

Abstract
Exosomes are cell-derived extracellular vesicles and play important roles in mediating intercellular communications. Due to their unique advantages in transporting a variety of biomolecules, exosomes have been emerging as a new class of nanocarriers with great potential for therapeutic applications. Despite advancements in loading chemotherapeutics and interfering RNAs into exosomes, active incorporation of protein molecules into exosomes remains challenging owing to their distinctive physicochemical properties and/or a lack of knowledge of cargo sorting during exosome biogenesis. Here we report the generation of a novel type of engineered exosomes with actively incorporated membrane proteins or soluble protein cargos, named genetically infused functionally tailored exosomes (GIFTed-Exos). Through genetic fusion with exosome-associated tetraspanin CD9, transmembrane protein CD70 and glucocorticoid‐induced tumor necrosis factor receptor family‐related ligand (GITRL) could be displayed on exosome surface, resulting in GIFTed-Exos with excellent T-cell co-stimulatory activities. By genetically linking to a CD9-photocleavable protein fusion, fluorescent protein mCherry, apoptosis-inducing protein apoptin, and antioxidant enzyme catalase could be effectively packed into exosomes for light-controlled release. The generated GIFTed-Exos display notable in vitro and in vivo activities for delivering distinct types of protein cargos to target cells. As a possibly general approach, GIFTed-Exos provide new opportunities to create exosomes with new functions and properties for biomedical research.
Keywords: drug delivery, exosomes, nanotechnology, protein engineering, synthetic biology
Introduction
Exosomes are nanoscale extracellular vesicles secreted by various types of cells. In addition to a bilayer phospholipid membrane, exosome contains a substantial amount of contents from parental cells such as metabolites, nucleic acids, and proteins [1, 2]. As endogenous nanocarriers, exosomes play important roles in mediating cell-cell communications [3, 4]. Through interactions with cell surface receptors and ligands, membrane fusion, and intracellular cargo delivery, cell-derived exosomes can modulate biological processes of recipient cells by one or more different modes of action [5–7].
In comparison to synthetic and viral nanovesicles, exosomes are characterized by unique and valuable properties for therapeutic development, including efficient cytosolic delivery of payloads, high biocompatibility, and low immunogenicity [8–11]. Given their potentials on treatment of many human diseases, exosomes have been emerging as a new and increasingly important class of therapeutic modality [12–15]. Development of exosomes with endogenous and/or exogenous cargos for biomedical applications has drawn considerable interests [11, 16–18].
To load small-molecule chemotherapeutics and interfering RNAs into exosomes, several methods have been commonly adopted including electroporation, sonication, and freeze-thaw cycles [19–23]. But these physical methods are usually much less effective to pack exogenous protein molecules into exosomes. Moreover, due to limited understanding of cargo sorting mechanism during exosome biogenesis, endogenous or recombinantly expressed protein cargos reply on passive loading for exosome encapsulation, resulting in significantly low efficiency and consequently lack of biological activity. Active incorporation of functional proteins into exosomes thus remains challenging. Considering diverse functions and great importance of proteins in biological systems, exosomes carrying sufficient amounts of proteins of interest may represent new research and therapeutic tools with desired properties.
Here we develop a novel form of engineered exosomes for actively incorporating functional membrane proteins and soluble proteins, termed genetically infused functionally tailored exosomes (GIFTed-Exos) (Figure 1). This was achieved through genetic fusions of exosome-associated tetraspanin CD9 with membrane proteins for functional display on exosomal surface as well as with a photocleavable protein (PhoCl) and cargo proteins in tandem for light-controlled release and exosome-mediated intracellular delivery [24]. The resulting GIFTed-Exos with surface displayed CD70 or glucocorticoid‐induced tumor necrosis factor receptor family‐related ligand (GITRL) exhibit excellent T-cell costimulatory activities. And GIFTed-Exos encapsulated with mCherry, apoptin, or catalase demonstrate functional delivery of distinct types of protein cargos to target cells. The designed GIFTed-Exos provide a new approach for generating function-oriented exosomes for biomedical applications.
