

ABSTRACT
Many bacteria and archaea produce the redox cofactor F420. F420 is structurally similar to the cofactors FAD and FMN but is catalytically more similar to NAD and NADP. These properties allow F420 to catalyze challenging redox reactions, including key steps in methanogenesis, antibiotic biosynthesis and xenobiotic biodegradation. In the last 5 years, there has been much progress in understanding its distribution, biosynthesis, role and applications. Whereas F420 was previously thought to be confined to Actinobacteria and Euryarchaeota, new evidence indicates it is synthesized across the bacterial and archaeal domains, as a result of extensive horizontal and vertical biosynthetic gene transfer. F420 was thought to be synthesized through one biosynthetic pathway; however, recent advances have revealed variants of this pathway and have resolved their key biosynthetic steps. In parallel, new F420-dependent biosynthetic and metabolic processes have been discovered. These advances have enabled the heterologous production of F420 and identified enantioselective F420H2-dependent reductases for biocatalysis. New research has also helped resolve how microorganisms use F420 to influence human and environmental health, providing opportunities for tuberculosis treatment and methane mitigation. A total of 50 years since its discovery, multiple paradigms associated with F420 have shifted, and new F420-dependent organisms and processes continue to be discovered.
Keywords: cofactor 420, redox chemistry, enzymology, cofactor biosynthesis, redox cofactor, cofactor distribution
This review provides a comprehensive description of the distribution and biosynthesis of the redox cofactor F420, as well as its enzymology, physiological roles and biotechnological applications.
ABBREVIATIONS
- 2PL
2-phospho-L-lactate
- 3PG
3-phospho-D-glycerate
- Adf
F420-dependent secondary alcohol dehydrogenase
- ANME
anaerobic methanotrophic archaea
- AOA
ammonium-oxidizing archaea
- APDs
4-alkyl-L-proline derivatives
- BGC
biosynthetic gene cluster
- CoM
coenzyme M
- CoB
coenzyme B
- CoB-S-S-CoM
coenzyme B, coenzyme M heterodisulfide
- Ddn
deazaflavin-dependent nitroreductase
- DFTR
F420H2-dependent flavin-containing thioredoxin reductase
- DH-F420
dehydro-F420
- EPPG
enolpyruvyl-diphospho-5’-guanosine
- FAD
flavin adenine dinucleotide
- FDOR(-A/B)
flavin/deazaflavin oxidoreductase (subfamily A or B)
- Ffd
F420-reducing formate dehydrogenase
- Fgd
F420-reducing glucose-6-phosphate dehydrogenase
- fHMAD
F420-dependent hydroxymycolic acid dehydrogenase
- FMN
flavin mononucleotide
- Fno
F420H2-dependent NADP reductase
- FOP
FO-5′-phosphate
- fPKR
F420H2-dependent phthiodiolone ketoreductase
- Fpo
F420H2-dependent methanophenazine reductase
- FprA
F420H2-dependent oxidase
- Fqo
F420H2-dependent quinone reductase
- FRET
Förster resonance energy transfer
- Frh
F420-reducing hydrogenase
- Fsr
F420H2-dependent sulfite reductase
- G6P
glucose-6-phosphate
- GPPG
glyceryl-diphospho-5‘-guanosine
- H4MPT
tetrahydromethanopterin
- CHO-H4MPT
5-formyltetrahydromethanopterin
- CH≡H4MPT
5,10-methenyltetrahydromethanopterin
- CH2=H4MPT
5,10-methylenetetrahydromethanopterin
- CH3-H4MPT
5-methyltetrahydromethanopterin
- LUCA
last universal common ancestor
- LLHT
luciferase-like hydride transferase
- LPPG
lactyl-diphospho-5‘-guanosine
- MAGs
metagenome derived genomes
- MDR
multidrug-resistant tuberculosis
- Mer
methylene-H4MPT reductase
- Mtd
methylene-H4MPT dehydrogenase
- NADH
nicotinamide adenine dinucleotide
- NADPH
nicotinamide adenine dinucleotide phosphate
- NTR
nitroreductase
- OYE
Old Yellow Enzymes
- PBDs
pyrrolobenzodiazepines
- PDIM
phthiocerol dimycocerosates
- PEP
phosphoenolpyruvate
- XDR
extensively drug-resistant tuberculosis
INTRODUCTION
Cofactors play a fundamental role in biological chemistry. When bound to enzymes, they provide chemical reactivity and specificity that is otherwise unattainable via protein sidechain and backbone chemistry (Begley 2010). Cofactors that mediate redox reactions often contain heterocyclic ring structures, which can accept and donate electrons at physiologically relevant redox potentials (Eicher, Hauptmann and Speicher 2013). In addition to the important heterocyclic riboflavin cofactors FAD and FMN (Fig. 1A), bacteria and archaea produce the structurally related deazaflavin cofactor, F420 (Factor 420; Fig. 1B; Cheeseman, Toms-Wood and Wolfe 1972; Eirich, Vogels and Wolfe 1979; Walsh 1980; Joosten and van Berkel 2007; Ney et al. 2017a). While F420 structurally resembles FAD and FMN, it is chemically more similar to the nicotinamide cofactors NAD and NADP (Fig. 1C; Jacobson and Walsh 1984; Walsh 1986; de Poorter, Geerts and Keltjens 2005; Huang et al. 2012; Buckel and Thauer 2013). Like NAD(P), F420 functions as a cellular hydride carrier (Hendrickson and Leigh 2008). It is reduced by dedicated F420-reducing dehydrogenases, with low potential electrons provided by catabolic substrates or NADPH (Schauer and Ferry 1986; Purwantini and Daniels 1996; Berk and Thauer 1997; Warkentin et al. 2001; Bashiri et al. 2008; Allegretti et al. 2014). The resulting reduced cofactor, termed F420H2, is then utilized by diverse F420H2-dependent reductases to reduce substrates in both catabolic and anabolic pathways (Wang et al. 2013; Ahmed et al. 2015; Purwantini, Daniels and Mukhopadhyay 2016; Greening et al. 2017; Mascotti et al. 2018; Steiningerova et al. 2020).
Figure 1.
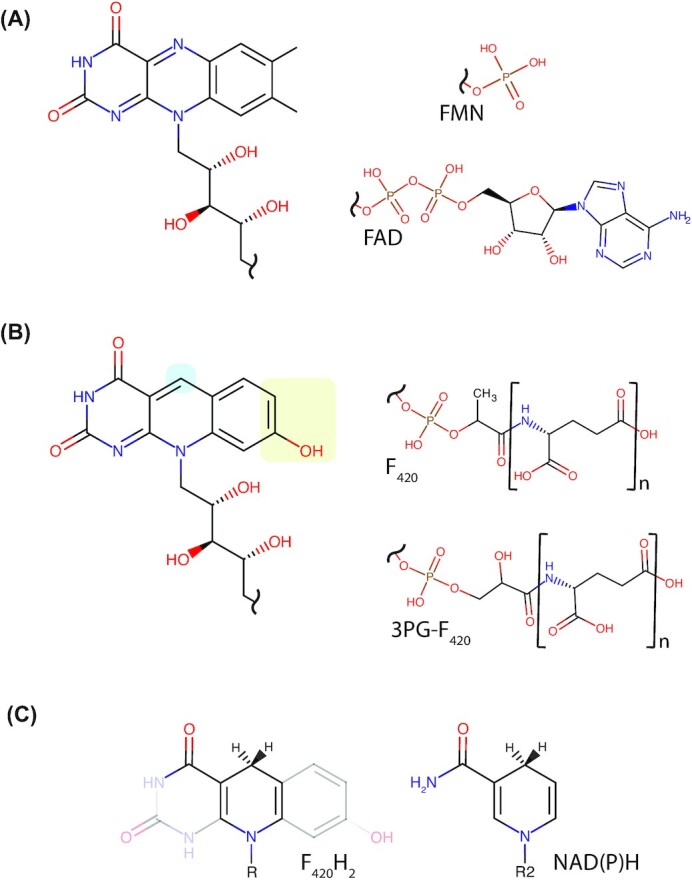
Structural comparison of F420 with flavins and nicotinamides. (A) Structures of the riboflavin head group and tail groups of the flavins FMN and FAD. (B) Structures of the 5-deazaflavin head group and tail groups of F420 and 3PG-F420. Locations of chemical substitutions between riboflavin and 5-deazaflavin are highlighted. N = 2–9 depending on the microbial species. (C) Structural similarity between the nicotinamides NAD(P)H and the central redox-active portion of F420H2. For F420H2, R represents the phospholactyl and oligoglutamate tail shown in panel B. For NAD(P)H, R2 represents the ribose-5-phosphate of the nicotinamide nucleotide and the adenosine nucleobase as shown in (Bogan and Brenner 2013).
F420 was first described in methanogenic archaea of the phylum Euryarchaeota (Cheeseman, Toms-Wood and Wolfe 1972; Tzeng, Bryant and Wolfe 1975) by the Wolfe group in 1971. Its production was subsequently shown to be universal among methanogenic Euryarchaeota and widespread among other members of this phylum (Eirich, Vogels and Wolfe 1979; van Beelen, Dijkstra and Vogels 1983; Lin and White 1986; De Wit and Eker 1987; Gorris and Voet 1991; Gorris 1994; Purwantini, Gillis and Daniels 1997). F420 biosynthesis genes are also encoded by diverse other archaea, including members of the TACK and Asgard archaeal superphyla (Evans et al. 2015; Kerou et al. 2016; Vanwonterghem et al. 2016; Ney et al. 2017a; Jay et al. 2018; Spang et al. 2019; Wang et al. 2019). Independent from its discovery in methanogens, F420 was isolated from antibiotic-producing streptomycetes belonging to the phylum Actinobacteria (Miller et al. 1960; McCormick and Morton 1982), and was then shown to be widely produced by members of this phylum, including all members of the genus Mycobacterium (Naraoka et al. 1984; Daniels, Bakhiet and Harmon 1985; Purwantini, Gillis and Daniels 1997). Outside of Actinobacteria, F420 biosynthesis genes have been detected in a diverse range of bacteria, and its production has been biochemically confirmed in both Proteobacteria and Chloroflexi (Ney et al. 2017a; Braga et al. 2019, 2020). Until recently, it was thought that the F420 biosynthesis pathway was identical in all producing organisms (Ney et al. 2017a). However, recent studies have uncovered variation in the substrates and enzymes utilized for F420 biosynthesis between bacteria and archaea, as well as a new variant of the mature cofactor in Proteobacteria (Bashiri et al. 2019; Braga et al. 2019; Grinter et al. 2020). This variability reflects the diversity of the organisms that produce F420, as well as the complex evolutionary history of the biosynthesis pathway, which is characterized by both vertical and horizontal gene transfer events (Weiss et al. 2016; Ney et al. 2017a).
In addition to its role in microbial physiology, F420 has garnered interest for its industrial, medical and environmental applications. The cofactor and its analogs have potential in industrial biocatalysis (Taylor, Scott and Grogan 2013; Greening et al. 2017; Bashiri et al. 2019; Drenth, Trajkovic and Fraaije 2019). The low redox potential and obligate hydride transfer chemistry of F420 enable reduction of otherwise recalcitrant organic molecules (Greening et al. 2017; Mathew et al. 2018; Martin et al. 2020). Numerous F420-dependent enzymes are present in microbial genomes, providing an inventory for industrial biocatalysis (Selengut and Haft 2010; Ahmed et al. 2015; Mascotti et al. 2018; Steiningerova et al. 2020). Some progress has been made towards use of F420-dependent enzymes in industrial catalysis, including the first heterologous production of the cofactor (Bashiri et al. 2019; Braga et al. 2019; Ney 2019), though further advances are required. With respect to medical applications, the F420-dependent enzyme deazaflavin-dependent nitroreductase (Ddn) from Mycobacterium tuberculosis activates the recently approved antitubercular drugs pretomanid and delamanid and F420 has been shown to play a role in antimicrobial resistance in mycobacterial pathogens (Hasan et al. 2010; Cellitti et al. 2012; Gurumurthy et al. 2013; Lee et al. 2020). Additionally, methanogenic archaea that reside in environments such as livestock rumen, rice paddies and waste landfill produce a significant portion of global methane emissions via a process that requires F420 (Kirschke et al. 2013; Greening et al. 2019). As such, inhibition of F420 biosynthesis or F420-dependent enzymes in livestock has been proposed as a strategy to reduce global greenhouse gas emissions (Attwood et al. 2011; Patra et al. 2017).
Significant progress has been made in understanding F420 in the five years since this topic was last reviewed comprehensively (Greening et al. 2016). We now have a much-improved understanding of the distribution, biosynthesis and roles of F420. These new findings have challenged several paradigms in the field, including the idea that F420 is restricted to a few microbial lineages and is synthesized through a universal pathway. This review provides a new synthesis of our understanding of F420, by integrating recent and historical literature while outlining remaining knowledge gaps. We also discuss how these fundamental advances facilitate applications, for example heterologous F420 production for biocatalysis.
CHEMISTRY, DISTRIBUTION AND ROLES OF F420
Chemical properties
Like the universal nicotinamide cofactors NAD(P) and flavin cofactors FMN/FAD, the primary role of F420 is to transfer electrons between compounds within the cell (Walsh 1986; Munro and McLean 2013). Chemically, F420 consists of three components: the redox-active isoalloxazine head group FO, a phospho-organic acid linker and a γ-linked polyglutamate tail of variable length (Fig. 1B; Eirich, Vogels and Wolfe 1978; Braga et al. 2019).
As a 5-deazaflavin moiety, the FO head group contains three chemical substitutions compared to flavins (Fig. 1A and B) that give F420 unique spectral and electrochemical properties (Fig. 2A and B). The key change is the substitution of the redox-active N-5 atom of the isoalloxazine ring for a carbon. In contrast to flavins, this substitution precludes F420 from forming a stable semiquinone, given unpaired electrons cannot delocalize through a C-5 isoalloxazine ring in low-energy states (O'Brien, Weinstock and Cheng 1967, 1970; Edmondson, Barman and Tollin 1972; Xia, Shen and Zhu 2015). As a result, F420 is an obligate hydride carrier similar to nicotinamides and does not readily undergo single-electron reactions such as autooxidation in air (Fisher, Spencer and Walsh 1976; Spencer, Fisher and Walsh 1976; Jacobson and Walsh 1984; Walsh 1986). In addition, when compared to flavins, C-7 and C-8 of F420 are demethylated and C-7 is hydroxylated, further altering the redox properties of the cofactor (Eirich, Vogels and Wolfe 1978). As a result of these three substitutions, F420 has a much lower standard redox potential (−340 mV) than riboflavin (−210 mV), FAD (−220 mV) or FMN (−190 mV; Thauer, Jungermann and Decker 1977; Walsh 1986). This redox potential is modulated by physiological conditions, resulting in a redox potential of −380 mV in hydrogenotrophic methanogens that maintain a 10:1 ratio of reduced to oxidized F420 (Jacobson and Walsh 1984; de Poorter, Geerts and Keltjens 2005). This makes F420 well suited to mediate the low potential reactions of anaerobic metabolism, as well as reductions that require a low potential electron donor (Thauer, Jungermann and Decker 1977; Hartzell et al. 1985).
Figure 2.

F420 protonaiton states, redox transitions and associated spectral shifts. (A) Changes in the protonation state of F420 and F420H2 as a result of the change in external pH. R = F420 tail group as depicted in Fig. 1B. (B) Spectral changes of F420 between pH 5.5 and 9.0 resulting from a change in protonation state depicted in panel A. Inset graph shows a change in absorbance at 420 nm. (C) Spectral change in F420H2 as in panel B. Inset graph shows changes in absorbance at 280 nm. Panels B and C are adapted from Mohamed et al. (2016a).
F420 can exist in a range of protonation states as summarized in Fig. 2. The resonance structure of the isoalloxazine ring of oxidized F420 lowers the pKa of the C-7 hydroxyl group to 6.3, favoring its deprotonation under basic conditions. Deprotonation of the F420 C-7 hydroxyl leads to delocalization of the resulting unbonded electron and the formation of a conjugated paraquinoid anion, which is the species that exhibits the classic F420 spectral properties of strong absorbance at 420 nm (Fig. 2A; Walsh 1980, 1986). In this paraquinoid state, F420− exhibits reduced electrophilicity, making it resistant to reduction via hydride acquisition at its C-5 carbon (de Poorter, Geerts and Keltjens 2005). Protonation of the F420 C-7 hydroxyl group results in a shift of its absorption maxima to ∼400 nm, as well as a decrease in the overall absorption coefficient (Fig. 2B; Mohamed et al. 2016a). During reduction in biological systems, F420 receives a hydride ion at its C-5 carbon with reductant derived from H2, glucose-6-phosphate (G6P), NADPH, or other low-potential electron donors, via the action of dedicated F420H2-dependent reductases (Fig. 2A; Aufhammer et al. 2004; Vitt et al. 2014; Le et al. 2015; Oyugi et al. 2018). N-1 of reduced F420 possesses an unbonded electron pair and a net negative charge, facilitating its protonation, hence the F420H2 nomenclature applied to the reduced compound (Jacobson and Walsh 1984; Walsh 1986). The pKa for the proton association with N-1 of reduced F420H2 is 6.9, meaning that the deprotonated reduced form, F420H−, may be the physiologically relevant state of this cofactor in many F420H2-dependent reductases, which has mechanistic implications as discussed below (Mohamed et al. 2016a). The changes to the bond structure of the isoalloxazine ring of F420H2 lead to a corresponding change in its optical properties (Fig. 2A and C; Eirich, Vogels and Wolfe 1979; Walsh 1986; Mohamed et al. 2016a). F420H2 exhibits weak absorbance at 320 nm, with deprotonation to F420H− causing minimal further changes to its absorption profile in the visible spectrum (Fig. 2C; Mohamed et al. 2016a). F420H2 formation interrupts conjugation across the isoalloxazine ring and isolates the benzenoid portion of the molecule, preventing deprotonation of the C-7 hydroxyl at physiological pH (pKa 9.7; Walsh 1980, 1986; Jacobson and Walsh 1984).
F420 is a fluorescent molecule, named for the absorbance of its oxidized FO head group at 420 nm, with corresponding fluorescence emission at 470 nm mediated by a π→π* transition upon photon absorption (Cheeseman, Toms-Wood and Wolfe 1972; Mohamed et al. 2016a). FO spectral properties are blue-shifted relative to flavin and give F420 a characteristic golden-yellow color and blue-green fluorescence (Cheeseman, Toms-Wood and Wolfe 1972; Eirich, Vogels and Wolfe 1978). The blue-shifted fluorescence of FO allows it to efficiently transfer photons to flavin via Förster resonance energy transfer (FRET). In addition to its incorporation into F420, FO is synthesized independently and its fluorescent properties are exploited by a class of DNA photolyases, which bind FO and FMN as cofactors to mediate the reductive cleavage of DNA pyrimidine dimers (Malhotra et al. 1992; Tamada et al. 1997). FO-utilizing DNA photolyases are present in cyanobacteria, unicellular algae and possibly higher eukaryotes including Drosophila (Mayerl et al. 1990; Sancar 1990; Glas et al. 2009). Like FO, F420 exhibits analogous autofluorescence and these properties can be used to identify F420-producing organisms such as methanogens and mycobacteria by fluorescence microscopy (Doddema and Vogels 1978; Maglica, Özdemir and McKinney 2015; Lambrecht et al. 2017), or sort them by flow cytometry. However, F420 is not used by DNA photolyases and its physiological role appears to be restricted to acting as a redox cofactor (Sancar 1990; Kiontke et al. 2014; Greening et al. 2016).
While the FO deazaflavin headgroup is solely responsible for F420 redox function, the phospho-organic acid linker and polyglutamate tail modulate cofactor functionality by imparting negative charge and mediating interactions with F420 dependent enzymes (Fig. 1B; Ney et al. 2017b). Bacterial F420-dependent enzymes from at least two superfamilies form electrostatic interactions with the phosphate group of the F420 linker via conserved motifs, enhancing their specificity for the cofactor (Ahmed et al. 2015; Purwantini, Daniels and Mukhopadhyay 2016). The polyglutamate tail of F420 varies in maximum length among producing organisms and exists as a population of different tail lengths from one to nine residues (Gorris and Voet 1991; Gorris 1994; Ney et al. 2017a, b). In archaea, the relative abundance of F420 with different tail lengths varies depending on culture conditions and growth phase, suggesting tail length may modulate F420 function (Peck 1989). Recently we investigated the effect of F420 polyglutamate tail length on the function of mycobacterial F420-dependent enzymes (Ney et al. 2017b). F420 containing both short (two) and long (five to eight) polyglutamate chains were compatible with these enzymes, though long-chain F420 bound these enzymes with six to 10-fold greater affinity. Chain length also significantly modulated the kinetics of the enzymes, with long-chain F420 increasing the substrate affinity (lower Km) but reducing the turnover rate (lower kcat). Molecular dynamics simulations indicated that F420-dependent enzymes make multiple dynamic electrostatic interactions with the F420-polyglutamate tail via conserved surface residues, likely explaining the observed differences in activity between short and long chain F420 (Ney et al. 2017b). These data suggest that variable F420 polyglutamate tail length may have evolved to modulate the activity of F420-dependent enzymes. Additionally, these findings have significant implications for the use of F420 in industrial applications, where a high catalytic turnover is likely to be desirable.
F420-dependent enzymes
F420-dependent enzymes are broadly classified as F420-reducing dehydrogenases or F420H2-dependent reductases based on the direction of the redox reaction they perform under physiological conditions (Greening et al. 2016). However, due to the relatively similar redox potentials of many F420-substrate pairs, some F420-dependent enzymes are bidirectional depending on the organism and physiological conditions (Eker, Hessels and Meerwaldt 1989; Berk and Thauer 1997; Afting, Hochheimer and Thauer 1998; Hendrickson and Leigh 2008). F420-dependent enzymes can be further divided into two classes based on their mechanism of electron transfer. In the first of these classes, bound F420 accepts or donates hydride directly to or from the enzyme substrate. In the second class, bound flavin (FAD or FMN) acts as an intermediate, either accepting a hydride from or donating a hydride to F420 (Shima et al. 2000; Ceh et al. 2009; Allegretti et al. 2014; Ahmed et al. 2015; Joseph et al. 2016; Oyugi et al. 2018). F420-dependent oxidoreductases of this second class often contain additional subunits with multiple iron-sulfur (FeS) clusters, which transfer electrons between the enzyme-substrate (i.e. H2 or formate) and F420, via FMN/FAD. In this role, the bound flavin acts as a modulator between the single-electron chemistry of the FeS clusters and the hydride chemistry of F420 (Wood, Haydock and Leigh 2003; Seedorf et al. 2007; Vitt et al. 2014).
F420-dependent enzymes are structurally diverse and can be classified into several families, which possess distinct folds and evolutionary histories (Greening et al. 2016). These families are often distributed in both F420-producing archaea and bacteria (Ney et al. 2017a) and have evolved to utilize F420 as a cofactor independently (Ahmed et al. 2015; Mascotti et al. 2018; Mascotti, Ayub and Fraaije 2020). F420-dependent enzyme families often include both F420-reducing and F420H2-oxidizing enzymes and are members of broader groups of oxidoreductases that utilize FMN, FAD, NAD(P)H, or heme as cofactors (Ahmed et al. 2015; Mascotti et al. 2018). Some of these groups contain multiple distinct lineages of enzymes that utilize F420, indicating that specificity for the cofactor arose on multiple occasions (Ahmed et al. 2015; Mascotti, Ayub and Fraaije 2020). The currently identified enzyme families that utilize F420 as a cofactor are summarized in Table 1, with functionally characterized F420-dependent dehydrogenases and F420H2-dependent reductases cataloged in Tables 2 and 3 respectively. The structures of representative examples of each family are shown in Figs 3 and 4. We have previously comprehensively reviewed the structure and function of these enzymes (Greening et al. 2016), and so we will not detail these aspects here.
Table 1.
F420-dependent enzyme families.
| F420-dependent protein family | Acronym | Protein fold | Mechanism of hydride transfer | Phylogenetic distribution | Characterized function(s) | Key references |
|---|---|---|---|---|---|---|
| Flavin/deazaflavin oxidoreductase | FDOR-A, FDOR-B | Split β-barrel | Direct F420-substrate | Actinobacteria, Chloroflexi | F420H2-dependent reduction of diverse substrates (e.g. menaquinone, tetracycline and biliverdin) with promiscuous activity often observed | Cellitti et al. (2012); Lapalikar et al. (2012); Ahmed et al. (2015); Greening et al. (2017) |
| Luciferase-like hydride transferase | LLHT | TIM-Barrel | Direct F420-substrate | Broadly found in F420 producing bacteria and archaea | F420-dependent oxidation or F420H2-dependent reduction of diverse substrates (e.g. G6P, mycolic acids and CH2=H4MPT) | Aufhammer et al.(2004, 2005); Bashiri et al. (2008); Nguyen et al. (2017); Mascotti et al. (2018) |
| F420-dependent NADPH oxidoreductase | Fno | Rossmann fold | Direct F420-substrate | Broadly found in F420 producing bacteria and archaea | Hydride transfer between F420/F420H2 and NADP/NADPH | Berk and Thauer (1997); Warkentin et al. (2001); Le et al. (2015); Joseph et al. (2016); Kumar et al. (2017) |
| F420-dependent H4MPT oxidoreductase | Mtd | Rossmann fold | Direct F420-substrate | Euryarchaeota: methanogens, ANME, Archaeoglobales | Hydride transfer between F420/F420H2 and CH≡H4MPT/CH2=H4MPT | Shima et al. (2000); Hagemeier et al. (2003a); Warkentin et al. (2005); Ceh et al. (2009) |
| F420H2-dependent flavodiiron oxidase | FprA | β-lactamase/flavodoxin | Indirect F420-flavin-2Fe-O2 | Methanogenic archaea | Reduction of dioxygen (O2) to water (2 H2O) to detoxify O2 | Seedorf et al. (2004,2007) |
| F420-dependent redox coupling oxidoreductase | FDRC | α/β/α-sandwich fold | Indirect F420-flavin-FeS-substrate | Euryarchaeota: methanogens, ANME, Archaeoglobales | Couples the reduction/oxidation of F420/F420H2 to that of formate, H2, methanophenazine, quinone or sulfite, via association with structurally diverse protein subunits. | Baron and Ferry (1989); Bäumer et al. (2000); Brüggemann, Falinski and Deppenmeier (2000); Johnson and Mukhopadhyay (2005,2008b); Welte and Deppenmeier (2011a); Allegretti et al. (2014); Vitt et al. (2014) |
| Deazaflavin-dependent thioredoxin reductase | DFTR | Thioredoxin reductase fold | Indirect F420-flavin-disulfide | Euryarchaeota: Methanococcales | Couples F420H2 oxidation to the reduction of thioredoxin | Susanti, Loganathan and Mukhopadhyay (2016) |
| F420-dependent bifurcating reductase | HdrA2 | HdrA-like fold | Indirect F420-flavin-FeS-substrate | Euryarchaeota: Methanosarcinales | Couples F420H2 oxidation to the reduction of CoM-S-S-CoB and ferredoxin via bifurcation | Yan, Wang and Ferry (2017) |
Table 2.
Functionally characterized F420-reducing dehydrogenases. This table updates and expands upon the enzymes previously summarized and reviewed by Greening et al. (2016).
| Oxidoreductase and domain | Physiological role | Taxonomic distribution | Family | EC no. | PDB ID | References |
|---|---|---|---|---|---|---|
| Archaea | ||||||
| Frh: F420-reducing hydrogenase | Methanogenic growth on H2. Couples oxidation of H2 to the reduction of F420. May be physiologically reversible. | All orders of methanogens | FDRC | 1.12.98.1 | 4OMF, 4CI0, 3ZFS, 6QGT | Tzeng, Wolfe and Bryant (1975); Jacobson et al. (1982); Muth, Morschel and Klein (1987); Kulkarni et al. (2009); Mills et al. (2013); Allegretti et al. (2014); Vitt et al. (2014); Ilina et al. (2019) |
| Ffd: F420-reducing formate dehydrogenase | Methanogenic growth on formate. Couples oxidation of formate to the reduction of F420. May be part of electron-bifurcating complex. | Many Euryarchaeota (e.g. Methanobacteriales, Methanococcales, Methanopyrales, Methanomicrobiales and Methanocellales) | FDRC | 1.2.99.9 | Jones and Stadtman (1981); Schauer and Ferry (1986); Costa et al. (2010); Tzeng, Wolfe and Bryant (1975); Wood, Haydock and Leigh (2003) | |
| Adf: F420-reducing secondary alcohol dehydrogenase | Growth on secondary alcohols. Couples oxidation of secondary alcohols (e.g. isopropanol) to the reduction of F420. | Some Euryarchaeota (Methanomicrobiales and Methanocellales) | LLHT | 1.1.98.5 | 1RHC | Widdel and Wolfe (1989); Bleicher and Winter (1991); Aufhammer et al. (2004); Martin et al. (2020) |
| Bacteria | ||||||
| Fno: F420-reducing NADPH dehydrogenase | Exchanges electrons between NADP and F420. F420 reduction direction dominant in bacteria, as F420 is the secondary cofactor. | Many Actinomycetales (e.g. Streptomyces, Thermobifida, Rhodococcus, Nocardiaand Nocardioides), Chloroflexi?, Alphaproteobacteria?, Betaproteobacteria? | Fno | 1.5.1.40 | 5N2I | Eker, Hessels and Meerwaldt (1989); Heiss et al. (2002); Kumar et al. (2017) |
| Fgd: F420-reducing glucose-6-phosphate dehydrogenase | Heterotrophic growth. Couples oxidation of glucose-6-phosphate to the reduction of F420 via the pentose phosphate pathway. | Many Actinomycetales (e.g. Mycobacterium, Actinoplanes, MicrobacteriumandAmycolatopsis), Chloroflexi, Alphaproteobacteria?, Thaumarchaeota? | LLHT | 1.1.98.2 | 3B4Y | Bashiri et al. (2008); Oyugi et al. (2018) |
| Fsd: F420-reducing sugar-6-phosphate dehydrogenase | Heterotrophic growth. Couples oxidation of glucose-, fructose- or mannose-6-phosphate to the reduction of F420. Similar to Fgd, with a catalytic preference for glucose-6-phosphate, but an expanded substrate specificity. | Some Actinomycetales (e.g. Nocardioides and Cryptosporangium) | LLHT | 1.1.98.2 | Mascotti et al. (2018) | |
| fHMAD: F420-reducing hydroxymycolic acid dehydrogenase | Cell wall biosynthesis. Catalyzes F420-dependent oxidation of hydroxymycolic acids to ketomycolic acids. | Few Mycobacterium (primarily pathogenic species) | LLHT | Bashiri et al. (2012); Purwantini and Mukhopadhyay (2013) | ||
| Amm4: F420-dependent ammosamide dehydrogenase | Putative dehydrogenase involved in primary amide formation in the pyrroloquinoline alkaloid ammosamide. Details of reaction mediated and the product formed are unresolved. | Few Actinomycetales (e.g. Streptomyces and Amycolatopsis) | FDOR-B | Jordan and Moore (2016) |
Table 3.
Functionally characterized F420H2-dependent reductases. This table updates and expands upon the enzymes previously summarized and reviewed by Greening et al. (2016).
| Oxidoreductase and domain | Physiological role | Taxonomic distribution | Family | EC no. | PDB ID | References |
|---|---|---|---|---|---|---|
| Archaea | ||||||
| Mtd: F420-reducing methylene-H4MPT dehydrogenase | Reduces CH≡H4MPT to CH2=H4MPT with F420H2 during CO2-reducing methanogenesis. Performs the opposite reaction during methylotrophic methanogenesis and anaerobic methane/alkane oxidation. | Various Euryarchaeota including: all orders of methanogens, Archaeoglobales, ANME and Halobacteriales; various TACK and Asgard archaea | Mtd | 1.5.98.1 | 1QV9, 1U6I, 3IQF, 3IQE | Hartzell et al. (1985); Te Brömmelstroet et al. (1991a,b); Hagemeier et al. (2003a,b); Ceh et al. (2009) |
| Mer: F420H2-dependent methylene-H4MPT reductase | Reduces CH2=H4MPT to CH3-H4MPT with F420H2 during CO2-reducing methanogenesis. Performs the opposite reaction during methylotrophic methanogenesis and anaerobic methane/alkane oxidation. | Various Euryarchaeota including: all orders of methanogens, Archaeoglobales, ANME and Halobacteriales; various TACK and Asgard archaea | LLHT | 1.5.98.2 | 1F07, 1EZW, 1Z69 | Te Brömmelstroet et al. (1991b); Shima et al. (2000); Aufhammer et al. (2005); Ceh et al. (2009) |
| Fpo: F420H2-dependent methanophenazine reductase | Proton-translocating primary dehydrogenase in respiratory chain transferring electrons from F420H2 to heterodisulfide. | Methanosarcinales | FDRC | 1.1.98.4 | Bäumer et al. (1998); Deppenmeier, Lienard and Gottschalk (1999); Ide, Bäumer and Deppenmeier (1999); Bäumer et al. (2000); Welte and Deppenmeier (2011a) | |
| Fqo: F420H2-dependent quinone reductase | Proton-translocating primary dehydrogenase in respiratory chain transferring electrons from F420H2 to sulfate. | Archaeoglobales and ANME | FDRC | 1.1.98.4 | KUNOW et al. (1994); Brüggemann, Falinski and Deppenmeier (2000); Hallam et al. (2004); Hocking et al. (2014) | |
| Fpr: F420H2-dependent oxidase | Detoxifies O2 by mediating the four-electron reduction of O2 to H2O with F420H2. | Methanobacteriales, Methanococcales, Methanomicrobiales and Methanocellales | FprA | 1.5.3.22 | 2OHH, 2OHI, 2OHJ | Seedorf et al. (2004,2007) |
| Fsr: F420H2-dependent sulfite reductase | Detoxifies sulfite by mediating the six electron reduction of sulfite to sulfide with F420H2. Also enables the use of sulfite as an S source. | Methanobacteriales and Methanococcales | FDRC | 1.8.98.3 | Johnson and Mukhopadhyay (2005, 2008a) | |
| Fno: F420H2-dependent NADP+ reductase | Exchanges electrons between NADP and F420. NADP+ reduction direction dominant in archaea, as NADP is the secondary cofactor. | Various Euryarchaeota including: all orders of methanogens, Archaeoglobales and ANME; various TACK and Asgard archaea | Fno | 1.5.1.40 | 1JAY, 1JAX | Tzeng, Wolfe and Bryant (1975); Kunow et al. (1993); Berk and Thauer (1997); Warkentin et al. (2001) |
| HdrA2B2C2: F420H2-dependent, electron-bifurcating, heterodisulfide reductase | The HdrA2 subunit of this complex oxidizes F420H2, with subunits HdrB2 and HdrC2 bifurcating the resulting electrons to ferredoxin and CoM-S-S-CoB (heterodisulfide). Thought to mediate energy conservation during acetoclastic methanogenesis. | Methanosarcinales | HdrA2 | Yan, Wang and Ferry (2017) | ||
| DFTR: F420H2-dependent thioredoxin reductase | Recycling of the thioredoxin disulfide through reduction by electrons transferred from F420H2, via a low potential FMN and disulfide redox center. | Methanococcales | DFTR | 1.8.1.9 | Susanti, Loganathan and Mukhopadhyay (2016) | |
| Bacteria | ||||||
| Ddn: F420H2-dependent menaquinone reductase | Reduction of the respiratory cofactor menaquinone for energy conservation and possibly to mitigate redox stress. Also catalyzes the promiscuous activation nitroimidazole prodrugs. FDOR-A1 family. | Most Actinomycetales (e.g., Mycobacterium, Streptomyces, Rhodococcus), Chloroflexi?, Methanosarcinales? | FDOR-A | 1.1.98.- | 3H96, 4Y9I, 3R5R, 3R57 | Taylor et al. (2010); Cellitti et al. (2012); Gurumurthy et al. (2013); Ahmed et al. (2015); Lee et al. (2020) |
| Fbr: F420H2-dependent biliverdin reductase | Reduction of the heme degradation product biliverdin to bilirubin. May also reduce mycobillins. FDOR-B3 and FDOR-B4 family. | Most Actinomycetales (e.g., Mycobacterium, Streptomyces, Rhodococcus), Chloroflexi? | FDOR-B | 2ASF, 4QVB, 1W9A | Canaan et al. (2005); Biswal et al. (2006); Ahmed et al. (2015, 2016); Mashalidis et al. (2015) | |
| Fts: F420H2-dependent tetracycline synthase | Reduction of dehydrotetracyclines to tetracyclines during streptomycete antibiotic synthesis. Role in mycobacteria unknown. FDOR-B1 family. | Most Actinomycetales (e.g., Mycobacterium, StreptomycesandRhodococcus), Chloroflexi?, Thaumarchaeota? | FDOR-B | Taylor et al. (2010); Wang et al. (2013); Ahmed et al. (2015) | ||
| TpnL: F420H2–dependent dehydropiperidine reductase | Reduction of the dehydropiperidine moiety to piperidine during the synthesis of thiopeptins antibiotics. FDOR-B family. | Some Actinomycetales (Streptomyces, Amycolatopsis, Micromonospora and Actinoalloteichus) | FDOR-B | Ichikawa, Bashiri and Kelly (2018) | ||
| GupA: F420H2–dependent dihydropyrazine reductase | Reduction of the dihydropyrazine ring to piperzine during the synthesis of guanipiperazines. FDOR-B family. | Some Actinomycetales (Streptomyces) | FDOR-B | Shi et al. (2021) | ||
| Other F420H2-dependent flavin/deazaflavin oxidoreductases (FDORs) | Physiological substrates of A2-A4, B1, B2, B5, B6, AA1- AA5 families unknown. Promiscuous reductase activity observed towards multiple chemical classes that may facilitate detoxification. AA1s may be fatty acid saturases. | Most Actinomycetales (e.g., Mycobacterium, Streptomyces and Rhodococcus), Chloroflexi?, Halobacteriales? | FDOR-A/B | 3F7E, 1RFE, 4ZKY | Lapalikar et al. (2012); Ahmed et al. (2015); Jirapanjawat et al. (2016); Greening et al. (2017) | |
| Fht: F420H2-dependent picrate reductase | Reduces 2,4,6-trinitrophenol (picrate) for use as a C and N source through hydride transfer to the nitroaromatic ring. | Few Actinomycetales (Rhodococcus, Nocardia, Nocardioides) | LLHT | Ebert, Rieger and Knackmuss (1999); Heiss et al. (2002) | ||
| Fps/Adp6: F420H2-dependent 4-alkyl-L-proline derivative reductases | Reduction of 4-alkyl-L-proline derivatives (APDs) in the final step in the biosynthesis of this compound. Different enzymes of this class impart structural diversity by reducing either the endocyclic imine or the exocyclic double bond of APDs. | Some Actinomycetales (Streptomyces, Micrococcus and Streptosporangium) | LLHT | Li et al. (2009a,b); Steiningerova et al. (2020) | ||
| fPKR: F420H2-dependent phthiodiolone ketoreductase | Reduction of phthiodiolone keto intermediates during the synthesis of phthiocerol dimycocerosates (PDIM), a class of mycobacterial cell surface apolar lipids. | Few Mycobacterium (primarily pathogenic species) | LLHT | Purwantini, Daniels and Mukhopadhyay (2016) | ||
| LxmJ: F420H2-dependent 2,3-didehydroalanine reductase | Stereospecific reduction of the 2,3-didehydroalanine reductase to D-alanine during class V lanthipeptide biosynthesis | Few Streptomyces | LLHT | Tao et al. (2020) | ||
| Other H2-dependent luciferase-like hydride transferases (LLHTs) | Unknown. Likely to have diverse roles in endogenous and exogenous redox metabolism of organic compounds. | Most Actinomycetales (e.g., Mycobacterium, StreptomycesandRhodococcus) | LLHT |
Figure 3.

