

Summary
Protein N-terminal acetylation is predominantly a ribosome-associated modification, with NatA-E serving as the major enzymes. NatC is the most unusual of these enzymes, containing one Naa30 catalytic subunit and two auxiliary subunits, Naa35 and Naa38; and substrate selectivity profile that overlaps with NatE. Here, we report the Cryo-EM structure of S. pombe NatC with a NatE/C-type bisubstrate analogue and inositol hexaphosphate (IP6), and associated biochemistry studies. We find that the presence of three subunits is a prerequisite for normal NatC acetylation activity in yeast and that IP6 binds tightly to NatC to stabilize the complex. We also describe the molecular basis for IP6-mediated NatC complex stabilization and the overlapping yet distinct substrate profiles of NatC and NatE.
Graphical Abstract
eTOC Blurb
NatC is an unusual member of protein N-terminal acetyltransferases in requiring a small Naa38 subunit for activity. Deng et al. finds that Naa38 is required for normal NatC acetylation, Inositol hexaphosphate binding contributes to yeast NatC complex stability and NatC adopts a unique NAT architecture with a distinct substrate profile.
Introduction
Protein N-terminal acetylation is one of the most common protein modifications, occurring on 50–85% of eukaryotic proteins and 10–30% of bacterial and archaea proteins (Aksnes et al., 2016; Arnesen et al., 2009; Deng and Marmorstein, 2021). N-terminal acetylation changes the local chemical properties of protein N-termini and impacts diverse protein functions including stability, half-life, folding, aggregation, degradation, complex formation, and localization (Aksnes et al., 2016; Arnesen, 2011; Deng and Marmorstein, 2021; Dikiy and Eliezer, 2014; Forte et al., 2011; Holmes et al., 2014; Scott et al., 2011; Shemorry et al., 2013; Yang et al., 2013). Aberrant N-terminal acetylation can also lead to cellular dysfunction including drought-stress signaling in plants(Linster and Wirtz, 2018), human diseases (Dorfel and Lyon, 2015; Myklebust et al., 2015), and cancer(Kalvik and Arnesen, 2013).
Protein N-terminal acetylation is an irreversible modification that is catalyzed by a family of enzymes called N-terminal acetyltransferases (NATs). Dozens of NATs are present, varying in subunit composition, substrate specificity, and regulatory mechanisms (Deng and Marmorstein, 2021). Included amongst them are the three bacterial (RimI, RimJ, and RimL)(Pathak et al., 2016; Vetting et al., 2008; Vetting et al., 2005) homologues and a single archaeal NATs homologue(Liszczak and Marmorstein, 2013; Mackay et al., 2007); all of which tend to harbor relatively flexible N-terminal substrate specificity. Eukaryotes, however, possess more NATs (NatA-H) for more specialized activities and functions. Among them, five NATs (NatA, B, C, D, E) are conserved from yeast to human (Rathore et al., 2016; Starheim et al., 2012), associating with the ribosome to co-translationally acetylate nascent protein chains in a co-translational manner (Magin et al., 2017; Polevoda et al., 2008). NatA, B, C, and E are multi-subunit complexes, each consisting of at least one catalytic subunit and one auxiliary subunit, with the auxiliary subunits serving to modulate the activities of the respective catalytic subunits and act as an anchor to the ribosome, while the rest of the NATs function independently of auxiliary subunits. In addition to NatA, the NatE complex contains an additional catalytic subunit, Naa50, responsible for modifying NatE-type substrates (Deng et al., 2019; Deng et al., 2020a).
Substrate recognition by eukaryotic NATs is usually dictated by the first few residues of the substrate via the active site structure (Deng et al., 2019; Deng and Marmorstein, 2021; Deng et al., 2020b; Goris et al., 2018; Hong et al., 2017; Liszczak et al., 2013; Magin et al., 2015; Stove et al., 2016). The active site of NatA contains a relatively small binding pocket to accommodate a small, uncharged residue at position 1 such as alanine, valine or threonine residues that remains following initiator methionine (iMet) – cleavage (Liszczak et al., 2013). NatB has a hydrophobic pocket for a retained iMet and uses a histidine residue to recognize sequences with D/E/N/Q at position 2 via hydrogen bonding (Arnesen et al., 2009; Deng and Marmorstein, 2021). Interestingly, NatC and NatE share similar and relatively versatile substrate profiles toward substrates containing a retained iMet followed by residues other than D/E/N/Q (Arnesen et al., 2009; Van Damme et al., 2016). NatD is a highly selective enzyme with histones H4 and H2A as its only known substrates(Hole et al., 2011; Song et al., 2003), arising from extensive and specific interactions with each of the first four residues of its substrate (Magin et al., 2015). Although NATs vary in terms of their modes of substrate recognition, their mechanism of catalysis usually involves the use of dedicated residues to serve as general base and acid residues (Deng and Marmorstein, 2021).
The activity of some NATs is further regulated by other protein modulators. For example, within the human NatE complex, there is catalytic crosstalk between the two NAA10 and NAA50 catalytic subunits (Deng et al., 2019). Moreover, the activities of both NatA and NatE are inhibited to a different extent by a small protein binding partner called Huntingtin-interacting protein K (HYPK)(Arnesen et al., 2010; Deng et al., 2020a; Gottlieb and Marmorstein, 2018; Weyer et al., 2017). Inositol hexaphosphate (IP6, also named inositol hexakisphosphate or phytic acid), a small molecule, usually reserved for cellular signaling, appears to act as a stabilizing ligand for both NatA and NatE, by binding in a pocket between NAA15 and NAA10, and, thus, plays an indirect role in promoting NatA and NatE acetylation activity (Cheng et al., 2019; Deng et al., 2019; Deng et al., 2020a; Gottlieb and Marmorstein, 2018).
NatC is distinct from other NATs since it contains two eukaryotic conserved auxiliary subunits – Naa38 and Naa35 – that act together with the Naa30 catalytic subunit(Ochaya et al., 2019; Polevoda and Sherman, 2001; Starheim et al., 2009). Initial studies reported that NatC activity requires the interaction of all three subunits, since knock out of any single subunit produced a similar phenotype in yeast that was accompanied by diminished N-terminal acetylation of a cognate N-terminal substrate, MIRLK-(Polevoda and Sherman, 2001). However, it was subsequently reported that the Naa38 subunit is not required for in vivo acetylation of an Arl3p substrate with an N-terminal protein sequence of MPHLV- (Setty et al., 2004). In plants, knockout of AtNAA35 did not lead to similar phenotypic effects as the deletion of AtNAA30(Pesaresi et al., 2003), and the single subunit of the A. thaliana analogue AtNAA30 can rescue the knockout of yeast NatC complex subunits (Pesaresi et al., 2003). Similarly, recombinant human NAA30 (hMak3) was shown to have substrate-specific acetylation activity, even in the absence of its auxiliary subunits(Starheim et al., 2009). Thus, the functional roles of the NatC auxiliary subunits are not clear, particularly with respect to Naa38, which is not well-conserved across species. Although the recently reported crystal structures and associated biochemical studies of S. cerevisiae NatC (ScNatC) by Grunwald et al. have provided some important insights into NatC substrate selectivity and its catalytic mechanism (Grunwald et al., 2020), several questions remain unanswered. In particular, those regarding the mechanisms dictating the overlapping yet distinct substrate profiles of NatC and NatE and the influence of IP6 on NatC.
In this study, we report that yeast NatC complex formation of all three subunits is a prerequisite for normal NatC acetylation activity and that, like NatA and NatE, IP6 binds tightly to and stabilizes NatC. We also report the Cryo-EM structure of ~100 kDa S. pombe NatC (SpNatC) bound to a NatE/C-type bisubstrate analogue and IP6, with related biochemistry, to reveal the molecular basis for IP6-mediated stabilization of the complex and the similar substrate profiles of NatE and NatC. Comparison of these studies with the recently published structural and functional studies of the ScNatC complex(Grunwald et al., 2020) reveal evolutionarily conserved and divergent features of NatC.
Results
Naa38 is required for normal NatC acetylation activity in yeast.
We found that overexpression of the SpNatC complex containing full-length SpNaa30 (residues 1–150), N-terminally truncated SpNaa35 (residues 31–708, with deletion of divergent residues, 1–30) and SpNaa38 (residues 48–116, with deletion of divergent residues, 1–47) in E. coli produced soluble protein that could be purified to homogeneity (Figure 1a). To evaluate the activity of the recombinant ternary SpNatC, we used an in-vitro acetyltransferase activity assay with different peptide substrates. Consistent with previous studies, the recombinant SpNatC is active toward both its own canonical substrate - “MLRF peptide” and the canonical NatE substrate – “MLGP peptide”, but with preference for the “MLRF peptide” (Grunwald et al., 2020) (Figure 1b, see methods section for full peptide sequences). To investigate the catalytic roles of Naa38, we could readily purify both binary (ScNaa301−161 and ScNaa3519−733) and ternary ScNatC complexes (ScNaa301−161 and ScNaa3519−733, and ScNaa381−70) from Saccharomyces cerevisiae to compare their activities. Consistent with the studies by Grunwald et al (Grunwald et al., 2020), evaluation of the activities of the binary and ternary ScNatC complexes toward the canonical NatC substrate (MLRF peptide) revealed that the ternary complex showed robust activity, while the binary complex showed compromised activity (Figure 1b). Thus, it appears that Naa38 is required for optimal NatC activity in yeast.
Figure 1. Binary NatC displays deficiency in acetylation activity and inositol hexaphosphate (IP6) binding stabilizes NatC complex formation.
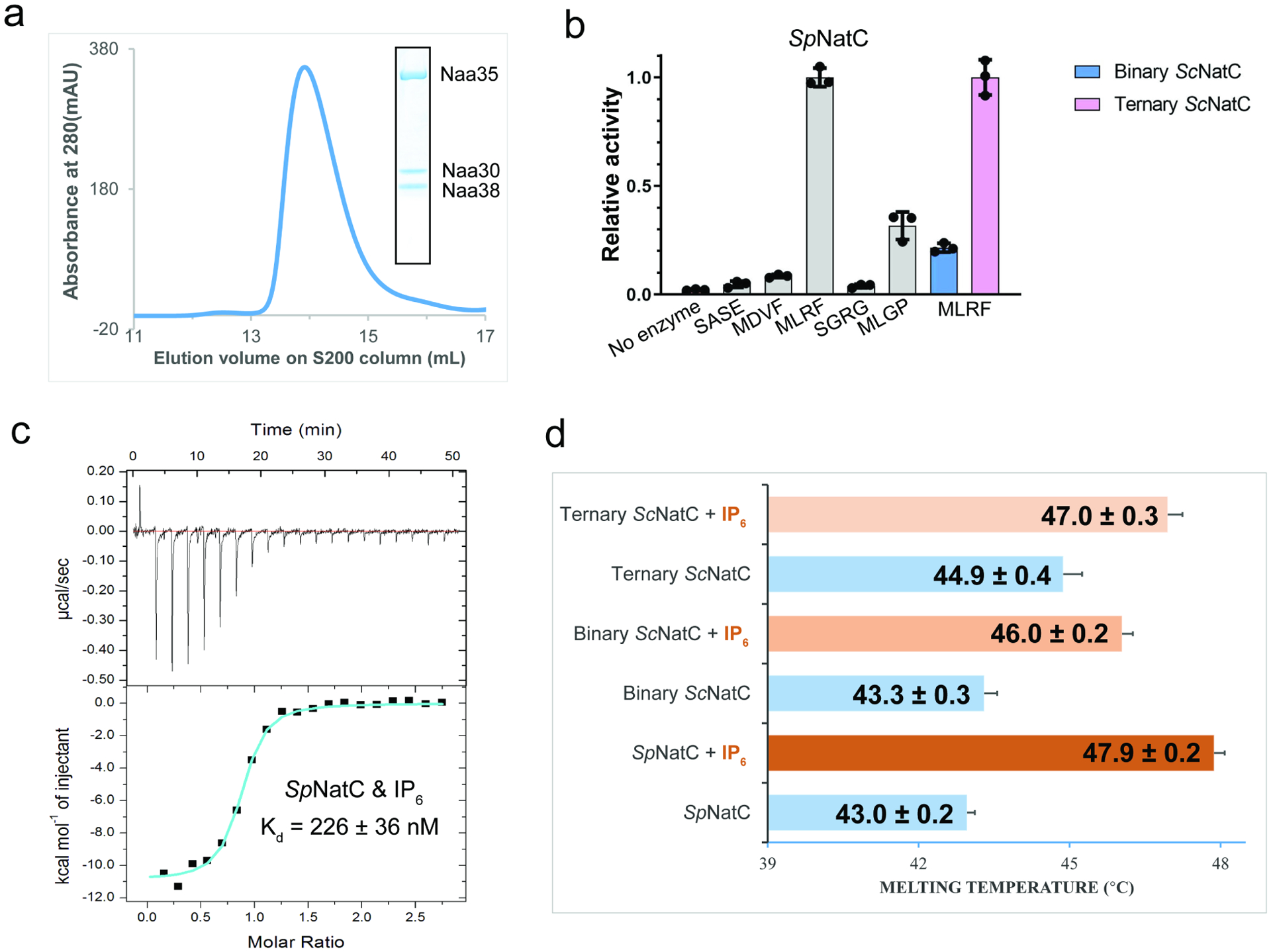
(a) Gel filtration elution profile of the ternary SpNatC complex using a Superdex S200 column. Coomassie-stained SDS–PAGE of elution peak is shown in the box, with bands for corresponding SpNatC subunits labeled.
