

Abstract
Obesity results from an imbalance between caloric intake and energy expenditure, and it is highly associated with colorectal carcinogenesis and therapeutic resistance in patients with colorectal cancer (CRC). Dysregulation of adipokine production in obesity has been reported to cause malignant behaviors in CRC. Leptin, which is the principal hormone secreted by adipocytes and an obesity-associated adipokine, is significantly overexpressed in CRC tissues. However, the effect of leptin on chemoresistance in CRC is unclear. Therefore, the aim of this study was to clarify the role of leptin and the underlying mechanisms in mediating 5-fluorouracil (5-FU) resistance in CRC. We used palmitate to artificially generate obese adipocytes. As expected, lipid accumulation was significantly increased in obese adipocytes. We demonstrated that CRC cells incubated with conditioned media (CM) harvested from obese adipocytes were associated with increased resistance to 5-FU. Notably, this increase in resistance to 5-FU was through the elevated production and secretion of leptin. Leptin could further stimulate the expression of AXL and activate its downstream signaling molecule, PLCγ, thereby resulting in an increased expression of p-glycoprotein (P-gp) in CRC cells. Mechanistically, leptin induced AXL expression via the inhibition of AMPK and subsequent increase in YAP activation and nuclear translocation. In addition, nuclear YAP interacted with TEAD and promoted the occupancy of TEAD on the AXL promoter, thereby stimulating AXL promoter activity after leptin treatment. Furthermore, leptin neutralization rescued the sensitivity of CRC tumors to 5-FU in mice fed on a high-fat diet (HFD). These results indicated that leptin mediated 5-FU resistance through YAP-dependent AXL overexpression in CRC.
Keywords: Chemoresistance, colorectal cancer, obesity, oncogene, receptor tyrosine kinase
Introduction
Colorectal cancer (CRC) is one of the most common gastrointestinal malignant cancers worldwide. Epidemiological investigations have demonstrated a strong positive association between obesity and the incidence and mortality of CRC [1,2]. For example, obese individuals have been reported to have a 20-40% increase in the risk of CRC compared to normal-weight individuals, with an increase in the risk by 2-3% per body mass index (BMI) unit [1]. In addition, a 30-year follow-up study found that BMI was a crucial risk factor for long-term CRC mortality, and that overweight and obese patients had higher rates of mortality compared to those with normal weight [2]. Previous studies have also demonstrated that obesity can decrease the efficacy of anti-cancer drugs, and therefore result in poorer prognosis and higher recurrence rates in CRC patients [3,4]. Moreover, a high BMI has been associated with shorter progression-free survival (PFS) and overall survival (OS) among CRC patients after receiving fluoropyrimidine-based combination therapy [5].
Obesity is caused by an imbalance between caloric intake and energy expenditure, thereby resulting in excessive accumulation of nutrients in adipocytes [6]. The excess nutrients are converted to triglycerides (TGs) in the adipocytes causing them to become enlarged (hypertrophy), which may be the pathological lesions of obesity [7]. In vitro studies have demonstrated that adipocytes can enhance the tumorigenic properties of CRC cells, including proliferation, migration and invasion [8,9]. Furthermore, diet-induced obesity (DIO) has been shown to lead to increased tumor growth in mice subcutaneously injected with CRC cells and azoxymethane (AOM)-induced CRC formation [10,11]. The chemically induced CRC tumorigenesis could be then reduced by caloric restriction [11]. These observations suggest the tumor-promoting effect of obesity on CRC. However, preclinical evidence and underlying mechanisms regarding the effect of obesity on drug resistance in CRC have yet to be elucidated.
Previous study has reported associations between an increased risk of CRC malignancy and increasing amounts of biologically active substances that secreted by hypertrophic adipocytes, such as leptin [12]. Leptin is the principal hormone secreted by adipocytes and has been positively correlated with increased adiposity during obesity development [13]. It was originally regarded to be a neurohormone controlling energy homeostasis and feeding behavior [14]. However, an increasing number of studies have also shown that high levels leptin in obesity are involved in increasing cell proliferation, angiogenesis and anti-apoptosis in CRC [15]. Clinically, previous studies have reported that circulating serum levels of leptin are remarkably higher in CRC patients with obesity, and that this could serve as an indicator of a poor prognosis [16]. However, little is known about the involvement of leptin and its underlying mechanisms in drug resistance of CRC cells.
The aim of this study was to evaluate the effect of obesity-associated adipokines, and especially leptin, on the sensitivity of CRC cells to 5-fluorouracil (5-FU), which is the standard first-line treatment for CRC. Our results demonstrated that obese adipocyte-secreted leptin increased resistance to 5-FU by inducing the expression of AXL in CRC cells.
Materials and methods
Cell lines and cell culture
H3347 and HCT116 cells (both kindly provided by Dr. Chen, Joanne Jeou-Yuan, Institute of Biomedical Sciences, Academia Sinica, Taipei, Taiwan) were cultured in RPMI medium (GIBCO, Eggenstein, Germany) containing 10% fetal bovine serum (FBS) (GIBCO, Eggenstein, Germany) and 1% Penicillin-Streptomycin-Amphotericin B (PSA) (Invitrogen Life Technologies, Waltham, MA, USA). MC-38 cells from Dr. Mi-Hua Tao, Institute of Biomedical Sciences, Academia Sinica, Taipei, Taiwan, were cultured in DMEM medium (GIBCO, Eggenstein, Germany) containing 10% FBS (GIBCO, Eggenstein, Germany) and 1% PSA (Invitrogen Life Technologies, Waltham, MA, USA).
Adipogenic differentiation
Simpson-Golabi-Behmel syndrome (SGBS) human pre-adipocyte cells were kindly provided by Dr. Martin Wabitsch from Ulm University Medical Center, Ulm, Germany. SGBS pre-adipocytes were propagated and differentiated to mature adipocytes as previously described [17]. Mature adipocytes were used for the following experiments at 21 days after induction of adipogenic differentiation.
Drugs and antibodies
5-FU and 5-aminoimidazole carboxamide ribonucleotide (AICAR) were purchased from Sigma-Aldrich, St. Louis, MO, USA. Leptin recombinant protein was obtained from PeproTech Inc., Rocky Hill, NJ, USA. The primary antibodies were as follows, anti-AXL (MyBioSourse, San Diego, CA, USA), anti-phospho-PLCγ (Tyr783) (Cell Signaling, Beverly, MA, USA), anti-PLCγ (Cell Signaling, Beverly, MA, USA), anti-phospho-AMPK (Thr172) (Cell Signaling, Beverly, MA, USA), anti-AMPK (Abcam, Cambridge, MA, USA), anti-phospho-LATS1 (Ser909), anti-LATS1 (Cell Signaling, Beverly, MA, USA), anti-phospho-YAP (Ser127) (Cell Signaling, Beverly, MA, USA), anti-YAP (Cell Signaling, Beverly, MA, USA) and anti-TEAD (Cell Signaling, Beverly, MA, USA). The HRP-conjugated secondary antibodies were obtained from Jackson ImmunoResearch Laboratories Inc., West Grove, PA, USA.
Palmitate preparation and conditioned media collection
Palmitate (Sigma-Aldrich, St. Louis, MO, USA) was used as an inducer to artificially generate hypertrophic mature SGBS adipocytes as previously described [18,19]. Briefly, palmitate was prepared in NaOH by heating to 80°C to obtain 100 mM stock solution. After complete dissolution of palmitate, the palmitate stock solution was then added to 5% bovine serum albumin (BSA) (Sigma-Aldrich, St. Louis, MO, USA) in serum-free DMEM/F12 medium to generate 5 mM palmitate working solution. Following fresh preparation, the SGBS mature adipocytes were treated with 0.6 mM palmitate for 24 hours. Cells incubated with DMEM/F12 medium containing 5% BSA served as controls. After treatment, the medium was replaced with serum-free medium. The conditioned media (CM) were harvested, centrifuged to remove cell debris and stored at -80°C before use.
Cell cytotoxicity assay
Cell viability was determined using MTT assay. H3347 and HCT116 cells were seeded at 3×103 cells per well in a 96-well culture plate and incubated overnight. The next day, the cells were treated with either DMSO, which served as the control, or IC50 of 5-FU for 48 hours. The IC50 values for H3347 and HCT116 cells were 14 μM and 4 μM, respectively (data not shown). After treatment, the cells were incubated with MTT solution for 2 hours at 37°C. The newly formed purple crystals were dissolved in DMSO, and absorbance was measured at 570 nm using an ELISA plate reader.
Triglyceride assay
The intracellular and serum contents of TGs were determined using a TG colorimetric assay kit (Cayman Chemical, Ann Arbor, MI, USA) according to the manufacturer’s instructions. Briefly, the cells were rinsed with 1× PBS and then harvested with the provided 1× standard diluent assay reagent. The samples were then sonicated at 4°C using a sonicator (Qsonica Q700, Newtown, CT, USA). Before assaying, the samples from SGBS cells and mice serum were further diluted with 1× standard diluent assay reagent. Ten microliters of the diluted sample was then mixed with 150 μl of the enzyme solution supplied and incubated for 15 minutes at room temperature. The absorbance at 540 nm was measured using an ELISA plate reader.
