeled with antibodies for p62/SQSTM1 and HA (Scale bar = 10 µm).
- Confocal images of biotinylated proteins colocalized with HA‐ATG9A‐BirA*. HA‐ATG9A‐BirA*‐expressing cells were grown in full DMEM media with or without biotin incubation (12 h), fixed, and labeled with Alexa Fluor 488‐conjugated streptavidin and HA antibody (Scale bar=10 µm). ns P > 0.05, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
To pursue BioID proteomics, we generated cell lines stably expressing either HA‐ATG9A‐BirA* or, as a control, HA‐BirA* alone. These cells were supplemented with biotin, followed by detergent lysis and capture of biotinylated proteins on streptavidin resin. An initial evaluation of captured proteins by Coomassie staining suggested an overall lower level of biotinylation by HA‐ATG9A‐BirA* compared with HA‐BirA* alone, as perhaps expected given the anchored, transmembrane nature of ATG9A (Fig 1B). Therefore, we proceeded with BioID proteomics following the experimental schematic outlined in Fig 1C. LC‐MS/MS data from these experiments are available in Appendix Table S1. Quantitative LC‐MS/MS of biological triplicates of the experiment in Fig 1C revealed 283 proteins that were significantly enriched (≥ 2‐fold increase, ≤ 0.05 P‐value; Dataset EV1) in the HA‐ATG9A‐BirA* samples versus HA‐BirA* alone (see volcano plot, Fig 1D, Appendix Fig S1A and B). These spanned an array of autophagy and trafficking regulators, including multiple components of EARP/GARP, AP‐1, AP‐3, AP‐4, Retromer, TRAPP, and SNARE complexes and all components of the canonical ULK1 complex (Fig 1D), a subset of which were validated by immunoblot (Fig 1E). Several of these HA‐ATG9A‐BirA*‐biotinylated proteins are already known to interact with ATG9A, including STX16, Arfaptin‐1, TBC1D5, AP‐1, AP‐2, and AP‐4, which increased our confidence in the BioID data (Orsi et al, 2012; Popovic & Dikic, 2014; Imai et al, 2016; Lamb et al, 2016; Mattera et al, 2017; Zhou et al, 2017; Aoyagi et al, 2018; Davies et al, 2018; Soreng et al, 2018; Judith et al, 2019). Furthermore, this proximity‐based ATG9A interactome was highly enriched for proteins associated with the organelles where ATG9A is known to reside, including the ER, TGN, ERGIC, and endosomal systems (Fig 1F and G).
ATG9A interacts with an ULK1‐independent ATG13 subcomplex that includes ATG101
Among the BioID proteomics data (Dataset EV1), our attention was drawn to members of the ULK1 complex, which emerged as top hits (Fig 1D). Of the ULK1 complex proteins, ATG13 showed the highest fold‐change increase in signal across all of the HA‐ATG9A‐BirA* replicates and we had previously observed interaction between ATG9A with ATG13 by co‐IP (Kannangara and Andersen, unpublished).
To investigate the interaction between ATG9A and ATG13 further, we generated ATG13 KO cells and then stably reconstituted them with WT ATG13 or one of two mutants of ATG13: ATG13 Δ2AA, which lacks a C‐terminal 2‐amino acid segment required for ULK1 binding (Alers et al, 2011; Hieke et al, 2015); or ATG13 ΔHORMA, which lacks the HORMA domain required for interaction with ATG101 and reported in yeast to be essential for recruiting Atg9 vesicles to the PAS (Fig 2A) (Jao et al, 2013; Qi et al, 2015; Suzuki et al, 2015). We verified that the ATG13 Δ2AA indeed fails to interact with ULK1 (Fig EV3A). Likewise, we found that the ATG13 ΔHORMA mutant is defective in interacting with endogenous ATG9A in mammalian cells (Fig EV3B).
Figure 2. ATG9A interacts with an ULK1‐independent ATG13 complex that includes ATG101.
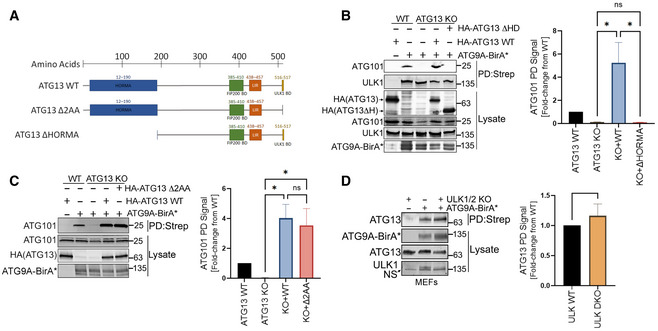
- Schematic representation of ATG13 mutations used in the study.
- HA‐ATG9A‐BirA* was expressed in HCT‐116 ATG13 WT, ATG13 KO, or ATG13 KO cells reconstituted with WT ATG13 or ATG13 ΔHORMA. Cells were grown in full DMEM media, treated with 50 µM biotin for 12 h, followed by detergent lysis and incubation with streptavidin resin. The graph on right shows quantification of normalized ATG101 infrared signal. Mean ± SEM, n = 3 (biological replicates). Significance measured using RM one‐way ANOVA test followed by Fisher’s LSD tests.
- Cells were treated as in panel B but included reconstitution with ATG13 Δ2AA mutant. The graph on right shows quantification of normalized ATG101 infrared signal. Mean ± SEM, n = 3 (biological replicates). Significance measured using RM one‐way ANOVA test followed by Fisher’s LSD tests (right).
- HA‐ATG9A‐BirA* was overexpressed in WT and ULK1/2 Double KO MEFs. Cells were subjected to streptavidin pulldown and immunoblotting with indicated antibodies. The graph on right shows quantification of normalized ATG13 infrared signal. Mean ± SEM. n = 3 (biological replicates). Significance measured using one‐sample t‐test compared with hypothetical mean of 1 (right). ns P > 0.05, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
Figure EV3. ATG9A interacts with an ULK1‐independent ATG13‐ATG101 subcomplex via the ATG13 HORMA domain.
- The ULK1 binding‐deficient mutant of ATG13 (ATG13 ∆2AA) was validated by immunoprecipitating HA‐ATG13 or HA‐ATG13 ∆2AA and immunoblotting for Myc‐ULK1 in HCT‐116 ATG13 KO cells.
- HCT‐116 parental or HCT‐116 ATG9A‐HA KI ATG13 KO cells reconstituted with 3X FLAG‐ATG13 WT or 3X FLAG‐ATG13 ΔHORMA were grown in full DMEM media and subjected to size exclusion chromatography. Fractions with high levels of ATG9A‐HA were pooled to facilitate immunoprecipitation. ATG9A‐HA was immunoprecipitated from pooled fractions followed by immunoblotting with indicated antibodies.
- Co‐IP of overexpressed HA‐ATG9A from HCT‐116 ATG13 KO cells followed by immunoblotting with indicated antibodies.
- Co‐IP of HA‐ULK1 from HCT‐116 ATG13 WT and KO cells followed by immunoblotting with indicated antibodies.
- Co‐IP of endogenous ATG9A from HCT‐116 ATG9A‐HA KI ATG101 KO cells followed by immunoblotting with indicated antibodies.
- Co‐IP of endogenous ATG9A from HCT‐116 ATG9A‐HA KI FIP200 KO cells followed by immunoblotting with indicated antibodies.
- Co‐IP of overexpressed HA‐ATG13 from HEK293T ATG9A KO cells followed by immunoblotting with indicated antibodies.
Importantly, in HA‐ATG9A‐BirA*‐expressing cells, the loss of ATG13 had no effect on streptavidin capture of ULK1, but completely abrogated the capture of ATG101 (Fig 2B), suggesting that ATG13 is required for the interaction of ATG9A with ATG101, but not ULK1. Also, the streptavidin capture of ATG101 was rescued by reconstituting the ATG13 KO cells with WT ATG13, while reconstitution with ATG13 ΔHORMA did not recover ATG101 binding (Fig 2B), which is consistent with a model wherein ATG9A interacts with ATG101 via ATG13. In contrast to ATG13 ΔHORMA, the reconstitution of ATG13 KO cells with the ULK1 binding‐defective ATG13 Δ2AA completely restored HA‐ATG9A‐BirA*‐mediated biotinylation of ATG101 (Fig 2C). Furthermore, in mouse embryonic fibroblasts, HA‐ATG9A‐BirA* biotinylates ATG13 regardless of the presence or absence of ULK1/2 (Fig 2D). Reciprocal co‐IP experiments also demonstrated that ATG9A‐ULK1 binding was not affected by loss of ATG13 (Fig EV3C and D).
To confirm our biochemical observations of the ATG9A–ATG13 interaction, we used CRISPR/Cas9 to knock‐in an HA affinity tag in‐frame on the C‐terminal end of genomic ATG9A in HCT‐116 cells (Appendix Fig S2A). Deep sequencing of multiple clones verified that the HA sequence was inserted correctly. To further validate the knock‐in, we detected an HA signal at the predicted molecular weight of ATG9A. We then verified that this signal was indeed ATG9A‐HA by knocking out the ATG9A locus with CRISPR/Cas9 and measuring the corresponding loss of HA signal (Appendix Fig S2B). We were also able to detect a strong ATG9A‐specific signal by immunostaining for HA and confocal imaging, which was lost upon CRISPR/Cas9 targeting of ATG9A (Appendix Fig S2C).
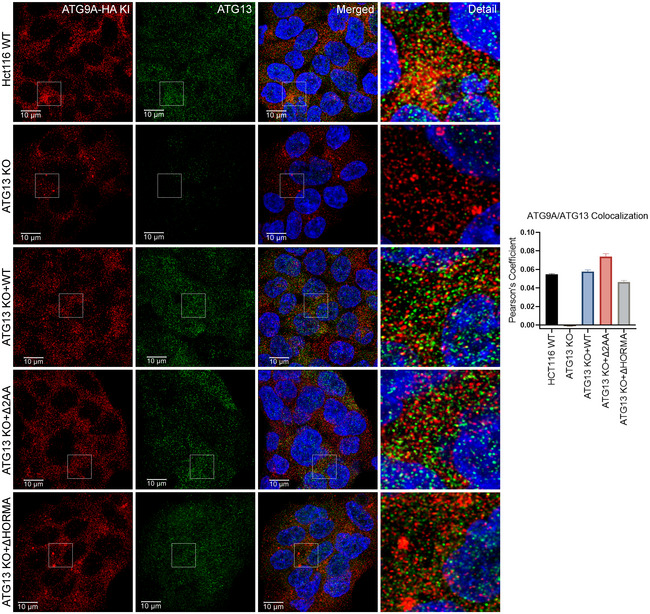
Using these HA KI cells (ATG9A‐HA KI), we found that endogenous ATG13 and ATG9A colocalize in discrete semi‐perinuclear puncta. Importantly, the colocalization of ATG9A and ATG13 is abrogated by loss of ATG101 but only marginally affected by loss of FIP200 (Fig EV4), which we also confirmed by direct co‐IP of ATG9A‐HA and ATG13 (Fig EV3E and F). Conversely, as we suspected, the interaction between ATG13 and ATG101 did not require ATG9A (Fig EV3G), suggesting that ATG9A is not an integral part of the ATG13‐ATG101 complex and that they may only transiently interact. Furthermore, in ATG13 KO reconstitution experiments, ATG9A showed increased colocalization with the ULK1 binding‐defective ATG13 Δ2AA and decreased colocalization with ATG13 ΔHORMA compared with WT ATG13 (Fig EV5). Taken together with the co‐IP and BirA* experiments in Fig 2, these data suggest that, aside from the canonical ULK1 complex, ATG9A interacts with an ULK1‐independent ATG13 subcomplex that includes ATG101.
