

ABSTRACT
Although largely overlooked compared to bacterial infections, fungal infections pose a significant threat to the health of humans and other organisms. Many pathogenic fungi, especially Candida species, are extremely versatile and flexible in adapting to various host niches and stressful situations. This leads to high pathogenicity and increasing resistance to existing drugs. Due to the high level of conservation between fungi and mammalian cells, it is hard to find fungus-specific drug targets for novel therapy development. In this respect, it is vital to understand how these fungi function on a molecular, cellular as well as organismal level. Fluorescence imaging allows for detailed analysis of molecular mechanisms, cellular structures and interactions on different levels. In this manuscript, we provide researchers with an elaborate and contemporary overview of fluorescence techniques that can be used to study fungal pathogens. We focus on the available fluorescent labelling techniques and guide our readers through the different relevant applications of fluorescent imaging, from subcellular events to multispecies interactions and diagnostics. As well as cautioning researchers for potential challenges and obstacles, we offer hands-on tips and tricks for efficient experimentation and share our expert-view on future developments and possible improvements.
Keywords: mycology, fluorescence imaging, molecular cell biology, fungal pathogenesis, microscopy
Fungal infections such as those caused by Candida species impose serious health problems to patients, and using state-of-the-art fluorescence techniques, it is possible to pave the way towards novel, highly-needed, druggable targets.
INTRODUCTION
Fungal infections pose a serious problem to human health and welfare. Where some fungal pathogens cause mild superficial infections of skin and mucosal tissue, others can cause severe disease and high lethality through systemic dissemination (Brown et al. 2012). Apart from the very few therapies available, most of these drug strategies suffer from inefficacy or host toxicity. Furthermore, as is the case for bacterial pathogens, both acquired and inherent resistance against antifungal drugs are on the rise. To contain infections and safeguard the health of infected patients, novel drug strategies have to be developed (Roemer and Krysan 2014). In order to keep expanding our knowledge on the fundamental processes of microbial infections, it is essential to keep track of novel techniques and use the proper tools for our research questions. Candida species are among the fungi that are most often isolated from infections. Being opportunistic pathogens, they possess commensal as well as pathogenic traits, taking advantage of a weakened host immune system to make the transition between states (Cauchie, Desmet and Lagrou 2017). Candida research differs significantly from other eukaryotic organisms because general genomics, life cycle, metabolism, etc. are quite distinct even from its close relative and model system baker's yeast. Several of these differences hamper the heterologous expression and easy adaptation of techniques within the Candida research community. For example, Candida albicans is generally encountered as an obligate diploid, therefore adaptations to the genome must often be applied to both alleles. Baker's yeast shows a very clear and distinct life cycle with sexual mating between haploid a and α mating types, allowing easy genetic experimentation. On the other hand, in C. albicans and Candida glabrata no sexual cycle seems to be apparent (Magee and Magee 2000; Boisnard et al. 2015). Additionally, the CTG clade containing many Candida species such as C. albicans, Candida parapsilosis, Candida tropicalis and the recently emerged and multidrug resistant Candida auris translate the CUG codon as serine instead of leucine, further complicating the heterologous expression of systems adapted for Saccharomyces cerevisiae or mammalian cells (Butler et al. 2009; Miranda et al. 2013). Due to these significant differences, specific optimization and adaptation steps are required in the development of tools and techniques for this human fungal pathogen.
Fluorescence-based techniques possess the unique quality to visualize processes in real time and in a dynamic manner inside cells and communities. They allow us to image interactions between molecules, cells and organisms, provide information on subcellular and organismal localization, report on niche-specific parameters such as pH and oxygen level and can even be used to assess enzymatic activity inside the cell. It is thus evident that fluorescence investigation of microbial and specifically fungal pathogenesis allows for rapid progress in the field and paves the way to the elucidation of novel druggable targets. In this review, we will focus on the fluorescence-based techniques applied to fungal pathogenesis and infections, from a micro to a macro scale, as depicted in Fig. 1.
Figure 1.

Schematic overview of fluorescent applications in fungal pathogenesis. Fluorescence techniques can be applied in mycology on several levels. Molecular interactions and activity, cellular phenomena, multicellular connections and organismal events can be investigated with the wide array of fluorescent techniques presented in this manuscript.
WHAT IS AVAILABLE?—FLUOROPHORES
Being able to perform the fluorescence-based techniques depicted in Fig. 1 across the different scales of imaging requires an extensive library of fluorophores. So far, the available fluorophores are fluorescent proteins, organic dyes or bioluminescent enzymes, but as science continues to progress and only a fraction of life on earth has been described, additional classes will be discovered. Because fluorophores are the basic requirement for the tools and techniques described in this review, we will start by exploring them before applying them to C. albicans tools.
Fluorescent proteins
At the advent of fluorescence—a Candida optimized GFP
The discovery of the wild type Green Fluorescent Protein (GFP) from Aequorea victoria and the practicality of using a genetically encoded tag to fluorescently label targets-of-interest with low toxicity, led to an enormous boost in life sciences research. Until now, all fluorescent proteins (FPs) for Candida albicans are structurally homologous, containing an 11-stranded β-barrel with an α-helical strand in the middle of the barrel enclosing the chromophore (Shimomura, Johnson and Saiga 1962; Prasher et al. 1992). Five years after the initial attempts to heterologously express GFP in C. albicans, the first of many species-specific modifications were made to the protein. The first adaptation consisted of replacing the CTG codons, which encode serines in C. albicans instead of leucines. As detailed in the Challenges section at the end of this manuscript, it is essential to avoid CTG codons in heterologous expression, to ensure proper translation, folding and functionality of the protein. Apart from codon optimization, two residues were mutated, S65G and S72A (Cormack et al. 1997). These mutations were shown to increase fluorescence intensity 20-fold compared to enhanced GFP in Escherichia coli due to a shift in absorption maximum towards 488 nm (Cormack, Valdivia and Falkow 1996). This mutant, called yeast enhanced GFP3 or YeGFP3, was also inserted in a set of plasmids for use in C. glabrata (Zordan et al. 2013). Independently from this first study, researchers from another laboratory replaced the CTG codons as well, but also introduced two sets of mutations consisting of either S65T or a combination of S65A, V68L, S72A and Q60R. The latter mutant was termed GFPmut2 (Ullah et al. 2013a). These mutants outperformed the wild type chromophore (Ene et al. 2016). As is the case for many FPs, specific properties of GFP, such as quantum yield, photostability and expression levels were continuously upgraded in other fields, such as mammalian, plant and bacterial research. By translating certain mutations obtained in E. coli to C. albicans, Zhang and colleagues were able to construct a significantly improved monomeric GFP version called γmGFP (Crameri et al. 1996; Zhang and Konopka 2010). Recently, we showed that further codon optimization using different dedicated algorithms does not improve the in vivo brightness of γmGFP, leading us to state that this variant is currently recommended as green FP in C. albicans research (Van Genechten et al. 2020).
Over the rainbow—expansion of the palette
After initial optimization of the green FP, an expansion of colors, based on mutagenesis of GFP, took place (Fig. 2A). By combining mutations S65G, V68L and S72A, which improve the folding of GFP (Cormack, Valdivia and Falkow 1996), with T203Y, a significantly red-shifted GFP variant was obtained. This variant, termed Yellow Fluorescent Protein (YFP), is spectrally distinguishable from GFP (Ormo et al. 1996). On the other hand, a blue-shifted version of GFP, Cyan Fluorescent Protein (CFP), was obtained by a single mutation of tyrosine 66 to tryptophan inside the chromophore (Heim and Tsien 1996). Versions of YFP and CFP were engineered in PCR-based cassettes for convenient cloning and transformation in C. albicans (Gerami-Nejad, Berman and Gale 2001). Other Candida species such as Candida glabrata are able to stably maintain plasmids and therefore use a different set of YFP and CFP expression vectors (Yanez-Carrillo et al. 2015). The original YFP and CFP were improved by the development of so-called next generation FPs. Introducing a single F46L mutation into the YFP template, leads to an acceleration of the oxidation step in the chromophore formation. This resulted in the development of Venus which has an enhanced maturation speed at 37°C (Nagai et al. 2002). In FRET-pairs, as introduced later, often circular permuted (cp) Venus is used. A circular permutated protein contains a different order of amino acids but a very similar structure as the original one (Kostyuk et al. 2019). The N- and C-terminus of a protein are linked together while new termini are positioned close to the chromophore, forming the cp variant of the protein. These variants are generally more flexible thereby allowing optimization of FRET efficiency. mTurquoise2 is another next generation FP that is the result of many incremental improvements of mVenus to SCFP1, -2 and -3 (Kremers et al. 2006). Within this SCFP3 template one single mutation, T65S, was able to increase brightness of the FP by 50%. This version, called monomeric Turquoise or mTurquoise, has a quantum yield of 0.84 and was then subsequentially improved via a I146F mutation to promote better packing of the bulky chromophore. The quantum yield is the ratio of the number of emitted photons to the number of absorbed photons, and is thus a measurement of the efficiency of the fluorescent protein. Table 1 provides a list of terminology related to fluorescence. The resulting mutant, mTurquoise2, has improved stabilization of the chromophore which leads to less non-radiative decay and thus an improved quantum yield of 0.93 (Goedhart et al. 2012). These two next generation FPs, mTurquoise2 and Venus, were codon-optimized and characterized for C. albicans (Van Genechten et al. 2020). Other FPs based on GFP are pHluorin and its successor pHluorin2, with the latter having an eightfold increase in fluorescence due to the F64L folding enhancer mutation, available for C. glabrata and C. albicans. Both are pH-sensitive FPs that can be used to estimate the pH in the direct environment of the FP by ratiometric measurement of emission upon excitation at 395 and 475 nm (Ullah et al. 2013b; Liu and Kohler 2016; Tournu et al. 2017). The pHluorin2 codon sequence was optimized using a guided random algorithm based on the codon biases of five highly expressed genes in C. albicans, RPL29, RPL32, RPL39, ACT1 and ENO1. This optimization method was also applied to other FPs, such as YemVenus, mTurquoise2 and mScarlet, but did not result in a significant improvement of the brightness (Tournu et al. 2017).
Figure 2.

Schematic overview of available Candida-optimized fluorophores. Panel A provides a schematic overview of the available mechanisms and incremental enhancements of the fluorescent proteins available for Candida research. From the optimization of GFP to an extensive color expansion and subsequent development of ‘Smart’ and smaller FPs. Panel B depicts the common excitation wavelengths available in a fluorescence microscope setup as vertical lines. A combination of gas lasers such as a krypton, argon and helium-neon laser provide 405, 488, 515, 568 and 640 nm as possible excitation wavelengths. A simplified depiction of emission spectra of Candida-specific fluorescent proteins indicates the range available for research and the possible spectral overlap. C represents the palette of available luciferases for Candida research. Substrate, oxygen and/or ATP requirements are color-coded according to their respective luciferase. Panel D are examples of three common organelle dyes used in C. albicans research. In the topmost panel NucblueTM is an example of a nuclear staining, with a singular lobe within each cell. Middle panel contains the fungal cell wall staining Calcofluor White. Bottom panel depicts the linear structures obtained after staining the mitochondria using Mitotracker Red.
Table 1.
Definitions of commonly used terminology related to fluorescence.
| Term | Explanation |
|---|---|
| Codon Adaptation Index (CAI) | Measure for codon usage bias. When CAI is 1, only high frequency codons are utilized. |
| Chromophore | Part of the fluorophore that is responsible for the emission |
| Point Spread Function (PSF) | Describes the pattern acquired after a single emitter is visualized through the objective |
| Circular permutation | Relationship between proteins in which the order of amino acids is different but the structure the same |
| Quantum Yield | Ratio of emitted photons over the absorbed photons. Thus, a measurement of efficiency of this conversion process. |
| Maturation speed | Relative time it takes to form a functional fluorescent protein |
| Extinction Coefficient | Measure for the absorption efficiency of light by the object |
| Photostability | Describes the stability or restistance of degradation when the object is illuminated with light |
| Phototoxicity | Laser illumination leads to the formation of Reactive Oxygen Species (ROS) which are toxic to the cell. |
| Autofluorescence | Fluorescence from inherent natural structures. |
| Photoconversion | Process of spectral changes under illumination |
| Scattering | Light rays deviate from their central path by interaction with an object |
| Resolution | Minimal disctance between two seperate points that can be distinguished by the optical device |
| Differential Interference Contrast (DIC) | Microscope technique utilizing the refractive index differences to visualize the object. |
Apart from the well-known jellyfish-derived FPs, coral-based FPs were rapidly discovered and adapted for C. albicans. RFP and mCherry from the Cnidarian Discosoma sp. were codon-optimized for C. albicans and C. glabrata (Gerami-Nejad, Dulmage and Berman 2009; Yanez-Carrillo et al. 2015). Altering 218 codons by replacing C or G at the third position by A or T and replacing CTG codons produced a yeast-enhanced mRFP (YemRFP) (Keppler-Ross, Noffz and Dean 2008). The advantage of these RFPs is the nearly complete lack of overlap in emission spectra with GFP and CFP. Recently, a synthetically engineered RFP called mScarlet was published. This synthetic construct was based on a template of mCherry, but gave rise to a significantly improved quantum yield and maturation speed (Bindels et al. 2017). One version of mScarlet, mScarlet-I, was independently adapted for C. albicans by the research groups of Van Dijck and Bennett (Frazer, Hernday and Bennett 2019; Van Genechten et al. 2020). This mScarlet-I was preferred over the mScarlet-H version because it has an improved maturation speed due to the T74I mutation, with a small decrease in quantum yield (Bindels et al. 2017). Further expansion of red FPs is ongoing with the recent optimization of mKate2 for C. albicans and its application within a heme biosensor (Weissman et al. 2020). A blue FP or BFP, was similarly obtained from the Anthozoan sea-anemone Entacmaea quadricolor and optimized for C. albicans. Interestingly, its emission spectrum can be distinguished completely from GFP with Fluorescence Activated Cell Sorting (FACS) (Loll-Krippleber et al. 2015). The most recently developed FP from a non-jellyfish origin is mNeongreen, a yellow-green FP, derived from Branchiostoma lanceolatum which also proved to be more photostable than γmGFP (Frazer, Hernday and Bennett 2019). So far, all the FPs that have been described, have emission spectra within the visible area. Infra-red FPs, however, have the added advantage of allowing deep tissue imaging of cells, because of the transparency of tissue to this type of light. These FPs originate from bacterial phytochrome photoreceptors and after several rounds of random mutagenesis iRFP702 was obtained. Further directed mutagenesis of residues at positions 180, 202, 203 and 254 led to spectral shifts resulting in iRFP670 with an emission maximum at 670 nm (Shcherbakova and Verkhusha 2013). This collection of iRFPs requires bilirubin as a co-factor which acts as the phytochrome required for fluorescence. An iRFP670 has been developed for deep tissue imaging of C. albicans in zebrafish (Bergeron et al. 2017).
Specialized FPs overcoming practical limitations
While all previously described FPs show classical behavior, i.e. they bleach mono-exponentially, a novel realm of smart FPs is rapidly developing with non-classical behaviors such as reversible and irreversible on-off switching, emission spectrum shifts, etc. Fast folding (ff)Dronpa is the first smart FP optimized for C. albicans (Van Genechten et al. 2020). It is a negative photoconvertible FP (PCFP) which can be switched to the on-state using UV light and reverted to the off-state using blue light as depicted in Fig. 2A. This photochromic behavior makes it suitable for super-resolution localization microscopy such as Stochastical Optical Fluctuation Imaging (SOFI) or Optical Lock-in Detection (OLID) to reduce the background autofluorescence and thus improve the signal-to-noise ratio (Marriott et al. 2008; Dedecker et al. 2012).
A drawback of all earlier described FPs is that the chromophore within the beta-barrel is formed by a reaction that requires molecular oxygen. These FPs can thus not be used in hypoxic conditions. This problem is circumvented by the C. albicans-adapted FMN-based FP (Candida-optimized FbFP), a FP that does not require molecular oxygen during maturation, but requires flavin as a co-factor. Even though FbFP requires flavin, which is only present at low concentrations in the nucleus, a targeted localization to the nucleus is functional in C. albicans in both normoxic and hypoxic conditions (Eichhof and Ernst 2016). Furthermore, CaFbFP is applied in an oxygen biosensor, which is further elucidated in part 3.3 on monitoring cellular activity. Functionality of this Candida-optimized FbFP is based on the light-oxygen-voltage-sensing domains of photoreceptors from Bacillus subtilis and Pseudomonas putida (Drepper et al. 2007; Tielker et al. 2009). The emission spectra for Candida-optimized FbFP and all other previously described FPs are depicted in Fig. 2B and the state-of-the art FPs for C. albicans and C. glabrata for each color are represented in Table 2.
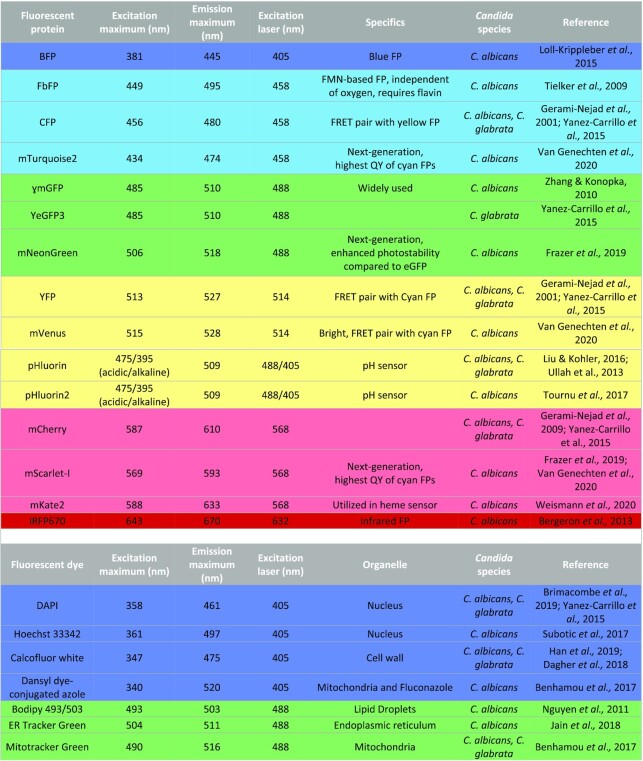
Table 2.
Overview of fluorescent proteins, dyes and biosensors available for use in Candida species. Several fluorescent proteins, dyes and biosensors are available for experimentation in Candida species. FP, fluorescent protein; FMN, flavin mononucleotide; FRET, fluorescence resonance energy transfer; QY, quantum yield.
|
Heading towards smaller FPs
Because of the relatively large size of all presently-available Candida-optimized FPs, each FP is approximately 250 amino acids long, a fusion construct with a protein-of-interest may impose a significant strain on the folding of this protein. Smaller FPs are, therefore, an interesting lead to follow up on (Fig. 2A). One such option is Y-FAST, this protein is approximately half the size of GFP-like fluorescent proteins. Y-FAST is functional as an extracellularly attached protein in S. cerevisiae and intracellularly in mammalian cells. Y-FAST in itself is not fluorescent, but it binds a fluorogen 4-hydroxybenzylidene-rhodanine (HBR) or 4-hydroxy-3-methylbenzylidene-rhodanine (HMBR), which stabilizes upon binding, leading to an increase in the quantum yield and a red-shift of the fluorogen (Plamont et al. 2016). Another option for a smaller FP in Candida research is UnaG, a 139 amino acid long protein isolated from Japanese eel. It is already utilized in mammalian cell lines and the mouse brain. In contrast to GFP, it is functional in anaerobic environments, but just like iRFP it requires bilirubin as a co-factor (Kumagai et al. 2013).
Self-labeling proteins
When you decide to fuse a fluorescent tag to your protein of interest, the color of your tag is often the determining factor. Self-labeling proteins offer a solution by providing flexibility in color even after the genetic fusing. Self-labeling proteins are genetic tags to which an exogenous substrate containing a fluorophore can bind covalently. These exogenous substrates have multiple fluorophores available, allowing quick and easy adaptations for multi-colour imaging. Three systems of the same principle are available so far. SNAP-, CLIP- and Halo-tag and have been optimized in S. cerevisiae (Keppler et al. 2003; Gautier et al. 2008; Los et al. 2008; Stagge et al. 2013). Optimization was required to allow the entrance of the labeling substrate into the cell. This required an additional electroporation step. These optimizations will also be necessary if the system were to be adapted for C. albicans research. These self-labeling protein systems can be used simultaneously allowing the researcher to perform multi-colour imaging.
Do-it-yourself
Selecting an optimal FP for your experiments
It is clear that the selection of FPs available for use in C. albicans and C. glabrata is growing. For each color, the optimal FP is represented in Table 2. Fluorescent proteins in the complete range of the visual spectrum as well as the infra-red region, are available, allowing for researchers to pick any FP with desirable traits or several FPs for multi-color imaging. When choosing fluorescent proteins to answer a specific research question, it is always useful to check all characteristics of the preferred FP, such as the possible cofactors, oxygen requirements and subcellular location. Each experimental condition has a particular set of physical properties which require specific FPs. Some FPs will have different maturation times at 30°C or 37°C and, as will be discussed in later chapters, some infection models require a specific type of FP for visualization. As presented above, FPs in a wide range of colors and with specific adaptations are available. Of note is that N-terminal tags often mask the signal sequences present near the N-terminus of a protein-of-interest and thus hamper the correct localization of the protein (Palmer and Freeman 2004).
Bioluminescence
Bioluminescence is the emission of light upon enzymatic processing of a substrate that is administered to the cells. The enzymes that catalyze the reactions are called luciferases and are generally encoded genetically. It is a common phenomenon in nature which can observed in fireflies or the jellyfish Aequorea victoria (Shimomura, Johnson and Saiga 1962). One of the advantages of bioluminescence compared to fluorescence is the lack of the need for excitation, which can be a limiting factor when your sample is prone to scattering of shorter wavelengths. The sensitivity of each luciferase differs, yet, in general, they provide a more sensitive reporter system with a better signal-to-noise ratio compared to fluorescent proteins and would even allow for monitoring of transcriptional regulation (Hirose et al. 2002).
There are several Candida-optimized and validated systems available, each with their own advantages and disadvantages. The first attempt to apply bioluminescence in C. albicans was based on the green-yellow firefly luciferase (fLuc). However, the system suffered from an extremely low signal (Srikantha, Chandrasekhar and Soll 1995). It appears that a single CTG codon within the firefly luciferase gene was sufficient to completely block its function. Because of this issue, another luciferase from the sea pansy Renilla reniformis (rLuc) was introduced successfully in C. albicans as a reporter system for white-opaque switching (Srikantha et al. 1996). This luciferase emits blue light upon administration of coelenterazine in the presence of oxygen.