F 420-dependent enzyme families that reduce or oxidize substrates via direct hydride transfer. Representative structures are shown of families of F420-dependent oxidoreductases in complex with F420 and substrate, inhibitor, or substrate analog. Inhibitors or substrate analogs are indicated with *. The secondary structural elements are highlighted (blue = α-helix, yellow = β-sheet or coil). (A) FDOR-A family F420H2-dependent menaquinone reductase (Ddn) from M. tuberculosis docked with menadione (PDB ID = 3R5R; Cellitti et al. 2012). (B) FDOR-B family enzyme of unknown function MSMEG_6526 from M. smegmatis in complex with 2-methyl-2,4-pentanediol (MPD; PDB ID = 4ZKY; Ahmed et al. 2015). (C) Rossmann-A fold enzyme NADPH:F420 oxidoreductase (Fno) from A. fulgidus in complex with NADPH (PDB ID = 1JAY; Warkentin et al. 2001). (D) LLHT family F420-reducing glucose-6-phosphate dehydrogenase (Fgd) from M. tuberculosis in complex with citrate (PDB ID = 3B4Y; Bashiri et al. 2008). The region of protein capping the active site is rendered transparent for clarity. (E) Rossmann-B fold enzyme F420-dependent CH2=H4MPT dehydrogenase (Mtd) from Methanopyrus kandleri in complex with CH2=H4MPT (PDB ID = 3IQE; Ceh et al. 2009).
Figure 4.

F420-dependent enzymes that mediate oxidation or reduction indirectly via flavin. Representative structures or models of families F420-dependent oxidoreductases that mediate hydride transfer via a bound flavin cofactor. Structures generated via homology modeling using Phyre2 (Kelley et al. 2015) are indicated with *. The secondary structural elements are highlighted (blue = α-helix, yellow = β-sheet or coil), FMN or FAD colored red and FeS clusters and metal ions are shown as spheres. (A) FDFO family F420H2-dependent flavodiiron oxidase (FprA) from Methanothermobacter thermautotrophicus responsible for the reduction of O2 to H2O (PDB ID = 2OHJ; Seedorf et al. 2007). (B) FDRC domain-containing F420-reducing NiFe hydrogenase (Frh) from Methanothermobacter marburgensis (PDB ID = 4CI0; Allegretti et al. 2014). (C) F420H2-dependent thioredoxin reductase (DFTR) from M. jannaschii (homology model; Susanti, Loganathan and Mukhopadhyay 2016).
Taxonomic distribution
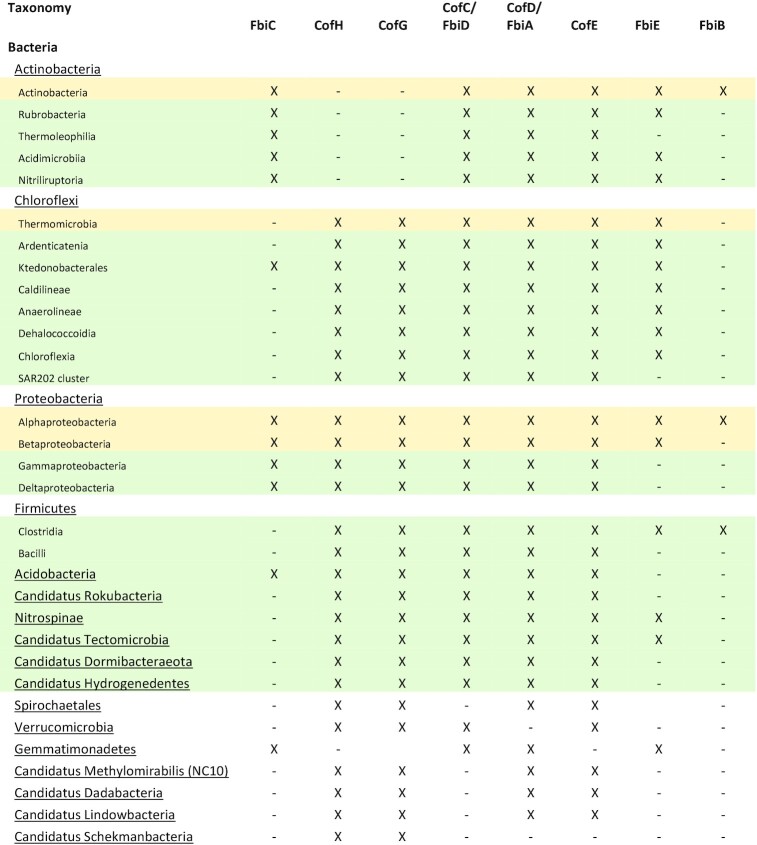
Until recently F420 was thought to be a rare cofactor, taxonomically restricted to the members of archaeal phylum Euryarchaeota and the bacterial phylum Actinobacteria (Ney et al. 2017a). However, recent studies applying genomic, spectroscopic and biochemical analysis have demonstrated that F420 is much more widely distributed among bacteria and archaea than previously thought (Kerou et al. 2016; Lackner et al. 2017; Ney et al. 2017a; Braga et al. 2019, 2020). Prior to these studies, it was assumed that FO production was more widespread than F420. Yet genomic analysis shows that, in the majority of organisms, the genes required for FO biosynthesis co-occur with those required for its conversion to F420, indicating that FO is generally produced as a precursor for F420 biosynthesis, with a possible secondary role as a chromophore in some F420 producers (Kiontke et al. 2014; Ney et al. 2017a). A phylogenetic tree and accompanying table outlining microbial lineages biochemically demonstrated to produce FO and F420, as well as those predicted to produce these cofactors based on genomic data, is presented in Fig. 5 and Table 4. There is currently no evidence that F420 is synthesized as a redox cofactor by eukaryotes. The distribution of F420 biosynthesis genes among bacteria and archaea appears to be widespread in some lineages (i.e. Euryarchaeota and Actinobacteria; Cheeseman, Toms-Wood and Wolfe 1972; Eirich, Vogels and Wolfe 1979; Lin and White 1986; Bair, Isabelle and Daniels 2001), but variable among others (i.e. TACK lineages of Archaea and Proteobacteria; Kerou et al. 2016; Ney et al. 2017a). F420 biosynthesis genes are highly abundant in metagenomes from diverse soil, marine and some host-associated ecosystems, further indicating that F420 biosynthesis is a widespread trait (Ney et al. 2017a).
Figure 5.

Phylogenetic distribution of FO and F420 producing organisms. A simplified two-domain tree of life depicted the organisms shown or predicted to produce the 5-deazaflavins FO or F420. This is based on currently available data from published work (Greening et al. 2016; Ney et al. 2017a), and genomic and metagenomic data in the NCBI database (as of October 2020). Tree topography is based on Hug et. al. (Hug et al. 2016) and Castelle and Banfield (2018), with additional reference to Zhou et al. (2020), Wang et al. (2019) and Momper, Aronson and Amend (2018). * = F420 biosynthesis genes detected only in multiple metagenome-assembled genomes (MAGs) or single-amplified genomes (SAGs) from these archaea and bacteria, rather than genomes derived from pure culture.
Table 4.
Confirmed and predicted F420-producing organisms. Experimentally confirmed F420 producers are highlighted in yellow, while predicted F420 producers with a full complement of F420 biosynthesis genes based on analysis of assembled pure culture genomes or multiple MAGs/SAGs, are highlighted in green.
|

|
Table 5.
Purified F420H2-dependent reductases that have been explored for substrate specificity and range.
| In vitro activity | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Enzyme nme | Originating organism | Sequence ID | Physiological substrate | Enzyme class | PDB ID | Quinones | Coumarins | Enones | Enals | Pyrones | Pyrans | Triarylmethanes | Secondary alcohols | Refs |
| MSMEG_5998 | M. smegmatis | ABK71916 | Menaquinone | FDOR-A1 | ++++ | +++ | + | ND | - | - | ++ | ND | Greening et al. (2017) | |
| MSMEG_2027 | M. smegmatis | ABK75334 | Menaquinone | FDOR-A1 | 4Y9I | +++ | + | + | ND | ++ | - | ++ | ND | Greening et al. (2017) |
| MSMEG_2850 | M. smegmatis | AWT53773 | FDOR-A1 | ++++ | +++ | + | ND | - | - | ++ | ND | Greening et al. (2017) | ||
| MSMEG_3356 | M. smegmatis | ABK75759 | FDOR-A1 | 3H96 | ND | ++ | ND | ND | ND | ND | ND | ND | Taylor et al. (2010) | |
| MSMEG_3004 | M. smegmatis | ABK74167 | FDOR-A1 | ND | ++ | ND | ND | ND | ND | ND | ND | Taylor et al. (2010); Lapalikar et al. (2012) | ||
| MSMEG_5030 | M. smegmatis | ABK74375 | FDOR-A2 | +++ | + | - | ND | + | + | ++ | ND | Greening et al. (2017) | ||
| MSMEG_3380 | M. smegmatis | ABK72884 | FDOR-B1 | 3F7E | +++ | ++ | + | ND | - | - | ++ | ND | Greening et al. (2017) | |
| MSMEG_0048 | M. smegmatis | ABK73917 | FDOR-B1 | ++ | + | + | ND | + | - | + | ND | Greening et al. (2017) | ||
| MSMEG_6325 | M. smegmatis | ABK73368 | FDOR-A3 | + | + | ++ | ND | - | - | ++ | ND | Greening et al. (2017) | ||
| MSMEG_5170 | M. smegmatis | ABK72943 | FDOR-B3 | +++ | + | - | ND | - | - | + | ND | Greening et al. (2017) | ||
| MSMEG_6848 | M. smegmatis | ABK75254 | LPOR-like/FDOR-B1 | +++ | + | - | ND | + | - | + | ND | (Greening et al. 2017) | ||
| MSMEG_6526 | M. smegmatis | ABK76173 | FDOR-B2 | 5JV4, 4ZKY | + | - | - | ND | - | - | + | ND | Greening et al. (2017) | |
| MSMEG_3880 | M. smegmatis | ABK75472 | Biliverdin | FDOR-B4 | + | - | - | ND | - | - | + | ND | Greening et al. (2017) | |
| MSMEG_5717 | M. smegmatis | ABK72164 | FDOR-B | ND | - | ND | ND | ND | ND | ND | ND | Greening et al. (2017) | ||
| FDR-Rh1 | Rhodococcus jostii | ABG96463 | FDOR-A | +++ | ND | + | +++ | ND | ND | ND | ND | Mathew et al. (2018) | ||
| FDR-Rh2 | Rhodococcus jostii | ABG97172 | FDOR-A | +++ | ND | + | ++ | ND | ND | ND | ND | Mathew et al. (2018) | ||
| FDR-Mha | Mycobacterium hassicum | WP_005623184 | FDOR-A | +++ | ND | + | ++ | ND | ND | ND | ND | Mathew et al. (2018) | ||
| Adf | M. thermophilicus | CAA77275 | LLHT | 1RHC | ND | ND | ND | ND | ND | ND | ND | +++ | Martin et al. (2020) |
F420 production and roles within archaea
Within archaea, F420 production has only been biochemically confirmed in Euryarchaeota and much of our understanding of the physiological roles of the cofactor is derived from these organisms (Jacobson and Walsh 1984; Schmitz et al. 1991; Vaupel and Thauer 1995; Berk and Thauer 1997; Thauer 1998; Brüggemann, Falinski and Deppenmeier 2000). Currently, available genomic and metagenomic datasets show that a complete complement of genes necessary for F420 biosynthesis is also present in members of two other major archaeal groups, the TACK and Asgard archaea (Kerou et al. 2016; Ney et al. 2017a; Jay et al. 2018; Spang et al. 2019). The majority of these putative F420-producing archaea remain uncultured, with most detected through metagenome-assembled genomes (MAGs) and single-amplified genomes (SAGs; Spang, Caceres and Ettema 2017; Williams et al. 2017; Ney et al. 2017a; Jay et al. 2018). Genomes assembled by these methods often exhibit low coverage and completeness and suffer from sampling bias due to their often low relative abundance in the community (Albertsen et al. 2013). As such, the current list of F420 producing archaea compiled for this review, and shown in Table 4, is an underestimation of the actual distribution of the cofactor. Growing evidence indicates that F420-dependent redox metabolism of one-carbon units is widespread in archaea, enabling the processes of methanogenesis, acetogenesis and alkane oxidation (Laso-Pérez et al. 2016; Adam, Borrel and Gribaldo 2019; Evans et al. 2019; Orsi et al. 2020). The central role of F420 in this pathway likely goes some way to explain its widespread production by the archaeal domain. However, the role of F420 goes well beyond one-carbon metabolism and the diversity of F420-producing archaea indicates that many additional functions likely remain to be discovered (Kozubal et al. 2013; Kerou et al. 2016; Susanti, Loganathan and Mukhopadhyay 2016; Ney et al. 2017a; Jay et al. 2018).
Roles in Euryarchaeota
Methanogenic Euryarchaeota
Methanogens are a diverse group of obligately anaerobic archaea that produce methane as the end product of their energy generation pathways (Liu and Whitman 2008). Methanogens encompass at least seven orders within the Euryarchaeota (Brochier, Forterre and Gribaldo 2004; Bapteste, Brochier and Boucher 2005; Brochier-Armanet, Forterre and Gribaldo 2011; Borrel et al. 2013; Evans et al. 2019), though genome-resolved metagenomics indicates there are potentially methanogenic archaea outside this phylum (Vanwonterghem et al. 2016; Sorokin et al. 2017; Spang and Ettema 2017; Berghuis et al. 2019). All cultured methanogens synthesize F420, which serves as a central redox cofactor for two of the three major routes of methanogenesis: the CO2-reducing and methylotrophic pathways (Cheeseman, Toms-Wood and Wolfe 1972; Edwards and McBride 1975; Doddema and Vogels 1978; Eirich, Vogels and Wolfe 1979; van Beelen, Dijkstra and Vogels 1983; Dolfing and Mulder 1985). As such, it is often present at high concentrations (up to 2.0 µmol per g dry weight) in these methanogens (Eirich, Vogels and Wolfe 1979; Isabelle, Simpson and Daniels 2002).
In the CO2-reducing pathway, CO2 is progressively reduced to methane using electrons derived from exogenous substrates such as H2, formate and less commonly secondary alcohols (Tzeng, Bryant and Wolfe 1975; Widdel and Wolfe 1989; Fig. 6). F420H2 donates a hydride for two of these reaction steps, after first being reduced by F420-reducing dehydrogenases utilizing H2 (Frh; Jacobson et al. 1982; Muth, Morschel and Klein 1987; Fiebig and Friedrich 1989; de Poorter, Geerts and Keltjens 2005; Allegretti et al. 2014), formate (Ffd; Schauer and Ferry 1986; Shuber et al. 1986; Baron and Ferry 1989), or secondary alcohols (Adf; Widdel and Wolfe 1989; Bleicher and Winter 1991). In this pathway, CO2 is first condensed with the cofactor methanofuran, before being transferred to tetrahydromethanopterin (H4MPT) to form 5-formyltetrahydromethanopterin (CHO-H4MPT). CHO-H4MPT then undergoes enzymatically mediated intramolecular condensation to form 5,10-methenyltetrahydromethanopterin (CH≡H4MPT; Thauer 2012), which is progressively reduced by F420H2 via methylene-H4MPT dehydrogenase (Mtd) to form 5,10-methylenetetrahydromethanopterin (CH2=H4MPT) and methylene-H4MPT reductase (Mer) to form N5-methyltetrahydromethanopterin (CH3-H4MPT; Hendrickson and Leigh 2008). The CO2-derived methyl group resulting from these reactions is then transferred to coenzyme M (CoM), before it is substituted by coenzyme B (CoB), forming the heterodisulfide CoB-S-S-CoM and releasing methane (Fig. 6; Thauer 1998). The methyl transfer from CH3-H4MPT to CoM is mediated by the MtrA-H membrane protein complex, which conserves energy through the pumping of sodium ions out of the cell (Welander and Metcalf 2005; Thauer et al. 2008). In addition to the MtrA-H complex, energy is also conserved through respiratory reduction of CoB-S-S-CoM in methanogens with cytochromes (i.e. Methanosarcinales) or by electron bifurcation in methanogens without cytochromes (Thauer et al. 2008; Kaster et al. 2011; Welte and Deppenmeier 2014). In the case of methanogens with cytochromes, F420H2 can serve as a direct electron donor to the respiratory chain; this depends on the activity of F420H2-dependent methanophenazine reductase (Fpo), a 14-subunit complex similar to bacterial complex I that directly pumps protons using the energy released from electron transfer from F420H2 to the membrane-diffusible cofactor methanophenazine (Deppenmeier et al. 1990; Abken and Deppenmeier 1997; Bäumer et al. 1998, 2000; Welte and Deppenmeier 2011b).
Figure 6.