(b) Comparison of SpNatC activity toward different peptide substrates. The activities shown in gray are normalized to the activity of SpNatC toward MLRF peptide. The pink and blue columns show the activity of binary and ternary ScNatC toward the MLRF peptide, respectively, and are normalized to the pink column. Errors are shown in SD with n = 3.
(c) Representative isothermal titration calorimetry (ITC) curve of IP6 titrated into SpNatC. The ITC fitting information is N = 0.830 ± 0.00943 sites, ΔH = −1.091×104 ± 177.0 cal mol−1, ΔS = −6.79 cal mol−1 deg−1.
(d) Differential scanning fluorimetry assays of NatC alone or with IP6. Average calculated melting temperature transitions are indicated. Numbers following the plus/minus sign and error bars in the figure indicate the SD of each sample, n = 3.
Inositol hexaphosphate (IP6) binding contributes to yeast NatC complex stability.
Earlier reports demonstrated that IP6 interacts with and stabilizes both the yeast and human NatA and NatE complexes (Cheng et al., 2019; Deng et al., 2019; Deng et al., 2020a; Gottlieb and Marmorstein, 2018), but does not appear to interact with human NatB (Deng et al., 2020b). To determine if IP6 could bind to the recombinant NatC complex, we employed isothermal titration calorimetry (ITC). We found that IP6 binds to SpNatC with a Kd of ~225 nM (with a stoichiometry of ~1) (Figure 1c), similar to its Kd value for yeast NatA(Deng et al., 2019). Differential scanning fluorimetry (DSF) was used to evaluate a potential role of IP6 binding in NatC complex stability. We found that addition of IP6 increased the thermal stability of ternary SpNatC, binary ScNatC, and ternary ScNatC, by ~4.9°C, 2.7°C, and 2.1°C, respectively. Thus, IP6 can provide additional thermal stability to the yeast NatC complexes (Figure 1d).
NatC adopts a distinct NAT architecture.
In order to understand the molecular details underlying the ternary NatC complex, its overlapping yet distinct substrate selectivity with NatE, and the mode of IP6 stabilization, we performed single particle Cryo-EM analysis using ternary SpNatC prepared in the presence of the CoA-Ac-MLGP bisubstrate conjugate and IP6. The MLGP sequence was selected for synthesis of the conjugate inhibitor as it is a predicted substrate for both NatC and NatE. A 3.16 Å-resolution Cryo-EM three-dimensional (3D) map was determined from 607,131 particles, which were selected from 5,397 raw electron micrographs (Supplementary Figure 1). Most areas of the EM map contained excellent sidechain density, with a local resolution of ~3 Å, suggesting the relative rigidity of the recombinant complex (Supplementary Figure 1B and 2). The atomic model was built de novo based on the EM map and details for the model refinement statistics can be found in Table 1 (Supplementary Figure 2).
Table 1 |.
Cryo-EM data collection, refinement and validation statistics
|
SpNatC with Bisubstrate and IP6 EMD-23110 PDB: 7L1K |
|
|---|---|
| Data collection and processing | |
| Magnification | 105,000 |
| Voltage (keV) | 300 |
| Electron exposure (e/Å2) | 42 |
| Defocus range (μm) | −1.0 to −3.0 |
| Pixel size (Å) | 0.84 |
| Symmetry imposed | C1 |
| Initial particles (no.) | 1,860,276 |
| Final particles (no.) | 607,131 |
| Map resolution (Å) | 3.16 |
| FSC threshold | 0.143 |
| Map resolution range (Å) | 2.5–3.5 |
| Refinement | |
| Initial model used (PDB code) | - |
| Model resolution (Å) | 3.4 |
| FSC threshold | 0.5 |
| Model resolution range (Å) | - |
| Map sharpening B factor (Å2) | 159.1 |
| Model composition | |
| Non-hydrogen atoms | 7216 |
| Protein residues | 883 |
| Ligands | 2 |
| B factors (Å2) | |
| Protein | 26.97/110.48/71.91 |
| Ligand | 86.46/102.51/93.10 |
| R.M.S. deviations | |
| Bonds lengths (Å) | 0.006 |
| Bond angles (°) | 1.036 |
| Validation | |
| MolProbity score | 1.76 |
| Clash score | 4.10 |
| Poor rotamers (%) | 0.38 |
| Ramachandran plot | |
| Favored (%) | 89.55 |
| Allowed (%) | 10.22 |
| Disallowed (%) | 0.23 |
The ternary SpNatC complex contains three proteins, the catalytic Naa30 subunit, the large auxiliary Naa35 subunit, and the small auxiliary Naa38 subunit. Upon superficial inspection of the complex, it appears as though it is only formed by two proteins, as Naa38 is embedded within the Naa35 fold to form a “composite” auxiliary subunit (Figure 2a and 2b). While Naa30 displays a canonical NAT fold with mixed α/β secondary structure, the Naa35/38 complex forms a clamp that pinches Naa30 on opposite sides. This clamp-like structure is distinct from the architecture of the large auxiliary subunits of NatA and NatB, which surround the base of their respective catalytic subunits (Deng et al., 2020b; Gottlieb and Marmorstein, 2018; Hong et al., 2017; Liszczak et al., 2013). The Naa35 architecture is mostly helical, with 25 helices, three short β-strands in its N-terminal region and a single β-hairpin at its C-terminal region that are unique to the NatC complex (Figure 2b and Supplementary Figure 3). Naa38 adopts an Sm-like fold, with an N-terminal α-helix, followed by a sharply bent β-sheet consisting of 5 five antiparallel β-strands (Figure 2b and Supplementary Figure 4). The Naa35/Naa38 clamp contains α4-α13 from Naa35 at its base, and α14-α25 and the C-terminal β-hairpin of Naa35 flanking one side of Naa30. The opposing side of Naa30 is flanked by a composite of the N-terminal end of Naa35 (α2, α 3, β1- β3) and the entirety of Naa38 (Figure 2a and 2b). Consistent with the composite nature of the Naa35-Naa38 interaction, the buried surface area between Naa38 and Naa35 is ~1925 Å2, which is notably higher than the area buried between Naa30 and Naa35 (~1735 Å2), and between Naa30 and Naa38 (~197 Å2).
Figure 2. Overall structure of SpNatC reveals intimate interactions among all subunits.
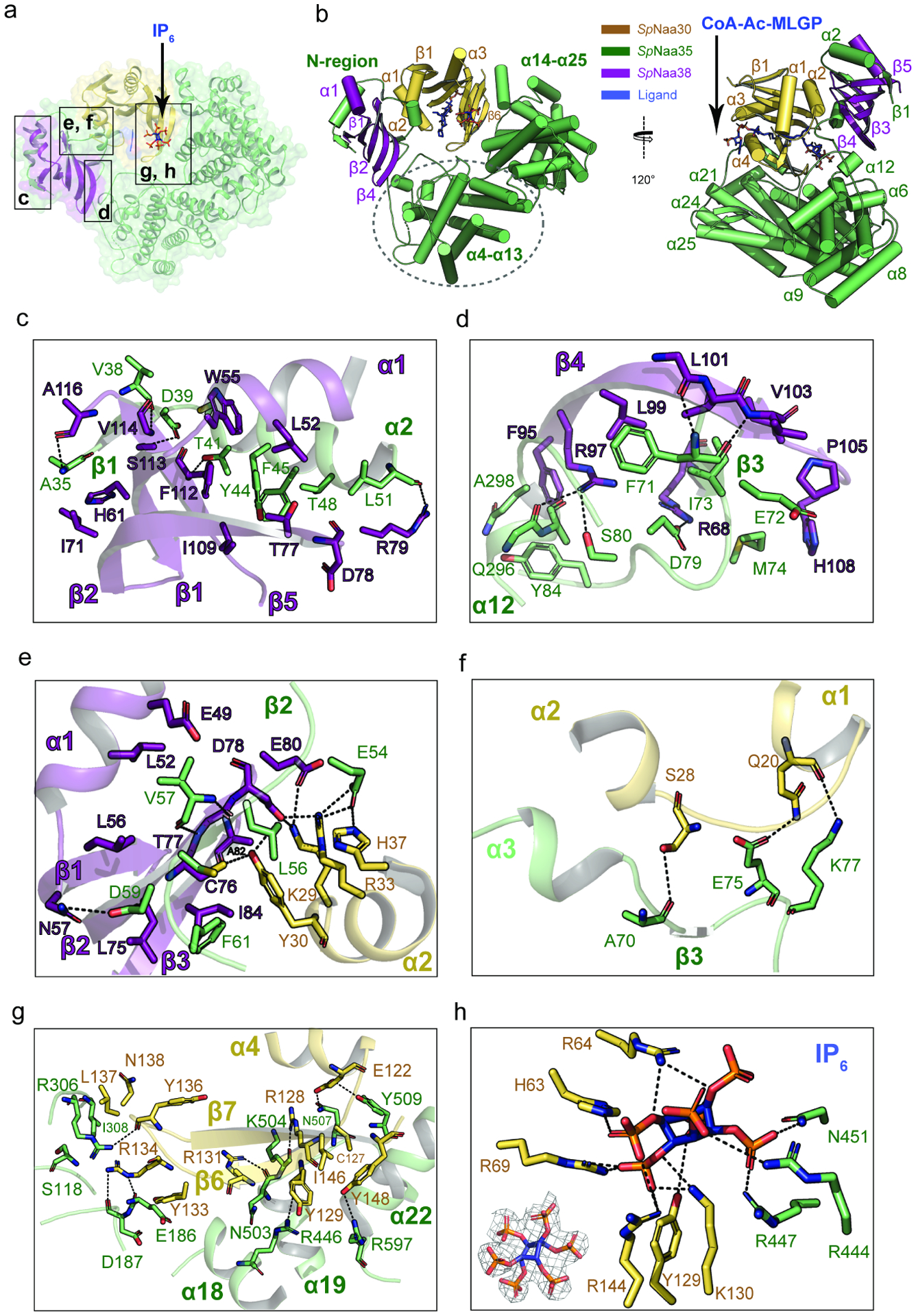
(a) Naa30 (bright orange), Naa38 (dark purple) and Naa35 (lime green) are shown in transparent surface and cartoon, with the boxed area labeled.
