

Abstract
Because disease-associated microglia (DAM) and disease-associated astrocytes (DAA) are involved in the pathophysiology of Alzheimer's disease (AD), we systematically identified molecular networks between DAM and DAA to uncover novel therapeutic targets for AD. Specifically, we develop a network-based methodology that leverages single-cell/nucleus RNA sequencing data from both transgenic mouse models and AD patient brains, as well as drug-target network, metabolite-enzyme associations, the human protein–protein interactome, and large-scale longitudinal patient data. Through this approach, we find both common and unique gene network regulators between DAM (i.e., PAK1, MAPK14, and CSF1R) and DAA (i.e., NFKB1, FOS, and JUN) that are significantly enriched by neuro-inflammatory pathways and well-known genetic variants (i.e., BIN1). We identify shared immune pathways between DAM and DAA, including Th17 cell differentiation and chemokine signaling. Last, integrative metabolite-enzyme network analyses suggest that fatty acids and amino acids may trigger molecular alterations in DAM and DAA. Combining network-based prediction and retrospective case-control observations with 7.2 million individuals, we identify that usage of fluticasone (an approved glucocorticoid receptor agonist) is significantly associated with a reduced incidence of AD (hazard ratio [HR] = 0.86, 95% confidence interval [CI] 0.83–0.89, P < 1.0 × 10−8). Propensity score–stratified cohort studies reveal that usage of mometasone (a stronger glucocorticoid receptor agonist) is significantly associated with a decreased risk of AD (HR = 0.74, 95% CI 0.68–0.81, P < 1.0 × 10−8) compared to fluticasone after adjusting age, gender, and disease comorbidities. In summary, we present a network-based, multimodal methodology for single-cell/nucleus genomics-informed drug discovery and have identified fluticasone and mometasone as potential treatments in AD.
Alzheimer's disease (AD) is expected to double in incidence by 2050 (Hebert et al. 2001), affecting upward of 16 million Americans and 90 million people worldwide (Alzheimer's Association 2016). Without new treatments, this will represent an unprecedented crisis of human suffering and financial cost. The attrition rate for AD clinical trials (2002–2012) is estimated at 99.6% (Cummings et al. 2014), and improved methods of drug discovery are therefore needed. The underlying pathophysiology of AD is especially poorly understood and appears to involve a complex, polygenic, and pleiotropic genetic architecture (Tasaki et al. 2018). Recent studies strongly implicate a crucial role of neuroinflammation in the pathophysiology of AD (Cao and Zheng 2018). However, broad anti-inflammatory therapies have not been clinically efficacious against AD. We believe this suggests a pressing need to better understand the heterogeneity of immune cells in AD, which could translate to identification of novel drug targets.
Recent single-cell/nucleus RNA sequencing (scRNA-seq or snRNA-seq) studies have suggested essential roles for microglia and astrocytes, such as determining the distribution of immune cell subpopulations in AD (Keren-Shaul et al. 2017; Habib et al. 2020). For example, disease-associated microglia (DAM) have been identified as a unique microglia subtype associated with AD (Keren-Shaul et al. 2017), and disease-associated astrocytes (DAA) have been identified as becoming increasingly abundant with progression of AD (Habib et al. 2020). Astrocytic release of cytokines, the primary immune messenger, influence the microglial activation state (e.g., CCL2 and ORM2) and also help microglia modulate astrocytic phenotype and function (e.g., IL1A and TNF) (Jha et al. 2019). A growing body of evidence suggests that both microglia and astrocytes are exquisitely sensitive to their environment and are affected by dysregulation of multiple biochemical pathways, such as abnormal lipid metabolism, in AD pathogenesis (Desale and Chinnathambi 2020). Systematic identification of the underlying molecular mechanisms linking DAM and DAA and AD could thus advance understanding of the underlying biology and offer potential novel drug targets.
Existing data resources, including transcriptomics and interactomics (protein–protein interactions [PPIs]), have not yet been fully exploited in pursuit of understanding the causal disease pathways in AD (Fang et al. 2020). With this in mind, integrative analyses of genomics, transcriptomics, and other omics can enable us to elucidate the cascade of molecular events contributing to complex neuro-inflammatory mechanisms, including microglia and astrocytes. We show how these analyses can accelerate the translation of high-throughput single-cell/nucleus omics findings into innovative therapeutic approaches for AD centered on the interactions of microglia and astrocytes.
Results
Network-based methodology pipeline
In this study, we presented an integrative multi-omics, network-based methodology to uncover molecular networks of DAM and DAA and to prioritize drug candidates for AD. We integrated sc/snRNA-seq data from both AD transgenic mouse models and AD patient brains, drug-target networks, enzyme-metabolite associations, PPIs, along with large-scale patient database validation (Fig. 1). The whole procedure is divided into four components: (1) We first assembled the five recent sc/snRNA-seq data sets (Supplemental Table S1) covering both microglia and astrocytes from either AD transgenic mouse models or human brains; (2) we performed standard bioinformatics analysis for sc/snRNA-seq data, including quality control, cell/nucleus clustering, and differential expression analysis; (3) we built the molecular network for DAM and DAA using the state-of-the-art network-based algorithm by integrating sc/snRNA-seq data into the human protein–protein interactome (Methods); (4) we prioritized repurposed drugs for potential treatment of AD by identifying those that specifically reverse dysregulated gene expression of microglia and astrocytes; and (5) we validated top drug candidates using the state-of-the-art pharmacoepidemiologic observations of a large-scale, longitudinal patient data (Fig. 1).
Figure 1.

A diagram illustrating the network-based framework. A standard single-cell/nucleus RNA sequencing (sc/snRNA-seq) data analysis pipeline includes quality control, clustering analysis, and differentially expressed gene (DEG) analysis. We built the molecular network using the state-of-the-art network-based algorithm (termed GPSnet) by integrating sc/snRNA-seq data into the human protein–protein interactome (Methods). Next, we prioritized repurposed drugs for potential treatment of Alzheimer's disease (AD) by identifying those that specifically reverse dysregulated gene expression for molecular networks of disease-associated microglia (DAM) or astrocyte (DAA): if drug-induced up- or down-related genes are significantly enriched in the dysregulated molecular networks, these drugs will be prioritized as potential candidates for treatment of AD. Finally, top drug candidates were validated further using a large-scale, longitudinal patient database. (GSEA) Gene set enrichment analysis; (CMap) connectivity map.
Discovery of DAM-specific molecular networks
We compared expression of cell marker genes (Cst7, Lpl, P2ry12, and Cx3cr1) for DAM among all cell/nucleus clusters (Fig. 2A,B; Supplemental Fig. S1A,B). Here, we used homeostasis-associated microglia (HAM) (Ginhoux and Prinz 2015) as control groups. We found a higher abundance of DAM nuclei in 5XFAD mice compared to wild-type (WT) mice (P = 0.048, t-test) (Supplemental Table S2; Supplemental Fig. S2A); yet, there was no nucleus abundance difference for HAM between 5XFAD and WT mice (P = 0.786) (Supplemental Fig. S2A). We observed a similar pattern when considering the scRNA-seq profile, in that the cell abundance percentage of the DAM in 5XFAD mice was much higher than in WT mice (P = 9.11 × 10−10) (Supplemental Table S3; Supplemental Fig. S2B). Altogether, both scRNA-seq and snRNA-seq profiles show significantly elevated abundance of DAM in 5XFAD compared to WT mice.
Figure 2.

Discovery of DAM-specific molecular networks for the transgenic mouse model of AD. (A) Uniform manifold approximation and projection (UMAP) plot of clustering 4389 microglia cells: the blue cluster denotes the homeostasis-associated microglia (HAM), and the green cluster denotes the DAM. (B) Expression levels (heatmap) of representative marker genes (up-regulation in DAM: Cst7 and Lpl; and down-regulation in DAM: P2ry12 and Cx3cr1) in different microglia subclusters. (C) A predicted DAM-specific molecular network contains 227 protein–protein interactions (PPIs) connecting 72 proteins. Node sizes are proportional to their corresponding |log2FC| during differential expression analysis. (FC) Fold-change. Node (gene/protein) color is coded by known immune pathways from the Kyoto Encyclopedia of Genes and Genomes (KEGG) database. Edge color is coded by known experimental evidences of PPIs (Methods). Key immune modulators related to AD are highlighted by bold text.