F420-dependent reactions of one-carbon metabolism in archaea. F420 is a cofactor involved in key steps in hydrogenotrophic methanogenesis, methylotrophic methanogenesis, anaerobic methanotrophy and anaerobic alkane oxidation in archaea. Hydride transfer reactions involving F420-dependent enzymes are indicated as is the enzyme responsible. F420H2 reduced through the oxidation of formate (Ffd), H2 (Frh), or secondary alcohols (Adf) can be utilized for reactions mediated by Mtd, Mer, or for other cellular processes. Only reactions mediated by F420-dependent enzymes are shown in detail. For a full outline of methanogenesis pathways, refer to the following reviews on the subject (Deppenmeier 2002; Thauer et al. 2008; Timmers et al. 2017; Evans et al. 2019).
F420 has distinct roles in the methylotrophic and acetoclastic methanogenesis pathways. In the methylotrophic methanogenesis pathway, methyl groups (from methanol, methylated amines and methylated sulfides) are alternatively converted into CH4 (reductive route) and CO2 (oxidative route; Fig. 6; Krzycki et al. 1987; Ferry and Kastead 2007). The oxidative route likely occurs through the reverse direction of the CO2-reducing pathway, with the methyl group first transferred to CoM, then H4MPT, before being oxidized sequentially by Mer and Mtd, yielding F420H2. The reductive route proceeds from CH3-S-CoM in the same fashion as the CO2-reducing pathway (Fig. 6; Deppenmeier 2002; Thauer et al. 2008). Methanogens with cytochromes use F420H2 generated through the oxidative arm of the methylotrophic pathway as an input to the respiratory chain via Fpo (Welte and Deppenmeier 2011b). Acetocastic methanogenesis is F420-independent, producing CO2 and CH4 from acetate utilizing a largely distinct set of enzymes to the hydrogenotrophic or methylotrophic pathways (Smith and Mah 1978; Barber et al. 2011). However, despite not being required for this process, F420 is present in facultatively acetoclastic Methanosarcina when grown solely on acetate and in the obligately acetoclastic genus Methanothrix, indicating that the cofactor has roles in methanogen physiology beyond those of methanogenesis (Baresi and Wolfe 1981; Barber et al. 2011; Zhu et al. 2012). In support of this, a potential electron-bifurcating heterodisulfide reductase that uses ferredoxin and F420H2 as electron donors has been identified in Methanosarcina acetivorans (Yan, Wang and Ferry 2017).
Dedicated F420-dependent enzymes have been shown to mediate other diverse reactions in methanogens, as detailed in Tables 2 and 3. These include reduction of the redox cofactors NADP+ (F420H2-dependent NADP reductase; Fno) and thioredoxin (F420H2-dependent flavin-containing thioredoxin reductase; DFTR; Spaans et al. 2015; Susanti, Loganathan and Mukhopadhyay 2016), mobilization of sulfite as a sulfur source (F420H2-dependent sulfite reductase; Fsr; Johnson and Mukhopadhyay 2008a) and detoxification of atmospheric O2 (F420H2-dependent oxidase; FprA; Seedorf et al. 2007).
Methane-, ethane- and butane-oxidizing Euryarchaeota
Anaerobic methanotrophy is a biogeochemically significant process in which methane of biological or abiotic origin is oxidized to CO2, with nitrate, sulfate, or transition metal ions as terminal electron acceptors (Bhattarai, Cassarini and Lens 2019). Up to 90% of the methane produced by marine sediment is estimated to be internally recycled by this process, thereby moderating methane release into the atmosphere (Reeburgh 2007; Conrad 2009; Knittel and Boetius 2009). There is strong evidence that this process is mediated by uncultured methanotrophic Euryarchaeota (ANME; Haroon et al. 2013; Cai et al. 2018). These archaea form at least three phylogenetically distinct groups, which are closely related to Methanomicrobiales (ANME-1) and Methanosarcinales (ANME-2a/b/c and ANME-3; Wang et al. 2014). ANME have not been propagated in pure culture. However, genetic, transcriptomic and biochemical evidence indicates they oxidize methane using an F420-dependent pathway analogously to methylotrophic methanogenesis (Wang et al. 2014; Timmers et al. 2017). Metagenomic and metatranscriptomic analysis of the nitrate-reducing methanotroph Methanoperedens nitroreducens (part of the ANME-2 lineage) in an enriched bioreactor showed that it expresses a complete reverse methanogenesis pathway, including Mtd and Mer, as well as F420 biosynthesis genes and a putative respiratory F420H2-dependent quinone reductase (Fqo) complex (Arshad et al. 2015). Many ANME appear to perform methanotrophy syntrophically, forming associations with sulfate, nitrite, or nitrate-reducing bacteria, which likely explains the inability to isolate them in pure culture (Boetius et al. 2000; Beal, House and Orphan 2009; Haroon et al. 2013). Similar to anaerobic methane oxidizers, enrichment cultures of novel Euryarchaeota lineages have recently been shown to be capable of anaerobically oxidizing short-chain alkanes. Members of candidate genera Argoarchaeum and Syntrophoarchaeum are capable of anaerobically oxidizing ethane and butane respectively (Laso-Pérez et al. 2016; Chen et al. 2019). They are predicted to produce F420, and likely utilize the reverse methanogenesis pathway, combined with β-oxidation, to oxidize these compounds (Laso-Pérez et al. 2016).
Sulfate-reducing and halophilic Euryarchaeota
Archaeoglobi are a class of sulfate-reducing archaea that appear to have evolved from a methanogenic ancestor but have developed a non-methanogenic lifestyle (Stetter et al. 1987; Klenk et al. 1997). Archaeoglobi are primarily heterotrophic sulfate-reducing hyperthermophiles that inhabit deep-sea vents (Stetter et al. 1987; Nercessian et al. 2005). The well-characterized isolate Archaeoglobus fulgidus uses F420 as its central redox cofactor (Möller-Zinkhan, Börner and Thauer 1989; Gorris and Voet 1991). F420 is reduced through distinct routes depending on whether the growth substrate is H2/CO2 or lactate (Möller-Zinkhan and Thauer 1990). Lactate is converted to three molecules of CO2, through a process analogous to the oxidative methylotrophic pathway of methanogens, generating F420H2 via the action of Mer and Mtd (Schmitz et al. 1991; Schwörer et al. 1993). A. fulgidus does not possess the F420-reducing hydrogenase Frh, and it remains unresolved how it generates F420H2 during hydrogenotrophic growth; possible routes include electron transfer from reduced ferredoxin, quinols (via reverse electron transfer), or NADPH (via Fno; Möller-Zinkhan, Börner and Thauer 1989; Klenk et al. 1997; Hocking et al. 2014). F420H2 produced by substrate oxidation then donates electrons to a sulfate-reducing respiratory chain via the proton-translocating F420H2-dependent quinone reductase (Fqo; Kunow et al. 1994; Brüggemann, Falinski and Deppenmeier 2000). Outside of central metabolism, little is known about the role of F420 in Archaeoglobi. However, A. fulgidus possesses Fno, which is thought to be the sole route for NADP reduction (Kunow et al. 1993; Warkentin et al. 2001). F420 production has also been experimentally determined in the halophiles Halobacterium and Halococcus, though its physiological role remains undetermined (Lin and White 1986; De Wit and Eker 1987).
Roles in other Archaea
TACK lineages of Archaea
The TACK lineage represents a major grouping of archaea originally containing the phyla Thaumarchaeota, Aigarchaeota, Crenarchaeota and Korarchaeota, but now expanded to contain several other recently identified phyla (Guy and Ettema 2011; Spang, Caceres and Ettema 2017; Wang et al. 2019). Diverse members of the TACK group contain a full complement of genes for F420 (Spang et al. 2012; Zhalnina et al. 2014; Evans et al. 2015; Kerou et al. 2016; Vanwonterghem et al. 2016; Ney et al. 2017a; Jay et al. 2018; Berghuis et al. 2019; Table 4), though no definitive experimental evidence confirming the production and roles of F420 has been presented. Putative F420 producing species adopt diverse aerobic and anaerobic lifestyles (Jay et al. 2018; Yu et al. 2018; Berghuis et al. 2019). F420 production appears to be a common trait in Thaumarchaeota (Tourna et al. 2011; Kozlowski et al. 2016; Ren et al. 2019; Reji and Francis 2020), including ammonium-oxidizing archaea (AOA) that mediate nitrification in soil and marine ecosystems (Kuypers, Marchant and Kartal 2018). Genomic analysis and fluorescence microscopy indicate both Nitrososphaera gargensis and Nitrososphaera viennensis synthesize F420 in significant quantities (Spang et al. 2012; Kerou et al. 2016), though the presence and role of F420 in this phylum has not been biochemically confirmed. Given Nitrososphaera are aerobes that cannot perform methanogenesis, the cofactor is unlikely to play a role in one-carbon transformations (Kerou et al. 2016; Ren et al. 2019). Proteomic analysis indicates that Fno and putative F420-dependent oxidoreductases of the luciferase-like hydride transferase (LLHT) and flavin/deazaflavin oxidoreductase (FDOR) families are produced at high levels, suggesting a role for the cofactor in biosynthetic or biodegradative processes (Kerou et al. 2016).
Several other TACK phyla also encode F420 biosynthesis genes. Marsarchaeota and Geoarchaeota, two closely related aerobic chemoheterotrophic phyla recently discovered in thermophilic iron-rich microbial mats, also encode F420 biosynthesis genes and F420-dependent oxidoreductases. Metatranscriptomic analysis indicates that F420-dependent oxidoreductases are highly expressed by Marsarchaeota living in microbial mats. These enzymes were hypothesized to play a role in the metabolism of extracellular sulfonates, although there is limited phylogenetic or biochemical evidence to support this (Jay et al. 2018). The candidate phyla Bathyarchaeota and Verstraetearchaeota are also predicted to produce F420 (Table 4; Evans et al. 2015; Vanwonterghem et al. 2016; Zhou et al. 2018). Based on the analysis of metagenome derived genomes (MAGs) from these species, Verstraetearchaeota are predicted to be capable of F420-dependent hydrogenotrophic methanogenesis (Fig. 6), the first example of an archaeon capable of this process to be discovered outside of the Euryarchaeota (Berghuis et al. 2019; Evans et al. 2019). Based on the presence of genes homologous to the methyl-CoM reducing complex Mcr, it was originally suggested that Bathyarchaeota are also capable of methylotrophic methanogenesis (Evans et al. 2015). However, the phylogenetic grouping of the Mcr genes present in Bathyarchaeota indicates that they utilize this complex for F420-dependent anaerobic alkane oxidation (Fig. 6), rather than methanogenesis, similarly to the recently identified candidate genus Syntrophoarchaeum and potentially the candidate phylum Helarchaeota (Laso-Pérez et al. 2016; Chen et al. 2019; Evans et al. 2019; Seitz et al. 2019).
Asgard archaea
The Asgard archaea are a recently discovered archaeal superphylum that includes the Lokiarchaeota, Thorarchaeota, Odinarchaeota, Heimdallarchaeota, Helarchaeota and Hermodarchaeota (Bulzu et al. 2019; Seitz et al. 2019; Spang et al. 2019). Phylogenetically, the Asgard archaea are the closest archaeal relatives of eukaryotes (López-García and Moreira 2019; Spang et al. 2019), and it has been proposed that eukaryotes evolved from a metabolic symbiosis between an Asgard archaeon and an Alphaproteobacterium that gave rise to the mitochondrion (López-García and Moreira 2019; Imachi et al. 2020). The reconstruction of metabolic networks of Asgard archaea from metagenome-assembled genomes indicates that they exhibit high metabolic diversity both within and between different phyla with respect to energy source, electron donor, carbon source and electron acceptor preferences (Bulzu et al. 2019; Seitz et al. 2019; Spang et al. 2019; Orsi et al. 2020). Members of Loki-, Heimdall- and Hel-archaeota possess all genes required for F420 biosynthesis, while available MAGs for Odin- and Thor-archaeota contain several of these genes (Table 4). Like members of the Euryarcheota and TACK lineages of Archaea, members of the Asgard archaea likely utilize F420-dependent pathways for carbon fixation and short-chain alkane oxidation, as well as potentially additional unknown processes (Sousa et al. 2016; MacLeod et al. 2019; Seitz et al. 2019; Spang et al. 2019; Orsi et al. 2020).
F420 production and roles within bacteria
In bacteria, F420 has been primarily studied in Actinobacteria. It has been biochemically identified in members of the genera Mycobacterium, Streptomyces, Rhodococcus, Nocardia and Nocardioides (Daniels, Bakhiet and Harmon 1985; Eker, Hessels and Meerwaldt 1989; Purwantini, Gillis and Daniels 1997; Ebert, Rieger and Knackmuss 1999; Selengut and Haft 2010), the majority of which are soil saprophytes. F420 is not essential for central metabolism in Actinobacteria, though the cofactor is used for a wide range of purposes that provide a growth or survival advantage (Ebert, Rieger and Knackmuss 1999; Hasan et al. 2010; Taylor et al. 2010; Wang et al. 2012; Gurumurthy et al. 2013; Greening et al. 2017; Lee et al. 2020). In addition to Actinobacteria, recent biochemical evidence indicates that F420 is produced by members of the phylum Chloroflexi and the classes Alphaproteobacteria and Betaproteobacteria (Ney et al. 2017a; Braga et al. 2019). Spectroscopic analysis suggests members of the candidate phylum Tectomicrobia also produce the cofactor (Lackner et al. 2017). The genes required for F420 biosynthesis are also encoded in multiple genomes from the cultivated phyla Acidobacteria, Firmicutes and Nitrospinae and the candidate phyla Rokubacteria, Tectomicrobia and Dadabacteria (Wilson et al. 2014; Hug et al. 2016; Becraft et al. 2017; Lackner et al. 2017; Ney et al. 2017a). Presently, no experimental studies have been performed investigating its biochemical and physiological role in bacterial species outside of Actinobacteria.
Roles in Actinobacteria
Mycobacteria
The genetic complement for F420 biosynthesis is present in all cultured environmental and pathogenic mycobacteria. F420 production has been experimentally confirmed in many Mycobacterium species including M. tuberculosis, M. smegmatis, M. phlei, M. bovis and M. avium (Bair, Isabelle and Daniels 2001). Two fast-growing saprophytic members of the genus, M. smegmatis and M. phlei, produce F420 in large quantities (0.3–0.6 µmol per g dry weight; Isabelle, Simpson and Daniels 2002), indicating it plays a significant role in mycobacterial physiology. In addition, F420 is produced by the obligate pathogens M. tuberculosis and M. leprae (Purwantini, Gillis and Daniels 1997; Bair, Isabelle and Daniels 2001), which suggests a conserved physiological function for the cofactor among mycobacteria, as well as a role in survival in the host. A further indication of its significance is that all mycobacterial species contain numerous enzymes known or predicted to utilize F420 as a cofactor (Selengut and Haft 2010; Ahmed et al. 2015). M. smegmatis is predicted to encode 75 F420 dependent enzymes (30 of FDOR family, 45 of LLHT family), while M. tuberculosis is predicted to encode 33 F420-dependent enzymes (15 of FDOR family, 17 of LLHT family; Selengut and Haft 2010; Ahmed et al. 2015). In addition to these known classes of F420-dependent enzymes, further F420-dependent enzymes may be present in mycobacteria, which belong to novel enzyme families and thus cannot be readily identified based on amino acid sequence homology (Kumar 2018). While the function of the majority of F420-dependent enzymes in mycobacteria remains poorly understood, recent phenotypic and biochemical studies have shed light on some of their physiological roles (Hasan et al. 2010; Bashiri et al. 2012; Gurumurthy et al. 2013; Ahmed et al. 2015; Jirapanjawat et al. 2016; Purwantini, Daniels and Mukhopadhyay 2016; Lee et al. 2020).
F420 is not essential for mycobacterial growth, with mutants deficient in its synthesis or reduction successfully generated in M. smegmatis (Purwantini and Mukhopadhyay 2009; Taylor et al. 2010; Grinter et al. 2020), M. tuberculosis (Darwin et al. 2003; Manjunatha et al. 2006; Gurumurthy et al. 2013) and M. bovis (Choi, Kendrick and Daniels 2002). However, several studies indicate that F420 contributes to the ability of mycobacteria to persist in response to oxygen deprivation, oxidative stress, nitrosative stress, or treatment with antimicrobial compounds (Purwantini and Mukhopadhyay 2009; Gurumurthy et al. 2013; Jirapanjawat et al. 2016; Lee et al. 2020; Rifat et al. 2020). F420 reduction in the cytoplasm of Mycobacterium appears to be solely mediated by the F420-reducing glucose 6-phosphate dehydrogenase (Fgd), rather than by Fno, which is employed by most other Actinobacteria (Purwantini, Gillis and Daniels 1997; Bashiri et al. 2008; Jirapanjawat et al. 2016). In mycobacteria, Fgd is one of two entry points into the reductive pentose phosphate pathway, in addition to the canonical NADP+-reducing enzyme. The metabolic coupling of F420 reduction of G6P oxidation represents a significant portion of the flux through the pentose phosphate pathway in mycobacteria, with Fgd activity in cell lysates roughly equivalent to NADP-dependent G6P dehydrogenase (Purwantini, Gillis and Daniels 1997, 1998). G6P levels are 100-fold higher in M. smegmatis than E. coli grown under comparable conditions and may serve as a store of reductant that is mobilized through F420 to combat oxidative stress (Hasan et al. 2010). Consistent with this hypothesis, mycobacteria use G6P when challenged with redox cycling agents (e.g. menadione), rapidly reduce such compounds using F420H2-dependent reductases and are hypersusceptible to challenge in strains unable to make or reduce F420 (Hasan et al. 2010; Gurumurthy et al. 2013; Jirapanjawat et al. 2016). Mycobacteria unable to produce or reduce F420 are also hypersusceptible to nitrosative stress, including from NaNO2 and NO (Darwin et al. 2003; Purwantini and Mukhopadhyay 2009). In a chemical assay, isolated F420H2 readily reduces NO2, leading to the suggestion that the cofactor may directly quench reactive nitrogen species (Purwantini and Mukhopadhyay 2009). However, the biochemical mechanism of F420 dependent oxidative and nitrosative stress resistance in Mycobacterium remains to be fully elucidated.
Emerging evidence suggests that F420H2 may also serve as a respiratory electron donor for mycobacteria. The FDOR-A family enzyme deazaflavin nitroreductase (Ddn) from M. tuberculosis, as well as its homologs from M. smegmatis, can reduce menaquinone at physiologically relevant rates (Fig. 7A; Lee et al. 2020). Furthermore, heterologous expression of Ddn stimulated the O2 consumption of isolated M. smegmatis membranes in an F420H2-dependent fashion, indicating it supplies F420 derived reductant to the respiratory chain. An M. tuberculosis mutant lacking this enzyme is impaired in its ability to recover from hypoxia-induced dormancy (Lee et al. 2020). However, more systematic studies are required to unravel the contribution of F420H2 compared to other electron donors in maintaining energy and redox homeostasis in mycobacterial cells.
Figure 7.

Physiological reactions proposed to be mediated by F420-dependent enzymes in mycobacteria. The bond oxidized or reduced is highlighted in orange for each substrate, with the enzyme responsible for the reaction indicated. For the reactions shown in A, B and D, F420H2 is generated by Fgd through oxidation of G6P. For the reaction shown in C, F420 oxidizes hydroxymycolic acid to ketomycolic acid at the extracellular face of the cytoplasmic membrane, yielding F420H2.
F420 also plays a role in the biosynthesis of the complex lipids that compose the outer envelope of Mycobacterium, thereby contributing to the virulence and intrinsic antibiotic resistance of the genus (Cox et al. 1999; Dubnau et al. 2000; Jain et al. 2007; Purwantini and Mukhopadhyay 2013; Purwantini, Daniels and Mukhopadhyay 2016). The outer envelope of pathogenic mycobacteria contains ketomycolic acids, which are important virulence factors (Yuan et al. 1998; Dubnau et al. 2000; Sambandan et al. 2013). Ketomycolic acids are produced by the oxidation of hydroxymycolic acids, after their transport to the extracellular side of the cellular membrane by fHMAD, an F420-reducing dehydrogenase of the LLHT family (Fig. 7B; Purwantini and Mukhopadhyay 2013). fHMAD is secreted from the cell via the TAT secretion system in complex with F420. As a dehydrogenase, fHMAD does not require a pool of reduced F420 to mediate ketomycolic acid formation, allowing it to function extracytoplasmically (Bashiri et al. 2012). Phthiocerol dimycocerosates (PDIM) are another family of lipids prevalent in the cell envelope of pathogenic mycobacteria. While likely absent from saprophytic species like M. smegmatis (Bansal-Mutalik and Nikaido 2014), in M. tuberculosis PDIM constitutes 46% of the total lipids (Wang et al. 2020), contributing to cell envelope impermeability and phagosomal escape from host cells (Quigley et al. 2017). In M. bovis, conversion of phthiodiolone dimycocerosates to PDIM is dependent on reduced F420H2 provided either enzymatically by Fgd or added exogenously to cell lysates. Based on sequence analysis, it is predicted that F420H2-dependent LLHT (phthiodiolone ketoreductase, fPKR) is responsible for the reduction of phthiodiolone dimycocerosates to phthiotriol dimycocerosates, the penultimate step in PDIM synthesis (Fig. 7C; Siméone et al. 2007; Purwantini, Daniels and Mukhopadhyay 2016). Suggestive of further roles for F420-dependent enzymes in lipid biosynthesis, proteomic analysis of the FDOR-AA family in mycobacteria indicates these enzymes are membrane-associated and their genetic context suggests they play a role in lipid synthesis (Ahmed et al. 2015). Synthesis of the complex lipid-rich mycobacterial outer envelope requires a high level of biosynthetic complexity (Kolattukudy et al. 1997; Bansal-Mutalik and Nikaido 2014; Marrakchi, Lanéelle and Daffé 2014), which may be provided by F420-dependent enzymes, thereby helping to explain their abundance and diversity in mycobacterial species.
F420H2-dependent reductases also provide a reductive detoxification system in mycobacteria, providing the ability to inactivate a range of exogenous compounds with antimicrobial activity (Jirapanjawat et al. 2016). M. smegmatis ΔfbiC and Δfgd strains are hypersensitive to a range of antimicrobial compounds, including quinone analogs (e.g. menadione), coumarin derivatives (e.g. methoxsalen), arylmethane dyes (e.g. malachite green) and quinolones (e.g. oxolinic acid; Guerra-Lopez, Daniels and Rawat 2007; Hasan et al. 2010; Jirapanjawat et al. 2016). The intrinsic resistance of wild-type M. smegmatis to these compounds is attributed to the large number of FDORs it uses. Numerous purified FDORs from M. smegmatis have been shown to promiscuously reduce members of the above chemical classes to varying degrees (Jirapanjawat et al. 2016; Greening et al. 2017). In support of the role of FDORs in reductive detoxification, wildtype M. smegmatis can reduce methoxsalen, malachite green and methyl violet added to cultures, but ΔfbiC and Δfgd strains cannot (Guerra-Lopez, Daniels and Rawat 2007; Jirapanjawat et al. 2016). Importantly, M. smegmatis ΔfbiC and Δfgd strains only display a modest increase in sensitivity to the clinically utilized antimycobacterials, including rifampicin, isoniazid and clofazimine, suggesting it lacks F420-dependent enzymes capable of reducing them (Jirapanjawat et al. 2016). While some FDORs from M. tuberculosis also promiscuously reduce exogenous compounds (Taylor et al. 2010; Cellitti et al. 2012), it remains to be determined whether F420H2-dependent reductases provide an analogous detoxification system in obligately pathogenic mycobacteria.
Despite our growing understanding of the general role of F420 dependent processes in Mycobacterium, few F420-dependent enzymes have a defined physiological function (Selengut and Haft 2010; Ahmed et al. 2015). In addition to those discussed above, an FDOR-B enzyme purified from M. tuberculosis is proposed to be an F420H2-dependent biliverdin reductase; the enzyme reductively converts biliverdin to bilirubin, a potent antioxidant that may play a role in resisting host-induced oxidative stress, though it remains to be established if this activity occurs physiologically (Fig. 7D; Biswal et al. 2006; Ahmed et al. 2015). To fully understand the role of F420 in Mycobacterium, further work is required to systematically characterize the phenotypes associated with this cofactor, including its role in resistance to antimicrobials, redox stress and hypoxia. Additionally, while F420 plays a role in mycobacterial physiology, the extent to which the cofactor is required for the long-term persistence of M. tuberculosis in the host is unclear. To reconcile the physiology with biochemical mechanisms, the role of specific F420-dependent enzymes in mediating the reactions behind these phenotypes needs to be determined through genetic and biochemical analysis.
Streptomycetes
In Streptomyces species, F420 plays an important role as a cofactor for enzymes involved in the synthesis of structurally diverse antibiotics and secondary metabolites (Wang et al. 2013; Ichikawa, Bashiri and Kelly 2018; Steiningerova et al. 2020; Tao et al. 2020). While it was not formally identified at the time, one of the earliest instances of F420 isolation was from Streptomyces aureofaciens, where it was shown to mediate the final hydrogenation step in chlorotetracycline biosynthesis (McCormick et al. 1958; Miller et al. 1960). More recently, it was shown that an F420H2-dependent FDOR-B family enzyme catalyzes the final reduction of the C5a-C11a double bond of the dehydrooxytetracycline precursor of several tetracycline variants (Fig. 8A; Wang et al. 2013). These enzymes are designated OxyR, CtcR and DacO4 in the oxytetracycline/tetracycline, chlorotetracycline and dactylocycline biosynthesis pathways respectively (Wang et al. 2012, 2013).
Figure 8.
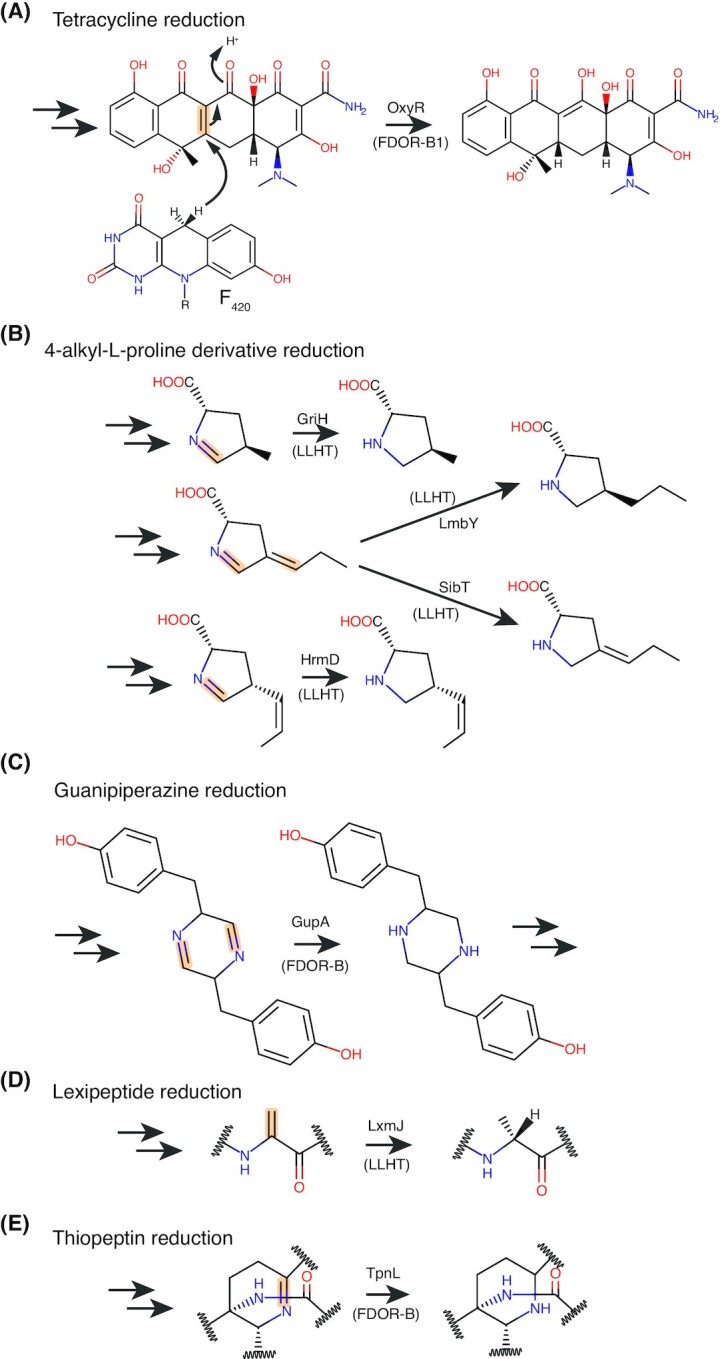
Reactions proposed to be mediated by F420-dependent reductases in streptomycetes. The bond reduced is highlighted in orange for each substrate, with the enzyme responsible for the reaction indicated. F420H2 for the reactions shown is generated by the enzyme Fno via the oxidation of NADPH.
A group of F420H2-dependent reductases from the LLHT superfamily contribute to the biosynthesis of 4-alkyl-L-proline derivatives (APDs) in various streptomycetes (Steiningerova et al. 2020). APDs are biosynthetic precursors for lincosamide and griselimycin antibiotics (Peschke et al. 1995; Lukat et al. 2017), several pyrrolobenzodiazepines (PBDs) with antitumorigenic and antibiotic properties (e.g. tomaymycin, sibiromycin, anthranmycin; Li et al. 2009a, b; Steiningerova et al. 2020), and the quorum-sensing peptide hormaomycin (Höfer et al. 2011). These F420H2-dependent LLHT reductases, named Apd6s, are present in the biosynthetic gene clusters (BGCs) for these secondary metabolites and perform the final reduction step in APD biosynthesis (Fig. 8B; Steiningerova et al. 2020). These Adp6 enzymes strikingly differ in the reduction reactions they perform. Apd6s from PBD and hormaomycin biosynthesis only reduce the endocyclic imine double bond of the ADP precursor, whereas the Adp6 enzyme associated with lincomycin biosynthesis also reduces the more inert exocyclic double bond of its 4-substituted Δ1-pyrroline-2-carboxylic acid substrate (Fig. 8B; Steiningerova et al. 2020). These differences in Adp6 specificity lead to variably saturated APD moieties that help mediate the biological function of the final compound that contains them (Steiningerova et al. 2020). Bioinformatic analysis indicates that Adp6 homologs are widely distributed within several bacterial phyla and often associated with BGCs of unknown function, suggesting they mediate the formation of novel APD-containing molecules (Steiningerova et al. 2020).
An F420H2-dependent reductase of the FDOR-B superfamily from Streptomyces chrestomyceticus, designated GupA, forms part of the BGC for guanipiperazines A and B. These compounds are formed through the condensation of two L-tyrosine molecules forming a dihydropyrazine ring that is reduced by GupA to form the piperazine ring found in the final product (Fig. 8C). While the function of these compounds is unknown, homologues of GupA and other components of the guanipiperazine BGC are widespread in Streptomyces species (Shi et al. 2021).
Other F420-dependent enzymes form part of the biosynthetic pathways of diverse posttranslationally modified peptide antibiotics. The BGC of the recently discovered lexapeptide, a class V lanthipeptide produced by Streptomyces rochei, contains the F420H2-dependent LLHT LxmJ that catalyzes the reduction of a lexapeptide dehydroalanine moiety to D-Ala (Fig. 8D). This reduction increases the potency of lexapeptide towards a panel of Gram-positive bacteria (Tao et al. 2020). Additionally, an F420H2-dependent FDOR-B family reductase designated TpnL is present in the BGC of the thiopeptide thiopeptin produced by Streptomyces tateyamensis. TpnL mediates the reduction of an imine within the dehydropiperidine moiety of thiopeptin, yielding a modified piperidine-containing product (Fig. 8E; Ichikawa, Bashiri and Kelly 2018). TpnL homologs are present in many known or predicted thiopeptide BGCs and form a distinct clade from other FDOR-B sequences (Ahmed et al. 2015; Ichikawa, Bashiri and Kelly 2018). Also of note is the BGC of the pyrroloquinoline alkaloid ammosamide from Streptomyces sp. CNR-698, which contains the putative F420-dependent FDOR-B protein Amm4. The authors predict Amm4 is an oxidase involved in primary amide biosynthesis, based on the accumulation of an ammosamaic acid shunt product in an Δamm4 producing strain (Jordan and Moore 2016).
The chemically diverse nature of the secondary metabolites with biosynthetic pathways containing F420-dependent enzymes, as well as the involvement of the structurally unrelated FDOR and LLHT enzyme families, demonstrates that F420-dependent enzymes are versatile biosynthetic tools for streptomycetes. An abundance of BCGs containing predicted F420-dependent enzymes indicates that F420 is likely to be utilized in the biosynthesis of many more secondary metabolites than those currently identified experimentally. For example, putative F420-dependent LLHTs are also encoded in the BGCs for a coronafacoyl phytotoxin produced by the plant pathogen Streptomyces scabiei (Bown et al. 2016), the aminoglycoside kasugamycin produced by Streptomyces kasugaensis (Ikeno et al. 2006), and mitomycin C produced by Streptomyces lawendulae (Mao, Varoglu and Sherman 1999). However, further experimentation is required to support the F420-dependence of these enzymes and their specific role in the biosynthesis of these compounds. At odds with our increasing knowledge of the role of F420 in secondary metabolism in streptomycetes, virtually nothing is known about its role in primary metabolism, or whether it shares some physiological roles to those described for its fellow actinobacteria genus Mycobacterium.
Other Actinobacteria
F420 is widely produced by other Actinobacteria including Rhodococcus, Nocardia and Nocardioides (Daniels, Bakhiet and Harmon 1985; Purwantini, Gillis and Daniels 1997; Ebert, Rieger and Knackmuss 1999). Bacteria from these genera utilize F420H2-dependent reductases from the LLHT superfamily to mobilize the explosive picrate (2,4,6-trinitrophenol) and related compounds (e.g., 2,4-dinitrophenol, 2,4-dinitroanisole) for degradation (Ebert, Fischer and Knackmuss 2001; Fida et al. 2014). Due to this capability, several actinobacterial strains such as Rhodococcus opacus and Nocardioides simplex can grow on picrate as their sole carbon and nitrogen source (Lenke et al. 1992; Ebert, Fischer and Knackmuss 2001). Other than its role in the remediation of nitroaromatic xenobiotics (Ebert, Rieger and Knackmuss 1999; Ebert, Fischer and Knackmuss 2001; Fida et al. 2014), little is known about the physiological roles of F420 in these Actinobacteria. Consistent with its role in picrate degradation, F420 likely contributes to the well-documented ability of soil actinomycetes to biodegrade a wide variety of complex organic compounds, including polycyclic aromatic hydrocarbons (McCarthy and Williams 1992; Schrijver and Mot 1999).
Roles in other bacteria
The production of F420 by bacteria outside of the phylum actinobacteria was only recently experimentally verified (Ney et al. 2017a; Braga et al. 2019,2020). As such, no F420H2-dependent reductases or F420-reducing dehydrogenases have been investigated experimentally in these bacteria. However, based on the levels of F420 production and analysis of the genomes of F420-producing bacteria, some inferences can be made regarding the role of F420 in these species. Of the species shown experimentally to produce F420, Thermomicrobium roseum (Chloroflexi) and Paraburkholderia rhizoxinica (Betaproteobacteria) are abundant producers, suggesting F420 plays a significant role in the physiology of these organisms (Braga et al. 2019). In contrast, F420 was only detected in Oligotropha carboxidovorans and Paracoccus denitrificans in trace quantities, indicating a minor or niche-specific role for the cofactor in these Alphaproteobacteria (Ney et al. 2017a). F420 producers within Betaproteobacteria and Tectomicrobia only encode Fno among known F420-reducing dehydrogenases, suggesting that NADPH is the major or only compound utilized for F420 reduction in these bacteria (Ney et al. 2017a). Predicted alphaproteobacterial F420-producers encode Fgd in addition to Fno, suggesting that G6P is also utilized for F420 reduction. In Chloroflexi, Fgd is the most prevalent F420-reducing dehydrogenase, but Adf and Fno homologs are also present, suggesting diverse substrates enable F420 reduction (Ney et al. 2017a).
The ecological niche and physiology of recently identified F420 producing bacteria suggest the cofactor is employed in diverse roles, which are at least partially analogous to those identified in Actinobacteria. The phylum Tectomicrobia (Candidatus Entotheonella spp.) includes uncultured bacteria that produce diverse bioactive secondary metabolites associated with marine sponges (Lackner et al. 2017). These bacteria possess large genomes (>9 Mbps) and are predicted to be F420 producers, given they are autofluorescent, possess a full set of F420 biosynthetic genes (Table 4) and encode multiple putative F420H2-dependent reductases (Wilson et al. 2014; Lackner et al. 2017; Ney et al. 2017a; Mori et al. 2018). F420 likely plays a role in secondary metabolite biosynthesis in Tectomicrobia similar to that of streptomycetes discussed above. Chloroflexi are one of the dominant bacterial phyla found in soils and are reputed for their biodegradative capacities (Björnsson et al. 2002; Speirs et al. 2019). They generally encode numerous F420H2-dependent reductases (Ney et al. 2017a), suggesting members of the phylum may use F420 as a cofactor for biodegradative reductases in a similar manner to M. smegmatis or Nocardia spp. The betaproteobacterial fungal endosymbiont P. rhizoxinica produces a chemically distinct F420 variant, 3PG-F420 (see section 3.1.2). In P. rhizoxinica, 3PG-F420 production is greatly enhanced when growing inside its fungal symbiont, suggesting the cofactor facilitates symbiosis. Based on the presence of genes encoding a putative ABC transporter adjacent to the 3PG-F420 BCG, P. rhizoxinica possibly exports the cofactor to be used by its fungal partner (Braga et al. 2019). Interestingly, the genome of P. rhizoxinica lacks genes with homology to known F420-dependent enzymes, suggesting that distinct mechanisms of cofactor utilization and cycling occur in this bacterium (Braga et al. 2019).
Experimental work is required to establish the role of F420 in these bacteria. Metagenomic analysis indicates that F420 producers are prevalent among aerobic soil bacteria (Ney et al. 2017a), suggesting it is widely used for its versatile biosynthetic and biodegradative properties. As a result, F420-dependent enzymes may directly affect the microbial and chemical composition of soils, by providing bacteria with the ability to both synthesize and degrade antimicrobial compounds, in an ongoing arms race.
THE MECHANISM AND EVOLUTION OF F420 BIOSYNTHESIS
Diversity within the F420 biosynthetic pathway
Figure 9 presents the steps in the F420 biosynthesis pathway and the enzymes that mediate them. Consistent with the initial discovery of F420 in methanogenic Euryarchaeota (Cheeseman, Toms-Wood and Wolfe 1972), initial investigation of the structure and biosynthesis of the cofactor was performed in these organisms (Eirich, Vogels and Wolfe 1978; Jacobson and Walsh 1984; Li et al. 2003a). The F420 biosynthesis pathway in methanogenic Euryarchaeota was established and was assumed to be universal to all F420 producing organisms (Greening et al. 2016; Bashiri et al. 2019). However, recent investigation of F420 biosynthesis in bacteria has revealed that divergent pathways for F420 biosynthesis are employed in different organisms. These differences originate from variation in the substrate compound utilized to link the FO head group of F420 to its poly-glutamate tail (Bashiri et al. 2019; Braga et al. 2019; Grinter et al. 2020). Based on current experimental evidence, the F420 biosynthesis pathway occurs via three variant pathways, found in Euryarchaeota, Actinobacteria and Proteobacteria, respectively (Fig. 9). However, future investigation may reveal further diversity in the F420 biosynthesis pathway.
Figure 9.

