

Abstract
Tourette’s Disorder (TD) is a neurodevelopmental disorder that affects about 0.7% of the population and is one of the most heritable neurodevelopmental disorders. Nevertheless, because of its polygenic nature and genetic heterogeneity, the genetic etiology of TD is not well understood. In this study, we combined the segregation information in 13 TD multiplex families with high-throughput sequencing and genotyping to identify genes associated with TD. Using whole-exome sequencing and genotyping array data, we identified both small and large genetic variants within the individuals. We then combined multiple types of evidence to prioritize candidate genes for TD, including variant segregation pattern, variant function prediction, candidate gene expression, protein-protein interaction network, candidate genes from previous studies, etc. From the 13 families, 71 strong candidate genes were identified, including both known genes for neurodevelopmental disorders and novel genes, such as HTRA3, CDHR1, and ZDHHC17. The candidate genes are enriched in several gene ontology categories, such as dynein complex and synaptic membrane. Candidate genes and pathways identified in this study provide biological insight into TD etiology and potential targets for future studies.
Introduction
Tourette’s Disorder (TD) is a neurodevelopmental disorder (NDD) that affects up to 1% of the global population 1, 2. TD is characterized by chronic motor and vocal tics and is often diagnosed in early childhood. Some patients do not present the full spectrum of syndrome for TD and are characterized as TD-related chronic tic disorders, such as chronic motor tic disorder or chronic vocal tic disorder 1, 2. TD has a high comorbidity with several psychiatric disorders: about 60% of patients are also diagnosed with attention-deficit hyperactivity disorder (ADHD) 3–6 and 40%−60% with obsessive-compulsive disorder (OCD) 7–10. Autism spectrum disorder (ASD) is also overrepresented in TD, and around 20–40% of individuals with ASD experience tics 11. It is estimated that the lifetime prevalence rate of comorbid psychiatric disorders for TD patients is up to 90% 9.
TD is a highly heritable polygenic neuropsychiatric disorder (population-based-h2 = 0.77; SNP-h2 = 0.58) 12, 13. The empirical recurrence risk estimates for TD and other chronic tic disorders in first-degree relatives is about 30% 14. Because of the high heritability of TD, many studies have been conducted to identify the genetic etiology of TD (reviewed in 15). Early TD genetic studies focused on mutations in single genes under the assumption of a monogenic inheritance model. Although some of the candidate genes were reported, mutations in these genes only explain a few of the TD cases (reviewed in 15). TD is now believed to have a complex multigenic allelic architecture, similar to other NDDs 16. More recently, several large studies were performed to identify genes associated with TD, including whole-exome sequencing (WES) studies of simplex families focusing on de novo mutations 17, 18 and a meta-analysis of Genome-Wide Association Studies (GWAS) with about 5,000 TD patients 13. However, only a few susceptibility genes/loci were identified in the WES and GWAS studies.
Due to its high heritability, one effective approach to identify TD candidate genes is to study large multiplex families. Children of TD patients are between 10 and 100 times more likely to be affected 14, 19, indicating shared genetic susceptibility among family members. Therefore, variant segregation pattern in multiplex families can help identify inherited rare variants with strong effects and provide additional information compared to simplex trios. The Tourette International Collaborative Genetics Study (TIC Genetics) is a collaboration that recruits TD affected families for studying the genetic factors of TD 20. In the current study, we generated WES and genotyping array data for individuals from 13 TIC Genetics multiplex families. We then prioritized risk genes with rare mutations in these families to identify potential risk genes for TD.
Subjects and Methods
Human subjects
Multiplex families were recruited through the TIC Genetics study 20 and the New Jersey Center for Tourette Syndrome (NJCTS) 21 with informed consent from all participants. The study protocol was locally approved at all TIC Genetics sites. All sequenced individuals were categorized as “white” race, except for the sperm donor (4001) in FAM4, whose ancestry is unknown (Table 1). The clinical assessment and definition of TD and chronic tic disorders were described in detail previously 20, and were based on the Diagnostic and Statistical Manual of Mental Disorders – Fourth edition, Text Revision (DSM-IV-TR) or Fifth edition (DSM-5) 22, 23. In this study, a TD-affected individual is defined as an individual who was diagnosed with TD or other tic disorders (i.e., chronic motor or vocal tic disorder, a combined subtype, transient tic disorder or tic disorder-Not Otherwise Specified 20). A multiplex family is defined as a family with at least three TD-affected individuals.
Table 1.
Summary of the multiplex families
| Family | Total Ind | TD | TD with OCD | TD with ADHD | WES (TD) | SNV/Indel | SNV/Indel Filtered | pVAAST (d) | pVAAST (r) | Candidate | Genotyping (TD) | CNV | CNV exonic |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FAM1 | 9 | 8 | 5 | 2 | 6 (5) | 103,621 | 14,911 | 35 | 5 | 34 | 6 (5) | 276 | 134 |
| FAM2 | 9 | 7 | 4 | 5 | 6 (6) | 101,590 | 14,917 | 199 | 8 | 81 | 7 (6) | 170 | 56 |
| FAM3 | 15 | 7 | 1 | 0 | 12 (7) | 136,028 | 27,180 | 89 | 10 | 26 | 12 (6) | 261 | 85 |
| FAM4 | 18 | 11 | 5 | 2 | 17 (11) | 167,676 | 43,915 | 10 | 25 | 33 | 0 | NA | NA |
| FAM5 | 12 | 8 | 4 | 1 | 10 (7) | 140,006 | 28,164 | 1 | NA | 1 | 11 (7) | 240 | 44 |
| FAM6 | 9 | 5 | 1 | 0 | 6 (4) | 124,489 | 22,345 | 50 | NA | 40 | 6 (4) | 142 | 30 |
| FAM7 | 8 | 4 | 0 | 0 | 8 (4) | 124,813 | 22,209 | 35 | NA | 32 | 8 (4) | 184 | 37 |
| FAM8 | 5 | 3 | 3 | 1 | 4 (3) | 114,025 | 17,741 | 136 | NA | 125 | 4 (3) | 121 | 62 |
| FAM9 | 4 | 3 | 1 | 3 | 4 (3) | 105,502 | 15,191 | 93 | 11 | 91 | 4 (3) | 116 | 45 |
| FAM10 | 5 | 3 | 2 | 1 | 5 (3) | 110,102 | 17,159 | 51 | 10 | 50 | 0 | NA | NA |
| FAM11 | 18 | 9 | 3 | 2 | 13 (8) | 133,278 | 27,134 | 18 | NA | 16 | 14 (9) | 289 | 57 |
| FAM12 | 16 | 8 | 2 | 0 | 11 (6) | 129,089 | 23,991 | 14 | NA | 13 | 0 | NA | NA |
| FAM13 | 23 | 6 | 0 | 0 | 8 (5) | 123,730 | 21,873 | 218 | NA | 50 | 0 | NA | NA |
| Total | 151 | 82 | 31 | 17 | 110 (72) | 280,363 | 139,092 | 949 | 69 | 543 | 72 (47) | 1,799 | 550 |
Total Ind: individuals surveyed with phenotypes in each family.
TD: individuals diagnosed as TD (Tourette Disorder, Chronic Tic Disorder-Motor subtype, Chronic Tic Disorder-Vocal subtype, Chronic Tic Disorder-Combined subtype, transient tic disorder or tic disorder-Not Otherwise Specified. see Methods for diagnosis detail).
TD with OCD: individuals with both TD and OCD diagnosis.
TD with ADHD: individuals with both TD and ADHD diagnosis.
WES (TD): individuals subjected to whole exome sequencing in each family, the number of individuals diagnosed with TD are shown in parenthesis.
SNV/Indel: SNVs (single nucleotide variants) or Indels (insertion and deletions) in each family in the variant call results.
SNV/Indel Filtered: variants after removing those with AF ≥ 10% in the 1000 Genomes project or ≥ 5% in ExAC.
pVAAST genes (d)/ pVAAST genes (r): candidate gene counts reported by pVAAST running in the dominant/recessive (d/r) mode.
Candidate: candidate gene counts after filtering.
Genotyping (TD): the number of individuals subjected to microarray analysis for CNV detection and the number of individuals diagnosed with TD are shown in parenthesis.
CNV: count of candidate CNVs in the copy number variants analysis.
CNV exonic: count of candidate CNVs with overlap with exon regions of gene models.
Whole-exome sequencing, variant calling and annotation
SureSelect Human All Exon V4, Human All Exon V4+UTR (Agilent Technologies, Santa Clara, CA, USA), or NimbleGen SeqCap EZ Exome V2 (Roche, Wilmington, MA, USA) kits were used for the WES library preparation. Sequencing was performed on the Illumina HiSeq platform with 100 PE or 150 PE format (Illumina, San Diego, CA, USA). Variant calling was performed using the Genome Analysis Toolkit (GATK) following the best practice pipeline 24 and variant annotation was performed using ANNOVAR 25. To control for the batch effect, joint variant calling was performed on all samples and only variants that are in the smaller set of the enrichment regions (SeqCap EZ Exome V2) among the multiple exome capture kits were considered (see Supplemental Methods for detailed steps and commands). The sequencing data is available at dbGaP under study accession phs001423.v2.p2.
Candidate gene prioritization, annotation, and filtering
pVAAST (pedigree Variant Annotation, Analysis and Search Tool) was used to identify candidate genes in each pedigree 26, 27. pVAAST is a likelihood-based tool that prioritize candidate genes using several types of variant information in each gene, including the segregation pattern, the predicted functional impact, and the allele frequency (AF) in general populations. pVAAST was run under dominant mode of inheritance for all families and under recessive mode of inheritance for families where the recessive mode of inheritance cannot be ruled out (Table 1, see Supplemental Methods for details).