RNA interference
Short interference RNAs (siRNAs) were designed and purchased from Dharmacon (Lafayette, CO, USA). The sequences of siRNA used for targeting AXL and YAP were 5’-GGAACUGCAUGCUGAAUGA-3’ and 5’-GGUCAGAGAUACUUCUUAA-3’, respectively. The RNAi interference processes were performed using DharmaFECT 1 transfection reagent (Dharmacon, Lafayette, CO, USA) according to manufacturer’s protocols. The sequence of non-targeting siRNA, which served as the negative control, was 5’-UAGCGACUAAACACAUCAA-3’. H3347 and HCT116 cells were seeded at a density of 2×105 in 6-well culture plates and incubated overnight. Subsequently, 25 nM of siRNA was transfected into the CRC cells for 24 hours. The transfected cells were then used in the following experiments.
RNA extraction and quantitative reverse transcription polymerase chain reaction (RT-PCR)
Total RNA was extracted, purified, and converted to cDNA using a high-capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA, USA). In the PCR step, 10 μM each of sense and antisense primers were used. The primer sequences are shown in Table 1. SYBR Green I Master mix (Bioline Inc., Boston, MA, USA) and a Roche LightCycler 480 RT-PCR system (Roche, Basel, Switzerland) were used for quantitative RT-PCR.
Table 1.
The primer sequences used for quantitative RT-PCR
| Gene | Primer | Sequences |
|---|---|---|
| Leptin | Forward | TTTGGCCCTATCTTTTCTATGTCC |
| Reverse | TGGAGGAGACTG ACTGCGTG | |
| AXL | Forward | ATCAGCTTCGGCTAGGCAG |
| Reverse | TCCGCGTAGCACTAATGTTCT | |
| YAP | Forward | TGAACA AACGTCCAGCAAGATAC |
| Reverse | CAGCCCCCAAAATGAACAGTAG | |
| P-gp | Forward | GCTGTCAAGGAAGCCAATGCCT |
| Reverse | TGCAATGGCGATCCTCTGCTTC | |
| GAPDH | Forward | CCACATCGCTCAGACACCAT |
| Reverse | TGACCAGGCGCCCAATA |
Enzyme-linked immunosorbent assay
Enzyme-linked immunosorbent assay (ELISA) was carried out on supernatants of SGBS adipocytes and mice serum using kits for human leptin (Invitrogen Life Technologies, Waltham, MA, USA) and mouse leptin (R&D Systems, Minneapolis, MN, USA), respectively, according to the manufacturer’s instructions.
Oil Red O staining
Lipid accumulation in SGBS cells was examined using Oil Red O staining via both visualization under a microscopy and quantification using an ELISA plate reader. Oil Red O stock solution (Sigma-Aldrich, St. Louis, MO, USA) was prepared in isopropanol. Cells were cultured in 6-well plates and fixed with 10% formalin for 1 hour. After washing with 60% isopropanol, the cells were stained for 10 minutes in Oil Red O solution freshly diluted with distilled water. The plates were visualized using a microscope and photographed using a camera. For quantitative analysis, Oil Red O was eluted with isopropanol. The absorbance of eluted Oil Red O was measured at 500 nm using an ELISA plate reader and isopropanol as a blank.
Co-immunoprecipitation
Protein extracts were incubated with anti-YAP or anti-TEAD antibodies at 4°C overnight. Anti-rabbit IgG was used as the negative control. Subsequently, protein G magnetic beads (GE Healthcare, Piscataway, NJ, USA) were added into each tube and reacted with rotation at 4°C for an additional 4 hours. The immuoprecipitated proteins were harvested by adding 20 μl of 1× sample dye followed by heated at 100°C for 10 minutes. The proteins were finally subjected to Western blot analysis with anti-YAP and anti-TEAD antibodies.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) was performed using a Pierce Magnetic ChIP kit (Thermo Fisher, Pittsburgh, PA, USA) according to the manufacturer’s protocols. Briefly, after the indicated treatments, H3347 and HCT116 cells were fixed with 1% formaldehyde at room temperature for 10 minutes followed by the addition of glycine to quench formaldehyde and stop crosslinking reactions. The nuclear extracts were isolated and sonicated at 4°C using a sonicator (Qsonica Q700, Newtown, CT, USA) to generate small DNA fragments. The sonicated chromatin solution was immunoprecipitated with anti-TEAD antibodies or anti-rabbit IgG with rotation at 4°C overnight. Anti-rabbit IgG was used as the negative control. The immunoprecipitated complexes were then captured using the supplied ChIP grade protein A/G magnetic beads. Chromatin was eluted with IP elution buffer, and the crosslinks were reversed using IP elution buffer containing 6 μl of 5 M NaCl and 2 μl of 20 mg/ml Proteinase K. The purified chromatin was used as a template for PCR amplification with designed primers, and the PCR products were detected using DNA agarose gel electrophoresis. The primer sequences were as follows: AXL1-F: 5’-CCTCCACGTGCCAAACACACAG-3’, AXL1-R: 5’-GAACGCTTGAGTCCAAGAGATCG-3’; AXL2-F: 5’-GTGTGTCCTTGTCCGAGGAG-3’, AXL2-R: 5’-GTGTCTCTCTATCCGGGCTCTG-3’.
Luciferase assay
The AXL promoter region containing four putative TEAD binding sites was amplified by PCR with the designed primers, F: 5’-GGACTAGTGAAGGCAGGATCAGACG-3’, R: 5’-CGCGGATCCGGGTGCCAAACTTTC-3’. The PCR products and pMCS-Cypridina Luc Vector (Thermo Fisher, Pittsburgh, PA, USA) were digested with SpeI and BamH1 restriction enzymes (New England Biolabs, Beverly, MA, USA) followed by ligation using T4 DNA ligase (Thermo Fisher, Pittsburgh, PA, USA). H3347 and HCT116 cells were co-transfected with pAXL-cypridina luc plasmid and internal control pTK-red firefly luc plasmid (Thermo Fisher, Pittsburgh, PA, USA) using polyjet transfection reagent (SignaGen Laboratories, Ijamsville, MD, USA) and then subjected to 100 ng/ml leptin recombinant protein with/without AICAR treatment. After treatment, the relative luciferase activity was measured using a dual luciferase assay system (Thermo Fisher, Pittsburgh, PA, USA).
Multidrug resistance assay
The activity of P-gp was evaluated using a multidrug resistance assay kit (Cayman Chemical, Ann Arbor, MI, USA) according to the manufacturer’s instructions. Briefly, after the indicated treatments, cells were incubated with calcein AM solution for 30 minutes at 37°C. The calcein AM used in this study was a substrate of P-gp, and it could be used to detect the activity of P-gp by measuring the cellular fluorescence retention. The cellular fluorescence intensity was measured using a fluorescent plate reader with excitation and emission wavelengths of 485 nm and 535 nm, respectively.
Immunohistochemistry
Immunohistochemical staining was performed using a Novolink polymer detection system (Leica Biosystems Newcastle Ltd., Newcastle, UK). In brief, formalin-fixed, paraffin-embedded tumor sections were deparaffinized in xylene and rehydrated through graded ethanol. Epitope retrieval was carried out by boiling for 30 minutes in Tris-EDTA buffer (pH 9.0) containing 10 mM Tris-base, 1 mM EDTA solution and 0.05% Tween-20. Endogenous peroxidase in tumor sections was neutralized by incubation with the provided peroxidase block at room temperature for 5 minutes. Non-specific binding to the tumor tissues was prevented by incubation with the supplied protein block at room temperature for an additional 5 minutes. The slides were incubated with primary antibodies against phospho-YAP (Ser127), AXL and P-gp at 4°C overnight followed by reaction with horseradish peroxidase (HRP)-polymer conjugates. Finally, the immunoreactivity was detected using 3,3’-diaminobenzidine (DAB) working solution and counterstaining with hematoxylin.
Western blot analysis
Whole cell lysates were prepared with lysis buffer containing RIPA buffer, protease inhibitor (Biological industries, Cromwell, CT, USA) and phosphatase inhibitor (Biological industries, Cromwell, CT, USA) for 40 minutes. Cytoplasmic and nuclear protein fractions were isolated using NE-PER nuclear and cytoplasmic extraction reagents (Thermo Fisher, Pittsburgh, PA, USA) according to the manufacturer’s instructions. A BCA assay kit (Thermo Fisher, Pittsburgh, PA, USA) was applied to quantify the protein concentration. The proteins were electrophoretically separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and then transferred onto polyvinylidenedifluoride membranes (PVDF membranes) (Merck Millipore, Darmstadt, Germany). After blocking with 5% non-fat milk, the membranes were probed with the indicated primary antibodies overnight at 4°C. Subsequently, the membranes were washed and incubated with HRP-conjugated secondary antibodies for 1 hour at room temperature. Bands were visualized using an ECL chemiluminescent substrate reagent kit (Merck Millipore, Darmstadt, Germany) and detected using a blot imager (UVP Inc., San Gabriel, CA, USA).