(b) The CoA-MLGP bisubstrate conjugate and inositol hexaphosphate (IP6) are shown in sticks and colored in blue.
(c) Zoom-in view of the first sub-interface between Naa38 and Naa35. Hydrogen bonds are indicated by dashed black lines. For simplicity, only some of the hydrophobic interactions are shown.
(d) Zoom-in view of the second sub-interface between Naa38 and Naa35.
(e) Zoom-in view of the third sub-interface between Naa38 and Naa35, which also involves some residues from Naa30 (light orange).
(f) Zoom-in view of the interface between Naa35 and Naa30 α1-α2 segment.
(g) Zoom-in view of the interface between Naa35 and Naa30 β6-β7 segment.
(h) Zoom-in view depicting key residues involved in interactions with IP6. The lower left corner shows the fit of the IP6 molecule in the EM density map. The contour level is 5.0 sigma.
Naa38 plays a key role in the NatC complex.
The structure of SpNatC was consistent with our biochemical data suggesting that Naa38 plays important structural and functional roles within the NatC complex. A more detailed view of the Naa35-Naa38 interface reveals that it is nucleated by the formation of three anti-parallel β-sheets between Naa35 and Naa38, resulting in extensive hydrogen bonding and van der Waals interactions, which also involve the α2 and α3 helices of Naa35. These interactions can roughly be divided into three regions according to the relative position of the β-sheets (Figure 2).
The first sub-interface involves the Naa35 β1-α2 segment (Figure 2c). Hydrogen bonds are formed between the backbone nitrogen of Naa35-Ala35 and the backbone carbonyl of Naa38-Ala116, the backbone carbonyl of Naa35-Val38 and the backbone nitrogen of Naa38-Val114, the sidechains of Naa35-Asp39 and of Naa38-Ser113, the sidechain of Naa35-Thr41 and the backbone nitrogen of Naa38-Phe112, and the backbone carbonyl of Naa35-Leu51 and the sidechain of Naa38-Arg79. This region also features a large area of van der Waals interactions involving residues Ala35, Gly36, Tyr37, Tyr44, Phe45, Ala47, and Thr48 from Naa35 and residues Gly48, Leu52, Trp55, His 61, Ile71, Thr77, Asp78, Ile109, Arg115, and Ala116 from Naa38.
The second sub-interface is mediated by Naa35-β3 and α12 (Figure 2d). Hydrogen bonds are formed between the backbone nitrogen of Naa35-Phe71 and the backbone carbonyl of Naa38-Leu101, the backbone carbonyl of Naa35-Phe71 and the backbone nitrogen of Naa38-Leu103, the sidechains of Naa35-Asp79 and Naa38-Arg68, the sidechains of Naa35-Ser80 and Naa38-Arg97, and the backbone carbonyl of Naa35-Gln296 and the sidechain of Naa38-Arg97. Residues involved in van der Waals interactions include Ala70, Glu72, Ile73, Met74, Tyr84, Ala 297, Gln 298, Val30, and Ala301, from Naa35, and residues Phe95, Ala98, Leu99, Val102, Ile104, Pro105, and His108 from Naa38.
The third sub-interface is mediated by Naa35-β2 but also involves extensive interactions with the catalytic Naa30 subunit (Figure 2e). The hydrogen bonds between Naa35 and Naa38 are formed between the sidechains of Naa35-Asp59 and Naa38-Asn57, the backbone nitrogen of Naa35-Val57 and the backbone carbonyl of Naa38-Thr77, and the backbone carbonyl of Naa35-Val57 and the backbone nitrogen of Naa38-Thr77. Naa38 engages with the α2 helix of the Naa30 subunit to form an extensive hydrogen bonding network via the Naa30-Tyr30 sidechain to the backbone carbonyl of Naa38-Ala82, and the sidechains of Naa38-Cys76 and Asp78; the sidechain of Naa30-Lys29 hydrogen bonds to the sidechains of Naa38-Asp78 and Glu80; and between the sidechains of Naa30-Arg33 and Naa38-Asp78. As the α1- α 2 loop of Naa30 plays a key role in protein N-terminal substrate recognition, these Naa38-Naa30 interactions likely play a key role in substrate recognition by NatC (see discussion). Finally, the Naa30- α2 helix also interacts with Naa35 where Naa35-Glu54 forms hydrogen bonds with Naa30-Arg33 and His37. Within this sub-interface, the hydrophobic interactions are primarily mediated by residues Glu49, Leu52, Leu56, Leu75, Cys76, Asp78, and Ile84 from Naa38 and residues Leu56, Cys58, Asp59 and Phe62 from Naa35. Taken together, it appears that Naa38 plays key structural and, likely, functional roles in NatC.
The substrate binding α1-α2 loop and β6-β7 segments of Naa30 are buried within Naa35.
Naa30 uses its α1-α2 loop (Figure 2f) and β6-β7 segments to interact with Naa35 (Figure 2g). Analogous segments in the NatA and NatB complexes are used for N-terminal substrate recognition. In these complexes, however, the β6-β7 segment is exposed to solvent (Deng et al., 2020b; Gottlieb and Marmorstein, 2018; Hong et al., 2017; Liszczak et al., 2013). By contrast, this interaction between Naa30 and Naa35 is facilitated by hydrogen bonds between the Naa30 α1-α2 loop and Naa35 α3-β3 segment: the sidechain of Naa30-Ser28 and the backbone carbonyl of Naa35-Ala70; between the sidechains of Naa30-Gln20 and Naa35-Glu75; and the backbone carbonyl of Naa30-Gln20 and the sidechain of Naa35-Lys77.
Notably, the interactions between Naa30 β6-β7 and Naa35 are significantly more extensive than those found in the NatA and NatB complexes (Figure 2g). Here, hydrogen bonds are formed between the sidechain of Naa30-Glu122 and the sidechains of Naa35-Gln507 and Tyr509, the sidechain of Naa30-Arg128 and the backbone carbonyl of Naa35-Lys504, the backbone carbonyl of Naa30-Tyr129 and the sidechain of Naa35-Arg446, the sidechain of Naa30-Arg131 and the backbone carbonyl of Naa35-Gln503, the sidechain of Naa30-Arg134 and the backbone carbonyls of both Naa35-Glu186 and Asp187, the backbone carbonyl of Naa30-Tyr136 and the sidechain of Naa35-Arg306, and the sidechains of Naa30-Tyr148 and Naa35-Arg597. Van der Waals interactions are observed between residues Cys127, Leu132, Tyr133, Leu137, Asn138, Phe143, Ile146, and Tyr148 from Naa30, and residues Ile308, Asn441, Cys443, Leu505, Phe581, Ser590, Tyr591, and Ala594 from Naa35. Together, it appears that the Naa35 auxiliary subunit makes extensive contacts with the protein N-terminal substrate binding loops of Naa30 to influence substrate recognition.
IP6 is bound at the interface between Naa30 and Naa35 in close proximity to the peptide substrate binding site.
EM density corresponding to a bound IP6 molecule is well-resolved, revealing a IP6 binding pocket at the interface between the Naa30 and Naa35 subunits (Figure 2h). Notably, the IP6 binding region is distinct from its binding sites in the NatA and NatE complexes(Deng et al., 2019; Deng et al., 2020a; Gottlieb and Marmorstein, 2018). In SpNatC, IP6 engages in polar interactions with Naa30 residues His63, Arg64, Arg69, Tyr129, Lys130, and Arg144, and Naa35 residues Arg444, Arg447, and Asn451. Interestingly, although the IP6 binding pocket of NatC features a similar chemical environment to the one found in the NatA and NatE complexes, the location in the architecture of these NAT complexes differs. In NatC, IP6 is located close (~8 Å) to the peptide substrate entrance site, while the pocket found in NatA and NatE is close to the Ac-CoA entrance site (Figure 3). Therefore, the NatC-bound IP6 molecule could be in position to alter NatC activity. To evaluate this possibility, we assayed the influence of IP6, added at saturating levels of 10 x Kd, on NatC activity and found that incubation of the NatC complex IP6 produced a relatively modest inhibitory effect, as it also did for SpNatA and hNatB, (Supplementary Figure 5). Based on this data, it appears that IP6 plays a predominantly structural role in NatC.
Figure 3. Comparison of NatC with NatA and NatB reveals divergent NatC architecture.
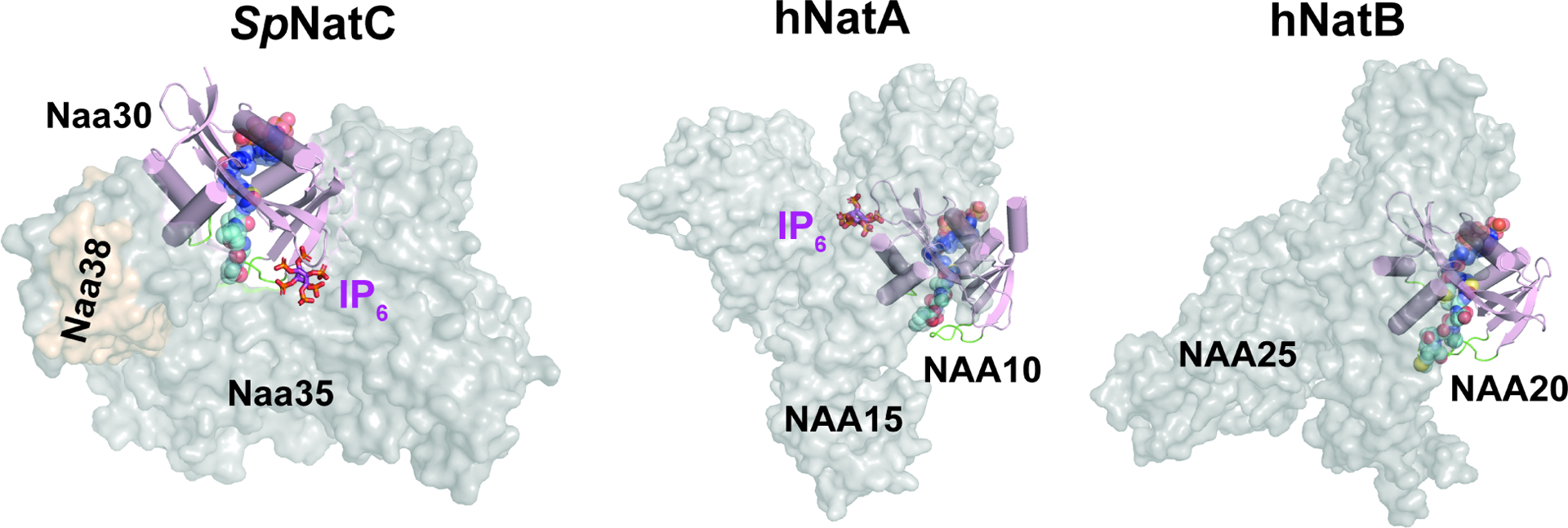
The auxiliary subunits are shown as transparent surface in grey or brown, while the catalytic subunits are shown as cartoon in light pink (catalytic subunits are aligned in the same orientation). The blue spheres, cyan spheres, and magenta sticks represent Ac-CoA, peptide substrate, and IP6, respectively. The highlighted green loops shown are α1-α2 and β6-β7 substrate binding loops of the catalytic subunits. The PDB models for generating this figure: HsNatA with IP6, PDB: 6C9M (the bisubstrate analogue shown is aligned from SpNatA, PDB:4KVM); HsNatB with the bisubstrate analogue, PDB: 6VP9; model of SpNatC with the bisubstrate analogue and IP6, PDB: 7L1K. As NatE is a complex with dual catalytic subunits and shares the same auxiliary subunit with NatA, for simplicity, it is not compared in this figure.
