

Abstract
Annotating cell identities is a common bottleneck in the analysis of single-cell genomics experiments. Here, we present scNym, a semisupervised, adversarial neural network that learns to transfer cell identity annotations from one experiment to another. scNym takes advantage of information in both labeled data sets and new, unlabeled data sets to learn rich representations of cell identity that enable effective annotation transfer. We show that scNym effectively transfers annotations across experiments despite biological and technical differences, achieving performance superior to existing methods. We also show that scNym models can synthesize information from multiple training and target data sets to improve performance. We show that in addition to high accuracy, scNym models are well calibrated and interpretable with saliency methods.
Single-cell genomics allows for simultaneous molecular profiling of thousands of diverse cells and has advanced our understanding of development (Trapnell 2015), aging (Angelidis et al. 2019; Kimmel et al. 2019; Ma et al. 2020), and disease (Tanay and Regev 2017). To derive biological insight from these data, each single-cell molecular profile must be annotated with a cell identity, such as a cell type or state label. Traditionally, this task has been performed manually by domain expert biologists. Manual annotation is time-consuming, somewhat subjective, and error prone. Annotations influence the results of nearly all downstream analyses, motivating more robust algorithmic approaches for cell type annotation.
Automated classification tools have been proposed to transfer annotations across data sets (Kiselev et al. 2018; Abdelaal et al. 2019; Alquicira-Hernandez et al. 2019; de Kanter et al. 2019; Pliner et al. 2019; Tan and Cahan 2019; Zhang et al. 2019). These existing tools learn relationships between cell identity and molecular features from a training set with existing labels without considering the unlabeled target data set in the learning process. However, results from the field of semisupervised representation learning suggest that incorporating information from the target data during training can improve the performance of prediction models (Kingma et al. 2014; Oliver et al. 2018; Berthelot et al. 2019; Verma et al. 2019). This approach is especially beneficial when there are systematic differences—a domain shift—between the training and target data sets. Domain shifts are commonly introduced between single-cell genomics experiments when cells are profiled in different experimental conditions or with different sequencing technologies.
A growing family of representation learning techniques encourages classification models to provide consistent interpolations between data points as an auxiliary training task to improve performance (Berthelot et al. 2019; Verma et al. 2019). In the semisupervised setting, the MixMatch approach implements this idea by “mixing” observations and their labels with simple weighted averages. Mixed observations from the training and target data sets form a bridge in feature space, encouraging the model to learn a smooth interpolation across the domains. Another family of techniques seeks to improve classification performance in the presence of domain shifts by encouraging the model to learn a representation in which observations from different domains are embedded nearby, rather than occupying distinct regions of a latent space (Wilson and Cook 2020). One successful approach uses a “domain adversary” to encourage the classification model to learn a representation that is invariant to data set–specific features (Ganin et al. 2016). Both interpolation consistency and domain invariance are desirable in the single-cell genomic setting, in which domain shifts are common and complex gene expression boundaries separate cell types.
Here, we introduce a cell type classification model that uses semisupervised and adversarial machine learning techniques to take advantage of both labeled and unlabeled single-cell data sets. We show that this model offers superior performance to existing methods and effectively transfers annotations across different animal ages, perturbation conditions, and sequencing technologies. Additionally, we show that our model learns biologically interpretable representations and offers well-calibrated metrics of annotation confidence that can be used to make new cell type discoveries.
Results
scNym
In the typical supervised learning framework, the model touches the target unlabeled data set to predict labels only after training has concluded. In contrast, our semisupervised learning framework trains the model parameters on both the labeled and unlabeled data in order to leverage the structure in the target data set, the measurements of which may have been influenced by myriad sources of biological and technical bias and batch effects. Although our model uses observed cell profiles from the unlabeled target data set, at no point does the model access ground truth labels for the target data. Ground truth labels on the target data set are used exclusively to evaluate model performance. Some single-cell classification methods require manual marker gene specification before model training. scNym requires no prior manual specification of marker genes but rather learns relevant gene expression features from the data.
scNym uses the unlabeled target data through a combination of MixMatch semisupervision (Berthelot et al. 2019) and by training a domain adversary (Ganin et al. 2016) in an iterative learning process (Methods) (Fig. 1A). The MixMatch semisupervision approach combines MixUp data augmentations (Zhang et al. 2018; Thulasidasan et al. 2019) with pseudolabeling of the target data (Lee 2013; Verma et al. 2019) to improve generalization across the training and target domains. At each training iteration, we “pseudolabel” unlabeled cells using predictions from the classification model and then augment each cell profile using a biased weighted average of gene expression and labels with another randomly chosen cell (Fig. 1B). The resulting mixed profiles are dominated by a single cell, adjusted modestly to more closely resemble another. As part of MixMatch, we mix profiles across the training and unlabeled data so that some of the resulting mixed profiles are interpolations between the two data sets. We fit the model parameters to minimize cell type classification error on these mixed profiles, encouraging the model to learn a general representation that allows for interpolation between observed cell states.
Figure 1.

scNym combines semisupervised and adversarial training to learn performant single-cell classifiers. (A) scNym takes advantage of target data during training by estimating “pseudolabels” for each target data point using model predictions. Training and target cell profiles and their labels are then augmented using weighted averages in the MixMatch procedure. An adversary is also trained to discriminate training and target observations. We train model parameters using a combination of supervised classification, interpolation consistency, and adversarial objectives. Here, we use H(·,·) to represent the cross-entropy function. (B) Training and target cell profiles are separated by a domain shift in gene expression space. scNym pseudolabels target profiles and generates mixed cell profiles (arrows) by randomly pairing cells. Mixed profiles form a bridge between training and target data sets. (C) scNym models learn a discriminative representation of cell state in a hidden embedding layer. Train and target cell profiles initially segregate in this representation. During training, adversarial gradients (colored arrows) encourage cells of the same type to mix in the scNym embedding.
The scNym classifier learns a representation of cell identity in the hidden neural network layers where cell types are linearly separable. Alongside, we train an adversarial model to predict the domain of origin for each cell (e.g., training set, target set) from this learned embedding. We train the scNym classifier to compete with this adversary, updating the classifier's embedding to make domain prediction more difficult. At each iteration, the adversary's gradients highlight features in the embedding that discriminate the different domains. We update the scNym classifier using the inverse of the adversarial gradients, reducing the amount of domain-specific information in the embedding as training progresses. This adversarial training procedure encourages the classification model to learn a domain-adapted embedding of the training and target data sets that improves classification performance (Fig. 1C). In inference mode, scNym predictions provide a probability distribution across all cell types in the training set for each target cell.
scNym transfers cell annotations across biological conditions
We evaluated the performance of scNym, transferring cell identity annotations in 11 distinct tasks. These tasks were chosen to capture diverse kinds of technological and biological variation that complicate annotation transfer. Each task represents a true cell type transfer across different experiments, in contrast to some efforts that report within-experiment hold-out accuracy.
We first evaluated cell type annotation transfer between animals of different ages. We trained scNym models on cells from young rats (5 mo old) from the Rat Aging Cell Atlas (Ma et al. 2020) and predicted on cells from aged rats (27 mo old) (Fig. 2A; Methods). We found that predictions from our scNym model trained on young cells largely matched the ground truth annotations (92.2% accurate) on aged cells (Fig. 2B,C).
Figure 2.

scNym transfers cell identity annotations between young and aged rat cells. (A) Young and aged cells from a rat aging cell atlas displayed in a UMAP projection (Ma et al. 2020). Some cell types show a domain shift between young and aged cells. scNym models were trained on young cells in the atlas and used to predict labels for aged cells. (B) Ground truth cell type annotations for the aged cells of the Rat Aging Cell Atlas shown in a UMAP projection. (C) scNym predicted cell types in the target aged cells. scNym predictions match ground truth annotation in the majority (>90%) of cases. (D) Accuracy (left) and κ-scores (right) for scNym and other state of the art classification models. scNym yields significantly greater accuracy and κ-scores than baseline methods (P < 0.01, Wilcoxon rank sums). Note that multiple existing methods could not complete this large task. (E) Aged skeletal muscle cells labeled with ground truth annotations (left) and the relative accuracy of scNym and scmap-cell (right) projected with UMAP. scNym accurately predicts multiple cell types that are confused by scmap-cell (arrows).