Animal studies
The animal experiments were performed following the guidelines established by Institutional Animal Care and Use Committee at National Defense Medical Center (NDMC). C57BL/6J mice were housed under 12/12 h light/dark cycles with free access to food and water. High-fat diet (HFD; 60% fat) and low-fat diet (LFD; 10% fat) (Research Diets, Inc., New Brunswick, NJ, USA) were used to generate obese and lean mice, respectively, as previously described [20]. The mice were fed on a HFD or LFD from 5 weeks of age until the end of the experiments. Food intake and body weight were recorded. Serum leptin and TG levels were detected by ELISA (R&D Systems, Minneapolis, MN, USA) and a TG colorimetric assay kit (Cayman Chemical, Ann Arbor, MI, USA), respectively. MC-38 cells (1×106) were subcutaneously implanted into the right flank of mice 10 weeks after initiating the diet, and treatment started when the tumor burden reached about 100 mm3. 5-FU (25 mg/kg) was injected intraperitoneally daily, and leptin neutralizing antibodies (50 μg per mouse) (R&D Systems, Minneapolis, MN, USA) were injected intraperitoneally every 3 days. Goat IgG (R&D Systems, Minneapolis, MN, USA) was used as a control. During the course of treatment, tumor volume was recorded using Vernier calipers and calculated using the formula: V = (width2 × length)/2. At the end of the treatment, the mice were sacrificed, and the excised tumors were weighted.
Statistical analysis
Statistical comparisons between two groups or among multiple groups were determined using the Student’s t test or one-way ANOVA followed by Bonferroni’s post hoc test, respectively. SigmaPlot (Systat Software Inc., San Jose, CA, USA) was used for statistical calculations and graphic representations. A P value less than 0.05 was considered to indicate a significant difference. All data were expressed as the mean ± SEM.
Results
Obese adipocytes led to 5-FU resistance in CRC cells
Hypertrophic adipocytes with dysregulated secretion of adipokines in obesity have been reported to promote CRC malignant behaviors [21]. In order to evaluate the effect of adipokines from hypertrophic adipocytes on 5-FU resistance of CRC cells, SGBS cells were used in this study as a replicable adipocyte-differentiating cell line suitable for the in vitro study of obesity and cancer [22,23]. SGBS pre-adipocytes were differentiated into mature adipocytes as previously described and regarded as being non-obese adipocytes [17]. Mature adipocytes incubated with palmitate were used to artificially generate hypertrophic mature adipocytes which were defined as obese adipocytes [24]. As shown in Figure 1A-C, lipid accumulation and intracellular TG content were significantly increased in the obese adipocytes compared to the non-obese adipocytes. These results showed that palmitate efficiently induced hypertrophy, which is the pathological feature of adipocytes during obesity development, in SGBS adipocytes. To examine the effect of obese adipocyte-secreted adipokines on 5-FU resistance in CRC cells, H3347 and HCT116 cells were pre-incubated with CM harvested from either non-obese adipocytes (M-CM) or obese adipocytes (P-CM) followed by treatment with 5-FU. The results demonstrated that cell viability was higher in the cells incubated with P-CM compared to those incubated with M-CM after 5-FU treatment (Figure 1D). Furthermore, 5-FU treatment caused a decrease in the expressions of the apoptotic molecules, cleaved caspase3 and Bax, and an increase in the expression of the anti-apoptotic molecule, Bcl-2, in P-CM-incubated cells compared to M-CM-incubated cells (Figure 1E). These results indicated that obese adipocyte-derived adipokines could cause 5-FU resistance in CRC.
Figure 1.

Obese adipocytes promoted 5-FU resistance in CRC cells. SGBS pre-adipocytes were differentiated into mature adipocytes (non-obese adipocytes) using an adipogenic differentiation process as previously described. Mature adipocytes treated with 0.6 mM palmitate for 24 hours were used to artificially generate hypertrophic adipocytes (obese adipocytes). (A) Lipid accumulation was determined using Oil Red O staining. (B) Oil Red O was eluted with isopropanol and measured using an ELISA plate reader at 500 nm. (C) Intracellular triglyceride (TG) content was analyzed using a TG colorimetric assay. (D and E) H3347 and HCT116 cells pre-incubated with M-CM or P-CM for 48 hours were subjected to 5-FU treatment for 48 hours. Cell viability of H3347 (D, left panel) and HCT116 (D, right panel) cells was analyzed by MTT assay. The expressions of apoptosis-related molecules, cleaved caspase3, Bax and Bcl-2, were examined by Western blot analysis (E). M-CM, non-obese adipocyte-derived conditioned media. P-CM, obese adipocyte-derived conditioned media. GAPDH served as the loading control. Data are represented as the mean ± SEM. SEM, error bars. *P<0.05 by Student’s t test or one-way ANOVA followed by Bonferroni’s post hoc test.
Obese adipocytes promoted 5-FU resistance in CRC cells through increased production and secretion of leptin
Previous studies have reported significant increases in leptin in human CRC tissues, and that this is a crucial mediator of CRC malignant behaviors [25-27]. Therefore, we investigated whether leptin is involved in obesity-mediated 5-FU resistance in CRC cells. Given that leptin is the principal hormone secreted by adipocytes and significantly increased in obese individuals [13], the production and secretion of leptin were analyzed in non-obese and obese adipocytes. The mRNA and protein expression levels of leptin were significantly increased in obese adipocytes compared to non-obese adipocytes (Figure 2A and 2B). P-CM was also enriched with leptin (Figure 2C). These results demonstrated that the expression and secretion of leptin were elevated in obese adipocytes. To further examine the role of leptin in obesity-mediated 5-FU resistance in CRC, leptin neutralizing antibodies were added to P-CM to neutralize the potential effect of leptin on CRC cells. As shown in Figure 2D and 2E, the inhibitory effect of P-CM on 5-FU-induced cytotoxicity was suppressed by leptin neutralization. In addition, leptin exposure significantly increased cell viability and decreased cell apoptosis of CRC cells in response to 5-FU treatment (Figure 2F and 2G). These results indicated the involvement of leptin in obesity-mediated 5-FU resistance of CRC cells.
Figure 2.

Leptin was involved in obese adipocyte-induced 5-FU resistance in CRC cells. (A and B) mRNA and protein levels of leptin in non-obese and obese adipocytes were analyzed by quantitative RT-PCR (A) and Western blot analysis (B), respectively. (C) Amounts of leptin in M-CM and P-CM were measured by ELISA. (D and E) H3347 and HCT116 cells were pre-incubated with M-CM or P-CM with/without 2 μg/ml leptin neutralizing antibodies for 48 hours followed by 5-FU treatment for 48 hours. Cell viability was evaluated by MTT analysis (D). The protein expressions of apoptosis-related molecules, cleaved caspase3, Bax and Bcl-2, were analyzed by Western blot analysis (E). (F and G) H3347 and HCT116 cells pre-treated with 100 ng/ml leptin recombinant protein for 48 hours were subjected to 5-FU treatment for another 48 hours. MTT assay and Western blot analysis were used to detect cell viability (F) and the expressions of apoptosis-related molecules (G), respectively. M-CM, non-obese adipocyte-derived conditioned media. P-CM, obese adipocyte-derived conditioned media. GAPDH served as the loading control. Data are expressed as the mean ± SEM. SEM, error bars. *P<0.05 by Student’s t test or one-way ANOVA followed by Bonferroni’s post hoc test.
Leptin increased 5-FU resistance in CRC cells by inducing the expression of AXL
AXL belongs to the TAM family of receptor tyrosine kinases, and it has been shown to mediate resistance to 5-FU in CRC [28]. It has also been proposed to be involved in the regulation of cell survival, proliferation, migration, invasion and drug resistance through its downstream pathways, including PLCγ signaling, in which activation of PLCγ has been shown to be detected by the increased expression of phospho-PLCγ at Tyr783 [29,30]. To investigate whether AXL is involved in leptin-induced 5-FU resistance, we first examined the regulatory role of leptin in Axl signaling. As shown in Figure 3A, P-CM incubation increased AXL expression and its downstream signaling molecule, PLCγ, activation in H3347 and HCT116 cells. However, these effects were abrogated by leptin neutralization. In addition, leptin treatment significantly increased AXL expression and PLCγ activation in CRC cells (Figure 3B). These results suggested the regulatory role of leptin in AXL expression and its downstream PLCγ activation. To further clarify the role of AXL in mediating 5-FU resistance under leptin treatment, CRC cells with/without depletion of AXL expression were subjected to leptin exposure followed by treatment with 5-FU. The mRNA and protein expressions of AXL were markedly decreased in cells transfected with AXL siRNA compared to those with non-targeting siRNA (Figure 3C and 3D). The effect of leptin on AXL expression and PLCγ activation was inhibited in AXL-silenced cells (Figure 3E). Depletion of AXL expression increased the susceptibility of leptin-exposed cells to 5-FU (Figure 3F and 3G). These results indicated that AXL signaling was involved in leptin-induced 5-FU resistance in CRC.
