

Abstract
Single-cell RNA sequencing has enabled to capture the gene activities at single-cell resolution, thus allowing reconstruction of cell-type-specific gene regulatory networks (GRNs). The available algorithms for reconstructing GRNs are commonly designed for bulk RNA-seq data, and few of them are applicable to analyze scRNA-seq data by dealing with the dropout events and cellular heterogeneity. In this paper, we represent the joint gene expression distribution of a gene pair as an image and propose a novel supervised deep neural network called DeepDRIM which utilizes the image of the target TF-gene pair and the ones of the potential neighbors to reconstruct GRN from scRNA-seq data. Due to the consideration of TF-gene pair’s neighborhood context, DeepDRIM can effectively eliminate the false positives caused by transitive gene–gene interactions. We compared DeepDRIM with nine GRN reconstruction algorithms designed for either bulk or single-cell RNA-seq data. It achieves evidently better performance for the scRNA-seq data collected from eight cell lines. The simulated data show that DeepDRIM is robust to the dropout rate, the cell number and the size of the training data. We further applied DeepDRIM to the scRNA-seq gene expression of B cells from the bronchoalveolar lavage fluid of the patients with mild and severe coronavirus disease 2019. We focused on the cell-type-specific GRN alteration and observed targets of TFs that were differentially expressed between the two statuses to be enriched in lysosome, apoptosis, response to decreased oxygen level and microtubule, which had been proved to be associated with coronavirus infection.
Keywords: single-cell RNA sequencing, gene regulatory network, deep neural network, transitive interactions
1 Introduction
Reconstruction of gene regulatory networks (GRNs) is critical to understand the mechanisms of synergic gene effects and context-specific transcriptional dynamics. High-throughput technologies such as chromatin immunoprecipitation (ChIP)-chip and ChIP-seq can directly capture the transcription factor (TF) binding sites of targeted genes; however, these techniques are costly and TF-specific, and are therefore unsuitable for use on a whole-genome scale [1]. As a consequential observation, the fact that the co-expression of TFs and their target genes has been adopted to reconstruct GRNs [2–6]. In the last two decades, microarrays and bulk RNA sequencing (RNA-seq) have been the two mainstream technologies used to capture gene expression profiles from diverse tissues. Both techniques have been widely applied to identify differentially expressed genes and reconstruct GRNs [7–9]. However, microarrays and RNA-seq inappropriately assume that gene expression is homogeneous among cells and ignore cellular heterogeneity. Indeed, tissue consists of a diverse range of cell types with distinct GRNs [10] and biological functions [11]. Several studies have sought to reconstruct GRNs using bulk gene expression data [12, 13], but the cell-type-specific GRNs remain largely unexplored. Single-cell RNA sequencing (scRNA-seq) offers an opportunity to capture cell-specific gene expression, which in turn could provide deeper insights into the cellular heterogeneity and cell-type-specific gene activities[14].
Most of the available algorithms for GRN reconstruction are designed for bulk gene expression, and function by resolving two computational challenges. In this context, unique difficulties arise if scRNA-seq data are adopted instead. First, putative TF-gene interactions are derived by examining their co-expression. Bulk gene expression data are commonly normalized to a standard Gaussian distribution, such that the TF-gene correlation can be quantified by methods such as mutual information (MI) [15], Pearson correlation coefficient (PCC) [16, 17]. The scRNA-seq gene expression data are zero-inflated due to the imbalanced transcript sampling. Although it is possible to impute zero entries before calculating the TF-gene co-expression, this may introduce unpredictable noise and bias [18], given that most of the imputation algorithms make use of gene–gene co-expression. Second, the TF-gene pairs with strong co-expression due to transitive interactions (e.g. those bridged by one or more intermediate genes) should be eliminated (Supplementary Figure S1). Several strategies have been designed to remove these transitive interactions by conditioning on the other confounding genes; examples include the Gaussian graphical model [19], conditional MI [20], context-based normalization and edge removal [3] and tree-based ensemble methods [5]. Unfortunately, these algorithms were originally developed to analyze bulk gene expression data, and are unsuitable for modeling scRNA-seq data [21]. Many algorithms have recently been proposed to cater for the unique characteristics of scRNA-seq for GRN reconstruction. SCODE [22] infers cell-specific pseudo-time and reconstructs the GRN by solving ordinary differential equations. PIDC [23] adopts partial information decomposition to break down the TF-gene correlation into redundant, synergistic and unique effects. SINCERITIES [24] utilizes regularized linear regression to infer GRNs from time-stamped scRNA-seq data by referring to temporal changes in the gene expression distributions. GENIE3 [5] is a tree-based ensemble method that was initially developed for bulk gene expression data. Aibar et al. later applied GENIE3 to reconstruct the global GRN for scRNA-seq and developed AUCell to score the active gene signatures for each cell [25]. Although these dedicated strategies have been designed to deal with the inherent issues in scRNA-seq data, none of them yield acceptable results benchmarked by cell-type-specific ChIP-seq data, and some are even close to random guessing [26].
CNNC [27] is a supervised deep neural network that represents the joint expression of a gene pair as an image and uses convolutional neural networks (CNNs) to predict gene–gene co-expression from scRNA-seq data. CNNC is robust to dropouts and can infer the interaction causalities using the information from cell-type-specific ChIP-seq data. We generated synthetic GRNs and their corresponding gene expression data (Methods and Figure 1) to examine whether CNNC could effectively distinguish direct and transitive interactions. We noted that a substantial number of the false positives obtained with CNNC were centered in the gene pairs with strong Pearson correlations (Figure 1A).
Figure 1 .
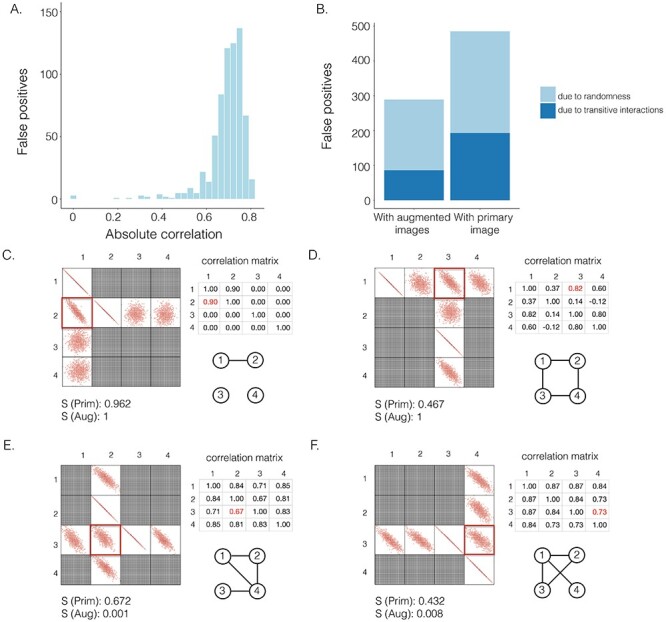
The effectiveness of neighbor images in reconstructing GRNs on the simulated data. A. The distribution of false positives from CNNC. B. The false positives of the two models with primary ( ) and augmented (
) and augmented ( ) images as inputs due to randomness and transitive interactions. C and D. Two examples that demonstrate both of the models can correctly identify the direct interactions (C:
) images as inputs due to randomness and transitive interactions. C and D. Two examples that demonstrate both of the models can correctly identify the direct interactions (C:  , D:
, D: 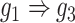 ). E and F. Two examples that demonstrate the model trained by augmented images can recognize and eliminate the false positives caused by the transitive edges (E:
). E and F. Two examples that demonstrate the model trained by augmented images can recognize and eliminate the false positives caused by the transitive edges (E: 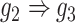 , F:
, F: 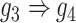 ).
). 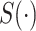 denotes the confidence scores from CNNC with primary (
denotes the confidence scores from CNNC with primary (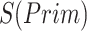 ) or augmented images (
) or augmented images (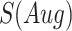 ) as inputs. The values in the correlation matrices are Pearson correlation coefficients for the gene pairs in the corresponding entries. The primary images are highlighted in the red squares.
) as inputs. The values in the correlation matrices are Pearson correlation coefficients for the gene pairs in the corresponding entries. The primary images are highlighted in the red squares.
Yet considering the image of the target TF-gene pair (primary image) as the only input for the prediction is insufficient (Figure 1A). Inspired by an approach named context likelihood of relatedness (CLR) [3] which has been used to remove the transitive interactions by normalizing the MI of the target TF-gene pairs to z-scores with their corresponding neighborhood, one can in fact consider both the target TF-gene pair (primary image) and the images from the gene pairs that share one gene with the target pair (neighbor images) as the input to the model (Figures 1 and 2).
Figure 2 .