We compared scNym performance on this task to state-of-the-art single-cell identity annotation methods (Kiselev et al. 2018; Abdelaal et al. 2019; Alquicira-Hernandez et al. 2019; de Kanter et al. 2019; Tan and Cahan 2019. We also compared scNym to state-of-the-art unsupervised data harmonization methods (Korsunsky et al. 2019; Stuart et al. 2019; Tran et al. 2020; Xu et al. 2021) followed by supervised classification with a support vector machine, for a total of 10 baseline approaches (Methods). scNym produced significantly improved labels over these methods, some of which could not complete this large task on our hardware (256 GB RAM; Wilcoxon rank sums on accuracy or κ-scores, P < 0.01) (Fig. 2D; Table 1). scNym runtimes were competitive with baseline methods (Supplemental Fig. S1). We found that some of the largest differences in accuracy between scNym and the commonly used scmap-cell method were in the skeletal muscle. scNym models accurately classified multiple cell types in the muscle that were confused by scmap-cell (Fig. 2E), showing that the increased accuracy of scNym is meaningful for downstream analyses.
Table 1.
Comparison of model performance across tasks
We next tested the ability of scNym to classify cell identities after perturbation. We trained on unstimulated human peripheral blood mononuclear cells (PBMCs) and predicted on PBMCs after stimulation with IFNB1 (Fig. 3A; Kang et al. 2018). scNym achieved high accuracy (>91%), superior to baseline methods (Fig. 3C; Table 1). The common scmap-cluster method frequently confused monocyte subtypes, whereas scNym did not (Fig. 3B).
Figure 3.

scNym transfers annotations from unstimulated immune cells to stimulated immune cells. (A) UMAP projection of unstimulated PBMC training data and stimulated PBMC target data with stimulation condition labels. (B) UMAP projections of ground truth cell type labels (left), scmap-cluster predictions (center), and scNym predictions for both CD14+ and FCGR3A+ monocytes. scmap-cluster confuses these populations (arrow). (C) Classification accuracy for scNym and baseline cell identity classification methods. scNym is significantly more accurate than other approaches (P < 0.01, Wilcoxon rank sums). (D) Integrated gradient analysis reveals genes that drive correct classification decisions. We recover known marker genes of many cell types (e.g., CD79A for B cells, PPBP for megakaryocytes). (E) Cell type specificity of the top salient genes in a UMAP projection of gene expression (log normalized counts per million). (F) Integrated gradient analysis reveals genes that drive incorrect classification of some FCGR3A+ monocytes as CD14+ monocytes. Several of the top 15 salient genes for misclassification are CD14+ markers that are up-regulated in incorrectly classified FCGR3A+ cells.
Cross-species annotation transfer is another context in which distinct biology creates a domain shift across training and target domains. To evaluate if scNym could transfer labels across species, we trained on mouse cells with either rat or human cells as target data and observed high performance (Supplemental Fig. S2).
scNym models learn biologically meaningful cell type representations
To interpret the classification decisions of our scNym models, we developed integrated gradient analysis tools to identify genes that influence model decisions (Methods) (Sundararajan et al. 2017). The integrated gradient method attributes the prediction of a deep network to its input features while satisfying desirable axioms of interpretability that simpler methods like raw gradients do not. For the PBMC cross-stimulation task, we found that salient genes included known markers of specific cell types such as CD79A for B cells and GNLY for NK cells. Integrated gradient analysis also revealed specific cell type marker genes that may not have been selected a priori, such as NCOA4 for megakaryocytes (Fig. 3D,E; Supplemental Fig. S3). We also performed integrated gradient analysis for a cross-technology mouse cell atlas experiment (described below) and found that marker genes chosen using scNym-integrated gradients were superior to markers chosen using SVM feature importance scores based on Gene Ontology enrichment (Supplemental Fig. S4). These results suggest that our models learned biologically meaningful representations that are more generalizable to unseen cell profiles, regardless of condition or technology.
We also used integrated gradient analysis to understand why the scNym model misclassified some FCGR3A+ monocytes as CD14+ monocytes in the PBMC cross-stimulation task (Methods). This analysis revealed genes driving these incorrect classifications, including some CD14+ monocyte marker genes that are elevated in a subset of FCGR3A+ monocytes (Fig. 3F). Domain experts may use integrated gradient analysis to understand and review model decisions for ambiguous cells.
scNym transfers annotations across single-cell sequencing technologies
To evaluate the ability of scNym to transfer labels across different experimental technologies, we trained on single-cell profiles from 10 mouse tissues in the Tabula Muris captured using the 10x Chromium technology and predicted labels for cells from the same compendium captured using Smart-seq2 (The Tabula Muris Consortium 2018). We found that scNym predictions were highly accurate (>90%) and superior to baseline methods (Supplemental Fig. S5A–C). scNym models accurately classified monocyte subtypes, whereas baseline methods frequently confused these cells (Supplemental Fig. S5D,E).
In a second cross-technology task, we trained scNym on mouse lung data from the Tabula Muris and predicted on lung data from the Mouse Cell Atlas, a separate experimental effort that used microwell-seq technology (Han et al. 2018). We found that scNym yielded high classification accuracy (>90%), superior to baseline methods, despite experimental batch effects and differences in the sequencing technologies (Supplemental Fig. S6). We also trained scNym models to transfer regional identity annotations in spatial transcriptomics data and found performance competitive with baseline methods (Supplemental Fig. S7). Together, these results show that scNym models can effectively transfer cell type annotations across technologies and experimental environments.
Multidomain training allows integration of multiple reference data sets
The number of public single-cell data sets is increasing rapidly (Svensson et al. 2018). Integrating information across multiple reference data sets may improve annotation transfer performance on challenging tasks. The domain adversarial training framework in scNym naturally extends to training across multiple reference data sets. We hypothesized that a multidomain training approach would allow for more general representations that improve annotation transfer. To test this hypothesis, we evaluated the performance of scNym to transfer annotations between single-cell and single-nucleus RNA-seq experiments in the mouse kidney. These data contained six different single-cell preparation methods and three different single-nucleus methods, capturing a range of technical variation in nine distinct domains (Fig. 4A,B; Denisenko et al. 2020).
Figure 4.

Multidomain training improves cross-technology annotation transfer in the mouse kidney. Cell type (A) and sequencing protocol annotations (B) in a UMAP projection of single-cell and nucleus RNA-seq profiles from the mouse kidney (Denisenko et al. 2020). Each protocol represents a unique training domain that captures technical variation. (C) Performance of scNym and baseline approaches on single-cell–to–nucleus and single-nucleus–to–cell annotation transfer. Methods are rank ordered by performance across tasks. scNym is superior to each baseline method on at least one task (Wilcoxon rank sums, P < 0.05). (D) Single-nucleus target data labeled with true cell types (left) or the relative accuracy of scNym and baseline methods (right) for the single-cell–to–single-nucleus task. scNym achieves more accurate labeling of mesangial cells and tubule cell types (arrows). (E) Kidney tubule cells from D visualized independently with true and predicted labels. scNym offers the closest match to true annotations. All methods make notable errors on this difficult task. (F) Comparison of scNym performance when trained on individual training data sets (1-domain) versus multidomain training across all available data sets. We found that multidomain training improves performance on both the cells-to-nuclei and nuclei-to-cells transfer tasks (Wilcoxon rank sums, P = 0.073 and P < 0.01, respectively).
scNym achieved significantly greater accuracy than baseline methods transferring labels from single-nucleus to single-cell experiments using multidomain training. This result was also achieved for the inverse transfer task, transferring annotations from single-cell to single-nucleus experiments (tied with best baseline) (Fig. 4C; Table 1). We found that scNym delivered more accurate annotations for multiple cell types in the cell-to-nucleus transfer task, including mesangial cells and tubule cell types (Fig. 4D,E). These improved annotations highlight that the performance advantages of scNym are meaningful for downstream analysis and biological interpretation. We found that multidomain scNym models achieved greater accuracy than any single-domain model on both tasks and effectively synthesized information from single-domain training sets of varying quality (Fig. 4F; Supplemental Fig. S8). We performed a similar experiment using data from mouse cortex nuclei profiled with four distinct single-cell sequencing methods, training on three methods at a time and predicting annotations for the held-out fourth method for a total of four unique tasks. scNym was the top-ranked method across tasks (Supplemental Fig. S9).