Diverse routes to F420 biosynthesis employed by bacteria and archaea. 1 = classical archaeal pathway (Euryarchaeota), 2 = bacterial pathway a (Actinobacteria, Chloroflexi), 3 = bacterial pathway b (Betaproteobacteria). The substrates and mechanisms for FO biosynthesis are shared between all identified pathways. Abbreviated compounds are as follows: PEP, phosphoenolpyruvate; 2PL, 2-phospho-L-lactate; 3PG, 3-phosphoglycerate; EPPG, enolpyruvyl-diphospho-5’-guanosine; LPPG, lactyl-diphospho-5’-guanosine; GPPG, 3-guanosine-5’-disphospho-D-glycerate. The enzymes involved in each biosynthesis step are indicated.
A note on nomenclature
Different nomenclature has been applied to F420 biosynthetic enzymes from archaea and bacteria. This reflects the incremental nature of the advance in our understanding of F420 biosynthesis, as well as the distant relationships between these domains. However, the continued use of different nomenclature is now justified with the recent discovery that F420 is synthesized through distinct routes in these domains. Nevertheless, the nomenclature has become increasingly convoluted in light of these discoveries, together with recent evidence demonstrating multiple horizontal gene transfer of F420 biosynthetic genes and gene fusion events. For simplicity, in this review, we will generally refer to F420 biosynthesis proteins mediating the archaeal pathway with the prefix ‘Cof’ and those mediating the bacterial pathway with the prefix ‘Fbi’.
In methanogens, the enzymes CofG and CofH mediate FO biosynthesis, CofC and CofD mediate F420-0 biosynthesis, and CofE is responsible for the formation of mature F420 via the addition of a γ-linked polyglutamate tail. In mycobacteria, the prefix ‘Fbi’ is utilized, with a different lettering system, where the following enzymes are analogous: FbiC is derived from a fusion of CofG and CofH, FbiD and FbiA are similar to CofC and CofD respectively, and FbiB is derived from a fusion of CofE and the nitroreductase (NTR) superfamily protein FbiE. It should be noted that some bacteria possess individual enzymes homologous to CofG, CofH, or CofE of archaeal F420 producers; we refer to these by the ‘Cof’ designation, as they are distinct from the corresponding ‘Fbi’ fusion enzymes. Likewise, at least some archaea possess some homologs of ‘Fbi’ fusion enzymes. Finally, the FbiA and FbiD variants from some Betaproteobacteria produce the chemically distinct variant 3PG-F420 (Braga et al. 2019). The subscript ‘3PG’ is applied to these enzymes.
Comparison of pathways
In the F420 biosynthetic pathway of Euryarchaeota, 2-phospho-L-lactate (2PL) links the FO head group of F420 to its polyglutamate tail (Fig. 9; Grochowski, Xu and White 2008). It has been proposed that 2PL is synthesized by the unidentified lactate kinase CofB, using lactate produced from L-lactaldehyde (Graupner and White 2001; Graupner, Xu and White 2002; Grochowski, Xu and White 2006). 2PL is conjugated to FO via the action of the enzymes CofC and CofD to create F420-0 (i.e. F420 with no glutamate tail). CofC activates 2PL through condensation with GTP to form the intermediate compound lactyl-diphospho-5‘-guanosine (LPPG; Grochowski, Xu and White 2008). CofD subsequently transfers 2PL from LPPG to FO to form F420-0 (Graupner, Xu and White 2002). The activity of CofC is contingent on the presence of CofD, suggesting that these enzymes form a catalytic complex to regulate the production to the LPPG intermediate, which is unstable (Bashiri et al. 2019; Braga et al. 2019). F420-0 is then converted to mature F420 via the addition of a variable-length γ-linked glutamate tail by the enzyme CofE (Li et al. 2003a; Nocek et al. 2007). In archaea, the length of the glutamate tail varies from 4 to 5 in methanogens with cytochromes or 2–3 in those without (Gorris 1994). In some Euryarchaeota, an additional terminal α-linked glutamate is added by the α-L-glutamate ligase CofF (Li et al. 2003b).
In mycobacteria, the central metabolic intermediate phosphoenolpyruvate (PEP), rather than 2PL, is utilized as the precursor for F420 biosynthesis in this genus (Bashiri et al. 2019; Grinter et al. 2020). The incorporation of PEP into F420 follows an analogous route to that of 2PL in archaea, with the enzymes FbiD and FbiA (homologs of CofC and CofD respectively) first converting PEP into the intermediate enolpyruvyl-diphospho-5’-guanosine (EPPG) and then condensing it with FO to form dehydro-F420-0 (DH-F420-0), in which the enol group of PEP remains oxidized (Fig. 9; Grinter et al. 2020). DH-F420-0 is then modified to form mature F420 by the dual-functional enzyme FbiB. FbiB possesses an N-terminal domain homologous to CofE, which adds a variable-length γ-linked polyglutamate tail of 2–8 residues (Bashiri et al. 2019). The C-terminal domain of FbiB (FbiBC-term) reduces the enol group of DH-F420 converting it into mature F420 (Bashiri et al. 2016, 2019). The reduction of DH-F420 improves the stability of the molecule by removing the high-energy phosphoenol bond (Braga et al. 2020). The Chloroflexi strain T. roseum utilizes an independent homolog of FbiBC-term to reduce DH-F420, herein referred to as FbiE (Braga et al. 2020). Genomic analysis indicates that independent FbiE homologs are present in the genomes of several predicted bacterial and archaeal F420 producers, and putative F420-producing members of the archaeal phylum Lokiarchaeota possess a dual functional FbiB homolog (Braga et al. 2020), suggesting that diverse bacteria and archaea also employ a PEP dependent pathway for F420 biosynthesis (Fig. 10B).
Figure 10.

Genetic organization of F420 biosynthetic genes in bacteria and archaea. A schematic of the generalized the genetic context of F420 biosynthetic genes in experimentally confirmed (panel A) and predicted (panel B) F420-producing bacteria (left) and archaea (right). F420 biosynthetic genes are labeled and color-coded. Additional F420 related genes are colored grey and labeled as follows: Fgd, F420-reducing glucose 6-phosphate dehydrogenase; Fno, F420-reducing NADPH dehydrogenase; FDOR, predicted F420H2-dependent reductase; LLHT, predicted F420H2-dependent luciferase-like hydride transferase; Mer, F420H2-dependent CH2=H4MPT reductase. Hypothetical genes or those with no known F420-related function are shown and colored white. Black tilde symbols designate undefined intergenomic space. Gene context is adapted from Ney et al. (2017a) or determined directly from available genome sequences.
A third route for F420 biosynthesis is employed by the betaproteobacterium P. rhizoxinica, in which 3-phospho-D-glycerate (3PG) is utilized in place of 2PL or PEP for F420 biosynthesis. This leads to the formation of the chemically distinct species 3PG-F420 and depends on the action of FbiD3PG and FbiA3PG, homologs of CofC/FbiD and CofD/FbiA respectively (Fig. 9; Braga et al. 2019). The specificity for 3PG over PEP or 2PL appears to originate from FbiD3PG, which preferentially mediates the incorporation of 3PG into 3-guanosine-5′-disphospho-D-glycerate (GPPG; Braga et al. 2019). A homolog of CofE then adds a variable-length γ-linked polyglutamate tail of 1–6 residues to form mature 3PG-F420 (Braga et al. 2019).
The selective pressures underlying the rerouting of the F420 biosynthesis pathways remain unclear. Considering that CofC/FbiD/FbiD3PG evolved from a common F420-producing ancestral enzyme (Ney et al. 2017a; Bashiri et al. 2019; Braga et al. 2019), a substrate switch must have occurred in at least two lineages to yield the three observed biosynthesis pathways. This switch likely occurred to reconcile the precursor utilized for F420 production with its presence or level of availability in the metabolite pool of the F420-producing organism.
Structural and biochemical basis
Recently, considerable progress has been made in understanding F420 biosynthesis in both bacteria and archaea. Except for CofG/CofH and FbiC, crystal structures have been determined for all enzymes in the F420 biosynthetic pathway, with biochemical analysis revealing considerable detail on the catalytic mechanisms employed during F420 biosynthesis.
Synthesis of FO by CofG/CofH and FbiC
In all studied F420 producing organisms, FO is synthesized through a universal mechanism involving two SAM-radical domain enzymes (Decamps et al. 2012). In archaea and some bacteria, these domains exist as two separate proteins CofG and CofH, while in Actinobacteria and Proteobacteria they are present in the single fusion protein FbiC (Ney et al. 2017a; Fig. 11A). No structures for these enzymes have been determined to date, likely due to the difficulty of working with these oxygen-sensitive proteins (Imlay 2006; Philmus et al. 2015). However, mass spectrometric analysis of CofG and CofH reaction products combined with substrate deuteration has provided considerable insight into the catalytic mechanism behind FO synthesis (Decamps et al. 2012; Philmus et al. 2015). FO is formed by the condensation of L-tyrosine with pyrimidine ribityldiaminouracil (5-amino-6-ribitylamino-2,4 [1H,3H]-pyrimidinedione), which is also a substrate for riboflavin biosynthesis (Bacher et al. 2000; Decamps et al. 2012). In the first step of this two-step reaction, the 5′-deoxyadenosyl radical generated by CofH abstracts a hydrogen atom from the tyrosine amine, which causes the tyrosine to fragment to form a p-hydroxybenzyl radical. This radical then undergoes addition to the double bond of pyrimidine ribityldiaminouracil, with this compound subsequently oxidized by the [4Fe4S] of CofH to yield an intermediate product (Fig. 11B). This product is then accepted by CofG, where the 5′-deoxyadenosyl radical formed by this enzyme extracts a further hydrogen, creating a radical intermediate that undergoes cyclization followed by oxidation by the [4Fe4S] cluster of CofG to yield FO (Fig. 11C; Philmus et al. 2015). Despite existing as a fusion protein, the two domains of FbiC appear to function independently, with diffusion rather than direct substrate transfer responsible for the transfer of the product of the FbiC C-terminal domain to the N-terminal domain to complete FO biosynthesis (Philmus et al. 2015).
Figure 11.

Production of the deazaflavin FO is catalyzed by dual SAM-radical domains. (A) Structural model of the SAM radical domains that mediate Fo synthesis, consisting of the two separate proteins CofG and CofH (in archaea and some bacteria) or a single fusion protein FbiC (in bacteria and eukaryotes). Structural models constructed based on homology modeling using Phyre2 based on the structure of MqnE from Pedobacter heparinus (PDB ID = 6XI9; Kelley et al. 2015). (B) A summary of the proposed reaction performed by CofH. (C) A summary of the proposed reaction performed by CofG. For the full reaction scheme summarized in panels B and C refer to Philmus et al. (2015). R = The FO ribose tail as shown in Fig. 9.
Synthesis of LPPG, EPPG and GPPG by CofC, FbiD and FbiD3PG
The FO is linked to the polyglutamate tail of mature F420 via either a 2PL for F420 or 3PG for 3PG-F420 (Graupner, Xu and White 2002; Braga et al. 2019; Grinter et al. 2020). In order to activate them for condensation with FO, 2PL, PEP or 3PG are condensed with GTP to form LPPG, EPPG and GPPG, respectively (Fig. 9). Despite the differences in their preferred substrate CofC, FbiD and FbiD3PG are homologs thought to share a common catalytic mechanism (Bashiri et al. 2019; Braga et al. 2019). The crystal structure of Apo-CofC from Methanosarcina mazei (PDB ID: 2I5E) was first determined by a structural genomics consortium (2006), demonstrating that the enzyme possesses a nucleotide-binding Rossmann fold, though the lack of substrate in this crystal structure limited the insight of the catalytic mechanism provided by this structure. Recently, the structure of FbiD from M. tuberculosis was determined in complex with PEP and two catalytic Mg2+ ions (Fig. 12A; Bashiri et al. 2019). Based on this structure, key substrate-binding residues were identified (Fig. 12C; Bashiri et al. 2019). Further, structural comparison between FbiD and the distantly related bifunctional acetyltransferase/uridyltransferase GlmU reveals a putative GTP-binding pocket (Fig. 12B). This pocket is occluded in the crystal structure of FbiD, which is consistent with the observation that neither purified CofC nor FbiD are active in the absence of their partner enzyme CofD and FbiA (Bashiri et al. 2019; Braga et al. 2019). This suggests that conformational activation of CofC/FbiD occurs in the presence of CofD/FbiA, likely to prevent the futile production of an unstable product (Bashiri et al. 2019).
Figure 12.
Crystal structure of FbiD from M. tuberculosis in complex with PEP substrate. (A) Cartoon view of the crystal structure of FbiD in complex with PEP (PDB ID = 6BWH). (B) A surface model of the FbiB active site showing the region predicted to bind GTP adopts an occluded conformation. (C) Key residues of FbiD involved in the coordination of the PEP substrate and catalytic Mg2+ ions. Atomic distances less than 3.2 Å are shown as dashed yellow lines.
Synthesis of F420-0, DH-F420-0 and 3PG-F420-0 by CofD, FbiA and FbiA3PG
CofD, FbiA and FbiA3PG are homologous enzymes that mediate the transfer of 2PL, PEP or 3PG from the diphospho-5‘-guanosine intermediate produced by CofC/FbiD to FO. This results in the formation of the intermediates F420-0, DH-F420-0 and 3PG-F420-0, with no glutamate moieties (Graupner, Xu and White 2002). The crystal structures CofD from Methanosarcina mazei and FbiA from M. smegmatis have been determined in the presence of their substrates (Forouhar et al. 2008; Grinter et al. 2020). These enzymes require a divalent cation for activity, which was absent from the crystal structure of CofD, meaning the catalytically important portions of the FO and GDP substrates were disordered in this structure (Forouhar et al. 2008). However, we recently determined the structure of FbiA in complex with FO, GDP and Ca2+, providing a clear picture of the catalytic complex of this enzyme (Fig. 13A; Grinter et al. 2020). The catalytic metal ion represented by Ca2+ in this structure is coordinated by aspartates 45 and 57. Aspartate 46 in CofD, which is equivalent to aspartate 57 in FbiA, is important for catalytic activity, suggesting it is also involved in catalytic metal ion coordination in this enzyme (Forouhar et al. 2008). In addition to the two aspartate residues, the catalytic Ca2+ ion is further coordinated by a H2O molecule, the terminal hydroxyl of FO and the β-phosphate of EPPG (Fig. 13B). This coordination positions the EPPG β-phosphate for nucleophilic attack by the FO terminal hydroxyl, leading to the transfer of PEP and the creation of the DH-F420-0 product (Grinter et al. 2020). To mediate this nucleophilic attack, FO requires activation, with likely candidate bases being the carboxyl or β-phosphate group of EPPG. A proposed reaction mechanism based on proton subtraction by the former is presented in Fig. 13C.
Figure 13.

Crystal structure of the FbiA-substrate complex from M. smegmatis. (A) Crystal structure of the FbiA dimer [PDB ID = 6UW5] in complex with GDP in Mol. A and Fo and EPPG (modeled in place of co-crystallized GDP) in Mol. B. FbiA is shown as a cartoon model, substrate molecules are shown as sticks and a Ca2+ ion (likely Mg2+ in the active enzyme) is shown as a sphere. (B) A cross-eye stereo view of the active site of FbiA Mol. B from panel A, showing key residues for coordinating the FbiA substrate complex as sticks and coordination distances as dotted lines. (C) A proposed reaction mechanism for synthesis DH-F420-0 by FbiA, in which the carboxyl group of EPPG donates an electron to FO, activating it to perform nucleophilic attack on the EPPG β-phosphate.
Addition of the poly-glutamate tail to F420 by CofE and FbiBN-term
CofE and the N-terminal domain of FbiB (FbiBN-term) are non-ribosomal peptide synthases that perform the final step in F420 biosynthesis, adding a variable number of γ-linked glutamate residues to form the F420 tail. The crystal structure of CofE from A. fulgidus revealed that the protein possesses a novel fold, consisting of an intertwined butterfly-shaped dimer (Fig. 14A; Nocek et al. 2007). The GDP and catalytic Mn2+ ion bound version of this structure revealed the location of a Y-shaped active site with grooves hypothesized to be responsible for binding F420-0 and L-glutamate in addition to GTP (Fig. 14B; Nocek et al. 2007). A proposed catalytic mechanism for CofE and FbiBN-term, based on that of the nucleotide-dependent tetrahydrofolate:L-glutamate γ-ligase (FPGS; Sheng et al. 2000) and UDP-N-acetylmuramoyl-L-alanine:glutamate ligase (MurD; Bertrand et al. 1997), involves the activation of the terminal carboxyl of F420-0 by the addition of a phosphate group from GTP. Subsequently, the carbonyl carbon of the resulting acyl phosphate undergoes a nucleophilic attack by the glutamate amine, leading to the formation of a tetrahedral intermediate, which breaks down to the final F420 product and inorganic phosphate (Fig. 14C; Forouhar et al. 2008). Biochemical and genetic evidence indicates that CofE and FbiBN-term are responsible for the addition of both the initial glutamate to FO and extension of the poly-glutamate chain (Bashiri et al. 2016,2019). Further research is required to resolve how the active site of these enzymes can perform both the initial and subsequent glutamate additions.
Figure 14.
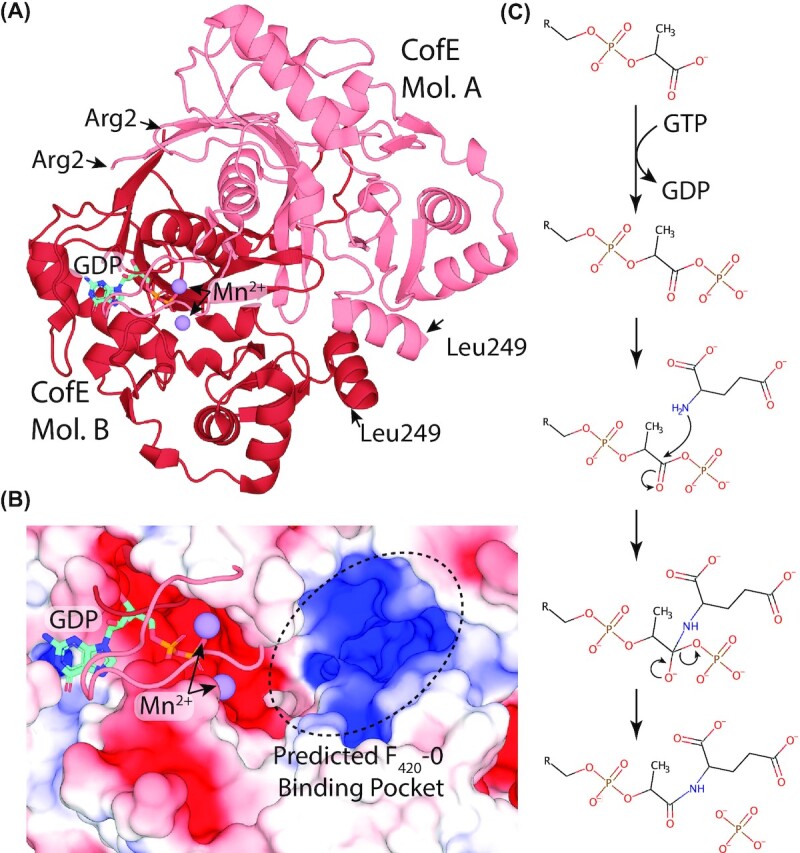
Crystal structure of CofE from A. fulgidus in complex with GDP and Mn2+. (A) Cartoon view of the crystal structure of the functional dimer of CofE [PDB ID = 2PHN]. CofE subunits are shown in pink (Mol. A) and red (Mol. B). Bound GDP and catalytic Mn2+ ions are shown for Mol. B only, as stick and sphere representation respectively. (B) Electrostatic surface view of the CofE active site showing bound GDP, catalytic Mn2+ ions and predicted F420-0 binding pocket. (C) Proposed catalytic mechanism for the first γ-linked glutamate addition mediated by CofE, R = FO minus terminal hydroxyl.
Reduction of DH-F420 by FbiBC-term and FbiE
The reduction of DH-F420 is performed by FbiBC-term in mycobacteria or FbiE in Chloroflexi, homologs that belong to the FMN-dependent NTR superfamily. The crystal structure of the isolated FbiBC-term from mycobacteria has been determined, revealing an intertwined dimer, in complex with either FMN or F420 in distinct binding sites (Fig 15A; Bashiri et al. 2016). The relative positions of FMN and DH-F420-0 were modeled based on these structures, providing a plausible active site and catalytic mechanism for this enzyme (Bashiri et al. 2019), where N-5 of FMNH2 is ideally positioned for hydride transfer to the DH-F420-0 enol group (Fig. 15B).
Figure 15.