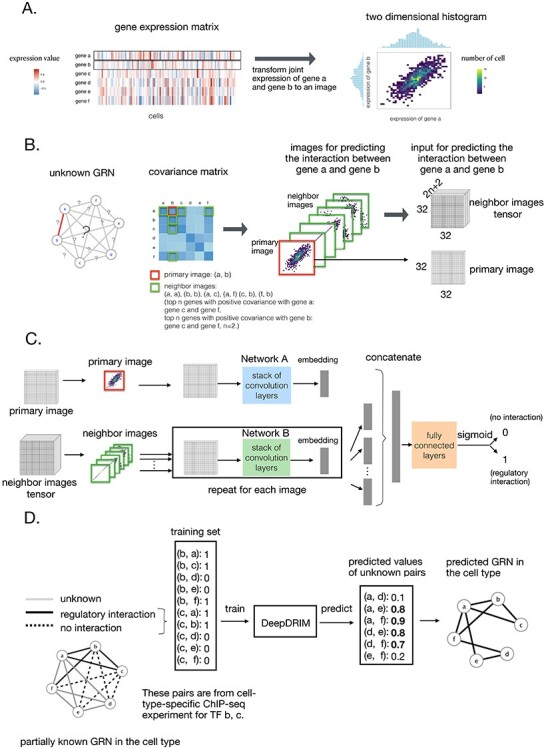
Overview of DeepDRIM. A. Representation of the joint gene expression of gene a and gene b as a primary image. B. The 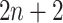 neighbor images are generated from the genes with strong positive covariance with gene a or gene b. C. The network architecture of DeepDRIM, including Network A and Network B, which are two stacked convolutional embedding structures designed to process the primary and neighbor images, respectively. Detailed network structures are shown in Supplementary Figure S3. D. An example for the prediction of a cell-type-specific GRN using DeepDRIM.
neighbor images are generated from the genes with strong positive covariance with gene a or gene b. C. The network architecture of DeepDRIM, including Network A and Network B, which are two stacked convolutional embedding structures designed to process the primary and neighbor images, respectively. Detailed network structures are shown in Supplementary Figure S3. D. An example for the prediction of a cell-type-specific GRN using DeepDRIM.
Here we propose DeepDRIM (deep learning-based direct regulatory interaction model), a supervised deep neural network that can reconstruct highly accurate cell-type-specific GRNs from scRNA-seq data by considering both primary and neighbor images. The rationale and workflow of DeepDRIM are shown in Figure 2. DeepDRIM first transforms the primary and neighbor images (Figure 2A and B) into low-dimensional embeddings using multiple convolutional layers, where their embeddings are then concatenated as the input to a multiple-layer perceptron to calculate the regulatory confidence scores (Figure 2C). We compared the effectiveness of DeepDRIM with PCC, MI, GENIE3 and CNNC for the analysis of eight real scRNA-seq datasets. Our results demonstrated that DeepDRIM yielded the best performance with respect to both the area under the receiver operating characteristic curve (AUROC) and the area under the precision-recall curve (AUPRC), and significantly outperformed CNNC (Figure 3A-D). We also compared DeepDRIM with six effective algorithms that were recently highlighted for reconstructing GRN on scRNA-seq data [26]. The results demonstrated that DeepDRIM substantially outperformed these algorithms on the five scRNA-seq datasets with the pseudotime-ordered cells (Figure 3E-F). Further simulation demonstrated that the performance of DeepDRIM could be improved by involving more neighbor images, and was robust to the dropout rate, the cell number and the size of the training set (Figure 4A–D).
Figure 3 .
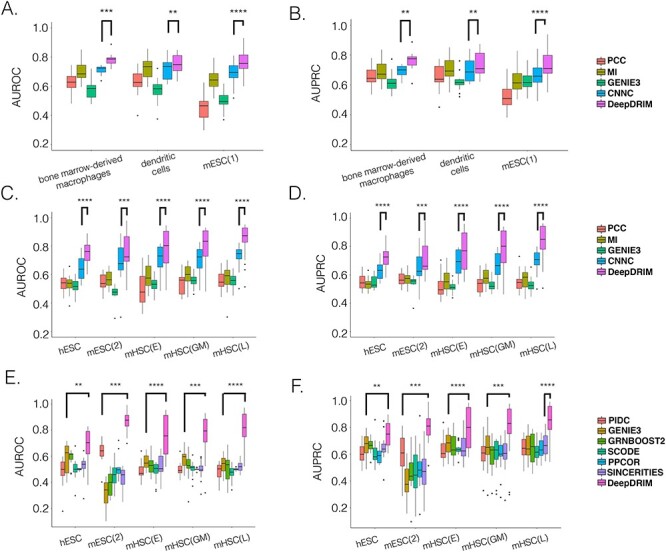
Comparison of DeepDRIM with the existing algorithms for GRN reconstruction on the scRNA-seq data from eight cell lines. A, B, C and D: 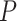 -values were calculated between CNNC and DeepDRIM. E and F: The
-values were calculated between CNNC and DeepDRIM. E and F: The 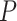 -values were calculated between DeepDRIM (the best performer) and the second best algorithms (Supplementary Tables S3-S4). We separated the eight datasets into two panels (A-B and C-F) because the three datasets in A and B did not include pseudo-time information, which was required for the methods PIDC, SCODE and SINCERITIES in E-F.
-values were calculated between DeepDRIM (the best performer) and the second best algorithms (Supplementary Tables S3-S4). We separated the eight datasets into two panels (A-B and C-F) because the three datasets in A and B did not include pseudo-time information, which was required for the methods PIDC, SCODE and SINCERITIES in E-F.
Figure 4 .
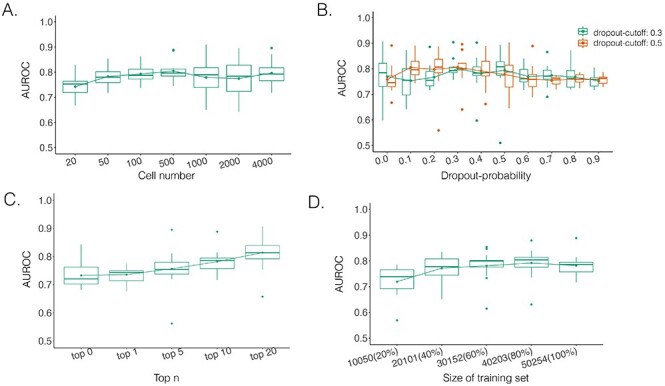
Performance of DeepDRIM with a wide range of the qualities of scRNA-seq data (cell numbers and dropout rates), the number of involved neighbor images and the size of training set.
We applied DeepDRIM to the scRNA-seq data collected from the bronchoalveolar lavage fluid of patients with mild and severe symptoms of coronavirus disease 2019 (COVID-19) [28] to discover the changes in B cell-specific GRNs. As a result, we observed that a large number of differentially expressed TFs (DETFs) were ‘activated’ in patients with severe disease (Figure 5A and B). Furthermore, in patients with severe COVID-19 symptoms, the functions of the target genes were enriched in lysosome, apoptosis, response to decreased oxygen levels and microtubules (Figure 5C and D, Figure 6A and B), all of which have been previously shown to be associated with COVID-19 [29, 30] and virus infection [31].
Figure 5 .
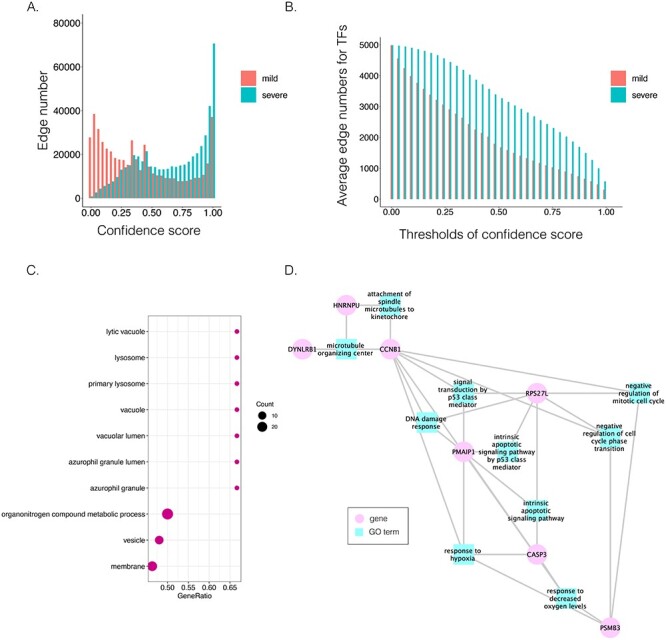
Comparison of B cell-specific GRNs for patients with mild and severe COVID-19. A. The distribution of the confidence scores of the differentially expressed transcription factors and their target genes. B. The average target numbers of differentially expressed TFs (DETFs) given different confidence score thresholds. C. The significant GO terms after Benjamini–Hochberg correction. The dot size implies the number of genes of the GO term. The x-axis refers to the GeneRatio, which is the proportion of genes in the provided list belong to the particular GO terms. D. The GO modules and the involved key transcription factors/genes related to COVID-19 symptoms.
Figure 6 .