Figure EV4. ATG13 colocalization with ATG9A is independent of FIP200/ULK1 complex.
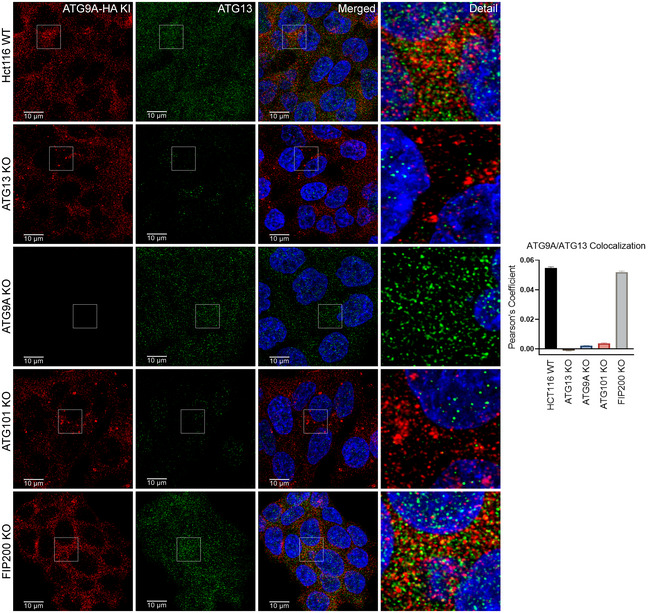
HCT‐116 ATG9A‐HA KI ATG13 WT, ATG13 KO, ATG101 KO, ATG9A KO, and FIP200 KO cells were grown in full DMEM media, fixed, labeled with antibodies for HA and ATG13, and imaged (Scale bar = 10 µm) (left). Quantification of ATG9A colocalization with ATG13. Mean ± SEM, n = 1 independent experiment with 30 technical replicates. Images are derived from the same experiment as Fig EV5 and Appendix Fig S3.
Figure EV5. ATG13 colocalization with ATG9A requires the ATG13 HORMA domain, but occurs independently of ULK1 binding.
-
A, BHCT‐116 ATG9A‐HA KI ATG13 WT, ATG13 KO or ATG13 KO cells reconstituted with ATG13 WT, ATG13 Δ2AA, and ATG13 ΔHORMA. Cells were grown in full DMEM media, fixed, labeled with antibodies for HA and ATG13, and imaged. (Scale bar = 10 µm).
- C
The loss of ATG13 and ATG101 results in an accumulation of ATG9A at large clusters of p62/SQSTM1
To understand what role ATG13 may play in regulating ATG9A under basal conditions, we analyzed the effect of ATG13 KO on endogenous ATG9A in the ATG9A‐HA KI cells. Confocal imaging of these cells revealed a striking accumulation of ATG9A in large spherical puncta. We then questioned whether these large accumulations of ATG9A in ATG13 KO cells were a result of a defective ULK1 complex or could be attributed to an ULK1‐independent function of ATG13 and potentially ATG101. Thus, in addition to ATG13 KO lines, we used our ATG101 and FIP200 KO versions of the ATG9A‐HA KI cell line as well as an additional panel of HEK293T lines. Of note, our attempts to disrupt ULK1 by CRISPR/Cas9 were unsuccessful (no viable cells recovered), despite using multiple sgRNAs and cell lines, so we relied on FIP200 as a surrogate for ULK1 complex KO, given that loss of FIP200 has been shown to disrupt the ULK1 complex (Hara et al, 2008; Gammoh et al, 2013). We found that loss of ATG101 resulted in the same distinctly large ATG9A puncta (Appendix Fig S3A), while FIP200 KOs showed no significant increase in ATG9A puncta size by confocal imaging. Furthermore, the increase in ATG9A puncta size in ATG13 KOs was rescued to a normal ATG9A distribution by reconstitution with WT or ATG13 Δ2AA, but not ATG13 ΔHORMA (Appendix Fig S3B).
To identify where in the cell these large ATG9A structures reside, we co‐stained the cells for markers of various organelles or proteins known to colocalize with ATG9A, including the ER, Golgi, endosomes, lysosomes, p62/SQSTM1, and IM/PAS. While the majority of organelle or protein markers decreased or showed no significant change in colocalization with ATG9A, we found that the large accumulations of ATG9A in ATG13 KO cells were almost entirely colocalized with p62/SQSTM1 (Fig 3A–G, quantification in panel H).
Figure 3. The loss of ATG13 causes a shift in colocalization of ATG9A with organelle and autophagy markers and triggers an accumulation of ATG9A with p62/SQSTM1.

-
A–GRepresentative images of ATG9A colocalization with different organelle and autophagy markers. HCT‐116 ATG9A‐HA KI ATG13 WT or ATG13 KO cells were grown in full DMEM media, fixed and labeled with antibodies for HA, and indicated organelle/cellular markers and imaged (Scaler bar = 10 µm).
-
HQuantification of ATG9A colocalization with indicated organelle markers of golgi (GOLGIN97), mitochondria (TUFM), ER (PDIA3), endosomal system (VPS26A), early endosome (EEA1), lysosome (LAMP1), autophagy adaptor (p62), and autophagosome markers (ATG16L1, WIPI4, ATG2A, LC3II, WIPI2), respectively. Mean ± SEM, n = 1 independent experiment with 30 technical replicates. Images for p62 colocalization (G) are from the same single independent experiment as Figs 4C and 5C.
By immunoblot, we found that p62/SQSTM1 levels were significantly elevated in the ATG13, ATG101, and ATG9A KO lines, while FIP200 KOs showed a marginal increase in p62/SQSTM1, depending on the cell type (Fig 4A and B). In agreement with these data, confocal imaging revealed a high level of p62/SQSTM1 accumulation in ATG13, ATG9A, and ATG101 KO cells (Fig 4C and D). Similarly, loss of ATG101 phenocopied the effect of ATG13 KO (Fig 4C and D) by inducing a large accumulation of ATG9A at the p62/SQSTM1 puncta. Interestingly, although FIP200 KO cells showed no significant increase in ATG9A puncta size overall, we did see increased ATG9A‐p62/SQSTM1 colocalization in these cells, perhaps reflecting a subtle p62/SQSTM1 build‐up that we were unable to detect by imaging, but could see by immunoblot (Fig 4C and E).
Figure 4. The loss of ATG13 and ATG101 result in an accumulation of ATG9A at large clusters of p62/SQSTM1.
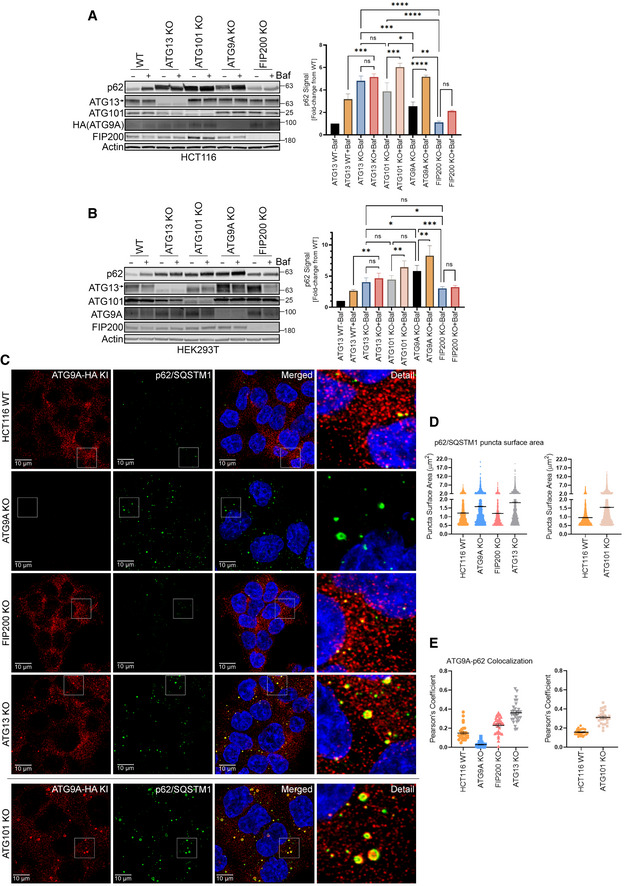
- Endogenous p62/SQSTM1 level in HCT‐116 ATG9A‐HA KI ATG13 WT, ATG13 KO, ATG101 KO, ATG9A KO, and FIP200 KO clones was measured by immunoblotting with indicated antibodies. Cells were grown in full DMEM media, treated with or without 100 nM Bafilomycin for 24 h and whole‐cell lysates were subjected to immunoblotting (left). The graph on right shows quantification of normalized p62 infrared signal. Mean ± SEM, n = 3 (biological replicates). Significance measured using RM one‐way ANOVA test followed by Fisher’s LSD tests (right).
- Endogenous p62/SQSTM1 level in HEK293T ATG13 WT, ATG13 KO, ATG101 KO, ATG9A KO, and FIP200 KO clones was measured by immunoblotting with indicated proteins. Cells were grown in full DMEM media, treated with or without 100 nM Bafilomycin for 24 h, and whole‐cell lysates were subjected to immunoblotting. Mean ± SEM, n = 3 (biological replicates). Significance measured using RM one‐way ANOVA test followed by Fisher’s LSD tests (right).
- Confocal images of ATG9A colocalization with p62/SQSTM1. HCT‐116 ATG9‐HA KI ATG13 WT, ATG13 KO, ATG101 KO, ATG9A KO, and FIP200 KO cells were grown in full DMEM media, fixed, labeled with antibodies for HA and p62/SQSTM1, and imaged (Scale bar 10 µm). ATG101 KO was stained and quantified separately. Images are from the same single independent experiment as Figs 3G and 5C.
- Quantification of average surface area of p62/SQSTM1 puncta in C. Mean ± SEM, n = 1 independent experiment with 30 technical replicates. ATG101 KO was stained and quantified separately. Mean ± SEM, n = 1 independent experiment with 30 technical replicates. A break was inserted in the Y axis to accommodate the wide range of p62/SQSTM1 puncta sizes.
- Quantification of ATG9A colocalization with p62/SQSTM1. Mean ± SEM, n = 1 independent experiment with 30 technical replicates. ATG101 KO was stained and quantified separately. Mean ± SEM, n = 1 independent experiment with 30 technical replicates.
