

Abstract
Spore-forming pathogens like Clostridioides difficile depend on germination to initiate infection. During gemination, spores must degrade their cortex layer, which is a thick, protective layer of modified peptidoglycan. Cortex degradation depends on the presence of the spore-specific peptidoglycan modification, muramic-∂-lactam (MAL), which is specifically recognized by cortex lytic enzymes. In C. difficile, MAL production depends on the CwlD amidase and its binding partner, the GerS lipoprotein. To gain insight into how GerS regulates CwlD activity, we solved the crystal structure of the CwlD:GerS complex. In this structure, a GerS homodimer is bound to two CwlD monomers such that the CwlD active sites are exposed. Although CwlD structurally resembles amidase_3 family members, we found that CwlD does not bind Zn2+ stably on its own, unlike previously characterized amidase_3 enzymes. Instead, GerS binding to CwlD promotes CwlD binding to Zn2+, which is required for its catalytic mechanism. Thus, in determining the first structure of an amidase bound to its regulator, we reveal stabilization of Zn2+ co-factor binding as a novel mechanism for regulating bacterial amidase activity. Our results further suggest that allosteric regulation by binding partners may be a more widespread mode for regulating bacterial amidase activity than previously thought.
Author summary
Spore germination is essential for many spore-forming pathogens to initiate infection. In order for spores to germinate, they must degrade a thick, protective layer of cell wall known as the cortex. The enzymes that digest this layer selectively recognize the spore-specific cell wall modification, muramic-∂-lactam (MAL). MAL is made in part through the activity of the CwlD amidase, which is found in all spore-forming bacteria. While Bacillus subtilis CwlD appears to have amidase activity on its own, Clostridioides difficile CwlD activity depends on its binding partner, the GerS lipoprotein. To understand why C. difficile CwlD requires GerS, we determined the X-ray crystal structure of the CwlD:GerS complex and discovered that GerS binds to a site distant from CwlD’s active site. We also found that GerS stabilizes CwlD binding to its co-factor, Zn2+, indicating that GerS allosterically activates CwlD amidase. Notably, regulation at the level of Zn2+ binding has not previously been described for bacterial amidases, and GerS is the first protein to be shown to allosterically activate an amidase. Since binding partners of bacterial amidases were only first discovered 15 years ago, our results suggest that diverse mechanisms remain to be discovered for these critical enzymes.
Introduction
Many spore-forming pathogens, such as Clostridioides difficile, rely on germination to initiate infection. C. difficile is a toxin-producing bacterium that is the leading cause of healthcare-associated infections in many developed countries [1,2]. As with many spore-forming bacteria, successful germination depends on the degradation of the thick protective layer of modified peptidoglycan known as the cortex. The cortex functions to maintain spore metabolic dormancy [3] by keeping spores in a partially dehydrated state [3], so its removal is critical for metabolism to resume during germination. Cortex degradation is mediated by cortex lytic enzymes that recognize a cortex-specific peptidoglycan modification known as muramic-δ-lactam (MAL). This strict substrate specificity ensures that cortex lytic enzymes degrade the cortex and not the germ cell wall [3,4].
MAL residues are generated once the cortex layer is synthesized during sporulation [5]. The sequential action of two conserved enzymes, the CwlD amidase and PdaA deacetylase, produce MAL. CwlD removes the peptide side chain from N-acetylmuramic acid (NAM) [6–8] so that PdaA can deacetylate NAM and catalyze lactam ring formation to form MAL [6,9,10]. Mutants that lack MAL due to loss of either of these cortex-modifying enzymes exhibit severe germination defects in both Bacillus subtilis and C. difficile [6,8–11]. In the case of C. difficile, MAL-deficient mutants also exhibit virulence defects in animal models of infection [9,12], highlighting the critical importance of this process.
While the functions of CwlD and PdaA are conserved between B. subtilis and C. difficile [6,8–10], we recently showed that C. difficile CwlD amidase activity depends on the GerS lipoprotein [6,12]. Notably, GerS is unique to the Peptostreptococcaceae family, suggesting that CwlD amidase activity in this family may be differentially regulated relative to other spore-forming organisms [6,12]. Indeed, B. subtilis CwlD does not appear to require additional partners for its function [7]. These observations raise the question as to why C. difficile CwlD function depends on GerS. Given that GerS and CwlD directly interact [6], GerS binding to CwlD presumably regulates CwlD activity.
CwlD is a member of the amidase_3 family (PF01520), which play critical roles during cell division in many bacteria (e.g AmiC) [13]. Amidase_3 family members are Zn2+-dependent N-acetylmuramoyl amidases that use a Zn2+ ion as part of their catalytic mechanism [14]. The activity of these amidases is tightly regulated in many bacteria because their aberrant cleavage of the peptidoglycan layer can result in cell lysis [15,16]. Studies in the past ~10 years have revealed novel mechanisms for regulating amidase activity, with binding partners appearing to allosterically regulate amidase function [17]. In many Gram-negative bacteria, the highly conserved amidases, AmiA, AmiB, and AmiC, are activated upon binding regulators like EnvC, NlpD, and the recently discovered ActS [16,18–20]. These regulators likely displace an auto-inhibitory loop unique to this subset of amidases from the amidase active site [21,22]. In addition, the activity of the Staphylococcus aureus LytH amidase was recently shown to depend on the transmembrane protein, ActH [15]. While the mechanism by which ActH licenses LytH’s ability to denude peptidoglycan remains unclear, LytH’s ability to cleave stem peptides may be restricted to uncrosslinked peptidoglycan [15]. Taken together, these data suggest that amidase activation by binding partners may be a more widespread phenomenon that initially thought.
To gain insight into the molecular mechanism by which GerS regulates CwlD amidase activity, we solved the crystal structure of the CwlD:GerS complex. This is the first structure of an amidase bound to its regulator, and it reveals that GerS forms a homodimer that binds two CwlD monomers at a site opposite from CwlD’s active site. Since C. difficile CwlD adopts a typical amidase_3 fold in the complex, GerS binding likely impacts CwlD amidase activity at a distance. Indeed, our biochemical analyses indicate that C. difficile CwlD does not bind Zn2+ stably on its own in contrast with previously characterized amidase_3 family members. Instead, GerS binding to CwlD appears to promote CwlD binding to Zn2+, suggesting a novel mechanism for allosterically regulating amidase_3 family member function. Taken together, our analyses expand our understanding of the diverse mechanisms used by bacteria to regulate the activity of peptidoglycan amidases via protein-protein interactions.
Results
Analyzing the dimerization of CwlD and GerS and the stoichiometry of the CwlD:GerS complex
To investigate why CwlD activity depends on GerS, we sought to biochemically characterize the CwlD:GerS complex and identify residues required to mediate this interaction using X-ray crystallography. Constructs encoding the soluble domains of CwlD and GerS were over-expressed in Escherichia coli and a co-affinity purification strategy was used to purify untagged GerS bound to His-tagged CwlDΔ1–25 as previously described [6]. These domains correspond to CwlD and GerS lacking their predicted transmembrane (residues 1–25) and signal peptide/lipobox (residues 1–22) domains, respectively. The stability of this complex was analyzed using size exclusion chromatography (SEC) by comparing the elution profile of the complex relative to its individual CwlD and GerS proteins, which were both purified as His-tagged variants. CwlDΔ25-His6 alone primarily eluted as a monomer in solution, with a major peak corresponding to an apparent molecular weight (MW) of 33 kDa (Fig 1A) and a second less prominent peak corresponding to an apparent MW of 67 kDa, suggesting that CwlD can dimerize. Coomassie staining of the elution fractions confirmed that CwlD was the major protein detected in both peaks (Fig 1A). GerSΔ22-His6 alone eluted in 3 separate peaks, with apparent MWs of 23, 45 and 81 kDa (Fig 1B), strongly suggesting that GerS has the capacity to multimerize. The 23 kDa peak likely represents the monomeric form of the protein, since it corresponds to the expected MW; the 45 kDa peak likely corresponds to a dimer; and the 81 kDa peak likely represents a trimer or tetramer of GerS (Fig 1B). Coomassie staining of the three peaks confirmed that they contained predominantly GerS. Together, these results show that CwlD and GerS are mostly monomeric in solution but that GerS readily forms higher order complexes.
Fig 1. Size Exclusion Chromatography analyses of CwlD, GerS, and the CwlD:GerS complex.

Purified CwlDΔ25-His6 (A), GerSΔ22-His6 (B), and the CwlDΔ25-His6:GerSΔ22 complex (C) were analyzed by size exclusion chromatography. mAU corresponds to the UV absorbance measurements (A280) during the protein elution. The estimated molecular weights of the protein species are indicated at the top of each peak. The input sample is derived from the co-affinity purification high imidazole elution fraction. This input sample as well as the fractions (0.5 mL) from the indicated elution volumes were analyzed by SDS-PAGE and Coomassie staining. Dashed grey lines indicate the fractions collected. The data shown are from one biological replicate, which is representative of three biological replicates performed independently. The molecular weight (MW) marker is shown on the left-hand side of the gel.
SEC analysis of the CwlD:GerS complex revealed that it primarily eluted as a complex with a calculated MW of 72 kDa (1st peak, Fig 1C). This result would be consistent with a ternary complex of a GerS dimer (46 kDa) with a CwlD monomer (28 kDa), or vice versa. However, Coomassie staining of the SEC fractions revealed that the two proteins are present at a 1:1 ratio, suggesting that they bind stoichiometrically. In addition to this major peak, a smaller elution peak was observed that corresponds to an apparent MW of 27 kDa. Coomassie staining indicated that this peak primarily contained His-tagged CwlDΔ25 alone (Fig 1C). Since the input sample shows more CwlD than GerS, this band likely corresponds to free His-tagged CwlDΔ25 that is not bound to GerSΔ22.
Crystal structure of the CwlD:GerS complex
Overall complex structure
To gain atomic-level insight into how GerS activates CwlD activity, we crystallized the SEC-purified CwlD:GerS complex and determined its structure. The asymmetric unit contained one molecule of CwlD and one monomer of GerS with the CwlD structure being typical of the amidase superfamily. GerS is largely anti-parallel beta sheet with the exception of an N-terminal alpha helix and forms a cupped hand-like shape similar to the LolA family of lipid-associated proteins [23]. While the members of the LolA family are typically monomeric [24], completion of the beta fold for GerS required two molecules such that the N-terminal alpha helix through the first 2 beta strands comes from one molecule in the crystal and the remainder of the GerS fold comes from a symmetry mate (Fig 2A and 2B). Notably, both GerS molecules provide contacts to a single CwlD. This crossover-dependent assembly of the GerS fold is the same as was observed for the domain swapped assembly structure of the F47E mutant of LolA (PDBID 6FHM) [25]. Thus, the likely biological assembly for the CwlD:GerS complex contains two molecules of GerS and two CwlD and is in agreement with both dynamic light scattering (DLS) data (S1 Fig) and size exclusion chromatography (Fig 1). The combined buried surface area upon binding of CwlD to the two GerS molecules is 1415 Å2 and contains 10 hydrogen bonds and 6 salt bridges as calculated using PISA [26]. This interface is analogous to the interface observed for the C-terminal domain of the anti-anti-sigma factor MucB from P. aeruginosa (PDB ID 6IN8) [27] and the stress response factor RseB from E. coli (PDB ID 3M4W) [28].
