

Abstract
Background
Glutamergic system dysfunction has been implicated in the pathophysiology of bipolar depression. This is an update of the 2015 Cochrane Review for the use of glutamate receptor modulators for depression in bipolar disorder.
Objectives
1. To assess the effects of ketamine and other glutamate receptor modulators in alleviating the acute symptoms of depression in people with bipolar disorder. 2. To review the acceptability of ketamine and other glutamate receptor modulators in people with bipolar disorder who are experiencing depressive symptoms.
Search methods
We searched the Cochrane Central Register of Controlled Trials (CENTRAL), Ovid MEDLINE, Embase and PsycINFO all years to July 2020. We did not apply any restrictions to date, language or publication status.
Selection criteria
RCTs comparing ketamine or other glutamate receptor modulators with other active psychotropic drugs or saline placebo in adults with bipolar depression.
Data collection and analysis
Two review authors independently selected studies for inclusion, assessed trial quality and extracted data. Primary outcomes were response rate and adverse events. Secondary outcomes included remission rate, depression severity change scores, suicidality, cognition, quality of life, and dropout rate. The GRADE framework was used to assess the certainty of the evidence.
Main results
Ten studies (647 participants) were included in this review (an additional five studies compared to the 2015 review). There were no additional studies added to the comparisons identified in the 2015 Cochrane review on ketamine, memantine and cytidine versus placebo. However, three new comparisons were found: ketamine versus midazolam, N‐acetylcysteine versus placebo, and riluzole versus placebo. The glutamate receptor modulators studied were ketamine (three trials), memantine (two), cytidine (one), N‐acetylcysteine (three), and riluzole (one). Eight of these studies were placebo‐controlled and two‐armed. In seven trials the glutamate receptor modulators had been used as add‐on drugs to mood stabilisers. Only one trial compared ketamine with an active comparator, midazolam. The treatment period ranged from a single intravenous administration (all ketamine studies), to repeated administration for riluzole, memantine, cytidine, and N‐acetylcysteine (with a follow‐up of eight weeks, 8 to 12 weeks, 12 weeks, and 16 to 20 weeks, respectively). Six of the studies included sites in the USA, one in Taiwan, one in Denmark, one in Australia, and in one study the location was unclear. All participants had a primary diagnosis of bipolar disorder and were experiencing an acute bipolar depressive episode, diagnosed according to the Diagnostic and Statistical Manual of Mental Disorders fourth edition (IV) or fourth edition text revision (IV‐TR).
Among all glutamate receptor modulators included in this review, only ketamine appeared to be more efficacious than placebo 24 hours after infusion for response rate (odds ratio (OR) 11.61, 95% confidence interval (CI) 1.25 to 107.74; P = 0.03; participants = 33; studies = 2; I² = 0%, low‐certainty evidence). Ketamine seemed to be more effective in reducing depression rating scale scores (MD ‐11.81, 95% CI ‐20.01 to ‐3.61; P = 0.005; participants = 32; studies = 2; I2 = 0%, very low‐certainty evidence). There was no evidence of ketamine's efficacy in producing remission over placebo at 24 hours (OR 5.16, 95% CI 0.51 to 52.30; P = 0.72; participants = 33; studies = 2; I2 = 0%, very low‐certainty evidence).
Evidence on response, remission or depression rating scale scores between ketamine and midazolam was uncertain at 24 hours due to very low‐certainty evidence (OR 3.20, 95% CI 0.23 to 45.19). In the one trial assessing ketamine and midazolam, there were no dropouts due to adverse effects or for any reason (very low‐certainty evidence).
Placebo may have been more effective than N‐acetylcysteine in reducing depression rating scale scores at three months, although this was based on very low‐certainty evidence (MD 1.28, 95% CI 0.24 to 2.31; participants = 58; studies = 2). Very uncertain evidence found no difference in response at three months (OR 0.82, 95% CI 0.32 to 2.14; participants = 69; studies = 2; very low‐certainty evidence). No data were available for remission or acceptability.
Extremely limited data were available for riluzole vs placebo, finding only very‐low certainty evidence of no difference in dropout rates (OR 2.00, 95% CI 0.31 to 12.84; P = 0.46; participants = 19; studies = 1; I2 = 0%).
Authors' conclusions
It is difficult to draw reliable conclusions from this review due to the certainty of the evidence being low to very low, and the relatively small amount of data usable for analysis in bipolar disorder, which is considerably less than the information available for unipolar depression. Nevertheless, we found uncertain evidence in favour of a single intravenous dose of ketamine (as add‐on therapy to mood stabilisers) over placebo in terms of response rate up to 24 hours, however ketamine did not show any better efficacy for remission in bipolar depression. Even though ketamine has the potential to have a rapid and transient antidepressant effect, the efficacy of a single intravenous dose may be limited. We did not find conclusive evidence on adverse events with ketamine, and there was insufficient evidence to draw meaningful conclusions for the remaining glutamate receptor modulators.
However, ketamine's psychotomimetic effects (such as delusions or delirium) may have compromised study blinding in some studies, and so we cannot rule out the potential bias introduced by inadequate blinding procedures. To draw more robust conclusions, further methodologically sound RCTs (with adequate blinding) are needed to explore different modes of administration of ketamine, and to study different methods of sustaining antidepressant response, such as repeated administrations.
Plain language summary
Ketamine and other glutamate receptor modulators for bipolar depression
Why is this review important?
Bipolar disorder is one of the most severe mental health conditions characterised by episodes of mania (abnormally high mood or irritability amongst other symptoms for a short time), or hypomania (the same symptoms lasting for a shorter time) and major depression (low mood). The depressive phase of the illness is linked with a greatly increased risk of self‐harm and suicide. Current treatments for depression in bipolar disorder are not always effective and can be slow to work. Among the most promising new and alternative treatments are drugs called glutamate receptor modulators. These drugs work in a different way to the drugs usually used, such as antidepressants. This is an update of a review published in 2015. As more clinical studies have been published since then, it is important to update this review with the most recent evidence.
Who will be interested in this review?
‐ People with bipolar disorder, their friends and families.
‐ General practitioners, psychiatrists, psychologists, and pharmacists.
‐ Professionals working in adult mental health services.
What questions does this review aim to answer?
1. Are ketamine and other glutamate receptor modulators better at treating bipolar depression than placebo (dummy pill) or other antidepressants? 2. Do patients prescribed ketamine or other glutamate receptor modulators experience fewer side effects than people who take placebo or other antidepressants?
Which studies were included in the review?
We searched medical databases to find all relevant studies completed up to 30 July 2020. These studies had to be randomised controlled trials (where people in the study are randomly assigned to receive either the drug being tested or a different drug or placebo to compare the results). To be included in the review, studies had to compare ketamine or other glutamate receptor modulators with placebo or other medicines in adults with bipolar depression. We included 10 studies (647 participants). The studies investigated five different glutamate receptor modulator drugs: ketamine (three trials), memantine (two trials), cytidine (one trial), N‐acetylcysteine (three trials), and riluzole (one trial). Nine studies compared glutamate receptor modulators with placebo, and one study compared ketamine with another drug. Most of the trials in the review included participants who were also receiving another medication (either lithium, valproate, or lamotrigine). We rated the certainty of the evidence 'very low' to 'low' across different comparisons, meaning that we cannot be confident that the results are a close representation of the truth.
What does the evidence from the review tell us?
The effectiveness of glutamate receptor modulators was measured primarily as the number of patients whose symptoms of depression were reduced by 50% with treatment. A single dose of ketamine injected into a vein proved to be better than placebo, but this was based on very limited evidence (two studies with 33 participants), and its effect only lasted for up to 24 hours. No differences were found in side effects between ketamine and placebo, despite common reports of trance‐like states or dissociation (a dream‐like state in which body and mind are experienced separately). The small number of participants included in this review means that we cannot say for certain whether ketamine or glutamate receptor modulators work better than other antidepressants. No differences were found between memantine, cytidine, N‐acetylcysteine and placebo for numbers of people who responded to treatment or who experienced side effects, and no data were available for riluzole.
What should happen next?
Ketamine may or may not be an effective medication as an add‐on treatment to mood stabilisers in people with bipolar depression, but because the amount of data was small, we are unable to draw any firm conclusions. The data suggests that ketamine may work very quickly in bipolar depression, but that the effects only last for a short amount of time. All trials examined the effectiveness of ketamine when injected, which is less practical than other options such as taking a pill. Future research should focus on longer‐term use of ketamine compared with placebo and other drugs, so that we can draw confident conclusions about which treatments are more effective. More research is needed on the long‐term side effects, as some studies have shown that long‐term ketamine use is linked to memory problems.
Summary of findings
Background
Description of the condition
Bipolar disorder is a severe and recurrent mood disorder with a lifetime prevalence in the order of 2.4% (Merikangas 2011; Joseph 2021). Symptoms usually appear in late adolescence or early adulthood and can blight both education and early employment opportunities, with lifelong implications. The disorder is characterised by manic symptoms (abnormally elevated mood or irritability, increased energy and related symptoms, which might include psychosis, and all of which confer severe functional impairment) that usually manifest as periods of mania, or in less degree with similar but milder symptoms in the absence of severe functional impairment that is sometimes referred to as hypomania. However, even though diagnosis is predicated on the occurrence of manic symptoms patients with bipolar disorder almost invariably experience episodes of major depression and indeed these often precede the emergence of mania (APA 2013; WHO 2018). Previous studies have shown that depressive symptomatology (syndromal and subsyndromal) dominates the longitudinal course of both bipolar I and II disorder (Judd 2002; Judd 2003), and that clinically there is considerable overlap between the clinical symptomatology of bipolar depression and unipolar (major) depression. For example, both syndromes are characterised by low mood, feelings of guilt, lack of motivation and enjoyment, anxiety, and suicidal thoughts. However, it has been suggested that psychomotor retardation, early morning waking and psychotic features are more common in patients with bipolar disorder (Mitchell 2011). In addition, in bipolar disorder depressive symptoms can co‐occur with manic symptoms, and depressive episodes can be followed immediately by manic episodes. Switches from depression to mania (and vice versa) are recognised features of the disorder but may also be precipitated by antidepressant drug treatment (Salvadore 2010; Malhi 2021). Bipolar disorder carries an increased risk of suicide and self‐harm (Malhi 2018). In a World Health Organization survey, between 20% and 25% of patients reported a history of suicide attempts (Merikangas 2011); this risk is greatest during the depressive phase. The risk of completed suicides among adults with bipolar disorder is between 20 to 30 times greater than the general population (Pompili 2013).
Description of the intervention
Treatment of bipolar depression usually involves medicines and may include psychological therapies (Geddes 2013; McIntyre 2020). There are important differences in the pharmacological management of unipolar and bipolar depression with conventional antidepressant medicines playing a much more limited role in the treatment of bipolar depression (Malhi 2020a). Even with currently recommended pharmacological treatments, clinical response in bipolar depression is often slow and incomplete (Cohen 2019). Currently approved medications for bipolar depression include lithium, quetiapine, and the combination of olanzapine and fluoxetine. In addition to these, the anticonvulsant lamotrigine, and new second‐generation antipsychotics (such as lurasidone and cariprazine) are also prescribed. Understanding of the mechanisms of action of these medicines in bipolar depression is not well‐developed, but is thought to involve a number of different neurotransmitters including serotonin, dopamine, and norepinephrine. Combination treatment with olanzapine and fluoxetine are recommended as a first‐line treatment for depression in bipolar disorder (Taylor 2014). There is emerging evidence that glutamatergic system dysfunction might play a role in the pathophysiology of bipolar depression. Glutamate, one of the most important brain neurotransmitters, is involved in memory, learning, and cognition. However, investigating glutamate neurotransmission in humans is challenging and as yet there are no clearly established biomarkers of abnormal glutamate activity in bipolar depression. One observation that has aroused interest is a possible increase in glutamate activity in the prefrontal cortex measured by magnetic resonance spectroscopy (MRS). This has been reported by two meta‐analyses examining glutamate levels in patients with bipolar disorder (Gigante 2012; Chitty 2013) and is interesting because it contrasts with findings from studies in unipolar depression where a potential decrease in this measure is noted (Moriguchi 2019). Nevertheless, it is possible that these findings in bipolar patients may reflect effects of medication rather than illness (Li 2018). Additionally, some of the drugs used to treat bipolar depression are likely to influence glutamatergic mechanisms. For example, in animal studies, lamotrigine lowers neuronal glutamate release (Cunningham 2000). However, in an MRS study in bipolar depressed patients, Godlewska 2019 found no effect of lamotrigine treatment to lower cortical glutamate. In fact, in patients who responded clinically during lamotrigine treatment, glutamate levels were increased relative to baseline after several weeks of therapy. Another drug effective in the treatment of bipolar depression is the glutamate NMDA receptor antagonist, ketamine. Ketamine has been most widely studied in resistant unipolar depression and nasal esketamine has obtained a license for this indication. However, limited data suggest that a single intravenous administration of ketamine is also effective in relieving bipolar depression, although as in unipolar depression the effect is somewhat transient and continued administration is necessary to sustain any initial effect.
How the intervention might work
The mode of action of ketamine in treating depression is not yet clarified, especially as other drugs with a similar action at the NMDA receptor, such as memantine, seem to lack ketamine’s striking antidepressant effects (Zarate 2006). Therefore, other factors must be involved in ketamine’s antidepressant action. The currently favoured hypothesis is that blockade of NMDA receptors on inhibitory GABA neurones leads to a ‘surge’ in glutamate release which then activates 2‐amino‐3‐ (5‐methyl‐3‐oxo‐1,2‐oxazol‐4‐yl) propanoic acid (AMPA) receptors. Simulation of AMPA receptors leads to increased neuroplasticity with elevated levels of brain derived neurotrophic factor (BDNF) and phosphorylation of tropomyosin receptor kinase B (TrkB) (Wilkinson 2019). Another suggested downstream effector of ketamine is the mammalian target of rapamycin (mTOR) pathway (Li 2010). Activation of mTOR pathway by ketamine in a rat model has resulted in both an antidepressant effect and formation of spine synapses in the prefrontal cortex, whereas blockade of this pathway abolished this response (Li 2010). In depressed patients, however, blockade of mTOR with rapamycin enhanced the antidepressant response to ketamine (Abdallah 2018). Ketamine also has some effects on opiate receptors and one study has shown that pre‐treatment with the opiate receptor blocker, naltrexone, prevented the antidepressant effect of ketamine, suggesting a role for opiate mechanisms in its antidepressant action (Williams 2018). Thus, the precise way in which ketamine relieves depressive symptoms is not clear. It seems likely that its mode of action in bipolar depression and unipolar depression will be similar. Nevertheless, the potential role of glutamate mechanisms in the successful treatment of bipolar depression has led to trials of other glutamatergic modifying agents, such as riluzole and memantine.
Why it is important to do this review
Bipolar disorder is one of the most severe psychiatric disorders and ranks in the top 10 causes of medical disability worldwide (Murray 2014). It has an early age of onset and is characterised by a chronic pattern of relapse into mania and depression. In addition to the effects of symptoms (both syndromal and subsyndromal) on functioning and quality of life; the depressive phase of the illness is associated with a greatly increased risk of self harm and suicide (Witt 2020). Current treatments for depressive symptoms are of limited efficacy and onset of action is generally slow (Kendall 2014). Even though lithium seems to be effective in reducing the risk of suicide in people with mood disorders (Cipriani 2013a), there are no fast‐acting treatments proven to reduce suicidal ideation or behaviour, and therefore current practice is careful assessment and close monitoring of those at risk. Consequently, there is an urgent need to identify effective treatments for bipolar depression that are fast‐acting and reduce the risk of self‐harm and suicide. As for bipolar depression, notwithstanding concerns about potential adverse events, there is some evidence that ketamine and other glutamate receptor modulators might provide rapid relief of severe depression, but also concerns about potential adverse events (McCloud 2015).
This review is an update of the previous Cochrane Review (McCloud 2015) and is one of a pair, the other of which focuses on ketamine and other glutamate receptor modulators for depression in unipolar disorder in adults (Dean 2021). Reliable information about ketamine and other glutamate receptor modulators in bipolar depression (including modes of administration, comparative efficacy, duration of effect, and safety) is not only clinically useful (Schwartz 2016), but also urgently needed because such evidence can improve patients’ outcomes in the treatment of depression and provide a basis for future clinical research and treatment guidelines (Malhi 2016; Malhi 2020b).
Objectives
To assess the effects of ketamine and other glutamate receptor modulators in alleviating the acute symptoms of depression in people with bipolar disorder.
To review the acceptability of ketamine and other glutamate receptor modulators in comparison with placebo or other antidepressant agents in people with bipolar disorder who are experiencing acute depressive symptoms.
Methods
Criteria for considering studies for this review
Types of studies
We included only double‐blind or single‐blind randomised controlled trials (RCTs) (either published or unpublished) comparing ketamine, memantine, or other glutamate receptor modulators with other active psychotropic drugs or saline placebo in people with bipolar depression.
For trials that have a cross‐over design, we only considered results from the first period prior to cross‐over.
We planned to include cluster randomised trials (CRTs) if the effect of clustering could be accounted for in the statistical analysis.
We excluded quasi‐randomised trials, such as those allocating by using alternate days of the week, as well as trials that did not explicitly describe the method of allocation as randomised.
Types of participants
Participant characteristics
We considered for inclusion people of both sexes aged 18 years or older with a primary diagnosis of bipolar disorder (currently experiencing a depressive episode) according to any of the following standard operational criteria: Feighner criteria (Feighner 1972), Research Diagnostic Criteria (Spitzer 1978), DSM‐III (APA 1980), DSM‐III‐R (APA 1987), DSM‐IV (APA 1994), DSM‐IV‐TR (APA 2000), DSM‐5 (APA 2013), or ICD‐10 (WHO 1992). We included studies using operational diagnostic criteria essentially similar to the above.
We excluded studies using ICD‐9, as it has only disease names and no diagnostic criteria. We also excluded studies that defined depression as scoring above a certain cut‐off on a screening questionnaire.
if identified, we would have included studies recruiting participants with treatment‐resistant bipolar depression, and had planned to examine these in a sensitivity analysis.
Comorbidities
We would have included studies in which less than 20% of participants were suffering from unipolar depression, and planned to examine the validity of this decision in a sensitivity analysis. We did not consider concurrent secondary diagnosis of another psychiatric disorder an exclusion criterion. However, we excluded studies in which all participants had a concurrent primary diagnosis of another Axis I or II disorder. We also excluded participants with a serious concomitant medical illness or with postpartum depression.
Setting
We applied no restriction on setting.
Subset data
We included studies with a subset of participants that met the review inclusion criteria in the analysis, provided we could extract data for this subset from the study report.
Types of interventions
Experimental Interventions
Ketamine: any dose and pattern of administration
Riluzole: any dose and pattern of administration
Amantadine: any dose and pattern of administration
Dextromethorphan: alone or in combination with quinidine
Quinolinic acid: any dose and pattern of administration
Memantine: any dose and pattern of administration
Atomoxetine: any dose and pattern of administration
Tramadol: any dose and pattern of administration
Lanicemine: any dose and pattern of administration
MK‐0657: any dose and pattern of administration
Any other glutamate receptor modulators (for example, D‐cycloserine, GLYX‐13)
Comparator interventions
Placebo (or saline placebo)
Any pharmacologically active agent (either conventional, e.g. midazolam, or nonconventional, e.g. scopolamine or Hypericum) or agent included to mimic the psychotropic side effects of the glutamate agent.
All interventions could be delivered either as monotherapy or as combined with other treatments. We applied no restrictions on dose, frequency, intensity, route, or duration. We included trials that allowed rescue medications (as required, short term, infrequent use of medications aimed at emergent symptom relief only, for example short‐term use of hypnotics) as long as these medications were equally distributed among the randomised arms.
We did not include lamotrigine among the list of comparisons because the randomised evidence about this drug has been synthesised elsewhere (Thomas 2010; Zavodnick 2012).
Types of outcome measures
We included studies that met the above inclusion criteria regardless of whether they reported on the following outcomes.
Primary outcomes
Efficacy outcome (dichotomous): number of participants who respond to treatment, where treatment response is defined as (1) a reduction of at least 50% compared to baseline on the Hamilton Rating Scale for Depression (HRSD) (Hamilton 1960), Montgomery‐Åsberg Depression Rating Scale (MADRS) (Montgomery 1979), or any other depression scale, depending on the study authors' definition or (2) 'much or very much improved' (score 1 or 2) on the Clinical Global Impression‐Improvement (CGI‐I) scale (Guy 1976). Where both scales were provided, we preferred the former criteria for judging response. We used the response rate instead of a continuous symptom score for the primary efficacy analysis to make the interpretation of results easier for clinicians (Guyatt 1998). To avoid possible outcome reporting bias, we did not use the original authors' definitions of response or remission, if different from above, in this review (Furukawa 2007a).
-
Adverse events outcome (dichotomous): We evaluated adverse events using the following outcome measures.
Total number of participants experiencing at least one side effect.
-
Total number of participants experiencing the following specific side effects:
agitation/anxiety;
constipation;
delusions;
diarrhoea;
dissociative symptoms;
dizziness;
dry mouth;
hallucinations;
headache;
hypo/hypertension;
insomnia;
mania/hypomania;
nausea;
seizure;
sleepiness/drowsiness;
urination problems;
vomiting;
tremor.
In order to avoid missing any relatively rare or unexpected, yet important, side effects (for instance sexual side effects), in the data extraction phase we collected information on all side effects data reported in the studies and discussed ways to summarise them post hoc. We extracted descriptive data regarding adverse‐effect profiles from all available studies. Due to a lack of consistent reporting of adverse effects, which came primarily from the study authors' descriptions, we combined terms describing similar side effects. For example, we combined 'dry mouth', 'reduced salivation', and 'thirst' into 'dry mouth'. We then grouped all adverse effect categories by organ system, such as neuropsychiatric, gastrointestinal, respiratory, sensory, genitourinary, dermatological, and cardiovascular.
Secondary outcomes
Efficacy outcome (dichotomous): number of participants who achieve remission. Remission is defined as (1) a score of less than 7 on the HRSD‐17 (Furukawa 2007b), or less than 8 for all the other longer versions of the HRSD, or less than 11 on the MADRS (Bandelow 2006), or less than 6 on the Quick Inventory of Depressive Symptomatology (16‐Item) (QIDS) (http://www.ids-qids.org/); or (2) participants who were 'not ill or borderline mentally ill' (score 1 or 2) on the Clinical Global Impression‐Severity score out of the total number of randomised participants. Where both are provided, we used the former criterion for judging remission.
Efficacy outcome (continuous): mean endpoint scores or mean change scores in depression severity (on HRSD, MADRS, Clinical Global Impression‐Severity (CGI‐S) or Inventory of Depressive Symptomatology (IDS)) from baseline to the time point in question (we allowed a looser form of intention‐to‐treat (ITT) analysis, whereby all the participants with at least one post‐baseline measurement were represented by their last observations carried forward (LOCF), but in any pooled analysis we examined the impact of the LOCF in a sensitivity analysis).
Suicidality, including suicidal ideation, suicide attempts (nonfatal self‐harm), and deaths by suicide. We examined suicidality and suicide ideation according to the outcome measures reported in the original studies (either as spontaneously reported or as a score on a standardised rating scale).
Cognition. We examined this according to the outcome measures reported in the original studies.
Loss of hope and other health‐related quality of life measures. We included data on the following validated quality of life instruments: SF‐12 (Ware 1998), SF‐36 (Ware 1992), Health of the Nation Outcome Scales (Wing 1998), and the WHO‐QOL (WHOQOL Group 1998).
Costs to healthcare services. We collected data according to what was reported in the original studies:
-
Acceptability (dichotomous), evaluated using the following outcome measures.
overall number of participants who dropped out during the trial as a proportion of the total number of randomised participants;
number of participants who dropped out due to lack of efficacy during the trial as a proportion of the total number of randomised participants;
number of participants who dropped out due to side effects during the trial as a proportion of the total number of randomised participants.
Timing of outcome assessment
As study authors report response rates at various time points of trials, we decided a priori to subdivide the treatment indices as follows.
Ultra‐rapid response: at 24 hours, ranging between 12 and 36 hours (primary efficacy outcome).
Rapid response: at 72 hours, ranging between 37 and less than 96 hours.
Early response: at one week, ranging between four and 10 days.
Acute response: at two weeks, ranging between 11 days and less than three weeks.
Medium response: at four weeks, ranging between three and six weeks.
Long‐term response: at three months, ranging between seven weeks and six months.
Hierarchy of outcome measures
When several possible outcome measures are reported for the same outcome, we used the primary outcome according to the original study.
Search methods for identification of studies
Electronic searches
1. Bibliographic databases
For the second version of this review (first published in September 2015 (McCloud 2015)), the Information specialist with the Cochrane Common Mental Disorders Group (CCMD) conducted update searches (30 July 2020) directly on the core bibliographic databases, from 2015 onwards (Appendix 1):
Cochrane Central Register of Controlled Trials (CENTRAL; 2020, Issue 7) in the Cochrane Library (searched 30 July 2020);
MEDLINE Ovid (2015 to July 28 2020);
Embase Ovid (2015 to 2020 Week 30);
PsycINFO Ovid (2015 to July Week 3).
Earlier searches of these databases was conducted via the Cochrane Common Mental Disorders Controlled Trials Register (CCMDCTR) (all years to 9 January 2015) (Appendix 2).
2. International trial registries
International trial registries were searched via CENTRAL on the Cochrane Library and directly via the World Health Organization's trials portal (ICTRP) and ClinicalTrials.gov to identify unpublished or ongoing studies (30 July 2020).
3. Adverse events search
The information Specialist with CCMD also conducted a companion search for adverse events data (30 July 2020) on Ovid MEDLINE, Embase and PsycINFO (Appendix 3), although we have not incorporated these data into this version of the review.
We applied no restrictions on language or publication status to the searches.
Searching other resources
Grey literature
We conducted complementary searches on the websites of the following drug regulatory authorities for additional unpublished data: the US Food and Drug Administration (FDA), the Medicines and Healthcare products Regulatory Agency in the UK, the European Medicines Agency in the EU, the Pharmaceuticals and Medical Devices Agency in Japan, and the Therapeutic Goods Administration in Australia (July 2020).
Reference lists
We checked the reference lists of all included studies and relevant systematic reviews and major textbooks of affective disorder written in English to identify additional studies missed from the original electronic searches (for example unpublished or in‐press citations).
Correspondence
We contacted trialists and subject experts for information on unpublished or ongoing studies or to request additional trial data.
Data collection and analysis
Selection of studies
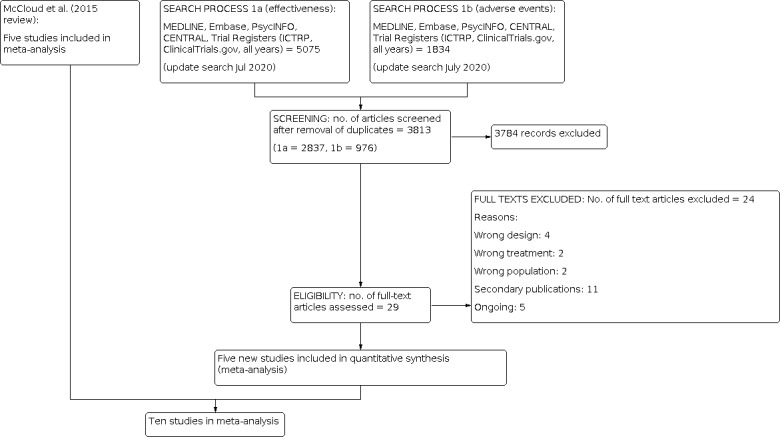
Two review authors (from RD, AB, CH, RS, and SS) independently screened titles and abstracts for inclusion of all the potential studies we identified as a result of the search and coded them as 'retrieve' (eligible or potentially eligible/unclear) or 'do not retrieve'. We retrieved the full‐text study reports/publication, and two review authors (from RD, TM, AB, RS, and SS) independently screened the full text and identified studies for inclusion, and identified and recorded reasons for exclusion of the ineligible studies. Any disagreement was resolved through discussion or, if required, by consulting a third person (AC). We identified and removed duplicate records and collated multiple reports that related to the same study so that each study, rather than each report, was the unit of interest in the review. We recorded the selection process in sufficient detail to complete a PRISMA (Moher 2009) flow diagram (Figure 1) and Characteristics of excluded studies table.
1.
Study flow diagram.
Data extraction and management
We used a data collection form to extract study characteristics and outcome data that had been piloted on at least one study in the review. Two review authors (RD, TM) extracted study characteristics and outcome data from included studies, with both authors independently extracting data from each study. We extracted the following study characteristics.
Participant characteristics (age, sex, depression diagnosis, comorbidity, depression severity, antidepressant treatment history for the index episode, study setting).
Intervention details (intended dosage range, mean daily dosage actually prescribed, cointervention if any, ketamine as investigational drug or as comparator drug, sponsorship).
Outcome measures of interest from the included studies.
We noted in the Characteristics of included studies table if outcome data were not reported in a usable way. We resolved disagreements by consensus or by involving a third person (AC). Two review authors (RD, TM) transferred data into the Review Manager 5 (RevMan 2014) file. We double‐checked that data were entered correctly by comparing the data presented in the systematic review with the study reports. Two review authors (RD, TM) spot‐checked study characteristics for accuracy against the trial report.
Main comparisons
Ketamine versus placebo
Ketamine versus other glutamate moderators
Ketamine versus other pharmacologically active agents (either conventional, e.g. midazolam, or nonconventional, e.g. scopolamine or Hypericum)
Other glutamate receptor modulators versus placebo
Other glutamate receptor modulators versus other pharmacologically active agents (either conventional, e.g. midazolam, or nonconventional, e.g. scopolamine or Hypericum)
All interventions could be delivered either as monotherapy or combined with other treatments. We applied no restrictions on dose, frequency, intensity, route, or duration.
Assessment of risk of bias in included studies
Two review authors (RD, TM) independently assessed risk of bias for each study, using the criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011b). Any disagreements were resolved by discussion or by involving another review author (AC). We assessed the risk of bias according to the following domains.
Random sequence generation
Allocation concealment
Blinding of participants and personnel
Blinding of outcome assessment
Incomplete outcome data
Selective outcome reporting
Other bias
We judged each potential source of bias as high, low, or unclear and provided a supporting quotation from the study report together with a justification for our judgement in the risk of bias table. We summarised the risk of bias judgements across different studies for each of the domains listed. We considered blinding separately for different key outcomes where necessary (for example, for unblinded outcome assessment, risk of bias for all‐cause mortality may be very different than for a participant‐reported mood scale). Where information on risk of bias relates to unpublished data or correspondence with a trialist, we noted this in the risk of bias table.
When considering treatment effects, we took into account the risk of bias for the studies that contributed to that outcome.
Measures of treatment effect
Dichotomous data
We calculated the odds ratio (OR) with corresponding 95% confidence interval (95% CI) for dichotomous or event‐like outcomes. We calculated response rates out of the total number of randomised participants. We applied intention‐to‐treat (ITT) analysis whereby all dropouts not included in the analysis were considered non‐responders. For statistically significant results, we calculated the number needed to treat for an additional beneficial outcome (NNTB) and the number needed to treat for an additional harmful outcome (NNTH).
Continuous data
We calculated the mean difference (MD) with 95% CIs where the same scale was used to measure an outcome. We planned to use the standardised mean difference (SMD) along with corresponding 95% CI if different scales were used.
For both continuous and dichotomous data, we undertook meta‐analyses only where this was meaningful, that is if the treatments, participants, and the underlying clinical question were similar enough for pooling to make sense. We narratively described skewed data reported as medians and interquartile ranges.
Where multiple trial arms were reported in a single trial, we planned to include only the relevant arms. However, this did not apply to any of the included studies.
Unit of analysis issues
Cluster‐randomised trials (CRTs)
We planned to include CRTs if either of the two methods below were possible.
When the CRT was correctly analysed in the original report, we planned to enter the effect estimate and standard error using the generic inverse variance method in RevMan 2014.
-