We next performed differential expression analyses between DAM and HAM. As expected, 35 AD genes and microglia markers were differentially expressed in DAM compared to HAM in 5XFAD mice, including Apoe, Trem2, Cst7, Lpl, P2ry12, and Cx3cr1 (Supplemental Fig. S3A,B). We next reconstructed molecular networks (Fig. 2C; Supplemental Fig. S1C) for DAM based on snRNA-seq (snDAMnet) and scRNA-seq (scDAMnet) data sets, using the GPSnet algorithm (Cheng et al. 2019b). The snDAMnet includes 227 PPIs connecting 72 human gene products (e.g., BIN1, HCK, HSP90AA1, IL6ST, PAK1, PRKCD, and SYK) (Supplemental Table S4). We assembled AD-associated genes from multiple sources, including the GWAS catalog (Buniello et al. 2019) and experimental evidences from animal models and human studies (Piñero et al. 2017). We found that genes in snDAMnet were significantly enriched in AD-association (adjusted P-value [q] = 5.44 × 10−11, Fisher's exact test) (Supplemental Table S4), such as Adam10, Bin1, Cd33, and Mapk14. The scDAMnet contains 69 gene products (e.g., Axl, Cst7, Lyn, Mertk, and P2ry12) (Supplemental Table S5) involving 97 human PPIs. The scDAMnet is significantly enriched by 27 AD-associated genes (e.g., Apoe, Ccl3, Ctsd, Inpp5d, and Marcks, q = 1.56 × 10−8) (Supplemental Table S5) as well. We found that genes in DAMnets are significantly enriched in immune pathways (Supplemental Tables S4, S5), including multiple key immune modulators related to AD (Fig. 2C; Supplemental Fig. S1C). Last, we illustrated snDAMnet and scDAMnet across three selected immune pathways: fragment crystallizable (Fc) gamma receptor (R)-mediated phagocytosis, the chemokine signaling pathway, and Th17 cell differentiation (Supplemental Fig. S3C,D), as discussed below.
Fc gamma R-mediated phagocytosis
We identified 15 genes (such as Bin1, Prkcd, Syk, Inpp5d, and Hck) in the Fc gamma R-mediated phagocytosis pathway enriched by either snDAMnet or scDAMnet (Supplemental Tables S4, S5). Bridging integrator 1 (BIN1), a well-established risk gene for AD by the International Genomics of Alzheimer's Project, contains a microglia-specific enhancer and promoter encoded by a genome-wide significant AD variant rs6733839 (Medway and Morgan 2014). One possible role for BIN1 in DAM function may be gene regulation as a microglia-specific enhancer and promoter altered by rs6733839 (Corces et al. 2020). Spleen-associated tyrosine kinase (SYK) has also already been shown to play a role in AD pathological lesions and has been proposed as a possible drug target for AD (Schweig et al. 2017). Inositol polyphosphate-5-phosphatase D (INPP5D), identified as one of the genetic risk factors for late-onset AD, also affects AD pathology by regulating microglia (Rosenthal and Kamboh 2014).
Chemokine signaling pathway
Chemokine signaling is enriched in both snDAMnet and scDAMnet, and these two networks contain 13 genes, including Pak1, Ccl3, Ccl4, Ccr5, and Lyn (Supplemental Tables S4, S5). p21 (RAC1) activated kinase 1 (PAK1) is dysregulated in AD, and targeting the PAK signaling pathway has been proposed as a therapeutic strategy for AD (Ma et al. 2012). C-C motif chemokine ligand 3 and 4 (CCL3 and CCL4) and C-C motif chemokine receptor 5 (CCR5) (Guedes et al. 2018) have been shown to be up-regulated in adult human microglia or in mouse microglia exposed to the amyloid-β (Aβ) peptide. A recent study observed elevated activity of LYN proto-oncogene, Src family tyrosine kinase (LYN) in AD patients, and inhibiting LYN expression prevents Aβ-induced neuronal cell death, suggesting LYN as a potential therapeutic target for AD (Gwon et al. 2019).
Th17 cell differentiation
The T helper type 17 (Th17) cells are CD4+ T cells that promote a cell-mediated immune response against invading bacteria and fungi. We identified six genes (Ppp3ca, Hsp90aa1, Mapk14, Hif1a, Tgfbr2, and Il6st) in the Th17 cell differentiation pathway enriched by snDAMnet (Supplemental Table S4). With respect to mitogen-activated protein kinase 14 (MAPK14), a mouse model study suggested that inhibiting MAPK14 mitigates AD pathology (Alam and Scheper 2016). The transcription factor hypoxia inducible factor 1 subunit alpha (HIF1A) was involved in a variety of neurodegenerative diseases, including AD (Zhang et al. 2011). Heat shock protein 90 (HSP90), a chaperone protein, regulates tau pathology by forming macromolecular complexes with co-chaperones and inhibiting HSP90-mitigated tau pathology by proteasomal degradation (Campanella et al. 2018).
Discovery of DAA-specific molecular networks
We compared gene expression of 13 DAA cell markers among all nuclei clusters (Fig. 3A,B; Supplemental Fig. S4A). We found that a normalized nucleus abundance of DAA in 5XFAD mice is higher than that in WT mice (P = 9.79 × 10−3, t-test) (Supplemental Table S6; Supplemental Fig. S2C). The mDAAnet (Fig. 3C) includes 407 PPIs connecting 116 proteins (Supplemental Table S7). The mDAAnet contains 56 AD-associated genes (q = 1.84 × 10−22, Fisher's exact test) (Supplemental Table S7). A t-distributed stochastic neighbor embedding (t-SNE) plot of DAA and non-DAA nuclei are presented in Figure 4A (Supplemental Fig. S5A). The hDAAnet contains 16 PPIs connecting 10 proteins (Fig. 4B), including six AD-associated proteins (JUND, MAP1B, FOS, MFGE8, JUNB, and JUN, q = 8.69 × 10−4, Fisher's exact test) (Supplemental Table S8).
Figure 3.

Discovery of DAA-specific molecular networks in transgenic mouse model of AD. (A) T-distributed stochastic neighbor embedding (t-SNE) plot of clustering 7748 astrocyte nuclei. Red cluster denotes the DAA. (B) Stacked violin plot displaying the expression patterns of four representative genes (with the remaining nine genes in Supplemental Fig. S4A) across different astrocyte subclusters. (C) A predicted DAA-specific molecular network contains 407 protein–protein interactions (PPIs) connecting 116 gene products (proteins). Node sizes are proportional to their corresponding |log2FC| during differential expression analysis. Node color is coded by known immune pathways from the Kyoto Encyclopedia of Genes and Genomes (KEGG) database. Edge color is coded by experimental evidences of PPIs. Key immune modulators related to AD are highlighted by bold text.
Figure 4.

Discovery of DAA-specific molecular networks from single-nucleus RNA sequencing data of human brains with AD. (A) T-distributed stochastic neighbor embedding (t-SNE) plot of clustering 2119 astrocyte nuclei between AD patients and healthy controls. (B) An identified DAA-specific molecular network contains 16 protein–protein interactions (PPIs) connecting 10 gene products (proteins). (C) UMAP plot for 5599 astrocyte nuclei clustering analysis of brain entorhinal cortex (EC) regions among AD patients with different Braak stages. (D) UMAP plot of clustering 8348 astrocyte nuclei for brain superior frontal gyrus (SFG) regions among AD patients with different Braak stages. (E) An identified DAA-specific molecular network containing 43 protein–protein interactions (PPIs) connecting 26 gene products (proteins) for EC. (F) An identified DAA-specific molecular network containing 22 PPIs connecting 13 genes/proteins for SFG. Node sizes are proportional to their corresponding |log2FC|. Node color is coded by known immune pathways from the KEGG database. Edge color is coded by experimental evidences of PPIs. Key immune modulators related to AD are highlighted by bold text.