Figure 3.

AXL was involved in leptin-induced 5-FU resistance in CRC cells. (A) Protein levels of AXL, phospho-PLCγ (Tyr783) and PLCγ were determined by Western blot analysis in H3347 and HCT116 cells incubated with M-CM or P-CM with/without 2 μg/ml leptin neutralizing antibodies for 48 hours. (B) The expressions of AXL, phospho-PLCγ (Tyr783) and PLCγ in cells treated with 100 ng/ml leptin recombinant protein for 48 hours were analyzed by Western blot analysis. (C and D) mRNA and protein levels of AXL in H3347 and HCT116 cells transfected with non-targeting siRNA or AXL siRNA were examined by quantitative RT-PCR (C) and Western blot analysis (D). (E) CRC cells transfected with non-targeting siRNA or AXL siRNA were treated with 100 ng/ml leptin recombinant protein for 48 hours. The expressions of AXL, phospho-PLCγ (Tyr783) and PLCγ were detected by Western blot analysis. (F and G) H3347 and HCT116 cells transfected with non-targeting siRNA or AXL siRNA were treated with 100 ng/ml leptin recombinant protein for 48 hours followed by 5-FU treatment for another 48 hours. Cell viability was determined by MTT assay (F). The expressions of apoptosis-related molecules, including cleaved caspase3, Bax and Bcl-2, were analyzed by Western blot analysis (G). M-CM, non-obese adipocyte-derived conditioned media. P-CM, obese adipocyte-derived conditioned media. SC siRNA, non-targeting siRNA. GAPDH was used as the loading control. Data are expressed as the mean ± SEM. SEM, error bars. *P<0.05 by Student’s t test or one-way ANOVA followed by Bonferroni’s post hoc test.
Leptin induced AXL expression through AMPK-mediated YAP-TEAD transcriptional complex activation
The YAP-TEAD complex has been shown to induce AXL expression in hepatocellular carcinoma and lung cancer [31,32]. YAP acts as a transcriptional cofactor to mediate the DNA binding ability and transcriptional activity of TEAD protein [33]. YAP is regulated by phosphorylation-dependent nucleocytoplasmic shuttling. When YAP is phosphorylated, it binds to 14-3-3 proteins, resulting in its cytoplasmic retention. However, upon dephosphorylation, YAP can translocate into the nucleus and form a complex with TEAD to initiate the transcription of downstream target genes [34]. AMPK, which is the downstream target suppressed by leptin, has been shown to regulate the activation of the YAP-TEAD complex. AMPK can inhibit YAP through two mechanisms: directly by phosphorylating YAP, and indirectly via activation of the LATS1 kinase [35,36]. Therefore, we hypothesized that leptin stimulates AXL expression in CRC cells by inhibiting AMPK followed by activation of the YAP-TEAD complex. To test this hypothesis, we examined the phosphorylation status of AMPK, LATS1 and YAP in CRC cells after treatment with leptin with/without the AMPK activator, AICAR. As shown in Figure 4A, leptin significantly decreased AMPK and LATS1 activity, as indicated by decreased levels of phospho-AMPK (Thr172) and phospho-LATS1 (Ser909). In addition, a decrease in the expression of phospho-YAP (Ser127) and an increase in the expression of AXL were observed in CRC cells after leptin treatment. However, these effects were reversed by treating the cells with a combination of leptin and AICAR (Figure 4A). Since YAP dephosphorylation results in its nuclear translocation and increased interaction with TEAD, subcellular localization of YAP and its interaction with TEAD were investigated. Treatment of H3347 and HCT116 cells with leptin resulted in increases in YAP nuclear distribution and YAP interaction with TEAD, however these effects were reversed by AICAR treatment (Figure 4B and 4C). These results indicated that AMPK inhibition was involved in leptin-induced YAP dephosphorylation and nuclear translocation in CRC cells.
Figure 4.

AMPK was the downstream regulator of leptin-induced activation of the YAP-TEAD complex in CRC cells. H3347 and HCT116 cells were treated with 100 ng/ml leptin recombinant protein with/without 0.5 mM of the AMPK activator, AICAR, for 48 hours. A. Protein levels of phospho-AMPK (Thr172), AMPK, phospho-LATS1 (Ser909), LATS1, phospho-YAP (Ser127), YAP and AXL were analyzed by Western blot analysis. B. Subcellular distributions of YAP and TEAD were investigated by Western blot analysis of the cytoplasmic and nuclear fractions of CRC cells after the indicated treatments. Lamin B and GAPDH served as nuclear and cytoplasmic controls, respectively. C. The interaction between YAP and TEAD was evaluated using co-immunoprecipitation. Protein extracts of CRC cells were immunoprecipitated with anti-YAP or anti-TEAD antibodies. The expressions of YAP and TEAD in the precipitated proteins were detected by Western blot analysis. D. The association between TEAD and the AXL promoter was examined by ChIP assay. The ChIP assay was carried out on the AXL promoter using anti-TEAD antibodies followed by PCR to amplify the DNA region containing TEAD binding sites. E. pAXL-cypridina luc plasmid and internal control pTK-red firefly luc plasmid were co-transfected into H3347 and HCT116 cells followed by treatment with 100 ng/ml leptin recombinant protein with/without 0.5 mM AICAR for 48 hours. The relative promoter activity of AXL was subsequently detected using a dual luciferase assay. GAPDH served as the loading control. Anti-rabbit IgG was used as the isotype control. Data are expressed as the mean ± SEM. SEM, error bars. *P<0.05 by one-way ANOVA followed by Bonferroni’s post hoc test.
Due to the regulatory role of YAP in the DNA binding ability of TEAD, we then investigated the association between TEAD and the AXL promoter in CRC cells after treatment with leptin with/without AICAR. TEAD contains a conserved N-terminal DNA-binding domain, the transcriptional enhancer activator (TEA) domain, which is responsible for recognizing sequence motif 5’-GGAATG-3’ within the promoter region of target genes [37]. Four putative TEAD-binding sites have been located upstream of the transcriptional start site within the AXL promoter. Therefore, we designed two pairs of primers to amplify the two different promoter regions (AXL promoter-1 and AXL promoter-2). Each amplified promoter region contained two of the four TEAD binding sites on the AXL promoter. As demonstrated in Figure 4D, the associations of TEAD with AXL promoter-1 and AXL promoter-2 were significantly increased in response to leptin treatment, while AICAR exposure disrupted these interactions in CRC cells. Due to the involvement of leptin in regulating the binding ability of TEAD to the AXL promoter, we investigated the promoter activity of AXL in cells after treatment with leptin with/without AICAR. The leptin-treated cells displayed increased promoter activity of AXL, however the effect was reversed by AICAR treatment (Figure 4E). These results indicated that leptin could regulate DNA binding ability and transactivation capability of the YAP-TEAD complex by suppressing AMPK in CRC cells.
To further evaluate the involvement of the YAP-TEAD complex in leptin-induced AXL expression, H3347 and HCT116 cells with/without knockdown of YAP expression were subjected to leptin treatment. The mRNA and protein expressions of YAP were markedly decreased in cells transfected with YAP siRNA compared to those transfected with non-targeting siRNA (Figure 5A and 5B). YAP silencing significantly diminished leptin-induced AXL expression in CRC cells (Figure 5C). These results demonstrated the involvement of the YAP-TEAD complex in leptin-induced AXL expression.
Figure 5.

The YAP-TEAD complex was involved in the leptin-induced AXL expression in CRC cells. (A and B) The mRNA and protein expressions of YAP in H3347 and HCT116 cells transfected with either non-targeting siRNA or YAP siRNA were examined by quantitative RT-PCR (A) and Western blot analysis (B). (C) H3347 and HCT116 cells transfected with non-targeting siRNA or YAP siRNA were treated with 100 ng/ml leptin recombinant protein for 48 hours. The expressions of phospho-YAP (Ser127), YAP and AXL were analyzed by Western blot analysis. SC siRNA, non-targeting siRNA. GAPDH served as the internal control. Data are expressed as the mean ± SEM. SEM, error bars. *P<0.05 by Student’s t test.
AXL/PLCγ axis mediated leptin-induced P-gp expression and activity in CRC cells
As our results demonstrated that the AXL/PLCγ axis was a crucial mediator of leptin-induced 5-FU resistance in CRC cells, we investigated how the AXL/PLCγ axis regulates 5-FU resistance. Previous study has shown that PLCγ activation can induce the expression of P-gp [38]. P-gp is a transmembrane ATP-binding cassette (ABC) transporter which functions as a drug transporter. It has been associated with 5-FU resistance in cancer [39]. In addition, we had demonstrated that PLCγ activation was significantly increased in CRC cells after exposure to leptin, as evidenced by the increased expression of phospho-PLCγ at Tyr783, and that this effect was suppressed by knockdown of AXL expression (Figure 3E). Hence, we hypothesized that the AXL/PLCγ axis may mediate 5-FU resistance by inducing the expression of P-gp in CRC cells. To test this hypothesis, CRC cells transfected with non-targeting siRNA or AXL siRNA were treated with leptin, and the expression and activity of P-gp were subsequently analyzed. As shown in Figure 6A and 6B, mRNA and protein expression levels of P-gp were both significantly elevated upon leptin exposure. However, these effects were reversed by depleting AXL expression. Furthermore, the activity of P-gp was significantly increased in response to leptin treatment, as evidenced by a decrease in the cellular retention of the P-gp substrate, calcein (Figure 6C). Leptin-induced calcein retention was suppressed in AXL-silenced cells (Figure 6C). These results suggested the involvement of P-gp in AXL/PLCγ axis-mediated 5-FU resistance upon leptin exposure.