NatC architecture displays significant differences from NatA, NatE and NatB.
Overall, there are three notable differences between NatC and the other multi-subunit NATs – NatA, NatE and NatB. First, the auxiliary subunits of NatA, NatE and NatB contain only helical secondary structure, forming 10–13 conserved tetratricopeptide repeat (TPR) motifs(Deng et al., 2019; Deng et al., 2020a; Deng et al., 2020b; Gottlieb and Marmorstein, 2018; Hong et al., 2017; Liszczak et al., 2013). In contrast, the NatC auxiliary subunits (Naa35 and Naa38) do not contain TPR repeats and but do contain several beta strands that make key interactions in the complex (Figure 2b). Secondly, the auxiliary subunits of NatA, NatE, and NatB form a closed ring-like cradle to completely wrap around their corresponding catalytic subunit. In contrast, NatC auxiliary subunits arrange themselves into a clamp-like structure to only wrap roughly half-way around the Naa30 catalytic subunit (Figure 3). Thirdly, the Naa30 β6-β7 peptide substrate recognition loop is buried within the auxiliary subunit, while the corresponding loops in NatA, NatE, and NatB are largely exposed to solvent (Figure 3). Thus, the relative orientation of the catalytic subunit to the auxiliary subunit (s) is different in NatC relative to NatA and NatB. In turn, this has implications in altering the NatC substrate binding mode: the NatC substrate binding tunnel is roughly perpendicular to the Naa30-Naa35 interface, while the substrate binding tunnel of NatA, NatE, and NatB is parallel to the catalytic-auxiliary subunit-interface (Figure 3). Taken together, the NatC complex forms an architecture that is distinct from other multi-subunit NATs.
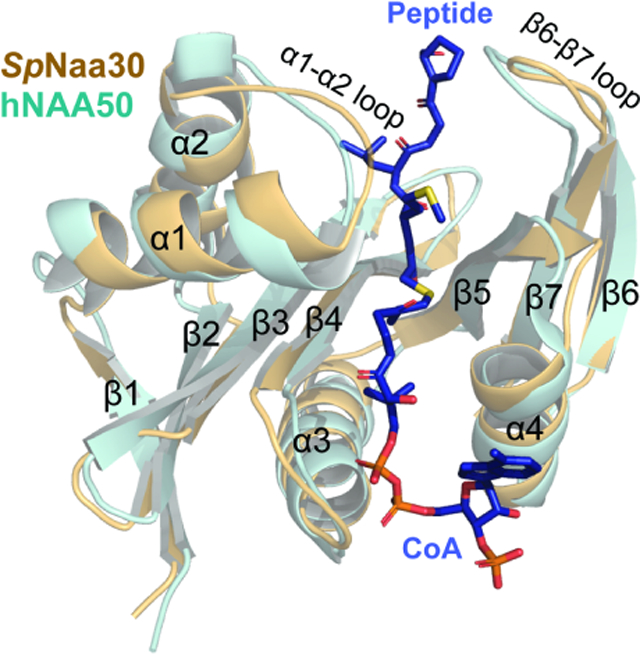
Substrate recognition by Naa30 shows similarity to NAA50.
Overall, Naa30 displays a typical NAT fold containing four α-helices and seven β-strands with similar substrate binding modes: Ac-CoA enters the catalytic active site through a groove formed by α3 and α4 of the catalytic subunit, while the peptide substrate enters the active site on the opposite side of the catalytic subunit flanked by the α1–α2 and β6-β7 peptide substrate binding loops (Deng and Marmorstein, 2021) (Figure 4a and 4b). In the Cryo-EM map, density for the CoA-Ac-MLGP conjugate bisubstrate is well-resolved, allowing us to confidently model both the CoA portion and all four residues of the peptide portion (Figure 4c).
Figure 4. Substrate recognition by Naa30 is similar to NAA50.
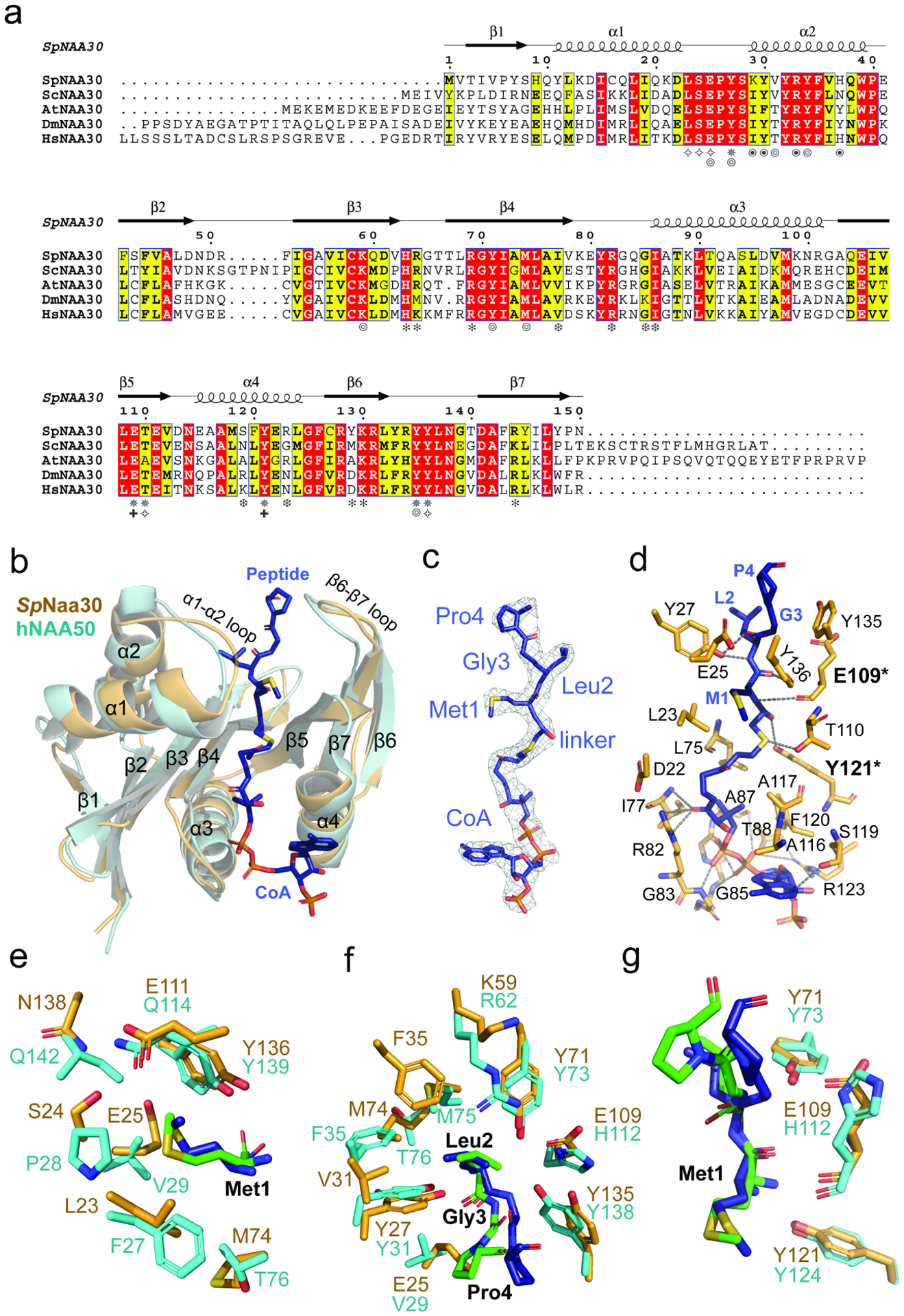
(a) Sequence alignment of Naa30 orthologs from S. pombe (Sp), S. cerevisiae (Sc), A. thaliana (At), D. melanogaster (Dm), and H. sapiens (Hs). Numbering and secondary structure elements for SpNaa30 are indicated above the sequence alignment. Residues of SpNaa30 that contact the peptide backbone (✵), CoA (❆), Met1 (✧), Leu2 (⦾), Naa38 (⦿), IP6 (✽), and catalytic residues (✚) are indicated.
(b) Structural alignment of SpNaa30 (bright orange) with HsNAA50 (cyan), with secondary structure elements indicated.
(C) The fit of the bisubstrate inhibitor in the EM density map. The contour level is 5.0 sigma.
(d) Highlighted polar and hydrophobic interactions between CoA-Ac-MLGP and SpNaa30 are depicted in the 3D view.
(e) Residues forming a hydrophobic pocket surrounding the substrate peptide Met1 sidechain are shown in sticks (Orange, SpNaa30; Cyan, HsNAA50)
(f) Residues forming a hydrophobic pocket surrounding the substrate peptide Leu2 sidechain are shown in sticks. (Orange, SpNaa30; Cyan, HsNAA50)
(g). Residues proposed as catalytic residues are shown in sticks. (Orange, SpNaa30 E109 and Y121; Cyan, HsNAA50 Y73 and H112).
Naa30 harbors a conserved Ac-CoA binding motif R82XXG85XA87 where Ac-CoA binding is mediated by a series of hydrogen bonds, mainly to the pyrophosphate group. Specifically, the sidechains of Arg82, Ser119, Arg123 and backbone atoms from Ile77, Gly85, Ile86, and Ala87 contribute to Ac-CoA hydrogen bonding (Figure 4d).
In our NatC model, Naa30 binds the peptide substrate mainly through peptide backbone hydrogen bonding and hydrophobic pockets for the sidechains of the first two peptide residues. Direct hydrogen bonds are formed between the backbone amide groups of the peptide residues 1 and 2 to Naa30 residues Tyr27, Glu109, Thr110, Tyr121, and Tyr136 (Figure 4d). The binding pocket for the peptide Met1 is surrounded by residues Leu23, Ser24, Glu25, Glu111, and Tyr136 (Figure 4e), while the binding pocket for peptide Leu2 is half-open and surrounded by Tyr27, Val31, Tyr34, Phe35, Lys59, Tyr71, and Met74 (Figure 4f). Beyond the first two residues, there are no direct contacts between the peptide substrate and Naa30, with the exception of distant contacts between the backbones of peptide Gly3 and peptide Pro4 with Naa30-Tyr135 (4–4.5 Å) and Naa30-Glu25 (~4 Å), respectively. These two Naa30 residues likely play a role in recognizing positions 3 and 4 of high-affinity NatC peptide substrates, as supported by the recent report by Grunwald et al.(Grunwald et al., 2020)
Previous studies have described the overlapping substrate profile of NatC and NAA50. Since our NatC model contains the canonical NatE (NAA50)-type peptide substrate (Met2-Leu2-Gly3-Pro4) in the bisubstrate inhibitor, we were able to evaluate this relationship by visual inspection of the inhibitor-bound SpNaa30 subunit from the model. The SpNaa30 model aligns well with the HsNAA50 structure (PDB: 3TFY) bound to the same MLGP peptide fragment, with a root-mean-square deviation (RMSD) of 1.23 Å (over 105 common Cα atoms) (Figure 4b). A more detailed view of the peptide substrate binding site reveals that Met1 sits in similar binding pockets in both structures (Figure 4e), consistent with their high specificity for Met1. Specificity for Leu at position 2 is also similar. Notably, residues NatC-Tyr27 and Tyr71, which are responsible for Leu2 backbone recognition align well with the corresponding residues in NAA50 (Figure 4f). For Leu2 sidechain recognition, the residues, Naa30-Val31 and Lys59 are replaced by bulkier residues, NAA50-Phe35 and Arg62 (Figure 4f), respectively. However, Naa30-Phe35, Ala73 and Met74 are replaced by NAA50-Phe35, Met75 and Thr76 (Figure 4f), perhaps explaining the greater tolerance of NAA50 for other residues at position 2. In both models, the peptide substrates begin to diverge from the architecture of their respective models at residue 3, where the Cα atoms of the corresponding peptide Gly3 residues of both models are ~3 Å apart. This also seems to be correlated with a shift in the p-hydroxyl group of a nearby conserved Tyr residue (Naa30-Tyr135 and NAA50-Tyr138) by about 2.5 Å (Figure 4g). The corresponding peptide Pro4 residues also point in different directions (Figure 4g). We propose that these differences in peptide substrate positioning beyond residue 2 is mediated by the different active site environments of Naa30 and NAA50 in this region, thus allowing these two enzymes to harbor varying activities toward substrates with the same residues at positions 1 and 2 but differing residues at position 3 and beyond (See discussion). Taken together, this comparison explains the overlapping yet distinct substrate profiles of NatC and NatE.
