

Abstract
The essential eukaryotic chaperone Hsp90 regulates the form and function of diverse client proteins, many of which govern thermotolerance, virulence, and drug resistance in fungal species. However, use of Hsp90 inhibitors as antifungal therapeutics has been precluded by human host toxicities and suppression of immune responses. We recently described resorcylate aminopyrazoles (RAPs) as the first class of Hsp90 inhibitors capable of discriminating between fungal (Cryptococcus neoformans, Candida albicans) and human isoforms of Hsp90 in biochemical assays. Here, we report an iterative structure-property optimization toward RAPs capable of inhibiting C. neoformans growth in culture. In addition, we report the first X-ray crystal structures of C. neoformans Hsp90 nucleotide binding domain (NBD), as the apoprotein and in complexes with the non-species-selective Hsp90 inhibitor NVP-AUY922 and three RAPs revealing unique ligand-induced conformational rearrangements which reaffirm the hypothesis that intrinsic differences in protein flexibility can confer selective inhibition of fungal versus human Hsp90 isoforms.
Graphical Abstract

INTRODUCTION
Invasive fungal diseases impose a major economic and public health burden worldwide, killing over 1.5 million each year.1-3 Approximately 90% of deaths due to fungal infection are caused by Candida, Aspergillus, and Cryptococcus species.4 Of these, Cryptococcus is now recognized as one of the most significant fungal threats to human health, with incidence increasing in both immunocompromised and immunocompetent hosts.1, 5 Recent data from the WHO place the annual global burden of cryptococcal meningitis, the major clinical manifestation of the disease, in excess of 223,000 cases annually, causing more than 150,000 attributable mortalities and approximately 30% of all AIDS-associated deaths.6, 7 Cryptococcal meningitis has a 100% mortality rate if left untreated, and mortality rates remain high at 30% in resource-rich and 70% in resource-poor contexts.8 Of the meager four drug classes approved for treatment of systemic fungal infections, only three are effective against Cryptococcus. Azoles and polyenes, which can each be used as single agents, both act via distinct mechanisms to deplete ergosterol, the major component of fungal membranes.9, 10 Resistance to each class has emerged, particularly for the azoles, with fungal infections becoming increasingly difficult to cure. A third class, antimetabolite pyrimidines, are only used in combination with other antifungals due to rapid emergence of resistance.11 The fourth antifungal class, the cell wall-targeting echinocandins, have limited activity against Cryptococcus and no CNS penetration.12-15 Given the limited array of antifungal drug classes and targets and the dramatically increasing incidence of resistance to current agents, the discovery of new chemotypes that act in ways that are mechanistically distinct from the available armamentarium are desperately needed.
Targeting core hubs of stress response circuitry is a powerful, as-yet unexploited strategy to cripple fungal pathogens. A prime example is the eukaryotic molecular chaperone Hsp90.16-19 Hsp90 is essential in all fungi, and in addition to enabling survival, Hsp90 client proteins are also critical to virulence and drug-resistance mechanisms.20 Although profound compromise of Hsp90 expression or function is lethal to fungi, more modest compromise via chemical inhibitors or genetic reduction prevents and reverses resistance to antifungals in culture.21-23 In Cryptococcus neoformans, recent studies have implicated Hsp90 in its thermotolerance, capsule assembly and sensitivity to antifungals, strongly influencing its virulence in a nematode model.24, 25 Beyond an intrinsic potential for broad-spectrum single agent antifungal activity, the unique effects on fungal biology inherent to Hsp90 inhibitors also render them strong candidates for development of combination treatment regimens that would actively impede mechanisms of drug resistance.
Due in large part to its prominent role in supporting the function of numerous oncoproteins, Hsp90 has been studied extensively as an anticancer target.26 Unfortunately, all Hsp90 inhibitors currently being developed as anticancer drugs exert mammalian host toxicities, especially suppression of innate and acquired immune mechanisms, that preclude their use as antifungals.27 In fact, although numerous drug candidates have progressed into clinical trials over the past decade, none have been approved due to limited anticancer efficacy at their maximally tolerated doses.28 One approach to reducing toxicity and improving therapeutic index for cancer and other indications has been the pursuit of paralog-selective inhibitors across the four Hsp90-family members in humans: Hsp90α, Hsp90β, Trap1 and Grp94.29, 30 The resorcylate scaffold, one of several privileged Hsp90-inhibitory chemotypes, has been modified to confer human paralog selectivity with applications in oncology and glaucoma.31-38 In addition, selective purine mimetics, such as TAS-11639 and modified analogs of BIIB021 selectively targeting Trap1,30 have been described. Paralog selectivity has also been reported for modified benzamides resembling SNX-2112.40, 41
We previously described efforts to develop Hsp90 inhibitors that are selective for fungal isoforms, initially templated from the resorcylic acid macrolactone natural products radicicol and monocillin.42 Building on successes in achieving Candida albicans selectivity with semisynthetic radicicol and monocillin oximes, as well as the strong preclinical track record of non-macrocyclic resorcylate drug candidates such as AT13387 (onalespib),43-46 NVP-AUY922 (luminespib),47-52 and STA-9090 (ganetespib),53-61 we more recently disclosed the design, synthesis and characterization of resorcylate aminopyrazoles (RAPs). RAPs are the first class of inhibitors capable of selectively inhibiting fungal isoforms of Hsp90 (specifically, C. neoformans and C. albicans) over their human orthologues Hsp90α/β, TRAP1, and Grp94.62 Our development of increasingly potent and fungal selective RAPs, as well as structure-activity relationships (SAR) for 112 early members of the series against C. neoformans and C. albicans culminated in our first generation of fungal selective lead RAPs (1, Figure 1), which were all N-methylated at R1 and C-arylated at R2. Interestingly, species-specific divergences in potency and fungal selectivity (FS) occurred with ortho/para positioning of R2 aryl ring substituents, while activity and selectivity generally converged with meta-substituents. However, despite this interesting biochemical activity profile, whole cell permeance of RAPs was an unsolved problem; of the 94 compounds from this study that showed potent cryptococcal Hsp90 binding (EC50 ≤ 1 μM in fungal lysate), only six had whole cell minimal inhibitory concentrations (MICs) ≤ 25 μM and only one of these six fit the desired profile of high biochemical potency and high fungal selectivity.
Figure 1.

Generic structure of resorcylate aminopyrazoles (RAPs, left) and summary of activity and selectivity profile for previously reported first-generation cryptococcal-selective RAP series of type 1, which are N-methylated at R1 and bear a monosubstituted phenyl ring at R2 to increase fungal selectivity.
In fungi, the cell wall presents an inherent impediment to passive small molecule permeability that is not present in mammalian cells and other microorganisms which lack this strong protective barrier.63 As a key virulence factor, C. neoformans also elaborates a dense polysaccharide capsule, which in addition to the plasma membrane and semi-permeable fibrillary network of the cell wall contributes to limiting the accumulation of passively permeable compounds within this organism.64,65, 66 In C. neoformans as in other fungi, a network of efflux pumps also prevents accumulation of many xenobiotics.67 In this report, we thus designate “permeant” molecules as having the combined features of active or passive cell penetration and evasion of efflux in C. neoformans so as to accumulate within the fungus and engage the target.67
Among the six cryptococcal-permeant analogues in our first-generation RAP library, we identified unique structural features which appeared to distinguish these compounds from non-permeant near-neighbors. Here, we now report significant achievements toward improving whole cell anticryptococcal activity via a rational structure-property approach. In addition, we describe the first X-ray structures of the C. neoformans Hsp90 nucleotide binding domain (NBD), crystallized as the apoprotein and in co-complexes with three fungal selective RAPs. Together, these new developments define the binding mode of this novel Hsp90 inhibitor class, providing insights into the most plausible chemo-structural origins of their observed fungal selectivity. Equally important from the therapeutic perspective, they advance our understanding of the structure-activity and structure-property relationships critical to conferring selective, whole cell anticryptococcal activity.
RESULTS AND DISCUSSION
Rational optimization of RAPs towards the combined features of potent, fungal selective Hsp90 inhibition and improved whole cell activity.
To arrive at rationally-designed, second-generation RAPs with the combined qualities of fungal accumulation and fungal selectivity, we sought to merge the structural features found to confer biochemical selectivity for fungal Hsp90 with nascent features which appeared to track with permeance. To enable efficient elaboration of new second-generation analogues, the RAP scaffold (Figure 1) was synthesized in a convergent manner using an alternate protecting group strategy from our published first-generation set,62 allowing for modular variation of the aminopyrazole and amide portion of the molecule. By synthesizing bespoke 5-aminopyrazoles, various N-alkyl groups at R1 as well as differential substitution patterns on the R2 aromatic ring could be incorporated (Scheme 1). Starting from commercially available mono- and disubstituted methyl benzoate derivatives (2a-ab), acylation of deprotonated acetonitrile afforded α-cyanoketone products 3a-ab. Condensation of 3a-ab with a variety of N-alkyl-hydrazines incorporated the R1 alkyl group of the 5-aminopyrazole building blocks (4a-bb).
Scheme 1:

Preparation of building block aminopyrazoles 4.
Conditions: (a) MeCN, n-BuLi, THF, −78 °C to rt; (b) R1NH-NH2, MeOH, 120 °C, microwave or R1NH-NH2·HCl, NEt3, MeOH, 120 °C, microwave.
The resorcinol core was prepared by the formylation of 3,5-dimethoxybromobenzene (5) and subsequent demethylation with BBr3 (Scheme 2). Bis-benzyl protection of 6 gave stable common intermediate (7) which could be elaborated with various amide groups. Pinnick oxidation of the aldehyde (7), followed by HATU-mediated amide coupling with isoindoline hydrochloride or substituted fluoroisoindoline derivatives, gave resorcylate amide bromides (8a-c). The synthesis was completed by Buchwald-Hartwig cross-coupling of aryl bromides 8a-c with 5-aminopyrazole (4a-bb) followed by global deprotection with palladium on carbon to give the targeted RAP compounds (9a-bi).
Scheme 2:
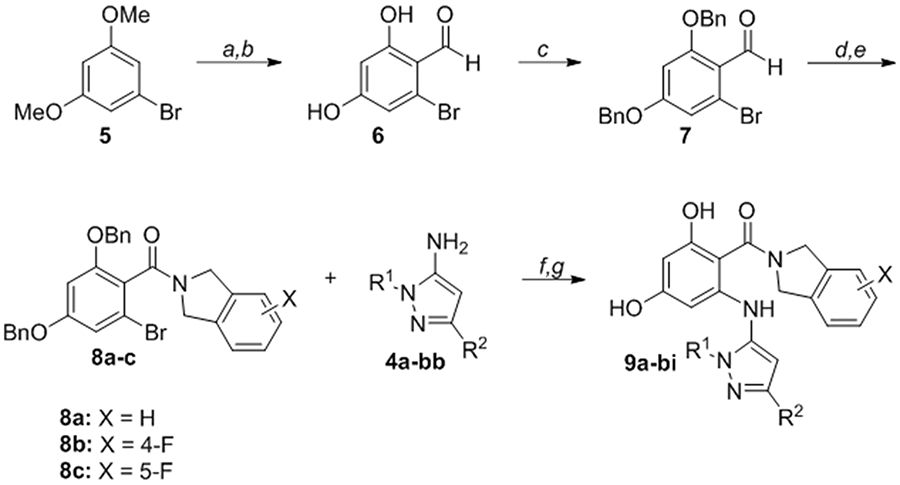
Convergent synthesis of RAPs 9a-bi from resorcylate amides 8a-c and aminopyrazoles 4a-bb
Conditions: (a) POCl3, DMF, 0 °C to 100 °C; (b) BBr3, CH2Cl2, 0 °C to rt; (c) BnBr, K2CO3, MeCN, reflux; (d) NaOCl2, NaH2PO4·H2O, 2-methyl-2-butene, THF/t-BuOH/H2O; (e) isoindoline·HCl (for 8a), or 4-fluoroisoindoline·HCl (for 8b), or 5-fluoroisoindoline·HCl (for 8c), HATU, Et3N, THF/CH2Cl2; (f) Pd(OAc)2 (10 mol%), Xantphos (20 mol%), Cs2CO3, toluene, 130 °C or Pd2(dba)3 (4 mol%), Xantphos (10 mol%), NaOPh, dioxane, 170 °C, microwave; (g) Pd/C (cat.), H2, MeOH or Pd(OH)2/C (cat.), H2, EtOAc.
To determine target binding affinity, we used a fluorescence polarization (FP)-based competitive binding assay.42, 62 Use of whole-cell lysates enables assessment of binding to Hsp90 while complexed with relevant cochaperones and, in the case of human lysate, to the entire repertoire of Hsp90 family members expressed in human cells. Given these factors, FP in lysate is highly relevant to selectivity in the whole cell context but can only provide relative quantification of binding affinity because the absolute concentration of the protein targets is unknown. In comparison, FP using purified NBD proteins permits measurement of assay-independent ligand dissociation constants.68 In our published first-generation set, the most potent and selective of the inhibitors were meta-methoxy substituted RAPs 10-12 (Figure 2A), which disappointingly all failed to inhibit cryptococcal growth at concentrations up to 25 μM. In addition to five non-selective whole-cell-active RAPs (13-17, Figure 2B), the only RAP exhibiting the combined features of >5-fold cryptococcal selectivity plus a cryptococcal MIC ≤ 25 μM was the R2 ortho-tolyl-substituted compound 18. This compound was one of only three examples of a RAP bearing an ortho-substituted R2 aryl ring, and was the single instance among these three with an isoindoline amide. Notably, despite a roughly equal proportion of isoindoline, pyridopyrrolidine and pyrazolopyrrolidine amides within the early SAR set (Figure 1), whole cell activity was limited solely to isoindoline or fluoroisoindolines at this site.
Figure 2.

(A) RAPs with meta-substituted R2 aryl rings exhibiting high fungal selectivity (FS) for cryptococcal Hsp90 over human isoforms; (B) Among 94 RAPs with fungal target affinity (H99 EC50) ≤ 1 μM, only 6 demonstrate whole cell antifungal activity (MIC ≤ 25 μM). Values for compounds with either fold-selectivity >5 or MIC80 ≤12.5 μM are highlighted in blue. Values for compounds with both fold-selectivity >5 and MIC80 ≤12.5 μM are highlighted in red.
Based on these observations, we postulated that lipophilicity at the amide may be a key driver for permeance, and opted to explore other ortho-substituents in combination with isoindoline (Table 1, Entries 1-3) to discern whether compound 18 represented an outlier or was suggestive of a potential structural trend. Gratifyingly, improved whole cell activity was also observed in the biochemically potent ortho-fluorinated (9a), ortho-chlorinated (9b), and ortho-trifluoromethylated (9c) analogues. However, this improvement was offset by a disappointing drop in fungal selectivity for all three compounds.
Table 1.
Early exploration of the impact of (1) ortho-substituents at the R2 aryl ring and (2) increased aliphatic bulk at R1 on biochemical potency and selectivity in binding cryptococcal Hsp90, and in growth inhibition of C. neoformans in culture.
| |||||||
|---|---|---|---|---|---|---|---|
| Entry | Compound | Amide (“X”) |
R1 | R2 |
C. neoformans EC50a (μM) |
C. neoformans Fold- selectivityb |
C. neoformans MIC80c (μM) |
| 1 | 9a | A | Me |

|
0.180 | 2.0 | <6 |
| 2 | 9b | A | Me |

|
0.105 | 0.9 | 6.25 |
| 3 | 9c | A | Me |
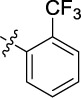
|
0.055 | 0.8 | 3.25 |
| 4 | 9d | A | tBu |
|
0.421 | 11.1 | 12.5 |
| 5 | 9e | C | Me | 0.199 | 5.0 | <25 | |
| 6 | 9f | A | Me |
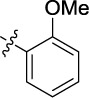
|
0.137 | 1.4 | <25 |
| 7 | 9g | A | iPr | 0.189 | 2.5 | 12.5 | |
| 8 | 9h | A | 0.655 | 2.1 | 6.25 | ||
| 9 | 9i | B | cyHex | 0.248 | 4.3 | 12.5 | |
| 10 | 9j | C | 0.137 | 5.8 | 12.5 | ||
| 11 | 9k | A | cyPent | 0.626 | 1.7 | 12.5 | |
| 12 | 9l | A | iBu | 0.431 | 3.5 | 12.5 | |
| 13 | 9m | A | tBu | 0.066 | 0.7 | 25 | |
| 14 | 9n | A | Me |

|
0.041 | 0.3 | 0.5 |
| 15 | 9o | A | tBu | 0.107 | 6.8 | 1.8 | |
| 16 | 9p | A | tBu |
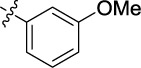
|
2.993 | 2.9 | 12.5 |
| 17 | 9q | A | tBu |

|
4.206 | >2.8 | 6.25 |
| 18 | 9r | A | tBu |

|
10.000 | 0.8 | 6.25 |
| 19 | 9s | A | Me |
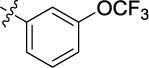
|
4.951 | 0.7 | >25 |
| 20 | 9t | A | tBu | 7.591 | 0.4 | 3.13 | |
| 21 | 9u | A | Me |
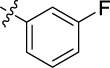
|
0.023 | 37.3 | 25 |
| 22 | 9v | A | tBu | 0.782 | 10.5 | 12.5 | |
| 23 | 9w | A | Me |

|
1.001 | 8.0 | 25 |
| 24 | 9x | A | tBu | 1.897 | >6.3 | 25 | |
| 25 | 9y | A | cyHex | 0.976 | 2.3 | 12.5 | |
EC50 values were determined by fluorescence polarization (FP)-based equilibrium competition assay performed in 384-well format using whole cell lysates prepared from C. neoformans and serial compound dilutions. All determinations were performed in duplicate.
To calculate fold-selectivity, EC50 values determined by FP using human HepG2 cell lysate were divided by the EC50 value determined using C. neoformans lysate. The resulting ratio was then normalized by the ratio of values determined for the Hsp90 inhibitor geldanamycin using lysate of each cell type. Results for key selective compounds were confirmed by repeat assay in lysates, as well as by measuring ligand dissociation constants (Ki) using purified NBDs in FP assays (see Supplemental Tables 1-2 and Supplemental Figures S1-S3).
Concentration of compound resulting in >80% inhibition of fungal cell growth compared to vehicle control. Values for compounds with either fold-selectivity >5 or MIC80 ≤12.5 μM are highlighted in blue. Values for compounds with both fold-selectivity >5 and MIC80 ≤12.5 μM are highlighted in red.
Given the comparatively higher selectivity observed with the ortho-methylated parent compound, we next designed analogues in which this substituent was retained, and instead paired with other potentially permeance-enhancing features. Revisiting the cohort of permeant compounds in Figure 2B, another structural commonality that we deemed unique to these compounds as compared to the larger set was the presence of relatively large, lipophilic groups at both R1 and R2, again in the presence of an isoindoline or fluoroisoindoline amide. Analogues 9d and 9e were prepared as examples of such features in combination with the ortho-tolyl group at R2. The introduction of an N-tert-butyl group (9d) at R1 was sufficient to retain both fungal selectivity (11.1-fold) and whole cell activity (MIC 12.5 μM) that was comparable to the N-Me parent 18 (14.1-fold and MIC 10 μM, respectively). Isoindoline fluorination of 18, however, was detrimental to both whole cell activity and selectivity (compound 9e).
In our prior study,62 similar biochemical potencies and fungal selectivities were observed among compounds bearing a meta-tolyl or meta-methoxyphenyl group at R2. In contrast, at the ortho site we found that new compound 9f, the methoxy analogue of 18, showed a precipitous loss of selectivity and no measurable whole cell activity, despite high binding affinity for Hsp90. Compounds 9g-9l were next prepared to again test the strategy of increased bulk and lipophilicity at R1 to improve permeance; for these analogues whole cell activities were again improved, but gains in fungal selectivity were only modest. Interestingly, converting the methoxy to trifluoromethoxy delivered mixed results, with the N-methylated compound 9n exhibiting no selectivity and sub-micromolar whole cell activity, while the N-tert-butyl compound 9o showed modest 6.8-fold selectivity in combination with an impressive MIC of 1.8 μM.
With initial confirmation that the whole cell activity of R2-arylated RAPs could be improved via increased bulk and lipophilicity at R1, we next sought to deploy this strategy on R2 meta-substituted scaffolds, which had trended toward the highest cryptococcal Hsp90 selectivity in our previous study. Compounds 9p, 9q and 9r were each prepared as N-tert-butyl analogues of earlier N-methylated inhibitors, which had exhibited fungal selectivities ranging from low (e.g. meta-trifluoromethyl) to the highest observed in the study at 12- or 33-fold (meta-methyl or meta-methoxy). Unfortunately, although this modification was again successful in achieving sufficient permeance for whole cell activity, biochemical potencies dropped to the low micromolar range and a concomitant drop in selectivity was also observed. Similar trends were also observed with meta-trifluoromethoxylated compounds 9s and 9t. In contrast, improved selectivity and potency was observed with meta-fluorination at R2 (compounds 9u and 9v) with measurable, albeit high MIC values against whole cells. Replacement of the fluorine with chlorine in these analogues (9w and 9x), plus an N-cyclohexyl variant (9y) reduced biochemical potencies from nanomolar to low micromolar range. Again, with these chlorinated analogues there was an apparent tradeoff between fungal selectivity >5-fold and whole cell activity < 25 μM, with no compounds espousing both features.
In sum, this initial cohort of second-generation RAPs confirmed our prior observations: while selectivity and permeance were most often orthogonal qualities, the combination of key structural features could in some cases (e.g. compounds 9d, 9u, 9v) provide an inroad to optimized inhibitors.
Although the trend was not consistent across all analogues, we did note that in several instances, conversion of the R1 N-methyl to N-tert-butyl dampened fungal selectivity in addition to improving whole cell activity. In response, we next pursued an alternative method of pairing the apparent permeance-inducing ortho substituent with a selectivity-inducing meta-substituent, on scaffolds which retain the R1 N-methylation (Table 2). Here, we observed improved fungal selectivities with the 2,3-dimethyl series 9z-9ab, but this R2 group generally did not render the inhibitors permeant. Interestingly, this set also exhibited an extreme sensitivity to positioning of the isoindoline fluorine substituent with respect to biochemical potency. In contrast, we had significant success with R2-fluorinated analogues. Compounds 9ac-9ae, with an R2 2,5-substitution pattern embodying one of two possible hybrids of monosubstituted compounds 18 and 9u, showed dramatically improved potencies and selectivities, in concert with good whole cell MICs ranging from 6.25-14.1 μM. Similarly, improved activity profiles were observed among 2-fluoro-3-methyl analogues 9af-9ah. Inspired by these promising results, we next examined an array of disubstituted aryl rings at R2, paired with isoindoline amide A (compounds 9ai-9au). While selectivity and permeance sometimes diverged, we identified a number of ortho/meta pairings (again in both 2,3- and 2,5-relationships) which conferred high selectivity, and in select cases (9ak, 9am, 9an and 9ar) good concomitant whole cell activity (MIC values 12.5 μM or lower). Of note, the meta-chlorination present in analogues 9ak and 9ao again appeared to dampen biochemical potency, as was observed in 9w-9y (Table 1). This reduced cryptococcal potency in turn lowered fungal selectivity in comparison to other near-neighbor analogues. In contrast, meta-fluorination appeared to consistently yield biochemical potencies below 100 nM, fungal selectivities greater than 15-fold, and whole cell activity at or below MIC 12.5 μM when paired with either a suitable ortho-substituent (9ac-9ae, 9am, 9an) or a bulky aliphatic group at R1 (9v, Table 1). Compound 9as, bearing two ortho-fluorine substituents in a 2,6 arrangement, exhibited moderate potency and whole cell activity, but a complete loss of selectivity. Lastly, ortho substitution (9at, 9au) was also sufficient to achieve permeance in combination with additional para-substituents, but consistent with our previously published SAR, these para-substituted analogues were not found to confer any cryptococcal selectivity.
Table 2.
Efforts to improve whole-cell anticryptococcal activities and selectivities for inhibitors N-methylated at R1, via a paring of ortho/meta substituents on the R2 aryl ring.

| ||||||
|---|---|---|---|---|---|---|
| Entry | Compound | Amide (“X”) |
R2 |
C. neoformans EC50a (μM) |
C. neoformans fold-selectivityb |
C. neoformans MIC80c (μM) |
| 1 | 9z | A |

|
0.227 | 30.0 | >25 |
| 2 | 9aa | B | 1.096 | 9.6 | >25 | |
| 3 | 9ab | C | 0.020 | 169.5 | 25 | |
| 4 | 9ac | A |
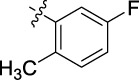
|
0.034 | 38.8 | 6.25 |
| 5 | 9ad | B | 0.017 | 168.4 | 12.5 | |
| 6 | 9ae | C | 0.267 | 10.0 | 14.1 | |
| 7 | 9af | A |
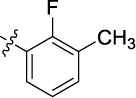
|
0.108 | 29.5 | 12.5 |
| 8 | 9ag | B | 0.020 | 233.9 | 25 | |
| 9 | 9ah | C | 0.259 | 11.5 | 12.5 | |
| 10 | 9ai | A |
|
0.190 | 9.6 | 25 |
| 11 | 9aj | A |
|
2.343 | 5.0 | >25 |
| 12 | 9ak | A |
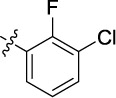
|
0.406 | 7.3 | 9.4 |
| 13 | 9al | A |

|
0.391 | 4.2 | 25 |
| 14 | 9am | A |

|
0.115 | 6.5 | 12.5 |
| 15 | 9an | A |

|
0.117 | 25.4 | 6.3 |
| 16 | 9ao | A |
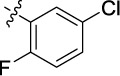
|
0.921 | 4.3 | 15.7 |
| 17 | 9ap | A |
|
0.202 | 10.4 | 25 |
| 18 | 9aq | A |

|
0.078 | 18.1 | >25 |
| 19 | 9ar | A |

|
0.334 | 7.6 | 12.5 |
| 20 | 9as | A |
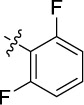
|
0.239 | 0.3 | 12.5 |
| 21 | 9at | A |
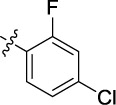
|
0.468 | 3.2 | 18.8 |
| 22 | 9au | A |

|
0.039 | 1.2 | 9.4 |
EC50 values were determined by FP-based equilibrium competition assay performed in 384-well format using whole cell lysates prepared from C. neoformans and serial compound dilutions. All determinations were performed in duplicate.
To calculate fold-selectivity, the EC50 value determined in human HepG2 cell lysate was divided by the EC50 value determined in C. neoformans lysate. The resulting ratio was then normalized by the ratio of values determined for the Hsp90 inhibitor geldanamycin using lysate of each cell type. Results for key selective compounds were confirmed by repeat assay in lysates, as well as by measuring ligand dissociation constants (Ki) using purified NBDs in FP assays (see Supplemental Tables 1 and 2).
Concentration of compound resulting in >80% inhibition of fungal cell growth compared to vehicle control. Values for compounds with either fold-selectivity >5 or MIC80 ≤12.5 μM are highlighted in blue. Values for compounds with both fold-selectivity >5 and MIC80 ≤12.5 μM are highlighted in red.
With two successful permeance-enhancing strategies identified, we next examined the combination of both approaches (Table 3). Gratifyingly, merging our optimal disubstitution patterns at R2 with a variety of bulky aliphatic moieties at R1 (tert-butyl, isopropyl, cyclopentyl, and cyclohexyl) afforded inhibitors 9av-9bi with almost universally improved activity profiles. Notably, all compounds in this series had measurable MICs at or below 25 μM, with several compounds achieving the best cryptococcal whole cell growth inhibition observed for fungal selective RAPs to-date at 3.13 μM (9bb, 9bd, 9bf, and 9bi). Among this series, the strongest whole cell potencies appeared to track to the N-tert-butyl and N-cyclohexyl substitutions at R1, with the corresponding N-isopropyl (9ba, 9be) and N-cyclopentyl (9bc, 9bh) congeners espousing slightly higher MICs at 6.25 μM. Interestingly, while substitution of the isoindoline amide of 9bf for 4-fluoroisoindoline (compound 9bg) depressed potency to increase the lysate EC50 to the low-micromolar range, this compound still exhibited selectivity and whole cell activity comparable to near neighbor analogues with lysate EC50s in the 100-400 nM range.
Table 3.
Strategic merging of bulky aliphatic moieties at R1 and optimal disubstituted aromatics at R2 result in advanced leads with significantly improved activity profiles.
| |||||||
|---|---|---|---|---|---|---|---|
| Entry | Compound | Amide (“X”) |
R1 | R2 |
C. neoformans EC50a (μM) |
C. neoformans fold-selectivityb |
C. neoformans MIC80 (μM) |
| 1 | 9av | A | tBu |

|
0.945 | >12.7 | 25 |
| 2 | 9aw | A | iPr |
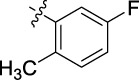
|
0.337 | 18.7 | 25 |
| 3 | 9ax | A | tBu | 0.634 | 13.0 | 12.5 | |
| 4 | 9ay | A | cyPent | 0.571 | 3.1 | 6.25 | |
| 5 | 9az | A | cyHex | 0.469 | 5.9 | 6.25 | |
| 6 | 9ba | A | iPr |
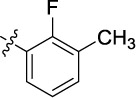
|
0.117 | 59.7 | 6.25 |
| 7 | 9bb | A | tBu | 0.156 | 40.7 | 3.13 | |
| 8 | 9bc | A | cyPent | 0.316 | 10.8 | 6.25 | |
| 9 | 9bd | A | cyHex | 0.130 | 9.3 | 3.13 | |
| 10 | 9be | A | iPr |