Figure 1.
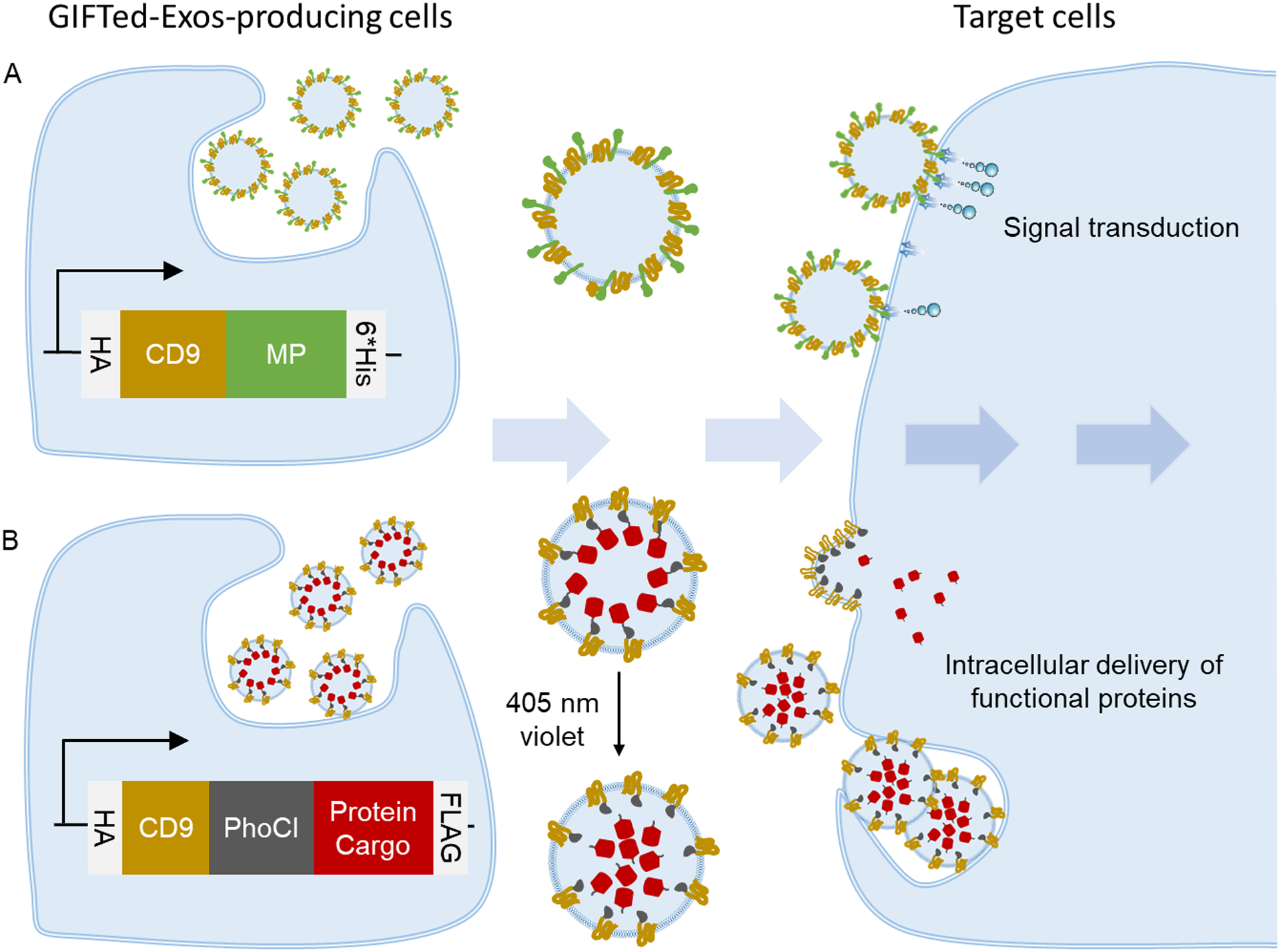
A schematic for design and generation of GIFTed-Exos for modulating cell functions. (A) GIFTed-Exos expressing a membrane protein (MP) of interest. Genetic fusion of a MP with CD9 facilitates display on exosome surface. (B) GIFTed-Exos carrying a cargo protein of interest. Genetic fusion of a cargo protein with CD9 and a photocleavable protein (PhoCl) in tandem allows active loading into exosomes and subsequent optical release for intracellular delivery.
