

SUMMARY
Interferon-γ (IFN-γ)-producing CD4+ T helper-1 (Th1) cells are critical for protection from microbes that infect the phagosomes of myeloid cells. Current understanding of Th1 cell differentiation is based largely on reductionist cell culture experiments. We assessed Th1 cell generation in vivo by studying antigen-specific CD4+ T cells during infection with the phagosomal pathogen Salmonella enterica (Se), or influenza A virus (IAV), for which CD4+ T cells are less important. Both microbes induced T follicular helper (Tfh) and interleukin-12 (IL-12)-independent Th1 cells. During Se infection, however, the Th1 cells subsequently outgrew the Tfh cells via an IL-12-dependent process and formed subsets with increased IFN-γ production, ZEB2-transcription factor-dependent cytotoxicity, and capacity to control Se infection. Our results indicate that many infections induce a module that generates Tfh and poorly differentiated Th1 cells, which is followed in phagosomal infections by an IL-12-dependent Th1 cell amplification module that is critical for pathogen control.
Keywords: Th1, Tfh, IL-12, ZEB2, infection
eTOC
CD4+ T cells are critical for protection from phagosomal bacteria but not acute viruses. Krueger et al. find that although both infections drive formation of weakly protective Tfh and IL-12 independent Th1 cells, the phagosomal pathogen stimulated the outgrowth of IL-12 dependent Th1 cells with superior protective capacity.
Graphical Abstract
INTRODUCTION
CD4+ T cells use T cell antigen receptors (TCRs) to recognize major histocompatibility complex II (MHCII)-bound microbial peptides (p:MHCII) on host cells (Rudolph et al., 2006). TCR signaling causes naïve CD4+ T cells to proliferate and differentiate into T helper-1 (Th1), Th2, Th9, Th17, T follicular (Tfh), or peripheral regulatory T (Treg) cells depending on cytokines from cells of the innate immune system (Sun and Zhang, 2014). These specialized subsets are thought to protect their host from different infections. Th1 cells are critical for host protection from microbes that infect the phagosomes of myeloid cells (Tubo and Jenkins, 2014).
In vitro experimentation shows that Th1 cells form by a step-wise process in which TCR, IFN-γ receptor, and STAT1 signaling causes expression of T-bet (Afkarian et al., 2002; Lighvani et al., 2001; Mullen et al., 2001; Schulz et al., 2009), the master transcription factor of Th1 cells (Szabo et al., 2000). As TCR signaling wanes (Schulz et al., 2009), T-bet induces the IL-12 receptor and IL-12 receptor signaling activates STAT4 (Thieu et al., 2008), which drives proliferation of the Th1 cells and enhances their T-bet expression, interferon-γ (IFN-γ) production capacity, and commitment to the Th1 cell lineage (Mullen et al., 2001; Murphy and Reiner, 2002; O’Shea and Paul, 2010).
In vivo formation of antigen-specific Th1 cells has been studied most intensively in acute infections. These infections drive microbial p:MHCII-specific CD4+ naïve T cells to proliferate and form Th1 cells and two Tfh effector cell populations - CXCR5hi BCL6hi germinal center T follicular helper cells (GC-Tfh) and their CXCR5 intermediate (int) BCL6lo precursors (Ballesteros-Tato et al., 2012; Choi et al., 2011; Johnston et al., 2009; Kunzli et al., 2020; Marshall et al., 2011; Pepper and Jenkins, 2011; Yu et al., 2009). This heterogeneity is driven by STAT5 signals from IL-2 receptors favoring Th1 cells on the one hand and STAT3 signals from IL-6 and IL-21 receptors promoting Tfh cells on the other (Ballesteros-Tato et al., 2012; Johnston et al., 2012; Pepper et al., 2011; Ray et al., 2014). Ninety percent of the effector cells die after the infections are cleared leaving Th1 and Tfh-like CXCR5int memory cells (Hale et al., 2013;Kunzli et al., 2020; Marshall et al., 2011; Pepper et al., 2011).
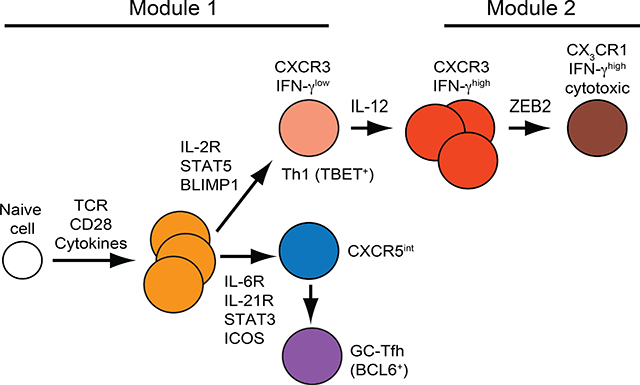
Although these studies provide basic information on in vivo Th1 formation, their relevance to immunity is less clear because acute infections can be controlled by hosts lacking CD4+ T cells (Allan et al., 1990; Baldridge et al., 1990; Ladel et al., 1994; Matloubian et al., 1994). Th1 cells have therefore also been studied in vivo under conditions where they are critical for pathogen control as is the case of Salmonella enterica serovar Typhimurium (Se) and Mycobacterium tuberculosis (Mtb) infections of the macrophage phagosome (Tubo and Jenkins, 2014). In these situations, microbial p:MHCII-specific CD4+ T cell populations contain very few Tfh cells and are dominated by Th1 cells (Goldberg et al., 2018; Sallin et al., 2017). Here, we compared the CD4+ T cell response to acute infection with influenza A virus (IAV) to that with the phagosomal pathogen Se to better understand how Th1 cells are formed in vivo in a context where they are critical for immunity. We found that early on, both infections induced an equal mix of Tfh and relatively undifferentiated IL-12-independent Th1 cells that were maintained indefinitely in the case of IAV. In contrast, during Se infection, an IL-12- and ZEB2-dependent process caused the Th1 cells to outgrow the Tfh cells and acquire a more differentiated phenotype associated with enhanced IFN-γ production, terminal differentiation, cytotoxicity, and phagosomal pathogen control. These results suggest that many infections activate a common module that generates mixed Th1 and Tfh effector cell populations, which in some infections is followed by a later IL-12-dependent module that amplifies the Th1 cell component critical for control of phagosomal pathogens.
RESULTS
Se infection drives greater expansion of antigen-specific CD4+ T cells than IAV infection
We studied Th1 cell formation during IAV infection, a case where the infection can be cleared without CD4+ T cells (Allan et al., 1990), and Se infection, where CD4+ T cells play a critical role in pathogen control (Goldberg et al., 2018; Johanns et al., 2010). We chose IAV and Se bacteria engineered to express 2W (Rees et al., 1999), a well-studied immunogenic I-Ab-binding peptide that is recognized by a relatively large population of naive CD4+ T cells in mice that express I-Ab (Moon et al., 2007). Focus on the same pre-immune population eliminated variables related to epitope stability or T cell repertoire size. Se-2W bacteria were given orally or intravenously, while IAV-2W was given intranasally. Antigens from both infections become systemic in all cases, however, as indicated by profound T cell activation in the spleen (Goldberg et al., 2018; Turner et al., 2013).
Initial experiments were performed to assess the tempo of the CD4+ T cell response to each infection. Cells from spleens and lymph nodes of infected C57BL/6 (B6) mice were stained with fluorochrome-labeled 2W:I-Ab tetramer and fluorochrome antibody–labeled magnetic beads and enriched on magnetized columns as previously described (Moon et al., 2007). 2W:I-Ab tetramer–binding conventional (FOXP3−) T cells were identified by flow cytometry among the CD4+ cells that bound to the columns (Supp. Fig. 1). The secondary lymphoid organs of uninfected B6 mice contained about 300 2W:I-Ab–specific CD4+ T cells (Moon et al., 2007) (Fig. 1A). By day four, the number of these cells in Se-2W-infected mice had already increased but remained at the starting number in IAV-2W-infected mice. By day seven, however, the number of 2W:I-Ab–specific CD4+ T cells in IAV-2W-infected mice had also increased above the baseline. The number of 2W:I-Ab–specific CD4+ T cells peaked at day seven for IAV-2W infection at 40,000 cells, and at day 10 for Se-2W infection at a 800,000 cells. In both cases, the number of 2W:I-Ab–specific CD4+ T cells then contracted and stabilized over the next several months in both cases as previously described for other infections (Marshall et al., 2011; Pepper et al., 2011). Thus, clonal expansion of 2W:I-Ab–specific CD4+ T cells started earlier and was larger for Se-2W than IAV-2W infection.
Fig. 1. Expansion and differentiation of naïve 2W:I-Ab-specific T cells after infection with IAV-2W or Se-2W.
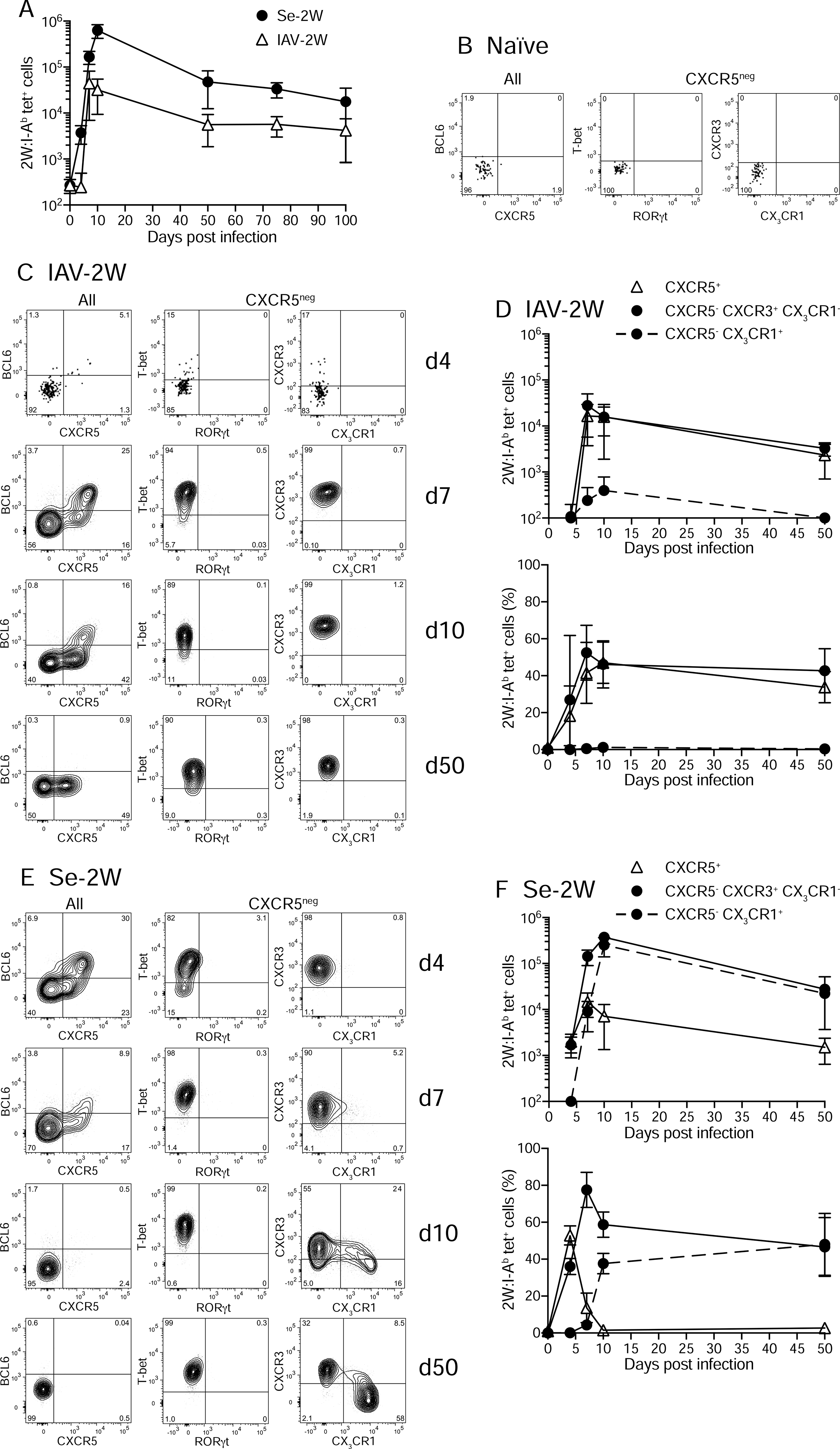
(A) Mean numbers (± SD) of 2W:I-Ab tetramer-binding cells in the spleen and lymph nodes of B6 mice at the indicated times after infection. (B) Flow cytometry plots of the indicated molecules for 2W:I-Ab tetramer+ (left) or CXCR5− 2W:I-Ab tetramer+ cells (middle and right) from tetramer-enriched spleen and lymph node samples from an unimmunized B6 mouse. (C) Flow cytometry plots of the indicated molecules for 2W:I-Ab tetramer+ (left) or CXCR5− 2W:I-Ab tetramer+ cells (middle and right) from tetramer-enriched spleen and lymph node samples of B6 mice four, seven, 10, or 50 days after infection with IAV-2W. (D) Mean numbers (± SD) (top) or percentages (bottom) of 2W:I-Ab tetramer-binding cells with the indicated phenotypes from individual mice at the indicated times after IAV-2W infection. (E) Flow cytometry plots of the indicated molecules for 2W:I-Ab tetramer+ (left) or CXCR5− 2W:I-Ab tetramer+ cells (middle and right) from tetramer-enriched spleen and lymph node samples of B6 mice four, seven, 10, or 50 days after infection with Se-2W. (F) Mean numbers (± SD) (top) or percentages (bottom) of 2W:I-Ab tetramer-binding cells with the indicated phenotypes from individual mice at the indicated times after Se-2W infection. A-F, n ≥ three mice per group/time point. See also Fig. S1 and S2.