The discovery that the CTG codon in C. albicans is translated as serine instead of leucine led to the codon optimization of several FPs, as previously discussed, but also of the firefly luciferase (fLuc). The firefly luciferase is preferred over the R. reniformis luciferase because the substrate of rLuc, coelenterazine, is less stable than the substrate of the firefly luciferase, luciferin. Nine CTG codons within the firefly luciferase were exchanged for TTG codons, rendering the construct completely functional and thus emitting green-yellow light upon addition of luciferin (Doyle et al. 2006b). One of the drawbacks of luciferin is its poor ability to penetrate the remodeled hyphal cell wall (Doyle et al. 2006a). The combination of poor diffusion of luciferin with the requirement of intracellular ATP, hampers the applicability of fLuc. Another bioluminescent reporter is the Gaussia princeps luciferase (gLuc), which, much like the rLuc system, catalyzes the reaction of coelenterazine to produce light. It has the advantage that it does not require ATP, yet is still oxygen-dependent. To circumvent the issue of poor penetration of its substrate, gLuc was expressed extracellularly by attaching it to a GPI-linked cell wall protein (Enjalbert et al. 2009). This problem highlights the main drawback of bioluminescent systems, the necessary optimization of substrate administration and its poor diffusion across the cell membrane (Gaur et al. 2017).
As mentioned before, an advantage of bioluminescence is that it does not require excitation, thereby circumventing the inefficient penetration of light through tissues. Especially light of shorter wavelengths is unable to penetrate tissue, due to scattering and absorption by blood hemoglobin, collagen, etc. (Avci et al. 2013). Emitted light from the luciferase reaction is also severely attenuated by tissue depth, therefore a more red-shifted luciferase would allow for deeper tissue imaging. By introducing S284T, L295F, T214A and A215L mutations in the gLuc optimized gene, a red-shifted thermostable mutant was acquired. This luciferase is significantly more sensitive in vivo in a systemic and oropharyngeal candidiasis (OPC) model compared to the original gLuc (Dorsaz, Coste and Sanglard 2017). Another red- and green-emitting luciferase system was adapted from the click beetle Pyrophorus plagiophtalamus. These click beetle luciferases (cbLuc) use luciferin as a substrate in the presence of ATP and oxygen. Because these luciferases use the same substrate as the firefly enzymes, it is possible to spectrally separate both luciferases after activation with one substrate. However, they have not been applied in an in vivo infection model yet (Kapitan et al. 2016).
As is the case for fluorescent proteins, luciferases are larger proteins, ranging from 311 to 550 amino acids. Overexpression of genes encoding such proteins can diminish the amount of tRNAs and amino acids available for efficient translation of essential proteins. It can also perturb cellular processes, such as folding and degradation processes in the endoplasmic reticulum. A smaller 19 kDa luciferase, called NanoLuc, has been reported and adapted for C. albicans. It uses the commercial Nano-Glo containing furimazine as a substrate and is ATP-independent (Luna-Tapia et al. 2015; Masser et al. 2016). The emission maximum of NanoLuc, 460 nm, does not make it suitable for in vivo imaging. An overview of the luciferases with their requirements and substrate is presented in Fig. 2C.
None of the luciferases presented above are optimal for every situation. Therefore, when selecting a luciferase for an experiment, the sensitivity, thermostability and emission wavelength should be taken into account. Immediate further improvements would be an increase of signal strength of the red-shifted luciferases so that they have a similar performance as the green-emitting luciferases. Decreasing their size would be an added advantage. Another significant improvement to every luciferase system would be the engineering of C. albicans to make the substrate itself, thereby omitting the need for external administration and circumventing the poor diffusion of the substrate into the cell.
Dyes
In fungal research, dyes are mainly used to provide cellular context or to investigate certain characteristics of a cell, such as ploidy, live/dead ratios and cell wall remodeling. In this section, we discuss the dyes that perform their function as such and do not need to be conjugated to an antibody. Nuclear dyes, such as DAPI (4’,6-diamidino-2-phenylindole) and Hoechst 33342, bind AT-rich regions of DNA. These are blue dyes that are excited with UV light. Because of the direct binding to the DNA, the dyes are toxic. They have been applied successfully in C. albicans and C. glabrata and are ideal for multi-color imaging with any green and/or red FP (Subotić et al. 2017; Brimacombe et al. 2019; Yanez-Carrillo et al. 2015). An example of a Hoechst 33342 staining (Nucblue࣪) is shown in Fig. 2D and an overview of the dyes discussed in this review, is given in Table 2.
The cell wall is an organelle that differentiates Candida from mammalian cells and is therefore a preferred target for antifungal agents. The echinocandins inhibit beta-glucan synthase and thereby limit the production of this cell wall component. Another component of the cell wall is chitin. Even though a cell wall staining often uses antibodies or lectins to bind the carbohydrates that make up the brunt of the cell wall, a specific dye is available to perform this without any need for conjugation and extra washing or blocking steps. Calcofluor White is a dye specifically binding glucans and chitin, which are prominent in the fungal cell wall. Similar to the nuclear dyes described above, it is a blue dye that is excited with UV light. It has been utilized in the study of cell wall composition and estimation of chitin levels of C. albicans (Ballou et al. 2016; Dagher et al. 2018; Han et al. 2019).
The target of fluconazole is lanosterol 14α-demethylase encoded by ERG11. This target is localized to the endoplasmic reticulum. The endoplasmic reticulum is therefore an important cell organelle in antifungal drug research. Using a combination of fluorescence or Förster resonance energy transfer (FRET) and the ER Tracker staining, the interaction between Ras1 and Gpi1 was proven to be localized to the ER (Jain et al. 2018). ER Tracker specifically binds to K+-channels which are present on the ER membrane and the plasma membrane. Two different colors of BODIPY® ER Tracker dyes, red and green, have been applied in C. albicans (Shah et al. 2015; Jain et al. 2018).
The last organelle that can be specifically stained without using antibodies or lectins is the mitochondrion. This organelle is stained by the Mitotracker dyes, which encompass green, orange, red and deep red colors, allowing researchers to pick one that is compatible with their setup. Mitotracker dyes diffuse passively over membranes and sequester into the mitochondria, where they are oxidized and retained because of their reaction with active thiol-groups (Keij, Bell-Prince and Steinkamp 2000). Using Mitotracker Red, localization of several proteins to the mitochondria was shown (Ferrari et al. 2011; Dana et al. 2019). We show the localization pattern of Mitotracker Red in Fig. 2D. Two other dyes were engineered that localize to the mitochondria. These dyes are based on an azole backbone to which a Dansyl or Cy5 is attached, emitting green and far-red light, respectively. They were shown to co-localize significantly with Mitotracker green proving that azoles are sequestered in mitochondria and that these azole-based dyes can be used as a mitochondrial stain (Benhamou et al. 2017).
Finally, BODIPY 493/503 is a specific stain for lipid droplets. Although initially lipid droplets were not considered to be a true classical organelle, their extensive role in lipid trafficking and metabolism indicated their role as a conserved organelle. Lipid droplets are shown to be necessary for full virulence of C. parapsilosis in a murine infection model (Nguyen et al. 2011).
Other cell organelles do not have a specific dye that can be applied in Candida research. There are, as far as we know, no dyes available to stain the Golgi apparatus. The dyes presented above can be applied in live cell imaging, but have some toxicity that could lead to imaging artefacts such as highly fluorescent vesicles. Organic dyes show low overall cell toxicity and are a preferred option if available. When dyes are utilized in experiments, control experiments should be performed to check the possible harmful effects.
WHAT IS POSSIBLE?—SUBCELLULAR IMAGING
The basic application of fluorescence in fungal research is subcellular imaging of proteins or structures. To achieve information on the presence and localization of certain components, the target-of-interest should be fused to a fluorescent label through appropriate linkers. Whilst many methods are available to monitor the fluorescent signal coming from fluorophores within Candida species, microscopy remains the most prominent technique. Further technical advancements have made it possible to image in a time- and space-resolved manner. Apart from cellular structures, also protein-protein interactions and cellular activity can be imaged.
Imaging subcellular structures
Design of linker sequences
Several types of linkers that connect proteins to the above-mentioned fluorescent tags exist, such as flexible, rigid and cleavable linkers. Dependent on the type of study, the linker sequence should maintain a certain level of flexibility (Kerppola 2009). Generally, linkers are composed of small, polar or non-polar amino acids, such as the widely used (Gly-Gly-Gly-Gly-Ser)n linkers (Ene et al. 2013). Glycine residues are small, allowing for flexibility, while serine aids in maintaining the stability by forming hydrogen bonds with the surrounding water molecules, thereby reducing unfavorable interactions with the protein moiety.
Widefield and confocal microscopy
Widefield microscopy is one of the basic methods of microscopy and therefore widely applied in fungal research. However, one needs to understand the basic principle of fluorescence widefield microscopy in order to make further improvements and adaptations. In widefield microscopy the whole sample, a fluorescent Candida cell, is illuminated evenly by the excitation light passing through the objective. The entire excited sample emits light which passes through the objective and is focused on the camera. This basic microscopy setup was originally used to visualize the C. albicans optimized GFP, YFP, CFP, mCherry and is still applied regularly due to its relatively low cost and high effectiveness (Cormack et al. 1997; Gerami-Nejad, Berman and Gale 2001; Gerami-Nejad, Dulmage and Berman 2009). The resolution of widefield microscopy is limited to approximately half of the wavelength of the excitation light, being about 200nm. However, it is possible to image the subcellular localization of proteins using this setup because a Candida cell is around 4–10 μm in size and hyphae can easily stretch out to 30–50 μm. Since the whole sample is illuminated, out-of-focus emission light is also captured by the objective, causing background signal. Furthermore, this background signal is further increased by autofluorescent molecules such as riboflavin, causing again a drop in the effective signal-to-noise ratio (Demuyser et al. 2020).
Using confocal microscopy can partially solve the issue of background noise, by specifically illuminating a certain area in a single plane of the sample. This is done by focusing excitation light onto a single area in the sample and consequently scanning the entire sample to construct the complete image. The emitted light from a single pixel area passes through a pinhole which reflects out-of-focus light. The selected photons are captured by a photomultiplier tube (PMT) or an avalanche photon detector (APD) instead of the CCD or CMOS cameras used for widefield microscopy. Additionally, it is possible to change the plane of illumination, therefore allowing to image at different depths within the sample. A reconstruction of several images at different depths can result in a 3D-model of the sample, which is particularly interesting when studying biofilm structures (Tsui, Kong and Jabra-Rizk 2016). Even though scanning speeds can often be adjusted in software, it remains a rather slow method. By using spinning disk confocal microscopy one can increase the speed at which a confocal image can be acquired. This technique uses a spinning disk containing multiple holes instead of one pinhole. Since excitation light is allowed to pass through multiple holes of the spinning disk, an image can be reconstructed much faster (Davidovits and Egger 1971). Since confocal microscopy is able to reject much of the out-of-focus autofluorescence background of Candida species, it is the preferred method for subcellular imaging and colocalization studies of proteins-of-interest.
Super-resolution microscopy
The methods described above are limited by Abbe's law, which states that the resolution is controlled by diffraction according to:
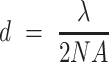 |
Where d is the smallest distance between two resolvable objects, λ is the wavelength of the emitted light and NA is the numerical aperture of the objective. In practice, this results in a resolution limit of near 200 nm. As already mentioned earlier, this resolution is sufficient for many applications in Candida research, but further enhancements would potentially increase our understanding of certain molecular mechanisms.
In the chapter on fluorophores, we already discussed ffDronpa, a smart fluorescent protein. This protein can switch reversibly between a bright and a dark state. This specific behavior is a prerequisite for single molecule localization microscopy (SMLM). This method is based on the fact that if two emitters are in close proximity, they cannot be discerned due to the overlap of their PSFs. However, if two images are obtained sequentially where only one of the emitters is in the bright state, it is possible to pinpoint the center of the emitter using the gaussian nature of the PSF. Several SMLM techniques are available, PALM, (d)STORM and pcSOFI, each requiring a certain induced behavior of the fluorescent protein (Betzig et al. 2006; Rust, Bates and Zhuang 2006; Dertinger et al. 2009). The codon-optimized ffDronpa is particularly suitable for pcSOFI and this would be a robust method able to enhance the localization of fluorophores and reduce the significant background autofluorescence of C. albicans (Dedecker et al. 2012; Moeyaert et al. 2014).
Another way to provide sub-diffraction resolution, however not performed with Candida species yet, is structured illumination microscopy (SIM). Here, the sample is illuminated with a specific sine pattern which, in combination with Fourier transformation, leads to a diffraction limit of approximately 100 nm. This technique has been applied on yeast species to image the spindle pole body. SIM was even combined with light-sheet-based fluorescence microscopy (LSFM) yielding csiLSFM by which the resolution of a GFP-tagged ER structure was significantly enhanced (Burns et al. 2015; Chang, Perez Meza and Stelzer 2017). Both studies utilized classical FPs of which optimized versions are available for C. albicans suggesting that these methods can be applied on this pathogenic yeast as well. Much like SIM imaging, stimulated emission depletion microscopy (STED) requires a specific microscope setup that produces a carefully constructed illumination pattern. Similar to laser scanning microscopy, one specific area in the sample is excited. Around this excitation area a ‘donut’ shaped depletion ring reduces the excitation point spread function (PSF), the function describing the blurring of a point by the optics of the system, by switching off the fluorophores on the outer side of the excitation area. This results in a smaller excitation area and thus an increase in resolution (Klar et al. 2000). A combination of STED and the Snap, Halo and CLIP-labeling system was successfully applied in S. cerevisiae and resolved details in the cell cortex that were not distinguishable using confocal microscopy (Stagge et al. 2013).
Light-sheet microscopy
Even though confocal microscopy is a significant improvement over widefield microscopy and the excitation light is focused onto a small area of the sample, out-of-focus laser light is still able to excite and bleach dyes and FPs outside of the focal plane. This leads to a decreased signal when another plane of the sample is imaged. Light-sheet-based fluorescence microscopy (LSFM) offers a solution for this issue. The advantages of this technique for deep-tissue imaging will be further discussed in the section on host-pathogen imaging, but the implementation of LSFM can also be useful for imaging single yeast cells. As the name implies, a sheet of excitation light is created by focusing a collimated laser beam onto a thin plane by a cylindrical lens (Greger, Swoger and Stelzer 2007). This cylindrical lens is orthogonal to the objective lens which focuses the emitted signal from the sample onto the detector. Since only a small part of the sample is illuminated, there is no need for a pinhole to remove out-of-focus excitation light. The method is able to capture images much faster than a confocal microscope since the sample is not completely scanned, but all pixels are excited and imaged in parallel by the same type of camera as in widefield microscopy (Reynaud et al. 2008). Using this setup, embryos of zebrafish, tumor cells and amoebae have been visualized (Huisken and Stainier 2007; Lorenzo et al. 2011; Takao et al. 2012). It has also been successfully applied to image organic dyes in baker's yeast, indicating that this technique could also be applied on Candida species (Reynaud et al. 2008).
Do-it-yourself
Selecting the best microscopy technique for your research
The limiting factor in subcellular imaging of Candida species is often the fluorescent probe instead of the microscopy setup. Choosing your fluorescent protein or dye wisely is thus of uttermost importance. Nevertheless, some critical differences between a widefield and a confocal microscope should also be considered when choosing the best method for a certain experiment. If your target-of-interest is a highly mobile protein, a confocal microscope will not be able to construct an image fast enough. Therefore, the movement of this protein will cause a smear of fluorescent signal in your final image. Even parts of the cytoskeleton such as alpha-tubulin are continuously degraded and re-assembled, and therefore highly mobile in live cells. Since the speed of widefield microscopy relies on the camera acquisition rate, mobile targets can be imaged using such a setup. Fixating the cells is another option to image mobile proteins, yet comes at a cost of introducing artefacts, decreasing fluorescence from fluorescent proteins and losing the capacity to perform time-lapse experiments.
As mentioned earlier, many stress factors introduce elevated autofluorescence in the fungal cell, these experiments would benefit from confocal microscopy to reject most of the out-of-focus fluorescence. Especially when the signal from the sample is sparse, reducing the out-of-focus fluorescence is of utmost importance to improve your signal-to-noise ratio. Confocal imaging is also the method of choice in imaging C. albicans biofilms or other thicker samples, because these are more prone to scattering. Another way to resolve this, but at a greater cost, is light-sheet microscopy, which is able to combine the fast image acquisition with selective illumination to reduce out-of-focus fluorescence.
If your signal is sparse, increasing excitation power can overcome this issue, but is not always beneficial due to phototoxicity and increased photobleaching as discussed later. In confocal microscopy or widefield microscopy, respectively decreasing the scanning speed or increasing the exposure time of the camera and thus acquiring more signal from all single points within the sample is another way to boost the signal obtained from the fluorophore. Furthermore, utilizing low-fluorescent medium based on yeast nitrogen base where folic acid and riboflavin are omitted, diminishes the amount of autofluorescence and is thus an enhancement for both widefield and confocal imaging.
Imaging protein-protein interactions
When studying an organism's signaling pathways and metabolism in order to discover novel antifungal drug targets, it is essential to take into account the numerous protein-protein interactions (PPIs). However, for C. albicans, and even more so for other Candida species, protein-protein interactions have been largely neglected in the drug discovery process. Moreover, very few of the documented interactions have been gathered in accessible databases. Recently, colleagues from our laboratory have performed an exhaustive literature search of all known PPIs in C. albicans, thereby greatly updating the existing databases (Schoeters and Van Dijck 2019). Depending on the research question, several techniques can be used for the study of PPIs in C. albicans.
For high-throughput screening of PPIs, the yeast two-hybrid (Y2H) technique is mostly used. However, since many genes contain CTG codons, the use of S. cerevisiae as model system for C. albicans PPI studies is complicated. To circumvent this issue, the Vesicular Capture Interaction system was developed in C. albicans in 2009 (Boysen et al. 2009). In this assay, the bait consists of one protein-of-choice, fused to Vps32, a subunit of the endosomal sorting complex required for transport (ESCRT). In the prey construct, another protein-of-choice is fused to GFP. By using a vps4 mutant strain, in which vesicular accumulation of Vps32 is stimulated, a positive interaction between both proteins-of-interest will be visible as bright fluorescent spots. It is, however, still questionable to which extent this assay can be used in high-throughput setups. For this reason, colleagues from our laboratory developed a novel Candida two-hybrid system to be able to perform high-throughput PPI screenings directly in the pathogenic organism (Stynen, Van Dijck and Tournu 2010; Schoeters et al. 2018). Alternatively, when not so specifically interested in high-throughput screenings, yet more in the specific interaction between multiple proteins, techniques such as co-immunoprecipitation (co-IP) or tandem-affinity purification can be used (Alkafeef et al. 2018; Sellam et al. 2019). The major disadvantage of such techniques is the lack of an in vivo, cellular context and any spatial information on the native location of the interaction inside the cell.
BiFC
Bimolecular Fluorescence Complementation or BiFC and Fluorescence/Förster Resonance Energy Transfer or FRET are in vivo techniques that can provide information on the native localization of the interaction. BiFC is a technique whereby a fluorescent protein is split into two complementary parts, an N- and C-terminal fragment. Both parts are fused to two proteins of which interaction is under investigation (Kerppola 2009). When the proteins interact, the fluorescent protein fragments are brought together irreversibly and a fully functional and excitable FP is reconstituted. A few years ago, we developed the first BiFC system, based on YemVenus, optimized it for use in C. albicans and showed its functionality by visualizing interaction of Snf4-Kis1, Bcy1-Tpk1/2 and Gpr1-Gpa2 protein pairs (Subotić et al. 2017). When performing PPI studies, it is important to confirm an interaction with more than one technique. Subotić et al. therefore used the established Candida two-hybrid system from our laboratory to confirm the interactions earlier observed by BiFC. In the same year, a similar BiFC assay was optimized for C. albicans, using the S. cerevisiae system as described in Sung and Huh 2007 (Sung and Huh 2007; Mamouei et al. 2017). The non-yeast optimized fluorescent protein Venus was used, in which all CTG codons were mutated to TTG by site-directed mutagenesis. The physical interaction between several ferroxidases and permeases was assessed by means of this setup. Very recently, a novel Tet-on-BiFC system was established in which mCherry is used as a fluorescent protein (Lai, Sun and Shieh 2020). Researchers validated the system by showing interaction between the rho-type GTPase Cdc42 and the putative rho GDP dissociation factor Rdi1. In this system dominant markers were used, whereas the system we developed, is based on the use of auxotrophic markers. Furthermore, the Tet-on system allows for regulated expression of both genes-of-interest. The main disadvantage, however, is the usage of mCherry, which has a lower quantum yield in C. albicans, compared to YemVenus and is therefore not always suitable for endogenous expression of tagged genes. Based on the S. cerevisiae system, researchers also used a BiFC assay to detect PPIs in C. glabrata (Sung and Huh 2007). Heterodimer formation between mating type proteins Cga1 and Cgalpha2 as well as Cgalpha3 homodimer formation were detected using a Venus-based BiFC assay (Robledo-Márquez et al. 2016). Positive signals were detected and quantified by flow cytometry. Both interactions were confirmed by co-IP using Flag-tagged and cMyc-tagged proteins. However, not all co-IP detected interactions could be confirmed by BiFC analysis. Likely certain interactions that are detected in vitro, are less relevant under in vivo conditions, where also other factors influence the interaction. This indicates that assessing interactions in a cellular context is always preferable to evaluate the relevance of the PPI.
FRET
Due to the irreversible character of FP complementation, BiFC-based methods lack the potential to report on the PPI in a dynamic, more physiologically-relevant way. FRET, on the contrary, holds the unique quality to display an interaction in vivo and in real-time in the cell, while providing information on spatiotemporal dynamics. The process is based on the non-radiative transfer of energy between two fluorophores which are in close proximity. Excitation of one FP (donor FP) leads to emission of this FP. If another FP (acceptor FP) is nearby, part of the excitation energy from the donor FP, is transferred to the acceptor FP, eventually leading to emission of this latter FP. The theoretical FRET efficiency (EFRET) can be calculated as follows: EFRET = 1/(1+(r/R0)6), where r represents the distance between the fluorophores and R0 the distance at which the FRET efficiency is 50% (Bajar et al. 2016). The most common FPs to be combined as a FRET pair are cyan ECFP—yellow EYFP or green EGFP—red mCherry, as the spectral overlap between the donor emission spectrum and the acceptor excitation spectrum is considerably high(Bajar et al. 2016).
Several methods can be used to monitor FRET inside a living cell and are categorized, based on whether EFRET or a change in EFRET, for instance upon addition of an inducer, is measured. A first method to directly measure EFRET is acceptor photobleaching. If energy transfer happens from the donor to the acceptor, photobleaching the acceptor will cause the donor to retain more energy, and an increase in donor emission will be observed (Bajar et al. 2016). Using this technique, researchers showed interaction between the GTPase Ras1 and the GPI-N-acetylglucosaminyl transferase Gpi2 in the endoplasmic reticulum of C. albicans (Jain et al. 2018). Another technique to measure FRET is fluorescence lifetime imaging (FLIM)-FRET, in which the fluorescence lifetime (which is the time a FP spends in the excited state before emitting a photon and returning to the ground state) of the donor is measured in the absence and presence of the acceptor. The fluorescence lifetime of the donor will decrease if FRET occurs (Piston and Kremers 2007; Bajar et al. 2016). A second category of methods is mostly used to measure relative changes in FRET efficiency, for example upon addition of an inducer, rather than the absolute FRET efficiency. In sensitized emission-FRET, the donor is excited and emission of both donor and acceptor is monitored (van Rheenen, Langeslag and Jalink 2004). Ratiometric FRET is derived from sensitized emission-FRET. It calculates the change in EFRET over the change in donor emission. This is often the preferred technique when unimolecular FRET-based biosensors are used, as illustrated in the section ‘Monitoring cellular activity’. Finally, certain commercial assays also make use of FRET as a readout for specific biochemical or biological processes. One example is the LIVE/DEAD® FungaLightTM Yeast Viability Kit from ThermoFisher, in which two fluorescent probes are used, SYTO®9 and propidium iodide. SYTO®9 labels nucleic acids fluorescently green while PI probes nucleic acid fluorescently red. Both stains, however, differ in their membrane permeability. While SYTO®9 can penetrate both living and dead cells, PI only penetrates cells with damaged membranes. In cells with intact cell membranes, as a result, only green fluorescence can be observed, while cells with damaged membranes fluoresce mainly red due to a FRET-related decrease in green fluorescence if SYTO®9. Using this technique, researchers measured the anti-Candida effect of Psd1, an antimicrobial peptide isolated from Pisum sativum seeds (Gonçalves et al. 2017).