For gene function predictions, the pLI (probability of being loss-of-function intolerant) score and the missense Z score were extracted from gnomAD for each gene 28. Brain developmental gene expression data were obtained from the Gene Tissue Expression project (GTEx) 29, 30, the BrainSpan Atlas of the Developing Human Brain project 31, and the Human Developmental Biology Resource (HDBR) 32 (see Supplementary Methods for detail). Diseases related to NDDs were extracted and curated from the DISEASES database 33 (https://diseases.jensenlab.org). The gene knock-out mouse behavior were downloaded from the International Mouse Phenotyping Consortium (IMPC) 34 (ftp://ftp.ebi.ac.uk/pub/databases/impc/latest/).
Gene lists from previous NDD studies
Risk genes for several NDDs were collected from previous studies, including TD, OCD, ADHD, ASD, intellectual disability, epileptic encephalopathies, and schizophrenia. The gene sets and the genes in the gene sets are summarized in Table S1.
Protein–Protein interaction network, Gene Ontology, pathway, and protein complex enrichment analysis
Three databases were used to investigate Protein-Protein Interaction (PPI) networks among candidate genes, including STRING 35, ConsensusPathDB 36, and GIANT_v2 37, 38 (see Supplementary Methods for detail). These databases were shown to have the best performance in a recent benchmark paper 39. The benchmark paper also showed that interactions present in at least two databases provide higher confidence than those specific to one database.
Enrichment analyses were performed for all NDD genes with over-representation analysis provided by ConsensusPathDB 36. An enriched term (i.e., Gene Ontology (GO), pathway, or protein complex) was selected for further analysis if: 1) the total number of genes belong to the term is ≤ 200; 2) the term includes more than one gene from TD_multiplex gene list and more than two genes from all TD genes (TD_multiplex + TD_simplex + TD_CNV); and 3) the term includes genes from more than one multiplex family. Enrichment p-value for each gene list was calculated with Fisher’s exact test (see Supplementary Methods for detail).
Copy Number Variant (CNV) analysis
Genotyping was performed at the Keck Biotechnology Resource Laboratory at Yale University School of Medicine and at RUCDR Infinite Biologics® at Rutgers University using the Illumina HumanOmni1-Quad or OmniExpressExome BeadChip (Illumina, San Diego, CA). Samples from each pedigree were genotyped on the same platform. Genotype calling and CNV detection were performed as previously described 40 (see Supplementary Methods for detail). A pCNV threshold of <=0.05 was used for initial selection of CNVs. CNV annotation was performed by CNVision, AnnotSV 41, and a custom program for inheritance pattern analysis. CNVs that are smaller than 1,000 base pairs or larger than 2 million base pairs were excluded.
Results
Families and phenotypes
Figure 1 describes the overall design of the project. A total of 13 multiplex families were included in this study. Family size ranged from 4 to 23, with a total of 151 individuals (Table 1, Figure S1). The percentage of individuals diagnosed with TD and other chronic tic disorders was 60% (51/85) and 47% (31/66) for males and females, respectively. Details of individual phenotypes are summarized in Table S2.
Figure 1. Overall study design.
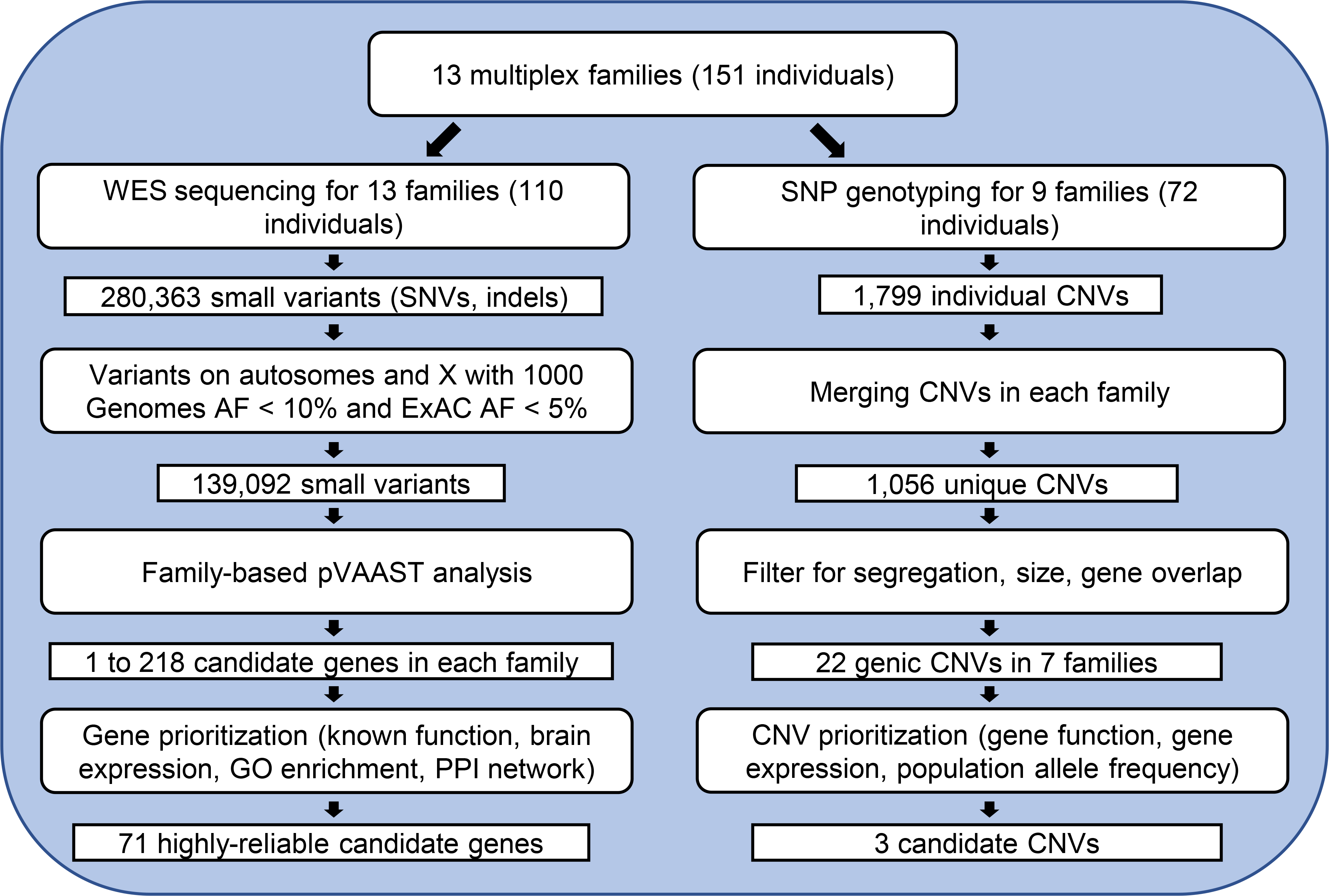
WES: whole-exome sequencing; SNP: single nucleotide polymorphism; AF: allele frequency; CNV: copy number variant; GO: gene ontology; PPI: protein-protein interaction.
Whole-exome sequencing and candidate gene prioritization
Among the 13 families, we obtained DNA samples from 110 individuals for WES, including 72 individuals with TD and 38 non-TD family members. At least three individuals with TD were sequenced in each family (Table 1). Overall, the 110 individuals sequenced had an average mean depth of coverage of 42.5x and an average median coverage of 34.9x (Table S2). Using the sequencing data, we identified single nucleotide variants (SNVs) and small insertions and deletions (indels). The total number of variants that passed quality control was (6.64 ± 0.72) × 104 in each individual. After removing common variants (see Methods and Supplemental Methods), the number of variants in each individual ranged from 3,970 to 8,673, with an average of 6,710 (Table S2).
Next, we used the candidate gene prioritization tool pVAAST to identify candidate genes in each pedigree. The number of candidate genes reported among the 13 families varied from 1 to 218, with a total of 1,018 genes (Table 1). Because the variant segregation pattern is a major factor for candidate gene prioritization in multiplex families, small families (e.g., FAM9) and families where all individuals were affected (e.g., FAM2) have less power to distinguish variant segregation patterns, thus producing a higher number of candidate genes than the larger families. To reduce the number of candidate genes, we applied additional filters based on variant segregation pattern (true positive events ≥ 2 and false rate < 0.3), gene expression level in brain (max TPM > 5), and AF in general populations (gnomAD 2.1.1 AF < 0.05) (see Methods and Supplemental Methods for detail). After filtering, the number of candidate genes ranged from 1 to 125 among the 13 families, with a total of 543 unique genes (Table 1). Hereafter we refer to these genes as “TD_multiplex” genes (Table S3). Among the 543 unique genes, 25 were identified in more than one family, most of which were identified in families with many candidate genes (Table S2).
Fourteen of the TD_multiplex genes were identified in a previous study of TD simplex trio families 18 (Table S4). In addition, two genes were reported in previous TD studies: DNAJC13 (DnaJ heat shock protein family (Hsp40) member C13) 42 and SLC6A4 (solute carrier family 6 member 4) 43. Some of these genes are also reported to be associated with other NDDs (Table 2, Table S1B). For example, ANK3 (ankyrin 3) has been reported to be associated with ADHD and ASD, in addition to TD.
Table 2.