If the original report failed to adjust for cluster effects, we could still include such a trial in the meta‐analysis if we could extract the following information:
number of clusters randomised to each intervention or the average size of each cluster;
outcome data ignoring the cluster design for the total number of participants;
estimate of the intracluster correlation coefficient (ICC).
The ICC may be borrowed from similarly designed studies when such are available. We planned to then conduct the approximately correct analysis following the procedures described in section 16.3.4 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011c). However, no CRTs met the inclusion criteria.
Cross‐over trials
A major concern of cross‐over trials is the potential of carry‐over effects, which occur if an effect (for example, pharmacological, physiological, or psychological) of the treatment in the first phase is carried over to the second phase. As a consequence, on entry to the second phase, the participants can differ systematically from their initial state, despite a washout phase. For the same reason, cross‐over trials are not appropriate if the condition of interest is unstable (Elbourne 2002). As both effects are very likely in bipolar depression, we only used data from the first phase of cross‐over studies. However, we are aware that cross‐over trials for which only first period data are available should be considered to be at risk of bias (Higgins 2011c).
Studies with multiple treatment groups
Where a study involved more than two treatment arms, we planned to include all relevant treatment arms in the comparisons. If data were binary, we would have simply combined them into one group or divided the comparison arm into two (or more) groups as appropriate. If data were continuous, we planned to combine data following the formula in section 7.7.3.8 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011d). However, this was not the case for any of the included studies.
Dealing with missing data
Dichotomous data
We calculated treatment responders and treatment remitters on a strict ITT basis; we included dropouts in the analysis. Where participants were excluded from the trial before the endpoint, we assumed that they experienced a negative outcome (for example, failure to respond to treatment). We planned to examine the validity of this decision in sensitivity analyses by applying worst‐ and best‐case scenarios (that is, we assumed missing data to be responders or non‐responders in the corresponding sensitivity analyses). When dichotomous outcomes were not reported but baseline mean, endpoint mean, and corresponding standard deviations (SDs) of the HRSD (or other depression scale) were reported, we converted continuous outcome data expressed as mean and SD into the number of responding and remitted participants, based on a validated imputation method (Furukawa 2005). When the more sophisticated and arguably more valid imputation method (for example, mixed‐effects model, multiple imputation) was reported in the original study, we used these numbers to impute the number of responders. We planned to examine the validity of this imputation in sensitivity analyses.
Continuous data
When there were missing continuous data and the method of LOCF was used to perform an ITT analysis, we used the LOCF data.
Missing data
We contacted the original study authors for missing data.
Missing statistics
When only the standard error or t‐test or P values were reported, we calculated SDs as suggested by Altman 1996. Where SDs were not reported, we contacted trial authors and asked them to supply the data. In the absence of a response from the trial authors, we borrowed SDs from other studies in the review (Furukawa 2006). We planned to examine the validity of this imputation in sensitivity analyses.
Assessment of heterogeneity
We first investigated heterogeneity between studies by visual inspection of the forest plots. If the 95% CIs of the ORs for each study in the pooled analysis did not include means of other studies, we investigated potential sources of heterogeneity. We also calculated the I2 statistic (Higgins 2003). We used the Cochrane Handbook for Systematic Reviews of Interventions’ rough guide to its interpretation as follows: 0% to 40% might not be important; 30% to 60% may represent moderate heterogeneity; 50% to 90% may represent substantial heterogeneity; and 75% to 100% considerable heterogeneity. We also kept in mind that the importance of the observed value of I2 depends on (i) the magnitude and direction of effects and (ii) the strength of evidence for heterogeneity (for example, P value from the Chi2 test, or a CI for I2). If the I2 value is below 50% but the direction and magnitude of treatment effects were suggestive of important heterogeneity, we investigated the potential sources of heterogeneity. Finally, we performed subgroup analyses to investigate heterogeneity.
Assessment of reporting biases
We planned to enter data from included studies into a funnel plot (trial effect against trial variance) to investigate small‐study effects (Sterne 2000), but none of our analyses contained sufficient studies to allow this. In future updates of this review, we plan to use the test for funnel plot asymmetry only when at least 10 studies are included in the meta‐analysis, as per protocol. In the event of using a funnel plot, we will interpret results cautiously, with visual inspection of the funnel plots (Higgins 2011b). If we identify evidence of small‐study effects, we will investigate possible reasons for funnel plot asymmetry, including publication bias (Egger 1997).
Data synthesis
For the primary analysis, we calculated the pooled OR with corresponding 95% CI for dichotomous outcomes. We calculated the pooled MD with corresponding 95% CI for continuous outcomes. We presented any skewed data and non‐quantitative data descriptively. An outcome that has a minimum score of zero could be considered skewed when the mean is smaller than twice the SD. However, the skewness of change scores is difficult to depict as the possibility of negative values exists. We therefore used change scores for meta‐analysis of MDs. We considered a P value of less than 0.05 and a 95% CI that does not cross the line of no effect statistically significant. In forest plots with two or more studies we used a random‐effects model for both dichotomous and continuous variables. We adopted the random‐effects model under these circumstances because it has the highest generalisability for empirical examination of summary effect measures in meta‐analyses (Furukawa 2002). However, as recommended by the Cochrane Handbook for Systematic Reviews of Interventions (10.4.4.1), when concerned about the influence of small‐study effects on the results of a meta‐analysis with between‐study heterogeneity, we routinely examined the robustness by comparing the fixed‐effect model and the random‐effects model. We reported any material differences between the models.
Subgroup analysis and investigation of heterogeneity
As multiple analyses lead to false‐positive and false‐negative conclusions, subgroup analyses should be performed and interpreted with caution (Brookes 2001; Brookes 2004). We planned to perform the following subgroup analyses where possible for the following variables; however this was not necessary.
Depression severity (severe major depression, moderate or mild major depression): 'severe major depression' was defined by a threshold baseline severity score for entry of 25 or more for the 17‐item HRSD (Dozois 2004) and 31 or more for MADRS (Muller 2003).
Treatment settings (psychiatric inpatients, psychiatric outpatients, primary care): as bipolar depressive episodes in primary care may have a different profile than that of psychiatric inpatients or outpatients (Suh 1997), it is possible that results obtained from either of these settings may not be applicable to the other settings (Arroll 2009).
Older people (greater than 65 years of age), separately from other adult participants: older people may be more vulnerable to adverse effects associated with antidepressants, and a decreased dosage is often recommended. We pooled groups whose mean age was more than 65 years.
Sensitivity analysis
We planned the following sensitivity analyses for primary outcomes a priori.
Excluding trials with unclear allocation concealment or unclear double‐blinding.
Excluding studies that included participants with unipolar depression or psychotic features.
Excluding studies that recruited participants with treatment‐resistant bipolar depression.
Excluding studies with unfair dose comparisons (Cipriani 2009).
Excluding trials with a dropout rate greater than 20%.
Excluding trials for which the response rates had to be calculated based on an imputation method (Furukawa 2005), and for which the SD had to be borrowed from other trials (Furukawa 2006).
Our routine comparisons of random‐effects and fixed‐effect models, as well as our secondary outcomes of remission rates and continuous severity measures, may be considered additional forms of sensitivity analyses.
Summary of findings and assessment of the certainty of the evidence
We constructed a summary of findings table for each new comparison (ketamine versus midazolam, N‐acetylcysteine versus placebo, riluzole versus placebo), with regard to the following five outcomes. Where possible, we presented data at 24 hours, as this was considered the most clinically relevant, and presented the data closest to this time point only.
Response.
Total dropouts.
Remission.
Severity of depression at end of trial.
Dropouts due to adverse effects.
Summary of finding tables constructed in Caddy 2015 were also included for comparisons without new data.
In the 'Summary of findings' tables we used GRADEproGDT software (GRADEproGDT 2015) and the principles of the GRADE approach (Atkins 2004), which assess the quality of a body of evidence based on the extent to which there can be confidence that the obtained effect estimate reflects the true underlying effect. The quality of a body of evidence is judged on the basis of the included studies’ risks of bias, the directness of the evidence, unexplained heterogeneity, imprecision, and the risk of publication bias. We used the average rate in all the arms of the included trials as the 'assumed risk' for each outcome because we did not expect salient differences in such risks among different agents. We therefore did not target any particularly high‐ or low‐risk populations; all the tables were for medium‐risk populations.
Results
Description of studies
Results of the search
The first version of this review on this topic (McCloud 2015) retrieved five articles that met the criteria for inclusion.
CCDAN's Information Specialist ran searches in 2020 using two separate strategies: one for effectiveness (MEDLINE, Embase, PsycINFO, CENTRAL (2015 to 30 July 2020); Trial Registers (ICTRP, clinicaltrials.gov) (all years to 30 July 2020)) (n = 5075); and one for adverse effects data (MEDLINE, Embase, PsycINFO (2014 to 30 July 2020)) (n = 1834).
From a total of 6909 records retrieved from the searches, we removed 3096 duplicate records and excluded a further 3784 on the basis of the title and abstract. We retrieved full‐text articles for 29 records, yielding five new studies. Thus 10 studies in total were included.
Included studies
See: Characteristics of included studies; Figure 1.
The first version of this review (McCloud 2015) retrieved five studies (Anand 2012; Diazgranados 2010; Lee 2014; Yoon 2009; Zarate 2012). For this updated review we identified five additional studies that met the inclusion criteria (Bauer 2019; Berk 2019; Ellegaard 2019; Grunebaum 2017; Park 2017). Grunebaum 2017 assessed the efficacy of ketamine against an active comparator, midazolam, using a parallel design. Bauer 2019, Berk 2019 and Ellegaard 2019 investigated N‐acetylcysteine against placebo in a parallel design. Park 2017 investigated riluzole versus placebo in a parallel design, but the trial ended prematurely due to futility.
Two of the studies identified in the previous review assessed the efficacy of ketamine (Diazgranados 2010; Zarate 2012); two assessed the efficacy of memantine (Anand 2012; Lee 2014); and one assessed the efficacy of cytidine (Yoon 2009). All of these studies were two‐arm, placebo‐controlled trials. The former review did not find any head‐to‐head trials (i.e. active drug versus active drug), so the publication of a midazolam‐controlled trial is a significant addition (Grunebaum 2017).
Design
Nine of the 10 included studies were double‐blind, randomised, placebo‐controlled trials (Anand 2012; Bauer 2019; Berk 2019; Diazgranados 2010; Ellegaard 2019; Lee 2014; Park 2017; Yoon 2009; Zarate 2012). One was a double‐blind, randomised, midazolam‐controlled trial (Grunebaum 2017). Eight out of the 10 studies had a parallel design (Anand 2012 and Lee 2014, investigating memantine; Yoon 2009, investigating cytidine; Grunebaum 2017, investigating ketamine; Bauer 2019, Berk 2019 and Ellegaard 2019, investigating N‐acetylcysteine; Park 2017, investigating riluzole), whilst the remaining two studies, both of which investigated ketamine, used a cross‐over design (Diazgranados 2010; Zarate 2012).
The treatment period ranged from a single administration for ketamine (Diazgranados 2010; Zarate 2012; Grunebaum 2017) to eight weeks for riluzole (Park 2017), eight to 12 weeks for memantine (Anand 2012; Lee 2014), 12 weeks for cytidine (Yoon 2009), and 16 to 20 weeks for N‐acetylcysteine (Bauer 2019; Berk 2019; Ellegaard 2019). Ketamine was administered intravenously in all three of the included studies investigating this drug, whilst the remaining interventions were all administered orally. In six cases, the glutamate receptor modulators were given as an add‐on to mood stabilisers (valproate, lithium, lamotrigine) (Anand 2012; Bauer 2019; Diazgranados 2010; Lee 2014; Yoon 2009; Zarate 2012). In three studies, participants were required to have been taking these previously (either continuously or in another trial) and have shown "inadequate response"; either valproate or lithium in Diazgranados 2010 and Zarate 2012, and lamotrigine in the case of Anand 2012. In one case (Lee 2014), participants started taking valproate at the beginning of the study, and in the final case it is unclear whether patients were selected based on mood stabiliser status (though they were required to take valproate throughout; Yoon 2009).
Sample sizes
The total number of participants from the 10 included studies was 647, with a minimum sample size of 15 (Zarate 2012) and a maximum sample size of 232 (Lee 2014).
Setting
Three of the trials treated patients on an inpatient basis (Diazgranados 2010; Grunebaum 2017; Zarate 2012), and three on an outpatient basis (Anand 2012; Bauer 2019; Berk 2019). In the remaining four studies the setting was unclear (Ellegaard 2019; Lee 2014; Park 2017; Yoon 2009). The majority of trials took place in the USA (Anand 2012; Bauer 2019; Diazgranados 2010; Grunebaum 2017; Park 2017; Zarate 2012), one took place in Taiwan (Lee 2014), one in Australia (Berk 2019), and one in Denmark (Ellegaard 2019); the location of Yoon 2009 was unknown. Two of the studies (Diazgranados 2010; Zarate 2012) were conducted by the same research team at the National institute for mental health (NIMH) Mood Disorders Research Unit, in Bethesda, Maryland and followed the same protocol (NCT00088699). Five of the nine trials were single‐centre studies (Anand 2012; Bauer 2019; Diazgranados 2010; Grunebaum 2017; Zarate 2012), two were multi‐centre studies (Berk 2019; Ellegaard 2019), and in the remaining three it was unclear whether the trials were single‐centred or multi‐centred (Lee 2014; Park 2017; Yoon 2009).
Participants
All studies reported demographic and/or clinical characteristics of participants. The proportion of women randomised ranged from 32% (Park 2017) to 67% (Diazgranados 2010). No studies recruited participants under 18 years, and only two studies recruited people over 65 years (Berk 2019; Park 2017). Mean ages ranged from 31.8 years to 47.9 years.
In all the included studies, all patients had a primary diagnosis of bipolar disorder, according to the DSM‐IV or DSM‐IV‐TR (and this was confirmed through clinical interview), and defined an inclusion criterion of a current depressive phase, specifying the severity of the depression as at least moderate, with the exception of three studies (Anand 2012; Grunebaum 2017; Park 2017), which had a HRSD score more than or equal to 15 and 16, or MADRS score more than or equal to 20, respectively as an inclusion criterion. One study recruited participants experiencing a depressive or mixed episode, however only data from those experiencing a depressive episode are included in our data (Bauer 2019). One trial recruited only patients with bipolar II depression (Lee 2014), whilst all of the remaining trials recruited both types of the disorder. Three studies included only participants who had an ‘inadequate response so far’ to an open‐label mood stabiliser, with no further definition provided (Anand 2012; Diazgranados 2010; Zarate 2012), and no studies defined 'treatment‐resistant' patients as an inclusion criterion.
Interventions
Of the two studies which compared ketamine with placebo, both used ketamine as the experimental intervention and administered it intravenously; one with a single dose (Zarate 2012), and the other with two doses (Diazgranados 2010), two weeks apart. One study comparing ketamine with midazolam administered one single fixed intravenous dose of the allocated intervention (Grunebaum 2017). Of the two studies that used memantine as the experimental intervention, one administered a fixed dose of 5 mg orally per day (Lee 2014), while the other titrated the dose weekly from 5 mg to 20 mg according to tolerability (Anand 2012). Cytidine was administered at 1g twice a day (Yoon 2009). N‐acetylcysteine was administered orally in three studies at either 2000 mg/day (Bauer 2019; Berk 2019) or 3 g (Ellegaard 2019). Riluzole was orally administered at flexible doses starting from 50 mg up to 200 mg daily.
Seven of the 10 trials required participants to receive concomitant mood stabiliser medication as an add‐on (Anand 2012; Bauer 2019; Berk 2019; Diazgranados 2010; Lee 2014; Yoon 2009; Zarate 2012). In two of these studies, participants were required to have been taking either valproate or lithium for at least four weeks with inadequate response, and then continued doing so throughout the trial (Diazgranados 2010; Zarate 2012). Anand 2012 used the same criteria, with the drug lamotrigine. Two studies (Lee 2014; Yoon 2009) treated all participants with open‐label valproate throughout the trial. Six studies allowed patients to receive other concomitant medication for their depression (Anand 2012; Bauer 2019; Berk 2019; Ellegaard 2019; Grunebaum 2017; Lee 2014), whilst four studies specified washout periods (Diazgranados 2010; Park 2017; Yoon 2009; Zarate 2012).
Outcomes
We managed to include dichotomous efficacy outcomes (response and remission rates) for at least one time point in seven out of the 10 included studies. In two cases, we imputed these from the available continuous data (Grunebaum 2017; Lee 2014). In another case, we calculated data for missing time points using the graph provided (Anand 2012). There was no remission data available for the N‐acetylcysteine comparison (Bauer 2019; Ellegaard 2019), and no response or remission data available for the riluzole comparison (Park 2017). The continuous efficacy outcome in all included studies was measured on the MADRS or HRSD.
Adverse events data were unavailable for phase 1 (before cross‐over) in the two ketamine studies (Diazgranados 2010; Zarate 2012), so we have included adverse events data from across both phases for completeness. All other data were from either phase 1 of cross‐over trials or from parallel design trials. We found no data for three of the prespecified secondary outcomes: cognition, quality of life, and cost to healthcare services.
Excluded studies
See: Characteristics of excluded studies; Figure 1
We excluded 12 studies. The main reason for exclusion was incorrect diagnosis (six studies: Berk 2008; Chen 2014; Cocchi 1977; Ehrensing 1978; Lee 2012; Luckenbaugh 2014).
Ongoing studies
See: Characteristics of ongoing studies
We identified five ongoing studies, through screening retrieved records and online database information (Figure 1).
Studies awaiting classification
There are no studies which were awaiting classification.
Risk of bias in included studies
For details of the risk of bias judgements for each study, see Characteristics of included studies. A graphical representation of the overall risk of bias in included studies can be seen in Figure 2 and Figure 3.
2.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
We cannot rule out the potential bias introduced by inadequate blinding procedures. For instance, saline infusion does not necessarily provide adequate blinding for ketamine, as both patients and personnel could possibly guess which treatment a patient has received based on differences during the infusion, for example psychotomimetic side effects. The assessment of bias reported below is based on the adequacy of blinding attempts as described in the methods section of the individual papers, not on the actual degree of blinding achieved. We rated studies as 'low risk' when all measures used to blind study participants and personnel from knowledge of which intervention a participant received were described. Studies were rated as 'unclear risk' when there was a lack of information on blinding procedures. Neither of the two included studies assessing the efficacy of ketamine versus placebo tested the blinding or provided any information relating to whether the intended blinding was effective.
Allocation
Random sequence generation
We classified eight of the 10 studies (Anand 2012; Bauer 2019; Berk 2019; Diazgranados 2010; Ellegaard 2019; Grunebaum 2017; Park 2017; Zarate 2012) as 'low risk' for selection bias, having described the method of random sequence generation in details. The remaining two studies (Lee 2014; Yoon 2009) reported only that the trials were "randomised", with no information on the method used, and so we classified them as 'unclear risk'.
Allocation concealment
Three studies were rated as 'low risk' for allocation concealment (Bauer 2019; Berk 2019; Ellegaard 2019). The remaining seven studies reported no details on allocation concealment, and so we classified them as 'unclear risk' (Anand 2012; Diazgranados 2010; Grunebaum 2017; Lee 2014; Park 2017; Yoon 2009; Zarate 2012).
Blinding
Blinding of participants and personnel
We rated three studies as 'low risk' with reference to blinding of participants and personnel (Berk 2019; Diazgranados 2010; Zarate 2012). We classified six studies as 'unclear risk', having not reported sufficient detail on the blinding of participants and personnel (Anand 2012; Bauer 2019; Ellegaard 2019; Lee 2014; Park 2017; Yoon 2009). One study was classified as high risk due to lack of detail on blinding procedures, and large numbers of participants and personnel guessing the allocated groups (Grunebaum 2017).
Blinding of outcome assessment
One study provided details of the methods used in blinding of outcome assessment, and was rated as 'low risk' (Berk 2019). Eight studies were classified them as 'unclear risk' (Anand 2012; Bauer 2019; Diazgranados 2010; Ellegaard 2019; Lee 2014; Park 2017; Yoon 2009; Zarate 2012), and one was rated as high risk due to large numbers of clinical assessors guessing the allocated groups (Grunebaum 2017).
Incomplete outcome data
We classified two studies as being at 'high risk' with regards to attrition bias (Lee 2014; Yoon 2009), owing to a lack of information on dropout rates. We considered the remaining eight studies to be of 'low risk' as sufficient dropout detail was provided (Anand 2012; Bauer 2019; Berk 2019; Diazgranados 2010; Ellegaard 2019; Grunebaum 2017; Park 2017; Zarate 2012).
Selective reporting
We considered three of the included studies to be at 'high risk' of reporting bias (Anand 2012; Grunebaum 2017; Park 2017), as a result of missing primary outcome data and a lack of supplemental information. Three studies were rated as 'low risk' (Bauer 2019; Berk 2019; Ellegaard 2019). We classified all other studies as 'unclear risk' (Diazgranados 2010; Lee 2014; Yoon 2009; Zarate 2012), having reported data graphically but not in tables. We contacted all study authors for missing and unpublished data. We were able to obtain supplementary information two of the new studies included in the review (Bauer 2019; Grunebaum 2017) (see Acknowledgements).
Other potential sources of bias
We identified one other potential source of bias, relating to one of the included studies (Anand 2012). The authors stated that "blind was opened after ten subjects completed the study to examine the side‐effect and tolerability profile of active memantine". We rated all the remaining studies as 'unclear'.
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5; Table 6
Summary of findings 1. Ketamine compared to placebo for adults with depression in bipolar disorder.
| Ketamine compared to placebo for adults with depression in bipolar disorder | ||||||
| Patient or population: adults (aged 18 years+) with depression in bipolar disorder Setting: any setting (outpatient, inpatient, or both) Intervention: ketamine Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with ketamine | |||||
| Efficacy: number of participants who respond to treatment ‐ at 24 hours | Study population | OR 11.61 (1.25 to 107.74) | 33 (2 RCTs) | ⊕⊕⊝⊝ LOW1,2 | ||
| 1 per 1,000 | 10 more per 1000 (0 fewer to 96 more) | |||||
| Efficacy: number of participants who achieve remission ‐ at 1 week | Study population | OR 3.35 (0.12 to 93.83) | 18 (1 RCT) | ⊕⊝⊝⊝ VERY LOW1,3 | ||
| 1 per 1,000 | 2 more per 1,000 (1 fewer to 85 more) | |||||
| Depression rating scale score ‐ at 1 week | ‐ | MD 0.88 lower (5.88 lower to 4.12 higher) | ‐ | 28 (2 RCTs) | ⊕⊝⊝⊝ VERY LOW1,3 | |
| Acceptability: total dropouts | Study population | OR 3.48 (0.56 to 21.74) | 33 (2 RCTs) | ⊕⊝⊝⊝ VERY LOW1,3 | ||
| 18 per 1000 | 318 per 1000 (71 to 741) | |||||
| Acceptability: dropouts due to adverse effects | ‐ | ‐ | ‐ | ‐ | ‐ | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; HDRS: Hamilton depression rating scale; OR: Odds ratio. | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect. | ||||||
1 Downgraded by one point because no studies described the outcome assessment as masked. 2 Downgraded by one point because of small sample size overall. Although wide, the confidence interval does exclude no effect and so we have not downgraded a second level for imprecision.
3 Downgraded by two points because of small sample size overall and wide confidence intervals across the line of no difference.
Summary of findings 2. Ketamine compared to midazolam for adults with depression in bipolar disorder.
| Ketamine compared to midazolam for adults with depression in bipolar disorder | ||||||
| Patient or population: adults (aged 18 years+) with depression in bipolar disorder Setting: any setting (outpatient, inpatient, or both) Intervention: ketamine Comparison: midazolam | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with midazolam | Risk with ketamine | |||||
| Efficacy: number of participants who respond to treatment ‐ at 24 hours | Study population | OR 3.20 (0.23 to 45.19) | 16 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 | ||
| 111 per 1,000 | 286 per 1,000 (28 to 850) | |||||
| Efficacy: number of participants who achieve remission ‐ at 24 hours | Study population | OR 1.33 (0.07 to 25.91) | 16 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 | ||
| 111 per 1,000 | 143 per 1,000 (9 to 764) | |||||
| Depression rating scale score ‐ at 24 hours | ‐ | MD 5.85 lower (12.13 lower to 0.43 higher) | ‐ | 16 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 | |
| Acceptability: dropouts due to adverse effects at 24 hours | Study population | not estimable | 16 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 | ||
| not estimable | not estimable | |||||
| Acceptability: total dropouts | Study population | not estimable | 16 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 | ||
| not estimable | not estimable | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; HDRS: Hamilton depression rating scale; OR: Odds ratio. | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect. | ||||||
1 Downgraded by two points due to unclear method of allocation concealment and selective reporting bias.
2 Downgraded by two points due to the very low number of participants available for this outcome and the associated width of the confidence intervals.
Summary of findings 3. Memantine compared to placebo for adults with depression in bipolar disorder.
| Mematine compared to placebo for adults with depression in bipolar disorder | ||||||
| Patient or population: adults (aged 18 years+) with depression in bipolar disorder Setting: any setting (outpatient, inpatient, or both) Intervention: memantine Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with mematine | |||||
| Efficacy: number of participants who respond to treatment ‐ at 1 week | Study population | OR 1.08 (0.06 to 19.05) | 29 (1 RCT) | ⊕⊝⊝⊝ VERY LOW1,2 | ||
| 67 per 1000 | 72 per 1000 (4 to 576) | |||||
| Efficacy: number of participants who respond to treatment ‐ at 2 weeks | Study population | OR 4.88 (0.78 to 30.29) | 29 (1 RCT) | ⊕⊝⊝⊝ VERY LOW1,2 | ||
| 133 per 1000 | 429 per 1000 (107 to 823) | |||||
| Efficacy: number of participants who respond to treatment ‐ at 4 weeks | Study population | OR 5.33 (1.02 to 27.76) | 29 (1 RCT) | ⊕⊝⊝⊝ VERY LOW1,2 | ||
| 200 per 1000 | 571 per 1000 (203 to 874) | |||||
| Efficacy: number of participants who respond to treatment ‐ at 3 months | Study population | OR 1.66 (0.69 to 4.03) | 261 (2 RCTs) | ⊕⊕⊝⊝ LOW1,3 | ||
| 326 per 1000 | 445 per 1000 (250 to 661) | |||||
| Efficacy: number of participants who achieve remission ‐ at 1 week | Study population | OR 1.08 (0.06 to 19.05) | 29 (1 RCT) | ⊕⊝⊝⊝ VERY LOW1,2 | ||
| 67 per 1000 | 72 per 1000 (4 to 576) | |||||
| Depression rating scale score ‐ at 3 months | ‐ | MD 0.6 lower (2.63 lower to 1.43 higher) | ‐ | 157 (1 RCT) | ⊕⊕⊝⊝ LOW1 | |
| Acceptability: total dropouts | Study population | OR 0.77 (0.45 to 1.31) | 261 (2 RCTs) | ⊕⊕⊝⊝ LOW1,3 | ||
| 33 per 1000 | 278 per 1000 (184 to 396) | |||||
| Moderate | ||||||
| 275 per 1000 | 226 per 1000 (146 to 332) | |||||
| Acceptability: dropouts due to adverse effects | Study population | OR 0.34 (0.01 to 8.34) | 232 (1 RCT) | ⊕⊕⊝⊝ LOW1,3 | ||
| 9 per 1000 | 3 per 1000 (0 to 67) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; HDRS: Hamilton depression rating scale; OR: Odds ratio. | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect. | ||||||
1 Downgraded by one point because no studies described the outcome assessment as masked. 2 Downgraded by two points because of the very small sample size and the wide confidence interval. 3 Downgraded by one point because of wide confidence intervals.
Summary of findings 4. Cytidine compared to placebo for adults with depression in bipolar disorder.
| Cytidine compared to placebo for adults with depression in bipolar disorder | ||||||
| Patient or population: adults (aged 18 years+) with depression in bipolar disorder Setting: any setting (outpatient, inpatient, or both) Intervention: cytidine Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with cytidine | |||||
| Efficacy: number of participants who respond to treatment ‐ at 3 months | Study population | OR 1.13 (0.30 to 4.24) | 35 (1 RCT) | ⊕⊝⊝⊝ VERY LOW1,2 | ||
| 471 per 1000 | 501 per 1000 (211 to 790) | |||||
| Efficacy: number of participants who achieve remission | ‐ | ‐ | ‐ | ‐ | ‐ | |
| Depression rating scale score | ‐ | ‐ | ‐ | ‐ | ‐ | |
| Acceptability: total dropouts | Study population | OR 0.94 (0.12 to 7.52) | 35 (1 RCT) | ⊕⊝⊝⊝ VERY LOW1,2 | ||
| 118 per 1000 | 111 per 1000 (16 to 501) | |||||
| Acceptability: dropouts due to adverse effects | ‐ | ‐ | ‐ | ‐ | ‐ | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; HDRS: Hamilton depression rating scale; OR: Odds ratio. | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect. | ||||||
1 Downgraded by one point because no studies described the outcome assessment as masked. 2 Downgraded by two points because of the very small sample size and the wide confidence interval.
Summary of findings 5. N‐acetylcysteine compared to placebo for adults with depression in bipolar disorder.
| N‐acetylcysteine compared to placebo for adults with depression in bipolar disorder | ||||||
| Patient or population: adults (aged 18 years+) with depression in bipolar disorder Setting: any setting (outpatient, inpatient, or both) Intervention: N‐acetylcysteine Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with N‐acetylcysteine | |||||
| Efficacy: number of participants who respond to treatment ‐ at 3 months | 559 per 1,000 | 509 per 1,000 (288 to 731) | OR 0.82 (0.32 to 2.14) |
69 (2 studies) |
⊕⊕⊝⊝ LOW1 | |
| Efficacy: number of participants who achieve remission ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ | |
| Depression rating scale score ‐ at 3 months | ‐ | MD 1.28 higher (0.24 higher to 2.31 higher) | ‐ | 58 (2 studies) |
⊕⊕⊝⊝ LOW1 | |
| Acceptability: total dropouts ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ | |
| Acceptability: dropouts due to adverse effects ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval. | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect. | ||||||
1 Downgraded by two points because of the very small sample size and the wide confidence interval.
Summary of findings 6. Riluzole compared to placebo for adults with depression in bipolar disorder.
| Riluzole compared to placebo for adults with depression in bipolar disorder | ||||||
| Patient or population: Adults (aged 18 years+) with depression in bipolar disorder Setting: Any setting (outpatient, inpatient, or both) Intervention: riluzole Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with riluzole | |||||
| Efficacy: number of participants who respond to treatment ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ | |
| Efficacy: number of participants who achieve remission ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ | |
| Depression rating scale scores ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ | |
| Acceptability: total dropouts | Study population | OR 2.00 (0.31 to 12.84) | 19 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 | ||
| 455 per 1,000 | 625 per 1,000 (205 to 915) | |||||
| Acceptability: dropouts due to adverse effects ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio. | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect. | ||||||
1 Downgraded by two points due to trial ending prematurely.
2 Downgraded by two points because of small sample size overall and wide confidence intervals across the line of no difference.
Our included studies evaluated only ketamine and four drugs classified in the prespecified category 'other glutamate receptor modulators'; memantine, cytidine, N‐acetylcysteine, and riluzole. These drugs were compared with placebo in nine of the studies (Anand 2012; Bauer 2019; Berk 2019; Diazgranados 2010; Ellegaard 2019; Lee 2014; Park 2017; Yoon 2009; Zarate 2012), and one used a pharmacologically active agent as a comparator (Grunebaum 2017).
We found data for the efficacy outcome data at all time points up until two weeks for ketamine versus placebo (Diazgranados 2010; Zarate 2012). For the memantine versus placebo comparison,
data were only available for time points from one week onwards (Anand 2012; Yoon 2009). For cytidine, data were only available at the three‐month time point (Lee 2014). For N‐acetylcysteine versus placebo, response data was only available at 3 months, whilst AE data was available at two weeks and three months (Bauer 2019; Berk 2019; Ellegaard 2019). For ketamine versus midazolam data was only available for 24 hours due to non‐responders in the placebo arm being given open‐label ketamine treatments (Grunebaum 2017). The riluzole versus placebo comparison only had data available for withdrawals due to the study ending prematurely (Park 2017). For adverse events, we reported all findings in the tables and forest plots, but in the text below we only mentioned results that were statistically significant (all analyses here below used a fixed‐effect model, unless otherwise specified).
1. Ketamine versus placebo
Two studies contributed to this comparison, providing outcome data on 33 participants (Diazgranados 2010; Zarate 2012). We obtained data at 24 hours, three days, one week and two weeks, for the outcome measures: response, remission, and change scores from baseline. We also obtained data on adverse events and acceptability, but no data were available on other prespecified outcomes. In both of the included studies, ketamine was given as an add‐on to valproate or lithium (depending on what the participant had taken previously).
Primary outcomes
1.1 Efficacy: number of participants who respond to treatment
A single intravenous dose of ketamine appeared to be more efficacious than placebo at 24 hours (odds ratio (OR) 11.61, 95% confidence interval (CI) 1.25 to 107.74; P = 0.03, I² = 0%, 2 studies, 33 participants, number needed to treat for an additional beneficial outcome (NNTB) = 3, 95% CI 2 to 10 ‐ Analysis 1.1, Figure 4). At 72 hours there were only five events in the ketamine arm and zero events in the placebo arm, and confidence intervals were very large so no difference could be determined (OR 8.24, 95% CI 0.84 to 80.61; P = 0.07, I² = 0%, 2 studies, 33 participants). We found no difference in response between ketamine and placebo at one week, although this was based on very low certainty‐evidence with small sample sizes and wide confidence intervals (OR 4.00, 95% CI 0.33 to 48.66; P = 0.28, 1 study, 18 participants). We note that no responders were found in either group by Zarate 2012 at the one‐week time point, or by either of the included studies after two weeks.
1.1. Analysis.