Fig 2. Structure of the CwlD:GerS complex.

(A) Shown is the cartoon representation of the CwlD:GerS complex within the crystal with CwlD in blue tones and GerS in grey. The crystallographic asymmetric unit contains a single protomer of each with the 2 GerS/2 CwlD assembly formed by domain swapping of the GerS monomers. (B) The assembly is shown in space filling representation. (C) Least squares superposition of CwlD (cyan throughout) and its homologs performed using Superpose [44] in CCP4. (D) Close-up view of the metal binding site for CwlD with GerS from a fortuitous crystal packing (grey) providing coordination of the ion. (E) CwlD homolog structures (CwlD Paenibacillus polymyxa PDBID 1JWQ, unpublished; C. difficile CD2761 amidase_3 family member, 4RN7, unpublished; Nostoc punctiforme, AmiC2, 5EMI [29]; and C. difficile Cwp6, 5J72 [52]) in the active form have the Zn2+ atom accessible to the solvent. They also have water molecules serving as 1–2 ligands (blue spheres), one of which is coordinated by a conserved glutamate. (F) CwlD homolog structures (Bartonella henselae AmiB PDBID 3NE8 [22] and E. coli AmiC 4BIN [21]) in the inactive forms have residues from their respective inhibitory helices providing Zn2+ ligands.
Both CwlD and GerS have N-terminal sequences used for membrane association that were omitted for the crystallization constructs though neither would be occluded in the complex and would be free to extend to the membrane. For GerS, this would be an extension of the alpha helix on the periphery of the complex. For CwlD, the extension sits surface-exposed adjacent to the complex interface. Importantly, the binding orientation for CwlD relative to GerS leaves the CwlD Zn2+-containing active site solvent accessible to substrate rather than at the complex interface.
The CwlD structure
The structure of CwlD is the mixed alpha-beta fold expected for the N-acetylmuramoyl-L-alanine amidase superfamily (Fig 2C), with 3 conserved glutamate residues and 2 histidines coordinating the Zn2+ ion (Fig 2D). His49 (positioned by Glu53), His133, and the carboxylate of Glu65 provide four of the ligands of the near octahedral ion coordination with distances of 2.2 Å. Zn2+ coordination falls into two classes for the amidase family: (i) the active form where water molecules provide the 2 additional ion ligands, one of which coordinated by the conserved Glu199 position in CwlD (Fig 2E) and (ii) the autoinhibited form where a glutamate or glutamine from the inhibitory helix conserved in Ami family amidases [21,22] is liganded (Fig 2F). While CwlD does not have an autoinhibitory helix, the N-terminus of a GerS symmetry molecule provides the additional liganding for the Zn2+. As this symmetry molecule is not one from the 2 GerS monomers used to bind CwlD, the interaction is not physiologically relevant. Instead, it is a fortuitous crystal packing event resulting in the displacement of the water molecules expected to coordinate the Zn2+ in the active form of CwlD in solution.
The GerS structure and CwlD interface
The heavy atom-phased solutions revealed that the functional folding of GerS for binding to CwlD requires two molecules of GerS. The LolA-like conserved anti-parallel beta sheet of GerS folds into a pseudo half beta barrel utilizing 11 strands where CwlD sits. The topology for the strands of the beta fold is strand numbers 6-5-4-3-2-1-11-10-9-8-7 where strands 1 and 2 at the core of the fold come from the second molecule of GerS. The interaction of the N-terminal alpha helix and the embedding of beta stands 1 and 2 into the symmetry mate generates an extensive buried surface area of 3766 Å2 and is composed of 57 hydrogen bonds and 2 salt bridges involving residues 22 through 71 at which point crossover occurs. Alpha helix 1 (residues 65–82) and the C-terminal alpha helix (residues 217–235) of CwlD constitute the most significant interface with GerS contacting stands 2 through 6. A series of hydrophobic residues from strands 4–6 provide complementary packing for Trp233, Ile227, Tyr226 and Leu79 of CwlD. Numerous other interactions of interest occur at the CwlD:GerS interface. Arg169 from CwlD forms a salt bridge with Asp106 from strand 6 of GerS while His84 from the loop connecting strands 3 and 4 of GerS creates a communication network via Glu78 of helix 1 of CwlD to His61 of strand 2 of GerS.
Zn2+ stabilizes the CwlD:GerS complex
Our finding that CwlD in the co-crystal structure strongly resembles active amidase_3 family members raises the question as to whether GerS binding to CwlD induces conformational changes in CwlD and whether this binding event licenses CwlD activity. Our attempts to gain insight into this former question failed because we were unable to crystallize C. difficile CwlDΔ25-His6 on its own (Fig 1A) even though CwlDΔ25-His6 was stably folded in these DLS analyses (S1 Fig). Interestingly, DLS analyses of the SEC-purified CwlD:GerS complex revealed two peaks, one with a predicted radius of 120 kDa (which would correspond to a dimer of dimers of CwlD:GerS) and one that appeared to be aggregated protein. This latter peak may represent GerS that has dissociated from the SEC-purified complex.
To identify factors that might affect the stability of GerS binding to CwlD, we tested the effect of disrupting (i) CwlD binding to Zn2+ and (ii) CwlD’s Zn2+-dependent catalytic mechanism on the CwlD:GerS interaction. To disrupt CwlD’s ability to bind Zn2+, we generated constructs encoding alanine mutations of CwlD’s Zn2+-binding residues, His49 and Glu65. To disrupt CwlD’s catalytic mechanism, we mutated the putative catalytic Glu199 residue to alanine. We then co-produced these His-tagged CwlD variants in E. coli and measured their ability to pull-down untagged GerS using the co-affinity purification assay.
CwlD-His6 variants carrying the Zn2+-binding mutations (H49A and E65A) reduced the amount of untagged GerS that co-purified, although it was still possible to purify the mutant CwlD:GerS complexes (Fig 3A). In contrast, wild-type levels of GerS co-purified with the E199A variant. When the co-affinity purifications were analyzed by SEC, the CwlDH49A:GerS and CwlDE65A:GerS complexes (Fig 3B and 3C) eluted later than complexes containing either wild-type CwlD or CwlDE199A (Fig 3D). CwlDH49A:GerS and CwlDE65A:GerS complexes eluted with a calculated MW of 55 and 60 kDa, respectively, while the wild-type CwlD:GerS complex eluted with a predicted MW of 72 kDa (Fig 3B and 3C). Since the elution volume of proteins by SEC depends on both their shape and MW, these observations suggest that the shape and/or stoichiometry of CwlD:GerS complexes changes when CwlD is able to bind Zn2+. However, Coomassie staining of the different fractions revealed that both proteins are present at ratios close to 1:1, irrespective of the CwlD point mutations (Fig 3E). Based on these observations, loss of Zn2+-binding likely de-stabilizes the CwlD:GerS complex, although we cannot rule out the possibility that the CwlD Zn2+ binding mutations affect the stoichiometry of CwlD binding to GerS.
Fig 3. Mutation of CwlD Zn2+-binding residues, His49 and Glu65, decreases the stability of the CwlD:GerS complex.

(A) Coomassie stain of wild-type CwlD-His6, H49A, E65A or E199A His-tagged CwlD variants co-affinity purified with untagged GerS. The indicated proteins were produced in E. coli and purified using Ni2+- affinity resin. Cleared lysate and eluate fractions were analyzed using Coomassie staining. Comparison of the size exclusion chromatography traces of the wild-type CwlD:GerS complex with CwlDH49A:GerS (B), CwlDE65A:GerS (C) and CwlDE199A:GerS (D) complexes. mAU corresponds to the UV absorbance measurements (A280) during the protein elution. The 1st peak of each trace corresponds to the protein complex; the estimated molecular weight (MW) in kDa of the complex is given on top of this first peak. (E) Coomassie stain of fractions corresponding to the 1st and 2nd peak of each trace. The numbers at the bottom correspond to the apparent MW of each peak. The data shown are from one biological replicate, which is representative of three biological replicates performed independently.
To directly assess the effect of Zn2+ on the stability of the CwlD:GerS complex, co-affinity purified CwlD:GerS complex was incubated for one hour in the presence of either a five-fold excess of Zn2+ or 1 mM EDTA and then analyzed by SEC (Fig 4A). In the presence of Zn2+, the wild-type CwlD:GerS complex primarily eluted at a higher predicted MW (83 kDa), while incubation with EDTA resulted in three peaks that corresponded to predicted MWs of 65, 45, and 28 kDa (Fig 4A). Although Coomassie staining indicated that the complex was still present in the 65 and 45 kDa peaks, GerS appeared to be present at higher levels in the 45 kDa peak than CwlD, suggesting that this peak may comprise GerS dimers along with CwlD alone. Since the 28 kDa peak was primarily comprised of His-tagged CwlD, these analyses strongly suggest that removing Zn2+ from the CwlD:GerS complex via EDTA treatment promotes the dissociation of the complex. Notably, when the CwlDH49A:GerS complex was incubated with Zn2+ or EDTA, no changes in the SEC trace were observed (Fig 4B). Collectively, these results reinforce the notion that Zn2+ coordination affects the stability of the CwlD:GerS complex.
Fig 4. Zn2+ stabilizes the CwlD:GerS complex.

Purified wild-type CwlD:GerS (A) and CwlDH49A:GerS (B) complexes were analyzed by size exclusion chromatography after treatment with a five-fold excess of Zn2+Cl2 (green traces) or 1 mM EDTA (red traces) for 1 hr. mAU corresponds to the UV absorbance measurements (A280) during the protein elution. Fractions (0.5 mL) corresponding to the indicated elution volumes were analyzed by SDS-PAGE. The numbers at the bottom of the gels correspond to the apparent MW of each peak. The data shown are from one biological replicate, which is representative of three biological replicates performed independently.