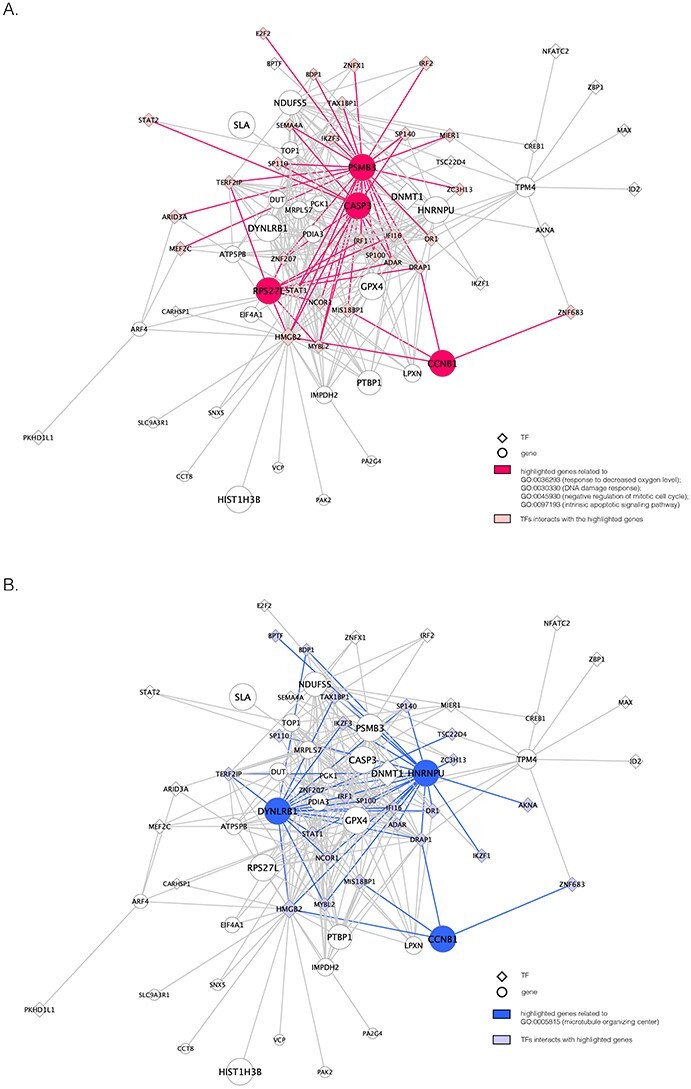
The unique GRNs of DETFs from the patients with severe COVID-19. A. GRNs related to response to a decreased oxygen level (GO:0036293); DNA damage response (GO:0030330); negative regulation of the mitotic cell cycle (GO:0045930); and the intrinsic apoptotic signaling pathway (GO:0097193). B. GRNs related to the microtubule organizing center (GO:0005851). The edges are shown if their absolute Pearson correlation coefficients are larger than 0.4. DETFs: differentially expressed transcription factors.
2 Results
2.1 Effectiveness of neighbor images in removing transitive interactions
We generated simulated data and attempted to train CNNC using the two types of input, one with only the primary images and the other with the augmented images (combined primary and neighbor images, Methods). We observed that the overall proportion of false positives and those due to transitive interactions were remarkably decreased by 40.4% and 55.4%, when considering the neighbor images in the model (Figure 1B). The rationale behind this observation can be regarded as taking a ‘normalization’ on the primary image over their neighborhood to alleviate the overestimation of the strength of interaction. In addition, Figure 1C and D clearly illustrate that the consideration of neighbor images will not undermine the power in predicting the direct interactions (e.g. gene 1 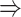 gene 2 in Figure 1C, and gene 1
gene 2 in Figure 1C, and gene 1 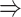 gene 3 in Figure 1D). In Figure 1E, gene 2 connects to gene 3 via the indirect edges gene 2
gene 3 in Figure 1D). In Figure 1E, gene 2 connects to gene 3 via the indirect edges gene 2 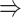 gene 4
gene 4 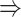 gene 3. Furthermore, we noticed that the correlations of both {gene 2, gene 4} (|PCC| = 0.81) and {gene 4, gene 3} (|PCC| = 0.83) were stronger than the target {gene 2, gene 3} (|PCC| = 0.67), which provided explicit evidence that {gene 2, gene 3} should be marked as a false positive. By considering neighbor images, the model reduce the predicted confidence score of {gene 2, gene 3} from 0.672 to 0.001, with a similar situation observed in Figure 1F. These findings consolidate the importance of considering the local neighborhood in GRN construction to eliminate false positives due to transitive interactions.
gene 3. Furthermore, we noticed that the correlations of both {gene 2, gene 4} (|PCC| = 0.81) and {gene 4, gene 3} (|PCC| = 0.83) were stronger than the target {gene 2, gene 3} (|PCC| = 0.67), which provided explicit evidence that {gene 2, gene 3} should be marked as a false positive. By considering neighbor images, the model reduce the predicted confidence score of {gene 2, gene 3} from 0.672 to 0.001, with a similar situation observed in Figure 1F. These findings consolidate the importance of considering the local neighborhood in GRN construction to eliminate false positives due to transitive interactions.
2.2 Overview of DeepDRIM
DeepDRIM is proposed to reconstruct cell-type-specific GRNs from scRNA-seq data with high precision and a low false positive rate. Figure 2 illustrates how DeepDRIM can be used to predict the interaction between gene 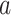 and gene
and gene 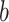 . First, DeepDRIM converts the joint gene expression of gene
. First, DeepDRIM converts the joint gene expression of gene 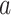 and gene
and gene 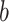 into a two-dimensional histogram with 32 by 32 bins (primary image, Figure 2A), where the intensity of each bin refers to the number of cells falling within it. Second, DeepDRIM constructs
into a two-dimensional histogram with 32 by 32 bins (primary image, Figure 2A), where the intensity of each bin refers to the number of cells falling within it. Second, DeepDRIM constructs 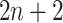 neighbor images, where the
neighbor images, where the 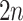 images that refer to the
images that refer to the 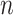 genes have top positive covariance with gene
genes have top positive covariance with gene 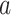
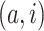 or gene
or gene 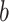
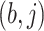 and the
and the  images represent the self-images
images represent the self-images 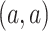 and
and 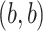 . These neighbor images are given to the model to capture the neighborhood context of the primary image (Figure 2B), which provides the key information required to distinguish the direct and transitive interactions. We organize the neighbor images as a tensor rather than an augmented image to achieve better performance on real data (Supplementary Figure S2). Third, two CNNs were used to process the primary image (Network A) and the neighbor image tensor (32 by 32 by 2
. These neighbor images are given to the model to capture the neighborhood context of the primary image (Figure 2B), which provides the key information required to distinguish the direct and transitive interactions. We organize the neighbor images as a tensor rather than an augmented image to achieve better performance on real data (Supplementary Figure S2). Third, two CNNs were used to process the primary image (Network A) and the neighbor image tensor (32 by 32 by 2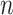 +2) (Network B), respectively (Figure 2C, Methods and Supplementary Figure S3). Network A follows VGGnet [32], which is similar to CNNC. Network B is a siamese-like neural network which is designed for processing multiple neighbor images. The neural networks are trained by known TF-gene interactions taken from publicly available cell-type-specific ChIP-seq data. Finally, the unknown interactions are predicted by the directed edges with confidence scores (between 0 and 1, Figure 2D).
+2) (Network B), respectively (Figure 2C, Methods and Supplementary Figure S3). Network A follows VGGnet [32], which is similar to CNNC. Network B is a siamese-like neural network which is designed for processing multiple neighbor images. The neural networks are trained by known TF-gene interactions taken from publicly available cell-type-specific ChIP-seq data. Finally, the unknown interactions are predicted by the directed edges with confidence scores (between 0 and 1, Figure 2D).
2.3 DeepDRIM outperforms the existing algorithms for reconstructing cell-type-specific GRNs
We collected the scRNA-seq datasets from eight cell lines (see Methods for the definitions of their abbreviations) and their corresponding ChIP-seq data from two sources [26, 27] to compare DeepDRIM with the existing methods (Table 1) using TF-aware 3fold cross-validation (Methods). We first assessed DeepDRIM with PCC, MI, CNNC and GENIE3; GENIE3 is one of the best algorithms for reconstructing GRNs on scRNA-seq [26] and bulk gene expression data [33, 34].
Table 1.
scRNA-seq datasets from the eight cell lines used in the experiments
| Cell lines | Genes | Cells | Size of training set | Number of TFs | Pseudo-time |
|---|---|---|---|---|---|
| Bone marrow-derived macrophages [35] | 20 463 | 6283 | 50 254 | 13 | N |
| Dendritic cells [35] | 20 463 | 4126 | 28 046 | 16 | N |
| mESC(1) [60] | 24 175 | 2717 | 154 931 | 38 | N |
| hESC [61] | 17 735 | 758 | 100 720 | 18 | Y |
| mESC(2) [62] | 18 385 | 421 | 94 332 | 18 | Y |
| mHSC(E) [63] | 4762 | 1071 | 49 114 | 18 | Y |
| mHSC(GM) [63] | 4762 | 889 | 43 712 | 18 | Y |
| mHSC(L) [63] | 4762 | 847 | 48 884 | 18 | Y |
Abbreviation and sources in footnote1.