Mutational analysis reveals that Naa30 Glu-109 and Tyr-121 play key catalytic roles.
In order to evaluate key Naa30 residues identified in our model and their functional roles in substrate binding and catalysis, we used a radioactive in vitro acetyltransferase activity assay in conjunction with the MLRF peptide as the peptide substrate to kinetically characterize WT and mutant SpNatC proteins. Each mutant was purified to homogeneity and displayed identical gel filtration chromatography elution profiles (data not shown), consistent with their native folding and complex formation. Consistent with our structural observations, mutation of a majority of residues involved in peptide substrate recognition resulted in an increase in Km (Table 2 and Supplementary Figure 6). As the “YY motif” is shown to be conserved in most NATs (Stove et al., 2016) (except NatD/NAA40), single mutation of either Naa30-Tyr135 or Tyr136 to an alanine resulted in a significant loss in activity, with the Y136A mutant displaying almost no detectable activity. In previous studies, NAA50 residues Tyr73 and His112 were proposed to contribute to catalysis (Liszczak et al., 2011). The corresponding residues in Naa30, Tyr71 and Glu109, however, had an unexpected effect on catalysis. Naa30-Y71A or Y71F had a relatively modest effect on catalysis, while mutation of Naa30-Glu109 to either alanine or glutamine led to a more significant loss of catalytic efficiency, impacting both Km and kcat. NAA50-Tyr124 and hNatB-Tyr123 were recently proposed to play analogous catalytic roles (Deng et al., 2020b), and the corresponding mutation of Naa30-Tyr121 to alanine demonstrated that NatC activity was significantly compromised (Table 2 and Supplementary Figure 6). While Glu109 is positioned to function as a general base to deprotonate the α-amino group of the peptide substrate, Tyr121 is positioned to function as a general acid to re-protonate the CoA leaving group. This is consistent with the proposed roles of the analogous Glu118 and Tyr130 residues in ScNatC (Grunwald et al., 2020), as well as the conserved nature of these residues (Figure 4a). In addition, Naa30-Asn114 is highly conserved among all NATs (data not shown) and it was recently suggested to play a structural role in orienting the catalytic tyrosine residues (SpNaa30-Tyr135 and Tyr136) by ensuring the proper position of the active site helix (α4) for interaction with Ac-CoA (Deng et al., 2020b). Consistent with the importance of Asn114, we observed that Naa30-N114A exhibited an ~50% loss in kcat. Taken together, the conserved Naa30-Glu109 and Tyr121 residues likely play key catalytic roles in NatC complex activity (Figure 4g).
Table 2.
SpNatC complex (WT and mutant SpNaa30) catalytic parameters
| Substrate | Protein (SpNatC) | kcat* (min−1) | Relative kcat (normalized to WT) | Km* (μM) | Relative Km (normalized to WT) | kcat/Km (normalized to WT) |
|---|---|---|---|---|---|---|
|
Gag Peptide (MLRF-) |
WT | 19.7 ± 3.6 | 1.0 | 7.8 ± 1.4 | 1.0 | 1.0 |
| S24A | 19.1 ± 2.8 | 0.97 | 8.9 ± 1.0 | 1.1 | 0.85 | |
| E25A | 33.6 ± 11 | 1.7 | 11.1 ± 6.1 | 1.42 | 1.2 | |
| Y27A | 8.7 ± 1.6 | 0.44 | 2.5 ± 0.4 | 0.32 | 1.4 | |
| Y71A | 32.7 ± 8.5 | 1.7 | 10.7 ± 3.8 | 1.4 | 1.2 | |
| Y71F | 18.4 ± 8.3 | 0.94 | 13.9 ± 11 | 1.8 | 0.52 | |
| M74A | 13.3 ± 1.9 | 0.68 | 11.4 ± 5.2 | 1.5 | 0.46 | |
| E109A | 10.4 ± 0.6 | 0.53 | 15.2 ± 9.6 | 1.95 | 0.27 | |
| E109Q | 2.4 ± 0.4 | 0.12 | 72.1 ± 95 | 9.25 | 0.013 | |
| N114A | 10.9 ± 1.9 | 0.55 | 4.4 ± 0.9 | 0.56 | 0.99 | |
| Y121A | 1.8 ± 0.7 | 0.093 | 9.7 ± 5.0 | 1.2 | 0.075 | |
| Y121F | 1.8 ± 0.5 | 0.090 | 5.3 ± 2.0 | 0.68 | 0.13 | |
| Y135A | 4.1 ± 1.3 | 0.21 | 4.0 ± 3.9 | 0.51 | 0.41 | |
| Y136A | 0.2 ± 0.1 | 0.011 | 0.7 ± 0.4 | 0.091 | 0.13 |
values indicated mean ± S.D. of three independent kinetic curves fitting results.
See Supplementary Figure.6, for the raw curves.
Discussion
Here, we report that the notably small SpNaa38 auxiliary subunit cooperates with the large Naa35 auxiliary subunit to stabilize the active N-terminal acetyltransferase NatC complex for its robust N-terminal acetylation activity. Through structure/function studies, we observed that Naa38 interacts with Naa30-α2, a region that directly contributes to recognition of peptide substrate backbone and sidechain residues. Based on these observations and similar ScNatC complex biochemical and structural findings described by Grunwald et al.(Grunwald et al., 2020), it appears that Naa38 is essential for normal NatC acetylation activity. Therefore, we propose that Naa38 plays a conserved role in yeast. Future studies are needed to further interrogate this in the plant or human systems, as some reports suggest that Naa38 may not be as important in higher eukaryotes (Pesaresi et al., 2003; Starheim et al., 2009).
In S. pombe, we have found that the ternary SpNatC complex displays acetylation activity toward both canonical NatC and NatE-type peptide substates, but with a preference for NatC-type substrates. While recognition of residues 1 and 2 of the cognate substrate appears highly homologous between the Naa30 and NAA50 catalytic subunits of NatC and NatE, respectively, the substrate binding pockets responsible for recognizing substrate residues beyond the penultimate sidechain diverge significantly. This likely explains the divergence in substrate profiles observed between these two NATs.
IP6 was previously found to play a stabilizing role at the interface between the NAA15 and NAA10 subunits of the NatA and NatE complexes (Deng et al., 2019; Deng et al., 2020a; Gottlieb and Marmorstein, 2018),(Cheng et al., 2019). Here, we find that IP6 also appears to play a stabilizing role in both ScNatC and SpNatC, and that the SpNatC structure reveals that IP6 binds at the Naa35-Naa30 interface located near the peptide substrate entrance site, although it does not appear to play a major direct role in modulating NatC activity. Given that only 4 of the 6 phosphate groups of IP6 are engaged in protein interactions in the SpNatC structure (Supplementary Figure. 7), we note that it is also possible that other phosphorylated forms of inositol such as IP4 or IP5 might also bind SpNatC in a physiological setting.
Grunwald et al. recently reported X-ray crystal structures of ScNatC in several liganded states along with associated biochemical studies (Grunwald et al., 2020). Overall, their findings are in agreement with our findings on SpNatC reported here. There are, however, some notable differences when comparing NatC from S. pombe and S. cerevisiae, which have implications for the conserved and unique features of NatC.
The ScNatC structures bound to cognate NatC-type peptide substrates shows specificity for the first four residues, which is consistent with their reported peptide substrate mutation and binding data. In contrast, our structure, featuring SpNatC bound to a peptide sequence that is optimal for NatE (NAA50) acetylation, shows SpNaa30’s specificity for only the first two residues. This mode of recognition is consistent with recognition for these same residues by NAA50, while there is significant divergence in the regions of the peptide binding grooves of Naa30 and NAA50 that are responsible for interaction with substrates residues beyond residue 2. This comparison suggests that discrimination between Naa30 and NAA50 substrates is largely dictated by the identities of the third and fourth residues in an N-terminal substrate. As NatC-type and NatE-type substrates can include many different hydrophobic/amphipathic N-termini (ML- MI-, MF, MW-, MV-, MM-, MH-, and MK-) (Ree et al., 2018), it may be difficult for a single enzyme to cover acetylation of such a broad repertoire of protein N-termini. Given that Naa50 is inactive in yeast (Deng et al., 2019), we propose that the overlapping substrate profile for residues at positions 1 and 2 by NatC and NatE may have evolved in higher eukaryotic cells to fully cover the acetylation on these types of N-terminal substrates.
Grunwald et al. identified several electropositive regions (EPR) on ScNatC that are implicated in ribosome association. Of these regions, labeled EPR1-EPR4 (Grunwald et al., 2020) (Figure 5), mutation of only EPR2 was found to influence ribosome association. Comparison with the electrostatic surface potential of SpNatC shows notable differences with ScNatC (Figure 5). Specifically, EPR2 is not present in SpNatC, since the ScNatC “ScNaa35 tip” is missing in SpNatC. While EPR1 is conserved, this electropositive patch harbors the IP6-binding pocket in SpNatC and, therefore, is likely the IP6 binding site in ScNatC. In addition, the surface region of SpNatC corresponding to EPR3 is broader compared to the corresponding surface of ScNatC and is located close to where ScNatC-EPR2 would be. It is therefore possible that SpNatC utilizes EPR3 to compensate for the absence of EPR2. Taken together, this comparison suggests that the mode of ribosome association utilized by SpNatC and ScNatC and, by extension, NatC complexes from other species, may differ. Given the similar overall sequence homology between human NatC with SpNatC (34.3% similarity for NAA30+NAA35+NAA38) and ScNatC (33.5% overall sequence similarity for NAA30+NAA35+NAA38), it is unclear if human NatC will be more similar to ScNatC or SpNatC.
Figure 5. Divergent structural features exist between NatC in S. pombe and S. cerevisiae.