Next, we used our panel of ATG13 mutants to more definitively assess the ULK1‐indpendence of ATG13 in these experiments. We found that reconstitution of ATG13 KO cells with the ULK1 binding‐defective ATG13 Δ2AA mutant completely rescued the accumulation of p62/SQSTM1 and restored a more normal distribution of ATG9A in the cell (Fig 5A–E; Appendix Fig S3B). The ATG13 ΔHORMA mutant failed to rescue the defect in ATG9A accumulation at large p62/SQSTM1 puncta (Fig 5A–E; Appendix Fig S3B). We did not see any exacerbation of these p62/SQSTM1 puncta in starved conditions (Fig 5B). These data support the idea that interaction with ATG101, but not ULK1, is essential to promote the basal autophagy function of ATG13.
Figure 5. ATG13‐mediated rescue of ATG9A accumulation at p62/SQSTM1 clusters requires the ATG13 HORMA domain but is independent of ULK1.

- Endogenous p62/SQSTM1 level was measured in HCT‐116 ATG9A‐HA KI ATG13 WT, ATG13 KO or ATG13 KO cells reconstituted with ATG13 WT, ATG13 ΔHORMA, and ATG13 Δ2AA by immunoblotting with indicated proteins (left). Cells were grown in full DMEM media, and whole‐cell lysates were subjected to immunoblotting (left). Quantification of normalized p62 infrared signal. Mean ± SEM, n = 3 (biological replicates). Significance measured using RM one‐way ANOVA test followed by Fisher’s LSD tests (right).
- HCT‐116 cells in A were incubated in EBSS for 4 h then processed and analyzed as in A.
- Confocal images of ATG9A colocalization with p62/SQSTM1 in HCT‐116 ATG9A‐HA KI‐ ATG13 WT, ATG13 KO, or ATG13 KO cells reconstituted with ATG13 WT, ATG13 Δ2AA, and ATG13 ΔHORMA. Cells were grown in full DMEM media, fixed, labeled with antibodies for HA and p62/SQSTM1, and imaged (Scale bar = 10 µm). Images are from the same single independent experiment as Figs 3G and 4C.
- Quantification of average surface area of p62/SQSTM1 puncta in C. Mean ± SEM, n = 1 independent experiment with 30 technical replicates. A break was inserted in the y‐axis to accommodate the wide range of p62/SQSTM1 puncta sizes.
- Quantification of ATG9A colocalization with p62/SQSTM1 in C. Mean ± SEM, n = 1 independent experiment with 30 technical replicates.
Source data are available online for this figure.
The loss of ATG13 results in increased ATG9A recruitment to stalled clusters of p62/SQSTM1
To explore more fully the mechanism of the accumulation of ATG9A in ATG13 KO, we observed the dynamics of ATG9A and p62/SQSTM1 in live cells using EGFP‐p62/SQSTM1 and mRuby2‐ATG9A stably expressed in ATG13 KO cells. We found that trafficking of ATG9A seemed to increase with the size of the p62/SQSTM1 cluster, which was particularly evident in the ATG13 KO cells (Fig 6A and Movies EV1 and EV2). We also found that these stalled p62/SQSTM1 clusters colocalized almost entirely with ubiquitin, consistent with these clusters being accumulations of ubiquitinated cellular material (Fig 6B). To confirm that these clusters of p62/SQSTM1 and ubiquitin were a result of stalled degradation in the ATG13 KO, we performed a modified pulse chase by using HA‐ATG9A‐BirA* to pulse‐label (using a pulse of biotin) p62/SQSTM1, which we could then track over time by purifying any residual biotinylated p62/SQSTM1 on streptavidin resin. As expected, the loss of ATG13 results in delayed degradation of the biotinylated p62/SQSTM1 (Fig 6C).
Figure 6. Loss of ATG13 results in increased ATG9A recruitment to stalled clusters of p62/SQSTM1.
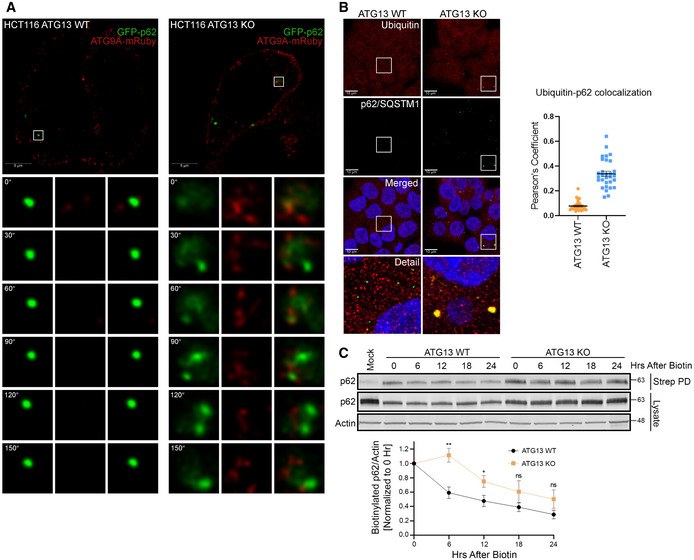
- Live‐cell imaging of HCT‐116 ATG13 WT and HCT‐116 ATG13 KO cells stably expressing mRuby2‐ATG9A and EGFP‐p62/SQSTM1. Representative montages show mRuby2‐ATG9A recruitment to EGFP‐p62/SQSTM1 puncta (Scale bar = 5 µm).
- Confocal images of ubiquitin colocalization with p62/SQSTM1 in HCT‐116 ATG13 WT and HCT‐116 ATG13 KO cells. Cells were grown in full DMEM media, fixed, labeled with antibodies for ubiquitin and p62/SQSTM1, and imaged (Scale bar = 10 µm) (left). Quantification of ubiquitin colocalization with p62/SQSTM1 (right). Mean ± SEM, n = 1 independent experiment with 30 technical replicates.
- Modified pulse chase experiment. HEK293T ATG13 WT and HEK293T ATG13 KO cells stably expressing HA‐ATG9A‐BirA* were pulse labeled with biotin for 24 h, media was replaced with full DMEM and streptavidin pulldown was performed at indicated time points. Quantification of normalized p62 infrared signal. Mean ± SEM, n = 3 (biological replicates). Significance measured using Student’s t‐test (bottom). ns P > 0.05, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
A split‐mVenus approach captures an ATG13‐ATG101 interaction with ATG9A
To characterize the function and cellular localization of the ULK1‐independent ATG13‐ATG101 complex more fully, we created a bimolecular fluorescence complementation (BiFC) assay with split‐mVenus halves fused to ATG13 and ATG101 (Fig 7A). We found that the ATG13‐ and ATG101‐fused split‐mVenus halves expressed well were able to dimerize/reconstitute as measured by FACS and that the ATG13 ΔHORMA mutant abrogated this reconstitution (Appendix Fig S4A and B). We also verified that the split‐mVenus‐fused ATG13 and ATG101 were functional, because when expressed together, they were able to rescue defective autophagy in an ATG13‐ATG101 double KO cell line (Appendix Fig S4C). We then took advantage of a GFP‐TRAP nanobody resin to capture the intact ATG13‐ATG101 dimer for co‐IP immunoblotting (Fig 7A and B). We found that both WT and Δ2AA mutant ATG13‐ATG101 dimers interact with ATG9A, supporting the idea AT9A interacts with an ULK1‐independent ATG13‐ATG101 dimer. We also found that FIP200 coimmunoprecipitates with the ATG13 Δ2AA mutant, suggesting that ATG13 can also interact with FIP200 in an ULK1‐independent manner (Shi et al, 2020).
Figure 7. A split‐mVenus approach captures an ATG13‐ATG101 interaction with ATG9A.
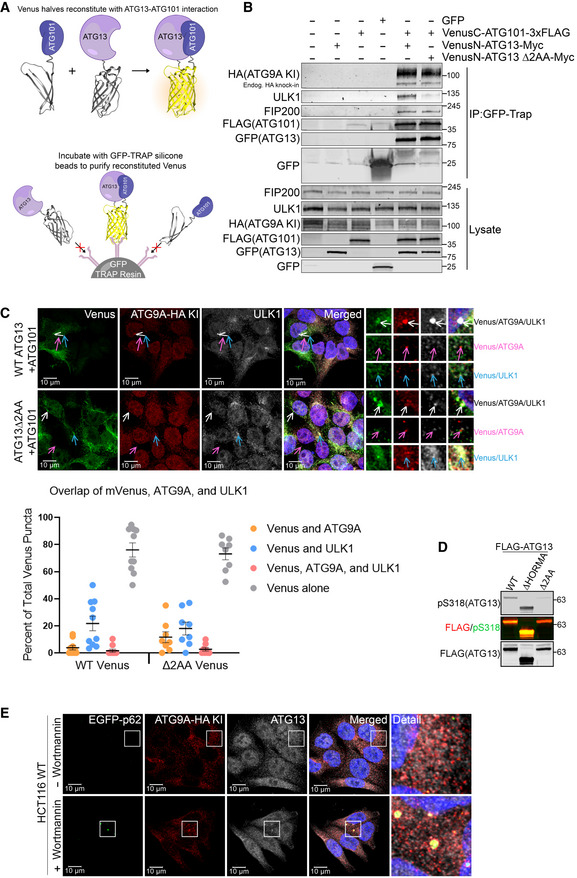
- Schematic representation of bimolecular fluorescence complementation (BiFC) assay by using split‐mVenus approach.
- HCT‐116 ATG9A‐HA KI cells with overexpressed GFP only, VenusC‐ATG101‐3X FLAG only, VenusN‐ATG13‐Myc only, VenusN‐ATG13 Δ2AA only, or both Venus N and C halves together were subjected to immunoprecipitation with GFP‐trap nano body resin. Immunoblotted for indicated proteins.
- Confocal images of Venus colocalization with ATG9A and ULK1 in HCT‐116 ATG9A‐HA KI ATG13‐ATG101 double KO cells stably expressing VenusC‐ATG101‐3X FLAG and VenusN‐ATG13‐Myc or VenusN‐ATG13 Δ2AA‐Myc. Cells were grown in full DMEM media, fixed, labeled with antibodies for HA and ULK1, and imaged (Scale bar = 10 µm). Quantification of overlap shown as percent of total Venus puncta (bottom). Mean ± SEM, n = 1 independent experiment with 10 technical replicates.
- HCT‐116 ATG9A‐HA KI ATG13 KO cells reconstituted with stably expressing ATG13 WT, ATG13 Δ2AA, and ATG13 ΔHORMA grown in full DMEM media, lysed, and immunoblotted with indicated proteins.
- Confocal images of EGFP‐p62/SQSTM1 colocalization with HA‐ATG9A and ATG13 in HCT‐116 ATG9A‐HA KI cells stably expressing EGFP‐p62/SQSTM1. Cells were grown in full DMEM media with or without 1 μM Wortmannin for time 4 h, fixed, labeled with antibodies for HA and ATG13, and imaged (Scale bar = 10 µm).