Comparison 1: Ketamine versus placebo, Outcome 1: Response rate
4.

Forest plot of comparison: 1 Ketamine versus placebo, outcome: 1.1 Response rate.
1.2 Adverse events
We found no differences in any adverse events between a single infusion of ketamine and placebo, although many outcomes were based on very small participant numbers (Table 7).
1. Adverse events.
| Adverse event | Study | Glutamate receptor modulator | Comparator | Odds ratio, random‐effects (95% CI) | ||
| Events | Total | Events | Total | |||
| Ketamine versus placebo | ||||||
| Neuropsychiatric | ||||||
| Agitation/anxiety | Zarate 2012 | 1 | 14 | 2 | 12 | 0.38 [0.03 to 4.87] |
| Cognitive impairments | Diazgranados 2010 | 1 | 17 | 1 | 16 | 0.94 [0.05 to 16.37] |
| Concentration difficulties | Zarate 2012 | 1 | 14 | 1 | 12 | 0.85 [0.05 to 15.16] |
| Difficulty speaking | Zarate 2012 | 1 | 14 | 0 | 12 | 2.78 [0.10 to 74.70] |
| Dissociative symptoms | Diazgranados 2010 | 1 | 17 | 0 | 16 | 3.00 [0.11 to 79.13] |
| Dizziness | Diazgranados 2010; Zarate 2012 | 4 | 31 | 3 | 28 | 1.22 [0.25 to 5.94] |
| Fearful | Zarate 2012 | 1 | 14 | 0 | 12 | 2.78 [0.10 to 74.70] |
| Feeling spacey | Diazgranados 2010 | 1 | 17 | 2 | 16 | 0.44 [0.04 to 5.36] |
| Feeling strange/weird/bizarre | Diazgranados 2010 | 0 | 17 | 1 | 16 | 0.30 [0.01 to 7.79] |
| Insomnia | Zarate 2012 | 9 | 14 | 5 | 12 | 2.52 [0.52 to 12.30] |
| Noise sensitivity | Zarate 2012 | 1 | 14 | 0 | 12 | 2.78 [0.10 to 74.70] |
| Sleepiness/drowsiness | Diazgranados 2010; Zarate 2012 | 7 | 31 | 5 | 28 | 1.33 [0.37 to 4.80] |
| Slowed | Zarate 2012 | 0 | 14 | 1 | 12 | 0.26 [0.01 to 7.12] |
| Vivid dreams | Diazgranados 2010; Zarate 2012 | 4 | 31 | 1 | 28 | 3.06 [0.44 to 21.01] |
| Gastrointestinal | ||||||
| Appetite decrease | Zarate 2012 | 1 | 14 | 0 | 12 | 2.78 [0.10 to 74.70] |
| Diarrhoea | Zarate 2012 | 1 | 14 | 0 | 12 | 2.78 [CI 0.10 to 74.70] |
| Dry mouth | Zarate 2012 | 1 | 14 | 0 | 12 | 2.78 [0.10 to 74.70] |
| Flatulence | Zarate 2012 | 2 | 14 | 0 | 12 | 5.00 [0.22 to 115.05] |
| Nausea | Diazgranados 2010 | 1 | 17 | 0 | 16 | 3.00 [0.11 to 79.13] |
| Stomach/abdominal discomfort | Zarate 2012 | 1 | 14 | 1 | 12 | 0.85 [CI 0.05 to 15.16] |
| Stool discolouration | Zarate 2012 | 1 | 14 | 0 | 12 | 2.78 [0.10 to 74.70] |
| Weight loss | Zarate 2012 | 1 | 14 | 0 | 12 | 2.78 [0.10 to 74.70] |
| Respiratory | ||||||
| Coughing | Zarate 2012 | 1 | 14 | 0 | 12 | 2.78 [0.10 to 74.70] |
| Somatic | ||||||
| Breast pain/swelling | Zarate 2012 | 1 | 14 | 0 | 12 | 2.78 [0.10 to 74.70] |
| Decreased body temperature | Zarate 2012 | 1 | 14 | 0 | 12 | 2.78 [0.10 to 74.70] |
| Flushed | Zarate 2012 | 1 | 14 | 0 | 12 | 2.78 [0.10 to 74.70] |
| Increased body temperature | Zarate 2012 | 0 | 14 | 1 | 12 | 0.26 [0.01 to 7.12] |
| Leg cramping | Zarate 2012 | 1 | 14 | 0 | 12 | 2.78 [0.10 to 74.70] |
| Muscle/bone/joint pain | Zarate 2012 | 0 | 14 | 4 | 12 | 0.07 [0.00 to 1.36] |
| Sweating | Zarate 2012 | 1 | 14 | 0 | 12 | (OR 2.78, 95% CI 0.10 to 74.70) |
| Genitourinary | ||||||
| Decreased libido | Zarate 2012 | 0 | 14 | 1 | 12 | 0.26 [0.01 to 7.12] |
| Increased libido | Zarate 2012 | 1 | 14 | 0 | 12 | 2.78 [0.10 to 74.70] |
| Dermatological | ||||||
| Dermatological/skin irritation/lesions | Zarate 2012 | 1 | 14 | 0 | 12 | 2.78 [0.10 to 74.70] |
| Red blotching | Zarate 2012 | 1 | 14 | 0 | 12 | 2.78 [0.10 to 74.70] |
| Cardiovascular | ||||||
| Tachycardia | Zarate 2012 | 1 | 14 | 0 | 12 | 2.78 [0.10 to 74.70] |
| Neurological | ||||||
| Headache | Zarate 2012 | 3 | 14 | 3 | 12 | 0.82 [0.13 to 5.08] |
| Tremor | Zarate 2012 | 1 | 14 | 0 | 12 | 2.78 [0.10 to 74.70] |
| Endocrine | ||||||
| Menstrual irregulation | Zarate 2012 | 1 | 14 | 0 | 12 | 2.78 [0.10 to 74.70] |
| Memantine versus placebo | ||||||
| Neuropsychiatric | ||||||
| Dizziness | Lee 2014 | 0 | 115 | 1 | 117 | 0.34 [0.01 to 8.34] |
| Mania/hypomania | Anand 2012 | 2 | 14 | 3 | 15 | 0.67 [0.09 to 4.73] |
| Gastrointestinal | ||||||
| Gastrointestinal problems | Anand 2012 | 5 | 14 | 3 | 15 | 2.22 [0.42 to 11.83] |
| Respiratory | ||||||
| Respiratory problems | Anand 2012 | 1 | 14 | 1 | 15 | 1.08 [CI 0.06 to 19.05] |
| Somatic | ||||||
| Hair loss | Lee 2014 | 0 | 115 | 1 | 117 | 0.34 [0.01 to 8.34] |
| Genitourinary | ||||||
| Sexual issues | Anand 2012 | 1 | 14 | 0 | 15 | 3.44 [0.13 to 91.79] |
| Urination problems | Anand 2012 | 0 | 14 | 1 | 15 | 0.33 [0.01 to 8.88] |
| Cardiovascular | ||||||
| Cardiovascular problems | Anand 2012 | 1 | 14 | 3 | 15 | 0.31 [0.03 to 3.38] |
| Endocrine | ||||||
| Endocrine problems | Anand 2012 | 1 | 14 | 0 | 15 | 3.44 [0.13 to 91.79] |
| Miscellaneous | ||||||
| Central nervous system issues | Anand 2012 | 10 | 14 | 11 | 15 | 0.91 [0.18 to 4.64] |
| Immunological issues | Anand 2012 | 0 | 14 | 1 | 15 | 0.33, [0.01 to 8.88] |
| Cytidine versus placebo | ||||||
| Neuropsychiatric | ||||||
| Agitation/anxiety | Yoon 2009 | 1 | 18 | 0 | 17 | 3.00 [CI 0.11 to 78.81] |
| Dizziness | Yoon 2009 | 0 | 18 | 1 | 17 | 0.30 [0.01 to 7.81] |
| Sleepiness/drowsiness | Yoon 2009 | 2 | 18 | 1 | 17 | 2.00 [0.16 to 24.33] |
| Gastrointestinal | ||||||
| Dry mouth | Yoon 2009 | 0 | 18 | 1 | 17 | 0.30 [0.01 to 7.81] |
| Gastrointestinal problems | Yoon 2009 | 2 | 18 | 2 | 17 | 0.94 [0.12 to 7.52] |
| Weight gain | Yoon 2009 | 1 | 18 | 0 | 17 | 3.00 [0.11 to 78.81] |
| Neurological | ||||||
| Headache | Yoon 2009 | 3 | 18 | 2 | 17 | 1.50 [0.22 to 10.30] |
| Tremor | Yoon 2009 | 2 | 18 | 2 | 17 | 0.94 [0.12 to 7.52] |
Secondary outcomes
1.3 Efficacy: number of participants who achieve remission
We found no evidence that a single infusion of ketamine was more effective than placebo for remission at any time point, although this may be affected by wide confidence intervals and low sample sizes (Analysis 1.2). We note that there were no remitters in either group, in either study at the two‐week time point.
1.2. Analysis.

Comparison 1: Ketamine versus placebo, Outcome 2: Remission rate
1.4 Change scores on depression scale from baseline
A single intravenous infusion of ketamine appeared to be more effective than placebo at 24 hours (mean difference (MD) ‐11.81, 95% CI ‐20.01 to ‐3.61; P = 0.005, I² = 0%, 2 studies, 32 participants; Analysis 1.3, Figure 5), and at 72 hours (MD ‐9.10, 95% CI ‐16.00 to ‐2.21; P = 0.010, I² = 0%, 2 studies, 31 participants). However, this effect seemed to disappear after one week (MD ‐0.88, 95% CI ‐5.88 to 4.12; P = 0.73, I² = 0%, 2 studies, 28 participants). The evidence suggests that there may be no difference between ketamine and placebo at two weeks (MD ‐1.14, 95% CI ‐6.30 to 4.01; P = 0.66, I² = 0%, 2 studies, 26 participants).
1.3. Analysis.

Comparison 1: Ketamine versus placebo, Outcome 3: Depression rating scale score
1.5 Suicidality
No data were available for this outcome.
1.6 Cognition
No data were available for this outcome.
1.7 Loss of hope or other health‐related quality of life measures
No data were available for this outcome.
1.8 Costs to healthcare services
No data were available for this outcome.
1.9 Acceptability: total dropouts and dropouts due to adverse effects
We found no difference between a single intravenous infusion of ketamine and placebo in acceptability, either in terms of total dropouts (Analysis 1.4), or in relation to lack of efficacy (Analysis 1.5). This was based on very low‐certainty evidence with small sample sizes and wide confidence intervals.
1.4. Analysis.

Comparison 1: Ketamine versus placebo, Outcome 4: Acceptability ‐ total dropouts
1.5. Analysis.

Comparison 1: Ketamine versus placebo, Outcome 5: Acceptability ‐ lack of efficacy
2. Ketamine versus active comparator
One new study was included in this comparison, providing outcome data on 16 participants (Grunebaum 2017). We obtained data at 24 hours for outcome measures: response, remission, and change scores from baseline. We also obtained data on adverse events and acceptability, but no data were available on other prespecified outcomes. In this study, ketamine or midazolam was given in addition to current psychotropic medications (except benzodiazepines). Table 2.
Primary outcomes
2.1 Efficacy: number of participants who respond to treatment
The evidence suggests there may be no difference in response between ketamine and midazolam at 24 hours, based on small participant numbers and wide confidence intervals (OR 3.20, 95% CI 0.23 to 45.19; P = 0.39, 1 study, 16 participants; very low‐certainty evidence) (Analysis 2.1; Figure 5).
2.1. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 1: Response rate
5.

Forest plot of comparison: 2 Ketamine versus Midazolam, outcome: 2.1 Response rate.
2.2 Adverse events
No adverse events were reported for both ketamine and midazolam at 24 hours; very low‐certainty evidence (Analysis 2.4).
2.4. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 4: Acceptability: adverse effects
Secondary outcomes
2.3 Efficacy: number of participants who achieve remission
Uncertain evidence suggested no difference in remission rates for ketamine and midazolam groups at 24 hours (OR 1.33, 95% CI 0.07 to 25.91, P = 0.85, 1 study, 16 participants; very low‐certainty evidence) (Analysis 2.2).
2.2. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 2: Remission rate
2.4 Depression rating scale score
There was unclear evidence about the effect of ketamine over midazolam on depression rating scale scores 24 hours after infusion. Data were only available for 16 participants and confidence intervals were wide (MD ‐5.85, 95% CI ‐12.13 to 0.43; P = 0.07, 1 study; very low‐certainty evidence) (Analysis 2.3).
2.3. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 3: Depression rating scale score
2.5 Suicidality
Scale for Suicidal Ideation (SSI) scores did not appear to differ between the ketamine and midazolam groups at 24 hours (MD ‐5.86, 95% CI ‐15.76 to 4.04; P = 0.25, 1 study, 16 participants; Analysis 2.6).
2.6. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 6: Suicidality rating scale
2.6 Cognition
No data were available for this outcome.
2.7 Loss of hope or other health‐related quality of life measures
No data were available for this outcome.
2.8 Costs to healthcare services
No data were available for this outcome.
2.9 Acceptability: total dropouts or dropouts due to adverse effects
There were no dropouts due to adverse effects or for any reason in Grunebaum 2017 (Analysis 2.4; Analysis 2.5).
2.5. Analysis.

Comparison 2: Ketamine versus Midazolam, Outcome 5: Acceptability: total dropouts
3. Memantine versus placebo
Two studies contributed to this comparison, providing outcome data on 261 participants (Anand 2012; Lee 2014). We obtained outcome data at one week, two weeks, four weeks and three months for the measures response and remission rate. For change scores from baseline, we obtained data for the three‐month time point only. We also obtained information on adverse events, suicidality and acceptability, but no data were available on the other outcomes we prespecified in the review protocol (Rendell 2015). In the Anand 2012 study, both arms received lamotrigine throughout (and had already been taking it), whilst in the Lee 2014 study all participants began taking valproate for the study.
Primary outcomes
3.1 Efficacy: number of participants who respond to treatment
There was no difference between memantine and placebo in response at one week (OR 1.08, 95% CI 0.06 to 19.05; P = 0.96, 1 study, 29 participants; Analysis 3.1, Figure 6), and at two weeks (OR 4.88, 95% CI 0.78 to 30.29; P = 0.09, 1 study, 29 participants), based on uncertain evidence with wide confidence intervals and small sample sizes. A marginal difference was found in favour of memantine at four weeks (OR 5.33, 95% CI 1.02 to 27.76; P = 0.05; 1 study, 29 participants, NNTB = 3, 95% CI 2 to 25). No effect was present at the three‐month time point (OR 1.66, 95% CI 0.69 to 4.03; P = 0.26, I² = 36%, 2 studies, 26 participants).
3.1. Analysis.

Comparison 3: Memantine versus placebo, Outcome 1: Response rate
6.

Forest plot of comparison: 2 Memantine versus placebo, outcome: 2.1 Response rate.
3.2 Adverse events
We found no difference between memantine and placebo in any adverse events (Analysis 3.2; Table 7).
3.2. Analysis.

Comparison 3: Memantine versus placebo, Outcome 2: Adverse events: Young Mania Rating Scale (12 weeks)
Secondary outcomes
3.3 Efficacy: number of participants who achieve remission
There was uncertain evidence of no difference between memantine and placebo in remission rate at one week, two weeks, and three months (Analysis 3.3). At four weeks, the data were limited by a small sample size and wide confidence intervals (OR 3.67, 95% CI 0.77 to 17.43; P = 0.10; I2 = 0%, 1 study, 29 participants).
3.3. Analysis.

Comparison 3: Memantine versus placebo, Outcome 3: Remission rate
3.4 Change scores on depression scale from baseline
Change scores on depression scale from baseline did not appear to differ between ketamine and placebo groups (Analysis 3.4).
3.4. Analysis.

Comparison 3: Memantine versus placebo, Outcome 4: Depression rating scale score
3.5 Suicidality
A suicidality measure showed no difference between memantine and placebo (OR 0.34, 95% CI 0.01 to 8.34; P = 0.51, 1 study, 232 participants; Analysis 3.5). This was defined by the authors as number of participants who dropped out of the study as a result of attempted suicide within the duration of the trial.
3.5. Analysis.

Comparison 3: Memantine versus placebo, Outcome 5: Suicidality: suicide attempts
3.6 Cognition
No data were available for this outcome.
3.7 Loss of hope or other health‐related quality of life measures
No data were available for this outcome.
3.8 Costs to healthcare services
No data were available for this outcome.
3.9 Acceptability: total dropouts or dropouts due to adverse effects
There were not enough data to be able to determine a difference in dropout rate between the memantine and placebo groups, either as overall dropout rate (Analysis 3.6), due to lack of efficacy (Analysis 3.7), or due to adverse effects (Analysis 3.8).
3.6. Analysis.