The Th1 cell population induced by Se infection improves its IFN-γ production capacity and gains a CX3CR1+ subset over time
We next examined the differentiation of 2W:I-Ab–specific CD4+ T cells during each infection. As expected for naive T cells (Jenkins et al., 2010), the 2W:I-Ab–specific cells in the secondary lymphoid organs of uninfected mice expressed low amounts of CD44 and did not express T-bet, RORγt - the lineage-defining transcription factor for Th17 cells (Ivanov et al., 2006), BCL6, CXCR5, CXCR3 - the chemokine receptor for CXCL9 and CXCL10 (Groom and Luster, 2011), or CX3CR1 - the chemokine receptor for fractalkine (Imai et al., 1997) (Fig. 1B). After IAV-2W infection, the 2W:I-Ab–specific T cells expanded enough in the secondary lymphoid organs by day seven to allow an accurate assessment of phenotype (Fig. 1C). About half of the cells in the population at this time were CXCR5hi BCL6hi GC-Tfh or CXCR5int BCL6lo Tfh-like cells and the other half were CXCR5− cells that expressed T-bet, but not RORγt, and CXCR3, but not CX3CR1 (Fig. 1C and 1D). The IAV-2W-induced 2W:I-Ab–specific population maintained this composition into the later effector period on day 10 and into the post-contraction memory phase on day 50 (Fig. 1D) although the GC-Tfh population lost expression of BCL6 as previously described (Fig. 1C) (Hale et al., 2013; Kunzli et al., 2020; Pepper et al., 2011; Tubo et al., 2016). On day four, Se-2W-infected mice also contained an expanded 2W:I-Ab–specific population in the secondary lymphoid organs consisting of about 50 percent CXCR5hi BCL6hi GC-Tfh or CXCR5int BCL6lo cells, and 50 percent CXCR5− T-bet+ RORγt− CXCR3+ CX3CR1− cells (Fig. 1F) as in IAV-2W infected mice. The CXCR5+ populations were rapidly outnumbered after day four, however, by an expanding population of CXCR5− T-bet+ Th1 cells, which contained a CX3CR1+ subset as early as day seven and that peaked at 50 percent of the population by day 50. Thus, Th1 cells came to dominate the 2W:I-Ab–specific T cell population in Se-2W-infected mice because of proliferation at a faster rate than Tfh cells after day four.
We also examined the liver because Se infection spreads to this location (Monack et al., 2004). Day 10 was chosen for this analysis because it is near the time of peak clonal expansion of 2W:I-Ab–specific T cells for both infections (Fig. 1D, 1F). 2W:I-Ab–specific effector T cells were present in the livers of IAV-2W and Se-2W infected mice, although many more were in the latter group (Supp. Fig. S2A and 2B). In IAV-2W infection, the 2W:I-Ab–specific T cell population was composed exclusively of T-bet+ (Supp. Fig. S2C) Th1 cells most expressing CXCR3 (Supp. Fig. S2A) and lacking CX3CR1 (Supp. Fig. S2A and S2D). Although all the cells in the livers of Se-2W infected mice were also T-bet+ Th1 cells (Supp. Fig. S2C), about 40 percent of the cells expressed CX3CR1+ (Supp. Fig. S2D). Thus, the livers of IAV-2W or Se-2W infected mice lacked Tfh cells and contained the Th1 populations that were found in the secondary lymphoid organs.
We then measured the in vivo IFN-γ production potential of the Th1 cells in the secondary lymphoid organs. As shown in Fig. 2A and 2B, IFN-γ was not detected by ex vivo intracellular staining in 2W:I-Ab–specific CD4+ T cells four to 16 days after either infection, presumably because very few of the T cells were being stimulated in vivo in this time frame. An intravenous injection of 2W peptide was then used to recall IFN-γ production (Khoruts et al., 1998). About 20 percent of the T-bet+ 2W:I-Ab–specific T cells in the early effector phase of each infection (day four-five for Se-2W, day seven for IAV-2W) produced IFN-γ two hours after the 2W peptide injection (Fig. 2A, 2B). A similar fraction of the T-bet+ 2W:I-Ab–specific T cell population in the later effector phase of the IAV-2W infection (day 10–16) had IFN-γ production potential. In contrast, the fraction of the T-bet+ 2W:I-Ab–specific T cell population that was capable of producing IFN-γ increased to 80 percent by day seven of Se-2W-infection and remained at this fraction on days 10–16. Notably, like the T-bet+ 2W:I-Ab–specific T cells in Se-2W-infected mice on day four, the cells on day seven were almost all CXCR3+ CX3CR1− (Fig. 1E) indicating that this population had improved its IFN-γ production potential between days four and seven. The fact that the day 10–16 population, which contained 80 percent IFN-γ producing cells, was a mixture of CXCR3+ CX3CR1− and CX3CR1+ Th1 cells indicated that the majority of cells in both subsets could produce IFN-γ. Together, the results indicated that only a minority of the epitope-specific CD4+ effector Th1 cells induced at early times by the two different infections could produce IFN-γ. While this pattern was maintained into the later effector phase in IAV-2W infection, the Th1 cell population that emerged in Se-2W infection during this period had increased IFN-γ production potential.
Fig. 2. IFN-γ production by Th1 cells in IAV-2W or Se-2W infections.
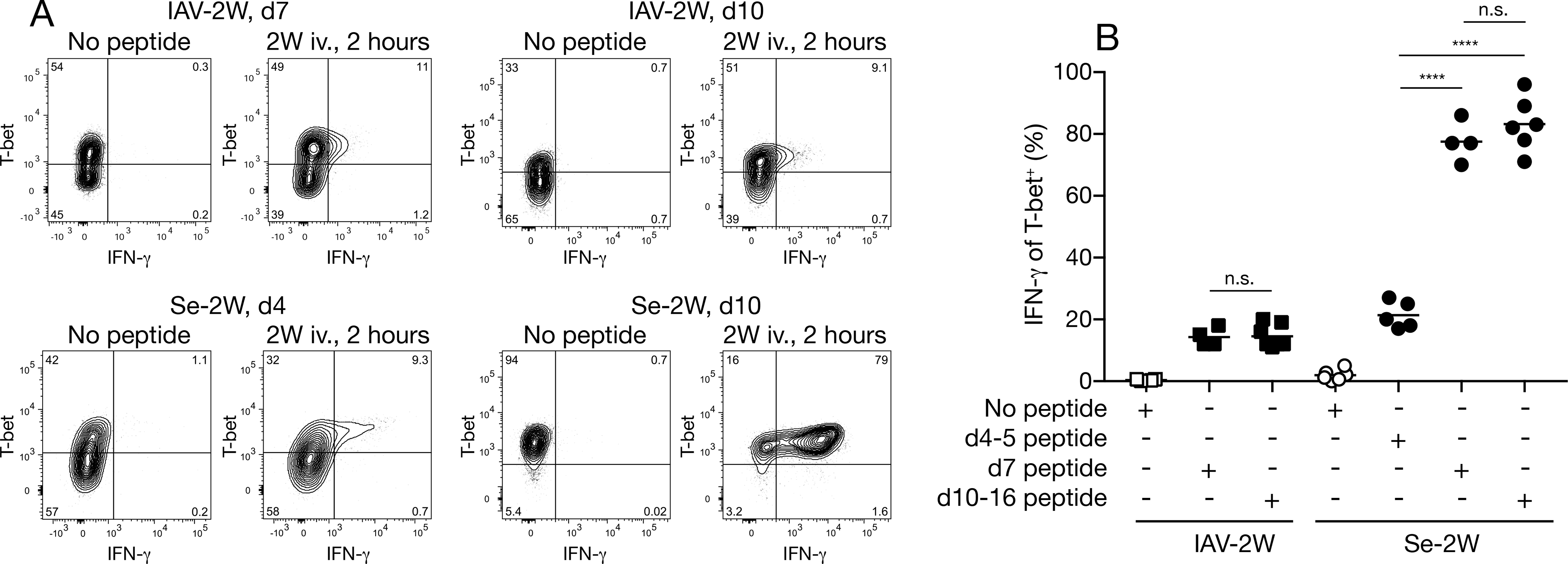
(A) Flow cytometry plots of 2W:I-Ab tetramer-binding cells from mice with the indicated infections that were not or were injected with 2W peptide two hours before analysis. IAV-2W day seven, Se-2W day four, and the day 10 pairs were analyzed on different days. (B) Percent of IFN-γ+ cells among T-bet+ 2W:I-Ab tetramer-binding cells from individual mice (n ≥ four from two independent experiments) with the indicated infections for the indicated times, two hours after injection of 2W peptide or not. Mean values on scatter plots are indicated with a horizontal bar. Values were compared using one-way ANOVA. ****p < 0.0001; n.s., not significant.
The Th1 cell population induced by Se infection becomes more IL-12-dependent over time
In vitro studies show that Th1 cell differentiation begins with IL-12-independent T-bet induction followed by IL-12- and STAT4-dependent T-bet enhancement and commitment to the Th1 cell lineage (Mullen et al., 2001; Murphy et al., 2000; Schulz et al., 2009; Thieu et al., 2008). Other studies indicate that type I IFN receptor (encoded by the Ifnar1 gene) (Longhi et al., 2009), IFN-γ receptor (Hsieh et al., 1993; Lighvani et al., 2001; Scott, 1991), and IL-18 receptor signaling (Kaplanski, 2018) can also influence Th1 cell differentiation.
We explored the role of these factors in Th1 cell differentiation beginning with IAV-2W infection. As shown in Fig. 3A, 2W:I-Ab–specific T cells accumulated by days seven-10 of IAV-2W infection to the same degree in mice lacking IL-12, IL-18, or IFNAR, or wild-type or IFNAR-deficient mice treated with neutralizing IL-12 or IFN-γ antibodies, as they did in wild-type mice. Similarly, IL-12- (Fig. 3B, 3C) or IL-18-deficiency or neutralization of IL-12 or IFN-γ had no effect on the formation of T-bet+ cells (Fig. 3C), the amount of T-bet per cell (Fig. 3D), or the capacity of T-bet+ cells to produce IFN-γ after peptide challenge (Fig. 3E). In contrast, IFNAR-deficiency led to a two-fold reduction in the percentage of T-bet+ cells in the 2W:I-Ab–specific T cell population in IAV-2W-infected mice (Fig. 3B, 3C), and addition of IL-12 or IFN-γ antibodies did not increase the magnitude of this defect (Fig. 3C). The T-bet+ population in IAV-2W-infected IFNAR-deficient mice expressed the same amount of T-bet (Fig. 3D) and contained the same 20 percent frequency of IFN-γ producing cells as the population in wild-type mice (Fig. 3E). Thus, the formation of the Th1 cell population with a 20 percent component of IFN-γ-producing cells did not involve IL-12, IL-18, or IFN-γ. About half of the cells in this population, however, required IFNAR to form.
Fig. 3. Molecular requirements for Th1 cell formation during IAV-2W infection.
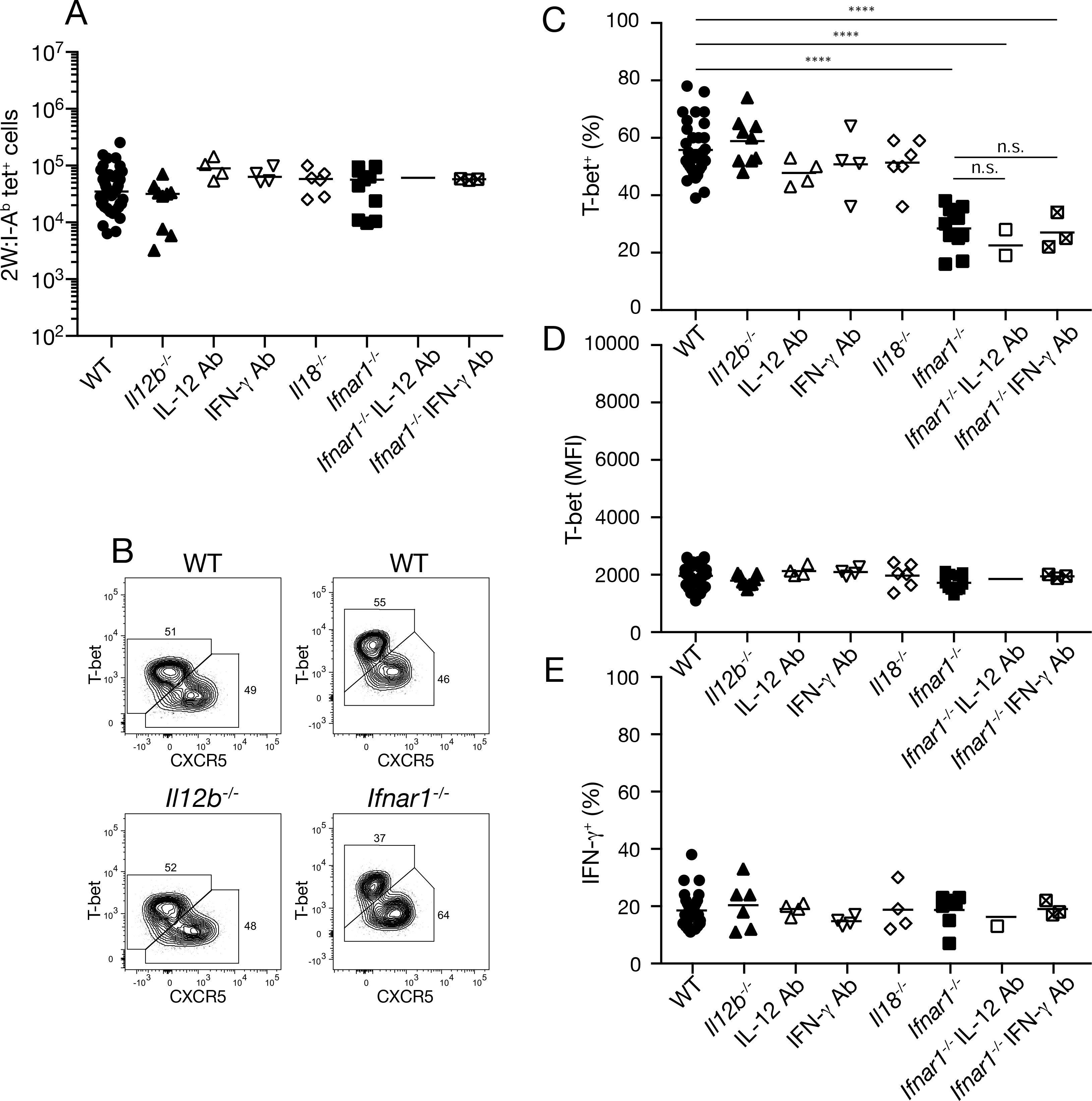
(A) Number of 2W:I-Ab tetramer-binding cells from individual mice (n ≥ three from two independent experiments) of the indicated genotypes on days seven-10 of IAV-2W infection. (B) Flow cytometry plots of 2W:I-Ab tetramer-binding cells from IAV-2W-infected mice of the indicated genotypes. (C-E) Percent T-bet+ (C), mean fluorescence intensity of T-bet+ cells (D), or percent IFN-γ+ cells among T-bet+ 2W:I-Ab tetramer-binding cells two hours after peptide challenge (E) from individual mice (n ≥ three from two independent experiments) of the indicated genotypes. Mean values on scatter plots are indicated with a horizontal bar. Values in A, C-E were compared using one-way ANOVA. ****p < 0.0001; n.s., not significant.