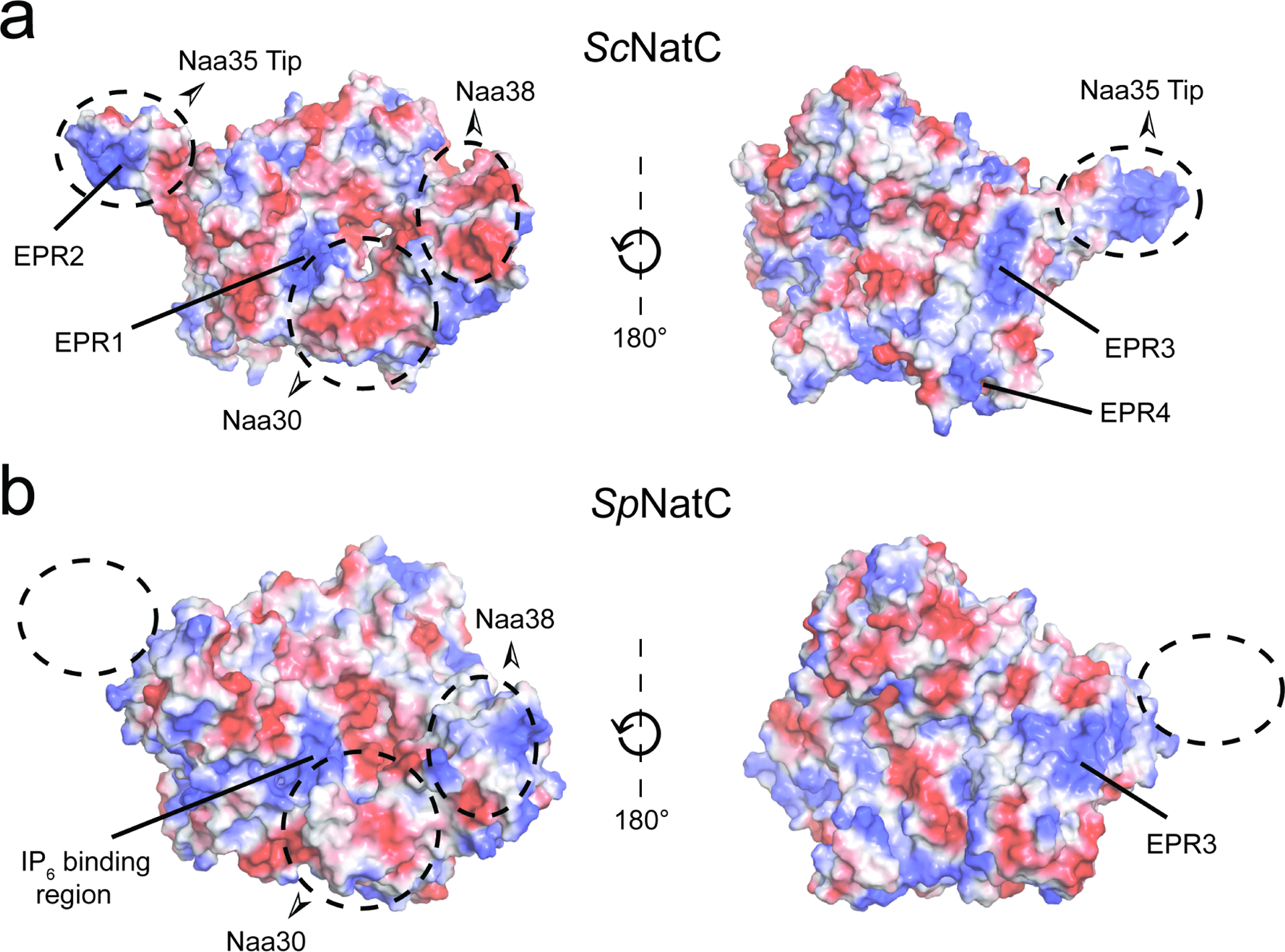
(a) Electrostatic surface potential of the ScNatC structure (PDB: 6YGB) with EPRs indicated.
(b) Electrostatic surface potential of SpNatC (PDB: 7L1K) aligned with the orientations shown in (a).
NatC has been shown to be important for proper chloroplast(Pesaresi et al., 2003) and mitochondrial(Van Damme et al., 2016) function, essential for embryonic development(Wenzlau et al., 2006), cell viability and p53-dependent apoptosis(Starheim et al., 2009), and as a potential therapeutic target in cancer(Mughal et al., 2015). Interestingly, a whole-exome sequencing analysis of cerebral palsy indentified significant genetic heterogeneity including a W532C mutation in human NAA35 (McMichael et al., 2015). Although this study does not implicate any causal role of the hNAA35-W532C mutation, it is interesting to note that hNAA35-W532 corresponds to F522 in helix α20 of SpNAA35 and is conserved as a non-polar residue in other NAA35 orthologs (Supplementary Figure 3). Analysis of the SpNatC structure reveals that this residue sits in a hydrophobic pocket formed with non-polar residues in helix α21. We therefore would predict that a hNAA35-W532C mutation would destabilize this interaction and in turn reduce stability of hNAA35 and activity of hNatC. In light of these important connections to NatC function, the studies reported here have important implications for health and disease.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ronen Marmorstein (marmor@upenn.edu).
Materials Availability
Primary data are available upon reasonable request from the corresponding author.
Data and Code Availability
The cryo-electron microscopy map for the SpNatC – Bisubstrate analogue -IP6 and the atomic coordinate have been deposited in the EMDataBank and ProteinDataBank, with accession codes EMD-23110 and PDB: 7L1K, respectively.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
We used E.coli Rosetta (DE3)pLysS cells for recombinant expression of ternary SpNatC and binary and ternary ScNatC complexes for biochemical biophysical and Cryo-EM experiments. The cells were cultured using standard practices in LB media. See Methods Details - SpNatC Complex Expression and Purification.
We used Spodoptera frugiperda (Sf9) cells cultured in SFM II medium for the recombinant expression of hNatA and hNatB for biochemical experiments. See Methods Details - NatA and NatB Expression and Purification.
METHOD DETAILS
SpNatC Complex Expression and Purification.
SpNaa3531−708 (with truncation of the N-terminal 30 residues) and full-length SpNaa30FL were cloned into a modified pET DUET vector containing an N-terminal His6-SUMO tag. SpNaa3848−116 (with truncation of the N-terminal 47 residues) was cloned into a pCDF vector with an N-terminal His6 tag. To obtain the heterotrimeric NatC complex (WT ternary SpNatC), these two plasmids were used to co-transform Rosetta (DE3)pLysS competent E. coli cells, which was then cultured to grow at 37 °C in the presence of both ampicillin (100 μg/mL) and streptomycin (50 μg/mL). When the absorbance OD600 reached ~ 0.7, protein expression was induced at 16°C with 0.5 mM isopropyl 1-thio-β-galactopyranoside (IPTG) overnight. Cells were harvested by centrifugation, resuspended, and lysed by sonication in lysis buffer containing 25 mM Tris, pH 8.0, 300 mM NaCl, 10 mg/ml PMSF (phenylmethanesulfonylfluoride). The lysate was clarified by centrifugation and passed over Nickel-NTA resin (Thermo Scientific), which was subsequently washed with ~10 column volumes of wash buffer containing 25 mM Tris, pH 8.0, 300 mM NaCl, 20 mM imidazole, 10 mM 2-mercaptoethanol (βME). The protein was eluted in batches with buffer containing 25 mM Tris, pH 8.0, 300 mM NaCl, 200 mM imidazole, 10 mM βME. After elution, His6-tagged Ulp1 protease was added to the eluate to cleave the His6-SUMO tag and dialyzed further into buffer containing 25 mM sodium citrate monobasic, pH 5.5, 10 mM NaCl and 10 mM βME. Protein was purified with a 5-mL HiTrap SP cation-exchange column (GE Healthcare) and eluted with a salt gradient (10–1000 mM NaCl). Peak fractions were concentrated to ~0.5 mL with a 50 kDa concentrator (Amicon Ultra, Millipore), and loaded onto an S200 gel-filtration column (GE Healthcare) in a buffer containing 25 mM HEPES, pH 7.0, 200 mM NaCl, 1 mM dithiothreitol (DTT). Peak fractions were pooled and concentrated to ~15 mg/ml as measured by UV280 using a Nanodrop and flash-frozen for storage at −80 °C until use. All protein mutants were generated using the QuikChange protocol from Stratagene and obtained following the expression and purification protocols described above. The primers used to generate the mutants are listed in Supplementary Table 1.
Binary and Ternary ScNatC Expression and Purification.
ScNaa301−161 and ScNaa3519−733 were cloned into a pET DUET vector containing an TEV cleavable N-terminal His6-tag. Binary complex (ScNaa301−161/ScNaa3519−733) was obtained by transforming this plasmid in ScarabXpress T7lac (Scarab Genomics) competent E. coli cells, which were grown in terrific broth media (DOT Scientific) supplemented with ampicillin (100 μg mL−1) at 37°C to an OD600 of ~0.9 and induced by addition of 0.5 mM IPTG at 17°C for 16 hr. All subsequent purification steps were carried out at 4°C. Cells were isolated by centrifugation, lysed by sonication in lysis buffer containing 25 mM Tris, pH 8.0, 300 M NaCl, 10 mM β-ME and 10 mg/mL PMSF. The lysate was clarified by centrifugation and passed over nickel resin, which was subsequently washed with >20 CV of lysis buffer supplemented with 25 mM imidazole. The protein was eluted in lysis buffer supplemented with 200 mM imidazole by batch elution. TEV protease (~1 mg/ml) was added to the eluted protein and dialyzed into buffer containing 25 mM Tris, pH 8.0, 300 mM NaCl, 5 mM Imidazole, 10 mM β-ME. This solution was passed through an additional nickel column to remove TEV protease as well as any uncut binary ScNatC. The resin was then washed with approximately 3 CV of dialysis buffer supplemented with 25 mM imidazole, which was then pooled with the initial flowthrough. This solution was dialyzed into ion exchange buffer containing 25 mM HEPES, pH 7.5, 50 mM NaCl and 10 mM β-ME and loaded onto a 5 ml HiTrap SP anion exchange column (GE Healthcare). The binary complex ScNatC was then eluted using a salt gradient (50–750 mM NaCl). Peak fractions were concentrated using a 50-kDa MWCO concentrator (Amicon) and further purified Superdex 200 Increase 10/300 GL gel filtration column (GE Healthcare) in a buffer containing 25 mM HEPES, pH 7.0, 200 mM NaCl, 1 mM TCEP. Peak fractions were concentrated to a UV280 of ~4–5 mg mL−1 as measured by nanodrop. The protein was then flash-frozen and stored at −80°C until use.
To obtain the ternary ScNatC complex, ScNaa381−70 was cloned into a pRSF vector with a TEV-cleavable N-terminal STREP-tag. This plasmid was co-transformed with the pET DUET plasmid containing ScNaa301−161 and ScNaa3519−733 and cultured as described above except with the addition of kanamycin (50 μg mL−1) to select for pRSF ScNaa381−70 plasmid. The purification of the ternary ScNatC complex was the same as described for the binary ScNatC complex.
NatA and NatB Expression and Purification.
NatA and NatB were expressed and purified from Spodoptera frugiperda (Sf9) cells cultured in SFM II medium as previously reported (Deng et al., 2020b; Gottlieb and Marmorstein, 2018).
Acetyltransferase Activity Assays.
All acetyltransferase assays were carried out at room temperature in a reaction buffer containing 75 mM HEPES, pH 7.0, 120 mM NaCl, 1 mM DTT as described previously (Deng et al., 2019). The full sequence of peptide substrates are listed below: “SASE” peptide (NatA-type): NH2-SASEAGVRWGRPVGRRRRP-COOH; “MDVF” peptide (NatB-type): NH2-MDVFMKGRWGRPVGRRRRP-COOH; “MLRF” peptide (NatC-type): NH2-MLRFVTKRWGRPVGRRRRP-COOH; “MLGP” peptide (NatE-type): NH2-MLGPEGGRWGRPVGRRRRP-COOH; “SGRG”/H4 peptide (NatD-type): NH2-SGRGKGGKG LGKGGAKRHR-COOH). All peptides were purchased from GenScript. To evaluate SpNatC activity against these peptides, 50 nM SpNatC was mixed with 50 μM Ac-CoA and 500 μM peptide, and allowed to react for 10 min. To determine steady-state catalytic parameters of SpNatC (WT or mutants) with respect to the peptide substrate, 50 nM SpNatC (WT or mutants) was mixed with 50 μM Ac-CoA (14C-labeled, 4 mCi mmol−1; PerkinElmer Life Sciences) and varying peptide concentrations (ranging from 0.98 μM to 500 μM, 10-data points) for 5-minute reactions. P81 paper was purchased from SVI (St. Vincent’s Institute Medical Research). All radioactive count values were converted to molar units with a standard curve created with known concentrations of radioactive Ac-CoA added to scintillation fluid. GraphPad Prism (version 5.01) was used for all data fitting to the Michaelis– Menten equation. The errors in Table 2 represent the standard deviation of the best fit values of three independent kinetic curves.