We further inspected human brain region–specific molecular networks for DAA (Supplemental Fig. S6). The uniform manifold approximation and projection (UMAP) plots of DAA and non-DAA nuclei are presented for two brain regions of AD patients, including entorhinal cortex (EC) and superior frontal gyrus (SFG) (Fig. 4C,D). The hDAAECnet contains 43 human PPIs connecting 26 proteins (Fig. 4E), including 11 AD-associated proteins (q = 3.77 × 10−4) (Supplemental Table S9). The hDAASFGnet contains 22 PPIs connecting 13 proteins (Fig. 4F), including eight AD-associated proteins (q = 1.22 × 10−4). Molecular networks (hDAAECnet and hDAASFGnet) between EC and SFG share nine proteins: DCLK2, HPSE2, HSP90AA1, HSPA1A, HSPA1B, HSPB1, ID2, JUN, and TNC (Fig. 4E,F). For two brain regions, there are no apparent differences of nucleus abundance percentage across different Braak stages for both DAA and non-DAA (Supplemental Tables S10–S12; Supplemental Fig. S2D–F).
We next performed functional pathway enrichment analysis and found that genes identified in DAA molecular networks were significantly enriched in multiple immune pathways (Supplemental Figs. S4B, S5B, S7A,B). We next turned to investigate gene functions using the two most significant immune pathways as examples: IL17 signaling pathway and antigen processing and presentation. We identified seven genes (NFKB1, CEBPB, MAPK1, HSP90AA1, FOS, JUND, and JUN) in the IL17 signaling pathway jointly enriched by all four DAA networks from both mouse models and AD patient brains (Supplemental Tables S7–S9). Nuclear factor kappa B subunit 1 (NFKB1) and NFKB inhibitor alpha (NFKBIA) control transcription of cytokines and chemokines in astrocytes and they commonly result in cellular damage or accelerate the production of Aβ in astrocytes (González-Reyes et al. 2017). Fos proto-oncogene, AP-1 transcription factor subunit (FOS), and Jun proto-oncogene, AP-1 transcription factor subunit (JUN) are transcriptional factors mediating functional roles in AD pathobiology (Anderson et al. 1994). There are three genes (HSP90AA1, HSPA1A, and HSPA1B) in the antigen processing and presentation pathway enriched in either hDAAECnet or hDAASFGnet (Supplemental Table S9). Heat shock protein 90 alpha family class A member 1 (HSP90AA1) has been previously linked to AD (Campanella et al. 2018). Both heat shock protein family A (Hsp70) member 1A (HSPA1A) (Evgen'ev et al. 2017) and heat shock protein family A (Hsp70) member 1B (HSPA1B) have been shown to regulate oxidative stress in either mouse model or human AD brains (Clarimón et al. 2003), suggesting their crucial roles in AD biology and possible treatment approaches.
Alzheimer's conserved molecular networks between microglia and astrocytes
We next compared the network relationship between DAM and DAA under the human interactome model (Methods). We only investigated DAM and DAA in transgenic mouse models because there is a lack of well-defined DAM in human AD brains. Using a network proximity measure (Methods), we found a statistically significant network-based relationship between DAM and DAA (Fig. 5A; Supplemental Table S13): (1) scDAMnet and mDAAnet (Z-score = −1.9, P = 0.029, permutation test), and (2) snDAMnet and mDAAnet (Z-score = −4.07, P < 0.001, permutation test). Mechanistically, we found eight overlapped genes (APOE, CALM2, CD9, CD63, CTSB, CTSD, IQGAP1, and LGALS3BP) and 11 commonly enriched immune pathways between DAM and DAA, such as B cell and T cell receptor signaling and Th17 cell differentiation (Supplemental Tables S4, S5, S7). For example, Cd9 and Lgals3bp are differentially expressed in both DAM and DAA of mouse models (Fig. 5B). Galectin 3 binding protein (LGALS3BP), a secreted glycoprotein, has been reported as a potential marker in aging (Costa et al. 2020). Two immune pathways (Fc gamma R-mediated phagocytosis and chemokine signaling) are also enriched in both DAMnets and mDAAnet. Except for LGALS3BP and CD9 (Fig. 5B), another seven proteins (AXL, CKB, CSF1R, FGR, HIF1A, INPP5D, and RPLP2) are also shared between scDAMnet and snDAMnet (Fig. 5A). The immune pathway platelet activation is uniquely enriched in snDAMnet (Supplemental Table S4); yet, IL17 signaling pathway and Th1 and Th2 cell differentiation are exclusively enriched in mDAAnet (Supplemental Table S7). In summary, microglia and astrocytes may trigger neuroinflammation in AD by a specific molecular network manner.
Figure 5.

Comparison of molecular networks between DAA and microglia (DAM). (A) Visualization of interplays between DAM and DAA molecular networks in the human protein–protein interactome network model. (B) Expression levels of Lgals3bp and Cd9 for homeostatic associated microglia (HAM) versus DAM and non-DAA versus DAA. The adjusted P-value (q) is computed using the MAST R package (Supplemental Material). All details for gene differential expression analyses are provided in Supplemental Tables S4, S5, and S7.
Metabolites trigger molecular networks between astrocyte and microglia
AD is a pervasive metabolic disorder associated with altered immune responses (Mahajan et al. 2020). We found that metabolic genes from the KEGG (Kanehisa et al. 2017) have a closer network relationship with DAM and DAA networks in the human interactome (Supplemental Table S13). We next investigated whether metabolites trigger network perturbation between DAM and DAA under the human protein–protein interactome model. We constructed a network with 373,320 edges (26,990 metabolite-enzyme associations and 346,330 PPIs). We assembled 155 AD-related metabolites supported by experimental evidences (Supplemental Table S14) and then reconstructed a subnetwork consisting of 266 AD-related metabolites and enzymes (Fig. 6A; Supplemental Fig. S8A).
Figure 6.

A metabolite-triggered molecular network between DAA and microglia (DAM). (A) A highlighted subnetwork of the metabolite-enzyme network between DAM and DAA in the human protein–protein interactome network model. (FC) Fold-change. (B,C) Expression of Ctsb is significantly elevated in DAM (GSE98969) (B) and DAA (GSE143758) (C) compared to homeostatic associated microglia (HAM) and non-DAA, respectively. (D) Expression of Spp1 is significantly elevated in DAM (GSE98969) compared with HAM. Each dot represents one cell/nucleus. (E) Spearman's correlation analysis shows that Spp1 and Pld3 have a slight coordinated change trends in DAM. Gene expression is counted by the average unique molecular identifier (UMI) count.
We found 77 enzymes involved in the AD-related metabolites: (1) 50 enzymes from DAM; (2) 30 enzymes from DAA, and (3) three enzymes (CTSB, CTSD and APOE) shared between DAM and DAA (Supplemental Fig. S8B; Supplemental Table S14). Ctsb, encoding cathepsin B in catabolism and immune resistance (Wu et al. 2017), has elevated expression (Fig. 6B,C) in both DAM (fold-change [FC] = 2.48, q = 8.89 × 10−84) and DAA (FC = 2.14, q = 3.95 × 10−43) of mouse models. Pathway enrichment analysis revealed that 77 enzymes were enriched in metabolic homeostasis (e.g., glycolysis and gluconeogenesis) and immune signaling pathways (including IL3 and IL5) (Supplemental Fig. S8C).
Using a betweenness centrality measure (Supplemental Table S14), we found that fatty acids and amino acids (Fig. 6A) were two primary types of metabolites involved in molecular networks between DAM and DAA. For example, Spp1 (Shan et al. 2012) and Fos (Sun et al. 2017), two cellular molecules that promote chronic inflammatory diseases, are significantly more expressed in both DAM (FC = 5.35, q = 5.51 × 10−56) (Fig. 6D) and DAA (FC = 1.92, q = 1.09 × 10−49) compared to HAM and non-DAA, respectively. Elaidic acid shows the largest centrality among all metabolites and is connected with SPP1 and CD44 through involvement in fatty acid metabolism, including phospholipase D family member 3 (PLD3) and galactosidase beta 1 (GLB1) (Kim et al. 2010; Hsieh et al. 2017). Coexpression analysis reveals a slight correlation of Spp1 and Pld3 in DAM (Spearman's correlation r = 0.48, P = 0.06, t-test) (Fig. 6E). Meanwhile, arachidonic acid and palmitic acid, two long-chain fatty acids that have well-documented effects in inducing inflammatory responses (Freigang et al. 2013), are also involved in both DAA and DAM (Fig. 6A). In summary, these findings suggest functional roles of cellular metabolites (including fatty acids and amino acids) in the immune interplay of astrocyte and microglia in AD. Further experimental validations are warranted to verify network-based astrocyte-/microglia-associated metabolism findings.