We then took advantage of the ATG13‐ATG101 BiFC signal to see the cellular localization of the intact ATG13‐ATG101 dimer. In support of the mVenus‐capture co‐IP in Fig 7B, we found that ATG9A interacts with the ATG13‐ATG101 dimer in small puncta that do not include ULK1 (Fig 7C). Interestingly, we also found that the WT and Δ2AA mutant ATG13‐ATG101‐ATG9A complexes show colocalization with ULK1 in the largest clusters, but some of these clusters only partially overlapped with ULK1 or contained no ULK1 at all (Fig 7C), supporting the idea that ATG13 and ULK1 arrive independently at structures destined for degradation (Shi et al, 2020). Immunoblotting for phospho‐S318 of ATG13 confirmed the lack of functional interaction between ULK1 and ATG13 Δ2AA (Fig 7D), despite their occasional colocalization.
To ask whether the interaction between ATG9A and endogenous ATG13 occurs at stalled clusters of p62/SQSTM1, we used the PI3K inhibitor Wortmannin to force the accumulation of p62/SQSTM1. Indeed, ATG13 and ATG9A colocalize at large p62/SQSTM1 clusters (Fig 7E). Taken together, these data, along with the protein–protein interaction data in previous figures, support the model that the ATG13‐ATG101 complex interacts with ATG9A, independently of ULK1, at p62/SQSTM1 clusters, likely in an effort to dispose of these clusters through basal autophagy. All together, these data support a model in which the ATG13‐ATG101 complex, independent of its physical interaction with ULK1, interacts with ATG9A to promote the basal turnover of p62/SQSTM1‐associated clusters of ubiquitinated cellular material (Fig 8).
Figure 8. An ULK1‐independent ATG13‐ATG101 complex regulates basal ATG9A function and p62/SQSTM1 turnover.
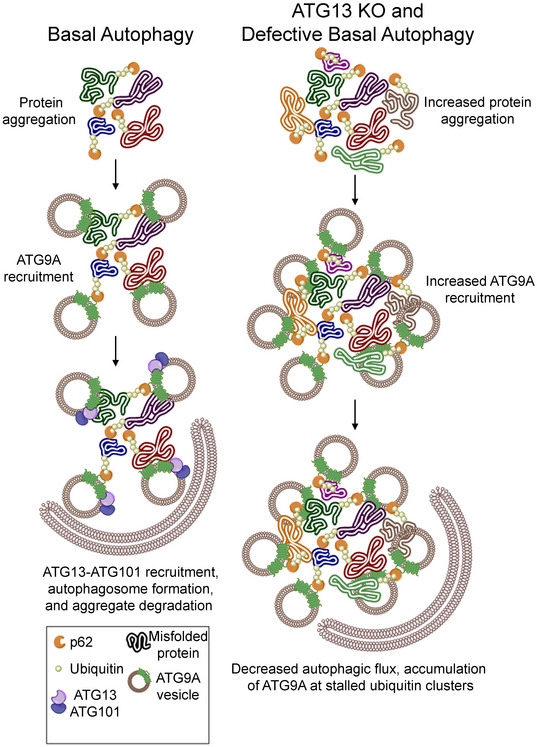
During adaptor‐mediated basal autophagy, an ULK1‐inpependent ATG13‐ATG101 complex interacts with ATG9A at p62/SQSTM1 clusters and promotes the ATG9A‐mediated turnover of these clusters. Upon loss of the ULK1‐indpendent ATG13‐ATG101 complex, or disruption of the ATG13‐ATG101‐ATG9A interaction, the p62/SQSTM1 clusters accumulate, resulting in an accumulation of ATG9A at p62/SQSTM1 clusters.
Discussion
In this study, we began with an interest in mapping the proximity‐based interactome of ATG9A under conditions of basal autophagy (Dataset EV1). These data revealed an ATG9A interactome heavily weighted toward vesicle‐trafficking pathways, including entire (or nearly entire) complexes of trafficking regulators such as the EARP, GARP, TRAPP, AP‐1, AP‐4, AP‐3, SNARE, and Retromer complexes (Fig 1G). In addition, all components of the ULK1 complex were prominent in the ATG9A BioID interactome (Fig 1D). We initially focused on two members of the ULK1 complex, ATG13 and ATG101, given that their interaction with ATG9A seemed the most robust and direct. The prevailing model for ATG13 and ATG101, based primarily on studies done from the perspective of ULK1 during nutrient stress, attributes their function in autophagy to supporting the ULK1 complex. Indeed, ATG13 is required for nutrient stress‐induced ULK1 kinase activity, which, in turn, is necessary to activate downstream autophagic machinery and inhibit mTORC1 (Cheong et al, 2008; Kawamata et al, 2008; Ganley et al, 2009; Hosokawa et al, 2009a; Jung et al, 2009; Yamamoto et al, 2016). However, the role of ATG13 and ATG101 in basal autophagy, whether strictly within or outside the ULK1 complex, is poorly understood.
Some evidence suggests that ATG13 can act, at least to some degree, independently of ULK1. First, ATG13 and ULK1 do not overlap completely in phenotype. Mice lacking ULK1, and its semi‐redundant homologue ULK2, succumb to a neonatal lethality during the weening period, similar to the loss of other core autophagy genes (Chan et al, 2007; Kundu et al, 2008; Cheong et al, 2011; Lee & Tournier, 2011). In contrast, mice lacking ATG13 die in utero, suggesting that ATG13 may have additional functions outside the ULK1 complex (Kaizuka & Mizushima, 2016). In addition, Hurley and colleagues demonstrated that in order for Atg1 (yeast homologue of ULK1) to promote phagophore expansion via its early autophagy targeting and tethering (EAT) domain, it must exist in an Atg13‐free state (Lin et al, 2018). Furthermore, super resolution microscopy in yeast demonstrated markedly different stoichiometry and localization patterns for Atg1 and Atg13 at the PAS, as well as Atg13‐independent localization of Atg1 to the PAS (Lin et al, 2018). Moreover, while ATG13 and ULK1 both associate with membrane via their own lipid binding motifs, they show different biochemical properties, with ULK1, but not ATG13, showing tight, detergent‐resistant association with membrane fractions (Chan et al, 2009).
Previous studies on ATG13 mutants that fail to bind ULK1 also suggest a function for ATG13 that does not require physical interaction with ULK1. Stork and colleagues demonstrated, via a knockout‐reconstitution approach in MEFs, that cells expressing ULK1 binding‐deficient ATG13 mutants have near‐WT levels of autophagy. In contrast, cells expressing HORMA domain mutants of ATG13, which are incapable of binding ATG101, are severely autophagy impaired (Alers et al, 2011; Hieke et al, 2015; Wallot‐Hieke et al, 2018), suggesting that ATG13 and ATG101 cooperate to promote autophagy independently of ULK1. In addition, recent work suggests that ATG13 functions independently of ULK1 in mitophagy (Zachari et al, 2019). Our results build on these data by showing an ULK1‐independent ATG13‐ATG101 complex interacts with ATG9A in basal autophagy and is sufficient to promote the basal autophagic turnover of p62/SQSTM1.
Work by Ohsumi and colleagues demonstrated that the HORMA domain of yeast Atg13 interacts with Atg9 (Suzuki et al, 2015). We were able to support this observation by showing that deletion of the HORMA domain from ATG13 impairs its interaction with ATG9A by co‐IP in mammalian cells (Fig EV3B) and also results in the same defects in basal autophagy that we observed with ATG101 KO or ATG9A KO cells (p62/SQSTM1 accumulation) (Figs 4 and 5). We also found that ATG13 was required for the interaction between ATG9A and ATG101; while vice versa, ATG101 was required for the interaction between ATG9A and ATG13 (Fig 2B and C, Appendix Fig S5E). These data support the model that an intact ATG13‐ATG101 subcomplex interacts with ATG9A to promote basal ATG9A function in autophagy.
Our data raise additional questions about the dynamics of interactions between the ATG13‐ATG101 dimer and ATG9A and what additional roles ATG13‐ATG101 plays outside the ULK1 complex. Ktistakis and colleagues found that ATG13 translocates, independently of ULK1/2, to mitophagy structures and that ATG13 also colocalizes with a pool of ATG9A (Karanasios et al, 2016; Zachari et al, 2019). This pool of ATG13 was presumed to mark intact ULK1 complex. However, using the split‐mVenus system, we were able to visualize an ULK1‐independent ATG13‐ATG101 dimer in complex with ATG9A, which often occurred in small vesicles, suggesting that this ATG13‐ATG101 dimer may traffic with ATG9A. In addition, the ATG13‐ATG101‐ATG9A complex accumulated at large clusters of p62/SQSTM1 and ubiquitin (Fig 7). Interestingly, ULK1 also appeared at some of these large clusters, even in cells expressing the ULK1 binding‐defective ATG13, supporting the idea that components of the ULK1 complex arrive independently at pre‐autophagosomal structures (Itakura et al, 2012; Shi et al, 2020). This idea is also supported by an ULK1‐independent interaction between the ATG13‐ATG101 dimer and FIP200 (Fig 7B) and our binary co‐IP data showing that ATG13 does not require FIP200 or ULK1 to interact with ATG9A; and vice versa, ULK1 does not require ATG13 to interact with ATG9A (Fig 2D, Appendix Fig S5C, D and F).
Another intriguing question relates to how ATG9A gets recruited to clusters of ubiquitin and p62/SQSTM1. Perhaps in the absence of ATG13 or ATG101, the accumulation of ubiquitin and/or p62/SQSTM1 sends a persistent, interminable signal to recruit ATG9A. Indeed, the concept of p62/SQSTM1 recruiting active signaling molecules to autophagy structures has been demonstrated previously (Komatsu et al, 2010; Duran et al, 2011; Goodall et al, 2016) and could help explain why the accumulation of p62/SQSTM1, caused by loss of ATG13 or other core autophagy regulators, resulted in an apparent “dead‐end” recruitment of ATG9A to these structures. Alternatively, ATG9A itself may somehow act as a sensor of ubiquitin build‐up. This would be an elegant way for a cell to calibrate its basal autophagic response to whatever level of cellular debris needs recycling (Yamasaki et al, 2020).
Lastly, what additional proteins associate with the ULK1‐independent ATG13‐ATG101 complex in basal autophagy? Gel filtration studies show co‐elution of ATG13 and ATG101 below the 3–4 MDa ULK1 complex, but within a range that suggests other components of the complex and/or higher order oligomerization of ATG13/ATG101 (Hosokawa et al, 2009a; Hosokawa et al, 2009b). ULK1 binding‐deficient mutants of ATG13 and the split‐mVenus ATG13‐ATG101 system will be useful tools to answer this question.
In conclusion, our study uncovers the first BioID‐based interactome for ATG9A, which includes an array of vesicle‐trafficking complexes along the ER‐Golgi‐endosomal axis, and autophagy regulatory proteins that include ATG13 and ATG101. From these data, we discovered an ULK1‐independent role for the ATG13‐ATG101 in regulating ATG9A‐mediated turnover of p62/SQSTM1‐tethered clusters of ubiquitin. These data expand our understanding of basal autophagy and have implications for the development of therapeutic strategies aimed at degenerative diseases in which defective basal autophagy plays a critical role.