Comparison 3: Memantine versus placebo, Outcome 6: Acceptability ‐ total dropouts
3.7. Analysis.

Comparison 3: Memantine versus placebo, Outcome 7: Acceptability ‐ lack of efficacy
3.8. Analysis.

Comparison 3: Memantine versus placebo, Outcome 8: Acceptability ‐ adverse events
4. Cytidine versus placebo
One study contributed to this comparison, providing outcome data on 35 participants (Yoon 2009). Data were available on response rate at the three‐month time point only, and on the outcome measures: adverse events and acceptability. No other prespecified outcome data were available. Both arms of the study also took valproate throughout, though it is unclear whether participants had been taking this previously or not.
Primary outcomes
4.1 Efficacy: number of participants who respond to treatment
There was no difference between cytidine and placebo in response rate at three months (OR 1.13, 95% CI 0.30 to 4.24; P = 0.86, 1 study, 35 participants; Analysis 4.1; Figure 7).
4.1. Analysis.

Comparison 4: Cytidine versus placebo, Outcome 1: Response rate
7.

Forest plot of comparison: 4 Cytidine versus placebo, outcome: 4.1 Response rate.
4.2 Adverse events
We found no difference between the cytidine and placebo groups in adverse events experienced (Table 7).
Secondary outcomes
4.3 Efficacy: number of participants who achieve remission
No data were available for this outcome.
4.4 Depression rating scale score
No data were available for this outcome.
4.5 Suicidality rating scale
No data were available for this outcome.
4.6 Cognition
No data were available for this outcome.
4.7 Loss of hope or other health‐related quality of life measures
No data were available for this outcome.
4.8 Costs to healthcare services
No data were available for this outcome.
4.9 Acceptability: total dropouts
No difference in overall acceptability (total dropouts) between cytidine and placebo was identified (OR 0.94, 95% CI 0.12 to 7.52; P = 0.95, 1 study, 35 participants; Analysis 4.2).
4.2. Analysis.

Comparison 4: Cytidine versus placebo, Outcome 2: Acceptability ‐ total dropouts
5. N‐acetylcysteine versus placebo
Three studies contributed to this comparison, consisting of data from 278 participants (Bauer 2019; Berk 2019; Ellegaard 2019). Response data were only available for Bauer 2019 and Ellegaard 2019 for long‐term response. Long‐term adverse event data were available for both studies in the form of Young Mania Rating Scale scores; however, were was only short‐term data for Ellegaard 2019. Table 5.
Primary outcomes
5.1 Efficacy: number of participants who respond to treatment
There was no difference between N‐acetylcysteine and placebo in response rate at three months (OR 0.82, 95% CI 0.32 to 2.14; P = 0.69; participants = 69; studies = 2; I2 = 0%; Analysis 5.1; Figure 8).
5.1. Analysis.

Comparison 5: N‐acetylcysteine versus placebo, Outcome 1: Response rate
8.

Forest plot of comparison: 5 N‐acetylcysteine versus placebo, outcome: 5.1 Response rate.
5.2 Adverse Events
The Young Mania Rating Scale (YMRS) was used to assess adverse events relating to mania at two weeks (Ellegaard 2019) and three months (Berk 2019; Ellegaard 2019). There was a difference favouring N‐acetylcysteine over placebo at both two weeks (MD ‐0.90, 95% CI ‐1.11 to ‐0.69; P < 0.001; 1 study; 80 participants; Analysis 5.2) and three months (MD ‐0.84, 95% CI ‐1.08 to ‐0.60; P< 0.001; 2 studies, 121 participants; Analysis 5.2).
5.2. Analysis.

Comparison 5: N‐acetylcysteine versus placebo, Outcome 2: Adverse events: Young Mania Rating Scale
Secondary outcomes
5.3 Efficacy: number of participants who achieve remission
No data were available for this outcome.
5.4 Depression rating scale score
Placebo was more effective in reducing depression rating scale scores over N‐acetylcysteine at three months (MD 1.28, 95% CI 0.24 to 2.31; P = 0.02; participants = 58; studies = 2; I2 = 0%; Analysis 5.3).
5.3. Analysis.

Comparison 5: N‐acetylcysteine versus placebo, Outcome 3: Depression rating scale score change
5.5 Suicidality rating scale
There was no difference in suicidality rating scale scores between N‐acetylcysteine and placebo (MD 0.10, 95% CI ‐0.02 to 0.22; 1 study; 41 participants; Analysis 5.4).
5.4. Analysis.

Comparison 5: N‐acetylcysteine versus placebo, Outcome 4: Suicidality rating scale
5.6 Cognition
No data were available for this outcome.
5.7 Loss of hope or other health‐related quality of life measures
No data were available for this outcome.
5.8 Costs to healthcare services
No data were available for this outcome.
5.9 Acceptability: total dropouts or dropouts due to adverse effects
No data were available for this outcome.
6. Riluzole versus placebo
One study contributed to this comparison, providing data on nineteen participants (Park 2017). Data were only available for acceptability (participant withdrawal) due to the trial ending prematurely due to futility. Table 6.
Primary outcomes
6.1 Efficacy: number of participants who respond to treatment
No data were available for this outcome.
6.2 Adverse events
No data were available for this outcome.
Secondary outcomes
6.3 Efficacy: number of participants who achieve remission
No data were available for this outcome.
6.4 Change scores on depression scale from baseline
No data were available for this outcome.
6.5 Suicidality
No data were available for this outcome.
6.6 Cognition
No data were available for this outcome.
6.7 Loss of hope or other health‐related quality of life measures
No data were available for this outcome.
6.8 Costs to healthcare services
No data were available for this outcome.
6.9 Acceptability: total dropouts
There was no difference in dropout rate between those receiving riluzole and placebo, based on very low quality evidence (OR 2.00, 95% CI 0.31 to 12.84; P = 0.46; participants = 19; studies = 1; I2 = 0%) (Analysis 6.1).
6.1. Analysis.