Figure 6.

Leptin induced p-glycoprotein expression and activity via the AXL/PLCγ axis in CRC cells. H3347 and HCT116 cells transfected with non-targeting siRNA or AXL siRNA were treated with 100 ng/ml leptin recombinant protein for 48 hours. (A and B) mRNA and protein levels of p-glycoprotein (P-gp) were analyzed by quantitative RT-PCR (A) and Western blot analysis (B). (C) The activity of P-gp was determined using a multidrug resistance assay. SC siRNA, non-targeting siRNA. GAPDH served as the loading control. Data are expressed as the mean ± SEM. SEM, error bars. *P<0.05 by one-way ANOVA followed by Bonferroni’s post hoc test.
Leptin inhibition improved the response of CRC cells to 5-FU in obese mice
To further validate the effect of leptin on 5-FU resistance in CRC in vivo, we evaluated the sensitivity of CRC cells to 5-FU in tumor-bearing mice fed on a HFD (Figure 7A). Although there was no significant difference in food intake between the mice fed on a HFD or LFD (Figure 7B), a HFD was significantly associated with increased body weight (Figure 7C). Serum TG (Figure 7D) and leptin (Figure 7E) levels were also significantly elevated in the mice fed with a HFD. In addition, the growth inhibitory effect of 5-FU on CRC tumors was less effective in the obese mice (Figure 7F and 7G). Of note, leptin neutralization sufficiently rescued the sensitivity of CRC tumors in obese mice to 5-FU treatment (Figure 7F and 7G). To validate the in vitro findings, the expressions of phospho-YAP (Ser127), AXL and P-gp in tumor tissues were determined. As shown in the representative images of immunohistochemical staining, dephosphorylation of YAP and increased expressions of AXL and P-gp were observed in tumor tissues from obese mice compared to those from lean mice (Figure 7H). The administration of leptin neutralizing antibodies reversed the expressions of these molecules in tumor tissues from obese mice (Figure 7H). These results further elucidated the role of leptin in obesity-induced 5-FU resistance in CRC.
Figure 7.
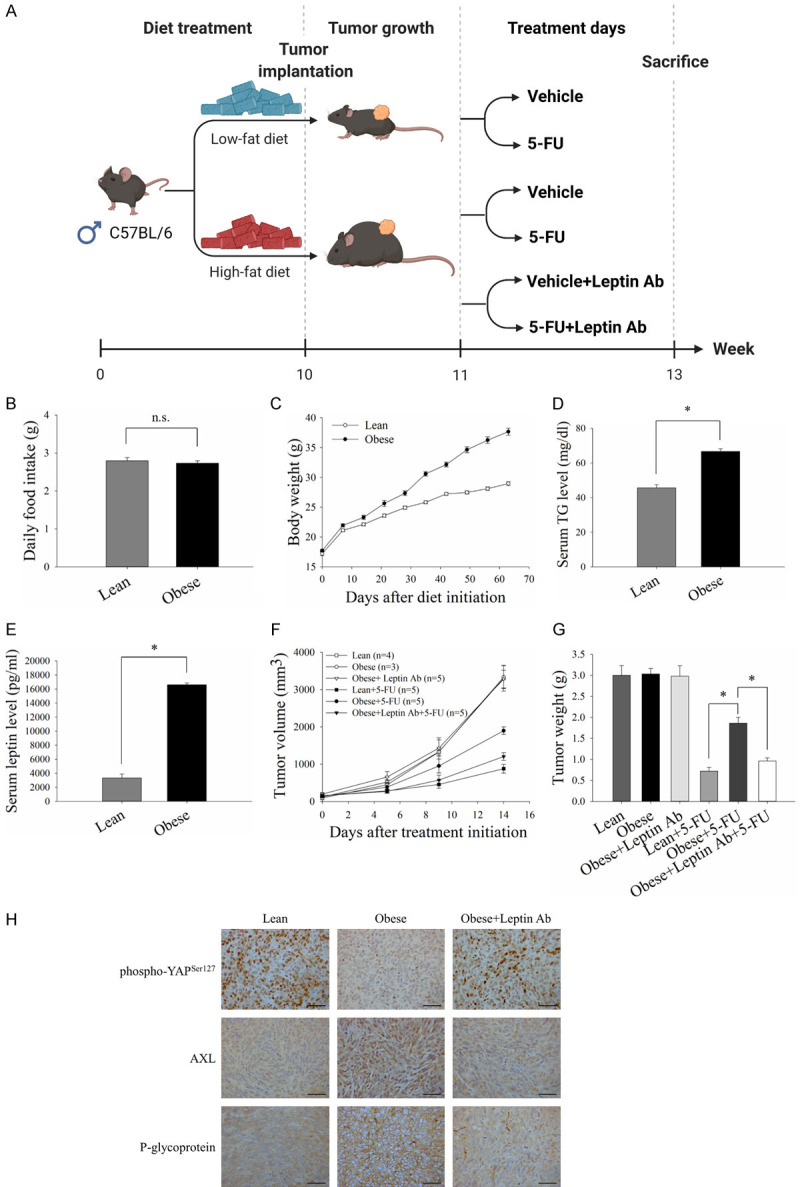
Leptin inhibition sensitized CRC cells to 5-FU in obese mice. C57BL/6 mice were fed on a HFD or LFD from 5 weeks of age until the end of the experiments. MC-38 cells (1×106) were subcutaneously implanted 10 weeks after initiating the diet, and treatment was started when the tumor burden reach about 100 mm3. 5-FU (25 mg/kg) and neutralizing antibodies against leptin (50 μg per mouse) were injected intraperitoneally into tumor-bearing mice for 2 weeks. Goat IgG antibody was served as the control. The tumor volume was calculated using the formula: V = (width2 × length)/2. A. Timeline of the animal experiments and treatment schedule for the tumor-bearing mice. B and C. Food intake and body weight changes of the mice fed on a HFD or LFD. D. Triglyceride (TG) levels in mice serum were detected using a TG colorimetric assay. E. ELISA was used to measure the serum level of leptin in the mice. F and G. Average tumor growth curves and tumor weights of CRC tumors in the mice with the indicated treatments. Tumor growth was monitored every 3-4 days. H. Representative immunohistochemistry images for phospho-YAP (Ser127), AXL and P-gp staining in CRC tumor sections (Scale bar, 50 μm). Data are expressed as the mean ± SEM of at least three mice in each group. SEM, error bars. n.s., not significant. *P<0.05 by Student’s t test or one-way ANOVA followed by Bonferroni’s post hoc test.
Discussion
Obesity has been strongly associated with tumor malignancy and therapeutic resistance in CRC [1,2,5]. However, the preclinical evidence and underlying mechanisms in obesity-induced drug resistance are not well-established in CRC. In this study, we demonstrated that leptin from obese adipocytes contributed to 5-FU resistance in CRC via increased AXL expression and PLCγ activation. Of note, AMPK inhibition and the subsequent activation of the YAP-TEAD complex were involved in leptin-stimulated AXL expression. Our findings are the first to demonstrate that YAP-dependent AXL overexpression is involved in leptin-induced 5-FU resistance in CRC.
Previous study has reported that drug resistance is a major cause of a poor prognosis and high recurrence rates among CRC patients with obesity [40]. In the present study, we showed that obese adipocytes could promote 5-FU resistance in CRC. This finding is consistent with previous studies which reported that obesity decreases the efficacy of anti-cancer drugs in CRC [9,11], however other studies have reported contradictory results regarding the regulatory role of obesity in CRC [41,42]. Simkens et al. reported that a high BMI was positively associated with longer OS in patients with advanced CRC after receiving chemotherapy [41]. Furthermore, overweight and obese patients with metastatic CRC have been reported to have a prolonged OS compared to normal-weight patients after receiving anti-cancer therapies [42]. One explanation for these discrepancies may be differences in the influence of obesity on early- and late-stage CRC. A low BMI is also a negative prognostic factor reflecting cachexia and malnutrition, which are surrogate markers of advanced stage and aggressive tumors [43]. Hence, obese individuals may have better nutritional status to tolerate the destructive effects of anti-cancer therapies and cancer cells at a later stage of CRC.