Materials and Methods
Cell culture, transfection, and viral transduction
HEK293T cells, HCT‐116 cells, and their derivatives were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, 11965‐092) supplemented with 10% fetal bovine serum (FBS; Genesee Scientific, 25‐514) at 37°C in a 5% CO2 incubator. ULK1/2 knockout Mouse Embryonic Fibroblasts (MEFs), kindly provided by Sharon Tooze, London Research Institute, United Kingdom, were cultured in DMEM supplemented with 10% FBS at 37°C in a 5% CO2 incubator.
HEK293T and HCT‐116 cells were transiently transfected using polyethylenimine (PEI) (Polyscience, Inc.) or transporter 5 (Polysciences, 2600‐8‐5) according to the manufacturer’s protocols. MEF cells were transiently transfected using Lonza Amaxa™ Nucleofector™ kit (VPD1004) with Nucleofector II device (Amaxa Biosystems) Original and manipulated plasmids used for transient transfection listed in plasmids section.
To stably express 3xFLAG‐ATG13 WT, 3xFLAG‐ATG13 Δ2AA, and 3xFLAG‐ATG13 ΔHORMA in ATG13 KO HCT‐116 cell line (HCT‐116 ATG9A‐HA KI ATG13 KO), cDNA constructs were cloned into pLenti‐puro (Addgene, 39481) vector backbone (primer sequences are mentioned below in plasmids section). For virus generation, LentiX‐293T cells were plated to 20% confluency the day before transfection in 15 cm tissue culture dishes. The next day, cells were transfected with pLenti‐puro vector containing ATG13 constructs, viral packaging (psPAX2), viral envelope (pMD2.G) at 4:2:1 DNA ratio with 14 µg total DNA, 600 µl of serum free media, and 42 µl PEI. Supernatant was removed from LentiX‐293T cells after 72 h, centrifuged at 400 g for 5 min and then syringe filtered using a 0.45 µm filter (Millipore). Polybrene was then added to a final concentration of 8 µg/ml, and HCT‐116 ATG9A‐HA KI ATG13 KO cells were infected overnight. Cells were then allowed to recover for 24 h in DMEM/10% FBS before being selected with 2 µg/ml puromycin for 72 h.
HA‐BirA* stably expressing HEK293T cell line was generated by using pLEX‐uORF‐HA‐BirA*‐STOP‐IRES‐Puro plasmid according to the above protocol. To generate HA‐ATG9A‐BirA* stably expressing HEK293T cell line, HA‐ATG9A‐BirA* was cloned to pLenti‐puro plasmid and introduced with viral transduction according to above protocol. ATG9A WT and ΔC were also cloned onto pLenti plasmid for generation of stable expression in ATG9A KO HeLa cells according to the above protocol. Cell lines used for live‐cell imaging were derived from HCT‐116 ATG9A‐HA KI and HCT‐116 ATG9A‐HA KI ATG13 KO cells. EGFP‐p62/SQSTM1‐3xFLAG and HA‐mRuby2‐ATG9A were cloned into pLenti‐puro vector backbone and induced with viral transduction according to the above protocol. Cell lines used for split‐mVenus BiFC confocal images were derived from HCT‐116 ATG9A‐HA KI and HCT‐116 ATG9A‐HA KI ATG13‐ATG101 double KO cells. VN173‐ATG13‐Myc (N‐term mVenus half) and VC155‐ATG101‐3xFLAG (C‐term mVenus half) were cloned into pLenti‐puro vector backbone and induced with viral transduction according to the above protocol. Cells used for BiFC histograms, expression blot, and immunoprecipitation were transiently transfected using the same plasmids with PEI transfection.
Antibodies and chemicals
The following antibodies and chemicals were used. HA‐Tag Mouse monoclonal (microscopy diltion‐1:500, Cell Signaling Technology, 2367S), HA‐Tag Rabbit monoclonal (microscopy dilution‐1:500, Cell Signaling Technology, 3724S), ATG101 Rabbit monoclonal (Cell Signaling Technology, 13492S), FIP200 Rabbit monoclonal (Cell Signaling Technology, 12436S), ATG13 Rabbit monoclonal (Abcam, ab201467), ULK1 Rabbit monoclonal (Abcam, ab128859), ULK1 Rabbit monoclonal (microscopy dilution‐1:500, Cell Signaling Technology, 8054S), ATG9A Rabbit monoclonal (Abcam, ab108338), ATG9A Rabbit monoclonal (Cell Signaling Technology, 13509S), AP4M1 (Abcam, ab96306), AP3B1 (ProteinTech, 13384‐1‐AP), TBC1D5 (ProteinTech, 17078‐1‐AP), FLAG monoclonal M2 (Sigma Aldrich, F1804‐200 UG), ATG16L1 Rabbit monoclonal (microscopy dilution‐1:500, Cell Signaling Technology, 8089S), VSP26A Mouse monoclonal (microscopy dilution‐1:500, Millipore Sigma, AMAB90967), TUFM Mouse monoclonal (microscopy dilution‐1:500, Millipore Sigma, AMAB90964), PDIA3 Mouse monoclonal (microscopy dilution‐1:500, Millipore Sigma, AMAB90988), Golgin97 Mouse monoclonal (microscopy dilution‐1:500, Cell Signaling Technology, 97537S), p62/SQSTM1 Mouse monoclonal (Abcam, ab56416), p62/SQSTM1 Rabbit monoclonal (microscopy dilution‐1:10,000, Abcam, ab109012), LAMP1 Mouse monoclonal (microscopy dilution‐1:50, Developmental Studies Hybridoma Bank, H4A3), WDR45/WIPI4 Rabbit polyclonal (microscopy dilution‐1:250, Abcam, ab240905), LC3B Rabbit monoclonal (microscopy dilution‐1:500, Abcam, ab192890), EEA1 Chicken polyclonal (microscopy dilution‐1:250, Millipore Sigma, GW21443A), Actin (Cell Signaling Technology, 4970S), Streptavidin Alexa Fluor™ 488 Conjugate (Thermo Fisher Scientific, S32354) Biotin (Sigma, B4639‐1G), Pierce™ High Capacity Streptavidin Agarose (Thermo Fisher Scientific, 20357), Pierce™ Anti‐HA Agarose (Thermo Fisher Scientific, 26182), and Bafilomycin A1 (Cayman Chemical Company, 11038).
Plasmids
HA‐ATG9A plasmid was kindly provided by Sharon Tooze, London Research Institute, United Kingdom. HA‐ATG9A‐BirA* plasmid was kindly provided by Dr. Daniel Simmons, Brigham Young University. pLEX‐uORF‐HA‐BirA*‐STOP‐IRES‐Puro plasmid was a gift from Paul Khavari (Addgene Plasmid #120558). HA‐hATG13 plasmid was a gift from Do‐Hyung Kim (Addgene Plasmid #31967). p3xFLAG‐CMV10‐hAtg101 plasmid was a gift from Noboru Mizushima (Addgene Plasmid 22877). HA‐p62 plasmid was a gift from Qing Zhong (Addgene Plasmid 28027). pEGFP‐N1‐FLAG plasmid was a gift from Patrick Calsou (Addgene Plasmid). pLenti‐puro plasmid was a gift from Ie‐Ming Shih (Addgene Plasmid 39481). pCE‐BiFC‐VN173, pCE‐BiFC‐VC155, pBiFC‐VN173, and pBiFC‐VC155 plasmids were a gift from Chang‐Deng Hu (Addgene Plasmids # 22019, 22020, 22010, and 22011, respectively). Viral assembly plasmids were psPAX2 and pMD2.G, a gift from Didier Trono (Addgene Plasmids # 12260 and 12259, respectively).
For generation of ATG9A and ATG13 stable addback plasmids, we used Twist Bioscience to clone into the pLenti‐puro plasmid. We inserted WT, C terminus truncated, and mRuby2 fusion ATG9A into the plasmid between MluI and AgeI restriction sites. ATG13 WT was also inserted between MluI and AgeI. ATG13 WT was then further processed into ATG13 Δ2AA using primers TAAGCGGCCGCTAAGTAAG (forward) and GGTTTCCACAAAGGCATCAAAC (reverse) and into ATG13 ΔHORMA using primers ATTAACTTGGCATTCATGTC (forward) and CTTTCTGTCCTGGGAATTG (reverse).
Mutant ATG9A and ATG13 DNA constructs were created using Q5 reagents and protocol from New England Biolabs (NEB, E0554). Primer sets used for mutagenesis are: ATG9A ΔC TACGTCTATCTAGTCCTTACAATC (forward) and AAGAGGGCATTTTCAGGG (reverse), ATG13 ΔHORMA ATTAACTTGGCATTCATGTC (forward) CTTTCTGTCCTGGGAATTG (reverse), and ATG13 Δ2AA TAAGCGGCCGCTAAGTAAG (forward) GGTTTCCACAAAGGCATCAAAC (reverse). HA‐ATG9‐mVenus constructs were created by insertion of EcoRV and KpnI restriction enzyme cut‐sites onto the HA‐ATG9A plasmid using Q5 mutagenesis and primer set: AAGATATCAGACAAGGCTGAGCAGG (forward) (EcoRV) and GGTACCATACCTTGTGCACCTGAG (reverse) (Kpn1). Both HA‐ATG9A and BiFC‐mVenus plasmids were then digested and ligated using T4 ligase (NEB, M0202) to create HA‐ATG9A‐VC155 and HA‐ATG9A‐VN173. ATG13 and ATG101 mVenus constructs were created by NEB HiFi Assembly reagents and protocol from New England Biolabs (NEB, E2621) combining pLenti ATG13 with pBiFC‐VN173 and pLenti puro with p3xFLAG‐CMV10‐hAtg101 and pBiFC‐VC155, respectively. pLenti EGFP‐p62/SQSTM1‐3xFLAG was created by NEB HiFi Assembly combining HA‐p62, pEGFP‐N1‐FLAG, and pLenti‐puro plasmids.
CRISPR‐Cas9
ATG9A C‐terminal 1xHA‐tagged HCT‐116 cells were generated at the Genome Engineering and iPSC Center at Washington University School of Medicine (St. Louis, MO). The 1xHA tag was introduced using CRISPR‐Cas9 ribonucleoproteins (RNPs) using following guide RNA and a single‐stranded oligonucleotide with 60 bp homology arms.
5′‐TCTCCCCACAGGTATAGACA‐3′
5′gcaggtatcaaccagaagctgaagactatctccattaccaaccctttctccccacaggtaTACCCATACGACGTACCAGATTACGCTtagacaaggctgagcagggttcctgtggcccaggatggaggcc accgctgccctgccatc‐3′.
Targeted deep‐sequencing was used to validate reagents and genotype single cell‐derived clones as previously described (Sentmanat et al, 2018) with following tailed PCR primers.
Fwd 5′‐TCAGGTGCACAAGGTAAGGGCCCCG‐3′.
Rev 5′‐GCCAGGGAACACTCAGAGGAGCCGT‐3′.
Cells were maintained according to ATCC guidelines with McCoy's 5a Modified Medium (Cat. No. 16600108) and 10% fetal bovine serum supplemented with GlutaMax (Gibco, Cat. No. 35050061) and penicillin‐streptomycin (Gibco, Cat. No.15070063).