We performed a similar analysis for 2W:I-Ab–specific T cells present in the later effector phase of Se-2W infection. IL-12-deficient but not IFNAR-deficient or IL-18-deficient mice had five-fold fewer 2W:I-Ab–specific T cells than wild-type mice on day 10 after Se-2W infection (Fig. 4A) indicating that IL-12, but not IFNAR or IL18 was involved in the outgrowth of this population. The smaller population of 2W:I-Ab–specific T cells in IL-12-deficient mice consisted of a mixture of CXCR5− T-bet+ cells and CXCR5+ Tfh cells, whereas the population in wild-type mice consisted mainly of CXCR5− T-bet+ cells (Fig. 4B, 4C). The T-bet+ population in IL-12-deficient mice also expressed less T-bet per cell (Fig. 4D) and lacked the CX3CR1+ subset that was prominent in wild-type mice (Fig. 4E). Only about 10 percent of the T-bet+ 2W:I-Ab–specific T cells in IL-12-deficient Se-2W-infected mice produced IFN-γ two hours after in vivo peptide challenge compared to 90 percent in wild-type mice (Fig. 4F). The 2W:I-Ab–specific T cell populations that formed in IFNAR- or IL-18-deficient Se-infected mice were indistinguishable from the T-bethigh population with IFN-γ producing cells and a CX3CR1+ subset that formed in wild-type mice (Figs. 4C–F). These results show that the amplification of a T-bethigh population with enhanced IFN-γ production potential and a CX3CR1+ subset in Se-2W-infected mice depends on IL-12 but not IFNAR or IL-18.
Fig. 4. Molecular requirements for Th1 cell formation during Se-2W infection.
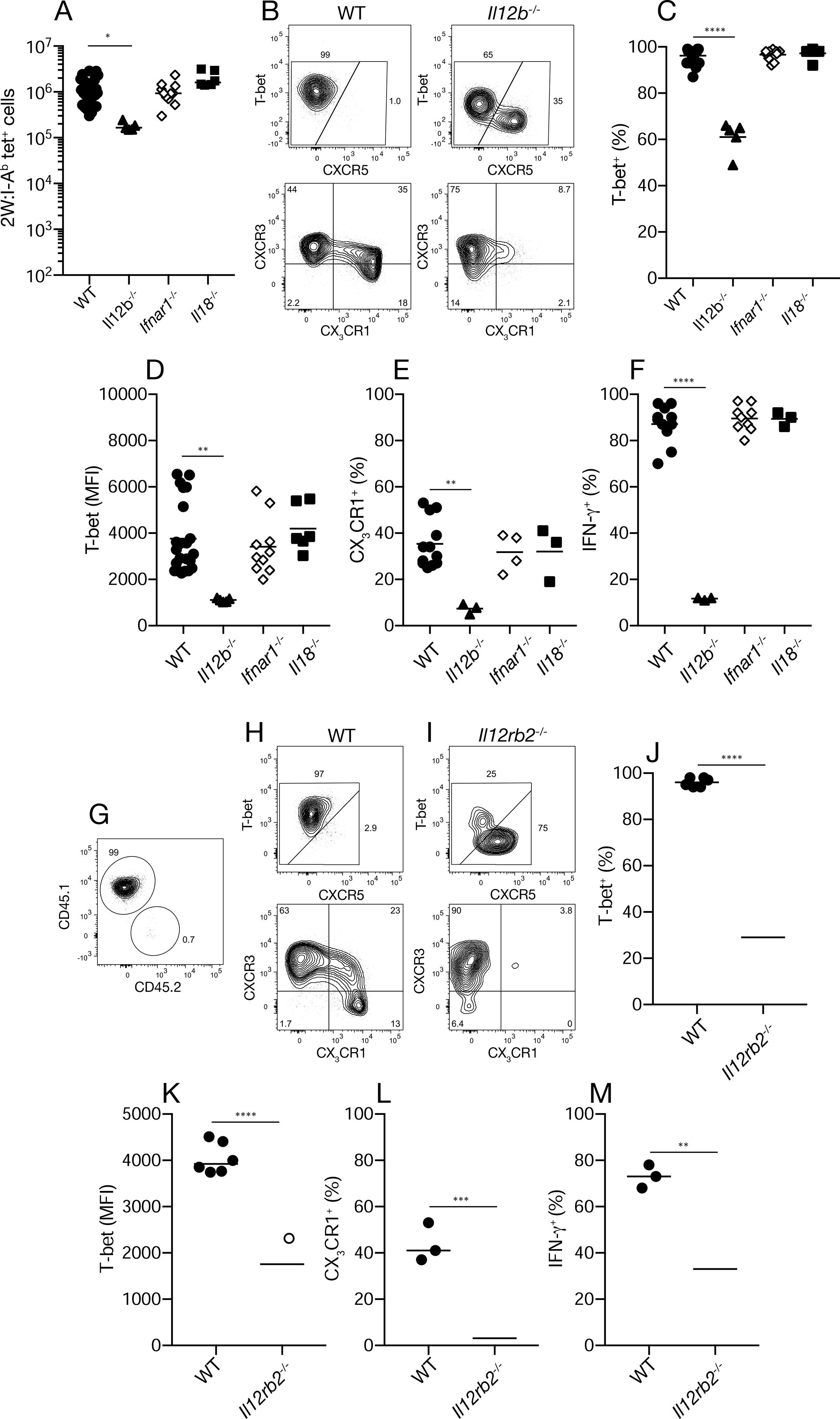
(A) Number of 2W:I-Ab tetramer-binding cells from individual mice (n ≥ five from two independent experiments) of the indicated genotypes on day 10 of Se-2W infection. (B) Flow cytometry plots of 2W:I-Ab tetramer-binding cells from Se-2W-infected mice of the indicated genotypes. (C-F) Percent T-bet+ (C), mean fluorescence intensity of T-bet+ cells (D), percent CX3CR1+ cells (E), or IFN-γ+ cells among T-bet+ 2W:I-Ab tetramer-binding cells two hours after peptide challenge (F) from individual mice (n ≥ three from two independent experiments) of the indicated genotypes. (G) Flow cytometry plot used to identify 2W:I-Ab tetramer-binding cells of recipient (CD45.1) or donor (CD45.2) origin from Se-2W-infected B6 mice that received CD4+ T cells from IL-12 receptor-deficient mice. (H, I) Flow cytometry plots of 2W:I-Ab tetramer-binding cells of recipient (wild-type) or donor (IL-12 receptor-deficient) origin from Se-2W-infected B6 mice that received CD4+ T cells from IL-12 receptor-deficient mice. (J-M) Percent T-bet+ (J), mean fluorescence intensity of T-bet+ cells (K), percent CX3CR1+ cells (L), or percent IFN-γ+ cells two hours after peptide challenge (M) among wild-type or IL-12 receptor-deficient T-bet+ 2W:I-Ab tetramer-binding cells from individual mice (n ≥ three from two independent experiments). Values in A, C-F were compared using one-way ANOVA. Values in J-M were compared using an unpaired Student’s t-test. p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. Mean values on scatter plots are indicated with a horizontal bar.
We then used an adoptive transfer approach to determine whether the defects in Th1 cell differentiation observed in Se-infected IL-12-deficient mice were intrinsic to the T cells. Congenically-marked CD4+ T cells from uninfected IL-12 receptor-deficient mice were injected into wild-type mice, which were then infected with Se-2W bacteria. The phenotypes of effector T cells derived from the transferred IL-12 receptor-deficient naïve cells or the wild-type naïve cells of the recipient (Fig. 4G) were then assessed on day 10. As expected, the wild-type population of 2W:I-Ab–specific T cells of recipient origin was composed of T-bethigh CXCR3+ CX3CR1− and CX3CR1+ Th1 cells, most of which produced IFN-γ after 2W peptide injection (Fig. 4H, 4J–M). In contrast, the IL-12 receptor-deficient donor-derived population contained a mixture of Tfh cells (Fig. 4I) and a T-bet+ Th1 cell population (Fig. 4I, 4J) that had less T-bet per cell (Fig. 4K), expressed CXCR3 but not CX3CR1 (Fig. 4I and 4L), and had fewer cells that made IFN-γ after peptide challenge (Fig. 4M) than the comparable wild-type population. These results show that a CD4+ T cell intrinsic capacity to respond to IL-12 is required for all the features of the Th1 cell amplification process that occurs during Se infection.
Th1 cell amplification accelerates after day four of Se infection (Fig. 1F), indicating that this is a critical period of IL-12 production. Mice were treated with IL-12 antibody beginning on day two of Se-2W infection to neutralize IL-12 during this interval and then analyzed on day seven as a test of this possibility. The 2W:I-Ab–specific T cell population in IL-12 antibody-treated mice was three times smaller than that in untreated mice (Fig. 5A) and consisted of an equal mixture of CXCR5+ Tfh and T-bet+ Th1 cells while the population in untreated mice was dominated by T-bet+ Th1 cells (Fig. 5B). In addition, the T-bet+ cells in IL-12 antibody-treated mice expressed less T-bet per cell (Fig. 5C) and a smaller fraction produced IFN-γ after peptide injection than the comparable population in untreated mice (Fig. 5D). The T-bet+ population in IL-12 antibody-treated mice also contained only CXCR3+ CX3CR1− cells, while the population in untreated mice contained this subset plus a few CX3CR1+ cells (Fig. 5E) as expected for the day seven time point (Fig. 1F). Thus, neutralization of IL-12 beginning on day two of Se-2W infection prevented Th1 cell amplification and yielded an epitope-specific population similar to the one generated by IAV infection.
Fig. 5. Effects of IL-12 neutralization or addition on epitope-specific CD4+ T cells during infections.