Network-based discovery of repurposable drugs
We next turned to identify drug candidates by specifically targeting molecular networks of DAM and DAA. As shown in Figure 1, we assembled drug-gene signatures in human cell lines from the connectivity map (CMap) database (Lamb et al. 2006). We posited that if a drug significantly reverses dysregulated gene expression of DAM or DAA, this drug may have potential in treating AD. For gene set enrichment analysis (GSEA), we used enrichment score (ES) > 0 and q < 0.05 as a cutoff to prioritize drug candidates. For 1309 drugs from the CMap (Lamb et al. 2006), we obtained 27, 53, 28, 33, and 94 candidate drugs (ES > 0 and q < 0.05) for snDAMnet, scDAMnet, hDAAECnet, hDAASFGnet, and hDAAnet, respectively (Supplemental Table S15). As shown in Figure 7A, we found that network-predicted drugs parsed into seven pharmacological categories: anti-inflammatory, immunosuppressive, adrenergic beta receptor agonists, adrenergic alpha-antagonists, antihypertensive, antineoplastic, and others. Tretinoin, also known as all-trans retinoic acid (ATRA), an FDA-approved drug for acute promyelocytic leukemia (APL) (Warrell et al. 1991), is one of our highest predictions (Supplemental Table S15). Treatment with tretinoin reduced microglia and astrocyte activities and enhanced cognitive capabilities (Ding et al. 2008) in a mouse model. Mechanistically, tretinoin directly targets mitogen-activated protein kinase 1 (MAPK1), LYN, and FGR in the scDAMnet (Fig. 7B). Salbutamol, a selective beta2-adrenergic receptor agonist in treating asthma, is a highly predicted candidate on snDAMnet (Supplemental Table S15). In vitro studies showed that salbutamol was a direct inhibitor of tau filament formation (Townsend et al. 2020). As shown in Figure 7C, salbutamol interacts with three immune gene products (PRKCD, GRB2, and MAPK14) in snDAMnet, consistent with mechanistic observations in AD (Russo et al. 2002). Altogether, these network-predicted drugs (Supplemental Table S15) offer potential candidate compounds to be tested in nonclinical models or clinical trials in the future.
Figure 7.
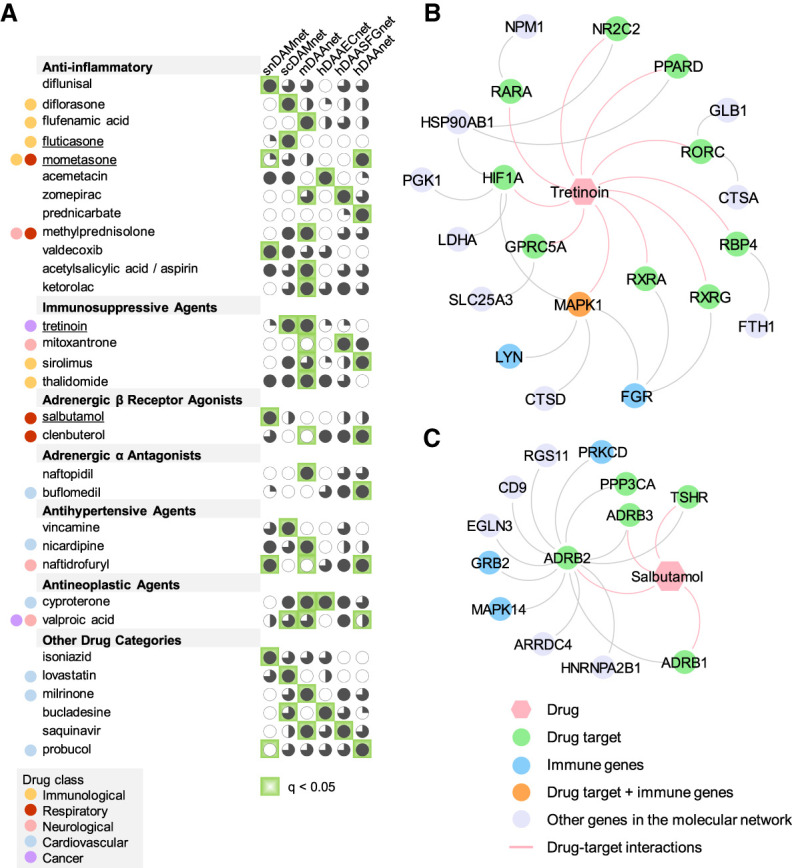
Network-based discovery of repurposable drug candidates for AD by specifically reversing gene expression of DAM and DAA. (A) Selected drugs that specifically target five different DAM or DAA molecular networks. Drug are grouped by five different classes (immunological, respiratory, neurological, cardiovascular, and cancer) (Supplemental Table S12) defined by the first-level of the Anatomical Therapeutic Chemical (ATC) codes. Four high-confidence drugs (fluticasone, mometasone, salbutamol, and tretinoin) were highlighted. (B,C) Proposed mechanism-of-actions for two selected drugs (tretinoin [B] and salbutamol [C]) by drug-target network analysis.
Validating likely causal drug-AD associations in patient data
We next selected drug candidates using subject matter expertise based on a combination of factors: (1) strength of the predicted associations; (2) novelty of the predicted associations with established mechanisms-of-action; (3) literature-based evidence in support of prediction; and (4) availability of sufficient patient data for meaningful evaluation (exclusion of infrequently used medications). Applying these criteria resulted in fluticasone, an approved glucocorticoid receptor (NR3C1) agonist for several inflammation-related indications (Lumry 1999). As shown in Figure 7A, we found anti-inflammatory agents are the biggest network-predicted drug class. We thus evaluated fluticasone on AD by analyzing 7.23 million US commercially insured individuals from the MarketScan Medicare supplemental database. We conducted two cohort analyses to evaluate the predicted association using state-of-the-art pharmacoepidemiologic analysis: (1) fluticasone versus a matched control population (non-fluticasone user), and (2) fluticasone versus mometasone (a stronger NR3C1 agonist) (Lumry 1999). For each comparison, we estimated the unstratified Kaplan–Meier curves and conducted propensity score–stratified log-rank tests using the Cox regression model.
We found that individuals taking fluticasone were at significantly decreased risk for development of AD (hazard ratio [HR] = 0.86, 95% confidence interval [CI] 0.83–0.89, P < 1.0 × 10−8) (Fig. 8A,C). Propensity score–stratified cohort studies confirmed that usage of mometasone (a stronger NR3C1 agonist) are significantly associated with reduced risk of AD compared to fluticasone (HR = 0.74, 95% CI 0.68-0.81, P < 1.0 × 10−8) (Fig. 8B,C). Another independent database, FDA MedWatch Adverse Events Database, revealed that the combination of fluticasone and ibuprofen could be a therapeutic option for AD (Lehrer and Rheinstein 2018). Fluticasone and mometasone are approved steroids to treat asthma and various allergies with anti-inflammatory, antipruritic, and vasoconstrictive properties (Lumry 1999). Previous studies showed crucial roles of NR3C1 in AD (de Quervain et al. 2004; Canet et al. 2018), suggesting possible protective effects of fluticasone and mometasone on AD (Fig. 8A–C) via modulating the glucocorticoid signaling.
Figure 8.