Crystal structure of FbiBC-term from M. tuberculosis. (A) Cartoon view of the crystal structure of the functional dimer of the FbiB C-terminal domain responsible for the FMNH2 mediated reduction of DH-F420-0 [PDB IDs = 4XOO, 4XOQ]. FbiBC-term is shown in green and FMN and DH-F420-0 (modeled based on the co-crystal structure of FO) are shown as sticks. (B) A zoomed view of the FbiBC-term active site in complex with FMN and DH-F420-0 as in panel A, with a cartoon and transparent atomic surface of the FbiBC-term shown.
Evolution of the F420 biosynthesis pathway
While it is now clear that F420 is widely distributed in bacteria and archaea, it is not universally distributed like redox cofactors FMN/FAD and NAD/NADP (Daniels, Bakhiet and Harmon 1985; Ney et al. 2017a; Braga et al. 2019, 2020). This distribution poses the question of how F420 biosynthesis originated and how the genes responsible were disseminated across bacteria and archaea. It has been proposed that the capacity to synthesize F420 was present in the last universal common ancestor (LUCA) and was selectively retained by a subset of bacterial and archaeal lineages (Weiss et al. 2016). However, current evidence suggests that the F420 biosynthesis pathway evolved in a stepwise fashion in archaea and bacteria, with horizontal gene transfer mediating assembly of the complete biosynthesis pathway (Ney et al. 2017a). Such inferences are supported by the variable distribution (Fig. 5), genetic organization (Fig. 10) and phylogenetic analysis of the F420 biosynthesis genes (Ney et al. 2017a). In Fig. 16, we present a schematic of the gene transfer events that potentially occurred during the evolution of F420 biosynthesis. By necessity, this model focuses on well-studied F420 producers, and the direction of several gene transfer events remains unresolved.
Figure 16.

Schematic showing possible events in the evolution of F420 and its acquisition by different bacterial and archaeal lineages. Arrows with solid lines indicate potential horizontal transfer of ancestral F420 biosynthetic genes. Dashed lines with bidirectional arrows indicate a likely gene transfer event of unknown directionality. Phyla labels are simplified for clarity. Note the figure is a speculative model drawn based on data from sources discussed in the main text and other models are also consistent with these data.
The synthesis of FO, as the catalytically active headgroup of F420, almost certainly evolved first. FO has near-identical redox properties to F420 and can function as a cofactor for F420-dependent enzymes in vitro (Jacobson and Walsh 1984; Drenth, Trajkovic and Fraaije 2019). However, the uncharged aromatic structure of FO allows it to readily diffuse across lipid membranes (Bashiri et al. 2010), limiting its usefulness as a redox cofactor due to its metabolically costly loss from the cell (Shah et al. 2019). The problem of diffusive loss is less acute when FO functions as a chromophore, through its role in DNA repairing photolyase (Malhotra et al. 1992; Sancar 1994), given in this case it is tightly associated with its enzyme (Kim and Sancar 1993). Phylogenetic analysis of the enzymes responsible for FO biosynthesis (CofG and CofH) suggests they may have arisen in a deep-branching archaeon (Ney et al. 2017a), and were then horizontally acquired by bacteria and certain other archaea. In Actinobacteria, a fusion of the cofG and cofH genes created fbiC (Choi, Kendrick and Daniels 2002; Philmus et al. 2015), which was subsequently acquired by several other F420-producing bacteria (Ney et al. 2017a).
The next stage in the evolution of F420 biosynthesis was the addition of a phospho-carboxylic acid group to FO to form F420-0, a catalytic intermediate of the current biosynthesis pathway (Bashiri et al. 2019; Grinter et al. 2020). This modification imparts a negative charge, preventing its diffusion across cellular membranes (Bashiri et al. 2010), while not affecting the redox properties of the molecule. Phylogenetic evidence suggests that the ancestors of CofC/FbiD/FbiD3PG and CofD/FbiA/FbiA3PG potentially originated in an actinobacterial ancestor before being laterally acquired by other bacteria and archaea (Nelson-Sathi et al. 2015; Ney et al. 2017a). This suggests that F420 was first employed as a redox cofactor in Actinobacteria, before being horizontally acquired by other bacteria and archaea, including Euryarchaeota. The final stage in the evolution of F420 biosynthesis was the addition of the variable-length γ-linked polyglutamate tail to F420-0 by the enzyme CofE (Li et al. 2003a; Bashiri et al. 2019). The polyglutamate tail greatly enhances the affinity and specificity of interactions between F420 and F420-dependent oxidoreductases, possibly explaining why it arose (Ney et al. 2017b; Drenth, Trajkovic and Fraaije 2019). The evolutionary origin of CoE is unclear (Nocek et al. 2007; Ney et al. 2017a), though the polyglutamate tail synthesized by CofE is present in F420 from all currently investigated producing species (Gorris 1994; Li et al. 2003a; Bashiri et al. 2016; Greening et al. 2016; Ney et al. 2017a; Braga et al. 2019), indicating it is universally important for F420 function and has thus been co-inherited with other F420 biosynthesis genes. In Actinobacteria, cofE underwent a fusion with the DH-F420-0 reductase gene fbiE to produce the bifunctional fbiB.
APPLICATIONS OF F420 BIOSYNTHESIS
Progress and challenges for the use of F420 in industrial catalysis
The hydrogenation reactions performed by F420H2-dependent reductases are of interest for biocatalysis due to their regio- and enantioselectivity, which can generate up to two chiral centers (Greening et al. 2017; Mathew et al. 2018). Further, the low redox potential of F420 allows it to mediate the reduction of otherwise recalcitrant bonds, including alkenes, enamines, enones, enoates and cyclic imines (Taylor et al. 2010; Jirapanjawat et al. 2016; Greening et al. 2017; Ichikawa, Bashiri and Kelly 2018; Mathew et al. 2018; Steiningerova et al. 2020). F420H2-dependent reductases provide an alternative to the nicotinamide-dependent Old Yellow Enzymes (OYEs) for performing these reactions (Stuermer et al. 2007; Winkler, Faber and Hall 2018), with some evidence suggesting that F420H2-dependent reductases can generate reaction products with the opposite stereochemistry than OYEs (Mathew et al. 2018). However, while recent work has addressed a number of the challenges associated with utilizing F420-dependent enzymes for industrial applications, several further challenges must be addressed before their potential for chemical synthesis can be realized.
Two plausible scenarios exist for the utilization of F420H2-dependent reductases in the production of industrially relevant compounds. Purified F420H2-dependent reductases can be utilized, in conjunction with an F420-reducing regeneration system, to perform the biocatalytic reduction of the desired substrate. Alternatively, a synthetic biology approach could be employed, utilizing microbes engineered to produce F420 and express F420-dependent biosynthetic pathways to produce compounds via microbial cell culture. In this section, we will discuss challenges and recent developments relating to the heterologous production of F420 and the development of suitable F420-dependent enzymes for compound synthesis. These advances apply both to systems utilizing purified F420-dependent enzymes and the development of synthetic biological systems utilizing F420.
Development of high yield F420 production
One of the major challenges in the development and utilization of F420-dependent enzymes for biotechnological applications is the low yield of F420 obtained when purified from native sources (Isabelle, Simpson and Daniels 2002; Mathew et al. 2018; Bashiri et al. 2019). However, recent advances in our understanding of the F420 biosynthesis pathway, as well as successful heterologous production, have improved the prospects of obtaining the cofactor in industrially relevant quantities (Bashiri et al. 2019; Grinter et al. 2020).
F420 was initially purified from methanogenic archaea, such as Methanobacterium thermoautotrophicum (Eirich, Vogels and Wolfe 1978, 1979; Isabelle, Simpson and Daniels 2002). However, the relative technical difficulty in culturing these obligate anaerobes led to the identification and optimization of the aerobic actinomycete M. smegmatis as an alternative source of F420 (Isabelle, Simpson and Daniels 2002). Despite producing F420 at levels 5-fold lower than methanogens, the ease of culture and high cell densities achieved by this bacterium led to M. smegmatis being largely adopted as the preferred organism for F420 production, except in cases where short-chain F420-2 is required (Isabelle, Simpson and Daniels 2002; Ney et al. 2017b). Heterologous plasmid-based expression of FbiA, FbiB and FbiC in M. smegmatis by Bashiri et al. increased production of F420 from this bacterium 10-fold to levels greater than those produced by methanogens (Bashiri et al. 2010). This augmented F420 production in M. smegmatis is the currently preferred method of F420 production (Lapalikar et al. 2012; Ahmed et al. 2015; Mashalidis et al. 2015; Ney et al. 2017a; Oyugi et al. 2018; Drenth, Trajkovic and Fraaije 2019; Steiningerova et al. 2020). However, yields from this method are still unlikely to be compatible with economically viable production on an industrial scale. The estimated maximum yield of F420 from this process is ∼3 g/kg dry weight (∼0.9 g/kg wet cell weight; Isabelle, Simpson and Daniels 2002; Bashiri et al. 2010). Therefore, considerable further optimization of this system or alternative processes for F420 production are required.
One option for large-scale production of F420 is the use of a heterologous expression system, which is amenable to optimization through metabolic engineering. Until recently, a perceived bottleneck for heterologous production was the use of 2PL as a substrate in the F420 biosynthetic pathway, as it is not produced in significant quantities by bacteria (Graupner and White 2001; Graupner, Xu and White 2002; Bashiri et al. 2019). However, the discovery that mycobacterial F420 biosynthesis utilizes the abundant metabolite PEP paved the way for its heterologous production (Bashiri et al. 2019; Grinter et al. 2020). Concurrently, Ney and Greening first successfully produced F420 in E. coli through the heterologous expression of FbiC, FbiA, CofD and FbiB (Ney 2019). However, the yields for F420 produced in E. coli were lower than that achieved for purification from M. smegmatis or methanogens (Isabelle, Simpson and Daniels 2002; Bashiri et al. 2019; Shah et al. 2019), meaning considerable further engineering is required to make the system compatible with industrial production. Through the course of their discovery of 3PG-F420 production by P. rhizoxinica, Braga et. al. independently heterologously produced this chemical F420 variant in E. coli (Braga et al. 2019). While 3PG-F420 was only produced in low quantities, it is compatible with F420 dependent enzymes from organisms producing the classical version of the cofactor (Braga et al. 2019). As such, this work provides an alternative set of enzymes and precursor substrates for heterologous F420 production, which may assist in increasing production levels.
Synthetic or semisynthetic synthesis of F420-like cofactors is a promising alternative strategy for obtaining large quantities of cofactor compatible with industrial applications. FO can be produced synthetically in large quantities and is catalytically compatible with some F420-dependent enzymes (Hossain et al. 2015). However, it generally exhibits much lower catalytic efficiency, making it a less than ideal cofactor for industrial applications (Drenth, Trajkovic and Fraaije 2019). Recently, Drenth et. al. utilized a biosynthetic approach to enzymatically phosphorylate the terminal hydroxyl of FO, yielding the F420 analog FO-5′-phosphate (FOP). FOP was functional as a cofactor for both F420-reducing dehydrogenases and F420H2-dependent reductases with a higher catalytic efficiency than FO. However, a reduction in catalytic efficiency compared to F420 was observed depending on the enzyme employed (2- to 22-fold reduction; Drenth, Trajkovic and Fraaije 2019; Martin et al. 2020). This reduction in efficiency reinforces the importance of the F420 polyglutamate tail in protein-cofactor interactions (Ney et al. 2017b) and suggests that FOP use will be limited to compatible enzymes or those which have been engineered to suit this cofactor. Currently, no definitive solution exists to cheaply and efficiently produce F420 or a suitable analog that can be utilized by enzymes with a high level of catalytic efficiency. However, the recent progress discussed above can likely be built upon to provide a solution to this bottleneck in the near future.
Development of an efficient F420 reduction system
To harness their potential for asymmetric hydrogenation on an industrial scale, F420H2-dependent reductases require a source of reduced F420H2. Ideally, F420H2 would be regenerated from enzymatically oxidized F420 via a mechanism integral to the reaction system, providing a high F420H2/F420 ratio and a sustained source of the reduced cofactor to maximize reaction yields. While it may be possible to directly reduce F420 by electrochemical or photochemical means (Wichmann and Vasic-Racki 2005), this has not been comprehensively investigated. As such, enzymatic regeneration of F420 using existing F420-reducing dehydrogenases is currently the most practical means of cofactor regeneration.
F420-reducing dehydrogenases that utilize a number of diverse substrates have been identified (Table 2). In their physiological context, these dehydrogenases produce a free pool of reduced cytoplasmic F420H2, which is bound by F420H2-dependent reductases and utilized to directly transfer hydride to their substrate (Ahmed et al. 2015, 2016; Greening et al. 2017; Mathew et al. 2018). This system differs mechanistically from OYEs, which tightly bind a FMN molecule that is first reduced by NAD(P)H before substrate reduction (Stuermer et al. 2007; Toogood, Gardiner and Scrutton 2010). The independent nature of F420-reducing dehydrogenases and F420H2-dependent reductases provides flexibility compared to OYEs, allowing for the use of different substrates for cofactor regeneration by F420-reducing dehydrogenases. Several F420-reducing dehydrogenases have been produced recombinantly and structurally characterized (Table 2; Warkentin et al. 2001; Aufhammer et al. 2004,2005; Bashiri et al. 2008; Allegretti et al. 2014), facilitating enzyme production and optimization via structure-guided protein engineering. However, to provide an economically viable solution to F420 reduction for industrial catalysis, the enzyme employed must be stable and readily producible in large quantities. Additionally, it must reduce F420 with reasonable catalytic efficiency and its substrate must be cheap and readily obtainable. Considering these criteria, some F420-reducing dehydrogenases are more attractive targets than others for industrial cofactor regeneration.
Enzymes originating from methanogenic archaea, which utilize H2 (Frh) or formate (Ffd) for F420 reduction, are superficially attractive targets due to the low cost of their substrates and the lack of contaminating solutes resulting from their oxidation (Shah et al. 2019). However, both of these enzymes are multi-subunit proteins that utilize complex transition metal cofactors for substrate oxidation and transfer the resulting electrons to F420 via multiple iron-sulfur clusters (Schauer and Ferry 1986; Vitt et al. 2014). The complexity of these enzymes, combined with the oxygen sensitivity of their metal-containing functional groups, means they are unlikely to be a practical solution for F420-reduction (Baron and Ferry 1989; Vitt et al. 2014). F420 reduction utilizing NADPH as a hydride donor, via the enzyme Fno, represents a more attractive means of cofactor regeneration (Berk and Thauer 1997; Kumar et al. 2017). Fno is a small single subunit protein, which is produced by a wide range of bacteria and archaea, providing numerous homologs from which to select an industrially compatible enzyme (Eirich and Dugger 1984; Kunow et al. 1993; Le et al. 2015; Kumar et al. 2017). Representative crystal structures of Fno from thermophilic species have been determined, providing the basis for optimization via protein engineering (Kunow et al. 1993; Kumar et al. 2017). Drawbacks for the use of Fno for cofactor regeneration include the relative expense of NADPH, which is required in stoichiometric quantities to the target for reduction (unless an additional enzyme and substrate is added for NADP+ regeneration), as well as the presence of contaminating NADP+ in the final reaction mix. The use of G6P for F420 regeneration via the enzyme Fgd is another option that has similar advantages and disadvantages to NADPH. Fgd is a single-chain protein that can be recombinantly produced and for which a crystal structure has been determined (Purwantini and Daniels 1996; Bashiri et al. 2008). However, G6P is relatively expensive and its use in F420 reduction leads to the generation of the by-product 6-phosphoglucono-D-lactone. Recently homologs of Fgd with significant activity towards other sugar phosphates were identified (Mascotti et al. 2018). These enzymes, named F420-reducing sugar-6-phosphate dehydrogenases (Fsd), also exhibit low levels of F420 reductase activity with non-phosphorylated sugars (Mascotti et al. 2018). While the rates of F420 reduction with these sugars were too low to be catalytically useful, they suggest that through protein engineering Fsd could be adapted to utilize more economical non-phosphorylated sugars as substrates (Mascotti et al. 2018).
The F420-reducing secondary alcohol dehydrogenase Adf is the most promising target for F420H2 regeneration on an industrial scale. Adf is a relatively small (37 kDa) single chain enzyme produced by thermostable organisms, which can utilize inexpensive secondary alcohols like isopropanol to reduce F420 with a reasonable catalytic efficiency (Bleicher and Winter 1991). The product of this reaction is a volatile ketone (e.g. acetone), which can be readily separated from the reaction product. The standard potential for the reduction of acetone to isopropanol is −290 mV, higher than that of F420/F420H2 (−340 mV; Thauer, Jungermann and Decker 1977; Jacobson and Walsh 1984), meaning that a relatively high concentration of substrate (>100 mM) would be needed to ensure efficient F420 reduction. However, Adf has been shown experimentally to tolerate isopropanol up to 100 mM with no significant reduction in activity (Martin et al. 2020). Relatively high concentrations of secondary alcohols are often well tolerated by enzymes and this tolerance could be improved for F420H2-dependent reductases by protein engineering (Doukyu and Ogino 2010). Moreover, the addition of secondary alcohols could increase the solubility of enzyme substrates with poor solubility. In summary, Adf is a simple protein with high stability, that can be produced recombinantly, has good catalytic efficiency, an inexpensive substrate and produces a volatile product. As such, of the currently characterized F420 reducing enzymes, it is the only candidate that fulfills all the above criteria for industrial applications.
Discovery and engineering of F420H2-dependent reductases
The range of biological reduction reactions performed by F420-dependent enzymes discussed above indicates their potential for performing reduction reactions. The diversity of molecules identified as physiological substrates for F420H2-dependent reductases, encompassing large and small soluble molecules, as well as hydrophobic molecules and lipids, also reflects their versatility as biocatalysts (Figs 7 and 8; Wang et al. 2013; Purwantini, Daniels and Mukhopadhyay 2016; Greening et al. 2017; Lee et al. 2020; Steiningerova et al. 2020; Tao et al. 2020). Furthermore, the abundance of predicted F420-dependent enzymes encoded in microbial genomes indicates that currently-characterized F420-mediated reactions represent a small subset of those that exist in nature (Ahmed et al. 2015; Ney et al. 2017a; Mascotti et al. 2018). This indicates that there is a wealth of F420-dependent enzymes with potential for use in industrial catalysis.
The use of F420H2-dependent reductases to perform industrially important reduction reactions currently represents the most promising application of F420 to industrial processes (Greening et al. 2017; Mathew et al. 2018; Drenth, Trajkovic and Fraaije 2019). F420H2-dependent reductases can reduce a range of activated alkenes (quinones, coumarins, enones, enals, pyrones and pyrans), unsaturated nitrogen-containing compounds (imines, enamines and nitrobenzenes) and secondary alcohols (Figs 7, 8 and 18; Taylor et al. 2010; Jirapanjawat et al. 2016; Greening et al. 2017; Mathew et al. 2018; Martin et al. 2020). Additionally, reactions performed by F420H2-dependent reductases tend to be stereoselective, particularly for larger, more complex substrates (Mathew et al. 2018; Martin et al. 2020), leading to the creation of chiral products that are often essential for use as pharmaceutical drugs or agricultural pesticides (Patel 2001; Nguyen, He and Pham-Huy 2006; Sekhon 2009). In addition to the use of F420 for substrate reduction, F420-reducing dehydrogenases may also prove useful for performing oxidation during chemical synthesis. The use of fHMAD by pathogenic mycobacteria to oxidatively generate ketomycolic acids provides proof of concept for this (Fig. 7C; Purwantini and Mukhopadhyay 2013). F420-reducing dehydrogenases could potentially be employed for substrate oxidation, coupled with F420H2-dependent reductases, to yield useful oxidation and reduction products from a single process.
Figure 18.
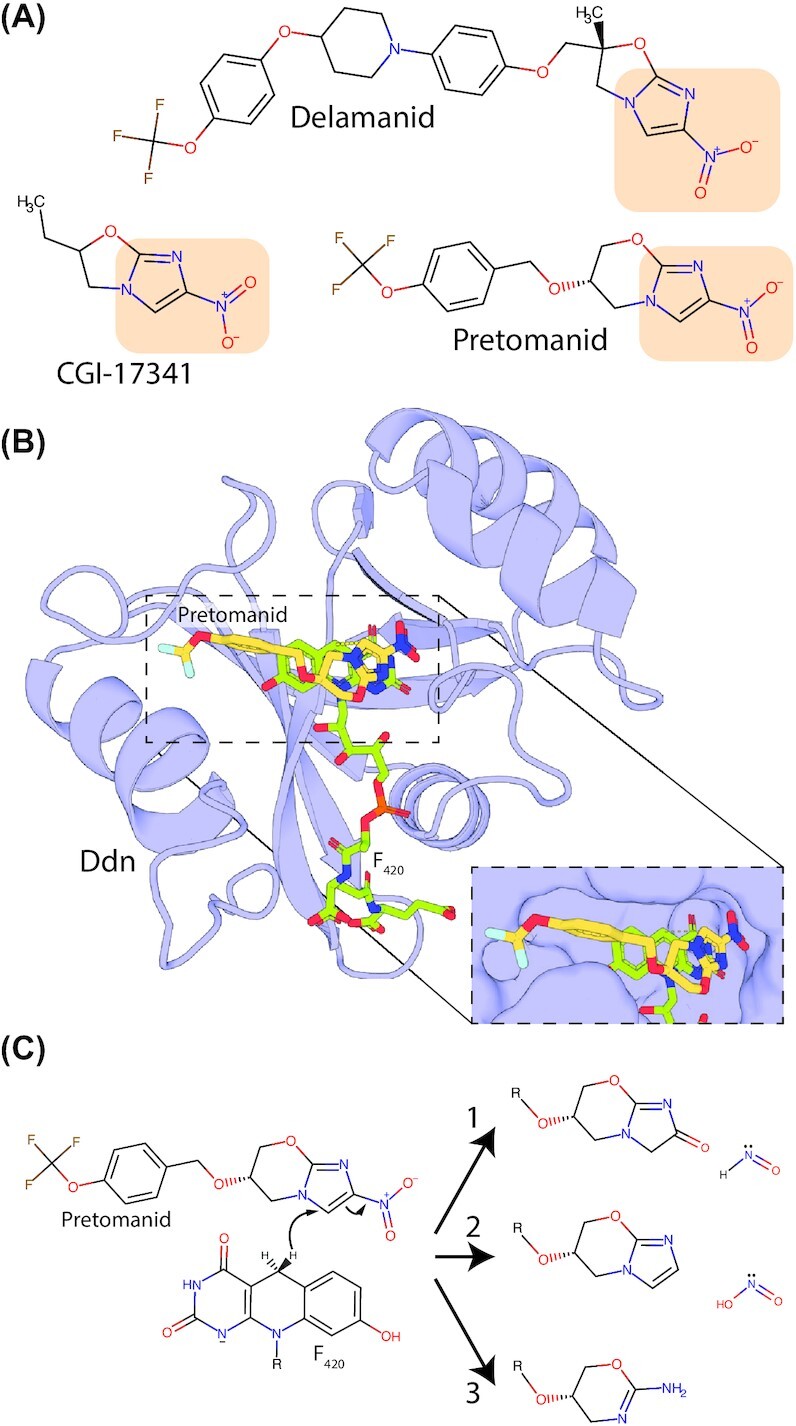
Nitroimidazole prodrugs effective against M. tuberculosis and their activation by Ddn. (A) Structure of nitroimidazole-containing prodrugs developed for tuberculosis treatment. Delamanid and pretomanid were recently approved for the treatment of M. tuberculosis infection, while CGI-17341 was abandoned due to toxicity concerns. The nitroimidazole functional group is highlighted in orange. (B) The complex between pretomanid and its activating enzyme Ddn from M. tuberculosiswas generated by molecular docking using AutoDock Vina (Trott and Olson 2010). The proximity between the nitroimidazole group of pretomanid and the hydride transferring C5 carbon of F420 is shown in the inset panel. (C) Proposed products for the breakdown of pretomanid following reduction by Ddn, full reaction schemes leading to product generation refer to Singh et al. (2008).
Some work has been performed characterizing the activity, substrate specificity and product stereochemistry of actinobacterial F420H2-dependent reductases from the diverse FDOR-A and FDOR-B families (Ahmed et al. 2015; Greening et al. 2017; Mathew et al. 2018). FDORs are small single-chain proteins that are relatively easy to produce recombinantly, making them good targets for industrial catalysis (Ahmed et al. 2015, 2016; Greening et al. 2017; Mathew et al. 2018). Initial work characterizing these enzymes showed that they exhibit significant levels of promiscuous activity towards aflatoxins and some other coumarin derivates, leading to the reductive hydrolysis of these compounds (Taylor et al. 2010; Lapalikar et al. 2012). Subsequent work showed that purified FDORs are capable of reducing a range of quinones, coumarins, enones, enals, pyrones, pyrans and triarylmethane dyes, with activity and substrate specificity varying widely between enzymes (Fig. 17 and Table 2; Greening et al. 2017; Mathew et al. 2018). FDOR-A family enzymes investigated displayed relatively high levels of activity towards quinones, in line with their proposed physiological role as menaquinone reductases (Ahmed et al. 2015; Lee et al. 2020). Mathew et al. showed that three diverse actinobacterial FDOR-A enzymes exhibited high levels of enantioselectivity towards a panel of α/β-unsaturated ketones and aldehydes, as well as regioselectivity towards a benzyl-denial compound (Mathew et al. 2018). The observed enantioselectivity of these enzymes towards several substrates was the opposite of that observed when the substrates were reduced by OYEs, indicating that FDORs could provide stereochemical flexibility for enzymatic catalysis (Mathew et al. 2018). However, the catalytic rates of purified FDORs towards these substrates, where reported, are low, meaning that significant engineering is required to render them suitable for industrial catalysis (Jirapanjawat et al. 2016; Greening et al. 2017; Mathew et al. 2018). Interestingly, a panel of diverse FDORs showed no activity against compounds containing functional groups (nitroimidazoles and imines) known to be reduced by F420H2-dependent FDORs in a physiological context (Greening et al. 2017). This observation, combined with the vastly different activities observed for FDORs towards different substrates with the same functional group, indicates that protein-substrate interactions are important for determining enzyme activity and significant engineering will be required to adapt the substrate binding sites of these enzymes to industrially relevant substrates.
Figure 17.