Important disadvantages of using FRET to detect PPIs in living Candida cells are the variability in expression of both proteins-of-interest and concomitantly, the donor and acceptor, the high level of autofluorescence and specific cellular context (pH, O2, presence of quenchers), which often complicate straightforward analysis. Other systems operating in a simpler environment, such as an alternative host organism or an in vitro setup, can be used, yet generally at the expense of biological relevance. Researchers recently showed that an antifungal peptide inhibits cell wall biosynthesis in Candida tropicalis by binding to β-1,6-glucanase Kre9 using E. coli as an alternative host (Li et al. 2020). Using an in vitro setup, other researchers illustrated the role of critical amino acid residues in ATP binding and hydrolysis by the drug efflux pump Cdr1 in C. albicans (Rai et al. 2008). They exploited the intrinsic fluorescence of a tryptophan residue and labelled a cysteine residue in the active site with a fluorescent sulfhydryl probe. Proximity of both residues was analyzed under several conditions and their role in Mg2+ and ATP binding and hydrolysis was specified. Using another in vitro single molecule FRET-based setup, researchers monitored binding of the C. glabrata telomere-ending binding protein complex or CST and one of its subunits, Cdc13, to higher order G-tail structures of the telomere ending (Lue and Chan 2013). Using a biotinylated PEG-coated surface, they immobilized biotinylated DNA by binding to neutravidin. The DNA strand was labelled with two fluorescent dyes, Cy3 and Cy5. Upon binding of the CST or Cdc13 proteins to the DNA sequence, the fluorophores set apart and energy transfer between both diminishes.
Improvements for fluorescent PPI assays
Using new techniques or better probes, a number of the shortcomings of standard fluorescent PPI techniques can be overcome. Development of new fluorophores in certain organisms, has led to specific improvements of FRET and BiFC experiments in the respective research fields. A few examples of new adaptations are FRET or BiFC with far-red and/or infrared fluorophores, especially interesting for imaging interactions in deeper tissues; FRET based on FPs with large Stokes shifts, to minimize cross-talk between channels; FRET pairs with a dark acceptor, as such reducing bleed-through and phototoxicity; and multi-color FRET or BiFC, to allow visualization of multiple interactions or processes at the same time (Filonov and Verkhusha 2013; Bajar et al. 2016; Fujii, Yoshimura and Kodama 2018). Specifically for BiFC, overcoming the irreversibility of the interaction would be a great improvement. One option here is to replace the GFP-like fluorophore by IFP1.4, an infrared fluorescent protein which is not yet optimized for Candida species (Shu et al. 2009). Upon reassembly of the two IFP protein parts and addition of the chromophore, biliverdin, fluorescence can be detected, which is reported to be reversible (Tchekanda, Sivanesan and Michnick 2014). The fluorescent protein can also be replaced by a split fluorescence-activating and absorption shifting tag or FAST, which allows for binding and stabilization of the fluorogen HBR. Upon complementation of the FAST parts, and binding of HBR, the fluorescence of the latter increases significantly. Most interestingly, the splitFAST system is fully reversible which is a major advantage over the classical FP-based fluorescence complementation (Tebo and Gautier 2019). Both for FRET and BiFC, a fluorescent protein could be replaced by a luciferase enzyme. Since no excitation light is needed here, problems such as photo-toxicity, autofluorescence, photobleaching and cross-excitation are avoided to a large extent (Sun et al. 2016). BiFC analysis could be performed using a luciferase enzyme instead of a fluorescent protein, in this case the assay is called a split-luciferase system (Fukutani et al. 2017). BRET or bioluminescence resonance energy transfer resembles FRET, with the exception that the donor fluorophore is replaced by a bioluminescent protein, a luciferase. This leads to an increase in signal-to-noise ratio and thus sensitivity of the measurements. As mentioned earlier, quite a few bioluminescent proteins have been established for Candida species, yet BRET has never been reported. In S. cerevisiae the system has proven to be efficient in screening PPI inhibitors in the context of anticancer drug development (Corbel et al. 2017).
Other more recent advances making use of resonance energy transfer are single molecule FRET (smFRET), in which total internal reflection microscopy is integrated to overcome the limited spatial resolution, and sequential RET, in which a combination of BRET and FRET allows for assessment of the physical interaction between three proteins (Carriba et al. 2008). A similar combination between FRET or BRET and BiFC can be made to monitor formation of multiprotein binding complexes (Cui et al. 2019). Finally, also colocalization analysis can be used to assess interaction between proteins (Cui et al. 2019). The main limitation here is spatial resolution, restricted among others by the resolution of the light microscope and the wavelength of the light. One option is to use super-resolution microscopy, as detailed in the previous section, in dual color imaging, by STED, PALM or STORM. Alternatively, also 3D imaging can improve the accuracy of interaction assessment through colocalization. Mapping PPIs in Candida species can be of utmost importance to unravel signaling cascades and discover novel antifungal drug targets (Schoeters et al. 2018). Due to increased automatization and establishment of high-content imaging, many of the above-mentioned techniques could be used in a high-throughput setup as is already the case for other organisms (Song, Madahar and Liao 2011; Miller et al. 2015; Corbel et al. 2017).
Monitoring cellular activity
Apart from proteins or interactions, also cellular activity can be visualized, using fluorescence microscopy, or quantified, using FACS. Biosensors are molecules that allow for monitoring of either chemicals, conditions, enzymatic activity or physicochemical properties. An overview of all discussed biosensors, is given in Table 2.
pH
An important biochemical characteristic for cells is the pH, both intracellular as well as extracellular. Candida albicans is able to adapt to completely different niches with the pH ranging from 4 to 8 and monitor changes in the external pH. One of the pathways involved in monitoring acidification or alkalization of the environment is the Rim101 signaling pathway. Alkalization of the environment is sensed at the plasma membrane by Rim21 and signaled intracellularly, leading to filamentation (Obara and Kihara 2014). On the other hand, the intracellular pH is an important factor of niche adaptation. A first version of the ratiometric pH sensor, pHluorin, was adapted for C. albicans in 2016. pHluorin is a pH-sensitive fluorescent protein which undergoes a change in excitation maximum upon acidification or alkalization, with a fixed emission maximum at 509 nm. Depending on the acidity of the cellular environment, excitation at 475 nm and 395 nm increases and decreases or vice versa. The ratio of the emission intensity at these two excitation peaks is used to calculate the intracellular pH using a calibration curve. Using this assay, the group of Köhler has shown that, upon administration of omeprazole to the cells, the cytosolic pH dropped due to the inhibition of the plasma membrane proton ATPase, Pma1 (Liu and Kohler 2016). Similar experiments were performed in C. glabrata where the pHluorin from S. cerevisiae proved to be functional, albeit in a tandem copy setup, to improve brightness. In these experiments, it was shown that C. glabrata maintains a higher intracellular pH compared to S. cerevisiae and that the administration of fluconazole to the cells did not affect the intracellular pH, while echinocandins and amphotericin B did (Ullah et al. 2013a). An enhanced version of this pH-sensitive FP is pHluorin2. This version has improved fluorescence at 37°C due to the folding enhancing mutation F64L (Mahon 2011). Attaching pHluorin2 to Cpy1 or Pga59 of C. albicans, localized this sensor to the vacuole or the external surface of the cell, respectively. Using this sensor, it was shown that there is a significant difference in vacuolar pH across clinical isolates and that azoles further acidify the vacuole. Contradictory, in a previous study it was shown that the cytoplasmic pH did not change upon administration of fluconazole (Tournu et al. 2017). Beside pHluorin, also other pH probes or dyes can be used. Many were already used in S. cerevisiae, such as the fluorescent proteins RaVC and pHRed and dyes Lysosensor Yellow/blue, C-fluorescein and C-SNARF-4, as reviewed elsewhere (Valkonen et al. 2013). Using the latter dye, of which the emission maximum wavelength changes upon altering the pH, several fungal biofilms were visualized and pH analyzed (Schlafer, Kamp and Garcia 2018).
Oxygen
Another biochemical parameter that has an influence on C. albicans virulence is oxygen (Grahl et al. 2012). The availability of oxygen is inversely linked to CO2 in eukaryotic cells, since metabolism consumes O2 and produces CO2. This leads to the generation of a hypoxic environment in the direct vicinity of the cells. CO2 has a well-known effect on the virulence of C. albicans, since it induces hyphae through activation of the PKA pathway (Klengel et al. 2005). During infection, oxygen levels drop due to metabolic activity and damage to epithelial cells. Candida albicans is able to withstand these low oxygen conditions and continues growing, albeit at a lower rate. Low oxygen availability also induces hyphal formation through the transcriptional regulator Ace2 (Mulhern, Logue and Butler 2006). This illustrates that, next to the pH, oxygen is another important biochemical parameter sensed by C. albicans to adapt to host niches. The group of Ernst developed a reporter for molecular oxygen, utilizing the oxygen-requirements of FbFPs and classical GFP-like fluorescent proteins. When cells are grown in a hypoxic environment, the chromophores of classical FPs, such as YFP, are unable to mature and are therefore not fluorescent. FbFP, however, does not require oxygen and is able to emit light under these conditions. A tandem of YFP and two copies of Candida-optimized FbFP form a functional FRET pair that is able to transfer energy from Candida-optimized FbFP to YFP under normoxic conditions, whilst no fluorescence from YFP is detected under hypoxic conditions. The presented fusion construct can thus be used as an in vivo oxygen sensor, termed YFOS for yeast fluorescent oxygen sensor (Eichhof and Ernst 2016).
cAMP-PKA activity
In 2018, we established the first FRET-based biosensors in C. glabrata (Demuyser et al. 2018). Based on the AKAR3 and EPAC2 biosensors developed for mammalian cells and S. cerevisiae, we created the C. glabrata variants. AKAR3 is an A-type kinase activity reporter which measures phosphorylation activity by protein kinase A (PKA). ECFP is used as donor and cpVenus as acceptor. As mentioned earlier, circular permutations generally render an FP more flexible. It was shown that certain circular permutations of Venus resulted in significantly increased FRET efficiencies (van der Krogt et al. 2008). The cp173Venus variant was selected in this respect (Allen and Zhang 2006). Upon activation of PKA, the sensor domain is phosphorylated, bringing together donor and acceptor. EPAC2 is an intracellular cAMP sensor with ECFP-EYFP as FRET pair. Upon binding of cAMP, both FPs are set apart, leading to a reduction in FRET. For both sensors, ratiometric FRET was used to show a concomitant increase in cAMP and activation of PKA upon addition of glucose to starved C. glabrata cells.
Heme iron
Recently, the group of Prof Dr James Kronstad developed a heme sensor (HS1) for use in the fungus Cryptococcus neoformans (Bairwa et al. 2020). This sensor fuses the red FP, mKate2 to an EGFP in which a heme-binding domain is embedded. When heme binds the sensor, the EGFP fluorescence is quenched and a decrease in FRET signal is perceived. This sensor was also adapted for C. albicans where it was utilized to investigate iron homeostasis and the role of heme oxygenase in extracellular heme toxicity(Weissman et al. 2020). This heme sensor will enable more in-depth analysis of the function of iron and heme in azole susceptibility (Demuyser et al. 2017).
Outlook on biosensors
Many biosensors have been developed over the past years, although mainly in mammalian cell systems (Cui et al. 2019). Their applications are very diverse and can be, roughly, divided in three categories. An overview of several types of biosensors is given by Terai et al. 2019; Miyawaki and Niino 2015 and Skruzny et al. 2019 specifically for S. cerevisiae (Miyawaki and Niino 2015; Skruzny, Pohl and Abella 2019; Terai et al. 2019). Several biosensors measure the presence of small molecules, such as ions, secondary metabolites and metabolic intermediates. Especially interesting for examination of fungal virulence factors, and signal transduction thereof, would be the Ca2+, Zn2+ and Cu+ sensors, as all of these ions have been shown to be involved in pathogenesis or drug resistance, as reviewed by Li et al. 2018 (Li et al. 2018). Measuring sterol levels would be of particular interest to the investigation of azole sensitivity in Candida species, as both have been extensively studied, yet never in a dynamic and visual manner as is possible with FRET biosensors (Song et al. 2015; Aron et al. 2016; Chauhan, Jentsch and Menon 2019). Secondary messenger cAMP, plays an essential role in the PKA pathway and related pathogenesis (Hogan and Sundstrom 2009). A cAMP biosensor, EPAC2, was developed for use in C. glabrata by our laboratory, although novel types exist and could be tested as well, in several Candida species (Surdo et al. 2017; Demuyser et al. 2018; Botman, van Heerden and Teusink 2020). Nutrient sensors that are of particular interest in Candida metabolism, are those detecting glucose, sucrose and lactate (Höfig et al. 2018; Lager et al. 2006; San Martín et al. 2013; Van Ende, Wijnants and Van Dijck 2019). Trehalose and trehalose-6-phosphate have been reported to play an important role in fungal metabolism and have been put forward as potential antifungal drug targets (Arguëlles 2017). FRET-based sensors have been generated to measure levels of both sugars in mammalian cells and S. cerevisiae (Peroza et al. 2015; Kikuta et al. 2016). A second category of biosensors are those monitoring enzyme activity, such as the previously described AKAR3 for PKA activity (Demuyser et al. 2018). The options here are numerous. Biosensors have been developed for measurement of the activity of GPCRs, proteases, kinases, phosphatases, small GTPases and many others (Miyawaki and Niino 2015; Komatsu et al. 2018; Skruzny, Pohl and Abella 2019). Finally, a relatively recent class of FRET-based biosensors that were developed, can measure the physicochemical properties of cells, such as pH, redox state, protein crowding and tension (Oku et al. 2013; Morikawa et al. 2016; Freikamp et al. 2017; Burgstaller et al. 2019). Translating these technologies to a Candida-directed setup would allow for detailed investigation of several virulence attributes. As an example, FRET-based tension sensors allow for investigation of cell-cell adhesion and cell-matrix adhesion possibly interesting in biofilm formation and multispecies interactions (Freikamp et al. 2017).
Apart from the numerous applications of FRET-based biosensors one can think of, several advances have been made recently that aim to lift these technologies to the next level. A first one is monitoring activity of several signaling enzymes in one assay by multiplexing sensors. Lifetime detection of multiple FRET donors at the same time has allowed for simultaneous detection of PKA and ERK kinase activities in the cell (Demeautis et al. 2017). In FRET-BRET hybrid biosensors, advantages of both FRET and BRET sensors are combined by simply fusing a bioluminescent protein to a FRET biosensor (Komatsu et al. 2018). The main advantages that these hyBRET sensors owe to their BRET character are their applicability in whole-body imaging in model organisms and the ability to read out the signals in standard luminescence microplate readers. However, the HyBRET sensors generally also offer higher signal intensities compared to BRET sensors. Another option to combine FRET-based biosensors with in vivo imaging in a multicellular host organism, is the application of intravital imaging. In this setup, host cells, or infecting pathogens, expressing genetically engineered biosensors can be imaged in deeper layers of host tissue, thereby providing unique insights in the processes happening in the otherwise hidden places of the host body (Hirata and Kiyokawa 2016). Finally, it must be said that lately FRET-based biosensors have to compete more and more with single fluorescent protein (SP)-based sensors. Generally, these biosensors need less data analysis and allow for faster recording of the desired signal (Terai et al. 2019).
Do-it-yourself
Selection of FPs and linkers for PPI and biosensor assays
Optimization of EFRET can be achieved by using a donor FP with a high quantum yield and an acceptor with a high extinction coefficient. Increased overlap between the donor emission and acceptor excitation spectra will also enhance the efficiency of energy transfer (Piston and Kremers 2007). A very convenient tool to select proper FPs for FRET is fpbase (www.fpbase.org/fret/), where spectra can be displayed and matched and relevant parameters can be calculated. For Candida species, we recommend the use of a cyan FP, such as the novel mTurquoise2 and a yellow variant, such as YemVenus. Depending on the research question, a different method for calculation of EFRET should be chosen. For intermolecular FRET measurements, acceptor photobleaching, FRET-FLIM or sensitized emission FRET is preferable, while biosensors can be also monitored by ratiometric FRET. The main difference between both classes is that in intramolecular FRET, the donor and acceptor are always present in equimolecular concentrations, while this is not the case for intermolecular FRET. It is recommended to always confirm the positive FRET signal using at least two different methods.
Importantly, when using BiFC or FRET for the assessment of an interaction, one has to consider the conformation. For BiFC, the chance of reconstituting the fully functional fluorescent protein during an interaction, depends on the orientation of the non-fluorescent fragments. To decrease the chance of false negative results, a few measures have to be taken. First, both C- and N-terminal tagging combinations should be tested, bearing in mind the potential presence of cryptic signaling sites. Secondly, choosing the appropriate linker sequence is vital to allow for flexibility. Similarly, for FRET, the maximal EFRET is reached when the FPs are aligned in parallel (Day and Davidson 2012). Since the orientation of these FP dipole moments can hardly be predicted, the best way to deal with this is to insert a flexible linker between the two FPs or between the protein-of-interest and the FP to allow multiple conformations. In the section on ‘Imaging cellular structures’, some linkers were already proposed. It was shown that, although serine residues are important for flexibility, FRET efficiency in a simple ECFP-linker-EYFP probe increases with higher Gly/Ser ratios in the linker(van Rosmalen, Krom and Merkx 2017). Other linkers, containing alternative residues, can be used as well (Kerppola 2006). Apart from the sequence, also linker length should be considered. It was shown that EFRET of the ECFP-linker-EYFP probe decreased with longer linker lengths (Evers et al. 2006). Komatsu and coworkers also optimized their linker sequence and length, specifically for their GTPase activity FRET biosensor (Komatsu et al. 2011). They show that linkers with more Ser-Ala-Gly-Gly repeats make the sensor orientation-independent and lead to an increase in gain. However, implicating extremely long linkers (above 116 amino acids), did not further cause an increase in FRET, confirming the delicate equilibrium between length and flexibility. It is thus of pivotal importance to maintain adequate flexibility and protein functionality, without creating too much distance between the interacting domains. In our C. albicans BiFC system, a simple (Gly-Gly-Gly-Gly-Ser)2 linker proved to be efficient (Subotić et al. 2017).
Selection of proper controls for PPI and biosensor assays
Both for BiFC and FRET analyses, it is essential to include the proper controls to exclude false positive and minimize false negative results. In BiFC, nonspecific complementation of the fluorophore fragments often happens to a certain extent. The best negative control, for both BiFC and FRET, is mutation of the interaction domain (Kerppola 2006; Subotić et al. 2017). It is essential, however, to verify stability of the fusion proteins under these circumstances. Only when the signal is significantly reduced in the absence of the interacting domain, it likely represents a real interaction. FRET-biosensors can also be verified using such control. For instance, one can mutate the phosphorylation site of the AKAR probe (as explained in the ‘Monitoring cellular activity’ section) and verify absence of FRET even under PKA activating conditions. In the case where no interacting residues are known, one can screen for mutations that decrease the BiFC or FRET efficiency. When this is also not an option, you could assess the interaction between a protein-tag fusion and the complementary free tag, which should not produce any significant fluorescent or FRET signal (Subotić et al. 2017). In any case, it is important to verify the expression levels of the gene fusions as well as controls. In C. albicans, integration of plasmids is highly variable (Demuyser et al. 2017). The number of integration events should be the same for all constructs. As a positive control, a known interaction can be assessed. Specifically for FRET measurements, extra controls are necessary depending on the method used (Piston and Kremers 2007). For instance, when acceptor photobleaching is applied, one has to be certain that bleaching the acceptor does not affect donor fluorescence. Using sensitized emission, although the procedure itself is rather simple, many controls need to be tested and integrated in the calculation of EFRET. The main issue here is crosstalk or bleed-through, more precisely the direct excitation of the acceptor using donor excitation light and the emission spectrum of the donor bleeding through in the acceptor emission channel. In all cases, the background effect should be taken into account when calculating EFRET.
WHAT IS POSSIBLE?—SINGLE CELL PHENOMENA
Imaging of cells on a subcellular level allows researchers to investigate localization of proteins, detection of interactions and other molecular targets using fluorescent proteins. On a slightly larger scale, using fluorescent techniques to analyze complete or even many cells, researchers are able to draw conclusions about phenomena averaged over individual cells and accumulating to entire populations.
Techniques based on fluorescent proteins
Loss of heterozygosity
Candida albicans has a very dynamic and unstable genome. With genomic duplications, chromosome loss and other genomic rearrangements occurring frequently (Hickman et al. 2013). Loss of Heterozygosity (LOH) is one of these genomic rearrangements which appear in in vivo infection models and are able to render strains resistant to antifungals (Coste et al. 2006; Forche et al. 2009). A LOH reporter using BFP and GFP was developed to rapidly assess the amount of LOH events in a Candida population. This reporter system makes use of an additional locus that contains GFP on one chromosome and BFP on another chromosome. Fluorescence of the cells can be easily and rapidly measured using Fluorescence Activated Cell Sorting. Using appropriate gating settings, the number of cells expressing only BFP or GFP are an assessment for the amount of LOH events at that locus within the population. The combination of FACS and the fluorescent marker to assess LOH was verified using the markers that are also integrated at the locus. However, these LOH events are only measurable if they happen at the specific locus where GFP and BFP are inserted (Loll-Krippleber et al. 2015).
Phenotypic switching
Next to being genetically flexible, C. albicans is also phenotypically flexible. It is able to undergo a switch between a white and an opaque state. These cell states are visually distinguishable by the color and shape of the respective colonies on a rich nutrient medium. White cells form semi-spherical colonies, whilst opaque cells form a grey, flatter colony. This difference in phenotype can also be observed in single cells, where opaque-type cells are larger and bean-shaped compared to white-type cells (Slutsky et al. 1987). Using FPs as reporters for gene expression, it is possible to assess the phenotypic state of a single cell. This assay has been altered and optimized by using the next-generation FPs mNeonGreen and mScarlet placed behind the promotors of respectively WOR4 or WH11. If cells are in the opaque state, they fluoresce yellow-green based on expression of mNeonGreen, whilst in the white state they express mScarlet. The proportion of cells in either state can be assessed using FACS and appropriate filter sets. Using FACS in this setting allows for a rapid analysis of the phenotypic state of individual cells, instead of scoring colonies on plates (Frazer, Hernday and Bennett 2019).
Techniques based on organic dyes
Some organic dyes provide cellular context on a subcellular level, these have been discussed in the first chapter. However, these are not the only organic dyes useful for research on the pathogens C. albicans or C. glabrata. Organic dyes can be a powerful tool when combined with specific antibodies or lectins and provide insight into single cell characteristics within a population.