Top candidate genes in each family
| Variant Annotation | Gene Annotation | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Family | Gene | Confidence | Chr | Position | AF | mutation | Effect | Segregation | pLI | mis_z | Other NDDs | TPM1 | TPM2 | TPM3 | IMPC | Interaction |
| FAM1 | CASP8 | H | 2 | 202,134,324 | 1.33E-05 | I->V | DT | 0|6 | 0.00 | 1.01 | 3 | 4 | 7 | 59 | ||
| FAM1 | GAK | H | 4 | 862,363 | 7.30E-03 | D->Y | DD | 0|6 | 0.01 | 0.02 | 145 | 44 | 22 | 103 | ||
| FAM1 | PNKD | H | 2 | 219,204,814 | 3.33E-05 | Q->stop | .. | 0|6 | 0.00 | 0.32 | 167 | 254 | 52 | 4 | ||
| FAM1 | ABCA7 | M | 19 | 1,048,950 | 6.40E-05 | G->R | DD | 0|5 | 0.00 | −1.47 | ASD | 66 | 45 | 19 | F | 5 |
| FAM2 | CX3CL1 | H | 16 | 57,413,660 | 5.10E-03 | A->V | TD | 1|6 | 0.61 | 0.84 | TD_simplex | 124 | 1165 | 43 | T | 36 |
| FAM2 | DNAH7 | H | 2 | 196,866,433 | 1.07E-02 | M->R | DD | 1|6 | 0.00 | −1.09 | 6 | 6 | 30 | 5 | ||
| FAM2 | SYT17 | H | 16 | 19,194,929 | 4.00E-04 | E->D | DD | 1|6 | 0.00 | 1.18 | ASD | 47 | 150 | 24 | 3 | |
| FAM2 | AKAP9 | M | 7 | 91,631,542 | 9.57E-05 | A->T | TT | 1|6 | 0.00 | −0.13 | ASD | 42 | 99 | 86 | 88 | |
| FAM2 | NIPA1 | M | 15 | 23,048,912 | 1.10E-05 | V->M | .. | 1|6 | 0.01 | 1.94 | TD_CNV, ASD | 149 | 293 | 49 | F | 4 |
| FAM2 | SLC6A2 | L | 16 | 55,725,894 | 9.55E-05 | T->M | TD | 1|6 | 0.08 | 2.02 | ADHD | 0 | 1 | 9 | F | 27 |
| FAM3 | CDH23 | H | 10 | 73,270,925 | 3.00E-04 | A->T | TD | 2|12 | 0.00 | 0.71 | ADHD | 42 | 97 | 38 | T | 10 |
| FAM3 | CDHR1 | H | 10 | 85,972,932 | 4.50E-03 | N->S | DT | 1|12 | 0.00 | −0.70 | 79 | 150 | 45 | 1 | ||
| FAM3 | ESR1 | H | 6 | 152,265,352 | 5.00E-04 | R->C | DD | 2|12 | 1.00 | 1.61 | 3 | 9 | 4 | 163 | ||
| FAM3 | OPRM1 | H | 6 | 154,411,110 | 3.60E-03 | S->C | DD | 2|12 | 0.00 | −0.62 | ADHD | 3 | 13 | 7 | 51 | |
| FAM3 | ABCC1 | L | 16 | 16,138,519 | 8.88E-05 | S->F | DT | 2|12 | 0.00 | 1.77 | 19 | 36 | 37 | 12 | ||
| FAM3 | MAPK3 | L | 16 | 30,128,265 | 1.90E-03 | E->K | DD | 3|12 | 0.04 | 1.74 | ASD | 404 | 531 | 64 | 180 | |
| FAM4 | CENPJ | H | 13 | 25,458,540 | 3.18E-05 | E->stop | DD | 2|12 | 0.00 | 0.22 | 42 | 41 | 105 | T | 48 | |
| FAM4 | ZDHHC17 | H | 12 | 77,243,235 | 3.20E-05 | I->T | TD | 1|12 | 1.00 | 2.57 | 91 | 134 | 165 | 31 | ||
| FAM4 | ADM | M | 11 | 10,327,330 | . | S->W | DD | 1|12 | 0.04 | −0.11 | 58 | 332 | 117 | 38 | ||
| FAM4 | DCAF1 | M | 3 | 51,475,633 | 8.03E-06 | K->R | .T | 0|12 | 1.00 | 4.84 | 36 | 87 | 47 | 46 | ||
| FAM5 | TADA3 | H | 3 | 9,831,246 | 1.00E-03 | R->C | DT | 1|10 | 0.00 | 2.12 | 142 | 218 | 72 | 20 | ||
| FAM6 | KCNH5 | H | 14 | 63,246,495 | 1.00E-04 | A->G | DD | 0|5 | 0.02 | 2.51 | 9 | 83 | 11 | 20 | ||
| FAM6 | RAB11FIP3 | H | 16 | 569,754 | 3.00E-04 | E->Q | TD | 0|5 | 1.00 | 1.27 | TD_simplex | 182 | 158 | 86 | T | 12 |
| FAM6 | RIMS1 | H | 6 | 73,017,069 | 2.00E-04 | S->Y | DD | 1|6 | 0.99 | 2.01 | ASD | 169 | 282 | 34 | 19 | |
| FAM6 | AEBP1 | L | 7 | 44,144,407 | 3.19E-05 | E->G | DT | 1|6 | 0.00 | 1.00 | 86 | 182 | 106 | F | 22 | |
| FAM7 | CADPS2 | H | 7 | 122,033,277 | 2.00E-04 | S->L | DT | 1|8 | 0.37 | 1.09 | ASD | 332 | 679 | 83 | F | 4 |
| FAM7 | NINL | H | 20 | 25,457,679 | 6.00E-04 | A->S | DD | 0|8 | 0.00 | −0.98 | ASD | 63 | 61 | 68 | F | 40 |
| FAM7 | POFUT1 | H | 20 | 30,816,172 | 3.18E-05 | E->K | TT | 0|8 | 0.96 | 0.95 | 18 | 46 | 35 | 4 | ||
| FAM7 | AHCY | L | 20 | 32,880,242 | 3.60E-03 | G->R | DD | 0|8 | 0.04 | 1.55 | 109 | 361 | 263 | F | 51 | |
| FAM8 | ANK3 | H | 10 | 61,833,414 | 3.40E-03 | S->S | DD | 0|4 | 1.00 | 2.79 | TD_simplex, ASD, ADHD | 107 | 218 | 96 | 24 | |
| FAM8 | DNAH5 | H | 5 | 13,807,706 | 9.56E-05 | M->I | TT | 0|4 | 0.00 | −0.77 | TD_simplex | 1 | 6 | 20 | F | 8 |
| FAM8 | GABRB3 | H | 15 | 27,018,841 | 3.20E-03 | P->S | TT | 0|4 | 0.95 | 3.39 | TD_CNV, ASD, OtherNeuro | 58 | 182 | 81 | 15 | |
| FAM8 | GPLD1 | H | 6 | 24,429,341 | 4.69E-03 | Q deletion | .. | 0|4 | 0.00 | −0.30 | TD_simplex | 36 | 42 | 14 | F | 17 |
| FAM8 | IGF2BP1 | H | 17 | 47,117,430 | 6.70E-03 | H->Q | TT | 0|4 | 1.00 | 3.52 | TD_simplex | 0 | 113 | 290 | 27 | |
| FAM8 | NLGN3 | H | X | 70,389,354 | 1.90E-03 | T->A | TT | 0|4 | 0.98 | 4.21 | ASD | 90 | 315 | 109 | T | 21 |
| FAM8 | KIDINS220 | M | 2 | 8,928,870 | . | A->V | DD | 0|4 | 0.07 | 2.26 | 99 | 792 | 376 | 20 | ||
| FAM8 | NDUFB1 | M | 14 | 92,583,927 | 4.30E-03 | M->L | TT | 0|4 | 0.00 | −0.21 | 249 | 667 | 218 | 12 | ||
| FAM8 | VPS13A | M | 9 | 79,938,036 | 3.80E-03 | R->C | DT | 0|4 | 0.00 | 1.76 | 22 | 33 | 26 | F | 26 | |
| FAM8 | CAPN6 | L | X | 110,495,596 | 9.12E-05 | K->M | DD | 0|4 | 0.82 | 1.52 | 0 | 13 | 151 | 1 | ||
| FAM8 | SUGP1 | L | 19 | 19,414,217 | . | R->G | DD | 0|4 | 1.00 | 1.21 | 76 | 132 | 39 | F | 26 | |
| FAM9 | AGRN | H | 1 | 979,748 | 3.21E-02 | E->V | DD | 1|4 | 0.00 | 0.23 | 127 | 185 | 149 | 53 | ||
| FAM9 | DNAJC13 | H | 3 | 132,221,143 | 2.20E-03 | R->H | DD | 0|4 | 1.00 | 2.28 | 28 | 47 | 50 | 16 | ||
| FAM9 | UNC13C | H | 15 | 54,306,012 | 3.20E-03 | D->E | DD | 0|4 | 0.00 | −0.14 | 77 | 327 | 16 | F | 1 | |
| FAM9 | VGF | H | 7 | 100,806,657 | . | P->A | DD | 0|3 | 0.57 | 0.25 | 455 | 801 | 83 | T | 21 | |
| FAM9 | SORT1 | M | 1 | 109,910,100 | 2.20E-03 | I->V | TT | 0|4 | 1.00 | 2.10 | 440 | 544 | 102 | F | 28 | |
| FAM9 | SYNJ2 | M | 6 | 158,492,660 | 3.40E-03 | T->M | DD | 0|4 | 0.00 | 0.72 | 230 | 125 | 47 | F | 34 | |
| FAM9 | ALDH7A1 | L | 5 | 125,880,710 | 4.50E-03 | T->A | DD | 0|4 | 0.00 | 0.37 | 96 | 138 | 139 | 9 | ||