For the comparison of ternary and binary ScNatC complex activity, 10 nM of either binary ScNatC or ternary ScNatC was mixed with 100 μM of 14C-labeled Ac-CoA and “MLRF” peptide, for a 12-minute reaction in buffer containing 50 mM HEPES, pH 7.5, 150 mM NaCl and 1 mM DTT.
For the activity comparison of ternary SpNatC and other NATs in the presence or absence of IP6, 50 nM of SpNatC, SpNatA or hNatB was mixed with 50 μM of 14C-labeled Ac-CoA, 50 μM of their peptide substrate, in reaction buffer containing 75 mM HEPES, pH 7.0, 120 mM NaCl, 1 mM DTT, supplemented with either 0 or 2 μM IP6 (Sigma-Aldrich), for a 5-minute reaction. The peptide substates for SpNatA and hNatB are SASE peptide and MDVF peptide, respectively. SpNatA(Liszczak et al., 2013) and hNatB(Deng et al., 2020b) are purified as previously described.
ITC measurements.
ITC measurements were carried out using a MicroCal iTC200 at 20 °C. Samples were dialyzed into buffer containing 25 mM HEPES pH 7.0, 200 mM NaCl, and 1 mM DTT. 15 μM of SpNatC in the cell and 300 μM of IP6 in the syringe were used. The raw data were analyzed with the MicroCal ITC analysis software.
Differential Scanning Fluorimetry Assays.
Sypro Orange (50,000X stock, ThermoFisher Scientific) was diluted 1:200, and 4 μL was mixed with 16 μL solution with 0.1 mg/ml of various NatC samples in pH 7.0, 200 mM NaCl, and 1 mM DTT, with or without 10 μM IP6. Fluorescent readings were recorded using a qPCR (ABI 7900 RealTime PCR) with a 1% ramp rate, while heated from 20 °C to 95 °C. Melting curves were generated from these readings and melting temperatures were determined by taking the first derivative of the curves. DSF scans of all samples were performed in triplicate as technical replicates. Error bars in the figure indicates the Standard Deviation (SD) of each sample.
Cryo-EM data Collection.
For initial sample screening, 0.05 mg/ml fresh SpNatC sample was prepared with 3-molar excess of both bisubstrate and IP6. SpNatC particles on cryo grids exhibited a severe preferred orientation, which generated an incorrect initial 3D model (data not shown). Further screening by addition of detergent NP-40 in sample did not improve this situation. To solve this issue, 1 μL of 80 mM CHAPSO was mixed with 20 μL of 12 mg/mL SpNatC. 3 μL of this mixed sample was applied to Quantifoil R1.2/1.3 holey carbon support grids, blotted and plunged into liquid ethane, using an FEI Vitrobot Mark IV. An FEI TF20 was used for screening the grids and data collection was performed with a Titan Krios equipped with a K3 Summit direct detector (Gatan), at a magnification of 105,000 ×, with defocus values from −0.1 to −3.0 μm. Each stack was exposed and counted in super-resolution mode with a total dose of 42 e−/Å2, resulting in 35 frames per stack. Image stacks were automatically collected with EPU. A full description of the Cryo-EM data collection parameters can be found in Table 1.
Cryo-EM Data Processing.
Original image stacks were summed and corrected for drift and beam-induced motion at the micrograph level using MotionCor2 (Zheng et al., 2017), and binned twofold, resulting in a pixel size of 0.84 A° /pixel. After motion correction in Relion 3.0(Kimanius et al., 2016), corrected micrographs were imported into CryoSPARC(Punjani et al., 2017) to perform defocus estimation, the resolution range of each micrograph with Gctf(Zhang, 2016) and all the subsequent data analysis. 2D classifications were performed on the particles auto-picked by “blob picker” with particle diameter of 100 – 200 Å. The representative 2D classes were used as templates to further auto-pick 1,860,276 particles from 5514 micrographs. After bad particles were removed by two runs of 2D classification, 849,844 particles were used to generate four Ab-Initio models and two rounds of heterogeneous refinement were performed using the four models. 508, 298 particles were used for auto refinement and per particle CTF refinement. The final map was refined to an overall resolution of 3.20 Å, with local resolution estimated in Cryo-SPARC (Punjani et al., 2017).
Cryo-EM Model Building and Refinement.
The SpNatC atomic model was manually built de novo using the program COOT (Emsley and Cowtan, 2004) according to the Cryo-EM map, with the guidance of predicted secondary structure and bulky residues such as Phe, Tyr, Trp and Arg. The complete model was then refined by real-space refinement in PHENIX(Adams et al., 2010). All representations of Cryo-EM density and structural models were performed with Chimera(Pettersen et al., 2004) and PyMol (Schrodinger, 2015) (https://pymol.org/2/). The sequence alignments with secondary structure display were created by ESPript 3.0 (Robert and Gouet, 2014a). The surface area calculation was performed using PDBePISA (Krissinel and Henrick, 2007) (Proteins, Interfaces, Structures and Assemblies) (http://www.ebi.ac.uk/pdbe/pisa/).
Quantification and statistical analysis
In Figure 1b, acetylation activity data shows standard deviation (SD) errors, n = 3.
In Figure 1c, ITC data shows a representative curve with standard deviation (SD) fitting errors. ITC measurements were repeated at least twice.
In Figure 1d, differential scanning fluorimetry (DSF) assay data shows standard deviation (SD) errors, n = 3.
In Table. 2, acetyltransferase kinetic errors were generated by GraphPad Prism with Michaelis–Menten equation fitting. Errors indicate SD, with n = 3.
In Supplementary Figure. 5, acetylation activity data shows standard deviation (SD) errors, n = 3.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| E. coli Rosetta (DE3)pLysS | EMD Millipore | Cat#70956-3 |
| ScarabXpress T7lac | (Scarab Genomics) | Cat#C-1709-05K |
| Spodoptera frugiperda (Sf9) cells | Invitrogen | Cat#11496015 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| SpNatC (Naa3531−708 /SpNaa30FL /SpNaa3848−116) | (Liszczak et al., 2011) This Paper | N/A |
| Binary ScNatC (ScNaa301−161 /ScNaa3519−733) | (Liszczak et al., 2013) This Paper | N/A |
| Ternary ScNatC (ScNaa301−161 /ScNaa3519−733/ScNaa381−70) | This Paper | N/A |
| SpNatA (SpNaa15/Naa101−156) | (Liszczak et al., 2013) | N/A |
| hNatB (hNAA251−972/hNAA201−163) | (Deng et al., 2020b) | N/A |
| Nickel Resin | Thermo Scientific | Cat#88223 |
| HiTrap SP HP, 5 mL | GE Healthcare | Cat#17115201 |
| Superdex 200 10/300 GL | GE Healthcare | Cat#17517501 |
| [14C]Acetyl-CoA (4 mCi/mmol) | PerkinElmer Life Sciences | Cat#NEC313050UC |
| P81 Phosphocellulose squares | St. Vincent’s Institute Medical Research | N/A |
| MLGP Peptide (NH2-MLGPEGGRWGRPVGRRRRP-COOH) | Genscript | N/A |
| SASE Peptide (NH2-SASEAGVRWGRPVGRRRRP-COOH) | Genscript | N/A |
| MDVF Peptide (NH2-MDVFMKGRWGRPVGRRRRP-COOH) | Genscript | N/A |
| SGRG Peptide (NH2-SGRGKGGKGLGKGGAKRHR-COOH) | Genscript | N/A |
| MLRF Peptide (NH2-MLRFVTKRWGRPVGRRRRP-COOH) | Genscript | N/A |
| Deposited Data | ||
| Atomic coordinates and structure factors (ternary SpNatC) | This paper | PDB: 7L1K |
| Atomic coordinates and structure factors (hNaa50) | (Liszczak et al., 2011) | PDB: 3TFY |
| Atomic coordinates and structure factors (hNatA) | (Gottlieb and Marmorstein, 2018) | PDB: 6C9M |
| Atomic coordinates and structure factors (SpNatA) | (Liszczak et al., 2013) | PDB: 4KVM |
| Atomic coordinates and structure factors (hNatB) | (Deng et al,, 2020) | PDB: 6VP9 |
| Cryo-EM map (ternary SpNatC) | This paper | EMD-23110 |
| Oligonucleotides | ||
| Primers for recombinant NatC mutants | This paper | Supplementary Table 1 |
| Recombinant DNA | ||
| pET DUET vector- His6-SUMO- | This paper | N/A |
| pET DUET vector- His6-TEV- | This paper | N/A |
| Software and Algorithms | ||
| Phenix | (Adams et al., 2010) | https://www.phenix-online.org/documentation/reference/refinement.html |
| RELION | (Zivanov et al., 2018) | N/A |
| MotionCor2 | (Zheng et al., 2017) | N/A |
| CryoSPARC | (Punjani et al., 2017) | N/A |
| Coot | (Emsley and Cowtan, 2004) | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| PyMOL | Schrodinger LLC | http://www.pymol.org |
| Prism 5.0 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Clustal Omega | (Sievers et al., 2011) | https://www.ebi.ac.uk/Tools/msa/clustalo/ |
| ESPript 3.0 | (Robert and Gouet, 2014b) | http://espript.ibcp.fr/ESPript/ESPript/ |
| LigPlot+ | (Laskowski and Swindells, 2011) | https://www.ebi.ac.uk/thornton-srv/software/LigPlus/ |
Highlights.
Naa38 is required for normal NatC acetylation activity in yeast
Inositol hexaphosphate (IP6) binding contributes to yeast NatC complex stability
S. pombe NatC adopts a unique NAT architecture, distinct from NatA and NatB
Naa30 shows an overlapping yet distinct substrate profile with Naa50
ACKNOWLEDGEMENTS.
This work was supported by NIH grant R35 GM118090 awarded to R.M and R01 NS103873 awarded to E.J.P. We thank Zuo Biao and Sudheer Molugu from the University of Pennsylvania Electron Microscopy Resource Lab for help with initial cryo-grids screening; and Stefan Steimle from the Beckman Center for Cryo-EM at the University of Pennsylvania for technical assistance on data collection. We also thank Elaine Zhou for help with analyzing the DSF data.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests
The authors declare no competing interests.