Our results demonstrate that DeepDRIM outperformed all four methods in the eight cell types, and was significantly better than the second best CNNC (Figure 3A–D, Supplementary Tables S1 and S2) with respect to both AUROC (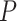 -values
-values 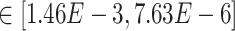 ) and AUPRC (
) and AUPRC (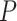 -values
-values 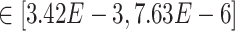 ). We also showed that DeepDRIM efficiently eliminated false positives from CNNC in all the eight scRNA-seq datasets (Supplementary Figure S4).
). We also showed that DeepDRIM efficiently eliminated false positives from CNNC in all the eight scRNA-seq datasets (Supplementary Figure S4).
To further evaluate the effectiveness of DeepDRIM, we collected six algorithms that have been recently identified with the highest median AUPRC in synthetic networks and Boolean models from BEELINE [26]. Because some of these algorithms require pseudotime-ordered cells, we selected five eligible cell types (Table 1) and found the six algorithms perform differently for each of them (Supplementary Tables S3 and S4). We compared the efficiency of DeepDRIM to these algorithms and found that DeepDRIM significantly outperformed all six tested algorithms (Figure 3E-F). DeepDRIM achieved an average median AUROC of 0.789 and an AUPRC of 0.809 across the five cell types, while the second best methods only achieved an AUROC of 0.591 (Supplementary Table S3) and an AUPRC of 0.657 (Supplementary Table S4). The TF-specific AUROC and AUPRC are shown in Supplementary Tables S5-S7.
2.4 DeepDRIM is robust to the quality of scRNA-seq data and the size of the training set
The performance of DeepDRIM can be affected by the quality of scRNA-seq data (the dropout rate and cell number), the number of involved neighbor images and the size of the training set. To evaluate the robustness of DeepDRIM toward these factors, we first selected the scRNA-seq data from bone marrow-derived macrophages [35] as a template and simulated a series of scRNA-seq data with a range of parameters (Methods). Seven scRNA-seq gene expression datasets were generated by subsampling the involved cell numbers (from 20 to 4000 cells), which in turn changed the resolution of both the primary and neighbor images. We found DeepDRIM to be robust to the low-resolution images when the number of cells was greater than 100 (Figure 4A). Next, we imputed the dropouts in the template using MAGIC [36] and then randomly masked the entries as dropouts with a range of dropout rates (Methods). As shown in Figure 4B, DeepDRIM demonstrates stable performance in diverse dropout configurations. Third, we compared the performance of DeepDRIM by varying the number of neighbor images input into the model. As a result, we found that the more neighbor images that were involved, the better the performance of DeepDRIM (Figure 4C). In practice, involving more images would be more computationally costly. In our study, we chose the top 10 genes with the strongest positive covariance with the target TF or gene, thus involving a total of 22 neighbor images (if not specified) to balance the two factors. In addition, to evaluate the effect of the size of the training set, we subsampled 20%, 40%, 60%, 80% and 100% of the benchmarked TF-gene pairs for training. Our results revealed that the size of the training set did not significantly affect the performance of DeepDRIM (Figure 4D), and almost reached a plateau when 40% of the training set (including 20 101 TF-gene pairs) was applied.
2.5 Uncovering the variation of B cell-specific GRNs between the patients with mild and severe COVID-19
Patients diagnosed with COVID-19 can have mild or severe acute respiratory distress syndrome, although the underlying molecular mechanisms responsible for these differences remain unknown. We performed a case study to elucidate the differences in B cell-specific GRNs between the patients with mild and severe COVID-19, because the immune responses have been reported to be distinct between the two situations [37]. To this end, we downloaded scRNA-seq data from the bronchoalveolar lavage fluid of six patients with severe symptoms, three patients with mild symptoms and three healthy controls [28]. The cell type clusters were obtained by SC3[38] and the one belonged to B cells was recognized according to the marker genes provided by the original paper [28]. We extracted validated TF-gene pairs in B cells from the Gene Transcription Regulation Database [39] as the positive pairs, and combined them with the negative pairs from the same TFs and the gene expression from the healthy controls as the training set (Methods).
We observed a clear difference in the GRNs between the two types of patients, and also found that the target genes of the DETFs were highly correlated with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection. First, we observed that DETFs had significantly more targets (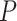 -values =
-values = 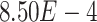 , Wilcoxon rank sum test) in the patients with severe symptoms, suggesting that these DETFs are more ‘active’ in working with their target genes (Methods and Figure 5A-B). Indeed, the DETFs in the patients with severe symptoms had 1.9 times more targets with high confidence (confidence scores
, Wilcoxon rank sum test) in the patients with severe symptoms, suggesting that these DETFs are more ‘active’ in working with their target genes (Methods and Figure 5A-B). Indeed, the DETFs in the patients with severe symptoms had 1.9 times more targets with high confidence (confidence scores 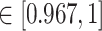 ; the last bar in Figure 5A) than the patients with mild symptoms. Next, we focused on the GRNs of DETFs that were unique to the patients with severe symptoms (Figure 5D, Figure 6A and B). The informative target genes were selected based on the following two criteria: (1) They should belong to the top 5000 genes with the highest expression variance in B cells; (2) they should be ranked in the top 0.1% of the confidence scores of the patients with severe symptoms. The eligible genes were annotated with PageRank scores [40] (Methods and Supplementary Table S8) and gene ontology (GO) modules by gene set enrichment analysis (GSEA) [41](Methods and Supplementary Table S9).
; the last bar in Figure 5A) than the patients with mild symptoms. Next, we focused on the GRNs of DETFs that were unique to the patients with severe symptoms (Figure 5D, Figure 6A and B). The informative target genes were selected based on the following two criteria: (1) They should belong to the top 5000 genes with the highest expression variance in B cells; (2) they should be ranked in the top 0.1% of the confidence scores of the patients with severe symptoms. The eligible genes were annotated with PageRank scores [40] (Methods and Supplementary Table S8) and gene ontology (GO) modules by gene set enrichment analysis (GSEA) [41](Methods and Supplementary Table S9).
We identified the selected 138 genes were significantly enriched in three GO terms after Benjamini–Hochberg adjustment; they were vesicle (GO:0031982), lysosome (GO:0005764) and vacuole (GO:0005773) (p.adjust¡0.05) (Figure 5C). The lysosome and vacuole have been approved to be associated with SARS-Cov-2 host cell infection [42]. The endosomal entry route of the virus binds to the host membrane and then it can enter the endocytic pathway, from early endosomes via late endosomes to endolysosomes, and finally lysosomes, which is accompanied by vacuolar acidification. In late lysosomes, the protein complex is cleaved by cathepsin L, resulting in the fusion of the viral and host cell membrane [42]. Besides lysosomes and vacuole, vesicles has also been reported to be related to the spread of COVID-19 by acting in the exosomal pathway and influencing intercellular communication [43].
In addition, we identified four suggestive GO modules that were associated with two common symptoms in patients with COVID-19, hypoxemia and lymphopenia (Figure 5D, Figure 6A): (1) response to decreased oxygen levels (GO:0036293; 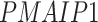 ,
, 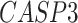 ,
, 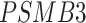 ,
, 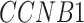 ,
, 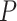 -values=
-values=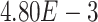 ); (2) DNA damage response (GO:0030330;
); (2) DNA damage response (GO:0030330; 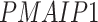 ,
, 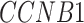 ,
, 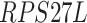 ,
, 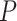 -values=
-values=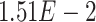 ); (3) negative regulation of the mitotic cell cycle (GO:0045930;
); (3) negative regulation of the mitotic cell cycle (GO:0045930; 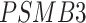 ,
, 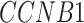 ,
, 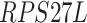 ,
, 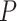 -values=
-values=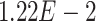 ); and (4) the intrinsic apoptotic signaling pathway (GO:0097193;
); and (4) the intrinsic apoptotic signaling pathway (GO:0097193; 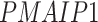 ,
, 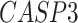 ,
, 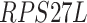 ,
, 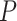 -values=
-values=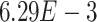 ). The patients were reported to have low oxygen levels or hypoxemia without dyspnea [44, 45], both of which were strongly correlated with the GO modules ‘response to decreased oxygen level’ and associated with ‘the intrinsic apoptotic signaling pathway’ [46]. Cao et al. [29] reported that genes related to apoptosis could lead to lymphopenia in patients with COVID-19. Xiong et al. [30] identified differentially expressed genes in peripheral blood mononuclear cells of patients with COVID-19 and healthy controls. These genes were enriched in apoptosis and p53 signaling pathways, both of which could lead to lymphopenia. Among the genes in these four GO modules,
). The patients were reported to have low oxygen levels or hypoxemia without dyspnea [44, 45], both of which were strongly correlated with the GO modules ‘response to decreased oxygen level’ and associated with ‘the intrinsic apoptotic signaling pathway’ [46]. Cao et al. [29] reported that genes related to apoptosis could lead to lymphopenia in patients with COVID-19. Xiong et al. [30] identified differentially expressed genes in peripheral blood mononuclear cells of patients with COVID-19 and healthy controls. These genes were enriched in apoptosis and p53 signaling pathways, both of which could lead to lymphopenia. Among the genes in these four GO modules, 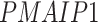 [47, 48],
[47, 48], 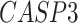 [49, 50],
[49, 50], 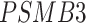 [51] and
[51] and 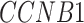 [30] have been reported to be associated with COVID-19 individually (Supplementary Table S10).