| FAM9 | DHDDS | L | 1 | 26,764,735 | 2.50E-03 | R->Q | TT | 0|3 | 0.25 | 1.09 | 185 | 145 | 25 | F | 1 | |
| FAM10 | MAP1LC3B | H | 16 | 87,432,453 | 7.10E-03 | E->Q | TD | 1|5 | 0.00 | 0.29 | 291 | 511 | 180 | 42 | ||
| FAM10 | SLC6A4 | H | 17 | 28,538,374 | 6.00E-04 | I->V | TT | 1|5 | 0.25 | 1.93 | ASD, ADHD | 1 | 8 | 19 | 25 | |
| FAM10 | GPR37 | M | 7 | 124,404,617 | 2.00E-04 | N->K | TT | 1|5 | 0.00 | 0.84 | ASD | 377 | 541 | 56 | 44 | |
| FAM10 | ROBO3 | M | 11 | 124,740,144 | 3.80E-03 | D->N | DD | 1|5 | 0.00 | 0.61 | 26 | 38 | 688 | 2 | ||
| FAM10 | VWC2 | M | 7 | 49,951,764 | 4.00E-04 | E->Q | DD | 1|5 | 0.97 | 1.67 | 43 | 137 | 19 | 1 | ||
| FAM11 | CNTNAP5 | M | 2 | 125,504,806 | 4.00E-04 | D->G | TT | 2|13 | 0.99 | 0.39 | ASD | 3 | 94 | 35 | F | 0 |
| FAM11 | HTRA3 | M | 4 | 8,293,193 | 7.40E-03 | V->M | DD | 1|13 | 0.00 | 0.50 | 3 | 44 | 43 | F | 14 | |
| FAM11 | NCKAP5L | M | 12 | 50,189,151 | . | G->V | TT | 1|13 | 0.63 | 0.74 | 49 | 40 | 33 | 3 | ||
| FAM11 | DNAH11 | L | 7 | 21,737,777 | . | S->R | DD | 3|13 | 0.00 | −5.61 | 1 | 15 | 33 | 4 | ||
| FAM11 | NRCAM | L | 7 | 107,836,262 | 3.00E-04 | N->S | TT | 3|13 | 0.02 | 1.91 | ASD | 243 | 675 | 309 | T | 21 |
| FAM12 | TNN | H | 1 | 175,097,769 | 2.50E-03 | V->I | TT | 0|11 | 0.00 | −0.54 | 0 | 11 | 13 | 8 | ||
| FAM12 | COL4A2 | M | 13 | 111,082,914 | 4.00E-04 | H->L | TT | 1|11 | 0.00 | 2.19 | 19 | 925 | 439 | F | 62 | |
| FAM12 | NCL | M | 2 | 232,325,406 | 6.09E-03 | DE deletion | .. | 0|11 | 1.00 | 0.85 | 386 | 660 | 1028 | 95 | ||
| FAM12 | DYNC2H1 | L | 11 | 103,339,392 | 1.16E-05 | V->M | TD | 0|11 | 0.00 | 0.91 | 9 | 19 | 70 | F | 13 | |
| FAM12 | NAALAD2 | L | 11 | 89,896,570 | 6.00E-04 | R->W | DD | 0|11 | 0.00 | 0.47 | 22 | 24 | 53 | 3 | ||
| FAM12 | STARD9 | L | 15 | 42,983,572 | 1.10E-03 | G->S | TT | 0|11 | 0.00 | 2.50 | 36 | 26 | 30 | F | 1 | |
| FAM13 | DNAH3 | H | 16 | 21,098,323 | 5.70E-03 | R->S | DD | 1|8 | 0.00 | 0.03 | ASD | 1 | 5 | 22 | 9 | |
| FAM13 | ETS1 | H | 11 | 128,426,219 | . | E->K | DD | 1|8 | 0.78 | 2.52 | TD_simplex | 16 | 70 | 219 | 32 | |
| FAM13 | GRIN2A | H | 16 | 9,858,173 | 3.70E-03 | N->K | TD | 1|8 | 1.00 | 2.83 | ASD, ADHD, OtherNeuro | 45 | 281 | 17 | F | 38 |
| FAM13 | TUBGCP5 | H | 15 | 22,846,897 | 7.50E-03 | V->I | .. | 1|8 | TD_CNV, ASD | 23 | 32 | 24 | ||||
| FAM13 | EPHB2 | M | 1 | 23,233,346 | 2.90E-03 | D->N | DT | 1|8 | 1.00 | 2.45 | ASD | 11 | 200 | 161 | 50 | |
| FAM13 | KCNK1 | M | 1 | 233,802,497 | 7.10E-03 | R->R | TD | 1|8 | 0.03 | 0.72 | 261 | 501 | 154 | 22 | ||
| FAM13 | PCDH12 | L | 5 | 141,336,635 | 3.30E-03 | T->M | DD | 1|8 | 0.00 | 0.78 | 13 | 24 | 20 | F | 1 | |
Confidence: confidence of genes as causal for TD based on evidence collected. H, high; M, medium; L: low.
AF: allele frequency in gnomAD. If a variant does not exist in gnomAD, the ExAC AF is used. Variants not found in both databases are empty.
Effect: deleterious effect predicted by SIFT and Pholyphen-2. D for deleterious, T for tolerated, “.” for data not available.
Segregation: the inconsistency between the observation and the expected mutation segregation in individuals. The text is: (false positive + false negative) | (total - unknown). Genes with empty values are candidate genes from pVAAST run using recessive mode.
pLI: probability of being loss-of-function intolerant score. Typically, pLI ≥ 0.9 is a cutoff for extremely loss of function intolerant.
mis_z: Z score for the deviation of observed counts from the expected number of missense mutations. Positive Z scores indicate increased intolerance to variation.
Other NDDs: overlap with other neurodevelopmental disorder gene lists in Table S1.
TPM1, TPM2, TPM3: max TPM values of brain tissues in GTEx, BrainSpan, and HDBR, respectively.
IMPC: mouse knock-out model results from IMPC. T: genes with “behavior/neurological phenotype” or “nervous system phenotype”; F: genes without “behavior/neurological phenotype” or “nervous system phenotype”; Empty: data not available.
Interaction: Number of interactions with NDD_all genes (excluding risk genes from the same family) defined by STRING, GIANT_v2 or ConsensusPathDB.
Candidate genes in individual families
To understand the functional impact of TD_multiplex candidate genes, we collected several types of evidence for each gene, including their expression pattern in brain, known disease association, tolerance to mutations, knock-out mouse phenotypes, and their interaction with other genes (Figure 2, see Methods and Supplemental Methods for detail). We favored candidate genes that contain variants with good segregation patterns, low AF in the generation population, and genes that were identified in previous studies of TD or other NDDs, with nervous system related functions, with higher or tissue-specific expression in brain, and genes that causes a related-behavior change in mouse knockout models. Using these annotations, we further refined the TD_multiplex candidate genes and selected a final list of 71 candidate genes (Table 2). Below we briefly describe the high-confidence top candidate genes in several families.
Figure 2. Information used for variant and gene prioritization.
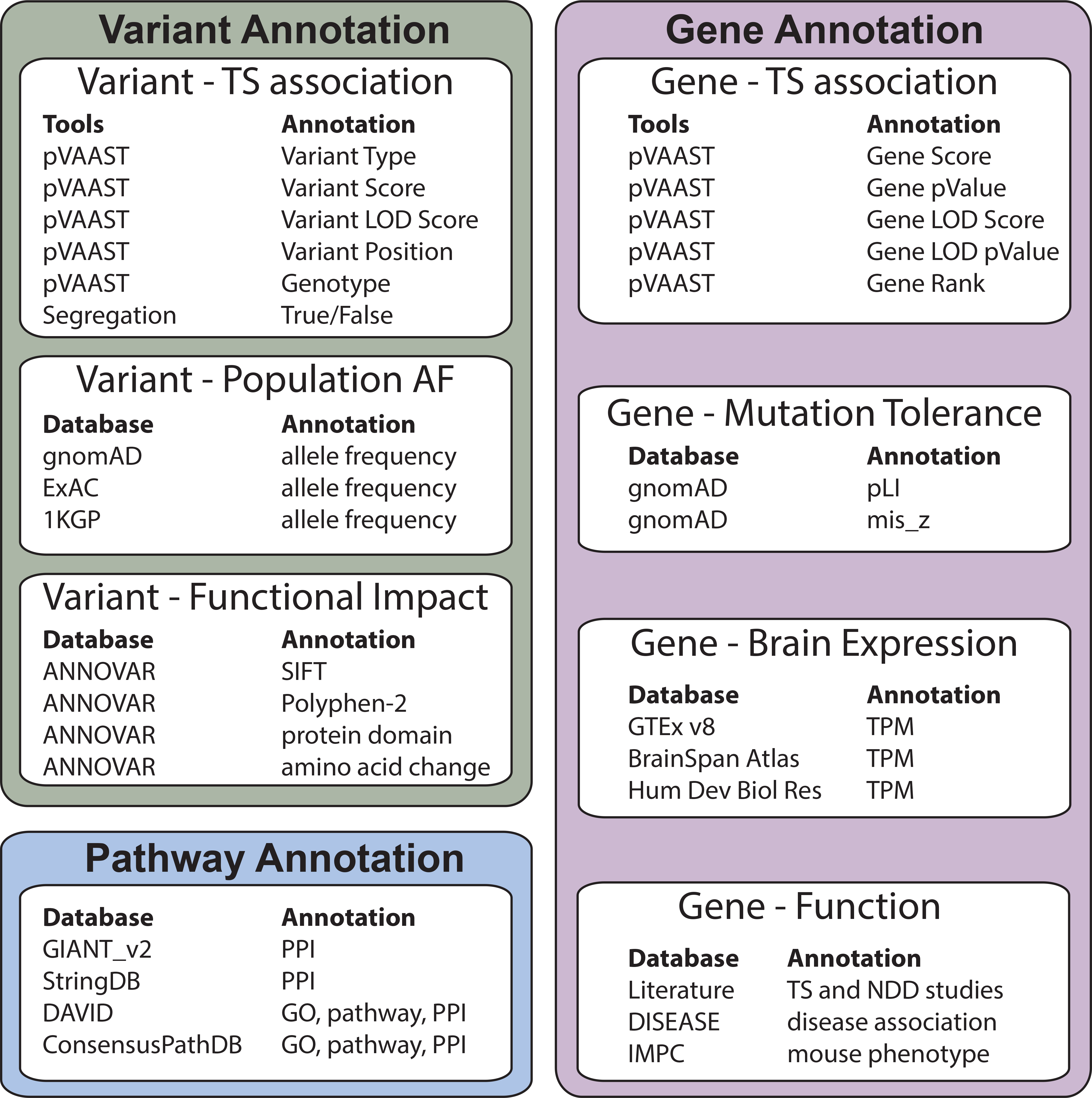
Detailed description of the “Annotation” fields can be found in the Supplemental Methods and Table 2. PPI: Protein-protein interaction; GO: Gene Ontology; pLI: probability of being loss-of-function intolerant score; mis_z: Z score for the deviation of observed counts from the expected number of missense mutations; TPM: Transcript Per Million.