To confirm that the CwlD Zn2+-binding residue mutations disrupt binding to Zn2+, we analyzed the Zn2+ content of SEC-purified wild-type CwlD:GerS and CwlDH49A:GerS complexes using inductively coupled plasma mass spectroscopy (ICP-MS). An average of about 500 ng Zn2+/ mg protein was detected in the wild-type CwlD:GerS, while only background levels of Zn2+ were detected in the CwlDH49A:GerS complex (Fig 5). The same result was observed for a CwlD:GerS complex harboring an alanine mutation in the CwlD Zn2+-binding residue, Glu65 (S1 Table). To validate the effects of these mutations on the individual proteins, we purified His-tagged wild-type CwlD and CwlDH49A on their own (S3 Fig), as well as His-tagged GerS alone (Fig 1B), and quantified the Zn2+ levels in these individual proteins. Intriguingly, no Zn2+ was detected in any of these samples, including with wild-type CwlD alone (Fig 5). This latter result was unexpected because Zn2+ is present in the crystal structures of many amidase_3 family members even though exogenous Zn2+ was not added to the expression or purification buffers in these studies [21,22,29]. Because of this surprising result, we purified another C. difficile amidase_3 homolog, CD2761, which binds Zn2+ in X-ray crystallography analyses (PBD ID 4RN7, unpublished). Even more Zn2+ was detected bound to CD2761 (~1100 ng Zn2+/mg protein) than to WT CwlD:GerS (~450 ng Zn2+/mg protein). Thus, if CwlD was able to bind Zn2+ stably on its own, we would expect to detect it using this method. Taken together, our results indicate that C. difficile CwlD behaves differently from all previously characterized amidase_3 family members to our knowledge in that it does not stably bind to Zn2+. Instead, GerS appears to promote CwlD binding to Zn2+. Conversely, CwlD binding to Zn2+ stabilizes the CwlD:GerS complex (compare CwlDH49A and CwlDE65A to CwlDWT), Figs 3 and 5), suggesting a mutual dependence between CwlD, Zn2+, and GerS binding at least in vitro.
Fig 5. Elemental analyses of CwlD and CwlD:GerS complex.
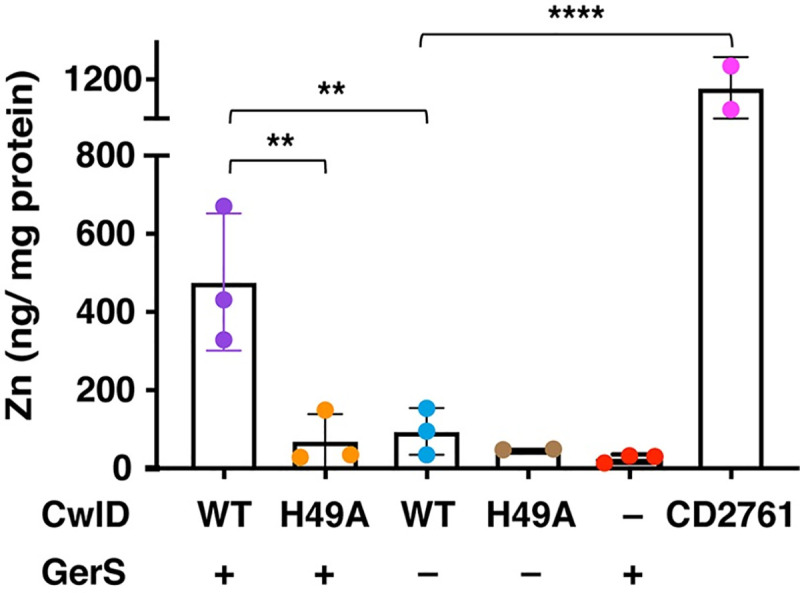
Elemental analyses of Zn2+ in CwlD:GerS complexes corresponding to His-tagged wild-type CwlD:GerS and CwlDH49A:GerS, and individual His-tagged proteins (wild-type CwlD, GerS and CD2761 and CwlDH49A). Elemental analyses were performed on affinity-purified proteins produced in E. coli in the absence of Zn2+ supplementation that were further purified using size exclusion chromatography (also in the absence of Zn2+ supplementation). Each dot represents data from a given biological replicate. Three independent protein purifications were analyzed with the exception for CwlDH49A and CD2761, for which two independent protein purifications were performed. Bars represent the mean of the three (or two) independent experiments. Standard deviation is shown. Statistical significance was determined using a one-way ANOVA and Tukey’s test. **, p < 0.005; **** p < 0.0001.
CwlD binding to Zn2+ is required for amidase activity and stabilizes both CwlD and GerS in mature spores
We next wanted to confirm that the Zn2+-binding residues and the presumed catalytic glutamate 199 are required for CwlD function in C. difficile. To this end, we designed cwlD constructs encoding mutations in the Zn2+-coordinating residues, His49 and Glu65, and in the putative catalytic Glu199 residue. We then assessed the ability of these constructs to complement the germination defect of a C. difficile ΔcwlD strain. All complementation constructs used in this study were integrated into the ectopic pyrE locus using allele-coupled exchange [30] and expressed from their native promoters. To analyze germination, we used an optical density-based assay that measures the decrease in optical density of a population of germinating spores over time due to cortex hydrolysis and rehydration of the spore cytosol [31]. While the optical density of wild-type spores decreased by around 40%, the optical density of the cwlD H49A, E65A and E199A complementation strains did not change, similar to the parental ΔcwlD strain (Fig 6A). Taken together, these results indicate that His49, Glu65 and Glu199 are all required for CwlD function.
Fig 6. Mutations in Zn2+ binding residues disrupt CwlD function and affect the stability of the CwlD:GerS complex in the spore.

(A) Spore germination as measured by the change in the OD600 in response to germinant in cwlD mutants spores relative to wild-type spores. Purified spores were resuspended in BHIS, and germination was induced by adding taurocholate (1% final concentration). The ratio of the OD600 of each strain at a given time point relative to the OD600 at time zero is plotted. The mean OD600 determined from three biological replicates from three independent spore preparations are shown. Shading represents the standard deviation as the area between error bars for each time point measured. Statistical significance relative to wild-type was determined using two-way ANOVA and Tukey’s test. **** p < 0.0001. (B) Western blot analysis of CwlD and GerS levels in sporulating cells from wild type (WT), ΔcwlD, and ΔcwlD strains complemented with constructs encoding the CwlD variants H49A, E65A and E199A. His49 and Glu65 directly bind Zn2+ in the structure, while Glu199 is part of the amidase catalytic mechanism. CwlD-c and GerS-c indicate the cleaved form of both proteins. Δspo0A (Δ0A) was used as negative control for sporulating cells, and SpoIVA was used as a loading control. (C) Western blot analysis of CwlD and GerS levels in spores from the strains used in the experiments described in (A). SleC was used as a loading control.
Interestingly, the Zn2+-binding mutations, H49A and E65A, decreased both CwlD and GerS levels in spores, while disruption of the putative catalytic glutamate E199A did not (Fig 6C). Notably, the CwlDH49A and CwlDE65A variants were detected at wild-type levels in sporulating cells, indicating that these Zn2+-binding mutations do not affect the production or stability of CwlD (Fig 6B). Instead, they appear to affect CwlD incorporation or stability in mature spores. This result is consistent with our previous observation that loss of CwlD decreases GerS levels in mature spores, whereas loss of GerS does not affect CwlD levels [6]. Since the Zn2+-binding mutations in CwlD did not affect the stability of recombinant His-tagged CwlD in SEC analyses (S3 Fig), our findings in C. difficile reinforce our biochemical data indicating that CwlD binding to Zn2+ stabilizes CwlD:GerS complex formation, which apparently affects CwlD stability or incorporation in mature spores.
Identification of a salt bridge critical for GerS binding to CwlD
To directly test whether GerS binding to CwlD stabilizes CwlD binding to Zn2+, we sought to identify mutations that disrupt CwlD:GerS complex formation separate from the Zn2+-binding mutations. Guided by the crystal structure of the complex, we identified two potential salt bridges that might promote CwlD:GerS binding: (i) CwlD arginine 169 (R169)-GerS aspartic acid 106 (D106) and (ii) CwlD glutamate 78 (E78)-GerS histidine 61 (H61) (Fig 7A). To assess the importance of these salt bridges in mediating binding between CwlD and GerS, we generated constructs encoding His-tagged CwlD variants carrying the following substitutions: arginine 169 to aspartic acid (R169D) or glutamine (R169Q) and glutamate 78 to glutamine (E78Q). We also generated untagged expression constructs encoding cognate salt bridge mutations in GerS, namely aspartate 106 to arginine (D106R) or alanine (D106A) and histidine 61 to alanine (H61A). We first co-produced the His-tagged CwlD variants in E. coli and measured their ability to pull-down untagged wild-type GerS using the co-affinity purification assay. CwlD variants carrying substitutions in Arg169 reduced GerS co-purification by 90% (Figs 7B, 7C and S4). Similarly, GerS variants carrying substitutions in Asp106 reduced GerS co-purifications with CwlD by ~90% also. Swapping the charges on the CwlD Arg169: GerS Asp106 interaction by combining the CwlD R169D and GerS D106R substitutions partially restored CwlD:GerS complex formation (Fig 7B and 7C), further highlighting the importance of this salt bridge in mediating complex formation. In contrast, substitutions in residues comprising the second salt bridge did not strongly affect CwlD or GerS binding, since mutation of the CwlDE78 or GerSH61 did not affect the levels of GerS that co-affinity purified with His-tagged CwlD (Fig 7D). Taken together, these results implicate the CwlDR169:GerSD106 salt bridge in stabilizing the interaction between recombinant CwlD amidase and its binding partner, GerS.
Fig 7. A salt bridge between CwlDR169 and GerSD106 is important for CwlD:GerS binding.

(A) Specific interactions between CwlD and GerS observed in the structure. Salt bridge formation between CwlD Arg169 (R169) and GerS Asp106 (D106) is highlighted in the solid boxes and between CwlD Glu78 (E78) and GerS His61 (H61) in the dotted boxes. The light blue mesh shows the heavy atom phased electron density after density modification and building contoured at 1 sigma. The yellow mesh represents the anomalous signal at 3 sigma for the selenomethionine derivative and the maroon represents the sodium iodide derivative after scaling, outlier rejection and phasing within Autosharp (Global Phasing Limited). (B) Coomassie stain of co-affinity purifications of wild-type or R169D CwlD-His6, with wild-type GerS or the D106R variant. The indicated proteins were produced in E. coli and purified using Ni2+- affinity resin. Cleared lysate (CL) and eluate (E) fractions were analyzed using Coomassie staining. (C) Percentage of GerS pulled down with CwlD relative to the amount detected in the wild-type CwlD:GerS complex, which was set at 100%. The mean of two assays from two independent co-affinity purifications is shown, and dots represent each of those co-affinity purifications. Statistical significance relative to the wild-type complex was determined using a one-way ANOVA and Tukey’s test. **** p < 0.0001. (D) Coomassie stain of cleared lysate and elution fractions from co-affinity purifications of His-tagged CwlD:GerS, CwlDE78Q:GerS, or CwlD:GerSH61A.