Compounds reduced by purified F420-dependent enzymes. (A) Classes of compounds shown to be reduced by purified F420-dependent enzymes. The reduced bond is highlighted in orange for each compound. The enzymes tested, as well as their substrate range and relative activity levels, are provided in Table 5. (B) Proposed reaction schemes for the reduction of menaquinone and malachite green by F420H2-dependent reductases of the FDOR-A superfamily. In both cases, initial hydride transfer to one carbon atom of an activated alkene is followed by tautomerization yielding the final reaction product.
Further investigation into the mechanisms of substrate binding and reduction by FDORs will assist in engineering enzymes with higher catalytic efficiency and predicting product stereochemistry. It has been proposed that proton donation to the F420-substrate after initial hydride transfer from F420 occurs from N1 of F420H2 (Fig. 1B; Shah et al. 2019). If true, both hydrogenation events occur on the same face of the activated alkene substrate, leading to cis-hydrogenation when a disubstituted alkene is reduced by the enzyme. This is in contrast to OYEs where trans-hydrogenation products are formed (Hollmann, Opperman and Paul 2020). However, cis-hydrogenation by FDORs has not been demonstrated experimentally, and spectroscopic and computational analysis suggests that F420H− (in which N1 is deprotonated) is the form of the cofactor utilized by the FDOR-A Ddn (Mohamed et al. 2016a). If F420H− is the general physiological form of the cofactor used by FDORs, then substrate protonation must instead proceed from solvent or an enzyme sidechain (Mohamed et al. 2016a,b; Greening et al. 2017). A better understanding of the structural and biochemical basis for the binding of physiological and industrially relevant substrates to FDORs is also required. No crystal structures of FDOR-substrate complexes are available, though some docking analysis has been performed, indicating residues important for substrate binding (Ahmed et al. 2015; Greening et al. 2017; Lee et al. 2020). Systematic structural analysis of diverse FDORs in complex with catalytically relevant substrates will facilitate the identification of key residues involved in substrate binding and catalysis.
LLHTs, another major family of F420-dependent enzymes, are also a promising source of enzymes for industrial applications. Fgd and Adf, F420-reducing dehydrogenases from this family, have been utilized for F420H2 generation at a laboratory scale (Jirapanjawat et al. 2016; Mascotti et al. 2018; Drenth, Trajkovic and Fraaije 2019). Recently, Martin et al. utilized Adf for the stereoselective reduction of diverse secondary ketones, with F420H2 generated by Fgd using G6P as a substrate, or using Adf to drive F420 both reduction and oxidation by including high concentrations of isopropanol in the reaction. While this study provides an important proof of concept for the generation of chiral alcohols using F420-dependent enzymes, Adf exhibited low apparent activity towards these substrates (Martin et al. 2020). Other F420-dependent LLHT reductases have yet to be investigated for their catalytic potential. However, LLHTs are utilized by bacteria in the biosynthesis of diverse molecules, including mycolic acids, 4-alkyl-L-proline intermediates and lexapeptides, and in the reductive degradation of picrate (Purwantini and Mukhopadhyay 2013; Purwantini, Daniels and Mukhopadhyay 2016; Steiningerova et al. 2020; Tao et al. 2020). The ability of LLHTs to perform these diverse reactions, both on large polar and hydrophobic molecules, suggests their biosynthetic potential. Notably, the TIM-barrel fold of LLHTs is unrelated to that of FDORs, possessing a structurally divergent substrate-binding pocket (Fig. 4; Mascotti et al. 2018). As such, LLHTs are likely to provide complementary substrate specificity and stereoselectivity to FDORs. Like FDORs, sequenced microbial genomes contain a wealth of putative F420-dependent LLHTs with unknown function, many of which are contained within hypothetical secondary metabolite BCGs (Selengut and Haft 2010; Mascotti et al. 2018; Steiningerova et al. 2020)
In summary, while our current understanding of F420-dependent enzymes sets the stage for the use of F420 in industrial catalysis, considerable further work is required to develop enzymes suitable for such processes. To realize this goal these enzymes will require a high level of activity towards economically relevant substrates, with favorable kinetic parameters. Also, they will likely need to display high levels of stereoselectivity, yielding a commercially relevant enantiomer. Further, for use in synthetic chemistry, the enzymes will need to be robust, able to withstand relatively harsh extremes of temperature, pH, ionic strength and concentrations of non-polar solvents. Currently, no enzymes with these properties have been described or developed. A possibly promising strategy for the development of such enzymes would be to identify commercially relevant substrates, with chemistry amenable to reduction or oxidation by F420-dependent enzymes, which are recalcitrant to currently available synthetic chemical processes. The wealth of F420-dependent enzymes present in microbial genomes could be screened for enzymes capable of reducing these substrates to some degree. These enzymes could be then subjected to rigorous structural and biochemical characterization, combined with concerted protein engineering efforts, to produce enzymes suitable for the reduction of these substrates on an industrial scale.
Ramifications and applications of microbial utilization of F420
In addition to the importance of F420 in microbial physiology and its potential applications for chemical synthesis, our understanding of the diverse role of the cofactor has wider significance for improving human health and sustainability. As discussed below, hydrogenotrophic methanogens that rely on F420 are an important source of global greenhouse gases, while in M. tuberculosis F420 is important for the activation of nitroimidazole prodrugs and plays an insufficiently characterized role in survival in the host. Our recently acquired knowledge on the biosynthesis and roles of F420 can be utilized to develop methanogenesis inhibitors and antitubercular drugs, as well as to predict and alleviate nitroimidazole resistance.
Methane mitigation through inhibition of F420 dependent enzymes
F420-dependent hydrogenotrophic methanogens, notably Methanobrevibacter gottschalkii and Methanobrevibacter ruminantium, are core members of the foregut microbiota of ruminants (Leahy et al. 2010; Henderson et al. 2015). Methane emitted by ruminants is a significant driver of global warming, with ruminant methane emissions accounting for approximately 40% of global methane emissions and 5% of global greenhouse gas emissions (Greening et al. 2019). Bacteria and archaea that utilize alternative acetogenic or respiratory pathways for H2 oxidation, independently of F420, are abundant in the rumen (Greening et al. 2019). Thus, strategies to inhibit or outcompete hydrogenotrophic methanogens should foster a microbial community that produces less methane, reducing the greenhouse gas emissions from livestock farming (Morgavi et al. 2010; Greening et al. 2019). Potent methyl-CoM reductase inhibitors, such as 3-nitrooxypropanol, reduce methane production without compromising animal health and productivity (Hristov et al. 2015; Duin et al. 2016). Given hydrogenotrophic methanogenesis requires F420, strategies that inhibit F420 production or its use by F420-dependent enzymes will also inhibit the growth of these archaea. While specific strategies for the inhibition of F420 biosynthesis or dependent enzymes in the rumen have not been reported, several studies indicate this may be possible. The pterin lumazine inhibits methanogen growth and methane formation in pure culture, possibly through its structural similarity to F420 (Nagar-Anthal et al. 1996), although this effect was less significant in mixed culture (Ungerfeld et al. 2004). Recently, in silico screening identified inhibitors of Fno from the methanogen Methanobrevibacter smithii, some of which bind the enzyme with affinity in the nM range. Some of these inhibitors are non-toxic dietary supplements and could be readily investigated for their ability to reduce methane emissions by inhibiting F420-dependent enzymes (Cuccioloni et al. 2020). If effective, inhibitors of F420 dependent pathways could be provided as dietary supplements, possibly in conjunction with other strategies to inhibit the growth of hydrogenotrophic methanogens or promote the growth of other hydrogenotrophs in the rumen.
Nitroimidazole prodrug activation in M. tuberculosis
It has been recognized since the late 1980s that compounds containing the nitroimidazole functional group can display potent antitubercular activity (Nagarajan et al. 1989; Liu et al. 2018). Early attempts to develop nitroimidazole-containing molecules for tuberculosis treatment, including the compound CGI-17341 (Ashtekar et al. 1993; Fig. 18A), failed due to toxicity concerns. However, concerted drug development efforts have yielded two compounds with potent antitubercular activities and acceptable safety profiles. These compounds, named delamanid and pretomanid (Fig. 18A), were both recently approved for the treatment of multidrug-resistant tuberculosis (MDR) as a combination therapy (Liu et al. 2018; Keam 2019). Delamanid was approved in 2014 by the European Medicines Agency for use in an appropriate combination regimen for MDR tuberculosis treatment and is subject to ongoing safety and efficacy studies (Ryan and Lo 2014; von Groote-Bidlingmaier et al. 2019). Pretomanid was approved in 2019 by the U.S. Food & Drug Administration for treatment of MDR or extensively drug-resistant tuberculosis (XDR), as part of a three-drug ‘BPaL’ regime, also including bedaquiline and linezolid (Keam 2019). Safety and efficacy trials indicate pretomanid, administered in combination with bedaquiline, moxifloxacin and pyrazinamide, is also effective in the treatment of MDR and XDR tuberculosis, although this regime is awaiting regulatory approval (Tweed et al. 2019).
Nitroimidazole-containing compounds act as prodrugs in the treatment of tuberculosis and are reductively activated in the M. tuberculosis cell by the aforementioned FDOR-A enzyme Ddn (Fig. 18B; Cellitti et al. 2012; Fujiwara et al. 2018). The activation of delamanid and pretomanid occurs through the promiscuous activity of Ddn, which appears to play a physiological role in the reduction of menaquinone (Lee et al. 2020). The reduction of the nitroimidazole functional group by Ddn is thought to lead to several reaction products, including the release of HNO and HNO2 and the formation of des-nitro forms of the drugs (Fig. 18C; Singh et al. 2008). While the precise details of the mechanism of action of delamanid and pretomanid towards M. tuberculosis remain to be elucidated, evidence supports a role for the release of reactive nitrogen species in their toxicity (Manjunatha et al. 2006; Singh et al. 2008; Manjunatha, Boshoff and Barry 2009). Pretomanid also selectively inhibits the synthesis of ketomycolic acids in the M. tuberculosis, potentially through the direct or indirect inhibition of the F420-reducing dehydrogenase fHMAD (Stover et al. 2000; Purwantini and Mukhopadhyay 2013). Further, global metabolomic analysis of pretomanid treated M. smegmatis identified the accumulation of the toxic metabolite methylglyoxal, indicating that metabolic poisoning may also play a role in the antitubercular activity of nitroimidazole drugs (Baptista et al. 2018). As nitroimidazole drugs are effective against M. tuberculosis under both aerobic and anaerobic conditions, and their activation by Ddn leads to several reactive intermediates and reaction products, the mechanism of action of these compounds is likely complex and multifaceted (Singh et al. 2008; Mukherjee and Boshoff 2011).
Resistance to delamanid and pretomanid in M. tuberculosis is imparted by mutations that prevent the production of F420 (via loss of functional FbiA, FbiB, FbiC or FbiD) or its reduction to F420H2 (via loss of functional Fgd; Choi et al. 2001; Manjunatha et al. 2006; Jing et al. 2019; Lee et al. 2020). Additionally, mutations that result in either the complete loss of Ddn function or loss of its promiscuous activity towards the prodrugs also result in resistance (Lee et al. 2020). The latter finding is of concern for the longevity of delamanid and pretomanid for treatment of tuberculosis. Particularly concerning is that clinical M. tuberculosis isolates with mutations in Ddn that abolish its activity towards delamanid and pretomanid, but not its physiological substrate menaquinone, have been identified (Lee et al. 2020; Rifat et al. 2020). A number of these isolates were from patients that had not been treated with delamanid and pretomanid, suggesting that inherently resistant strains of M. tuberculosis exist (Yang et al. 2018; Lee et al. 2020). Interestingly, some mutations in Ddn result in resistance to pretomanid but not delamanid (Lee et al. 2020), likely due to the differences in their interactions with the Ddn substrate-binding pocket. Based on this observation, a robust understanding of Ddn substrate binding may allow for the deployment of patient-tailored prodrug variants less susceptible to polymorphisms in Ddn. As discussed in the next section, the effect of the loss of F420 on the virulence and transmissibility of M. tuberculosis as a result of nitroimidazole treatment remains uncertain.
Targeting F420 for the development of antitubercular drugs
The role of F420 in activating pretomanid and delamanid has rekindled interest in the role of F420 in mycobacterial physiology (Cellitti et al. 2012; Haver et al. 2015; Fujiwara et al. 2018; Lee et al. 2020; Rifat et al. 2020). However, despite recent progress in this area, we lack a comprehensive understanding of the importance of F420 in mycobacterial physiology. This is especially true for mycobacterial pathogens like M. tuberculosis, in part due to the difficulty in working with this slow-growing and highly pathogenic bacterium (Cole et al. 1998). However, phenotypes associated with the loss of F420 production discussed above strongly suggest that the cofactor plays a role in the ability of M. tuberculosis to cause disease (Gurumurthy et al. 2013; Jirapanjawat et al. 2016; Lee et al. 2020; Rifat et al. 2020). These observations, together with the ubiquity of F420 in mycobacteria, the abundance of F420-dependent enzymes in M. tuberculosis and the absence of the cofactor from human cells, make the processes that produce or use F420 potential targets for the development of antimicrobial compounds. One method of targeting F420 would be the development of compounds that inhibit the enzymes responsible for its biosynthesis (i.e. FbiA, FbiB, FbiC and FbiD) or reduction (Fgd; Bashiri et al. 2008, 2019). Crystal structures are available for the majority of these enzymes, facilitating inhibitor design (Bashiri et al. 2008, 2019; Grinter et al. 2020). An alternative approach would be the development of F420 analogs that inhibit F420-dependent enzymes (Eirich, Vogels and Wolfe 1978). However, to make the considerable effort required for the identification and optimization of F420 biosynthesis inhibitors attractive, a better understanding of the importance of the cofactor for virulence is required.
The role of F420-dependent enzymes in reductively detoxifying antimicrobial compounds is also of interest for M. tuberculosis treatment (Jirapanjawat et al. 2016; Rifat et al. 2020). Profiling the antibiotic sensitivity of F420-deficient mutants of mycobacterial pathogens may identify antibiotics that selectively display activity against these strains. There is some evidence that loss of F420 leads to a heightened sensitivity to the antibiotics isoniazid, moxifloxacin and clofazimine in M. tuberculosis (Gurumurthy et al. 2013; Rifat et al. 2020). Given that loss of F420 production is known to mediate resistance to pretomanid and delamanid (Haver et al. 2015; Jing et al. 2019; Lee et al. 2020), antibiotics that are more effective against F420 deficient mutants may be useful in combination with these drugs to reduce or mitigate the development of resistance. Preliminary analysis of F420-dependent LLHTs in pathogenic mycobacteria demonstrates they play an important role in outer-envelope lipid biosynthesis. Outer envelope lipids, like PDIMs and ketomycolic acids, are important for mycobacterial virulence (Purwantini and Mukhopadhyay 2013; Purwantini, Daniels and Mukhopadhyay 2016). Growing evidence indicates that these outer envelope lipids constitute a second outer membrane in mycobacteria (Hoffmann et al. 2008; Bansal-Mutalik and Nikaido 2014). In M. tuberculosis this membrane contains high concentrations of PDIM, which contribute to the impermeability of this barrier and the antibiotic resistance of this species (Wang et al. 2020). As such, a systematic understanding of the role of F420-dependent enzymes in outer-envelope lipid biosynthesis will inform future efforts to combat pathogenic mycobacteria through inhibition of this process.
OUTLOOK
When we first reviewed this topic five years ago (Greening et al. 2016), we noted multiple knowledge gaps that have since been addressed. These included: resolving the chemical steps and structural basis of F420 biosynthesis (Bashiri et al. 2019; Braga et al. 2019; Grinter et al. 2020); surveying the distribution of F420 across different taxa and ecosystems (Ney et al. 2017a); investigating the chemistry of the F420 headgroup and tail to catalysis (Mohamed et al. 2016a; Ney et al. 2017b); and enabling F420-dependent industrial biocatalysis through achieving heterologous cofactor production (Bashiri et al. 2019; Braga et al. 2019; Ney 2019) and characterizing promising F420-dependent oxidoreductases (Greening et al. 2017; Mascotti et al. 2018; Mathew et al. 2018; Drenth, Trajkovic and Fraaije 2019; Martin et al. 2020). Some of these lines of investigation resulted in unexpected findings, most notably that F420 biosynthesis genes are extremely widely distributed (Ney et al. 2017a; Table S2, Supporting Information), the biosynthesis pathway has multiple variants and was misannotated in bacteria (Bashiri et al. 2019; Braga et al. 2019, 2020; Grinter et al. 2020) and some bacteria produce entirely novel variants of this cofactor (Braga et al. 2019).
Despite these important insights, we still lack a systematic understanding of the physiological role of the cofactor. Unanswered questions include why mycobacteria encode a multitude of predicted FDORs and LLHTs, and why newly identified F420 producers such as Proteobacteria and Chloroflexi synthesize this cofactor. Further research is needed to resolve whether lineages such as the TACK and Asgard archaea, Firmicutes and Tectomicrobia do produce F420 as predicted, and if so, which variants do they make and through which pathways? Other knowledge gaps that could be addressed in coming years include a more detailed understanding of the evolution of the F420 biosynthetic pathway, structural resolution of FO biosynthesis and newly discovered F420-dependent methanogen enzymes, as well as the long-standing question of which enzyme mediates 2-phospholactate production in methanogens. As we detailed in the final section of this review, there is also ample potential to translate this fundamental knowledge to address medical, environmental and industrial challenges to improve human health and sustainability. Exactly 50 years since its discovery by Wolfe and colleagues, it is increasingly clear that F420 is a widespread and versatile cofactor, fundamental to the physiology of many bacteria and archaea.
Contributor Information
Rhys Grinter, Department of Microbiology, Monash Biomedicine Discovery Institute, Monash University, Clayton, VIC 3800, Australia.
Chris Greening, Department of Microbiology, Monash Biomedicine Discovery Institute, Monash University, Clayton, VIC 3800, Australia.
FUNDING
This work was supported by an NHMRC EL2 Investigator Grant (APP1178715; awarded to C.G.), an NHMRC New Investigator Grant (APP5191146; awarded to C.G.), an NHMRC grant (APP1139832; awarded to C.G.) and an NHMRC EL1 Investigator Grant (APP1197376; Awarded to R.G.).
Conflicts of Interest
None declared.
REFERENCES
- Abken H-J, Deppenmeier U. Purification and properties of an F420H2 dehydrogenase from Methanosarcina mazei Gö1. FEMS Microbiol Lett. 1997;154:231–7. [Google Scholar]
- Adam PS, Borrel G, Gribaldo S. An archaeal origin of the Wood–Ljungdahl H4MPT branch and the emergence of bacterial methylotrophy. Nat Microbiol. 2019;4:2155–63. [DOI] [PubMed] [Google Scholar]
- Afting C, Hochheimer A, Thauer R. Function of H2-forming methylenetetrahydromethanopterin dehydrogenase from Methanobacterium thermoautotrophicum in coenzyme F420 reduction with H2. Arch Microbiol. 1998;169:206–10. [DOI] [PubMed] [Google Scholar]
- Ahmed FH, Carr PD, Lee BMet al. Sequence–structure–function classification of a catalytically diverse oxidoreductase superfamily in Mycobacteria. J Mol Biol. 2015;427:3554–71. [DOI] [PubMed] [Google Scholar]
- Ahmed FH, Mohamed AE, Carr PDet al. Rv2074 is a novel F420H2-dependent biliverdin reductase in Mycobacterium tuberculosis. Protein Sci. 2016;25:1692–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albertsen M, Hugenholtz P, Skarshewski Aet al. Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat Biotechnol. 2013;31:533–8. [DOI] [PubMed] [Google Scholar]
- Allegretti M, Mills DJ, McMullan Get al. Atomic model of the F420-reducing [NiFe] hydrogenase by electron cryo-microscopy using a direct electron detector. Elife. 2014;3:e01963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arshad A, Speth DR, de Graaf RMet al. A metagenomics-based metabolic model of nitrate-dependent anaerobic oxidation of methane by methanoperedens-like archaea. Front Microbiol. 2015;6:e1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashtekar DR, Costa-Perira R, Nagrajan Ket al. In vitro and in vivo activities of the nitroimidazole CGI 17341 against Mycobacterium tuberculosis. Antimicrob Agents Chemother. 1993;37:183–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwood G, Altermann E, Kelly Wet al. Exploring rumen methanogen genomes to identify targets for methane mitigation strategies. Anim Feed Sci Technol. 2011;166-167:65–75. [Google Scholar]
- Aufhammer SW, Warkentin E, Berk Het al. Coenzyme binding in F420-dependent secondary alcohol dehydrogenase, a member of the bacterial luciferase family. Structure. 2004;12:361–70. [DOI] [PubMed] [Google Scholar]
- Aufhammer SW, Warkentin E, Ermler Uet al. Crystal structure of methylenetetrahydromethanopterin reductase (Mer) in complex with coenzyme F420: architecture of the F420/FMN binding site of enzymes within the nonprolyl cis-peptide containing bacterial luciferase family. Protein Sci. 2005;14:1840–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacher A, Eberhardt S, Fischer Met al. Biosynthesis of vitamin B2 (riboflavin). Annu Rev Nutr. 2000;20:153–67. [DOI] [PubMed] [Google Scholar]
- Bair TB, Isabelle DW, Daniels L. Structures of coenzyme F420 in Mycobacterium species. Arch Microbiol. 2001;176:37–43. [DOI] [PubMed] [Google Scholar]
- Bansal-Mutalik R, Nikaido H. Mycobacterial outer membrane is a lipid bilayer and the inner membrane is unusually rich in diacyl phosphatidylinositol dimannosides. Proc Natl Acad Sci. 2014;111:4958–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bapteste É, Brochier C, Boucher Y. Higher-level classification of the Archaea: evolution of methanogenesis and methanogens. Archaea. 2005;1:353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baptista R, Fazakerley DM, Beckmann Met al. Untargeted metabolomics reveals a new mode of action of pretomanid (PA-824). Sci Rep. 2018;8:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber RD, Zhang L, Harnack Met al. Complete genome sequence of Methanosaeta concilii, a specialist in aceticlastic methanogenesis. J Bacteriol. 2011;193:3668–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baresi L, Wolfe RS. Levels of coenzyme F420, coenzyme M, hydrogenase, and methylcoenzyme M methylreductase in acetate-grown Methanosarcina. Appl Environ Microbiol. 1981;41:388–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron SF, Ferry JG. Reconstitution and properties of a coenzyme F420-mediated formate hydrogenlyase system in Methanobacterium formicicum. J Bacteriol. 1989;171:3854–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashiri G, Antoney J, Jirgis ENet al. A revised biosynthetic pathway for the cofactor F420 in prokaryotes. Nat Commun. 2019;10:1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashiri G, Perkowski EF, Turner APet al. Tat–dependent translocation of an F420–binding protein of Mycobacterium tuberculosis. PLoS One. 2012;7:e45003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashiri G, Rehan AM, Greenwood DRet al. Metabolic engineering of cofactor F420 production inMycobacterium smegmatis. PLoS One. 2010;5:e15803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashiri G, Rehan AM, Sreebhavan Set al. Elongation of the poly-γ-glutamate tail of F420 requires both domains of the F420: γ-glutamyl ligase (FbiB) of Mycobacterium tuberculosis. J Biol Chem. 2016;291:6882–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashiri G, Squire CJ, Moreland NJet al. Crystal structures of F420-dependent glucose-6-phosphate dehydrogenase FGD1 involved in the activation of the anti-tuberculosis drug candidate PA-824 reveal the basis of coenzyme and substrate binding. J Biol Chem. 2008;283:17531–41. [DOI] [PubMed] [Google Scholar]
- Beal EJ, House CH, Orphan VJ. Manganese-and iron-dependent marine methane oxidation. Science. 2009;325:184–7. [DOI] [PubMed] [Google Scholar]
- Becraft ED, Woyke T, Jarett Jet al. Rokubacteria: genomic giants among the uncultured bacterial phyla. Front Microbiol. 2017;8:2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begley TP. 7.01 - Overview and introduction. In: Liu H-W, Mander L (eds.) Comprehensive Natural Products II. Oxford: Elsevier, 2010, 1–2. [Google Scholar]
- Berghuis BA, Yu FB, Schulz Fet al. Hydrogenotrophic methanogenesis in archaeal phylum Verstraetearchaeota reveals the shared ancestry of all methanogens. Proc Natl Acad Sci. 2019;116:5037–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berk H, Thauer RK. Function of coenzyme F420-dependent NADP reductase in methanogenic archaea containing an NADP-dependent alcohol dehydrogenase. Arch Microbiol. 1997;168:396–402. [DOI] [PubMed] [Google Scholar]
- Bertrand JA, Auger G, Fanchon Eet al. Crystal structure of UDP-N-acetylmuramoyl-L-alanine: d-glutamate ligase from Escherichia coli. EMBO J. 1997;16:3416–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattarai S, Cassarini C, Lens PNL. Physiology and distribution of archaeal methanotrophs that couple anaerobic oxidation of methane with sulfate reduction. Microbiol Mol Biol Rev. 2019;83:e00074–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswal BK, Au K, Cherney MMet al. The molecular structure of Rv2074, a probable pyridoxine 5'-phosphate oxidase from Mycobacterium tuberculosis, at 1.6 A resolution. Acta Crystallogr Sect F. 2006;62:735–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björnsson L, Hugenholtz P, Tyson GWet al. Filamentous Chloroflexi (green non-sulfur bacteria) are abundant in wastewater treatment processes with biological nutrient removal. Microbiology. 2002;148:2309–18. [DOI] [PubMed] [Google Scholar]
- Bleicher K, Winter J. Purification and properties of F420- and NADP+-dependent alcohol dehydrogenases of Methanogenium liminatans and Methanobacterium palustre, specific for secondary alcohols. Eur J Biochem. 1991;200:43–51. [DOI] [PubMed] [Google Scholar]
- Boetius A, Ravenschlag K, Schubert CJet al. A marine microbial consortium apparently mediating anaerobic oxidation of methane. Nature. 2000;407:623–6. [DOI] [PubMed] [Google Scholar]
- Bogan K, Brenner C. Biochemistry: Niacin/NAD (P). 2013. [Google Scholar]
- Borrel G, O'Toole PW, Harris HMBet al. Phylogenomic data support a seventh order of methylotrophic methanogens and provide insights into the evolution of methanogenesis. Genome Biol Evol. 2013;5:1769–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bown L, Altowairish MS, Fyans JKet al. Production of the Streptomyces scabies coronafacoyl phytotoxins involves a novel biosynthetic pathway with an F420-dependent oxidoreductase and a short-chain dehydrogenase/reductase. Mol Microbiol. 2016;101:122–35. [DOI] [PubMed] [Google Scholar]
- Braga D, Hasan M, Kröber Tet al. Redox coenzyme F420 biosynthesis in thermomicrobia involves reduction by stand-alone nitroreductase superfamily enzymes. Appl Environ Microbiol. 2020;86:e00457–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braga D, Last D, Hasan Met al. Metabolic pathway rerouting in Paraburkholderia rhizoxinica evolved long-overlooked derivatives of coenzyme F420. ACS Chem Biol. 2019;14:2088–94. [DOI] [PubMed] [Google Scholar]
- Brochier-Armanet C, Forterre P, Gribaldo S. Phylogeny and evolution of the Archaea: one hundred genomes later. Curr Opin Microbiol. 2011;14:274–81. [DOI] [PubMed] [Google Scholar]