FAM3
CDHR1 (Cadherin Related Family Member 1) was the only candidate gene with a perfectly segregating variant within the pedigree (ENST00000372117.3:p.Asn623Ser). The mutation is in the Cadherin domain which was predicted to be deleterious by SIFT 44. CDHR1 shows specific high expression in brain regions including caudate, hypothalamus, and nucleus accumbens 29, 30. CDHR1 is a calcium-dependent cell-cell adhesion membrane protein and is involved in vision related diseases such as cone-rod dystrophy 45.
FAM5
TADA3 (Transcriptional Adaptor 3) was the top candidate gene as ranked by pVAAST. The candidate mutation is a nonsynonymous mutation (ENST00000301964.2:pArg171Cys) that is predicted to be deleterious by SIFT and damaging by Polyphen-2 46. The variant was present in all seven affected individuals in the three-generation pedigree, but also present as a heterozygous variant in the unaffected grandfather. This gene is a component of the histone acetyl transferase (HAT) coactivator complex and a regulator of p53 and it is important for chromatin modulation and cell cycle progression 47. TADA3 is universally highly expressed in brain regions (> 70 TPM in the GTEx data) 30. As a multifaceted protein, TADA3 plays an important role in chromatin remodeling, cellular proliferation, cellular senescence, DNA damage response, and embryonic development 47.
FAM11
HTRA3 (HtrA Serine Peptidase 3) had a nonsynonymous mutation (ENST00000307358.2:p.Val269Met) in the protease domain, predicted to be deleterious by SIFT and Polyphen-2. The mutation was present in all eight affected individuals and an unaffected grandchild. HTRA3 is highly expressed (TPM > 10) during some of the early brain developmental stages 31, 32. HTRA3 is a secreted protein with four domains, which is highly conserved in vertebrates 48. Several mouse models for Wolf–Hirschhorn syndrome with deletions of Htra3 cause significant phenotypic changes including seizures, a behavior similar to tics 49.
FAM12
NCL (Nucleolin) exhibited a perfectly segregating in-frame deletion (ENST00000322723.4:p.Asp261del). NCL is involved in the synthesis and maturation of ribosomes and is LoF intolerant (pLI = 1) 50. This gene is highly expressed in brain (Table S3). In a GWAS study, NCL was associated with ADHD 51. Disrupting the function of NCL causes nucleolar stress, which is believed to be one of the pathogenic mechanisms for neurodegenerative diseases including polyglutamine (polyQ) diseases and Parkinson’s diseases 52, 53. In Huntington disease, the polyQ mutation in the HTT gene functionally disrupts the normal ribosomal interaction with NCL 54.
FAM4
Pedigree FAM4 includes a single sperm donor, six mothers, and nine offspring, among which eight are TD cases. As four of the mothers were unaffected, it is highly likely that there are one or a few dominant mutation(s) from the sperm donor which were passed to the affected children. In the pVAAST dominant run, 10 genes remained after filtering. Among those genes, DCAF1 (DDB1 And CUL4 Associated Factor 1, also known as VPRBP) contains a perfectly segregating variant. DCAF1 is a Serine/threonine-protein kinase which is also a component of E3 ubiquitin-protein ligase complexes and plays key roles in various biological processes including cell cycle, telomerase regulation, and histone modification 55.
Besides DCAF1, several other genes also contain variants with one genotype/ phenotype discrepancy (Table 2). ZDHHC17 (Zinc Finger DHHC-Type Palmitoyltransferase 17) contains a rare nonsynonymous variant (ENST00000426126.2:p.Ile582Thr), which is predicted to be possibly damaging by Polyphen-2. The variant is present in the sperm donor, five affected children, and absent in one affected child and all unaffected individuals. ZDHHC17 was previously named Huntingtin interacting protein 14, based on its interaction with the Huntington disease gene, HTT. It is a membrane protein with palmitoyl-transferase activity which is specific for several critical neuronal proteins including HTT 56 and GRIN2B (Glutamate Ionotropic Receptor NMDA Type Subunit 2B) 57. A mouse model for Huntington disease, Zdhhc17Gt(RRJ233)Byg/Zdhhc17Gt(RRJ233)Byg shows a series of phenotypes in the nervous system and behavior/neurological changes, including increased neuronal apoptosis, decreased brain weight, abnormal striatum morphology, and hyperactivity. ZDHHC17 showed significant interactions with genes in TD_simplex, ADHD, and ASD_high lists (Table S3), suggesting it plays important roles in multiple NDDs.
Other small families
Because smaller families contain less information in variant segregation, these families typically present more candidate genes than larger ones and it is more difficult to rank the genes. However, several candidate genes are known to associate with other NDDs or were identified in previous TD genetic studies (Table 2). For example, CNTNAP5 (Contactin Associated Protein Family Member 5) in FAM11 is a known NDD gene and variants in CNTNAP5 have been reported to be associated with ASD and other NDDs (https://gene.sfari.org/database/human-gene/CNTNAP5). ANK3 (ankyrin 3) in FAM8 is a high-confidence ASD candidate gene and has also been reported in TD_simplex family studies 18. The overlap between our top TD_multiplex family genes with NDD candidate genes suggest a shared genetic etiology for TD and other NDDs.
Individual family CNV analysis
For nine families, we also obtained CNVs using genotyping array (Table 1). A total of 1,799 high quality CNVs were identified, with an average of 200 CNVs per family (ranging from 116 to 289). The majority of the CNVs were unique to one individual. After filtering on the segregation pattern, gene overlap, and the CNV size (See Methods for filtering detail), we obtained 22 genic CNVs, including 16 exonic CNVs (Table S5). Risk genes predicted by CNVs did not overlap candidate genes from the SNV analysis (Table 3). We further prioritized the 22 CNVs based on their population prevalence (AF <1%), encompassed candidate gene brain expression level, and candidate gene function. After prioritization, three candidate CNVs remained. The first CNV is a heterozygous deletion in FAM1 (g.chr5:73980065–73992881) overlapping the first several exons of HEXB (Hexosaminidase Subunit Beta). This deletion was perfectly segregating with the phenotype and the genomic region only contains two rare CNVs in human populations in the gnomAD structure variation database (https://gnomad.broadinstitute.org/region/5–73980065-73992881?dataset=gnomad_sv_r2_1). HEXB encodes a subunit of the lysosomal enzyme beta-hexosaminidase, and mutations in HEXB has been associated with neurodegenerative Sandhoff disease 58. The second CNV is a heterozygous deletion in FAM9 (g.chr20:5281253–5289644). The deletion is rare in populations and overlaps the last exon of gene PROKR2 (Prokineticin Receptor 2). PROKR2 has increased brain expression and has been associated with Kallman syndrome 59. The third CNV, from FAM5, is in the intron of ZNF385D (Zinc Finger Protein 385D). This heterozygous deletion segregated in five out of eight affected individuals in the family and is rare in general populations. ZNF385D has been associated with other neuropsychiatric disorders, such as schizophrenia 60 and reading disability 61, supporting ZNF385D as a candidate gene for TD.
Pathways implicated in multiple families
Because of the apparently high genetic heterogeneity of TD, it is likely that each multiplex family have different underlying genetic risk factors. Although only 25 of the 543 TD_multiplex genes were present in more than one family, the families could share the underlying TD genetic etiology beyond the single gene level, such as pathways and gene groups. In support of the latter, a polygenic risk score has been associated with TD 12. Therefore, we examined the interaction for the top 71 candidate genes (Table 2), as well as their interactions with other NDD genes. Our result showed strong interactions among our top candidate genes: 35 of the 71 genes showed interactions and form a single PPI network (Figure 3A). Many of the remaining genes showed extensive interaction with other NDD genes (Figure 3B), suggesting the shared genetic etiology among NDDs. Interestingly, submodules emerging from the PPI network include potassium voltage-gated channel genes, and axonemal dynein genes (Figure 3B).
Figure 3. Protein-protein interaction (PPI) networks.