[30] have been reported to be associated with COVID-19 individually (Supplementary Table S10).
In addition to these main findings, we also noted that there were four genes with top PageRank scores in the patients with severe symptoms in which unique GRNs could be related to SARS-CoV-2 infection. Three of them (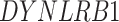 ,
, 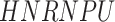 and
and 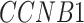 ) belong to GO:0005815 (microtubule organizing center,
) belong to GO:0005815 (microtubule organizing center, 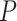 -values=
-values=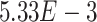 ), which has been reported to be a major facilitator of virus infection [31] due to its ability to provide invading pathogens with directed transport (Figure 6B). The other gene
), which has been reported to be a major facilitator of virus infection [31] due to its ability to provide invading pathogens with directed transport (Figure 6B). The other gene 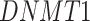 is related to
is related to 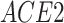 [52], which is a known co-receptor for the SARS-CoV-2 [53].
[52], which is a known co-receptor for the SARS-CoV-2 [53].
3 Discussion
Understanding the GRNs is fundamental to the advancement of molecular biology research. Gene expression profiles from high-throughput sequencing enable computational algorithms to reconstruct GRNs by examining TF-gene co-expression. Bulk RNA-seq hides the gene activities at single-cell resolution and will be replaced by scRNA-seq in the near future. However, the gene expression distribution from scRNA-seq data is not consistent with the assumptions made by most of the existing methods, which leads to their poor performance in reconstructing GRNs on the scRNA-seq data [54]. In addition, the widely spread dropouts cause bias in calculating gene–gene co-expression, even after imputation [55].
In this study, we propose DeepDRIM, a supervised deep neural network, to reconstruct GRNs on scRNA-seq data. Comprehensive evaluation of the performance of DeepDRIM on different cell types demonstrated that it outperformed the existing algorithms designed for either bulk or scRNA-seq gene expression data. It is inadvisable to calculate TF-gene interactions on scRNA-seq data using classical correlation-based methods due to the ubiquitous cellular heterogeneity and dropouts (Figure 3A–D). To avoid these limitations, DeepDRIM converts the numerical representation of TF-gene expression to an image and applies a CNN to embed it into a lower dimension. This strategy also avoids data normalization and does not presume any distribution. DeepDRIM requires validated TF-gene pairs for use as a training set to highlight the key areas in the embedding space that can distinguish the direct interactions and false positives.
We trained and tested DeepDRIM using data from the same cell type. As there is sometimes an insufficient number of cells or validated TF-gene pairs in the training set, we were interested in training the model using one cell type and then applying it to another. We trained DeepDRIM using bone marrow-derived macrophages and then applied it to mESC(1) and vice versa (Supplementary Figure S5). The results suggest that it is necessary to apply DeepDRIM to matched cell types in training and test sets; thus, ideas such as transfer learning between cell types are not applicable to this supervised model.
The neighborhood context of the target TF-gene pairs has been widely applied to remove false positives in GRN reconstruction from bulk gene expression data via z-score normalization [3], conditional MI [20, 56] and graphical lasso [57]. However, these methods commonly assume that the gene expression profiles follow a Gaussian distribution, which violates our observation in scRNA-seq data. Most of the existing algorithms designed for scRNA-seq are unsupervised and require pseudotime-ordered cells, making them inapplicable to bone marrow-derived macrophages, dendritic cells and mESC(1), as illustrated in Table 1. DeepDRIM uses the neighborhood context with respect to neighbor images, and consists of two parts: (1) images from the genes that positively correlate with the TF or gene from the target pair, and (2) two self-images. In the current model, we adopted covariance to select the top correlated genes. Although such linear correlation is not resistant to outliers and dropouts, similar method has shown its effectiveness in discovering gene–gene co-expression from scRNA-seq data [58]. The two self-images can highlight the variance of single gene expression.
Because the neighbor images are constructed by selecting the most ‘relevant’ genes with the target pairs, we compared four gene selection strategies, positive covariance (current implementation), PCC, MI and randomness on hESC and mHSC(GM) (Supplementary Figure S6A-B). We observed that our current strategy was the best and the worst one is random selection, suggesting the neighbor images should involve the local context of the target image as much as possible. We also tried to compare the current strategy to the other three based on selecting (1)top negative covariance genes, (2) top absolute covariance genes and (3) random genes on hESC, mHSC(GM) and mHSC(L) (Supplementary Figure S6C-D). We observed the neighbor genes from top absolute covariance and random genes were always the worst two, but the best one was not stable. Current strategy outperforms the other three in hESC and mHSC(L), but worse than top negative genes in mHSC(GM). We implemented a function in DeepDRIM to allow the users to choose the appropriate strategy for neighbor gene selection.
The running time and memory would be influenced by the number of neighbor images and the size of the training set. Taking hESC as an example, DeepDRIM would spend 51.57GB memory and 11.01 GPU h (2.70GHz 4 x NVIDIA Tesla V100S) if 22 neighbor images were involved (10 neighbor genes). If memory and time permits, 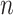 (number of neighbor genes) can be set to a larger value to include more neighbor information.
(number of neighbor genes) can be set to a larger value to include more neighbor information.
DeepDRIM can not only predict the existence of TF-gene interactions, but also determine their causalities (edge directions). This task is not given much attention by the unsupervised algorithms, despite it being an important consideration if regulatory interactions exist between two TFs. For this particular task, DeepDRIM does not surpass CNNC, because CNNC only focuses on the primary image and it is easier to capture the causalities by learning the regulatory directions from the validated TF-gene pairs. We generated a combined model from DeepDRIM and CNNC (Supplementary Notes) and found that it can effectively reduce the false positives without losing any accuracy in the prediction of causality (Supplementary Figure S7).
As same as CNNC, DeepDRIM can also make use of the sequence knowledge and be extended to work on the time-course data [27, 59]. Supplementary Figure S8 demonstrates the network structure of DeepDRIM to tackle with the motif position weight matrix.
Many studies have been proposed with the aim to identify all of the cell types in the human tissues, with the ultimate goal of creating a human cell atlas to facilitate interpretation of the gene activities in individual cell types. DeepDRIM bridges the gap between cell types and gene functions, and will serve to increase our understanding of the activities of key TFs. We believe that as the cell-type-specific ChIP-seq data accumulate, DeepDRIM will attract increased attention in the scRNA-seq research community, and will shed light on drug target discovery and precision medicine in the future.
4 Conclusion
We propose DeepDRIM, a supervised deep neural network model, to predict GRNs from scRNA-seq data. DeepDRIM converts the joint expression of a TF–gene pair into a primary image and considers the neighbor images as the neighborhood context of the primary image to remove false positives due to transitive interactions. DeepDRIM also utilizes the training set to capture the key areas in the CNN embeddings that can recognize the TF-gene interactions and causalities. Our findings demonstrate that DeepDRIM outperforms nine existing algorithms on the eight cell types tested and is robust to the quality of scRNA-seq data. DeepDRIM can also identify the GRNs of B cells that are different between patients with mild and severe COVID-19 symptoms. We believe that DeepDRIM can fill the gaps in reconstructing cell-type-specific GRNs on scRNA-seq data and contributes to the rapidly growing single-cell research community.
5 Methods
5.1 Representation of gene pair joint expression
The scRNA-seq gene expression profiles are represented as a two-dimensional matrix 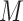 , where
, where 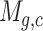 represents the expression of gene
represents the expression of gene 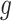 in cell
in cell 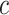 . We added a small pseudo-count to
. We added a small pseudo-count to 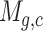 to avoid empty entries before applying log-normalization:
to avoid empty entries before applying log-normalization:
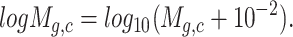 |
(1) |
The joint histogram of genes 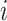 and
and 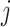 (
(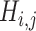 ) is generated by splitting
) is generated by splitting 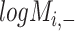 and
and 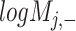 (‘-’: across all of the cells) into 32 bins, respectively. The value of each bin is derived from the number of cells that falls in the corresponding slot; this value is further log-normalized to avoid extreme values:
(‘-’: across all of the cells) into 32 bins, respectively. The value of each bin is derived from the number of cells that falls in the corresponding slot; this value is further log-normalized to avoid extreme values:
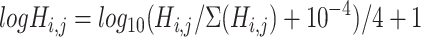 |
(2) |
We generated an image (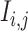 ) for genes
) for genes 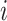 and
and 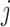 of 32 by 32 pixels, where the intensity of each pixel is the corresponding value in
of 32 by 32 pixels, where the intensity of each pixel is the corresponding value in 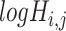 . DeepDRIM requires two image sets to predict the direct interaction between genes
. DeepDRIM requires two image sets to predict the direct interaction between genes 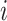 and
and 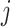 , namely (1) the primary image
, namely (1) the primary image 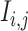 and (2) the neighbor images. The neighbor images consist of (1)
and (2) the neighbor images. The neighbor images consist of (1) 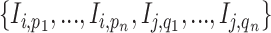 , where (
, where (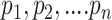 ) and (
) and (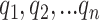 ) are the top
) are the top 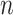 genes that have strong positive covariance with gene
genes that have strong positive covariance with gene 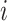 and gene
and gene 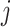 , respectively; and (2) two self-images
, respectively; and (2) two self-images 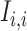 and
and 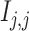 . The default value of
. The default value of 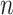 was 10 in the experiments.
was 10 in the experiments.