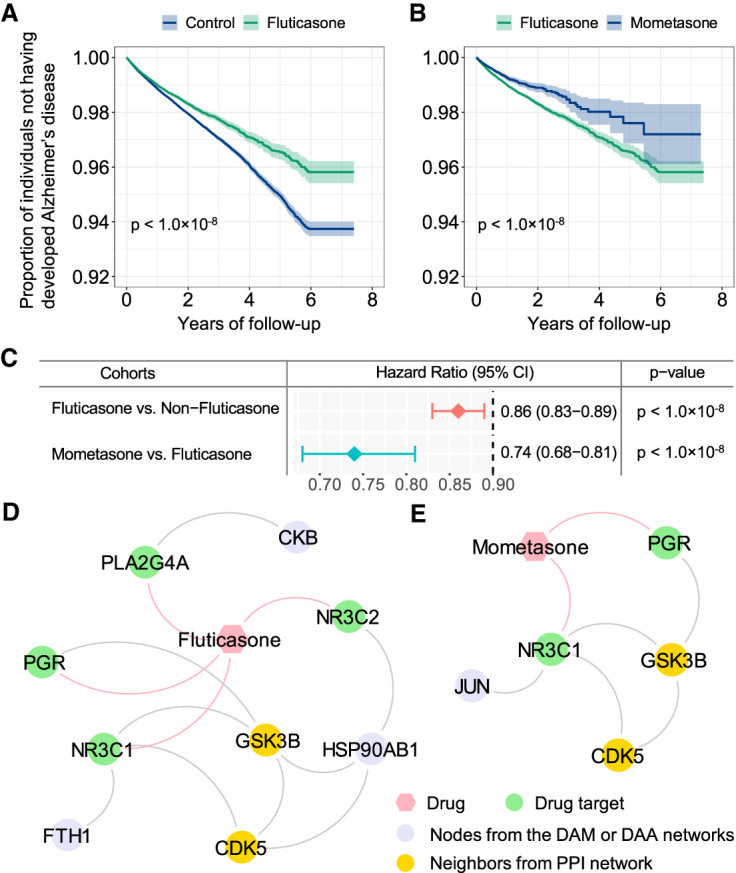
Retrospective case-control analysis reveals that usage of fluticasone and mometasone is significantly associated with reduced likelihood of AD in a longitudinal patient database with 7.23 million subjects. Two comparison analyses were conducted: (A) fluticasone (a glucocorticoid receptor agonist) versus a matched control population (non-fluticasone users), and (B) mometasone (a stronger glucocorticoid receptor agonist) versus fluticasone. For each comparator, we estimated the unstratified Kaplan–Meier curves, conducted propensity score–stratified (n strata = 10) rank test and applied Cox models after adjusting all possible confounding factors, including age, gender, race, and disease comorbidities (Supplemental Table S16). (C) Hazard ratios (HRs) and 95% confidence interval (CI) for two drug cohort studies. Propensity score–stratified Cox-proportional hazards models were used to conduct statistical inference for the hazard ratios. (D,E) Proposed mechanism-of-action for treatment of AD by fluticasone and mometasone using drug-target network analysis.
To further infer the potential mechanisms-of-action of fluticasone and mometasone in AD, we next integrated networks from drug-target interactions, predicted networks of DAM and DAA, and human PPIs. Network analysis shows that fluticasone and mometasone indirectly target glycogen synthase kinase 3 beta (GSK3B) and cyclin-dependent kinase 5 (CDK5) via PPIs in molecular networks of DAM and DAA (Fig. 8D,E). Lipopolysaccharide-stimulation increased inflammatory responses in microglia by activating phosphorylation of CDK5 (Na et al. 2015). CDK5R1 signaling plays a crucial role in microglial phagocytosis of Aβ (Ma et al. 2013). GSK3B inhibitors reduce microglial migration, inflammation, and inflammation-induced neurotoxicity (Huang and Mucke 2012). Altogether, these observations suggest that fluticasone and mometasone have potentially protective effects on AD by reducing glucocorticoid signaling and CDK5/GSK3B-mediated inflammation on microglia or astrocytes (Fig. 8). Further experimental validation on the network-inferred mechanism-of-action is warranted.
Discussion
We acknowledged several potential limitations in the current study. Although two snRNA-seq and scRNA-seq data sets of DAM present consistent expression patterns (Supplemental Tables S4, S5), snDAMnet and scDAMnet showed a small overlap of differentially expressed genes. There are several possible explanations. Single-cell and single-nucleus may generate different cell abundances during cell processing. The procedure for preparing single-cell suspensions from fresh samples may alter the gene expression profiles of individual cells and change the derived cell type proportions because some cells are more vulnerable to cell dissociation protocols (Lake et al. 2016).
The network proximity analyses show significant network-based relationships between DAM and DAA (Supplemental Table S13), including immune pathways enriched by both DAM and DAA. These findings provide insights into intercellular communication between microglia and astrocytes; yet, systematic identification of ligand-receptor interactions connecting cell surface proteins of DAM and DAA may identify previously unrecognized mechanisms regarding intercellular communication between microglia and astrocytes in AD and offer novel drug targets for development of anti-inflammatory treatments. There was less significant association between human and mouse molecular networks (DAM vs. DAA) (Supplemental Table S13), consistent with different immune responses of AD brains between human and mouse models (Hemonnot et al. 2019). Another study reported distinct gene signatures of DAM between 5XFAD mouse model and human AD brains (Keren-Shaul et al. 2017); furthermore, up-regulation of two mouse DAM marker genes (Lpl and Cst7) cannot be detected in human AD brains (Zhou et al. 2020b). In addition, divergence of mouse and human cortex may influence network-based findings presented here (Hodge et al. 2019). Development of advanced network-based methodologies to identify conserved cell types and the underlying molecular networks between human and animal models from evolutionary perspectives is needed in the future. Finally, potential literature biases regarding PPIs, incompleteness of networks, and small sample size of sn/scRNA-seq data sets (Supplemental Table S1) may influence our network-based findings as well.
In summary, we presented a network-based methodology that incorporates large-scale snRNA-seq and scRNA-seq data from either mouse models or AD patient brains, human PPIs, enzyme-metabolite associations, and drug target networks, along with large-scale patient-level data observation. We showed that molecular networks derived from DAM and DAA are significantly enriched for various well-known immune pathways and AD-related pathobiological pathways. We showed that the identified molecular networks from DAM and DAA offer potential targets for drug repurposing, and we validated two network-predicted drugs (fluticasone and mometasone) in reducing risk of AD using large-scale, longitudinal patient data. In summary, we believe that the network-based methodology presented here, if broadly applied, would significantly catalyze innovation in AD drug discovery by utilizing the large-scale single-cell/nucleus omics data.
Methods
Resources of single-cell/nucleus RNA sequencing data
The complete sc/snRNA-seq data sets used in this study (Supplemental Table S1) are available from the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) database under accession numbers GSE98969 (Keren-Shaul et al. 2017), GSE140511 (Zhou et al. 2020b), GSE143758 (Habib et al. 2020), GSE147528 (Leng et al. 2021), and GSE138852 (Grubman et al. 2019). One scRNA-seq data set (GSE98969) contains C57BL/6 (whole brain, n = 16) and 5XFAD (n = 16) mice, including 12,288 sequenced cells (Keren-Shaul et al. 2017). Two of four snRNA-seq data sets were collected from mouse samples as well (GSE140511 and GSE143758). Data set GSE140511 (Zhou et al. 2020b) contained four types of transgenic male mouse models, including C57BL/6, 5XFAD, Trem2 knockout C57BL/6, and Trem2 knockout 5XFAD. In this study, we considered the 7-mo mouse models, which in total sequenced 90,647 nuclei. The second mouse nucleus data set (GSE143758) contains two transgenic mouse models (C57BL/6 and 5XFAD) from both hippocampus and cortex regions (Habib et al. 2020). We utilized in total 55,367 nuclei data from the 7-mo hippocampus mouse models: (a) 5XFAD (n = 5), and (b) C57BL/6 (n = 5) (Habib et al. 2020). A human snRNA-seq data set (Leng et al. 2021) contains 10 male frozen postmortem human brain tissues for both superior frontal gyrus (63,608 nuclei) and entorhinal cortex (42,528 nuclei), including astrocytes, excitatory neurons, inhibitory neurons, microglia, oligodendrocytes, oligodendrocyte progenitor cells (OPCs), and endothelial cells (GSE147528) (Leng et al. 2021). A new human snRNA-seq data set (Grubman et al. 2019) containing 12 frozen postmortem human brain tissues (n = 6 AD case and n = 6 healthy controls [GSE138852]) from entorhinal cortex regions was further used, which covers astrocyte, microglia, neuron, oligodendrocyte, OPC, and endothelial cell types. All statistical analyses were conducted in R (R Core Team 2020), and the details for bioinformatics analysis of each data set were provided in the Supplemental Material.
Building human protein–protein interactome
To build the comprehensive human interactome from the most contemporary data available, we assembled 18 commonly used PPI databases with experimental evidence: (1) binary PPIs tested by high-throughput yeast-two-hybrid (Y2H) systems (Luck et al. 2020); (2) kinase-substrate interactions; (3) signaling networks; (4) binary PPIs from three-dimensional protein structures; (5) protein complexes data; and (6) carefully literature-curated PPIs. In total, 351,444 PPIs connecting 17,706 unique proteins were used in this study (Supplemental Material) and are freely available at https://alzgps.lerner.ccf.org.