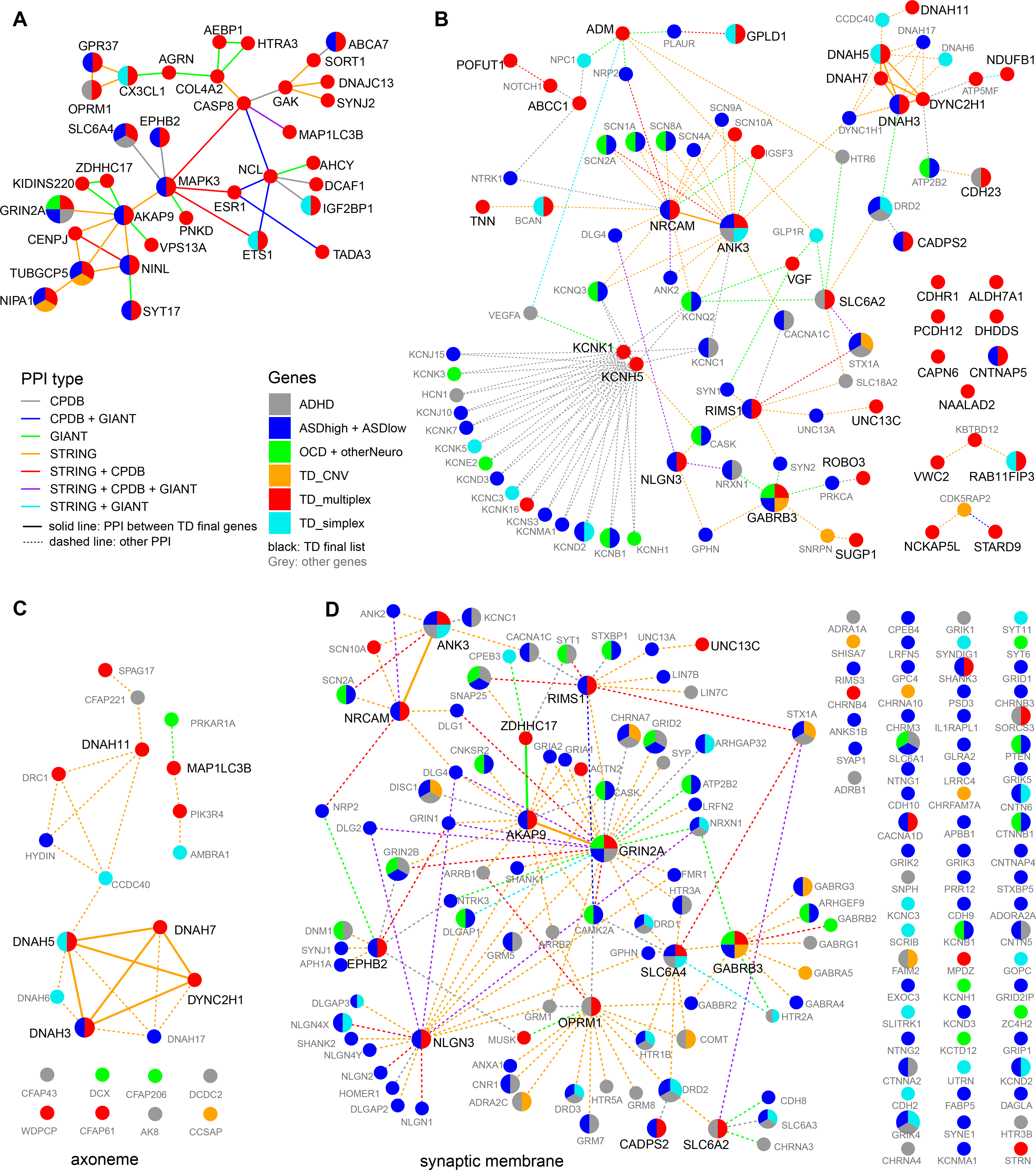
(A) PPI network of the 71 TD top candidate genes. Only genes that can be connected are shown. (B) PPI networks of the 71 TD top candidate genes not in (A). Other NDD_all genes were added as intermediate nodes if they interact with more than one TD top candidate genes. For intermediate nodes, only interactions with top candidate genes were included. (C) PPI network formed by NDD_all genes identified in axoneme (GO:0005930). (D) PPI networks formed by NDD_all genes in synaptic membrane (GO:0097060). To simplify the network, interactions between non-candidate genes were removed. PPI networks were defined by three databases, ConsensusPathDB, STRING, and GIANT_v2. Genes were colored by the gene lists (see Methods for details).
To further explore relationships among TD candidate genes and to identify potential shared mechanisms between TD and different NDDs, we performed enrichment analysis for all 2,345 NDD genes (NDD_all, Table S1) using ConsensusPathDB, and calculated the significance of enrichment for each gene list using Fisher’s exact test (Table S6). NDD_all, TD_multiplex, as well as several other gene lists were highly enriched in GO terms and pathways related to brain function and developmental processes, highlighting that many genes in this group are involved in brain development (Table S6). Among GO terms enriched in TS_multiplex gene list, “dynein light intermediate chain” (GO:0051959, 29 genes) is the top molecular function term and “axoneme part” (GO:0044447, 37 genes) is the top cellular component term. Seven genes from five multiplex families are defined under the dynein light intermediate chain term, including DNAH3, DNAH5, DNAH7, DNAH11, RAB11FIP3, DYNC2H1, and CCDC88B. Interestingly, four genes in the TD_simplex gene set (DNAH5, DNAH6, DNAH10, and RAB11FIP3) and four genes in the ASD gene set (DNAH3, DNAH10, DNAH17, and CCDC88C) are also present in the “dynein light intermediate chain” gene set and the NDD_all genes showed significant enrichment for the term as well (q = 5.1×10−4, Table S6). These genes encode dynein motor proteins that are parts of the cellular cytoskeleton and showed extensive interaction in the PPI network (Figure 3C). The overlapping candidate genes within the dynein complex in both TD and other NDDs suggests that microtubule function might be an important factor for NDDs and TD in particular. In the enrichment analysis of all NDD genes, the most enriched GO terms included synaptic vesicle cycle and presynaptic membrane (hypergeometric test, q = 9.2×10−23 and 2.7×10−20, respectively, Table S6). Most of these genes were also under the GO term “synaptic membrane” and showed extensive interaction in the PPI network (Fig. 3D). In addition to their enrichment in overall gene list, genes in the “synaptic vesicle cycle” and “presynaptic membrane” GO terms were also enriched in most individual gene lists (excluding TD_CNV and OCD), indicating the importance of genes in these two categories in multiple NDDs.
Discussion
TD is a highly heritable and frequently occurring neurodevelopmental disorder that can cause significant burden in patients and families 14. As a complex disease with heterogeneous etiology, it is difficult to study its mechanism with classic candidate gene methods and only a few genes have been reported from single gene studies (Reviewed in 15). Multiplex families provide additional information compared to simplex trio studies: as multiple affected individuals are identified in a single family, the variant segregation pattern within the family can help identify inherited rare variants with strong effects. In this study, we examined 13 multiplex families to identify potential TD candidate genes.
Among our multiplex families, the co-occurrence of OCD and ADHD is consistent with previous studies 3–5, 7–9: a total of 37.8% (31/82) of individuals with TD and other tic disorders were also diagnosed with OCD, and 20.7% (17/82) had comorbid ADHD (Table S2). On the other hand, the sex ratio between male and female is 1.3, smaller than the ratio in population studies where boys are affected three to five times more often than girls 1. It has been proposed that the lower affectation rate in girls could be explained by a “female protective effect” which protects females from many variants with small effects. This increased rate of affected females in multiplex families may be consistent with the segregation of large-effect variants that are needed to affect females in these families.
Our initial analysis of the 13 families identified hundreds of candidate genes across the genome. The large number of candidate genes could be due to both the complex genetic etiology of TD and the lack of segregation information in small families. Therefore, to facilitate candidate gene prioritization, we considered multiple types of information for variants and genes, including mutation functional impact prediction, segregation pattern, gene function, gene known disease association(s), and gene expression patterns in brain (Figure 2). By combining the evidence, we prioritized candidate risk genes and generated a top candidate gene list of 71 genes. We also found three segregating genic CNVs that could also contribute to TD etiology in three families. Some of the top candidate genes are well known NDD genes, such as GRIN2B, CNTNAP5, GABRB3, and ANK3. For four patients with diagnosis for ADHD, their exomes contain variants in reported genes associated with ADHD (SLC6A2, ANK3). We believe these genes are also TD risk genes in these families because the variants showed strong segregating patterns with TD patients. Fourteen genes contained likely damaging de novo mutations in previous studies of TD simplex trios (Table S4), such as BCAN, DNAH5, and RAB11FIP3, providing additional support for their involvement in TD etiology. More importantly, we identified several novel candidate genes in the multiplex families that have not been previously linked to TD or NDDs. For example, TADA3 is the single candidate gene in FAM5. The evidence from this study warrants close examination of these genes in future TD studies.
The candidate genes also allowed us to identify potential pathways and biological processes that are important for TD etiology. One of the strongest signals in our dataset is associated with the components of dynein pathways, the motor proteins for cellular cytoskeleton (Figure 3C). Dyneins are important for protein transport within neurons, especially for signaling proteins that function in axons that are far distal to the cell body 62. This result suggests that the cytoskeleton might play an important role in TD development. One interesting observation is that although the GO term and pathway enrichments of many NDD gene lists are related to neuronal and brain function, many enrichments in TD_multiplex and TD_simplex gene lists are different from enrichments in other NDD lists (Table S6). This suggests that TD might have distinct genetic risk factors compared to other NDDs. We also observed shared enrichment in molecular functions and biological pathways among all NDD genes. For all NDD genes, the most enriched GO terms and pathways showed strong relationships with brain functions, such as presynaptic membrane, synaptic vesicle cycle, neurotransmitter secretion, dopamine metabolic process, glutamate receptor signaling pathway, and GABAergic synapse (Table S6). Therefore, studies from other NDDs may also contribute to understanding the genetic bases of TD.
As a complex disorder with genetic heterogeneity 18, risk genes in TD are likely to work together in networks. Our PPI network analysis supports this hypothesis: many PPIs were predicted within the top TD candidate genes and with genes from other NDD lists by the three PPI databases (Fig. 3). For these disorders, some of the most enriched terms are related to the proper function of neurons, such as synaptic membrane, synaptic vesicle cycle, and voltage−gated ion channel activity.
In summary, by leveraging the segregation information in multiplex families, the power of whole-exome sequencing and genome-wide CNV analysis, we identified new candidate genes as well as biological processes and pathways that could contribute to the development of TD. Future studies of TD with larger sample sizes are needed to provide further support for the candidate genes. Furthermore, functional studies of these candidate genes, either in cell-culture systems or animal models, will provide a better understanding of their roles in TD, eventually leading to improved diagnosis and treatment.