Dead/Live assays
A first example of such an organic dye is fluorescein diacetate, which is a cell-permeant chemical that is broken down by intracellular esterases into fluorescein, which is a green dye that can be excited with 488-nm light. This Dead/Live assay is thus an alternative for the commercial LIVE/DEAD® FungaLightTM Yeast Viability Kit from ThermoFisher that was discussed earlier. Using fluorescein diacetate it was shown that a C. albicans biofilm treated with high concentrations of amphotericin B still contains some living persister cells, with esterase activity, within a large complex of dead cells (Wuyts, Van Dijck and Holtappels 2018). The dye was also used in a screening assay for antifungal agents against C. albicans, where the absence of fluorescence from fluorescein is linked to cell death. Comparison of this technique to the standard method according to CLSI, revealed that it is suitable for detecting antifungal activity of amphotericin B, miconazole and fluconazole (Brouwer et al. 2006). Unfortunately, it was shown that Cdr1, a multidrug transporter, is able to extrude fluorescein diacetate, thereby limiting fluorescence, even from metabolically active cells (Yang et al. 2001). Whilst fluorescein diacetate specifically stains living cells, propidium iodide stains dead cells. It is a red dye which undergoes a red-shift in excitation and emission upon intercalating between the bases of DNA. It is only able to penetrate the cells if their integrity is disturbed. Therefore, it has been applied in an antifungal screen for compounds that target the membrane structure and thus lead to membrane permeabilization (Menzel et al. 2017). Another study utilized propidium iodide to assess pheromone-induced death of C. albicans and showed that approximately 20% of opaque SC5314 cells die upon administration of α pheromone (Alby et al. 2010). Both studies harvest the power and simplicity of a simple staining with propidium iodide, counting 10 to 105 cells with FACS for rapid data acquisition. Even though propidium iodide is a DNA intercalator and should therefore mainly be localized to the nucleus, significant portions of the dye accumulate in the cytoplasm, which makes it unsuitable for localization microscopy.
DNA content assay
Propidium iodide is often applied in S. cerevisiae to assess ploidy by intercalating within the DNA. Similarly, SybrGreen is also a DNA intercalating dye that is widely used in gel electrophoresis to visualize DNA bands under blue light and can also be used to investigate ploidy. Fixed cells that are exposed overnight to SybrGreen can be analysed using FACS by excitation with 488 nm and emission captured through a 520/30-nm bandpass filter. The fluorescence intensity is related to the DNA content, with haploid strains having a mean fluorescence intensity nearly 50% lower compared to diploid strains. Using this simple assay, it was proven that C. albicans is not an obligate diploid and that all isolates of the novel emerging pathogen Candida auris are haploid (Hickman et al. 2013; Bravo Ruiz et al. 2019).
Cell wall assays
One of the most significant differences between Candida species and mammalian cells is the fungal cell wall. This structure consists of an outer layer of α- and β-mannans and phosphomannans, with an interwoven matrix of chitins and β-glucan underneath. A common staining method for the mannans is based on Concanavalin A linked to FITC, Texas red, Alexa Fluor dyes, etc. Concanavalin A is a lectin, thus binding to sugars, such as α- and β-mannans in the cell wall. This lectin-based staining is a common ‘bread and butter’ method and was recently applied to stain cells in a amphotericin B treated biofilm (Wuyts, Van Dijck and Holtappels 2018). The staining is compatible with flow cytometry allowing a rapid assessment of the amount of mannans present in the cell wall in various clinical isolates and under different treatment conditions (Warolin, Essmann and Larsen 2005). A lectin-based assay was also developed to assess the amount of β-glucan masking. This β-glucan assay utilizes the mammalian β-glucan receptor, Dectin-1 as a tool to specifically target the beta-glucans within the cell wall. Soluble Dectin-1 binds to the β-glucans after which an antibody selective for this Dectin-1 and crosslinked with a dye, such as Alexa Fluor 488 or FITC, is able to stain the β-glucan, allowing for localization microscopy or whole cell beta-glucan content analysis using flow cytometry (Graham et al. 2006). Using this assay, it was shown that Candida strains grown on L-lactate mask their β-glucan to evade the host-immune response (Ballou et al. 2016).
Drug efflux pump assays
Research on Candida species often revolves around gaining knowledge about this pathogen as a way to develop novel antifungal drugs or to understand the mechanisms in which the pathogen becomes resistant to the antifungals that are available. One of these mechanisms of antifungal resistance is to lower the amount of intracellular antifungal, by means of sequestration or actively removing the drugs using drug efflux pumps. Activity of these drug efflux pumps in C. albicans is similar to the pumps of multidrug-resistant tumor cells. To assess the activity of these tumor cell efflux pumps, a yellow mitochondrial dye called Rhodamine 6G has been proven effective. When administered to the cells, it is selectively pumped out of the cytosol and therefore unable to stain the mitochondria of living mammalian cells (Kutushov and Gorelik 2013). Even though Rhodamine 6G was not shown to stain the mitochondria in C. albicans, it can be used to assess the activity of fungal drug efflux pumps. Using this assay, it was shown that Cdr1 is able to remove Rhodamine 6G, and that a CDR1 overexpressing strain, resistant to azoles, showed increased dye extrusion (Maesaki et al. 1999; Sharma et al. 2009). Another dye shown to be exported by the multidrug pumps Cdr1, Cdr2 and Mdr1, is Nile red. Nile red becomes highly fluorescent in a lipid-rich environment. It is usually excited using 488-nm light in a flow cytometry setup. Rhodamine 6G and Nile red are complementary drug efflux assays since Nile red is exported by all three drug efflux pumps in C. albicans, whilst Rhodamine 6G is not exported by Mdr1. This allows for discrimination of the mode-of-action of chemosensitizing compounds in a high-throughput screening setup (Ivnitski-Steele et al. 2009).
Combining multiple assays
All assays presented above use simple but clever techniques to exploit the power of fluorescent proteins and dyes to gain insights in the virulence and characteristics of Candida species. Each method has its limitations, upon which improvements can be made. It is clear that a combination of two assays is not always possible due to spectral overlap or other practical incompatibilities. When two dye-assays need to be combined, one can easily select a secondary antibody or lectin with alternative conjugated dyes. However, in the case of fluorescent proteins-based assays, such as the pH and oxygen sensing systems, simply choosing another fluorescent protein is not possible. Combining assays within a single strain is thus not always possible. Apart from the obvious option to evaluate the two parameters-of-interest sequentially or in parallel, one can also assess the possibility to acquire a spectral shift in one of the fluorescent proteins, for instance, by random mutagenesis. One could achieve this by error-prone or site-directed PCR on the FP. So far, most of these mutagenesis approaches expressed the mutagenized FP in E. coli and read out the fluorescence parameters in this organism. It would be even more relevant, however, to transform the mutagenized set of plasmids directly into Candida after which the transformed colonies are screened on plates or in a 96 well format. This way one integrates the possible effects of the organism and even organelle micro-environment in the assay.
WHAT IS POSSIBLE?—MULTISPECIES INTERACTIONS
Apart from visualizing molecular interactions of C. albicans on a subcellular level, fluorescence microscopy can also be used to detect and describe multi-species interactions. One can image the interaction between the fungus and its host organism or between the fungus and alternative micro-organisms, such as bacteria or other fungi. Both types of interactions are essential in fungal virulence and the study thereof indispensable for full understanding of pathogenesis and infection.
Host-pathogen interactions
The most relevant way of analyzing mechanisms and processes of fungal pathogens, is during infection of the host species, in many cases human beings. It is needless to say, however, what the difficulties are of such an endeavor. The use of model host organisms can be seen as a valid alternative for studying fungal infections. Fig. 3 represents an overview of model hosts which have been used for in vivo imaging of fungal infections. Care should be taken while choosing the model organism for your study, since not all imaging techniques can be used for all species. It goes without saying that the model organism should be relevant for the process under study. Aspects to keep in mind are body temperature, immune system, natural microbiota, etc. The host species should resemble the target host, in many cases human beings, as closely as possible. Furthermore, the animal should allow easy experimentation. Some practical aspects to consider, are ease of breading, housing and cost.
Figure 3.
Exemplification of fluorescence application in fungal infections of model organisms. A and B represent yolk infection of zebrafish larvae with GFP-labelled wild type C. albicans cells. C and D display co-infection of C. elegans by C. albicans and E. faecalis. E-H show biofilm-related subcutaneous infection of mice with C. glabrata, imaged with bioluminescence over time. I and J represent images of GFP-labelled C. albicans intradermal infection of the mouse ear, 30 min and 24 h post infection, respectively. K and L show a C. albicans infection of Galleria mellonella using bioluminescence. M, N and O represent invasion of C. albicans hyphae in epithelial cells. Actin filaments of Caco2 cells were stained with phalloidin-Alexa Fluor 596, C. albicans was stained with an antibody linked to Alexa Fluor 488 before and Calcofluor White after permeabilization of the epithelial cells. More information as well as references are given in the main text.
Using animal models for fluorescence imaging, however, implies some extra features to reflect on. Most importantly, the organism must be transparent for the type of excitation and emission light used during imaging. Certain biological molecules omnipresent in animal tissues, such as hemoglobin, melanin, water, collagen and nucleic acids, absorb photons in the visible and near-visible region, thereby decreasing visualization efficiency of exogenous chromophores (Bachmann et al. 2002). Secondly, some host molecules are highly fluorescent on their own, leading to, what is called, autofluorescence. Flavins and NAD(P)H are main causes of such native fluorescence in biological systems, yet also other molecules, such as fatty acids and proteins, emit light upon excitation at a particular wavelength (Croce and Bottiroli 2014). Lastly, biological tissue scatters light, meaning it causes diversion of photons from a straight path (Ntziachristos 2010). All these factors complicate simple and efficient imaging in animal tissues and organisms. Studying an infection in a host environment has several advantages. First, the conditions, comprising amongst others interaction with microbiota and host cells, the available nutrients and physical environment, mimic reality far closer to what is possible in vitro. Secondly, using imaging techniques allows the researcher to follow the same sample over time, thereby reducing variation of data which could possibly hide biologically interesting trends. Furthermore, it reduces the number of animals that is used in one study. From an ethical point of view, imaging processes in a living host rather than in postmortem tissue sections, is a great improvement. In the following section, we will describe a number of interesting animal models that can be and are used for fluorescent imaging of fungal infections.
Zebrafish
Multiple model organisms are easy to breed, allow simple housing and come at a relatively low cost, yet of high importance for imaging are the optical properties of the animal-of-choice. Apart from few pigmented cells, zebrafish larvae of 2–3 weeks of age are completely transparent, allowing for direct imaging of fungal infections (Kimmel et al. 1995). While pigmentation increases gradually towards adulthood, transparent adult zebrafish can also be obtained. One can chemically treat the embryo, for instance using 1-phenyl-2-thiourea, to inhibit melanogenesis (Karlsson, von Hofsten and Olsson 2001). Important side-effects of such treatment are impairment of normal behavior and survival of the adult fish (Antinucci and Hindges 2016). Multiple pigmentation mutant zebrafish strains have been generated. The best-known example is the casper mutant. This strain combines two mutations, the nacre mutation causes absence of melanocytes and the roy mutation causes lack of iridophores (White et al. 2008). Introducing a third mutation causes, what the researchers describe as, a crystal-clear phenotype because these mutant fish also lack retinal pigmentation (Antinucci and Hindges 2016).
Because of the lack of general pigmentation, virtually any fluorophore can be used to visualize either microbial or host cells. The lab of Prof Dr Robert Wheeler is specialized in imaging fungal infections in zebrafish larvae as a model host. Simply labeling the C. albicans strain fluorescently allows for analysis of dissemination and morphology in vivo during the infection process, as exemplified in Fig. 3A and B (Brothers, Newman and Wheeler 2011; Seman et al. 2018). Researchers have also developed fungal reporter strains, that report on the stresses encountered in the host niche. One such reporter allows for monitoring of oxidative stress and uses a fluorescence marker placed behind an oxidative stress-induced promoter (Brothers, Newman and Wheeler 2011; Brothers et al. 2013). Another example is a transcriptional arginine starvation reporter, where the fluorophore is placed behind the ARG1 or ARG3 promoter (Jiménez-López et al. 2013). By using zebrafish as a model system, one can determine the virulence constituents of the pathogen as well as the host immune factors necessary for efficiently preventing or reducing infection. The innate immune system of zebrafish is very similar to that of humans. The adaptive part, which differs to a larger extent, takes longer to develop in the model organism, allowing, however, for more in depth analysis of the initial immune response upon infection (Tobin, May and Wheeler 2012). One can study phagocyte recruitment by fluorescently labeling the cells-of-interest. Very recently, it was shown that the cytolytic peptide toxin, candidalysin, which is secreted by the fungal hyphae upon infection, does not only induce cell damage, as visualized by the fluorescent nuclear SytoxOrange dye, but also triggers an immune response in the host (Moyes et al. 2016). Using a transgenic zebrafish line with GFP-labelled neutrophils and red dTomato-labelled fungal cells, they showed that the epidermal growth factor is necessary for efficient recruitment of immune cells to the site of infection (J. Ho et al. 2019). Using a switchable fluorophore, such as Kaede, allows for specifically activating a subset of immune cells and following their movement upon infection (Brothers et al. 2013). Obviously, these zebrafish infection models can also be used for studying non-albicans Candida infections (Johnson et al. 2018; Archambault et al. 2019). The team of Prof Dr Jeniel Nett showed recently that contrary to what is seen for C. albicans, C. auris recruits significantly less neutrophils to the site of infection and that no observable NETs or neutrophil extracellular traps are formed, potentially explaining to some extent the particularly severe character of C. auris infections (Johnson et al. 2018).
Apart from its transparency, zebrafish are extremely suitable for live imaging because of the ability to immobilize them for a longer period of time and obtain high resolution images. Furthermore, their small size allows for high-content imaging, which could be especially interesting for screening compounds as potential new antifungal drugs or toxicity analyses (Oldach and Zhang 2014; Martinez et al. 2015). There are plenty of zebrafish infection models available, each exploiting a different route of infection and resembling an alternative niche in the human host (Rosowski et al. 2018; Lim et al. 2020). The ability to visualize the entire organism allows for unbiased mapping of pathogen distribution and effects at the host level. Apart from normal fluorescence imaging, zebrafish are also suitable for alternative microscopy techniques. One example is light sheet microscopy, during which only a thin plane of the sample is excited. It also allows for imaging of the entire organism for an extended period of time, due to the relatively low phototoxicity and solid immobilization (Kaufmann et al. 2012; Taormina et al. 2012). Disadvantages of using zebrafish as a model system, are their lack of certain organs compared to humans and the absence of a proper adaptive immune system. Furthermore, these fish are best kept at a temperature between 22 and 33°C, which is significantly lower compared to the human body temperature at which most fungal pathogens thrive best (Rosowski et al. 2018).
Caenorhabditis elegans
Another model organism that meets the criteria of imaging, is Caenorhabditis elegans, a nematode worm. Apart from being very small, easy to breed and maintain, and low in cost, importantly, C. elegans is also transparent. Furthermore, it is easy to immobilize, using agarose, and obtain high resolution images. Similar to zebrafish, the immune system of C. elegans is limited to an elaborate innate part. The nematode feeds on bacteria and also infections are mainly initiated by ingestion of pathogens. Therefore, the intestinal epithelium is an essential barrier against pathogenic micro-organisms (Pukkila-Worley and Ausubel 2012). Luckily, many of the mechanisms involved in this intestinal immunity are conserved and can thus be studied, using this model organism (Aballay and Ausubel 2002). When C. albicans cells are fed to the worms, they are ingested and produce hyphae. This results in the disruption of tissue and eventually in the death of the animal (Pukkila-Worley et al. 2009). Using fluorescent reporter constructs in C. elegans, Pukkila-Worley et al. showed that certain nematode genes are specifically upregulated upon bacterial infections and others upon fungal infection, with quite some genes showing opposite expression patterns in both situations (Pukkila-Worley, Ausubel and Mylonakis 2011).
To assess effectiveness of potential antifungal compounds, C. elegans can be used as a model host. Using this approach, it was shown that pyridoxatin, a small compound isolated from another fungus, efficiently inhibits virulence of C. albicans. One can also setup a high-throughput screening system to identify promising antifungal drugs. Combining this with high-content imaging, either using a fluorescently-labelled pathogen or a live/dead staining, would allow to screen for compounds that reduce virulence and/or mortality of the pathogen (Anastassopoulou, Fuchs and Mylonakis 2011). Using such as setup, researchers found novel compounds that show promising activity in the nematode as well as murine model of candidiasis (Breger et al. 2007). One aspect to keep in mind when using C. elegans as a model host for fluorescence analysis of fungal infections, is the relatively high autofluorescence in the blue/green region of the spectrum. Upon stress or ageing, the worm produces lipofuscin, which emits light at the same wavelength as GFP, thereby hampering visualization of weak signals (Pincus, Mazer and Slack 2016). Although the use of certain filters can overcome this issue, often red fluorophores form a safer option (Elkabti, Issi and Rao 2018; Teuscher and Ewald 2018).
Bioluminescence imaging in mice
As discussed at the beginning of this paragraph, certain molecules present in living tissue absorb light, for example melanin and hemoglobin. This is the main reason why using normal excitable fluorophores is commonly not advisable in non-transparent animals. Bioluminescence is a process by which proteins, named luciferases, produce light by oxidation of a substrate, often in the presence of O2 and/or ATP. As there is no need for excitation, no energy is lost. However, emission light can still be absorbed by the tissue. Since hemoglobin is the major cause of photon absorption in most tissues and it mainly absorbs light below 600 nm, it is advisable to opt for luciferases that emit light above this threshold wavelength (Zhao et al. 2005). Multiple luciferase enzymes were already discussed in section 1.2. Bioluminescence. Here, we will specifically link the enzymes to in vivo experimentation.
The firefly luciferase (fLuc) originates from beetle species and needs luciferin as a substrate (Wood, Lam and McElroy 1989). ATP is also required, thereby linking bioluminescence to the physiological state of the cell. Emission of the firefly luciferase occurs at wavelengths above 600 nm, which limits absorption by biological molecules such as hemoglobin (Brock 2012). The firefly luciferase system was applied to a vaginal infection model in mice, where a proper correlation was shown with colony forming unit counts. Systemically, a significant signal could not be detected. The authors suggested several reasons for this shortcoming, such as limited access of the substrate to the infected body site, restricted light penetration and lower permeability to the substrate of hyphal cells compared to yeast cells. Researchers of the HKI in Jena completely codon-optimized the luciferase for use in C. albicans and the peroxisomal targeting sequence, which is known to impair emission, was removed (Jacobsen et al. 2014). Using the optimized gene, these researchers were able to obtain a strong signal in a systemic infection by C. albicans. Upon infection of mice via the lateral tail vein, bioluminescent patches became visible in the kidneys, the brain and urinary bladder. Interestingly, using this platform, it became appreciable that, upon treatment of a systemic infection with caspofungin, some fungal cells persisted in the gall bladder, functioning here as a cryptic host niche. This example shows how imaging-based research can offer more insights than originally envisaged in the research question. Recently, the firefly luciferase was further improved (Dorsaz, Coste and Sanglard 2017). Researchers adapted the codon-optimized version (Jacobsen et al. 2014) and mutated certain residues to obtain a shift towards the red side of the spectrum and improve thermostability and enhanced light emission. The resulting luciferase showed improved sensitivity of C. albicans detection in in vitro and in vivo setups.
Another relevant luciferase enzyme originates from the sea pansy, Gaussia princeps—gLuc, which requires coelenterazine as a substrate. Notably, the emission of light is independent of ATP (Brock 2012). Drawbacks are the very poor distribution of the substrate in living organisms and the auto-oxidation that the substrate tends to undergo, leading to a high background signal. Furthermore, the emission at wavelengths below 600 nm causes high absorbance. Remarkably, the luciferase enzyme is secreted, which allows for improved accessibility of the substrate, yet limits localization of the fungal cells. To circumvent this limitation, other researchers adapted the luciferase for it to become attached to the fungal cell wall and thus more readily available for its substrate in both yeast and hyphal forms (Enjalbert et al. 2009). The applicability of this system in imaging of disease progression, was shown in a murine model of vaginal candidiasis where effectivity of a β-glucan-conjugate vaccine was confirmed (Pietrella et al. 2010). Although the system proved fit for use in a mouse model of oropharyngeal candidiasis as well, systemic infections still appeared impossible to image (Mosci et al. 2013; Gabrielli et al. 2015).
Both the firefly and the Gaussia princeps luciferase have advantages and shortcomings. While the optimized firefly luciferase is optimal for systemic and deep-seeded infections, the Gaussia princeps luciferase can be used for superficial infections. Colleagues from our laboratory have used the latter luciferase to establish and optimize a platform to visualize biofilm-related C. albicans infections in a subcutaneous mouse model (Vande Velde et al. 2014; Kucharíková et al. 2015). The system outperforms the standard methods used for assessing biofilm biomass as it allows to monitor the fungal burden from the start of the experiment onwards. It was shown that the established platform enables efficient evaluation of antifungal therapies over a long period of time, both in vitro and in vivo (Vande Velde et al. 2018). Very recently, we have established a similar system for monitoring C. glabrata biofilm infections, using an optimized firefly luciferase (Fig. 3E, F, G and H) (Persyn et al. 2019). Bioluminescence imaging is not restricted to rodents. In 2015, a C. albicans strain expressing the optimized firefly luciferase was used in the mini host Galleria mellonella (Fig. 3K and L) (Delarze et al. 2015). This model organism, although quite rudimentary in anatomy, has a number of advantages over rodent model systems. It is small, cheap and experimentation is ethically less regulated. This makes it better suited for high-throughput screening setups. The researchers also successfully used a novel, water-soluble, formulation of coelenterazine, which is not toxic and thus allows for in vivo kinetic studies of the infection.
Alternative imaging options for mice
Contrary to luciferases, standard fluorophores need to be excited by light. This limits their use in in vivo model systems because of high absorption of light by animal tissue. Two ways to circumvent this issue, are to image alternative host areas or to use fluorophores that are excited at wavelengths that are not readily absorbed by tissue. In 2010, Mitra et al. published an alternative in vivo model system in which GFP-tagged C. albicans cells can be imaged during a systemic infection in mice (Mitra et al. 2010). Injecting a fungal suspension intradermally into mouse ears, allowed to visualize the fungal cells at high resolution at this location by normal confocal microscopy (Fig. 3I and J). Infection of the kidneys, as determined post-mortally, confirmed the systemic character of the infection. The second option to use standard excitable fluorophores in in vivo non-transparent model systems, is to use FPs with a longer excitation wavelength. One such FP has already been presented when discussing the possibilities of working in the transparent zebrafish larvae, namely iRFP670. This FP is excited at 643 nm and emits at 670 nm (Shcherbakova and Verkhusha 2013). Light of such high wavelength is only minimally absorbed by hemoglobin and melanin. Furthermore, this spectral area is interesting due to its low light scattering and autofluorescence properties. Shcherbakova et al. show that injection of iRFP670-expressing tumors caused remarkable fluorescence in the intact animal (Shcherbakova and Verkhusha 2013). Using such labels for fungal pathogens could possibly also yield satisfactory results and would, most interestingly, omit the need for addition of exogenous substrates.