To confirm the importance of the salt bridge in C. difficile, we analyzed CwlD:GerS binding using co-immunoprecipitation analyses that employed FLAG-tagged CwlD as bait. To this end, we generated complementation constructs encoding either wild-type or R169D FLAG-tagged CwlD (CwlDFLAG). We then measured their ability to bind untagged GerS in sporulating C. difficile lysates. We also tested the effect of Zn2+-binding (H49A) and catalytic residue (E199A) mutations on CwlDFLAG:GerS complex formation by generating complementation constructs encoding FLAG-tagged variants carrying these mutations. When wild-type FLAG-tagged CwlD was immunoprecipitated from sporulating cell lysates, untagged GerS readily co-purified. In contrast, immunoprecipitation of CwlDR169D and CwlDH49A resulted in a 5-fold reduction in the amount of GerS pulled-down via FLAG-tagged CwlD (Fig 8A and 8B), consistent with the co-affinity purification analyses in E. coli (Fig 7B). CwlDE199A retained the ability to bind GerS, although this interaction decreased ~40% relative to the wild-type CwlD:GerS complex (Fig 8A and 8B). This finding could suggest that decreasing Zn2+ coordination by a water molecule in the active site decreases the affinity of CwlDE199A for GerS in sporulating cells.
Fig 8. Validation of the CwlD:GerS salt bridge by co-immunoprecipitation.

(A) Western blot of GerS in co-immunoprecipitation fractions obtained from pulling down FLAG-tagged proteins produced in sporulating cells for wild-type CwlD, CwlDR169D, CwlDH49A, or CwlDE199A. CwlD was detected using an anti-FLAG antibody, while untagged GerS was detected using an anti-GerS antibody. CwlD-c and GerS-c indicate cleaved form of both proteins. (B) Percentage of GerS pulled down with FLAG-tagged CwlD relative to the amount detected in pull-downs with FLAG-tagged wild-type CwlD. A mean of three independent assays is shown, except for CwlDE199A-FLAG, for which two independent biological replicates were performed. Dots represent the % of GerS pulled-down in each biological replicate. Statistical significance relative to the wild-type complex was determined using one-way ANOVA and Tukey’s test. **, p< 0.005; **** p < 0.0001.
Interestingly, the primary form of GerS detected in co-immunoprecipitations with FLAG-tagged CwlD was the cleaved form of GerS (Fig 8A). As a lipoprotein, GerS is secreted across the outer membrane by its N-terminal signal peptide in sporulating cells [12]. Following secretion, GerS gets lipidated and then its signal peptide is removed through the action of the lipoprotein signal peptidase, Lsp [32]. Notably, while full-length GerS and cleaved GerS are detectable in sporulating cells, only cleaved GerS is detected in mature spores [12] consistent with its maturation as a lipoprotein [32]. These observations strongly suggest that GerS and CwlD interact after they are transported across the outer membrane. Altogether, our data indicate that stable CwlD:GerS complex formation depends on a salt bridge formed between CwlDR169 and GerSD106 and that stable binding between CwlD and GerS likely happens in the outer forespore membrane.
Functional analyses of the salt bridge in C. difficile
To test whether the CwlD:GerS interaction is critical for the CwlD function in C. difficile, we analyzed the germination phenotype of the cwlD complementation strain producing FLAG-tagged CwlDR169D. This strain was used in the co-immunoprecipitation assays described above (Fig 8A). We also assessed the effect of a GerS D106R substitution on spore germination by complementing ΔgerS with a construct encoding FLAG-tagged GerSD106R. The germination phenotypes of the ΔcwlD/cwlDR169D-FLAG and ΔgerS/gerS D106R-FLAG strains were analyzed using the optical density-based spore germination assay. While the optical density of the parental ΔcwlD and ΔgerS spores did not significantly decrease over the course of 90 min [6], the optical density of the ΔcwlD/cwlDR169D-FLAG and ΔgerS/gerS D106R-FLAG spores decreased by >40% over this time. ΔcwlD/cwlDR169D-FLAG mutant spores germinated like wild-type spores and the wild-type complementation spores, ΔcwlD/cwlDFLAG and ΔgerS/gerSFLAG spores (Figs 9A and S5A), whereas ΔgerS/gerSD106R-FLAG mutant spores germinated more slowly than wild-type and wild-type complementation spores (p < 0.001, Fig 9A).
Fig 9. A GerSD106R mutation affects the stability of GerS protein and leads to slower germination.

(A) Spore germination as measured by the change in OD600 in response to germinant in gerSD106R and cwlDR169D mutant spores relative to wild-type spores. Purified spores were resuspended in BHIS, and germination was induced by adding taurocholate (1% final concentration). The ratio of the OD600 of each strain at a given time point relative to the OD600 at time zero is plotted. The mean of three assays from 3 independent spore preparations are shown. Shading represents the standard deviation as the area between error bars for each time point measured. Statistical significance relative to wild-type was determined using two-way ANOVA and Tukey’s test. ***, p< 0.0005; **** p < 0.0001. (B) Germination efficiency of gerSD106R and cwlDR169D point mutant spores. The number of colony forming units (CFUs) produced by germinating spores is shown. The mean OD600 determined from three independent spore preparations is shown along with the associated standard deviations. Statistical analyses relative to the wild-type were performed using one-way ANOVA and Tukey’s test. **, p < 0.005; ****, p < 0.0001. (C) Western blot analysis of CwlD and GerS levels in sporulating cells from the wild-type (WT), ΔgerS, ΔgerS complemented with either gerS, or gerS encoding either wild-type or D106R FLAG-tagged GerS variants as well as ΔcwlD, ΔcwlD complemented with either cwlD encoding either wild-type or R169D GerS variants. CwlD-c and GerS-c indicate cleaved form of both wild-type proteins; asterisks indicate the cleaved form of FLAG-tagged. Δspo0A (Δ0A) was used as negative control for sporulating cells, and SpoIVA was used as a loading control. GerS and CwlD levels were analyzed using GerS, CwlD and FLAG antibodies. (B) Western blot analysis of CwlD and GerS levels in spores from the strains used in the experiments described in (C). SleC was used as a loading control.
To better understand the slower germination observed in this optical density-based assay for ΔgerS/gerSD106R-FLAG spores, we visually monitored their germination using phase-contrast microscopy. One hour after inducing germination, all wild-type and ΔgerS/gerSFLAG spores lost their phase-bright appearance (S6A Fig) as the cortex is degraded and the core hydrates [33]. In contrast, ΔgerS/gerSD106RFLAG spores germinated more heterogeneously, with ~30% remaining phase-bright during this time period (S6A and S6B Fig). ΔgerS spores remained phase-bright during this time consistent with their inability to degrade the cortex, although some spore germination was detectable at the 90’ timepoint [12].
We next analyzed the germination efficiency of the cwlD and gerS point mutants by plating spores on media containing germinant. While the ΔcwlD and ΔgerS parental strains exhibited a ~2 log decrease in spore germination efficiency relative to the wild-type strain (Fig 9B), no statistically significant defect in spore germination was observed for cwlDR169D-FLAG and gerSD106R-FLAG mutant spores relative to wild-type or the ΔcwlD/cwlDFLAG and ΔgerS/gerSFLAG complementation strain spores (Fig 9B).
Since CwlD enhances the levels of GerS in mature spores [6], and CwlD Zn2+-binding mutations affect both CwlD and GerS levels in mature spores (Fig 6), we analyzed the levels of CwlD and GerS in the cwlDR169D-FLAG and gerSD106R-FLAG mutant spores. Interestingly, both the cwlDR169D-FLAG and gerSD106R-FLAG mutations led to lower levels of GerS in spores, while CwlD levels were unaffected by these mutations (Fig 9D). The CwlDR169D-FLAG variant also ran at a higher apparent MW than wild-type CwlDFLAG. To ensure that the point mutations did not affect the stability of these proteins in sporulating cells, we analyzed the levels of these proteins in the different complementation strains using western blotting. CwlDR169D-FLAG levels were similar to those detected in the wild-type ΔcwlD/cwlDFLAG complementation strain. Even though lower levels of GerSD106R-FLAG were detected using the GerS antibody in sporulating cells, no differences were found at the level of this protein with the anti-FLAG antibody (Fig 9C). This discrepancy suggests that our anti-GerS antibody binds the GerSD106R variant less efficiently than wild-type GerS. Consistent with the GerSD106R substitution causing a conformational change in GerS, GerSD106R-FLAG ran at a different apparent MW than wild-type GerSFLAG in sporulating cells. Taken together, these results indicate that disruption of the CwlD:GerS salt bridge decreases the stability of CwlD binding to GerS in both E. coli and C. difficile (Figs 7 and 8). In the case of the GerSD106R substitution, decreasing GerS binding to CwlD reduces CwlD function partially in optical density and phase-contrast-based germination assays (Figs 9A and S6). However, since the salt bridge mutations did not markedly impair spore germination despite severely decreasing CwlD:GerS binding, our results suggest that low levels of GerS are sufficient to license CwlD activity.
Discussion
The activity of cell wall hydrolases like amidases must be tightly regulated during bacterial growth to avoid perturbations that can lead to cell lysis [34]. A wide array of mechanisms control cell wall hydrolase activity, including transcription, protein localization, stability, peptidoglycan modification, and protein-protein interactions [17]. The regulation of cell wall amidases by protein-protein interactions was first reported only 12 years ago and was previously thought to be largely restricted to the AmiA/B/C amidases of Gram-negative bacteria [16,22] until the activities of the LytH amidase of Staphylococcus aureus [15] and CwlD amidase of C. difficile (6) were found to depend upon binding partners. However, in the absence of structures of the amidases bound to their protein regulators, it remained unclear whether these amidases were allosterically regulated by their binding partners.