- Brochier C, Forterre P, Gribaldo S. Archaeal phylogeny based on proteins of the transcription and translation machineries: tackling the Methanopyrus kandleri paradox. Genome Biol. 2004;5:R17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brüggemann H, Falinski F, Deppenmeier U. Structure of the F420H2:quinone oxidoreductase of Archaeoglobus fulgidus. Eur J Biochem. 2000;267:5810–4. [DOI] [PubMed] [Google Scholar]
- Buckel W, Thauer RK. Energy conservation via electron bifurcating ferredoxin reduction and proton/Na+ translocating ferredoxin oxidation. Biochimica et Biophysica Acta (BBA) Bioenergetics. 2013;1827:94–113. [DOI] [PubMed] [Google Scholar]
- Bulzu P-A, Andrei A-Ş, Salcher MMet al. Casting light on Asgardarchaeota metabolism in a sunlit microoxic niche. Nat Microbiol. 2019;4:1129–37. [DOI] [PubMed] [Google Scholar]
- Bäumer S, Ide T, Jacobi Cet al. The F420H2 dehydrogenase from Methanosarcina mazei Is a redox-driven proton pump closely related to NADH dehydrogenases. J Biol Chem. 2000;275:17968–73. [DOI] [PubMed] [Google Scholar]
- Bäumer S, Murakami E, Brodersen Jet al. The F420H2: heterodisulfide oxidoreductase system from Methanosarcina species: 2-hydroxyphenazine mediates electron transfer from F420H2 dehydrogenase to heterodisulfide reductase. FEBS Lett. 1998;428:295–8. [DOI] [PubMed] [Google Scholar]
- Cai C, Leu AO, Xie G-Jet al. A methanotrophic archaeon couples anaerobic oxidation of methane to Fe(III) reduction. ISME J. 2018;12:1929–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canaan S, Sulzenbacher G, Roig-Zamboni Vet al. Crystal structure of the conserved hypothetical protein Rv1155 from Mycobacterium tuberculosis. FEBS Lett. 2005;579:215–21. [DOI] [PubMed] [Google Scholar]
- Castelle CJ, Banfield JF. Major new microbial groups expand diversity and alter our understanding of the tree of life. Cell. 2018;172:1181–97. [DOI] [PubMed] [Google Scholar]
- Ceh K, Demmer U, Warkentin Eet al. Structural basis of the hydride transfer mechanism in F420-dependent methylenetetrahydromethanopterin dehydrogenase. Biochemistry. 2009;48:10098–105. [DOI] [PubMed] [Google Scholar]
- Cellitti SE, Shaffer J, Jones DHet al. Structure of Ddn, the deazaflavin-dependent nitroreductase from Mycobacterium tuberculosis involved in bioreductive activation of PA-824. Structure. 2012;20:101–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheeseman P, Toms-Wood A, Wolfe R. Isolation and properties of a fluorescent compound, Factor 420, from Methanobacterium strain MoH. J Bacteriol. 1972;112:527–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S-C, Musat N, Lechtenfeld OJet al. Anaerobic oxidation of ethane by archaea from a marine hydrocarbon seep. Nature. 2019;568:108–11. [DOI] [PubMed] [Google Scholar]
- Choi K-P, Bair TB, Bae Y-Met al. Use of transposon Tn5367 mutagenesis and a nitroimidazopyran-based selection system to demonstrate a requirement for fbiA and fbiB in coenzyme F420 biosynthesis by Mycobacterium bovis BCG. J Bacteriol. 2001;183:7058–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi K-P, Kendrick N, Daniels L. Demonstration that fbiC is required by Mycobacterium bovis BCG for coenzyme F420 and FO biosynthesis. J Bacteriol. 2002;184:2420–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole S, Brosch R, Parkhill Jet al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–44. [DOI] [PubMed] [Google Scholar]
- Conrad R. The global methane cycle: recent advances in understanding the microbial processes involved. Environ Microbiol Rep. 2009;1:285–92. [DOI] [PubMed] [Google Scholar]
- Costa KC, Wong PM, Wang Tet al. Protein complexing in a methanogen suggests electron bifurcation and electron delivery from formate to heterodisulfide reductase. Proc Natl Acad Sci. 2010;107:11050–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox JS, Chen B, McNeil Met al. Complex lipid determines tissue-specific replication of Mycobacterium tuberculosis in mice. Nature. 1999;402:79–83. [DOI] [PubMed] [Google Scholar]
- Cuccioloni M, Bonfili L, Cecarini Vet al. Structure/activity virtual screening and in vitro testing of small molecule inhibitors of 8-hydroxy-5-deazaflavin: NADPH oxidoreductase from gut methanogenic bacteria. Sci Rep. 2020;10:e13150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels L, Bakhiet N, Harmon K. Widespread distribution of a 5-deazaflavin cofactor in Actinomyces and related bacteria. Syst Appl Microbiol. 1985;6:12–7. [Google Scholar]
- Darwin KH, Ehrt S, Gutierrez-Ramos J-Cet al. The proteasome of Mycobacterium tuberculosis is required for resistance to nitric oxide. Science. 2003;302:1963–6. [DOI] [PubMed] [Google Scholar]
- Decamps L, Philmus B, Benjdia Aet al. Biosynthesis of F0, precursor of the F420 cofactor, requires a unique two radical-SAM domain enzyme and tyrosine as substrate. J Am Chem Soc. 2012;134:18173–6. [DOI] [PubMed] [Google Scholar]
- de Poorter LMI, Geerts WJ, Keltjens JT. Hydrogen concentrations in methane-forming cells probed by the ratios of reduced and oxidized coenzyme F420. Microbiology. 2005;151:1697–705. [DOI] [PubMed] [Google Scholar]
- Deppenmeier U, Blaut M, Mahlmann Aet al. Reduced coenzyme F420: heterodisulfide oxidoreductase, a proton-translocating redox system in methanogenic bacteria. Proc Natl Acad Sci. 1990;87:9449–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deppenmeier U, Lienard T, Gottschalk G. Novel reactions involved in energy conservation by methanogenic archaea. FEBS Lett. 1999;457:291–7. [DOI] [PubMed] [Google Scholar]
- Deppenmeier U. The unique biochemistry of methanogenesis. Progress in Nucleic Acid Research and Molecular Biology. Vol. 71, Academic Press, 2002, 223–83. [DOI] [PubMed] [Google Scholar]
- De Wit L, Eker A. 8-Hydroxy-5-deazaflavin-dependent electron transfer in the extreme halophile Halobacterium cutirubrum. FEMS Microbiol Lett. 1987;48:121–5. [Google Scholar]
- Doddema HJ, Vogels GD. Improved identification of methanogenic bacteria by fluorescence microscopy. Appl Environ Microbiol. 1978;36:752–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolfing J, Mulder J-W. Comparison of methane production rate and coenzyme F420 content of methanogenic consortia in anaerobic granular sludge. Appl Environ Microbiol. 1985;49:1142–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doukyu N, Ogino H. Organic solvent-tolerant enzymes. Biochem Eng J. 2010;48:270–82. [Google Scholar]
- Drenth J, Trajkovic M, Fraaije MW. Chemoenzymatic synthesis of an unnatural deazaflavin cofactor that can fuel F420-dependent enzymes. ACS Cat. 2019;9:6435–43. [Google Scholar]
- Dubnau E, Chan J, Raynaud Cet al. Oxygenated mycolic acids are necessary for virulence of Mycobacterium tuberculosis in mice. Mol Microbiol. 2000;36:630–7. [DOI] [PubMed] [Google Scholar]
- Duin EC, Wagner T, Shima Set al. Mode of action uncovered for the specific reduction of methane emissions from ruminants by the small molecule 3-nitrooxypropanol. Proc Natl Acad Sci. 2016;113:6172–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert S, Fischer P, Knackmuss H-J. Converging catabolism of 2,4,6-trinitrophenol (picric acid) and 2,4-dinitrophenol by Nocardioides simplex FJ2-1A. Biodegradation. 2001;12:367–76. [DOI] [PubMed] [Google Scholar]
- Ebert S, Rieger P-G, Knackmuss H-J. Function of coenzyme F420 in aerobic catabolism of 2,4,6-trinitrophenol and 2, 4-dinitrophenol by Nocardioides simplex FJ2-1A. J Bacteriol. 1999;181:2669–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmondson DE, Barman B, Tollin G. Importance of the N-5 position in flavine coenzymes. Properties of free and protein-bound 5-deaza analogs. Biochemistry. 1972;11:1133–8. [DOI] [PubMed] [Google Scholar]
- Edwards T, McBride BC. New method for the isolation and identification of methanogenic bacteria. Appl Microbiol. 1975;29:540–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eicher T, Hauptmann S, Speicher A. The Chemistry of Heterocycles: Structures, Reactions, Synthesis, and Applications: John Wiley & Sons, 2013. [Google Scholar]
- Eirich LD, Dugger RS. Purification and properties of an F420-dependent NADP reductase from Methanobacterium thermoautotrophicum. Biochimica et Biophysica Acta (BBA) Gen Sub. 1984;802:454–8. [Google Scholar]
- Eirich LD, Vogels G, Wolfe R. Distribution of coenzyme F420 and properties of its hydrolytic fragments. J Bacteriol. 1979;140:20–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eirich LD, Vogels GD, Wolfe RS. Proposed structure for coenzyme F420 from Methanobacterium. Biochemistry. 1978;17:4583–93. [DOI] [PubMed] [Google Scholar]
- Eker A, Hessels J, Meerwaldt R. Characterization of an 8-hydroxy-5-deazaflavin: NADPH oxidoreductase from Streptomyces griseus. Biochimica et Biophysica Acta (BBA) Gen Sub. 1989;990:80–6. [DOI] [PubMed] [Google Scholar]
- Evans PN, Boyd JA, Leu AOet al. An evolving view of methane metabolism in the Archaea. Nat Rev Microbiol. 2019;17:219–32. [DOI] [PubMed] [Google Scholar]
- Evans PN, Parks DH, Chadwick GLet al. Methane metabolism in the archaeal phylum Bathyarchaeota revealed by genome-centric metagenomics. Science. 2015;350:434–8. [DOI] [PubMed] [Google Scholar]
- Ferry JG, Kastead KA. Methanogenesis. Archaea: Mol Cell Biol. 2007:288–314.. DOI: 10.1128/9781555815516.ch13. [Google Scholar]
- Fida TT, Palamuru S, Pandey Get al. Aerobic biodegradation of 2, 4-dinitroanisole by Nocardioides sp. strain JS1661. Appl Environ Microbiol. 2014;80:7725–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiebig K, Friedrich B. Purification of the F420-reducing hydrogenase from Methanosarcina barkeri (strain Fusaro). Eur J Biochem. 1989;184:79–88. [DOI] [PubMed] [Google Scholar]
- Fisher J, Spencer R, Walsh C. Enzyme-catalyzed redox reactions with the flavin analogues 5-deazariboflavin, 5-deazariboflavin 5'-phosphate, and 5-deazariboflavin 5'-diphosphate, 5'→ 5'-adenosine ester. Biochemistry. 1976;15:1054–64. [DOI] [PubMed] [Google Scholar]
- Forouhar F, Abashidze M, Xu Het al. Molecular insights into the biosynthesis of the F420 coenzyme. J Biol Chem. 2008;283:11832–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara M, Kawasaki M, Hariguchi Net al. Mechanisms of resistance to delamanid, a drug for Mycobacterium tuberculosis. Tuberculosis. 2018;108:186–94. [DOI] [PubMed] [Google Scholar]
- Glas AF, Maul MJ, Cryle Met al. The archaeal cofactor F0 is a light-harvesting antenna chromophore in eukaryotes. Proc Natl Acad Sci. 2009;106:11540–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorris L, Voet A. Structural characteristics of methanogenic cofactors in the non-methanogenic archaebacterium Archaeoglobus fulgidus. Biofactors. 1991;3:29–35. [PubMed] [Google Scholar]
- Gorris L. Cofactor contents of methanogenic bacteria reviewed. Biofactors. 1994;4:139–45. [PubMed] [Google Scholar]
- Graupner M, White RH. Biosynthesis of the phosphodiester bond in coenzyme F420 in the methanoarchaea. Biochemistry. 2001;40:10859–72. [DOI] [PubMed] [Google Scholar]
- Graupner M, Xu H, White RH. Characterization of the 2-phospho-L-lactate transferase enzyme involved in coenzyme F420 biosynthesis in Methanococcus jannaschii. Biochemistry. 2002;41:3754–61. [DOI] [PubMed] [Google Scholar]
- Greening C, Ahmed FH, Mohamed AEet al. Physiology, biochemistry, and applications of F420-and FO-dependent redox reactions. Microbiol Mol Biol Rev. 2016;80:451–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greening C, Geier R, Wang Cet al. Diverse hydrogen production and consumption pathways influence methane production in ruminants. ISME J. 2019;13:2617–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greening C, Jirapanjawat T, Afroze Set al. Mycobacterial F420H2-dependent reductases promiscuously reduce diverse compounds through a common mechanism. Front Microbiol. 2017;8:1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinter R, Ney B, Brammananth Ret al. Cellular and structural basis of synthesis of the unique intermediate dehydro-F420-0 in mycobacteria. Msystems. 2020;5:e00389–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grochowski LL, Xu H, White RH. Identification and characterization of the 2-phospho-L-lactate guanylyltransferase involved in coenzyme F420 biosynthesis. Biochemistry. 2008;47:3033–7. [DOI] [PubMed] [Google Scholar]
- Grochowski LL, Xu H, White RH. Identification of lactaldehyde dehydrogenase in Methanocaldococcus jannaschii and its involvement in production of lactate for F420 biosynthesis. J Bacteriol. 2006;188:2836–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra-Lopez D, Daniels L, Rawat M. Mycobacterium smegmatis mc2155 fbiC and MSMEG_2392 are involved in triphenylmethane dye decolorization and coenzyme F420 biosynthesis. Microbiology. 2007;153:2724–32. [DOI] [PubMed] [Google Scholar]
- Gurumurthy M, Rao M, Mukherjee Tet al. A novel F420-dependent anti-oxidant mechanism protects Mycobacterium tuberculosis against oxidative stress and bactericidal agents. Mol Microbiol. 2013;87:744–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy L, Ettema TJ. The archaeal ‘TACK'superphylum and the origin of eukaryotes. Trends Microbiol. 2011;19:580–7. [DOI] [PubMed] [Google Scholar]
- Hagemeier CH, Shima S, Thauer RKet al. Coenzyme F420-dependent methylenetetrahydromethanopterin dehydrogenase (Mtd) from Methanopyrus kandleri: a methanogenic enzyme with an unusual quarternary structure. J Mol Biol. 2003;332:1047–57. [DOI] [PubMed] [Google Scholar]
- Hagemeier CH, Shima S, Warkentin Eet al. Coenzyme F420-dependent methylenetetrahydromethanopterin dehydrogenase from Methanopyrus kandleri: the selenomethionine-labelled and non-labelled enzyme crystallized in two different forms. Acta Crystallogr Sect D Biol Crystallogr. 2003;59:1653–5. [DOI] [PubMed] [Google Scholar]
- Hallam SJ, Putnam N, Preston CMet al. Reverse methanogenesis: testing the hypothesis with environmental genomics. Science. 2004;305:1457–62. [DOI] [PubMed] [Google Scholar]
- Haroon MF, Hu S, Shi Yet al. Anaerobic oxidation of methane coupled to nitrate reduction in a novel archaeal lineage. Nature. 2013;500:567–70. [DOI] [PubMed] [Google Scholar]
- Hartzell PL, Zvilius G, Escalante-Semerena JCet al. Coenzyme F420 dependence of the methylenetetrahydromethanopterin dehydrogenase of Methanobacterium thermoautotrophicum. Biochem Biophys Res Commun. 1985;133:884–90. [DOI] [PubMed] [Google Scholar]
- Hasan MR, Rahman M, Jaques Set al. Glucose 6-phosphate accumulation in mycobacteria implications for a novel F420-dependent anti-oxidant defense system. J Biol Chem. 2010;285:19135–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haver HL, Chua A, Ghode Pet al. Mutations in genes for the F420 biosynthetic pathway and a nitroreductase enzyme are the primary resistance determinants in spontaneous in vitro-selected PA-824-resistant mutants of Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2015;59:5316–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiss G, Hofmann KW, Trachtmann Net al. npd gene functions of Rhodococcus (opacus) erythropolis HL PM-1 in the initial steps of 2, 4, 6-trinitrophenol degradation. Microbiology. 2002;148:799–806. [DOI] [PubMed] [Google Scholar]
- Henderson G, Cox F, Ganesh Set al. Rumen microbial community composition varies with diet and host, but a core microbiome is found across a wide geographical range. Sci Rep. 2015;5:14567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickson EL, Leigh JA. Roles of coenzyme F420-reducing hydrogenases and hydrogen-and F420-dependent methylenetetrahydromethanopterin dehydrogenases in reduction of F420 and production of hydrogen during methanogenesis. J Bacteriol. 2008;190:4818–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hocking WP, Stokke R, Roalkvam Iet al. Identification of key components in the energy metabolism of the hyperthermophilic sulfate-reducing archaeon Archaeoglobus fulgidus by transcriptome analyses. Front Microbiol. 2014;5:e95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann C, Leis A, Niederweis Met al. Disclosure of the mycobacterial outer membrane: cryo-electron tomography and vitreous sections reveal the lipid bilayer structure. Proc Natl Acad Sci. 2008;105:3963–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollmann F, Opperman DJ, Paul CE. Enzymatic reductions-A chemist's perspective. Angew Chem Int Ed. 2020;60:5644–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossain MS, Le CQ, Joseph Eet al. Convenient synthesis of deazaflavin cofactor FO and its activity in F420-dependent NADP reductase. Org Biomol Chem. 2015;13:5082–5. [DOI] [PubMed] [Google Scholar]
- Hristov AN, Oh J, Giallongo Fet al. An inhibitor persistently decreased enteric methane emission from dairy cows with no negative effect on milk production. Proc Natl Acad Sci. 2015;112:10663–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Wang S, Moll Jet al. Electron bifurcation involved in the energy metabolism of the acetogenic bacterium Moorella thermoacetica growing on glucose or H2 plus CO2. J Bacteriol. 2012;194:3689–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hug LA, Thomas BC, Sharon Iet al. Critical biogeochemical functions in the subsurface are associated with bacteria from new phyla and little studied lineages. Environ Microbiol. 2016;18:159–73. [DOI] [PubMed] [Google Scholar]
- Höfer I, Crüsemann M, Radzom Met al. Insights into the biosynthesis of hormaomycin, an exceptionally complex bacterial signaling metabolite. Chem Biol. 2011;18:381–91. [DOI] [PubMed] [Google Scholar]
- Ichikawa H, Bashiri G, Kelly WL. Biosynthesis of the thiopeptins and identification of an F420H2-dependent dehydropiperidine reductase. J Am Chem Soc. 2018;140:10749–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ide T, Bäumer S, Deppenmeier U. Energy conservation by the H2: heterodisulfide oxidoreductase from Methanosarcina mazei Gö1: identification of two proton-translocating segments. J Bacteriol. 1999;181:4076–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeno S, Aoki D, Hamada Met al. DNA sequencing and transcriptional analysis of the kasugamycin biosynthetic gene cluster from Streptomyces kasugaensis M338-M1. J Antibiot (Tokyo). 2006;59:18–28. [DOI] [PubMed] [Google Scholar]
- Ilina Y, Lorent C, Katz Set al. X-ray crystallography and vibrational spectroscopy reveal the key determinants of biocatalytic dihydrogen cycling by [NiFe] hydrogenases. Angew Chem Int Ed. 2019;58:18710–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imachi H, Nobu MK, Nakahara Net al. Isolation of an archaeon at the prokaryote–eukaryote interface. Nature. 2020;577:519–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imlay JA. Iron-sulphur clusters and the problem with oxygen. Mol Microbiol. 2006;59:1073–82. [DOI] [PubMed] [Google Scholar]
- Isabelle D, Simpson DR, Daniels L. Large-scale production of coenzyme F420-5, 6 by using Mycobacterium smegmatis. Appl Environ Microbiol. 2002;68:5750–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson F, Daniels L, Fox Jet al. Purification and properties of an 8-hydroxy-5-deazaflavin-reducing hydrogenase from Methanobacterium thermoautotrophicum. J Biol Chem. 1982;257:3385–8. [PubMed] [Google Scholar]
- Jacobson F, Walsh C. Properties of 7, 8-didemethyl-8-hydroxy-5-deazaflavins relevant to redox coenzyme function in methanogen metabolism. Biochemistry. 1984;23:979–88. [Google Scholar]
- Jain M, Petzold CJ, Schelle MWet al. Lipidomics reveals control of Mycobacterium tuberculosis virulence lipids via metabolic coupling. Proc Natl Acad Sci. 2007;104:5133–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay ZJ, Beam JP, Dlakić Met al. Marsarchaeota are an aerobic archaeal lineage abundant in geothermal iron oxide microbial mats. Nat Microbiol. 2018;3:732–40. [DOI] [PubMed] [Google Scholar]
- Jing W, Zhang T, Zong Zet al. Comparison of in vitro activity of the nitroimidazoles delamanid and pretomanid against multidrug-resistant and extensively drug-resistant tuberculosis. Eur J Clin Microbiol Infect Dis. 2019;38:1293–6. [DOI] [PubMed] [Google Scholar]
- Jirapanjawat T, Ney B, Taylor MCet al. The redox cofactor F420 protects mycobacteria from diverse antimicrobial compounds and mediates a reductive detoxification system. Appl Environ Microbiol. 2016;82:6810–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson EF, Mukhopadhyay B. A new type of sulfite reductase, a novel coenzyme F420-dependent enzyme, from the methanarchaeon Methanocaldococcus jannaschii. J Biol Chem. 2005;280:38776–86. [DOI] [PubMed] [Google Scholar]
- Johnson EF, Mukhopadhyay B. A novel coenzyme F420 dependent sulfite reductase and a small sulfite reductase in methanogenic archaea. In Microbial Sulfur Metabolism: Springer, 2008b, 202–16. [Google Scholar]
- Johnson EF, Mukhopadhyay B. Coenzyme F420-dependent sulfite reductase-enabled sulfite detoxification and use of sulfite as a sole sulfur source by Methanococcus maripaludis. Appl Environ Microbiol. 2008;74:3591–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones J, Stadtman T. Selenium-dependent and selenium-independent formate dehydrogenases of Methanococcus vannielii. Separation of the two forms and characterization of the purified selenium-independent form. J Biol Chem. 1981;256:656–63. [PubMed] [Google Scholar]
- Joosten V, van Berkel WJH. Flavoenzymes. Curr Opin Chem Biol. 2007;11:195–202. [DOI] [PubMed] [Google Scholar]
- Jordan PA, Moore BS. Biosynthetic pathway connects cryptic ribosomally synthesized posttranslationally modified peptide genes with pyrroloquinoline alkaloids. Cell Chem Biol. 2016;23:1504–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph E, Le CQ, Nguyen Tet al. Evidence of negative cooperativity and half-site reactivity within an F420-dependent enzyme: kinetic analysis of F420H2: NADP+ oxidoreductase. Biochemistry. 2016;55:1082–90. [DOI] [PubMed] [Google Scholar]
- Kaster A-K, Moll J, Parey Ket al. Coupling of ferredoxin and heterodisulfide reduction via electron bifurcation in hydrogenotrophic methanogenic archaea. Proc Natl Acad Sci. 2011;108:2981–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keam SJ. Pretomanid: first approval. Drugs. 2019;79:1797–803. [DOI] [PubMed] [Google Scholar]
- Kelley LA, Mezulis S, Yates CMet al. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10:845–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerou M, Offre P, Valledor Let al. Proteomics and comparative genomics of Nitrososphaera viennensis reveal the core genome and adaptations of archaeal ammonia oxidizers. Proc Natl Acad Sci. 2016;113:E7937–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim ST, Sancar A. Photochemistry, photophysics, and mechanism of pyrimidine dimer repair by DNA photolyase. Photochem Photobiol. 1993;57:895–904. [DOI] [PubMed] [Google Scholar]
- Kiontke S, Gnau P, Haselsberger Ret al. Structural and evolutionary aspects of antenna chromophore usage by class II photolyases. J Biol Chem. 2014;289:19659–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschke S, Bousquet P, Ciais Pet al. Three decades of global methane sources and sinks. Nat Geosci. 2013;6:813–23. [Google Scholar]
- Klenk H-P, Clayton RA, Tomb J-Fet al. The complete genome sequence of the hyperthermophilic, sulphate-reducing archaeon Archaeoglobus fulgidus. Nature. 1997;390:364–70. [DOI] [PubMed] [Google Scholar]
- Knittel K, Boetius A. Anaerobic oxidation of methane: progress with an unknown process. Annu Rev Microbiol. 2009;63:311–34. [DOI] [PubMed] [Google Scholar]
- Kolattukudy P, Fernandes ND, Azad Aet al. Biochemistry and molecular genetics of cell-wall lipid biosynthesis in mycobacteria. Mol Microbiol. 1997;24:263–70. [DOI] [PubMed] [Google Scholar]
- Kozlowski JA, Stieglmeier M, Schleper Cet al. Pathways and key intermediates required for obligate aerobic ammonia-dependent chemolithotrophy in bacteria and Thaumarchaeota. ISME J. 2016;10:1836–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozubal MA, Romine M, deM Jennings Ret al. Geoarchaeota: a new candidate phylum in the Archaea from high-temperature acidic iron mats in Yellowstone National Park. ISME J. 2013;7:622–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzycki JA, Kenealy WR, DeNiro MJet al. Stable carbon iotope fractionation by Methanosarcina barkeri during methanogenesis from acetate, methanol, or carbon dioxide-hydrogen. Appl Environ Microbiol. 1987;53:2597–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni G, Kridelbaugh DM, Guss AMet al. Hydrogen is a preferred intermediate in the energy-conserving electron transport chain of Methanosarcina barkeri. Proc Natl Acad Sci. 2009;106:15915–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar H, Nguyen Q-T, Binda Cet al. Isolation and characterization of a thermostable F420: NADPH oxidoreductase from Thermobifida fusca. J Biol Chem. 2017;292:10123–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar H. Exploring Deazaflavoenzymes as Biocatalysts: University of Groningen, 2018. [Google Scholar]
- Kunow J, Linder D, Stetter KOet al. F420H2: quinone oxidoreductase from Archaeoglobus fulgidus. Eur J Biochem. 1994;223:503–11. [DOI] [PubMed] [Google Scholar]
- Kunow J, Schwörer B, Stetter KOet al. A F420-dependent NADP reductase in the extremely thermophilic sulfate-reducing Archaeoglobus fulgidus. Arch Microbiol. 1993;160:199–205. [Google Scholar]
- Kuypers MM, Marchant HK, Kartal B. The microbial nitrogen-cycling network. Nat Rev Microbiol. 2018;16:263–76. [DOI] [PubMed] [Google Scholar]
- Lackner G, Peters EE, Helfrich EJet al. Insights into the lifestyle of uncultured bacterial natural product factories associated with marine sponges. Proc Natl Acad Sci. 2017;114:E347–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrecht J, Cichocki N, Hübschmann Tet al. Flow cytometric quantification, sorting and sequencing of methanogenic archaea based on F420 autofluorescence. Microb Cell Fact. 2017;16:180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapalikar GV, Taylor MC, Warden ACet al. F420H2-dependent degradation of aflatoxin and other furanocoumarins is widespread throughout the actinomycetales. PLoS One. 2012;7:e30114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laso-Pérez R, Wegener G, Knittel Ket al. Thermophilic archaea activate butane via alkyl-coenzyme M formation. Nature. 2016;539:396–401. [DOI] [PubMed] [Google Scholar]
- Leahy SC, Kelly WJ, Altermann Eet al. The genome sequence of the rumen methanogen Methanobrevibacter ruminantium reveals new possibilities for controlling ruminant methane emissions. PLoS One. 2010;5:e8926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le CQ, Joseph E, Nguyen Tet al. Optimization of expression and purification of recombinant Archeoglobus fulgidus F420H2: NADP+ Oxidoreductase, an F420 cofactor dependent enzyme. Protein J. 2015;34:391–7. [DOI] [PubMed] [Google Scholar]
- Lee BM, Harold LK, Almeida DVet al. Predicting nitroimidazole antibiotic resistance mutations in Mycobacterium tuberculosis with protein engineering. PLoS Pathog. 2020;16:e1008287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenke H, Pieper D, Bruhn Cet al. Degradation of 2,4-dinitrophenol by two Rhodococcus erythropolis strains, HL 24-1 and HL 24-2. Appl Environ Microbiol. 1992;58:2928–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Graupner M, Xu Het al. CofE catalyzes the addition of two glutamates to F420-0 in F420 coenzyme biosynthesis in Methanococcus jannaschii. Biochemistry. 2003;42:9771–8. [DOI] [PubMed] [Google Scholar]
- Li H, Xu H, Graham DEet al. Glutathione synthetase homologs encode α-L-glutamate ligases for methanogenic coenzyme F420 and tetrahydrosarcinapterin biosyntheses. Proc Natl Acad Sci. 2003;100:9785–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, White R. Occurrence of coenzyme F420 and its gamma-monoglutamyl derivative in nonmethanogenic archaebacteria. J Bacteriol. 1986;168:444–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Matsumoto M, Ishida Het al. Delamanid: from discovery to its use for pulmonary multidrug-resistant tuberculosis (MDR-TB). Tuberculosis. 2018;111:20–30. [DOI] [PubMed] [Google Scholar]
- Liu Y, Whitman WB. Metabolic, phylogenetic, and ecological diversity of the methanogenic archaea. Ann N Y Acad Sci. 2008;1125:171–89. [DOI] [PubMed] [Google Scholar]
- Li W, Chou S, Khullar Aet al. Cloning and characterization of the biosynthetic gene cluster for tomaymycin, an SJG-136 monomeric analog. Appl Environ Microbiol. 2009;75:2958–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Khullar A, Chou Set al. Biosynthesis of sibiromycin, a potent antitumor antibiotic. Appl Environ Microbiol. 2009;75:2869–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukat P, Katsuyama Y, Wenzel Set al. Biosynthesis of methyl-proline containing griselimycins, natural products with anti-tuberculosis activity. Chem Sci. 2017;8:7521–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-García P, Moreira D. Eukaryogenesis, a syntrophy affair. Nat Microbiol. 2019;4:1068–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod F, Kindler GS, Wong HLet al. Asgard archaea: diversity, function, and evolutionary implications in a range of microbiomes. AIMS Microbiol. 2019;5:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maglica Ž, Özdemir E, McKinney JD. Single-cell tracking reveals antibiotic-induced changes in mycobacterial energy metabolism. MBio. 2015;6: e02236–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra K, Kim S-T, Walsh Cet al. Roles of FAD and 8-hydroxy-5-deazaflavin chromophores in photoreactivation by Anacystis nidulans DNA photolyase. J Biol Chem. 1992;267:15406–11. [PubMed] [Google Scholar]