|
0.234 | 23.5 | 6.25 |
| 11 | 9bf | A | tBu | 0.347 | 20.2 | 3.13 | |
| 12 | 9bg | B | tBu | 1.150 | >10.5 | 6.25 | |
| 13 | 9bh | A | cyPent | 0.533 | 3.9 | 6.25 | |
| 14 | 9bi | A | cyHex | 0.295 | 7.0 | 3.13 | |
EC50 values were determined by FP-based equilibrium competition assay performed in 384-well format using whole cell lysates prepared from C. neoformans and serial compound dilutions. All determinations were performed in duplicate.
To calculate fold-selectivity, the EC50 value determined in human HepG2 cell lysate was divided by the EC50 value determined in C. neoformans lysate. The resulting ratio was then normalized to the ratio of values determined for the nonselective inhibitor geldanamycin using lysate of each cell type. Results for key selective compounds were confirmed by repeat assay in lysates, as well as by measuring ligand dissociation constants (Ki) using purified NBDs in FP assays (see Supplemental Tables 1 and 2).
Concentration of compound resulting in >80% inhibition of fungal cell growth compared to vehicle control. Values for compounds with either fold-selectivity >5 or MIC80 ≤12.5 μM are highlighted in blue. Values for compounds with both fold-selectivity >5 and MIC80 ≤12.5 μM are highlighted in red.
With a larger cohort of whole cell-active, fungal selective compounds in hand, we next sought to further evaluate these compounds for potential use in animals. In our prior study, poor metabolic stability was identified as a significant liability among several of our most promising first-generation RAPs, with many compounds showing very high microsomal metabolism (t1/2 <10 min), and only one RAP with intermediate (CLint ~20-70, T1/2 20-60 min) metabolic stability. In this study, we examined a wider array of RAPs from our second-generation set, including all of the most promising compounds summarized in Table 4, to better understand the metabolic liabilities present and attempt rational modifications to minimize them. Starting from first-generation RAP 18 (T1/2 = 6 min), we found that a number of structural modifications conferred a net stabilizing effect, with microsomal half-lives for nearly half of the compounds (11 of 24) extended to >20 minutes. Such modifications include conversion of the R1 methyl to tert-butyl (9ax) and cyclohexyl (9az), fluorination of the isoindoline at the 4- (9ad) and 5- position (9ae), and conversion of the R2 ortho-methyl substituent to fluorine (9am) and chlorine (9an). However, matched-pair analysis for other members of the cohort (not shown) showed that these same types of single structural modification impart divergent effects on microsomal stability, depending on the structure of the parent RAP. While it is difficult to discern clear structure-property relationships or pinpoint exact sites of metabolism from this data, we postulate that the observed trends are consistent with either metabolism that is largely occurring at the isoindoline with extreme sensitivity to CYP450 binding effects imparted by the entire aminopyrazole subunit, or more likely that several potential metabolic soft spots exist on the RAP scaffold, and that access to these potential hot-spots by CYP450 enzymes is strongly governed by each site’s positioning relative to other substituents exerting distal effects on CYP450 binding. While the extension of half-lives from the low to intermediate range represents a key advancement, there is clearly room for further optimization of metabolic stability.
Table 4.
Mammalian cytotoxicity and microsomal stability assessments for 24 select RAPs, including 22 RAPs with biochemical FS >5 and C. neoformans MIC ≤ 12.5 μM. All whole cell testing results are representative of two independent experiments, each performed with technical triplicates. Compounds with NIH 3T3 therapeutic index >2 are highlighted in green. Compounds with intermediate microsomal stability (CLint ~20-70 μL/min/mg)69 are highlighted in orange.
| Entry | Compound |
C. neoformans fold- selectivity |
C. neoformans MIC80 (μM) |
HepG2 Cytotoxicity IC50 (μM) |
NIH 3T3 cytotoxicity IC50 (μM) |
NIH 3T3 Therapeutic Index (TI) |
Microsomal stability |
|
|---|---|---|---|---|---|---|---|---|
| T1/2 (min) |
CLint (μL/min/ mg) |
|||||||
| 1 | 9d | 11.1 | 12.5 | 5.9 | 8.0 | 0.6 | 19.8 | 70.0 |
| 2 | 9j | 5.8 | 12.5 | 0.4 | 1.3 | 0.1 | 21.7 | 63.8 |
| 3 | 9o | 6.8 | 1.8 | 0.7 | 5.7 | 3.2 | 33.8 | 41.0 |
| 4 | 9v | 10.5 | 12.5 | 3.6 | 18.8 | 1.5 | 40.8 | 34.0 |
| 5 | 9ac | 38.8 | 6.25 | 0.3 | 2.8 | 0.4 | 5.0 | 278.3 |
| 6 | 9ad | 168.4 | 12.5 | 0.4 | 5.8 | 0.5 | 14.4 | 96.0 |
| 7 | 9ae | 10.0 | 14.1 | 0.5 | 1.9 | 0.1 | 24.5 | 56.6 |
| 8 | 9af | 29.5 | 12.5 | 0.1 | 5.8 | 0.5 | 36.3 | 38.2 |
| 9 | 9ah | 11.5 | 12.5 | 0.1 | 1.4 | 0.1 | 33.5 | 41.4 |
| 10 | 9ak | 7.3 | 9.4 | 0.5 | 4.4 | 0.5 | 23.9 | 58.0 |
| 11 | 9am | 6.5 | 12.5 | 0.3 | 1.8 | 0.1 | 22.4 | 61.8 |
| 12 | 9an | 25.4 | 6.3 | 0.56 | 2.9 | 0.5 | 17.6 | 78.6 |
| 13 | 9ar | 7.6 | 12.5 | 0.7 | 1.8 | 0.1 | 28.8 | 48.2 |
| 14 | 9av | >12.7 | 25 | 9.9 | >10 | -- | 17.9 | 77.4 |
| 15 | 9ax | 13.0 | 12.5 | 11.3 | >10 | -- | 5.7 | 243.2 |
| 16 | 9az | 5.9 | 6.25 | 2.2 | 4.6 | 0.7 | 14.4 | 96.6 |
| 17 | 9ba | 59.7 | 6.25 | 2.7 | 7.8 | 1.2 | 5.2 | 268.6 |
| 18 | 9bb | 40.7 | 3.13 | 0.6 | 7.1 | 2.3 | 12.7 | 109.0 |
| 19 | 9bc | 10.8 | 6.25 | 2.4 | 5.8 | 0.9 | 18.4 | 75.2 |
| 20 | 9bd | 9.3 | 3.13 | 0.8 | 1.4 | 0.4 | 16.5 | 84.0 |
| 21 | 9be | 23.5 | 6.25 | 1.9 | 3.1 | 0.5 | 20.9 | 66.4 |
| 22 | 9bf | 20.2 | 3.13 | 1.2 | 4.0 | 1.3 | 22.5 | 61.6 |
| 23 | 9bg | >10.5 | 6.25 | 3.1 | 11.2 | 1.8 | 18.8 | 73.8 |
| 24 | 9bi | 7.0 | 3.13 | 1.5 | 3.0 | 1.0 | 11.6 | 119.6 |
Next, we examined the downstream translatability of potency and selectivity in lysates to the context of whole cells. Strikingly, a plot of biochemical vs. whole cell potency (Figure 3A) clearly shows that these values are non-correlative. The lack of correlation suggests that although barriers to cryptococcal permeance have been lowered with this series, as-yet-undefined mechanisms still reduce compound accumulation in Cryptococcus, and as a result dampen whole cell potency to levels well below what would be expected based on biochemical target engagement. This point is perhaps best exemplified by direct comparison of compounds 9bg and 9ac (Figure 3A); despite a >30-fold difference in biochemical potency in cryptococcal H99 lysate (1.15 μM vs. 34 nM, respectively), both compounds were equally effective in H99 whole cells (MIC 6.25 μM).
Figure 3.

Scatter plot of the top 22 most fungal selective, whole-cell active RAPs shows (A) biochemical potency (H99 lysate pEC50, x-axis) and whole cell antifungal activity (C. neoformans MIC, μM, y-axis) are poorly correlated. Similarly, no correlation is seen between fungal selectivity and therapeutic index (TI, mammalian IC50 ÷ C. neoformans MIC) in (B) NIH 3T3 cells or (C) HepG2 cells. For Panels A-C, scatter points are colored based on relative H99 selectivity in cellular lysates. Panel (D) depicts the structures of key highlighted compounds.
To investigate whether permeance limitations in Cryptococcus extended to mammalian cells, we proceeded to evaluate the cytotoxicity of our most promising compounds, again mainly focused on those with FS >5 and MIC ≤ 12.5 μM, in mammalian systems (Table 4). These RAPs were tested in 9-point dose response for cytotoxic activity against human liver cancer (HepG2) and mouse fibroblast (NIH 3T3) cell lines (Table 4, Supplemental Tables 3 and 4, Supplemental Figures S4 and S5). The disparity in these cell lines between whole cell activity and biochemical potency for our RAPs was not as great as in Cryptococcus. As a result, we found that high fungal target selectivity in lysates did not translate to a useful therapeutic index (TI = mammalian IC50 ÷ C. neoformans MIC80) in most cases. Using the non-cancerous mouse fibroblast cell line NIH 3T3, known to be less sensitive to Hsp90 inhibition than cancer cells such as HepG2,43 only a handful of compounds showed a modest therapeutic window (TI 1.5-3.1). Somewhat surprisingly, we observed a near-inverse relationship between cellular TI and lysate fold-selectivity; the most selective compound in mouse fibroblasts (9o) was among the least selective by FP, whereas the RAP exhibiting the highest lysate selectivity (9ad) was among the least selective in NIH 3T3 cells (Figures 3B and 3D). In HepG2 cells, which due to their oncogenic nature are known to be hypersensitive to Hsp90 inhibition, none of the compounds exhibited cytotoxicity less than the minimum concentration required to suppress C. neoformans growth (all TIs < 1, Figures 3C and 3D). From these data, we conclude that although progress has been made, achieving adequate fungal permeance remains a challenge, a well-recognized hurdle for many small molecule xenobiotics.70
Structural Insights into Fungal Selectivity of RAPs
In addition to pursuing a property-driven optimization of our RAP series, we performed structural studies to better understand the binding mode of these inhibitors and the basis of their selectivity. We determined the crystal structures (Supplemental Tables 5 and 6) of the C. neoformans NBD domain in the apo state, in complex with the nonselective Hsp90 inhibitor NVP-AUY922 (Luminespib),71 and in complex with the fungal selective RAPs 10, 18 (Figure 2), and 19 (Figure 4).62 These structures provide the first insights into the mode of RAP binding and also reveal a plausible structural rationale for their fungal selectivity.
Figure 4.
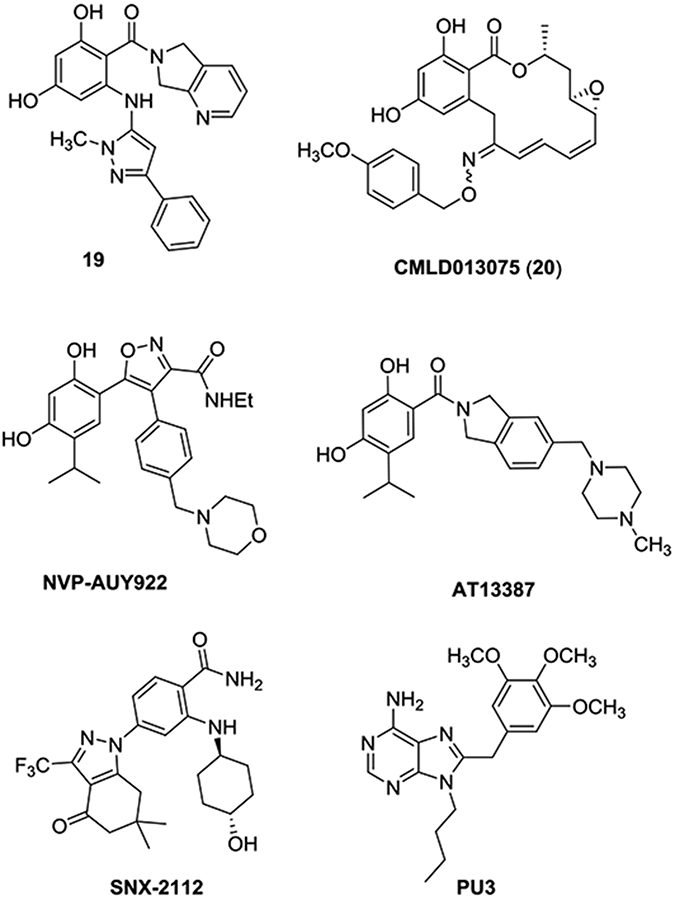
Chemical structures of ligands including RAP 19,62 previously-reported C. albicans-selective inhibitor CMLD013075,42 clinical inhibitors NVP-AUY922,71 AT13387 and SNX-2112, and purine inhibitor PU3.72
Multiple copies of the protein were present in the asymmetric units of the crystals in all cases (three in apo, six in complex with AUY922 and two with each of 10, 18, and 19), providing independent views of the protein in different states (Supplemental Figure S6). Excluding the flexible lid domain that spans residues 86-124 and includes helices α4-α6, the core backbone structure of the C. neoformans NBD in the apo state and in complex AUY922 aligns well with the previously determined human and C. albicans structures (Figure 5A). The conservation of this core, which includes the entire β sheet and α1, across three distantly related species in both apo and ligand-bound states demonstrates that this core is rigid and not easily deformed. In the more flexible lid domain, two of the three C. neoformans apo chains resemble the “closed” (catalytically active) state of the human structure (PDB: 1YER73), as defined by the inward positioning of the catalytic loop between helices α4 and α5, while the third chains adopts the “open” state, similar to that seen in the human structure (1YES73). In the apo and AUY922 structures, the lid adopts a range of conformational states and the catalytic loop between helices α4 and α5 ranges in length from 2 to 8 residues. The lid region in the six RAP structures, however, are similar to each other but differ from the previously described structures. Notably, helices α4 and α5 are fused into a single continuous helix from residues 86-110 that curves around the nucleotide binding site (Supplemental Figure S6). The structure of this lid region is similar in all six RAP complex structures, however there are minor differences in the conformation of the fused helix, including a bulge74 at residue at L93 in 3 of 6 cases. In addition, and in contrast to the C. neoformans apo and AUY922 structures, the six RAP structures show consistent changes in the conserved core of the domain. Notably, strand β1 could not be modelled due to disorder, while α1 and the following loop connecting to α2 are shifted relative to the canonical structure (Figure 5B). Similar N-terminal changes involving β1 and α1 have been observed in structures of C. albicans with AUY922 and the C. albicans-selective Hsp90 inhibitor CMLD013075 (20, Figure 4), although the specific details differ (Figure 5C).42
Figure 5.

Overlay of human, C. albicans and C. neoformans Hsp90 NBD structures, viewed from the “back” side of the domain. In this view, the ligand-binding pocket and lid helices α4-α6 are behind the protein, away from the viewer, and are deemphasized in order to accentuate the changes in the β-sheet and α1. (A) Superposition of Cα traces from 21 structures that share a similar core. Traces are colored black, with β-strands in blue with blue labels. The lid is in light gray behind strands β5-β4-β7. Bound ligands in the nucleotide binding pocket are shown in pale orange behind strands β4-β7. The structures include the human protein in the apo state (PDB ID 1YER and 1YES, 2 chains) and in complex with radicicol (4EGK, 1chain), SNX-2112 (4NH7, 2 chains), AUY922 (2VCI, 1 chain), AT13387 (2XJX, 1 chain), and PU3 (1UY6, 1 chain); C. albicans apo (6CJI, 1 chain) and in complex with ADP (6CJJ, 1 chain), radicicol (6CJL, 2 chains), and SNX-2112 (6CJR, 1 chain)); and C. neoformans apo (7K9R, 3 chains) and in complex with AUY922 (7K9S, 6 chains). (B) Same as A, but with the addition of structures from C. neoformans with compounds 10 (7K9W, 2 chains), 18 (7K9V, 2 chains), and 19 (7K9U, 2 chains), shown in red. This set shows consistent changes in β5, β6 and α1 and a disordering of strand β1 (not modeled in the crystal structures). (C) Same as A, but with the addition of chains from C. albicans in complex with AUY922 (6CJS, 1 chain) and CMLD013075 (20) (6CJP, 2 chain), shown in magenta. Changes in the core structure are seen in β6, β1 and α1. (D) Individual structures from the box in panel B showing C. neoformans in complex with AUY922 (7K9S, left) and 18 (7K9V, right).
Overall, five regions of the NBDs make contact with the ligands: residues from α2, the loop that precedes α4, the α4- α5 segment of the lid, helix α7, and residues on the inward-facing strands of β4, β5, β7 of the main sheet (Supplemental Figure S7). With the exception of the lid, there is little structural variability in the backbone of these elements, and most of the side chains adopt similar rotamers irrespective of the species or bound ligand. As previously described, the vast majority of the Hsp90 residues that are in direct contact with ligands are identical between the three species, with the exception of positions S38/A41/S52 on α2 and M173/L176/V186 on β7 (C. neoformans/C. albicans/human residue numbering). These side chains occupy similar volumes in all structures and are roughly isosteric, but they do make contributions to the surface of the pocket that accepts the resorcinol ring, among others. These positions may modulate ligand binding affinity and thus may contribute to species specificity.
A comparison of the C. neoformans complex with 10 and the human complex with AT13387 shows excellent overlap between the common resorcinol and isoindoline rings (Figure 6A). In comparison to other liganded Hsp90 structures in the PDB, the RAP aminopyrazole ring is positioned in an induced pocket that is similar to, but slightly deeper than, the pocket induced by the indazole ring of nonselective inhibitor SNX-2112.41, 42 While originally designed to mimic the CMLD013075 oxime, the aminopyrazole instead sits in an orthogonal position, with a π-stacking interaction between the R2 aryl ring and Hsp90 F124.
Figure 6.

Comparison with previously reported complexes. (A) Overlay of the human Hsp90/AT13387 complex (2XJX, cyan carbon atoms) with the C. neoformans/10 complex (7K9W, orange carbon atoms). The surface of the fungal protein is shown, and the AT13387 piperazine ring is omitted for clarity. (B) Staggered view of aligned ligands from four complexes: the two chains from the C. neoformans/10 crystal structure (7K9W), human/SNX2112 (4NH7), and human/PU3 (1UY6). The gray oval represents the deep hydrophobic pocket of Hsp90, indicated in the left part of panel A.
The R2 aryl rings in the RAP series introduce an important feature that distinguishes these compounds from previously reported inhibitors.62 A comparison of the structures of the human and two fungal NBDs in complex with AUY922 reveal highly similar structures, with the exception of moderate shifts in the α4-α5 lid region (Figure 7A). However, in all six RAP structures, the lid is displaced outward in response to the R2 rings (Figure 7B). Hydrophobic residues L93, F124 and W148 are in close contact in all of the apo and AUY922 complexes (Figure 7C, D) but are displaced in the RAP structures in order to accommodate the aryl rings (Figure 7E, F). The aromatic residues F124 and W148 in the NBD are on the relatively well-fixed α7 and β5 strand elements, while L93 is on the more malleable lid and is more easily displaced in order to accept the aryl ring. The disruption of this packing and outward displacement of L93 promotes the adoption of an α-helical conformation in the loop between α4 and α5, resulting in the formation of a continuous helix that spans the N-terminus of α4 to the C terminus of α5 (residues 86-110) (Supplemental Figure S6). Equivalent L93/F124/W148 packing is observed in the majority of apo and complex NBD structures from human and C. albicans, and similar disruptions of this core have been observed by the aminopyrazole ring of the non-selective inhibitor SNX2112 in human41and C. albicans,42 and the methoxy-substituted aryl ring of PU3 in human (Figure 6B).72, 75 Notably, the RAP R2 aryl ring penetrates more deeply into this site and engages F124 in π-π stacking interaction (Figures 6, 7), potentially offsetting the energetic cost of repacking the hydrophobic core.
Figure 7.

Ligand-driven reorganization of the C. neoformans binding pocket. (A) Overlay of the human (light blue, 2VCI), C. albicans (red, 6CJS) and C. neoformans (orange, 7K9S) complexes with AUY922. (B) Overlay of the C. neoformans NBD in complex with AUY422 (orange, 7K9S) and RAP compounds 10 (7K9W), 18 (7K9V) and 19 (7K9U) (dark blue). (C-F) Residues L93, F125 and W128 from helices α4, α7 and strand β5, respectively, are in packing contact in the apo (6CJI, C) and AUY922 (7K9S, D) complexes, but are disrupted by the R2 aryl ring of the RAPs (7K9W, E/F). The two chains in the co-crystals of the C. neoformans NBD with 10 reveal different rotamers of the meta-methoxy aryl ring, shown in panels E and F.
The C. neoformans structures reveal two packing arrangements of the RAP R2 aryl groups (Figure 6B, Supplemental Figure S6). In four of the complexes (chain A of 10, chains A and B of 18 and chain A of 19), the aryl ring is in the position shown in Figure 7E, while in the other two (chain B of 10 and 19), the conformer is as in Figure 7F. Thus, the pocket can adjust to two rotamers of the ligand aryl ring relative to the aminopyrazole ring. These structures, in combination with the SNX-2112 and AT13387 structures, demonstrate the plasticity of this induced binding pocket in both fungal and human Hsp90s. While we expect that the RAPs can induce similar rearrangements in the human protein, the weaker Kds strongly suggests that the energetics of repacking differs between the two species. However, there is no obvious trend to show how the aryl ring substituents may favor different binding states, but these structures clearly show that ring substitution is a viable strategy to modulate the binding affinity within this series of inhibitors.
The insertion of RAP aryl rings displaces and remodels the α4-α5 lid, but also has smaller but significant effects on β5 due to backbone shifts at residue W148 (Figures 7E-F, 8). This has a domino effect that affects neighboring strand β6 by displacing F156, ultimately resulting in the loss of the N-terminal β1 strand of the sheet (Figure 8). In turn, the disruption of stand β1 results in the shift in α1 since the N-terminus of this helix is no longer anchored to the protein (Figures 5, 8).
Figure 8.

RAP binding disrupts the β-sheet. (A) The overlay of the C. neoformans and human structures from Figure 5 were divided into two sets. Set A includes all of the eight human structures [1YER, 1YES, 4EGK, 4NH7 (2 chains), 2VCI, 2XJX, and 1UY6] and the nine C. neoformans apo and AUY922 structures [7K9R (3 chains), and 7K9S (6 chains)], and is shown in black Cα trace. Set B includes the six fungal RAP complexes [7K9U (2 chains), 7K9V (2 chains) and 7K9W (2 chains)] and is shown in red Cα trace. Selected side chains from set A are show in stick representation with van der Waals surface and fungal/human numbering. (B) The same backbone traces are represented, but with the sidechains from set B as well as the RAP inhibitor 10 in space filling representation. Note that strand β1 is present only in the structures from set A.
Each of the three major ligand-induced conformational changes seen in the C. neoformans RAP complexes (disruption of the L93/F125/W128 core, fusion of the lid helices and disruption of the N-terminus) have been observed in other NBD complexes, but not in the same structures (Supplemental Table 7). Given the current data, it is challenging to propose a single unifying model to explain how these changes are correlated to each other and to ligand binding. We propose, however, that the fusion of the lid helices is not directly correlated with the other observed changes and is most likely driven by direct interactions with the ligands. Support for this model can be found in the structures with AUY922, which are known for all three species under consideration. The binding of AUY922 to the C. albicans NBD results in a fused lid helix and disrupted N-terminus but does not disrupt the core, while none of these changes are present in the human AUY922 complex. In addition, a fused lid helix is seen in two of the six AUY922 complexes with the C. neoformans NBDs, but all have an intact L93/F125/W128 core and an ordered N-terminus. The second pattern that emerges from the set of complexes is that the disruption of the N-terminal region co-occurs with the disruption of the W148/F156 stack between strands β5 and β6. This stack is disrupted in some, but not all, of the ligand complexes that disrupt the L93/F125/W128 core. These include the six C. neoformans/RAP structures presented here and the C. albicans complexes with AUY922 and CMLD013075.42 Complexes with SNX2112 (human, C. albicans) and PU3 (human) are able to disrupt the core packing, but do not reach deeply enough into the induced pocket to affect the position of W148, and consequently, these ligands do not disrupt the β1/α1 N-terminus. It is notable that N-terminal remodeling has been observed in approximately half of the available C. neoformans and C. albicans structures, but never in a human structure. This may simply reflect the selection of structures that are currently available, or it may be because the fungal NBDs are more prone to structural rearrangements in regions outside of the lid. Overall, these results reflect the complex interplay between the ligands and the proteins, and supports the idea that Hsp90 inhibitors can be designed with species-specific properties.
Summary of Structure-Activity Relationships and Insights from Modeling
With X-ray crystallographic structures providing an improved understanding of the binding modes of R1-methylated, R2 monosubstituted RAPs, we next attempted to further rationalize the observed structure-activity and structure-property relationships across different RAP structural subtypes. Figure 9 shows a scatter plot summarizing the cryptococcal fold-selectivity (Y-axis), biochemical potency (color), and whole cell activity (symbol) of the cohort of inhibitors from Table 4, binned on the X-axis based on their patterns of substitution at R1 (methyl vs. bulky aliphatic substituents) as well as at the R2 phenyl ring (2-substituted, 3-substituted, 2,3-disubstituted and 2,5-disubstituted). From this plot, we can glean generalized SAR trends through head-to-head comparisons of different cohorts. For example, in comparing groups A to B and E to F, it is clear that ortho-monosubstituted compounds in group A generally have very good (low nM) biochemical potencies but are limited in their fungal selectivity. In contrast meta- monosubstitutions (groups B and F) generally lead to suppression of biochemical potency, in some cases to the μM range. Interestingly, for the meta-monosubstituted subset B, selectivity does not appear to track as cleanly with biochemical activity as compared to the ortho-monosubstituted subset A. Also apparent is the general overall improvement in biochemical potency and selectivity that is achieved when incorporating a second substituent at R2 (groups C/D/G/H as compared to groups A/B/E/F). It is also notable that group to group, these improvements in selectivity do sometimes come at a cost: for example, the most potent and selective N-methylated, 2,3-disubstituted inhibitors (group D) tend to have poor whole cell activity, and highly selective inhibitors bearing bulky R1 substituents are generally less potent than their N-methylated counterparts (cf. groups G/H and groups C/D).
Figure 9.

Scatter plots of cryptococcal selectivity, biochemical potency, and whole cell activity for Table 4 RAPs, binned by aminopyrazole substructure class as defined by the R1 and R2 substitution patterns. The Y-axis depicts cryptococcal fold-selectivity on a logarithmic scale. Points are colored by H99 lysate EC50 on a logarithmic scale, on a gradient ranging from cyan (10 μM) to red (1 nM). Point symbols indicate whole cell activity class, with circles corresponding to compounds with little to no whole cell activity (H99 MIC ≥25 μM), and stars indicating whole cell active compounds with a H99 MIC <25 μM.
In judging these structural subclasses by their full complement of relevant parameters, two clear "optimal" themes emerge (Figure 10A): first, for compounds that are N-methylated at R1, overall whole cell activity is best among R2 disubstituted compounds where the substituents have a 2,5 relationship (group C); in contrast, compounds bearing bulky aliphatic groups at R1 generally exhibit improved potency and selectivity when R2 is 2,3-disubstituted (group H). Interestingly, these two orthogonal subsets also diverge somewhat in the nature of their "optimal" properties; while group C is exemplary in biochemical potency and selectivity, group H is stronger in terms of whole cell anti-cryptococcal activity; this feature is even more pronounced when considering the relative whole cell potencies, which for simplicity are not represented here. In Figure 10B we provide representative leading members of each of these two classes, as starting points for future optimizations of potency, selectivity, whole cell activity, and additional ADME parameters including metabolic stability.
Figure 10.

(A) Two predominant structural classes among RAPs with optimal combined biochemical potency/fungal selectivity/whole cell activity profile, each with slightly divergent activity profiles; (B) Representative structures and activities of top inhibitors 9ac, 9ad, 9ba and 9bb from these subclasses.
We next attempted to model these various structural subclasses into our existing X-ray structures to rationalize structure-activity trends observed among the R2 disubstituted inhibitors. In doing so, it is important to consider that due to the inherent plasticity of the Hsp90 NBD and the number and scope of remodeling events which have so far been documented across different species and in co-complex with different ligand structural subclasses, any efforts to model other RAP structural subclasses into these rigid, RAP-induced receptors is of only limited and speculative utility. Nonetheless, in each RAP-liganded X-ray structure the R1-methyl substituent is projected outward and solvent exposed; we posit that expansion of this group to a larger lipophilic moiety is likely to create entropic penalties due bulk water disruption, which may in part explain the general overall trend of lowered biochemical EC50s as the size and lipophilicity of this substituent increases. Given the proximity of this solvent-exposed group to a polar region of the binding pocket periphery, it is also possible that incompatible sterics and polarities also compound this issue. In addition, since the overall structural disruptions of the NBD appear to be induced by the R2 aryl ring, we postulate that the added steric demands imposed by a second substituent on this ring may be the source of further improvements to selectivity. While we are reluctant to definitively propose binding modes for the R2-disubstituted RAPs, we have performed computational (Glide) docking76, 77 of the 24 RAPs from Table 4 to explore whether the pockets induced by compounds 10, 18 and 19 are large enough to accommodate larger R1 and R2 substituents in similar binding poses without any further protein structure remodeling (Figure 11 and Supplemental Table 8). Glide docking with positioning restricted to the reference RAP ligand core produced excellently scored poses (−14.3 to −15.5 kcal/mol), for 20 out of the 24 compounds into at least one of the six independent chains with a core atom RMSD to the native ligand of < 0.1 Å. Notably, each compound also produced at least one high-scored docking pose for each of the two types of R2 rotamers that were observed for compound 10 in the independent chains of the 7K9W structure, underscoring the flexibility of this pocket to accommodate disubstituted R2 aryl rings in multiple orientations. Select docking poses of compounds 9an, 9ba, and 9bc which illustrate this phenomenon are depicted in Figure 11B-G.
Figure 11.