Our results demonstrated that obese adipocytes led to 5-FU resistance in CRC through increased production and secretion of leptin. In addition, leptin neutralization rescued the sensitivity to 5-FU in cells pre-incubated with P-CM and in CRC tumors grown in obese mice. Moreover, leptin recombinant protein significantly contributed to 5-FU resistance in CRC cells. Our results are consistent with previous findings that adipocyte-secreted leptin conferred resistance to chemotherapy and targeted therapy in melanoma cells [44]. In addition, Strong et al. reported that adipose stromal cells isolated from obese women (ObASCs) displayed higher leptin expressions compared to those from lean individuals. Furthermore, breast cancer cells co-cultured with ObASCs demonstrated increased cell proliferation and metastasis, while those co-cultured with leptin knockdown-ObASCs did not display similar effects [45]. Hence, the development of a leptin antagonist may effectively antagonize drug resistance in cancer [26]. These findings indicate the crucial role of leptin in obesity-mediated drug resistance in cancer.
Furthermore, our results demonstrated that leptin contributed to 5-FU resistance in CRC by upregulating AXL expression, and that AMPK was involved in this process. Although our results indicated that leptin could induce AXL expression through suppression of AMPK activity, a paradoxical tissue-specific effect of leptin on AMPK activation has been reported in previous studies [14,46,47]. In breast cancer cells, leptin has been observed to increase cell proliferation through AMPK inhibition [47]. In addition, leptin has been shown to decrease activation of AMPK in the hypothalamus, thus reducing appetite and leading to weight loss [14]. On the other hand, leptin has been shown to stimulate AMPK activity in skeletal muscle, liver, pancreas and adipose tissues to achieve metabolic homeostasis [46]. However, the mechanisms underlying the distinct responses of AMPK activation to leptin exposure in different tissues have not been well defined. AMPK is a heterotrimeric complex composed of one catalytic α-subunit and two regulatory β- and γ-subunits [48]. The well-established mechanisms of AMPK activation are binding of AMP and/or ADP to the γ-subunit and phosphorylation of the α-subunit at Thr172 [48]. In addition to the kinases and phosphatases that directly phosphorylate or dephosphorylate Thr172 of the α-subunit, several other regulatory mechanisms for Thr172 phosphorylation have also been reported [49,50]. In cancer, Akt can phosphorylate the α-subunit of AMPK at Ser485/491, thereby preventing upstream kinases from phosphorylating AMPK at the activating site, Thr172 [49]. In response to lipolytic signals, protein kinase A (PKA) can phosphorylate the α-subunit of AMPK at Ser173, resulting in inhibition of liver kinase B1 (LKB1)-mediated Thr172 phosphorylation in adipocytes [50]. These findings imply the context-dependent regulation of AMPK phosphorylation and activation, and this may explain the distinct responses of AMPK activation in different tissues upon leptin treatment. Therefore, the detailed mechanisms for the suppression of AMPK by leptin need to be further investigated in CRC.
Our results also demonstrated that leptin induced AXL expression in CRC cells by inhibiting AMPK activity followed by increasing YAP activation and nuclear localization. Emerging evidence has shown that dysregulation of the YAP-TEAD complex is the crucial mechanism underlying resistance to anti-cancer therapies [51,52]. For example, YAP can mediate resistance of lung cancer cells to RAF- and MEK-targeted therapies through TEAD-associated transactivation of the anti-apoptotic protein, Bcl-xL [53]. Clinically, YAP activation has been associated with shorter OS and disease-free survival (DFS) in CRC patients after receiving 5-FU-based neoadjuvant chemotherapy [54]. We further observed that nuclear YAP interacted with TEAD and subsequently promoted TEAD occupancy on the AXL promoter and increased AXL promoter activity after leptin treatment. Consistent with previous studies [55,56], we showed that YAP was an upstream transcriptional cofactor together with TEAD in the regulation of AXL expression, however crosstalk between AXL and YAP has also been reported in recent studies [57,58]. AXL, as the downstream target of YAP, has been shown to interact with and directly phosphorylate YAP at Try391 and Try407. AXL-induced YAP phosphorylation at these residues then results in YAP activation and increased YAP-mediated transcriptional activity [57]. In addition, Li et al. showed that AXL-induced STAT3 activation can mediate crosstalk between AXL and YAP via competitive binding to LATS1, thereby causing YAP dephosphorylation and nuclear translocation [58]. These findings imply the role of YAP in amplifying AXL signaling through a feed-forward mechanism. Given that we demonstrated that the YAP-TEAD complex is involved in leptin-induced AXL expression, further studies are warranted to investigate the regulatory role of AXL in YAP phosphorylation under leptin treatment.
Our results further showed that Axl/PLCγ signalling might confer 5-FU resistance in CRC cells through inducing P-gp expression. P-gp has been demonstrated to be widely expressed in human tissues, including intestine [39]. Apart from 5-FU, several anti-cancer drugs have been reported to be the substrates of P-gp, such as doxorubicin, cisplatin, gefitinib and tamoxifen [39]. Pharmacological inhibition of P-gp has been shown to increase the cellular retention of anti-cancer drugs, thereby resulting in the decreased cell survival of cancer cells [59]. The stimulatory effect of PLCγ on P-gp expression has been shown to be mediated by Raf-MAPK pathway, and that treated the cells with MAPK inhibitor suppresses PLCγ-induced P-gp expression [38]. In addition, leptin has been shown to activate Raf-MAPK pathway in cancer [60]. Therefore, the potential involvement of Raf-MAPK pathway in PLCγ-induced P-gp expression in leptin-treated CRC cells can be further evaluated.
In conclusion, we found that the obesity-associated adipokine, leptin, caused 5-FU resistance of CRC cells through AXL/PLCγ signaling, which could further increase P-gp expression. Mechanistically, leptin upregulated AXL expression by suppressing AMPK activity followed by stimulation of the YAP-TEAD transcriptional complex. Of note, 5-FU was less effective in suppressing tumor growth in obese mice, while leptin neutralization could rescue the sensitivity of CRC tumors to 5-FU (Figure 8). Hence, our findings provide mechanistic insights into the effect of obesity on chemoresistance in CRC, and may lead to improvements in the therapeutic efficacy and disease outcomes of CRC patients with obesity.
Figure 8.
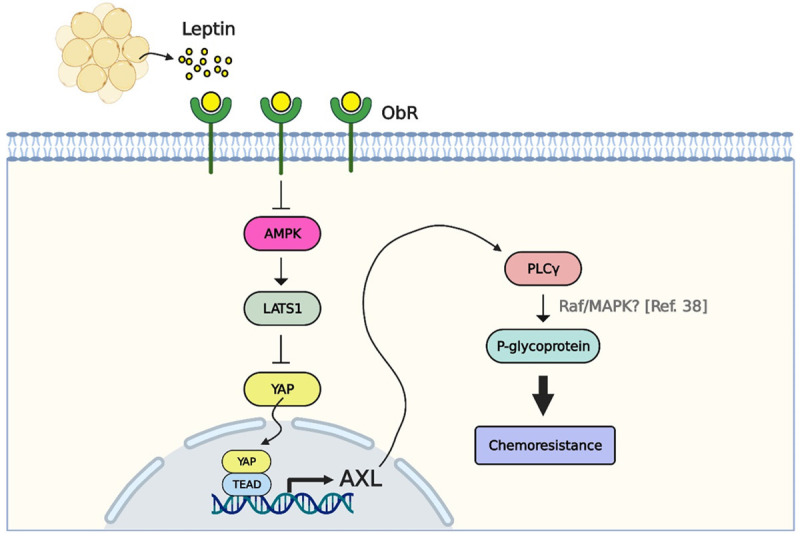
Obese adipocyte-associated leptin increased 5-FU resistance through YAP-dependent AXL upregulation. Leptin secreted by obese adipocytes resulted in 5-FU resistance in CRC cells by increasing AXL expression and PLCγ activation. Mechanistically, leptin induced AXL expression by suppressing AMPK and LATS1 followed by stimulation of YAP activation and nuclear translocation. Nuclear YAP further interacted with TEAD, thereby promoting the association between TEAD and the AXL promoter and eventually increasing AXL promoter activity. In addition, the AXL/PLCγ axis led to 5-FU resistance by inducing P-gp expression in CRC cells after leptin treatment.
Acknowledgements
This work was supported by research grants from the Ministry of science and technology (MOST 108-2314-B-016-003-MY3), the National Defense Medical Center (MND-MAB-110-101), the Tri-Service General Hospital (TSGH-C04-109029, TSGH-C03-110023), and the Chi Mei Medical Center (CMNDMC10901), Taiwan. We would like to acknowledge the Laboratory Animal Center of National Defense Medical Center (NDMC-LAC), Taiwan, for support in animal husbandry and medical care. The illustrations were created with BioRender.com.
Disclosure of conflict of interest
None.