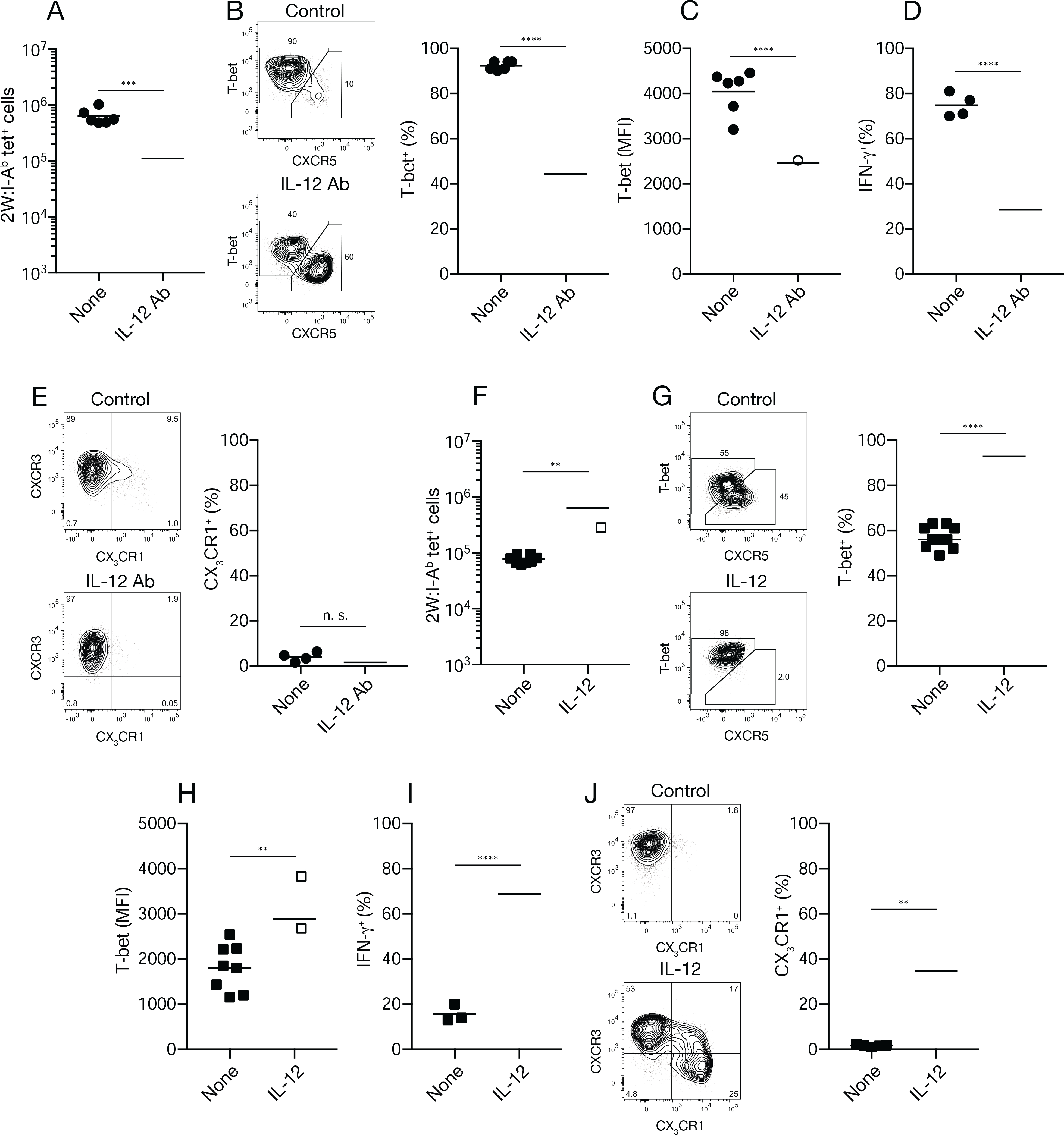
(A) Number of 2W:I-Ab tetramer-binding cells from individual Se-2W-infected mice (n ≥ six from two independent experiments) given nothing or IL-12 antibody (Ab) on days two and four of infection and analyzed on day seven. (B) Flow cytometry plots of 2W:I-Ab tetramer-binding cells from Se-2W-infected mice given nothing (top) or IL-12 antibody (bottom) on days two and four of infection and analyzed on day seven, with a scatter plot of the percent of T-bet+ cells among 2W:I-Ab tetramer-binding cells. (C, D) Mean fluorescence intensity of T-bet+ cells (C) or IFN-γ+ cells among T-bet+ 2W:I-Ab tetramer-binding cells two hours after peptide challenge (D) from individual mice (n ≥ four from two independent experiments) of the indicated treatment groups. (E) Flow cytometry plots of 2W:I-Ab tetramer-binding cells from Se-2W-infected mice given nothing (top) or IL-12 antibody (bottom) on days two and four of infection and analyzed on day seven, with a scatter plot of the percent of CX3CR1+ cells among 2W:I-Ab tetramer-binding cells from the two groups. (F) Number of 2W:I-Ab tetramer-binding cells from individual IAV-2W-infected mice (n = nine from three independent experiments) given nothing or recombinant IL-12 on days three-six of infection and analyzed on day 10. (G) Flow cytometry plots of 2W:I-Ab tetramer-binding cells from IAV-2W-infected mice given nothing (top) or recombinant IL-12 (bottom) on days three-six of infection and analyzed on day 10, with a scatter plot of the percent T-bet+ cells among 2W:I-Ab tetramer-binding cells. (H, I) Mean fluorescence intensity of T-bet+ cells (H) or IFN-γ+ cells among T-bet+ 2W:I-Ab tetramer-binding cells two hours after peptide challenge (I) from individual mice (n ≥ three from two independent experiments) of the indicated treatment groups. (J) Flow cytometry plots of T-bet+ 2W:I-Ab tetramer-binding cells from Se-2W-infected mice given nothing (top) or recombinant IL-12 (bottom) on days three-six of infection and analyzed on day 10, with a scatter plot of the percent of CX3CR1+ cells among T-bet+ 2W:I-Ab tetramer-binding cells. Values were compared using an unpaired Student’s t-test. **p < 0.01; ***p < 0.001; ****p < 0.0001; n.s., not significant. Mean values on scatter plots are indicated with a horizontal bar.
These results raised the possibility that Th1 cell amplification does not occur during IAV-2W infection because IL-12 is limited after the early phase of the infection. We tested this possibility by injecting IL-12 into mice beginning on day three of IAV-2W infection. The 2W:I-Ab–specific T cell population in IL-12-treated mice was three times larger than that in untreated mice (Fig. 5F) and was dominated by T-bet+ cells while the population in untreated mice consisted of an equal mixture of CXCR5+ Tfh and T-bet+ cells (Fig. 5G). In addition, the T-bet+ cells in IL-12-treated mice expressed more T-bet per cell (Fig. 5H) and a larger fraction produced IFN-γ after peptide injection than the comparable population in untreated mice (Fig. 5I). The T-bet+ population in IL-12-treated mice also contained CX3CR1+ cells, while the population in untreated mice lacked this subset (Fig. 5J). Together, the results of IL-12 blockade in Se infection and IL-12 addition in IAV infection indicate that the presence of IL-12 after the early phase of infection is necessary and sufficient to generate the Th1 cell amplification program.
CX3CR1+ Th1 cells are cytotoxic and depend on the ZEB2 transcription factor
We performed bulk RNA sequencing on tetramer-purified CXCR3+ CX3CR1− and CX3CR1+ Th1 cells to get clues about the origin and function of the CX3CR1 Th1 cell subset. B6 × 129 F1 mice, another I-Ab-expressing strain, and an I-Ab tetramer containing a peptide from the Se LpdA protein (LpdAp) (Karunakaran et al., 2017) were used in this case. The LpdAp:I-Ab-specific T cell population in Se-infected 129 mice contains the same CXCR3+ CX3CR1− and CX3CR1+ Th1 cell subsets (Goldberg et al., 2018) as the 2W:I-Ab-specific T cell population in Se-2W-infected B6 mice.
About 1,000 differentially expressed genes were identified (Fig. 6A). CXCR3+ CX3CR1− cells preferentially expressed Cxcr3, Il23r, Pecam1, Btla, and Tnfrsf4 mRNA. In contrast, CX3CR1+ cells expressed more Cx3cr1, S1p5r, and Klrg1 mRNA than CXCR3+ CX3CR1− cells. CX3CR1+ cells also expressed Gzma, Prf1, and Fasl encoding granzyme A, perforin, and Fas ligand, which have cytotoxic activities (Golstein and Griffiths, 2018). We therefore considered the possibility that CX3CR1+ cells belonged to the cytotoxic CD4+ T cell subset (Marshall and Swain, 2011; Weiskopf et al., 2015). We tested it using an in vivo assay, which we developed to measure 2W:I-Ab-specific cytotoxic CD4+ T cells (Kotov et al., 2018), and Cd4cre Cx3cr1DTR mice (Diehl et al., 2013), which express the simian diphtheria toxin (DT) receptor in CX3CR1+ T cells and can be used to deplete this population. Splenocytes from uninfected mice were labeled with high or low concentrations of the vital dye CellTrace Violet (CTV) (Quah et al., 2013) and the cells labeled with the low concentration were pulsed with the 2W peptide. The CTVhigh and CTVlow splenocytes were then mixed and injected into uninfected wild-type mice or day 30 Se-2W-infected wild-type or Cd4cre Cx3cr1DTR mice that had been treated with DT. About 60 percent of the 2W peptide-pulsed CTVlow target cells were preferentially eliminated in DT-treated Se-2W-infected wild-type mice compared to the uninfected controls indicating that cytotoxic CD4+ T cells were present in the former group (Fig. 6B, 6C). In contrast, only 25 percent of the 2W peptide-pulsed CTVlow target cells were eliminated in DT-treated Se-2W-infected Cd4cre Cx3cr1DTR mice, which lacked CX3CR1+ T cells (Fig. 6D). Thus, the CX3CR1+ 2W:I-Ab-specific CD4+ T cells in Se-2W-infected mice have cytotoxic activity.
Fig. 6. Formation of CX3CR1+ cytotoxic cells is ZEB2-dependent.
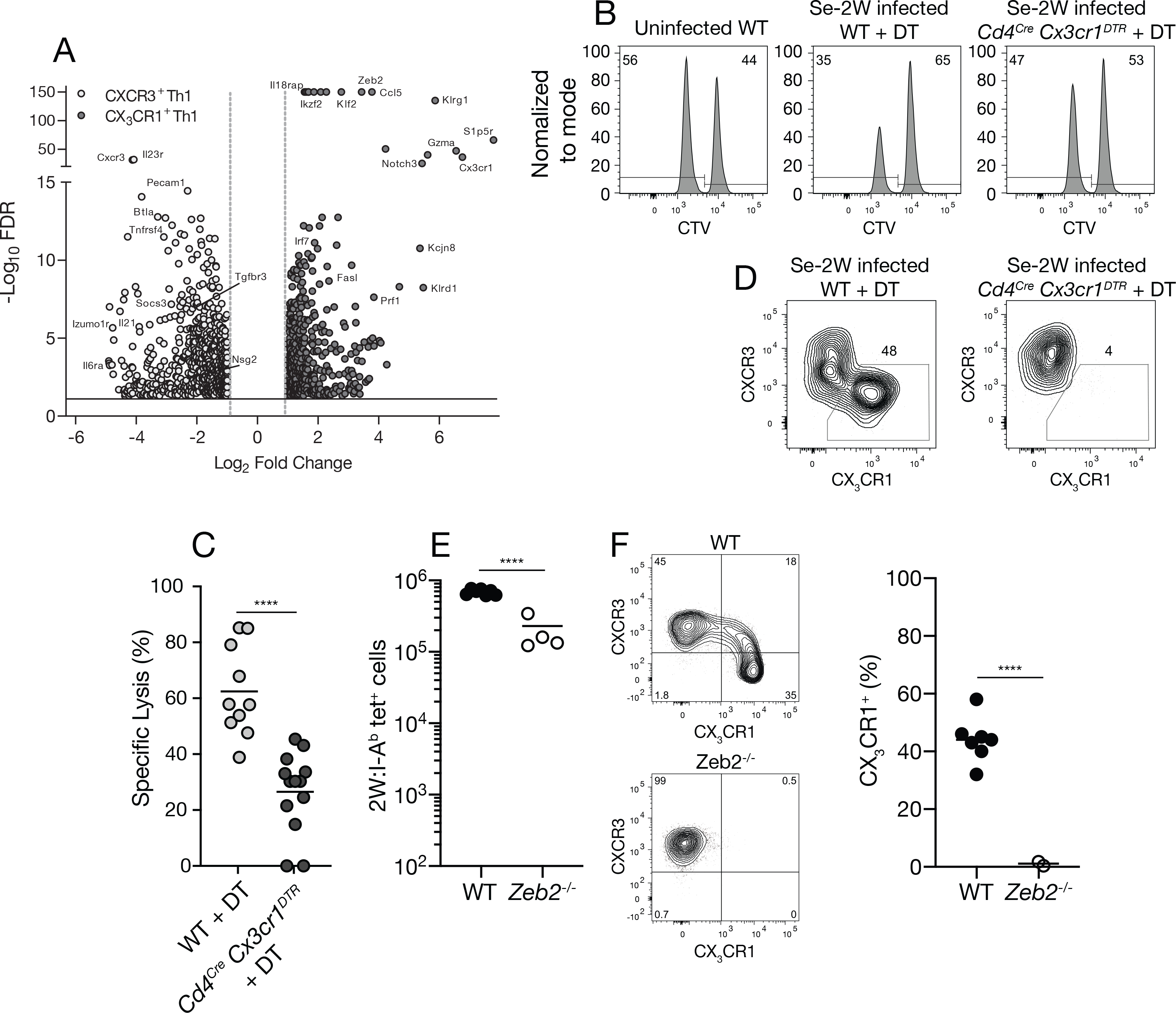
(A) Volcano plot of RNA sequencing results from LpdAp:I-Ab tetramer-binding CXCR3+ (open) or CX3CR1+ (closed) cells from day 30 Se-infected B6 × 129 F1 mice. (B) Flow cytometric analysis of an in vivo cytotoxicity assay using CTV to identify CTVlo splenic B cells pulsed with 2W peptide and CTVhi unpulsed B cells 20 hours after injection into the indicated mice. (C) Specific cell lysis of 2W peptide-pulsed B cells in individual mice (n ≥ 10 per group from three independent experiments) from the indicated groups. (D) Flow cytometry plots of T-bet+ 2W:I-Ab tetramer-binding cells from day 30 Se-2W-infected mice from the indicated groups demonstrating depletion of CX3CR1+ cells from DT-treated Cd4Cre Cx3cr1DTR mice. (E) Number of 2W:I-Ab tetramer-binding cells of wild-type (Zeb2fl/fl) or ZEB-deficient (Cd4cre Zeb2fl/fl) origin in day 10 Se-2W-infected chimeric mice. (F) Flow cytometry plots of T-bet+ 2W:I-Ab tetramer-binding cells of wild-type (Zeb2fl/fl) or ZEB2-deficient (Cd4cre Zeb2fl/fl) origin in day 10 Se-2W-infected chimeric mice, with a scatter plot of the percent of CX3CR1+ cells among T-bet+ 2W:I-Ab tetramer-binding cells. Values were compared using an unpaired Students t-test (****p < 0.0001) and were derived from two-three independent experiments. The horizontal lines represent the mean for each group.
The RNA sequencing experiments also showed that Se-induced CX3CR1+ T cells preferentially express the ZEB2 transcription factor (Fig. 6A), which was reported to control terminal differentiation of CD8+ cytotoxic T cells (Dominguez et al., 2015; Omilusik et al., 2015). We tested the role of ZEB2 in the formation of CX3CR1+ CD4+ T cells using a radiation chimera approach. Bone marrow cells from congenically-marked Zeb2fl/fl mice or Cd4cre Zeb2fl/fl mice (Omilusik et al., 2015) were injected into lethally irradiated recipients. The recipients were rested for two months to allow for development of ZEB2-deficient T cells from the Cd4cre Zeb2fl/fl bone marrow and wild-type T cells from the Zeb2fl/fl bone marrow. The chimeric mice were then infected with Se-2W bacteria and analyzed 14 days later. The ZEB2-deficient 2W:I-Ab–specific T cell population was about three times smaller than the wild-type one (Fig. 6E). This difference in size was associated with the preferential loss of CX3CR1+ T cells (Fig. 6F). Thus, the component of the Th1 amplification pathway that forms CX3CR1+ cytotoxic cells depends on ZEB2.