Results of computational modeling using RAP-liganded X-ray crystal structures. (A) X-ray crystal structure (7K9V) with compound 18 bound at Chain A; (B, C) comparative view of two rotamers of compound 9an docked into 7K9V-Chain A. Docking scores are −15.3 kcal/mol and −14.5 kcal/mol, respectively; (D, E) comparative view of two rotamers of compound 9ba docked into structure 7K9V-Chain A. Docking scores are −14.7 kcal/mol and −14.5 kcal/mol, respectively; (F, G) comparative view of two rotamers of compound 9bc docked into structure 7K9V-Chain A. Docking scores are −14.2 kcal/mol and −14.4 kcal/mol, respectively.
Out of the set of RAPs docked, only the R1-cyclohexyl substituted compounds (9j, 9az, 9bd and 9bi) failed to produce any poses in any of the six chains under the applied constraints, which we ascribe to the entropic and steric factors described above. For the remaining 20 compounds, very high docking scores were obtained across compounds with widely varying biochemical potencies and selectivities; as such, it is difficult to fully rationalize the divergent activities on the basis of this structural modeling. However, this limited computational study does confirm an overall compatibility of the R2-disubstituted RAPs with the RAP-induced binding pocket. In addition, the accommodation of multiple R2 substituents and in multiple orientations suggests that the potency gains observed for disubstituted compounds may in part arise due to additional beneficial van der Walls interactions and an improved occupancy of the deep hydrophobic cavity in the R2-induced pocket.78, 79 What is not clear from this modeling, however, is whether these added R1/R2 substituents may induce further changes to the protein structure, nor do they explain what role the size and nature of the R1/R2 groups play in other important improvements such as whole cell permeance and reduced mammalian cytotoxicity. Given the number of unexpected ligand-induced conformations that have been observed in fungal Hsp90 structures to date, further crystallographic work is needed to fully understand the binding modes of the optimized R2-disubstituted compounds described herein.
CONCLUSION
Continuing in our pursuit of fungal selective, resorcylate inhibitors of the protein-folding chaperone Hsp90, and building on early leads from our prior study of resorcylate aminopyrazoles (RAPs), we have used a rational structure-property approach to improve the whole-cell fungal permeance of a lead series which exhibits high selectivity for cryptococcal Hsp90. Towards this goal, we have achieved synthesis of 22 new fungal selective RAPs that effectively inhibit growth of C. neoformans whole cells at low-to-sub micromolar concentrations. Further pharmacological and biological characterization of these RAPs showed that despite moderate improvements in both microsomal stability and whole cell anti-cryptococcal activity, further optimization of fungal permeance is still needed to achieve an adequate therapeutic window for use in infection-relevant contexts. We have also solved the first X-ray crystal structures of C. neoformans Hsp90 in various states, including structures in complex with three fungal selective RAPs. Conformational rearrangements induced by RAPs, while unique within the direct binding site, create downstream conformational reorganizations that are consistent with other changes induced by a resorcylate compound that selectively binds C. albicans Hsp90. Further efforts to parlay these foundational findings into improved fungal selective Hsp90 inhibitors for use as chemical biological probes and eventually therapeutics are ongoing.
EXPERIMENTAL SECTION
Fungal Strains and Culture Conditions.
The strain used in this study was C. neoformans H99.80 Archive of this strain was maintained at −80 °C in 25% glycerol. Active cultures were maintained on solid (2% agar) yeast extract peptone (YPD, 1% yeast extract, 2% bacopeptone, 2% glucose) at 4 °C for no more than 1 month.
Antifungal Sensitivity Testing.
Minimum inhibitory concentrations (MICs) were determined in flat bottom, 96-well plate format using RPMI medium (Gibco SKU no. 318000-089, 3.5% MOPS, 2% glucose, pH 7.0). A modified broth microdilution protocol was used as previously described,23, 81 except that relative viable cell number was monitored by standard dye reduction assay after a 3 h incubation with resazurin at 37 °C. All compounds were formulated in dimethyl sulfoxide (DMSO, Sigma-Aldrich Co.). Each compound was tested in duplicate in at least two independent experiments.
Mammalian cell culture and cytotoxicity testing.
The cell lines NIH 3T3 (CRL-1658) and HepG2 (HB-8065) were purchased from the American Type Culture Collection (ATCC). Cells were maintained in RPMI medium supplemented with 10% fetal bovine serum at 37 °C under 5% CO2. Experiments were performed using cells within 10 passages post recovery from low-passage stocks maintained in liquid nitrogen and confirmed negative for mycoplasma contamination by PCR-based assay. Cells were plated in 384-well format at a density of 2000 per well (HepG2) or 2500 per well (NIH 3T3) in RPMI supplemented with 10% fetal bovine serum (FBS). Following overnight adherence, two-fold dilutions of test inhibitors were added to wells and plates incubated for 72h. Subsequent determination of relative viable cell number was performed using standard resazurin dye-reduction assay with incubation at 37 °C for 3h and measurement in a Tecan Spark microplate reader using SparkControl software (version 2.2). Nonlinear four-parameter curve fitting of raw dose-response data was performed in GraphPad Prism 7 to determine IC50 values. Results reported are representative of two independent experiments, each performed with technical triplicates.
FP Assays.
FP assays performed with whole cell lysates and purified C. neoformans Hsp90 NBD were performed as previously described.62 In calculating fold-selectivity for compounds a normalization factor of 0.84 was applied based on the EC50 values determined in human and fungal cell lysate for geldanamycin, the same Hsp90 inhibitor used to generate the FP probe itself.
NBD Expression and Purification.
Recombinant Hsp90 NBDs were expressed and purified as previously described.42, 62
Analytical LC-MS/MS conditions.
Compound levels for in vitro metabolic stability assays were monitored by LC-MS/MS using an AB Sciex (Framingham, MA) 4000 QTRAP® mass spectrometer coupled to a Shimadzu (Columbia, MD) Prominence LC. The parent ion and the two most prominent daughter ions were followed to confirm compound identity, although only the most abundant daughter was used for quantitation. Analytes were detected with the mass spectrometer in negative MRM (multiple reaction monitoring) mode for the following compounds: 9i 541.3-404.0; 9j 541.2-404.0; 9n 477.1-358.0; 9o 551.3-432.0; 9ac 457.1-337.9; 9ad 475.2-338.1; 9ae 475.1-337.9; 9ah 475.2-338.0; 9ak 477.1-357.9. The following compounds were detected in positive MRM mode: 9d 483.2-308.0; 9v 487.2-312.0; 9af 459.2-340.1; 9am 463.1-344.1; 9ar 529.1-410.1; 9av 497.2-322.0; 9ax 501.2-326.0; 9az 527.2-326.0; 9ba 487.2-326.1; 9bb 501.2-326.0; 9bc 513.2-326.0; 9bd 527.2-326.1; 9be 491.2-330.1; 9bf 505.2-330.0; 9bg 523.2-330.1; 9bi 531.2-330.1. An Agilent (Santa Clara, CA) C18 XDB column (5 micron, 50 X 4.6 mm) was used for chromatography for compounds detected in negative mode with the following conditions: Buffer A: dH2O + 0.1% formic acid, Buffer B: methanol + 0.1% formic acid, 0 - 1.0 min 5% B, 1.0 - 2 min gradient to 100% B, 2 - 3.5 min 100% B, 3.5 - 3.6 min gradient to 5% B, 3.6 - 4.5 5% B. Tolbutamide (transition 269.1-169.9) from Sigma (St. Louis, MO) was used as an internal standard (IS). Conditions for compounds evaluated in positive mode were nearly identical except the gradient conditions were as follows: 0 - 1.0 min 5% B, 1.0 – 1.5 min gradient to 100% B, 1.5 - 3.0 min 100% B, 3.0 - 3.2 min gradient to 5% B, 3.2 - 4.5 5% B and N-benzylbenzamide (Sigma) was used as the internal standard (transition 212.1-91.1). Solvents (methanol and water) and formic acid were all LC-MS/MS grade (“Optima”) and purchased from Fisher Scientific (Waltham, MA).
Mouse liver microsome stability.
Female ICR/CD-1 mouse microsome fractions were purchased from BioIVT (Westbury, NY), and the protocol provided with the microsomes for assessment of compound stability was followed as detailed herein. All chemical reagents aside from the aforementioned solvents were purchased from Sigma (St. Louis, MO). Microsome protein (0.5 mg/mL) was added to a glass screw cap tube then 50 mM Tris, pH 7.5 solution, containing the compound of interest in DMSO (final DMSO concentration 0.1%) was added on ice. The final concentration of compound after addition of all reagents was 2 μM. An NADPH-regenerating system (1.7 mg/ml NADP, 7.8 mg/mL glucose-6-phosphate, 6 U/mL glucose-6-phosphate dehydrogenase in 2% w/v NaHCO3/10 mm MgCl2) was added for analysis of Phase I metabolism after both the microsome/compound mixture and regenerating system were warmed to 37 °C for 5 min. The tube was then placed in a 37 °C shaking water bath. At varying time points after addition of the NADPH regenerating system, the reaction was stopped by the addition of 0.5 mL of methanol containing an internal standard (IS) and formic acid such that the final concentration of tolbutamide IS was 50 ng/mL, N-benzylbenzamide IS was 100 ng/mL and acid was 0.1%. Time 0 samples were quenched with methanol prior to addition of compound. The samples were incubated 10’ at room temperature and then spun at 16,100 x g for 5 min in a microcentrifuge at 4 °C. The supernatant was analyzed by LC-MS/MS. The method described in McNaney, et al.69 was used with modification for determination of metabolic stability half-life by substrate depletion. A “% remaining” value was used to assess metabolic stability of a compound over time. The LC/MS/MS peak area of the incubated sample at each time point was divided by the LC/MS/MS peak area of the time 0 (T0) sample and multiplied by 100. The natural Log (LN) of the % remaining of compound was then plotted versus time (in min) and a linear regression curve plotted going through y-intercept at LN(100). The metabolism of some compounds failed to show linear kinetics at later time point, so those time points were excluded. The half-life (T ½) was calculated as T½ = −0.693/slope. To determine intrinsic clearance,82-85 which allows for comparison of data obtained using different volumes and protein concentrations, the following calculations were employed:
Statistical Methods.
For FP experiments in support of SAR studies, GraphPad Prism 5.0 was used to perform curve fitting and calculate the concentrations of compounds resulting in 50% reduction in maximal polarization signal (EC50). All curve fits demonstrated a correlation coefficient (R2) > 0.95 The number of independent experiments performed and the number of technical replicates in each experiment are provided in the legends of figures and tables characterizing the biochemical and biological activities of compounds. In calculating the error of selectivity determinations, the fractional error of measurements in each species was summed to yield a composite error for the derived ratio.
Protein crystallization and structure determination.
Aliquots of purified C. neoformans NBD (residues 1-211 or 1-215) were crystallized either alone (apo) or in the presence of AUY922 or RAPs 10, 18 and 19. Crystals were obtained by mixing one part of protein solution at 10–15 mg/mL (20 mM Hepes pH 7.5 , 500 mM NaCl, 8% glycerol, 10% DMSO with or without 2 mM ligand) with one part of reservoir solution. Crystals were obtained by sitting-drop vapor-diffusion at 21 °C. The best diffracting crystals were produced with a reservoir containing 20-25% PEG6K, 0.1 M Hepes pH 7, and 1 M LiCl2.
Crystals were cryo-protected by passage through paratone before flash cooling in liquid nitrogen. Diffraction data were collected at the APS or NSLS II synchrotrons as indicated in Supplementary Tables 5 and 6. Data were processed and integrated with XDS86 and Aimless87 and structures were determined by molecular replacement using C. albicans Hsp90 NBD (PDB ID 6CJI) as a search model for the apo structure. The apo structure was then used to solve the RAP complexes by molecular replacement. Model building was done with Coot88 and the structures were refined with Phenix.89 Software used in this project was curated by SBGrid.90
Computational Docking.
Molecular docking of the 24 compounds listed in Table 4 was performed using six separate docking grids (7K9U Chain A, 7K9U Chain B, 7K9V Chain A, 7K9V Chain B, 7K9W Chain A, and 7K9W Chain B) using the Glide docking program (Schrödinger Maestro Version 12.4.079). Proteins were treated using Schrödinger’s Protein Preparation Wizard with retention of waters, optimization of hydrogen bonds, and restrained minimization converging heavy atoms to RMSD 0.3 Å. Protein structures were kept rigid during docking experiments. Docking grids were prepared using a receptor-based box centered at the native ligand for each chain. Ligands were prepared using Schrodinger’s LigPrep and kept at a neutral ionization state. Docking was run at Standard precision with flexible ligands, with the docking restricted to a reference position (identified using Maximum Common Substructure) defined by the 5-((1H-pyrazol-5-yl)amino)benzene-1,3-diol core substructure from the native ligand of each chain. The output was set to report up to five poses per ligand, and Epik state penalties were included the calculated docking score. All of the output docking poses fell into one of the two possible R2 aryl ring rotamers as originally observed for compound 10 in the 7K9W X-ray structure. Where two R2 aryl ring rotamers were present, the highest-scored poses for each of the two rotamers was kept for analysis; otherwise, only the highest-scored pose for each compound into each chain was kept. Tabular results for docking each compound into each chain are reported in Supplemental Table 8.
Chemistry Methods
General Methods
1H NMR spectra were recorded at 400 or 500 MHz at ambient temperature. 13C NMR spectra were recorded at 101 or 126 MHz at ambient temperature. 19F NMR spectra were recorded at 470 MHz at ambient temperature. Chemical shifts are reported in parts per million. Data for 1H NMR are reported as follows: chemical shift, multiplicity (app = apparent, br = broad, s = singlet, d = doublet, t = triplet, q = quartet, sxt = sextet, hept = heptet, m = multiplet,), coupling constants, and integration. All 13C NMR spectra were recorded with complete proton decoupling. Analytical thin layer chromatography was performed using 0.25 mm silica gel 60-F plates. Silica flash chromatography was performed using prepacked columns (SI-HC, puriFlash or Premium Universal, Yamazen) on either an Interchim puriFlash450 or Yamazen Smart Flash EPCLC W-Prep2XY system. All mass-guided preparative HPLC was performed using an acetonitrile/water gradient (mobile phase modified with 0.01% formic acid) on a Waters FractionLynx system equipped with a 600 HPLC pump, a micromass ZQ quadrupole, Waters 996 diode array, and Sedere Sedex 75 ELS detectors, using an XBridge Prep C18 5 μM OBD 19 mm diameter column of either 100 mm or 250 mm length. Isolated yields refer to chromatographically and spectroscopically pure compounds, unless otherwise stated. All reactions were carried out in oven-dried glassware under a nitrogen atmosphere unless otherwise noted. Analytical UPLC–MS experiments were performed using a Waters Acquity (ultraperformance liquid chromatography) with a binary solvent manager, SQ mass spectrometer, Waters 2996 PDA (photodiode array) detector, and evaporative light scattering detector (ELSD). All microwave experiments were performed on a CEM Discover microwave reactor, using a sealed 10 or 35 mL vessel with temperatures monitored by an external sensor. All compounds tested in biological assays were determined to be >95% pure by UPLC–MS-ELSD analysis
α-Cyanoketone synthesis
α-Cyanoketones 3a, 3c, 3e, 3g, 3h, 3i, 3k, and 3l were obtained from commercial sources.
General Procedure A
To a flame-dried nitrogen flushed round bottom flask equipped with a magnetic stir bar was added anhydrous THF (4 mL) and nBuLi solution (3 mmol, 1.2 equiv). The solution was cooled to −78 °C in a dry ice/acetone bath. To the reaction flask was added a solution of MeCN (0.26 mL, 5 mmol, 2 equiv, 2 M in THF) dropwise over 2 minutes. The mixture was stirred for 1 hour at −78 °C. A solution of methyl ester 2 (2.5 mmol, 2 M in THF) was prepared and added to the reaction flask dropwise over 2 minutes. The reaction was stirred at −78 °C for 30 minutes then at room temperature for 2 hours. The reaction was quenched with 1 M HCl (30 mL), extracted with EtOAc (3 x 50 mL), washed with brine, dried with Na2SO4 and concentrated under reduced pressure. Purification by silica flash chromatography (2-40% EtOAc in hexanes) afforded the desired α-cyanoketone product 3.
3-Oxo-3-(2-(trifluoromethyl)phenyl)propanenitrile (3b) was prepared from methyl 3-(trifluoromethyl)benzoate (0.78 mL, 5 mmol) according to General Procedure A. 3b (799 mg, 3.75 mmol, 74% yield). 1H NMR (400 MHz, CDCl3) δ 7.82 – 7.75 (m, 1H), 7.73 – 7.64 (m, 2H), 7.54 – 7.48 (m, 1H), 3.95 (s, 2H).
3-Oxo-3-(2-(trifluoromethoxy)phenyl)propanenitrile (3e) was prepared from methyl 2-(trifluoromethoxy)benzoate (1.10 g, 5 mmol) according to General Procedure A. 3e (1.08 g, 4.73 mmol, 94% yield). 1H NMR (400 MHz, CDCl3) δ 7.88 (dd, J = 7.9, 1.8 Hz, 1H), 7.70 – 7.63 (m, 1H), 7.49 – 7.41 (m, 1H), 7.41 – 7.33 (m, 1H), 4.05 (s, 2H).
3-Oxo-3-(3-(trifluoromethoxy)phenyl)propanenitrile (3i) was prepared from methyl 3-(trifluoromethoxy)benzoate (1.10 g, 5 mmol) according to General Procedure A. 3i (979 mg, 4.27 mmol, 85% yield). 1H NMR (400 MHz, CDCl3) δ 7.86 – 7.81 (m, 1H), 7.79 – 7.75 (m, 1H), 7.62 – 7.55 (m, 1H), 7.53 – 7.48 (m, 1H), 4.13 (s, 2H).
3-(2,3-Dimethylphenyl)-3-oxopropanenitrile (3l) was prepared from methyl 2,3-dimethylbenzoate (985 mg, 6 mmol) according to General Procedure A. 3l (647 mg, 3.74 mmol, 62% yield). 1H NMR (400 MHz, CDCl3) δ 7.37 (d, J = 7.7 Hz, 2H), 7.21 (t, J = 7.7 Hz, 1H), 3.99 (s, 2H), 2.40 (s, 3H), 2.34 (s, 3H).
3-(5-Fluoro-2-methylphenyl)-3-oxopropanenitrile (3m) was prepared from methyl 5-fluoro-2-methyl-benzoate (1.01 g, 5 mmol) according to General Procedure A. 3m (706 mg, 3.98 mmol, 66% yield). 1H NMR (400 MHz, CDCl3) δ 7.34 – 7.27 (m, 2H), 7.20 (td, J = 8.1, 2.6 Hz, 1H), 4.02 (s, 2H), 2.52 (s, 3H).
3-(2-Fluoro-3-methylphenyl)-3-oxopropanenitrile (3n) was prepared from methyl 2-fluoro-3-methyl-benzoate (1.01 g, 6 mmol) according to General Procedure A. 3n (1020 mg, 5.76 mmol, 95% yield). 1H NMR (400 MHz, CDCl3) δ 7.76 (t, J = 7.3 Hz, 1H), 7.51 – 7.43 (m, 1H), 7.22 – 7.13 (m, 1H), 4.12 – 4.05 (m, 2H), 2.34 (s, 3H).
3-(2,3-Difluorophenyl)-3-oxopropanenitrile (3o) was prepared from methyl 2,3-difluorobenzoate (1.03 g, 6 mmol) according to General Procedure A. 3o (694 mg, 3.83 mmol, 63% yield). 1H NMR (400 MHz, CDCl3) δ 7.76 – 7.68 (m, 1H), 7.53 – 7.43 (m, 1H), 7.30 – 7.21 (m, 1H), 4.11 (d, J = 2.4 Hz, 2H).
3-(2-Methyl-3-(trifluoromethyl)phenyl)-3-oxopropanenitrile (3p) was prepared from methyl 2-methyl-3-(trifluoromethyl)benzoate (501 mg, 2.3 mmol) according to General Procedure A. 3p (449 mg, 1.98 mmol, 85% yield). 1H NMR (400 MHz, CDCl3) δ 7.84 (d, J = 7.9 Hz, 1H), 7.67 (d, J = 7.8 Hz, 1H), 7.44 (t, J = 7.9 Hz, 1H), 4.00 (s, 2H), 2.57 (s, 3H).
3-(3-Chloro-2-fluorophenyl)-3-oxopropanenitrile (3q) was prepared from methyl 3-chloro-2-fluoro-benzoate (471 mg, 2.5 mmol) according to General Procedure A. 3q (443 mg, 2.24 mmol, 89% yield). 1H NMR (400 MHz, CDCl3) δ 7.89 – 7.81 (m, 1H), 7.73 – 7.64 (m, 1H), 7.30 – 7.19 (m, 1H), 4.11 (s, 2H).
3-(2-Fluoro-3-(trifluoromethoxy)phenyl)-3-oxopropanenitrile (3r) To a 20 mL scintillation vial was added 2-fluoro-3-(trifluoromethoxy)benzoic acid (504 mg, 2.25 mmol), concentrated sulfuric acid (0.2 mL) and methanol (6 mL, 0.38 M). The vial was sealed, and the reaction was heated at 75 °C for 18 h. The reaction was cooled to room temperature, diluted with ethyl acetate (50 mL), washed with saturated sodium bicarbonate solution (3 x 50 mL), washed with brine (50 mL), dried with Na2SO4 and concentrated under reduced pressure. The crude methyl ester was carried on without further purification and treated according to General Procedure A. 3r (445 mg, 1.80 mmol, 80% yield). 1H NMR (400 MHz, CDCl3) δ 7.96 – 7.88 (m, 1H), 7.66 – 7.58 (m, 1H), 7.39 – 7.31 (m, 1H), 4.12 (d, J = 2.7 Hz, 2H).
3-(2,5-Difluorophenyl)-3-oxopropanenitrile (3s) was prepared from methyl 2,5-difluorobenzoate (516 mg, 3 mmol) according to General Procedure A. 3s (248 mg, 1.37 mmol, 45% yield). 1H NMR (400 MHz, CDCl3) δ 7.72 – 7.60 (m, 1H), 7.40 – 7.28 (m, 1H), 7.25 – 7.14 (m, 1H), 4.10 (d, J = 2.7 Hz, 2H).
3-(2-Chloro-5-fluorophenyl)-3-oxopropanenitrile (3t) was prepared from methyl 2-chloro-5-fluoro-benzoate (471 mg, 2.5 mmol) according to General Procedure A. 3t (441 mg, 2.23 mmol, 89% yield). 1H NMR (400 MHz, CDCl3) δ 7.53 – 7.45 (m, 1H), 7.42 – 7.36 (m, 1H), 7.31 – 7.22 (m, 1H), 4.19 (s, 2H).
3-(5-Chloro-2-fluorophenyl)-3-oxopropanenitrile (3u) was prepared from methyl 5-chloro-2-fluoro-benzoate (471 mg, 2.5 mmol) according to General Procedure A. 3u (445 mg, 2.25 mmol, 90% yield). 1H NMR (400 MHz, CDCl3) δ 7.94 (m, 1H), 7.59 (m, 1H), 7.18 (m, 1H), 4.08 (d, J = 2.6 Hz, 2H).
3-(2-Fluoro-5-methylphenyl)-3-oxopropanenitrile (3v) was prepared from methyl 2-fluoro-5-methyl-benzoate (505 mg, 3 mmol) according to General Procedure A. 3v (323 mg, 1.82 mmol, 60% yield). 1H NMR (400 MHz, CDCl3) δ 7.74 (dd, J = 7.1, 2.4 Hz, 1H), 7.45 – 7.34 (m, 1H), 7.08 (dd, J = 11.3, 8.4 Hz, 1H), 4.08 (dd, J = 2.5, 0.6 Hz, 2H), 2.38 (s, 3H).
3-(2-Fluoro-5-(trifluoromethyl)phenyl)-3-oxopropanenitrile (3w) was prepared from methyl 2-fluoro-5-(trifluoromethyl)benzoate (511 mg, 2.3 mmol) according to General Procedure A. 3w (489 mg, 2.11 mmol, 91% yield). 1H NMR (400 MHz, CDCl3) δ 8.29 – 8.21 (m, 1H), 7.94 – 7.86 (m, 1H), 7.42 – 7.33 (m, 1H), 4.14 – 4.10 (m, 2H).
3-(2-Fluoro-5-(trifluoromethoxy)phenyl)-3-oxopropanenitrile (3x) To a 20 mL scintillation vial was added 2-fluoro-5-(trifluoromethoxy)benzoic acid (504 mg, 2.25 mmol), concentrated sulfuric acid (0.2 mL) and Methanol (6 mL, 0.38 M). The vial was sealed, and the reaction was heated at 75 °C for 18 h. The reaction was cooled to room temperature, diluted with ethyl acetate (50 mL), washed with saturated sodium bicarbonate solution (3 x 50 mL), washed with brine (50 mL), dried with Na2SO4 and concentrated under reduced pressure. The crude methyl ester was carried on without further purification and treated according to General Procedure A. 3x (414 mg, 1.68 mmol, 74% yield). 1H NMR (400 MHz, CDCl3) δ 7.83 (dd, J = 5.9, 2.9 Hz, 1H), 7.53 – 7.46 (m, 1H), 7.31 – 7.25 (m, 1H), 4.13 – 4.09 (m, 2H).
3-(2,6-Difluorophenyl)-3-oxopropanenitrile (3y) was prepared from methyl 2,6-difluorobenzoate (258 mg, 1.5 mmol) according to General Procedure A. 3y (169 mg, 0.93 mmol, 62% yield). 1H NMR (400 MHz, CDCl3) δ 7.60 – 7.50 (m, 1H), 7.09 – 6.99 (m, 2H), 4.02 – 3.94 (m, 2H).
3-(4-Chloro-2-fluorophenyl)-3-oxopropanenitrile (3z) was prepared from methyl 4-chloro-2-fluoro-benzoate (1.13 g, 6 mmol) according to General Procedure A. 3z (1.08 g, 5.46 mmol, 91% yield). 1H NMR (400 MHz, CDCl3) δ 8.00 – 7.87 (m, 1H), 7.34 – 7.28 (m, 1H), 7.28 – 7.20 (m, 1H), 4.10 – 4.03 (m, 2H).
3-(2-Chloro-4-fluorophenyl)-3-oxopropanenitrile (3aa) was prepared from methyl 2-chloro-4-fluoro-benzoate (1.13 g, 6 mmol) according to General Procedure A. 3aa (1.11 g, 5.62 mmol, 93% yield). 1H NMR (400 MHz, CDCl3) δ 7.79 – 7.70 (m, 1H), 7.25 – 7.19 (m, 1H), 7.16 – 7.06 (m, 1H), 4.15 (s, 2H).