scNym confidence scores enable expert review and allow new cell type discoveries
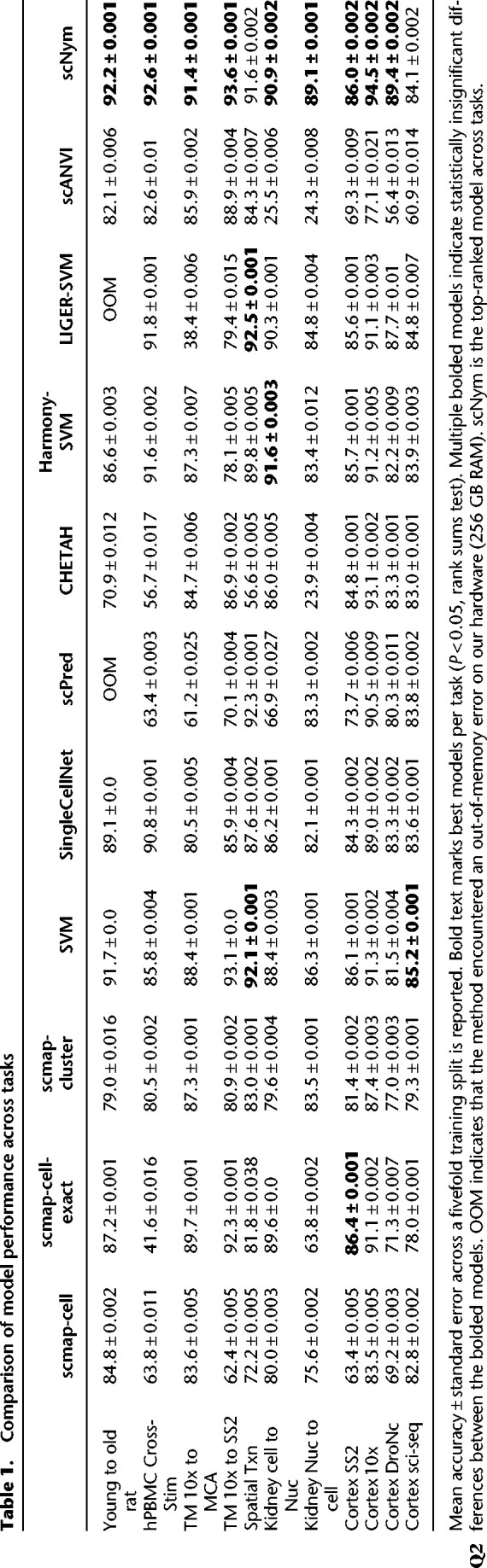
Calibrated predictions, in which the classification probability returned by the model precisely reflects the probability it is correct, enable more effective interaction of the human researcher with the model output. We investigated scNym calibration by comparing the prediction confidence scores to prediction accuracy (Methods). We found that semisupervised adversarial training improved model calibration, such that high-confidence predictions are more likely to be correct (Fig. 5A,B; Supplemental Figs. S10A,B, S11). scNym confidence scores can therefore be used to highlight cells that may benefit from manual review (Supplemental Figs. S10C, S11B), further improving the annotation exercise when it contains a domain expert in the loop.
Figure 5.
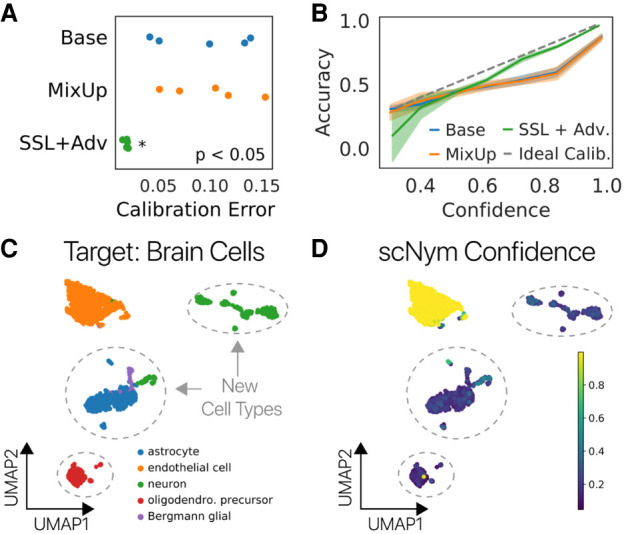
scNym confidence scores highlight unseen cell types. (A) scNym calibration error for models trained on the human PBMC cross-stimulation task. Semisupervised and adversarial training significantly reduced calibration error relative to models trained with only supervised methods (Base, MixUp). (B) Calibration curves capturing the relationship between model confidence and empirical accuracy for models in A. (C) scNym models were trained to transfer annotations from a mouse atlas without brain cell types to data from mouse brain tissue. We desire a model that provides low-confidence scores to the new cell types and high-confidence scores for endothelial cells seen in other tissues. (D) scNym confidence scores for target brain cells. New cell types receive low-confidence scores as desired (dashed outlines).
scNym confidence scores can also highlight new, unseen cell types in the target data set using an optional pseudolabel thresholding procedure during training, inspired by FixMatch (Methods) (Sohn et al. 2020). The semisupervised and adversarial components of scNym encourage the model to find a matching identity for cells in the target data set. Pseudolabel thresholding allows scNym to exclude cells with low-confidence pseudolabels from the semisupervised and adversarial components of training, stopping these components from mismatching unseen cell types and resulting in correctly uncertain predictions.
To test this approach, we simulated two experiments in which we “discover” multiple cell types by predicting annotations on the Tabula Muris brain cell data using models trained on nonbrain tissues (Methods) (Fig. 5A,B). We first used pretrained scNym models to predict labels for new cell types not present in the original training or target sets, and scNym correctly marked these cells with low-confidence scores (Supplemental Fig. S12). In the second experiment, we included new cell types in the target set during training and found that scNym models with pseudolabel thresholding correctly provided low-confidence scores to new cell types, highlighting these cells as potential cell type discoveries for manual inspection (Fig. 5C,D; Supplemental Fig. S13).
We found that scNym embeddings capture cell type differences even within the low-confidence cell population, such that clustering these cells in the scNym embedding can provide a hypothesis for how many new cell types might be present (Supplemental Fig. S14). We also found that putative new cell types could be discriminated from other low-confidence cells, like prediction errors on a cell type boundary (Supplemental Fig. S15). These results show that scNym confidence scores can highlight target cell types that were absent in the training data, potentially enabling new cell type discoveries.
Semisupervised and adversarial training components improve annotation transfer
We ablated different components of scNym to determine which features were responsible for high performance. We found that semisupervision with MixMatch and training with a domain adversary improved model performance across multiple tasks (Fig. 6B; Supplemental Fig. S16). We hypothesized that scNym models might benefit from domain adaptation through the adversarial model by integrating the cells into a latent space more effectively. Supporting this hypothesis, we found that training and target domains were significantly more mixed in scNym embeddings (Supplemental Fig. S17). These results suggest that semisupervision and adversarial training improve the accuracy of cell type classifications.
Figure 6.