5.2 Network structure of DeepDRIM
The network structure of DeepDRIM consists of two components, Network A and Network B, which process the primary and neighbor images, respectively (Figure 2C and Supplementary Figure S3). Network A is inspired by VGGnet [32], which contains the stacked convolutional and maxpooling layers, and uses the rectified linear activation function (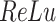 ) as the activation function. The structure of Network B is similar to that of Network A, and is a siamese-like neural network, where the weights are shared among all of the subnetworks. Each image is embedded into a vector of size 512, and a total of
) as the activation function. The structure of Network B is similar to that of Network A, and is a siamese-like neural network, where the weights are shared among all of the subnetworks. Each image is embedded into a vector of size 512, and a total of 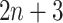 images (1 primary image and
images (1 primary image and 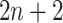 neighbor images) are converted into a vector of size
neighbor images) are converted into a vector of size 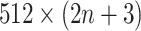 . This vector is then condensed by two stacked fully connected layers, and is processed for binary classification using the sigmoid function. Moreover, the network structure of DeepDRIM is shown in Supplementary Figure S3 (including hyperparameter values) and its weights are randomly initialized. DeepDRIM was trained by mini-batched stochastic gradient descent with batch size of 32. It runs a maximum of 200 epochs with an early stop if the validation accuracy does not improve in 10 epochs.
. This vector is then condensed by two stacked fully connected layers, and is processed for binary classification using the sigmoid function. Moreover, the network structure of DeepDRIM is shown in Supplementary Figure S3 (including hyperparameter values) and its weights are randomly initialized. DeepDRIM was trained by mini-batched stochastic gradient descent with batch size of 32. It runs a maximum of 200 epochs with an early stop if the validation accuracy does not improve in 10 epochs.
5.3 Simulation of scRNA-seq data to examine the effect of neighbor images
We simulated 2500 small datasets, each with four genes and 1000 cells. The ground truth network for each dataset was represented by a sparse precision matrix 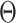 , where each entry had a
, where each entry had a 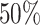 chance of being non-zero and drawn from
chance of being non-zero and drawn from 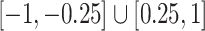 , or otherwise was assigned zero. We simulated the gene expression profiles from a multivariate normal distribution
, or otherwise was assigned zero. We simulated the gene expression profiles from a multivariate normal distribution 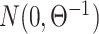 [64]. Next, we randomly chose two gene pairs from each dataset, one involving a direct interaction (
[64]. Next, we randomly chose two gene pairs from each dataset, one involving a direct interaction (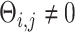 ) as a positive case, and the other involving an independent pair (
) as a positive case, and the other involving an independent pair (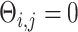 ) as a negative case. For each case, we prepared two types of images, a primary image of 32 by 32 pixels, and an augmented image by concatenating the primary and six neighbor images (Figure 1C–F) of 96 by 96 pixels. We generated two training sets with 5000 primary and 5000 augmented images, respectively. These images were used to train CNNC and the performance was evaluated using the AUROC from the 5-fold cross-validation.
) as a negative case. For each case, we prepared two types of images, a primary image of 32 by 32 pixels, and an augmented image by concatenating the primary and six neighbor images (Figure 1C–F) of 96 by 96 pixels. We generated two training sets with 5000 primary and 5000 augmented images, respectively. These images were used to train CNNC and the performance was evaluated using the AUROC from the 5-fold cross-validation.
5.4 scRNA-seq data from eight cell lines
We prepared the real scRNA-seq data from eight cell lines and the corresponding cell-type-specific ChIP-seq data as the benchmarks (Table 1) to compare DeepDRIM with the existing algorithms for GRN reconstruction. The eight cell lines comprised bone marrow-derived macrophages [35], dendritic cells [35], IB10 mouse embryonic stem cells (mESC(1)) [60], human embryonic stem cells (hESC) [61] and 5G6GR mouse embryonic stem cells (mESC(2)) [62], as well as three mouse hematopoietic stem cell lines [63] of erythroid lineage (mHSC(E)), granulocyte-macrophage lineage (mHSC(GM)) and lymphoid lineage (mHSC(L)). All scRNA-seq data were preprocessed and normalized according to the descriptions in [26, 27]. In practice, GENIE3 is slow if too many genes or cells are involved; thus, we removed the less informative cells and genes using the strategies described in [25].
We extracted the validated TF targets from the ChIP-seq data as positive cases, and randomly selected the balanced nontarget genes as negative cases. As training sets that are too large and are computationally insolvable in terms of generating images, we randomly selected 18 TFs and their validated targets as positive cases in the training data for hESC, mESC(2), mHSC(E), mHSC(GM) and mHSC(L) to alleviate the computational burden (Table 1).
To improve the performance of the unsupervised methods in Figure 3E-F, only the overlap between top-varying 500 genes and the TFs/genes in the training set were selected from the scRNA-seq data of hESC, mESC(2), mHSC(E), mHSC(GM) and mHSC(L). In cross-validation (see Supplementary Note), we trained CNNC and DeepDRIM using 2/3 TF-gene pairs in the training set and evaluated their performance on the overlap between the TFs/genes in the remaining 1/3 test set and top-varying 500 genes. This could guarantee all the supervised and unsupervised were evaluated on the same TF-gene pairs.
5.5 Comparison of DeepDRIM to existing algorithms for GRN reconstruction
We compared DeepDRIM with the nine existing algorithms using their default parameters. The nine algorithms were PCC, MI, CNNC [27], PIDC [23], GENIE3[5], GRNBOOST2 [65], SCODE [22], PPCOR [66] and SINCERITIES [24]. With the exception of PCC, MI and CNNC, the other six methods were performed using the interfaces provided by BEELINE [26]. The AUROC and AUPRC for each TF were collected to calculate the 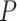 -values between two algorithms using the Wilcoxon signed rank test. Given that CNNC and DeepDRIM are supervised models, the TFs from the ChIP-seq data were divided into three independent parts for cross-validation (Supplementary Note).
-values between two algorithms using the Wilcoxon signed rank test. Given that CNNC and DeepDRIM are supervised models, the TFs from the ChIP-seq data were divided into three independent parts for cross-validation (Supplementary Note).
5.6 Simulation of scRNA-seq data to evaluate robustness
The simulated datasets were transferred from the scRNA-seq of bone marrow-derived macrophages [35] to preserve the characteristics of scRNA-seq data. We simulated gene expression profiles with various cell numbers and sizes of training sets via sub-sampling from the total 6283 cells and 50 254 validated TF-gene pairs from the ChIP-seq data. We applied MAGIC [36] to impute the missing values in the raw gene expression matrix, and subsequently masked the corresponding entries according to the ‘dropout step’ in BoolODE [26]. BoolODE has two parameters, 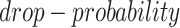 and
and 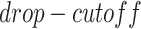 , which are used to control the number of entries to be masked. The entries have a probability of
, which are used to control the number of entries to be masked. The entries have a probability of 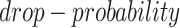 to be masked if their gene expression values are at the bottom
to be masked if their gene expression values are at the bottom 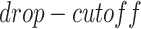 . We set the
. We set the 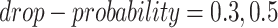 and the
and the 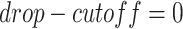 to
to 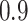 .
.
5.7 Generation of validated TF-gene pairs for B cells in patients with COVID-19
We extracted the ChIP-seq data with the keyword ‘human B cell’ in the Gene Transcription Regulation Database [39] and determined the TF target genes as those with high confidence peaks (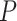 -value
-value 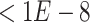 ) in the promoter regions of these genes. The promoter regions were defined as the 10 kb upstream and 1 kb downstream regions of the transcript start sites. To generate a balanced training set, we extracted an equal number of negative pairs by randomly selecting the nontarget genes of the selected TFs.
) in the promoter regions of these genes. The promoter regions were defined as the 10 kb upstream and 1 kb downstream regions of the transcript start sites. To generate a balanced training set, we extracted an equal number of negative pairs by randomly selecting the nontarget genes of the selected TFs.