In determining the structure of the C. difficile CwlD amidase bound to its protein regulator, the GerS lipoprotein, and demonstrating that GerS binds CwlD at a site distant from CwlD’s active site (Fig 2), we show in this study that the C. difficile CwlD amidase is allosterically activated by the Peptostreptococcaceae-specific GerS lipoprotein. Our biochemical data strongly suggest that GerS binding to CwlD promotes CwlD binding to Zn2+, which directly participates in the catalytic mechanism of amidase_3 family members. Unlike all previously characterized Amidase_3 family members studied to our knowledge [15,17,21,22,29], SEC-purified CwlD does not bind Zn2+ stably on its own in ICP-MS analyses, whereas Zn2+ is readily detected in the SEC-purified CwlD:GerS complex (Fig 5). In addition, the stability of the CwlD:GerS complex depends on CwlD binding to Zn2+ in both E. coli and C. difficile (Figs 3, 4 and 6), and disrupting CwlD Zn2+ binding appears to destabilize CwlD in mature C. difficile spores (Fig 5). Thus, Zn2+ plays a structural role in regulating CwlD activity beyond its direct role in catalysis. Consistent with this hypothesis, mutation of a salt bridge at the CwlD:GerS interface (CwlDR169:GerSD106) destabilizes the complex in E. coli and C. difficile (Figs 7 and 8), reduces GerS levels in mature spores, and impairs (albeit subtly) CwlD function in germination analyses of ΔgerS/gerSD106R spores (Figs 9 and S6). Notably, this salt bridge is conserved across Peptostreptococcaceae family members, suggesting that this interaction plays a similar role in related GerS homologs.
The salt bridge mutations provided further insight into when and where CwlD and GerS interact, since these mutations greatly reduced GerS levels in mature spores without affecting GerS levels in sporulating cells (Fig 9). Given that the co-immunoprecipitation data revealed that only the secreted, cleaved form of GerS binds full-length, FLAG-tagged CwlD (Fig 8), CwlD:GerS complex formation apparently stabilizes GerS in mature spores. Taken together, these results imply that the allosteric conduit connecting GerS to CwlD’s active site functions as a two-way street, with CwlD binding to GerS affecting GerS’s conformational stability, and GerS binding to CwlD controlling CwlD’s ability to bind Zn2+.
Based on these observations, we propose the following model. GerS binds CwlD only after GerS is secreted across mother cell-derived membranes and completes its lipoprotein maturation process. CwlD is also transported across mother cell-derived membranes via its predicted N-terminal transmembrane domain [35], but only its full-length, membrane-bound form binds lipidated GerS. This binding event likely triggers a conformational change in CwlD that promotes Zn2+ binding, since Zn2+ is only found when CwlD is complexed with GerS (Fig 5). This interaction may also help GerS orient CwlD’s active site towards its cortex peptidoglycan substrate close to the membrane, since only the membrane-bound forms of these proteins interact.
Despite these conclusions, our study raises several questions regarding the regulation of CwlD amidase activity. Does GerS regulate CwlD amidase activity outside of GerS’s effect on CwlD binding to Zn2+? Does GerS need to remain bound to CwlD for CwlD to function as an amidase? Why does C. difficile CwlD require a binding partner when the B. subtilis CwlD can remodel peptidoglycan on its own when heterologously produced in E. coli [7]? Testing whether B. subtilis CwlD alone can bind Zn2+ and whether the structures of these two CwlD homologs differ will provide important insight into these questions, although it is also possible that B. subtilis (or other organisms lacking GerS homologs) encode an unknown regulator that modulates CwlD activity. Finally, what is the signaling conduit that allows GerS binding to an allosteric site on CwlD to stabilize CwlD active site binding to Zn2+?
Addressing this latter question would be greatly facilitated by structural characterization of CwlD alone. While we were unable to crystallize only CwlD, crystallographic analyses of Ami family amidases prior to their activation by protein regulators indicate that E. coli AmiC and Bartonella henselae AmiB are auto-inhibited by a regulatory α-helix that blocks access to their active sites [21,22]. Notably, CwlD homologs lack this regulatory helix (Fig 2), and amidases that lack this helix, such as Nostoc punctiforme AmiC2 and Bacillus polymyxa CwlV are active on their own [29,36]. While the precise mechanism by which the binding partners of Ami family amidases, EnvC, NlpD, and the recently discovered ActS, activate these amidases [20,37] has yet to be characterized, the mechanism that allosterically activates C. difficile CwlD almost certainly differs from the Ami family amidases.
Another question to be resolved is how CwlD binding to GerS affects GerS structure and thus stability. In the co-crystal structure, two GerS subunits intimately dimerize in a hand-shake like manner (Figs 1 and 2). Given the extensive interactions between the two monomers, GerS dimerization is likely critical for GerS to complex with CwlD. GerS dimerization may also help position CwlD’s active site towards its peptidoglycan substrate, since GerS binds close to CwlD’s N-terminal membrane-associated region in the co-crystal structure (Fig 2).
One way to address the importance of GerS dimerization would be by targeting amino acid residues in the cross-over arm. However, this may be challenging because those mutations could affect the stability of GerS itself. Interestingly, when the β-strand exchange between GerS subunits is eliminated by creating a GerS pseudo-monomer, GerS exhibits structural homology to the LolA/LolB family of proteins, which are responsible for the transport of lipoproteins across the periplasm of Gram-negative bacteria during assembly of the outer membrane [23]. The significance of this structural similarity remains unclear, since most LolA superfamily structures are monomeric. Intriguingly, a single mutation in LolA (F47E) has been reported to convert LolA from a monomer into a domain-swapped homodimer, and the homodimer binds LolA’s interacting partner, LolC, more efficiently [25]. The equivalent residue in C. difficile GerS is an aspartate, raising the possibility that GerS could be converted into a monomer like LolA by mutating to phenylalanine. While future work will address the importance of GerS dimerization to its function, it is interesting to note that proteins with structural homology to GerS, LolA/LolB and RseB/MucB, bind lipids [24,38]. Thus, GerS may also bind lipids, which could impact its localization in sporulating cells.
While we cannot rule out the possibility that GerS regulates multiple aspects of CwlD function, e.g. localization, our results collectively suggest a new mechanism for activating amidases via stabilization of Zn2+ co-factor binding and provide novel structural insight into how cell wall hydrolase activity can be regulated by protein binding partners. Indeed, stabilizing co-factor binding to enzymes could be a general strategy used by other peptidoglycan hydrolases. Also, given the diversity of hydrolases and regulators, distinct structures and regulatory mechanisms exist likely remain to be identified [17]. Finally, since both GerS lipoprotein and PdaA deacetylase impact C. difficile infection [9,12], our study suggests that inhibiting CwlD:GerS binding during spore formation could impair C. difficile spore germination and thus reduce disease transmission and recurrence.
Material and methods
Bacterial strains and growth conditions
C. difficile strain construction was performed using 630ΔermΔcwlDΔpyrE and 630ΔermΔgerSΔpyrE as the parental strains via pyrE-based allelic-coupled exchange (ACE ([30]). This system allows for single-copy complementation of the ΔcwlD and ΔgerS parental mutants, from an ectopic locus. The C. difficile strains used are listed in S2 Table. They were grown on brain heart infusion media (BHIS) supplemented with taurocholate (TA, 0.1% wt/vol; 1.9 mM), thiamphenicol (10–15 μg /mL), kanamycin (50 μg/ml), cefoxitin (8 μg/ml) and L-cysteine (0.1% w/v; 8.25 mM) as needed. For ACE, C. difficile defined medium (CDDM) [39] was supplemented with 5-fluoroorotic acid (5-FOA) at 2 mg/ml and uracil at 5 μg/ml. Cultures were grown at 37°C under anaerobic conditions using a gas mixture containing 85% N2, 5% CO2 and 10% H2.
The Escherichia coli strains used for BL21(DE3)-based protein production and for HB101/pRK24-based conjugations are listed in S2 Table. E. coli strains were grown at 37°C with shaking at 225 rpm in Luria-Bertani broth (LB). The media was supplemented with chloramphenicol (20 μg/ml), ampicillin (50 μg/ml), or kanamycin (30 μg/ml) as indicated.
E. coli strain construction
All primers used for cloning are listed in S3 Table. Details of E. coli strain construction are provided in S1 Text. Plasmid constructs were confirmed by sequencing using Genewiz and transformed into either the HB101/pRK24 conjugation strain (used with C. difficile) or BL21(DE3) expression strain.
C. difficile strain construction and complementation
Allele-couple exchange (ACE) [30] was used to construct all complementation strains. Complementations were performed as previously described by conjugating HB101/ pRK24 carrying pMTL-YN1C plasmids into ΔpyrE-based strains [40] using allele-coupled exchange.
Protein purification for recombinant E. coli analyses
Starter cultures were grown in 20 mL LB broth with 100 μg/mL ampicillin and, in some cases, 30 μg/mL of kanamycin. Terrific broth (TB) with ampicillin and kanamycin was inoculated with the starter culture (1:1000) and incubated for ~60 hr at 20°C with 225 rpm shaking. The purifications were carried out in two steps: nickel bead-based affinity purification followed by size exclusion chromatography (SEC). Affinity purification was carried out as stated above: the cells were pelleted, resuspended in lysis buffer (500 mM NaCl, 50 mM Tris [pH 7.5], 15 mM imidazole, 10% [vol/vol] glycerol, 2 mM ß-mercaptoethanol), flash frozen in liquid nitrogen, thawed and then sonicated. The insoluble material was pelleted and the soluble fraction was incubated with Ni-NTA agarose beads (Qiagen) for 2 hours to capture the His6 tagged proteins. Washes were carried out three times with low imidazole buffer (500mM NaCl, 10mM Tris-HCl pH 7.5, 10% (v/v) glycerol, 15 mM imidazole, 2 mM ß -mercaptoethanol) to decrease non-specific binding to the beads. Proteins were eluted using high-imidazole buffer (500 mM NaCl, 50 mM Tris [pH 7.5], 200 mM imidazole, 10% [vol/vol] glycerol, 2 mM ß-mercaptoethanol) after nutating the sample for 5 to 10 min. Pooled protein elutions were concentrated to 20 mg/ml or less in a gel filtration buffer consisting of 200 mM NaCl, 10 mM Tris HCl pH 7.5, 5% glycerol and 1 mM DTT. SEC was carried out using a Superdex 200 Increase 10/300 GL (GE Healthcare) column. Proteins were resolved on 14% SDS-PAGE gels, and the gels were stained using GelCode Blue according to the manufacturer’s instructions (ThermoFisher Scientific). The purified proteins were concentrated, aliquoted, flash frozen, and stored at—20°C for future analysis.
For EDTA and Zn2+Cl2 analyses, 50 uM of Ni-affinity purified protein complexes were incubated for 1h at 4°C on gel filtration buffer with either 1 mM of EDTA or fivefold excess of Zn2+Cl2. Complex samples were then injected on the SEC column primed with gel filtration buffer containing 1 mM of EDTA or 50 uM of Zn2+Cl2.