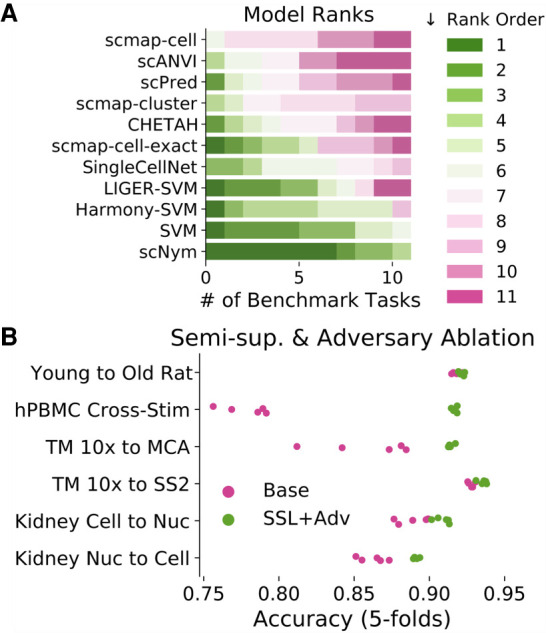
Comparison of semisupervised scNym to other single-cell classification methods and ablated scNym variants. (A) We assign each method a rank order (rank 1 is best) based on performance for each benchmark task. scNym is the top-ranked method across tasks and ranks highly on all tasks. A support vector machine (SVM) baseline is the next best method, consistent with a previous benchmarking study (Abdelaal et al. 2019). (B) Ablation experiments comparing simplified supervised scNym models (Base) against the full scNym model with semisupervised and adversarial training (SSL + Adv.). We found that semisupervised and adversarial training significantly improved scNym performance across diverse tasks (all tasks shown, Wilcoxon rank sums, P < 0.05).
scNym is robust to hyperparameter selection
Hyperparameter selection can be an important determinant of classification model performance. In all tasks presented here, we have used the same set of default scNym parameters derived from past recommendations in the representation learning literature (Methods). To determine how sensitive scNym performance is to these hyperparameter choices, we trained scNym models on the hPBMC cross-stimulation task across a grid of hyperparameter values. We found that scNym is robust to hyperparameter changes within an order of magnitude of the default values, showing that our defaults are not “overfit” to the benchmark tasks presented here (Supplemental Fig. S18). We also performed hyperparameter optimization using reverse fivefold cross-validation for the top three baseline methods (SVM, SingleCellNet, scmap-cell-exact) to determine if an optimized baseline was superior to scNym across four benchmarking tasks (Methods). We found that scNym performance using default parameters was superior to the performance of baseline methods after hyperparameter tuning (Supplemental Table S3; Supplemental Fig. S19).
Discussion
Single-cell genomics experiments have become more accessible owing to commercial technologies, enabling a rapid increase in the use of these methods (Svensson et al. 2020). Cell identity annotation is an essential step in the analysis of these experiments, motivating the development of high-performance, automated annotation methods that can take advantage of diverse data sets. Here, we introduced a semisupervised adversarial neural network model that learns to transfer annotations from one experiment to another, taking advantage of information in both labeled training sets and an unlabeled target data set.
Our benchmark experiments show that scNym models provide high performance across a range of cell identity classification tasks, including cross-age, cross-perturbation, and cross-technology scenarios. scNym performs better in these varied conditions than 10 state-of-the-art baseline methods, including three unsupervised data integration approaches paired with supervised classifiers (Fig. 6A; Table 1). The superiority of scNym is consistent across diverse performance metrics, including accuracy, Cohen's κ-score, and the multiclass receiver operating characteristic (MCROC) (Supplemental Fig. S20; Supplemental Tables S1, S2).
The key idea that differentiates scNym from previous cell classification approaches is the use of semisupervised (Berthelot et al. 2019) and adversarial training (Ganin et al. 2016) to extract information from the unlabeled, target experiment we wish to annotate. Through ablation experiments, we showed that these training strategies improve the performance of our models. Performance improvements were most pronounced when there were large, systematic differences between the training and target data sets (Fig. 3). Semisupervision and adversarial training also allow scNym to integrate information across multiple training and target data sets, improving performance (Fig. 4). As large-scale single-cell perturbation experiments become more common (Dixit et al. 2016; Srivatsan et al. 2020) and multiple cell atlases are released for common model systems, our method's ability to adapt across distinct biological and technical conditions will only increase in value.
Most downstream biological analyses rely upon cell identity annotations, so it is important that researchers are able to interpret the molecular features that drive model decisions. We showed that backpropagation-based saliency analysis methods are able to recover specific cell type markers, confirming that scNym models learn interpretable, biologically relevant features of cell type. In future work, we hope to extend upon these interpretability methods to infer perturbations that alter cell identity programs using the informative representations learned by scNym.
Methods
scNym model
Our scNym model consists of a neural network with an input layer; two hidden layers, each with 256 nodes; and an output layer with a node for each class. The first three layers are paired with batch normalization (Ioffe and Szegedy 2015), rectified linear unit activation, and dropout (Srivastava et al. 2014). The final layer is paired with a softmax activation to transform real number outputs of the neural network into a vector of class probabilities. The model maps cell profile vectors x to probability distributions p(y|x) over cell identity classes y:
We train scNym to map cell profiles in a gene expression matrix x ∈ XCells×Genes to paired cell identity annotations y ∈ y. Transcript counts in the gene expression matrix are normalized to counts per million (CPM) and log-transformed after addition of a pseudocount (log(CPM + 1)). During training, we randomly mask 10% of genes in each cell with zero values and then renormalize to obtain an augmented profile.
We use the Adadelta adaptive stochastic gradient descent method (Zeiler 2012) with an initial learning rate of η = 1.0 to update model parameters on minibatches of cells, with batch sizes of 256. We apply a weight decay term of λWD = 10−4 for regularization. We train scNym models to minimize a standard cross-entropy loss function for supervised training:
where y(k) is an indicator variable for the membership of x in class k, and k ∈ K represents class indicators.
We fit all scNym models for a maximum of 400 epochs and selected the optimal set of weights using early stopping on a validation set consisting of 10% of the training data. We initiate early stopping after training has completed at least 5% of the total epochs to avoid premature termination.
Before passing each minibatch to the network, we perform dynamic data augmentation with the “MixUp” operation (Zhang et al. 2018). MixUp computes a weighted average of two samples x and x′ where the weights λ are randomly sampled from a beta distribution with a symmetric shape parameter α:
For all experiments here, we set α = 0.3 based on performance in the natural image domain (Zhang et al. 2018). Forcing models to interpolate predictions smoothly between samples shifts the decision boundary away from high-density regions of the input distribution, improving generalization. This procedure has been shown to improve classifier performance on multiple tasks (Zhang et al. 2018). Model calibration—the correctness of a model's confidence scores for each class—is generally also improved by this augmentation scheme (Thulasidasan et al. 2019).
Semisupervision with MixMatch
We train semisupervised scNym models using the MixMatch framework (Berthelot et al. 2019), treating the target data set as unlabeled data . At each iteration, MixMatch samples minibatches from both the labeled data set and unlabeled data set . We generate “pseudolabels” (Lee 2013) using model predictions for each observation in the unlabeled minibatch (Supplemental Methods):
We next “sharpen” the pseudolabels using a “temperature scaling” procedure (Hinton et al. 2015; Guo et al. 2017) with the temperature parameter T = 0.5 as a form of entropy minimization (Supplemental Methods). This entropy minimization encourages unlabeled examples to belong to one of the described classes.
We then randomly mix each observation and label/pseudolabel pair in both the labeled and unlabeled minibatches with another observation using MixUp (Zhang et al. 2018). We allow labeled and unlabeled observations to mix together during this procedure (Supplemental Methods):
where (wi, qi) is either a labeled observation and ground truth label (xi, yi) or an unlabeled observation and the pseudolabel (ui, zi). This procedure yields a minibatch X′ of mixed labeled observations and a minibatch U′ of mixed unlabeled observations.
We introduce a semisupervised interpolation consistency penalty during training in addition to the standard supervised loss. For observations and pseudolabels in the mixed unlabeled minibatch U′, we penalize the mean squared error (MSE) between the mixed pseudolabels and the model prediction for the mixed observation (Supplemental Methods):
This encourages the model to provide smooth interpolations between observations and their ground truth or pseudolabels, generalizing the decision boundary of the model. We weight this unsupervised loss relative to the supervised cross-entropy loss using the weighting function λSSL (t) → [0, 1]. We initialize this coefficient to λSSL = 0 and increase the weight to a final value of λSSL = 1 over 100 epochs using a sigmoid schedule (Supplemental Methods):
Domain adaptation with domain adversarial networks
We use domain adversarial networks (DANs) as an additional approach to incorporate information from the target data set during training (Ganin et al. 2016). The DAN method encourages the classification model to embed cells from the training and target data set with similar coordinates, such that training and target data sets are well mixed in the embedding. By encouraging the training and target data set to be well mixed, we take advantage of the inductive bias that cell identity classes in each data set are similar, despite technical variation or differences in conditions (Supplemental Methods).