5.8 Identification of differentially expressed TFs
We applied SCDE [67] to determine the differentially expressed TFs if the expression fold changes 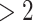 or
or 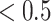 , and the
, and the 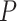 -values to be
-values to be 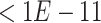 after multiple testing correction.
after multiple testing correction.
5.9 Gene PageRank score and functional annotation
We calculated gene PageRank scores using ‘networkx’ [40] (Additional file 5) and applied GSEA to annotate the enriched GO modules with 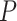 -value
-value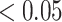 [41]. The genes were ordered by their PageRank scores in GSEA analysis.
[41]. The genes were ordered by their PageRank scores in GSEA analysis.
6 Data availability
DeepDRIM is available at https://github.com/jiaxchen2-c/DeepDRIM. Gene expression and ChIP-Seq data of bone marrow-derived macrophages, dendritic cells, mESC(1) are available at https://github.com/xiaoyeye/CNNC. Gene expression and ChIP-Seq data of hESC, mESC(2), mHSC(E), mHSC(GM), mHSC(L) are available at https://doi.org/10.5281/zenodo.3378975. Gene expression profiles from the bronchoalveolar lavage fluid of COVID-19 patients and healthy controls are available at GSE145926.
Key Points
We developed DeepDRIM, a novel supervised deep neural network that represents the transcription factor-gene joint expression as an image and considers both the target pair and the pairs of its potential neighbors.
DeepDRIM can efficiently eliminate the false positives due to transitive interactions by considering neighborhood context and is resistant to the dropout events in scRNA-seq.
We compared DeepDRIM with nine existing GRN reconstruction algorithms designed for either bulk or single-cell RNA-seq data. It achieves evidently better performance on the eight cell lines.
DeepDRIM was applied to B cells scRNA-seq from the bronchoalveolar lavage fluid of the COVID-19 patients with mild and severe symptoms. We observed the differently expressed TFs had significantly more targets in the data from severe patients. The functions of their target genes were found to be enriched in several functional modules, which had been proved to be associated with SARS-CoV-2 infection.
Supplementary Material
Acknowledgments
We also thank Research Grants Council of Hong Kong, Hong Kong Baptist University and HKBU Research Committee for their kind support of this project.
Jiaxing Chen is currently a postdoctoral scholar of Computer Science with Hong Kong Baptist University. Her research interests include bioinformatics and computational biology.
ChinWang Cheong is currently a PhD candidate of Computer Science with Hong Kong Baptist University. His research interests include artificial intelligence, healthcare informatics and bioinformatics.
Liang Lan is currently an Assistant Professor of Computer Science with Hong Kong Baptist University. His research interests include data mining and machine learning.
Xin Zhou is an Assistant Professor at the Departments of Biomedical Engineering and Computer Science, and a core faculty member of the Data Science Institute, Vanderbilt University. Her main research interests include computational genomics, bioinformatics, computational neuroscience, and machine learning.
Jiming Liu is currently the Chair Professor of Computer Science with Hong Kong Baptist University. His research interests include data analytics, data mining and machine learning, complex network analytics, data driven complex systems modeling, and health informatics.
Aiping Lyu is currently the Dean and Chair Professor of School of Chinese Medicine with Hong Kong Baptist University. His research interests include drug discovery, herbal medicine, network pharmacology and bioinformatics.
William K. Cheung is currently the Head and Associate Professor of the Department of Computer Science, Hong Kong Baptist University, Hong Kong. His current research interests include artificial intelligence, data mining, collaborative information filtering, social network analysis, and healthcare informatics.
Lu Zhang is currently an Assistant Professor of Computer Science with Hong Kong Baptist University. His research interests include bioinformatics and computational biology.
Footnotes
Bone marrow-derived macrophages [35], Dendritic cells [35], mESC(1): IB10 mouse embryonic stem cells [60], hESC: human embryonic stem cells [61], mESC(2): 5G6GR mouse embryonic stem cells [62], mHSC(E), mHSC(GM) and mHSC(L): three mouse hematopoietic stem cell lines of erythroid lineage, granulocyte-macrophage lineage and lymphoid lineage [63].
Contributor Information
Jiaxing Chen, Department of Computer Science, Hong Kong Baptist University, Waterloo Road, Kowloon Tong, Hong Kong.
ChinWang Cheong, Department of Computer Science, Hong Kong Baptist University, Waterloo Road, Kowloon Tong, Hong Kong.
Liang Lan, Department of Computer Science, Hong Kong Baptist University, Waterloo Road, Kowloon Tong, Hong Kong.
Xin Zhou, Department of Biomedical Engineering, Vanderbilt University, Vanderbilt Place Nashville, 37235, TN, USA.
Jiming Liu, Department of Computer Science, Hong Kong Baptist University, Waterloo Road, Kowloon Tong, Hong Kong.
Aiping Lyu, School of Chinese Medicine, Hong Kong Baptist University, Waterloo Road, Kowloon Tong, Hong Kong.
William K Cheung, Department of Computer Science, Hong Kong Baptist University, Waterloo Road, Kowloon Tong, Hong Kong.
Lu Zhang, Department of Computer Science, Hong Kong Baptist University, Waterloo Road, Kowloon Tong, Hong Kong.
9 Author contributions statement
LZ, WKC conceived the study; LZ, JXC designed DeepDRIM; JXC implemented the algorithm and analyzed the results. JXC, CWC conducted the experiments. JXC, LZ, WKC wrote the article. JML, APL and ZX reviewed the paper. All authors read and approved the final manuscript.
Funding
This research is partially supported by Hong Kong Research Grant Council Early Career Scheme (HKBU 22201419), HKBU Start-up Grant Tier 2(RC-SGT2/19-20/SCI/007), HKBU’s Interdisciplinary Research Clusters Matching Scheme (IRCRC/IRCs/17-18/04) and Guangdong Basic and Applied Basic Research Foundation (2019A1515011046). XZ is partially supported by Vanderbilt university development funds (FF_300033).
Conflicts of Interest
The authors declare that they have no conflicts of interest.
References
- 1. Park PJ. Chip–seq: advantages and challenges of a maturing technology. Nat Rev Genet 2009; 10(10): 669–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Haury A-C, Mordelet F, Vera-Licona P, et al. . Tigress: trustful inference of gene regulation using stability selection. BMC Syst Biol 2012;6(1):145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Faith JJ, Hayete B, Thaden JT, et al. . Large-scale mapping and validation of escherichia coli transcriptional regulation from a compendium of expression profiles. PLoS Biol 2007;5(1):e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Küffner R, Petri T, Tavakkolkhah P, et al. . Inferring gene regulatory networks by anova. Bioinformatics 2012;28(10):1376–82. [DOI] [PubMed] [Google Scholar]
- 5. Irrthum A, Wehenkel L, Geurts P, et al. . Inferring regulatory networks from expression data using tree-based methods. PloS one 2010;5(9):e12776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Xing L, Guo M, Liu X, et al. . An improved bayesian network method for reconstructing gene regulatory network based on candidate auto selection. BMC Genomics 2017;18(9):17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Oshlack A, Robinson MD, Young MD. From rna-seq reads to differential expression results. Genome Biol 2010;11(12):220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Huang S, Eichler G, Bar-Yam Y, et al. . Cell fates as high-dimensional attractor states of a complex gene regulatory network. Phys Rev Lett 2005;94(12):128701. [DOI] [PubMed] [Google Scholar]
- 9. Delgado FM, Gómez-Vela F. Computational methods for gene regulatory networks reconstruction and analysis: A review. Artif Intell Med 2019;95:133–45. [DOI] [PubMed] [Google Scholar]
- 10. Grubman A, Chew G, Ouyang JF, et al. . A single-cell atlas of entorhinal cortex from individuals with alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat Neurosci 2019;22(12):2087–97. [DOI] [PubMed] [Google Scholar]
- 11. Boyd NF, Martin LJ, Bronskill M, et al. . Breast tissue composition and susceptibility to breast cancer. J Natl Cancer Inst 2010;102(16):1224–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huang J, Zheng J, Yuan H, et al. . Distinct tissue-specific transcriptional regulation revealed by gene regulatory networks in maize. BMC Plant Biol 2018;18(1):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Talukdar HA, Asl HF, Jain RK, et al. . Cross-tissue regulatory gene networks in coronary artery disease. Cell systems 2016;2(3):196–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Siebert S, Farrell JA, Cazet JF, et al. . Stem cell differentiation trajectories in hydra resolved at single-cell resolution. Science 2019;365(6451):eaav9314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Margolin AA, Nemenman I, Basso K, et al. . Aracne: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. In: BMC bioinformatics, Vol. 7. BioMed Central, 2006, S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Salleh FHM, Arif SM, Zainudin S, et al. . Reconstructing gene regulatory networks from knock-out data using gaussian noise model and pearson correlation coefficient. Comput Biol Chem 2015;59:3–14. [DOI] [PubMed] [Google Scholar]
- 17. Munsky B, Neuert G, Van Oudenaarden A. Using gene expression noise to understand gene regulation. Science 2012;336(6078):183–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Andrews TS, Hemberg M. False signals induced by single-cell imputation. F1000Research 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Allen GI, Liu Z. A log-linear graphical model for inferring genetic networks from high-throughput sequencing data. In: 2012 IEEE International Conference on Bioinformatics and Biomedicine. IEEE, 2012, 1–6. [Google Scholar]
- 20. Zhang X, Zhao X-M, He K, et al. . Inferring gene regulatory networks from gene expression data by path consistency algorithm based on conditional mutual information. Bioinformatics 2012;28(1):98–104. [DOI] [PubMed] [Google Scholar]
- 21. Chen S, Mar JC. Evaluating methods of inferring gene regulatory networks highlights their lack of performance for single cell gene expression data. BMC bioinformatics 2018;19(1):1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Matsumoto H, Kiryu H, Furusawa C, et al. . Scode: an efficient regulatory network inference algorithm from single-cell rna-seq during differentiation. Bioinformatics 2017;33(15): 2314–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chan TE, Stumpf MPH, Babtie AC. Gene regulatory network inference from single-cell data using multivariate information measures. Cell systems 2017;5(3):251–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gao NP, Ud-Dean SMM, Gandrillon O, et al. . Sincerities: inferring gene regulatory networks from time-stamped single cell transcriptional expression profiles. Bioinformatics 2018;34(2):258–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Aibar S, González-Blas CB, Moerman T, et al. . Scenic: single-cell regulatory network inference and clustering. Nat Methods 2017;14(11):1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pratapa A, Jalihal AP, Law JN, et al. . Benchmarking algorithms for gene regulatory network inference from single-cell transcriptomic data. Nat Methods 2020;17(2):147–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yuan Y, Bar-Joseph Z. Deep learning for inferring gene relationships from single-cell expression data. Proc Natl Acad Sci 2019;116(52):27151–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liao M, Yang L, Yuan J, et al. . Single-cell landscape of bronchoalveolar immune cells in patients with covid-19. Nat Med 2020;1–3. [DOI] [PubMed] [Google Scholar]
- 29. Cao W, Li T. Covid-19: towards understanding of pathogenesis. Cell Res 2020;30(5):367–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xiong Y, Liu Y, Cao L, et al. . Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in covid-19 patients. Emerging microbes & infections 2020;9(1):761–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Greber UF, Way M. A superhighway to virus infection. Cell 2006;124(4):741–54. [DOI] [PubMed] [Google Scholar]
- 32. Simonyan K, Zisserman A. Very deep convolutional networks for large-scale image recognition arXiv preprint arXiv:1409.1556. 2014.