Comparison 6: Riluzole versus placebo, Outcome 1: Acceptability
Subgroup analyses
Due to the small number of included studies per comparison, we could not perform any of the pre‐planned subgroup analyses.
Discussion
Summary of main results
In this systematic review, we sought to appraise both the efficacy and acceptability of ketamine and other glutamate receptor modulators for the treatment of depressive symptoms in bipolar disorder. We identified five new randomised controlled trials (RCTs) (Bauer 2019; Berk 2019; Ellegaard 2019; Grunebaum 2017; Park 2017), which gave us a total of 10 RCTs with 647 participants and assessing five different interventions. Nine of these studies compared the experimental intervention with placebo (Anand 2012; Bauer 2019; Berk 2019; Diazgranados 2010; Ellegaard 2019; Lee 2014; Park 2017; Yoon 2009; Zarate 2012). One study compared ketamine with an active drug, midazolam, in addition to current psychotropic medication (Grunebaum 2017).
The previous version of the review (McCloud 2015) found that whilst the certainty of evidence ranged between low and very low, there was evidence of the efficacy of a single infusion of ketamine over placebo in terms of the primary outcome (response rate) at time points up to 24 hours. There was evidence that a single intravenous dose of ketamine was more effective than placebo in terms of the continuous efficacy outcome (mean change or endpoint severity score) at time points up to three days, with this effect disappearing at one week. However, these results indicated that any rapid antidepressant effects of ketamine are not sustained or long‐lasting. For the secondary efficacy outcome of remission rate, there was no difference between a single infusion ketamine and placebo at any time point, with no patients remitting after two weeks. Finally, there were not any significant differences between a single infusion of ketamine and placebo in terms of adverse effects, but this was likely due to the small amount of data available for this outcome.
The new trial examining ketamine versus midazolam found that there was no effect of ketamine over an active comparator for response rate or depression score at 24 hours, further supporting the results of previous trials (Grunebaum 2017). However, this was a pilot study with a small sample size of 16, so there is likely to have been insufficient power to find a statistically significant result.
These findings, demonstrating a rapid antidepressant effect of ketamine are quite similar to what we found in other Cochrane Reviews on unipolar depression (Caddy 2015; Dean 2021). However, the present review suggests that the antidepressant effect may be shorter in bipolar depression. Owing to the delayed onset of many other antidepressants (Berton 2006), these preliminary results of ketamine (among all other glutamate receptor modulators) may provide proof of principle for a new class of antidepressants with more rapid efficacy than currently achieved using monoaminergic modulators (Wang 2015).
Three new studies were included in this review which investigated N‐acetylcysteine versus placebo. There were extremely limited data available for all outcomes. We found no evidence for the efficacy of N‐acetylcysteine over placebo; moreover, placebo was found to be more effective at decreasing depression ratings than N‐acetylcysteine. N‐acetylcysteine was found to result in increased manic symptoms over placebo.
There was not enough evidence available to draw any reliable conclusions regarding the efficacy of memantine, cytidine, or riluzole.
Overall completeness and applicability of evidence
Although we carried out a thorough search, the overall completeness of evidence is limited. We obtained data on only 10 studies which met our inclusion criteria, and these investigated only five glutamate receptor modulators. We did not obtain data for seven of the prespecified interventions, and only one of the included studies involved an active comparator. For the main intervention (ketamine versus placebo) data were only available on five of nine predefined outcomes, on a total of 33 participants. This review is therefore limited by the very preliminary evidence in this area, although what is available suggests that further research is warranted to better inform clinical practice.
Several factors restrict the applicability of the evidence presently reviewed. Although all participants had received a DSM‐IV or DSM‐IV‐TR diagnosis of bipolar disorder, the baseline level of depression varied across participants, with one study including some patients within the ‘mild’ range according to the Hamilton Rating Scale for Depression (HRSD). Some studies attempted to define a ‘treatment‐resistant’ population for recruitment (only as having had an 'inadequate response so far' to open‐label mood stabilisers), whilst others treated patients who had not been prescribed psychotropic drugs before. One study included only those with a bipolar II diagnosis, whilst the remaining studies recruited a mixture. Almost all included studies allowed concomitant medications, but the majority of studies did not specify which mood stabilisers could be used as add‐on treatments. This heterogeneity did not translate into significant heterogeneity in the statistical analyses, however, the differences among the samples of patients studied in this review limited the applicability of this evidence to the wider population of patients with bipolar disorder. Moving towards a universally agreed upon definition of ‘treatment‐resistant’ depressive episodes in bipolar disorder would also be beneficial, in line with the focus on this in the unipolar literature (Kubitz 2013; Hidalgo‐Mazzei 2019). Seven of the studies did not mention the efficacy of previous treatments in the inclusion criteria, and the remaining three stated that participants were required to have had an 'inadequate response so far' to open‐label mood stabilisers.
Only one study included in this review used an active drug, midazolam, as a comparator. The majority of studies used placebo as a comparator, rather than the mood‐stabilising drugs which are more frequently used in practice, limiting the applicability of the evidence.
It should also be noted that the included ketamine studies all administered the drug as a single intravenous dose; adverse effects are likely to differ with intranasal administration or multiple doses.
Quality of the evidence
The certainty of the included studies was difficult to ascertain, owing to the fact that the majority of the risk of bias judgements were deemed ‘unclear’. This is a result of problems in study reporting but introduces the potential for bias within this review. In particular, ‘selection bias’ was deemed unclear for all of the included studies.
Although we attempted to reduce the risk of reporting bias by contacting all authors of included trials, many studies are also missing data for key time points. For example, the cytidine versus placebo comparison contains efficacy data at three months only, despite the tendency for other glutamate receptor modulators to have a rapid, short‐lived effect.
Overall, sample sizes were on average very small (more than one study had less than 10 participants per arm), which makes it difficult to draw meaningful conclusions. This resulted in wide confidence intervals, which lowered our confidence in the results for many of our outcomes by two levels according to GRADE. The lower limit for the confidence interval of the effect of ketamine on response at 24 hours when compared with placebo was compatible with a reasonably beneficial effect, so we considered this to warrant downgrading by one level rather than two in view of the small sample size (Table 2). It is also problematic to make comparisons between ketamine and the two other drugs, owing to the indirectness of this evidence.
An important factor to take into consideration is the bias that may have occurred in blinding procedures. Given the profile of ketamine and its psychotomimetic side effects, participants and personnel may not have remained blinded to treatment arm allocation, despite attempts to blind them. Neither of the two included studies assessing the efficacy of ketamine versus placebo tested the blinding or provided any information relating to whether the intended blinding was effective, but Diazgranados 2010 recognised the possibility that the dissociative effects might compromise study blinding. The one study assessing ketamine versus midazolam used a lower dose for safety and to minimise sedation that could unblind participants; however the study authors' testing of the blind revealed that 75% of participants guessed their intervention group correctly (Grunebaum 2017). Clinical assessors also had their blinding tested, and correctly guessed the groups of more than half of participants. This should be considered a major limitation for all ketamine studies, which is likely to result in a biased assessment of the intervention effect.
The retrieved data were also limited in their scope owing to study limitations. Substantial variation among the included studies was seen regarding concomitant medications. Four studies allowed other psychotropic medications to be taken throughout the trial (Anand 2012; Bauer 2019; Berk 2019; Grunebaum 2017), whilst others had strict washout periods (excepting relevant mood stabilisers) which varied in length. Eight studies required participants to receive mood stabilisers alongside the glutamate receptor modulator, but some participants were already taking these (and showing 'inadequate response'), whilst others began doing so after screening. This is a particular problem when it is considered that several studies only assessed participants against inclusion criteria at screening, rather than before the start of treatment. This could mean that an observed response for some participants was a result of the new mood stabiliser rather than the experimental drug. Dosages and titration schedules also differed, an issue which may have caused some conflicting results in the memantine studies.
The certainty of the evidence in the present review ranged from low to very low according to the GRADE approach and this information should be taken into account when interpreting results from this study.
Potential biases in the review process
We contacted the original study authors and were able to obtain supplemental data for the majority of included studies with unpublished information. Notwithstanding this, there are still outcome data missing from several of the pre‐planned analyses, which could have made an important contribution to this review with an impact on the final results. In order to include as much data as possible, we also imputed some dichotomous efficacy outcomes, using a validated method which has been employed in previous Cochrane Reviews (Cipriani 2010; Cipriani 2012; Cipriani 2013b; Guaiana 2010; Magni 2013; Purgato 2014). All imputed data were sent to the study authors for confirmation before we entered them into Review Manager 5 (RevMan 2014) for the statistical analyses. In the two ketamine studies (Diazgranados 2010; Zarate 2012), there were no data for adverse events from before cross‐over, so we included data from across both phases in order to include as much information as possible when assessing the tolerability of ketamine. The small number of included studies made it impossible to formally evaluate the potential for publication bias (i.e. with funnel plots). Whilst every effort was made to identify all relevant trials, we cannot rule out the possibility that unpublished trials remain unknown to us.
Agreements and disagreements with other studies or reviews
Other recently published reviews in the field have found that ketamine exerts a rapid effect that diminishes in efficacy around one to two weeks after infusion (Alberich 2017; Grady 2017; Kraus 2017; Kryst 2020). These reviews, though, have generally collated findings from both major depressive disorder and bipolar disorder, which is problematic owing to their likely differences in both biological basis and symptom presentation. Moreover, all previous reviews considered cumulative data from cross‐over studies. To overcome these limitations, we tried to be as rigorous as possible in our review, including only double‐blind or single‐blind randomised studies in bipolar depression and considering only data before crossing over in cross‐over trials (we did this according to Higgins 2011a, in order to reduce the risk of a 'carry over' treatment effect). Other reviews have found differing effect sizes for unipolar depression and bipolar disorder, where the effect at 24 hours was significantly larger for the former and at seven days was significantly larger for the latter (Coyle 2015). Our findings were different and, according to our results, ketamine could represent a treatment which is efficacious only in a very short time window and probably for a selected sample of patients.
As reported in other recent reviews, in terms of adverse events we did not manage to find very informative data (Coyle 2015; Naughton 2014; Niciu 2014). This is a relevant issue most of all for long‐term treatment. Some observational studies reported persisting reduction in frequent ketamine users compared to other groups in spatial working memory and pattern recognition memory, a trend for poorer performance in verbal recognition memory and a reduction in the percentage correct on the pattern recognition memory task, with a greater number of errors on the spatial working memory task (Morgan 2010). Cognitive impairment is particularly important in patients with bipolar disorder (Bauer 2014). It is important to highlight, however, that the same tasks did not show an impairment in healthy volunteers following an acute dose of ketamine (Honey 2003), so it is likely that these adverse events arise only after long‐term treatment.
Authors' conclusions
Implications for practice.
Overall, this review provides very limited evidence for an antidepressant effect of acute administration of ketamine (as an add‐on therapy to mood stabilisers) compared with placebo in the treatment of bipolar depression. Our confidence in the findings of the review is limited by the low number of trials overall and contributing data to the meta‐analysis for each comparison (Efthimiou 2019). The largest body of evidence included in a single forest plot incorporated only two studies (see the ketamine versus placebo and N‐acetylcysteine versus placebo comparisons). We found no evidence to support the use of other glutamate receptor modulators in bipolar depression.
The effect of ketamine was found to have a quick onset, which may be promising for clinical practice, but the effect was not long‐lasting. An important clinical implication for ketamine in bipolar depression would be in cases where a rapid response is crucial, for instance in patients at high risk of self‐harm or suicide (Smith 2018). However, the studies included in this review did not report adequate data about such important outcomes.
The three trials included in this review that studied ketamine administered the drug intravenously, which poses problems in clinical application (Goodwin 2016). The practicalities of the equipment, time and staff requirements limit the access and widespread clinical application. However, there may be potential for other methods of administration which would not pose as many challenges clinically, such as intranasal esketamine. A further important consideration is ketamine's psychotomimetic profile, which leads to question the abuse potential and liability in prescribing this drug to clinical populations (Bonaventura 2021).
In the present review, there was inconclusive information found on the side‐effect profile of ketamine, with the only available data being from both phases of cross‐over trials. The adverse events documented from long‐term ketamine abuse include cognitive impairment and bladder dysfunction (Malhi 2020c). It is therefore important that both short‐ and long‐term side effects are thoroughly evaluated in considering the clinical application of ketamine.
Implications for research.
We assessed the certainty of evidence in the present review as low to very low, according to GRADE. There were very few trials included overall as well as in each comparison, and sample sizes for each data point were usually very small. In order for robust conclusions to be drawn regarding the antidepressant effects of this drug in bipolar disorder, studies that are of a high methodological standard are required, with larger sample sizes and longer follow‐up periods. In order to generate high‐quality trials, future research should also focus on adequate blinding methods by using an active comparator. Additionally, there is a need for bipolar disorder studies which compare glutamate receptor modulators (and most of all, ketamine) with other active interventions, or as a monotherapy, in order to draw reliable conclusions about comparative efficacy between treatments (Cipriani 2020). Active intervention comparators should include mood‐stabilisers that are used in practice.
Long‐term adverse effects, particularly of repeated exposure to ketamine, remain a major concern in this area. The present review did not find conclusive evidence on the primary outcome of adverse events in ketamine, and it is therefore difficult to draw conclusions of the risk/benefit profile of the drug. Furthermore, the included studies involved only a single intravenous infusion. Morgan 2010 noted that frequent recreational users of the drug are more likely to show some cognitive impairments (such as impaired spatial working memory), dissociative and delusional symptoms, and even, interestingly, elevated depression scores. Therefore, further research is needed in order to assess the short‐ and long‐term side‐effect profile of ketamine.
In the present review the included ketamine studies administered the drug as a single intravenous dose, of which the practical limitations are outlined above. Preliminary evidence has suggested potential efficacy of other methods of administration, such as intranasal and intramuscular. It is, however, clear that further high‐quality research is needed to explore the efficacy and side‐effect profile of other forms of administration.
The longest trial included in this review examining the efficacy of ketamine was two weeks, which emphasises the short‐term nature of the trials to date. There may be potential to sustain ketamine's antidepressant effects through repeated administrations or combination treatment regimens, such as the delivery of psychotherapy or other medications following ketamine administration (McMullen 2021). Future research should therefore focus on conducting longer‐term trials and study ways in sustaining ketamine's antidepressant effects.
It would be beneficial for future research to assess whether (and how) glutamate receptor modulator efficacy would differ between bipolar I and bipolar II patients, which is an important factor that has not yet been considered. More research addressing the factors which distinguish bipolar depression from unipolar depression is necessary. The difference between individual diagnoses is an area which still requires consideration, as the role that bipolar versus unipolar diagnosis can play in treatment response to ketamine is still unclear. In fact, conventional antidepressants are generally not very efficacious in the bipolar disorder population (Taylor 2014), and some studies have found more success in patients with a family and/or personal history of alcohol dependence (Phelps 2009), which is promising given that this addiction is commonly comorbid with bipolar disorder.
In the presently reviewed studies, there is inconsistency regarding the allowance of concomitant medication. This is something worth focusing on in future bipolar research, owing to the frequent use of mood stabilisers in clinical practice. In particular, researchers should ensure that any observed effects cannot be attributed to mood stabilisers by only recruiting patients who have failed to show an adequate response to their current mood stabiliser (as in Zarate 2012 and Diazgranados 2010), and should move towards an operational definition for this.
Future research should use digital technology to better capture on a daily basis the variability of mood and its clinical implications (including sleep), using self‐reported measures on validated scales and remote monitoring systems (Stanislaus 2020).
What's new
| Date | Event | Description |
|---|---|---|
| 6 October 2021 | New search has been performed | Five new studies identified and added in this update. |
| 6 October 2021 | New citation required but conclusions have not changed | The review has been updated. |
History
Protocol first published: Issue 4, 2015 Review first published: Issue 9, 2015
Acknowledgements
We would like to thank Michael Grunebaum, Isabelle Bauer and Marsal Sanches for their help in retrieving and providing additional information on two included studies (Grunebaum 2017; Bauer 2019).
The National Institute for Health Research (NIHR) is the largest single funder of the Cochrane Common Mental Disorders (CCMD) Group. Andrea Cipriani is supported by the National Institute for Health Research (NIHR) Oxford Cognitive Health Clinical Research Facility, by an NIHR Research Professorship (grant RP‐2017‐08‐ST2‐006), by the NIHR Oxford and Thames Valley Applied Research Collaboration and by the NIHR Oxford Health Biomedical Research Centre (grant BRC‐1215‐20005). RM is supported by the Oxford Health Biomedical Research Council. KH and JRG are a NIHR Emeritus Senior Investigators.
The authors and the CCMD Editorial Team are also grateful to the following peer reviewers for their time and comments: Nuala Livingstone, Mrityunjai Kumar and S Rees. They would also like to thank Cochrane Copy Edit Support for the team's help
Disclaimer The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the NIHR, the National Health Service (NHS), or the Department of Health and Social Care.
Appendices
Appendix 1. Search strategies (2015‐2020)
Ovid MEDLINE databases
Ovid MEDLINE(R) Epub Ahead of Print, In‐Process & Other Non‐Indexed Citations, Ovid MEDLINE(R) Daily and Ovid MEDLINE(R) <1946 to July 28 2020> [Date limited 2015 onwards] Search Strategy: ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 depression/ 2 depressive disorder/ or depressive disorder, major/ or depressive disorder, treatment‐resistant/ or dysthymic disorder/ 3 *Mood Disorders/ or *Affective Symptoms/ 4 "bipolar and related disorders"/ or bipolar disorder/ 5 (depression or depressive? or MDD or dysthymi*).ti,ab,kf. 6 depressed.ti. or (depress* adj2 (mood? or bipolar or unipolar or adult? or clinical* or current* or chronic* or individuals or inpatients or outpatients or patients or participants or people or persons or population? or residents or subjects or symptoms or men or males or females or women or elders or elderly or seniors or veterans or volunteers)).ab,kf. 7 (affective disorder* or affective spectrum disorder* or affective state* or affective symptom* or mixed state* or mood disorder*).ti,ab,kf. 8 or/1‐7 9 Amantadine/ or Memantine/ 10 Atomoxetine Hydrochloride/ 11 Acetylcysteine/tu 12 Cycloserine/ 13 Dextromethorphan/ 14 *Excitatory Amino Acid Antagonists/tu 15 ((glutamate* or glutamin* or glutathione* or glycin*) adj2 (modulat* or inhibit* or system?)).ti,ab,kf,hw. 16 Ketamine/ 17 N‐Methylaspartate/ 18 Quinolines/tu 19 Riluzole/ 20 Sarcosine/ 21 Tramadol/ 22 *receptors, glutamate/ or *receptors, ionotropic glutamate/ or *receptors, ampa/ or *receptors, kainic acid/ or *receptors, n‐methyl‐d‐aspartate/ 23 receptors, glutamate/de, ai or receptors, ionotropic glutamate/de, ai or receptors, ampa/de, ai or receptors, kainic acid/de, ai or receptors, n‐methyl‐d‐aspartate/de, ag, ai 24 Glycine Plasma Membrane Transport Proteins/ai 25 (amantadin* or atomoxetin* or cycloserin* or d‐cycloserin* or DCS or dextromethorphan or (GLYX 13 or GLYX13 or rapastinel) or "MK 0657" or MK0657 or (ketamin* or ketalar or ketaject or ketanest) or (lanicemin* or AZD6765 or AZD 6765) or memantin* or quinolin* or rellidep or riluzol* or (tramadol* or ETS6103 or ETS 6103 or viotra) or ampa or cerc 301 or cerc301 or d‐serin* or GluN2B or mGlu* or N acetyl cysteine* or N acetylcysteine or N methyl D aspartate or NMDA? or nrx 1074 or nrx1074 or kainite or NR2B or sarcosin* or NAC).ti,ab,kf. 26 (Org 26576 or Org26576 or CP‐101,606 or CP101606).ti,ab,kf. 27 Cytidine.ti,ab,kf,hw. 28 or/9‐27 29 controlled clinical trial.pt. 30 randomized controlled trial.pt. 31 (randomi#ed or randomi#ation or randomi#ing).ti,ab,kf. 32 (RCT or "at random" or (random* adj3 (administ* or allocat* or assign* or class* or control* or crossover or cross‐over or design* or determine* or divide* or division or distribut* or expose* or fashion or number* or place* or recruit* or split or subsitut* or treat*))).ti,ab,kf. 33 placebo*.ab,ti,kf. 34 trial.ab,ti,kf. 35 groups.ab. 36 (control* and (trial or study or group*) and (placebo or waitlist* or wait* list* or ((treatment or care) adj2 usual))).ti,ab,kf,hw. 37 ((single or double or triple or treble) adj2 (blind* or mask* or dummy)).ti,ab,kf. 38 random allocation/ or single‐blind method/ or double‐blind method/ 39 or/29‐38 40 exp animals/ not humans.sh. 41 39 not 40 42 8 and 28 and 41 43 (2015* or 2016* or 2017* or 2018* or 2019* or 2020*).yr,dp,dt,ep,ez. 44 42 and 43 45 8 and (26 or 27) and 41 46 44 or 45 *************************** Ovid Embase <1980 to 2020 Week 30> [Date limited 2015 onwards] Search Strategy: 1 *depression/ or depression/dt or agitated depression/ or atypical depression/ or chronic depression/ or depressive psychosis/ or endogenous depression/ or involutional depression/ or late life depression/ or major depression/ or masked depression/ or "mixed anxiety and depression"/ or reactive depression/ or recurrent brief depression/ or treatment resistant depression/ 2 bipolar disorder/ or bipolar depression/ or bipolar i disorder/ or bipolar ii disorder/ or "mixed mania and depression"/ 3 mood disorder/ or major affective disorder/ or minor affective disorder/ 4 (depression or depressive? or MDD or TRD or dysthymi*).ti,ab,kw. 5 depressed.ti. or (depress* adj2 (mood? or bipolar or unipolar or adult? or clinical* or current* or chronic* or individuals or inpatients or outpatients or patients or participants or people or persons or population? or residents or subjects or symptoms or men or males or females or women or elders or elderly or seniors or veterans or volunteers)).ab,kw. 6 (affective spectrum disorder* or affective state* or mixed state*).ti,ab,kw. 7 or/1‐6 8 amantadine/ 9 memantine/ 10 atomoxetine/ 11 acetylcysteine/ 12 cycloserine/ 13 dextromethorphan/ 14 *glutamic acid/ 15 (glutamate* adj2 (modulat* or inhibit* or system?)).ti,ab,kw. 16 ketamine/ 17 Esketamine/ or Norketamine/ 18 n methyl dextro aspartic acid/ 19 *n methyl dextro aspartic acid receptor/ 20 quinoline/ 21 riluzole/ 22 Sarcosine/ 23 Tramadol/ 24 AZD 6765/ or "mk 0657"/ 25 *n methyl dextro aspartic acid receptor blocking agent/ or n methyl dextro aspartic acid receptor stimulating agent/ 26 *excitatory amino acid receptor/ or *glutamate receptor/ or exp *ionotropic receptor antagonist/ 27 AMPA receptor positive allosteric modulator/ 28 (amantadin* or atomoxetin* or cycloserin* or d‐cycloserin* or DCS or dextromethorphan or (GLYX 13 or GLYX13 or rapastinel) or "MK 0657" or MK0657 or (ketamin* or ketalar or ketaject or ketanest) or (lanicemin* or AZD6765 or AZD 6765) or memantin* or quinolin* or rellidep or riluzol* or (tramadol* or ETS6103 or ETS 6103 or viotra) or ampa or cerc 301 or cerc301 or d‐serin* or GluN2B or mGlu* or N acetyl cysteine* or N acetylcysteine or N methyl D aspartate or NMDA? or nrx 1074 or nrx1074 or kainite or NR2B or sarcosin* or NAC).ti,ab,kw. 29 (Org 26576 or Org26576 or CP‐101,606 or CP101606).ti,ab,kw,hw. 30 Cytidine.ti,ab,kw,hw. 31 or/8‐30 32 randomized controlled trial/ 33 randomization.de. 34 controlled clinical trial/ and (Disease Management or Drug Therapy or Prevention or Rehabilitation or Therapy).fs. 35 *clinical trial/ 36 placebo.de. 37 placebo.ti,ab. 38 trial.ti. 39 (randomi#ed or randomi#ation or randomi#ing).ti,ab,kw. 40 (RCT or "at random" or (random* adj3 (administ* or allocat* or assign* or class* or control* or determine* or divide* or division or distribut* or expose* or fashion or number* or place* or recruit* or split or subsitut* or treat*))).ti,ab,kw. 41 ((singl$ or doubl$ or trebl$ or tripl$) adj3 (blind$ or mask$ or dummy)).mp. 42 (control* and (trial or study or group) and (placebo or waitlist* or wait* list* or ((treatment or care) adj2 usual))).ti,ab,kw,hw. 43 or/32‐42 44 ((animal or nonhuman) not (human and (animal or nonhuman))).de. 45 43 not 44 46 7 and 31 and 45 47 (2015* or 2016* or 2017* or 2018* or 2019* or 2020*).yr,dc,dp 48 46 and 47 49 7 and (29 or 30) and 45 50 48 or 49 51 (review.ab. and review.pt.) not trial.ti. 52 50 not 51 *************************** Ovid PsycINFO <1806 to July Week 3> [Date limited 2015 onwards] Search Strategy: ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 major depression/ or anaclitic depression/ or dysthymic disorder/ or endogenous depression/ or late life depression/ or reactive depression/ or recurrent depression/ or treatment resistant depression/ 2 exp "Depression (Emotion)"/ or Atypical Depression/ 3 bipolar disorder/ 4 *Affective Disorders/ 5 (depression or depressive? or MDD or TRD or dysthymi*).ti,ab,id. 6 depressed.ti. or (depress* adj2 (mood? or bipolar or unipolar or adult? or clinical* or current* or chronic* or individuals or inpatients or outpatients or patients or participants or people or persons or population? or residents or subjects or symptoms or men or males or females or women or elders or elderly or seniors or veterans or volunteers)).ab,id. 7 (affective disorder* or affective spectrum disorder* or affective state* or affective symptom* or mixed state* or mood disorder*).ti,ab,id. 8 or/1‐7 9 amantadine/ 10 atomoxetine/ 11 glutamate receptors/ or glutamic acid/ 12 ketamine/ 13 n‐methyl‐d‐aspartate/ 14 tramadol/ 15 (amantadin* or atomoxetin* or cycloserin* or d‐cycloserin* or DCS or dextromethorphan or (GLYX 13 or GLYX13 or rapastinel) or "MK 0657" or MK0657 or (ketamin* or ketalar or ketaject or ketanest) or (lanicemin* or AZD6765 or AZD 6765) or memantin* or quinolin* or rellidep or riluzol* or (tramadol* or ETS6103 or ETS 6103 or viotra) or ampa or cerc 301 or cerc301 or d‐serin* or GluN2B or mGlu* or N acetyl cysteine* or N acetylcysteine or N methyl D aspartate or NMDA? or nrx 1074 or nrx1074 or kainite or NR2B or sarcosin* or NAC).ti,ab,id,hw. 16 (Org 26576 or Org26576 or CP‐101,606 or CP101606).ti,ab,id. 17 Cytidine.ti,ab,id. 18 or/9‐17 19 clinical trials.sh. 20 (randomi#ed or randomi#ation or randomi#ing).ti,ab,id. 21 (RCT or at random or (random* adj3 (assign* or allocat* or control* or crossover or cross‐over or design* or divide* or division or number))).ti,ab,id. 22 (control* and (trial or study or group) and (placebo or waitlist* or wait* list* or ((treatment or care) adj2 usual))).ti,ab,id,hw. 23 ((single or double or triple or treble) adj2 (blind* or mask* or dummy)).ti,ab,id. 24 trial.ti. 25 placebo.ti,ab,id,hw. 26 treatment outcome.md. 27 treatment effectiveness evaluation.sh. 28 mental health program evaluation.sh. 29 or/19‐28 30 8 and 18 and 29 31 (2015* or 2016* or 2017* or 2018* or 2019* or 2020*).yr,an. 32 30 and 31 33 8 and (16 or 17) and 29 34 32 or 33 *************************** Ovid XSearch: Esketamine MEDLINE, Embase, PsycINFO (all years, searched 30 July 2020) Search Strategy: ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 esketamine.mp. 2 (depression or depressive? or MDD or dysthymi*).mp. 3 depressed.ti. 4 (depress* adj2 (mood? or bipolar or unipolar or adult? or clinical* or current* or chronic* or individuals or inpatients or outpatients or patients or participants or people or persons or population? or residents or subjects or symptoms or men or males or females or women or elders or elderly or seniors or veterans or volunteers)).mp. 5 2 or 3 or 4 6 1 and 5 7 (randomi#ed or randomi#ation or randomi#ing or (RCT or "at random" or (random* adj3 (administ* or allocat* or assign* or class* or control* or crossover or cross‐over or design* or determine* or divide* or division or distribut* or expose* or fashion or number* or place* or recruit* or split or subsitut* or treat*)))).ti,ab,kf,kw,id. 8 (placebo* or trial).ab,ti,kf,kw,id. or groups.ab. 9 (control* and (trial or study or group*) and (placebo or waitlist* or wait* list* or ((treatment or care) adj2 usual))).ti,ab,kf,kw,id,hw. 10 ((single or double or triple or treble) adj2 (blind* or mask* or dummy)).ti,ab,kf,kw,id. 11 (controlled clinical trial or randomized controlled trial).pt. 12 random allocation/ or single‐blind method/ or double‐blind method/ 13 randomized controlled trial/ 14 randomization.de. 15 controlled clinical trial/ and (Disease Management or Drug Therapy or Prevention or Rehabilitation or Therapy).fs. 16 *clinical trial/ 17 placebo.de. 18 treatment outcome.md. or treatment effectiveness evaluation.sh. or mental health program evaluation.sh. 19 or/7‐18 20 6 and 19 21 remove duplicates from 20