- Manjunatha U, Boshoff HI, Barry CE. The mechanism of action of PA-824: novel insights from transcriptional profiling. Commun Integr Biol. 2009;2:215–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manjunatha UH, Boshoff H, Dowd CSet al. Identification of a nitroimidazo-oxazine-specific protein involved in PA-824 resistance in Mycobacterium tuberculosis. Proc Natl Acad Sci. 2006;103:431–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Y, Varoglu M, Sherman DH. Molecular characterization and analysis of the biosynthetic gene cluster for the antitumor antibiotic mitomycin C from Streptomyces lavendulae NRRL 2564. Chem Biol. 1999;6:251–63. [DOI] [PubMed] [Google Scholar]
- Marrakchi H, Lanéelle M-A, Daffé M. Mycolic acids: structures, biosynthesis, and beyond. Chem Biol. 2014;21:67–85. [DOI] [PubMed] [Google Scholar]
- Martin C, Tjallinks G, Trajkovic Met al. Facile stereoselective reduction of prochiral ketones by using an F420-dependent alcohol dehydrogenase. ChemBioChem. 2020;22:156–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascotti ML, Ayub MJ, Fraaije M. On the diversity of F420-dependent oxidoreductases: a sequence-and structure-based classification. bioRxiv. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascotti ML, Kumar H, Nguyen Q-Tet al. Reconstructing the evolutionary history of F420-dependent dehydrogenases. Sci Rep. 2018;8:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashalidis EH, Gittis AG, Tomczak Aet al. Molecular insights into the binding of coenzyme F420 to the conserved protein Rv1155 from Mycobacterium tuberculosis. Protein Sci. 2015;24:729–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew S, Trajkovic M, Kumar Het al. Enantio-and regioselective ene-reductions using F420H2-dependent enzymes. Chem Commun. 2018;54:11208–11. [DOI] [PubMed] [Google Scholar]
- Mayerl F, Piret J, Kiener Aet al. Functional expression of 8-hydroxy-5-deazaflavin-dependent DNA photolyase from Anacystis nidulans in Streptomyces coelicolor. J Bacteriol. 1990;172:6061–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy AJ, Williams ST. Actinomycetes as agents of biodegradation in the environment—a review. Gene. 1992;115:189–92. [DOI] [PubMed] [Google Scholar]
- McCormick J, Morton GO. Identity of cosynthetic factor I of Streptomyces aureofaciens and fragment FO from coenzyme F420 of Methanobacterium species. J Am Chem Soc. 1982;104:4014–5. [Google Scholar]
- McCormick J, Sjolander NO, Miller PAet al. The biological reduction of 7-chloro-5a (11a)-dehydrotetracycline to 7-chloro-tetracycline by Streptomyces aureofaciens. J Am Chem Soc. 1958;80:6460–1. [Google Scholar]
- Miller PA, Sjolander NO, Nalesnyk Set al. Cosynthetic factor I, a factor involved in hydrogen-transfer in Streptomyces aureofaciens. J Am Chem Soc. 1960;82:5002–3. [Google Scholar]
- Mills DJ, Vitt S, Strauss Met al. De novo modeling of the F420-reducing [NiFe]-hydrogenase from a methanogenic archaeon by cryo-electron microscopy. eLife. 2013;2:e00218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed A, Ahmed F, Arulmozhiraja Set al. Protonation state of F420H2 in the prodrug-activating deazaflavin dependent nitroreductase (Ddn) from Mycobacterium tuberculosis. Mol Biosyst. 2016a;12:1110. [DOI] [PubMed] [Google Scholar]
- Mohamed AE, Condic-Jurkic K, Ahmed FHet al. Hydrophobic shielding drives catalysis of hydride transfer in a family of F420H2-dependent enzymes. Biochemistry. 2016b;55:6908–18. [DOI] [PubMed] [Google Scholar]
- Momper L, Aronson HS, Amend JP. Genomic description of ‘Candidatus Abyssubacteria,’a novel subsurface lineage within the candidate phylum Hydrogenedentes. Front microbiol. 2018;9:1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgavi D, Forano E, Martin Cet al. Microbial ecosystem and methanogenesis in ruminants. Animal. 2010;4:1024. [DOI] [PubMed] [Google Scholar]
- Mori T, Cahn JK, Wilson MCet al. Single-bacterial genomics validates rich and varied specialized metabolism of uncultivated Entotheonella sponge symbionts. Proc Natl Acad Sci. 2018;115:1718–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee T, Boshoff H. Nitroimidazoles for the treatment of TB: past, present and future. Fut Med Chem. 2011;3:1427–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro AW, McLean KJ. Electron transfer cofactors. In: Roberts GCK (ed.) Encyclopedia of Biophysics, Berlin, Heidelberg: Springer Berlin Heidelberg, 2013,601–6. [Google Scholar]
- Muth E, Morschel E, Klein A. Purification and characterization of an 8-hydroxy-5-deazaflavin-reducing hydrogenase from the archaebacterium Methanococcus voltae. Eur J Biochem. 1987;169:571–7. [DOI] [PubMed] [Google Scholar]
- Möller-Zinkhan D, Börner G, Thauer RK. Function of methanofuran, tetrahydromethanopterin, and coenzyme F420 in Archaeoglobus fulgidus. Arch Microbiol. 1989;152:362–8. [Google Scholar]
- Möller-Zinkhan D, Thauer RK. Anaerobic lactate oxidation to 3CO2 by Archaeoglobus fulgidus via the carbon monoxide dehydrogenase pathway: demonstration of the acetyl-CoA carbon-carbon cleavage reaction in cell extracts. Arch Microbiol. 1990;153:215–8. [Google Scholar]
- Nagar-Anthal KR, Worrell VE, Teal Ret al. The pterin lumazine inhibits growth of methanogens and methane formation. Arch Microbiol. 1996;166:136–40. [Google Scholar]
- Nagarajan K, Shankar RG, Rajappa Set al. Nitroimidazoles XXI 2, 3-dihydro-6-nitroimidazo [2, 1-b] oxazoles with antitubercular activity. Eur J Med Chem. 1989;24:631–3. [Google Scholar]
- Naraoka T, Momoi K, Fukasawa Ket al. Isolation and identification of a naturally occurring 7, 8-didemethyl-8-hydroxy-5-deazariboflavin derivative from Mycobacterium avium. Biochimica et Biophysica Acta (BBA) Gen Sub. 1984;797:377–80. [Google Scholar]
- Nelson-Sathi S, Sousa FL, Roettger Met al. Origins of major archaeal clades correspond to gene acquisitions from bacteria. Nature. 2015;517:77–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nercessian O, Bienvenu N, Moreira Det al. Diversity of functional genes of methanogens, methanotrophs and sulfate reducers in deep-sea hydrothermal environments. Environ Microbiol. 2005;7:118–32. [DOI] [PubMed] [Google Scholar]
- Ney B, Ahmed FH, Carere CRet al. The methanogenic redox cofactor F420 is widely synthesized by aerobic soil bacteria. ISME J. 2017a;11:125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ney B, Carere CR, Sparling Ret al. Cofactor tail length modulates catalysis of bacterial F420-dependent oxidoreductases. Front Microbiol. 2017b;8:1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ney B. Characterisation and Industrial Application of Mycobacterial F420 Biosynthesis Volume Bachelor of Science (Honours. ): Australian National University, 2019. [Google Scholar]
- Nguyen LA, He H, Pham-Huy C. Chiral drugs: an overview. Int J Biomed Sci IJBS. 2006;2:85. [PMC free article] [PubMed] [Google Scholar]
- Nguyen Q-T, Trinco G, Binda Cet al. Discovery and characterization of an F420-dependent glucose-6-phosphate dehydrogenase (Rh-FGD1) from Rhodococcus jostii RHA1. Appl Microbiol Biotechnol. 2017;101:2831–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nocek B, Evdokimova E, Proudfoot Met al. Structure of an amide bond forming F420: γγ-glutamyl ligase from Archaeoglobus fulgidus-a member of a new family of non-ribosomal peptide synthases. J Mol Biol. 2007;372:456–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien D, Weinstock L, Cheng C. 10-deazariboflavin. Chem Ind. 1967;48:2044. [PubMed] [Google Scholar]
- O'Brien DE, Weinslock LT, Cheng C. Synthesis of 10-deazariboflavin and related 2, 4-Dioxopyrimido [4, 5-b] quinolines. J Heterocycl Chem. 1970;7:99–105. [Google Scholar]
- Orsi WD, Vuillemin A, Rodriguez Pet al. Metabolic activity analyses demonstrate that Lokiarchaeon exhibits homoacetogenesis in sulfidic marine sediments. Nat Microbiol. 2020;5:248–55. [DOI] [PubMed] [Google Scholar]
- Oyugi MA, Bashiri G, Baker ENet al. Mechanistic insights into F420-dependent glucose-6-phosphate dehydrogenase using isotope effects and substrate inhibition studies. Biochimica et Biophysica Acta (BBA) Proteins Proteomics. 2018;1866:387–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel RN. Biocatalytic synthesis of intermediates for the synthesis of chiral drug substances. Curr Opin Biotechnol. 2001;12:587–604. [DOI] [PubMed] [Google Scholar]
- Patra A, Park T, Kim Met al. Rumen methanogens and mitigation of methane emission by anti-methanogenic compounds and substances. J Anim Sci Biotechnol. 2017;8:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peck MW. Changes in concentrations of coenzyme F420 analogs during batch growth of Methanosarcina barkeri and Methanosarcina mazei. Appl Environ Microbiol. 1989;55:940–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peschke U, Schmidt H, Zhang HZet al. Molecular characterization of the lincomycin-production gene cluster of Streptomyces lincolnensis 78-11. Mol Microbiol. 1995;16:1137–56. [DOI] [PubMed] [Google Scholar]
- Philmus B, Decamps L, Berteau Oet al. Biosynthetic versatility and coordinated action of 5′-deoxyadenosyl radicals in deazaflavin biosynthesis. J Am Chem Soc. 2015;137:5406–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purwantini E, Daniels L, Mukhopadhyay B. F420H2 is required for phthiocerol dimycocerosate synthesis in mycobacteria. J Bacteriol. 2016;198:2020–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purwantini E, Daniels L. Molecular analysis of the gene encoding F420-dependent glucose-6-phosphate dehydrogenase fromMycobacterium smegmatis. J Bacteriol. 1998;180:2212–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purwantini E, Daniels L. Purification of a novel coenzyme F420-dependent glucose-6-phosphate dehydrogenase from Mycobacterium smegmatis. J Bacteriol. 1996;178:2861–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purwantini E, Gillis TP, Daniels L. Presence of F420-dependent glucose-6-phosphate dehydrogenase in Mycobacterium and Nocardia species, but absence from Streptomyces and Corynebacterium species and methanogenic Archaea. FEMS Microbiol Lett. 1997;146:129–34. [DOI] [PubMed] [Google Scholar]
- Purwantini E, Mukhopadhyay B. Conversion of NO2 to NO by reduced coenzyme F420 protects mycobacteria from nitrosative damage. Proc Natl Acad Sci. 2009;106:6333–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purwantini E, Mukhopadhyay B. Rv0132c of Mycobacterium tuberculosis encodes a coenzyme F420-dependent hydroxymycolic acid dehydrogenase. PLoS One. 2013;8:e81985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley J, Hughitt VK, Velikovsky CAet al. The cell wall lipid PDIM contributes to phagosomal escape and host cell exit of Mycobacterium tuberculosis. MBio. 2017;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeburgh WS. Oceanic methane biogeochemistry. Chem Rev. 2007;107:486–513. [DOI] [PubMed] [Google Scholar]
- Reji L, Francis CA. Metagenome-assembled genomes reveal unique metabolic adaptations of a basal marine Thaumarchaeota lineage. ISME J. 2020:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren M, Feng X, Huang Yet al. Phylogenomics suggests oxygen availability as a driving force in Thaumarchaeota evolution. ISME J. 2019;13:2150–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rifat D, Li S-Y, Ioerger TRet al. Mutations in fbiD (Rv2983) as a novel determinant of resistance to pretomanid and delamanid in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2020;65:e01948–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan NJ, Lo JH. Delamanid: first global approval. Drugs. 2014;74:1041–5. [DOI] [PubMed] [Google Scholar]
- Sambandan D, Dao DN, Weinrick BCet al. Keto-mycolic acid-dependent pellicle formation confers tolerance to drug-sensitive Mycobacterium tuberculosis. mBio. 2013;4:e00222–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancar A. Structure and function of DNA photolyase. Biochemistry. 1994;33:2–9. [DOI] [PubMed] [Google Scholar]
- Sancar GB. DNA photolyases: physical properties, action mechanism, and roles in dark repair. Mut Res DNA Repair. 1990;236:147–60. [DOI] [PubMed] [Google Scholar]
- Schauer NL, Ferry JG. Composition of the coenzyme F420-dependent formate dehydrogenase from Methanobacterium formicicum. J Bacteriol. 1986;165:405–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz RA, Linder D, Stetter KOet al. N5,N10-Methylenetetrahydromethanopterin reductase (coenzyme F420-dependent) and formylmethanofuran dehydrogenase from the hyperthermophileArchaeoglobus fulgidus. Arch Microbiol. 1991;156:427–34. [Google Scholar]
- Schrijver AD, Mot RD. Degradation of pesticides by actinomycetes. Crit Rev Microbiol. 1999;25:85–119. [DOI] [PubMed] [Google Scholar]
- Schwörer B, Breitung J, Klein ARet al. Formylmethanofuran: tetrahydromethanopterin formyltransferase and N5,N10-methylenetetrahydromethanopterin dehydrogenase from the sulfate-reducing Archaeoglobus fulgidus: similarities with the enzymes from methanogenic Archaea. Arch Microbiol. 1993;159:225–32. [DOI] [PubMed] [Google Scholar]
- Seedorf H, Dreisbach A, Hedderich Ret al. F420H2 oxidase (FprA) from Methanobrevibacter arboriphilus, a coenzyme F420-dependent enzyme involved in O2 detoxification. Arch Microbiol. 2004;182:126–37. [DOI] [PubMed] [Google Scholar]
- Seedorf H, Hagemeier CH, Shima Set al. Structure of coenzyme F420H2 oxidase (FprA), a di-iron flavoprotein from methanogenic Archaea catalyzing the reduction of O2 to H2O. FEBS J. 2007;274:1588–99. [DOI] [PubMed] [Google Scholar]
- Seitz KW, Dombrowski N, Eme Let al. Asgard archaea capable of anaerobic hydrocarbon cycling. Nat Commun. 2019;10:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekhon BS. Chiral pesticides. J Pest Sci. 2009;34:1–12. [Google Scholar]
- Selengut JD, Haft DH. Unexpected abundance of coenzyme F420-dependent enzymes in Mycobacterium tuberculosis and other actinobacteria. J Bacteriol. 2010;192:5788–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah MV, Antoney J, Kang SWet al. Cofactor F420-dependent enzymes: an under-explored resource for asymmetric redox biocatalysis. Catalysts. 2019;9:868. [Google Scholar]
- Sheng Y, Sun X, Shen Yet al. Structural and functional similarities in the ADP-forming amide bond ligase superfamily: implications for a substrate-induced conformational change in folylpolyglutamate synthetase. J Mol Biol. 2000;302:425–38. [DOI] [PubMed] [Google Scholar]
- Shi J, Xu X, Liu PYet al. Discovery and biosynthesis of guanipiperazine from a NRPS-like pathway. Chem Sci. 2021;12:2925–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shima S, Warkentin E, Grabarse Wet al. Structure of coenzyme F420 dependent methylenetetrahydromethanopterin reductase from two methanogenic archaea. J Mol Biol. 2000;300:935–50. [DOI] [PubMed] [Google Scholar]
- Shuber AP, Orr EC, Recny MAet al. Cloning, expression, and nucleotide sequence of the formate dehydrogenase genes from Methanobacterium formicicum. J Biol Chem. 1986;261:12942–7. [PubMed] [Google Scholar]
- Siméone R, Constant P, Malaga Wet al. Molecular dissection of the biosynthetic relationship between phthiocerol and phthiodiolone dimycocerosates and their critical role in the virulence and permeability of Mycobacterium tuberculosis. FEBS J. 2007;274:1957–69. [DOI] [PubMed] [Google Scholar]
- Singh R, Manjunatha U, Boshoff HIet al. PA-824 kills nonreplicating Mycobacterium tuberculosis by intracellular NO release. Science. 2008;322:1392–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MR, Mah RA. Growth and methanogenesis by Methanosarcina strain 227 on acetate and methanol. Appl Environ Microbiol. 1978;36:870–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorokin DY, Makarova KS, Abbas Bet al. Discovery of extremely halophilic, methyl-reducing euryarchaea provides insights into the evolutionary origin of methanogenesis. Nat Microbiol. 2017;2:17081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa FL, Neukirchen S, Allen JFet al. Lokiarchaeon is hydrogen dependent. Nat Microbiol. 2016;1:1–3. [DOI] [PubMed] [Google Scholar]
- Spaans SK, Weusthuis RA, Van Der Oost Jet al. NADPH-generating systems in bacteria and archaea. Front Microbiol. 2015;6:742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spang A, Caceres EF, Ettema TJ. Genomic exploration of the diversity, ecology, and evolution of the archaeal domain of life. Science. 2017;357:eaaf3883. [DOI] [PubMed] [Google Scholar]
- Spang A, Ettema TJ. Archaeal evolution: the methanogenic roots of Archaea. Nat Microbiol. 2017;2:1–2. [DOI] [PubMed] [Google Scholar]
- Spang A, Poehlein A, Offre Pet al. The genome of the ammonia-oxidizing Candidatus Nitrososphaera gargensis: insights into metabolic versatility and environmental adaptations. Environ Microbiol. 2012;14:3122–45. [DOI] [PubMed] [Google Scholar]
- Spang A, Stairs CW, Dombrowski Net al. Proposal of the reverse flow model for the origin of the eukaryotic cell based on comparative analyses of Asgard archaeal metabolism. Nat Microbiol. 2019;4:1138–48. [DOI] [PubMed] [Google Scholar]
- Speirs L, Rice DT, Petrovski Set al. The phylogeny, biodiversity, and ecology of the Chloroflexi in activated sludge. Front Microbiol. 2019;10:2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer R, Fisher J, Walsh C. Preparation, characterization, and chemical properties of the flavin coenzyme analogues 5-deazariboflavin, 5-deazariboflavin 5'-phosphate, and 5-deazariboflavin 5'-diphosphate, 5'→ 5'-adenosine ester. Biochemistry. 1976;15:1043–53. [DOI] [PubMed] [Google Scholar]
- Steiningerova L, Kamenik Z, Gazak Ret al. Different reaction specificities of F420H2-dependent reductases facilitate pyrrolobenzodiazepines and lincomycin to fit their biological targets. J Am Chem Soc. 2020;142:3440–8. [DOI] [PubMed] [Google Scholar]
- Stetter KO, Lauerer G, Thomm Met al. Isolation of extremely thermophilic sulfate reducers: evidence for a novel branch of archaebacteria. Science. 1987;236:822–4. [DOI] [PubMed] [Google Scholar]
- Stover CK, Warrener P, VanDevanter DRet al. A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature. 2000;405:962–6. [DOI] [PubMed] [Google Scholar]
- Stuermer R, Hauer B, Hall Met al. Asymmetric bioreduction of activated C=C bonds using enoate reductases from the old yellow enzyme family. Curr Opin Chem Biol. 2007;11:203–13. [DOI] [PubMed] [Google Scholar]
- Susanti D, Loganathan U, Mukhopadhyay B. A Novel F420-dependent thioredoxin reductase gated by low potential FAD a tool for redox regulation in an anaerobe. J Biol Chem. 2016;291:23084–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamada T, Kitadokoro K, Higuchi Yet al. Crystal structure of DNA photolyase from Anacystis nidulans. Nat Struct Mol Biol. 1997;4:887–91. [DOI] [PubMed] [Google Scholar]
- Tao M, Xu M, Zhang Fet al. Functional genome mining reveals a novel class V lanthipeptide containing a D-amino acid introduced by an F420H2-dependent reductase. Angew Chem Int Ed. 2020. [DOI] [PubMed] [Google Scholar]
- Taylor M, Scott C, Grogan G. F420-dependent enzymes-potential for applications in biotechnology. Trends Biotechnol. 2013;31:63–4. [DOI] [PubMed] [Google Scholar]
- Taylor MC, Jackson CJ, Tattersall DBet al. Identification and characterization of two families of F420H2-dependent reductases from Mycobacteria that catalyse aflatoxin degradation. Mol Microbiol. 2010;78:561–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Te Brömmelstroet B, Hensgens CM, Keltjens JTet al. Purification and characterization of coenzyme F420-dependent 5, 10-methylenetetrahydromethanopterin dehydrogenase from Methanobacterium thermoautotrophicum strain ΔH. Biochimica et Biophysica Acta (BBA) Gen Sub. 1991;1073:77–84. [DOI] [PubMed] [Google Scholar]
- Te Brömmelstroet BW, Geerts WJ, Keltjens JTet al. Purification and properties of 5, 10-methylenetetrahydromethanopterin dehydrogenase and 5, 10-methylenetetrahydromethanopterin reductase, two coenzyme F420-dependent enzymes, from Methanosarcina barkeri. Biochimica et Biophysica Acta (BBA) Protein Struct Mol Enzymol. 1991;1079:293–302. [DOI] [PubMed] [Google Scholar]
- Thauer RK, Jungermann K, Decker K. Energy conservation in chemotrophic anaerobic bacteria. Bacteriol Rev. 1977;41:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thauer RK, Kaster A-K, Seedorf Het al. Methanogenic archaea: ecologically relevant differences in energy conservation. Nat Rev Microbiol. 2008;6:579–91. [DOI] [PubMed] [Google Scholar]
- Thauer RK. Biochemistry of methanogenesis: a tribute to Marjory Stephenson: 1998 Marjory Stephenson prize lecture. Microbiology. 1998;144:2377–406. [DOI] [PubMed] [Google Scholar]
- Thauer RK. The Wolfe cycle comes full circle. Proc Natl Acad Sci. 2012;109:15084–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmers PH, Welte CU, Koehorst JJet al. Reverse methanogenesis and respiration in methanotrophic archaea. Archaea, 2017;2017:1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toogood HS, Gardiner JM, Scrutton NS. Biocatalytic reductions and chemical versatility of the old yellow enzyme family of flavoprotein oxidoreductases. ChemCatChem. 2010;2:892–914. [Google Scholar]
- Tourna M, Stieglmeier M, Spang Aet al. Nitrososphaera viennensis, an ammonia oxidizing archaeon from soil. Proc Natl Acad Sci. 2011;108:8420–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tweed CD, Dawson R, Burger DAet al. Bedaquiline, moxifloxacin, pretomanid, and pyrazinamide during the first 8 weeks of treatment of patients with drug-susceptible or drug-resistant pulmonary tuberculosis: a multicentre, open-label, partially randomised, phase 2b trial. Lancet Respir Med. 2019;7:1048–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzeng S, Wolfe R, Bryant M. Factor 420-dependent pyridine nucleotide-linked hydrogenase system of Methanobacterium ruminantium. J Bacteriol. 1975;121:184–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzeng SF, Bryant MP, Wolfe RS. Factor 420-dependent pyridine nucleotide-linked formate metabolism of Methanobacterium ruminantium. J Bacteriol. 1975;121:192–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungerfeld E, Rust S, Boone Det al. Effects of several inhibitors on pure cultures of ruminal methanogens. J Appl Microbiol. 2004;97:520–6. [DOI] [PubMed] [Google Scholar]
- van Beelen P, Dijkstra AC, Vogels GD. Quantitation of coenzyme F420 in methanogenic sludge by the use of reversed-phase high-performance liquid chromatography and a fluorescence detector. Eur J Appl Microbiol Biotechnol. 1983;18:67–9. [Google Scholar]
- Vanwonterghem I, Evans PN, Parks DHet al. Methylotrophic methanogenesis discovered in the archaeal phylum Verstraetearchaeota. Nat Microbiol. 2016;1:1–9. [DOI] [PubMed] [Google Scholar]
- Vaupel M, Thauer RK. Coenzyme F420-dependent N5, N10-methylenetetrahydromethanopterin reductase (Mer) from Methanobacterium thermoautotrophicum strain Marburg: cloning, sequencing, transcriptional analysis, and functional expression in Escherichia coli of the mer gene. Eur J Biochem. 1995;231:773–8. [DOI] [PubMed] [Google Scholar]
- Vitt S, Ma K, Warkentin Eet al. The F420-reducing [NiFe]-hydrogenase complex from Methanothermobacter marburgensis, the first X-ray structure of a group 3 family member. J Mol Biol. 2014;426:2813–26. [DOI] [PubMed] [Google Scholar]
- von Groote-Bidlingmaier F, Patientia R, Sanchez Eet al. Efficacy and safety of delamanid in combination with an optimised background regimen for treatment of multidrug-resistant tuberculosis: a multicentre, randomised, double-blind, placebo-controlled, parallel group phase 3 trial. Lancet Respir Med. 2019;7:249–59. [DOI] [PubMed] [Google Scholar]
- Walsh C. Flavin coenzymes: at the crossroads of biological redox chemistry. Acc Chem Res. 1980;13:148–55. [Google Scholar]
- Walsh C. Naturally occurring 5-deazaflavin coenzymes: biological redox roles. Acc Chem Res. 1986;19:216–21. [Google Scholar]
- Wang F-P, Zhang Y, Chen Yet al. Methanotrophic archaea possessing diverging methane-oxidizing and electron-transporting pathways. ISME J. 2014;8:1069–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Bashiri G, Gao Xet al. Uncovering the enzymes that catalyze the final steps in oxytetracycline biosynthesis. J Am Chem Soc. 2013;135:7138–41. [DOI] [PubMed] [Google Scholar]
- Wang P, Kim W, Pickens LBet al. Heterologous expression and manipulation of three tetracycline biosynthetic pathways. Angew Chem. 2012;124:11298–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Boshoff HI, Harrison JRet al. PE/PPE proteins mediate nutrient transport across the outer membrane of Mycobacterium tuberculosis. Science. 2020;367:1147–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Wegener G, Hou Jet al. Expanding anaerobic alkane metabolism in the domain of Archaea. Nat Microbiol. 2019;4:595–602. [DOI] [PubMed] [Google Scholar]
- Warkentin E, Hagemeier CH, Shima Set al. The structure of F420-dependent methylenetetrahydromethanopterin dehydrogenase: a crystallographic ‘superstructure' of the selenomethionine-labelled protein crystal structure. Acta Crystallogr Sect D Biol Crystallogr. 2005;61:198–202. [DOI] [PubMed] [Google Scholar]
- Warkentin E, Mamat B, Sordel-Klippert Met al. Structures of F420H2:NADP+ oxidoreductase with and without its substrates bound. EMBO J. 2001;20:6561–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss MC, Sousa FL, Mrnjavac Net al. The physiology and habitat of the last universal common ancestor. Nat Microbiol. 2016;1:1–8. [DOI] [PubMed] [Google Scholar]
- Welander PV, Metcalf WW. Loss of the mtr operon in Methanosarcina blocks growth on methanol, but not methanogenesis, and reveals an unknown methanogenic pathway. Proc Natl Acad Sci. 2005;102:10664–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welte C, Deppenmeier U. Bioenergetics and anaerobic respiratory chains of aceticlastic methanogens. Biochimica et Biophysica Acta (BBA) Bioenerg. 2014;1837:1130–47. [DOI] [PubMed] [Google Scholar]
- Welte C, Deppenmeier U. Membrane-bound electron transport in Methanosaeta thermophila. J Bacteriol. 2011;193:2868–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welte C, Deppenmeier U. Re-evaluation of the function of the F420 dehydrogenase in electron transport of Methanosarcina mazei. FEBS J. 2011;278:1277–87. [DOI] [PubMed] [Google Scholar]
- Wichmann R, Vasic-Racki D. Cofactor regeneration at the lab scale. Technol Transf Biotechnol: Springer, 2005, 225–60. [DOI] [PubMed] [Google Scholar]
- Widdel F, Wolfe R. Expression of secondary alcohol dehydrogenase in methanogenic bacteria and purification of the F420-specific enzyme from Methanogenium thermophilum strain TCI. Arch Microbiol. 1989;152:322–8. [Google Scholar]
- Williams TA, Szöllősi GJ, Spang Aet al. Integrative modeling of gene and genome evolution roots the archaeal tree of life. Proc Natl Acad Sci. 2017;114:E4602–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MC, Mori T, Rückert Cet al. An environmental bacterial taxon with a large and distinct metabolic repertoire. Nature. 2014;506:58–62. [DOI] [PubMed] [Google Scholar]
- Winkler CK, Faber K, Hall M. Biocatalytic reduction of activated CC-bonds and beyond: emerging trends. Curr Opin Chem Biol. 2018;43:97–105. [DOI] [PubMed] [Google Scholar]
- Wood GE, Haydock AK, Leigh JA. Function and regulation of the formate dehydrogenase genes of the methanogenic archaeon Methanococcus maripaludis. J Bacteriol. 2003;185:2548–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia K, Shen G-B, Zhu X-Q. Thermodynamics of various F420 coenzyme models as sources of electrons, hydride ions, hydrogen atoms and protons in acetonitrile. Org Biomol Chem. 2015;13:6255–68. [DOI] [PubMed] [Google Scholar]
- Yang JS, Kim KJ, Choi Het al. Delamanid, bedaquiline, and linezolid minimum inhibitory concentration distributions and resistance-related gene mutations in multidrug-resistant and extensively drug-resistant tuberculosis in Korea. Ann Lab Med. 2018;38:563–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Wang M, Ferry JG. A ferredoxin-and F420H2-dependent, electron-bifurcating, heterodisulfide reductase with homologs in the domains bacteria and archaea. MBio. 2017;8:e02285–16.: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Zhu Y, Crane DDet al. The effect of oxygenated mycolic acid composition on cell wall function and macrophage growth in Mycobacterium tuberculosis. Mol Microbiol. 1998;29:1449–58. [DOI] [PubMed] [Google Scholar]
- Yu T, Wu W, Liang Wet al. Growth of sedimentary Bathyarchaeota on lignin as an energy source. Proc Natl Acad Sci. 2018;115:6022–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhalnina KV, Dias R, Leonard MTet al. Genome sequence of Candidatus Nitrososphaera evergladensis from group I. 1b enriched from Everglades soil reveals novel genomic features of the ammonia-oxidizing archaea. PLoS One. 2014;9:e101648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Liu Y, Xu Wet al. Genome-and community-level interaction insights into carbon utilization and element cycling functions of Hydrothermarchaeota in hydrothermal sediment. Msystems. 2020;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Pan J, Wang Fet al. Bathyarchaeota: globally distributed metabolic generalists in anoxic environments. FEMS Microbiol Rev. 2018;42:639–55. [DOI] [PubMed] [Google Scholar]
- Zhu J, Zheng H, Ai Get al. The genome characteristics and predicted function of methyl-group oxidation pathway in the obligate aceticlastic methanogens, Methanosaeta spp. PLoS One. 2012;7:e36756. [DOI] [PMC free article] [PubMed] [Google Scholar]