Novel in-host imaging techniques
Intravital microscopy (IVM) is an interesting novel tool that holds the promise of generating immense progress in the field of fungal pathogenesis. Using this technology, cell biology can be imaged in the living host at a relatively high resolution (Weigert et al. 2010). An image window is implanted in the tissue- or organ-of-interest, allowing for direct and real-time monitoring of cellular processes. Several advances made in the field of microscopy, such as two- and three-photon imaging, have improved the technology in terms of imaging depth and resolution. Due to limitations of intravital microscopy, fibered confocal fluorescence microscopy or FCFM provides some particular advantages (Choi, Kwok and Yun 2015). In this technique, the objective is replaced by a bundle of flexible, optical fibers, which can penetrate the tissue further, compared to what is possible with IVM. This technique has been used to image fungal infection of host organisms, for instance of pulmonary infections, caused by Aspergillus or Cryptococcus species (Vanherp et al. 2018).
As emphasized in a recent review on the matter, techniques combining whole-body imaging with a decent cellular resolution are still largely lacking from the mycologists’ toolbox (Van Dyck et al. 2020). Some promising technologies are, however, ready to be translated to fungal research. Selective plane illumination microscopy (SPIM), also referred to as light sheet microscopy, allows for imaging of larger, mesoscopic, samples, such as small organisms, organs or tissues with an intermediate to high resolution. Using a sheet of excitation light, planar illumination of a focal plane leads to optical sectioning of the 3D sample. Since only a thin plane of the sample is illuminated, the phototoxicity and background fluorescence is minimized. The immense potential of SPIM in host-pathogen imaging was recently illustrated by Amich et al. They used this technique to investigate the interaction between Aspergillus fumigatus and host immune cells in whole mouse lungs (Amich et al. 2020). The unique combination of whole-body analysis with high resolution imaging, yielded information on the spatial distribution patterns of immune cell populations in response to fungal infection. Alternative to SPIM, one can also use fluorescence optical projection tomography (OPT). In this technique, fluorescent images are captured while rotating the sample over an axis, generating multiple angular projections (McGinty et al. 2008). The resolution of the latter is limited in larger samples, yet it can work with fluorescent as well as non-fluorescent contrasts (A. Liu et al. 2019). Combining both SPIM and OPT in a hybrid technique, called OPTiSPIM, allows for fluorescent and non-fluorescent, high-resolution analysis of mesoscopic samples (Mayer et al. 2014). Both OPT and OPTiSPIM, have been used mainly in developmental biology with few examples of applications in infection biology (Ohtani et al. 2014; Schmidt et al. 2017).
Imaging microbe interaction with host cells
Working with living animals, although being most relevant, is not always easy nor possible in any laboratory context. Alternatively, one can examine the interaction between fungal pathogens and host cells or tissues in 2D or 3D setups. Here as well, fluorescence microscopy can help in visualizing the multispecies interaction and its characteristics.
Several cell types can come into contact with fungal pathogens, depending on the type and site of the infection. With alternative types of host cells, the type of interaction differs as well. When C. albicans colonizes the oral, vaginal epithelium or enterocytes, damage is induced by active penetration or induced endocytosis of host cells by the hyphae. To visualize the invasion process, researchers typically stain the fungal cells before and after permeabilization of the human cells. Staining the fungal cells when the host cells are still intact, will result in the visualization of the extracellular parts of the hyphae. Staining again, with a different dye, after permeabilization of the human cells, will result in visualization of the entire fungal cell (Dalle et al. 2010; Wächtler et al. 2012; Yang et al. 2018). As a primary stain, Alexa Fluor 488 can be used, fused to Concanavalin A, which will stain the fungal cell wall. As a secondary stain, Calcofluor White can be used, also staining the cell wall (Fig. 3M, N and O). Using this setup, researchers have confirmed that actin filaments of the epithelial cells reorganize at the point where fungal hyphae enter by induced endocytosis (Park et al. 2005; Dalle et al. 2010; Wöllert et al. 2012; Maza et al. 2017). Another possibility is to use immunocytochemistry with antibodies targeting C. albicans and fused (either directly or via a secondary antibody) to a fluorescent dye (Falgier et al. 2011). Instead of permeabilizing the host cells and applying a second staining, detailed analysis of differential interference contrast (DIC) images is also possible. To improve relevance of mammalian cell cultures, it is possible to use them in a transwell setup. Here, one can differentiate between both apical and basolateral sides of a host cell type. Different medium and supplements can be supplied, different microbes can be added, etc (Graf et al. 2019).
Another interaction model of interest for fungal infections, is one where fungal cells encounter immune cells, especially phagocytes. These cells can take up the fungal cells through phagocytosis. In order to visualize this process, researchers have established a system based on differential staining of internalized versus extracellular yeast cells (Carneiro et al. 2014). First, yeast cells are stained with SytoxGreen, a nuclear stain. After potential uptake by the phagocytes, another stain, propidium iodide, is used to quench the SytoxGreen stain. However, only those cells that are not taken up by the immune cells will be prone to this quenching. Eventually, cells that were taken up in the phagocytes will remain green while the extracellular ones will turn red. This differential staining can be used to visualize the cells using a microscope but can also allow quantitative analysis by flow cytometry. Researchers of the lab of Prof Dr Robin May have similarly used flow cytometry to investigate interaction of GFP-tagged Cryptococcus species with macrophages in an automated fashion (Voelz et al. 2010). Similar setups have also been used, employing different stains or genetic fluorescent markers. Examples of stains that can be used for yeast cells are FITC and Calcofluor White (Keppler-Ross et al. 2010; Dementhon, El-Kirat-Chatel and Noël 2012). Staining the host cells, for instance using calcein, which is an indicator of metabolism and membrane integrity, or a labelled antibody can be used to allow visualization of adhesion and interaction with the fungal cells (Dementhon, El-Kirat-Chatel and Noël 2012; de Turris et al. 2015). To overcome the resolution barrier set by normal fluorescence microscopy, researchers from Ghent University have established correlated fluorescence-atomic force microscopy (El-Kirat-Chatel and Dufrene 2012). Taking advantage of both worlds, this technique allows visualization of both interacting partners while still acquiring information about nanostructures of relevance to the interaction.
Apart from working with monolayers of mammalian cells, researchers can also investigate the interaction between microbes and host cells using ex vivo or in vitro 3D model systems, as such adding information and complexity to the interaction model. Different niches where fungal infections occur, can be mimicked. An example of how ex vivo systems can prove effective in studying a fungal infection is given by Wendland et al. They obtained a piece of the intestinal epithelium of female pigs and inoculated them with a C. albicans suspension. The surface layer where interaction had taken place, was scraped off and visualized using staining with Calcofluor White (Wendland et al. 2006). Similarly, ear skin from pigs was used to investigate effectivity of treatment against a mixed C. albicans—Staphylococcus aureus infection (Nair et al. 2016). In a model of teeth infections, premolar teeth extracted from humans were infected with C. albicans. After treatment, fungal viability was investigated using fluorescein diacetate (Gowri et al. 2016). Other human or animal materials that were already used for ex vivo investigation of Candida infection are saliva, tongues, oral or vaginal mucosa, corneas, blood, skin, etc. (Shin et al. 2013). Alternatively, it would also be possible to use organoid models. Organoids are self-organizing, 3D cultures of human or animal cells (Kim, Koo and Knoblich 2020). They are typically generated from pluripotent or adult stem cells and form mini organs mimicking the actual organ system in the host. A great advantage organoid-based infection models have, is the option to investigate patient-specific conditions, as the organoids can be generated from patient stem cells. Different conditions can be applied inside and outside of the organoid and also separate microbiomes can be established. As far as we know, no organoid systems have been used for Candida infections. On the contrary, various infections with bacteria have been investigated, such as Samonella, Helicobacter, etc. as reviewed by Bartfeld (Bartfeld 2016). Finally, to take one more step towards a fully relevant model system, it is possible to use organ-on-chip systems. In these devices, it is possible to integrate various tissue and cell types and interactions, flow of fluids, such as blood, and mechanical aspects into one compact system. In such a model for infection, one can assess evolution of the pathogen, the response of the host as well as heterogeneity of both (Tang et al. 2020). In this respect, many bacterial as well as viral infections have been investigated, while fungal infections are severally underrepresented (Bein et al. 2018). Maurer and coworkers used an intestine-on-chip model to show how Lactobacillus rhamnosus reduces tissue damage and translocation by C. albicans in the gut (Maurer et al. 2019). The whole process can be beautifully visualized by differential fluorescent tagging of all interaction partners.
Taken together, it can be acknowledged that many relevant ex vivo and in vitro systems exist that can replace animal models. Although the complexity of these systems will always be lower compared to an actual animal, the option to integrate human components and even personalized parts into these systems holds great promise for advancing infection research. As mentioned earlier, important aspects to keep in mind when visualizing thicker organic samples using fluorescence microscopy, are autofluorescence, transparency of the tissue and scattering of light. In some cases, it is more desirable to use a specific imaging setup such as intravital microscopy or appropriate fluorophores, such as infrared markers.
Microbe-microbe interactions
Apart from host-microbe interactions, fluorescently labeling microbes can also aid in elucidating relevance and mechanisms of microbe-microbe interactions. As microbes inhabit almost all niches in our body, it is fair to state that the interaction between pathogens and the rest of the microbiome is likely of utmost importance in determination of disease outcome. Recently, the Wheeler lab described how C. albicans and the bacterium Pseudomonas aeruginosa, enhance virulence compared to single-species infections, using the zebrafish larvae as host system (Bergeron et al. 2017). The main site of infection of P. aeruginosa is the lungs. Using zebrafish as a model system, the infection route showing the highest degree of homology is the swimbladder. By labelling the bacterium with a red fluorophore, dTomato, and the fungus with a far-red FP, iRFP670, these researchers were able to visualize how both species synergistically enhance virulence of one another, as well as mortality. Using a transgenic fish line with EGFP-labelled neutrophils allowed for concomitant visualization of immune cell recruitment upon mixed species infection. Multispecies interactions can also be studied using C. elegans as a model system. By using a C. albicans strain that constitutively expresses GFP, researchers visually showed that the pathogenic bacterium Acinetobacter baumannii reduces filamentation of the fungus and concomitant killing of the host organism to a large extent (Peleg et al. 2008). Similar research shows the same negative relation is true for C. albicans and the opportunistic intestinal pathogen Enterococcus faecalis (Cruz et al. 2013). In the latter case, an mCherry-expressing C. albicans strain and GFP-expressing E. faecalis strain were used, as shown in Fig. 3C and D.
Multiple microbial species often colocalize in biofilms. These are 3D-structures of microbial communities, where several micro-organisms can survive in the presence of an extracellular matrix. Whether pathogens, such as C. albicans, benefit from these interactions can be studied using fluorescence imaging. To label the different partners in a biofilm, one can use different methods. Apart from expression of a genetic label and using a chemical dye, another option is immunofluorescence, in which microbe-specific antibodies tagged with a fluorescent dye, such as FITC, are used. Further, FISH or fluorescence in situ hybridization makes use of fluorescent probes that specifically target a certain region in the species-specific DNA or RNA. Using combinations of several of these labels allows for visualization of integrative processes including multiple organisms. Diaz et al. showed that C. albicans and the bacterium Streptococcus oralis act synergistically in an in vitro model of the oral mucosa (Diaz et al. 2012). Candida cells were labelled by an antibody fused to FITC, S. oralis was labelled using a Streptococcus-specific FISH probe and the epithelial cells were stained using the Hoechst 33258 nuclear stain. Using the same tools, the researchers also showed the important role of glucosyltransferase in the biofilm-related interaction between Streptococcus gordonii and C. albicans (Ricker, Vickerman and Dongari-Bagtzoglou 2014). Apart from 2D images, also 3D models of biofilm structures can be generated. This is especially interesting for analysis of biofilm architecture and location-specific composition.
WHAT IS POSSIBLE?—DIAGNOSIS
One of the main hurdles in efficient treatment of fungal infections, is accurate and rapid detection of the infecting pathogen. Many of the procedures used today are still very time-consuming and not adequately sensitive or specific, which in turn leads to incorrect or inefficient treatment of the infected patient. The golden standard in clinical diagnostics of invasive candidiasis remains blood culture. Disadvantages are the low sensitivity and the long time needed to detect growth, on average 37 h (Fernandez et al. 2009). Furthermore, after the incubation period, the infecting species still needs to be identified, causing again a delay in the treatment procedure ranging from a few hours to several days (Ibáñez-Martínez, Ruiz-Gaitán and Pemán-García 2017). Standard identification procedures are based on carbohydrate assimilation or fermentation reactions, growth on chromogenic culture media, such as CHROMagar, induction and observation of hyphae formation, etc. Lately, the use of matrix assisted laser desorption/ionization—time of flight (MALDI-ToF) mass spectrometry after cultivation has gained popularity to identify fungi in a clinical setting and even to test drug susceptibility (Peng et al. 2019). These protocols typically take several days to be completed (El-Kholy et al. 2015). The most primitive way of detecting fungal cells in an infection using fluorescence, is by staining and macroscopic or microscopic evaluation. Using an aniline blue dye in the medium, researchers were able to differentiate between several Candida species and other fungi based on macroscopic observation of fluorescence of colonies under UV light irradiation (Goldschmidt et al. 1991). A major disadvantage, again, is the time required for growth of the Candida cells on this modified medium. In this respect, microscopic observation of fluorescent cells is a better alternative. Calcofluor White staining has been used most often. Slight differences in fluorescence pattern between different Candida species can be observed, with C. albicans showing the most distinctive pattern, i.e. hyphae and pseudohyphae (Safavieh et al. 2017; Yao et al. 2019). Other fluorescent whiteners, such as blankophor, can be used as well (Hamer, Moore and Denning 2006). Finally, since Candida species are known to be highly autofluorescent, this could also be used as a direct readout, without the need of staining (Graus, Neumann and Timlin 2017).
FISH
More recently, the use of alternative fluorescence techniques, has pushed the limits of these standard practices in terms of timing (from days to hours), sensitivity and specificity. FISH is used to detect the ribosomal RNA (rRNA, such as 18S or 26S rRNA) of a certain species by attachment of a fluorophore, such as FITC, to the sequence-specific probe, followed by fluorescence detection. In standard FISH, the probe backbone consists of a basic DNA phosphate ring structure. However, the intensity of these positive signals is often very low, due to electrostatic repulsion between the probe and target sequence. By replacing the DNA backbone by a non-charged polyamide backbone, the hybridization can be tighter and of higher specificity, causing higher intensity signals (Oliveira, Almeida and Azevedo 2020). Peptide nucleic acid fluorescent in situ hybridization or PNA FISH thus represents a promising alternative to the golden standard detection techniques (Rigby et al. 2002). Several studies have confirmed that this technique can indeed point out that an alternative therapy strategy is needed. Alexander et al. and Forrest et al. both show that PNA FISH was 100% sensitive and specific in the rapid identification of C. albicans in blood samples (Alexander et al. 2006; Forrest et al. 2006). In case C. albicans was detected, caspofungin treatment was replaced by fluconazole therapy, as this is the drug of choice for these infections. Due to this fast and reliable identification of the species type, the usage of caspofungin could be significantly reduced, along with the accompanying costs per patient. Since the initial description of PNA FISH in C. albicans in 2002, several adaptations were made. A second-generation two-color assay detects C. albicans as green fluorescence (FITC) and C. glabrata as red (Texas Red) allowing for simultaneous detection of both species. The commercially available Yeast Traffic Light system (AdvanDx, Woburn, MA), can be used to detect C. albicans and C. parapsilosis as green, C. glabrata and C. krusei as red and C. tropicalis as yellow (Fig. 4) (Radic et al. 2016).
Figure 4.
Identification of Candida species using PNA-FISH. The commercially available Yeast Traffic Light system (AdvanDx, OpGen, Woburn, MA), can be used to detect C. albicans and C. parapsilosis as green, C. glabrata and C. krusei as red and C. tropicalis as yellow.
qRT-PCR
Other diagnostic tools for fungal infections, are based on the detection and amplification of nucleic acids. Although these tests are not widely used in the clinic yet, they offer a substantial set of advantages over the currently-used culture-based diagnostics. The amplification of the target nucleic acid sequence can happen in an isothermal or non-isothermal fashion, with an example of the latter being PCR (Safavieh et al. 2017). The readout of the amplified nucleic acid sequence is often based on fluorescent labeling. A recently developed diagnostic tool that uses fluorescence as a readout, is quantitative, real-time PCR or qRT-PCR, where pathogenic DNA or RNA is amplified and fluorescently detected. One of the major advantages of direct pathogen detection in patient samples is the high sensitivity and the absence of a culturing step. Whereas normal PCR suffers from high levels of false positives, qRT-PCR is much more reliable (Bretagne and Costa 2005). A fully automated platform of extraction and detection could significantly decrease the processing time needed to obtain results. Several systems for the rapid identification of C. albicans by real-time PCR have been generated, with some of them allowing for simultaneous probing of several fungal as well as bacterial species in multiplex systems (Horváth et al. 2013). Most of the systems make use of the difference in melting temperatures of sequence-specific probes, targeting for instance ITS regions, or free dyes, such as SYBR Green, or combinations of both (Horváth et al. 2013). SYBR Green is a commonly used fluorescent dye that intercalates between the DNA double helix. The readout of amplification by sequence-specific probes can be based on FRET, with excitation of the donor probe transferring energy to the acceptor probe when bound to the adjacent nucleic acid sequences. Another option is a Taqman probe, where a specific nucleic acid sequence links together a fluorophore and a quencher molecule. Once the DNA or RNA is amplified, both molecules are separated upon which the fluorophore emits light. Researchers have established systems for specific and simultaneous detection of multiple clinically-relevant Candida species, such as C. albicans, C. glabrata, C. dubliniensis, C. tropicalis, C. parapsilosis, C. krusei, C. lusitaniae, C. guilliermondii and importantly C. auris, as well as some relevant bacterial species (Horváth et al. 2013; Ashrafi et al. 2015; Asadzadeh et al. 2018; Arastehfar et al. 2019; Nabili et al. 2013; Zhang et al. 2016). Apart from real-time detection, also DNA microarrays take advantage of a fluorescent readout. Thousands of potentially complementary sequences are attached to a surface and probed with a fluorescently labelled query sequence (Campa et al. 2008). Upon hybridization, the fluorescent pattern is visualized and analyzed for species specificity. Finally, another option to analyze amplification of pathogen-specific DNA or RNA, is through capillary electrophoresis-laser-induced fluorescence. Using fluorescently labelled primers, variability in length of, for instance, the ITS rRNA can be monitored during capillary electrophoresis (Obručová et al. 2016). A major disadvantage of using qRT-PCR or other nucleic acid-based methods for detection of pathogens, is the false-positive results that can be yielded from dead pathogens. As DNA is stable over a long period of time, a positive result in the assay does not necessarily mean that the pathogen is alive or viable (Keer and Birch 2003; Artz et al. 2006). In this sense, it is preferable to detect mRNA as the stability significantly less compared to DNA. Apart from fluorescence-based monitoring of Candida infection, obviously also other techniques can be used, such as nuclear magnetic resonance, gas-liquid chromatography, etc. (Safavieh et al. 2017). These are, however, not in the scope of this review.
CHALLENGES
Using fluorescence as a tool in research always imposes certain challenges. The success of simply tagging a protein-of-interest with a fluorophore depends on several factors, such as the type of fluorophore and whether it is the best option in these specific cellular conditions, the linker type and length, the effect of the tagging on protein function, the method used to monitor fluorescence, etc. No matter which research questions need to be answered or which tool is used to do so, some extent of optimization and quality control is always needed. Yet, apart from the general challenges that apply to fluorescence measurements in any given organism, certain additional obstacles are specific to working with Candida species. These complications can be subdivided in two categories, namely those related to the proper expression of fluorescent proteins or probes, and those related to fluorescence analysis.
Expression of FPs and probes
Candida albicans belongs to the Saccharomycetaceae family and more particularly to the CTG clade, which represents a subgroup in this family (McManus and Coleman 2014). Members of this clade frequently translate the CTG codon as a serine instead of a leucine, due to the recognition of the codon by an unorthodox seryl-tRNA with 5’-CAG-3’ anticodon (Ohama et al. 1993). Similar to C. albicans, most of the other clinically-relevant Candida species, such as C. auris, C. parapsilosis, C. tropicalis, C. dubliniensis, C. guilliermondii and C. lusitaniae belong the CTG clade, meaning that they all have an aberrant codon usage. It is reasonable to state that this slows down genetic manipulation and general experimentation in these fungi to a great extent. Most of the established tools or markers that are available for S. cerevisiae or mammalian systems, cannot be used directly in C. albicans or any of the other CTG clade members. In 1998, it was shown that presence of only one CTG codon was enough to completely inhibit translation of the GFP mRNA in C. albicans, indicating that codon optimization is indispensable (Morschhäuser, Michel and Hacker 1998). Even in case translation is permitted, presence of a CTG codon can lead to structural and thus functional variation in the protein (Cutfield, Sullivan and Cutfield 2000; Feketová et al. 2010; Miranda et al. 2013). Codon optimization is indeed essential in many cases. Some C. albicans optimized fluorescent proteins have been generated, in which the CTG codon has been replaced. Apart from specific CTG adaptations to the fluorescent protein, also optimization based on codon usage or preference has been performed and proved to further enhance expression and fluorescence intensity (Cormack et al. 1997; Gerami-Nejad, Berman and Gale 2001; Van Genechten et al. 2020). Other than C. albicans, C. glabrata, C. krusei and C. kefyr do not belong to the CTG clade and are more related to S. cerevisiae. Translation of established technologies has shown to be more straightforward in these species (Zordan et al. 2013; Ho and Haynes 2015; Demuyser et al. 2018). However, apart from aberrant codon usage by some species, many yeast species have AT-rich and highly plastic genomes, which also tend to complicate genetic manipulation (Massey et al. 2003). Translocations, truncations and ploidy changes are commonly observed. These processes allow for adaptation to new niches and contexts (Selmecki, Forche and Berman 2010). One practical implication is that integration of a plasmid in the genome of C. albicans happens at a highly variable frequency. In order to use Candida overexpression strains in research and compare expression or fluorescence intensity of tagged proteins, one should thus analyze the number of plasmid integration events that happened in each of the overexpression transformants (Demuyser et al. 2017; Demuyser et al. 2020; Van Genechten et al. 2020). This can simply be done by qPCR analysis on genomic DNA, using primers in the promoter region of the plasmid. Additionally, overexpression strains induce artifacts such as accumulation of the FP in the ER, vacuole and other terminal membranes, leading to misinterpretation of the localization results (Moore and Murphy 2009).
Fluorescence analysis
Apart from challenges in proper expression of the tagged construct, a few other peculiarities specific to one or more Candida species, are worth considering before starting fluorescence experimentation.