5-Aminopyrazole Synthesis
5-aminopyrazoles 4a, 4f, and 4s were obtained from commercial sources.
General Procedure B
To a microwave vial equipped with a magnetic stir bar was added α-cyanoketone 3 (1 equiv), methyl hydrazine (1.1 equiv) and anhydrous methanol (1 M). The vial was capped and heated in a microwave reactor maintaining 120 °C for 90 minutes. The vial was cooled to room temperature and the reaction was concentrated under reduced pressure. The crude residue was purified by silica flash chromatography (5-60% EtOAc in CH2Cl2) to afford the desired 5-aminopyrazole.
General Procedure C
To a microwave vial equipped with a magnetic stir bar was added α-cyanoketone 3 (1 equiv), N-alkyl-hydrazine hydrochloride (1.1 equiv), triethylamine (1.5 equiv) and anhydrous methanol (1 M). The vial was capped and heated in a microwave reactor maintaining 120 °C for 90 minutes. The vial was cooled to room temperature and the reaction was concentrated under reduced pressure. The crude residue was purified by silica flash chromatography (5-50% EtOAc in hexanes) to afford the desired 5-aminopyrazole.
3-(2-Chlorophenyl)-1-methyl-1H-pyrazol-5-amine (4b) was prepared from 2-chlorobenzoylacetonitrile 3a (898 mg, 5 mmol, 1 equiv) according to General Procedure B. 4b (508 mg, 2.45 mmol, 48 % yield). 1H NMR (400 MHz, CDCl3) δ 7.76 (dd, J = 7.8, 1.8 Hz, 1H), 7.41 (dd, J = 7.8, 1.5 Hz, 1H), 7.32 – 7.18 (m, 2H), 6.11 (s, 1H), 3.75 (s, 3H), 3.56 (s, 2H).
1-Methyl-3-(2-(trifluoromethyl)phenyl)-1H-pyrazol-5-amine (4c) was prepared from 3b (710 mg, 3.33 mmol, 1 equiv) according to General Procedure B. 4c (501 mg, 2.1 mmol, 62% yield). 1H NMR (400 MHz, CDCl3) δ 7.74 – 7.60 (m, 2H), 7.54 (t, J = 7.6 Hz, 1H), 7.42 (t, J = 7.7 Hz, 1H), 5.85 – 5.77 (m, 1H), 3.77 – 3.72 (m, 3H), 3.69 – 3.51 (m, 2H).
1-(tert-Butyl)-3-(o-tolyl)-1H-pyrazol-5-amine (4d) was prepared from o-toluoylacetonitrile 3c (239 mg, 1.5 mmol, 1 equiv) and tert-butylhydrazine hydrochloride (206 mg, 1.65 mmol, 1.1 equiv) according to General Procedure C. 4d (221 mg, 0.96 mmol, 64% yield). 1H NMR (400 MHz, CDCl3) δ 7.56 – 7.51 (m, 1H), 7.37 – 7.30 (m, 1H), 7.23 – 7.12 (m, 2H), 5.77 (s, 1H), 3.56 (s, 2H), 2.51 (s, 3H), 1.69 (s, 9H).
1-Methyl-3-(o-tolyl)-1H-pyrazol-5-amine (4e) was prepared from o-toluoylacetonitrile 3c (239 mg, 1.5 mmol, 1 equiv) according to General Procedure B. 4e (135 mg, 0.72 mmol, 47% yield). 1H NMR (400 MHz, CDCl3) δ 7.55 – 7.46 (m, 1H), 7.23 – 7.17 (m, 3H), 5.73 (s, 1H), 3.74 (s, 3H), 3.51 (s, 2H), 2.46 (s, 3H).
1-Isopropyl-3-(2-methoxyphenyl)-1H-pyrazol-5-amine (4g) was prepared from 2-methoxybenzoylacetonitrile 3d (175 mg, 1 mmol, 1 equiv) and isopropylhydrazine hydrochloride (122 mg, 1.1 mmol, 1.1 equiv) according to General Procedure C. 4g (191 mg, 0.83 mmol, 82% yield). 1H NMR (400 MHz, CDCl3) δ 8.00 – 7.86 (m, 1H), 7.26 – 7.24 (m, 1H), 7.08 – 6.87 (m, 2H), 6.13 (s, 1H), 4.50 – 4.32 (m, 1H), 3.88 (s, 3H), 3.45 (s, 2H), 1.52 (d, J = 6.7 Hz, 6H).
1-Cyclohexyl-3-(2-methoxyphenyl)-1H-pyrazol-5-amine (4h) was prepared from 2-methoxybenzoylacetonitrile 3d (175 mg, 1 mmol, 1 equiv) and cyclohexylhydrazine hydrochloride (166 mg, 1.1 mmol, 1.1 equiv) according to General Procedure C. 4h (231 mg, 0.85 mmol, 84% yield). 1H NMR (400 MHz, CDCl3) δ 7.99 – 7.84 (m, 1H), 7.26 – 7.18 (m, 1H), 7.08 – 6.89 (m, 2H), 6.12 (s, 1H), 4.06 – 3.91 (m, 1H), 3.88 (s, 3H), 3.45 (s, 2H), 2.12 – 1.86 (m, 6H), 1.79 – 1.67 (m, 1H), 1.49 – 1.20 (m, 3H).
1-Cyclopentyl-3-(2-methoxyphenyl)-1H-pyrazol-5-amine (4i) was prepared from 2-methoxybenzoylacetonitrile 3d (175 mg, 1.1 mmol, 1 equiv) and cyclopentylhydrazine hydrochloride (150 mg, 1.1 mmol, 1.1 equiv) according to General Procedure C. 4i (204 mg, 0.79 mmol, 79% yield). 1H NMR (400 MHz, CDCl3) δ 8.01 – 7.80 (m, 1H), 7.27 – 7.20 (m, 2H), 7.05 – 6.84 (m, 2H), 6.13 (s, 1H), 4.64 – 4.30 (m, 1H), 3.88 (s, 3H), 3.47 (s, 2H), 2.24 – 2.11 (m, 2H), 2.11 – 1.89 (m, 4H), 1.74 – 1.61 (m, 2H).
1-Isobutyl-3-(2-methoxyphenyl)-1H-pyrazol-5-amine (4j) was prepared from 2-methoxybenzoylacetonitrile 3d (175 mg, 1mmol, 1 equiv) and isobutylhydrazine hydrochloride (137 mg, 1.1mmol, 1.1 equiv) according to General Procedure C. 4j (206 mg, 0.84 mmol, 84% yield). 1H NMR (400 MHz, CDCl3) δ 7.93 – 7.85 (m, 1H), 7.30 – 7.22 (m, 1H), 7.05 – 6.90 (m, 2H), 6.12 (s, 1H), 3.88 (s, 3H), 3.80 (d, J = 7.4 Hz, 2H), 3.44 (s, 2H), 2.36 – 2.19 (m, 1H), 0.97 (d, J = 6.6 Hz, 6H).
1-(tert-Butyl)-3-(2-methoxyphenyl)-1H-pyrazol-5-amine (4k) was prepared from 2-methoxybenzoylacetonitrile 3d (168 mg, 0.96 mmol, 1 equiv) and tert-butylhydrazine hydrochloride (131 mg, 1.05 mmol, 1.1 equiv) according to General Procedure C. 4k (193 mg, 0.79 mmol, 81% yield). 1H NMR (400 MHz, CDCl3) δ 8.01 – 7.92 (m, 1H), 7.25 – 7.19 (m, 1H), 7.05 – 6.95 (m, 1H), 6.92 (d, J = 8.3 Hz, 1H), 6.18 (s, 1H), 3.88 (s, 3H), 3.56 (s, 2H), 1.69 (s, 9H).
1-Methyl-3-(2-(trifluoromethoxy)phenyl)-1H-pyrazol-5-amine (4l) was prepared from 3e (458 mg, 2 mmol, 1 equiv) according to General Procedure B. 4l (348 mg, 1.35 mmol, 67% yield). 1H NMR (400 MHz, CDCl3) δ 8.03 – 7.96 (m, 1H), 7.33 – 7.27 (m, 3H), 6.03 (s, 1H), 3.74 (s, 3H), 3.54 (s, 2H).
1-(tert-Butyl)-3-(2-(trifluoromethoxy)phenyl)-1H-pyrazol-5-amine (4m) was prepared from 3e (343 mg, 1.5 mmol, 1 equiv) and tert-butylhydrazine hydrochloride (206 mg, 1.65 mmol, 1.1 equiv) according to General Procedure C. 4m (303 mg, 1.01 mmol, 67% yield). 1H NMR (400 MHz, CDCl3) δ 8.10 – 8.00 (m, 1H), 7.37 – 7.22 (m, 4H), 6.06 (s, 1H), 3.56 (s, 2H), 1.74 – 1.65 (m, 9H).
1-(tert-Butyl)-3-(3-methoxyphenyl)-1H-pyrazol-5-amine (4n) was prepared from 3-methoxybenzoylacetonitrile 3f (219 mg, 1.25 mmol, 1 equiv) and tert-butylhydrazine hydrochloride (171 mg, 1.38 mmol, 1.1 equiv) according to General Procedure C. 4n (288 mg, 1.17 mmol, 93% yield). 1H NMR (400 MHz, CDCl3) δ 7.35 – 7.29 (m, 2H), 7.28 – 7.21 (m, 1H), 6.84 – 6.76 (m, 1H), 5.89 (s, 1H), 3.85 (s, 3H), 3.59 (s, 2H), 1.75 – 1.59 (m, 9H).
1-(tert-Butyl)-3-(m-tolyl)-1H-pyrazol-5-amine (4o) was prepared from m-toluoylacetonitrile 3g (199 mg, 1.25 mmol, 1 equiv) and tert-butylhydrazine hydrochloride (171 mg, 1.38 mmol, 1.1 equiv) according to General Procedure C. 4o (276 mg, 1.20 mmol, 96% yield). 1H NMR (400 MHz, CDCl3) δ 7.62 – 7.57 (m, 1H), 7.56 – 7.48 (m, 1H), 7.23 (t, J = 7.6 Hz, 1H), 7.10 – 7.01 (m, 1H), 5.89 (s, 1H), 3.58 (s, 2H), 2.37 (s, 3H), 1.69 (s, 9H).
1-(tert-Butyl)-3-(3-(trifluoromethyl)phenyl)-1H-py razol-5-amine (4p) was prepared from 3-(trifluoromethyl)benzoylacetonitrile 3h (266 mg, 1.25 mmol, 1 equiv) and tert-butylhydrazine hydrochloride (171 mg, 1.38 mmol, 1.1 equiv) according to General Procedure C. 4p (255 mg, 0.90 mmol, 71% yield). 1H NMR (400 MHz, CDCl3) δ 8.04 – 7.95 (m, 1H), 7.93 – 7.85 (m, 1H), 7.52 – 7.39 (m, 2H), 5.93 (s, 1H), 3.63 (s, 2H), 1.70 (s, 9H).
1-Methyl-3-(3-(trifluoromethoxy)phenyl)-1H-pyrazol-5-amine (4q) was prepared from 3i (344 mg, 1.5 mmol, 1 equiv) according to General Procedure B. 4q (246 mg, 0.96 mmol, 63% yield). 1H NMR (400 MHz, CDCl3) δ 7.70 – 7.60 (m, 1H), 7.60 – 7.55 (m, 1H), 7.37 (t, J = 8.0 Hz, 1H), 7.15 – 7.06 (m, 1H), 5.86 (s, 1H), 3.72 (s, 3H), 3.57 (s, 2H).
1-(tert-Butyl)-3-(3-(trifluoromethoxy)phenyl)-1H-pyrazol-5-amine (4r) was prepared from 3i (344 mg, 1.65 mmol, 1 equiv) and tert-butylhydrazine hydrochloride (206 mg, 1.65 mmol, 1.1 equiv) according to General Procedure C. 4r (248 mg, 0.83 mmol, 55% yield). 1H NMR (400 MHz, CDCl3) δ 7.74 – 7.63 (m, 1H), 7.62 – 7.57 (m, 1H), 7.35 (t, J = 8.0 Hz, 1H), 7.12 – 7.04 (m, 1H), 5.89 (s, 1H), 3.60 (s, 2H), 1.69 (s, 9H).
1-(tert-Butyl)-3-(3-fluorophenyl)-1H-pyrazol-5-amine (4t) was prepared from 3-fluorobenzoylacetonitrile 3j (245 mg, 1.5 mmol, 1 equiv) and tert-butylhydrazine hydrochloride (206 mg, 1.65 mmol, 1.1 equiv) according to General Procedure C. 4t (222 mg, 0.95 mmol, 63% yield). 1H NMR (400 MHz, CDCl3) δ 7.54 – 7.40 (m, 2H), 7.35 – 7.25 (m, 1H), 6.98 – 6.88 (m, 1H), 5.89 (s, 1H), 3.61 (s, 2H), 1.69 (s, 9H).
3-(3-Chlorophenyl)-1-methyl-1H-pyrazol-5-amine (4u) was prepared from 3-chlorobenzoylacetonitrile 3k (898 mg, 5 mmol, 1 equiv) according to General Procedure B. 4u (614 mg, 2.96 mmol, 59% yield). 1H NMR (400 MHz, CDCl3) δ 7.72 (s, 1H), 7.61 (d, J = 7.6 Hz, 1H), 7.33 – 7.21 (m, 2H), 5.86 (s, 1H), 3.75 (s, 3H), 3.60 (s, 2H).
1-(tert-Butyl)-3-(3-chlorophenyl)-1H-pyrazol-5-amine (4v) was prepared from 3-chorobenzoylacetonitrile 3k (224 mg, 1.25 mmol, 1 equiv) and tert-butylhydrazine hydrochloride (171 mg, 1.38 mmol, 1.1 equiv) according to General Procedure C. 4v (232 mg, 0.93 mmol, 74% yield). 1H NMR (400 MHz, CDCl3) δ 7.77 – 7.73 (m, 1H), 7.64 – 7.58 (m, 1H), 7.31 – 7.24 (m, 1H), 7.24 – 7.17 (m, 1H), 5.88 (s, 1H), 3.61 (s, 2H), 1.69 (s, 9H).
3-(3-Chlorophenyl)-1-cyclohexyl-1H-pyrazol-5-amine (4w) was prepared from 3-chlorobenzoylacetonitrile 3k (269 mg, 1.5 mmol, 1 equiv) and cyclohexylhydrazine hydrochloride (226 mg, 1.5 mmol, 1 equiv) according to General Procedure C. 4w (328 mg, 1.19 mmol, 79% yield). 1H NMR (400 MHz, CDCl3) δ 7.72 (t, J = 2.0 Hz, 1H), 7.61 – 7.54 (m, 1H), 7.26 – 7.20 (m, 1H), 7.20 – 7.15 (m, 1H), 5.81 (s, 1H), 3.95 – 3.81 (m, 1H), 3.52 (s, 2H), 2.00 – 1.82 (m, 6H), 1.70 (d, J = 12.0 Hz, 1H), 1.50 – 1.24 (m, 3H).
3-(2,3-Dimethylphenyl)-1-methyl-1H-pyrazol-5-amine (4x) was prepared from 3l (350 mg, 2 mmol, 1 equiv) according to General Procedure B. 4x (197 mg, 0.98 mmol, 48% yield). 1H NMR (400 MHz, CDCl3) δ 7.32 – 7.26 (m, 2H), 7.13 – 7.08 (m, 2H), 5.66 (s, 1H), 3.73 (s, 3H), 3.51 (s, 2H), 2.33 (s, 3H), 2.31 (s, 3H).
3-(5-Fluoro-2-methylphenyl)-1-methyl-1H-pyrazol-5-amine (4y) was prepared from 3m (298 mg, 1.68 mmol, 1 equiv) according to General Procedure B. 4y (194 mg, 0.95 mmol, 56% yield). 1H NMR (400 MHz, CDCl3) δ 7.32 – 7.22 (m, 1H), 7.18 – 7.10 (m, 1H), 6.93 – 6.84 (m, 1H), 5.72 (s, 1H), 3.72 (s, 3H), 3.54 (s, 2H), 2.41 (s, 3H).
3-(2-Fluoro-3-methylphenyl)-1-methyl-1H-pyrazol-5-amine (4z) was prepared from 3n (461 mg, 2.6 mmol, 1 equiv) according to General Procedure B. 4z (299 mg, 1.46 mmol, 55% yield). 1H NMR (400 MHz, CDCl3) δ 7.77 – 7.68 (m, 1H), 7.13 – 6.97 (m, 2H), 6.07 – 5.93 (m, 1H), 3.74 (s, 3H), 3.56 (s, 2H), 2.33 – 2.28 (m, 3H).
3-(2,3-Difluorophenyl)-1-methyl-1H-pyrazol-5-amine (4aa) was prepared from 3o (221 mg, 1.22 mmol, 1 equiv) according to General Procedure B. 4aa (142 mg, 0.68 mmol, 55% yield). 1H NMR (400 MHz, CDCl3) δ 7.74 – 7.62 (m, 1H), 7.11 – 6.99 (m, 2H), 6.05 (d, J = 4.0 Hz, 1H), 3.75 (s, 3H), 3.55 (s, 2H).
1-Methyl-3-(2-methyl-3-(trifluoromethyl)phenyl)-1H-pyrazol-5-amine (4ab) was prepared from 3p (284 mg, 1.25 mmol, 1 equiv) according to General Procedure B. 4ab (138 mg, 0.54 mmol, 43% yield). 1H NMR (400 MHz, CDCl3) δ 7.65 – 7.57 (m, 2H), 7.32 – 7.27 (m, 1H), 5.68 (s, 1H), 3.79 – 3.75 (m, 3H), 3.66 (s, 2H), 2.51 (d, J = 1.8 Hz, 3H).
3-(3-Chloro-2-fluorophenyl)-1-methyl-1H-pyrazol-5-amine (4ac) was prepared from 3q (247 mg, 1.25 mmol, 1 equiv) according to General Procedure B. 4ac (180 mg, 0.80 mmol, 63% yield). 1H NMR (400 MHz, CDCl3) δ 7.86 (t, J = 7.4 Hz, 1H), 7.30 (t, J = 7.3 Hz, 1H), 7.08 (m, 1H), 6.05 (d, J = 4.2 Hz, 1H), 3.76 (s, 3H), 3.71 – 3.49 (m, 2H).
3-(2-Fluoro-3-(trifluoromethoxy)phenyl)-1-methyl-1H-pyrazol-5-amine (4ad) was prepared from 3r (247 mg, 1 mmol, 1 equiv) according to General Procedure B. 4ad (202 mg, 0.73 mmol, 73% yield). 1H NMR (400 MHz, CDCl3) δ 7.92 – 7.85 (m, 1H), 7.25 – 7.09 (m, 2H), 6.09 – 5.99 (m, 1H), 3.74 (s, 3H), 3.60 (s, 2H).
3-(2,5-Difluorophenyl)-1-methyl-1H-pyrazol-5-amine (4ae) was prepared from 3s (248 mg, 1.37 mmol, 1 equiv) according to General Procedure B. 4ae (193 mg, 0.92 mmol, 67% yield). 1H NMR (400 MHz, CDCl3) δ 7.68 – 7.59 (m, 1H), 7.12 – 6.98 (m, 1H), 6.98 – 6.82 (m, 1H), 6.04 (d, J = 4.1 Hz, 1H), 3.74 (s, 3H), 3.54 (s, 2H).
3-(2-Chloro-5-fluorophenyl)-1-methyl-1H-pyrazol-5-amine (4af) was prepared from 3t (247 mg, 1.25 mmol, 1 equiv) according to General Procedure B. 4af (159 mg, 0.71 mmol, 56% yield). 1H NMR (400 MHz, CDCl3) δ 7.57 – 7.49 (m, 1H), 7.41 – 7.31 (m, 1H), 6.98 – 6.85 (m, 1H), 6.15 (s, 1H), 3.74 (s, 3H), 3.69 – 3.43 (m, 2H).
3-(5-Chloro-2-fluorophenyl)-1-methyl-1H-pyrazol-5-amine (4ag) was prepared from 3u (247 mg, 1.25 mmol, 1 equiv) according to General Procedure B. 4ag (183 mg, 0.81 mmol, 64% yield). 1H NMR (400 MHz, CDCl3) δ 7.98 – 7.88 (m, 1H), 7.20 – 7.13 (m, 1H), 7.08 – 6.93 (m, 1H), 6.03 (d, J = 4.1 Hz, 1H), 3.74 (s, 3H), 3.53 (s, 2H).
3-(2-Fluoro-5-methylphenyl)-1-methyl-1H-pyrazol-5-amine (4ah) was prepared from 3v (323 mg, 1.82 mmol, 1 equiv) according to General Procedure B. 4ah (271 mg, 1.32 mmol, 72% yield). 1H NMR (400 MHz, CDCl3) δ 7.81 – 7.64 (m, 1H), 7.14 – 6.80 (m, 2H), 6.13 – 5.91 (m, 1H), 3.81 – 3.66 (m, 3H), 3.66 – 3.46 (m, 2H), 2.33 (s, 3H).
3-(2-Fluoro-5-(trifluoromethyl)phenyl)-1-methyl-1H-pyrazol-5-amine (4ai) was prepared from 3w (289 mg, 1.25 mmol, 1 equiv) according to General Procedure B. 4ai (109 mg, 0.42 mmol, 33% yield). 1H NMR (400 MHz, CDCl3) δ 8.33 – 8.24 (m, 1H), 7.54 – 7.43 (m, 1H), 7.23 – 7.13 (m, 1H), 6.10 – 6.00 (m, 1H), 3.78 (s, 3H), 3.69 (s, 2H).
3-(2-Fluoro-5-(trifluoromethoxy)phenyl)-1-methyl-1H-pyrazol-5-amine (4aj) was prepared from 3x (247 mg, 1 mmol, 1 equiv) according to General Procedure B. 4aj (163 mg, 0.59 mmol, 59% yield). 1H NMR (400 MHz, CDCl3) δ 7.86 – 7.81 (m, 1H), 7.11 – 7.05 (m, 2H), 6.04 (dd, J = 4.1, 0.7 Hz, 1H), 3.75 (s, 3H), 3.59 (s, 2H).
3-(2,6-Difluorophenyl)-1-methyl-1H-pyrazol-5-amine (4ak) was prepared from 3y (169 mg, 0.93 mmol, 1 equiv) according to General Procedure B. 4ak (48 mg, 0.23 mmol, 24% yield). 1H NMR (400 MHz, CDCl3) δ 7.26 – 7.15 (m, 1H), 6.94 (t, J = 8.3 Hz, 2H), 5.90 (d, J = 1.9 Hz, 1H), 3.77 (s, 3H), 3.53 (s, 2H).
3-(4-Chloro-2-fluorophenyl)-1-methyl-1H-pyrazol-5-amine (4al) was prepared from 3z (296 mg, 1.5 mmol, 1 equiv) according to General Procedure B. 4al (172 mg, 0.76 mmol, 50% yield). 1H NMR (400 MHz, CDCl3) δ 7.90 (t, J = 8.3 Hz, 1H), 7.17 – 7.08 (m, 2H), 6.01 (d, J = 4.1 Hz, 1H), 3.75 (s, 3H), 3.70 – 3.51 (m, 2H).
3-(2-Chloro-4-fluorophenyl)-1-methyl-1H-pyrazol-5-amine (4am) was prepared from 3aa (296 mg, 1.5 mmol, 1 equiv) according to General Procedure B. 4am (135 mg, 0.60 mmol, 39% yield). 1H NMR (400 MHz, CDCl3) δ 7.80 – 7.69 (m, 1H), 7.21 – 7.09 (m, 1H), 7.09 – 6.95 (m, 1H), 6.06 (s, 1H), 3.74 (s, 3H), 3.60 (s, 2H).
1-(tert-Butyl)-3-(2,3-dimethylphenyl)-1H-pyrazol-5-amine (4ao) was prepared from 3l (260 mg, 1.5 mmol, 1 equiv) and tert-butylhydrazine hydrochloride (206 mg, 1.65 mmol, 1.1 equiv) according to General Procedure C. 4ao (122 mg, 0.50 mmol, 33% yield). 1H NMR (400 MHz, CDCl3) δ 7.33 – 7.27 (m, 1H), 7.12 – 7.03 (m, 2H), 5.69 (s, 1H), 3.60 (s, 2H), 2.37 (s, 3H), 2.30 (s, 3H), 1.69 (s, 9H).
3-(5-Fluoro-2-methylphenyl)-1-isopropyl-1H-pyrazol-5-amine (4ap) was prepared from 3m (177 mg, 1 mmol, 1 equiv) and isopropylhydrazine hydrochloride (82 mg, 1.1 mmol, 1.1 equiv) according to General Procedure C. 4ap (93 mg, 0.40 mmol, 40% yield). 1H NMR (400 MHz, CDCl3) δ 7.30 – 7.23 (m, 1H), 7.23 – 7.08 (m, 1H), 6.92 – 6.83 (m, 1H), 5.72 (s, 1H), 4.51 – 4.32 (m, 1H), 3.65 (s, 2H), 2.43 (s, 3H), 1.51 (d, J = 6.6 Hz, 6H).
1-(tert-Butyl)-3-(5-fluoro-2-methylphenyl)-1H-pyrazol-5-amine (4aq) was prepared from 3m (266 mg, 1.5 mmol, 1 equiv) and tert-butylhydrazine hydrochloride (206 mg, 1.65 mmol, 1.1 equiv) according to General Procedure C. 4aq (118 mg, 0.48 mmol, 31% yield). 1H NMR (400 MHz, CDCl3) δ 7.32 – 7.27 (m, 1H), 7.17 – 7.10 (m, 1H), 6.89 – 6.79 (m, 1H), 5.78 (s, 1H), 3.58 (s, 2H), 2.46 (s, 3H), 1.68 (s, 9H).
1-Cyclopentyl-3-(5-fluoro-2-methylphenyl)-1H-pyrazol-5-amine (4ar) was prepared from 3m (177 mg, 1 mmol, 1 equiv) and cyclopentylhydrazine hydrochloride (150 mg, 1.1 mmol, 1.1 equiv) according to General Procedure C. 4ar (202 mg, 0.78 mmol, 78% yield). 1H NMR (400 MHz, CDCl3) δ 7.34 – 7.24 (m, 1H), 7.18 – 7.10 (m, 1H), 6.88 (m, 1H), 5.75 (s, 1H), 4.67 – 4.53 (m, 1H), 2.43 (s, 3H), 2.23 – 1.87 (m, 6H), 1.67 (m, 2H).
1-Cyclohexyl-3-(5-fluoro-2-methylphenyl)-1H-pyrazol-5-amine (4as) was prepared from 3m (177 mg, 1 mmol, 1 equiv) and cyclohexylhydrazine hydrochloride (166 mg, 1.1 mmol, 1.1 equiv) according to General Procedure C. 4as (185 mg, 0.68 mmol, 68% yield). 1H NMR (400 MHz, CDCl3) δ 7.29 – 7.21 (m, 1H), 7.18 – 7.08 (m, 1H), 6.93 – 6.83 (m, 1H), 5.75 – 5.70 (m, 1H), 4.08 – 3.92 (m, 1H), 3.77 (s, 2H), 2.42 (s, 3H), 2.04 – 1.85 (m, 7H), 1.71 (d, J = 12.6 Hz, 1H), 1.50 – 1.29 (m, 2H).
3-(2-Fluoro-3-methylphenyl)-1-isopropyl-1H-pyrazol-5-amine (4at) was prepared from 3n (177 mg, 1 mmol, 1 equiv) and isopropylhydrazine hydrochloride (82 mg, 1.1 mmol, 1.1 equiv) according to General Procedure C. 4at (149 mg, 0.64 mmol, 64% yield). 1H NMR (400 MHz, CDCl3) δ 7.83 (td, J = 7.4, 2.0 Hz, 1H), 7.13 – 6.99 (m, 2H), 6.03 (d, J = 4.1 Hz, 1H), 4.46 (hept, J = 6.6 Hz, 1H), 3.73 (s, 1H), 2.30 (d, J = 2.3 Hz, 3H), 1.52 (d, J = 6.6 Hz, 6H).
1-(tert-Butyl)-3-(2-fluoro-3-methylphenyl)-1H-pyrazol-5-amine (4au) was prepared from 3n (266 mg, 1.5 mmol, 1 equiv) and tert-butylhydrazine hydrochloride (206 mg, 1.65 mmol, 1.1 equiv) according to General Procedure C. 4au (238 mg, 0.96 mmol, 64% yield). 1H NMR (400 MHz, CDCl3) δ 7.86 – 7.78 (m, 1H), 7.11 – 6.94 (m, 2H), 6.10 – 5.97 (m, 1H), 3.55 (s, 2H), 2.33 – 2.25 (m, 3H), 1.69 (s, 9H).
1-Cyclopentyl-3-(2-fluoro-3-methylphenyl)-1H-pyrazol-5-amine (4av) was prepared from 3n (177 mg, 1 mmol, 1 equiv) and cyclopentylhydrazine hydrochloride (150 mg, 1.1 mmol, 1.1 equiv) according to General Procedure C. 4av (208 mg, 0.80 mmol, 80% yield). 1H NMR (400 MHz, CDCl3) δ 7.91 – 7.79 (m, 1H), 7.20 – 6.99 (m, 2H), 6.09 – 6.02 (m, 1H), 4.74 – 4.49 (m, 1H), 2.37 – 2.23 (m, 3H), 2.24 – 1.88 (m, 6H), 1.80 – 1.56 (m, 2H).
1-Cyclohexyl-3-(2-fluoro-3-methylphenyl)-1H-pyrazol-5-amine (4aw) was prepared from 3n (266 mg, 1.5 mmol, 1 equiv) and cyclohexylhydrazine hydrochloride (226 mg, 1 mmol, 1 equiv) according to General Procedure C. 4aw (322 mg, 1.18 mmol, 78 % yield). 1H NMR (400 MHz, CDCl3) δ 7.82 – 7.72 (m, 1H), 7.10 – 6.97 (m, 2H), 6.02 (d, J = 4.2 Hz, 1H), 4.02 – 3.86 (m, 1H), 3.47 (s, 2H), 2.30 (d, J = 2.4 Hz, 3H), 2.07 – 1.83 (m, 6H), 1.78 – 1.68 (m, 1H), 1.49 – 1.22 (m, 3H).
3-(2,3-Difluorophenyl)-1-isopropyl-1H-pyrazol-5-amine (4ax) was prepared from 3o (181 mg, 1 mmol, 1 equiv) and isopropylhydrazine hydrochloride (82 mg, 1.1 mmol, 1.1 equiv) according to General Procedure C. 4ax (128 mg, 0.54 mmol, 54% yield). 1H NMR (400 MHz, CDCl3) δ 7.84 – 7.74 (m, 1H), 7.14 – 6.99 (m, 2H), 6.08 – 6.02 (m, 1H), 4.52 – 4.37 (m, 1H), 3.76 – 3.55 (m, 2H), 1.52 (d, J = 6.4 Hz, 6H).
1-(tert-Butyl)-3-(2,3-difluorophenyl)-1H-pyrazol-5-amine (4ay) was prepared from 3o (272 mg, 1.5 mmol, 1 equiv) and tert-butylhydrazine hydrochloride (206 mg, 1.65 mmol, 1.1 equiv) according to General Procedure C. 4ay (244 mg, 0.97 mmol, 64 % yield). 1H NMR (400 MHz, CDCl3) δ 7.83 – 7.71 (m, 1H), 7.13 – 6.96 (m, 2H), 6.12 – 6.04 (m, 1H), 3.78 – 3.51 (m, 2H), 1.69 (s, 9H).
1-Cyclopentyl-3-(2,3-difluorophenyl)-1H-pyrazol-5-amine (4az) was prepared from 3o (181 mg, 1 mmol, 1 equiv) and cyclopentylhydrazine hydrochloride (150 mg, 1.1 mmol, 1.1 equiv) according to General Procedure C. 4az (193 mg, 0.73 mmol, 73% yield). 1H NMR (400 MHz, CDCl3) δ 7.82 – 7.73 (m, 1H), 7.11 – 6.98 (m, 2H), 6.05 (d, J = 4.1 Hz, 1H), 4.55 (p, J = 7.4 Hz, 1H), 3.85 – 3.53 (m, 2H), 2.27 – 1.91 (m, 6H), 1.78 – 1.59 (m, 2H).
1-Cyclohexyl-3-(2,3-difluorophenyl)-1H-pyrazol-5-amine (4bb) was prepared from 3o (181 mg, 1.5 mmol, 1 equiv) and cyclohexylhydrazine hydrochloride (166 mg, 1.1 mmol, 1.1 equiv) according to General Procedure C. 4bb (237 mg, 0.85 mmol, 85% yield). 1H NMR (400 MHz, CDCl3) δ 7.92 – 7.83 (m, 1H), 7.10 (t, J = 6.1 Hz, 2H), 6.10 (d, J = 3.8 Hz, 1H), 4.24 – 4.04 (m, 1H), 2.22 – 2.05 (m, 1H), 2.03 – 1.86 (m, 5H), 1.74 – 1.66 (m, 1H), 1.51 – 1.26 (m, 3H).
Recorcylate Amide Bromide Synthesis
2-Bromo-4,6-dimethoxybenzaldehyde (S1):
To a 250 mL round-bottom flask equipped with a magnetic stir-bar was added 3,5-dimethoxybromobenzene (10.0 g, 46.1 mmol) and DMF (23 mL, 2 M). The mixture was cooled to 0 °C and POCl3 (12.8 mL, 138 mmol, 3 equiv) was added dropwise over 5 minutes. The reaction was warmed to room temperature then heated to 90 °C for 6 hours. The reaction was cooled to room temperature and poured into ice water (200 mL). The reaction was quenched with a slow addition of KOH (55 g) to reach pH 14. The slurry was warmed to room temperature and stirred for 16 hours. The aqueous phase was extracted with Et2O (3 x 200 mL), the combined organic extracts were washed with water (3 x 100 mL) and brine (150 mL), dried with Na2SO4 and concentrated under reduced pressure. No further purification was required affording aldehyde S1 as a brown solid (10.4g, 92% yield). 1H NMR (400 MHz, CDCl3) δ 10.31 (s, 1H), 6.78 (d, J = 2.2 Hz, 1H), 6.44 (d, J = 2.2 Hz, 1H), 3.95 – 3.80 (m, 6H). UPLC/MS [M+H]=245.359, TR=1.41 min.
2-Bromo-4,6-dihydroxybenzaldehyde (6):
To a flamed-dried 250 mL round-bottom flask equipped with a magnetic stir-bar was added 2-bromo-4,6-dimethoxybenzaldehyde S1 (10 g, 40.8 mmol). The flask was fitted with a rubber septum, evacuated and backfilled with N2. Anhydrous CH2Cl2 (150 mL, 0.27 M) was added and the mixture was cooled to 0 °C. In a separate round-bottom flask, a solution of BBr3 (11.6 mL, 122 mmol, 3 equiv.) in anhydrous CH2Cl2 (30 mL, 4 M) was prepared. The BBr3 solution was added dropwise via cannula over 15 minutes. The reaction was slowly warmed to room temperature as the ice bath melted and stirred for 18 hours. The reaction was poured into ice water (300 mL), extracted with EtOAc (3 x 200 mL), washed with brine (300 mL), dried with Na2SO4 and concentrated under reduced pressure. The crude product was purified by silica flash chromatography (5-50% acetone in hexanes) affording aldehyde 6 (6.61g, 75% yield) as a light purple solid. 1H NMR (400 MHz, DMSO-d6) δ 12.19 (s, 1H), 9.97 (s, 1H), 6.71 (d, J = 2.1 Hz, 1H), 6.30 (d, J = 2.0 Hz, 1H). UPLC/MS [M+H]=216.956, TR=1.29 min.
2,4-Bis(benzyloxy)-6-bromobenzaldehyde (7).
To a 200 mL round-bottom flask equipped with a magnetic stir-bar was added 2-bromo-4,6-dihydroxybenzaldehyde 6 (3.33 g, 15.3 mmol), K2CO3 (5.3 g, 38.3 mmol, 2.5 equiv), benzyl bromide (4.6 mL, 38.3 mmol, 2.5 equiv) and MeCN (45 mL, 0.33 M). The flask was fitted with a reflux condenser and the reaction was heated at reflux in a 90 °C oil bath for 16 hours. The reaction was cooled to room temperature and the salts were removed by vacuum filtration, washing with EtOAc (100 mL). The filtrate was concentrated under reduced pressure and purified by silica flash chromatography (2-40% EtOAc in hexanes) affording aldehyde 7 (5.0 g, 82% yield) as a colorless solid. 1H NMR (400 MHz, CDCl3) δ 10.39 (s, 1H), 7.49 – 7.29 (m, 10H), 6.89 (d, J = 2.1 Hz, 1H), 6.57 (d, J = 2.2 Hz, 1H), 5.13 (s, 2H), 5.07 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 188.86, 163.47, 162.92, 135.66, 135.43, 128.94, 128.87, 128.71, 128.40, 127.76, 127.18, 126.64, 117.63, 113.07, 100.33, 70.96, 70.76. UPLC/MS [M+H]=397.337, TR=2.30 min.
General Procedure D
To a 100 mL round-bottom flask equipped with a magnetic stir-bar was added 2,4-bis(benzyloxy)-6-bromobenzaldehyde 7 (1.59 g, 4 mmol), t-BuOH (12 mL) and THF (10 mL). In a separate flask sodium chlorite (1.09 g, 12 mmol, 3 equiv) and sodium monobasic phosphate monohydrate (4.14 g, 30 mmol, 7.5 equiv) were dissolved in H2O (10 mL). The aqueous solution was added to the reaction portion-wise over 2 minutes turning the reaction bright yellow. 2-Methyl-2-butene (5.1 mL, 48 mmol, 12 equiv) was added and the reaction was stirred for 30 minutes until the yellow color dissipated and the reaction returned to colorless. The reaction was quenched with 3 M HCl (30 mL) and extracted with EtOAc (3 x 50 mL). The combined organic extracts were washed with saturated NH4Cl solution (2 x 50 mL), washed with brine (50 mL), dried with Na2SO4 and concentrate under reduced pressure. The resulting benzoic acid was immediately used without purification.