To generate ATG13 knockout cells, two independent single‐guide RNAs #1: 5′‐GGACAGCTGCCTGCAGTCGGG‐3′, #2: 5′‐ACACGGTGTACAACAGACTG‐3′, were designed against human ATG13 (ENSG00000175224). CRISPR design tools available at www.atum.bio and crispr.mit.edu were used. The gRNAs were cloned into the pSpCas9(BB)‐2A‐Puro (PX459) plasmid. PX459 was a gift from Feng Zhang (Addgene, 48139). Cells expressing the gRNA constructs were separated by serial dilution and monoclonal lines were isolated manually under puromycin selection. Knockout efficiency was measured by Western blotting. ATG9A knockout cells were generated using single‐guide RNA 5′‐CTGTTGGTGCACGTCGCCGAG‐3′ against human ATG9A (ENSG00000198925). ATG101 knockout cells were generated using single‐guide RNA 5′‐ACCTACTCCATTGGCACCGT‐3′ against human ATG101 (ENSG00000123395). FIP200 knockout cells were generated using single‐guide RNA 5′‐CAGGTGCATCTAGAAGACCC‐3′ against human FIP200 (ENSG00000023287). All gRNA’s were designed at crispr.mit.edu or www.atum.bio and cloned/propagated according to the above protocol.
Immunoprecipitation and immunoblotting
To prepare whole‐cell extracts, cells were washed twice and harvested with ice‐cold phosphate‐buffered saline (PBS). Cell pellets were resuspended in RIPA lysis buffer (25 mM Tris–HCl [pH 7.5], 75 mM NaCl, 0.5% [wt/vol] Triton X‐100, 2.5 mM EDTA, 0.05% [wt/vol] SDS, and 0.25% [wt/vol] Deoxycholate) supplemented with protease and phosphatase inhibitors and incubated for 15 min on ice or at 4°C with gentle rotation. Lysates were syringed through a 25‐gauge needle 10 times and centrifuged at 21,000 g for 10 min at 4°C.
For co‐immunoprecipitation, cells were transfected with HA‐ATG9A or HA‐ATG13 for 48 h or HCT‐116 ATG9A‐HA KI cells expressing endogenous ATG9A‐HA were used. Cells were lysed with ATG9A lysis buffer (20 mM Tris–HCl [pH 7.5], 150 mM NaCl, 0.3% [wt/vol] Triton X‐100, and 5 mM EDTA) supplemented with protease and phosphatase inhibitors, and lysates were incubated with anti‐HA–agarose beads for 1h at 4°C with gentle rotation. The beads were then washed once with lysis buffer and three times with cold PBS. The co‐immunoprecipitated proteins were eluted with modified Laemmli sample buffer by boiling at 100°C for 5 min. The proteins were analyzed, followed by immunoblotting using infrared fluorescent secondary antibodies and a Li‐Cor Odyssey imaging system.
For proximity‐dependent biotin ligase assay‐based protein co‐precipitation, cells were transiently transfected with biotin ligase constructs for 48 h or stably expressing biotin ligase constructs, treated with 5 μM Biotin for 12 h, and lysed in ATG9A lysis buffer as mentioned above. Cleared lysates were incubated with streptavidin agarose resin for 1 h at 4°C with gentle rotation. The resin was then washed twice with lysis buffer and three times with cold PBS. The precipitated proteins were eluted with modified Laemmli sample buffer by boiling at 100°C for 5 min. The proteins were analyzed, followed by immunoblotting using infrared fluorescent secondary antibodies and a LI‐COR Odyssey imaging system.
For the quantification of immunoblots, infrared fluorescent signal was normalized to a reference control to obtain fold‐change ratios for all other lanes. The fold‐change differences were then compared using repeated measures (paired) one‐way ANOVA followed by uncorrected Fisher’s LSD tests excluding the reference control (no standard deviation). Significance is indicated by asterisks according to the following scale: nsP > 0.05, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001. Immunoblots with only two samples (reference control and one other) were normalized the same way, and significance was determined using a one‐sample t‐test compared with a hypothetical mean of 1 (reference control fold change). Significance was indicated by the same scale. Graphs and statistics were completed using GraphPad Prism 9 software.
BioID‐coupled Mass spectrometry
HEK293T cells stably expressing HA‐BirA* or HA‐ATG9A‐BirA* cells were lysed in ATG9A lysis buffer and proteins were co‐precipitated as mentioned above. Precipitated proteins by streptavidin resin were eluted with modified Laemmli sample buffer by boiling at 100°C for 5 min. The following steps were performed at Duke Proteomics Core Facility. Samples in loading buffer were supplemented with SDS for a final concentration of 5% for digestion and spiked with undigested casein at a total of either 200 or 400 fmol. Samples were then reduced with 10 mM dithiothreitol for 30 min at 80C and alkylated with 25 mM iodoacetamide for 30 min at room temperature. Next, they were supplemented with a final concentration of 1.2% phosphoric acid and 765 μl of S‐Trap (ProtiFi) binding buffer (90% MeOH/100mM TEAB). Proteins were trapped on the S‐Trap, digested using 20 ng/ul sequencing grade trypsin (Promega) for 1 h at 47C, and eluted using 50 mM TEAB, followed by 0.2% FA, and lastly using 50% ACN/0.2% FA. All samples were then lyophilized to dryness and resuspended in 240 μl 1%TFA/2% acetonitrile containing 12.5 fmol/μl yeast alcohol dehydrogenase (ADH_YEAST). A QC Pool was created by taking 3 μl from each sample, which was run periodically throughout the acquisition period.
Quantitative LC‐MS/MS data analysis
The MS/MS data were searched against the SwissProt homo sapiens database (downloaded in Nov 2019) appended with a contaminant database, and an equal number of reversed‐sequence “decoys” for false discovery rate determination. Mascot Distiller and Mascot Server (v 2.5, Matrix Sciences) were utilized to produce fragment ion spectra and to perform the database searches. Database search parameters included fixed modification on Cys (carbamidomethyl) and variable modifications on Meth (oxidation) and Asn and Gln (deamidation). Peptide Validator and Protein FDR Validator nodes in Proteome Discoverer were used to annotate the data at a maximum 1% protein false discovery rate.
Additional data filtering was accomplished using the following strategy: Missing values were imputed after sample loading and total intensity normalization in the following manner. If a peptide had less than two quantitated values across all of the samples, the entire peptide entry was removed. If less than half of the values are missing in a treatment group, values are imputed with an intensity derived from a normal distribution defined by measured values within the same intensity range (20 bins). If greater than half values are missing for a peptide in a group and a peptide intensity is > 5e6, then it was concluded that peptide was misaligned and its measured intensity is set to 0. All remaining missing values are imputed with the lowest 5% of all detected values. Please note that all subsequent analyses were from these normalized protein levels.
The overall dataset had 38,797 peptide matches. Additionally, 740,677 MS/MS spectra were acquired for peptide sequencing by database searching. Following database searching and peptide scoring using Proteome Discoverer validation, the data were annotated at a 1% protein false discovery rate, resulting in identification of 38,797 peptides and 4,014 proteins. After data filtering and normalization, 38,472 peptides and 3,994 proteins were quantitated (the processed list of interactors is in Dataset EV1).
Statistical analysis of LC‐MS/MS data
BirA* vs HA‐ATG9A‐BirA* were compared for statistical analysis. Fold changes and a two‐tailed heteroscedastic t‐test on log2‐transformed data for each of these comparisons were calculated. Briefly, proteins were filtered to include those with a greater than twofold expression and a P‐value of < 0.05. A total of 283 proteins passed this filter, including ATG9A. For each of these proteins, the log2 values of the normalized proteins were plotted against the ‐log10 of the P‐value. Relevant GO categories were projected onto the significant 283 proteins for illustration purposes.
Confocal microscopy
HCT‐116 and derivative cell lines were used for confocal microscopy colocalization experiments. Cells were seeded onto acid‐etched coverslips and cultured for 36 h before fixation. Cells were fixed for 20 min with 2% paraformaldehyde (PFA) and permeabilized with 0.1% Triton X‐100/PBS for 5 min (HEK293T cells were permeabilized for 15 min to increase immunostaining). Samples were then blocked with 10% FBS/SEA BLOCK Blocking Buffer (Thermo Scientific) and incubated with indicated antibodies (microscopy dilutions listed in antibodies and chemicals section) at 4°C overnight. Cells were washed with 0.1% Tween/PBS (PBS‐T) and incubated with Alexa Fluor‐conjugated secondary antibodies (Abcam ab150105, Thermo Scientific A21206 and A10034, or Millipore Sigma SAB4600238 for Alexa Fluor 488) (Thermo Scientific A10037 or A11036 for Alexa Fluor 568) (Thermo Scientific A21071 or Invitrogen A‐21126 for Alexa Fluor 633) at 1:500 dilution in SEA BLOCK‐blocking buffer. Cells were washed with PBS‐T and counter‐stained with 1.43 µM DAPI for 5 min. Coverslips were then mounted with ProLong Diamond Antifade Mountant (Thermo Scientific P36961). Images were acquired on a LEICA TCS SP8 confocal microscope fitted with a HC PL APO 63×/1.40 Oil CS2 objective and a HyD detection system (Leica Microsystems).
Images taken for mVenus ATG13‐ATG101 and EGFP‐p62 were of cells stably expressing respective fluorescent fusion proteins and processed as described above with the indicated antibodies. mVenus constructs were expressed in an ATG13‐ATG101 double KO background to remove any endogenous dimerization while the p62/SQSTM1 construct was expressed with endogenous p62/SQSTM1 still intact.
ATG9A‐BirA* validation confocal experiments were performed in HEK293T cells stably expressing the ATG9A‐BirA* construct in a WT HEK293T background (endogenous ATG9A still intact). ATG9A‐BirA* was visualized using the HA mouse antibody previously described, p62/SQSTM1 was visualized using endogenous antibody, and biotinylated proteins were visualized using Alexa Fluor 488‐conjugated streptavidin.
Live‐cell microscopy was performed in HCT‐116 WT and ATG13 KO cells stably expressing EGFP‐p62/SQSTM1‐3xFLAG and HA‐mRuby2‐ATG9A. Cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum until seeded onto glass bottom microwell dishes (MatTek P35GC‐1.5‐10‐C) at which time cells were transferred to FluoroBrite DMEM (Fisher Scientific A1896701) supplemented with 10% fetal bovine serum. Cells were allowed to adhere and grow for 48 h prior to imaging. Images were taken every 1.524 s for 100 frames totaling 2.54 min. Frames were zoomed into p62 puncta and cropped for still‐frame images while videos show all frames full‐sized and then repeated zoomed into p62 puncta.
Confocal microscopy data analysis
Samples for each set were seeded, fixed, and stained on the same day with identical antibody concentrations, laser power, magnification, and image resolution by set. Global analysis sets were prepared and analyzed individually, but all parameters were maintained for each WT/KO pair and laser power adjustments were only made to maintain equal fluorescence between pairs.
All images were processed using Huygens Essential express deconvolution tool, and Pearson’s coefficient was calculated using the colocalization analyzer tool in the same software. Threshold intensity values for all sets were determined by multiplying the average intensity for each channel by the same factor to maintain consistency within each set. Puncta surface area was calculated using Leica 3D analysis software with a minimum threshold of 0.5 μm2. All puncta beneath this threshold were omitted in our analysis to remove background signal. Mean and standard error of the mean were calculated for both Pearson’s coefficient and puncta surface area by GraphPad Prism 9 software.