For elemental analyses, SEC-purified proteins were concentrated to ~ 6 mg/mL in 50 mM NaCl, 10 mM Tris HCl pH 7.5, 5% glycerol and 1 mM DTT, were diluted 10X in 0.1% HNO3 and analyzed by ICP-MS (8900, Agilent, Santa Clara, CA) in helium mode at the Dartmouth Trace Elemental Analyses Core.
Protein purification for crystallography
E. coli B834 (DE3) methionine auxotrophic strain 2593 was used to co-affinity purify the soluble domain of GerS (GerSΔ22) with the His-tagged soluble domain of CwlD (CwlDΔ25). Minimal medium consists of M9 medium (1X M9 salts, 2 mM MgSO4, 0.1mM CaCl2 and 5 g/L (0.5%) glucose (dextrose), 40 mg/L of all amino acids except methionine, 40 mg/L Selenomethionine, 2 mg/L Thiamine and 2 mg/L biotin. Briefly, a starter culture was grown in minimal media supplemented with 5% LB overnight at 37°C. Minimal media was then inoculated with started culture (1:100) and incubated at 37°C to OD600 of 0.6. Cells were induced with 200 uM isopropyl-β- D-1-thiogalactopyranoside (IPTG) for 5h and then harvested by centrifugation at 4000 rpm for 15 min. The complex was purified as described above. Protein purification was performed in the presence of the reducing agent TCEP (1 mM). The protein complex was then purified by SEC using a Superdex 200 Increase 10/300 GL (GE Healthcare) column. Finally, SEC fractions containing the CwlD:GerS complex were pooled, concentrated to 22 mg/mL, and flash frozen in liquid nitrogen.
Crystallization and structure determination
Purified GerS, CwlD, and CwlD:GerS complex were evaluated for crystallization potential using dynamic light scattering using a DynaPro Protein Solutions DLS instrument (S1 Fig). Purified GerS showed multimodal polydispersity regardless of buffer conditions, in agreement with the gel filtration profile, and was prone to aggregation in DLS analyses (S1 Fig). Numerous crystallization matrix screens were performed with only phase separation observed for CwlD but several hits using PEG 3350 for the CwlD:GerS complex. The optimized condition for the complex contained 50 mM BisTris at pH 7.5 with PEG 3350 from 12–18%, 5% glycerol, and 0.25 M ammonium nitrate. Crystals were grown by hanging drop vapor diffusion at 18°C with 30 mg/ml complex concentration mixed 1:1 with reservoir solution. Crystals were cryocooled in liquid nitrogen after soaking in crystallization reagent increased to 20% PEG 3350 and the addition of 14% ethylene glycol. The native complex and selenomethionine-substituted sample crystalized in the same condition. A derivative containing sodium iodide (NaI) was produced by soaking of native crystals with reagent containing 250 mM NaI for 10 minutes before cooling.
Data were collected at GM/CA Sector 23ID (Advanced Photon Source, Argonne National Laboratory) and data processed with the Proteum3 suite (Bruker AXS), Dials [41], and iMosflm [42]. Selenomethionine and NaI derivative data were collected at 12.659 kEv while the native data were collected at 12 kEv. Initial processing suggested space group P6122 or P6522 with one complex per asymmetric unit at a resolution of 2.4 Å (S4 Table). An initial solution was obtained by single anomalous dispersion (SAD) using the selenomethionine (SeMet/Se) incorporated crystal and the SAD routine from Crank2 [43]within CCP4 [44]and showed a preliminary C-alpha trace favoring the core beta sheet for GerS formed via a two-strand exchanged dimer. Data were subsequently integrated, scaled and merged with multiple programs and phasing trials repeated as Se-SAD using ShelX [45]within Crank2 to produce an initial solution using 6 Se sites (1 Se and 1 Zn2+ from CwlD and 4 Se from GerS) CCall/CCweak of 50.1/30.7 and CFOM of 80.9. Additional runs used the native, Se, NaI under multiple isomorphous replacement using Sharp/Autosharp (Global Phasing Limited) and produced near identical solutions to the SAD and summarized in S4 Table. The Rmerge relative to the native data for MIR of the SeMet was 31.2% and for the Iodide 43.0% due to changes in cell edge lengths. Anomalous signal for all three data sets at the conserved Zn2+ site for CwlD homologs was observed supporting the presence of the metal (S2 Fig). Automated iterative build and density modification cycles using Parrot and Buccaneer [44] typically achieved Rfree values near 30%. Manual building used Coot [46]with model refinement performed using Phenix with a final Rfree of 24.5%. Structural orthologs were identified and superimposed using the DALI protein structure comparison server [47]. All figures were prepared using PyMOL (Schrödinger Inc.).
Plate-based sporulation
C. difficile strains were inoculated from glycerol stocks overnight onto BHIS plates containing taurocholate. Colonies arising from these plates were used to inoculate liquid BHIS cultures, which were grown to early stationary phase, back-diluted 1:50 into BHIS, and grown until the cultures reached an OD600 between 0.35 and 0.75. Sporulation was induced on 70:30 agar plates as previously described [48] for 18–24 h, after which cells were analyzed by phase-contrast microscopy and harvested for western blot analyses.
Immunoprecipitation analyses
Immunoprecipitations were performed on lysates prepared from cultures induced to sporulate on 70:30 plates for 22 h. The samples were processed as previously described [49]. Cultures from three 70:30 plates per strain were scraped into 3 x 1 mL of FLAG IP buffer (FIB: 50 mM Tris pH 7.5, 150 mM NaCl, 0.02% sodium azide, 1X Halt protease inhibitor (ThermoScientific). The cultures were transferred into tubes containing a ~300 μL of 0.1 mm zirconia/silica beads (BioSpec). The cultures were lysed using a FastPrep-24 (MP Biomedicals) for 4 x 60 s at 5.5 M/s, with 5 min cooling on ice between lysis intervals. The tubes were pelleted at 3,000 x g for 2 min to pellet beads, unbroken cells/spores, and insoluble material. After washing the tubes with additional FIB, the lysates were pooled, and NP-40 detergent was added to a final concentration of 0.1%, to solubilize membrane-associated proteins. Then, the FLAG-conjugated magnetic resin was added. This magnetic resin was generated by incubating Dynabead Protein G (ThermoScientific) with anti-FLAG antibodies (Sigma Aldrich) at room temperature followed by washing. The lysates were incubated with the anti-FLAG resin for 1 hour at room temperature with rotation. After washing the beads with FIB, FLAG-tagged proteins were eluted using FIB containing 0.1 mg/mL FLAG peptide (Sigma Aldrich). After cell lysis, NP-40 detergent was added to the FIB at a concentration of 0.1%. All immunoprecipitations were performed on three independent biological replicates.
Western blot analysis
Samples for immunoblotting were prepared as previously described [50]. Briefly, sporulating cell pellets were resuspended in 100 μl of PBS, and 50 μl samples were freeze-thawed for three cycles and then resuspended in 100 μl EBB buffer (8 M urea, 2 M thiourea, 4% [wt/vol] SDS, 2% [vol/vol] ß-mercaptoethanol). C. difficile spores (~1 x 106) were resuspended in EBB buffer, which can extract proteins in all layers of the spore, including the core. Both sporulating cells and spores were incubated at 95°C for 20 min with vortex mixing. Samples were centrifuged for 5 min at 15,000 rpm, and 4 x sample buffer (40% [vol/vol] glycerol, 1 M Tris [pH 6.8], 20% [vol/vol] ß -mercaptoethanol, 8% [wt/vol] SDS, 0.04% [wt/vol] bromophenol blue) was added. Samples were incubated again at 95°C for 5 to 10 min with vortex mixing followed by centrifugation for 5 min at 15,000 rpm. The samples were resolved by the use of 14% SDS-PAGE gels and transferred to a Millipore Immobilon-FL polyvinylidene difluoride (PVDF) membrane. The membranes were blocked in Odyssey blocking buffer with 0.1% (vol/vol) Tween 20. Polyclonal rabbit anti-GerS and anti-CwlD [6] antibodies were used at a 1:1,000 dilution. Polyclonal mouse anti-SpoIVA [51] and mouse anti-SleC [40] antibodies were used at 1:2,500 dilution and 1:5,000 dilution, respectively. IRDye 680CW and 800CW infrared dye-conjugated secondary antibodies were used at 1:20,000 dilutions. Odyssey LiCor CLx was used to detect secondary antibody infrared fluorescence emissions.
Spore purification
Sporulation was induced on 70:30 agar plates for 2 to 3 days as previously described [48]. Spores were washed 6 times in ice-cold water, incubated overnight in water on ice, treated with DNase I (New England Biolabs) at 37°C for 60 min, and purified on a HistoDenz (Sigma- Aldrich) gradient. Phase-contrast microscopy was used to assess spore purity (>95% pure); the optical density of the spore stock was measured at OD600; and spores were stored in water at 4°C. Two 70:30 plates were used to induce sporulation for each strain; purified spores were resuspended in 200 μl, and the total optical density of this mixture was determined for each strain. At least three independent spore preparations were performed.
Germination assay
Germination assays were performed as previously described [12]. Approximately 1 x 107 (OD600 of 0.35) were resuspended in 100 μl of water, and 10 μl of this mixture was removed for 10-fold serial dilutions in PBS. The dilutions were plated on BHIS-TA, and colonies arising from germinated spores were enumerated at 20 h. Germination efficiencies were calculated by averaging the CFU produced by spores for a given strain relative to the number produced by wild-type spores for at least three biological replicates from two independent spore preparations. Statistical significance was determined by performing a one-way analysis of variance (ANOVA) on natural log-transformed data using Tukey’s test.
Germination OD600 kinetics assay and phase-contrast microscopy
For OD600 kinetics assays with taurocholate in 96-well plates, ~1.6 x 107 spores (0.55 OD600 units) for each condition tested were resuspended in BHIS and 180 μL were aliquoted into two wells of a 96 well flat bottom tissue culture plate (Falcon) for each condition tested. The spores were exposed to 1% taurocholate, or water (untreated) in a final volume of 200 μL. The OD600 of the samples was measured every 3 minutes in a Synergy H1 microplate reader (Biotek) at 37°C with constant shaking between readings. The change in OD600 over time was calculated as the ratio of the OD600 at each time point to the OD600 at time zero. These assays were performed on three independent spore preparations. Data shown are averages from three replicates.
For phase contrast microscopy, purified spores were resuspended to an OD600 of 0.5 in BHIS ± 1% taurocholate and incubated aerobically for 90 min at 37°C. Samples were spun down (5,000 x g, 10 min, 4°C) washes with PBS and fixed with 3.7% paraformaldehyde for 15 min at RT. Following fixation, samples were washed with PBS and mounted on agarose pads.