Keeping those cells from moving
When you want to monitor alterations in cellular fluorescence intensity over time, for instance upon addition of an inducer or inhibitor, time-lapse experiments are possible. Most types of microscopes allow for continuous imaging, either through a camera or programmed image acquisition. One of the main obstacles when performing time-lapse experiments, is the constant movement of cells. Cells often move around when in liquid and show aberrant fluorescence when left too long, as the liquid dries out. There are several options to immobilize cells for a longer period of time. First, one can opt to coat the microscopy slide or dish with Concanavalin A or polylysin. However, adding a compound or nutrient to the cells often changes the focus substantially, which is detrimental in a time-lapse setup. As an alternative, one can use a microfluidic system where the cells can be immobilized by physical restraining (Demuyser et al. 2018). The CellASIC® ONIX2 microfluidics device from Merck uses plates with microfluidic chambers where the cells are immobilized by an elastic ceiling. Different plates are available, designed for various organisms and cell types. For persistent focus shift, another option is to use a microscope with autofocus function, in which an internally reflected infrared laser automatically corrects the focus over a longer period of time (Bathe-Peters, Annibale and Lohse 2018).
Autofluorescence
It has been shown by other research groups as well as ours, that C. albicans produces a yellow pigment, termed riboflavin or vitamin B2 (Bai et al. 2011; Demuyser et al. 2020). Apart from its intense yellow color, this vitamin possesses the unique quality of fluorescence. Excitation is maximal at 450 nm with a smaller peek visible at 370 nm. The emission spectrum reaches its maximum at 530 nm. Importantly, many FPs are excited with light of the same range as riboflavin or emit light around the same maximum. As an example, the ECFP-EYFP FRET pair is excited at 434 nm and emits light at 477 and 527 nm. This indicates that extreme care should be taken when using these fluorophores in certain situations. We show that riboflavin production is augmented in C. albicans when PKA activity increases as well as in iron-limiting conditions (Demuyser et al. 2020). Using cyan and yellow FPs under these particular conditions might lead to artefacts in the signal due to riboflavin production. Proper controls should be analyzed to avoid misinterpretation of the signals. Other factors can cause autofluorescence in fungal cells as well, such as tryptophan metabolism or melanin production (Chaskes and Phillips 1974; Morris-Jones et al. 2005). In some cases, unique autofluorescence spectra by different Candida species can even be exploited in combination with hyperspectral fluorescence microscopy to diagnose the causing agent in infections (Graus, Neumann and Timlin 2017).
During an infection, fungal cells experience all sorts of stresses. It is therefore relevant to study stress response in the lab as well. Application of stress, however, often leads to increased autofluorescence levels and thus a decreased signal-to-noise ratio. This does not pose a problem when organic dyes are used, since they have high excitation coefficients. Yet, the in vivo brightness of fluorescent proteins is often much lower and the signal can be ‘lost’ within the elevated autofluorescent background. As most of the autofluorescent signal originating from riboflavin is excited with blue light and emits in the green channel, it is recommended to use a blue-shifted or red-shifted FP when performing experiments that could potentially increase autofluorescence levels.
Phototoxicity
Laser light, used in fluorescence experiments, can also cause phototoxicity in living cells (Icha et al. 2017). Excitation light is absorbed by organic molecules, such as flavins and porphyrins, which become degraded when reacting with oxygen, resulting in the production of reactive oxygen species (ROS). These damage the cell by oxidation of DNA, proteins and fatty acids, possibly leading to growth arrest or even cell death. Similarly, excited fluorophores can react with oxygen, upon which they bleach. The ROS resulting from this photobleaching likewise cause toxicity in the cells. One option to reduce phototoxicity, is to omit riboflavin or pyridoxal from the culture medium (Icha et al. 2017). LoFlo medium by Formedium is an example of such medium lacking folic acid and riboflavin. Such medium has been used in research on C. albicans fluorescence (Van Genechten et al. 2020). Otherwise, antioxidants or ROS scavengers can be added to the medium to minimize cellular ROS levels (Stockley et al. 2017). Using a lower illumination intensity or pulsed illumination will also reduce phototoxicity effects. Alternatively, selectively illuminating the focal plane by light sheet microscopy, TIRF microscopy or 2-photon microscopy or using excitation light with a longer wavelength is also possible (Icha et al. 2017). The sensitivity of C. albicans to phototoxicity has been exploited in treatment strategies. Such photodynamic therapy entails the addition of certain photosensitizers, or non-toxic dyes, which produce ROS under illumination with a harmless visible light. Specifically targeting the dyes to pathogenic cells allow for precise eradication of the infection without much chance of triggering resistance (Dougherty 2002; Dai et al. 2012). In the past, the unwanted toxicity of many compounds, such as cosmetics, have even been tested using C. albicans as a model organism, indicating its sensitivity to phototoxicity (Knudsen 1985; Faergemann and Larkö 1988; Horikawa and Miura 1988).
CONCLUSION
In this manuscript, we offer researchers a comprehensive overview of fluorescent techniques and applications in the field of fungal pathogenesis. We summarize the available fluorescent labels, based on fluorescent proteins, bioluminescent markers and dyes, and disclose how novel fluorophores could overcome several practical limitations researchers face today. Fluorescent imaging can contribute to insights in fungal pathogenesis on several levels. Subcellularly, structures can be visualised and protein-protein interactions can be detected. Information concerning the pH, oxygen level, enzyme activity and concentration of secondary metabolites can be obtained. The use of biosensors opens great possibilities for investigation of these physiologically relevant parameters. On the cellular level, fluorescence measurements can be used to report on phenomena such as viability, loss-of-heterozygosity, phenotypic switching and drug efflux. In the context of drug resistance and organismal niche adaptability, these characteristics are important to consider. Infections are often found to be polymicrobial in nature and the interaction between these micro-organisms affects the disease outcome. Differentially imaging these microbes offers unique insights in how fungi and bacteria influence each other and the host organism. Several animal models are available to image fungal cells in their relevant context. Transparency and low background fluorescence are important characteristics of model systems in a fluorescence setup. Recent improvements, however, allow standard animal models, such as rodents, to be used in an imaging setup as well. Finally, also diagnosis of fungal infections on the organismal level is possible using fluorescence. FISH and qRT-PCR are commonly used tools to identify the presence and cause of an infection. Taken together, fluorescence imaging offers great potential to advance our knowledge on fungal metabolism, virulence, interactions and infectability. Guided by this review, scientists will be able to explore the elaborate possibilities to optimize their specific experimental setup in function of the stated research question. It is important to keep investing time and funding in optimization of existing techniques and introduction of novel technologies as only in this manner unknown yet relevant aspects of fungal pathogenesis can be discovered.
ACKNOWLEDGEMENTS
We thank Nico Vangoethem for his assistance in production of the figures. We want to thank Prof Dr Dominique Sanglard (CHUV, Switzerland) for providing us with fluorescent images of infected G. mellonella. We thank Prof Dr Robert wheeler (University of Maine, USA) and the journal Infection and Immunity for permission to reuse a figure from their manuscript Seman, BG et al. Yeast and Filaments Have Specialized, Independent Activities in a Zebrafish Model of Candida albicans Infection. Infect Immun86, doi:10.1128/IAI.00415–18 (2018). We thank Prof Dr Michael Lorenz and Prof Dr Danielle Garsin (University of Texas, USA) and the journal Infection and Immunity for permission to reuse a figure from their manuscript Cruz MR, Graham CE, Gagliano BC, Lorenz MC & Garsin DA Enterococcus faecalis inhibits hyphal morphogenesis and virulence of Candida albicans. Infect Immun81, 189–200, doi:10.1128/IAI.00914–12 (2013). We also thank the journal Antimicrobial Agents and Chemotherapy for permission to reuse a figure from our manuscript Persyn, A. et al. Monitoring of Fluconazole and Caspofungin Activity against In Vivo Candida glabrata Biofilms by Bioluminescence Imaging. Antimicrob Agents Chemother63, doi:10.1128/AAC.01555–18 (2019). We express our gratitude to Prof Dr Melanie Wellington (University of Rochester, USA) and the Journal of Biomedical Optics Agents and Chemotherapy for permission to reuse a figure from the manuscript Mitra S, Dolan K, Foster TH & Wellington, M Imaging morphogenesis of Candida albicans during infection in a live animal. J Biomed Opt15, 010504, doi:10.1117/1.3290243 (2010). We thank Prof Dr Bernhard Hube (University of Jena, Germany) and the journal of Cellular Microbiology for permission to reuse a figure from the manuscript Dalle F et al. Cellular interactions of Candida albicans with human oral epithelial cells and enterocytes. Cell Microbiol12, 248–271, doi:10.1111/j.1462–5822.2009.01394.x (2010). We thank OpGen® for providing us permission to reuse the fluorescent image of the Yeast Traffic Light system.
Contributor Information
Wouter Van Genechten, VIB-KU Leuven Center for Microbiology, Kasteelpark Arenberg 31, 3001 Leuven-heverlee, Belgium; Laboratory of Molecular Cell Biology, Institute of Botany and Microbiology, KU Leuven, Kasteelpark Arenberg 31, 3001 Leuven-Heverlee, Belgium; Laboratory for Nanobiology, Department of Chemistry, KU Leuven, Celestijnenlaan 200g, 3001 Leuven-Heverlee, Belgium.
Patrick Van Dijck, VIB-KU Leuven Center for Microbiology, Kasteelpark Arenberg 31, 3001 Leuven-heverlee, Belgium; Laboratory of Molecular Cell Biology, Institute of Botany and Microbiology, KU Leuven, Kasteelpark Arenberg 31, 3001 Leuven-Heverlee, Belgium.
Liesbeth Demuyser, VIB-KU Leuven Center for Microbiology, Kasteelpark Arenberg 31, 3001 Leuven-heverlee, Belgium; Laboratory of Molecular Cell Biology, Institute of Botany and Microbiology, KU Leuven, Kasteelpark Arenberg 31, 3001 Leuven-Heverlee, Belgium.
FUNDING
We acknowledge funding by the FWO—Flanders Research Foundation (1S01817N and G062616N).
Conflicts of interest
None declared.
REFERENCES
- Aballay A, Ausubel FM. Caenorhabditis elegans as a host for the study of host-pathogen interactions. Curr Opin Microbiol. 2002;5:97–101. [DOI] [PubMed] [Google Scholar]
- Alby K, Schaefer D, Sherwood RKet al. Identification of a cell death pathway in Candida albicans during the response to pheromone. Eukaryot Cell. 2010;9:1690–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander BD, Ashley ED, Reller LBet al. Cost savings with implementation of PNA FISH testing for identification of Candida albicans in blood cultures. Diagn Microbiol Infect Dis. 2006;54:277–82. [DOI] [PubMed] [Google Scholar]
- Alkafeef SS, Yu C, Huang Let al. Wor1 establishes opaque cell fate through inhibition of the general co-repressor Tup1 in Candida albicans. PLos Genet. 2018;14:e1007176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen MD, Zhang J. Subcellular dynamics of protein kinase A activity visualized by FRET-based reporters. Biochem Biophys Res Commun. 2006;348:716–21. [DOI] [PubMed] [Google Scholar]
- Amich J, Mokhtari Z, Strobel Met al. Three-Dimensional light sheet fluorescence microscopy of lungs to dissect local host immune-aspergillus fumigatus interactions. mBio. 2020;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastassopoulou CG, Fuchs BB, Mylonakis E. Caenorhabditis elegans-based model systems for antifungal drug discovery. Curr Pharm Des. 2011;17:1225–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antinucci P, Hindges R. A crystal-clear zebrafish for in vivo imaging. Sci Rep. 2016;6:29490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arastehfar A, Fang W, Daneshnia Fet al. Novel multiplex real-time quantitative PCR detecting system approach for direct detection of Candida auris and its relatives in spiked serum samples. Future Microbiol. 2019;14:33–45. [DOI] [PubMed] [Google Scholar]
- Archambault LS, Trzilova D, Gonia Set al. Intravital imaging reveals divergent cytokine and cellular immune responses to Candida albicans and Candida parapsilosis. mBio. 2019;10:e00266–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arguëlles JC. Trehalose as antifungal target: The picture is still incomplete. Virulence. 2017;8:237–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aron AT, Loehr MO, Bogena Jet al. An endoperoxide reactivity-based FRET probe for ratiometric fluorescence imaging of labile iron pools in living cells. J Am Chem Soc. 2016;138:14338–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artz RR, Avery LM, Jones DLet al. Potential pitfalls in the quantitative molecular detection of Escherichia coli O157:H7 in environmental matrices. Can J Microbiol. 2006;52:482–8. [DOI] [PubMed] [Google Scholar]
- Asadzadeh M, Ahmad S, Al-Sweih Net al. Rapid and accurate identification of Candida albicans and Candida dubliniensis by real-time PCR and melting curve analysis. Med Princ Pract. 2018;27:543–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashrafi M, Nabili M, Shokohi Tet al. A real time PCR assay on blood for diagnosis of invasive candidiasis in immunocompromised patient. Curr Med Mycol. 2015;1:35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avci P, Gupta A, Sadasivam Met al. Low-level laser (light) therapy (LLLT) in skin: stimulating, healing, restoring. Semin Cutan Med Surg. 2013;32:41–52. [PMC free article] [PubMed] [Google Scholar]
- Bachmann SP, VandeWalle K, Ramage Get al. In vitro activity of caspofungin against Candida albicans biofilms. Antimicrob Agents Chemother. 2002;46:3591–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai C, Xu XL, Wang HSet al. Characterization of a hyperactive Cyr1 mutant reveals new regulatory mechanisms for cellular cAMP levels in Candida albicans. Mol Microbiol. 2011;82:879–93. [DOI] [PubMed] [Google Scholar]
- Bairwa G, Sánchez-León E, Do Eet al. A Cytoplasmic Heme Sensor Illuminates the Impacts of Mitochondrial and Vacuolar Functions and Oxidative Stress on Heme-Iron Homeostasis in Cryptococcus neoformans. mBio. 2020;11:e00986–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajar BT, Wang ES, Zhang Set al. A guide to fluorescent protein FRET pairs. Sensors (Basel). 2016;16:1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballou ER, Avelar GM, Childers DSet al. Lactate signalling regulates fungal beta-glucan masking and immune evasion. Nat Microbiol. 2016;2, 16238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartfeld S. Modeling infectious diseases and host-microbe interactions in gastrointestinal organoids. Dev Biol. 2016;420:262–70. [DOI] [PubMed] [Google Scholar]
- Bathe-Peters M, Annibale P, Lohse MJ. All-optical microscope autofocus based on an electrically tunable lens and a totally internally reflected IR laser. Opt Express. 2018;26:2359–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bein A, Shin W, Jalili-Firoozinezhad Set al. Microfluidic Organ-on-a-Chip Models of Human Intestine. Cell Mol Gastroenterol Hepatol. 2018;5:659–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benhamou RI, Bibi M, Steinbuch KBet al. Real-time imaging of the azole class of antifungal drugs in live candida cells. ACS Chem Biol. 2017;12:1769–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergeron AC, Seman BG, Hammond JHet al. Candida albicans and pseudomonas aeruginosa interact to enhance virulence of mucosal infection in transparent zebrafish. Infect Immun. 2017;85:e00475–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betzig E, Patterson GH, Sougrat Ret al. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313:1642–5. [DOI] [PubMed] [Google Scholar]
- Bindels DS, Haarbosch L, van Weeren Let al. mScarlet: a bright monomeric red fluorescent protein for cellular imaging. Nat Methods. 2017;14:53–56. [DOI] [PubMed] [Google Scholar]
- Boisnard S, Zhou Li Y, Arnaise Set al. Efficient mating-type switching in Candida glabrata induces cell death. PLoS One. 2015;10:e0140990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botman D, van Heerden JH, Teusink B. An improved ATP FRET sensor for yeast shows heterogeneity during nutrient transitions. ACS Sens. 2020;5:814–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boysen JH, Fanning S, Newberg Jet al. Detection of protein-protein interactions through vesicle targeting. Genetics. 2009;182:33–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo Ruiz G, Ross ZK, Holmes Eet al. Rapid and extensive karyotype diversification in haploid clinical Candida auris isolates. Curr Genet. 2019;65:1217–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breger J, Fuchs BB, Aperis Get al. Antifungal chemical compounds identified using a C. elegans pathogenicity assay. PLoS Pathog. 2007;3:e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretagne S, Costa JM. Towards a molecular diagnosis of invasive aspergillosis and disseminated candidosis. FEMS Immunol Med Microbiol. 2005;45:361–8. [DOI] [PubMed] [Google Scholar]
- Brimacombe CA, Burke JE, Parsa JYet al. A natural histone H2A variant lacking the Bub1 phosphorylation site and regulated depletion of centromeric histone CENP-A foster evolvability in Candida albicans. PLoS Biol. 2019;17:e3000331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brock M. Application of bioluminescence imaging for in vivo monitoring of fungal infections. Int J Microbiol. 2012;2012:956794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brothers KM, Gratacap RL, Barker SEet al. NADPH oxidase-driven phagocyte recruitment controls Candida albicans filamentous growth and prevents mortality. PLoS Pathog. 2013;9:e1003634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brothers KM, Newman ZR, Wheeler RT. Live imaging of disseminated candidiasis in zebrafish reveals role of phagocyte oxidase in limiting filamentous growth. Eukaryot Cell. 2011;10:932–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouwer N, Kohen J, Jamie Jet al. Modification of the fluorescein diacetate assay for screening of antifungal agents against Candida albicans: comparison with the NCCLS methods. J Microbiol Methods. 2006;66:234–41. [DOI] [PubMed] [Google Scholar]
- Brown GD, Denning DW, Gow NAet al. Hidden killers: human fungal infections. Sci Transl Med. 2012;4:165rv13. [DOI] [PubMed] [Google Scholar]
- Burgstaller S, Bischof H, Gensch Tet al. pH-Lemon a fluorescent protein-based pH reporter for acidic compartments. ACS Sens. 2019;4:883–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns S, Avena JS, Unruh JRet al. Structured illumination with particle averaging reveals novel roles for yeast centrosome components during duplication. Elife. 2015;4:e08586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler G, Rasmussen MD, Lin MFet al. Evolution of pathogenicity and sexual reproduction in eight Candida genomes. Nature. 2009;459:657–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campa D, Tavanti A, Gemignani Fet al. DNA microarray based on arrayed-primer extension technique for identification of pathogenic fungi responsible for invasive and superficial mycoses. J Clin Microbiol. 2008;46:909–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carneiro C, Vaz C, Carvalho-Pereira Jet al. A new method for yeast phagocytosis analysis by flow cytometry. J Microbiol Methods. 2014;101, 56–62. [DOI] [PubMed] [Google Scholar]
- Carriba P, Navarro G, Ciruela Fet al. Detection of heteromerization of more than two proteins by sequential BRET-FRET. Nat Methods. 2008;5:727–33. [DOI] [PubMed] [Google Scholar]
- Cauchie M, Desmet S, Lagrou K. Candida and its dual lifestyle as a commensal and a pathogen. Res Microbiol. 2017;168:802–10. [DOI] [PubMed] [Google Scholar]
- Chang BJ, Perez Meza VD, Stelzer EHK. csiLSFM combines light-sheet fluorescence microscopy and coherent structured illumination for a lateral resolution below 100 nm. Proc Natl Acad Sci USA. 2017;114:4869–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaskes S, Phillips AW. Pigmentation and autofluorescence of Candida species after growth on tryptophan media. Can J Microbiol. 1974;20:595–603. [DOI] [PubMed] [Google Scholar]
- Chauhan N, Jentsch JA, Menon AK. Measurement of Intracellular Sterol Transport in Yeast. Methods Mol Biol. 2019;1949:115–36. [DOI] [PubMed] [Google Scholar]
- Choi M, Kwok SJ, Yun SH. In vivo fluorescence microscopy: lessons from observing cell behavior in their native environment. Physiology (Bethesda). 2015;30:40–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbel C, Sartini S, Levati Eet al. Screening for protein-protein interaction inhibitors using a bioluminescence resonance energy transfer (BRET)-based assay in yeast. SLAS Discov. 2017;22:751–59. [DOI] [PubMed] [Google Scholar]
- Cormack BP, Bertram G, Egerton Met al. Yeast-enhanced green fluorescent protein (yEGFP): a reporter of gene expression in Candida albicans. Microbiology. 1997;143:303–11. [DOI] [PubMed] [Google Scholar]
- Cormack BP, Valdivia RH, Falkow S. FACS-optimized mutants of the green fluorescent protein (GFP). Gene. 1996;173:33–8. [DOI] [PubMed] [Google Scholar]
- Coste A, Turner V, Ischer Fet al. A mutation in Tac1p a transcription factor regulating CDR1 and CDR2, is coupled with loss of heterozygosity at chromosome 5 to mediate antifungal resistance in Candida albicans. Genetics. 2006;172:2139–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crameri A, Whitehorn EA, Tate Eet al. Improved green fluorescent protein by molecular evolution using DNA shuffling. Nat Biotechnol. 1996;14:315–9. [DOI] [PubMed] [Google Scholar]
- Croce AC, Bottiroli G. Autofluorescence spectroscopy and imaging: a tool for biomedical research and diagnosis. Eur J Histochem. 2014;58:2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz MR, Graham CE, Gagliano BCet al. Enterococcus faecalis inhibits hyphal morphogenesis and virulence of Candida albicans. Infect. Immun. 2013;81:189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y, Zhang X, Yu Met al. Techniques for detecting protein-protein interactions in living cells: principles limitations and recent progress. Sci China Life Sci. 2019;62:619–32. [DOI] [PubMed] [Google Scholar]
- Cutfield JF, Sullivan PA, Cutfield SM. Minor structural consequences of alternative CUG codon usage (Ser for Leu) in Candida albicans exoglucanase. Protein Eng. 2000;13:735–8. [DOI] [PubMed] [Google Scholar]
- Dagher Z, Xu S, Negoro PEet al. Fluorescent tracking of yeast division clarifies the essential role of spleen tyrosine kinase in the intracellular control of candida glabrata in macrophages. Front Immunol. 2018;9:1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai T, Gupta A, Murray CKet al. Blue light for infectious diseases: Propionibacterium acnes Helicobacter pylori and beyond?. Drug Resist. Updat. 2012;15:223–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalle F, Wächtler B, L'Ollivier Cet al. Cellular interactions of Candida albicans with human oral epithelial cells and enterocytes. Cell Microbiol. 2010;12:248–71. [DOI] [PubMed] [Google Scholar]
- Dana H, Sun Y, Mohar Bet al. High-performance calcium sensors for imaging activity in neuronal populations and microcompartments. Nat Methods. 2019;16:649–57. [DOI] [PubMed] [Google Scholar]
- Davidovits P, Egger MD. Scanning laser microscope for biological investigations. Appl Opt. 1971;10:1615–9. [DOI] [PubMed] [Google Scholar]