The ATG101 KO set in p62/SQSTM1 analysis was prepared and analyzed separately from other conditions in the same set and was therefore analyzed against a replicate wild‐type rather than being integrated into the same dataset. Antibody concentrations, magnification, and image resolution were maintained between ATG101 KO and the remaining p62/SQSTM1 analysis, but laser power was adjusted to maintain equal ATG9A‐p62/SQSTM1 staining between preparations. All other preparation and analysis protocols remained the same.
Flow cytometry
HCT‐116 cells were transfected with HA‐ATG9A‐mVenus155 and HA‐ATG9A‐mVenus173 or HA‐ATG9A as a control and allowed to incubate 48 h. Cells were harvested with trypsin and fixed with 2% PFA for 20 min. Samples were then washed three times with PBS‐T and resuspended in PBS. Cytometry data were attained on a Beckman Coulter Cytoflex Cytometer using 488 nm laser excitation and detection on 525/40 BP fluorescence channel. Positive and negative fluorescent cells were gated based on negative control fluorescence peak using FlowJo analysis software. Mean intensity of positive and negative populations was determined using all cells within respective gate.
For split‐mVenus flow data, HCT‐116 cells were transfected as previously described with pLenti VN173‐ATG13‐Myc WT/Δ2AA and pLenti VC155‐ATG101‐3xFLAG (overexpression not stably transduced) and incubated for 48 h. Cells were then washed with PBS and harvested with trypsin. Live cells were resuspended in PBS and separated using a sterile cell strainer (Thermo Scientific 22‐363‐548). Cytometry data were attained on a BD FACSAriaTM Fusion Cell Sorter using 488 nm laser excitation and detection on 525/50 FITC channel. Positive and negative fluorescent cells were gated based on negative control fluorescence peak.
Modified pulse chase experiment
HEK293T ATG13 WT and ATG13 KO cells stably expressing HA‐ATG9A‐BirA* were treated with 50uM Biotin for 24 h. Media was replaced with Full DMEM media after 24 h of biotin treatment. Cells were lysed with RIPA lysis buffer (25 mM Tris–HCl [pH 7.5], 75 mM NaCl, 0.5% [wt/vol] Triton X‐100, 2.5 mM EDTA, 0.05% [wt/vol] SDS, and 0.25% [wt/vol] Deoxycholate), and streptavidin pulldown was performed at 0, 6, 12, 18, and 24‐h time points. Streptavidin pulldown and Western blotting were performed as mentioned above. Mean ± SEM, n = 3 (biological replicates). Significance measured using Student’s t‐test (bottom). Significance is indicated by asterisks according to the following scale: ns P > 0.05, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
Author contributions
Experiment planning: ARK, DMP, CMM, and JLA; Experiments: ARK, DMP, ERB, EJS, JCY, and AMT; Unpublished control or preliminary experiments: VKW, LMO, and MTL; Critical reagents: AMT, MTL, LMO, JJL, and DLS; Manuscript writing: JLA, ARK, and DMP; All authors: Reading and approval of the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Movie EV1
Movie EV2
Source Data for Figure 5A,B
Acknowledgements
We thank the Fritz B. Burns Foundation for student fellowships and critical instrumentation. We thank the Duke University School of Medicine for use of the Proteomics and Metabolomics Shared resource and for generous guidance and assistance with proteomics. We thank the Simmons Center for Cancer Research for graduate fellowships to ARK, DMP, and CMM. JLA is supported by a generous gift from the Fritz B. Burns Foundation, an American Cancer Society Research Scholar Grant (133550‐RSG‐19‐006‐01‐CCG), and a National Cancer Institute/National Institutes of Health grant (2R15CA202618‐02).
EMBO reports (2021) 22: e51136.
Data availability
The BioID proteomics data in this study are publicly available in the MassIVE database (Massive.ucsd.edu) with the identifier MSV000087520. The data can be accessed directly at the following link: https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?task=4278c4fab099494d957df374433f4d55
References
- Alers S, Löffler AS, Paasch F, Dieterle AM, Keppeler H, Lauber K, Campbell DG, Fehrenbacher B, Schaller M, Wesselborg Set al (2011) Atg13 and FIP200 act independently of Ulk1 and Ulk2 in autophagy induction. Autophagy 7: 1423–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonucci L, Fagman JB, Kim JY, Todoric J, Gukovsky I, Mackey M, Ellisman MH, Karin M (2015) Basal autophagy maintains pancreatic acinar cell homeostasis and protein synthesis and prevents ER stress. Proc Natl Acad Sci USA 112: E6166–6174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoyagi K, Itakura M, Fukutomi T, Nishiwaki C, Nakamichi Y, Torii S, Makiyama T, Harada A, Ohara‐Imaizumi M (2018) VAMP7 regulates autophagosome formation by supporting Atg9a functions in pancreatic beta‐cells from male mice. Endocrinology 159: 3674–3688 [DOI] [PubMed] [Google Scholar]
- Chan EY, Kir S, Tooze SA (2007) siRNA screening of the kinome identifies ULK1 as a multidomain modulator of autophagy. J Biol Chem 282: 25464–25474 [DOI] [PubMed] [Google Scholar]
- Chan EY, Longatti A, McKnight NC, Tooze SA (2009) Kinase‐inactivated ULK proteins inhibit autophagy via their conserved C‐terminal domains using an Atg13‐independent mechanism. Mol Cell Biol 29: 157–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong H, Lindsten T, Wu J, Lu C, Thompson CB (2011) Ammonia‐induced autophagy is independent of ULK1/ULK2 kinases. Proc Natl Acad Sci USA 108: 11121–11126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong H, Nair U, Geng J, Klionsky DJ (2008) The Atg1 kinase complex is involved in the regulation of protein recruitment to initiate sequestering vesicle formation for nonspecific autophagy in Saccharomyces cerevisiae . Mol Biol Cell 19: 668–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloer EW, Siesser PF, Cousins EM, Goldfarb D, Mowrey DD, Harrison JS, Weir SJ, Dokholyan NV, Major MB (2018) p62‐dependent phase separation of patient‐derived KEAP1 mutations and NRF2. Mol Cell Biol 38: e00644‐17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies AK, Itzhak DN, Edgar JR, Archuleta TL, Hirst J, Jackson LP, Robinson MS, Borner GHH (2018) AP‐4 vesicles contribute to spatial control of autophagy via RUSC‐dependent peripheral delivery of ATG9A. Nat Commun 9: 3958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Pace R, Skirzewski M, Damme M, Mattera R, Mercurio J, Foster AM, Cuitino L, Jarnik M, Hoffmann V, Morris HDet al (2018) Altered distribution of ATG9A and accumulation of axonal aggregates in neurons from a mouse model of AP‐4 deficiency syndrome. PLoS Genet 14: e1007363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikic I, Elazar Z (2018) Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol 19: 349–364 [DOI] [PubMed] [Google Scholar]
- Duran A, Amanchy R, Linares JF, Joshi J, Abu‐Baker S, Porollo A, Hansen M, Moscat J, Diaz‐Meco MT (2011) p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol Cell 44: 134–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan D, Kim J, Shaw RJ, Guan KL (2011) The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy 7: 643–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gammoh N, Florey O, Overholtzer M, Jiang X (2013) Interaction between FIP200 and ATG16L1 distinguishes ULK1 complex‐dependent and ‐independent autophagy. Nat Struct Mol Biol 20: 144–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganley IG, du Lam H, Wang J, Ding X, Chen S, Jiang X (2009) ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem 284: 12297–12305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodall ML, Fitzwalter BE, Zahedi S, Wu M, Rodriguez D, Mulcahy‐Levy JM, Green DR, Morgan M, Cramer SD, Thorburn A (2016) The autophagy machinery controls cell death switching between apoptosis and necroptosis. Dev Cell 37: 337–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin JM, Dowdle WE, DeJesus R, Wang Z, Bergman P, Kobylarz M, Lindeman A, Xavier RJ, McAllister G, Nyfeler Bet al (2017) Autophagy‐independent lysosomal targeting regulated by ULK1/2‐FIP200 and ATG9. Cell Rep 20: 2341–2356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Chang C, Huang R, Liu B, Bao L, Liu W (2012) AP1 is essential for generation of autophagosomes from the trans‐Golgi network. J Cell Sci 125: 1706–1715 [DOI] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki‐Migishima R, Yokoyama M, Mishima K, Saito I, Okano Het al (2006) Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441: 885–889 [DOI] [PubMed] [Google Scholar]
- Hara T, Takamura A, Kishi C, Iemura S, Natsume T, Guan JL, Mizushima N (2008) FIP200, a ULK‐interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol 181: 497–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C, Baba M, Cao Y, Klionsky DJ (2008) Self‐interaction is critical for Atg9 transport and function at the phagophore assembly site during autophagy. Mol Biol Cell 19: 5506–5516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hieke N, Löffler AS, Kaizuka T, Berleth N, Böhler P, Drießen S, Stuhldreier F, Friesen O, Assani K, Schmitz Ket al (2015) Expression of a ULK1/2 binding‐deficient ATG13 variant can partially restore autophagic activity in ATG13‐deficient cells. Autophagy 11: 1471–1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S‐I, Natsume T, Takehana K, Yamada Net al (2009a) Nutrient‐dependent mTORC1 association with the ULK1‐Atg13‐FIP200 complex required for autophagy. Mol Biol Cell 20: 1981–1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa N, Sasaki T, Iemura S, Natsume T, Hara T, Mizushima N (2009b) Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy 5: 973–979 [DOI] [PubMed] [Google Scholar]
- Imai K, Hao F, Fujita N, Tsuji Y, Oe Y, Araki Y, Hamasaki M, Noda T, Yoshimori T (2016) Atg9A trafficking through the recycling endosomes is required for autophagosome formation. J Cell Sci 129: 3781–3791 [DOI] [PubMed] [Google Scholar]
- Itakura E, Kishi‐Itakura C, Koyama‐Honda I, Mizushima N (2012) Structures containing Atg9A and the ULK1 complex independently target depolarized mitochondria at initial stages of Parkin‐mediated mitophagy. J Cell Sci 125: 1488–1499 [DOI] [PubMed] [Google Scholar]
- Jakobi AJ, Huber ST, Mortensen SA, Schultz SW, Palara A, Kuhm T, Shrestha BK, Lamark T, Hagen WJH, Wilmanns Met al (2020) Structural basis of p62/SQSTM1 helical filaments and their role in cellular cargo uptake. Nat Commun 11: 440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jao CC, Ragusa MJ, Stanley RE, Hurley JH (2013) A HORMA domain in Atg13 mediates PI 3‐kinase recruitment in autophagy. Proc Natl Acad Sci USA 110: 5486–5491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judith D, Jefferies HBJ, Boeing S, Frith D, Snijders AP, Tooze SA (2019) ATG9A shapes the forming autophagosome through Arfaptin 2 and phosphatidylinositol 4‐kinase IIIbeta. J Cell Biol 218: 1634–1652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, Kim DH (2009) ULK‐Atg13‐FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 20: 1992–2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaizuka T, Mizushima N (2016) Atg13 is essential for autophagy and cardiac development in mice. Mol Cell Biol 36: 585–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakuta S, Yamaguchi J, Suzuki C, Sasaki M, Kazuno S, Uchiyama Y (2017) Small GTPase Rab1B is associated with ATG9A vesicles and regulates autophagosome formation. FASEB J 31: 3757–3773 [DOI] [PubMed] [Google Scholar]