Description of GPSnet
GPSnet (Cheng et al. 2019b) takes two inputs: node (gene) scores and a background PPI network. The node score was defined as follows: for differentially expressed genes (DEGs) with q ≤ 0.05, and the node scores denote absolute value of log2FC. To generate a network module, GPSnet starts with a randomly selected gene/protein (node). During each iteration, one of the candidate genes (first-order neighbor) that is satisfying the following two conditions at the same time will be added: (1) P-value of the connectivity significance P(i) (Eq. 1) is <0.01; and (2) the updated module score is greater than the current one (Eq. 2). We repeated steps (1) and (2) until no more genes (nodes) can be added to generate each raw module. In this study, we built ∼100,000 raw modules ranked by module scores. For each raw module, the corresponding module score can be computed (Eq. 2), and all raw modules are ranked in decreasing module score order. The protein frequency is defined based on truncated raw modules. We generated the final network modules by assembling top ranked raw modules (Supplemental Tables S4, S5, S7–S9).
| (1) |
| (2) |
where N denotes all proteins/genes in the PPI, n represents numbers of nodes in the module, dn is the numbers of neighbors of gene i in current module, di is the degree of gene i, MSn+1(i) denotes the updated module score if adding node i, s(i) denotes the score of node i, M denotes the current module, and μ is the average node score of all genes with respect to the PPI network.
Network proximity
To quantify the relationships of two molecular networks (DAM vs. DAA) in the human interactome, we adopted the shortest-based network proximity measure (Cheng et al. 2019a) as follows:
| (3) |
where d(x,y) is the shortest path length between gene x and y from gene sets X and Y, respectively. To evaluate whether such proximity was significant, the computed network proximity is transferred into Z-score form as shown in the following:
| (4) |
Here, μd and σd are the mean and standard deviation of permutation test with 1000 random experiments. In each random experiment, two random subnetworks Xr and Yr are constructed with the same numbers of nodes and degree distribution as the given two subnetworks X and Y, separately, in the PPI network.
Network analysis of metabolite–enzyme associations
We assembled 155 AD-related metabolites from 12 studies (Supplemental Table S14) and the Human Metabolome Database (HMDB) (Wishart et al. 2018). All metabolites were identified in AD-related human samples, including brain tissues, cerebrospinal fluid, and blood. All these results are freely available in our AlzGPS (Zhou et al. 2021) database (https://alzgps.lerner.ccf.org). We mapped 240 DAM and DAA disease module genes and the 155 AD-related metabolites to the network and computed the maximal subgraph. Finally, we computed the network paths connecting the DAM and DAA gene products on the network as well as the betweenness centrality of each vertex (Supplemental Material).
Gene set enrichment analysis (GSEA)
We assembled drug-gene signatures from the CMap database containing 6100 expression profiles relating 1309 compounds (Lamb et al. 2006). We utilized GSEA algorithm to predict drugs across each molecular network of DAM and DAA. Detailed descriptions of GSEA have been provided in our recent study (Zhou et al. 2020a) and the Supplemental Material.
Enrichment analysis
All pathway and disease enrichment analyses were conducted using either KEGG 2019 Mouse or KEGG 2019 Human and DisGeNET (Piñero et al. 2017) from Enrichr (Kuleshov et al. 2016), respectively.
Pharmacoepidemiologic validation
We used the MarketScan Medicare Claims database from 2012 to 2017 for the pharmacoepidemiologic analysis. This database includes individual-level procedure codes, diagnosis codes, and pharmacy claim data for 7.23 million patients. Pharmacy prescriptions of fluticasone and mometasone were identified using RxNorm and National Drug Code (NDC). For an individual exposed to fluticasone and mometasone, a drug episode was defined as from drug initiation to drug discontinuation. A control cohort was selected from patients who were not exposed to fluticasone. The disease outcome defined by the International Classification of Disease (ICD) codes (Supplemental Table S16) was time from drug initiation to diagnosis of AD. The survival curves for time to AD were estimated using a Kaplan–Meier estimator approach. We used the large number of covariates generated throughout the process to address clinical scenarios evaluated in each drug cohort. Propensity score–stratified survival analyses were conducted to investigate the risk of AD between fluticasone users and non-fluticasone users, as well as fluticasone users and mometasone users. Specifically, for each comparison, the propensity score of taking fluticasone was estimated by using a logistic regression model, in which the covariates included age, gender, geographical location, and disease comorbidities. Further, propensity score–stratified Cox-proportional hazards models were used to conduct statistical inference for the hazard ratios (HRs) of developing AD between two cohorts. All details are provided in the Supplemental Material.
Software availability
All codes written for and used in this study are available from GitHub (https://github.com/ChengF-Lab/alzGPSnet) and as Supplemental Code.
Supplementary Material
Acknowledgments
This work was supported by the National Institute on Aging (NIA) under Award Number R01AG066707 and 3R01AG066707-01S1 to F.C. This work was supported in part by the NIA under Award Number R56AG063870 to F.C., L.M.B., and J.B.L. A.A.P., L.M.B., J.C., J.B.L., and F.C. are supported together by the Translational Therapeutics Core of the Cleveland Alzheimer's Disease Research Center (National Institutes of Health/NIA: 1 P30 AGO62428-01). A.A.P. is also supported by the Brockman Foundation, American Heart Association Project 19PABH134580006-AHA/Allen Initiative in Brain Health and Cognitive Impairment, the Elizabeth Ring Mather and William Gwinn Mather Fund, S. Livingston Samuel Mather Trust, G.R. Lincoln Family Foundation, Wick Foundation, Gordon and Evie Safran, the Leonard Krieger Fund of the Cleveland Foundation, the Maxine and Lester Stoller Parkinson's Research Fund, and Louis Stokes Veterans Administration (VA) Medical Center resources and facilities. J.B.L. is supported by the Alzheimer's Drug Discovery Foundation, Cleveland Clinic Lerner Research Institute, Department of Defense, Douglas Herthel DVM Memorial Research Fund, Eisai, GE Healthcare, Jane and Lee Seidman Fund, Lewy Body Dementia Association, Michael J. Fox Foundation, National Institutes of Health (NIH)/NIA funds (P30 AG062428, UO1 NS100610, RO1 AG022304, RO1 AG0577552, RO3 AG063235, R21 AG064271, P20 AG068053), and Sanofi. J.C. is supported by Keep Memory Alive (KMA), National Institute of General Medical Sciences grant P20GM109025, National Institute of Neurological Disorders and Stroke grant U01NS093334, and NIA grant R01AG053798.
Author contributions: F.C. conceived the study. J.X., P.Z., and Y. Huang performed all experiments and data analysis. L.M.B., J.L., C.-W.C., L.L., Y. Hou, Y.Z., A.A.P., J.B.L., and J.C. discussed and interpreted all results. F.C., J.X., P.Z., Y. Huang, and J.C. wrote the manuscript. All authors critically revised the manuscript and gave final approval.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.272484.120.
Freely available online through the Genome Research Open Access option.
Competing interest statement
J.C. has provided consultation to Acadia, Actinogen, Alkahest, Alzheon, Annovis, Avanir, Axsome, Biogen, BioXcel, Cassava, Cerecin, Cerevel, Cortexyme, Cytox, EIP Pharma, Eisai, Foresight, GemVax, Genentech, Green Valley, Grifols, Karuna, Merck, Novo Nordisk, Otsuka, Resverlogix, Roche, Samumed, Samus, Signant Health, Suven, Third Rock, and United Neuroscience pharmaceutical and assessment companies. J.C. has stock options in ADAMAS, AnnovisBio, MedAvante, and BiOasis. J.B.L. has received consulting fees from Acadia, Biogen, Eisai, GE Healthcare, and Sunovion. The other authors have no competing interests.