In a 100 mL round-bottom flask equipped with a magnetic stir-bar, the crude benzoic acid was dissolved in CH2Cl2 (12 mL) and THF (12 mL). Amine (4.40 mmol, 1.1 equiv), DIPEA (1.74 mL, 10 mmol, 2.5 equiv) and HATU (1.83 g, 4.80 mmol, 1.2 equiv) were added. The reaction was stirred at room temperature for 3 hours. The reaction was quenched with saturated NaHCO3 solution (30 mL), extracted with CH2Cl2 (3 x 50 mL), washed with brine (75 mL), dried with Na2SO4 and concentrate under reduced pressure. The crude product was purified by silica flash chromatography (5-60% EtOAc in hexane) to afford the desired amide 8.
(2,4-Bis(benzyloxy)-6-bromophenyl)(isoindolin-2-yl)methanone (8a) was prepared according to General Procedure D from isoindoline hydrochloride (685 mg, 4.40 mmol, 1.1 equiv). 8a (1.67 g, 3.24 mmol) was obtained in 81% yield as a colorless solid. 1H NMR (400 MHz, CDCl3) δ 7.48 – 7.22 (m, 13H), 7.15 (d, J = 7.3 Hz, 1H), 6.85 (d, J = 2.1 Hz, 1H), 6.59 (d, J = 2.1 Hz, 1H), 5.16 – 5.05 (m, 2H), 5.02 (d, J = 7.1 Hz, 4H), 4.68 – 4.45 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 165.90, 160.46, 156.51, 136.54, 136.47, 136.16, 136.03, 128.86, 128.70, 128.48, 128.12, 127.86, 127.71, 127.62, 127.04, 123.27, 122.74, 122.19, 120.39, 110.63, 100.80, 70.75, 70.63, 53.32, 51.89. UPLC/MS [M+H]=514.231, TR=2.10 min.
(2,4-Bis(benzyloxy)-6-bromophenyl)(4-fluoroisoindolin-2-yl)methanone (8b) was prepared according to General Procedure D from 4-fluoroisoindoline (290 mg, 2.11 mmol). 8b (585 mg, 1.10 mmol) was obtained in 57% yield. 1H NMR (400 MHz, CDCl3) δ 7.47 – 7.21 (m, 11H), 7.15 – 6.87 (m, 2H), 6.86 – 6.80 (m, 1H), 6.68 – 6.54 (m, 1H), 5.09 – 4.99 (m, 6H), 4.67 – 4.40 (m, 2H). UPLC/MS [M+H]=532.195, TR=2.09 min.
(2,4-Bis(benzyloxy)-6-bromophenyl)(5-fluoroisoindolin-2-yl)methanone (8c) was prepared according to General Procedure D from 5-fluoroisoindoline hydrochloride (382 mg, 2.2 mmol). 8c (917 mg, 1.72 mmol) was obtained in 86% yield.
1H NMR (400 MHz, CDCl3) δ 7.55 – 7.32 (m, 5H), 7.33 – 7.19 (m, 6H), 7.13 – 6.92 (m, 2H), 6.87 – 6.78 (m, 1H), 6.58 (dd, J = 2.2, 1.2 Hz, 1H), 5.09 – 4.90 (m, 6H), 4.60 – 4.38 (m, 2H). UPLC/MS [M+H]=532.438, TR=2.08 min.
RAP Synthesis
General Procedure E
E1a: To a flame-dried 2D vial equipped with a magnetic stir-bar was added resorcylate amide bromide 8a-c, (0.15 mmol, 1 equiv), aminopyrazole 4a-bb (0.165 mmol, 1.1 equiv), Pd(OAc)2 (15 μmol, 0.1 equiv), xantphos (30 μmol, 0.2 equiv) and Cs2CO3 (0.3 mmol, 2 equiv). The vial was fitted with a rubber septum, evacuated, backfilled with N2 and anhydrous toluene (0.6 mL, 0.25 M) was added. The septum was replaced with a cap and the vial was sealed with PTFE tape. The reaction was heated at 130 °C for 16 hours in a heating block. The reaction was cooled to room temperature and a filtered through a silica plug eluting with EtOAc (12 mL). The filtrate was concentrated and used without further purification.
E1b: To a flame-dried microwave vial equipped with a magnetic stir-bar was added resorcylate amide bromide 8a-c (0.15 mmol, 1 equiv), aminopyrazole 4a-bb (0.165 mmol, 1.1 equiv), Pd2(dba)3 (6 μmol, 0.04 equiv), xanthphos (15 μmol, 0.1 equiv), and sodium phenoxide (0.225 mmol, 1.5 equiv). The vial was fitted with a rubber septum, evacuated, backfilled with N2 and anhydrous 1,4-dioxane (0.1 M reaction concentration) was added. The septum was replaced with a microwave cap and the reaction was heated in a microwave holding at 170 °C for 2 hours. The reaction was cooled to room temperature and a filtered through a silica plug eluting with EtOAc (8X reaction volume). The filtrate was concentrated and used without further purification.
E2a: In a 2D vial equipped with a magnetic stir-bar was added benzyl protected RAP (1 equiv), MeOH (2 mL) and Pd/C (20% w.t.). The vial was fitted with a rubber septum and a hydrogen balloon. Hydrogen was bubbled through the solution for 10 minutes then the reaction was stirred under hydrogen atmosphere for 16 hours or until full conversion was observed by UPLC/MS. The reaction was filtered through a celite plug eluting with MeOH (6 mL) and EtOAc (6 mL) and concentrated. The crude material was purified by mass-guided preparative HPLC.
E2b: In a 2D vial equipped with a magnetic stir-bar was added benzyl protected RAP (1 equiv), EtOAc (2 mL) and Pd(OH)2/C (20% w.t.). The vial was fitted with a rubber septum and a hydrogen balloon. Hydrogen was bubbled through the solution for 10 minutes then the reaction was stirred under hydrogen atmosphere for 16 hours or until full conversion was observed by UPLC/MS. The reaction was filtered through a celite plug eluting with MeOH (6 mL) and EtOAc (6 mL) and concentrated. The crude material was purified by mass-guided preparative HPLC.
(2-((3-(2-fluorophenyl)-1-methyl-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(isoindolin-2-yl)methanone (9a): Amide bromide 8a (45 mg, 0.087 mmol, 1 equiv) was coupled to 5-amino-3-(2-fluorophenyl)-1-methylpyrazole 4a (18.4 mg, 0.096 mmol, 1.1 equiv) according to General procedure E1a. The resulting product was deprotected according to General Procedure E2a. 9a (16.7 mg, 0.037 mmol) was obtained in a 43% yield over two steps. 1H NMR (500 MHz, DMSO-d6) δ 9.64 (s, 1H), 9.31 (s, 1H), 7.89 (dt, J = 1.90, 7.8 Hz, 1H), 7.29-7.35 (m, 3H), 7.24-7.29 (m, 2H), 7.22-7.24 (m, 1H), 7.21 (s, 2H), 7.20 (s, 1H), 6.36 (d, J = 3.9 Hz, 1H), 5.89 (d, J = 2.0 Hz, 1H), 5.62 (d, J = 2.0 Hz, 1H), 4.63-4.86 (m, 4H), 3.63 (s, 3H); 13C NMR (126 MHz, DMSO-d6) δ 166.5, 160.3, 159.4, 158.3, 155.7, 143.9, 142.8, 141.6, 141.6, 129.2, 129.1, 127.6, 127.4, 124.6, 123.0, 121.3, 121.2, 116.3, 116.1, 104.5, 99.8, 99.7, 94.6, 93.0, 40.6, 35.2.
(2-((3-(2-Chlorophenyl)-1-methyl-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(isoindolin-2-yl)methanone (9b): Amide bromide 8a (51 mg, 0.1 mmol, 1 equiv) was coupled to 4b (23 mg, 0.11 mmol, 1.1 equiv) according to General Procedure E1a. The resulting product was deprotected according to General Procedure E2a. 9b (3.4 mg, 0.007mmol) was obtained in 7% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.64 (s, 1H), 9.32 (s, 1H), 7.78 – 7.72 (m, 1H), 7.50 – 7.45 (m, 1H), 7.38 – 7.29 (m, 4H), 7.29 – 7.25 (m, 2H), 7.21 (s, 1H), 6.48 (s, 1H), 5.90 (d, J = 2.1 Hz, 1H), 5.65 (d, J = 2.1 Hz, 1H), 4.82 – 4.69 (m, 4H), 3.62 (s, 3H); 13C NMR (126 MHz, DMSO-d6): δ 166.34, 159.23, 155.52, 145.71, 143.74, 140.82, 132.16, 130.72, 130.20, 129.88, 128.88, 127.23, 127.14, 122.86, 104.28, 100.19, 94.50, 92.87, 35.04. UPLC/MS [M+H]=461.337, TR=2.36 min.
(2,4-Dihydroxy-6-((1-methyl-3-(2-(trifluoromethyl)phenyl)-1H-pyrazol-5-yl)amino)phenyl)(isoindolin-2-yl)methanone (9c): Amide bromide 8a (77 mg, 0.15 mmol, 1 equiv) was coupled to 4c (40 mg, 0.165 mmol, 1.1 equiv) according to General Procedure E1a. The resulting product was deprotected according to General Procedure E2a. 9c (13 mg, 0.026 mmol) was obtained in 17% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.63 – 9.59 (m, 1H), 9.32 (s, 1H), 7.78 (dd, J = 8.0, 1.3 Hz, 1H), 7.68 – 7.63 (m, 1H), 7.63 – 7.60 (m, 1H), 7.58 – 7.53 (m, 1H), 7.41 – 7.25 (m, 4H), 7.23 (s, 1H), 6.18 (s, 1H), 5.91 (d, J = 2.1 Hz, 1H), 5.64 (d, J = 2.1 Hz, 1H), 4.75 (s, 4H), 3.61 (s, 3H); 13C NMR (126 MHz, DMSO-d6): δ 166.32, 159.22, 155.48, 146.45, 143.68, 140.85, 137.21, 133.27, 132.20, 131.56, 128.06, 127.25, 126.09, 125.26, 122.88, 104.34, 99.67, 94.51, 92.87, 35.02. UPLC/MS [M+H]=495.601, TR=1.49 min.
(2-((1-(tert-Butyl)-3-(o-tolyl)-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(isoindolin-2-yl)methanone (9d): Amide bromide 8a (62 mg, 0.120 mmol, 1 equiv) was coupled to 4d (30mg, 0.132 mmol, 1.1 equiv) according to General Procedure E1b. The resulting product was deprotected according to General Procedure E2a. 9d (18.5 mg, 0.038 mmol) was obtained in 32% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.69 (s, 1H), 9.29 (s, 1H), 7.58 – 7.51 (m, 1H), 7.38 – 7.32 (m, 2H), 7.32 – 7.26 (m, 2H), 7.24 – 7.16 (m, 3H), 6.98 (s, 1H), 6.37 (s, 1H), 5.85 (d, J = 2.1 Hz, 1H), 5.70 (d, J = 2.1 Hz, 1H), 4.81 (s, 4H), 2.48 (s, 3H), 1.55 (s, 9H); 13C NMR (126 MHz, DMSO-d6): δ 167.36, 159.88, 156.00, 147.57, 145.54, 139.92, 137.13, 135.37, 133.63, 131.35, 128.69, 127.73, 127.45, 126.14, 123.31, 103.48, 103.20, 94.24, 92.56, 59.70, 29.89, 21.90 UPLC/MS [M+H]=483.575, TR=1.81 min.
(2,4-Dihydroxy-6-((1-methyl-3-(o-tolyl)-1H-pyrazol-5-yl)amino)phenyl)(5-fluoroisoindolin-2-yl)methanone (9e): Compound 9e was obtained according to the route and procedures described in 62. 2-Bromo-4,6-bis(methoxymethoxy)phenyl)(5-fluoroisoindolin-2-yl)methanone (78 mg, 0.177 mmol, 1 equiv), 2-methyl-5-(o-tolyl)pyrazol-3-amine (4e) (22 mg, 0.195 mmol, 1.1 equiv), Pd2(dba)3 (8.1 mg, 5 mol%), xantphos (10.3 mg, 10 mol%), sodium phenoxide (31 mg, 0.266 mmol, 1.5 equiv), and anhydrous dioxane (2 mL) were heated to 120 °C in a 2 dram vial under an atmosphere of nitrogen. The reaction was cooled to room temperature, diluted with ethyl acetate, and passed through a pre-wetted 3 mL hydromatrix cartridge eluting with ethyl acetate. The eluents were dried with sodium sulfate, filtered, and condensed by rotary evaporation. Purification on a short silica column with a gradient of 25% to 40% acetone in hexanes afforded a yellow gum, which was then dissolved in methanol (5 mL) and 2M aqueous HCl (0.53 mL). The reaction was heated to 50 °C overnight, cooled to room temperature, and the solvent was removed by rotary evaporation. The crude residue was purified by mass-guided preparative HPLC to afford 9e (24.2 mg, 28% yield over two steps). 1H NMR (500 MHz, CDCl3) δ 7.41 (d, J = 7.34 Hz, 1H), 7.06-7.17 (m, 4H), 6.92 (dt, J = 1.96, 8.56 Hz, 1H), 6.86 (br d, J = 8.31 Hz, 1H), 6.81 (s, 1H), 6.10 (s, 1H), 5.97 (d, J = 0.98 Hz, 1H), 5.78 (d, J = 1.47 Hz, 1H), 4.91 (br s, 2H), 4.72-4.86 (m, 2H), 3.64 (s, 3H), 2.33 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 168.8, 161.8, 160.5, 156.4, 150.4, 144.8, 140.6, 136.0, 132.9, 132.0, 130.9, 129.1, 128.0, 126.0, 124.2, 124.1, 115.1, 114.9, 110.2, 110.0, 100.0, 95.9, 94.0, 52.9, 52.4, 40.9, 21.2. UPLC/MS [M+H]=459.393 , TR=1.70 min.
(2,4-Dihydroxy-6-((3-(2-methoxyphenyl)-1-methyl-1H-pyrazol-5-yl)amino)phenyl)(isoindolin-2-yl)methanone (9f). Compound 9f was obtained from (2-bromo-4,6-bis(methoxymethoxy)phenyl)(isoindolin-2-yl)methanone62 and 5-(2-methoxyphenyl)-2-methyl-pyrazol-3-amine (4f) according to the same procedures described above for 9e. 9f (17.8 mg) was obtained in 35% yield over two steps. 1H NMR (500 MHz, CDCl3) δ 7.76 (dd, J = 1.47, 7.83 Hz, 1H), 7.21-7.25 (m, 3H), 7.17-7.21 (m, 3H), 6.90 (br t, J = 7.50 Hz, 1H), 6.85 (d, J = 8.31 Hz, 1H), 6.72 (s, 1H), 6.49 (s, 1H), 5.97 (d, J = 0.98 Hz, 1H), 5.76 (d, J = 1.96 Hz, 1H), 4.94-5.09 (m, 2H), 4.81-4.93 (m, 2H), 3.75 (s, 3H), 3.66 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 168.8, 160.5, 156.8, 156.4, 147.1, 145.3, 140.3, 136.7, 129.0, 128.3, 127.7, 122.9, 122.3, 120.9, 111.4, 103.1, 101.6, 95.6, 93.8, 77.5, 55.5, 53.0, 40.9, 35.1. UPLC/MS [M+H] = 457.409 , TR=1.56 min.
(2,4-Dihydroxy-6-((1-isopropyl-3-(2-methoxyphenyl)-1H-pyrazol-5-yl)amino)phenyl)(isoindolin-2-yl)methanone (9g): Amide bromide 8a (77 mg, 0.150 mmol, 1 equiv) was coupled to 4g (38 mg, 0.165 mmol, 1.1 equiv) according to General Procedure E1a. The resulting product was deprotected according to General Procedure E2a. 9g (22 mg, 0.046 mmol) was obtained in 37% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.58 (s, 1H), 9.25 (s, 1H), 7.91 (m, 1H), 7.42 – 7.21 (m, 5H), 7.12 – 6.94 (m, 3H), 6.46 (s, 1H), 5.84 (d, J = 2.1 Hz, 1H), 5.54 (d, J = 2.1 Hz, 1H), 4.77 (m, 4H), 4.50 – 4.37 (p, J = 6.6 Hz, 1H), 3.85 – 3.78 (m, 3H), 1.35 (d, J = 6.6 Hz, 6H); 13C NMR (126 MHz, DMSO-d6): δ 166.62, 159.35, 156.32, 155.64, 145.20, 145.08, 139.03, 136.90, 128.51, 127.40, 127.25, 123.03, 122.49, 120.49, 111.89, 103.84, 102.14, 94.11, 92.40, 55.49, 47.87, 22.45. UPLC/MS [M+H]=485.231 , TR=1.52 min.
(2-((1-Cyclohexyl-3-(2-methoxyphenyl)-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(isoindolin-2-yl)methanone (9h): Amide bromide 8a (77 mg, 0.150 mmol, 1 equiv) was coupled to 4h (45 mg, 0.165 mmol, 1.1 equiv) according to General Procedure E1a. The resulting product was deprotected according to General Procedure E2a. 9h (20 mg, 0.039 mmol) was obtained in 26% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.66 (s, 1H), 9.33 (s, 1H), 8.01 – 7.96 (m, 1H), 7.43 (t, J = 4.4 Hz, 2H), 7.40 – 7.31 (m, 3H), 7.17 – 7.09 (m, 2H), 7.09 – 7.02 (m, 1H), 6.56 (s, 1H), 5.94 (d, J = 2.1 Hz, 1H), 5.65 (d, J = 2.1 Hz, 1H), 4.86 (s, 4H), 4.15 – 4.05 (m, 1H), 3.89 (s, 3H), 1.99 – 1.81 (m, 6H), 1.69 – 1.65 (m, 1H), 1.36 – 1.19 (m, 3H); 13C NMR (126 MHz, DMSO-d6): δ 166.49, 159.16, 156.15, 155.46, 144.91, 144.86, 138.97, 136.70, 128.33, 127.25, 127.07, 122.84, 122.30, 120.32, 111.72, 103.76, 102.07, 94.01, 92.29, 55.32, 55.25, 32.34, 25.12, 24.91. UPLC/MS [M+H]=526.363 , TR=3.06 min.
(2-((1-Cyclohexyl-3-(2-methoxyphenyl)-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(4-fluoroisoindolin-2-yl)methanone (9i): Amide bromide 8b (48 mg, 0.090 mmol, 1 equiv) was coupled to 4h (27 mg, 0.099 mmol, 1.1 equiv) according to General Procedure E1b. The resulting product was deprotected according to General Procedure E2a. 9i (19 mg, 0.035 mmol) was obtained in 39% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.59 (s, 1H), 9.26 (s, 1H), 7.93 – 7.85 (m, 1H), 7.37 – 7.30 (m, 1H), 7.28 – 7.22 (m, 1H), 7.19 (d, J = 7.6 Hz, 1H), 7.10 (t, J = 8.8 Hz, 1H), 7.07 – 7.00 (m, 2H), 7.00 – 6.93 (m, 1H), 6.46 (s, 1H), 5.85 (d, J = 2.1 Hz, 1H), 5.57 (d, J = 2.1 Hz, 1H), 4.92 – 4.68 (m, 4H), 4.07 – 3.97 (m, 1H), 3.79 (s, 3H), 1.91 – 1.70 (m, 7H), 1.61 – 1.57 (m, 1H), 1.28 – 1.14 (m, 3H), 13C NMR (126 MHz, DMSO-d6): δ 166.43, 159.25, 156.11, 155.52, 144.96, 144.87, 138.97, 128.29, 127.01, 122.24, 120.29, 113.63 (d, J = 19.4 Hz), 111.67, 103.45, 102.04, 93.99, 92.49, 79.16, 55.27, 32.34, 25.11, 24.91. UPLC/MS [M+H]=543.598 , TR=1.79 min.
(2-((1-Cyclohexyl-3-(2-methoxyphenyl)-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(5-fluoroisoindolin-2-yl)methanone (9j): Amide bromide 8c (48 mg, 0.90 mmol, 1 equiv) was coupled to 4h (27 mg, 0.099 mmol, 1.1 equiv) according to General Procedure E1b. The resulting product was deprotected according to General Procedure E2a. 9j (19 mg, 0.034 mmol) was obtained in 38% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.57 (s, 1H), 9.24 (s, 1H), 7.91 – 7.86 (m, 1H), 7.43 – 7.32 (m, 1H), 7.27 – 7.22 (m, 1H), 7.22 – 7.18 (m, 1H), 7.11 – 7.06 (m, 1H), 7.04 – 7.01 (m, 2H), 6.45 (s, 1H), 5.84 (d, J = 2.1 Hz, 1H), 5.57 (d, J = 2.1 Hz, 1H), 4.73 (s, 6H), 3.80 (s, 3H), 1.93 – 1.71 (m, 6H), 1.66 – 1.55 (m, 1H), 1.31 – 1.13 (m, 3H); 13C NMR (126 MHz, DMSO-d6): δ 166.44, 159.20, 156.12, 155.48, 144.91, 144.88, 139.00, 128.30, 127.05, 124.54, 122.27, 120.30, 114.22, 111.68, 110.06, 103.62, 102.01, 94.00, 92.44, 79.17, 55.30, 48.58, 32.35, 25.12. UPLC/MS [M+H]=543.322 , TR=1.79 min.
(2-((1-Cyclopentyl-3-(2-methoxyphenyl)-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(isoindolin-2-yl)methanone (9k): Amide bromide 8a (77 mg, 0.150 mmol, 1 equiv) was coupled to 4i (42 mg, 0.165 mmol, 1.1 equiv) according to General Procedure E1a. The resulting product was deprotected according to General Procedure E2a. 9k (30.5 mg, 0.060 mmol) was obtained in 40% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.59 (s, 1H), 7.92 – 7.87 (m, 1H), 7.39 – 7.32 (m, 2H), 7.30 – 7.22 (m, 3H), 7.10 – 7.01 (m, 2H), 7.01 – 6.93 (m, 1H), 6.47 (s, 1H), 5.86 (d, J = 2.1 Hz, 1H), 5.55 (d, J = 2.1 Hz, 1H), 4.77 (s, 4H), 4.58 (p, J = 7.3 Hz, 1H), 3.81 (s, 3H), 2.04 – 1.78 (m, 6H), 1.67 – 1.47 (m, 2H); 13C NMR (126 MHz, DMSO-d6): δ 166.52, 159.21, 156.18, 155.48, 145.00, 144.83, 139.60, 136.76, 128.38, 127.26, 127.10, 122.86, 122.30, 120.37, 111.75, 103.72, 101.94, 94.04, 92.39, 56.91, 55.34, 32.25, 24.16. UPLC/MS [M+H]=511.295 , TR=1.74 min.
(2,4-Dihydroxy-6-((1-isobutyl-3-(2-methoxyphenyl)-1H-pyrazol-5-yl)amino)phenyl)(isoindolin-2-yl)methanone (9l): Amide bromide 8a (77 mg, 0.150 mmol, 1 equiv) was coupled to 4j (40 mg, 0.165 mmol, 1.1 equiv) according to General Procedure E1a. The resulting product was deprotected according to General Procedure E2a. 9l (20.7 mg, 0.0415 mmol) was obtained in 28% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.63 (s, 1H), 9.29 (s, 1H), 7.90 – 7.84 (m, 1H), 7.34 (s, 2H), 7.31 – 7.24 (m, 3H), 7.08 (s, 1H), 7.07 – 7.04 (m, 1H), 6.98 – 6.94 (m, 1H), 6.50 (s, 1H), 5.87 (d, J = 2.1 Hz, 1H), 5.67 (d, J = 2.1 Hz, 1H), 4.77 (s, 4H), 3.82 (s, 3H), 3.72 (d, J = 7.3 Hz, 2H), 2.14 – 2.03 (m, 1H), 0.78 (d, J = 6.7 Hz, 6H); 13C NMR (126 MHz, DMSO-d6): δ 167.03, 159.72, 156.66, 155.95, 145.55, 144.68, 140.56, 137.11, 128.92, 127.72, 127.56, 123.31, 122.57, 120.83, 112.24, 104.24, 101.43, 94.67, 92.89, 55.82, 55.00, 29.14, 20.25. UPLC/MS [M+H]=499.343 , TR=1.74 min.
(2-((1-(tert-Butyl)-3-(2-methoxyphenyl)-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(isoindolin-2-yl)methanone (9m): Amide bromide 8a (77 mg, 0.150 mmol, 1 equiv) was coupled to 4k (40 mg, 0.165 mmol, 1.1 equiv) according to General Procedure E1a. The resulting product was deprotected according to General Procedure E2a. 9m (12.4 mg, 0.025 mmol) was obtained in 12.5% yield over two steps.
1H NMR (500 MHz, DMSO-d6): δ 9.67 (s, 1H), 9.26 (s, 1H), 7.94 – 7.89 (m, 1H), 7.41 – 7.33 (m, 2H), 7.31 – 7.24 (m, 3H), 7.08 – 7.02 (m, 1H), 6.97 (s, 1H), 6.94 – 6.91 (m, 1H), 6.54 (d, J = 0.7 Hz, 1H), 5.84 (d, J = 2.1 Hz, 1H), 5.62 (d, J = 2.1 Hz, 1H), 4.81 (s, 4H), 3.81 (s, 3H), 1.55 (s, 9H); 13C NMR (126 MHz, DMSO-d6): δ 167.35, 159.83, 156.68, 155.97, 145.81, 143.75, 139.66, 137.12, 128.81, 127.73, 127.46, 123.31, 122.73, 120.82, 112.21, 105.28, 103.41, 94.12, 92.51, 59.69, 55.81, 29.91. UPLC/MS [M+H]=499.343, TR=1.62 min.
(2,4-Dihydroxy-6-((1-methyl-3-(2-(trifluoromethoxy)phenyl)-1H-pyrazol-5-yl)amino)phenyl)(isoindolin-2-yl)methanone (9n): Amide bromide 8a (77 mg, 0.15 mmol, 1 equiv) was coupled to 4l (42 mg, 0.165 mmol, 1.1 equiv) according to General Procedure E1b. The resulting product was deprotected according to General Procedure E2a. 9n (36 mg, 0.70 mmol) was obtained in 47% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.63 (s, 1H), 9.29 (s, 1H), 8.00 – 7.92 (m, 1H), 7.44 – 7.36 (m, 3H), 7.32 (s, 2H), 7.28 – 7.23 (m, 3H), 6.35 (s, 1H), 5.92 (d, J = 2.1 Hz, 1H), 5.62 (d, J = 2.1 Hz, 1H), 4.75 (s, 4H), 3.63 (s, 3H); 13C NMR (126 MHz, DMSO-d6): δ 166.34, 159.27, 155.52, 145.02, 143.73, 143.28, 141.43, 136.73, 128.92, 128.62, 127.64, 127.25, 126.95, 122.85, 121.53, 121.22, 119.18, 104.39, 99.57, 94.56, 92.88, 35.17. UPLC/MS [M+H]=511.162, TR=1.49 min.
(2-((1-(tert-Butyl)-3-(2-(trifluoromethoxy)phenyl)-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(isoindolin-2-yl)methanone (9o): Amide bromide 8a (64 mg, 0.125 mmol, 1 equiv) was coupled to 4m (41 mg, 0.138 mmol, 1.1 equiv) according to General Procedure E1b. The resulting product was deprotected according to General Procedure E2a. 9o (14.7 mg, 0.027 mmol) was obtained in 22% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.69 (s, 1H), 9.26 (s, 1H), 8.02 – 7.97 (m, 1H), 7.45 – 7.37 (m, 3H), 7.37 – 7.31 (m, 2H), 7.32 – 7.26 (m, 2H), 7.02 (s, 1H), 6.44 (s, 1H), 5.86 (d, J = 2.1 Hz, 1H), 5.62 (d, J = 2.1 Hz, 1H), 4.80 (s, 4H), 1.56 (s, 9H); 13C NMR (126 MHz, DMSO-d6): δ 166.78, 159.40, 155.50, 144.95, 141.74, 140.21, 136.60, 128.80, 128.69, 127.62, 127.27, 127.04, 122.83, 121.57, 121.28, 119.24, 103.26, 103.19, 93.90, 92.08, 59.77, 29.32.UPLC/MS [M+H]=553.609 , TR=1.87 min.
(2-((1-(tert-Butyl)-3-(3-methoxyphenyl)-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(isoindolin-2-yl)methanone (9p): Amide bromide 8a (77 mg, 0.150 mmol, 1 equiv) was coupled to 4n (40 mg, 0.165 mmol, 1.1 equiv) according to General Procedure E1b. The resulting product was deprotected according to General Procedure E2a. 9p (32.3 mg, 0.065 mmol) was obtained in 43% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.70 (s, 1H), 9.28 (s, 1H), 7.38 – 7.32 (m, 3H), 7.32 – 7.25 (m, 4H), 6.97 (s, 1H), 6.86 – 6.80 (m, 1H), 6.58 (d, J = 0.6 Hz, 1H), 5.86 (d, J = 2.1 Hz, 1H), 5.67 (d, J = 2.1 Hz, 1H), 4.99 – 4.66 (m, 4H), 3.78 (s, 3H), 1.56 (s, 9H); 13C NMR (126 MHz, DMSO-d6): δ 166.90, 159.49, 159.42, 155.54, 146.34, 145.08, 140.43, 136.66, 135.22, 129.55, 127.28, 122.86, 117.14, 112.72, 110.05, 103.03, 100.57, 93.82, 92.24, 59.36, 55.02, 29.41 UPLC/MS [M+H]=499.255 , TR=1.66 min.
(2-((1-(tert-butyl)-3-(m-tolyl)-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(isoindolin-2-yl)methanone (9q): Amide bromide 8a (77 mg, 0.150 mmol, 1 equiv) was coupled to 4o (38 mg, 0.165 mmol, 1.1 equiv) according to General Procedure E1b. The resulting product was deprotected according to General Procedure E2a. 9q (34.7 mg, 0.072 mmol) was obtained in 48% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.70 (s, 1H), 9.28 (s, 1H), 7.61 – 7.57 (m, 1H), 7.57 – 7.52 (m, 1H), 7.40 – 7.32 (m, 2H), 7.32 – 7.21 (m, 3H), 7.11 – 7.04 (m, 1H), 7.00 – 6.96 (m, 1H), 6.53 (s, 1H), 5.86 (d, J = 2.1 Hz, 1H), 5.69 (d, J = 2.1 Hz, 1H), 5.06 – 4.62 (m, 4H), 2.32 (s, 3H), 1.56 (s, 9H); 13C NMR (126 MHz, DMSO-d6): δ 166.92, 159.43, 155.55, 146.55, 145.06, 140.42, 137.50, 136.67, 133.72, 128.37, 127.82, 127.29, 125.19, 122.86, 121.91, 103.06, 100.22, 93.84, 92.26, 59.31, 29.44, 21.10. UPLC/MS [M+H]=483.247 , TR=1.85 min.
(2-((1-(tert-Butyl)-3-(3-(trifluoromethyl)phenyl)-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(isoindolin-2-yl)methanone (9r): Amide bromide 8a (77 mg, 0.150 mmol, 1 equiv) was coupled to 4p (46.7 mg, 0.165 mmol, 1.1 equiv) according to General Procedure E1b. The resulting product was deprotected according to General Procedure E2a. 9r (49.8 mg, 0.093 mmol) was obtained in 62% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.72 (s, 1H), 9.29 (s, 1H), 8.10 – 8.02 (m, 2H), 7.64 – 7.57 (m, 2H), 7.41 – 7.31 (m, 2H), 7.31 – 7.24 (m, 2H), 7.03 (s, 1H), 6.75 (s, 1H), 5.88 (d, J = 2.1 Hz, 1H), 5.70 (d, J = 2.1 Hz, 1H), 5.04 – 4.59 (m, 4H), 1.58 (s, 9H); 13C NMR (126 MHz, DMSO-d6): δ 167.33, 159.89, 156.01, 145.47, 145.35, 141.49, 137.11, 130.13, 129.91 (q, J = 30.8 Hz), 128.95, 127.73, 125.82, 124.14 – 123.94 (m), 123.65, 123.30, 121.23 – 121.03 (m), 103.64, 101.20, 94.43, 92.85, 60.13, 29.80. UPLC/MS [M+H]=537.270 , TR=1.94 min.
(2,4-Dihydroxy-6-((1-methyl-3-(3-(trifluoromethoxy)phenyl)-1H-pyrazol-5-yl)amino)phenyl)(isoindolin-2-yl)methanone (9s): Amide bromide 8a (77 mg, 0.15 mmol, 1 equiv) was coupled to 4q (42 mg, 0.165 mmol, 1.1 equiv) according to general procedure E1b. The resulting product was deprotected according to general procedure E2a. 9s (43 mg, 0.084 mmol) was obtained in 56% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.64 (s, 1H), 9.30 (s, 1H), 7.76 – 7.70 (m, 1H), 7.69 – 7.64 (m, 1H), 7.47 (t, J = 8.0 Hz, 1H), 7.34 – 7.29 (m, 2H), 7.29 – 7.23 (m, 3H), 7.22 (s, 1H), 6.58 (s, 1H), 5.91 (d, J = 2.1 Hz, 1H), 5.66 (d, J = 2.1 Hz, 1H), 4.92 – 4.51 (m, 4H), 3.63 (s, 3H); 13C NMR (126 MHz, DMSO-d6): δ 166.40, 159.27, 155.55, 148.90 – 148.76 (m), 146.67, 142.02, 136.68, 135.92, 130.55, 127.24, 123.63, 122.84, 121.15, 119.45, 119.11, 116.59, 104.27, 97.13, 94.52, 93.09, 35.10. UPLC/MS [M+H]=511.162, TR=1.62 min.