- Karanasios E, Walker SA, Okkenhaug H, Manifava M, Hummel E, Zimmermann H, Ahmed Q, Domart MC, Collinson L, Ktistakis NT (2016) Autophagy initiation by ULK complex assembly on ER tubulovesicular regions marked by ATG9 vesicles. Nat Commun 7: 12420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamata T, Kamada Y, Kabeya Y, Sekito T, Ohsumi Y (2008) Organization of the pre‐autophagosomal structure responsible for autophagosome formation. Mol Biol Cell 19: 2039–2050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, Guan KL (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13: 132–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishi‐Itakura C, Koyama‐Honda I, Itakura E, Mizushima N (2014) Ultrastructural analysis of autophagosome organization using mammalian autophagy‐deficient cells. J Cell Sci 127: 4089–4102 [DOI] [PubMed] [Google Scholar]
- Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou Y‐S, Ueno I, Sakamoto A, Tong KIet al (2010) The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol 12: 213–223 [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Yet al (2005) Impairment of starvation‐induced and constitutive autophagy in Atg7‐deficient mice. J Cell Biol 169: 425–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J‐I, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami Eet al (2006) Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441: 880–884 [DOI] [PubMed] [Google Scholar]
- Ktistakis NT, Tooze SA (2016) Digesting the expanding mechanisms of autophagy. Trends Cell Biol 26: 624–635 [DOI] [PubMed] [Google Scholar]
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N (2004) The role of autophagy during the early neonatal starvation period. Nature 432: 1032–1036 [DOI] [PubMed] [Google Scholar]
- Kundu M, Lindsten T, Yang CY, Wu J, Zhao F, Zhang J, Selak MA, Ney PA, Thompson CB (2008) Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood 112: 1493–1502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai LTF, Yu C, Wong JSK, Lo HS, Benlekbir S, Jiang L, Lau WCY (2020) Subnanometer resolution cryo‐EM structure of Arabidopsis thaliana ATG9. Autophagy 16: 575–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb CA, Nuhlen S, Judith D, Frith D, Snijders AP, Behrends C, Tooze SA (2016) TBC1D14 regulates autophagy via the TRAPP complex and ATG9 traffic. EMBO J 35: 281–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EJ, Tournier C (2011) The requirement of uncoordinated 51‐like kinase 1 (ULK1) and ULK2 in the regulation of autophagy. Autophagy 7: 689–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JW, Park S, Takahashi Y, Wang HG (2010) The association of AMPK with ULK1 regulates autophagy. PLoS One 5: e15394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MG, Schoneberg J, Davies CW, Ren X, Hurley JH (2018) The dynamic Atg13‐free conformation of the Atg1 EAT domain is required for phagophore expansion. Mol Biol Cell 29: 1228–1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mari M, Griffith J, Rieter E, Krishnappa L, Klionsky DJ, Reggiori F (2010) An Atg9‐containing compartment that functions in the early steps of autophagosome biogenesis. J Cell Biol 190: 1005–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattera R, Park SY, De Pace R, Guardia CM, Bonifacino JS (2017) AP‐4 mediates export of ATG9A from the trans‐Golgi network to promote autophagosome formation. Proc Natl Acad Sci USA 114: E10697–E10706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura T, Tamura N, Kono N, Shimanaka Y, Arai H, Yamamoto H, Mizushima N (2017) Autophagosome formation is initiated at phosphatidylinositol synthase‐enriched ER subdomains. EMBO J 36: 1719–1735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsi A, Razi M, Dooley HC, Robinson D, Weston AE, Collinson LM, Tooze SA (2012) Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol Biol Cell 23: 1860–1873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T (2007) p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 282: 24131–24145 [DOI] [PubMed] [Google Scholar]
- Popovic D, Dikic I (2014) TBC1D5 and the AP2 complex regulate ATG9 trafficking and initiation of autophagy. EMBO Rep 15: 392–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi S, Kim DJ, Stjepanovic G, Hurley JH (2015) Structure of the human Atg13‐Atg101 HORMA heterodimer: an interaction hub within the ULK1 complex. Structure 23: 1848–1857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees JS, Li XW, Perrett S, Lilley KS, Jackson AP (2015) Protein neighbors and proximity proteomics. Mol Cell Proteomics 14: 2848–2856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux KJ, Kim DI, Raida M, Burke B (2012) A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J Cell Biol 196: 801–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh T, Fujita N, Hayashi T, Takahara K, Satoh T, Lee H, Matsunaga K, Kageyama S, Omori H, Noda Tet al (2009) Atg9a controls dsDNA‐driven dynamic translocation of STING and the innate immune response. Proc Natl Acad Sci USA 106: 20842–20846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seibenhener ML, Babu JR, Geetha T, Wong HC, Krishna NR, Wooten MW (2004) Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol Cell Biol 24: 8055–8068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sentmanat MF, Peters ST, Florian CP, Connelly JP, Pruett‐Miller S (2018) A survey of validation strategies for CRISPR‐Cas9 editing. Sci Rep 8: 888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang L, Chen S, Du F, Li S, Zhao L, Wang X (2011) Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc Natl Acad Sci USA 108: 4788–4793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Yokom AL, Wang C, Young LN, Youle RJ, Hurley JH (2020) ULK complex organization in autophagy by a C‐shaped FIP200 N‐terminal domain dimer. J Cell Biol 219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soreng K, Munson MJ, Lamb CA, Bjorndal GT, Pankiv S, Carlsson SR, Tooze SA, Simonsen A (2018) SNX18 regulates ATG9A trafficking from recycling endosomes by recruiting Dynamin‐2. EMBO Rep 19: e44837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staudt C, Gilis F, Tevel V, Jadot M, Boonen M (2016) A conserved glycine residue in the C‐terminal region of human ATG9A is required for its transport from the endoplasmic reticulum to the Golgi apparatus. Biochem Biophys Res Commun 479: 404–409 [DOI] [PubMed] [Google Scholar]
- Sun D, Wu R, Zheng J, Li P, Yu L (2018) Polyubiquitin chain‐induced p62 phase separation drives autophagic cargo segregation. Cell Res 28: 405–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki SW, Yamamoto H, Oikawa Y, Kondo‐Kakuta C, Kimura Y, Hirano H, Ohsumi Y (2015) Atg13 HORMA domain recruits Atg9 vesicles during autophagosome formation. Proc Natl Acad Sci USA 112: 3350–3355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, Meyerkord CL, Hori T, Runkle K, Fox TE, Kester M, Loughran TP, Wang HG (2011) Bif‐1 regulates Atg9 trafficking by mediating the fission of Golgi membranes during autophagy. Autophagy 7: 61–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, Tsotakos N, Liu Y, Young MM, Serfass J, Tang Z, Abraham T, Wang HG (2016) The Bif‐1‐Dynamin 2 membrane fission machinery regulates Atg9‐containing vesicle generation at the Rab11‐positive reservoirs. Oncotarget 7: 20855–20868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang HW, Wang YB, Wang SL, Wu MH, Lin SY, Chen GC (2011) Atg1‐mediated myosin II activation regulates autophagosome formation during starvation‐induced autophagy. EMBO J 30: 636–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallot‐Hieke N, Verma N, Schlütermann D, Berleth N, Deitersen J, Böhler P, Stuhldreier F, Wu W, Seggewiß S, Peter Cet al (2018) Systematic analysis of ATG13 domain requirements for autophagy induction. Autophagy 14: 743–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weerasekara VK, Panek DJ, Broadbent DG, Mortenson JB, Mathis AD, Logan GN, Prince JT, Thomson DM, Thompson JW, Andersen JL (2014) Metabolic stress‐induced rearrangement of the 14‐3‐3zeta interactome promotes autophagy via a ULK1‐ and AMPK‐regulated 14‐3‐3zeta interaction with phosphorylated Atg9A. Mol Cell Biol 34: 4379–4388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winslow AR, Chen CW, Corrochano S, Acevedo‐Arozena A, Gordon DE, Peden AA, Lichtenberg M, Menzies FM, Ravikumar B, Imarisio Set al (2010) alpha‐Synuclein impairs macroautophagy: implications for Parkinson's disease. J Cell Biol 190: 1023–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi J, Suzuki C, Nanao T, Kakuta S, Ozawa K, Tanida I, Saitoh T, Sunabori T, Komatsu M, Tanaka Ket al (2018) Atg9a deficiency causes axon‐specific lesions including neuronal circuit dysgenesis. Autophagy 14: 764–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H, Fujioka Y, Suzuki S, Noshiro D, Suzuki H, Kondo‐Kakuta C, Kimura Y, Hirano H, Ando T, Noda Net al (2016) The intrinsically disordered protein Atg13 mediates supramolecular assembly of autophagy initiation complexes. Dev Cell 38: 86–99 [DOI] [PubMed] [Google Scholar]
- Yamamoto H, Kakuta S, Watanabe TM, Kitamura A, Sekito T, Kondo‐Kakuta C, Ichikawa R, Kinjo M, Ohsumi Y (2012) Atg9 vesicles are an important membrane source during early steps of autophagosome formation. J Cell Biol 198: 219–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki A, Alam JM, Noshiro D, Hirata E, Fujioka Y, Suzuki K, Ohsumi Y, Noda NN (2020) Liquidity is a critical determinant for selective autophagy of protein condensates. Mol Cell 77: 1163–1175 e1169 [DOI] [PubMed] [Google Scholar]
- Young AR, Chan EY, Hu XW, Kochl R, Crawshaw SG, High S, Hailey DW, Lippincott‐Schwartz J, Tooze SA (2006) Starvation and ULK1‐dependent cycling of mammalian Atg9 between the TGN and endosomes. J Cell Sci 119: 3888–3900 [DOI] [PubMed] [Google Scholar]
- Zachari M, Ganley IG (2017) The mammalian ULK1 complex and autophagy initiation. Essays Biochem 61: 585–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachari M, Gudmundsson SR, Li Z, Manifava M, Cugliandolo F, Shah R, Smith M, Stronge J, Karanasios E, Piunti Cet al (2019) Selective autophagy of mitochondria on a ubiquitin‐endoplasmic‐reticulum platform. Dev Cell 50: 627–643.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C, Ma K, Gao R, Mu C, Chen L, Liu Q, Luo Q, Feng D, Zhu Y, Chen Q (2017) Regulation of mATG9 trafficking by Src‐ and ULK1‐mediated phosphorylation in basal and starvation‐induced autophagy. Cell Res 27: 184–201 [DOI] [PMC free article] [PubMed] [Google Scholar]