References
- Alam J, Scheper W. 2016. Targeting neuronal MAPK14/p38α activity to modulate autophagy in the Alzheimer disease brain. Autophagy 12: 2516–2520. 10.1080/15548627.2016.1238555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alzheimer's Association. 2016. 2016 Alzheimer's disease facts and figures. Alzheimers Dement 12: 459–509. 10.1016/j.jalz.2016.03.001 [DOI] [PubMed] [Google Scholar]
- Anderson AJ, Cummings BJ, Cotman CW. 1994. Increased immunoreactivity for Jun- and Fos-related proteins in Alzheimer's disease: association with pathology. Exp Neurol 125: 286–295. 10.1006/exnr.1994.1031 [DOI] [PubMed] [Google Scholar]
- Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, McMahon A, Morales J, Mountjoy E, Sollis E, et al. 2019. The NHGRI-EBI GWAS catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res 47: D1005–D1012. 10.1093/nar/gky1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campanella C, Pace A, Caruso Bavisotto C, Marzullo P, Marino Gammazza A, Buscemi S, Palumbo Piccionello A. 2018. Heat shock proteins in Alzheimer's disease: role and targeting. IJMS 19: 2603. 10.3390/ijms19092603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canet G, Chevallier N, Zussy C, Desrumaux C, Givalois L. 2018. Central role of glucocorticoid receptors in Alzheimer's disease and depression. Front Neurosci 12: 739. 10.3389/fnins.2018.00739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao W, Zheng H. 2018. Peripheral immune system in aging and Alzheimer's disease. Mol Neurodegener 13: 51. 10.1186/s13024-018-0284-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng F, Kovács IA, Barabási AL. 2019a. Network-based prediction of drug combinations. Nat Commun 10: 1197. 10.1038/s41467-019-09186-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng F, Lu W, Liu C, Fang J, Hou Y, Handy DE, Wang R, Zhao Y, Yang Y, Huang J, et al. 2019b. A genome-wide positioning systems network algorithm for in silico drug repurposing. Nat Commun 10: 3476. 10.1038/s41467-019-10744-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarimón J, Bertranpetit J, Boada M, Tàrraga L, Comas D. 2003. HSP70-2 (HSPA1B) is associated with noncognitive symptoms in late-onset Alzheimer's disease. J Geriatr Psychiatry Neurol 16: 146–150. 10.1177/0891988703256051 [DOI] [PubMed] [Google Scholar]
- Corces MR, Shcherbina A, Kundu S, Gloudemans MJ, Frésard L, Granja JM, Louie BH, Eulalio T, Shams S, Bagdatli ST, et al. 2020. Single-cell epigenomic analyses implicate candidate causal variants at inherited risk loci for Alzheimer's and Parkinson's diseases. Nat Genet 52: 1158–1168. 10.1038/s41588-020-00721-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa J, Pronto-Laborinho A, Pinto S, Gromicho M, Bonucci S, Tranfield E, Correia C, Alexandre BM, de Carvalho M. 2020. Investigating LGALS3BP/90 K glycoprotein in the cerebrospinal fluid of patients with neurological diseases. Sci Rep 10: 5649. 10.1038/s41598-020-62592-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings JL, Morstorf T, Zhong K. 2014. Alzheimer's disease drug-development pipeline: few candidates, frequent failures. Alzheimer's Res Ther 6: 37. 10.1186/alzrt269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Quervain DJF, Poirier R, Wollmer MA, Grimaldi LME, Tsolaki M, Streffer JR, Hock C, Nitsch RM, Mohajeri MH, Papassotiropoulos A. 2004. Glucocorticoid-related genetic susceptibility for Alzheimer's disease. Hum Mol Genet 13: 47–52. 10.1093/hmg/ddg361 [DOI] [PubMed] [Google Scholar]
- Desale SE, Chinnathambi S. 2020. Role of dietary fatty acids in microglial polarization in Alzheimer's disease. J Neuroinflammation 17: 93. 10.1186/s12974-020-01742-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Qiao A, Wang Z, Goodwin JS, Lee ES, Block ML, Allsbrook M, McDonald MP, Fan GH. 2008. Retinoic acid attenuates β-amyloid deposition and rescues memory deficits in an Alzheimer's disease transgenic mouse model. J Neurosci 28: 11622–11634. 10.1523/JNEUROSCI.3153-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evgen'ev MB, Krasnov GS, Nesterova IV, Garbuz DG, Karpov VL, Morozov AV, Snezhkina AV, Samokhin AN, Sergeev A, Kulikov AM, et al. 2017. Molecular mechanisms underlying neuroprotective effect of intranasal administration of human Hsp70 in mouse model of Alzheimer's disease. J Alzheimers Dis 59: 1415–1426. 10.3233/JAD-170398 [DOI] [PubMed] [Google Scholar]
- Fang J, Pieper AA, Nussinov R, Lee G, Bekris L, Leverenz JB, Cummings J, Cheng F. 2020. Harnessing endophenotypes and network medicine for Alzheimer's drug repurposing. Med Res Rev 40: 2386–2426. 10.1002/med.21709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freigang S, Ampenberger F, Weiss A, Kanneganti TD, Iwakura Y, Hersberger M, Kopf M. 2013. Fatty acid–induced mitochondrial uncoupling elicits inflammasome-independent IL-1α and sterile vascular inflammation in atherosclerosis. Nat Immunol 14: 1045–1053. 10.1038/ni.2704 [DOI] [PubMed] [Google Scholar]
- Ginhoux F, Prinz M. 2015. Origin of microglia: current concepts and past controversies. Cold Spring Harb Perspect Biol 7: a020537. 10.1101/cshperspect.a020537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Reyes RE, Nava-Mesa MO, Vargas-Sánchez K, Ariza-Salamanca D, Mora-Muñoz L. 2017. Involvement of astrocytes in Alzheimer's disease from a neuroinflammatory and oxidative stress perspective. Front Mol Neurosci 10: 427. 10.3389/fnmol.2017.00427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubman A, Chew G, Ouyang JF, Sun G, Choo XY, McLean C, Simmons RK, Buckberry S, Vargas-Landin DB, Poppe D, et al. 2019. A single-cell atlas of entorhinal cortex from individuals with Alzheimer's disease reveals cell-type-specific gene expression regulation. Nat Neurosci 22: 2087–2097. 10.1038/s41593-019-0539-4 [DOI] [PubMed] [Google Scholar]
- Guedes JR, Lao T, Cardoso AL, El Khoury J. 2018. Roles of microglial and monocyte chemokines and their receptors in regulating Alzheimer's disease-associated amyloid-β and tau pathologies. Front Neurol 9: 549. 10.3389/fneur.2018.00549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwon Y, Kim SH, Kim HT, Kam TI, Park J, Lim B, Cha H, Chang HJ, Hong YR, Jung YK. 2019. Amelioration of amyloid β-FcγRIIb neurotoxicity and tau pathologies by targeting LYN. FASEB J 33: 4300–4313. 10.1096/fj.201800926R [DOI] [PubMed] [Google Scholar]
- Habib N, McCabe C, Medina S, Varshavsky M, Kitsberg D, Dvir-Szternfeld R, Green G, Dionne D, Nguyen L, Marshall JL, et al. 2020. Disease-associated astrocytes in Alzheimer's disease and aging. Nat Neurosci 23: 701–706. 10.1038/s41593-020-0624-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert LE, Beckett LA, Scherr PA, Evans DA. 2001. Annual incidence of Alzheimer disease in the United States projected to the years 2000 through 2050. Alzheimer Dis Assoc Disord 15: 169–173. 10.1097/00002093-200110000-00002 [DOI] [PubMed] [Google Scholar]
- Hemonnot AL, Hua J, Ulmann L, Hirbec H. 2019. Microglia in Alzheimer disease: well-known targets and new opportunities. Front Aging Neurosci 11: 233. 10.3389/fnagi.2019.00233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge RD, Bakken TE, Miller JA, Smith KA, Barkan ER, Graybuck LT, Close JL, Long B, Johansen N, Penn O, et al. 2019. Conserved cell types with divergent features in human versus mouse cortex. Nature 573: 61–68. 10.1038/s41586-019-1506-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh P, Hallmark B, Watkins J, Karafet TM, Osipova LP, Gutenkunst RN, Hammer MF. 2017. Exome sequencing provides evidence of polygenic adaptation to a fat-rich animal diet in indigenous Siberian populations. Mol Biol Evol 34: 2913–2926. 10.1093/molbev/msx226 [DOI] [PubMed] [Google Scholar]
- Huang Y, Mucke L. 2012. Alzheimer mechanisms and therapeutic strategies. Cell 148: 1204–1222. 10.1016/j.cell.2012.02.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha MK, Jo M, Kim JH, Suk K. 2019. Microglia-astrocyte crosstalk: an intimate molecular conversation. Neuroscientist 25: 227–240. 10.1177/1073858418783959 [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. 2017. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res 45: D353–D361. 10.1093/nar/gkw1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, et al. 2017. A unique microglia type associated with restricting development of Alzheimer's disease. Cell 169: 1276–1290.e17. 10.1016/j.cell.2017.05.018 [DOI] [PubMed] [Google Scholar]