(2-((1-(tert-Butyl)-3-(3-(trifluoromethoxy)phenyl)-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(isoindolin-2-yl)methanone (9t): Amide bromide 8a (77 mg, 0.150 mmol, 1 equiv) was coupled to 4r (49 mg, 0.165 mmol, 1.1 equiv) according to General Procedure E1b. The resulting product was deprotected according to General Procedure E2a. 9t (39.4 mg, 0.071 mmol) was obtained in 47% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.71 (s, 1H), 9.28 (s, 1H), 7.82 – 7.77 (m, 1H), 7.73 – 7.68 (m, 1H), 7.50 (t, J = 8.0 Hz, 1H), 7.40 – 7.32 (m, 2H), 7.32 – 7.21 (m, 3H), 7.02 (s, 1H), 6.69 (s, 1H), 5.87 (d, J = 2.1 Hz, 1H), 5.68 (d, J = 2.1 Hz, 1H), 5.05 – 4.58 (m, 4H), 1.57 (s, 9H); 13C NMR (126 MHz, DMSO-d6): δ 167.33, 159.89, 156.01, 149.38 – 149.25 (m), 145.44, 145.40, 141.38, 137.11, 136.64, 131.01, 127.73, 124.08, 121.61, 119.79, 119.57, 117.16, 103.60, 101.30, 94.40, 92.80, 60.10, 29.79. UPLC/MS [M+H]=553.278, TR=544.246 min.
(2-((3-(3-Fluorophenyl)-1-methyl-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(isoindolin-2-yl)methanone (9u): Amide bromide 8a (45 mg, 0.087mmol, 1 equiv) was coupled to 2-methyl-5-(3-fluorophenyl)pyrazol-3-amine 4s (18 mg, 0.096 mmol, 1.1 equiv) according to General Procedure E1a. The resulting product was deprotected according to General Procedure E2a. 9u (24.7 mg, 0.056 mmol) was obtained in 64% yield over two steps.
1H NMR (400 MHz, Methanol-d4) δ 7.45 – 7.39 (m, 1H), 7.38 – 7.26 (m, 2H), 7.26 – 7.21 (m, 4H), 7.01 – 6.89 (m, 1H), 6.38 (s, 1H), 5.96 (d, J = 2.1 Hz, 1H), 5.79 (d, J = 2.1 Hz, 1H), 4.95 – 4.72 (m, 4H), 3.71 (s, 3H); (500 MHz, DMSO-d6): δ 9.63 (s, 1H), 9.29 (s, 1H), 7.59 – 7.53 (m, 1H), 7.50 – 7.46 (m, 1H), 7.41 – 7.35 (m, 1H), 7.35 – 7.29 (m, 2H), 7.30 – 7.25 (m, 2H), 7.19 (s, 1H), 7.10 – 7.03 (m, 1H), 6.54 (s, 1H), 5.89 (d, J = 2.1 Hz, 1H), 5.64 (d, J = 2.1 Hz, 1H), 4.84 – 4.68 (m, 4H), 3.61 (s, 3H); 13C NMR (126 MHz, DMSO-d6): δ 166.83, 162.99 (d, J = 242.2 Hz), 159.70, 155.99, 147.48 (d, J = 2.4 Hz), 144.29, 142.26, 136.54 (d, J = 8.2 Hz), 130.93 (d, J = 8.5 Hz), 127.69, 123.30, 121.06, 114.28 (d, J = 21.7 Hz), 111.51 (d, J = 22.4 Hz), 104.64, 97.54, 94.90, 93.42, 35.51. UPLC/MS [M+H]=445.408, TR=2.31 min.
(2-((1-(tert-Butyl)-3-(3-fluorophenyl)-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(isoindolin-2-yl)methanone (9v): Amide bromide 8a (77 mg, 0.150 mmol, 1 equiv) was coupled to 4t (38.5 mg, 0.165 mmol, 1.1 equiv) according to General Procedure E1b. The resulting product was deprotected according to General Procedure E2a. 9v (32.8 mg, 0.067 mmol) was obtained in 45% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.71 (s, 1H), 9.28 (s, 1H), 7.64 – 7.59 (m, 1H), 7.59 – 7.51 (m, 1H), 7.44 – 7.38 (m, 1H), 7.37 – 7.32 (m, 2H), 7.32 – 7.25 (m, 2H), 7.11 – 7.04 (m, 1H), 7.00 (s, 1H), 6.65 (s, 1H), 5.87 (d, J = 2.1 Hz, 1H), 5.67 (d, J = 2.1 Hz, 1H), 4.97 – 4.71 (m, 4H), 1.56 (s, 9H); 13C NMR (126 MHz, DMSO-d6): δ 166.88, 162.58 (d, J = 242.3 Hz), 159.43, 155.56, 145.35 (d, J = 2.6 Hz), 144.99, 140.75, 136.67, 136.31 (d, J = 8.1 Hz), 130.50 (d, J = 8.6 Hz), 127.29, 122.86, 120.65 (d, J = 2.7 Hz), 113.75 (d, J = 21.0 Hz), 111.06 (d, J = 22.4 Hz), 103.11, 100.80, 93.91, 92.30, 59.58, 29.37. UPLC/MS [M+H]=487.501, TR=1.82 min.
(2-((3-(3-Chlorophenyl)-1-methyl-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(isoindolin-2-yl)methanone (9w): Amide bromide 8a (129 mg, 0.25 mmol, 1 equiv) was coupled to 4u (57 mg, 0.275 mmol, 1.1 equiv) according to General Procedure E1b. The resulting product was deprotected according to General Procedure E2b. 9w (20.4 mg, 0.044 mmol) was obtained in 18% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.43 (s, 1H), 9.07 (s, 1H), 7.49 (t, J = 1.9 Hz, 1H), 7.45 – 7.39 (m, 1H), 7.11 (t, J = 7.9 Hz, 1H), 7.09 – 7.00 (m, 5H), 6.95 (s, 1H), 6.30 (s, 1H), 5.65 (d, J = 2.1 Hz, 1H), 5.40 (d, J = 2.1 Hz, 1H), 4.65 – 4.33 (m, 4H), 3.36 (s, 3H); 13C NMR (126 MHz, DMSO-d6): δ 166.38, 159.25, 155.56, 146.72, 143.76, 141.87, 136.95, 135.67, 133.40, 130.40, 127.23, 126.92, 124.13, 123.14, 122.83, 104.20, 96.93, 94.49, 93.01, 35.06. UPLC/MS [M+H]=461.175, TR=1.54 min.
(2-((1-(tert-Butyl)-3-(3-chlorophenyl)-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(isoindolin-2-yl)methanone (9x): Amide bromide 8a (77 mg, 0.150 mmol, 1 equiv) was coupled to 4v (41 mg, 0.165 mmol, 1.1 equiv) according to General Procedure E1b. The resulting product was deprotected according to General Procedure E2b. 9x (6.4 mg, 0.013 mmol) was obtained in 9% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.70 (s, 1H), 9.27 (s, 1H), 7.79 (t, J = 1.9 Hz, 1H), 7.77 – 7.69 (m, 1H), 7.39 (t, J = 7.9 Hz, 1H), 7.37 – 7.33 (m, 2H), 7.33 – 7.27 (m, 3H), 7.00 (s, 1H), 6.66 (s, 1H), 5.85 (d, J = 2.1 Hz, 1H), 5.67 (d, J = 2.1 Hz, 1H), 4.93 – 4.69 (m, 4H), 1.56 (s, 9H); 13C NMR (126 MHz, DMSO-d6): δ 166.84, 159.40, 155.53, 145.03, 144.90, 140.81, 136.31, 135.90, 133.40, 130.43, 127.27, 126.88, 124.08, 123.20, 122.85, 103.11, 100.71, 93.90, 92.29, 59.59, 29.35. UPLC/MS [M+H]=503.236 , TR=1.86 min.
(2-((3-(3-Chlorophenyl)-1-cyclohexyl-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(isoindolin-2-yl)methanone (9y): Amide bromide 8a (77 mg, 0.150 mmol, 1 equiv) was coupled to 4w (46 mg, 0.165 mmol, 1.1 equiv) according to General Procedure E1b. The resulting product was deprotected according to General Procedure E2b. 9y (85 mg, 0.016 mmol) was obtained in 11% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.59 (s, 1H), 9.25 (s, 1H), 7.76 (t, J = 1.9 Hz, 1H), 7.71 – 7.65 (m, 1H), 7.36 (t, J = 7.9 Hz, 1H), 7.34 – 7.25 (m, 5H), 7.10 (s, 1H), 6.55 (s, 1H), 5.86 (d, J = 2.1 Hz, 1H), 5.61 (d, J = 2.1 Hz, 1H), 4.75 (s, 4H), 4.07 – 3.98 (m, 1H), 1.90 – 1.70 (m, 6H), 1.61 – 1.55 (m, 1H), 1.26 – 1.13 (m, 3H); 13C NMR (126 MHz, DMSO-d6): δ 166.42, 159.16, 155.49, 146.69, 144.42, 140.58, 136.65, 135.92, 133.37, 130.38, 127.24, 126.83, 124.07, 123.18, 122.82, 104.02, 97.97, 94.27, 92.75, 55.44, 40.43, 32.24, 24.85. UPLC/MS [M+H]=529.319 , TR=1.90 min.
(2-((3-(2,3-Dimethylphenyl)-1-methyl-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(isoindolin-2-yl)methanone (9z): Amide bromide 8a (77 mg, 0.15 mmol, 1 equiv) was coupled to 4x (33.2 mg, 0.165 mmol, 1.1 equiv) according to General Procedure E1a. The resulting product was deprotected according to General Procedure E2a. 9z (5.5 mg, 0.012 mmol) was obtained in 8% yield over two steps. 1H NMR (500 MHz, DMSO-d6): 9.60 (s, 1H), 9.30 (s, 1H), 7.34 (s, 2H), 7.31 – 7.26 (m, 2H), 7.24 – 7.18 (m, 1H), 7.17 (s, 1H), 7.13 – 7.09 (m, 1H), 7.06 (t, J = 7.5 Hz, 1H), 6.13 (s, 1H), 5.88 (d, J = 2.1 Hz, 1H), 5.66 (d, J = 2.1 Hz, 1H), 4.82 – 4.70 (m, 4H), 3.60 (s, 3H), 2.30 (s, 3H), 2.26 (s, 3H); 13C NMR (126 MHz, DMSO-d6): δ 166.40, 159.24, 155.50, 149.40, 143.88, 140.45, 136.76, 133.87, 133.85, 128.76, 127.25, 126.92, 125.06, 122.87, 104.09, 99.74, 94.34, 92.74, 34.85, 20.36, 16.59. UPLC/MS [M+H]=455.303, TR= 1.52 min.
(2-((3-(2,3-Dimethylphenyl)-1-methyl-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(4-fluoroisoindolin-2-yl)methanone (9aa): Amide bromide 8b (66.6 mg, 0.125 mmol, 1 equiv) was coupled to 4x (27.7 mg, 0.138 mmol, 1.1 equiv) according to General Procedure E1b. The resulting product was deprotected according to General Procedure E2a. 9aa (17.5 mg, 0.037mmol) was obtained in 30% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.66 (s, 1H), 9.34 (s, 1H), 7.40 – 7.30 (m, 1H), 7.25 – 7.14 (m, 3H), 7.15 – 7.08 (m, 2H), 7.05 (t, J = 7.5 Hz, 1H), 6.14 (s, 1H), 5.89 (d, J = 2.1 Hz, 1H), 5.68 (d, J = 2.1 Hz, 1H), 4.83 – 4.70 (m, 4H), 3.17 (s, 3H), 2.30 (s, 3H), 2.26 (s, 3H); 13C NMR (126 MHz, DMSO-d6): δ 166.42, 159.39, 157.13 (d, J = 241.2 Hz), 155.58, 149.41, 144.01, 140.43, 136.76, 133.84 (d, J = 4.3 Hz), 129.93, 128.77, 126.89, 125.06, 123.23, 119.19, 113.69 (d, J = 19.2 Hz), 103.74, 99.74, 94.34, 92.89, 34.86, 20.37, 16.59. UPLC/MS [M+H]=473.751, TR=1.54min.
(2-((3-(2,3-Dimethylphenyl)-1-methyl-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(5-fluoroisoindolin-2-yl)methanone (9ab): Amide bromide 8c (53.2 mg, 0.1 mmol, 1 equiv) was coupled to 4x (22.1 mg, 0.11 mmol, 1.1 equiv) according to General Procedure E1b. The resulting product was deprotected according to General Procedure E2a. 9ab (8.2 mg, 0.017 mmol) was obtained in 17 % yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.60 (s, 1H), 9.30 (s, 1H), 7.38 – 7.34 (m, 1H), 7.24 – 7.18 (m, 2H), 7.17 (s, 1H), 7.13 – 7.03 (m, 3H), 6.12 (s, 1H), 5.88 (d, J = 2.1 Hz, 1H), 5.66 (d, J = 2.0 Hz, 1H), 5.05 – 4.42 (m, 4H), 3.60 (s, 3H), 2.29 (s, 3H), 2.26 (s, 3H); 13C NMR (126 MHz, DMSO-d6): δ 166.35, 159.28, 155.51, 149.38, 143.93, 140.44, 138.77, 136.74, 133.83 (d, J = 2.8 Hz), 128.75, 126.89, 125.04, 110.01 (d, J = 24.7 Hz), 103.93, 99.74, 94.32, 92.81, 34.85, 20.35, 16.58. UPLC/MS [M+H]=473.670, TR=1.53 min.
(2-((3-(5-Fluoro-2-methylphenyl)-1-methyl-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(isoindolin-2-yl)methanone (9ac): Amide bromide 8a (77 mg, 0.15 mmol, 1 equiv) was coupled to 4y (34 mg, 0.165 mmol, 1.1 equiv) according to General Procedure E1a. The resulting product was deprotected according to General Procedure E2a. 9ac (9 mg, 0.020 mmol) was obtained in 13% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.60 (s, 1H), 9.29 (s, 1H), 7.36 – 7.22 (m, 6H), 7.20 (s, 1H), 7.01 (td, J = 8.4, 2.9 Hz, 1H), 6.34 (s, 1H), 5.89 (d, J = 2.0 Hz, 1H), 5.66 (d, J = 2.0 Hz, 1H), 4.78 – 4.70 (m, 4H), 3.61 (s, 3H), 2.41 (s, 3H); 13C NMR (126 MHz, DMSO-d6): δ 160.37, 154.39 (d, J = 240.2 Hz), 153.23, 149.51, 141.62, 137.82, 134.93, 130.69, 128.85 (d, J = 7.4 Hz), 126.51 (d, J = 7.8 Hz), 124.91 (d, J = 2.8 Hz), 121.23, 116.84, 108.17 (d, J = 21.8 Hz), 107.62 (d, J = 21.1 Hz), 98.17, 93.52, 88.41, 86.87, 29.00, 14.55. UPLC/MS [M+H]=459.452, TR=1.43 min.
(2-((3-(5-Fluoro-2-methylphenyl)-1-methyl-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(4-fluoroisoindolin-2-yl)methanone (9ad): Amide bromide 8b (66.6 mg, 0.125 mmol, 1 equiv) was coupled to 4y (28.2 mg, 0.138 mmol, 1.1 equiv) according to General Procedure E1b. The resulting product was deprotected according to General Procedure E2a. 9ad (27 mg, 0.056 mmol) was obtained in 45% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.66 (s, 1H), 9.34 (s, 1H), 7.36 – 7.30 (m, 1H), 7.30 – 7.26 (m, 1H), 7.26 – 7.21 (m, 2H), 7.20 – 7.15 (m, 1H), 7.08 (t, J = 8.8 Hz, 1H), 7.00 (td, J = 8.4, 2.9 Hz, 1H), 6.34 (s, 1H), 5.90 (d, J = 2.1 Hz, 1H), 5.68 (d, J = 2.1 Hz, 1H), 4.87 – 4.71 (m, 4H), 3.62 (s, 3H), 2.40 (s, 3H); 13C NMR (126 MHz, DMSO-d6): δ 166.39, 160.39 (d, J = 240.5 Hz), 159.37, 147.62 (d, J = 2.3 Hz), 143.98, 140.95, 134.80 (d, J = 7.9 Hz), 132.51 (d, J = 8.0 Hz), 130.88 (d, J = 2.9 Hz), 129.85, 119.14, 114.12 (d, J = 22.1 Hz), 113.71 (d, J = 7.1 Hz), 113.55 (d, J = 8.8 Hz), 103.85, 99.51, 94.43, 93.12, 89.17 (d, J = 104.5 Hz), 35.02, 20.57. UPLC/MS [M+H]=477.489, TR=1.53 min.
(2-((3-(5-Fluoro-2-methylphenyl)-1-methyl-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(5-fluoroisoindolin-2-yl)methanone (9ae): Amide bromide 8c (66.6 mg, 0.125 mmol, 1 equiv) was coupled to 4y (28.2 mg, 0.138 mmol, 1.1equiv) according to General Procedure E1b. The resulting product was deprotected according to General Procedure E2a. 9ae (19 mg, 0.40 mmol) was obtained in 32% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.61 (s, 1H), 9.30 (s, 1H), 7.37 – 7.31 (m, 1H), 7.29 – 7.25 (m, 1H), 7.25 – 7.22 (m, 1H), 7.22 – 7.16 (m, 2H), 7.12 – 7.04 (m, 1H), 7.04 – 6.98 (m, 1H), 6.32 (s, 1H), 5.89 (d, J = 2.1 Hz, 1H), 5.66 (d, J = 2.1 Hz, 1H), 4.80 – 4.65 (m, 4H), 3.62 (s, 3H), 2.40 (s, 3H); 13C NMR (126 MHz, DMSO-d6): δ 166.32, 160.35 (d, J = 240.5 Hz), 159.26, 155.54, 147.60 (d, J = 2.4 Hz), 143.90, 140.96, 134.80 (d, J = 7.7 Hz), 132.49 (d, J = 8.1 Hz), 130.87 (d, J = 2.9 Hz), 124.50 (d, J = 8.8 Hz), 114.22, 114.05, 113.67, 113.51, 109.96 (d, J = 23.2 Hz), 104.03, 99.51, 94.41, 93.04, 35.01, 20.56. UPLC/MS [M+H]=477.489, TR=1.52 min.
(2-((3-(2-Fluoro-3-methylphenyl)-1-methyl-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(isoindolin-2-yl)methanone (9af): Amide bromide 8a (77 mg, 0.150 mmol, 1 equiv) was coupled to 4z (33.8 mg, 0.165 mmol, equiv) according to General Procedure E1a. The resulting product was deprotected according to General Procedure E2a. 9af (13.7 mg, 0.030 mmol) was obtained in 20% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.61 (s, 1H), 9.29 (s, 1H), 7.74 – 7.67 (m, 1H), 7.35 – 7.29 (m, 2H), 7.29 – 7.23 (m, 2H), 7.21 (s, 1H), 7.20 – 7.15 (m, 1H), 7.08 (t, J = 7.6 Hz, 1H), 6.35 (d, J = 4.1 Hz, 1H), 5.89 (d, J = 2.1 Hz, 1H), 5.64 (d, J = 2.0 Hz, 1H), 4.81 – 4.67 (m, 4H), 3.62 (s, 3H), 2.24 (d, J = 2.1 Hz, 3H); 13C NMR (126 MHz, DMSO-d6): δ 166.33, 159.21, 157.64 (d, J = 246.4 Hz), 155.51, 143.78, 142.88, 141.33, 130.20, 127.19, 125.07 – 124.96 (m), 124.79 (d, J = 17.8 Hz), 123.88, 122.81, 120.73, 104.26, 99.46, 94.47, 35.04, 14.29 (d, J = 5.0 Hz). UPLC/MS [M+H]=459.496, TR=1.49 min.
(2-((3-(2-Fluoro-3-methylphenyl)-1-methyl-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(4-fluoroisoindolin-2-yl)methanone (9ag): Amide bromide 8b (66.6 mg, 0.125 mmol, 1 equiv) was coupled to 4z (28.2 mg, 0.138 mmol, 1.1 equiv) according to General Procedure E1b. The resulting product was deprotected according to General Procedure E2a. 9ag (39 mg, 0.082 mmol) was obtained in 66% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.68 – 9.64 (m, 1H), 9.34 (s, 1H), 7.73 – 7.66 (m, 1H), 7.35 – 7.27 (m, 1H), 7.25 (s, 1H), 7.20 – 7.13 (m, 2H), 7.10 – 7.03 (m, 2H), 6.33 (d, J = 4.1 Hz, 1H), 5.91 (d, J = 2.1 Hz, 1H), 5.68 (d, J = 2.0 Hz, 1H), 4.87 – 4.67 (m, 4H), 3.63 (s, 3H), 2.23 (d, J = 2.1 Hz, 3H); 13C NMR (126 MHz, DMSO-d6): δ 166.36, 159.34, 157.65 (d, J = 247.1 Hz), 155.61, 144.25, 142.85, 141.38, 130.21, 124.92 (d, J = 4.1 Hz), 124.76, 123.87 (d, J = 3.6 Hz), 120.73 (d, J = 12.5 Hz), 119.08, 113.59 (d, J = 19.1 Hz), 103.95, 99.49 (d, J = 10.2 Hz), 94.50, 93.29, 79.16, 35.05, 14.28 (d, J = 4.8 Hz). UPLC/MS [M+H]=477.489, TR=1.44 min.
(2-((3-(2-Fluoro-3-methylphenyl)-1-methyl-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(5-fluoroisoindolin-2-yl)methanone (9ah): Amide bromide 8c (53.2 mg, 0.1 mmol, 1 equiv) was coupled to 4z (22.6 mg, 0.11 mmol, 1.1equiv) according to General Procedure E1b. The resulting product was deprotected according to General Procedure E2a. 9ah (18 mg, 0.038 mmol) was obtained in 38% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.62 (s, 1H), 9.26 (s, 0H), 7.73 – 7.66 (m, 1H), 7.35 – 7.29 (m, 1H), 7.22 (s, 1H), 7.20 – 7.12 (m, 2H), 7.11 – 7.02 (m, 2H), 6.32 (d, J = 4.1 Hz, 1H), 5.89 (d, J = 2.1 Hz, 1H), 5.66 (d, J = 2.1 Hz, 1H), 4.84 – 4.56 (m, 4H), 3.63 (s, 3H), 2.24 (d, J = 2.1 Hz, 3H); 13C NMR (126 MHz, DMSO-d6): δ 166.30, 160.78, 159.24, 156.66 (d, J = 244.7 Hz), 155.55, 143.88, 142.11 (d, J = 181.7 Hz), 130.16, 125.03 – 124.87 (m), 124.75 (d, J = 7.8 Hz), 124.41, 123.85 (d, J = 4.3 Hz), 120.74 (d, J = 12.4 Hz), 114.34 (d, J = 24.9 Hz), 109.81 (d, J = 25.6 Hz), 104.12, 99.50 (d, J = 10.4 Hz), 94.48, 93.18, 35.05, 14.26 (d, J = 4.8 Hz). UPLC/MS [M+H]=477.445, TR=1.51 min.
(2-((3-(2,3-Difluorophenyl)-1-methyl-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(isoindolin-2-yl)methanone (9ai): Amide bromide 8a (77 mg, 0.150 mmol, 1 equiv) was coupled to 4aa (34.5 mg, 0.165 mmol, 1.1 equiv) according to General Procedure E1a. The resulting product was deprotected according to General Procedure E2a. 9ai (5.7 mg, 0.012 mmol) was obtained in 8% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.61 (s, 1H), 9.30 (s, 1H), 7.70 – 7.63 (m, 1H), 7.35 – 7.28 (m, 3H), 7.27 – 7.22 (m, 3H), 7.22 – 7.16 (m, 1H), 6.37 (d, J = 3.7 Hz, 1H), 5.90 (d, J = 2.0 Hz, 1H), 5.65 (d, J = 2.1 Hz, 1H), 4.79 – 4.66 (m, 4H), 3.64 (s, 3H); 13C NMR (126 MHz, DMSO-d6): δ 166.28, 159.19, 155.53, 143.71, 141.77, 136.38, 132.64, 127.16, 124.43, 122.90, 122.78, 104.41, 94.60, 93.20, 35.16. UPLC/MS [M+H]=463.241, TR=1.45 min.
(2,4-Dihydroxy-6-((1-methyl-3-(2-methyl-3-(trifluoromethyl)phenyl)-1H-pyrazol-5-yl)amino)phenyl)(isoindolin-2-yl)methanone (9aj): Amide bromide 8a (64 mg, 0.125 mmol, 1 equiv) was coupled to 4ab (35 mg, 0.138 mmol, 1.1 equiv) according to General Procedure E1b. The resulting product was deprotected according to General Procedure E2a. 9aj (3.4 mg, 0.007 mmol) was obtained in 6% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.60 (s, 1H), 9.30 (s, 1H), 7.66 (t, J = 7.8 Hz, 2H), 7.38 (t, J = 7.8 Hz, 1H), 7.34 – 7.31 (m, 3H), 7.31 – 7.25 (m, 2H), 7.23 (s, 1H), 6.27 (s, 1H), 5.89 (d, J = 2.1 Hz, 1H), 5.68 (d, J = 2.0 Hz, 1H), 4.83 – 4.69 (m, 4H), 3.62 (s, 3H), 2.49 (s, 3H); 13C NMR (126 MHz, DMSO-d6): δ 166.34, 159.22, 155.52, 147.64, 143.75, 140.96, 136.17, 133.85, 133.27, 127.23, 125.93, 122.84, 104.25, 100.08, 94.47, 93.01, 34.99, 16.38; 19F NMR (470 MHz, DMSO-d6): δ −59.28. UPLC/MS [M+H]=509.514 , TR=1.56 min.
(2-((3-(3-Chloro-2-fluorophenyl)-1-methyl-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(isoindolin-2-yl)methanone (9ak): Amide bromide 8a (64 mg, 0.125 mmol, 1 equiv) was coupled to 4ac (31 mg, 0.138 mmol, 1.1 equiv) according to General Procedure E1b. The resulting product was deprotected according to General Procedure E2b. 9ak (18 mg, 0.037 mmol) was obtained in 30% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.62 (s, 1H), 9.31 (s, 1H), 7.87 – 7.80 (m, 1H), 7.51 – 7.44 (m, 1H), 7.33 – 7.27 (m, 2H), 7.27 – 7.19 (m, 4H), 6.38 (d, J = 4.0 Hz, 1H), 5.90 (d, J = 2.1 Hz, 1H), 5.66 (d, J = 2.1 Hz, 1H), 4.87 – 4.60 (m, 4H), 3.64 (s, 3H); 13C NMR (126 MHz, DMSO-d6): δ 166.29, 159.20, 155.55, 154.13 (d, J = 250.7 Hz), 143.69, 141.84 – 141.68 (m), 136.63, 129.05, 127.16, 126.21 (d, J = 3.6 Hz), 125.29 (d, J = 4.3 Hz), 122.86, 122.77, 120.45 (d, J = 18.0 Hz), 104.41, 99.55 (d, J = 9.8 Hz), 94.61, 93.22, 35.18; 19F NMR (470 MHz, DMSO-d6) δ −116.80 – −124.29 (m). UPLC/MS [M+H]=479.219 , TR=1.46 min.
(2-((3-(2-Fluoro-3-(trifluoromethoxy)phenyl)-1-methyl-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(isoindolin-2-yl)methanone (9al): Amide bromide 8a (64 mg, 0.125 mmol, 1 equiv) was coupled to 4ad (38 mg, 0.138 mmol, 1.1 equiv) according to General Procedure E1b. The resulting product was deprotected according to General Procedure E2a. 9al (29 mg, 0.056 mmol) was obtained in 45% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.63 (s, 1H), 9.32 (s, 1H), 7.93 – 7.86 (m, 1H), 7.49 – 7.41 (m, 1H), 7.34 – 7.20 (m, 6H), 6.40 (d, J = 3.7 Hz, 1H), 5.91 (d, J = 2.1 Hz, 1H), 5.67 (d, J = 2.1 Hz, 1H), 4.81 – 4.64 (m, 4H), 3.65 (s, 3H); 13C NMR (126 MHz, DMSO-d6): δ 166.32, 159.23, 155.57, 150.83 (d, J = 254.4 Hz), 143.69, 141.93 (d, J = 1.9 Hz), 141.50, 136.65, 136.23 – 136.00 (m), 127.18, 126.41 (d, J = 3.3 Hz), 125.00 (d, J = 4.5 Hz), 123.32 (d, J = 9.5 Hz), 122.79, 122.19, 121.12, 119.07, 104.49, 99.50 (d, J = 9.2 Hz), 94.68, 93.28, 35.22; 19F NMR (470 MHz, DMSO-d6) δ −57.82 (d, J = 4.9 Hz), −134.22 – −134.33 (m). UPLC/MS [M+H]=529.562 , TR=1.54 min.
(2-((3-(2,5-Difluorophenyl)-1-methyl-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(isoindolin-2-yl)methanone (9am): Amide bromide 8a (77 mg, 0.150 mmol, 1 equiv) was coupled to 4ae (34.5 mg, 0.165 mmol, 1.1 equiv) according to General Procedure E1a. The resulting product was deprotected according to General Procedure E2a. 9am (14.6 mg, 0.032 mmol) was obtained in 21% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.62 (s, 1H), 9.30 (s, 1H), 7.62 – 7.56 (m, 1H), 7.34 – 7.23 (m, 6H), 7.19 – 7.11 (m, 1H), 6.38 (d, J = 4.0 Hz, 1H), 5.90 (d, J = 2.1 Hz, 1H), 5.63 (d, J = 2.1 Hz, 1H), 4.84 – 4.60 (m, 4H), 3.64 (s, 3H); 13C NMR (126 MHz, DMSO-d6): δ 166.28, 159.19, 157.26, 155.53, 154.23 (d, J = 243.9 Hz), 143.70, 141.85 (d, J = 2.4 Hz), 141.70, 127.19, 122.80, 118.04 – 117.56 (m), 115.52 – 114.98 (m), 113.10 – 112.60 (m), 104.44, 99.60 (d, J = 10.1 Hz), 94.61, 93.15, 35.17 (d, J = 241.7 Hz); 19F NMR (470 MHz, DMSO-d6) δ −118.66 – −118.78 (m), −121.71 – −121.84 (m) UPLC/MS [M+H]=463.241, TR=1.46 min.
(2-((3-(2-Chloro-5-fluorophenyl)-1-methyl-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(isoindolin-2-yl)methanone (9an): Amide bromide 8a (64 mg, 0.125 mmol, 1 equiv) was coupled to 4af (31 mg, 0.138 mmol, 1.1 equiv) according to General Procedure E1b. The resulting product was deprotected according to General Procedure E2b. 9an (18 mg, 0.038 mmol) was obtained in 30% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.62 (s, 1H), 9.32 (s, 1H), 7.56 – 7.48 (m, 2H), 7.33 – 7.29 (m, 2H), 7.28 – 7.22 (m, 3H), 7.22 – 7.15 (m, 1H), 6.56 (s, 1H), 5.91 (d, J = 2.1 Hz, 1H), 5.66 (d, J = 2.0 Hz, 1H), 4.80 – 4.69 (m, 4H), 3.64 (s, 3H); 13C NMR (126 MHz, DMSO-d6): δ 166.30, 160.66 (d, J = 243.7 Hz), 159.21, 155.54, 144.63 (d, J = 1.9 Hz), 143.70, 141.25, 136.60, 133.85 (d, J = 8.5 Hz), 132.11 (d, J = 8.7 Hz), 127.21, 125.85 (d, J = 3.0 Hz), 122.82, 115.74 (d, J = 22.7 Hz), 115.64 (d, J = 22.1 Hz), 104.43, 100.19, 94.61, 93.12, 35.15; 19F NMR (470 MHz, DMSO-d6) δ −115.27 – −115.40 (m). UPLC/MS [M+H]=479.259 , TR=1.53 min.
(2-((3-(5-Chloro-2-fluorophenyl)-1-methyl-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(isoindolin-2-yl)methanone (9ao): Amide bromide 8a (64 mg, 0.125 mmol, 1 equiv) was coupled to 4ag (31 mg, 0.138 mmol, 1.1 equiv) according to General Procedure E1b. The resulting product was deprotected according to General Procedure E2b. 9ao (18 mg, 0.037 mmol) was obtained in 30% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.63 (s, 1H), 9.32 (s, 1H), 7.89 – 7.82 (m, 1H), 7.39 – 7.33 (m, 1H), 7.33 – 7.21 (m, 6H), 6.38 (d, J = 4.0 Hz, 1H), 5.91 (d, J = 2.1 Hz, 1H), 5.65 (d, J = 2.1 Hz, 1H), 4.96 – 4.53 (m, 4H), 3.64 (s, 3H); 13C NMR (126 MHz, DMSO-d6): δ 166.30, 159.21, 157.66 (d, J = 248.4 Hz), 155.55, 143.69, 141.92 (d, J = 2.3 Hz), 141.36 (d, J = 2.1 Hz), 136.61, 128.57 – 128.40 (m), 127.19, 126.41 (d, J = 4.3 Hz), 122.87 (d, J = 13.3 Hz), 122.79, 118.19 (d, J = 24.2 Hz), 104.49, 99.62 (d, J = 9.9 Hz), 94.67, 93.25, 35.20; 19F NMR (470 MHz, DMSO-d6) δ −118.55 – −118.64 (m). UPLC/MS [M+H]=479.219 , TR=1.57 min.
(2-((3-(2-Fluoro-5-methylphenyl)-1-methyl-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(isoindolin-2-yl)methanone (9ap): Amide bromide 8a (77 mg, 0.150 mmol, 1 equiv) was coupled to 4ah (33.9 mg, 0.165 mmol, 1.1 equiv) according to General Procedure E1a. The resulting product was deprotected according to General Procedure E2a. 9ap (12.5 mg, 0.027 mmol) was obtained in 18% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.60 (s, 1H), 9.29 (s, 1H), 7.72 – 7.66 (m, 1H), 7.37 – 7.30 (m, 2H), 7.30 – 7.23 (m, 2H), 7.19 (s, 1H), 7.14 – 7.05 (m, 2H), 6.33 (d, J = 3.9 Hz, 1H), 5.89 (d, J = 2.1 Hz, 1H), 5.63 (d, J = 2.1 Hz, 1H), 4.82 – 4.67 (m, 4H), 3.62 (s, 3H), 2.30 (s, 3H); 13C NMR (126 MHz, DMSO-d6): δ 166.32, 159.20, 156.44 (d, J = 244.4 Hz), 155.49, 143.78, 142.78, 141.37 (d, J = 2.1 Hz), 133.37 (d, J = 3.2 Hz), 129.35, 127.55 (d, J = 4.1 Hz), 127.21, 120.56 (d, J = 11.9 Hz), 115.75 (d, J = 22.3 Hz), 104.31, 99.55, 94.48, 92.90, 35.03, 20.22. UPLC/MS [M+H]=459.256, TR=1.52 min.
(2-((3-(2-Fluoro-5-(trifluoromethyl)phenyl)-1-methyl-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(isoindolin-2-yl)methanone (9aq): Amide bromide 8a (64 mg, 0.125 mmol, 1 equiv) was coupled to 4ai (36 mg, 0.138 mmol, 1.1 equiv) according to General Procedure E1b. The resulting product was deprotected according to General Procedure E2a. 9aq (36 mg, 0.071 mmol) was obtained in 57% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.66 (s, 1H), 9.34 (s, 1H), 8.21 – 8.15 (m, 1H), 7.72 – 7.65 (m, 1H), 7.49 – 7.41 (m, 1H), 7.28 (q, J = 3.2 Hz, 3H), 7.25 – 7.17 (m, 2H), 6.42 (d, J = 4.1 Hz, 1H), 5.93 (d, J = 2.1 Hz, 1H), 5.68 (s, 1H), 4.72 (s, 4H), 3.67 (s, 3H); 13C NMR (126 MHz, DMSO-d6): δ 166.78, 161.24 (d, J = 254.5 Hz), 159.68, 156.06, 144.19, 142.61 (d, J = 2.3 Hz), 141.56, 137.13, 127.62, 126.55 – 125.77 (m), 125.39, 124.86 – 124.44 (m), 123.21 (d, J = 3.0 Hz), 122.48 (d, J = 13.2 Hz), 118.08 (d, J = 23.5 Hz), 105.03, 100.17 (d, J = 10.3 Hz), 95.20, 93.91, 49.04, 35.69. UPLC/MS [M+H]=513.212, TR=1.63 min.