Supplementary Material
Acknowledgements
We thank the families who have participated in and contributed to this study. We are also grateful to the NJCTS for facilitating the inception and organization of the TIC Genetics study. This study was supported by a grant from the National Institute of Mental Health (R01MH092293 to G.A.H. and J.A.T.) and by a grant from the New Jersey Center for Tourette Syndrome (to G.A.H. and J.A.T.). This study was also supported by grants from the National Institute of Mental Health (K08MH099424 to T.V.F) and the National Institute for Environmental Health Science (R01 ES021462 for Y.K. and B.L.L.). Dr. Mir has received grants from the Instituto de Salud Carlos III (PI10/01674, PI13/01461), the Consejería de Economía, Innovación, Ciencia y Empresa de la Junta de Andalucía (CVI-02526, CTS-7685), the Consejería de Salud y Bienestar Social de la Junta de Andalucía (PI-0741/2010, PI-0437-2012, PI-0471-2013), the Sociedad Andaluza de Neurología, the Fundación Alicia Koplowitz, the Fundación Mutua Madrileña and the Jaques and Gloria Gossweiler Foundation. Dr. Morer has received grants from the Fundacion Alicia Koplowitz and belongs to the research group of the Comissionat per Universitats i Recerca del Departmanent d’Innovacio (DIUE) 2009SGR1119. Dr. Münchau has received grants from the Deutsche Forschungsgemeinschaft (DFG: MU 1692/3-1, MU 1692/4-1 and FOR 2698). Dr. Willsey received a Young Investigator Award from Tourette Association of America. Dr. Isobel Heyman declares that all research at Great Ormond Street Hospital NHS Foundation Trust and UCL Great Ormond Street Institute of Child Health is made possible by the NIHR Great Ormond Street Hospital Biomedical Research Centre. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
Conflict of interest
Dr. Coffey received research support from NIMH, Emalex, and Teva/Nuvelution; honoraria from American Academy of Child and Adolescent Psychiatry, Harvard Medical School /Psychiatry Academy, and Partners Healthcare. Dr. Coffey served on the advisory board of Skyland Trail and Teva/Nuvelution, and the follow roles at the Tourette Association of America: Co-Chair, Medical Advisory Board; TAA-CDC Partnership. Dr. Roessner has served as advisor or consultancy role for Lilly, Novartis, and Shire, and received conference attendance support or was paid for public speaking by Actelion, Lilly, Medice, Novartis and Shire.
TIC Genetics author list (in alphabetical order)
Mohamed Abdulkadir1,2,3, Lawrence W. Brown12, Xiaolong Cao1, Barbara J. Coffey13, Li Deng1,2, Andrea Dietrich3, Thomas V. Fernandez4,5, Blanca Garcia-Delgar6, Donald L. Gilbert14, Julie Hagstrøm7, Tammy Hedderly15, Gary A. Heiman1,2, Isobel Heyman16, Pieter J. Hoekstra3, Chaim Huyser17, Eunjoo Kim18, Young-Shin Kim19, Robert A. King4, Justin Koesterich1,2, Yun-Joo Koh20, Samuel Kuperman8, Bennett L. Leventhal21, Marcos Madruga-Garrido22, Athanasios Maras23, Pablo Mir24,25, Astrid Morer6,9,10, Alexander Münchau26, Cara Nasello1,2, Kerstin J. Plessen7,11, Veit Roessner27, Dong-Ho Song28, Matthew W. State13, Joshua K. Thackray1,2, Jay A. Tischfield1,2, A. Jeremy Willsey13,29, Jinchuan Xing1,2, Yeting Zhang1,2, Lisheng Zhou1, and Samuel H. Zinner30.
1 Department of Genetics, Rutgers, The State University of New Jersey, Piscataway, NJ, USA
2 Human Genetic Institute of New Jersey, Rutgers, the State University of New Jersey, Piscataway, NJ, USA
3 Department of Child and Adolescent Psychiatry, University Medical Center Groningen, University of Groningen, Groningen, The Netherlands
4 Yale Child Study Center, Yale University School of Medicine, New Haven, CT, USA
5 Department of Psychiatry, Yale University School of Medicine, New Haven, CT, USA
6 Department of Child and Adolescent Psychiatry, and Psychology, Institute of Neurosciences, Hospital Clinic, Universitari, Barcelona, Spain
7 Child and Adolescent Mental Health Center, Mental Health Services, Capital Region of Denmark, Denmark
8 Department of Psychiatry, Carver College of Medicine, University of Iowa, Iowa City, IA, USA
9 Institut d’Investigacions Biomediques August Pi i Sunyer, (IDIPABS), Barcelona, Spain
10 Centro de Investigacion en Red de Salud Mental (CIBERSAM), Instituto Carlos III, Spain
11 Division of Child and Adolescent Psychiatry, University Hospital Lausanne, Switzerland
12Pediatric Neuropsychiatry Program, Division of Neurology, The Children's Hospital of Philadelphia, Philadelphia, PA, USA
13Department of Psychiatry and Behavioral Sciences, University of Miami Miller School of Medicine, Miami, FL, USA
14Cincinnati Children’s Hospital Medical Center, Cincinnati, OH, USA
15Evelina London Children’s Hospital GSTT, Kings Health Partners AHSC, London, UK.
16Tic Disorder Clinic, Great Ormond Street Hospital for Children NHS Trust, London, WC1N 3JH, UK
17Levvel, specialists for Youth and Families, Amsterdam, 1015 AZ, The Netherlands.
18Department of Psychiatry, Gangnam Severance Hospital, Yonsei University College of Medicine, Seoul, South Korea
19Department of Psychiatry & Behavioral Sciences, UCSF Weill Institute for Neurosciences, University of California, San Francisco, San Francisco, CA, USA.
20Korea Institute for Children’s Social Development and Rudolph Child Research Center, Seoul, South Korea
21University of California San Francisco Medical Center, San Francisco, CA, 94143, USA.
22Sección de Neuropediatría, Instituto de Biomedicina de Sevilla, Hospital Universitario Virgen del Rocío/CSIC/Universidad de Sevilla, Seville, Spain
23Yulius Academy, Yulius Mental Health Organization, Dordrecht, 3311 JG, The Netherlands
24Unidad de Trastornos del Movimiento. Instituto de Biomedicina de Sevilla (IBiS). Hospital Universitario Virgen del Rocío/CSIC/Universidad de Sevilla. Seville, Spain
25Centro de Investigación Biomédica en Red sobre Enfermedades Neurodegenerativas (CIBERNED), Madrid, Spain
26Institute of Systems Motor Science; University of Lübeck, Lübeck, Germany
27Department of Child and Adolescent Psychiatry, TU Dresden, Germany
28Yonsei University Severance Hospital, Seoul, South Korea
29Institute for Neurodegenerative Diseases, UCSF Weill Institute for Neurosciences, University of California San Francisco, San Francisco, CA, USA
30Department of Pediatrics, Division of Developmental Medicine, University of Washington School of Medicine, Seattle, WA, USA
Footnotes
Supplementary information
Supplementary information is available at MP’s website.
References
- 1.Robertson MM, Eapen V, Singer HS, Martino D, Scharf JM, Paschou P et al. Gilles de la Tourette syndrome. Nat Rev Dis Primers 2017; 3: 16097. [DOI] [PubMed] [Google Scholar]
- 2.Scharf JM, Miller LL, Gauvin CA, Alabiso J, Mathews CA, Ben-Shlomo Y. Population Prevalence of Tourette Syndrome: A Systematic Review and Meta-Analysis. Movement Disord 2015; 30(2): 221–228. [DOI] [PubMed] [Google Scholar]
- 3.Cross-Disorder Group of the Psychiatric Genomics C. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet 2013; 381(9875): 1371–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cross-Disorder Group of the Psychiatric Genomics C, Lee SH, Ripke S, Neale BM, Faraone SV, Purcell SM et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nature genetics 2013; 45(9): 984–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O’Rourke JA, Scharf JM, Platko J, Stewart SE, Illmann C, Geller DA et al. The familial association of tourette’s disorder and ADHD: the impact of OCD symptoms. American journal of medical genetics Part B, Neuropsychiatric genetics : the official publication of the International Society of Psychiatric Genetics 2011; 156B(5): 553–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hirschtritt ME, Lee PC, Pauls DL, Dion Y, Grados MA, Illmann C et al. Lifetime Prevalence, Age of Risk, and Genetic Relationships of Comorbid Psychiatric Disorders in Tourette Syndrome. Jama Psychiat 2015; 72(4): 325–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu D, Mathews CA, Scharf JM, Neale BM, Davis LK, Gamazon ER et al. Cross-disorder genome-wide analyses suggest a complex genetic relationship between Tourette’s syndrome and OCD. The American journal of psychiatry 2015; 172(1): 82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davis LK, Yu D, Keenan CL, Gamazon ER, Konkashbaev AI, Derks EM et al. Partitioning the heritability of Tourette syndrome and obsessive compulsive disorder reveals differences in genetic architecture. PLoS genetics 2013; 9(10): e1003864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pringsheim T, Holler-Managan Y, Okun MS. Comprehensive systematic review summary: Treatment of tics in people with Tourette syndrome and chronic tic disorders (vol 92, pg 907, 2019). Neurology 2019; 93(9): 415–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pinto R, Monzani B, Leckman JF, Ruck C, Serlachius E, Lichtenstein P et al. Understanding the Covariation of Tics, Attention-Deficit/Hyperactivity, and Obsessive-Compulsive Symptoms: A Population-Based Adult Twin Study. Am J Med Genet B 2016; 171(7): 938–947. [DOI] [PubMed] [Google Scholar]
- 11.Darrow SM, Grados M, Sandor P, Hirschtritt ME, Illmann C, Osiecki L et al. Autism Spectrum Symptoms in a Tourette’s Disorder Sample. Journal of the American Academy of Child and Adolescent Psychiatry 2017; 56(7): 610–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abdulkadir M, Mathews CA, Scharf JM, Yu D, Tischfield JA, Heiman GA et al. Polygenic Risk Scores Derived From a Tourette Syndrome Genome-wide Association Study Predict Presence of Tics in the Avon Longitudinal Study of Parents and Children Cohort. Biological psychiatry 2019; 85(4): 298–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu D, Sul JH, Tsetsos F, Nawaz MS, Huang AY, Zelaya I et al. Interrogating the Genetic Determinants of Tourette’s Syndrome and Other Tic Disorders Through Genome-Wide Association Studies. The American journal of psychiatry 2019; 176(3): 217–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heiman GA, Rispoli J, Seymour C, Leckman JF, King RA, Fernandez TV. Empiric Recurrence Risk Estimates for Chronic Tic Disorders: Implications for Genetic Counseling. Frontiers in Neurology 2020; 11(770). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qi Y, Zheng Y, Li Z, Xiong L. Progress in Genetic Studies of Tourette’s Syndrome. Brain Sci 2017; 7(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deng H, Gao K, Jankovic J. The genetics of Tourette syndrome. Nat Rev Neurol 2012; 8(4): 203–213. [DOI] [PubMed] [Google Scholar]