- Kim MJ, Wainwright HC, Locketz M, Bekker LG, Walther GB, Dittrich C, Visser A, Wang W, Hsu FF, Wiehart U, et al. 2010. Caseation of human tuberculosis granulomas correlates with elevated host lipid metabolism. EMBO Mol Med 2: 258–274. 10.1002/emmm.201000079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. 2016. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44: W90–W97. 10.1093/nar/gkw377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake BB, Ai R, Kaeser GE, Salathia NS, Yung YC, Liu R, Wildberg A, Gao D, Fung HL, Chen S, et al. 2016. Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science 352: 1586–1590. 10.1126/science.aaf1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN, et al. 2006. The connectivity map: using gene-expression signatures to connect small molecules, genes, and disease. Science 313: 1929–1935. 10.1126/science.1132939 [DOI] [PubMed] [Google Scholar]
- Lehrer S, Rheinstein PH. 2018. Alzheimer's disease and intranasal fluticasone propionate in the FDA medWatch adverse events database. J Alzheimers Dis Rep 2: 111–115. 10.3233/ADR-170033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng K, Li E, Eser R, Piergies A, Sit R, Tan M, Neff N, Li SH, Rodriguez RD, Suemoto CK, et al. 2021. Molecular characterization of selectively vulnerable neurons in Alzheimer's disease. Nat Neurosci 24: 276–287. 10.1038/s41593-020-00764-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luck K, Kim DK, Lambourne L, Spirohn K, Begg BE, Bian W, Brignall R, Cafarelli T, Campos-Laborie FJ, Charloteaux B, et al. 2020. A reference map of the human binary protein interactome. Nature 580: 402–408. 10.1038/s41586-020-2188-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumry WR. 1999. A review of the preclinical and clinical data of newer intranasal steroids used in the treatment of allergic rhinitis. J Allergy Clin Immunol 104: S150–S158. 10.1016/s0091-6749(99)70311-8 [DOI] [PubMed] [Google Scholar]
- Ma QL, Yang F, Frautschy SA, Cole GM. 2012. PAK in Alzheimer disease, Huntington disease and X-linked mental retardation. Cell Logist 2: 117–125. 10.4161/cl.21602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Bao J, Zhao X, Shen H, Lv J, Ma S, Zhang X, Li Z, Wang S, Wang Q, et al. 2013. Activated cyclin-dependent kinase 5 promotes microglial phagocytosis of fibrillar β-amyloid by up-regulating lipoprotein lipase expression. Mol Cell Proteomics 12: 2833–2844. 10.1074/mcp.M112.026864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan UV, Varma VR, Griswold ME, Blackshear CT, An Y, Oommen AM, Varma S, Troncoso JC, Pletnikova O, O'Brien R, et al. 2020. Dysregulation of multiple metabolic networks related to brain transmethylation and polyamine pathways in Alzheimer disease: a targeted metabolomic and transcriptomic study. PLoS Med 17: e1003012. 10.1371/journal.pmed.1003012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medway C, Morgan K. 2014. Review: The genetics of Alzheimer's disease; putting flesh on the bones. Neuropathol Appl Neurobiol 40: 97–105. 10.1111/nan.12101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Na YR, Jung D, Gu GJ, Jang AR, Suh YH, Seok SH. 2015. The early synthesis of p35 and activation of CDK5 in LPS-stimulated macrophages suppresses interleukin-10 production. Sci Signal 8: ra121. 10.1126/scisignal.aab3156 [DOI] [PubMed] [Google Scholar]
- Piñero J, Bravo À, Queralt-Rosinach N, Gutiérrez-Sacristán A, Deu-Pons J, Centeno E, García-García J, Sanz F, Furlong LI. 2017. DisGeNET: a comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res 45: D833–D839. 10.1093/nar/gkw943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2020. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna. https://www.R-project.org/. [Google Scholar]
- Rosenthal SL, Kamboh MI. 2014. Late-onset Alzheimer's disease genes and the potentially implicated pathways. Curr Genet Med Rep 2: 85–101. 10.1007/s40142-014-0034-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo C, Dolcini V, Salis S, Venezia V, Zambrano N, Russo T, Schettini G. 2002. Signal transduction through tyrosine-phosphorylated C-terminal fragments of amyloid precursor protein via an enhanced interaction with Shc/Grb2 adaptor proteins in reactive astrocytes of Alzheimer's disease brain. J Biol Chem 277: 35282–35288. 10.1074/jbc.M110785200 [DOI] [PubMed] [Google Scholar]
- Schweig JE, Yao H, Beaulieu-Abdelahad D, Ait-Ghezala G, Mouzon B, Crawford F, Mullan M, Paris D. 2017. Alzheimer's disease pathological lesions activate the spleen tyrosine kinase. Acta Neuropathol Commun 5: 69. 10.1186/s40478-017-0472-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan M, Yuan X, Song L, Roberts L, Zarinkamar N, Seryshev A, Zhang Y, Hilsenbeck S, Chang SH, Dong C, et al. 2012. Cigarette smoke induction of osteopontin (SPP1) mediates TH17 inflammation in human and experimental emphysema. Sci Transl Med 4: 117ra9. 10.1126/scitranslmed.3003041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Lin Z, Liu CH, Gong Y, Liegl R, Fredrick TW, Meng SS, Burnim SB, Wang Z, Akula JD, et al. 2017. Inflammatory signals from photoreceptor modulate pathological retinal angiogenesis via c-Fos. J Exp Med 214: 1753–1767. 10.1084/jem.20161645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasaki S, Gaiteri C, Mostafavi S, De Jager PL, Bennett DA. 2018. The molecular and neuropathological consequences of genetic risk for Alzheimer's dementia. Front Neurosci 12: 699. 10.3389/fnins.2018.00699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend DJ, Mala B, Hughes E, Hussain R, Siligardi G, Fullwood NJ, Middleton DA. 2020. Circular dichroism spectroscopy identifies the β-adrenoceptor agonist salbutamol as a direct inhibitor of tau filament formation in vitro. ACS Chem Neurosci 11: 2104–2116. 10.1021/acschemneuro.0c00154 [DOI] [PubMed] [Google Scholar]
- Warrell RP, Frankel SR, Miller WH, Scheinberg DA, Itri LM, Hittelman WN, Vyas R, Andreeff M, Tafuri A, Jakubowski A, et al. 1991. Differentiation therapy of acute promyelocytic leukemia with tretinoin (all-trans-retinoic acid). N Engl J Med 324: 1385–1393. 10.1056/NEJM199105163242002 [DOI] [PubMed] [Google Scholar]
- Wishart DS, Feunang YD, Marcu A, Guo AC, Liang K, Vázquez-Fresno R, Sajed T, Johnson D, Li C, Karu N, et al. 2018. HMDB 4.0: the human metabolome database for 2018. Nucleic Acids Res 46: D608–D617. 10.1093/nar/gkx1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Ni J, Liu Y, Teeling JL, Takayama F, Collcutt A, Ibbett P, Nakanishi H. 2017. Cathepsin B plays a critical role in inducing Alzheimer's disease-like phenotypes following chronic systemic exposure to lipopolysaccharide from Porphyromonas gingivalis in mice. Brain Behav Immun 65: 350–361. 10.1016/j.bbi.2017.06.002 [DOI] [PubMed] [Google Scholar]
- Zhang Z, Yan J, Chang Y, Yan SS, Shi H. 2011. Hypoxia inducible factor-1 as a target for neurodegenerative diseases. Curr Med Chem 18: 4335–4343. 10.2174/092986711797200426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Hou Y, Shen J, Huang Y, Martin W, Cheng F. 2020a. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov 6: 14. 10.1038/s41421-020-0153-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Song WM, Andhey PS, Swain A, Levy T, Miller KR, Poliani PL, Cominelli M, Grover S, Gilfillan S, et al. 2020b. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer's disease. Nat Med 26: 131–142. 10.1038/s41591-019-0695-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Fang J, Bekris LM, Kim YH, Pieper AA, Leverenz JB, Cummings J, Cheng F. 2021. AlzGPS: a genome-wide positioning systems platform to catalyze multi-omics for Alzheimer's drug discovery. Alzheimer's Res Ther 13: 24. 10.1186/s13195-020-00760-w [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.