We introduce this technique into scNym by adding an adversarial domain classification network, . We implement as a two-layer neural network with a single hidden layer of 256 units and a rectified linear unit activation, followed by a classification layer with one output per domain of origin and a softmax activation. This adversary attempts to predict the domain of origin d from the penultimate classifier embedding v of each observation. For each forward pass, it outputs a probability vector, , estimating the likelihood the observation came from the training or target domain.
We assign a one-hot encoded domain label, d, to each molecular profile based on the experiment of origin (Supplemental Methods). During training, we pass a minibatch of labeled observations, x ∈ X, and unlabeled observations, u ∈ U, through the domain adversary to predict domain labels:
where is the domain probability vector, and denotes the embedding of x from the penultimate layer of the classification model . We fit the adversary using a multiclass cross-entropy loss, as described above for the main classification loss (Supplemental Methods).
To make use of the adversary for training the classification model, we use the “gradient reversal” trick at each backward pass. We update the parameters ϕ of the adversary using standard gradient descent on the loss Ladv. At each backward pass, this optimization improves the adversarial domain classifier (Supplemental Methods). We update the parameters θ of the classification model using the inverse of the gradients computed during a backward pass from Ladv. Using the inverse gradients encourages the classification model to generate an embedding where it is difficult for the adversary to predict the domain (Supplemental Methods). Our update rule for the classification model parameters therefore becomes
We increase the weight of the adversary gradients from λadv → [0, 0.1] over the course of 20 epochs during training using a sigmoid schedule. We scale the adversarial gradients flowing to θ, rather than the adversarial loss term, so that full-magnitude gradients are used to train a robust adversary, (Supplemental Methods). Incorporating both MixMatch and the domain adversary, our full loss function becomes
Pseudolabel thresholding for new cell type discovery
Entropy minimization and domain adversarial training enforce an inductive bias that all cells in the target data set belong to a class in the training data set. For many cell type classification tasks, this assumption is valid and useful. However, it is violated in the case in which new, unseen cell types are present in the target data set. We introduce an alternative training configuration to allow for quantitative identification of new cell types in these instances.
We have observed that new cell types will receive low-confidence pseudolabels, as they do not closely resemble any of the classes in the training set (Supplemental Fig. S12). We wish to exclude these low-confidence pseudolabels from our entropy minimization and domain adversarial training procedures, as these methods incorrectly encourage these new cell types to receive both high-confidence predictions and embeddings for a known cell type. We therefore adopt a notion of “pseudolabel confidence thresholding” introduced in the FixMatch method (Sohn et al. 2020). To identify confident pseudolabels to use during training, we set a minimum pseudolabel confidence τ = 0.9 and assign all pseudolabels a binary confidence indicator, ci ∈ {0, 1} (Supplemental Methods).
We make two modifications to the training procedure to prevent low-confidence pseudolabels from contributing to any component of the loss function. First, we use only high-confidence pseudolabels in the MixUp operation of the MixMatch procedure. This prevents low-confidence pseudolabels from contributing to the supervised classification or interpolation consistency losses (Supplemental Methods). Second, we use only unlabeled examples with high-confidence pseudolabels to train the domain adversary. These low-confidence unlabeled examples can therefore occupy a unique region in the model embedding, even if they are easily discriminated from training examples. Our adversarial loss is slightly modified to penalize domain predictions only on confident samples in the pseudolabeled minibatch (Supplemental Methods).
We found that this pseudolabel thresholding configuration option was essential to provide accurate, quantitative information about the presence of new cell types in the target data set (Supplemental Fig. S13). However, this option does modestly decrease performance when new cell types are not present. We therefore enable this option when the possibility of new cell types violates the assumption that the training and target data share the same set of cell types. We have provided a simple toggle in our software implementation to allow users to enable or disable this feature.
scNym model embeddings
We generate gene expression embeddings from our scNym model by extracting the activations of the penultimate neural network layer for each cell. We visualize these embeddings using UMAP (Becht et al. 2019; McInnes et al. 2020) by constructing a nearest-neighbor graph (k = 30) in principal component space derived from the penultimate activations. We set min_dist = 0.3 for the UMAP minimum distance parameter.
We present single-cell experiments using a two-dimensional representation fit using the UMAP algorithm (Becht et al. 2019). For each experiment, we compute a PCA projection on a set of highly variable genes after log (CPM + 1) normalization. We construct a nearest-neighbor graph using the first 50 principal components and fit a UMAP projection from this nearest-neighbor graph.
Entropy of mixing
We compute the “entropy of mixing” to determine the degree of domain adaptation between training and target data sets in an embedding X. The entropy of mixing is defined as the entropy of a vector of class membership in a local neighborhood of the embedding:
where pLocal is a vector of class proportions in a local neighborhood, and k ∈ K are class indices. We compute the entropy of mixing for an embedding X by randomly sampling n = 1000 cells and computing the entropy of mixing on a vector of class proportions for the 100 nearest neighbors to each point.
Integrated gradient analysis
We interpreted the predictions of our scNym models by performing integrated gradient analysis (Sundararajan et al. 2017). Given a trained model, , and a target class, k, we computed an integrated gradient score IG as the sum of gradients on a class probability, , with respect to an input gene expression vector, x, at M = 100 points along a linear path between the zero vector and the input x. We then multiplied the sum of gradients for each gene by the expression values in the input x. Stated formally, we computed
In the original integrated gradient formalism, this is equivalent to using the zero vector as a baseline. We average the integrated gradients across ns cell input vectors x to obtain class-level maps IGk, where ns = min (300, nk), and nk is the number of cells in the target class. To identify genes that drive incorrect classifications, we computed integrated gradients with respect to some class k for cells with true class k′ that were incorrectly classified as class k.
Interpretability comparison
We compared the biological relevance of features selected by scNym and SVM as a baseline by computing cell type–specific Gene Ontology enrichments. We trained both scNym and an SVM to transfer labels from the Tabula Muris 10x Genomics data set to the Tabula Muris Smart-seq2 data set. We then extracted feature importance scores from the scNym model using integrated gradients and from the SVM model based on coefficient weights. We selected cell type markers for each model as the top k = 100 genes with the highest integrated gradient values or SVM coefficients.
For 19 cell types with corresponding Gene Ontology terms, we computed the enrichment of the relevant cell type–specific Gene Ontology terms in scNym-derived and SVM-derived cell type markers using Fisher's exact test (Supplemental Methods). We present a sample of the gene sets used (Supplemental Table S4). We compared the mean odds ratio from Fisher's exact test across relevant Gene Ontology terms between scNym-derived markers and SVM-derived markers. To determine statistical significance of a difference in these mean odds ratios, we performed a paired t-test across cell types. We performed the procedure above using k ∈ {50, 100, 150} to determine the sensitivity of our results to this parameter. We found that scNym-integrated gradients had consistently stronger enrichments for relevant Gene Ontology terms across cell types for all values of k.
Model calibration analysis
We evaluated scNym calibration by binning all cells in a query set based on the softmax probability of their assigned class ——which we term the “confidence score.” We grouped cells into M = 10 bins Bm of equal width from [0, 1] and computed the mean accuracy of predictions within each bin:
where 1(a ≡ b) denotes a binary equivalency operation that yields 1 if a and b are equivalent and 0 otherwise and denotes the arithmetic average.
We computed the “expected calibration error” as previously proposed (Thulasidasan et al. 2019):
We also computed the “overconfidence error”, which specifically focuses on high confidence but incorrect predictions:
where N is the total number of samples, and |Bm| is the number of samples in bin Bm.
We performed this analysis for each model trained in a fivefold cross-validation split to estimate calibration for a given model configuration. We evaluated calibrations for baseline neural network models, models with MixUp but not MixMatch, and models with the full MixMatch procedure.