*************************** Cochrane Central Register of Controlled Trials (CENTRAL) [All Years to Issue 7, 2020] IDSearch #1 MeSH descriptor: [Depression] this term only #2 MeSH descriptor: [Depressive Disorder] this term only #3 MeSH descriptor: [Depressive Disorder, Major] this term only #4 MeSH descriptor: [Depressive Disorder, Treatment‐Resistant] this term only #5 MeSH descriptor: [Dysthymic Disorder] this term only #6 MeSH descriptor: [Mood Disorders] this term only #7 MeSH descriptor: [Affective Symptoms] this term only #8 MeSH descriptor: [Bipolar and Related Disorders] explode all trees #9 (depress* or MDD or TRD or dysthymi*):ti,ab,kw (Word variations have been searched) #10 "affective disorder*" or "affective spectrum disorder*" or "affective state*" or "affective symptom*" or "mixed state*" or "mood disorder*":ti,ab,kw (Word variations have been searched) #11 #1 or #2 or #3 or #4 or #5 or #6 or #7 or #8 or #9 or #10 #12 MeSH descriptor: [Adamantane] explode all trees #13 MeSH descriptor: [Atomoxetine Hydrochloride] this term only #14 MeSH descriptor: [Acetylcysteine] this term only #15 MeSH descriptor: [Cycloserine] this term only #16 MeSH descriptor: [Dextromethorphan] this term only #17 MeSH descriptor: [Excitatory Amino Acid Antagonists] explode all trees #18 ((glutamate* or glutamin* or glutathione* or glycin*) near/2 (modulat* or inhibit* or system*)):ti,ab,kw (Word variations have been searched) #19 MeSH descriptor: [Ketamine] this term only #20 MeSH descriptor: [N‐Methylaspartate] this term only #21 MeSH descriptor: [Quinolines] this term only #22 MeSH descriptor: [Riluzole] this term only #23 MeSH descriptor: [Sarcosine] this term only #24 MeSH descriptor: [N‐substituted Glycines] this term only #25 MeSH descriptor: [Tramadol] this term only #26 MeSH descriptor: [Receptors, Glutamate] explode all trees #27 MeSH descriptor: [Glycine Plasma Membrane Transport Proteins] this term only #28 MeSH descriptor: [Glutamate Plasma Membrane Transport Proteins] explode all trees #29 (amantadin* or atomoxetin* or cycloserin* or d‐cycloserin* or DCS or dextromethorphan or ("GLYX 13" or GLYX13 or rapastinel) or "MK 0657" or MK0657 or (ketamin* or ketalar or ketaject or ketanest) or (lanicemin* or AZD6765 or "AZD 6765") or esketamine or memantin* or quinolin* or rellidep or riluzol* or (tramadol* or ETS6103 or "ETS 6103" or viotra) or ampa or "cerc 301" or cerc301 or d‐serin* or GluN2B or mGlu* or "acetyl cysteine*" or acetylcysteine or "N methyl D aspartate" or NMDA* or "nrx 1074" or nrx1074 or kainite or NR2B or sarcosin* or NAC):ti,ab,kw (Word variations have been searched) #30 "Org 26576" or Org26576 or CP‐101,606 or CP101606:ti,ab,kw (Word variations have been searched) #31 Cytidine:ti,ab,kw (Word variations have been searched) #32 MeSH descriptor: [Cytidine] this term only #33 #12 or #13 or #14 or #15 or #16 or #17 or #18 or #19 or #20 or #21 or #22 or #23 or #24 or #25 or #26 or #27 or #28 or #29 or #30 or #31 or #32 #34 #11 and #33 #35 MeSH descriptor: [Anesthesia and Analgesia] explode all trees #36 sedation or anesthe* or anaesthe*:ti (Word variations have been searched) #37 ((respiratory or respiration or myocardial) next depression) or (depressed blood pressure):ti,ab,kw (Word variations have been searched) #38 (depression next (co or si)):kw (Word variations have been searched) #39 analgesi*:ti (Word variations have been searched) #40 #34 not (#35 or #36 or #37 or #38 or #39) #41 SR‐DEPRESSN or HS‐DEPRESSN #42 #40 not #41 *************************** Trial Registers WHO International Clinical Trials Registry Platform 1. (depression AND acetylcysteine OR depression AND amantadine OR depression AND atomoxetine OR depression AND AZD6765 OR depression AND AZD 6765 OR depression AND cerc 301 OR depression AND cerc301 OR depression AND cycloserine OR depression AND d‐cycloserine OR depression AND dextromethorphan OR depression AND d‐serine OR depression AND ETS6103 OR depression AND ETS 6103 OR depression AND esketamine OR depression AND ketamine OR depression AND ketalar OR depression AND ketaject OR depression AND ketanest OR depression AND kainite OR depression AND lanicemine OR depression AND memantine OR depression AND norketamine OR depression AND MK 0657 OR depression AND MK0657 OR depression AND nrx 1074 OR depression AND nrx1074 OR depression AND N‐acetyl‐cysteinene OR depression AND N‐acetylcysteine OR depression AND N‐methyl‐D‐aspartate OR depression AND NMDA OR depression AND quinoline OR depression AND rapastinel OR depression AND rellidep OR depression AND GLYX 13 OR depression AND GLYX13 OR depression AND riluzole OR depression AND sarcosine OR depression AND Tramadol OR depression AND viotra) 2. (depression AND glutamic acid OR depression AND glutamatergic OR depression AND glutamate AND modulation OR depression AND ampa OR depression AND GluN2B OR depression AND mGlu* or depression AND NR2B) 3. (depressive AND acetylcysteine OR depressive AND amantadine OR depressive AND atomoxetine OR depressive AND AZD6765 OR depressive AND AZD 6765 OR depressive AND cerc 301 OR depressive AND cerc301 OR depressive AND cycloserine OR depressive AND d‐cycloserine OR depressive AND dextromethorphan OR depressive AND d‐serine OR depressive AND ETS6103 OR depressive AND ETS 6103 OR depressive AND esketamine OR depressive AND ketamine OR depressive AND ketalar OR depressive AND ketaject OR depressive AND ketanest OR depressive AND kainite OR depressive AND lanicemine OR depressive AND memantine OR depressive AND norketamine OR depressive AND MK 0657 OR depressive AND MK0657 OR depressive AND nrx 1074 OR depressive AND nrx1074 OR depressive AND N‐acetyl‐cysteinene OR depressive AND N‐acetylcysteine OR depressive AND N‐methyl‐D‐aspartate OR depressive AND NMDA OR depressive AND quinoline OR depressive AND rapastinel OR depressive AND rellidep OR depressive AND GLYX 13 OR depressive AND GLYX13 OR depressive AND riluzole OR depressive AND sarcosine OR depressive AND Tramadol OR depressive AND viotra) 4. (depressive AND glutamic acid OR depressive AND glutamatergic OR depressive AND glutamate AND modulation OR depressive AND ampa OR depressive AND GluN2B OR depressive AND mGlu* or depressive AND NR2B) 5. (bipolar AND acetylcysteine OR bipolar AND amantadine OR bipolar AND atomoxetine OR bipolar AND AZD6765 OR bipolar AND AZD 6765 OR bipolar AND cerc 301 OR bipolar AND cerc301 OR bipolar AND cycloserine OR bipolar AND d‐cycloserine OR bipolar AND dextromethorphan OR bipolar AND d‐serine OR bipolar AND ETS6103 OR bipolar AND ETS 6103 OR bipolar AND esketamine OR bipolar AND ketamine OR bipolar AND ketalar OR bipolar AND ketaject OR bipolar AND ketanest OR bipolar AND kainite OR bipolar AND lanicemine OR bipolar AND memantine OR bipolar AND norketamine OR bipolar AND MK 0657 OR bipolar AND MK0657 OR bipolar AND nrx 1074 OR bipolar AND nrx1074 OR bipolar AND N‐acetyl‐cysteinene OR bipolar AND N‐acetylcysteine OR bipolar AND N‐methyl‐D‐aspartate OR bipolar AND NMDA OR bipolar AND quinoline OR bipolar AND rapastinel OR bipolar AND rellidep OR bipolar AND GLYX 13 OR bipolar AND GLYX13 OR bipolar AND riluzole OR bipolar AND sarcosine OR bipolar AND Tramadol OR bipolar AND viotra) 6. (bipolar AND glutamic acid OR bipolar AND glutamatergic OR bipolar AND glutamate AND modulation OR bipolar AND ampa OR bipolar AND GluN2B OR bipolar AND mGlu* or bipolar AND NR2B) 7. or/1‐6 ClinicalTrials.gov depression OR depressive OR MDD OR bipolar AND acetylcysteine OR amantadine OR atomoxetine OR AZD6765 OR AZD 6765 OR cerc 301 OR cerc301 OR cycloserine OR d‐cycloserine OR dextromethorphan OR d‐serine OR ETS6103 OR ETS 6103 OR esketamine OR ketamine OR ketalar OR ketaject OR ketanest OR kainite OR lanicemine OR memantine OR norketamine OR MK 0657 OR MK0657 OR nrx 1074 OR nrx1074 OR N‐acetyl‐cysteinene OR N‐acetylcysteine OR N‐methyl‐D‐aspartate OR NMDA OR quinoline OR rapastinel OR rellidep OR GLYX 13 OR GLYX13 OR riluzole OR sarcosine OR Tramadol OR viotra OR glutamic acid OR glutamatergic OR glutamate modulation OR ampa OR GluN2B OR mGlu OR NR2B
Appendix 2. Searches to 2015 c/o Cochrane Common Mental Disorders Controlled Trials Register (CCMDCTR)
The information specialist with CCMD searched their specialised register (all years to 9 Jan 2015) using the following terms.
#1. (depress* or dysthymi* or "affective disorder*" or “affective spectrum disorder*” or “affective state*” or "affective symptom*" or "mixed state*" or "mood disorder*" or MDD or unipolar or bipolar):ti,ab,kw,ky,emt,mh,mc #2. (amantadin* or atomoxetin* or *cycloserin* or dextromethorphan or "GLYX 13" or "MK 0657" or (ketamin* or Ketalar or Ketaject or Ketanest) or (lanicemin* or AZD6765) or memantin* or quinolin* or rellidep or riluzol* or (tramadol* or ETS6103 or viotra) or ampa or “cerc 301” or “d serin*” or glun2b or glutamate or glutamin* or glutamatergic or glutathione* or glycin* or mglu* or "N acetyl cysteine*" or “N methyl D aspartate” or nmda or “nrx 1074” or kainite or nr2b or sarcosin* or NAC):ti,ab,kw,ky,emt,mh,mc #3. (#1 and #2)
[Key to field codes: ti:title; ab:abstract; kw:keywords: ky:additional keywords; emt:EMTREE headings; mh:MeSH headings; mc:MeSH checkwords]
Details of the CCMDCTR
The Cochrane Common Mental Disorders Group (CCMD) maintains two archived clinical trials registers at its editorial base in York, UK: a references register and a studies‐based register. The CCMDCTR‐References Register contains over 40,000 reports of RCTs in depression, anxiety and neurosis. Approximately 50% of these references have been tagged to individual coded trials. The coded trials are held in the CCMDCTR‐Studies Register and records are linked between the two registers through the use of unique Study ID tags. Coding of trials is based on the EU‐Psi coding manual, using a controlled vocabulary; (please contact the CCMD Information Specialists for further details). Reports of trials for inclusion in the Group's registers are collated from routine (weekly), generic searches of MEDLINE (1950 to 2016), Embase (1974 to 2016) and PsycINFO (1967 to 2016); quarterly searches of the Cochrane Central Register of Controlled Trials (CENTRAL) and review‐specific searches of additional databases. Reports of trials are also sourced from international trial registers via the World Health Organization's trials portal (the International Clinical Trials Registry Platform (ICTRP), pharmaceutical companies, the handsearching of key journals, conference proceedings and other (non‐Cochrane) systematic reviews and meta‐analyses.
Details of CCMD's generic search strategies (used to identify RCTs) can be found on the Group's website, (cmd.cochrane.org/specialised-register), with an example of the core MEDLINE search (used to inform the register) listed below. The Group’s Specialised Register has fallen out‐of‐date with the Editorial Group’s move from Bristol to York in the summer of 2016.
Core search strategy used to inform the Cochrane Common Mental Disorders Group's Specialised Register: OVID MEDLINE (to June 2016)
A weekly search alert based on condition + RCT filter only 1. [MeSH Headings]: eating disorders/ or anorexia nervosa/ or binge‐eating disorder/ or bulimia nervosa/ or female athlete triad syndrome/ or pica/ or hyperphagia/ or bulimia/ or self‐injurious behavior/ or self mutilation/ or suicide/ or suicidal ideation/ or suicide, attempted/ or mood disorders/ or affective disorders, psychotic/ or bipolar disorder/ or cyclothymic disorder/ or depressive disorder/ or depression, postpartum/ or depressive disorder, major/ or depressive disorder, treatment‐resistant/ or dysthymic disorder/ or seasonal affective disorder/ or neurotic disorders/ or depression/ or adjustment disorders/ or exp antidepressive agents/ or anxiety disorders/ or agoraphobia/ or neurocirculatory asthenia/ or obsessive‐compulsive disorder/ or obsessive hoarding/ or panic disorder/ or phobic disorders/ or stress disorders, traumatic/ or combat disorders/ or stress disorders, post‐traumatic/ or stress disorders, traumatic, acute/ or anxiety/ or anxiety, castration/ or koro/ or anxiety, separation/ or panic/ or exp anti‐anxiety agents/ or somatoform disorders/ or body dysmorphic disorders/ or conversion disorder/ or hypochondriasis/ or neurasthenia/ or hysteria/ or munchausen syndrome by proxy/ or munchausen syndrome/ or fatigue syndrome, chronic/ or obsessive behavior/ or compulsive behavior/ or behavior, addictive/ or impulse control disorders/ or firesetting behavior/ or gambling/ or trichotillomania/ or stress, psychological/ or burnout, professional/ or sexual dysfunctions, psychological/ or vaginismus/ or Anhedonia/ or Affective Symptoms/ or *Mental Disorders/ 2. [Title/ Author Keywords]: (eating disorder* or anorexia nervosa or bulimi* or binge eat* or (self adj (injur* or mutilat*)) or suicide* or suicidal or parasuicid* or mood disorder* or affective disorder* or bipolar i or bipolar ii or (bipolar and (affective or disorder*)) or mania or manic or cyclothymic* or depression or depressive or dysthymi* or neurotic or neurosis or adjustment disorder* or antidepress* or anxiety disorder* or agoraphobia or obsess* or compulsi* or panic or phobi* or ptsd or posttrauma* or post trauma* or combat or somatoform or somati#ation or medical* unexplained or body dysmorphi* or conversion disorder or hypochondria* or neurastheni* or hysteria or munchausen or chronic fatigue* or gambling or trichotillomania or vaginismus or anhedoni* or affective symptoms or mental disorder* or mental health).ti,kf. 3. [RCT filter]: (controlled clinical trial.pt. or randomized controlled trial.pt. or (randomi#ed or randomi#ation).ab,ti. or randomly.ab. or (random* adj3 (administ* or allocat* or assign* or class* or control* or determine* or divide* or distribut* or expose* or fashion or number* or place* or recruit* or subsitut* or treat*)).ab. or placebo*.ab,ti. or drug therapy.fs. or trial.ab,ti. or groups.ab. or (control* adj3 (trial* or study or studies)).ab,ti. or ((singl* or doubl* or tripl* or trebl*) adj3 (blind* or mask* or dummy*)).mp. or clinical trial, phase ii/ or clinical trial, phase iii/ or clinical trial, phase iv/ or randomized controlled trial/ or pragmatic clinical trial/ or (quasi adj (experimental or random*)).ti,ab. or ((waitlist* or wait* list* or treatment as usual or TAU) adj3 (control or group)).ab.) 4. (1 and 2 and 3) Records were screened for reports of RCTs within the scope of the Cochrane Common Mental Disorders Group. Secondary reports of RCTs were tagged to the appropriate study record. Similar weekly search alerts were also conducted on OVID Embase and PsycINFO, using relevant subject headings (controlled vocabularies) and search syntax, appropriate to each resource.
Appendix 3. Adverse events search
Ovid MEDLINE databases
Ovid MEDLINE(R) Epub Ahead of Print, In‐Process & Other Non‐Indexed Citations, Ovid MEDLINE(R) Daily and Ovid MEDLINE(R) <1946 to July 28 2020> [Date limited 2014 onwards] Search Strategy: ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 (adverse outcome* or complication* or drug fatalit* or drug hypersensitivity or drug reaction* or drug safety or drug tolerance or patient safety or safety or side effect* or contraindication*).ti,sh. 2 (safety or adverse or tolerability or tolerance or tolerat* or harm or harms or harmful or injur* or damage* or impair* complication* or risk or risks).ti,ab. 3 (side effect* or treatment emergent or undesirable effect*).ti,ab. 4 (suicid* or death*).mp. 5 (agitat* or constipat* or delusion* or diarrh* or dissociat* or dizz* or dry mouth or hallucinat* or headache* or hypoten* or hyperten* or insomni* or manic or mania or hypomani* or nause* or seizur* or sleep* or drows* or urin* or vomit* or temor*).ti,ab,sh. 6 ae.fs. 7 to.fs. 8 or/1‐7 9 (atomoxetine or "GLYX 13" or "MK 0657" or lanicemine or AZD6765 or rellidep).mp. 10 amantadine/ae, po, to 11 Ketamine/ae, po, to 12 Dextromethorphan/ae, po, to 13 Memantine/ae, po, to 14 Riluzole/ae, po, to 15 Cycloserine/ae, po, to 16 Quinidine/ae, po, to 17 Tramadol/ae, po, to 18 or/10‐17 19 (amantadine or Ketamine or Dextromethorphan or Memantine or Riluzole or Cycloserine or Quinidine or Tramadol).ti,sh. 20 (adverse outcome* or complication* or drug fatalit* or drug hypersensitivity* or drug reaction* or drug tolerance or safety or side effect* or contraindication* or tolerability or harm or harms or harmful or side effect* or treatment emergent or undesirable effect*).ti. 21 (depression or depressive or mood disorder* or affective disorder* or bipolar).ti,ab,sh. 22 exp animals/ not humans.sh. 23 exp Anesthesia/ 24 ((8 and 9 and 21) or ((18 or (19 and 20)) and 21)) not (22 or 23) 25 (2014* or 2015* or 2016* or 2017* or 2018* or 2019* or 2020*).yr,dp,dt,ep,ez. 26 24 and 25 27 (Org 26576 or Org26576 or CP‐101,606 or CP101606).mp. 28 Quinolinic Acid/ae, to [Adverse Effects, Toxicity] 29 Sarcosine/ae, to [Adverse Effects, Toxicity] 30 Cytidine/ae, to [Adverse Effects, Toxicity] 31 (cytidine or sarcosine or quinolinic acid).ti,sh. 32 8 and 27 33 ((21 and 28) or 29 or 30) not (22 or 23) 34 ((31 and 20) or (31 and 8 and 21)) not (22 or 23) 35 26 or 32 or 33 or 34 *************************** Ovid Embase <1980 to 2020 Week 30> [Date limited 2014 onwards] Search Strategy: ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 (adverse outcome* or complication* or drug fatalit* or drug hypersensitivity or drug reaction* or drug safety or drug tolerance or patient safety or safety or side effect* or contraindication*).ti,sh. 2 (safety or adverse or tolerability or tolerance or tolerat* or harm or harms or harmful or injur* or damage* or impair* complication* or risk or risks).ti,ab. 3 (side effect* or treatment emergent or undesirable effect*).ti,ab. 4 (suicid* or death*).mp. 5 (agitat* or constipat* or delusion* or diarrh* or dissociat* or dizz* or dry mouth or hallucinat* or headache* or hypoten* or hyperten* or insomni* or manic or mania or hypomani* or nause* or seizur* or sleep* or drows* or urin* or vomit* or temor*).ti,ab,sh. 6 ae.fs. 7 to.fs. 8 or/1‐7 9 ("GLYX 13" or "MK 0657" or lanicemine or AZD6765 or rellidep).mp. 10 (Org 26576 or Org26576 or CP‐101,606 or CP101606).mp. 11 *Ketamine/ae, to 12 *Atomoxetine/ae, to 13 *amantadine/ae, to 14 *Dextromethorphan/ae, to 15 *Memantine/ae, to 16 Riluzole/ae, to 17 *Cycloserine/ae, to 18 *Quinidine/ae, to 19 *Tramadol/ae, to 20 cytidine/ae, to or quinolinic acid/ae, to or sarcosine/ae, to 21 or/11‐20 22 (amantadine or atomoxetine or Ketamine or Dextromethorphan or Memantine or Riluzole or Cycloserine or Quinidine or Tramadol).ti,sh. 23 (cytidine or sarcosine or quinolinic acid).ti,sh. 24 (adverse outcome* or complication* or drug fatalit* or drug hypersensitivity* or drug reaction* or drug tolerance or safety or side effect* or contraindication* or tolerability or harm or harms or harmful or side effect* or treatment emergent or undesirable effect*).ti. 25 (depression or depressive or mood disorder* or affective disorder* or bipolar).ti,sh. 26 ((animals or nonhuman) not (humans and (animals or nonhuman))).sh. 27 exp *anesthesiological procedure/ 28 (8 and (9 or 10)) not (26 or 27) 29 (((or/11‐19) and 25) or (22 and 24 and 25)) not (26 or 27) 30 (2014* or 2015* or 2016* or 2017* or 2018* or 2019* or 2020*).yr,dc,dp. 31 29 and 30 32 ((20 and 25) or (23 and 24)) not (26 or 27) 33 28 or 31 or 32 *************************** Ovid PsycINFO <1806 to July Week 3> [Date limited 2014 onwards] Search Strategy: ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 (adverse outcome* or complication* or drug fatalit* or drug hypersensitivity or drug reaction* or drug safety or drug tolerance or safety or side effect* or contraindication* or toxicity).ti,id,sh,tm. 2 (safety or adverse or tolerability or tolerance or tolerat* or harm or harms or harmful or injur* or damage* or impair* complication* or risk or risks or toxicity).ti,id,ab. 3 (side effect* or treatment emergent or undesirable effect*).ti,id,ab. 4 (suicid* or death*).ti,ab,id,sh,tm. 5 (agitat* or constipat* or delusion* or diarrh* or dissociat* or dizz* or dry mouth or hallucinat* or headache* or hypoten* or hyperten* or insomni* or manic or mania or hypomani* or nause* or seizur* or sleep* or drows* or urin* or vomit* or temor*).ti,ab,id,sh,tm. 6 or/1‐5 7 (Ketamin* or Ketaject or Ketalar or Ketanest or Ketaset or Ketalean or Vetalar or amantadin* or atomoxetine or "GLYX 13" or "MK 0657" or lanicemine or AZD6765 or rellidep or dextromethorphan or memantine or riluzole or cycloserine or quinidine or tramadol).ti,ab,id,sh. 8 N‐Methyl‐D‐Aspartate/ 9 or/7‐8 10 (animal not ((human or inpatient or outpatient) and animal)).po. 11 (depression or depressive or mood disorder* or affective disorder* or bipolar).ti,id,sh,tm,ab. 12 (6 and 9 and 11) not 10 13 (2014* or 2015* or 2016* or 2017* or 2018* or 2019* or 2020*).yr,an. 14 12 and 13 15 (Org 26576 or Org26576 or CP‐101,606 or CP101606 or cytidine or sarcosine or quinolinic acid).ti,ab,id,sh 16 (15 and 6) not 10 17 14 or 16 ***************************
Adverse effects of ketamine and other glutamate receptor modulators (OVID databases to 11‐Nov‐2014) (Version 1)
OVID MEDLINE was searched using the following terms: 1. (adverse outcome* or complication* or drug fatalit* or drug hypersensitivity or drug reaction* or drug safety or drug tolerance or patient safety or safety or side effect* or contraindication*).ti,sh. 2. (safety or adverse or tolerability or tolerance or tolerat* or harm or harms or harmful or injur* or damage* or impair* complication* or risk or risks).ti,ab. 3. (side effect* or treatment emergent or undesirable effect*).ti,ab. 4. (suicid* or death*).mp. 5. (agitat* or constipat* or delusion* or diarrh* or dissociat* or dizz* or dry mouth or hallucinat* or headache* or hypoten* or hyperten* or insomni* or manic or mania or hypomani* or nause* or seizur* or sleep* or drows* or urin* or vomit* or temor*).ti,ab,sh. 6. ae.fs. [Floating Subheading: Adverse Effects ‐ MEDLINE] 7. to.fs. [Floating Subheading: Toxicity – MEDLINE] 8. ct.fs. [Floating Subheading: Contraindications ‐ MEDLINE] 9. or/1‐8 10. (atomoxetine or "GLYX 13" or "MK 0657" or lanicemine or AZD6765 or rellidep).mp. 11. *Amantadine/ae,to 12. *Cycloserine/ae,to 13. *Dextromethorphan/ae,to 14. *Ketamine/ae,to 15. *Memantine/ae,to 16. *Quinidine/ae,to 17. Riluzole/ae,to 18. *Tramadol/ae,to 19. or/11‐18 20. (amantadine or ketamine or dextromethorphan or memantine or riluzole or cycloserine or quinidine or tramadol).ti,sh. 21. (adverse outcome* or complication* or drug fatalit* or drug hypersensitivity* or drug reaction* or drug tolerance or safety or side effect* or contraindication* or tolerability or harm or harms or harmful or side effect* or treatment emergent or undesirable effect*).ti. 22. (depression or depressive or mood disorder* or affective disorder* or bipolar).ti,ab,sh. 23. exp animals/ not humans.sh. 24. exp *anesthesia 25. ((9 and 10 and 22) or ((19 or (20 and 21)) and 22)) not (23 or 24)
OVID EMBASE was searched using the following terms: 1. (adverse outcome* or complication* or drug fatalit* or drug hypersensitivity or drug reaction* or drug safety or drug tolerance or patient safety or safety or side effect* or contraindication*).ti,sh. 2. (safety or adverse or tolerability or tolerance or tolerat* or harm or harms or harmful or injur* or damage* or impair* complication* or risk or risks).ti,ab. 3. (side effect* or treatment emergent or undesirable effect*).ti,ab. 4. (suicid* or death*).mp. 5. (agitat* or constipat* or delusion* or diarrh* or dissociat* or dizz* or dry mouth or hallucinat* or headache* or hypoten* or hyperten* or insomni* or manic or mania or hypomani* or nause* or seizur* or sleep* or drows* or urin* or vomit* or temor*).ti,ab,sh. 6. ae.fs. [Floating Subheading: Adverse Drug Reaction ‐ EMBASE] 7. to.fs. [Floating Subheading: Drug Toxicity – EMBASE] 8. or/1‐7 9. ("GLYX 13" or "MK 0657" or lanicemine or AZD6765 or rellidep).mp. 10. *Amantadine/ae,to 11. *Atomoxetine/ae,to 12. *Cycloserine/ae,to 13. *Dextromethorphan/ae,to 14. *Ketamine/ae,to 15. *Memantine/ae,to 16. *Quinidine/ae,to 17. Riluzole/ae,to 18. *Tramadol/ae,to 19. or/10‐18 20. (amantadine or atomoxetine or ketamine or dextromethorphan or memantine or riluzole or cycloserine or quinidine or tramadol).ti,sh. 21. (adverse outcome* or complication* or drug fatalit* or drug hypersensitivity* or drug reaction* or drug tolerance or safety or side effect* or contraindication* or tolerability or harm or harms or harmful or side effect* or treatment emergent or undesirable effect*).ti. 22. (depression or depressive or mood disorder* or affective disorder* or bipolar).ti,sh. 23. ((animal*1 or nonhuman) not (human*1 and (animal*1 or nonhuman))).sh. 24. exp *anesthesiological procedure/ 25. ((8 and 9 and 22) or ((19 or (20 and 21)) and 22)) not (23 or 24)
OVID PsycINFO was searched using a more sensitive set of terms: 1. (adverse outcome* or complication* or drug fatalit* or drug hypersensitivity or drug reaction* or drug safety or drug tolerance or safety or side effect* or contraindication* or toxicity).ti,id,sh,tm. 2. (safety or adverse or tolerability or tolerance or tolerat* or harm or harms or harmful or injur* or damage* or impair* complication* or risk or risks or toxicity).ti,id,ab. 3. (side effect* or treatment emergent or undesirable effect*).ti,id,ab. 4. (suicid* or death*).ti,ab,id,sh,tm. 5. (agitat* or constipat* or delusion* or diarrh* or dissociat* or dizz* or dry mouth or hallucinat* or headache* or hypoten* or hyperten* or insomni* or manic or mania or hypomani* or nause* or seizur* or sleep* or drows* or urin* or vomit* or temor*).ti,ab,id,sh,tm. 6. or/1‐5 7. (ketamin* or ketaject or ketalar or ketanest or ketaset or ketalean or vetalar or amantadin* or atomoxetine or "GLYX 13" or "MK 0657" or lanicemine or AZD6765 or rellidep or dextromethorphan or memantine or riluzole or cycloserine or quinidine or tramadol).ti,ab,id,sh. 8. N‐Methyl‐D‐Aspartate/ or Tramadol/ 9. or/7‐8 10. (depression or depressive or mood disorder* or affective disorder* or bipolar).ti,ab,id,sh,tm. 11. (animal not ((human or inpatient or outpatient) and animal)).po. 12. (6 and 9 and 10) not 11
Data and analyses
Comparison 1. Ketamine versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 Response rate | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1.1 at 24 hours | 2 | 33 | Odds Ratio (M‐H, Random, 95% CI) | 11.61 [1.25, 107.74] |
| 1.1.2 at 3 days | 2 | 33 | Odds Ratio (M‐H, Random, 95% CI) | 8.24 [0.84, 80.61] |
| 1.1.3 at 1 week | 1 | 18 | Odds Ratio (M‐H, Random, 95% CI) | 4.00 [0.33, 48.66] |
| 1.2 Remission rate | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.2.1 at 24 hours | 2 | 33 | Odds Ratio (M‐H, Random, 95% CI) | 5.16 [0.51, 52.30] |
| 1.2.2 at 3 days | 2 | 33 | Odds Ratio (M‐H, Random, 95% CI) | 3.62 [0.34, 38.60] |
| 1.2.3 at 1 week | 1 | 18 | Odds Ratio (M‐H, Random, 95% CI) | 3.35 [0.12, 93.83] |
| 1.3 Depression rating scale score | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.3.1 at 24 hours | 2 | 32 | Mean Difference (IV, Fixed, 95% CI) | ‐11.81 [‐20.01, ‐3.61] |
| 1.3.2 at 3 days | 2 | 31 | Mean Difference (IV, Fixed, 95% CI) | ‐9.10 [‐16.00, ‐2.21] |
| 1.3.3 at 1 week | 2 | 28 | Mean Difference (IV, Fixed, 95% CI) | ‐0.88 [‐5.88, 4.12] |
| 1.3.4 at 2 weeks | 2 | 26 | Mean Difference (IV, Fixed, 95% CI) | ‐1.14 [‐6.30, 4.01] |
| 1.4 Acceptability ‐ total dropouts | 2 | 33 | Odds Ratio (M‐H, Random, 95% CI) | 3.48 [0.56, 21.74] |
| 1.5 Acceptability ‐ lack of efficacy | 2 | 33 | Odds Ratio (M‐H, Random, 95% CI) | 5.65 [0.76, 41.87] |
Comparison 2. Ketamine versus Midazolam.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 2.1 Response rate | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1.1 at 24 hours | 1 | 16 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.20 [0.23, 45.19] |
| 2.2 Remission rate | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.2.1 at 24 hours | 1 | 16 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.33 [0.07, 25.91] |
| 2.3 Depression rating scale score | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 2.3.1 at 24 hours | 1 | 16 | Mean Difference (IV, Fixed, 95% CI) | ‐5.85 [‐12.13, 0.43] |
| 2.4 Acceptability: adverse effects | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.4.1 at 24 hours | 1 | 16 | Odds Ratio (M‐H, Fixed, 95% CI) | Not estimable |
| 2.5 Acceptability: total dropouts | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2.6 Suicidality rating scale | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 2.6.1 at 24 hours | 1 | 16 | Mean Difference (IV, Fixed, 95% CI) | ‐5.86 [‐15.76, 4.04] |
Comparison 3. Memantine versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 3.1 Response rate | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1.1 at 1 week | 1 | 29 | Odds Ratio (M‐H, Random, 95% CI) | 1.08 [0.06, 19.05] |
| 3.1.2 at 2 weeks | 1 | 29 | Odds Ratio (M‐H, Random, 95% CI) | 4.88 [0.78, 30.29] |
| 3.1.3 at 4 weeks | 1 | 29 | Odds Ratio (M‐H, Random, 95% CI) | 5.33 [1.02, 27.76] |
| 3.1.4 at 3 months | 2 | 261 | Odds Ratio (M‐H, Random, 95% CI) | 1.66 [0.69, 4.03] |
| 3.2 Adverse events: Young Mania Rating Scale (12 weeks) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 3.3 Remission rate | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.3.1 at 1 week | 1 | 29 | Odds Ratio (M‐H, Random, 95% CI) | 1.08 [0.06, 19.05] |
| 3.3.2 at 2 weeks | 1 | 29 | Odds Ratio (M‐H, Random, 95% CI) | 1.77 [0.25, 12.60] |
| 3.3.3 at 4 weeks | 1 | 29 | Odds Ratio (M‐H, Random, 95% CI) | 3.67 [0.77, 17.43] |
| 3.3.4 at 3 months | 2 | 261 | Odds Ratio (M‐H, Random, 95% CI) | 1.74 [0.68, 4.46] |
| 3.4 Depression rating scale score | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 3.4.1 at 3 months | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 3.5 Suicidality: suicide attempts | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.6 Acceptability ‐ total dropouts | 2 | 261 | Odds Ratio (M‐H, Random, 95% CI) | 0.77 [0.45, 1.31] |
| 3.7 Acceptability ‐ lack of efficacy | 2 | 261 | Odds Ratio (M‐H, Random, 95% CI) | 0.61 [0.18, 2.02] |
| 3.8 Acceptability ‐ adverse events | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only |
Comparison 4. Cytidine versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 4.1 Response rate | 1 | 35 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.12 [0.30, 4.24] |
| 4.1.1 at 3 months | 1 | 35 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.12 [0.30, 4.24] |
| 4.2 Acceptability ‐ total dropouts | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only |
Comparison 5. N‐acetylcysteine versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 5.1 Response rate | 2 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5.1.1 At 3 months | 2 | 69 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.82 [0.32, 2.14] |
| 5.2 Adverse events: Young Mania Rating Scale | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 5.2.1 2 weeks | 1 | 80 | Mean Difference (IV, Fixed, 95% CI) | ‐0.90 [‐1.11, ‐0.69] |
| 5.2.2 3 months | 2 | 121 | Mean Difference (IV, Fixed, 95% CI) | ‐0.84 [‐1.08, ‐0.60] |
| 5.3 Depression rating scale score change | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 5.3.1 at 3 months | 2 | 58 | Mean Difference (IV, Fixed, 95% CI) | 1.28 [0.24, 2.31] |
| 5.4 Suicidality rating scale | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only |
Comparison 6. Riluzole versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 6.1 Acceptability | 1 | 19 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.00 [0.31, 12.84] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Anand 2012.
| Study characteristics | ||
| Methods | Double‐blind, randomised controlled trial | |
| Participants |
Diagnosis: DSM‐IV bipolar disorder; HRSD score ≥ 15; current depressed episode