(2-((3-(2-Fluoro-5-(trifluoromethoxy)phenyl)-1-methyl-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(isoindolin-2-yl)methanone (9ar): Amide bromide 8a (64 mg, 0.125 mmol, 1 equiv) was coupled to 4aj (38 mg, 0.138 mmol, 1.1 equiv) according to General Procedure E1b. The resulting product was deprotected according to General Procedure E2a. 9ar (33 mg, 0.063 mmol) was obtained in 50% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.64 (d, J = 1.3 Hz, 1H), 9.32 (s, 1H), 7.82 – 7.76 (m, 1H), 7.40 – 7.19 (m, 7H), 6.40 (d, J = 4.1 Hz, 1H), 5.91 (d, J = 2.1 Hz, 1H), 5.65 (d, J = 2.0 Hz, 1H), 4.84 – 4.62 (m, 4H), 3.65 (s, 3H); 13C NMR (126 MHz, DMSO-d6): δ 166.31, 159.22, 157.27 (d, J = 248.5 Hz), 155.58, 144.54 – 144.35 (m), 143.74, 142.08 (d, J = 2.3 Hz), 141.25 (d, J = 2.2 Hz), 136.71, 127.18, 122.78, 122.65, 121.59 (d, J = 8.6 Hz), 121.09, 119.21 (d, J = 5.1 Hz), 119.05, 118.12 (d, J = 24.9 Hz), 104.53, 99.71 (d, J = 10.5 Hz), 94.70, 93.34, 35.23; 19F NMR (470 MHz, DMSO-d6) δ −57.20, −117.44 – −117.53 (m). UPLC/MS [M+H]=529.529, TR=1.66 min.
(2-((3-(2,6-Difluorophenyl)-1-methyl-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(isoindolin-2-yl)methanone (9as): Amide bromide 8a (45 mg, 0.087 mmol, 1 equiv) was coupled to 4ak (20 mg, 0.096 mmol, 1.1 equiv) according to General Procedure E1a. The resulting product was deprotected according to General Procedure E2a. 9as (20.6 mg, 0.045 mmol) was obtained in 52% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.63 (s, 1H), 9.33 (s, 1H), 7.44 – 7.36 (m, 1H), 7.32 (s, 2H), 7.30 – 7.24 (m, 2H), 7.24 – 7.17 (m, 1H), 7.14 (t, J = 8.3 Hz, 2H), 6.27 – 6.23 (m, 1H), 5.89 (d, J = 2.1 Hz, 1H), 5.61 (d, J = 2.1 Hz, 1H), 4.87 – 4.56 (m, 4H), 3.62 (s, 3H); 13C NMR (126 MHz, DMSO-d6): δ 166.35, 159.73 (dd, J = 249.7, 7.3 Hz), 159.25, 155.53, 143.72, 140.91, 137.83, 129.40, 127.26, 112.02 (d, J = 25.9 Hz), 112.02 (d, J = 15.0 Hz), 111.43, 104.32, 101.85 – 100.61 (m), 94.52, 92.84, 35.13; 19F NMR (470 MHz, DMSO-d6) δ −112.05 – −112.16 (m). UPLC/MS [M+H]=463.225, TR=1.36 min.
(2-((3-(4-Chloro-2-fluorophenyl)-1-methyl-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(isoindolin-2-yl)methanone (9at): Amide bromide 8a (64 mg, 0.125 mmol, 1 equiv) was coupled to 4al (31 mg, 0.138 mmol, 1.1 equiv) according to General Procedure E1b. The resulting product was deprotected according to General Procedure E2b. 9at (7 mg, 0.014 mmol) was obtained in 11% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.61 (s, 1H), 9.30 (s, 1H), 7.89 (t, J = 8.5 Hz, 1H), 7.46 – 7.40 (m, 1H), 7.32 – 7.21 (m, 6H), 6.34 (d, J = 4.0 Hz, 1H), 5.89 (d, J = 2.1 Hz, 1H), 5.63 (d, J = 2.1 Hz, 1H), 4.87 – 4.52 (m, 4H), 3.63 (s, 3H); 13C NMR (126 MHz, DMSO-d6): δ 166.29, 159.20, 157.79 (d, J = 250.8 Hz), 155.53, 143.72, 141.72 (t, J = 2.5 Hz), 136.72, 132.15 (d, J = 10.5 Hz), 128.50 (d, J = 4.6 Hz), 127.18, 124.84 (d, J = 3.2 Hz), 122.80, 120.19 (d, J = 11.8 Hz), 116.65 (d, J = 26.1 Hz), 104.39, 99.42 (d, J = 9.7 Hz), 94.58, 93.11, 35.14; 19F NMR (470 MHz, DMSO-d6) δ −113.56 – −113.67 (m). UPLC/MS [M+H]=479.259 , TR=1.58 min.
(2-((3-(2-Chloro-4-fluorophenyl)-1-methyl-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(isoindolin-2-yl)methanone (9au): Amide bromide 8a (64 mg, 0.125 mmol, 1 equiv) was coupled to 4am (31 mg, 0.138 mmol, 1.1 equiv) according to General Procedure E1b. The resulting product was deprotected according to General Procedure E2b. 9au (18 mg, 0.037 mmol) was obtained in 30% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.62 (s, 1H), 9.31 (s, 1H), 7.80 – 7.74 (m, 1H), 7.45 (dd, J = 8.9, 2.7 Hz, 1H), 7.32 (s, 2H), 7.29 – 7.20 (m, 4H), 6.45 (s, 1H), 5.90 (d, J = 2.1 Hz, 1H), 5.66 (d, J = 2.0 Hz, 1H), 4.85 – 4.64 (m, 4H), 3.62 (s, 3H); 13C NMR (126 MHz, DMSO-d6): δ 166.33, 160.98 (d, J = 247.9 Hz), 159.22, 155.53, 144.92, 143.74, 140.96, 136.58, 131.46 (d, J = 10.5 Hz), 131.29 (d, J = 8.7 Hz), 128.94 (d, J = 3.4 Hz), 127.22, 122.84, 117.17 (d, J = 24.9 Hz), 114.56 (d, J = 21.1 Hz), 104.33, 99.96, 94.54, 93.00, 35.04; 19F NMR (470 MHz, DMSO-d6) δ −113.06 – −113.20 (m). UPLC/MS [M+H]=479.219 , TR=1.52 min.
(2-((1-(tert-Butyl)-3-(2,3-dimethylphenyl)-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(isoindolin-2-yl)methanone (9av): Amide bromide 8a (64 mg, 0.125 mmol, 1 equiv) was coupled to 4ao (33 mg, 0.138 mmol, 1.1 equiv) according to General Procedure E1b. The resulting product was deprotected according to General Procedure E2a. 9av (31 mg, 0.063 mmol) was obtained in 50% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.68 (s, 1H), 9.30 (s, 1H), 7.37 – 7.32 (m, 2H), 7.32 – 7.25 (m, 3H), 7.16 – 7.05 (m, 2H), 6.99 (s, 1H), 6.24 (s, 1H), 5.86 (d, J = 2.1 Hz, 1H), 5.72 (d, J = 2.1 Hz, 1H), 4.89 – 4.75 (m, 4H), 2.34 (s, 3H), 2.27 (s, 3H), 1.55 (s, 9H); 13C NMR (126 MHz, DMSO-d6): δ 166.92, 159.44, 147.76, 145.08, 139.31, 136.86, 136.70, 133.91, 133.87, 128.68, 127.27, 126.89, 125.09, 122.85, 103.17, 103.03, 93.79, 92.11, 79.17, 59.17, 29.48, 20.43, 16.60. UPLC/MS [M+H]=497.600, TR=1.80 min.
(2-((3-(5-Fluoro-2-methylphenyl)-1-isopropyl-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(isoindolin-2-yl)methanone (9aw): Amide bromide 8a (64 mg, 0.125 mmol, 1 equiv) was coupled to 4ap (32 mg, 0.138 mmol, 1.1 equiv) according to General Procedure E1b. The resulting product was deprotected according to General Procedure E2a. 9aw (40 mg, 0.083 mmol) was obtained in 66% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.63 (s, 1H), 9.30 (s, 1H), 7.38 – 7.29 (m, 3H), 7.30 – 7.22 (m, 3H), 7.13 (s, 1H), 7.05 – 6.97 (m, 1H), 6.35 (s, 1H), 5.88 (d, J = 2.1 Hz, 1H), 5.63 (d, J = 2.1 Hz, 1H), 4.87 – 4.69 (m, 4H), 4.51 – 4.40 (m, J = 6.9 Hz, 1H), 2.44 (s, 3H), 1.35 (d, J = 6.5 Hz, 6H); 13C NMR (126 MHz, DMSO-d6): δ 166.48, 160.43 (d, J = 240.5 Hz), 159.26, 155.56, 147.71 (d, J = 2.4 Hz), 144.58, 139.50, 136.72, 135.06 (d, J = 7.9 Hz), 132.59 (d, J = 8.0 Hz), 130.93 (d, J = 2.9 Hz), 127.25, 122.85, 114.15 (d, J = 22.0 Hz), 113.51 (d, J = 20.7 Hz), 103.90, 100.20, 94.21, 92.57, 47.90, 22.20, 20.66; 19F NMR (470 MHz, DMSO-d6) δ −117.99 – −118.09 (m). UPLC/MS [M+H]=487.545, TR=1.70 min.
(2-((1-(tert-Butyl)-3-(5-fluoro-2-methylphenyl)-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(isoindolin-2-yl)methanone (9ax): Amide bromide 8a (64 mg, 0.125 mmol, 1 equiv) was coupled to 4aq (34 mg, 0.138 mmol, 1.1 equiv) according to General Procedure E1b. The resulting product was deprotected according to General Procedure E2a. 9ax (35.5 mg, 0.067 mmol) was obtained in 54% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.70 (s, 1H), 9.29 (s, 1H), 7.40 – 7.32 (m, 3H), 7.32 – 7.22 (m, 3H), 7.05 – 6.97 (m, 2H), 6.47 (s, 1H), 5.86 (d, J = 2.1 Hz, 1H), 5.70 (d, J = 2.1 Hz, 1H), 4.95 – 4.62 (m, 4H), 3.35 (s, 3H), 2.46 (s, 3H), 1.56 (s, 9H); 13C NMR (126 MHz, DMSO-d6): δ 166.90, 160.47 (d, J = 240.3 Hz), 159.45, 155.56, 146.05 (d, J = 2.4 Hz), 145.01, 139.78, 136.68, 134.88 (d, J = 7.7 Hz), 132.67 (d, J = 7.9 Hz), 130.90 (d, J = 2.9 Hz), 127.28, 122.85, 114.10 (d, J = 22.0 Hz), 113.48 (d, J = 20.8 Hz), 103.08, 102.94, 93.86, 92.18, 59.47, 29.38, 20.82; 19F NMR (470 MHz, DMSO-d6) δ −117.90 – −118.08 (m). UPLC/MS [M+H]=501.569, TR=1.87 min.
(2-((1-Cyclopentyl-3-(5-fluoro-2-methylphenyl)-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(isoindolin-2-yl)methanone (9ay): Amide bromide 8a (64 mg, 0.125 mmol, 1 equiv) was coupled to 4ar (36 mg, 0.138 mmol, 1.1 equiv) according to General Procedure E1b. The resulting product was deprotected according to General Procedure E2a. 9ay (31 mg, 0.060 mmol) was obtained in 50% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.64 (s, 1H), 9.31 (s, 1H), 7.39 – 7.30 (m, 3H), 7.30 – 7.22 (m, 3H), 7.15 (s, 1H), 7.04 – 6.97 (m, 1H), 6.36 (s, 1H), 5.88 (d, J = 2.1 Hz, 1H), 5.63 (d, J = 2.1 Hz, 1H), 4.87 – 4.69 (m, 4H), 4.61 (p, J = 7.1 Hz, 1H), 2.44 (s, 3H), 1.92 (s, 4H), 1.87 – 1.76 (m, 2H), 1.59 – 1.50 (m, 2H); 13C NMR (126 MHz, DMSO-d6): δ 166.50, 160.44 (d, J = 240.3 Hz), 159.25, 155.56, 147.70 (d, J = 2.3 Hz), 144.43, 140.18, 136.71, 134.96 (d, J = 7.7 Hz), 132.62 (d, J = 8.0 Hz), 130.92 (d, J = 2.8 Hz), 127.26, 122.84, 114.17 (d, J = 21.9 Hz), 113.51 (d, J = 20.7 Hz), 103.93, 100.05, 94.28, 92.72, 57.03, 32.28, 24.18, 20.72; 19F NMR (470 MHz, DMSO-d6) δ −117.94 – −118.06 (m). UPLC/MS [M+H]=513.564, TR=1.83 min.
(2-((1-Cyclohexyl-3-(5-fluoro-2-methylphenyl)-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(isoindolin-2-yl)methanone (9az): Amide bromide 8a (64 mg, 0.125 mmol, 1 equiv) was coupled to 4as (38 mg, 0.138 mmol, 1.1 equiv) according to General Procedure E1b. The resulting product was deprotected according to General Procedure E2a. 9az (35 mg, 0.066 mmol) was obtained in 53% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.60 (s, 1H), 9.28 (s, 1H), 7.36 – 7.29 (m, 3H), 7.30 – 7.21 (m, 3H), 7.11 (s, 1H), 7.00 (td, J = 8.4, 2.9 Hz, 1H), 6.36 (s, 1H), 5.87 (d, J = 2.1 Hz, 1H), 5.64 (d, J = 2.1 Hz, 1H), 4.87 – 4.70 (m, 4H), 4.07 – 4.00 (m, 1H), 2.43 (s, 3H), 1.90 – 1.70 (m, 6H), 1.64 – 1.55 (m, 1H), 1.25 – 1.12 (m, 3H); 13C NMR (126 MHz, DMSO-d6): δ 166.47, 160.42 (d, J = 240.3 Hz), 159.20, 155.52, 147.59 (d, J = 2.1 Hz), 144.50, 139.62, 136.65, 135.02 (d, J = 7.7 Hz), 132.57 (d, J = 7.9 Hz), 130.91 (d, J = 2.9 Hz), 127.25, 122.82, 114.13 (d, J = 22.0 Hz), 113.47 (d, J = 20.6 Hz), 103.99, 100.25, 94.25, 92.65, 55.33, 32.27, 25.05, 24.89, 20.69; 19F NMR (470 MHz, DMSO-d6) δ −118.01 – −118.16 (m). UPLC/MS [M+H]=527.578, TR=1.90 min.
(2-((3-(2-Fluoro-3-methylphenyl)-1-isopropyl-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(isoindolin-2-yl)methanone (9ba): Amide bromide 8a (64 mg, 0.125 mmol, 1 equiv) was coupled to 4at (32 mg, 0.138 mmol, 1.1 equiv) according to General Procedure E1b. The resulting product was deprotected according to General Procedure E2a. 9ba (38 mg, 0.078 mmol) was obtained in 65% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.62 (s, 1H), 9.29 (s, 1H), 7.79 – 7.71 (m, 1H), 7.36 – 7.30 (m, 2H), 7.30 – 7.23 (m, 2H), 7.21 – 7.15 (m, 1H), 7.14 (s, 1H), 7.09 (t, J = 7.6 Hz, 1H), 6.34 (d, J = 4.1 Hz, 1H), 5.87 (d, J = 2.1 Hz, 1H), 5.61 (d, J = 2.1 Hz, 1H), 4.85 – 4.68 (m, 4H), 4.52 – 4.40 (m, 1H), 2.25 (d, J = 2.1 Hz, 3H), 1.36 (d, J = 6.2 Hz, 6H); 13C NMR (126 MHz, DMSO-d6): δ 166.46, 159.25, 157.71 (d, J = 247.1 Hz), 155.56, 144.57, 142.99, 139.93 (d, J = 2.2 Hz), 136.72, 130.21 (d, J = 5.1 Hz), 127.26, 125.17 (d, J = 3.8 Hz), 124.86 (d, J = 17.6 Hz), 123.91 (d, J = 3.8 Hz), 122.87, 121.09 (d, J = 12.4 Hz), 104.00, 100.52 (d, J = 10.1 Hz), 94.26, 92.62, 48.02, 22.23, 14.35 (d, J = 4.8 Hz); 19F NMR (470 MHz, DMSO-d6) δ −120.64 – −121.59 (m). UPLC/MS [M+H]=487.545, TR=1.70 min.
(2-((1-(tert-Butyl)-3-(2-fluoro-3-methylphenyl)-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(isoindolin-2-yl)methanone (9bb): Amide bromide 8a (77 mg, 0.150 mmol, 1 equiv) was coupled to 4au (41 mg, 0.165 mmol, 1.1 equiv) according to General Procedure E1b. The resulting product was deprotected according to General Procedure E2a. 9bb (41.8 mg, 0.084 mmol) was obtained in 56% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.70 (s, 1H), 9.29 (s, 1H), 7.80 – 7.73 (m, 1H), 7.38 – 7.32 (m, 2H), 7.31 – 7.25 (m, 2H), 7.22 – 7.15 (m, 1H), 7.10 (t, J = 7.6 Hz, 1H), 7.02 (s, 1H), 6.43 (d, J = 4.2 Hz, 1H), 5.87 (d, J = 2.1 Hz, 1H), 5.68 (d, J = 2.1 Hz, 1H), 4.86 – 4.71 (m, 4H), 2.26 (d, J = 2.1 Hz, 3H), 1.57 (s, 9H); 13C NMR (126 MHz, DMSO-d6): δ 166.87, 159.42, 157.76 (d, J = 246.9 Hz), 155.56, 144.96, 141.26, 140.21 (d, J = 2.3 Hz), 136.68, 130.19 (d, J = 4.8 Hz), 127.27, 125.09 (d, J = 3.8 Hz), 124.84 (d, J = 17.5 Hz), 123.93 (d, J = 3.8 Hz), 122.85, 121.03 (d, J = 12.6 Hz), 103.29 (d, J = 10.1 Hz), 103.18, 93.94, 92.22, 59.59, 29.37, 14.32 (d, J = 4.7 Hz); 19F NMR (470 MHz, DMSO-d6) δ −121.11 – −121.25 (m). UPLC/MS [M+H]=501.437 , TR=1.88 min.
(2-((1-Cyclopentyl-3-(2-fluoro-3-methylphenyl)-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(isoindolin-2-yl)methanone (9bc): Amide bromide 8a (64 mg, 0.125 mmol, 1 equiv) was coupled to 4av (36 mg, 0.138 mmol, 1.1 equiv) according to General Procedure E1b. The resulting product was deprotected according to General Procedure E2a. 9bc (43 mg, 0.085 mmol) was obtained in 68% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.63 (s, 1H), 9.30 (s, 1H), 7.77 – 7.70 (m, 1H), 7.36 – 7.30 (m, 2H), 7.26 (dd, J = 5.6, 3.2 Hz, 2H), 7.21 – 7.15 (m, 2H), 7.09 (t, J = 7.6 Hz, 1H), 6.35 (d, J = 4.1 Hz, 1H), 5.88 (d, J = 2.1 Hz, 1H), 5.62 (d, J = 2.1 Hz, 1H), 4.76 (s, 4H), 4.61 (p, J = 7.3 Hz, 1H), 2.25 (d, J = 2.1 Hz, 3H), 1.95 – 1.89 (m, 4H), 1.87 – 1.76 (m, 2H), 1.58 – 1.52 (m, 2H); 13C NMR (126 MHz, DMSO-d6): δ 166.50, 159.25, 157.72 (d, J = 247.0 Hz), 155.56, 144.44, 142.94, 140.62 (d, J = 2.3 Hz), 136.72, 130.23 (d, J = 5.0 Hz), 127.27, 125.17 (d, J = 3.9 Hz), 124.87 (d, J = 17.5 Hz), 123.94 (d, J = 3.8 Hz), 122.86, 121.06 (d, J = 12.4 Hz), 104.03, 100.43 (d, J = 10.0 Hz), 94.34, 92.75, 57.15, 32.22, 24.15, 14.35 (d, J = 4.7 Hz); 19F NMR (470 MHz, DMSO-d6) δ −119.76 – −122.97 (m). UPLC/MS [M+H]=513.564, TR=1.80 min.
(2-((1-Cyclohexyl-3-(2-fluoro-3-methylphenyl)-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(isoindolin-2-yl)methanone (9bd): Amide bromide 8a (77 mg, 0.150 mmol, 1 equiv) was coupled to 4aw (45 mg, 0.165 mmol, 1.1 equiv) according to General Procedure E1b. The resulting product was deprotected according to General Procedure E2a. 9bd (50.7 mg, 0.096 mmol) was obtained in 64% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.60 (s, 1H), 9.28 (s, 1H), 7.77 – 7.70 (m, 1H), 7.35 – 7.30 (m, 2H), 7.29 – 7.22 (m, 2H), 7.20 – 7.13 (m, 1H), 7.13 – 7.05 (m, 2H), 6.35 (d, J = 4.1 Hz, 1H), 5.87 (d, J = 2.1 Hz, 1H), 5.62 (d, J = 2.0 Hz, 1H), 4.84 – 4.64 (m, 4H), 4.10 – 4.00 (m, 1H), 2.24 (d, J = 2.0 Hz, 3H), 1.79 (d, J = 28.9 Hz, 6H), 1.61 – 1.57 (m, 1H), 1.26 – 1.11 (m, 3H); 13C NMR (126 MHz, DMSO-d6): δ 166.50, 159.23, 157.71 (d, J = 247.1 Hz), 155.55, 144.52, 142.90, 140.04 (d, J = 2.1 Hz), 136.68, 130.19 (d, J = 4.8 Hz), 127.27, 125.15 (d, J = 3.8 Hz), 124.86 (d, J = 17.5 Hz), 123.90 (d, J = 3.8 Hz), 122.85, 121.09 (d, J = 12.4 Hz), 104.08, 100.60 (d, J = 10.1 Hz), 94.32, 92.70, 55.49, 32.30, 25.12, 24.93, 14.35 (d, J = 4.7 Hz); 19F NMR (470 MHz, DMSO-d6) δ −120.94 – −121.05 (m). UPLC/MS [M+H]=527.589, TR=1.90 min.
(2-((3-(2,3-Difluorophenyl)-1-isopropyl-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(isoindolin-2-yl)methanone (9be): Amide bromide 8a (64 mg, 0.125 mmol, 1 equiv) was coupled to 4ax (33 mg, 0.138 mmol, 1.1 equiv) according to General Procedure E1b. The resulting product was deprotected according to General Procedure E2a. 9be (33 mg, 0.067 mmol) was obtained in 56% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.65 (s, 1H), 9.31 (s, 1H), 7.75 – 7.68 (m, 1H), 7.41 – 7.09 (m, 7H), 6.37 (d, J = 3.7 Hz, 1H), 5.89 (d, J = 2.1 Hz, 1H), 5.61 (d, J = 2.0 Hz, 1H), 4.76 – 4.71 (m, 4H), 4.53 – 4.43 (m, 1H), 1.38 – 1.33 (m, 6H); 13C NMR (126 MHz, DMSO-d6): δ 166.40, 159.22, 155.62, 150.30 (d, J = 244.6 Hz), 147.06 (d, J = 248.9 Hz), 144.44, 140.40, 127.20, 124.63, 123.59, 122.80, 122.50, 115.63 (d, J = 16.6 Hz), 104.14, 100.40 (d, J = 8.7 Hz), 94.41, 92.85, 48.16, 22.16. UPLC/MS [M+H]=491.531, TR=1.59 min.
(2-((1-(tert-Butyl)-3-(2,3-difluorophenyl)-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(isoindolin-2-yl)methanone (9bf): Amide bromide 8a (64 mg, 0.125 mmol, 1 equiv) was coupled to 4ay (34.6 mg, 0.138 mmol, 1.1 equiv) according to General Procedure E1b. The resulting product was deprotected according to General Procedure E2a. 9bf (33.4 mg, 0.066 mmol) was obtained in 53% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.74 (s, 1H), 9.32 (s, 1H), 7.77 – 7.70 (m, 1H), 7.37 – 7.18 (m, 6H), 7.05 (s, 1H), 6.48 (d, J = 3.8 Hz, 1H), 5.88 (d, J = 2.1 Hz, 1H), 5.67 (d, J = 2.1 Hz, 1H), 4.82 – 4.79 (m, 4H), 1.57 (s, 9H); 13C NMR (126 MHz, DMSO-d6): δ 166.86, 159.44, 155.62, 150.40 (dd, J = 244.2, 12.7 Hz), 147.11 (dd, J = 249.6, 13.8 Hz), 144.89, 140.68 (d, J = 1.9 Hz), 140.21 (d, J = 3.4 Hz), 136.68, 127.30, 124.87 – 124.61 (m), 123.55 (d, J = 8.3 Hz), 122.87, 122.52 (t, J = 3.0 Hz), 115.71 (d, J = 17.1 Hz), 103.35, 103.29 (d, J = 1.9 Hz), 94.06, 92.34, 59.91, 29.34; 19F NMR (470 MHz, DMSO-d6) δ −138.74 – −139.72 (m), −142.92 – −143.90 (m). UPLC/MS [M+H]=505.538, TR=1.83 min.
(2-((1-(tert-Butyl)-3-(2,3-difluorophenyl)-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(4-fluoroisoindolin-2-yl)methanone (9bg): Amide bromide 8b (67 mg, 0.125 mmol, 1 equiv) was coupled to 4ay (35 mg, 0.138 mmol, 1.1 equiv) according to General Procedure E1b. The resulting product was deprotected according to General Procedure E2a. 9bg (34 mg, 0.066 mmol) was obtained in 53% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.77 (s, 1H), 9.33 (s, 1H), 7.77 – 7.70 (m, 1H), 7.38 – 7.27 (m, 2H), 7.26 – 7.17 (m, 2H), 7.14 – 7.04 (m, 2H), 6.47 (d, J = 3.7 Hz, 1H), 5.88 (d, J = 2.1 Hz, 1H), 5.67 (d, J = 2.1 Hz, 1H), 4.82 (m, 4H), 1.58 (s, 9H); 13C NMR (126 MHz, DMSO-d6): δ 166.80, 159.53, 155.66, 149.70 (d, J = 423.9 Hz), 144.98, 140.66, 140.18, 129.92, 124.69, 123.51 (d, J = 8.5 Hz), 122.46, 119.14, 115.66 (d, J = 17.2 Hz), 114.03, 113.68 (d, J = 19.1 Hz), 103.38, 102.94, 94.01, 92.45, 59.92, 29.32. UPLC/MS [M+H]=523.576, TR=1.86 min.
(2-((1-cyclopentyl-3-(2,3-difluorophenyl)-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(isoindolin-2-yl)methanone (9bh): Amide bromide 8a (64 mg, 0.125 mmol, 1 equiv) was coupled to 4az (36 mg, 0.138 mmol, 1.1 equiv) according to General Procedure E1b. The resulting product was deprotected according to General Procedure E2a. 9bh (44 mg, 0.085 mmol) was obtained in 68% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.74 (s, 1H), 9.40 (s, 1H), 7.81 (t, J = 7.2 Hz, 1H), 7.46 – 7.25 (m, 7H), 6.48 (d, J = 3.7 Hz, 1H), 6.00 (d, J = 2.1 Hz, 1H), 5.72 (d, J = 2.0 Hz, 1H), 4.87 – 4.83 (m, 4H), 4.73 (p, J = 7.3 Hz, 1H), 2.04 (s, 4H), 1.96 – 1.88 (m, 2H), 1.70 – 1.61 (m, 2H); 13C NMR (126 MHz, DMSO-d6): δ 166.45, 159.24, 155.58, 150.22 (d, J = 248.2 Hz), 147.18 (d, J = 248.2 Hz), 144.33, 141.78, 141.07, 136.74, 127.22, 124.68, 123.55 (d, J = 8.6 Hz), 122.81, 122.52, 115.65 (d, J = 16.8 Hz), 104.18, 100.35 (d, J = 8.9 Hz), 94.49, 92.99, 57.27, 32.23, 24.14. UPLC/MS [M+H]=517.533, TR=1.75 min.
(2-((1-cyclohexyl-3-(2,3-difluorophenyl)-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(isoindolin-2-yl)methanone (9bi): Amide bromide 8a (64 mg, 0.125 mmol, 1 equiv) was coupled to 4bb (38 mg, 0.138 mmol, 1.1 equiv) according to General Procedure E1b. The resulting product was deprotected according to General Procedure E2a. 9bi (46 mg, 0.087 mmol) was obtained in 73% yield over two steps. 1H NMR (500 MHz, DMSO-d6): δ 9.64 (s, 1H), 9.30 (s, 1H), 7.74 – 7.66 (m, 1H), 7.34 – 7.09 (m, 7H), 6.37 (d, J = 3.8 Hz, 1H), 5.89 (d, J = 2.1 Hz, 1H), 5.63 (d, J = 2.1 Hz, 1H), 4.76 – 4.72 (m, 4H), 4.13 – 4.03 (m, 1H), 1.91 – 1.70 (m, 6H), 1.62 – 1.58 (m, 1H), 1.21 – 1.15 (m, 3H); 13C NMR (126 MHz, DMSO-d6): δ 166.87, 159.64, 156.03, 151.83, 146.30, 142.17, 140.97, 137.05, 127.65, 125.04, 123.99 (d, J = 8.6 Hz), 123.22, 122.93, 116.04 (d, J = 16.8 Hz), 104.68, 100.90 (d, J = 9.4 Hz), 94.92, 93.43, 56.01, 40.87, 25.51, 25.33. UPLC/MS [M+H]=531.547, TR=1.85 min.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by a grant from the National Institutes of Health (Grant R01AI120958 to L.E.C., L.E.B., and D.J.K). L.E.C. holds a Canada Research Chair in Microbial Genomics & Infectious Disease and is Co-Director of the CIFAR Program, Fungal Kingdom: Threats & Opportunities. G.G.P. was supported by grants from NSERC and the Samuel Waxman Cancer Research Foundation.
Funding Sources
NIH R01AI120958
ABBREVIATIONS USED
- Hsp90
heat shock protein 90
- Trap1
TNF receptor associated protein 1
- Grp94
94 kDa glucoseregulated protein
- NBD
nucleotide binding domain
- ATP
adenosine triphosphate
- Clint
intrinsic clearance
- DIPEA
N,N-diisopropylethylamine
- ELSD
evaporative light-scattering detector
- FBS
fetal bovine serum
- FP
fluorescence polarization
- FS
fungal selectivity
- HATU
hexafluorophosphate azabenzotriazole tetramethyl uranium
- IS
internal standard
- MRM
multiple reaction monitoring
- TI
therapeutic index
- UPLC
ultraperformance liquid chromatography
- WHO
World Health Organization
- YPD
yeast extract peptone dextrose
Footnotes
The authors declare the following competing financial interest(s): L.E.C. is a cofounder, Chief Scientific Officer, and shareholder of Bright Angel Therapeutics, a platform company for the development of novel antifungal therapeutics. L.E.C. is a consultant for Boragen, a small molecule development company focused on leveraging the unique chemical properties of boron chemistry for crop protection and animal health. L.W. is a co-founder and shareholder of Bright Angel Therapeutics. L.E.B., D.S.H., P.T.M., L.E.C. and L.W. are named as inventors on a patent application pertaining to findings reported here.
ACCESSION CODES
Coordinates have been deposited in the PDB with the following accession numbers: apo (7K9R), NVP-AUY922 (7K9S), compound 19 (7K9U), compound 18 (7K9V), and compound 10 (7K9W). Authors will release coordinates and experimental data upon article publication.
ASSOCIATED CONTENT
The Supporting Information is available free of charge on the ACS Publications website at DOI: https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c01777
Supplemental Tables 1-8, Supplemental Figures S1-S7 (PDF)
Molecular formula strings and associated biological data (CSV)
Coordinate files used to generate Figure 11B (PDB)
Coordinate files used to generate Figure 11C (PDB)
Coordinate files used to generate Figure 11D (PDB)
Coordinate files used to generate Figure 11E (PDB)
Coordinate files used to generate Figure 11F (PDB)
Coordinate files used to generate Figure 11G (PDB)
REFERENCES
- 1.Brown GD; Denning DW; Gow NA; Levitz SM; Netea MG; White TC, Hidden killers: human fungal infections. Sci. Transl. Med 2012, 4 (165), Article 165rv113. [DOI] [PubMed] [Google Scholar]
- 2.Fisher MC; Hawkins NJ; Sanglard D; Gurr SJ, Worldwide emergence of resistance to antifungal drugs challenges human health and food security. Science 2018, 360 (6390), 739–742. [DOI] [PubMed] [Google Scholar]
- 3.Brown GD; Denning DW; Levitz SM, Tackling human fungal infections. Science 2012, 336 (6082), 647. [DOI] [PubMed] [Google Scholar]
- 4.Pfaller MA; Diekema DJ, Epidemiology of invasive mycoses in North America. Crit. Rev. Microbiol 2010, 36 (1), 1–53. [DOI] [PubMed] [Google Scholar]
- 5.Kohler JR; Hube B; Puccia R; Casadevall A; Perfect JR, Fungi that infect humans. Microbiol. Spectr 2017, 5 (3), DOI: 10.1128/microbiolspec.FUNK-0014-2016. [DOI] [PubMed] [Google Scholar]
- 6.Park BJ; Wannemuehler KA; Marston BJ; Govender N; Pappas PG; Chiller TM, Estimation of the current global burden of cryptococcal meningitis among persons living with HIV/AIDS. AIDS 2009, 23 (4), 525–530. [DOI] [PubMed] [Google Scholar]
- 7.Rajasingham R; Smith RM; Park BJ; Jarvis JN; Govender NP; Chiller TM; Denning DW; Loyse A; Boulware DR, Global burden of disease of HIV-associated cryptococcal meningitis: an updated analysis. Lancet Infect. Dis 2017, 17 (8), 873–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Loyse A; Burry J; Cohn J; Ford N; Chiller T; Ribeiro I; Koulla-Shiro S; Mghamba J; Ramadhani A; Nyirenda R; Aliyu SH; Wilson D; Le T; Oladele R; Lesikari S; Muzoora C; Kalata N; Temfack E; Mapoure Y; Sini V; Chanda D; Shimwela M; Lakhi S; Ngoma J; Gondwe-Chunda L; Perfect C; Shroufi A; Andrieux-Meyer I; Chan A; Schutz C; Hosseinipour M; Van der Horst C; Klausner JD; Boulware DR; Heyderman R; Lalloo D; Day J; Jarvis JN; Rodrigues M; Jaffar S; Denning D; Migone C; Doherty M; Lortholary O; Dromer F; Stack M; Molloy SF; Bicanic T; van Oosterhout J; Mwaba P; Kanyama C; Kouanfack C; Mfinanga S; Govender N; Harrison TS, Leave no one behind: response to new evidence and guidelines for the management of cryptococcal meningitis in low-income and middle-income countries. Lancet Infect. Dis 2019, 19 (4), e143–e147. [DOI] [PubMed] [Google Scholar]