Baseline methods
As baseline methods, we used 10 cell identity classifiers: scmap-cell, scmap-cluster (Kiselev et al. 2018; Andrews and Hemberg 2019), scmap-cell-exact (scmap-cell with exact k-NN search), a linear SVM (Abdelaal et al. 2019), scPred (Alquicira-Hernandez et al. 2019), SingleCellNet (Tan and Cahan 2021), CHETAH (de Kanter et al. 2019), Harmony followed by an SVM (Korsunsky et al. 2019), LIGER followed by an SVM (Stuart et al. 2019), and scANVI (Lopez et al. 2018; Xu et al. 2021). For model training, we split data into fivefolds and trained five separate models, each using fourfolds for training and validation data. This allowed us to assess variation in model performance as a function of changes in the training data. No class balancing was performed before training, although some methods perform class balancing internally. All models, including scNym, were trained on the same fivefold splits to ensure equitable access to information. All methods were run with the best hyperparameters suggested by the investigators unless otherwise stated for our hyperparameter optimization comparisons (for full details, see Supplemental Methods).
We applied all baseline methods to all benchmarking tasks. If a method could not complete the task given 256 GB of RAM and 8 CPU cores, we reported the accuracy for that method as “undetermined.” Only the scNym and scANVI models required GPU resources. We trained models on Nvidia K80, GTX1080ti, Titan RTX, or RTX 8000 GPUs, using only a single GPU per model.
Performance benchmarking
For all benchmarks, we computed the mean accuracy across cells (“accuracy”), Cohen's κ-score, and the multiclass receiver operating characteristic (MCROC). We computed the MCROC as the mean of ROC scores across cell types, treating each cell type as a binary classification problem. We used external methods to generate probabilistic outputs for baseline methods where appropriate (Platt 1999). We performed quality-control filtering and preprocessing on each data set before training (Supplemental Methods).
For the Rat Aging Cell Atlas (Ma et al. 2020) benchmark, we trained scNym models on single-cell RNA-seq from young, ad libitum fed rats (5 mo old) and predicted on cells from aged rats (ad libitum fed or calorically restricted). For the human PBMC stimulation benchmark, we trained models on unstimulated PBMCs collected from multiple human donors and predicted on IFNB1-stimulated PBMCs collected in the same experiment (Kang et al. 2018).
For the Tabula Muris cross-technology benchmark, we trained models on the Tabula Muris 10x Genomics Chromium platform and predicted on data generated using Smart-seq2. For the Mouse Cell Atlas (MCA) (Han et al. 2018) benchmark, we trained models on single-cell RNA-seq from lung tissue in the Tabula Muris 10x Chromium data (The Tabula Muris Consortium 2018) and predicted on MCA lung data. For the spatial transcriptomics benchmark, we trained models on spatial transcriptomics from a mouse sagittal–posterior brain section and predicted labels for another brain section (data downloaded from https://www.10xgenomics.com/resources/datasets/).
For the single-cell to single-nucleus benchmark in the mouse kidney, we trained scNym models on all single-cell data from six unique sequencing protocols and predicted labels for single nuclei from three unique protocols (Denisenko et al. 2020). For the single-nucleus to single-cell benchmark, we inverted the training and target data sets above to train on the nuclei data sets and predict on the single-cell data sets. We set unique domain labels for each protocol during training in both benchmark experiments. To evaluate the impact of multidomain training, we also trained models on only one single-cell or single-nucleus protocol using the domains from the opposite technology as target data.
For the multidomain cross-technology benchmark in mouse cortex nuclei, we generated four distinct subtasks from data generated using four distinct technologies to profile the same samples (Ding et al. 2020). We trained scNym and baseline methods to predict labels on one technology given the remaining three technologies as training data for all possible combinations. We used each technology as a unique domain label for scNym.
For the cross-species mouse-to-rat demonstration, we selected a set of cell types with comparable annotations in the Tabula Muris and Rat Aging Cell Atlas (Ma et al. 2020) to allow for quantitative evaluation. We trained scNym with mouse data as the source domain and rat data as the target domain. We used the new identity discovery configuration to account for the potential for new cell types in a cross-species experiment. For the cross-species mouse-to-human demonstration, we similarly selected a set of cell types with comparable cell annotation ontologies in the Tabula Muris l0x lung data and human lung cells from the IPF Cell Atlas (Habermann et al. 2020). We trained an scNym model using mouse data as the source domain and human data as the target, as for the mouse-to-rat demonstration.
Runtime benchmarking
We measured the runtime of scNym and each baseline classification method using subsamples from the multidomain kidney single-cell and single-nuclei data set (Denisenko et al. 2020). We measured runtimes for annotation transfer from single cells to single-nuclei labels using subsamples of size n ∈ {1250, 2500, 5000, 10000, 20000, 40000} for each of the training and target data sets. All methods were run on four cores of a 2.1-GHz Intel Xeon Gold 6130 CPU and 64 GB of CPU memory. GPU-capable methods (scNym, scANVI) were provided with one Nvidia Titan RTX GPU (consumer grade CUDA compute device).
Hyperparameter optimization experiments
We performed hyperparameter optimization across four tasks for the top three baseline methods: the SVM, SingleCellNet, and scmap-cell-exact. For the SVM, we optimized the regularization strength parameter C at 12 values with and without class weighting. For class weighting, we set class weights as either uniform or inversely proportional to the number of cells in each class to enforce class balancing (, where wk is the weight for class k, and nk is the number of cells for that class). For scmap-cell-exact, we optimized (1) the number of nearest neighbors (k ∈ {5, 10, 30, 50, 100}), (2) the distance metric (d(·,·) ∈ {cosine, euclidean}), and (3) the number of features to select with M3Drop (nf ∈ {500, 1000, 2000, 5000}). For SingleCellNet, we optimized with nTopGenes ∈ {10, 20}, nRand ∈ {35, 70, 140}, nTrees ∈ {100, 1000, 2000}, and nTopGenePairs ∈ {12, 25}.
We optimized scNym for two of the four tasks, owing to computational expense and superiority of default parameters relative to baseline methods. For scNym, we optimized (1) weight decay (λw ∈ 10−5, 10−4, 10−3), (2) batch size (M ∈ {128, 256}), (3) the number of hidden units (h ∈ {256, 512}), (4) the maximum MixMatch weight (λSSL ∈ {0.01, 0.1, 1.0}), and (5) the maximum DAN weight (λadv ∈ {0.01, 0.1, 0.2}). We did not optimize weight decay for the PBMC cross-stimulation task. We performed a grid search for all methods.
Hyperparameter optimization is nontrivial in the context of a domain shift between the training and test set. Traditional optimization using cross-validation on the training set alone may overfit parameters to the training domain, leading to suboptimal outcomes. This failure mode is especially problematic for domain adaptation models, in which decreasing the strength of domain adaptation regularizers may improve performance within the training data while actually decreasing performance on the target data.
In light of these concerns, we adopted a procedure known as reverse cross-validation to evaluate each hyperparameter set (Zhong et al. 2010). Reverse cross-validation uses both the training and target data sets during training to account for the effect of hyperparameters on the effectiveness of transferring labels across domains. Formally, we first split the labeled training data into a training set, validation set, and held-out test set . We use 10% of the training data set for the validation set and 10% for the held-out test set. We then train a model, , to transfer labels from the training set to the target data . We use the validation set for early stopping with scNym and concatenate it into the training set for other methods that do not use a validation set. We treat the predictions as pseudolabels for the unlabeled data set and subsequently train a second model, , to transfer annotations from the “pseudolabeled” data set back to the labeled data set . We then evaluate the “reverse accuracy” as the accuracy of the labels for the held-out test portion of the labeled data set .
We performed this procedure using a standard fivefold split for each parameter set. We computed the mean reverse cross-validation accuracy as the performance metric for robustness. For each method that we optimized, we selected the optimal set of hyperparameters as the set with the top reverse cross-validation accuracy.