- Day RN, Davidson MW. Fluorescent proteins for FRET microscopy: monitoring protein interactions in living cells. Bioessays. 2012;34:341–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedecker P, Mo GCH, Dertinger Tet al. Widely accessible method for superresolution fluorescence imaging of living systems. Proc Natl Acad Sci USA. 2012;109:10909–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delarze E, Ischer F, Sanglard Det al. Adaptation of a Gaussia princeps Luciferase reporter system in Candida albicans for in vivo detection in the Galleria mellonella infection model. Virulence. 2015;6:684–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demeautis C, Sipieter F, Roul Jet al. Multiplexing PKA and ERK1&2 kinases FRET biosensors in living cells using single excitation wavelength dual colour FLIM. Sci Rep. 2017;7:41026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dementhon K, El-Kirat-Chatel S, Noël T. Development of an in vitro model for the multi-parametric quantification of the cellular interactions between Candida yeasts and phagocytes. PLoS One. 2012;7:e32621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demuyser L, Palmans I, Vandecruys Pet al. Molecular elucidation of riboflavin production and regulation in candida albicans toward a novel antifungal drug target. mSphere. 2020;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demuyser L, Swinnen E, Fiori Aet al. Mitochondrial cochaperone Mge1 is involved in regulating susceptibility to fluconazole in saccharomyces cerevisiae and candida species. MBio. 2017;8:e00201–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demuyser L, Van Genechten W, Mizuno Het al. Introducing fluorescence resonance energy transfer-based biosensors for the analysis of cAMP-PKA signalling in the fungal pathogen Candida glabrata. Cell Microbiol. 2018;20:e12863. [DOI] [PubMed] [Google Scholar]
- Dertinger T, Colyer R, Iyer Get al. Fast background-free, 3D super-resolution optical fluctuation imaging (SOFI). Proc Natl Acad Sci USA. 2009;106:22287–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Turris V, Teloni R, Chiani Pet al. Candida albicans targets a lipid Raft/Dectin-1 platform to enter human monocytes and induce antigen specific T cell responses. PLoS One. 2015;10:e0142531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz PI, Xie Z, Sobue Tet al. Synergistic interaction between Candida albicans and commensal oral streptococci in a novel in vitro mucosal model. Infect Immun. 2012;80:620–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsaz S, Coste AT, Sanglard D. Red-shifted firefly luciferase optimized for candida albicans in vivo bioluminescence imaging. Front Microbiol. 2017;8:1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty TJ. An update on photodynamic therapy applications. J Clin Laser Med Surg. 2002;20:3–7. [DOI] [PubMed] [Google Scholar]
- Doyle TC, Nawotka KA, Kawahara CBet al. Visualizing fungal infections in living mice using bioluminescent pathogenic Candida albicans strains transformed with the firefly luciferase gene. Microb Pathog. 2006a;40:82–90. [DOI] [PubMed] [Google Scholar]
- Doyle TC, Nawotka KA, Purchio AFet al. Expression of firefly luciferase in Candida albicans and its use in the selection of stable transformants. Microb Pathog. 2006b;40:69–81. [DOI] [PubMed] [Google Scholar]
- Drepper T, Eggert T, Circolone Fet al. Reporter proteins for in vivo fluorescence without oxygen. Nat Biotechnol. 2007;25:443–5. [DOI] [PubMed] [Google Scholar]
- Eichhof I, Ernst JF. Oxygen-independent FbFP: Fluorescent sentinel and oxygen sensor component in Saccharomyces cerevisiae and Candida albicans. Fungal Genet Biol. 2016;92:14–25. [DOI] [PubMed] [Google Scholar]
- El-Kholy AA, El-Rachidi NG, El-Enany MGet al. Impact of serology and molecular methods on improving the microbiologic diagnosis of infective endocarditis in Egypt. Infection. 2015;43:523–9. [DOI] [PubMed] [Google Scholar]
- El-Kirat-Chatel S, Dufrene YF. Nanoscale imaging of the Candida-macrophage interaction using correlated fluorescence-atomic force microscopy. ACS Nano. 2012;6:10792–9. [DOI] [PubMed] [Google Scholar]
- Elkabti AB, Issi L, Rao RP. Caenorhabditis elegans as a model host to monitor the candida infection processes. J Fungi (Basel). 2018;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ene IV, Cheng SC, Netea MGet al. Growth of Candida albicans cells on the physiologically relevant carbon source lactate affects their recognition and phagocytosis by immune cells. Infect Immun. 2013;81:238–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ene IV, Lohse MB, Vladu AVet al. Phenotypic profiling reveals that candida albicans opaque cells represent a metabolically specialized cell state compared to default white cells. MBio. 2016;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enjalbert B, Rachini A, Vediyappan Get al. A multifunctional, synthetic Gaussia princeps luciferase reporter for live imaging of Candida albicans infections. Infect Immun. 2009;77:4847–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evers TH, van Dongen EM, Faesen ACet al. Quantitative understanding of the energy transfer between fluorescent proteins connected via flexible peptide linkers. Biochemistry. 2006;45:13183–92. [DOI] [PubMed] [Google Scholar]
- Faergemann J, Larkö O. Phototoxicity of skin microorganisms tested with a new model. Arch Dermatol Res. 1988;280:168–70. [DOI] [PubMed] [Google Scholar]
- Falgier C, Kegley S, Podgorski Het al. Candida species differ in their interactions with immature human gastrointestinal epithelial cells. Pediatr Res. 2011;69:384–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feketová Z, Mašek T, Vopálenský Vet al. Ambiguous decoding of the CUG codon alters the functionality of the Candida albicans translation initiation factor 4E. FEMS Yeast Res. 2010;10:558–69. [DOI] [PubMed] [Google Scholar]
- Fernandez J, Erstad BL, Petty Wet al. Time to positive culture and identification for Candida blood stream infections. Diagn Microbiol Infect Dis. 2009;64:402–7. [DOI] [PubMed] [Google Scholar]
- Ferrari S, Sanguinetti M, Torelli Ret al. Contribution of CgPDR1-regulated genes in enhanced virulence of azole-resistant Candida glabrata. PLoS One. 2011;6:e17589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filonov GS, Verkhusha VV. A near-infrared BiFC reporter for in vivo imaging of protein-protein interactions. Chem Biol. 2013;20:1078–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forche A, Magee PT, Selmecki Aet al. Evolution in Candida albicans populations during a single passage through a mouse host. Genetics. 2009;182:799–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest GN, Mankes K, Jabra-Rizk MAet al. Peptide nucleic acid fluorescence in situ hybridization-based identification of Candida albicans and its impact on mortality and antifungal therapy costs. J Clin Microbiol. 2006;44:3381–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazer C, Hernday AD, Bennett RJ. Monitoring phenotypic switching in candida albicans and the use of next-gen fluorescence reporters. Curr Protoc Microbiol. 2019;53:e76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freikamp A, Mehlich A, Klingner Cet al. Investigating piconewton forces in cells by FRET-based molecular force microscopy. J Struct Biol. 2017;197:37–42. [DOI] [PubMed] [Google Scholar]
- Fujii Y, Yoshimura A, Kodama Y. A novel orange-colored bimolecular fluorescence complementation (BiFC) assay using monomeric Kusabira-Orange protein. BioTechniques. 2018;64:153–61. [DOI] [PubMed] [Google Scholar]
- Fukutani Y, Ishii J, Kondo Aet al. Split luciferase complementation assay for the analysis of G protein-coupled receptor ligand response in Saccharomyces cerevisiae. Biotechnol Bioeng. 2017;114:1354–61. [DOI] [PubMed] [Google Scholar]
- Gabrielli E, Roselletti E, Luciano Eet al. Comparison between bioluminescence imaging technique and CFU count for the study of oropharyngeal candidiasis in mice. Cytometry A. 2015;87:428–36. [DOI] [PubMed] [Google Scholar]
- Gaur S, Bhargava-Shah A, Hori Set al. Engineering intracellularly retained gaussia luciferase reporters for improved biosensing and molecular imaging applications. ACS Chem Biol. 2017;12:2345–53. [DOI] [PubMed] [Google Scholar]
- Gautier A, Juillerat A, Heinis Cet al. An engineered protein tag for multiprotein labeling in living cells. Chem Biol. 2008;15:128–36. [DOI] [PubMed] [Google Scholar]
- Gerami-Nejad M, Berman J, Gale CA. Cassettes for PCR-mediated construction of green, yellow, and cyan fluorescent protein fusions in Candida albicans. Yeast. 2001;18:859–64. [DOI] [PubMed] [Google Scholar]
- Gerami-Nejad M, Dulmage K, Berman J. Additional cassettes for epitope and fluorescent fusion proteins in Candida albicans. Yeast. 2009;26:399–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedhart J, von Stetten D, Noirclerc-Savoy Met al. Structure-guided evolution of cyan fluorescent proteins towards a quantum yield of 93%. Nat Commun. 2012;3:751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldschmidt MC, Fung DY, Grant Ret al. New aniline blue dye medium for rapid identification and isolation of Candida albicans. J Clin Microbiol. 1991;29:1095–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonçalves S, Silva PM, Felício MRet al. Psd1 effects on candida albicans planktonic cells and biofilms. Front Cell Infect Microbiol. 2017;7:249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowri M, Sofi Beaula W, Biswal Jet al. beta-lactam substituted polycyclic fused pyrrolidine/pyrrolizidine derivatives eradicate C. albicans in an ex vivo human dentinal tubule model by inhibiting sterol 14-alpha demethylase and cAMP pathway. Biochim Biophys Acta. 2016;1860:636–47. [DOI] [PubMed] [Google Scholar]
- Graf K, Last A, Gratz Ret al. Keeping Candida commensal: how lactobacilli antagonize pathogenicity of Candida albicans in an in vitro gut model. Dis Model Mech. 2019;12:dmm039719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham LM, Tsoni SV, Willment JAet al. Soluble Dectin-1 as a tool to detect beta-glucans. J Immunol Methods. 2006;314:164–9. [DOI] [PubMed] [Google Scholar]
- Grahl N, Shepardson KM, Chung Det al. Hypoxia and fungal pathogenesis: to air or not to air?. Eukaryot Cell. 2012;11:560–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graus MS, Neumann AK, Timlin JA. Hyperspectral fluorescence microscopy detects autofluorescent factors that can be exploited as a diagnostic method for Candida species differentiation. J Biomed Opt. 2017;22:16002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greger K, Swoger J, Stelzer EH. Basic building units and properties of a fluorescence single plane illumination microscope. Rev Sci Instrum. 2007;78:023705. [DOI] [PubMed] [Google Scholar]
- Hamer EC, Moore CB, Denning DW. Comparison of two fluorescent whiteners, Calcofluor and Blankophor, for the detection of fungal elements in clinical specimens in the diagnostic laboratory. Clin Microbiol Infect. 2006;12:181–4. [DOI] [PubMed] [Google Scholar]
- Han Q, Wang N, Pan Set al. Elevation of cell wall chitin via Ca(2+) -calcineurin-mediated PKC signaling pathway maintains the viability of Candida albicans in the absence of beta-1,6-glucan synthesis. Mol Microbiol. 2019;12:960–972. [DOI] [PubMed] [Google Scholar]
- Heim R, Tsien RY. Engineering green fluorescent protein for improved brightness, longer wavelengths and fluorescence resonance energy transfer. Curr Biol. 1996;6:178–82. [DOI] [PubMed] [Google Scholar]
- Hickman MA, Zeng G, Forche Aet al. The ‘obligate diploid’ Candida albicans forms mating-competent haploids. Nature. 2013;494:55–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata E, Kiyokawa E. Future Perspective of Single-Molecule FRET Biosensors and Intravital FRET Microscopy. Biophys J. 2016;111:1103–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirose F, Ohshima N, Kwon EJet al. Drosophila Mi-2 negatively regulates dDREF by inhibiting its DNA-binding activity. Mol Cell Biol. 2002;22:5182–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan DA, Sundstrom P. The Ras/cAMP/PKA signaling pathway and virulence in Candida albicans. Future Microbiol. 2009;4:1263–70. [DOI] [PubMed] [Google Scholar]
- Ho HL, Haynes K. Candida glabrata: new tools and technologies-expanding the toolkit. FEMS Yeast Res. 2015;15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho J, Yang X, Nikou SAet al. Candidalysin activates innate epithelial immune responses via epidermal growth factor receptor. Nat Commun. 2019;10:2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horikawa E, Miura T. Phototoxic index of drug determined by Candida growth inhibition. J Dermatol. 1988;15:523–6. [DOI] [PubMed] [Google Scholar]
- Horváth A, Pető Z, Urbán Eet al. A novel, multiplex, real-time PCR-based approach for the detection of the commonly occurring pathogenic fungi and bacteria. BMC Microbiol. 2013;13:300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huisken J, Stainier DY. Even fluorescence excitation by multidirectional selective plane illumination microscopy (mSPIM). Opt Lett. 2007;32:2608–10. [DOI] [PubMed] [Google Scholar]
- Höfig H, Otten J, Steffen Vet al. Genetically encoded forster resonance energy transfer-based biosensors studied on the single-molecule level. ACS Sens. 2018;3:1462–70. [DOI] [PubMed] [Google Scholar]
- Ibáñez-Martínez E, Ruiz-Gaitán A, Pemán-García J. Update on the diagnosis of invasive fungal infection. Rev Esp Quimioter. 2017;30:16–21. [PubMed] [Google Scholar]
- Icha J, Weber M, Waters JCet al. Phototoxicity in live fluorescence microscopy, and how to avoid it. Bioessays. 2017;39. [DOI] [PubMed] [Google Scholar]
- Ivnitski-Steele I, Holmes AR, Lamping Eet al. Identification of Nile red as a fluorescent substrate of the Candida albicans ATP-binding cassette transporters Cdr1p and Cdr2p and the major facilitator superfamily transporter Mdr1p. Anal Biochem. 2009;394:87–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen ID, Lüttich A, Kurzai Oet al. In vivo imaging of disseminated murine Candida albicans infection reveals unexpected host sites of fungal persistence during antifungal therapy. J Antimicrob Chemother. 2014;69:2785–96. [DOI] [PubMed] [Google Scholar]
- Jain P, Sethi SC, Pratyusha VAet al. Ras signaling activates glycosylphosphatidylinositol (GPI) anchor biosynthesis via the GPI-N-acetylglucosaminyltransferase (GPI-GnT) in Candida albicans. J Biol Chem. 2018;293:12222–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez-López C, Collette JR, Brothers KMet al. Candida albicans induces arginine biosynthetic genes in response to host-derived reactive oxygen species. Eukaryot Cell. 2013;12:91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson CJ, Muse Davis K, Huttenlocher Aet al. Emerging fungal pathogen candida auris evades neutrophil attack. mBio. 2018;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapitan M, Eichhof I, Lagadec Qet al. Click beetle luciferases as dual reporters of gene expression in Candida albicans. Microbiology. 2016;162:1310–20. [DOI] [PubMed] [Google Scholar]
- Karlsson J, von Hofsten J, Olsson PE. Generating transparent zebrafish: a refined method to improve detection of gene expression during embryonic development. Mar Biotechnol (NY). 2001;3:522–7. [DOI] [PubMed] [Google Scholar]
- Kaufmann A, Mickoleit M, Weber Met al. Multilayer mounting enables long-term imaging of zebrafish development in a light sheet microscope. Development. 2012;139:3242–7. [DOI] [PubMed] [Google Scholar]
- Keer JT, Birch L. Molecular methods for the assessment of bacterial viability. J Microbiol Methods. 2003;53:175–83. [DOI] [PubMed] [Google Scholar]
- Keij JF, Bell-Prince C, Steinkamp JA. Staining of mitochondrial membranes with 10-nonyl acridine orange, MitoFluor Green, and MitoTracker Green is affected by mitochondrial membrane potential altering drugs. Cytometry. 2000;39:203–10. [DOI] [PubMed] [Google Scholar]
- Keppler-Ross S, Douglas L, Konopka JBet al. Recognition of yeast by murine macrophages requires mannan but not glucan. Eukaryot Cell. 2010;9:1776–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keppler-Ross S, Noffz C, Dean N. A new purple fluorescent color marker for genetic studies in Saccharomyces cerevisiae and Candida albicans. Genetics. 2008;179:705–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keppler A, Gendreizig S, Gronemeyer Tet al. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat. Biotechnol. 2003;21:86–9. [DOI] [PubMed] [Google Scholar]
- Kerppola TK. Design and implementation of bimolecular fluorescence complementation (BiFC) assays for the visualization of protein interactions in living cells. Nat Protoc. 2006;1:1278–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerppola TK. Visualization of molecular interactions using bimolecular fluorescence complementation analysis: characteristics of protein fragment complementation. Chem Soc Rev. 2009;38:2876–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuta S, Hou BH, Sato Ret al. FRET sensor-based quantification of intracellular trehalose in mammalian cells. Biosci Biotechnol Biochem. 2016;80:162–5. [DOI] [PubMed] [Google Scholar]
- Kim J, Koo BK, Knoblich JA. Human organoids: model systems for human biology and medicine. Nat Rev Mol Cell Biol. 2020;21:571–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmel CB, Ballard WW, Kimmel SRet al. Stages of embryonic development of the zebrafish. Dev Dyn. 1995;203:253–310. [DOI] [PubMed] [Google Scholar]
- Klar TA, Jakobs S, Dyba Met al. Fluorescence microscopy with diffraction resolution barrier broken by stimulated emission. Proc Natl Acad Sci USA. 2000;97:8206–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klengel T, Liang WJ, Chaloupka Jet al. Fungal adenylyl cyclase integrates CO2 sensing with cAMP signaling and virulence. Curr Biol. 2005;15:2021–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen EA. The Candida phototoxicity test. The sensitivity of different strains and species of Candida, standardization attempts and analysis of the dose-response curves for 5- and 8-methoxypsoralen. Photodermatol. 1985;2:80–5. [PubMed] [Google Scholar]
- Komatsu N, Aoki K, Yamada Met al. Development of an optimized backbone of FRET biosensors for kinases and GTPases. Mol Biol Cell. 2011;22:4647–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu N, Terai K, Imanishi Aet al. A platform of BRET-FRET hybrid biosensors for optogenetics, chemical screening, and in vivo imaging. Sci Rep. 2018;8:8984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostyuk AI, Demidovich AD, Kotova DAet al. Circularly permuted fluorescent protein-based indicators: history, principles, and classification. Int J Mol Sci. 2019;20:4200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremers GJ, Goedhart J, van Munster EBet al. Cyan and yellow super fluorescent proteins with improved brightness, protein folding, and FRET Forster radius. Biochemistry. 2006;45:6570–80. [DOI] [PubMed] [Google Scholar]
- Kucharíková S, Vande Velde G, Himmelreich Uet al. Candida albicans biofilm development on medically-relevant foreign bodies in a mouse subcutaneous model followed by bioluminescence imaging. J Vis Exp. 2015:52239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai A, Ando R, Miyatake Het al. A bilirubin-inducible fluorescent protein from eel muscle. Cell. 2013;153:1602–11. [DOI] [PubMed] [Google Scholar]
- Kutushov M, Gorelik O. Low concentrations of Rhodamine-6G selectively destroy tumor cells and improve survival of melanoma transplanted mice. Neoplasma. 2013;60:262–73. [DOI] [PubMed] [Google Scholar]
- Lager I, Looger LL, Hilpert Met al. Conversion of a putative Agrobacterium sugar-binding protein into a FRET sensor with high selectivity for sucrose. J Biol Chem. 2006;281:30875–83. [DOI] [PubMed] [Google Scholar]
- Lai WC, Sun HS, Shieh JC. Establishment of tetracycline-regulated bimolecular fluorescence complementation assay to detect protein-protein interactions in Candida albicans. Sci Rep. 2020;10:2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim JY, Park YH, Pyon YHet al. The LAMMER kinase is involved in morphogenesis and response to cell wall- and DNA-damaging stresses in Candida albicans. Med Mycol. 2020;58:240–47. [DOI] [PubMed] [Google Scholar]
- Li R, Liu Z, Dong Wet al. The antifungal peptide CGA-N12 inhibits cell wall synthesis of Candida tropicalis by interacting with KRE9. Biochem J. 2020;477:747–62. [DOI] [PubMed] [Google Scholar]
- Liu A, Xiao W, Li Ret al. Comparison of optical projection tomography and light-sheet fluorescence microscopy. J Microsc. 2019;275:3–10. [DOI] [PubMed] [Google Scholar]
- Liu NN, Kohler JR. Antagonism of fluconazole and a proton pump inhibitor against Candida albicans. Antimicrob Agents Chemother. 2016;60:1145–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Sun L, Lu Cet al. Promising antifungal targets against candida albicans based on ion homeostasis. Front Cell Infect Microbiol. 2018;8:286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loll-Krippleber R, Feri A, Nguyen Met al. A FACS-optimized screen identifies regulators of genome stability in Candida albicans. Eukaryot Cell. 2015;14:311–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzo C, Frongia C, Jorand Ret al. Live cell division dynamics monitoring in 3D large spheroid tumor models using light sheet microscopy. Cell Div. 2011;6:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Los GV, Encell LP, McDougall MGet al. HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem Biol. 2008;3:373–82. [DOI] [PubMed] [Google Scholar]
- Lue NF, Chan J. Duplication and functional specialization of the telomere-capping protein Cdc13 in Candida species. J Biol Chem. 2013;288:29115–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luna-Tapia A, Encell LP, McDougall MGet al. ERG2 and ERG24 are required for normal vacuolar physiology as well as candida albicans pathogenicity in a murine model of disseminated but not vaginal candidiasis. Eukaryot Cell. 2015;14:1006–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maesaki S, Marichal P, Vanden Bossche Het al. Rhodamine 6G efflux for the detection of CDR1-overexpressing azole-resistant Candida albicans strains. J Antimicrob Chemother. 1999;44:27–31. [DOI] [PubMed] [Google Scholar]
- Magee BB, Magee PT. Induction of mating in Candida albicans by construction of MTLa and MTLalpha strains. Science. 2000;289:310–3. [DOI] [PubMed] [Google Scholar]
- Mahon MJ. pHluorin2: an enhanced, ratiometric, pH-sensitive green florescent protein. Adv Biosci Biotechnol. 2011;2:132–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamouei Z, Zeng G, Wang YMet al. Candida albicans possess a highly versatile and dynamic high-affinity iron transport system important for its commensal-pathogenic lifestyle. Mol Microbiol. 2017;106:986–98. [DOI] [PubMed] [Google Scholar]
- Marriott G, Mao S, Sakata Tet al. Optical lock-in detection imaging microscopy for contrast-enhanced imaging in living cells. Proc Natl Acad Sci USA. 2008;105:17789–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez NJ, Titus SA, Wagner AKet al. High-throughput fluorescence imaging approaches for drug discovery using in vitro and in vivo three-dimensional models. Expert Opin Drug Discov. 2015;10:1347–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masser AE, Kandasamy G, Kaimal JMet al. Luciferase NanoLuc as a reporter for gene expression and protein levels in Saccharomyces cerevisiae. Yeast. 2016;33:191–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massey SE, Moura G, Beltrão Pet al. Comparative evolutionary genomics unveils the molecular mechanism of reassignment of the CTG codon in Candida spp. Genome Res. 2003;13:544–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer M, Gresnigt MS, Last Aet al. A three-dimensional immunocompetent intestine-on-chip model as in vitro platform for functional and microbial interaction studies. Biomaterials. 2019;220:119396. [DOI] [PubMed] [Google Scholar]
- Mayer J, Robert-Moreno A, Danuser Ret al. OPTiSPIM: integrating optical projection tomography in light sheet microscopy extends specimen characterization to nonfluorescent contrasts. Opt Lett. 2014;39:1053–6. [DOI] [PubMed] [Google Scholar]
- Maza PK, Bonfim-Melo A, Padovan ACBet al. Candida albicans: The ability to invade epithelial cells and survive under oxidative stress is unlinked to hyphal length. Front Microbiol. 2017;8:1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinty J, Tahir KB, Laine Ret al. Fluorescence lifetime optical projection tomography. J Biophotonics. 2008;1:390–4. [DOI] [PubMed] [Google Scholar]
- McManus BA, Coleman DC. Molecular epidemiology, phylogeny and evolution of Candida albicans. Infect Genet Evol. 2014;21:166–78. [DOI] [PubMed] [Google Scholar]