- 17.Willsey AJ, Fernandez TV, Yu D, King RA, Dietrich A, Xing J et al. De Novo Coding Variants Are Strongly Associated with Tourette Disorder. Neuron 2017; 94(3): 486–499 e489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang S, Mandell JD, Kumar Y, Sun N, Morris MT, Arbelaez J et al. De Novo Sequence and Copy Number Variants Are Strongly Associated with Tourette Disorder and Implicate Cell Polarity in Pathogenesis. Cell reports 2018; 24(13): 3441–3454 e3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pauls DL, Fernandez TV, Mathews CA, State MW, Scharf JM. The Inheritance of Tourette Disorder: A review. J Obsessive Compuls Relat Disord 2014; 3(4): 380–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dietrich A, Fernandez TV, King RA, State MW, Tischfield JA, Hoekstra PJ et al. The Tourette International Collaborative Genetics (TIC Genetics) study, finding the genes causing Tourette syndrome: objectives and methods. European child & adolescent psychiatry 2015; 24(2): 141–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heiman GA, King RA, Tischfield JA. New Jersey Center for Tourette Syndrome sharing repository: methods and sample description. BMC medical genomics 2008; 1: 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.American Psychiatric Association. Diagnostic and statistical manual of mental disorders, 4th Edition Text Revision (DSM-IV-TR). American Psychiatric Association: Washington DC, 2000. [Google Scholar]
- 23.American Psychiatric Association. Diagnostic and statistical manual of mental disorders, Fifth Edition (DSM-5). American Psychiatric Association: Washington DC, 2013. [Google Scholar]
- 24.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature genetics 2011; 43(5): 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang H, Wang K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nature protocols 2015; 10(10): 1556–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yandell M, Huff C, Hu H, Singleton M, Moore B, Xing J et al. A probabilistic disease-gene finder for personal genomes. Genome research 2011; 21(9): 1529–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hu H, Roach JC, Coon H, Guthery SL, Voelkerding KV, Margraf RL et al. A unified test of linkage analysis and rare-variant association for analysis of pedigree sequence data. Nature biotechnology 2014; 32(7): 663–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020; 581(7809): 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aguet F, Brown AA, Castel SE, Davis JR, He Y, Jo B et al. Genetic effects on gene expression across human tissues. Nature 2017; 550(7675): 204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Consortium GTEx. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 2020; 369(6509): 1318–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miller JA, Ding SL, Sunkin SM, Smith KA, Ng L, Szafer A et al. Transcriptional landscape of the prenatal human brain. Nature 2014; 508(7495): 199-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lindsay SJ, Xu YB, Lisgo SN, Harkin LF, Copp AJ, Gerrelli D et al. HDBR Expression: A Unique Resource for Global and Individual Gene Expression Studies during Early Human Brain Development. Front Neuroanat 2016; 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pletscher-Frankild S, Palleja A, Tsafou K, Binder JX, Jensen LJ. DISEASES: Text mining and data integration of disease-gene associations. Methods 2015; 74: 83–89. [DOI] [PubMed] [Google Scholar]
- 34.Dickinson ME, Flenniken AM, Ji X, Teboul L, Wong MD, White JK et al. High-throughput discovery of novel developmental phenotypes. Nature 2016; 537(7621): 508-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic acids research 2017; 45(D1): D362–D368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Herwig R, Hardt C, Lienhard M, Kamburov A. Analyzing and interpreting genome data at the network level with ConsensusPathDB. Nature protocols 2016; 11(10): 1889–1907. [DOI] [PubMed] [Google Scholar]
- 37.Wong AK, Krishnan A, Troyanskaya OG. GIANT 2.0: genome-scale integrated analysis of gene networks in tissues. Nucleic acids research 2018; 46(W1): W65–W70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Greene CS, Krishnan A, Wong AK, Ricciotti E, Zelaya RA, Himmelstein DS et al. Understanding multicellular function and disease with human tissue-specific networks. Nature genetics 2015; 47(6): 569–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang JK, Carlin DE, Yu MK, Zhang W, Kreisberg JF, Tamayo P et al. Systematic Evaluation of Molecular Networks for Discovery of Disease Genes. Cell Syst 2018; 6(4): 484–495 e485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sanders SJ, He X, Willsey AJ, Ercan-Sencicek AG, Samocha KE, Cicek AE et al. Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron 2015; 87(6): 1215–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Geoffroy V, Herenger Y, Kress A, Stoetzel C, Piton A, Dollfus H et al. AnnotSV: an integrated tool for structural variations annotation. Bioinformatics 2018; 34(20): 3572–3574. [DOI] [PubMed] [Google Scholar]
- 42.Sundaram SK, Huq AM, Sun Z, Yu W, Bennett L, Wilson BJ et al. Exome sequencing of a pedigree with Tourette syndrome or chronic tic disorder. Annals of neurology 2011; 69(5): 901–904. [DOI] [PubMed] [Google Scholar]
- 43.Moya PR, Wendland JR, Rubenstein LM, Timpano KR, Heiman GA, Tischfield JA et al. Common and rare alleles of the serotonin transporter gene, SLC6A4, associated with Tourette’s disorder. Movement disorders : official journal of the Movement Disorder Society 2013; 28(9): 1263–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic acids research 2003; 31(13): 3812–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stingl K, Mayer AK, Llavona P, Mulahasanovic L, Rudolph G, Jacobson SG et al. CDHR1 mutations in retinal dystrophies. Scientific reports 2017; 7(1): 6992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Current protocols in human genetics 2013; Chapter 7: Unit7 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chand V, Nandi D, Mangla AG, Sharma P, Nag A. Tale of a multifaceted co-activator, hADA3: from embryogenesis to cancer and beyond. Open Biol 2016; 6(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bowden M, Drummond AE, Salamonsen LA, Findlay JK, Nie GY. Evolutionary Conservation of Mammalian HTRA3 and Its Developmental Regulation in the Rat Ovary. J Exp Zool Part B 2009; 312b(7): 701–713. [DOI] [PubMed] [Google Scholar]
- 49.Naf D, Wilson LA, Bergstrom RA, Smith RS, Goodwin NC, Verkerk A et al. Mouse models for the Wolf-Hirschhorn deletion syndrome. Human molecular genetics 2001; 10(2): 91–98. [DOI] [PubMed] [Google Scholar]
- 50.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016; 536(7616): 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang L, Neale BM, Liu L, Lee SH, Wray NR, Ji N et al. Polygenic transmission and complex neuro developmental network for attention deficit hyperactivity disorder: Genome-wide association study of both common and rare variants. Am J Med Genet B 2013; 162b(5): 419–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang Q, An Y, Chen ZS, Koon AC, Lau KF, Ngo JCK et al. A Peptidylic Inhibitor for Neutralizing (r)(GGGGCC)(exp) -Associated Neurodegeneration in C9ALS-FTD. Mol Ther-Nucl Acids 2019; 16: 172–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tsoi H, Lau TCK, Tsang SY, Lau KF, Chan HYE. CAG expansion induces nucleolar stress in polyglutamine diseases. Proceedings of the National Academy of Sciences of the United States of America 2012; 109(33): 13428–13433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tsoi H, Chan HYE. Expression of Expanded CAG Transcripts Triggers Nucleolar Stress in Huntington’s Disease. Cerebellum 2013; 12(3): 310–312. [DOI] [PubMed] [Google Scholar]
- 55.Schabla NM, Mondal K, Swanson PC. DCAF1 (VprBP): emerging physiological roles for a unique dual-service E3 ubiquitin ligase substrate receptor. J Mol Cell Biol 2019; 11(9): 725–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ducker CE, Stettler EM, French KJ, Upson JJ, Smith CD. Huntingtin interacting protein 14 is an oncogenic human protein: palmitoyl acyltransferase. Oncogene 2004; 23(57): 9230–9237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kang RJ, Wang L, Sanders SS, Zuo K, Hayden MR, Raymond LA. Altered Regulation of Striatal Neuronal N-Methyl-D-Aspartate Receptor Trafficking by Palmitoylation in Huntington Disease Mouse Model. Front Synaptic Neuro 2019; 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mahdieh N, Mikaeeli S, Tavasoli AR, Rezaei Z, Maleki M, Rabbani B. Genotype, phenotype and in silico pathogenicity analysis of HEXB mutations: Panel based sequencing for differential diagnosis of gangliosidosis. Clin Neurol Neurosurg 2018; 167: 43–53. [DOI] [PubMed] [Google Scholar]
- 59.Dode C, Teixeira L, Levilliers J, Fouveaut C, Bouchard P, Kottler ML et al. Kallmann syndrome: mutations in the genes encoding prokineticin-2 and prokineticin receptor-2. PLoS genetics 2006; 2(10): e175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xu C, Aragam N, Li X, Villla EC, Wang L, Briones D et al. BCL9 and C9orf5 are associated with negative symptoms in schizophrenia: meta-analysis of two genome-wide association studies. PloS one 2013; 8(1): e51674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Eicher JD, Powers NR, Miller LL, Akshoomoff N, Amaral DG, Bloss CS et al. Genome-wide association study of shared components of reading disability and language impairment. Genes Brain Behav 2013; 12(8): 792–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Maday S, Twelvetrees AE, Moughamian AJ, Holzbaur EL. Axonal transport: cargo-specific mechanisms of motility and regulation. Neuron 2014; 84(2): 292–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.