New cell type discovery experiments
New cell type discovery with pretrained models
We evaluated the ability of scNym to highlight new cell types, unseen in the training data by predicting cell type annotations in the Tabula Muris brain data (Smart-seq2) using models trained on the 10x Genomics data from the 10 tissues noted above with the Smart-seq2 data as a corresponding target data set. No neurons or glia were present in the training or target set for this experiment. This experiment simulates the scenario in which a pretrained model has been fit to transfer across technologies (10x to Smart-seq2) and is later used to predict cell types in a new tissue, unseen in the original training or target data.
We computed scNym confidence scores for each cell as ci = max pi, where pi is the model prediction probability vector for cell i as noted above. To highlight potential cell type discoveries, we set a simple threshold on these confidence scores, di = ci ≤ 0.5, where di ∈ {0, 1} is a binary indicator variable. We found that scNym assigned low confidence to the majority of cells from newly “discovered” types unseen in the training set using this method.
New cell type discovery with semisupervised training
We also evaluated the ability of scNym to discover new cell types in a scenario in which new cell types are present in the target data used for semisupervised training. We used the same training data and target data as the experiment above, but we now introduce the Tabula Muris brain data (Smart-seq2) into the target data set during semisupervised training. We performed this experiment using our default scNym training procedure, as well as the modified new cell type discovery procedure described above.
As above, we computed confidence scores for each cell and set a threshold of di = ci ≤ 0.5 to identify potential new cell type discoveries. We found that scNym models trained with the new cell type discovery procedure provided low-confidence scores to the new cell types, suitable for identification of these new cells. We considered all new cell type predictions to be incorrect when computing accuracy for the new cell type discovery task.
Clustering candidate new cell types
We used a community detection procedure in the scNym embedding to suggest the number of distinct cell states represented by low-confidence cells. First, we identify cells with a confidence score lower than a threshold tconf to highlight putative cell type discoveries, di = ci < tconf. We then extract the scNym penultimate embedding activations for these low-confidence cells and construct a nearest-neighbor graph using the k = 15 nearest neighbors for each cell. We compute a Leiden community detection partition for a range of different resolution parameters, r ∈ {0.1, 0.2, 0.3, 0.5, 1.0}, and compute the Calinski–Harabasz score for each partition (Calinski and Harabasz 1974). We select the optimal partition in the scNym embedding as the partition generated with the maximum Calinski–Harabasz score and suggest that communities in this partition may each represent a distinct cell state.
Discriminating candidate new cell types from other low-confidence predictions
Cells may receive low-confidence predictions for multiple reasons, including (1) a cell is on the boundary between two cell types, (2) a cell has very little training data for the predicted class, and (3) the cell represents a new cell type unseen in the training data set. To discriminate between these possibilities, we use a heuristic similar to the one we use for proposing a number of new cell types that might be present. First, we extract the scNym embedding coordinates from the penultimate layer activations for all cells and build a nearest-neighbor graph. We then optimize a Leiden cluster partition by scanning different resolution parameters to maximize the Calinksi–Harabasz score. We then compute the average prediction confidence across all cells in each of the resulting clusters. We also visualize the number of cells present in the training data for each predicted cell type.
We consider cells with low prediction scores within an otherwise high-confidence cluster to be on the boundary between cell types. These cells may benefit from domain expert review of the specific criteria to use when discriminating between very similar cell identities. We consider low-confidence cell clusters with few training examples for the predicted class to warrant further domain expert review. Low-confidence clusters that are predicted to be a class with ample training data may represent new cell types and also warrant further review.
Software availability
Open-source code for our software and preprocessed reference data sets analyzed in this study are available in the scNym repository (https://github.com/calico/scnym) and as Supplemental Code.
Supplementary Material
Acknowledgments
We thank Zhenghao Chen, Amoolya H. Singh, and Han Yuan for helpful discussions and comments. Funding for this study was provided by Calico Life Sciences, LLC.
Author contributions: J.C.K. conceived the study, implemented software, conducted experiments, and wrote the paper. D.R.K. conceived the study and wrote the paper.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.268581.120.
Freely available online through the Genome Research Open Access option.
Competing interest statement
J.C.K. and D.R.K. are paid employees of Calico Life Sciences, LLC.
References
- Abdelaal T, Michielsen L, Cats D, Hoogduin D, Mei H, Reinders MJT, Mahfouz A. 2019. A comparison of automatic cell identification methods for single-cell RNA sequencing data. Genome Biol 20: 194. 10.1186/s13059-019-1795-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alquicira-Hernandez J, Sathe A, Ji HP, Nguyen Q, Powell JE. 2019. scPred: accurate supervised method for cell-type classification from single-cell RNA-seq data. Genome Biol 20: 264. 10.1186/s13059-019-1862-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews TS, Hemberg M. 2019. M3Drop: dropout-based feature selection for scRNASeq. Bioinformatics 35: 2865–2867. 10.1093/bioinformatics/bty1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelidis I, Simon LM, Fernandez IE, Strunz M, Mayr CH, Greiffo FR, Tsitsiridis G, Ansari M, Graf E, Strom TM, et al. 2019. An atlas of the aging lung mapped by single cell transcriptomics and deep tissue proteomics. Nat Commun 10: 963. 10.1038/s41467-019-08831-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becht E, McInnes L, Healy J, Dutertre CA, Kwok IWH, Ng LG, Ginhoux F, Newell EW. 2019. Dimensionality reduction for visualizing single-cell data using UMAP. Nat Biotechnol 37: 38–44. 10.1038/nbt.4314 [DOI] [PubMed] [Google Scholar]
- Berthelot D, Carlini N, Goodfellow I, Papernot N, Oliver A, Raffel CA. 2019. MixMatch: a holistic approach to semi-supervised learning. Advances in Neural Information Processing Systems 32: 5049–5059. [Google Scholar]
- Calinski T, Harabasz J. 1974. A dendrite method for cluster analysis. Commun Stat 3: 1–27. 10.1080/03610927408827101 [DOI] [Google Scholar]
- de Kanter JK, Lijnzaad P, Candelli T, Margaritis T, Holstege FCP. 2019. CHETAH: a selective, hierarchical cell type identification method for single-cell RNA sequencing. Nucleic Acids Res 47: e95. 10.1093/nar/gkz543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denisenko E, Guo BB, Jones M, Hou R, de Kock L, Lassmann T, Poppe D, Clément O, Simmons RK, Lister R, et al. 2020. Systematic assessment of tissue dissociation and storage biases in single-cell and single-nucleus RNA-seq workflows. Genome Biol 21: 130. 10.1186/s13059-020-02048-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J, Adiconis X, Simmons SK, Kowalczyk MS, Hession CC, Marjanovic ND, Hughes TK, Wadsworth MH, Burks T, Nguyen LT, et al. 2020. Systematic comparison of single-cell and single-nucleus RNA-sequencing methods. Nat Biotechnol 38: 737–746. 10.1038/s41587-020-0465-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit A, Parnas O, Li B, Chen J, Fulco CP, Jerby-Arnon L, Marjanovic ND, Dionne D, Burks T, Raychowdhury R, et al. 2016. Perturb-Seq: dissecting molecular circuits with scalable single-cell RNA profiling of pooled genetic screens. Cell 167: 1853–1866.e17. 10.1016/j.cell.2016.11.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganin Y, Ustinova E, Ajakan H, Germain P, Larochelle H, Laviolette F, Marchand M, Lempitsky V. 2016. Domain-adversarial training of neural networks. J Mach Learn Res 17: 1–35. [Google Scholar]
- Guo C, Pleiss G, Sun Y, Weinberger KQ. 2017. On calibration of modern neural networks. In Proceedings of the 34th International Conference on Machine Learning 70: 1321–1330. JMLR.org. [Google Scholar]
- Habermann AC, Gutierrez AJ, Bui LT, Yahn SL, Winters NI, Calvi CL, Peter L, Chung MI, Taylor CJ, Jetter C, et al. 2020. Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci Adv 6: eaba1972. 10.1126/sciadv.aba1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Wang R, Zhou Y, Fei L, Sun H, Lai S, Saadatpour A, Zhou Z, Chen H, Ye F, et al. 2018. Mapping the mouse cell atlas by microwell-Seq. Cell 173: 1307. 10.1016/j.cell.2018.05.012 [DOI] [PubMed] [Google Scholar]
- Hinton G, Vinyals O, Dean J. 2015. Distilling the knowledge in a neural network. arXiv:1503.02531 [stat.ML].