Supporting information
The left panel shows the two CwlD / two GerS complex within the crystal with dimensions along each axis shown in nm. Dynamic light scattering results are shown for purified CwlD, CwlD:GerS complex, and GerS. The radius of the CwlD:GerS complex in solution of 5.6 nm agrees well with 5.8 nm as measured in the crystal. The 200 nm peak in the complex sample is excess GerS not in the complex as GerS in the absence of CwlD behaves multimodally with some aggregation. Purified GerS shows multiple peaks on SEC with the expected monomer peak redistributing back into multiple peaks upon storage. At low protein concentration (1 mg/mL) and high salt (500 mM NaCl), the DLS radius of 3.7 nm (diameter of 7.4 nm) is in agreement with the largest measurement of 7.8 nm for the GerS dimer structure when excluding the two CwlD molecules.
(TIF)
Left shows the two CwlD (Blue tones) / 2 GerS (grey tones) complex. The yellow mesh shows the anomalous signal at 3 sigma for the selenomethionine derivative and the maroon the sodium iodide derivative from Autosharp (Global Phasing Limited). Middle panels show Se-SAD phases in blue and Se/NaI with native MIR phases in orange contoured at 1 sigma and the beta strand crossover between the two GerS protomers (solid box) with views 90o apart. Right panel (dotted box) shows the anomalous peaks near the zinc binding site of CwlD.
(TIF)
Purified His-tagged CwlDWT, CwlDH49A, and CwlDE65A were analyzed by size exclusion chromatography. mAU corresponds to the UV absorbance measurements (A280) during the protein elution.
(TIF)
The indicated proteins were produced in E. coli and purified using Ni2+- affinity resin. Cleared lysate (CL) and eluate (E) fractions were analyzed using Coomassie staining.
(TIF)
(A) Change in the OD600 in response to germinant of ΔgerS and ΔcwlD spores complemented with gerSFLAG and cwlDFLAG constructs, respectively, relative to spores complementated with native gerS or cwlD, respectively. Purified spores were resuspended in BHIS, and germination was induced by adding taurocholate (1% final concentration). The ratio of the OD600 of each strain at a given time point relative to the OD600 at time zero is plotted. The mean of three assays from 3 independent spore preparations are shown. Shading represents the standard deviation as the area between error bars for each time point measured. Statistical significance relative to wild-type was determined using two-way ANOVA and Tukey’s test. **** p < 0.0001. (B) Spore germination efficiency of the strains relative complemented with FLAG-tagged vs. wild-type complementation constructs. The number of colony forming units (CFUs) produced by germinating spores is shown. Average of results from three independent spore preparations are shown along with the associated standard deviations. Statistical analyses relative to the wild-type were performed using one-way ANOVA and Tukey’s test. ***, p < 0.001; **** p < 0.0001.
(TIF)
(A) Purified spores were resuspended in BHIS ± germinant and incubated aerobically for 90 min at 37°C. At the indicated time points samples were fixed in paraformaldehyde and visualized using phase-contrast microscopy. Scale bar, 1 um. B) Percent spore germination over time as detected by phase-contrast microscopy. The percentage of phase-bright spores of each strain at a given time point relative to the percentage at time zero is plotted. The mean of three assays from 2 independent spore preparations are shown. Shading represents the standard deviation as the area between error bars for each time point measured. Statistical significance relative to wild-type was determined using two-way ANOVA and Tukey’s test. **** p < 0.0001.
(TIF)
(DOCX)
(PDF)
(XLSX)
(DOCX)
(DOCX)
(DOCX)
Acknowledgments
We would like to thank B. Jackson at the Dartmouth Trace Element Analysis Core for help with the elemental analyses; N. Minton (U. Nottingham) for generously providing us with access to the 630ΔermΔpyrE strain and pMTL-YN1C and pMTL-YN3 plasmids for allele-coupled exchange (ACE).
Data Availability
The crystallographic model and associated structure factors are available from the Protein Data Bank with PDBID 7RAG. All other relevant data are within the manuscript and its Supporting Information files.
Funding Statement
Research in this manuscript was funded by R01GM108684 and R01GM140361 from the National Institutes of General Medical Sciences (NIGMS) to A.S. A.S. is a Burroughs Wellcome Investigator in the Pathogenesis of Infectious Disease supported by the Burroughs Wellcome Fund. B.E.E. is supported by NIH grant R50 CA233185. Single crystal X-ray diffraction experiments were conducted at GM/CA@APS which has been funded in whole or in part with Federal funds from the National Cancer Institute (ACB-12002) and the National Institute of General Medical Sciences (AGM-12006). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. O.D. was funded by R25 GM066567-15 from the National Institutes of General Medical Sciences (NIGMS). The content is solely the responsibility of the author(s) and does not necessarily reflect the views of the Burroughs Wellcome, Department of Energy, NIGMS, or the National Institutes of Health. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.(CDC) CDC. Antibiotic Resistance Threats in the United States. https://wwwcdcgov/drugresistance/biggest-threatshtml. 2019.
- 2.Jones AM, Kuijper EJ, Wilcox MH. Clostridium difficile: a European perspective. J Infect. 2013;66(2):115–28. Epub 2012/10/30. doi: 10.1016/j.jinf.2012.10.019 . [DOI] [PubMed] [Google Scholar]
- 3.Popham DL, Bernhards CB. Spore Peptidoglycan. Microbiol Spectr. 2015;3(6). Epub 2016/06/24. doi: 10.1128/microbiolspec.TBS-0005-2012 . [DOI] [PubMed] [Google Scholar]
- 4.Popham DL, Helin J, Costello CE, Setlow P. Muramic lactam in peptidoglycan of Bacillus subtilis spores is required for spore outgrowth but not for spore dehydration or heat resistance. Proc Natl Acad Sci U S A. 1996;93(26):15405–10. doi: 10.1073/pnas.93.26.15405 ; PubMed Central PMCID: PMC26417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Setlow P, Wang S, Li YQ. Germination of Spores of the Orders Bacillales and Clostridiales. Annu Rev Microbiol. 2017;71:459–77. doi: 10.1146/annurev-micro-090816-093558 . [DOI] [PubMed] [Google Scholar]
- 6.Diaz OR, Sayer CV, Popham DL, Shen A. Clostridium difficile Lipoprotein GerS Is Required for Cortex Modification and Thus Spore Germination. mSphere. 2018;3(3). Epub 2018/06/29. doi: 10.1128/mSphere.00205-18 ; PubMed Central PMCID: PMC6021603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gilmore ME, Bandyopadhyay D, Dean AM, Linnstaedt SD, Popham DL. Production of muramic ∂-lactam in Bacillus subtilis spore peptidoglycan. J Bacteriol. 2004;186(1):80–9. Epub 2003/12/18. doi: 10.1128/JB.186.1.80-89.2004 ; PubMed Central PMCID: PMC303458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sekiguchi J, Akeo K, Yamamoto H, Khasanov FK, Alonso JC, Kuroda A. et al. Nucleotide sequence and regulation of a new putative cell wall hydrolase gene, cwlD, which affects germination in Bacillus subtilis. J Bacteriol. 1995;177(19):5582–9. Epub 1995/10/01. doi: 10.1128/jb.177.19.5582-5589.1995 ; PubMed Central PMCID: PMC177368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coullon H, Rifflet A, Wheeler R, Janoir C, Boneca IG, Candela T. et al. N-Deacetylases required for muramic-delta-lactam production are involved in Clostridium difficile sporulation, germination, and heat resistance. J Biol Chem. 2018;293(47):18040–54. Epub 2018/09/30. doi: 10.1074/jbc.RA118.004273 ; PubMed Central PMCID: PMC6254358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fukushima T, Yamamoto H, Atrih A, Foster SJ, Sekiguchi J. A polysaccharide deacetylase gene (pdaA) is required for germination and for production of muramic delta-lactam residues in the spore cortex of Bacillus subtilis. J Bacteriol. 2002;184(21):6007–15. doi: 10.1128/JB.184.21.6007-6015.2002 ; PubMed Central PMCID: PMC135383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fimlaid KA, Jensen O, Donnelly ML, Francis MB, Sorg JA, Shen A. et.al Identification of a Novel Lipoprotein Regulator of Clostridium difficile Spore Germination. PLoS Pathogens. 2015. Epub October 23, 2015. doi: 10.1371/journal.ppat.1005239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fimlaid KA, Jensen O, Donnelly ML, Francis MB, Sorg JA, Shen A. et.al Identification of a Novel Lipoprotein Regulator of Clostridium difficile Spore Germination. PLoS Pathog. 2015;11(10):e1005239. Epub 2015/10/27. doi: 10.1371/journal.ppat.1005239 ; PubMed Central PMCID: PMC4619724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heidrich C, Templin MF, Ursinus A, Merdanovic M, Berger J, Schwarz H, et al. Involvement of N-acetylmuramyl-L-alanine amidases in cell separation and antibiotic-induced autolysis of Escherichia coli. Mol Microbiol. 2001;41(1):167–78. Epub 2001/07/17. doi: 10.1046/j.1365-2958.2001.02499.x . [DOI] [PubMed] [Google Scholar]