IL-12 amplified Th1 cells are more protective
Earlier work has shown that CXCR3+ CX3CR1− Th1 cells with robust IFN-γ production capacity and CX3CR1+ Th1 cells can both protect naïve hosts from Se infection after adoptive transfer (Goldberg et al., 2018). The fact that these are the subsets produced by the Th1 cell amplification process raised the possibility that the purpose of this module is to produce T cells that control phagosomal pathogens. This possibility was tested by comparing the protective capacity of T cell populations that had undergone Th1 cell amplification to ones that had not due to IL-12 blockade. Mice were infected with Se-2W bacteria and some were treated with IL-12 antibody. CD4+ T cells were purified from the secondary lymphoid organs of both groups seven days after infection and small samples were stained with 2W:I-Ab tetramer to assess the effectiveness of the block. As expected, the 2W:I-Ab-specific T cells proliferated in unblocked mice and formed a large population dominated by T-bethigh CXCR3+ cells with a minority CX3CR1+ subset, while the population in IL-12 antibody-treated mice was smaller and contained Tfh cells and T-bet+ CXCR3+ Th1 cells with less T-bet per cell and very few CX3CR1+ cells (Fig. 5A–E). The frequency of 2W:I-Ab tetramer-binding cells in each sample was then used to transfer populations containing the same number of 2W:I-Ab tetramer-binding cells into separate groups of naïve mice, thereby correcting for the differences in 2W and presumably other Se epitope-specific T cells in the two groups. Some of the recipients of CD4+ T cells from IL-12-treated donors were also treated with IL-12 antibody. The recipient mice and controls that did not receive CD4+ T cells were then infected with Se and viable bacteria in the spleen and liver were measured three days later.
Control naïve mice that did not receive CD4+ T cells had about 105 Se-2W bacteria in their spleens and livers (Fig. 7A). In contrast, mice that received CD4+ T cells from day seven Se-2W-infected donors contained ten-fold fewer bacteria in both organs, indicating that the transferred T cells had reduced the bacterial burden. Mice that received CD4+ T cells from IL-12 antibody-treated Se-2W-infected donors reduced the bacteria in the spleen three-fold compared to the controls and not at all in the liver whether or not the recipient mice were treated with IL-12 antibody. Thus, Se epitope-specific populations that had undergone Th1 cell amplification protected the spleen and especially the liver better than Se epitope-specific populations that had not undergone Th1 cell amplification.
Fig. 7. Amplified Th1 cells provide superior protection from Se infection.
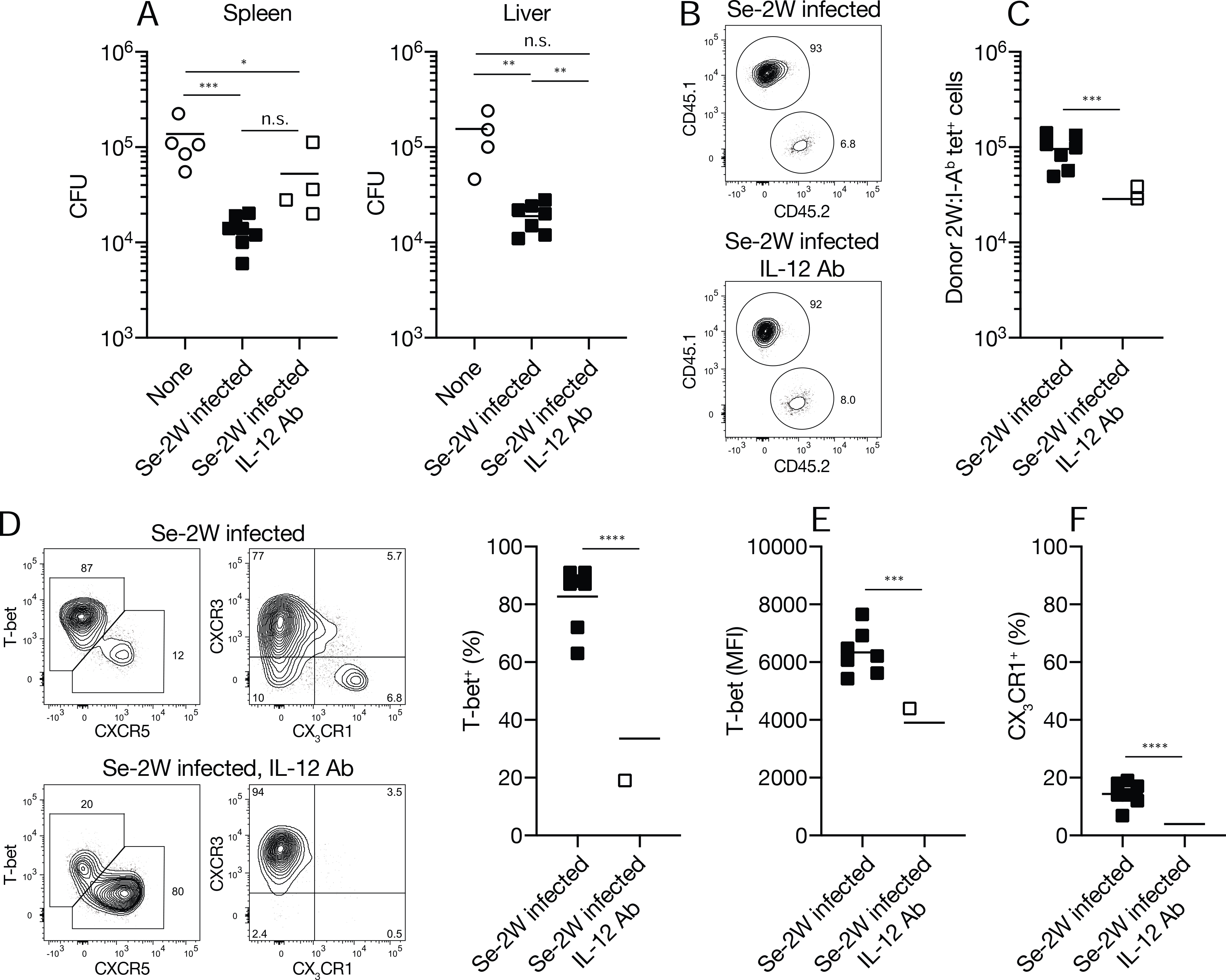
(A) CFU in the indicated organs from individual B6 mice (n = six per group from three independent experiments) that received CD4+ T cells from the indicated sources. (B) Flow cytometry plots used to identify 2W:I-Ab+ tetramer-binding T cells of donor (CD45.1) or recipient (CD45.2) origin in B6 mice that received CD4+ T cells from day seven Se-2W-infected (top) or IL-12 antibody-treated day seven Se-2W-infected mice (bottom). Some of the recipients of each type of donor T cell population were also treated with IL-12 antibody. IL-12 antibody treatment of the recipient mice had no discernable effect so the data for each donor population were pooled. (C) Number of 2W:I-Ab tetramer-binding T cells from individual day seven Se-2W-infected (top) or IL-12 antibody-treated day seven Se-2W-infected mice (bottom) (n = six per group from three independent experiments) after three days in Se-2W-infected recipients. (D) Flow cytometry plots of 2W:I-Ab tetramer-binding T cells from day seven Se-2W-infected or IL-12 antibody-treated day seven Se-2W-infected mice after three days in Se-2W-infected recipients, with scatter plots of percent T-bet+ cells. (E, F) Mean fluorescence intensity of T-bet+ cells (E) and percent CX3CR1+ cells (F) among T-bet+ 2W:I-Ab tetramer-binding from individual mice (n = six from three independent experiments). Values in A were compared using one-way ANOVA. Values in C-F were compared using an unpaired Student’s t-test. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns, not significant. Mean values on scatter plots are indicated with a horizontal bar.
The transferred T cell populations were monitored at the end of the three-day challenge period to see if their phenotype in the spleen had changed. The transferred populations were much larger than the recipient’s population, which had only begun a primary response (Fig. 7B). The transferred 2W:I-Ab-specific T cell population from unblocked mice was larger than the one from IL-12 blocked mice (Fig. 7C), indicating that the former proliferated more than the latter during the challenge period. Both populations, however, retained their pre-transfer phenotypes - the population from unblocked donors was dominated by T-bethigh CXCR3+ cells with some CX3CR1+ cells (Fig. 7D–F), while the population from IL-12 blocked donors contained Tfh cells (Fig. 7D), CXCR3+ Th1 cells with less T-bet per cell (Fig. 7E), and very few CX3CR1+ cells (Fig. 7F). Thus, the superior protective capacity of the population from unblocked Se-infected mice was associated with the presence of T-bethigh CXCR3+ cells and CX3CR1+ cells. These results demonstrate that the amplified Th1 cell population provides better protection from Se infection than the earlier mixed population of Th1 and Tfh cells.
DISCUSSION
The goal of this study was identification of the mechanisms that govern Th1 cell differentiation during infection with a phagosomal pathogen (Se) that is controlled by this lymphocyte subset. The epitope-tagged Se and IAV microbes studied here caused CD4+ naive T cells to form early effector cell populations composed of a balanced mixture of Tfh cells and Th1 cells as observed for Listeria monocytogenes, Lymphocytic Choriomeningitis Virus, and other IAV infections (Ballesteros-Tato et al., 2012; Choi et al., 2011; Johnston et al., 2009; Kunzli et al., 2020; Marshall et al., 2011; Pepper and Jenkins, 2011; Yu et al., 2009). Tfh and non-Tfh effector cells are also induced by Streptococcus pyogenes infection (Linehan et al., 2015) and immunization with house dust mite extract (Hondowicz et al., 2016) but in these cases the non-Tfh cells are Th17 and Th2 cells, respectively. Contemporaneous bifurcation of Th and non-Tfh cells thus appears to be a common activation module evolved to provide helper cells for both humoral and cell-mediated immunity. It results from a tug of war in which early effector cells that receive strong IL-2 receptor-mediated STAT5 signals induce BLIMP1, which represses BCL6, and become non-Tfh cells, while effector cells that receive weak IL-2 receptor-mediated STAT5 signals express BCL6 and become Tfh cells (Crotty, 2011). The fate of the non-Tfh cells is then determined by type 1, type 2, or type 17 innate immune system cytokines (Zhu et al., 2010). Although it is not totally clear what causes individual effector cells to become Tfh or non-Tfh cells, TCR signal strength is likely involved since it controls IL-2 receptor expression (Kotov et al., 2019; Krishnamoorthy et al., 2017).
Our results on in vivo generated Th1 cells are consistent with a two-step model of Th1 cell differentiation that emerges from cell culture experiments in the early 2000s (Mullen et al., 2001; Schulz et al., 2009). This model posited a first step in which TCR signaling stimulates IL-12-independent expression of T-bet and the IL-12 receptor, resulting in a Th1 cell population dominated by cells with weak IFN-γ production capacity. These cells then proceed to a second step in which IL-12 receptor signaling activates STAT4, which enhances T-bet expression, IFN-γ production capacity, and commitment to the Th1 lineage.
Our results indicate that the epitope-specific Th1 cells generated during IAV infection complete the first step of Th1 cell differentiation but do not proceed to the second. This contention is based on our findings that Th1 cells that formed during IAV infection did not depend on IL-12 as reported in studies of other viruses (Monteiro et al., 1998; Oxenius et al., 1999; Schijns et al., 1998; Xing, 2001), and only a minority had the potential to produce IFN-γ. Although the formation of about half of these Th1 cells depends on type 1 IFN as reported previously (Longhi et al., 2009), many other Th1 cells formed in the absence of IFNAR, IL-12, IL-18, or IFN-γ. These IFNAR-independent Th1 cells may form by default in response to TCR and IL-2 receptor-driven STAT5 signals when innate immune system cytokines that drive Th2 or Th17 cell differentiation are absent. The IFNAR-dependent and IFNAR-independent Th1 cells in IAV-infected mice may not undergo Th1 cell amplification because acute IAV infection does not sustain IL-12 production long enough. Our observation that Th1 cell amplification occurs in IAV-infected mice given exogenous IL-12 on day three and reports that certain chronic viral infections induce CX3CR1+ Th1 cells (Batista et al., 2020; De Giovanni et al., 2020; Weiskopf et al., 2015) are consistent with this possibility. Th1 cell populations containing only a minority of IFN-γ-producing cells may be advantageous to the host during IAV infection by providing a limited anti-viral effect without causing immunopathology. This contention is supported by reports that late IL-12 production actually increases viral burden (Ishikawa et al., 2016; Kostense et al., 1998; van der Sluijs et al., 2006), while IFN-γ deficiency reduces immunopathology during primary IAV infection (Califano et al., 2018).
Our studies show that Se infection induced the bifurcation module and generated early Tfh and Th1 effector cells. In this case, however, the Th1 cells underwent the IL-12-dependent second step of the Th1 cell differentiation process. A likely scenario for this progression is that sustained IL-12 production and IL-12 receptor signaling caused the CXCR3+ Th1 population generated during the bifurcation module to proliferate more than Tfh cells, increase expression of T-bet and IFN-γ production capacity, and generate CX3CR1+ progeny. The fact that Th1 cells proliferate longer than Tfh cells during the same infection has been described by Craft and colleagues and attributed to enhanced mTOR activation (Ray et al., 2015). The outgrowth of Th1 cells may contribute to poor germinal center formation during Se infection in addition to the directly suppressive effects of IL-12 on Tfh cell differentiation (Elsner and Shlomchik, 2019). Th1 outgrowth was associated with the formation of CX3CR1+ cells, which have been shown in other systems to arise from CXCR3+ precursors (Chu et al., 2016; Dominguez et al., 2015; Omilusik et al., 2015) by an IL-12-dependent mechanism (Sallin et al., 2017). Our results indicate that the generation of CX3CR1+ cells depends on the ZEB2 transcription factor, which is likely induced by STAT4 binding to the Zeb2 gene (Wei et al., 2010). ZEB2 turns on expression of several E-proteins (Omilusik et al., 2015), which may then stimulate production of CX3CR1, perforin, granzyme B, Fas ligand, and commit cells to the cytotoxic differentiation program.