- Ioffe S, Szegedy C. 2015. Batch normalization: accelerating deep network training by reducing internal covariate shift. In Proceedings of the 32th International Conference on Machine Learning 37: 448–456. JMLR.org. [Google Scholar]
- Kang HM, Subramaniam M, Targ S, Nguyen M, Maliskova L, McCarthy E, Wan E, Wong S, Byrnes L, Lanata CM, et al. 2018. Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat Biotechnol 36: 89–94. 10.1038/nbt.4042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmel JC, Penland L, Rubinstein ND, Hendrickson DG, Kelley DR, Rosenthal AZ. 2019. Murine single-cell RNA-seq reveals cell-identity- and tissue-specific trajectories of aging. Genome Res 29: 2088–2103. 10.1101/gr.253880.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingma DP, Mohamed S, Jimenez Rezende D, Welling M. 2014. Semi-supervised learning with deep generative models. In Advances in neural information processing systems (ed. Ghahramani Z, et al. ). Curran Associates, Red Hook, NY. [Google Scholar]
- Kiselev VY, Yiu A, Hemberg M. 2018. scmap: projection of single-cell RNA-seq data across data sets. Nat Methods 15: 359–362. 10.1038/nmeth.4644 [DOI] [PubMed] [Google Scholar]
- Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh PR, Raychaudhuri S. 2019. Fast, sensitive and accurate integration of single-cell data with harmony. Nat Methods 16: 1289–1296. 10.1038/s41592-019-0619-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DH. 2013. Pseudo-label: the simple and efficient semi-supervised learning method for deep neural networks. In International Conference on Machine Learning (ICML) 2013 Workshop: Challenges in Representation Learning (WREPL). ICML, Atlanta. [Google Scholar]
- Lopez R, Regier J, Cole MB, Jordan MI, Yosef N. 2018. Deep generative modeling for single-cell transcriptomics. Nat Methods 15: 1053–1058. 10.1038/s41592-018-0229-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S, Sun S, Geng L, Song M, Wang W, Ye Y, Ji Q, Zou Z, Wang S, He X, et al. 2020. Caloric restriction reprograms the single-cell transcriptional landscape of Rattus norvegicus aging. Cell 180: 984–1001. 10.1016/j.cell.2020.02.008 [DOI] [PubMed] [Google Scholar]
- McInnes L, Healy J, Melville J. 2020. UMAP: Uniform Manifold Approximation and Projection for dimension reduction. arXiv:1802.03426 [stat.ML].
- Oliver A, Odena A, Raffel CA, Cubuk ED, Goodfellow I. 2018. Realistic evaluation of deep semi-supervised learning algorithms. In Advances in neural information processing systems, Vol. 31 (ed. Bengio S, et al. ), pp. 3235–3246. Curran Associates, Red Hook, NY. [Google Scholar]
- Platt JC. 1999. Probabilistic outputs for support vector machines and comparisons to regularized likelihood methods. In Advances in large margin classifiers (ed. Smola AJ, et al. ), pp. 61–74. MIT Press, Cambridge, MA. [Google Scholar]
- Pliner HA, Shendure J, Trapnell C. 2019. Supervised classification enables rapid annotation of cell atlases. Nat Methods 16: 983–986. 10.1038/s41592-019-0535-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn K, Berthelot D, Li CL, Zhang Z, Carlini N, Cubuk ED, Kurakin A, Zhang H, Raffel C. 2020. FixMatch: simplifying semi-supervised learning with consistency and confidence. In Advances in neural information processing systems (ed. Larochelle H, et al. ), pp. 596–608. Curran Associates, Red Hook, NY. [Google Scholar]
- Srivastava N, Hinton GE, Krizhevsky A. 2014. Dropout: a simple way to prevent neural networks from overfitting. J Mach Learn Res 15: 1929–1958. [Google Scholar]
- Srivatsan SR, McFaline-Figueroa JL, Ramani V, Saunders L, Cao J, Packer J, Pliner HA, Jackson DL, Daza RM, Christiansen L, et al. 2020. Massively multiplex chemical transcriptomics at single cell resolution. Science 367: 45–51. 10.1126/science.aax6234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM III, Hao Y, Stoeckius M, Smibert P, Satija R. 2019. Comprehensive integration of single-cell data. Cell 177: 1888–1902.e1. 10.1016/j.cell.2019.05.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundararajan M, Taly A, Yan Q. 2017. Axiomatic attribution for deep networks. In Proceedings of the 34th International Conference on Machine Learning, Proceedings of Machine Learning Research, Sydney, Australia (ed. Precup D, Teh YW), Vol. 70, pp. 3319–3328. [Google Scholar]
- Svensson V, Vento-Tormo R, Teichmann SA. 2018. Exponential scaling of single-cell RNA-seq in the past decade. Nat Protoc 13: 599–604. 10.1038/nprot.2017.149 [DOI] [PubMed] [Google Scholar]
- Svensson V, da Veiga Beltrame E, Pachter L. 2020. A curated database reveals trends in single-cell transcriptomics. Database 2020: Baaa073. 10.1093/database/baaa073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Tabula Muris Consortium. 2018. Single-cell transcriptomics of 20 mouse organs creates a tabula muris. Nature 562: 367–372. 10.1038/s41586-018-0590-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Y, Cahan P. 2019. SingleCellNet: a computational tool to classify single cell RNA-Seq data across platforms and across species. Cell Syst 9: 207–213.e2. 10.1016/j.cels.2019.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanay A, Regev A. 2017. Scaling single-cell genomics from phenomenology to mechanism. Nature 541: 331–338. 10.1038/nature21350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thulasidasan S, Chennupati G, Bilmes JA, Bhattacharya T, Michalak S. 2019. On mixup training: improved calibration and predictive uncertainty for deep neural networks. Advances in Neural Information Processing Systems 32: 13888–13899. [Google Scholar]
- Tran HTN, Ang KS, Chevrier M, Zhang X, Lee NYS, Goh M, Chen J. 2020. A benchmark of batch-effect correction methods for single-cell RNA sequencing data. Genome Biol 21: 12. 10.1186/s13059-019-1850-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C. 2015. Defining cell types and states with single-cell genomics. Genome Res 25: 1491–1498. 10.1101/gr.190595.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma V, Lamb A, Kannala J, Bengio Y, Lopez-Paz D. 2019. Interpolation consistency training for semi-supervised learning. In Proceedings of the 28th International Joint Conference on Artificial Intelligence, IJCAI'19, pp. 3635–3641. AAAI Press, Palo Alto, CA. [DOI] [PubMed] [Google Scholar]
- Wilson G, Cook DJ. 2020. A survey of unsupervised deep domain adaptation. ACM Trans Intell Syst Technol 11: 1–46. 10.1145/3400066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Lopez R, Mehlman E, Regier J, Jordan MI, Yosef N. 2021. Probabilistic harmonization and annotation of single-cell transcriptomics data with deep generative models. Mol Syst Biol 17: e9620. 10.15252/msb.20209620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeiler MD. 2012. ADADELTA: an adaptive learning rate method. arXiv:1212.5701 [cs.LG].
- Zhang H, Cisse M, Dauphin YN, Lopez-Paz D. 2018. Mixup: beyond empirical risk minimization. In International Conference on Learning Representations, Stockholm. [Google Scholar]
- Zhang AW, O'Flanagan C, Chavez EA, Lim JLP, Ceglia N, McPherson A, Wiens M, Walters P, Chan T, Hewitson B, et al. 2019. Probabilistic cell-type assignment of single-cell RNA-seq for tumor microenvironment profiling. Nat Methods 16: 1007–1015. 10.1038/s41592-019-0529-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong E, Fan W, Yang Q, Verscheure O, Ren J. 2010. Cross validation framework to choose amongst models and datasets for transfer learning. In Machine learning and knowledge discovery in databases, lecture notes in computer science (ed. Balcázar JL, et al. ), pp. 547–562. Springer, Berlin. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.