- 9.Maertens JA, History of the development of azole derivatives. Clin. Microbiol. Infect 2004, 10, 1–10. [DOI] [PubMed] [Google Scholar]
- 10.Anderson TM; Clay MC; Cioffi AG; Diaz KA; Hisao GS; Tuttle MD; Nieuwkoop AJ; Comellas G; Maryum N; Wang S; Uno BE; Wildeman EL; Gonen T; Rienstra CM; Burke MD, Amphotericin forms an extramembranous and fungicidal sterol sponge. Nat. Chem. Biol 2014, 10 (5), 400–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cowen LE; Steinbach WJ, Stress, drugs, and evolution: the role of cellular signaling in fungal drug resistance. Eukaryot. Cell 2008, 7 (5), 747–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shapiro RS; Robbins N; Cowen LE, Regulatory circuitry governing fungal development, drug resistance, and disease. Microbiol. Mol. Biol. Rev 2011, 75 (2), 213–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Caplan T; Polvi EJ; Xie JL; Buckhalter S; Leach MD; Robbins N; Cowen LE, Functional genomic screening reveals core modulators of echinocandin stress responses in Candida albicans. Cell Rep. 2018, 23 (8), 2292–2298. [DOI] [PubMed] [Google Scholar]
- 14.Robbins N; Caplan T; Cowen LE, Molecular evolution of antifungal drug resistance. Annu. Rev. Microbiol 2017, 71, 753–775. [DOI] [PubMed] [Google Scholar]
- 15.Robbins N; Wright GD; Cowen LE, Antifungal Drugs: the Current Armamentarium and Development of New Agents. In The Fungal Kingdom, Heitman J; Howlett B; Crous P; Stukenbrock E; James T; Gow N, Eds. ASM Press: Washington DC., 2017; Vol. 4, pp 903–922. [DOI] [PubMed] [Google Scholar]
- 16.O'Meara TR; Veri AO; Polvi EJ; Li X; Valaei SF; Diezmann S; Cowen LE, Mapping the Hsp90 genetic network reveals ergosterol biosynthesis and phosphatidylinositol-4-kinase signaling as core circuitry governing cellular stress. PLoS Genet. 2016, 12 (6), e1006142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim SH; Iyer KR; Pardeshi L; Munoz JF; Robbins N; Cuomo CA; Wong KH; Cowen LE, Genetic analysis of Candida auris implicates Hsp90 in morphogenesis and azole tolerance and Cdr1 in azole resistance. mBio 2019, 10 (1), e02529–02518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Veri AO; Miao Z; Shapiro RS; Tebbji F; O'Meara TR; Kim SH; Colazo J; Tan K; Vyas VK; Whiteway M; Robbins N; Wong KH; Cowen LE, Tuning Hsf1 levels drives distinct fungal morphogenetic programs with depletion impairing Hsp90 function and overexpression expanding the target space. PLoS Genet. 2018, 14 (3), e1007270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O'Meara TR; Robbins N; Cowen LE, The Hsp90 chaperone network modulates Candida virulence traits. Trends Microbiol. 2017, 25 (10), 809–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leach MD; Klipp E; Cowen LE; Brown AJ, Fungal Hsp90: a biological transistor that tunes cellular outputs to thermal inputs. Nat. Rev. Microbiol 2012, 10 (10), 693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cowen LE; Lindquist S, Hsp90 potentiates the rapid evolution of new traits: drug resistance in diverse fungi. Science 2005, 309 (5744), 2185–2189. [DOI] [PubMed] [Google Scholar]
- 22.Singh SD; Robbins N; Zaas AK; Schell WA; Perfect JR; Cowen LE, Hsp90 governs echinocandin resistance in the pathogenic yeast Candida albicans via calcineurin. PLoS Pathog. 2009, 5 (7), e1000532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cowen LE; Singh SD; Kohler JR; Collins C; Zaas AK; Schell WA; Aziz H; Mylonakis E; Perfect JR; Whitesell L; Lindquist S, Harnessing Hsp90 function as a powerful, broadly effective therapeutic strategy for fungal infectious disease. Proc. Natl. Acad. Sci. USA 2009, 106 (8), 2818–2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chatterjee S; Tatu U, Heat shock protein 90 localizes to the surface and augments virulence factors of Cryptococcus neoformans. PLoS Negl. Trop. Dis 2017, 11 (8), e0005836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cordeiro Rde A; Evangelista AJ; Serpa R; Marques FJ; de Melo CV; de Oliveira JS; Franco Jda S; de Alencar LP; Bandeira Tde J; Brilhante RS; Sidrim JJ; Rocha MF, Inhibition of heat-shock protein 90 enhances the susceptibility to antifungals and reduces the virulence of Cryptococcus neoformans/Cryptococcus gattii species complex. Microbiology 2016, 162 (2), 309–317. [DOI] [PubMed] [Google Scholar]
- 26.Li L; Wang L; You QD; Xu XL, Heat shock protein 90 inhibitors: An update on achievements, challenges, and future directions. J. Med. Chem 2020, 63 (5), 1798–1822. [DOI] [PubMed] [Google Scholar]
- 27.Jaeger AM; Stopfer L; Lee S; Gaglia G; Sandel D; Santagata S; Lin NU; Trepel JB; White F; Jacks T; Lindquist S; Whitesell L, Rebalancing protein homeostasis enhances tumor antigen presentation. Clin. Cancer Res 2019, 25 (21), 6392–6405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jaeger AM; Whitesell L, HSP90: Enabler of cancer adaptation. Annu. Rev. Cancer Biol 2019, 3 (1), 275–297. [Google Scholar]
- 29.Gewirth DT, Paralog specific Hsp90 inhibitors - a brief history and a bright future. Curr. Top. Med. Chem 2016, 16 (25), 2779–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee C; Park HK; Jeong H; Lim J; Lee AJ; Cheon KY; Kim CS; Thomas AP; Bae B; Kim ND; Kim SH; Suh PG; Ryu JH; Kang BH, Development of a mitochondria-targeted Hsp90 inhibitor based on the crystal structures of human TRAP1. J. Am. Chem. Soc 2015, 137 (13), 4358–4367. [DOI] [PubMed] [Google Scholar]
- 31.Crowley VM; Khandelwal A; Mishra S; Stothert AR; Huard DJE; Zhao J; Muth A; Duerfeldt AS; Kizziah JL; Lieberman RL; Dickey CA; Blagg BSJ, Development of glucose regulated protein 94-selective inhibitors based on the BnIm and radamide scaffold. J. Med. Chem 2016, 59 (7), 3471–3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Patel PD; Yan P; Seidler PM; Patel HJ; Sun W; Yang C; Que NS; Taldone T; Finotti P; Stephani RA; Gewirth DT; Chiosis G, Paralog-selective Hsp90 inhibitors define tumor-specific regulation of HER2. Nat. Chem. Biol 2013, 9, 677–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Que NLS; Crowley VM; Duerfeldt AS; Zhao J; Kent CN; Blagg BSJ; Gewirth DT, Structure based design of a Grp94-selective inhibitor: Exploiting a key residue in Grp94 to optimize paralog-selective binding. J. Med. Chem 2018, 61 (7), 2793–2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stothert AR; Suntharalingam A; Huard DJ; Fontaine SN; Crowley VM; Mishra S; Blagg BS; Lieberman RL; Dickey CA, Exploiting the interaction between Grp94 and aggregated myocilin to treat glaucoma. Hum. Mol. Genet 2014, 23, 6470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stothert AR; Suntharalingam A; Tang X; Crowley VM; Mishra SJ; Webster JM; Nordhues BA; Huard DJE; Passaglia CL; Lieberman RL; Blagg BSJ; Blair LJ; Koren J; Dickey CA, Isoform-selective Hsp90 inhibition rescues model of hereditary open-angle glaucoma. Sci. Rep 2017, 7 (1), 17951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Muth A; Crowley V; Khandelwal A; Mishra S; Zhao J; Hall J; Blagg BSJ, Development of radamide analogs as Grp94 inhibitors. Bioorg. Med. Chem 2014, 22 (15), 4083–4098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Khandelwal A; Crowley VM; Blagg BSJ, Resorcinol-based Grp94-selective inhibitors. ACS Med. Chem. Lett 2017, 8 (10), 1013–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Khandelwal A; Kent CN; Balch M; Peng S; Mishra SJ; Deng J; Day VW; Liu W; Subramanian C; Cohen M; Holzbeierlein JM; Matts R; Blagg BSJ, Structure-guided design of an Hsp90β N-terminal isoform-selective inhibitor. Nat. Commun 2018, 9 (1), 425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ohkubo S; Kodama Y; Muraoka H; Hitotsumachi H; Yoshimura C; Kitade M; Hashimoto A; Ito K; Gomori A; Takahashi K; Shibata Y; Kanoh A; Yonekura K, TAS-116, a highly selective inhibitor of heat shock protein 90α and β, demonstrates potent antitumor activity and minimal ocular toxicity in preclinical models. Mol. Cancer Ther 2015, 14 (1), 14–22. [DOI] [PubMed] [Google Scholar]
- 40.Ernst JT; Neubert T; Liu M; Sperry S; Zuccola H; Turnbull A; Fleck B; Kargo W; Woody L; Chiang P; Tran D; Chen W; Snyder P; Alcacio T; Nezami A; Reynolds J; Alvi K; Goulet L; Stamos D, Identification of novel HSP90α/β isoform selective inhibitors using structure-based drug design. Demonstration of potential utility in treating CNS disorders such as Huntington’s Disease. J. Med. Chem 2014, 57 (8), 3382–3400. [DOI] [PubMed] [Google Scholar]
- 41.Ernst JT; Liu M; Zuccola H; Neubert T; Beaumont K; Turnbull A; Kallel A; Vought B; Stamos D, Correlation between chemotype-dependent binding conformations of HSP90α/β and isoform selectivity—Implications for the structure-based design of HSP90α/β selective inhibitors for treating neurodegenerative diseases. Bioorg. Med. Chem. Lett 2014, 24 (1), 204–208. [DOI] [PubMed] [Google Scholar]
- 42.Whitesell L; Robbins N; Huang DS; McLellan CA; Shekhar-Guturja T; LeBlanc EV; Nation CS; Hui R; Hutchinson A; Collins C; Chatterjee S; Trilles R; Xie JL; Krysan DJ; Lindquist S; Porco JA; Tatu U; Brown LE; Pizarro J; Cowen LE, Structural basis for species-selective targeting of Hsp90 in a pathogenic fungus. Nat. Commun 2019, 10 (1), 402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Woodhead AJ; Angove H; Carr MG; Chessari G; Congreve M; Coyle JE; Cosme J; Graham B; Day PJ; Downham R; Fazal L; Feltell R; Figueroa E; Frederickson M; Lewis J; McMenamin R; Murray CW; O'Brien MA; Parra L; Patel S; Phillips T; Rees DC; Rich S; Smith DM; Trewartha G; Vinkovic M; Williams B; Woolford AJA, Discovery of (2,4-Dihydroxy-5-isopropylphenyl)- 5-(4-methylpiperazin-1-ylmethyl)-1,3- dihydroisoindol-2-yl methanone (AT13387), a novel inhibitor of the molecular chaperone Hsp90 by fragment based drug design. J. Med. Chem 2010, 53 (16), 5956–5969. [DOI] [PubMed] [Google Scholar]
- 44.Do K; Speranza G; Chang L-C; Polley EC; Bishop R; Zhu W; Trepel JB; Lee S; Lee M-J; Kinders RJ; Phillips L; Collins J; Lyons J; Jeong W; Antony R; Chen AP; Neckers L; Doroshow JH; Kummar S, Phase I study of the heat shock protein 90 (Hsp90) inhibitor onalespib (AT13387) administered on a daily for 2 consecutive days per week dosing schedule in patients with advanced solid tumors. Invest. New Drug 2015, 33 (4), 921–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wagner AJ; Agulnik M; Heinrich MC; Mahadevan D; Riedel RF; von Mehren M; Trent J; Demetri GD; Corless CL; Yule M; Lyons JF; Oganesian A; Keer H, Dose-escalation study of a second-generation non-ansamycin HSP90 inhibitor, onalespib (AT13387), in combination with imatinib in patients with metastatic gastrointestinal stromal tumour. Eur. J. Cancer 2016, 61, 94–101. [DOI] [PubMed] [Google Scholar]
- 46.Canella A; Welker AM; Yoo JY; Xu J; Abas FS; Kesanakurti D; Nagarajan P; Beattie CE; Sulman EP; Liu J; Gumin J; Lang FF; Gurcan MN; Kaur B; Sampath D; Puduvalli VK, Efficacy of onalespib, a long-acting second-generation HSP90 inhibitor, as a single agent and in combination with temozolomide against malignant gliomas. Clin. Cancer Res 2017, 23 (20), 6215–6226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stühmer T; Zöllinger A; Siegmund D; Chatterjee M; Grella E; Knop S; Kortüm M; Unzicker C; Jensen MR; Quadt C; Chène P; Schoepfer J; García-Echeverría C; Einsele H; Wajant H; Bargou RC, Signalling profile and antitumour activity of the novel Hsp90 inhibitor NVP-AUY922 in multiple myeloma. Leukemia 2008, 22, 1604–1612. [DOI] [PubMed] [Google Scholar]
- 48.Jensen MR; Schoepfer J; Radimerski T; Massey A; Guy CT; Brueggen J; Quadt C; Buckler A; Cozens R; Drysdale MJ; Garcia-Echeverria C; Chène P, NVP-AUY922: a small molecule HSP90 inhibitor with potent antitumor activity in preclinical breast cancer models. Breast Cancer Res. 2008, 10 (2), R33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Doi T; Onozawa Y; Fuse N; Yoshino T; Yamazaki K; Watanabe J; Akimov M; Robson M; Boku N; Ohtsu A, Phase I dose-escalation study of the HSP90 inhibitor AUY922 in Japanese patients with advanced solid tumors. Cancer Chemother. Pharmacol 2014, 74 (3), 629–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Seggewiss-Bernhardt R; Bargou RC; Goh YT; Stewart AK; Spencer A; Alegre A; Bladé J; Ottmann OG; Fernandez-Ibarra C; Lu H; Pain S; Akimov M; Iyer SP, Phase 1/1B trial of the heat shock protein 90 inhibitor NVP-AUY922 as monotherapy or in combination with bortezomib in patients with relapsed or refractory multiple myeloma. Cancer 2015, 121 (13), 2185–2192. [DOI] [PubMed] [Google Scholar]
- 51.Renouf DJ; Hedley D; Krzyzanowska MK; Schmuck M; Wang L; Moore MJ, A phase II study of the HSP90 inhibitor AUY922 in chemotherapy refractory advanced pancreatic cancer. Cancer Chemother. Pharmacol 2016, 78 (3), 541–545. [DOI] [PubMed] [Google Scholar]
- 52.Bendell JC; Bauer TM; Lamar R; Joseph M; Penley W; Thompson DS; Spigel DR; Owera R; Lane CM; Earwood C; Burris HA, A phase 2 study of the Hsp90 inhibitor AUY922 as treatment for patients with refractory gastrointestinal stromal tumors. Cancer Invest. 2016, 34 (6), 265–270. [DOI] [PubMed] [Google Scholar]
- 53.Lin T-Y; Bear M; Du Z; Foley KP; Ying W; Barsoum J; London C, The novel HSP90 inhibitor STA-9090 exhibits activity against Kit-dependent and -independent malignant mast cell tumors. Exp. Hematol 2008, 36 (10), 1266–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ying WW; Du ZJ; Sun LJ; Foley KP; Proia DA; Blackman RK; Zhou D; Inoue T; Tatsuta N; Sang J; Ye SX; Acquaviva J; Ogawa LS; Wada Y; Barsoum J; Koya K, Ganetespib, a unique triazolone-containing Hsp90 inhibitor, exhibits potent antitumor activity and a superior safety profile for cancer therapy. Mol. Cancer Ther 2012, 11 (2), 475–484. [DOI] [PubMed] [Google Scholar]
- 55.Lock RB; Carol H; Maris JM; Kang MH; Reynolds CP; Kolb EA; Gorlick R; Keir ST; Billups CA; Kurmasheva RT; Houghton PJ; Smith MA, Initial testing (stage 1) of ganetespib, an Hsp90 inhibitor, by the pediatric preclinical testing program. Pediatr. Blood Cancer 2013, 60 (7), E42–E45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jhaveri K; Chandarlapaty S; Lake D; Gilewski T; Robson M; Goldfarb S; Drullinsky P; Sugarman S; Wasserheit-Leiblich C; Fasano J; Moynahan ME; D'Andrea G; Lim K; Reddington L; Haque S; Patil S; Bauman L; Vukovic V; El-Hariry I; Hudis C; Modi S, A phase II open-label study of ganetespib, a novel heat shock protein 90 inhibitor for patients with metastatic breast cancer. Clin. Breast Cancer 2014, 14 (3), 154–160. [DOI] [PubMed] [Google Scholar]
- 57.Jhaveri K; Wang R; Teplinsky E; Chandarlapaty S; Solit D; Cadoo K; Speyer J; D’Andrea G; Adams S; Patil S; Haque S; O’Neill T; Friedman K; Esteva FJ; Hudis C; Modi S, A phase I trial of ganetespib in combination with paclitaxel and trastuzumab in patients with human epidermal growth factor receptor-2 (HER2)-positive metastatic breast cancer. Breast Cancer Res. 2017, 19 (1), 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thakur MK; Heilbrun LK; Sheng S; Stein M; Liu G; Antonarakis ES; Vaishampayan U; Dzinic SH; Li X; Freeman S; Smith D; Heath EI, A phase II trial of ganetespib, a heat shock protein 90 Hsp90) inhibitor, in patients with docetaxel-pretreated metastatic castrate-resistant prostate cancer (CRPC)-a prostate cancer clinical trials consortium (PCCTC) study. Invest. New Drug 2016, 34 (1), 112–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Goyal L; Wadlow RC; Blaszkowsky LS; Wolpin BM; Abrams TA; McCleary NJ; Sheehan S; Sundaram E; Karol MD; Chen J; Zhu AX, A phase I and pharmacokinetic study of ganetespib (STA-9090) in advanced hepatocellular carcinoma. Invest. New Drug 2015, 33 (1), 128–137. [DOI] [PubMed] [Google Scholar]
- 60.Socinski MA; Goldman J; El-Hariry I; Koczywas M; Vukovic V; Horn L; Paschold E; Salgia R; West H; Sequist LV; Bonomi P; Brahmer J; Chen L-C; Sandler A; Belani CP; Webb T; Harper H; Huberman M; Ramalingam S; Wong K-K; Teofilovici F; Guo W; Shapiro GI, A multicenter phase II study of ganetespib monotherapy in patients with genotypically defined advanced non–small cell lung cancer. Clin. Cancer Res 2013, 19 (11), 3068–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Goldman JW; Raju RN; Gordon GA; El-Hariry I; Teofilivici F; Vukovic VM; Bradley R; Karol MD; Chen Y; Guo W; Inoue T; Rosen LS, A first in human, safety, pharmacokinetics, and clinical activity phase I study of once weekly administration of the Hsp90 inhibitor ganetespib (STA-9090) in patients with solid malignancies. BMC Cancer 2013, 13 (1), 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huang DS; LeBlanc EV; Shekhar-Guturja T; Robbins N; Krysan DJ; Pizarro J; Whitesell L; Cowen LE; Brown LE, Design and synthesis of fungal-selective resorcylate aminopyrazole Hsp90 inhibitors. J. Med. Chem 2020, 63 (5), 2139–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Martinez LR; Casadevall A, Susceptibility of Cryptococcus neoformans biofilms to antifungal agents in vitro. Antimicrob. Agents Chemother 2006, 50 (3), 1021–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Casadevall A; Nosanchuk JD; Williamson P; Rodrigues ML, Vesicular transport across the fungal cell wall. Trends Microbiol. 2009, 17 (4), 158–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zaragoza O, Multiple disguises for the same party: The concepts of morphogenesis and phenotypic variations in Cryptococcus neoformans. Front. Microbiol 2011, 2, 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.O'Meara TR; Alspaugh JA, The Cryptococcus neoformans capsule: a sword and a shield. Clin. Microbiol. Rev 2012, 25 (3), 387–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cannon RD; Lamping E; Holmes AR; Niimi K; Baret PV; Keniya MV; Tanabe K; Niimi M; Goffeau A; Monk BC, Efflux-mediated antifungal drug resistance. Clin. Microbiol. Rev 2009, 22 (2), 291–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rossi AM; Taylor CW, Analysis of protein-ligand interactions by fluorescence polarization. Nat. Protoc 2011, 6 (3), 365–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McNaney CA; Drexler DM; Hnatyshyn SY; Zvyaga TA; Knipe JO; Belcastro JV; Sanders M, An automated liquid chromatography-mass spectrometry process to determine metabolic stability half-life and intrinsic clearance of drug candidates by substrate depletion. Assay Drug Dev. Technol 2008, 6 (1), 121–129. [DOI] [PubMed] [Google Scholar]
- 70.Butts A; DiDone L; Koselny K; Baxter BK; Chabrier-Rosello Y; Wellington M; Krysan DJ, A repurposing approach identifies off-patent drugs with fungicidal cryptococcal activity, a common structural chemotype, and pharmacological properties relevant to the treatment of cryptococcosis. Eukaryot. Cell 2013, 12 (2), 278–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Brough PA; Aherne W; Barril X; Borgognoni J; Boxall K; Cansfield JE; Cheung K-MJ; Collins I; Davies NGM; Drysdale MJ; Dymock B; Eccles SA; Finch H; Fink A; Hayes A; Howes R; Hubbard RE; James K; Jordan AM; Lockie A; Martins V; Massey A; Matthews TP; McDonald E; Northfield CJ; Pearl LH; Prodromou C; Ray S; Raynaud FI; Roughley SD; Sharp SY; Surgenor A; Walmsley DL; Webb P; Wood M; Workman P; Wrightt L, 4,5-diarylisoxazole HSP90 chaperone inhibitors: Potential therapeutic agents for the treatment of cancer. J. Med. Chem 2008, 51 (2), 196–218. [DOI] [PubMed] [Google Scholar]
- 72.Taldone T; Chiosis G, Purine-scaffold Hsp90 inhibitors. Curr. Top. Med. Chem 2009, 9 (15), 1436–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Stebbins CE; Russo AA; Schneider C; Rosen N; Hartl FU; Pavletich NP, Crystal structure of an Hsp90–geldanamycin complex: targeting of a protein chaperone by an antitumor agent. Cell 1997, 89 (2), 239–250. [DOI] [PubMed] [Google Scholar]
- 74.Keefe LJ; Sondek J; Shortle D; Lattman EE, The alpha aneurism: a structural motif revealed in an insertion mutant of staphylococcal nuclease. Proc. Natl. Acad. Sci. USA 1993, 90 (8), 3275–3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wright L; Barril X; Dymock B; Sheridan L; Surgenor A; Beswick M; Drysdale M; Collier A; Massey A; Davies N; Fink A; Fromont C; Aherne W; Boxall K; Sharp S; Workman P; Hubbard RE, Structure-activity relationships in purine-based inhibitor binding to HSP90 isoforms. Chem. Biol. Drug. Des 2004, 11 (6), 775–785. [DOI] [PubMed] [Google Scholar]
- 76.Friesner RA; Banks JL; Murphy RB; Halgren TA; Klicic JJ; Mainz DT; Repasky MP; Knoll EH; Shelley M; Perry JK; Shaw DE; Francis P; Shenkin PS, Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem 2004, 47 (7), 1739–1749. [DOI] [PubMed] [Google Scholar]
- 77.Halgren TA; Murphy RB; Friesner RA; Beard HS; Frye LL; Pollard WT; Banks JL, Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem 2004, 47 (7), 1750–1759. [DOI] [PubMed] [Google Scholar]
- 78.Zurcher M; Diederich F, Structure-based drug design: exploring the proper filling of apolar pockets at enzyme active sites. J. Org. Chem 2008, 73 (12), 4345–4361. [DOI] [PubMed] [Google Scholar]
- 79.Bissantz C; Kuhn B; Stahl M, A medicinal chemist's guide to molecular interactions. J. Med. Chem 2010, 53 (14), 5061–5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Granger DL; Perfect JR; Durack DT, Virulence of Cryptococcus neoformans. Regulation of capsule synthesis by carbon dioxide. J. Clin. Invest 1985, 76 (2), 508–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.LaFayette SL; Collins C; Zaas AK; Schell WA; Betancourt-Quiroz M; Gunatilaka AA; Perfect JR; Cowen LE, PKC signaling regulates drug resistance of the fungal pathogen Candida albicans via circuitry comprised of Mkc1, calcineurin, and Hsp90. PLoS Pathog. 2010, 6 (8), e1001069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Houston JB, Utility of in vitro drug metabolism data in predicting in vivo metabolic clearance. Biochem. Pharmacol 1994, 47 (9), 1469–1479. [DOI] [PubMed] [Google Scholar]
- 83.Davies B; Morris T, Physiological parameters in laboratory animals and humans. Pharm. Res 1993, 10 (7), 1093–1095. [DOI] [PubMed] [Google Scholar]
- 84.Barter ZE; Bayliss MK; Beaune PH; Boobis AR; Carlile DJ; Edwards RJ; Houston JB; Lake BG; Lipscomb JC; Pelkonen OR; Tucker GT; Rostami-Hodjegan A, Scaling factors for the extrapolation of in vivo metabolic drug clearance from in vitro data: reaching a consensus on values of human microsomal protein and hepatocellularity per gram of liver. Curr. Drug Metab 2007, 8 (1), 33–45. [DOI] [PubMed] [Google Scholar]
- 85.Iwatsubo T; Suzuki H; Sugiyama Y, Prediction of species differences (rats, dogs, humans) in the in vivo metabolic clearance of YM796 by the liver from in vitro data. J. Pharmacol. Exp. Ther 1997, 283 (2), 462–469. [PubMed] [Google Scholar]
- 86.Kabsch W, XDS. Acta Crystallogr. D Biol. Crystallogr 2010, 66 (Pt 2), 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Winn MD; Ballard CC; Cowtan KD; Dodson EJ; Emsley P; Evans PR; Keegan RM; Krissinel EB; Leslie AG; McCoy A; McNicholas SJ; Murshudov GN; Pannu NS; Potterton EA; Powell HR; Read RJ; Vagin A; Wilson KS, Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr 2011, 67 (Pt 4), 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Emsley P; Lohkamp B; Scott WG; Cowtan K, Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr 2010, 66 (Pt 4), 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Liebschner D; Afonine PV; Baker ML; Bunkoczi G; Chen VB; Croll TI; Hintze B; Hung LW; Jain S; McCoy AJ; Moriarty NW; Oeffner RD; Poon BK; Prisant MG; Read RJ; Richardson JS; Richardson DC; Sammito MD; Sobolev OV; Stockwell DH; Terwilliger TC; Urzhumtsev AG; Videau LL; Williams CJ; Adams PD, Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D Struct. Biol 2019, 75 (Pt 10), 861–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Morin A; Eisenbraun B; Key J; Sanschagrin PC; Timony MA; Ottaviano M; Sliz P, Collaboration gets the most out of software. eLife 2013, 2, e01456. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.