The CD4+ T cell population in day seven Se-infected mice, which had begun the Th1 cell amplification process, provided better protection of the spleen and liver than a population stuck in the bifurcation model. This difference was likely related to the fact that the amplified Th1 cell population contained a greater number of IFN-γ-producing CXCR3+ cells and had a population of CX3CR1+ cytotoxic cells in both of these organs. In another study, we have shown that CXCR3+ Th1 cells are positioned around Se-containing granulomas in the spleen and liver and control the infection via an IFN-γ-dependent mechanism (Goldberg et al., 2018). Thus, one function of the Th1 cell amplification program is to generate cells that can position themselves at granuloma sites of persistent infection and produce the key cytokine needed for macrophages to kill the intracellular bacteria they harbor. Another function of this program is to generate CX3CR1+ cytotoxic Th1 cells, which also participate in control of Se infection, although to a lesser degree than CXCR3+ Th1 cells (Goldberg et al., 2018). CX3CR1+ Th1 cells may kill Se-infected macrophages harboring bacteria.
Although Th1 cell amplification benefits the host by controlling phagosomal pathogens, it may create a risk for more pathogenic viral infection as evidenced by the observation that people with tuberculosis had an increased risk of death during the 1918 H1N1 influenza pandemic (Noymer and Garenne, 2000). Phagosomal infections may produce a basal amount of IL-12, which could drive amplification of virus-specific Th1 cells capable of causing acute tissue damage and acute respiratory distress syndrome.
Limitations of Study
The fact that we only analyzed two pathogens is a limitation of our study. Studies of many other pathogens will be needed to establish that initial contemporaneous generation of Tfh and non-Tfh cells is a universal property of the CD4+ T cell response. Another limitation in our study is that it did not identify the mechanisms that generate IL-12 independent Th1 cells during module one. Lastly, additional studies independently targeting the two subsets of module two Th1 cells will be needed to determine their independent roles in protection against phagosomal pathogens.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Marc K. Jenkins (jenki002@umn.edu).
Materials Availability Statement
Mouse lines used in this study can be purchased, produced by breeding commercially available strains, or obtained from the donating lab(s) referenced. All additional reagents developed in the study will be made available from the Lead Contact.
Data and Code Availability Statement
The RNA sequencing results related to Fig. 6A have been deposited in GEO at the NCBI with accession number GSE168300.
EXPERIMENT MODEL AND SUBJECT DETAILS
Mice were housed in a specific pathogen-free facility at the University of Minnesota, and experiments were conducted according to federal and institutional guidelines and with the approval of the University of Minnesota Institutional Animal Care and Use Committee. 6- to 12-week-old, male and female, age and sex-matched mice were used for all experiments.
METHOD DETAILS
Mice
C57BL/6 (B6), B6.SJL-Ptprca Pep3b/BoyJ (CD45.1), B6.129S1-Il12btm1Jm/J (Il12b−/−) (Magram et al., 1996), B6.129S2-Ifnar1tm1Agt/Mmjax (Ifnar1−/−) (Muller et al., 1994), B6.129P2-Il18tm1Aki/J (Il18−/−) (Takeda et al., 1998), B6;129S1-Il12rb2tm1Jm/J (Il12rb2−/−) (Wu et al., 2000), B6N.129P2-Cx3cr1tm3(DTR)Litt/J (Diehl et al., 2013), Tg(Cd4-cre)1Cwi/BfluJ (Lee et al., 2001), and 129X1/SvJ (129) mice were used. B6 mice were bred to 129 mice to produce B6 × 129 F1 mice. B6N.129P2-Cx3cr1tm3(DTR)Litt/J mice were crossed to Tg(Cd4-cre)1Cwi/BfluJ, then crossed to 129 to produce Cd4Cre Cx3cr1DTR 129 mice. CD45.1.2 Cd4cre Zeb2fl/fl and Zeb2fl/fl bone marrow cells were provided by Kyla Omilusik and Ananda Goldrath at the University of California-San Diego. The data in all the figures came from mice on the B6 genetic background except for Figs. 6A and 6B–D, which contain data from B6 × 129 F1 and Cd4Cre Cx3cr1DTR 129 mice, respectively.
Infections
Mice were injected intravenously with 2 × 104 CFU Se-2W (AroA-deficient Se bacteria expressing 2W) (Benoun et al., 2018), or inoculated intranasally with 40 PFU of IAV-2W (influenza A/PR/8/34 virus expressing 2W) (Hemann et al., 2019). B6 × 129 F1 mice were given 108 CFU of Se-SL1344 (Goldberg et al., 2018) orally for the RNA sequencing experiments. B6 × 129 F1 and Cd4Cre Cx3cr1DTR 129 mice were infected orally with Se-SL1344–2W (Nelson et al., 2013) for in vivo cytotoxicity experiments.
Tetramers
Biotin-labeled I-Ab molecules containing the 2W (EAWGALANWAVDSA) or LpdA (HYFDPKVIPSI) peptides covalently attached to the I-Ab beta chain were produced with I-Ab alpha chains in Drosophila melanogaster S2 cells, then purified and tetramerized with streptavidin allophycocyanin (APC) as described previously (Moon et al., 2007).
Cell enrichment and flow cytometry
Tetramer-based cell enrichment was performed as previously described (Moon et al., 2007). Livers were perfused with Dulbecco’s PBS via the portal vein until fully blanched. Intrahepatic mononuclear cells were purified on a 21% Histodenz gradient after centrifugation at 1250 × g for 20 minutes without braking. Single cell suspensions of pooled spleens and lymph nodes or liver preparations were stained for one hour at room temperature with APC-conjugated peptide:I-Ab tetramers and BV650- or BUV395-conjugated CXCR5 antibodies, BV421-conjugated CXCR3 and BV605 conjugated CX3CR1 antibodies. Cells were enriched using EasySep Mouse APC Positive Selection kits according to the manufacturer’s instructions. Enriched samples were stained for 20 minutes at 4°C with fluorophore-conjugated antibodies specific for: CD4, CD90.2, B220, CD11b, CD11c, F4/80, CD44, or CD45.2, CD45.1. Cell viability was assessed using GhostDye Red 780. For analysis of the expression of transcription factors, stained cells were fixed and permeabilized with the Foxp3/Transcription Factor Staining Buffer Set according to the manufacturer’s instructions. Cells were stained overnight at 4°C with fluorophore-conjugated antibodies against: FOXP3, T-bet, RORγt or BCL6. Cells were counted and analyzed by flow cytometry with counting beads on a Fortessa (BD) flow cytometer. Data were analyzed using FlowJo software.
Intracellular cytokine detection
Infected mice were injected intravenously with 200 μg of 2W peptide in Dulbecco’s PBS for two hours before harvest. Spleen and lymph nodes were isolated in the presence of brefeldin A and stained according to methods described above. Cells were treated with Foxp3/Transcription Factor Staining Buffer Set according to the manufacturer’s instructions and stained overnight at 4°C with BV650-conjugated IFN-γ antibody.
Antibody neutralization and recombinant protein administration
IL-12p40 was neutralized in vivo by the intravenous injection of 1 mg of mAb C17.8 on days two and four post Se-2W infection or on days three and five post IAV-2W infection. IFN-γ was neutralized in vivo by the intravenous injection of 1 mg of mAb XMG1.2 on days three and five post IAV-2W infection. Recombinant mouse IL-12p70 was administered in vivo by intravenous injection of 60 ng each on days three, four, five and six post IAV-2W infection.
RNA-sequencing
Spleens and lymph nodes were harvested from B6 × 129 F1 mice 30 days after oral infection with 108 CFU Se-SL1344 bacteria. LpdAp:I-Ab-specific CD4+ T cells were purified using tetramer enrichment, and labeled with antibodies against mouse CD90.2, CD4, CD44, CXCR3, and CX3CR1 (all described above) for additional purification by cell sorting. Five thousand-50,000 LpdAp:I-Ab-specific CXCR3+ CX3CR1− and CXCR3− CX3CR1+ cells from four mice were sorted separately into RNAlater buffer and RNA was purified using the Qiagen RNeasy Plus micro spin column purification kit according to the manufacturer’s instructions. RNA integrity was confirmed using an Agilent Bioanalyzer and Dual-indexed Clontech Stranded Total RNA Pico Mammalian libraries were prepared by the University of Minnesota Genomics Center (UMGC). The libraries were then pooled and sequenced in 0.5 lanes of an Illumina HiSeq 2500 High-output 2×50-bp paired-end run using v4 chemistry. FastQ paired-end reads, averaging 16 million per sample, were trimmed using Trimmomatic v0.33 using a three-bp sliding-window from the 3’ end. Raw sequence data was quality controlled using FastQC and mapped to the GRCm38 (mm10) mouse genome assembly using Hisat2 v2.1.0. Differentially expressed genes between CXCR3+ CX3CR1− and CXCR3− CX3CR1+ populations were identified by applying a negative binomial distribution to the raw read counts using CLC Genomics Work Bench software (Qiagen). The list of differentially expressed genes was then filtered using an absolute fold change of two and an FDR corrected p-value < 0.05 as a minimal cut-off for further analysis.
In vivo cytotoxicity assay
Splenocytes were suspended in PBS (3 × 107 cells per milliliter) and incubated at 37°C for 10 minutes in 2 μM (hi) or 0.4 μM (lo) CTV, which was quenched by adding RPMI 1640 with 10% FBS. CTVlo cells were incubated for 1 hour at 37°C in RPMI 1640 with 10% FBS containing 10 μg/ml 2W peptide and the washed to remove free peptide. CTVhi and CTVlo cells were mixed at a 1:1 ratio, and 2 × 107 cells total cells were injected intravenously into individual mice. The recipient mice were sacrificed 20 hours later and flow cytometry was to determine the CTVlo/CTVhi ratio of B220+ B cells in the target cell population (Kotov et al., 2018). Specific lysis = 100 − (CTV ratio in experimental mice/CTV ratio in naive mice) × 100.
Se protection experiments
CD4+ T cells were purified from spleen and lymph node cell suspensions from B6 CD45.1+ mice seven days after Se-2W infection with or without IL-12 antibody treatment on days two and four, using CD4 negative selection. A small sample from each group was stained with fluorophore-conjugated 2W:I-Ab tetramer and CD4, CD44, CXCR5, CXCR3, and CX3CR1 antibodies and analyzed by flow cytometry to confirm the effectiveness of the IL-12 block and to determine the frequency of 2W:I-Ab tetramer-binding T cells. The frequency values were used to equalize the number of 2W:I-Ab tetramer-binding T cells in the two samples. Suspensions containing 2 × 105 2W:I-Ab tetramer-binding T cells were then injected into naïve B6 CD45.2+ mice. The recipient mice or controls that did not receive T cells were subsequently infected intravenously with Se-2W bacteria. Three days later, single cell suspensions of spleens and livers were plated on MacConkey Agar and bacterial colonies were counted the following day. A portion of the spleen samples was subjected to the 2W:I-Ab tetramer enrichment procedure described above with addition of CD45.1 and CD45.2 antibodies to the staining panel. The number of the 2W:I-Ab tetramer-binding cells and their phenotypes were determined as described above.
Bone marrow chimeras
Bone marrow cells were harvested from femurs and tibias. Five million each of CD45.1+ CD45.2+ Zeb2fl/fl or CD45.1+ CD45.2+ Cd4cre Zeb2fl/fl bone marrow cells were injected into lethally-irradiated (1,100 rads, split dose) CD45.1+ mice.
QUANTIFICATION AND STATISTICAL ANALYSIS
The statistical tests mentioned in the figure legends were performed using Prism software.
Supplementary Material
KEY RESOURCES TABLE (KRT).