N: 29 (outpatients) Age: memantine group M = 38 (SD = 15); placebo group M = 41 (SD = 14) Sex: memantine group 9 female + 5 male; placebo group 8 females + 7 males. Baseline depression severity: memantine group HRSD = 19 (SD = 4); placebo group HRSD = 19 (SD = 4) |
|
| Interventions | 8 weeks of treatment 100 mg/day lamotrigine in both arms, with either memantine or placebo as add‐on Memantine + lamotrigine ‐ week 1: 5 mg/day then increased weekly (depending on tolerability) to max 20 mg/day Placebo + lamotrigine ‐ capsules (Concomitant medication not mentioned) No washout period |
|
| Outcomes | Change in HRSD score Change in YMRS score Response rate (> 50% decrease in HRSD scores) Remission rate (final HRSD score < 8) Acceptability Adverse events Clinical global impression scores |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random number list generated by statistician sent to pharmacy |
| Allocation concealment (selection bias) | Unclear risk | Reported as double‐blind managed by pharmacy |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Matching active and placebo capsules |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not reported |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Withdrawals reported |
| Selective reporting (reporting bias) | High risk | Missing time points on HRSD. No continuous data available |
| Other bias | High risk | Quote:"Blind was opened after ten subjects completed the study to examine the side‐effect and tolerability profile of active memantine" |
Bauer 2019.
| Study characteristics | ||
| Methods | Double‐blind, randomised, placebo‐controlled trial | |
| Participants |
Diagnosis: DSM‐IV‐TR bipolar disorder; MADRS score ≥ 20; currently in a depressive or mixed episode.
N: 36 (22 depressed; 14 mixed) Age: placebo group M = 39.13 (SD = 9.99); aspirin + placebo group M = 49 (SD = 15.21); N‐acetylcysteine (NAC) + placebo group M = 36.38 (SD = 7.05); NAC + aspirin group M = 40 (SD = 17.64) Sex: placebo group 6/8 female; aspirin + placebo group 3/4 female; NAC + placebo group 5/8 female; NAC + aspirin group 1/4 female Baseline depression severity: placebo group MADRS M = 23.33 (SD = 4.719); aspirin + placebo MADRS M = 29.00 (11.314); NAC + placebo MADRS M = 19.50 (SD = 5.728); NAC + aspirin group M = 20.00 (SD =.000) ‐ data for depressive group only. |
|
| Interventions | Patients were randomly assigned to receive 1 of the following 4 treatments: aspirin (1000 mg/day) [500 mg twice daily], NAC (1000 mg/day [500 mg twice daily]), combined aspirin and NAC at same doses as
when administered separately, or placebo (sugar pill). Treatment with aspirin and/or NAC was adjunctive to patients’ ongoing treatment regimen (medications not specified) for a 16‐week period. |
|
| Outcomes | MADRS Response AEs IL‐6 CRP |
|
| Notes | Authors kindly provided supplementary data with results for patients experiencing a depressive episode only to separate this from the data of patients experiencing a mixed episode. Only data from patients experiencing a depressive episode are reported in this review. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation and group allocation was based on a computer‐generated allocation sequence |
| Allocation concealment (selection bias) | Low risk | Quote: “A researcher not otherwise involved in the trial and analysis carried out participant randomization and group allocation based on a computer‐generated allocation sequence” |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Stated but not tested. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Stated but not tested. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | CONSORT diagram included ‐ high number of people excluded, reasons all given. Similar withdrawal rates in all groups. Intention‐to‐treat analysis used. |
| Selective reporting (reporting bias) | Low risk | Trial registration available (NCT01797575). |
| Other bias | Unclear risk | None identified. |
Berk 2019.
| Study characteristics | ||
| Methods | Double‐blind, randomised, placebo‐controlled trial | |
| Participants |
Diagnosis: DSM‐IV‐TR bipolar disorder (I, II, or not otherwise specified) on MINI, MADRS score ≥ 20; current acute depressive episode
N: 181 Age: N‐acetylcysteine (NAC) group M = 44.9 (SD = 12.5), NAC+ combination of nutraceutical agents (CT) group M = 46.3 (SD = 12.7), placebo group M = 45.4 (SD = 11.9) Sex: NAC group = 61% female, NAC+CT group = 63.9% female, placebo group = 65.6% female Baseline depression severity: N‐acetylcysteine (NAC) group M = 28.8 (SD = 5.2), NAC+ combination of nutraceutical agents (CT) group M = 29.5 (SD = 5.6), placebo group M = 29.4 (SD = 5.6) |
|
| Interventions | 16 weeks treatment adjunctive to usual treatment (medications not specified) with 2000 mg/day NAC, 2000 mg/day NAC with the combination nutraceutical treatment, or placebo |
|
| Outcomes | MADRS HAM‐A BDRS YMRS CGI‐improvement CGI‐severity PGI‐I SOFAS LIFE‐RIFT Q‐LES‐Q‐SF |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “Participant number allocation to treatment arm was randomly assigned using permutated block randomisation. The computer‐generated randomisation plan was developed by an independent researcher utilising four‐to‐a‐block design. Participant numbers were sequentially allocated by trial clinicians.” (p2) |
| Allocation concealment (selection bias) | Low risk | Quote:“To facilitate the double‐blinding process, the trial medications (CT, NAC only, and placebo) were packed in the medicopacks and dispensed by an independent pharmacist in sealed containers. Medicopacks and capsules in all arms were identical, to conceal treatment allocation and blinding.” (p2) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote:“Medicopacks and capsules in all arms were identical, to conceal treatment allocation and blinding. The consultant statistician (SC), investigators, and participants were blinded to the group allocation.” (p2) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote:“The consultant statistician (SC), investigators, and participants were blinded to the group allocation”. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | CONSORT diagram included (p7) |
| Selective reporting (reporting bias) | Low risk | Protocol published, outcomes reported as expected. |
| Other bias | Unclear risk | None identified. |
Diazgranados 2010.
| Study characteristics | ||
| Methods | Randomised, double‐blind placebo‐controlled trial (cross‐over) | |
| Participants |
Diagnosis: DSM‐IV bipolar I or II depression without psychotic features; MADRS score ≥ 20; current major depressed episode for at least 4 weeks.
N: 18 randomised. Age: 47.9 years (SD = 13.1) Sex: 12 females, 6 males. Baseline depression severity: phase 1: Placebo group MADRS = 33.889 (SD = 4.833); ketamine group MADRS = 31.222 (SD = 4.410) |
|
| Interventions | Ketamine (9 in phase 1) vs placebo (9 in phase 1) as add‐on treatment to valproate or lithium, as mood stabilisers (continued taking as usual, but no other treatment allowed) 2 weeks (study duration) ketamine = 0.5 mg/kg single intravenous dose Intravenous saline solution as placebo 2‐week washout period |
|
| Outcomes | Change in MADRS scale HRSD‐17 score BDI Visual Analogue Scale Hamilton Anxiety Rating Scale BPRS Clinician Administered Dissociative Scale YMRS Response rate (50% improvement from BL in MADRS) Remission rate (MADRS score < 10) Dropout rate Adverse events |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: Patients were randomly assigned to the order in which they received the two infusions by a random number chart" |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | All staff, including the anaesthesiologist, were blind to whether placebo or drug was being administered. Study solutions were supplied in identical 50 mL syringes |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not reported |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Dropout rates reported and 'n' given for each time point |
| Selective reporting (reporting bias) | Unclear risk | No results tables available in original publication. All requested data received through correspondence |
| Other bias | Unclear risk | No other bias was identified in this study, but this possibility cannot be ruled out |
Ellegaard 2019.
| Study characteristics | ||
| Methods | Double‐blind, randomised, placebo‐controlled trial | |
| Participants |
Diagnosis: DSM‐IV bipolar disorder (I, II) on MINI, MADRS score ≥ 18; current acute depressive episode
N: 80 Age: N‐acetylcysteine (NAC) group M = 43.7 (SD = 10.0), placebo group M = 43.0 (SD = 10.2) Sex: NAC group = 65% female, placebo group = 52.5% female Baseline depression severity: N‐acetylcysteine (NAC) group MADRS M = 30.1 (SD = 7.9), placebo group M = 28.8 (SD = 7.1) |
|
| Interventions | Participants were randomised to receive 20 weeks of treatment with either NAC 3 mg/day or placebo in addition to treatment as usual (medications not specified). | |
| Outcomes | Response Remission Treatment emergent AEs MADRS Bech‐Rafaelsen Melancholia Scale YMRS WHO‐Five Well‐being index Global Assessment of Functioning scale Global Assessment of Symptoms scale CGI‐S |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were quote:“randomly allocated to NAC or placebo add‐on according to a pre‐constructed computer‐generated randomization list divided into blocks of eight.” |
| Allocation concealment (selection bias) | Low risk | Participants were Quote:“randomly allocated to NAC or placebo add‐on according to a pre‐constructed computer‐generated randomization list divided into blocks of eight.” |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No evidence of blind being tested |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not enough information provided. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | CONSORT diagram included, similar drop‐out rates in both arms. |
| Selective reporting (reporting bias) | Low risk | Protocol available, outcomes reported as expected. |
| Other bias | Unclear risk | None identified. |
Grunebaum 2017.
| Study characteristics | ||
| Methods | Controlled randomised, double‐blind, add‐on trial | |
| Participants |
Diagnosis: Bipolar disorder (DSM‐IV) with a current major depressive episode
N: 20 enrolled, 16 randomised Age: ketamine group mean = 39 (SD = 10.2); midazolam group mean = 43 (SD = 13.9) Sex: female = 10, male = 6 Baseline depression severity: ketamine group mean HDRS‐17 = 23.0 (SD = 5.1); midazolam group mean HDRS‐17 = 23.8 (SD = 4.1) |
|
| Interventions | Participants were randomised to receive double‐blinded treatment with either a single intravenous infusion over 40 minutes with racemic ketamine hydrochloride 0.5mg/kg or midazolam 0.02mg/kg in 100 mL of normal saline. Current medications were maintained except for benzodiazipines within 24 hours. Participants then received open‐label ketamine treatment for six months. | |
| Outcomes | Suicidal Ideation (SSI) HDRS‐17 BDI POMS WAIS‐III (reaction time, memory, language fluency, intelligence scale) Serum BDNF Cortisol CAR Systolic blood pressure Diastolic blood pressure Oxygen saturation Respiratory rate |
|
| Notes | Open‐label treatment not included in this review. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Permuted block design with 1:1 assignment between treatments and block size randomized between 4 and 6 with equal probability." |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment is not described. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Study was double‐blind, however, quote: "Of participants randomized to ketamine, five of seven correctly guessed their infusion drug during day 1 ratings versus seven of nine randomized to midazolam." |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: "Clinical assessors guessed correctly after four of seven ketamine and five of nine midazolam infusions.” |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Withdrawal rate is reported. No drop outs after randomisation, and outcome measures reported to have been completed by all participants. |
| Selective reporting (reporting bias) | High risk | No protocol available. Lots of outcome data is missing from the report. |
| Other bias | Unclear risk | None identified. |
Lee 2014.
| Study characteristics | ||
| Methods | Double‐blind randomised controlled trial | |
| Participants |
Diagnosis: DSM‐IV Bipolar II diagnosis, all with HRSD > 17 N: memantine group: 115 Placebo group: 117 Age: memantine group: 32.9 (SD = 12.02) Placebo group: 30.66 (SD = 11) Sex: memantine group: 53 males, 62 females Placebo group: 65 males, 52 females Baseline depression severity: memantine group: 19.20 (SD = 5.60) Placebo group: 19.22 (SD = 5.39) |
|
| Interventions | 13 weeks trial of memantine versus placebo as add‐on treatment to open‐label valproate continuation (500 mg and 1000 mg daily) Low dose memantine (5 mg/day) for 12 weeks Concomitant benzodiazepine medication (lorazepam < 8 mg) was used for night‐time sedation and to treat agitation and insomnia. Up to 20 mg daily fluoxetine was permitted for associated depressive symptoms Patients claimed to have never taken antidepressants/antipsychotics and had no history of taking memantine or mood stabilisers (no washout period) |
|
| Outcomes | Changes in depressive and manic symptoms (HRSD and YMRS scales) Adverse events Acceptability Effect of memantine on cytokine levels |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote:We conducted a double‐blind placebo‐controlled study |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not reported |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Does not specify when dropout occurred or whether LOCF is used |
| Selective reporting (reporting bias) | Unclear risk | Only baseline and endpoint continuous data reported in text (measured at weeks 1, 2, 4, 8 and 12), but all reported graphically |
| Other bias | Unclear risk | No other bias was identified in this study, but this possibility cannot be ruled out |
Park 2017.
| Study characteristics | ||
| Methods | Double‐blind placebo‐controlled pilot study | |
| Participants |
Diagnosis: DSM‐IV Bipolar disorder, MADRS score ≥20 N: riluzole N=8, placebo N=11 Age: Riluzole group: 45.25 (SD = 15.46) Placebo group: 47.64 (SD = 11.11) Sex: riluzole group: 7 males, 1 female Placebo group: 6 males, 5 females Baseline depression severity: data unavailable |
|
| Interventions | Participants were tapered off of any medications and were free of medications with central nervous system effects for seven days prior to the study and throughout the study, except for lorazepam as needed (up to 2 mg/day to manage agitation or anxiety). Riluzole (50 mg to 200mg/day) or placebo for eight weeks administered orally. Riluzole dosing began at 50 mg, twice daily, and was increased on a weekly basis by 50 mg, as tolerated, up to a maximum dose of 200 mg/day. |
|
| Outcomes | Dropout rate | |
| Notes | Trial ended prematurely due to futility. Limited data available. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote:"Randomized in a 1:1 allocation" |
| Allocation concealment (selection bias) | Unclear risk | Not reported. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No details given. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No details given. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Not significant. |
| Selective reporting (reporting bias) | High risk | Data from many outcomes not published. |
| Other bias | Unclear risk | None detected. |
Yoon 2009.
| Study characteristics | ||
| Methods | Randomised, double‐blind placebo‐controlled trial | |
| Participants |
Diagnosis: DSM‐IV Bipolar I or II diagnosis, all in depressed phase with HRSD > 18 N: cytidine group: 18 Placebo group: 17 Age: cytidine group: 33.5 (SD = 7.7) Placebo group: 36.8 (SD = 10.7) Sex: cytidine group: 9 males, 9 females Placebo group: 9 males, 8 females Baseline depression severity: cytidine group: 23.3 (SD = 2.3) Placebo group: 23.1 (SD = 2.0) |
|
| Interventions | 12‐week trial of cytidine vs placebo as add‐on treatment to valproate 1 mg twice per day of cytidine in capsules Placebo formulated as an inert fructose pill Valproate dosage changed until target plasma concentration achieved (50 mg to 100 mg/mL) over a 5‐day period Minimum 1 week washout period before randomisation (from all antimanic drugs or mood stabilisers other than valproate) Zolpidem (5 mg to 10 mg per day) for bedtime sedation and concomitant medications for stable medical conditions were permitted |
|
| Outcomes | Changes in HRSD scores from baseline Response rate (> 50% reduction in HRSD scores from baseline) Acceptability Adverse events Changes in cerebral glutamate/glutamine levels |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | 'double‐blind' |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not reported |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 2 participants in each condition dropped out, but no information available on whether LOCF was used, etc |
| Selective reporting (reporting bias) | Unclear risk | Measurements taken at weeks 1, 2, 3, 4 and 8 but only baseline reported in tables. All reported graphically |
| Other bias | Unclear risk | No other bias was identified in this study, but this possibility cannot be ruled out |
Zarate 2012.
| Study characteristics | ||
| Methods | Double‐blind randomised placebo‐controlled cross‐over study | |
| Participants |
Diagnosis: DSM‐IV bipolar I or II diagnosis without psychotic features, currently experiencing a major depressive episode of at least 4 weeks. MADRS > 19 at screening and at the start of each infusion N: 15 randomised. Age: 46.7 years (SD = 10.4) Sex: 8 females, 7 males. Baseline depression severity: ketamine group = 34.143 (SD = 5.429); placebo group = 35.625 (SD = 5.854) |
|
| Interventions | Ketamine (7 in phase 1) vs placebo (8 in phase 1) as add‐on treatment to either lithium or valproate within the specified range during the entirety of the study (levels obtained weekly) 0.5 mg/kg single dose intravenous ketamine infusions Placebo saline solution (0.9%) No concomitant treatment with psychotropic medications in 2 weeks before randomisation (5 weeks for fluoxetine) other than lithium or valproate (2‐week washout period) |
|
| Outcomes | MADRs scores HRSD scores BDI scores Visual Analogue Scale Hamilton Anxiety Rating Scale BPRS Clinician Administered Dissociative Scale YMRS Adverse events Response rates (50% improvement from baseline on MADRS) Remission rates (MADRS < 10) Effects on suicidal ideation |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomly assigned using a random number chart |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | All staff, including the anaesthesiologist, were blind to whether placebo or drug was being administered. Study solutions were supplied in identical 50 mL syringes |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not reported |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Dropout rates recorded and 'n' provided for each time point |
| Selective reporting (reporting bias) | Unclear risk | No results tables available in original publication. All requested data received through correspondence |
| Other bias | Unclear risk | No other bias was identified in this study, but this possibility cannot be ruled out |
AEs: adverse effects; BDI: Beck Depression Inventory; BDNF: Brain Derived Neurotrophic Factor; BDRS: Bipolar Depression Rating Scale; BL: Baseline;BL: Baseline; BPRS: Brief Psychiatric Rating Scale; CGI‐I: Clinical Global Impression;CRP: C‐reactive protein; DSM‐IV: Diagnostic and Statistical Manual of Mental Disorders, 4th Edition;HRSD: Hamilton Rating Scale for Depression; IL‐6; interleukin 6;LIFE‐RIFT: Range of Impaired Functioning Tool; LOCF: Last Observation Carried Forward; MADRS: Montgomery‐Asberg Depression Rating Scale; MINI: Mini International Neuropsychiatric Interview; NAC: N‐acetyl cysteine; PGI‐I: Patient Global Imression of Improvement; POMS: Profile of Mood States;Q‐LES‐Q: Quality of Life Enjoyment and Satisfaction Questionnaire; SD: standard deviation; SOFAS: Social and Occupational Functioning Assessment Scale; WAIS: Wechsler Adult Intelligence Scale; YMRS: Young Mania Rating Scale.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Alda 2017 | Incorrect intervention |
| Berk 2008 | Incorrect diagnosis |
| Berk 2010 | Incorrect population (no lower age limit) |
| Castillo 2017 | Incorrect intervention |
| Chen 2014 | Incorrect diagnosis (not all depressed) |
| Cocchi 1977 | Incorrect diagnosis (not all depressed) |
| Ehrensing 1978 | Incorrect diagnosis (mixed with unipolar) |
| Ellis 2014 | Wrong design |
| Kantrowitz 2015 | Wrong design |
| Lee 2012 | Incorrect diagnosis (not all depressed) |
| Lee 2017 | Wrong design |
| Luckenbaugh 2014 | Incorrect diagnosis (mixed with unipolar); secondary data |
Characteristics of ongoing studies [ordered by study ID]
ACTRN12612000830897.
| Study name | Mitochondrial agents in the treatment of bipolar disorder |
| Methods | Three‐arm, parallel randomised controlled trial |
| Participants | DSM‐IV bipolar disorder, current depressive phase (MADRS < 19), stable other therapy, 18+ |
| Interventions | 1. N‐acetylcysteine (NAC) capsules for 16 weeks (500 mg twice a day) 2. Acetyl L carnitine 500 mg + mitochondrial combination capsule + cardonutrient capsule for 16 weeks 3. Placebo treatment for 16 weeks |
| Outcomes | BL and every 4 weeks afterwards (6 visits) MADRS BDRS HAM‐A YMRS Impairment Functioning Tool SOFAS QLES‐Q CGI BP and CGI‐I Patient global impressions scale Change in blood oxidative and inflammatory markers |
| Starting date | 4/3/2013 |
| Contact information | Professor Michael Berk Mental Health Swanston Centre PO BOX 281 GEELONG VIC 3220 mikebe@barwonhealth.org.au |
| Notes | Recruiting |
ISRCTN14689382.
| Study name | Ketamine augmentation of ECT to improve outcomes in depression |
| Methods | Parallel RCT |
| Participants | Current DHRSD: SM‐IV diagnosis of a major depressive episode, moderate or severe as part of unipolar or bipolar disorder mood disorder 18+ years old Verbal IQ more than or equal to 85 |
| Interventions | Ketamine hydrochloride injection vs saline solution |
| Outcomes | HVLT‐R, AMI‐SD, COWAT MVG complex figure, GSE‐My MADRS more than or equal to 10 Number of ECT treatments to achieve response (50% MADRS decrease from baseline) CGI‐S, CGI‐I |
| Starting date | 1/5/2012 |
| Contact information | ian.anderson@manchester.ac.uk |
| Notes | Ongoing |
NCT01881763.
| Study name | Ketamine as an augmentation strategy for electroconvulsive therapy (ECT) in depression |
| Methods | Double‐blind, parallel randomised controlled trial |
| Participants | DSM‐IV unipolar or bipolar depression, 18‐70 years HRSD > 21 pre‐treatment MADRS > 19 at screening |
| Interventions | Ketamine versus methohexital (both IV) |
| Outcomes | Time to achieve remission (HRSD‐24) Cognitive side effects |
| Starting date | June 2010 |
| Contact information | Contact: Styliani Kaliora, M.D. skaliora@nshs.edu |
| Notes | Recruiting |
NCT03396068.
| Study name | RX‐101 for maintenance of remission from severe bipolar depression in patients with suicidal ideation (SBD‐ASIB) |
| Methods | Multi‐centre, randomised, stratified, double‐blind, parallel trial |
| Participants | DSM‐V and MINI bipolar depression, 18‐65 years Body mass index between 18‐35kg/m2 MADRS 30 at screening |
| Interventions | NRX‐101 (fixed =‐dose combination of D‐Cycloserine/lurasidone) versus lurasidone HCl (both oral) |
| Outcomes | MADRS C‐SSRS |
| Starting date | January 2019 |
| Contact information | Fred Grossman, D0 fgrossman@neurorxpharma.com |
| Notes | Not yet recruiting |
NCT03396601.
| Study name | NRX100 versus placebo for rapid stabilization of acute suicidal ideation and behavior in bipolar Depression (severe BD) |
| Methods | Multi‐centre, randomised, double‐blind, parallel trial |
| Participants | DSM‐V and MINI bipolar depression, 18 to 65 years Body mass index between 18 to 35kg/m2 MADRS 30 at screening |
| Interventions | Ketamine hydrochloride versus placebo |
| Outcomes | C‐SSRS |
| Starting date | January 2019 |
| Contact information | Fred Grossman, D0 fgrossman@neurorxpharma.com |
| Notes | Not yet recruiting |
AMI: alternate mark inversion; BDI: Beck Depression Inventory; BDRS: Bipolar Depression Rating Scale; BL: Baseline;CGI‐I: Clinical Global Impression – Global Improvement; CGI‐BP: Clinical Global Impression ‐ Bipolar;CGI‐S: Clinical Global Impression – Severity scale; C‐SSRS: Columbia‐Suicide Severity Rating Scale; DSM‐IV: Diagnostic and Statistical Manual of Mental Disorders, 4th Edition;ECT: ectroconvulsive therapy; HRSD: Hamilton Rating Scale for Depression;HVLT‐R: Hopkins Verbal Learning Test–Revised; IQ: intelligence quotient; IV: intravenous; MADRS: Montgomery‐Asberg Depression Rating Scale;MINI: Mini International Neuropsychiatric Interview; MMSE: Mini Mental State Examination;NAC: N‐acetyl cysteine; Q‐LES‐Q: Quality of Life Enjoyment and Satisfaction Questionnaire; SOFAS: Social and Occupational Functioning Assessment Scale; YMRS: Young Mania Rating Scale.
Differences between protocol and review
In order to address the comments of the peer reviewers, we decided to use a different threshold for depression severity (25 rather than 27 on HRSD‐17), and changed the references accordingly.
We removed the third objective, (’to investigate the adverse effects of ketamine and other glutamate receptor modulators in unipolar major depressive disorder, including general prevalence of adverse effects, compared with placebo or other antidepressant agents’) in order to make it clearer that whilst we did the search for adverse events data, in the end we only included data from RCTs.
Extra detail was added about the implementation of the random‐effects model in order to clarify methods used (see Data synthesis). The protocol stated: "We will use a random‐effects model because it has the highest generalisability for empirical examination of summary effect measures in meta‐analyses (Furukawa 2002). We will routinely examine the robustness of this summary measure by calculating the fixed‐effect model and random‐effects model ORs. We will report material differences between the models. We will calculate the pooled MD or SMD as appropriate with corresponding 95% CI for continuous outcomes. We will also use the random‐effects model for continuous outcomes. However, we will also routinely perform fixed‐effect analyses to investigate the effect of the choice of method on the effect estimates. We will report material differences between the models."
The following statement was added to the Types of interventions section: ’We did not include lamotrigine among the list of comparisons because the randomised evidence about this drug has been synthesised elsewhere (Thomas 2010; Zavodnick 2012)’.
We removed the statement: "We will also conduct a cited reference search on the Web of Science."
Contributions of authors
AC and KH conceived the review. RD, TM, SS, CH, AB and RS selected the studies, appraised their quality, and extracted data. RD, TM, SS, CH and RS entered the data into Review Manager 5 and RD carried out the analyses. RD drafted the manuscript and all other authors critically reviewed the text.
Sources of support
Internal sources
University of Oxford, UK
External sources
NIHR Oxford cognitive health Clinical Research Facility, UK
-
NIHR Oxford Health Biomedical Research Centre, UK
Grant BRC‐1215‐20005
-
NIHR Research Professorship, UK
Grant RP‐2017‐08‐ST2‐006
Declarations of interest
RD, TM, CH, SS, AB, RS, PJC, KH and JG report no competing interests.
RM runs NHS and self‐pay ketamine clinics for Oxford Health NHS Foundation Trust. RM has undertaken educational and scientific advisory board work for Janssen Pharmaceuticals to support educational and research activity, no funds are received personally. Janssen supported RM's attendance at the APA conference in New York in 2018. RM has undertaken scientific advisory board work for Sage pharmaceuticals, no funds are directly received. RM is supported by the NIHR Oxford Health Biomedical Research Centre.
GSM has received grant or research support from National Health and Medical Research Council, Australian Rotary Health, NSW Health, American Foundation for Suicide Prevention, Ramsay Research and Teaching Fund, Elsevier, AstraZeneca, Janssen‐Cilag, Lundbeck, Otsuka and Servier; and has been a consultant for AstraZeneca, Janssen‐Cilag, Lundbeck, Otsuka and Servier.
AC has received research and consultancy fees from INCiPiT (Italian Network for Paediatric Trials), CARIPLO Foundation and Angelini Pharma.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Anand 2012 {published data only}
- Anand A, Gunn AD, Barkay G, Karne HS, Nurnberger JI, Mathew SJ, et al. Early antidepressant effect of memantine during augmentation of lamotrigine inadequate response in bipolar depression: a double-blind, randomized, placebo-controlled trial. Bipolar Disorders 2012;14:64-70. [DOI] [PubMed] [Google Scholar]
Bauer 2019 {published and unpublished data}
- Bauer I, Green C, Coplo GD, Teixeira AL, Selvaraj, Durkin K, et al. A double-blind, randomized, placebo-controlled study of aspirin and N-Acetylcysteine as adjunctive treatments for bipolar depression. Journal of Clinical Psychiatry 2019;80(1):18m12200. [DOI] [PubMed] [Google Scholar]
Berk 2019 {published data only}
- Berk M, Turner A, Malhi GS, Ng C, Cotton SM, Dodd S, et al. A randomised controlled trial of a mitochondrial therapeutic target for bipolar depression: mitochondrial agents, N-acetylcysteine, and placebo. BMC Medicine 2019;17(1):18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Diazgranados 2010 {published and unpublished data}
- Diazgranados N, Ibrahim L, Brutsche NE, Newberg A, Kronstein P, Khalife S, et al. A randomized add-on trial of an N-methyl-D-aspartate antagonist in treatment-resistant bipolar depression. Archives of General Psychiatry 2010;67:793-802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lally N, Nugent AC, Luckenbaugh DA, Ameli R, Roiser JP, Zarate CA. Anti-anhedonic effect of ketamine and its neural correlates in treatment-resistant bipolar depression. Translational Psychiatry 2014;4:e469. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ellegaard 2019 {published data only}
- Ellegard PK, Licht RW, Nielson RE, Dean OM, Berk M, Poulsen HE, et al. The efficacy of adjunctive N-acetylcysteine in acute bipolar depression: a randomized placebo-controlled study. Journal of Affective Disorders 2019;245:1043-51. [DOI] [PubMed] [Google Scholar]
Grunebaum 2017 {published and unpublished data}
- Grunebaum MF, Ellis SP, Keilp JG, Moitra VK, Cooper TB, Marver JE, et al. Ketamine versus midazolam in bipolar depression with suicidal thoughts: a pilot midazolam-controlled randomized clinical trial. Bipolar Disorders March 2017;19:176-83. [DOI: 10.1111/bdi.12487] [DOI] [PubMed] [Google Scholar]
Lee 2014 {published data only}
- Lee SY, Chen SL, Chang YH, Chen PS, Huang SY, Tzeng NS, et al. The effects of add-on low-dose memantine on cytokine levels in bipolar II depression: a 12-week double-blind, randomized controlled trial. Journal of Clinical Psychopharmacology 2014;34:337-43. [DOI] [PubMed] [Google Scholar]
Park 2017 {published data only}
- Park LT, Lener MS, Luckenbaugh DA, Hopkins M, Iadorola N, Vieira Rodrigo M, et al. A double-blind, placebo-controlled, pilot study of riluzole monotherapy for acute bipolar depression. Journal of Clinical Psychopharmacology 2017;37(3):355-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Yoon 2009 {published data only}
- Yoon SJ, Lyoo IK, Haws C, Kim TS, Cohen BM, Renshaw PF. Decreased glutamate/glutamine levels may mediate cytidine's efficacy in treating bipolar depression: a longitudinal proton magnetic resonance spectroscopy study. Neuropsychopharmacology 2009;34(7):1810-8. [DOI] [PubMed] [Google Scholar]
Zarate 2012 {published and unpublished data}
- Lally N, Nugent AC, Luckenbaugh DA, Ameli R, Roiser JP, Zarate CA. Anti-anhedonic effect of ketamine and its neural correlates in treatment-resistant bipolar depression. Translational Psychiatry 2014;4:e469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarate CA, Brutsche NE, Ibrahim L, Franco-Chaves J, Diazgranados N, Cravchik A, et al. Replication of ketamine's antidepressant efficacy in bipolar depression: a randomized controlled add-on trial. Biological Psychiatry 2012;71:939-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies excluded from this review
Alda 2017 {published data only}