- Menzel LP, Chowdhury HM, Masso-Silva JAet al. Potent in vitro and in vivo antifungal activity of a small molecule host defense peptide mimic through a membrane-active mechanism. Sci Rep. 2017;7:4353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KE, Kim Y, Huh WKet al. Bimolecular Fluorescence Complementation (BiFC) analysis: advances and recent applications for genome-wide interaction studies. J Mo Biol. 2015;427:2039–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda I, Silva-Dias A, Rocha Ret al. Candida albicans CUG mistranslation is a mechanism to create cell surface variation. MBio. 2013;4:e00285–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra S, Dolan K, Foster THet al. Imaging morphogenesis of Candida albicans during infection in a live animal. J Biomed Opt. 2010;15:010504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyawaki A, Niino Y. Molecular spies for bioimaging–fluorescent protein-based probes. Mol Cell. 2015;58:632–43. [DOI] [PubMed] [Google Scholar]
- Moeyaert B, Nguyen Bich N, De Zitter Eet al. Green-to-red photoconvertible Dronpa mutant for multimodal super-resolution fluorescence microscopy. ACS Nano. 2014;8:1664–73. [DOI] [PubMed] [Google Scholar]
- Moore I, Murphy A. Validating the location of fluorescent protein fusions in the endomembrane system. Plant Cell. 2009;21:1632–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morikawa TJ, Fujita H, Kitamura Aet al. Dependence of fluorescent protein brightness on protein concentration in solution and enhancement of it. Sci Rep. 2016;6:22342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris-Jones R, Gomez BL, Diez Set al. Synthesis of melanin pigment by Candida albicans in vitro and during infection. Infect Immun. 2005;73:6147–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morschhäuser J, Michel S, Hacker J. Expression of a chromosomally integrated, single-copy GFP gene in Candida albicans, and its use as a reporter of gene regulation. Mol Gen Genet. 1998;257:412–20. [DOI] [PubMed] [Google Scholar]
- Mosci P, Pericolini E, Gabrielli Eet al. A novel bioluminescence mouse model for monitoring oropharyngeal candidiasis in mice. Virulence. 2013;4:250–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyes DL, Wilson D, Richardson JPet al. Candidalysin is a fungal peptide toxin critical for mucosal infection. Nature. 2016;532:64–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulhern SM, Logue ME, Butler G. Candida albicans transcription factor Ace2 regulates metabolism and is required for filamentation in hypoxic conditions. Eukaryot Cell. 2006;5:2001–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabili M, Ashrafi M, Janbabaie Get al. Quantification and optimization of Candida albicans DNA in blood samples using Real- Time PCR. Rep Biochem Mol Biol. 2013;2:42–7. [PMC free article] [PubMed] [Google Scholar]
- Nagai T, Ibata K, Park ESet al. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat Biotechnol. 2002;20:87–90. [DOI] [PubMed] [Google Scholar]
- Nair SV, Baranwal G, Chatterjee Met al. Antimicrobial activity of plumbagin, a naturally occurring naphthoquinone from Plumbago rosea, against Staphylococcus aureus and Candida albicans. Int J Med Microbiol. 2016;306:237–48. [DOI] [PubMed] [Google Scholar]
- Nguyen LN, Hamari Z, Kadereit Bet al. Candida parapsilosis fat storage-inducing transmembrane (FIT) protein 2 regulates lipid droplet formation and impacts virulence. Microbes Infect. 2011;13:663–72. [DOI] [PubMed] [Google Scholar]
- Ntziachristos V. Going deeper than microscopy: the optical imaging frontier in biology. Nat Methods. 2010;7:603–14. [DOI] [PubMed] [Google Scholar]
- Obara K, Kihara A. Signaling events of the Rim101 pathway occur at the plasma membrane in a ubiquitination-dependent manner. Mol Cell Biol. 2014;34:3525–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obručová H, Tihelková R, Kotásková Iet al. Evaluation of fluorescent capillary electrophoresis for rapid identification of candida fungal infections. J Clin Microbiol. 2016;54:1295–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohama T, Suzuki T, Mori Met al. Non-universal decoding of the leucine codon CUG in several Candida species. Nucleic Acids Res. 1993;21:4039–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtani M, Villumsen KR, Strøm HKet al. 3D visualization of the initial Yersinia ruckeri infection route in rainbow trout (Oncorhynchus mykiss) by optical projection tomography. PLoS One. 2014;9:e89672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oku M, Hoseki J, Ichiki Yet al. A fluorescence resonance energy transfer (FRET)-based redox sensor reveals physiological role of thioredoxin in the yeast Saccharomyces cerevisiae. FEBS Lett. 2013;587:793–8. [DOI] [PubMed] [Google Scholar]
- Oldach L, Zhang J. Genetically encoded fluorescent biosensors for live-cell visualization of protein phosphorylation. Chem Biol. 2014;21:186–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira R, Almeida C, Azevedo NF. Detection of microorganisms by fluorescence in situ hybridization using peptide nucleic acid. Methods Mol Biol. 2020;2105:217–30. [DOI] [PubMed] [Google Scholar]
- Ormo M, Cubitt AB, Kallio Ket al. Crystal structure of the Aequorea victoria green fluorescent protein. Science. 1996;273:1392–5. [DOI] [PubMed] [Google Scholar]
- Palmer E, Freeman T. Investigation into the use of C- and N-terminal GFP fusion proteins for subcellular localization studies using reverse transfection microarrays. Comp Funct Genomics. 2004;5:342–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Myers CL, Sheppard DCet al. Role of the fungal Ras-protein kinase A pathway in governing epithelial cell interactions during oropharyngeal candidiasis. Cell Microbiol. 2005;7:499–510. [DOI] [PubMed] [Google Scholar]
- Peleg AY, Tampakakis E, Fuchs BBet al. Prokaryote-eukaryote interactions identified by using Caenorhabditis elegans. Proc Natl Acad Sci USA. 2008;105:14585–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y, Zhang Q, Xu Cet al. MALDI-TOF MS for the rapid identification and drug susceptibility testing of filamentous fungi. Exp Ther Med. 2019;18:4865–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peroza EA, Ewald JC, Parakkal Get al. A genetically encoded Forster resonance energy transfer sensor for monitoring in vivo trehalose-6-phosphate dynamics. Anal Biochem. 2015;474:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persyn A, Rogiers O, Brock Met al. Monitoring of fluconazole and caspofungin activity against in vivo candida glabrata biofilms by bioluminescence imaging. Antimicrob Agents Chemother. 2019;63:e01555–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrella D, Rachini A, Torosantucci Aet al. A beta-glucan-conjugate vaccine and anti-beta-glucan antibodies are effective against murine vaginal candidiasis as assessed by a novel in vivo imaging technique. Vaccine. 2010;28:1717–25. [DOI] [PubMed] [Google Scholar]
- Pincus Z, Mazer TC, Slack FJ. Autofluorescence as a measure of senescence in C. elegans: look to red, not blue or green. Aging (Albany NY). 2016;8:889–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piston DW, Kremers GJ. Fluorescent protein FRET: the good, the bad and the ugly. Trends Biochem Sci. 2007;32:407–14. [DOI] [PubMed] [Google Scholar]
- Plamont MA, Billon-Denis E, Maurin Set al. Small fluorescence-activating and absorption-shifting tag for tunable protein imaging in vivo. Proc Natl Acad Sci USA. 2016;113:497–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasher DC, Eckenrode VK, Ward WWet al. Primary structure of the Aequorea victoria green-fluorescent protein. Gene. 1992;111:229–33. [DOI] [PubMed] [Google Scholar]
- Pukkila-Worley R, Ausubel FM, Mylonakis E. Candida albicans infection of Caenorhabditis elegans induces antifungal immune defenses. PLoS Pathog. 2011;7:e1002074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pukkila-Worley R, Ausubel FM. Immune defense mechanisms in the Caenorhabditis elegans intestinal epithelium. Curr Opin Immunol. 2012;24:3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pukkila-Worley R, Peleg AY, Tampakakis Eet al. Candida albicans hyphal formation and virulence assessed using a Caenorhabditis elegans infection model. Eukaryot Cell. 2009;8:1750–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radic M, Goic-Barisic I, Novak Aet al. Evaluation of PNA FISH(R) Yeast Traffic Light in identification of Candida species from blood and non-blood culture specimens. Med Mycol. 2016;54:654–8. [DOI] [PubMed] [Google Scholar]
- Rai V, Gaur M, Kumar Aet al. A novel catalytic mechanism for ATP hydrolysis employed by the N-terminal nucleotide-binding domain of Cdr1p, a multidrug ABC transporter of Candida albicans. Biochim Biophys Acta. 2008;1778:2143–53. [DOI] [PubMed] [Google Scholar]
- Reynaud EG, Krzic U, Greger Ket al. Light sheet-based fluorescence microscopy: more dimensions, more photons, and less photodamage. HFSP J. 2008;2:266–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricker A, Vickerman M, Dongari-Bagtzoglou A. Streptococcus gordonii glucosyltransferase promotes biofilm interactions with Candida albicans. J Oral Microbiol. 2014;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigby S, Procop GW, Haase Get al. Fluorescence in situ hybridization with peptide nucleic acid probes for rapid identification of Candida albicans directly from blood culture bottles. J Clin Microbiol. 2002;40:2182–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robledo-Márquez K, Gutiérrez-Escobedo G, Yáñez-Carrillo Pet al. Candida glabrata encodes a longer variant of the mating type (MAT) alpha2 gene in the mating type-like MTL3 locus, which can form homodimers. FEMS Yeast Res. 2016;16: fow082. [DOI] [PubMed] [Google Scholar]
- Roemer T, Krysan DJ. Antifungal drug development: challenges, unmet clinical needs, and new approaches. Cold Spring Harb Perspect Med. 2014;4:a019703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosowski EE, Knox BP, Archambault LSet al. The zebrafish as a model host for invasive fungal infections. J Fungi (Basel). 2018;4:136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rust MJ, Bates M, Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat Methods. 2006;3:793–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safavieh M, Coarsey C, Esiobu Net al. Advances in Candida detection platforms for clinical and point-of-care applications. Crit Rev Biotechnol. 2017;37:441–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- San Martín A, Ceballo S, Ruminot Iet al. A genetically encoded FRET lactate sensor and its use to detect the Warburg effect in single cancer cells. PLoS One. 2013;8:e57712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlafer S, Kamp A, Garcia JE. A confocal microscopy based method to monitor extracellular pH in fungal biofilms. FEMS Yeast Res. 2018;18. [DOI] [PubMed] [Google Scholar]
- Schmidt JG, Korbut R, Ohtani Met al. Zebrafish (Danio rerio) as a model to visualize infection dynamics of Vibrio anguillarum following intraperitoneal injection and bath exposure. Fish Shellfish Immunol. 2017;67:692–97. [DOI] [PubMed] [Google Scholar]
- Schoeters F, Munro CA, d'Enfert Cet al. A High-Throughput Candida albicans Two-Hybrid System. mSphere. 2018;3:e00391–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoeters F, Van Dijck P. Protein-protein interactions in Candida albicans. Front Microbiol. 2019;10:1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellam A, Chaillot J, Mallick Jet al. The p38/HOG stress-activated protein kinase network couples growth to division in Candida albicans. PLos Genet. 2019;15:e1008052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selmecki A, Forche A, Berman J. Genomic plasticity of the human fungal pathogen Candida albicans. Eukaryot Cell. 2010;9:991–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seman BG, Moore JL, Scherer AKet al. Yeast and filaments have specialized, independent activities in a zebrafish model of candida albicans infection. Infect Immun. 2018;86:IAI.00415–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah AH, Banerjee A, Rawal MKet al. ABC transporter Cdr1p harbors charged residues in the intracellular loop and nucleotide-binding domain critical for protein trafficking and drug resistance. FEMS Yeast Res. 2015;15:fov036. [DOI] [PubMed] [Google Scholar]
- Sharma M, Manoharlal R, Shukla Set al. Curcumin modulates efflux mediated by yeast ABC multidrug transporters and is synergistic with antifungals. Antimicrob Agents Chemother. 2009;53:3256–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbakova DM, Verkhusha VV. Near-infrared fluorescent proteins for multicolor in vivo imaging. Nat Methods. 2013;10:751–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimomura O, Johnson FH, Saiga Y. Extraction, purification and properties of aequorin, a bioluminescent protein from the luminous hydromedusan, Aequorea. J Cell Comp Physiol. 1962;59:223–39. [DOI] [PubMed] [Google Scholar]
- Shin SH, Lee YS, Shin YPet al. Therapeutic efficacy of halocidin-derived peptide HG1 in a mouse model of Candida albicans oral infection. J. Antimicrob Chemother. 2013;68:1152–60. [DOI] [PubMed] [Google Scholar]
- Shu X, Royant A, Lin MZet al. Mammalian expression of infrared fluorescent proteins engineered from a bacterial phytochrome. Science. 2009;324:804–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skruzny M, Pohl E, Abella M. FRET Microscopy in Yeast. Biosensors (Basel). 2019;9:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slutsky B, Staebell M, Anderson Jet al. “White-opaque transition”: a second high-frequency switching system in Candida albicans. J Bacteriol. 1987;169:189–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Madahar V, Liao J. Development of FRET assay into quantitative and high-throughput screening technology platforms for protein-protein interactions. Ann Biomed Eng. 2011;39:1224–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Yang M, Wegner SVet al. A genetically encoded FRET sensor for intracellular heme. ACS Chem Biol. 2015;10:1610–5. [DOI] [PubMed] [Google Scholar]
- Srikantha T, Chandrasekhar A, Soll DR. Functional analysis of the promoter of the phase-specific WH11 gene of Candida albicans. Mol Cell Biol. 1995;15:1797–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srikantha T, Klapach A, Lorenz WWet al. The sea pansy Renilla reniformis luciferase serves as a sensitive bioluminescent reporter for differential gene expression in Candida albicans. J Bacteriol. 1996;178:121–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stagge F, Mitronova GY, Belov VNet al. SNAP-, CLIP- and Halo-tag labelling of budding yeast cells. PLoS One. 2013;8:e78745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockley JH, Evans K, Matthey Met al. Surpassing light-induced cell damage in vitro with novel cell culture media. Sci Rep. 2017;7:849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stynen B, Van Dijck P, Tournu H. A CUG codon adapted two-hybrid system for the pathogenic fungus Candida albicans. Nucleic Acids Res. 2010;38:e184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subotić A, Swinnen E, Demuyser Let al. A bimolecular fluorescence complementation tool for identification of protein-protein interactions in Candida albicans. G3 (Bethesda). 2017;10:3509–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung MK, Huh WK. Bimolecular fluorescence complementation analysis system for in vivo detection of protein-protein interaction in Saccharomyces cerevisiae. Yeast. 2007;24:767–75. [DOI] [PubMed] [Google Scholar]
- Sun S, Yang X, Wang Yet al. In vivo analysis of protein-protein interactions with bioluminescence resonance energy transfer (BRET): Progress and prospects. Int J Mol Sci. 2016;17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surdo NC, Berrera M, Koschinski Aet al. FRET biosensor uncovers cAMP nano-domains at beta-adrenergic targets that dictate precise tuning of cardiac contractility. Nat Commun. 2017;8:15031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takao D, Taniguchi A, Takeda Tet al. High-speed imaging of amoeboid movements using light-sheet microscopy. PLoS One. 2012;7:e50846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, Abouleila Y, Si Let al. Human organs-on-chips for virology. Trends Microbiol. 2020;28:934–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taormina MJ, Jemielita M, Stephens WZet al. Investigating bacterial-animal symbioses with light sheet microscopy. Biol Bull. 2012;223:7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchekanda E, Sivanesan D, Michnick SW. An infrared reporter to detect spatiotemporal dynamics of protein-protein interactions. Nat. Methods. 2014;11:641–4. [DOI] [PubMed] [Google Scholar]
- Tebo AG, Gautier A. A split fluorescent reporter with rapid and reversible complementation. Nat Commun. 2019;10:2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terai K, Imanishi A, Li Cet al. Two decades of genetically encoded biosensors based on forster resonance energy transfer. Cell Struct Funct. 2019;44:153–69. [DOI] [PubMed] [Google Scholar]
- Teuscher AC, Ewald CY. Overcoming autofluorescence to assess GFP expression during normal physiology and aging in caenorhabditis elegans. Bio Protoc. 2018;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tielker D, Eichhof I, Jaeger KEet al. Flavin mononucleotide-based fluorescent protein as an oxygen-independent reporter in Candida albicans and Saccharomyces cerevisiae. Eukaryot Cell. 2009;8:913–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobin DM, May RC, Wheeler RT. Zebrafish: a see-through host and a fluorescent toolbox to probe host-pathogen interaction. PLoS Pathog. 2012;8:e1002349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tournu H, Luna-Tapia A, Peters BMet al. In vivo indicators of cytoplasmic, vacuolar, and extracellular pH using pHluorin2 in Candida albicans. mSphere. 2017;2:e00276–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsui C, Kong EF, Jabra-Rizk MA. Pathogenesis of Candida albicans biofilm. Pathog Dis. 2016;74:ftw018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulah A, Lopes MI, Brul Set al. Intracellular pH homeostasis in Candida glabrata in infection-associated conditions. Microbiology. 2013b;159:803–13. [DOI] [PubMed] [Google Scholar]
- Ullah A, Lopes MI, Brul Set al. Intracellular pH homeostasis in Candida glabrata in infection-associated conditions. Microbiology. 2013a;159:803–13. [DOI] [PubMed] [Google Scholar]
- Valkonen M, Mojzita D, Penttilä Met al. Noninvasive high-throughput single-cell analysis of the intracellular pH of Saccharomyces cerevisiae by ratiometric flow cytometry. Appl Environ Microbiol. 2013;79:7179–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Krogt GN, Ogink J, Ponsioen Bet al. A comparison of donor-acceptor pairs for genetically encoded FRET sensors: application to the Epac cAMP sensor as an example. PLoS One. 2008;3:e1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vande Velde G, Kucharíková S, Schrevens Set al. Towards non-invasive monitoring of pathogen-host interactions during Candida albicans biofilm formation using in vivo bioluminescence. Cell Microbiol. 2014;16:115–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vande Velde G, Kucharíková S, Van Dijck Pet al. Bioluminescence imaging increases in vivo screening efficiency for antifungal activity against device-associated Candida albicans biofilms. Int J Antimicrob Agents. 2018;52:42–51. [DOI] [PubMed] [Google Scholar]
- Van Dyck K, Rogiers O, Vande Velde Get al. Let's shine a light on fungal infections: A noninvasive imaging toolbox. PLoS Pathog. 2020;16:e1008257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Ende M, Wijnants S, Van Dijck P. Sugar Sensing and Signaling in Candida albicans and Candida glabrata. Front Microbiol. 2019;10:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Genechten W, Demuyser L, Dedecker Pet al. Presenting a codon-optimized palette of fluorescent proteins for use in Candida albicans. Sci Rep. 2020;10:6158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanherp L, Poelmans J, Hillen Aet al. Bronchoscopic fibered confocal fluorescence microscopy for longitudinal in vivo assessment of pulmonary fungal infections in free-breathing mice. Sci Rep. 2018;8:3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rheenen J, Langeslag M, Jalink K. Correcting confocal acquisition to optimize imaging of fluorescence resonance energy transfer by sensitized emission. Biophys J. 2004;86:2517–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rosmalen M, Krom M, Merkx M. Tuning the flexibility of glycine-serine linkers to allow rational design of multidomain proteins. Biochemistry. 2017;56:6565–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voelz K, Johnston SA, Rutherford JCet al. Automated analysis of cryptococcal macrophage parasitism using GFP-tagged cryptococci. PLoS One. 2010;5:e15968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warolin J, Essmann M, Larsen B. Flow cytometry of Candida albicans for investigations of surface marker expression and phagocytosis. Ann Clin Lab Sci. 2005;35:302–11. [PubMed] [Google Scholar]
- Weigert R, Sramkova M, Parente Let al. Intravital microscopy: a novel tool to study cell biology in living animals. Histochem Cell Biol. 2010;133:481–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissman Z, Pinsky M, Donegan RKet al. Using genetically encoded heme sensors to probe the mechanisms of heme uptake and homeostasis in Candida albicans. Cell Microbiol. 2020;23:e13282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendland J, Hellwig D, Walther Aet al. Use of the Porcine Intestinal Epithelium (PIE)-Assay to analyze early stages of colonization by the human fungal pathogen Candida albicans. J Basic Microbiol. 2006;46:513–23. [DOI] [PubMed] [Google Scholar]
- White RM, Sessa A, Burke Cet al. Transparent adult zebrafish as a tool for in vivo transplantation analysis. Cell Stem Cell. 2008;2:183–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood KV, Lam YA, McElroy WD. Introduction to beetle luciferases and their applications. J Biolumin Chemilumin. 1989;4:289–301. [DOI] [PubMed] [Google Scholar]
- Wuyts J, Van Dijck P, Holtappels M. Fungal persister cells: The basis for recalcitrant infections?. PLoS Pathog. 2018;14:e1007301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wächtler B, Citiulo F, Jablonowski Net al. Candida albicans-epithelial interactions: dissecting the roles of active penetration, induced endocytosis and host factors on the infection process. PLoS One. 2012;7:e36952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wöllert T, Rollenhagen C, Langford GMet al. Human oral keratinocytes: a model system to analyze host-pathogen interactions. Methods Mol Biol. 2012;845: 289–302. [DOI] [PubMed] [Google Scholar]
- Yanez-Carrillo P, Orta-Zavalza E, Gutiérrez-Escobedo Get al. Expression vectors for C-terminal fusions with fluorescent proteins and epitope tags in Candida glabrata. Fungal Genet Biol. 2015;80:43–52. [DOI] [PubMed] [Google Scholar]
- Yang DL, Zhang YQ, Hu YLet al. Protective effects of cis-2-Dodecenoic acid in an experimental mouse model of vaginal candidiasis. Biomed Environ Sci. 2018;31:816–28. [DOI] [PubMed] [Google Scholar]
- Yang HC, Mikami Y, Imai Tet al. Extrusion of fluorescein diacetate by multidrug-resistant Candida albicans. Mycoses. 2001;44:368–74. [DOI] [PubMed] [Google Scholar]
- Yao Y, Shi L, Zhang Cet al. Application of fungal fluorescent staining in oral candidiasis: diagnostic analysis of 228 specimens. BMC Microbiol. 2019;19:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Konopka JB. A photostable green fluorescent protein variant for analysis of protein localization in Candida albicans. Eukaryot Cell. 2010;9:224–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Hung GC, Nagamine Ket al. Development of candida-specific real-time PCR assays for the detection and identification of eight medically important candida species. Microbiol Insights. 2016;9:21–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Doyle TC, Coquoz Oet al. Emission spectra of bioluminescent reporters and interaction with mammalian tissue determine the sensitivity of detection in vivo. J Biomed Opt. 2005;10:41210. [DOI] [PubMed] [Google Scholar]
- Zordan RE, Ren Y, Pan SJet al. Expression plasmids for use in Candida glabrata. G3 (Bethesda). 2013;3:1675–86. [DOI] [PMC free article] [PubMed] [Google Scholar]