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD90.2 - Brilliant Ultraviolet 395 (clone 53-2.1) | BD Biosciences | Cat# 565257, RRID:AB_2739136 |
| CD4 - Briliant Ultraviolet 496 (clone GK1.5) | BD Biosciences | Cat# 564667, RRID:AB_2722549 |
| CD44 - Brilliant Violet 510 (clone IM7) | BD Biosciences | Cat# 563114, RRID:AB_2738011 |
| Bcl6 - PE (clone K112-91) | BD Biosciences | Cat# 561522, RRID:AB_10717126 |
| RORgT - Brilliant Biolet 786 (clone Q31-378) | BD Biosciences | Cat# 564723, RRID:AB_2738916 |
| CXCR3 - Brilliant Violet 421 (clone CXCR3-173) | BD Biosciences | Cat# 566283, RRID:AB_2739657 |
| IFNg - Brilliant Violet 650 (clone XMG11.2) | BD Biosciences | Cat# 563854, RRID:AB_2738451 |
| CXCR5 - Brilliant Ultrabiolet 395 (clone 2G8) | BD Biosciences | Cat# 563980, RRID:AB_2738521 |
| CXCR5 - Brilliant Violet 650 (clone L138D7) | Biolegend | Cat# 145517, RRID:AB_2562453 |
| CX3CR1 - Brilliant Violet 605 (clone SA011F11) | Biolegend | Cat# 149027, RRID: AB_2565937 |
| T-bet - PE-Cy7 (clone 4B10) | Biolegend | Cat# 644824, RRID:AB_2561761 |
| TER-119 - Biotin (clone TER-119) | Biolegend | Cat# 116204, RRID:AB_313705 |
| CD45.2 - Brilliant Violet 786 (clone 104) | Biolegend | Cat# 109839, RRID:AB_2562604 |
| F4/80 - Biotin (clone BM8) | Biolegend | Cat# 123106, RRID:AB_893501 |
| NK-1.1 - Biotin (clone PK136) | Biolegend | Cat# 108704, RRID:AB_313391 |
| CD62L - Biotin (MEL-14) | Biolegend | Cat# 104403, RRID:AB_313090 |
| CD45.1 - PerCP-Cy5.5 (clone A20) | ThermoFisher | Cat# 45-0453-82, RRID:AB_1107003 |
| CD45R (B220) - APC-eFluor780 (clone RA3-6B2) | ThermoFisher | Cat# 47-0452, RRID:AB_1518810 |
| CD11b - APC-eFlour 780 (clone M1/70) | ThermoFisher | Cat# 47-0112, RRID:AB_1603193 |
| CD11c - APC-eFlour 780 (clone N418) | ThermoFisher | Cat# 47-0114, RRID:AB_1548652 |
| F4/80 - APC-eFlour 780 (clone BM8) | ThermoFisher | Cat# 47-4801, RRID: AB_1548745 |
| Foxp3 - AF488 (clone FJK-16s) | ThermoFisher | Cat# 53-5773-82, RRID:AB_763537 |
| T-bet - PE (clone eBio4B10) | ThermoFisher | Cat# 12-5825-82, RRID:AB_925761 |
| CD45R (B220) - Biotin (clone RA3-6B2) | Tonbo Biosciences | Cat# 30-0452, RRID:AB_2621644 |
| CD8a - Biotin (clone 53-6.7) | Tonbo Biosciences | Cat# 30-0081, RRID:AB_2621638 |
| Ly-6G (GR1) - Biotin (clone RB6-8C5) | Tonbo Biosciences | Cat# 30-5931, RRID:AB_2621652 |
| CD11 c - Biotin (clone N418) | Tonbo Biosciences | Cat# 30-0114, RRID:AB_2621640 |
| CD11b - Biotin (clone M1/70) | Tonbo Biosciences | Cat# 30-0112, RRID:AB_2621639 |
| InVivoMAb anti-mouse FcγII/III CD16/CD32 (clone 2.4G2) | BioXcell | Cat# BE0307, RRID:AB_2736987 |
| InVivoMAb anti-mouse IL-12p40 (clone C17.8) | BioXcell | Cat# BE0051, RRID:AB_1107698 |
| InVivoMAb anti-mouse IFNγ (clone XMG1.2) | BioXcell | Cat# BE0055, RRID: AB_1107694 |
| Bacterial and Virus Strains | ||
| Influenza A/PR/8/34 virus 2W | Hemann et. al., 2019 | R. Langlois, Univ. of Minnesota |
| Salmonella enterica: serovar Typhimurium: strain SL1344 | Stocker & Campbell, 1959 | B. Cookson, Univ. of Washington |
| Salmonella enterica: serovar Typhimurium: strain SL1344-2W | Nelson et al., 2013 | M. Jenkins Univ. of Minnesota |
| Salmonella enterica: serovar Typhimurium: strain BRD509-2W | Benoun et at., 2018 | S. McSorley, Univ. of California-Davis |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Histodenz | Milipore sigma | Cat# 66108-95-0 |
| PhycoLink Streptavidin-Allophycocyanin | Prozyme | Cat# PJ27S |
| 123count eBeads Counting Beads | ThermoFisher | Cat# 01-1234-42 |
| UltraComp eBeads Compensation Beads | ThermoFisher | Cat# 01-2222-42 |
| Streptomycin Sulfate | ThermoFisher | Cat# 11860-038 |
| Kanamycin Sulfate | ThermoFisher | Cat# 11815-032 |
| Dulbecco’s PBS | ThermoFisher | Cat# 14190144 |
| RNAlater™ Stabilization Solution | ThermoFisher | Cat# AM7020 |
| Mouse IL-12 p70 Recombinant Protein | ThermoFisher | Cat# 14-8121-80 |
| RPMI1640, L-glutamine | ThermoFisher | Cat# 11875119 |
| Fetal Bovine Serum | Omega Scientific | Cat# FB-12 |
| 2W peptide (EAWGALANWAVDSA) | GenScript | NIA |
| MacConkey Agar, Granulated | HIMEDIA | Cat# GMH081 |
| EasySep Streptavidin RapidSpheres | STEMCELL Technologies | Cat# 50001 |
| Ghost Dye Red 780 | Tonbo | Cat# 13-0865 |
| Brefeldin A Solution (1,000X) | Biolegend | Cat# 420601 |
| Permeabilization Buffer | Tonbo | Cat# TNB-1213-L150 |
| 2W:I-Ab biotinylated monomer | Moon et. at., 2007 | NIA |
| LpdAp:I-Ab biotinylated monomer | Goldberg et. al., 2018 | NIA |
| Critical Commercial Assays | ||
| CellTrace™ Violet Cell Proliferation Kit | ThermoFisher | C34557 |
| EasySep Mouse APC Positive Selection Kit II | STEMCELL Technologies | Cat# 17681 |
| RNeasy Mini Kit | Qiagen | Cat# 74104 |
| FoxP3/Transcription Factor Staining Buffer Set | ThermoFisher | Cat# 00-5523-00 |
| Experimental Models: Cell Lines | ||
| Drosophila S2 cells | ThermoFisher | Cat# R690-07 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6NCr (B6) | NCIICharles River | Strain Code 556 |
| Mouse: B6.Ly5.1/Cr (CD45.1) | NCIICharles River | Strain Code 564 |
| Mouse: 129X1/SvJ (129) | The Jackson Laboratory | Cat# 000691 |
| Mouse: B6 × 129 F1 | This Paper | N/A |
| Mouse: B6.129S1-Il12btm1Jm/J | The Jackson Laboratory | Cat# 002693 |
| Mouse: B6.129S2-Ifnar1tm1Agt/Mmjax | The Jackson Laboratory | Cat# 32045-JAX |
| Mouse: B6.129P2-Il18tm1Aki/J | The Jackson Laboratory | Cat# 004130 |
| Mouse: B6:129S1-Il12rb2tm1Jm/J | The Jackson Laboratory | Cat# 003248 |
| Mouse: B6N.129P2-Cx3cr1tm3(DTR)Litt/J | The Jackson Laboratory | Cat# 025629 |
| Mouse: Tg(Cd4-cre)1CwiBfluJ | The Jackson Laboratory | Cat# 017336 |
| Mouse: Cd4cre CX3CR1dtr x 129 | This Paper | N/A |
| Mouse: CD45.1.2 Cd4cre Zeb2fl/fl bone marrow | Ananda Goldrath | N/A |
| Mouse: CD45.1.2 Zeb2fl/fl bone marrow | Ananda Goldrath | N/A |
| Software and Algorithms | ||
| FlowJo v. 10 | TreeStar | https://www.flowjo.com |
| Hisat2 v2.1.0 | Public domain | http://daehwankimlab.github.io/hisat2/download/ |
| CLC Genomics Workbench | Qiagen | https://digitalinsights.qiagen.com/ |
| Prism 8 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Trimmomatic | Public domain | http://www.usadellab.org/cms/?page=trimmomatic |
Highlights.
Influenza and Salmonella induce Tfh and IL-12-independent early Th1 effector cells
Salmonella but not influenza drives later outgrowth of IL-12-dependent Th1 cells
The Salmonella-induced late Th1 population has protective CXCR3+ and CX3CR1+ cells
Salmonella-induced CX3CR1+ cells are cytotoxic and depend on ZEB2
ACKNOWLEDGMENTS
Supported by grants from the US National Institutes of Health R01 AI039614 and R01 AI103760 to M.K.J., T32 AI83196 to D.I.K., and T32 HL007062 to P. D. K. and M.F.G. The authors acknowledge Jennifer Walter and Charles Elwood for their help with maintaining mice and Juan Abrahante Llorens for his help with analysis of RNA sequence data.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
INCLUSION AND DIVERSITY
We worked to ensure sex balance in the selection of non-human subjects.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Afkarian M, Sedy JR, Yang J, Jacobson NG, Cereb N, Yang SY, Murphy TL, and Murphy KM (2002). T-bet is a STAT1-induced regulator of IL-12R expression in naive CD4+ T cells. Nat. Immunol. 3, 549–557. [DOI] [PubMed] [Google Scholar]
- Allan W, Tabi Z, Cleary A, and Doherty PC (1990). Cellular events in the lymph node and lung of mice with influenza. Consequences of depleting CD4+ T cells. J. Immunol. 144, 3980–3986. [PubMed] [Google Scholar]
- Baldridge JR, Barry RA, and Hinrichs DJ (1990). Expression of systemic protection and delayed-type hypersensitivity to Listeria monocytogenes is mediated by different T-cell subsets. Infect. Immun. 58, 654–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros-Tato A, Leon B, Graf BA, Moquin A, Adams PS, Lund FE, and Randall TD (2012). Interleukin-2 inhibits germinal center formation by limiting T follicular helper cell differentiation. Immunity 36, 847–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista NV, Chang YH, Chu KL, Wang KC, Girard M, and Watts TH (2020). T cell-intrinsic CX3CR1 marks the most differentiated effector CD4(+) T cells, but Is largely dispensable for CD4(+) T cell responses during chronic viral infection. Immunohorizons 4, 701–712. [DOI] [PubMed] [Google Scholar]
- Benoun JM, Peres NG, Wang N, Pham OH, Rudisill VL, Fogassy ZN, Whitney PG, Fernandez-Ruiz D, Gebhardt T, Pham QM, et al. (2018). Optimal protection against Salmonella infection requires noncirculating memory. Proc. Natl. Acad. Sci. U S A 115, 10416–10421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Califano D, Furuya Y, Roberts S, Avram D, McKenzie ANJ, and Metzger DW (2018). IFN-gamma increases susceptibility to influenza A infection through suppression of group II innate lymphoid cells. Mucosal. Immunol. 11, 209–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YS, Kageyama R, Eto D, Escobar TC, Johnston RJ, Monticelli L, Lao C, and Crotty S (2011). ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity 34, 932–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu HH, Chan SW, Gosling JP, Blanchard N, Tsitsiklis A, Lythe G, Shastri N, Molina-Paris C, and Robey EA (2016). Continuous effector CD8(+) T cell production in a controlled persistent infection Is sustained by a proliferative intermediate population. Immunity 45, 159–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotty S (2011). Follicular helper CD4 T cells (TFH). Annu. Rev. Immunol. 29, 621–663. [DOI] [PubMed] [Google Scholar]
- De Giovanni M, Cutillo V, Giladi A, Sala E, Maganuco CG, Medaglia C, Di Lucia P, Bono E, Cristofani C, Consolo E, et al. (2020). Spatiotemporal regulation of type I interferon expression determines the antiviral polarization of CD4(+) T cells. Nat. Immunol. 21, 321–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl GE, Longman RS, Zhang JX, Breart B, Galan C, Cuesta A, Schwab SR, and Littman DR (2013). Microbiota restricts trafficking of bacteria to mesenteric lymph nodes by CX(3)CR1(hi) cells. Nature 494, 116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez CX, Amezquita RA, Guan T, Marshall HD, Joshi NS, Kleinstein SH, and Kaech SM (2015). The transcription factors ZEB2 and T-bet cooperate to program cytotoxic T cell terminal differentiation in response to LCMV viral infection. J. Exp. Med. 212, 2041–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsner RA, and Shlomchik MJ (2019). IL-12 blocks Tfh cell differentiation during Salmonella infection, thereby contributing to germinal center suppression. Cell Rep. 29, 2796–2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg MF, Roeske EK, Ward LN, Pengo T, Dileepan T, Kotov DI, and Jenkins MK (2018). Salmonella persist in activated macrophages in T cell-sparse granulomas but are contained by surrounding CXCR3 ligand-positioned Th1 cells. Immunity 49, 1090–1102 e1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golstein P, and Griffiths GM (2018). An early history of T cell-mediated cytotoxicity. Nat. Rev. Immunol. 18, 527–535. [DOI] [PubMed] [Google Scholar]
- Groom JR, and Luster AD (2011). CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol. Cell Biol. 89, 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale JS, Youngblood B, Latner DR, Mohammed AU, Ye L, Akondy RS, Wu T, Iyer SS, and Ahmed R (2013). Distinct memory CD4+ T cells with commitment to T follicular helper- and T helper 1-cell lineages are generated after acute viral infection. Immunity 38, 805–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemann EA, Green R, Turnbull JB, Langlois RA, Savan R, and Gale M Jr. (2019). Interferon-lambda modulates dendritic cells to facilitate T cell immunity during infection with influenza A virus. Nat. Immunol. 20, 1035–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hondowicz BD, An D, Schenkel JM, Kim KS, Steach HR, Krishnamurty AT, Keitany GJ, Garza EN, Fraser KA, Moon JJ, et al. (2016). Interleukin-2-dependent allergen-specific tissue-resident memory cells drive asthma. Immunity 44, 155–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh CS, Macatonia SE, O’Garra A, and Murphy KM (1993). Pathogen-induced Th1 phenotype development in CD4+ alpha beta-TCR transgenic T cells is macrophage dependent. Int. Immunol. 5, 371–382. [DOI] [PubMed] [Google Scholar]
- Imai T, Hieshima K, Haskell C, Baba M, Nagira M, Nishimura M, Kakizaki M, Takagi S, Nomiyama H, Schall TJ, and Yoshie O (1997). Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell 91, 521–530. [DOI] [PubMed] [Google Scholar]
- Ishikawa H, Ino S, Sasaki H, Fukui T, Kohda C, and Tanaka K (2016). The protective effects of intranasal administration of IL-12 given before influenza virus infection and the negative effects of IL-12 treatment given after viral infection. J. Med. Virol. 88, 1487–1496. [DOI] [PubMed] [Google Scholar]
- Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, and Littman DR (2006). The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126, 1121–1133. [DOI] [PubMed] [Google Scholar]
- Jenkins MK, Chu HH, McLachlan JB, and Moon JJ (2010). On the composition of the preimmune repertoire of T cells specific for peptide-major histocompatibility complex ligands. Annu. Rev. Immunol. 28, 275–294. [DOI] [PubMed] [Google Scholar]