- Alda M, McKinnon M, Blagdon R, Garnham J, MacLellan S, O'donovan C, et al. Methylene blue treatment for residual symptoms of bipolar disorder: randomised crossover study. British Journal of Psychiatry 2017;210(1):54-60. [DOI] [PubMed] [Google Scholar]
Berk 2008 {published data only}
- Magalhães PV, Dean OM, Bush AI, Copolov DL, Weisinger D, Malhi GS, et al. Systemic illness moderates the impact of N-acetyl cysteine in bipolar disorder. Progress in Neuropsychopharmacology and Biological Psychiatry 2012;37(1):132-5. [DOI] [PubMed] [Google Scholar]
- Berk M, Copolov DL, Dean O, Lu K, Jeavons S, Schapkaitz I, et al. N-acetyl cysteine for depressive symptoms in bipolar disorder-a double-blind randomized placebo-controlled trial. Biological Psychiatry 2008;64(6):468-75. [DOI] [PubMed] [Google Scholar]
- Dean O, Berk M, Cotton SM, Bush AI, Gama CS, Kapczinski F, et al. N-acetyl cysteine (NAC) as an adjunctive therapy for bipolar depression conference poster P35. In: Bipolar disorders. Abstracts of the 9th International Conference on Bipolar Disorder; 2011 June 9-11. Vol. 13. Pittsburgh [PA], 2011:38.
Berk 2010 {published data only}
- Berk M, Dodd S, Dean OM, Kohlmann K, Berk L, Malhi GS. The validity and internal structure of the Bipolar Depression Rating Scale: data from a clinical trial of N-acetylcysteine as adjunctive therapy in bipolar disorder. Acta Neuropsychiatrica 2010;22(5):237-42. [DOI] [PubMed] [Google Scholar]
Castillo 2017 {published data only}
- Castillo MF, Murata S, Schwarz M, Martin B, Halaris A. Celecoxib augmentation of escitalopram in treatment-resistant bipolar depression: effects on trycats. Neuropsychopharmacology 2017;43:S176-7. [Google Scholar]
Chen 2014 {published data only}
- Chen SL, Lee SY, Chang YH, Chen PS, Wang TY, Lu RB. Therapeutic effects of add-on low-dose dextromethorphan plus valproic acid in bipolar disorder. European Neuropsychopharmacology 2014;24(11):1753-9. [DOI] [PubMed] [Google Scholar]
Cocchi 1977 {published data only}
- Cocchi R, Fusari A, Lorini G, Roccia L. Acupunctura, "vital" drugs and psychopharmacological agents in the treatment of psychiatric patients with deep depressive disorders, with considerations on the probable neurophysiological mechanisms of the synergism of action [article in Italian]. Minerva Medica 1977;68(33):2309-14. [PubMed] [Google Scholar]
Ehrensing 1978 {published data only}
- Ehrensing RH, Kastin AJ. Dose-related biphasic effect of prolyl-leucyl-glycinamide (MIF-I) in depression. American Journal of Psychiatry 1978;135(5):562-6. [DOI] [PubMed] [Google Scholar]
Ellis 2014 {published data only}
- Ellis JS, Luckenbaugh DA, Zarate CA, Furey ML. Effects of ketamine versus scopolamine on individual depression and anxiety symptoms (abstract). Biological Psychiatry 2014;75:127S. [Google Scholar]
Kantrowitz 2015 {published data only}
- Kantrowitz JT, Halberstam B, Gangwisch J. Single-dose ketamine followed by daily D-Cycloserine in treatment-resistant bipolar depression. Journal of Clinical Psychiatry 2015;76(6):737-8. [DOI] [PubMed] [Google Scholar]
Lee 2012 {published data only}
- Lee SY, Chen SL, Chang YH, Chen SH, Chu CH, Huang SY, et al. The DRD2/ANKK1 gene is associated with response to add-on dextromethorphan treatment in bipolar disorder. Journal of Affective Disorders 2012;138:295-300. [DOI] [PubMed] [Google Scholar]
Lee 2017 {published data only}
- Lee SY, Chen SL, Wang TY, Chang YH, Chen PS, Huang SY, et al. The COMT Val158Met Polymorphism is associated with response to add-on dextromethorphan treatment in bipolar disorder. Journal of Clinical Pharmacology 2017;37(1):94-8. [DOI] [PubMed] [Google Scholar]
Luckenbaugh 2014 {published data only}
- Luckenbaugh DA, Niciu MJ, Ionescu DF, Nolan NM, Richards EM, Brutsche NE, et al. Do the dissociative side effects of ketamine mediate its antidepressant effects? Journal of Affective Disorders 2014;159:56-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to ongoing studies
ACTRN12612000830897 {published data only}
- ACTRN12612000830897. A double blind, placebo controlled, randomised trial to evaluate the effect of mitochondrial agents, N-acetyl cysteine or placebo on the depressive phase of bipolar disorder. www.anzctr.org.au/Trial/Registration/TrialReview.aspx?ACTRN=12612000830897 (first received 23 July 2012).
ISRCTN14689382 {published data only}14689382
- ISRCTN14689382. Ketamine augmentation of ECT to improve outcomes in depression. www.isrctn.org/ISRCTN14689382.
NCT01881763 {published data only}
- NCT01881763. Comparing therapeutic efficacy and cognitive side effects of electroconvulsive therapy (ECT) using ketamine versus methohexital anesthesia. www.clinicaltrials.gov/show/NCT01881763 (first received 20 June 2013).
NCT03396068 {unpublished data only}
- NCT03396068. NRX-101 for maintenance of remission from severe bipolar depression in patients with suicidal ideation. ClinicalTrials.gov/show/NCT03396068 (first received 10 January 2018).
NCT03396601 {unpublished data only}
- NCT03396601. NRX100 vs. placebo for rapid stabilization of acute suicidal ideation and behavior in bipolar depression (SevereBD). ClinicalTrials.gov/show/NCT03396601 (first received 11 January 2018).
Additional references
Abdallah 2018
- Abdallah C, Averill LA, Gueorguieva R, Goktas S, Purohit P, Ranganathan M, et al. Rapamycin, an immunosuppressant and mTORC1 inhibitor, triples the antidepressant response rate of ketamine at 2 weeks following treatment: a double-blind, placebo-controlled, cross-over, randomized clinical trial. bioRxiv 2018. [DOI: doi.org/10.1101/500959 ]
Alberich 2017
- Alberich S, Martínez-Cengotitabengoa M, López P, Zorrilla I, Núñez N, Vieta E, et al. Efficacy and safety of ketamine in bipolar depression: a systematic review. Revista de Psiquiatría y Salud Mental 2017;10(2):104-12. [DOI: 10.1016/j.rpsm.2016.05.005] [DOI] [PubMed] [Google Scholar]
Altman 1996
- Altman DG, Bland JM. Detecting skewness from summary information. BMJ 1996;313(7066):1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
APA 1980
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 3rd edition. Washington D.C.: American Psychiatric Association, 1980. [Google Scholar]
APA 1987
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 3rd edition. Washington D.C.: American Psychiatric Association, 1987. [Google Scholar]
APA 1994
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4th edition. Washington, D.C.: American Psychiatric Association, 1994. [Google Scholar]
APA 2000
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders - Text Revision (DSM-IV-TR). 4th edition. Washington, D.C.: American Psychiatric Association, 2000. [Google Scholar]
APA 2013
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th edition. Arlington (VA): American Psychiatric Association, 2013. [Google Scholar]
Arroll 2009
- Arroll B, Elley CR, Fishman T, Goodyear-Smith FA, Kenealy T, Blashki G, et al. Antidepressants versus placebo for depression in primary care. Cochrane Database of Systematic Reviews 2009, Issue 3. Art. No: CD007954. [DOI: 10.1002/14651858.CD007954] [DOI] [PMC free article] [PubMed] [Google Scholar]
Atkins 2004
- Atkins D, Best D, Briss PA, Eccles M, Falck-Ytter Y, Flottorp S, et al, GRADE Working Group. Grading quality of evidence and strength of recommendations. BMJ 2004;328(7454):1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bandelow 2006
- Bandelow B, Baldwin DS, Dolberg OT, Andersen HF, Stein DJ. What is the threshold for symptomatic response and remission for major depressive disorder, panic disorder, social anxiety disorder, and generalized anxiety disorder? Journal of Clinical Psychiatry 2006;67(9):1428–34. [DOI] [PubMed] [Google Scholar]
Bauer 2014
- Bauer IE, Pascoe MC, Wollenhaupt-Aguiar B, Kapczinski F, Soares JC. Inflammatory mediators of cognitive impairment in bipolar disorder. Journal of Psychiatric Research 2014;56:18-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
Berton 2006
- Berton O, Nestler EJ. New approaches to antidepressant drug discovery: beyond monoamines. Nature Reviews. Neuroscience 2006;7(2):137–51. [DOI] [PubMed] [Google Scholar]
Bonaventura 2021
- Bonaventura J, Lam S, Carlton M, Boehm MA, Gomez JL, Solís O, et al. Pharmacological and behavioral divergence of ketamine enantiomers: implications for abuse liability. Molecular Psychiatry 2021;15 April:Epub ahead of print. [DOI: 10.1038/s41380-021-01093-2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Brookes 2001
- Brookes ST, Whitley E, Peters TJ, Mulheran PA, Egger M, Davey Smith G. Subgroup analyses in randomized controlled trials: quantifying the risks of false-positives and false-negatives. Health Technology Assessment 2001;5(33):1–56. [DOI] [PubMed] [Google Scholar]
Brookes 2004
- Brookes ST, Whitely E, Egger M, Smith GD, Mulheran PA, Peters TJ. Subgroup analyses in randomized trials: risks of subgroup-specific analyses; power and sample size for the interaction test. Journal of Clinical Epidemiology 2004;57(3):229-36. [DOI] [PubMed] [Google Scholar]
Caddy 2015
- Caddy C, Amit BH, McCloud TL, Rendell JM, Furukawa TA, McShane R, et al. Ketamine and other glutamate receptor modulators for depression in adults. Cochrane Database of Systematic Reviews 2015, Issue 9. Art. No: CD011612. [DOI: 10.1002/14651858.CD011612.pub2] [DOI] [PubMed] [Google Scholar]
Chitty 2013
- Chitty KM, Lagopoulos J, Lee RS, Hickie IB, Hermens DF. A systematic review and meta-analysis of proton magnetic resonance spectroscopy and mismatch negativity in bipolar disorder. European Neuropsychopharmacology 2013;23(11):1348–63. [DOI: 10.1016/j.euroneuro.2013.07.007] [DOI] [PubMed] [Google Scholar]
Cipriani 2009
- Cipriani A, Furukawa TA, Salanti G, Geddes JR, Higgins JP, Churchill R, et al. Efficacy and acceptability of 12 new-generation antidepressants: a multiple-treatments meta-analysis. Lancet 2009;373(9665):746–58. [DOI] [PubMed] [Google Scholar]
Cipriani 2010
- Cipriani A, La Ferla T, Furukawa TA, Signoretti A, Nakagawa A, Churchill R, et al. Sertraline versus other antidepressive agents for depression. Cochrane Database of Systematic Reviews 2010, Issue 4. Art. No: CD006117. [DOI: 10.1002/14651858.CD006117.pub4] [DOI] [PMC free article] [PubMed] [Google Scholar]
Cipriani 2012
- Cipriani A, Purgato M, Furukawa TA, Trespidi C, Imperadore G, Signoretti A, et al. Citalopram versus other anti-depressive agents for depression. Cochrane Database of Systematic Reviews 2012, Issue 7. Art. No: CD006534. [DOI: 10.1002/14651858.CD006534.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Cipriani 2013a
- Cipriani A, Hawton K, Stockton S, Geddes JR. Lithium in the prevention of suicide in mood disorders: updated systematic review and meta-analysis. BMJ 2013;346:f3646. [DOI] [PubMed] [Google Scholar]
Cipriani 2013b
- Cipriani A, Koesters M, Furukawa TA, Nosè M, Purgato M, Omori IM, et al. Duloxetine versus other anti-depressive agents for depression. Cochrane Database of Systematic Reviews 2012, Issue 10. Art. No: CD006533. [DOI: 10.1002/14651858.CD006533.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Cipriani 2020
- Cipriani A, Ioannidis JPA, Rothwell PM, Glasziou P, Li T, Hernandez AF, et al. Generating comparative evidence on new drugs and devices after approval. Lancet 2020;395(10228):998-1010. [DOI: 10.1016/S0140-6736(19)33177-0] [DOI] [PubMed] [Google Scholar]
Cohen 2019
- Coehn, BM. Evidence-based drug treatment of acute depression in bipolar disorder. JAMA Psychiatry 2019;76(12):1314-5. [DOI] [PubMed] [Google Scholar]
Coyle 2015
- Coyle CM, Laws KR. The use of ketamine as an antidepressant: a systematic review and meta‐analysis. Human Psychopharmacology: Clinical and Experimental 2015;30(3):152-63. [DOI] [PubMed] [Google Scholar]
Cunningham 2000
- Cunningham MO, Jones RS. The anticonvulsant, lamotrigine decreases spontaneous glutamate release but increases spontaneous GABA release in the rat entorhinal cortex in vitro. Neuropharmacology 2000;39(11):2139-46. [DOI: 10.1016/s0028-3908(00)00051-4] [DOI] [PubMed] [Google Scholar]
Dean 2021
- Dean RL, Hurducas C, Hawton K, Spyridi S, Cowen PJ, Hollingsworth S, et al. Ketamine and other glutamate receptor modulators for depression in adults with unipolar major depressive disorder. Cochrane Database of Systematic Reviews 2021, Issue 9. Art. No: CD011612. [DOI: 10.1002/14651858.CD011612.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Dozois 2004
- Dozois DJ, Dobson KS. Depression. In: Antony MM, Barlow DH, editors(s). Handbook of Assessment and Treatment Planning for Psychological Disorders. New York: Guilford Press, 2004:259-99. [Google Scholar]
Efthimiou 2019
- Efthimiou O. Statistics in pills: meta-analysis of rare events. Evidence Based Mental Health 2019;22(3):102. [DOI: 10.1136/ebmental-2019-300103] [DOI] [PMC free article] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta-analysis detected by a simple, graphical test. BMJ 1997;315(7109):629–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne DR, Altman DG, Higgins JP, Curtin F, Worthington HV, Vail A. Meta-analyses involving crossover trials: methodological issues. International Journal of Epidemiology 2002;31(1):140–9. [DOI] [PubMed] [Google Scholar]
Feighner 1972
- Feighner JP, Robins E, Guze SB, Woodruff RA, Winokur G, Munoz R. Diagnostic criteria for use in psychiatric research. Archives of General Psychiatry 1972;26(1):57–63. [DOI] [PubMed] [Google Scholar]
Furukawa 2002
- Furukawa TA, Guyatt GH, Griffith LE. An empirical study of summary effect measures in meta-analyses. International Journal of Epidemiology 2002;31(1):72–6. [DOI] [PubMed] [Google Scholar]
Furukawa 2005
- Furukawa TA, Cipriani A, Barbui C, Brambilla P, Watanabe N. Imputing response rates from means and standard deviations in meta-analysis. International Clinical Psychopharmacology 2005;20(1):49–52. [DOI] [PubMed] [Google Scholar]
Furukawa 2006
- Furukawa TA, Barbui C, Cipriani A, Brambilla P, Watanabe N. Imputing missing standard deviations in meta-analyses can provide accurate results. Journal of Clinical Epidemiology 2006;59(1):7–10. [DOI] [PubMed] [Google Scholar]
Furukawa 2007a
- Furukawa TA, Watanabe N, Omori IM, Montori VM, Guyatt GH. Association between unreported outcomes and effect size estimates in Cochrane meta-analyses. JAMA 2007;297(5):468–70. [DOI] [PubMed] [Google Scholar]
Furukawa 2007b
- Furukawa TA, Akechi T, Azuma H, Okuyama T, Higuchi T. Evidence-based guidelines for interpretation of the Hamilton Rating Scale for Depression. Journal of Clinical Psychopharmacology 2007;27(5):531–4. [DOI] [PubMed] [Google Scholar]
Geddes 2013
- Geddes JR, Miklowitz DJ. Treatment of bipolar disorder. Lancet 2013;381(9878):1672-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
Gigante 2012
- Gigante AD, Bond DJ, Lafer B, Lam RW, Young LT, Yatham LN. Brain glutamate levels measured by magnetic resonance spectroscopy in patients with bipolar disorder: a meta-analysis. Bipolar Disorders 2012;14:478-87. [DOI: doi: 10.1111/j.1399-5618.2012.01033.x] [DOI] [PubMed] [Google Scholar]
Godlewska 2019
- Godlewska BR, Emir UE, Masaki C, Bargiotas T, Cowen PJ. Changes in brain Glx in depressed bipolar patients treated with lamotrigine: a proton MRS study. Journal of Affective Disorders 2019;246:418-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Goodwin 2016
- Goodwin GM, Haddad PM, Ferrier IN, Aronson JK, Barnes T, Cipriani A, et al. Evidence-based guidelines for treating bipolar disorder: revised third edition recommendations from the British Association for Psychopharmacology. Journal of Psychopharmacology 2016;30(6):495-553. [DOI: 10.1177/0269881116636545] [DOI] [PMC free article] [PubMed] [Google Scholar]
GRADEproGDT 2015 [Computer program]
- McMaster University (developed by Evidence Prime, Inc.) GRADEproGDT: GRADEpro Guideline Development Tool [www.guidelinedevelopment.org]. Version 2015. Hamilton: McMaster University (developed by Evidence Prime, Inc.), August 28th 2015.
Grady 2017
- Grady SE Marsh TA, Tenhouse A, Klein K. Ketamine for the treatment of major depressive disorder andbipolar depression: a review of the literature. Mental Health Clinician 2017;7(1):16-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
Guaiana 2010
- Guaiana G, Barbui C, Cipriani A. Hydroxyzine for generalised anxiety disorder. Cochrane Database of Systematic Reviews 2010, Issue 12. Art. No: CD006815. [DOI: 10.1002/14651858.CD006815.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Guy 1976
- Guy W. ECDEU Assessment Manual for Psychopharmacology. Rockville (MD): U.S. Department of Health and Human Services, 1976. [Google Scholar]
Guyatt 1998
- Guyatt GH, Juniper EF, Walter SD, Griffith LE, Goldstein RS. Interpreting treatment effects in randomised trials. BMJ 1998;316(7132):690–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hamilton 1960
- Hamilton M. A rating scale for depression. Journal of Neurology, Neurosurgery and Psychiatry 1960;23:56-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hidalgo‐Mazzei 2019
- Hidalgo-Mazzei D, Berk M, Cipriani A, Cleare AJ, Di Florio A, Dietch D, et al. Treatment-resistant and multi-therapy-resistant criteria for bipolar depression: consensus definition. British Journal of Psychiatry 2019;214(1):27-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta-analyses. BMJ 2003;327(7414):557–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011a
- Higgins JP, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions. Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane-handbook.org.
Higgins 2011b
- Higgins JP, Altman DG, Sterne JA (editors). Chapter 8: Assessing risk of bias in included studies. In: Higgins JP, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions. Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane–handbook.org.
Higgins 2011c
- Higgins JP, Deeks JJ, Altman DG (editors). Chapter 16: Special topics in statistics. In: Higgins JP, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions. Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane–handbook.org.
Higgins 2011d
- Higgins JP, Deeks JJ (editors). Chapter 7: Selecting studies and collecting data. In: Higgins JP, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions. Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane–handbook.org.
Honey 2003
- Honey RA, Turner DC, Honey GD, Sharar SR, Kumaran D, Pomarol-Clotet E. Subdissociative dose ketamine produces a deficit in manipulation but not maintenance of the contents of working memory. Neuropsychopharmacology 2003;28:2037–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
Joseph 2021
- Joseph B, Nandakumar AL, Ahmed AT, Gopal N, Murad MH, Frye MA, et al. Prevalence of bipolar disorder in multiple sclerosis: a systematic review and meta-analysis. Evidence-Based Mental Health 2021;24(2):88-94. [DOI: 10.1136/ebmental-2020-300207] [DOI] [PMC free article] [PubMed] [Google Scholar]
Judd 2002
- Judd LL, Akiskal HS, Schettler PJ, Endicott J, Maser J, Solomon DA, et al. The long-term natural history of the weekly symptomatic status of bipolar I disorder. Archives of General Psychiatry 2002;59(6):530-7. [DOI] [PubMed] [Google Scholar]
Judd 2003
- Judd LL, Akiskal HS, Schettler PJ, Coryell W, Endicott J, Maser JD, et al. A prospective investigation of the natural history of the long-term weekly symptomatic status of bipolar II disorder. Archives of General Psychiatry 2003;60(3):261-9. [DOI] [PubMed] [Google Scholar]
Kendall 2014
- Kendall T, Morriss R, Mayo-Wilson E, Marcus E, Guideline Development Group. Assessment and management of bipolar disorder: summary of updated NICE guidance. BMJ 2014;349:g5673. [DOI] [PubMed] [Google Scholar]
Kraus 2017
- Kraus C, Rabl U, Vanicek T, Carlberg L Popovic A. Administration of ketamine for unipolar and bipolar depression. International Journal of Psychiatry in Clinical Practice 2017;21(1):2-12. [DOI] [PubMed] [Google Scholar]
Kryst 2020
- Kryst J, Kawalec P, Mitoraj AM, Pilc A, Lasoń W, Brzostek T. Efficacy of single and repeated administration of ketamine in unipolar and bipolar depression: a meta-analysis of randomized clinical trials. Pharmacological Reports 2020;72:543-62. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kubitz 2013
- Kubitz N, Mehra M, Potluri RC, Garg N, Cossrow N Quinn TJ. Characterization of treatment resistant depression episodes in a cohort of patients from a US commercial claims database. PLOS One 2013;8(10):e76882. [DOI] [PMC free article] [PubMed] [Google Scholar]
Li 2010
- Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 2010;329(5994):959-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
Li 2018
- Li CT, Yang, KC, Lin, WC. Glutamatergic dysfunction and glutamatergic compounds for major psychiatric disorders: evidence from clinical neuroimaging studies. Frontiers in Psychiatry 2018;9:767. [DOI] [PMC free article] [PubMed] [Google Scholar]
Magni 2013
- Magni LR, Purgato M, Gastaldon C, Papola D, Furukawa TA, Cipriani A, et al. Fluoxetine versus other types of pharmacotherapy for depression. Cochrane Database of Systematic Reviews 2013, Issue 7. Art. No: CD004185. [DOI: 10.1002/14651858.CD004185.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Malhi 2016
- Malhi GS, Byrow Y, Cassidy F, Cipriani A, Demyttenaere K, Frye MA, et al. Ketamine: stimulating antidepressant treatment? BJ Psych Open 2016;11:e5-e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Malhi 2018
- Malhi GS, Outhred T, Das P, Morris G, Hamilton A, Mannie Z. Modeling suicide in bipolar disorders. Bipolar Disorders 2018;20(4):334-48. [DOI] [PubMed] [Google Scholar]
Malhi 2020a
- Malhi GS, Bell E, Boyce P, Bassett D, Berk M, Bryant R, et al. The 2020 Royal Australian and New Zealand College of psychiatrists clinical practice guidelines for mood disorders: bipolar disorder summary. Bipolar Disorders 2020;22(8):805-21. [DOI: 10.1111/bdi.13036] [DOI] [PubMed] [Google Scholar]
Malhi 2020b
- Malhi GS, Bell E, Singh AB, Bassett D, Berk M, Boyce P, et al. The 2020 Royal Australian and New Zealand College of Psychiatrists clinical practice guidelines for mood disorders: major depression summary. Bipolar Disorders 2020;22(8):788-804. [DOI: 10.1111/bdi.13035] [DOI] [PubMed] [Google Scholar]
Malhi 2020c
- Malhi GS, Bell E, Boyce P, Hopwood M, Murray G, Mulder R, et al. The 2020 mood disorders clinical practice guidelines: translating evidence into practice with both style and substance. Australian & New Zealand Journal of Psychiatry 2020;55(9):919-20. [DOI: 10.1177/00048674211010233] [DOI] [PubMed] [Google Scholar]
Malhi 2021
- Malhi GS, Bell E, Bassett D, Boyce P, Bryant R, Hazell P, et al. The 2020 Royal Australian and New Zealand College of Psychiatrists clinical practice guidelines for mood disorders. Australian & New Zealand Journal of Psychiatry 2021;55(1):7-117. [DOI: 10.1177/0004867420979353] [DOI] [PubMed] [Google Scholar]
McIntyre 2020
- McIntyre RS, Berk M, Brietzke E, Goldstein BI, López-Jaramillo C, Kessing LV, et al. Bipolar Disorders. Lancet 2020;396(10265):1841-56. [DOI] [PubMed] [Google Scholar]
McMullen 2021
- McMullen EP, Lee Y, Lipsitz O, Lui LM, Vinberg M, et al. Strategies to prolong ketamine's efficacy in adults with treatment-resistant depression. Advances in Therapy 2021;38(6):2795-820. [DOI: 10.1007/s12325-021-01732-8] [DOI] [PMC free article] [PubMed] [Google Scholar]
Merikangas 2011
- Merikangas KR, Jin R, He JP, Kessler RC, Lee S, Sampson NA, et al. Prevalence and correlates of bipolar spectrum disorder in the World Mental Health Survey Initiative. Archives of General Psychiatry 2011;68(3):241-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
Mitchell 2011
- Mitchell PB, Frankland A, Hadzi-Pavlovic D, Roberts G, Corry J, Wright A, et al. Comparison of depressive episodes in bipolar disorder and in major depressive disorder within bipolar disorder pedigrees. British Journal of Psychiatry 2011;199(4):303-9. [DOI] [PubMed] [Google Scholar]
Moher 2009
- Moher D, Liberati A, Tetzlaff J, Altman DG, The PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA Statement. BMJ 2009;339:2535. [PMC free article] [PubMed] [Google Scholar]
Montgomery 1979
- Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. British Journal of Psychiatry 1979;134:382–9. [DOI] [PubMed] [Google Scholar]
Morgan 2010
- Morgan CJA, Muetzelfeldt L, Curran HV. Consequences of chronic ketamine self-administration upon neurocognitive function and psychological wellbeing: a 1-year longitudinal study. Addiction 2010;105:121-33. [DOI] [PubMed] [Google Scholar]
Moriguchi 2019
- Moriguchi S, Takamiya A, Noda Y, Horita N, Wada M, Tsugawa S, et al. Glutamatergic neurometabolite levels in major depressive disorder: a systematic review and meta-analysis of proton magnetic resonance spectroscopy studies.. Molecular Psychiatry 2019;24(7):952-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
Muller 2003
- Muller MJ, Himmerich H, Kienzle B, Szegedi A. Differentiating moderate and severe depression using the Montgomery-Asberg depression rating scale (MADRS). Journal of Affective Disorders 2003;77(3):255–60. [DOI] [PubMed] [Google Scholar]
Murray 2014
- Murray CJ, Ortblad KF, Guinovart C, Lim SS, Wolock TM, Roberts DA, et al. Global, regional, and national incidence and mortality for HIV, tuberculosis, and malaria during 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 2014;384(9947):1005-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
Naughton 2014
- Naughton M, Clarke G, O'Leary OF, Cryan JF, Dinan TG. A review of ketamine in affective disorders: current evidence of clinical efficacy, limitations of use and pre-clinical evidence on proposed mechanisms of action. Journal of Affective Disorders 2014;156:24-35. [DOI] [PubMed] [Google Scholar]
Niciu 2014
- Niciu MJ, Henter ID, Luckenbaugh DA, Zarate CA Jr, Charney DS. Glutamate receptor antagonists as fast-acting therapeutic alternatives for the treatment of depression: ketamine and other compounds. Annual Review of Pharmacology and Toxicology 2014;54:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
Phelps 2009
- Phelps LE, Brutsche N, Moral JR, Luckenbaugh DA, Manji HK, Zarate CA Jr. Family history of alcohol dependence and initial antidepressant response to an N-methyl-D-aspartate antagonist. Biological Psychiatry 2009;65(2):181-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Pompili 2013
- Pompili M, Gonda X, Serafini G, Innamorati M, Sher L, Amore M, Rihmer Z, Girardi P. Epidemiology of suicide in bipolar disorders: a systematic review of the literature. Bipolar Disorders 2013;15(5):457-90. [DOI: 10.1111/bdi.12087] [DOI] [PubMed] [Google Scholar]
Purgato 2014
- Purgato M, Papola D, Gastaldon C, Trespidi C, Magni LR, Rizzo C, et al. Paroxetine versus other anti-depressive agents for depression. Cochrane Database of Systematic Reviews 2014, Issue 4. Art. No: CD006531. [DOI: 10.1002/14651858.CD006531.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
RevMan 2014 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Salvadore 2010
- Salvadore G, Quiroz JA, Machado-Vieira R, Henter ID, Manji HK, Zarate CA. The neurobiology of the switch process in bipolar disorder: a review. Journal of Clinical Psychiatry 2010;71(11):1488-501. [DOI] [PMC free article] [PubMed] [Google Scholar]
Schwartz 2016
- Schwartz J, Murrough JW, Iosifescu DV. Ketamine for treatment-resistant depression: recent developments and clinical applications.. Evidence-Based Mental Health. 2016;19(2):35-8. [DOI: doi: 10.1136/eb-2016-102355] [DOI] [PMC free article] [PubMed] [Google Scholar]
Smith 2018
- Smith KA, Cipriani A, Hawton KE. Modelling suicide in bipolar disorders: Limitations and opportunities. Bipolar Disorders 2018;20(6):566-7. [DOI: 10.1111/bdi.12683] [DOI] [PubMed] [Google Scholar]
Spitzer 1978
- Spitzer RL, Endicott J, Robins E. Research diagnostic criteria: rationale and reliability. Archives of General Psychiatry 1978;35(6):773–82. [DOI] [PubMed] [Google Scholar]
Stanislaus 2020
- Stanislaus S, Vinberg M, Melbye S, Frost M, Busk J, Bardram JE, et al. Daily self-reported and automatically generated smartphone-based sleep measurements in patients with newly diagnosed bipolar disorder, unaffected first-degree relatives and healthy control individuals. Evidence-Based Mental Health 2020;23(4):146-53. [DOI: 10.1136/ebmental-2020-300148] [DOI] [PMC free article] [PubMed] [Google Scholar]
Sterne 2000
- Sterne JA, Gavaghan D, Egger M. Publication and related bias in meta-analysis: power of statistical tests and prevalence in the literature. Journal of Clinical Epidemiology 2000;53(11):1119–29. [DOI] [PubMed] [Google Scholar]
Suh 1997
- Suh T, Gallo JJ. Symptom profiles of depression among general medical service users compared with specialty mental health service users. Psychological Medicine 1997;27(5):1051–63. [DOI] [PubMed] [Google Scholar]
Taylor 2014
- Taylor DM, Cornelius V, Smith L, Young AH. Comparative efficacy and acceptability of drug treatments for bipolar depression: a multiple-treatments meta-analysis. Acta Psychiatrica Scandinavica 2014;130(6):452-69. [DOI] [PubMed] [Google Scholar]
Thomas 2010
- Thomas SP, Nandhra HS, Jarvaraman A. Systematic review of lamotrigine augmentation of treatment resistant unipolar depression (TRD). Journal of Mental Health 2010;19(2):168-75. [DOI] [PubMed] [Google Scholar]
Wang 2015
- Wang J, Jing L, Toledo-Salas JC, Xu L. Rapid-onset antidepressant efficacy of glutamatergic system modulators: the neural plasticity hypothesis of depression. Neuroscience Bulletin 2015;31(1):75-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ware 1992
- Ware JE Jr, Sherbourne CD. The MOS 36-item short form health survey (SF-36). I. Conceptual framework and item selection. Medical Care 1992;30(6):473–83. [PubMed] [Google Scholar]
Ware 1998
- Ware JE, Kosinski M, Keller SD. SF-12: How to Score the SF-12. Physical and Mental Health Summary Scales. Lincoln (RI): QualityMetric Inc., 1998. [Google Scholar]
WHO 1992
- World Health Organization (WHO). The ICD-10 Classification of Mental and Behavioural Disorders. Geneva: WHO, 1992. [Google Scholar]
WHO 2018
- World Health Organization (WHO). International statistical classification of diseases and related health problems (11th Revision). Retrieved from https://icd.who.int/browse11/l-m/en.
WHOQOL Group 1998
- WHOQOL Group. The World Health Organization quality of life assessment (WHOQOL): Development and general psychometric properties. Social Science and Medicine 1998;46(12):1569–85. [DOI] [PubMed] [Google Scholar]
Wilkinson 2019
- Wilkinson ST, Sanacora G. A new generation of antidepressants: an update on the pharmaceutical pipeline for novel and rapid-acting therapeutics in mood disorders based on glutamate/GABA neurotransmitter systems. Drug Discovery Today 2019;24(2):606-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Williams 2018
- Williams NR, Heifets BD, Blasey C, Sudheimer K, Pannu J, Pankow H, et al. Attenuation of antidepressant effects of ketamine by opioid receptor antagonism.. American Journal of Psychiatry. 2018;175(12):1205-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wing 1998
- Wing JK, Beevor AS, Curtis RH, Park SB, Burns A. Health of the nation outcome scales (HoNOS). Research and development. British Journal of Psychiatry 1998;172:11-8. [DOI] [PubMed] [Google Scholar]
Witt 2020
- Witt K, Potts J, Hubers A, Grunebaum MF, Murrough JW, Loo C, Cipriani A, et al. Ketamine for suicidal ideation in adults with psychiatric disorders: A systematic review and meta-analysis of treatment trials.. Australian and New Zealand JournL of Psychiatry 2020;54(1):29-45. [DOI: <html><body id="body">doi: 10.1177/0004867419883341</body></html>] [DOI] [PubMed] [Google Scholar]
Zarate 2006
- Zarate CA Jr, Singh JB, Quiroz JA, De Jesus G, Denicoff KK, Luckenbaugh DA, et al. A double-blind, placebo-controlled study of memantine in the treatment of major depression. American Journal of Psychiatry 2006;163(1):153-5.. [DOI] [PubMed] [Google Scholar]
Zavodnick 2012
- Zavodnick AD, Ali R. Lamotrigine in the treatment of unipolar depression with and without comorbidities: a literature review. Psychiatric Quarterly 2012;83:371-83. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
McCloud 2015
- McCloud TL, Caddy C, Jochim J, Rendell JM, Diamond PR, Shuttleworth C, et al. Ketamine and other glutamate receptor modulators for depression in bipolar disorder in adults. Cochrane Database of Systematic Reviews 2015, Issue 9. Art. No: CD011611. [DOI: 10.1002/14651858.CD011611.pub2] [DOI] [PubMed] [Google Scholar]
Rendell 2015
- Rendell JM, Shuttleworth C, Jochim J, Diamond PR, Brett D, Amit BH, et al. Ketamine and other glutamate receptor modulators for depression in bipolar disorder in adults. Cochrane Database of Systematic Reviews 2015, Issue 4. Art. No: CD011611. [DOI: 10.1002/14651858.CD011611] [DOI] [PubMed] [Google Scholar]