- 33. Greenfield A, Madar A, Ostrer H, et al. . Dream4: Combining genetic and dynamic information to identify biological networks and dynamical models. PloS one 2010;5(10):e13397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Marbach D, Costello JC, Küffner R, et al. . Wisdom of crowds for robust gene network inference. Nat Methods 2012;9(8):796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Alavi A, Ruffalo M, Parvangada A, et al. . A web server for comparative analysis of single-cell rna-seq data. Nat Commun 2018;9(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Van Dijk D, Sharma R, Nainys J, et al. . Recovering gene interactions from single-cell data using data diffusion. Cell 2018;174(3):716–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Arunachalam PS, Wimmers F, Mok CKP, et al. . Systems biological assessment of immunity to mild versus severe covid-19 infection in humans. Science 2020;369(6508):1210–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kiselev VY, Kirschner K, Schaub MT, et al. . Sc3: consensus clustering of single-cell rna-seq data. Nat Methods 2017;14(5):483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yevshin I, Sharipov R, Valeev T, et al. . Gtrd: a database of transcription factor binding sites identified by chip-seq experiments. Nucleic Acids Res 2016;gkw951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hagberg A, Swart P, Chult DS. Exploring network structure, dynamics, and function using networkx. Technical report. Los Alamos, NM (United States): Los Alamos National Lab. (LANL), 2008. [Google Scholar]
- 41. Yu G, Wang L-G, Han Y, et al. . clusterprofiler: an r package for comparing biological themes among gene clusters. Omics: a journal of integrative biology 2012;16(5):284–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Blaess M, Kaiser L, Sauer M, et al. . Covid-19/sars-cov-2 infection: Lysosomes and lysosomotropism implicate new treatment strategies and personal risks. Int J Mol Sci 2020;21(14):4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hassanpour M, Rezaie J, Nouri M, et al. . The role of extracellular vesicles in covid-19 virus infection. Infect Genet Evol 2020;85:104422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tobin MJ, Laghi F, Jubran A. Why covid-19 silent hypoxemia is baffling to physicians. Am J Respir Crit Care Med 2020;202(3):356–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dhont S, Derom E, Van Braeckel E, et al. . The pathophysiology of ‘happy’ hypoxemia in covid-19. Respir Res 2020;21(1):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sendoel A, Hengartner MO. Apoptotic cell death under hypoxia. Phys Ther 2014;29(3):168–76. [DOI] [PubMed] [Google Scholar]
- 47. Khoury M, Cuenca J, Cruz FF, et al. . Current status of cell-based therapies for respiratory virus infections: applicability to covid-19. Eur Respir J 2020;55(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rao S, Lau A, So H-C. Exploring diseases/traits and blood proteins causally related to expression of ace2, the putative receptor of sars-cov-2: A mendelian randomization analysis highlights tentative relevance of diabetes-related traits. Diabetes Care 2020. [DOI] [PubMed] [Google Scholar]
- 49. Duan F, Guo L, Yang L, et al. . Modeling covid-19 with human pluripotent stem cell-derived cells reveals synergistic effects of anti-inflammatory macrophages with ace2 inhibition against sars-cov-2. 2020.
- 50. Ling X-Y, Tao J-L, Sun X, et al. . Exploring material basis and mechanism of lianhua qingwen prescription against coronavirus based on network pharmacology. Chin Trad Herbal Drugs 2020;1723–30. [Google Scholar]
- 51. Wauters E, Van Mol P, Garg AD, et al. . Discriminating mild from critical covid-19 by innate and adaptive immune single-cell profiling of bronchoalveolar lavages BioRxiv. 2020. [DOI] [PMC free article] [PubMed]
- 52. Sawalha AH, Zhao M, Coit P, et al. . Epigenetic dysregulation of ace2 and interferon-regulated genes might suggest increased covid-19 susceptibility and severity in lupus patients. Clin Immunol 2020;108410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ni W, Yang X, Yang D, et al. . Role of angiotensin-converting enzyme 2 (ace2) in covid-19. Crit Care 2020;24(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Li WV, Li JJ. An accurate and robust imputation method scimpute for single-cell rna-seq data. Nat Commun 2018;9(1):997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Crow M, Gillis J. Co-expression in single-cell analysis: Saving grace or original sin? Trends Genet 2018;34(11):823–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhang X, Zhao J, Hao J-K, et al. . Conditional mutual inclusive information enables accurate quantification of associations in gene regulatory networks. Nucleic Acids Res 2015;43(5):e31–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Danaher P, Wang P, Witten DM. The joint graphical lasso for inverse covariance estimation across multiple classes. J R Stat Soc Series B Stat Methodology 2014;76(2):373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Arisdakessian C, Poirion O, Yunits B, et al. . Deepimpute: an accurate, fast, and scalable deep neural network method to impute single-cell rna-seq data. Genome Biol 2019;20(1):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yuan Y, Bar-Joseph Z. Deep learning of gene relationships from single cell time-course expression data bioRxiv. 2020. [DOI] [PMC free article] [PubMed]
- 60. Klein AM, Mazutis L, Akartuna I, et al. . Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 2015;161(5):1187–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Chu L-F, Leng N, Zhang J, et al. . Single-cell rna-seq reveals novel regulators of human embryonic stem cell differentiation to definitive endoderm. Genome Biol 2016;17(1): 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hayashi T, Ozaki H, Sasagawa Y, et al. . Single-cell full-length total rna sequencing uncovers dynamics of recursive splicing and enhancer rnas. Nat Commun 2018;9(1):1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nestorowa S, Hamey FK, Sala BP, et al. . A single-cell resolution map of mouse hematopoietic stem and progenitor cell differentiation. Blood, The Journal of the American Society of Hematology 2016;128(8):e20–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Deng W, Zhang K, Liu S, et al. . Jrmgrn: joint reconstruction of multiple gene regulatory networks with common hub genes using data from multiple tissues or conditions. Bioinformatics 2018;34(20):3470–8. [DOI] [PubMed] [Google Scholar]
- 65. Moerman T, Santos SA, González-Blas CB, et al. . Grnboost2 and arboreto: efficient and scalable inference of gene regulatory networks. Bioinformatics 2019;35(12):2159–61. [DOI] [PubMed] [Google Scholar]
- 66. Kim S. ppcor: an r package for a fast calculation to semi-partial correlation coefficients. Communications for statistical applications and methods 2015;22(6):665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kharchenko PV, Silberstein L, Scadden DT. Bayesian approach to single-cell differential expression analysis. Nat Methods 2014;11(7):740–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.