References
- 1.Jochem C, Leitzmann M. Obesity and colorectal cancer. Recent Results Cancer Res. 2016;208:17–41. doi: 10.1007/978-3-319-42542-9_2. [DOI] [PubMed] [Google Scholar]
- 2.Shaukat A, Dostal A, Menk J, Church TR. BMI is a risk factor for colorectal cancer mortality. Dig Dis Sci. 2017;62:2511–2517. doi: 10.1007/s10620-017-4682-z. [DOI] [PubMed] [Google Scholar]
- 3.Guiu B, Petit JM, Bonnetain F, Ladoire S, Guiu S, Cercueil JP, Krause D, Hillon P, Borg C, Chauffert B, Ghiringhelli F. Visceral fat area is an independent predictive biomarker of outcome after first-line bevacizumab-based treatment in metastatic colorectal cancer. Gut. 2010;59:341–7. doi: 10.1136/gut.2009.188946. [DOI] [PubMed] [Google Scholar]
- 4.Faruk Aykan N, Yildiz I, Sen F, Kilic L, Keskin S, Ciftci R, Karabulut S, Sakar B, Disci R. Effect of increased body mass index (BMI) on time to tumour progression (TTP) in unresectable metastatic colorectal cancer (mCRC) patients treated with bevacizumab-based therapy. Med Oncol. 2013;30:679. doi: 10.1007/s12032-013-0679-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Artaç M, Korkmaz L, Coşkun HŞ, Dane F, Karabulut B, Karaağaç M, Çabuk D, Karabulut S, Aykan NF, Doruk H, Avcı N, Turhal NS. Bevacuzimab may be less effective in obese metastatic colorectal cancer patients. J Gastrointest Cancer. 2019;50:214–220. doi: 10.1007/s12029-017-0047-2. [DOI] [PubMed] [Google Scholar]
- 6.Bray GA. Medical consequences of obesity. J Clin Endocrinol Metab. 2004;89:2583–9. doi: 10.1210/jc.2004-0535. [DOI] [PubMed] [Google Scholar]
- 7.Rosen ED. Two paths to fat. Nat Cell Biol. 2015;17:360–1. doi: 10.1038/ncb3133. [DOI] [PubMed] [Google Scholar]
- 8.Wen YA, Xing X, Harris JW, Zaytseva YY, Mitov MI, Napier DL, Weiss HL, Mark Evers B, Gao T. Adipocytes activate mitochondrial fatty acid oxidation and autophagy to promote tumor growth in colon cancer. Cell Death Dis. 2017;8:e2593. doi: 10.1038/cddis.2017.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ko JH, Um JY, Lee SG, Yang WM, Sethi G, Ahn KS. Conditioned media from adipocytes promote proliferation, migration, and invasion in melanoma and colorectal cancer cells. J Cell Physiol. 2019;234:18249–18261. doi: 10.1002/jcp.28456. [DOI] [PubMed] [Google Scholar]
- 10.Rondini EA, Harvey AE, Steibel JP, Hursting SD, Fenton JI. Energy balance modulates colon tumor growth: interactive roles of insulin and estrogen. Mol Carcinog. 2011;50:370–82. doi: 10.1002/mc.20720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Olivo-Marston SE, Hursting SD, Perkins SN, Schetter A, Khan M, Croce C, Harris CC, Lavigne J. Effects of calorie restriction and diet-induced obesity on murine colon carcinogenesis, growth and inflammatory factors, and microRNA expression. PLoS One. 2014;9:e94765. doi: 10.1371/journal.pone.0094765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tabuso M, Homer-Vanniasinkam S, Adya R, Arasaradnam RP. Role of tissue microenvironment resident adipocytes in colon cancer. World J Gastroenterol. 2017;23:5829–5835. doi: 10.3748/wjg.v23.i32.5829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wauman J, Zabeau L, Tavernier J. The leptin receptor complex: heavier than expected? Front Endocrinol (Lausanne) 2017;8:30. doi: 10.3389/fendo.2017.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huynh MK, Kinyua AW, Yang DJ, Kim KW. Hypothalamic AMPK as a regulator of energy homeostasis. Neural Plast. 2016;2016:2754078. doi: 10.1155/2016/2754078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Candelaria PV, Rampoldi A, Harbuzariu A, Gonzalez-Perez RR. Leptin signaling and cancer chemoresistance: perspectives. World J Clin Oncol. 2017;8:106–119. doi: 10.5306/wjco.v8.i2.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.M.S T. Serum leptin hormone as an indicator of bad prognosis in colon cancer patients. Egypt J Hosp Med. 2014;55:184. [Google Scholar]
- 17.Fischer-Posovszky P, Newell FS, Wabitsch M, Tornqvist HE. Human SGBS cells-a unique tool for studies of human fat cell biology. Obes Facts. 2008;1:184–9. doi: 10.1159/000145784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yin J, Wang Y, Gu L, Fan N, Ma Y, Peng Y. Palmitate induces endoplasmic reticulum stress and autophagy in mature adipocytes: implications for apoptosis and inflammation. Int J Mol Med. 2015;35:932–40. doi: 10.3892/ijmm.2015.2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim JI, Huh JY, Sohn JH, Choe SS, Lee YS, Lim CY, Jo A, Park SB, Han W, Kim JB. Lipid-overloaded enlarged adipocytes provoke insulin resistance independent of inflammation. Mol Cell Biol. 2015;35:1686–99. doi: 10.1128/MCB.01321-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Incio J, Ligibel JA, McManus DT, Suboj P, Jung K, Kawaguchi K, Pinter M, Babykutty S, Chin SM, Vardam TD, Huang Y, Rahbari NN, Roberge S, Wang D, Gomes-Santos IL, Puchner SB, Schlett CL, Hoffmman U, Ancukiewicz M, Tolaney SM, Krop IE, Duda DG, Boucher Y, Fukumura D, Jain RK. Obesity promotes resistance to anti-VEGF therapy in breast cancer by up-regulating IL-6 and potentially FGF-2. Sci Transl Med. 2018;10:eaag0945. doi: 10.1126/scitranslmed.aag0945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Divella R, De Luca R, Abbate I, Naglieri E, Daniele A. Obesity and cancer: the role of adipose tissue and adipo-cytokines-induced chronic inflammation. J Cancer. 2016;7:2346–2359. doi: 10.7150/jca.16884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wabitsch M, Brenner RE, Melzner I, Braun M, Moller P, Heinze E, Debatin KM, Hauner H. Characterization of a human preadipocyte cell strain with high capacity for adipose differentiation. Int J Obes Relat Metab Disord. 2001;25:8–15. doi: 10.1038/sj.ijo.0801520. [DOI] [PubMed] [Google Scholar]
- 23.Allott EH, Oliver E, Lysaght J, Gray SG, Reynolds JV, Roche HM, Pidgeon GP. The SGBS cell strain as a model for the in vitro study of obesity and cancer. Clin Transl Oncol. 2012;14:774–82. doi: 10.1007/s12094-012-0863-6. [DOI] [PubMed] [Google Scholar]
- 24.Kim JI, Huh JY, Sohn JH, Choe SS, Lee YS, Lim CY, Jo A, Park SB, Han W, Kim JB. Lipid-overloaded enlarged adipocytes provoke insulin resistance independent of inflammation. Mol Cell Biol. 2015;35:1686–1699. doi: 10.1128/MCB.01321-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park J, Scherer PE. Leptin and cancer: from cancer stem cells to metastasis. Endocr Relat Cancer. 2011;18:C25–9. doi: 10.1530/ERC-11-0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Candelaria PV, Rampoldi A, Harbuzariu A, Gonzalez-Perez RR. Leptin signaling and cancer chemoresistance: perspectives. World J Clin Oncol. 2017;8:106–119. doi: 10.5306/wjco.v8.i2.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koda M, Sulkowska M, Kanczuga-Koda L, Surmacz E, Sulkowski S. Overexpression of the obesity hormone leptin in human colorectal cancer. J Clin Pathol. 2007;60:902–6. doi: 10.1136/jcp.2006.041004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dunne PD, McArt DG, Blayney JK, Kalimutho M, Greer S, Wang T, Srivastava S, Ong CW, Arthur K, Loughrey M, Redmond K, Longley DB, Salto-Tellez M, Johnston PG, Van Schaeybroeck S. AXL is a key regulator of inherent and chemotherapy-induced invasion and predicts a poor clinical outcome in early-stage colon cancer. Clin Cancer Res. 2014;20:164–75. doi: 10.1158/1078-0432.CCR-13-1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schoumacher M, Burbridge M. Key roles of AXL and MER receptor tyrosine kinases in resistance to multiple anticancer therapies. Curr Oncol Rep. 2017;19:19. doi: 10.1007/s11912-017-0579-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Axelrod H, Pienta KJ. AXL as a mediator of cellular growth and survival. Oncotarget. 2014;5:8818–8852. doi: 10.18632/oncotarget.2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu MZ, Chan SW, Liu AM, Wong KF, Fan ST, Chen J, Poon RT, Zender L, Lowe SW, Hong W, Luk JM. AXL receptor kinase is a mediator of YAP-dependent oncogenic functions in hepatocellular carcinoma. Oncogene. 