Reference
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aksnes H, Drazic A, Marie M, and Arnesen T (2016). First Things First: Vital Protein Marks by N-Terminal Acetyltransferases. Trends BiochemSci 41, 746–760. [DOI] [PubMed] [Google Scholar]
- Arnesen T (2011). Towards a functional understanding of protein N-terminal acetylation. PLoS biology 9, e1001074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnesen T, Starheim KK, Van Damme P, Evjenth R, Dinh H, Betts MJ, Ryningen A, Vandekerckhove J, Gevaert K, and Anderson D (2010). The chaperone-like protein HYPK acts together with NatA in cotranslational N-terminal acetylation and prevention of Huntingtin aggregation. Molecular and cellular biology 30, 1898–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnesen T, Van Damme P, Polevoda B, Helsens K, Evjenth R, Colaert N, Varhaug JE, Vandekerckhove J, Lillehaug JR, Sherman F, et al. (2009). Proteomics analyses reveal the evolutionary conservation and divergence of N-terminal acetyltransferases from yeast and humans. Proc Natl Acad Sci U S A 106, 8157–8162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Gottlieb L, Marchi E, Kleyner R, Bhardwaj P, Rope AF, Rosenheck S, Moutton S, Philippe C, Eyaid W, et al. (2019). Phenotypic and biochemical analysis of an international cohort of individuals with variants in NAA10 and NAA15. Hum Mol Genet 28, 2900–2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng S, Magin RS, Wei X, Pan B, Petersson EJ, and Marmorstein R (2019). Structure and Mechanism of Acetylation by the N-Terminal Dual Enzyme NatA/Naa50 Complex. Structure 27, 1057–1070 e1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng S, and Marmorstein R (2021). Protein N-Terminal Acetylation: Structural Basis, Mechanism, Versatility, and Regulation. Trends BiochemSci 46, 15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng S, McTiernan N, Wei X, Arnesen T, and Marmorstein R (2020a). Molecular basis for N-terminal acetylation by human NatE and its modulation by HYPK. Nat Commun 11, 818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng S, Pan B, Gottlieb L, Petersson EJ, and Marmorstein R (2020b). Molecular basis for N-terminal alpha-synuclein acetylation by human NatB. Elife 9, e57491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikiy I, and Eliezer D (2014). N-terminal acetylation stabilizes N-terminal helicity in lipid- and micelle-bound alpha-synuclein and increases its affinity for physiological membranes. J Biol Chem 289, 3652–3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorfel MJ, and Lyon GJ (2015). The biological functions of Naa10 - From amino-terminal acetylation to human disease. Gene 567, 103–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Forte GM, Pool MR, and Stirling CJ (2011). N-terminal acetylation inhibits protein targeting to the endoplasmic reticulum. PLoS biology 9, e1001073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goris M, Magin RS, Foyn H, Myklebust LM, Varland S, Ree R, Drazic A, Bhambra P, Stove SI, Baumann M, et al. (2018). Structural determinants and cellular environment define processed actin as the sole substrate of the N-terminal acetyltransferase NAA80. Proc Natl Acad Sci U S A 115, 4405–4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb L, and Marmorstein R (2018). Structure of Human NatA and Its Regulation by the Huntingtin Interacting Protein HYPK. Structure 26, 925–935 e928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunwald S, Hopf LVM, Bock-Bierbaum T, Lally CCM, Spahn CMT, and Daumke O (2020). Divergent architecture of the heterotrimeric NatC complex explains N-terminal acetylation of cognate substrates. Nat Commun 11, 5506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hole K, Van Damme P, Dalva M, Aksnes H, Glomnes N, Varhaug JE, Lillehaug JR, Gevaert K, and Arnesen T (2011). The human N-alpha-acetyltransferase 40 (hNaa40p/hNatD) is conserved from yeast and N-terminally acetylates histones H2A and H4. PLoS One 6, e24713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes WM, Mannakee BK, Gutenkunst RN, and Serio TR (2014). Loss of amino-terminal acetylation suppresses a prion phenotype by modulating global protein folding. Nat Commun 5, 4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong H, Cai Y, Zhang S, Ding H, Wang H, and Han A (2017). Molecular Basis of Substrate Specific Acetylation by N-Terminal Acetyltransferase NatB. Structure 25, 641–649 e643. [DOI] [PubMed] [Google Scholar]
- Kalvik TV, and Arnesen T (2013). Protein N-terminal acetyltransferases in cancer. Oncogene 32, 269–276. [DOI] [PubMed] [Google Scholar]
- Kimanius D, Forsberg BO, Scheres SH, and Lindahl E (2016). Accelerated cryo-EM structure determination with parallelisation using GPUs in RELION-2. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E, and Henrick K (2007). Inference of macromolecular assemblies from crystalline state. Journal of molecular biology 372, 774–797. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, and Swindells MB (2011). LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. Journal of chemical information and modeling 51, 2778–2786. [DOI] [PubMed] [Google Scholar]
- Linster E, and Wirtz M (2018). N-terminal acetylation: an essential protein modification emerges as an important regulator of stress responses. J Exp Bot 69, 4555–4568. [DOI] [PubMed] [Google Scholar]
- Liszczak G, Arnesen T, and Marmorstein R (2011). Structure of a ternary Naa50p (NAT5/SAN) N-terminal acetyltransferase complex reveals the molecular basis for substrate-specific acetylation. J Biol Chem 286, 37002–37010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liszczak G, Goldberg JM, Foyn H, Petersson EJ, Arnesen T, and Marmorstein R (2013). Molecular basis for N-terminal acetylation by the heterodimeric NatA complex. Nat Struct Mol Biol 20, 1098–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liszczak G, and Marmorstein R (2013). Implications for the evolution of eukaryotic amino-terminal acetyltransferase (NAT) enzymes from the structure of an archaeal ortholog. Proc Natl Acad Sci U S A 110, 14652–14657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay DT, Botting CH, Taylor GL, and Whiasssate MF (2007). An acetylase with relaxed specificity catalyses protein N-terminal acetylation in Sulfolobus solfataricus. Molecular microbiology 64, 1540–1548. [DOI] [PubMed] [Google Scholar]
- Magin RS, Deng S, Zhang H, Cooperman B, and Marmorstein R (2017). Probing the interaction between NatA and the ribosome for co-translational protein acetylation. PLoS One 12, e0186278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magin RS, Liszczak GP, and Marmorstein R (2015). The molecular basis for histone H4- and H2A-specific amino-terminal acetylation by NatD. Structure 23, 332–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMichael G, Bainbridge MN, Haan E, Corbett M, Gardner A, Thompson S, van Bon BW, van Eyk CL, Broadbent J, Reynolds C, et al. (2015). Whole-exome sequencing points to considerable genetic heterogeneity of cerebral palsy. Molecular psychiatry 20, 176–182. [DOI] [PubMed] [Google Scholar]
- Mughal AA, Grieg Z, Skjellegrind H, Fayzullin A, Lamkhannat M, Joel M, Ahmed MS, Murrell W, Vik-Mo EO, Langmoen IA, et al. (2015). Knockdown of NAT12/NAA30 reduces tumorigenic features of glioblastoma-initiating cells. Mol Cancer 14, 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myklebust LM, Stove SI, and Arnesen T (2015). Naa10 in development and disease. Oncotarget 6, 34041–34042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochaya S, Franzen O, Buhwa DA, Foyn H, Butler CE, Stove SI, Tyler KM, Arnesen T, Matovu E, Aslund L, et al. (2019). Characterization of Evolutionarily Conserved Trypanosoma cruzi NatC and NatA-N-Terminal Acetyltransferase Complexes. Journal of parasitology research 2019, 6594212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak D, Bhat AH, Sapehia V, Rai J, and Rao A (2016). Biochemical evidence for relaxed substrate specificity of Nalpha-acetyltransferase (Rv3420c/rimI) of Mycobacterium tuberculosis. Sci Rep 6, 28892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesaresi P, Gardner NA, Masiero S, Dietzmann A, Eichacker L, Wickner R, Salamini F, and Leister D (2003). Cytoplasmic N-terminal protein acetylation is required for efficient photosynthesis in Arabidopsis. Plant Cell 15, 1817–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. Journal of computational chemistry 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Polevoda B, Brown S, Cardillo TS, Rigby S, and Sherman F (2008). Yeast N(alpha)-terminal acetyltransferases are associated with ribosomes. J Cell Biochem 103, 492–508. [DOI] [PubMed] [Google Scholar]
- Polevoda B, and Sherman F (2001). NatC Nalpha-terminal acetyltransferase of yeast contains three subunits, Mak3p, Mak10p, and Mak31p. J Biol Chem 276, 20154–20159. [DOI] [PubMed] [Google Scholar]
- Punjani A, Rubinstein JL, Fleet DJ, and Brubaker MA (2017). cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nature methods 14, 290–296. [DOI] [PubMed] [Google Scholar]
- Rathore OS, Faustino A, Prudencio P, Van Damme P, Cox CJ, and Martinho RG (2016). Absence of N-terminal acetyltransferase diversification during evolution of eukaryotic organisms. Sci Rep 6, 21304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ree R, Varland S, and Arnesen T (2018). Spotlight on protein N-terminal acetylation. Exp Mol Med 50, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert X, and Gouet P (2014a). Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Research 42, W320–W324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert X, and Gouet P (2014b). Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res 42, W320–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrodinger LLC (2015). The PyMOL Molecular Graphics System, Version 1.8.
- Scott DC, Monda JK, Bennett EJ, Harper JW, and Schulman BA (2011). N-terminal acetylation acts as an avidity enhancer within an interconnected multiprotein complex. Science 334, 674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setty SR, Strochlic TI, Tong AH, Boone C, and Burd CG (2004). Golgi targeting of ARF-like GTPase Arl3p requires its Nalpha-acetylation and the integral membrane protein Sys1p. Nat Cell Biol 6, 414–419. [DOI] [PubMed] [Google Scholar]
- Shemorry A, Hwang CS, and Varshavsky A (2013). Control of protein quality and stoichiometries by N-terminal acetylation and the N-end rule pathway. Mol Cell 50, 540–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Molecular Systems Biology 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song OK, Wang X, Waterborg JH, and Sternglanz R (2003). An Nalpha-acetyltransferase responsible for acetylation of the N-terminal residues of histones H4 and H2A. J Biol Chem 278, 38109–38112. [DOI] [PubMed] [Google Scholar]
- Starheim KK, Gevaert K, and Arnesen T (2012). Protein N-terminal acetyltransferases: when the start matters. Trends BiochemSci 37, 152–161. [DOI] [PubMed] [Google Scholar]
- Starheim KK, Gromyko D, Evjenth R, Ryningen A, Varhaug JE, Lillehaug JR, and Arnesen T (2009). Knockdown of human N alpha-terminal acetyltransferase complex C leads to p53-dependent apoptosis and aberrant human Arl8b localization. Molecular and cellular biology 29, 3569–3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stove SI, Magin RS, Foyn H, Haug BE, Marmorstein R, and Arnesen T (2016). Crystal Structure of the Golgi-Associated Human Nalpha-Acetyltransferase 60 Reveals the Molecular Determinants for Substrate-Specific Acetylation. Structure 24, 1044–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Damme P, Kalvik TV, Starheim KK, Jonckheere V, Myklebust LM, Menschaert G, Varhaug JE, Gevaert K, and Arnesen T (2016). A Role for Human N-alpha Acetyltransferase 30 (Naa30) in Maintaining Mitochondrial Integrity. Molecular & cellular proteomics : MCP 15, 3361–3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetting MW, Bareich DC, Yu M, and Blanchard JS (2008). Crystal structure of RimI from Salmonella typhimurium LT2, the GNAT responsible for N(alpha)-acetylation of ribosomal protein S18. Protein Sci 17, 1781–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetting MW, de Carvalho LP, Roderick SL, and Blanchard JS (2005). A novel dimeric structure of the RimL Nalpha-acetyltransferase from Salmonella typhimurium. J Biol Chem 280, 22108–22114. [DOI] [PubMed] [Google Scholar]
- Wenzlau JM, Garl PJ, Simpson P, Stenmark KR, West J, Artinger KB, Nemenoff RA, and Weiser-Evans MC (2006). Embryonic growth-associated protein is one subunit of a novel N-terminal acetyltransferase complex essential for embryonic vascular development. Circulation research 98, 846–855. [DOI] [PubMed] [Google Scholar]
- Weyer FA, Gumiero A, Lapouge K, Bange G, Kopp J, and Sinning I (2017). Structural basis of HypK regulating N-terminal acetylation by the NatA complex. Nat Commun 8, 15726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Fang Q, Wang M, Ren R, Wang H, He M, Sun Y, Yang N, and Xu R-M (2013). Nα-acetylated Sir3 stabilizes the conformation of a nucleosome-binding loop in the BAH domain. Nature Structural & Molecular Biology 20, 1116–1118. [DOI] [PubMed] [Google Scholar]
- Zhang K (2016). Gctf: Real-time CTF determination and correction. Journal of structural biology 193, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng Y, and Agard DA (2017). MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nature methods 14, 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJ, Lindahl E, and Scheres SH (2018). New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7, e42166. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The cryo-electron microscopy map for the SpNatC – Bisubstrate analogue -IP6 and the atomic coordinate have been deposited in the EMDataBank and ProteinDataBank, with accession codes EMD-23110 and PDB: 7L1K, respectively.