- Johanns TM, Ertelt JM, Rowe JH, and Way SS (2010). Regulatory T cell suppressive potency dictates the balance between bacterial proliferation and clearance during persistent Salmonella infection. PLoS Pathog. 6:e1001043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston RJ, Choi YS, Diamond JA, Yang JA, and Crotty S (2012). STAT5 is a potent negative regulator of TFH cell differentiation. J. Exp. Med. 209, 243–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston RJ, Poholek AC, DiToro D, Yusuf I, Eto D, Barnett B, Dent AL, Craft J, and Crotty S (2009). Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science 325, 1006–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplanski G (2018). Interleukin-18: Biological properties and role in disease pathogenesis. Immunol. Rev. 281, 138–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karunakaran KP, Yu H, Jiang X, Chan Q, Goldberg MF, Jenkins MK, Foster LJ, and Brunham RC (2017). Identification of MHC-bound peptides from dendritic cells infected with Salmonella enterica strain SL1344: Implications for a nontyphoidal Salmonella vaccine. J. Proteome Res. 16, 298–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoruts A, Mondino A, Pape KA, Reiner SL, and Jenkins MK (1998). A natural immunological adjuvant enhances T cell clonal expansion through a CD28-dependent, interleukin (IL)-2-independent mechanism. J. Exp. Med. 187, 225–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostense S, Sun WH, Cottey R, Taylor SF, Harmeling S, Zander D, Small PA Jr., and Bender BS (1998). Interleukin 12 administration enhances Th1 activity but delays recovery from influenza A virus infection in mice. Antiviral. Res. 38, 117–130. [DOI] [PubMed] [Google Scholar]
- Kotov DI, Kotov JA, Goldberg MF, and Jenkins MK (2018). Many Th cell subsets have Fas ligand-dependent cytotoxic potential. J. Immunol. 200, 2004–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotov DI, Mitchell JS, Pengo T, Ruedl C, Way SS, Langlois RA, Fife BT, and Jenkins MK (2019). TCR affinity biases Th cell differentiation by regulating CD25, Eef1e1, and Gbp2. J. Immunol. 202, 2535–2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamoorthy V, Kannanganat S, Maienschein-Cline M, Cook SL, Chen J, Bahroos N, Sievert E, Corse E, Chong A, and Sciammas R (2017). The IRF4 gene regulatory module functions as a read-write integrator to dynamically coordinate T helper cell fate. Immunity 47, 481–497 e487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunzli M, Schreiner D, Pereboom TC, Swarnalekha N, Litzler LC, Lotscher J, Ertuna YI, Roux J, Geier F, Jakob RP, et al. (2020). Long-lived T follicular helper cells retain plasticity and help sustain humoral immunity. Sci. Immunol. 5, eaay5552. [DOI] [PubMed] [Google Scholar]
- Ladel CH, Flesch IE, Arnoldi J, and Kaufmann SH (1994). Studies with MHC-deficient knock-out mice reveal impact of both MHC I- and MHC II-dependent T cell responses on Listeria monocytogenes infection. J. Immunol. 153, 3116–3122. [PubMed] [Google Scholar]
- Lee PP, Fitzpatrick DR, Beard C, Jessup HK, Lehar S, Makar KW, Perez-Melgosa M, Sweetser MT, Schlissel MS, Nguyen S, et al. (2001). A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity 15, 763–774. [DOI] [PubMed] [Google Scholar]
- Lighvani AA, Frucht DM, Jankovic D, Yamane H, Aliberti J, Hissong BD, Nguyen BV, Gadina M, Sher A, Paul WE, and O’Shea JJ (2001). T-bet is rapidly induced by interferon-gamma in lymphoid and myeloid cells. Proc. Natl. Acad. Sci. U S A 98, 15137–15142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linehan JL, Dileepan T, Kashem SW, Kaplan DH, Cleary P, and Jenkins MK (2015). Generation of Th17 cells in response to intranasal infection requires TGF-beta1 from dendritic cells and IL-6 from CD301b+ dendritic cells. Proc. Natl. Acad. Sci. U S A 112, 12782–12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longhi MP, Trumpfheller C, Idoyaga J, Caskey M, Matos I, Kluger C, Salazar AM, Colonna M, and Steinman RM (2009). Dendritic cells require a systemic type I interferon response to mature and induce CD4+ Th1 immunity with poly IC as adjuvant. J. Exp. Med. 206, 1589–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magram J, Connaughton SE, Warrier RR, Carvajal DM, Wu CY, Ferrante J, Stewart C, Sarmiento U, Faherty DA, and Gately MK (1996). IL-12-deficient mice are defective in IFN gamma production and type 1 cytokine responses. Immunity 4, 471–481. [DOI] [PubMed] [Google Scholar]
- Marshall HD, Chandele A, Jung YW, Meng H, Poholek AC, Parish IA, Rutishauser R, Cui W, Kleinstein SH, Craft J, and Kaech SM (2011). Differential expression of Ly6C and T-bet distinguish effector and memory Th1 CD4(+) cell properties during viral infection. Immunity 35, 633–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall NB, and Swain SL (2011). Cytotoxic CD4 T cells in antiviral immunity. J. Biomed. Biotechnol. 2011, 954602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matloubian M, Concepcion RJ, and Ahmed R (1994). CD4+ T cells are required to sustain CD8+ cytotoxic T-cell responses during chronic viral infection. J. Virol. 68, 8056–8063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monack DM, Bouley DM, and Falkow S (2004). Salmonella typhimurium persists within macrophages in the mesenteric lymph nodes of chronically infected Nramp1+/+ mice and can be reactivated by IFNgamma neutralization. J. Exp. Med. 199, 231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro JM, Harvey C, and Trinchieri G (1998). Role of interleukin-12 in primary influenza virus infection. J. Virol. 72, 4825–4831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon JJ, Chu HH, Pepper M, McSorley SJ, Jameson SC, Kedl RM, and Jenkins MK (2007). Naive CD4(+) T cell frequency varies for different epitopes and predicts repertoire diversity and response magnitude. Immunity 27, 203–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen AC, High FA, Hutchins AS, Lee HW, Villarino AV, Livingston DM, Kung AL, Cereb N, Yao T-P, Yang SY, and Reiner SL (2001). Role of T-bet in committment of Th1 cells before IL-12-dependent selection. Science 292, 1907–1910. [DOI] [PubMed] [Google Scholar]
- Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, and Aguet M (1994). Functional role of type I and type II interferons in antiviral defense. Science 264, 1918–1921. [DOI] [PubMed] [Google Scholar]
- Murphy KM, Ouyang W, Farrar JD, Yang J, Ranganath S, Asnagli H, Afkarian M, and Murphy TL (2000). Signaling and transcription in T helper development. Annu. Rev. Immunol. 18, 451–494. [DOI] [PubMed] [Google Scholar]
- Murphy KM, and Reiner SL (2002). The lineage decisions of helper T cells. Nat. Rev. Immunol. 2, 933–944. [DOI] [PubMed] [Google Scholar]
- Nelson RW, McLachlan JB, Kurtz JR, and Jenkins MK (2013). CD4+ T cell persistence and function after infection are maintained by low-level peptide:MHC class II presentation. J. Immunol. 190, 2828–2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noymer A, and Garenne M (2000). The 1918 influenza epidemic’s effects on sex differentials in mortality in the United States. Popul. Dev. Rev. 26, 565–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Shea JJ, and Paul WE (2010). Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science 327, 1098–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omilusik KD, Best JA, Yu B, Goossens S, Weidemann A, Nguyen JV, Seuntjens E, Stryjewska A, Zweier C, Roychoudhuri R, et al. (2015). Transcriptional repressor ZEB2 promotes terminal differentiation of CD8+ effector and memory T cell populations during infection. J. Exp. Med. 212, 2027–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxenius A, Karrer U, Zinkernagel RM, and Hengartner H (1999). IL-12 is not required for induction of type 1 cytokine responses in viral infections. J. Immunol. 162, 965–973. [PubMed] [Google Scholar]
- Pepper M, and Jenkins MK (2011). Origins of CD4(+) effector and central memory T cells. Nat. Immunol. 131, 467–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepper M, Pagán AJ, Igyártó BZ, Taylor JJ, and Jenkins MK (2011). Opposing signals from the Bcl6 transcription factor and the interleukin-2 receptor generate T helper 1 central and effector memory cells. Immunity 35, 583–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quah BJ, Wijesundara DK, Ranasinghe C, and Parish CR (2013). Fluorescent target array T helper assay: a multiplex flow cytometry assay to measure antigen-specific CD4+ T cell-mediated B cell help in vivo. J. Immunol. Meth. 387, 181–190. [DOI] [PubMed] [Google Scholar]
- Ray JP, Marshall HD, Laidlaw BJ, Staron MM, Kaech SM, and Craft J (2014). Transcription factor STAT3 and type I interferons are corepressive insulators for differentiation of follicular helper and T helper 1 cells. Immunity 40, 367–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray JP, Staron MM, Shyer JA, Ho PC, Marshall HD, Gray SM, Laidlaw BJ, Araki K, Ahmed R, Kaech SM, and Craft J (2015). The Interleukin-2-mTORc1 kinase axis defines the signaling, differentiation, and metabolism of T helper 1 and follicular B helper T cells. Immunity 43, 690–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees W, Bender J, Teague TK, Kedl RM, Crawford F, Marrack P, and Kappler J (1999). An inverse relationship between T cell receptor affinity and antigen dose during CD4(+) T cell responses in vivo and in vitro. Proc. Natl. Acad. Sci. U S A 96, 9781–9786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph MG, Stanfield RL, and Wilson IA (2006). How TCRs bind MHCs, peptides, and coreceptors. Annu. Rev. Immunol. 24, 419–466. [DOI] [PubMed] [Google Scholar]
- Sallin MA, Sakai S, Kauffman KD, Young HA, Zhu J, and Barber DL (2017). Th1 differentiation drives the accumulation of intravascular, non-protective CD4 T cells during tuberculosis. Cell Rep. 18, 3091–3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schijns VE, Haagmans BL, Wierda CM, Kruithof B, Heijnen IA, Alber G, and Horzinek MC (1998). Mice lacking IL-12 develop polarized Th1 cells during viral infection. J. Immunol. 160, 3958–3964. [PubMed] [Google Scholar]
- Schulz EG, Mariani L, Radbruch A, and Hofer T (2009). Sequential polarization and imprinting of type 1 T helper lymphocytes by interferon-gamma and interleukin-12. Immunity 30, 673–683. [DOI] [PubMed] [Google Scholar]
- Scott P (1991). IFN-gamma modulates the early development of Th1 and Th2 responses in a murine model of cutaneous leishmaniasis. J. Immunol. 147, 3149–3155. [PubMed] [Google Scholar]
- Sun B, and Zhang Y (2014). Overview of orchestration of CD4+ T cell subsets in immune responses. Adv. Exp. Med. Biol. 841, 1–13. [DOI] [PubMed] [Google Scholar]
- Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, and Glimcher LH (2000). A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 100, 655–669. [DOI] [PubMed] [Google Scholar]
- Takeda K, Tsutsui H, Yoshimoto T, Adachi O, Yoshida N, Kishimoto T, Okamura H, Nakanishi K, and Akira S (1998). Defective NK cell activity and Th1 response in IL-18-deficient mice. Immunity 8, 383–390. [DOI] [PubMed] [Google Scholar]
- Thieu VT, Yu Q, Chang HC, Yeh N, Nguyen ET, Sehra S, and Kaplan MH (2008). Signal transducer and activator of transcription 4 is required for the transcription factor T-bet to promote T helper 1 cell-fate determination. Immunity 29, 679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubo NJ, Fife BT, Pagan AJ, Kotov DI, Goldberg MF, and Jenkins MK (2016). Most microbe-specific naive CD4(+) T cells produce memory cells during infection. Science 351, 511–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubo NJ, and Jenkins MK (2014). CD4+ T Cells: guardians of the phagosome. Clin. Microbiol. Rev. 27, 200–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner DL, Bickham KL, Farber DL, and Lefrancois L (2013). Splenic priming of virus-specific CD8 T cells following influenza virus infection. J. Virol. 87, 4496–4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Sluijs KF, van Elden LJ, Xiao Y, Arens R, Nijhuis M, Schuurman R, Florquin S, Jansen HM, Lutter R, and van der Poll T (2006). IL-12 deficiency transiently improves viral clearance during the late phase of respiratory tract infection with influenza A virus in mice. Antiviral. Res. 70, 75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L, Vahedi G, Sun HW, Watford WT, Takatori H, Ramos HL, Takahashi H, Liang J, Gutierrez-Cruz G, Zang C, et al. (2010). Discrete roles of STAT4 and STAT6 transcription factors in tuning epigenetic modifications and transcription during T helper cell differentiation. Immunity 32, 840–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiskopf D, Bangs DJ, Sidney J, Kolla RV, De Silva AD, de Silva AM, Crotty S, Peters B, and Sette A (2015). Dengue virus infection elicits highly polarized CX3CR1+ cytotoxic CD4+ T cells associated with protective immunity. Proc. Natl. Acad. Sci. U S A 112, 4256–4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Wang X, Gadina M, O’Shea JJ, Presky DH, and Magram J (2000). IL-12 receptor beta 2 (IL-12R beta 2)-deficient mice are defective in IL-12-mediated signaling despite the presence of high affinity IL-12 binding sites. J. Immunol. 165, 6221–6228. [DOI] [PubMed] [Google Scholar]
- Xing Z (2001). Breach of IL-12 monopoly in the initiation of type 1 immunity to intracellular infections: IL-12 is not required. Cell. Mol. Biol. (Noisy-le-grand) 47, 689–694. [PubMed] [Google Scholar]
- Yu D, Rao S, Tsai LM, Lee SK, He Y, Sutcliffe EL, Srivastava M, Linterman M, Zheng L, Simpson N, et al. (2009). The transcriptional repressor Bcl-6 directs T follicular helper cell lineage commitment. Immunity 31, 457–468. [DOI] [PubMed] [Google Scholar]
- Zhu J, Yamane H, and Paul WE (2010). Differentiation of effector CD4 T cell populations. Annu. Rev. Immunol. 28, 445–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA sequencing results related to Fig. 6A have been deposited in GEO at the NCBI with accession number GSE168300.