2011;30:1229–1240. doi: 10.1038/onc.2010.504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Choi JY, Lee H, Kwon EJ, Kong HJ, Kwon OS, Cha HJ. TGFβ promotes YAP-dependent AXL induction in mesenchymal-type lung cancer cells. Mol Oncol. 2020;15:679–696. doi: 10.1002/1878-0261.12857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huh HD, Kim DH, Jeong HS, Park HW. Regulation of TEAD transcription factors in cancer biology. Cells. 2019;8:600. doi: 10.3390/cells8060600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu FX, Zhao B, Guan KL. Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell. 2015;163:811–28. doi: 10.1016/j.cell.2015.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang W, Xiao ZD, Li X, Aziz KE, Gan B, Johnson RL, Chen J. AMPK modulates Hippo pathway activity to regulate energy homeostasis. Nat Cell Biol. 2015;17:490–9. doi: 10.1038/ncb3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mo JS, Meng Z, Kim YC, Park HW, Hansen CG, Kim S, Lim DS, Guan KL. Cellular energy stress induces AMPK-mediated regulation of YAP and the Hippo pathway. Nat Cell Biol. 2015;17:500–10. doi: 10.1038/ncb3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin KC, Park HW, Guan KL. Regulation of the Hippo pathway transcription factor TEAD. Trends Biochem Sci. 2017;42:862–872. doi: 10.1016/j.tibs.2017.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang JM, Vassil AD, Hait WN. Activation of phospholipase C induces the expression of the multidrug resistance (MDR1) gene through the Raf-MAPK pathway. Mol Pharmacol. 2001;60:674–80. [PubMed] [Google Scholar]
- 39.Silva R, Vilas-Boas V, Carmo H, Dinis-Oliveira RJ, Carvalho F, de Lourdes Bastos M, Remiao F. Modulation of P-glycoprotein efflux pump: induction and activation as a therapeutic strategy. Pharmacol Ther. 2015;149:1–123. doi: 10.1016/j.pharmthera.2014.11.013. [DOI] [PubMed] [Google Scholar]
- 40.Martinez-Useros J, Garcia-Foncillas J. Obesity and colorectal cancer: molecular features of adipose tissue. J Transl Med. 2016;14:21. doi: 10.1186/s12967-016-0772-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Simkens LH, Koopman M, Mol L, Veldhuis GJ, Ten Bokkel Huinink D, Muller EW, Derleyn VA, Teerenstra S, Punt CJ. Influence of body mass index on outcome in advanced colorectal cancer patients receiving chemotherapy with or without targeted therapy. Eur J Cancer. 2011;47:2560–7. doi: 10.1016/j.ejca.2011.06.038. [DOI] [PubMed] [Google Scholar]
- 42.Aparicio T, Ducreux M, Faroux R, Barbier E, Manfredi S, Lecomte T, Etienne PL, Bedenne L, Bennouna J, Phelip JM, François E, Michel P, Legoux JL, Gasmi M, Breysacher G, Rougier P, De Gramont A, Lepage C, Bouché O, Seitz JF for FFCD investigators. Overweight is associated to a better prognosis in metastatic colorectal cancer: a pooled analysis of FFCD trials. Eur J Cancer. 2018;98:1–9. doi: 10.1016/j.ejca.2018.03.031. [DOI] [PubMed] [Google Scholar]
- 43.Argilés JM, Busquets S, Stemmler B, López-Soriano FJ. Cancer cachexia: understanding the molecular basis. Nat Rev Cancer. 2014;14:754–62. doi: 10.1038/nrc3829. [DOI] [PubMed] [Google Scholar]
- 44.Chi M, Chen J, Ye Y, Tseng HY, Lai F, Tay KH, Jin L, Guo ST, Jiang CC, Zhang XD. Adipocytes contribute to resistance of human melanoma cells to chemotherapy and targeted therapy. Curr Med Chem. 2014;21:1255–67. doi: 10.2174/0929867321666131129114742. [DOI] [PubMed] [Google Scholar]
- 45.Strong AL, Ohlstein JF, Biagas BA, Rhodes LV, Pei DT, Tucker HA, Llamas C, Bowles AC, Dutreil MF, Zhang S, Gimble JM, Burow ME, Bunnell BA. Leptin produced by obese adipose stromal/stem cells enhances proliferation and metastasis of estrogen receptor positive breast cancers. Breast Cancer Res. 2015;17:112. doi: 10.1186/s13058-015-0622-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lim CT, Kola B, Korbonits M. AMPK as a mediator of hormonal signalling. J Mol Endocrinol. 2010;44:87–97. doi: 10.1677/JME-09-0063. [DOI] [PubMed] [Google Scholar]
- 47.El-Masry OS, Al-Sakkaf K, Brown BL, Dobson PR. Differential crosstalk between the AMPK and PI3K/Akt pathways in breast cancer cells of differing genotypes: leptin inhibits the effectiveness of AMPK activation. Oncol Rep. 2015;34:1675–80. doi: 10.3892/or.2015.4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–62. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hawley SA, Ross FA, Gowans GJ, Tibarewal P, Leslie NR, Hardie DG. Phosphorylation by Akt within the ST loop of AMPK-α1 down-regulates its activation in tumour cells. Biochem J. 2014;459:275–87. doi: 10.1042/BJ20131344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Djouder N, Tuerk RD, Suter M, Salvioni P, Thali RF, Scholz R, Vaahtomeri K, Auchli Y, Rechsteiner H, Brunisholz RA, Viollet B, Mäkelä TP, Wallimann T, Neumann D, Krek W. PKA phosphorylates and inactivates AMPKalpha to promote efficient lipolysis. EMBO J. 2010;29:469–81. doi: 10.1038/emboj.2009.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nguyen CDK, Yi C. YAP/TAZ signaling and resistance to cancer therapy. Trends Cancer. 2019;5:283–296. doi: 10.1016/j.trecan.2019.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim MH, Kim J. Role of YAP/TAZ transcriptional regulators in resistance to anti-cancer therapies. Cell Mol Life Sci. 2017;74:1457–1474. doi: 10.1007/s00018-016-2412-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lin L, Sabnis AJ, Chan E, Olivas V, Cade L, Pazarentzos E, Asthana S, Neel D, Yan JJ, Lu X, Pham L, Wang MM, Karachaliou N, Cao MG, Manzano JL, Ramirez JL, Torres JM, Buttitta F, Rudin CM, Collisson EA, Algazi A, Robinson E, Osman I, Muñoz-Couselo E, Cortes J, Frederick DT, Cooper ZA, McMahon M, Marchetti A, Rosell R, Flaherty KT, Wargo JA, Bivona TG. The Hippo effector YAP promotes resistance to RAF- and MEK-targeted cancer therapies. Nat Genet. 2015;47:250–6. doi: 10.1038/ng.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Touil Y, Igoudjil W, Corvaisier M, Dessein AF, Vandomme J, Monte D, Stechly L, Skrypek N, Langlois C, Grard G, Millet G, Leteurtre E, Dumont P, Truant S, Pruvot FR, Hebbar M, Fan F, Ellis LM, Formstecher P, Van Seuningen I, Gespach C, Polakowska R, Huet G. Colon cancer cells escape 5FU chemotherapy-induced cell death by entering stemness and quiescence associated with the c-Yes/YAP axis. Clin Cancer Res. 2014;20:837–46. doi: 10.1158/1078-0432.CCR-13-1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xu MZ, Chan SW, Liu AM, Wong KF, Fan ST, Chen J, Poon RT, Zender L, Lowe SW, Hong W, Luk JM. AXL receptor kinase is a mediator of YAP-dependent oncogenic functions in hepatocellular carcinoma. Oncogene. 2011;30:1229–40. doi: 10.1038/onc.2010.504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ghiso E, Migliore C, Ciciriello V, Morando E, Petrelli A, Corso S, De Luca E, Gatti G, Volante M, Giordano S. YAP-dependent AXL overexpression mediates resistance to EGFR inhibitors in NSCLC. Neoplasia. 2017;19:1012–1021. doi: 10.1016/j.neo.2017.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Saab S, Chang OS, Nagaoka K, Hung MC, Yamaguchi H. The potential role of YAP in Axl-mediated resistance to EGFR tyrosine kinase inhibitors. Am J Cancer Res. 2019;9:2719–2729. [PMC free article] [PubMed] [Google Scholar]
- 58.Li J, Shi C, Zhou R, Han Y, Xu S, Ma H, Zhang Z. The crosstalk between AXL and YAP promotes tumor progression through STAT3 activation in head and neck squamous cell carcinoma. Cancer Sci. 2020;111:3222–3235. doi: 10.1111/cas.14546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nanayakkara AK, Follit CA, Chen G, Williams NS, Vogel PD, Wise JG. Targeted inhibitors of P-glycoprotein increase chemotherapeutic-induced mortality of multidrug resistant tumor cells. Sci Rep. 2018;8:967. doi: 10.1038/s41598-018-19325-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Booth A, Magnuson A, Fouts J, Foster M. Adipose tissue, obesity and adipokines: role in cancer promotion. Horm Mol Biol Clin Investig. 2015;21:57–74. doi: 10.1515/hmbci-2014-0037. [DOI] [PubMed] [Google Scholar]