- 14.Vermassen A, Leroy S, Talon R, Provot C, Popowska M, Desvaux M. et al. Cell Wall Hydrolases in Bacteria: Insight on the Diversity of Cell Wall Amidases, Glycosidases and Peptidases Toward Peptidoglycan. Front Microbiol. 2019;10:331. Epub 2019/03/16. doi: 10.3389/fmicb.2019.00331 ; PubMed Central PMCID: PMC6403190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Do T, Schaefer K, Santiago AG, Coe KA, Fernandes PB, Kahne D, et al. Staphylococcus aureus cell growth and division are regulated by an amidase that trims peptides from uncrosslinked peptidoglycan. Nat Microbiol. 2020;5(2):291–303. Epub 2020/01/15. doi: 10.1038/s41564-019-0632-1 ; PubMed Central PMCID: PMC7046134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uehara T, Parzych KR, Dinh T, Bernhardt TG. Daughter cell separation is controlled by cytokinetic ring-activated cell wall hydrolysis. EMBO J. 2010;29(8):1412–22. Epub 2010/03/20. doi: 10.1038/emboj.2010.36 ; PubMed Central PMCID: PMC2868575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Do T, Page JE, Walker S. Uncovering the activities, biological roles, and regulation of bacterial cell wall hydrolases and tailoring enzymes. J Biol Chem. 2020;295(10):3347–61. Epub 2020/01/25. doi: 10.1074/jbc.REV119.010155 ; PubMed Central PMCID: PMC7062177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moll A, Dorr T, Alvarez L, Chao MC, Davis BM, Cava F, et al. Cell separation in Vibrio cholerae is mediated by a single amidase whose action is modulated by two nonredundant activators. J Bacteriol. 2014;196(22):3937–48. Epub 2014/09/04. doi: 10.1128/JB.02094-14 ; PubMed Central PMCID: PMC4248829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stohl EA, Lenz JD, Dillard JP, Seifert HS. The Gonococcal NlpD Protein Facilitates Cell Separation by Activating Peptidoglycan Cleavage by AmiC. J Bacteriol. 2015;198(4):615–22. Epub 2015/11/18. doi: 10.1128/JB.00540-15 ; PubMed Central PMCID: PMC4751805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gurnani Serrano CK, Winkle M, Martorana AM, Biboy J, More N, Moynihan P, et al. ActS activates peptidoglycan amidases during outer membrane stress in Escherichia coli. Mol Microbiol. 2021. Epub 2021/03/05. doi: 10.1111/mmi.14712 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rocaboy M, Herman R, Sauvage E, Remaut H, Moonens K, Terrak M, et al. The crystal structure of the cell division amidase AmiC reveals the fold of the AMIN domain, a new peptidoglycan binding domain. Mol Microbiol. 2013;90(2):267–77. Epub 2013/08/10. doi: 10.1111/mmi.12361 . [DOI] [PubMed] [Google Scholar]
- 22.Yang DC, Tan K, Joachimiak A, Bernhardt TG. A conformational switch controls cell wall-remodelling enzymes required for bacterial cell division. Mol Microbiol. 2012;85(4):768–81. Epub 2012/06/22. doi: 10.1111/j.1365-2958.2012.08138.x ; PubMed Central PMCID: PMC3418388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Konovalova A, Silhavy TJ. Outer membrane lipoprotein biogenesis: Lol is not the end. Philos Trans R Soc Lond B Biol Sci. 2015;370(1679). Epub 2015/09/16. doi: 10.1098/rstb.2015.0030 ; PubMed Central PMCID: PMC4632606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takeda K, Miyatake H, Yokota N, Matsuyama S, Tokuda H, Miki K. et al. Crystal structures of bacterial lipoprotein localization factors, LolA and LolB. EMBO J. 2003;22(13):3199–209. Epub 2003/07/04. doi: 10.1093/emboj/cdg324 ; PubMed Central PMCID: PMC165648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaplan E, Greene NP, Crow A, Koronakis V. Insights into bacterial lipoprotein trafficking from a structure of LolA bound to the LolC periplasmic domain. Proc Natl Acad Sci U S A. 2018;115(31):E7389–E97. Epub 2018/07/18. doi: 10.1073/pnas.1806822115 ; PubMed Central PMCID: PMC6077701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372(3):774–97. Epub 2007/08/08. doi: 10.1016/j.jmb.2007.05.022 . [DOI] [PubMed] [Google Scholar]
- 27.Li S, Lou X, Xu Y, Teng X, Liu R, Zhang Q, et al. Structural basis for the recognition of MucA by MucB and AlgU in Pseudomonas aeruginosa. FEBS J. 2019;286(24):4982–94. Epub 2019/07/13. doi: 10.1111/febs.14995 . [DOI] [PubMed] [Google Scholar]
- 28.Kim DY, Kwon E, Choi J, Hwang HY, Kim KK. Structural basis for the negative regulation of bacterial stress response by RseB. Protein Sci. 2010;19(6):1258–63. Epub 2010/06/01. doi: 10.1002/pro.393 ; PubMed Central PMCID: PMC2895250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Buttner FM, Faulhaber K, Forchhammer K, Maldener I, Stehle T. Enabling cell-cell communication via nanopore formation: structure, function and localization of the unique cell wall amidase AmiC2 of Nostoc punctiforme. FEBS J. 2016;283(7):1336–50. Epub 2016/02/03. doi: 10.1111/febs.13673 . [DOI] [PubMed] [Google Scholar]
- 30.Ng YK, Ehsaan M, Philip S, Collery MM, Janoir C, Collignon A, et al. Expanding the repertoire of gene tools for precise manipulation of the Clostridium difficile genome: allelic exchange using pyrE alleles. PLoS One. 2013;8(2):e56051. Epub 2013/02/14. doi: 10.1371/journal.pone.0056051 ; PubMed Central PMCID: PMC3566075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Donnelly ML, Fimlaid KA, Shen A. Characterization of Clostridium difficile Spores Lacking Either SpoVAC or Dipicolinic Acid Synthetase. J Bacteriol. 2016;198(11):1694–707. Epub 2016/04/06. doi: 10.1128/JB.00986-15 ; PubMed Central PMCID: PMC4959285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Charlton TM, Kovacs-Simon A, Michell SL, Fairweather NF, Tate EW. Quantitative Lipoproteomics in Clostridium difficile Reveals a Role for Lipoproteins in Sporulation. Chem Biol. 2015;22(11):1562–73. Epub 2015/11/21. doi: 10.1016/j.chembiol.2015.10.006 . [DOI] [PubMed] [Google Scholar]
- 33.Dembek M, Stabler RA, Witney AA, Wren BW, Fairweather NF. Transcriptional analysis of temporal gene expression in germinating Clostridium difficile 630 endospores. PLoS One. 2013;8(5):e64011. Epub 2013/05/22. doi: 10.1371/journal.pone.0064011 ; PubMed Central PMCID: PMC3655068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Typas A, Banzhaf M, Gross CA, Vollmer W. From the regulation of peptidoglycan synthesis to bacterial growth and morphology. Nat Rev Microbiol. 2011;10(2):123–36. Epub 2011/12/29. doi: 10.1038/nrmicro2677 ; PubMed Central PMCID: PMC5433867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8(10):785–6. Epub 2011/10/01. doi: 10.1038/nmeth.1701 . [DOI] [PubMed] [Google Scholar]
- 36.Shida T, Hattori H, Ise F, Sekiguchi J. Mutational analysis of catalytic sites of the cell wall lytic N-acetylmuramoyl-L-alanine amidases CwlC and CwlV. J Biol Chem. 2001;276(30):28140–6. Epub 2001/05/26. doi: 10.1074/jbc.M103903200 . [DOI] [PubMed] [Google Scholar]
- 37.Peters NT, Morlot C, Yang DC, Uehara T, Vernet T, Bernhardt TG. et al. Structure-function analysis of the LytM domain of EnvC, an activator of cell wall remodelling at the Escherichia coli division site. Mol Microbiol. 2013;89(4):690–701. Epub 2013/06/26. doi: 10.1111/mmi.12304 ; PubMed Central PMCID: PMC3923381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li T, He L, Li C, Kang M, Song Y, Zhu Y, et al. Molecular basis of the lipid-induced MucA-MucB dissociation in Pseudomonas aeruginosa. Commun Biol. 2020;3(1):418. Epub 2020/08/05. doi: 10.1038/s42003-020-01147-1 ; PubMed Central PMCID: PMC7400510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Karasawa T, Ikoma S, Yamakawa K, Nakamura S. A defined growth medium for Clostridium difficile. Microbiology (Reading). 1995;141 (Pt 2):371–5. Epub 1995/02/01. doi: 10.1099/13500872-141-2-371 . [DOI] [PubMed] [Google Scholar]
- 40.Donnelly ML, Li W, Li YQ, Hinkel L, Setlow P, Shen A. et al. A Clostridium difficile-Specific, Gel-Forming Protein Required for Optimal Spore Germination. mBio. 2017;8(1). Epub 2017/01/18. doi: 10.1128/mBio.02085-16 ; PubMed Central PMCID: PMC5241399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clabbers MTB, Gruene T, Parkhurst JM, Abrahams JP, Waterman DG. Electron diffraction data processing with DIALS. Acta Crystallogr D Struct Biol. 2018;74(Pt 6):506–18. Epub 2018/06/07. doi: 10.1107/S2059798318007726 ; PubMed Central PMCID: PMC6096487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Battye TG, Kontogiannis L, Johnson O, Powell HR, Leslie AG. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 4):271–81. Epub 2011/04/05. doi: 10.1107/S0907444910048675 ; PubMed Central PMCID: PMC3069742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Skubak P, Pannu NS. Automatic protein structure solution from weak X-ray data. Nat Commun. 2013;4:2777. Epub 2013/11/16. doi: 10.1038/ncomms3777 ; PubMed Central PMCID: PMC3868232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 4):235–42. Epub 2011/04/05. doi: 10.1107/S0907444910045749 ; PubMed Central PMCID: PMC3069738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sheldrick GM. A short history of SHELX. Acta Crystallogr A. 2008;64(Pt 1):112–22. Epub 2007/12/25. doi: 10.1107/S0108767307043930 . [DOI] [PubMed] [Google Scholar]
- 46.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 4):486–501. Epub 2010/04/13. doi: 10.1107/S0907444910007493 ; PubMed Central PMCID: PMC2852313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Holm L. DALI and the persistence of protein shape. Protein Sci. 2020;29(1):128–40. Epub 2019/10/14. doi: 10.1002/pro.3749 ; PubMed Central PMCID: PMC6933842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ribis JW, Ravichandran P, Putnam EE, Pishdadian K, Shen A. The Conserved Spore Coat Protein SpoVM Is Largely Dispensable in Clostridium difficile Spore Formation. mSphere. 2017;2(5). Epub 2017/09/30. doi: 10.1128/mSphere.00315-17 ; PubMed Central PMCID: PMC5607322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Touchette MH, Benito de la Puebla H, Ravichandran P, Shen A. SpoIVA-SipL Complex Formation Is Essential for Clostridioides difficile Spore Assembly. J Bacteriol. 2019;201(8). Epub 2019/01/30. doi: 10.1128/JB.00042-19 ; PubMed Central PMCID: PMC6436350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Putnam EE, Nock AM, Lawley TD, Shen A. SpoIVA and SipL are Clostridium difficile spore morphogenetic proteins. J Bacteriol. 2013;195(6):1214–25. Epub 2013/01/08. doi: 10.1128/JB.02181-12 ; PubMed Central PMCID: PMC3592010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kevorkian Y, Shirley DJ, Shen A. Regulation of Clostridium difficile spore germination by the CspA pseudoprotease domain. Biochimie. 2016;122:243–54. Epub 2015/08/02. doi: 10.1016/j.biochi.2015.07.023 ; PubMed Central PMCID: PMC4732931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Usenik A, Renko M, Mihelic M, Lindic N, Borisek J, Perdih A, et al. The CWB2 Cell Wall-Anchoring Module Is Revealed by the Crystal Structures of the Clostridium difficile Cell Wall Proteins Cwp8 and Cwp6. Structure. 2017;25(3):514–21. Epub 2017/01/31. doi: 10.1016/j.str.2016.12.018 . [DOI] [PubMed] [Google Scholar]