Results
Design of GIFTed-Exos for modulating cell functions.
Considering high abundance of tetraspanin CD9 in exosome membrane, we envisioned that genetic fusion of membrane proteins with CD9 may not only enrich their expressions in exosomes but also facilitate their functional display on exosome surface. Furthermore, fusing soluble protein cargos with CD9 may enable active loading into exosomes. To irreversibly liberate protein payloads from the CD9-based fusions via a non-invasive approach, a key step for subsequent exosome-mediated intracellular delivery, the PhoCl could be inserted between CD9 and protein cargos. Transient irradiation of exosomes expressing the CD9-PhoCl-protein cargo fusions with 405 nm violet light would then result in rapid cleavage at C-terminus of PhoCl and release of the protein cargo. To test this notion, we genetically linked two membrane proteins, CD70 and GITRL, with CD9 and attached three distinct protein cargos to CD9-PhoCl fusion, including a fluorescent protein (mCherry), an apoptosis-inducing protein (apoptin), and an antioxidant enzyme (catalase) (Figure 1).
CD70 and GITRL GIFTed-Exos act as T cell co-stimulatory nanovesicles.
CD70, a homotrimeric type II tumor necrosis factor (TNF)-related transmembrane molecule, binds to its receptor CD27 and then induces T-cell co-stimulation and promotes B- and natural killer (NK)-cell responses, which can contribute to tumor control [25–27]. Currently, soluble CD70, CD70-expressing dendritic cells (DCs), and agonistic antibodies against CD27 are suggested to promote cytotoxic T lymphocyte (CTL) effector responses and CTL memory to cancer [28, 29].
GITRL, a type II transmembrane protein belonging to the TNF superfamily, can interact with its receptor glucocorticoid‐induced tumor necrosis factor receptor family‐related (GITR) protein, leading to co-stimulation of both effector and regulatory T cells. Using soluble GITRL, GITRL-transfected cells, and anti-GITR agonist antibodies, previous studies revealed that turning on the GITR/GITRL signaling pathway can enhance anti-tumor immunity by inducing activation of CD4+ and/or CD8+ T cells [30–32].
Given that CD70 and GITRL play active roles in a homotrimeric or homodimeric form, exosomes expressing surface CD70 or GITRL may function as excellent T-cell co-stimulatory vesicles. To this end, we genetically fused human CD70 or GITRL to C-terminus of human CD9 using a flexible (GGGGS)2 linker. A hemagglutinin (HA) epitope tag and a 6×His tag were placed at N- and C-terminus of the fusion protein, respectively (Figure 2A). The designed CD70 GIFTed-Exos and GITRL GIFTed-Exos were prepared through transient transfection of human Expi293F suspension cells and purified as described previously [33]. In addition, the same CD70 and GITRL expression constructs without the fused CD9 (designated as CD70-Exos and GITRL-Exos) were also generated for comparison (Figure 2A). Western blots confirmed the expression of designed CD9-CD70 and CD9-GITRL fusions as well as exosomal markers CD9, CD81, and CD63 (Figure 2B). In contrast to CD70-Exos and GITRL-Exos, both CD70 GIFTed-Exos and GITRL GIFTed-Exos show significantly increased expression levels of CD70 and GITRL, supporting that CD9-based fusions improve expression of membrane proteins in exosomes. The yield of GITRL GIFTed-Exos from the transient transfection was estimated to be 65.05 ± 5.15% based on Ni-NTA affinity chromatography, which can be further improved by generating stable cell lines expressing the gene of interest. Nanoparticle tracking analysis (NTA) revealed that the GIFTed-Exos have an average size of around 120 nm, similar to that of native exosomes (Figure 2C).
Figure 2.
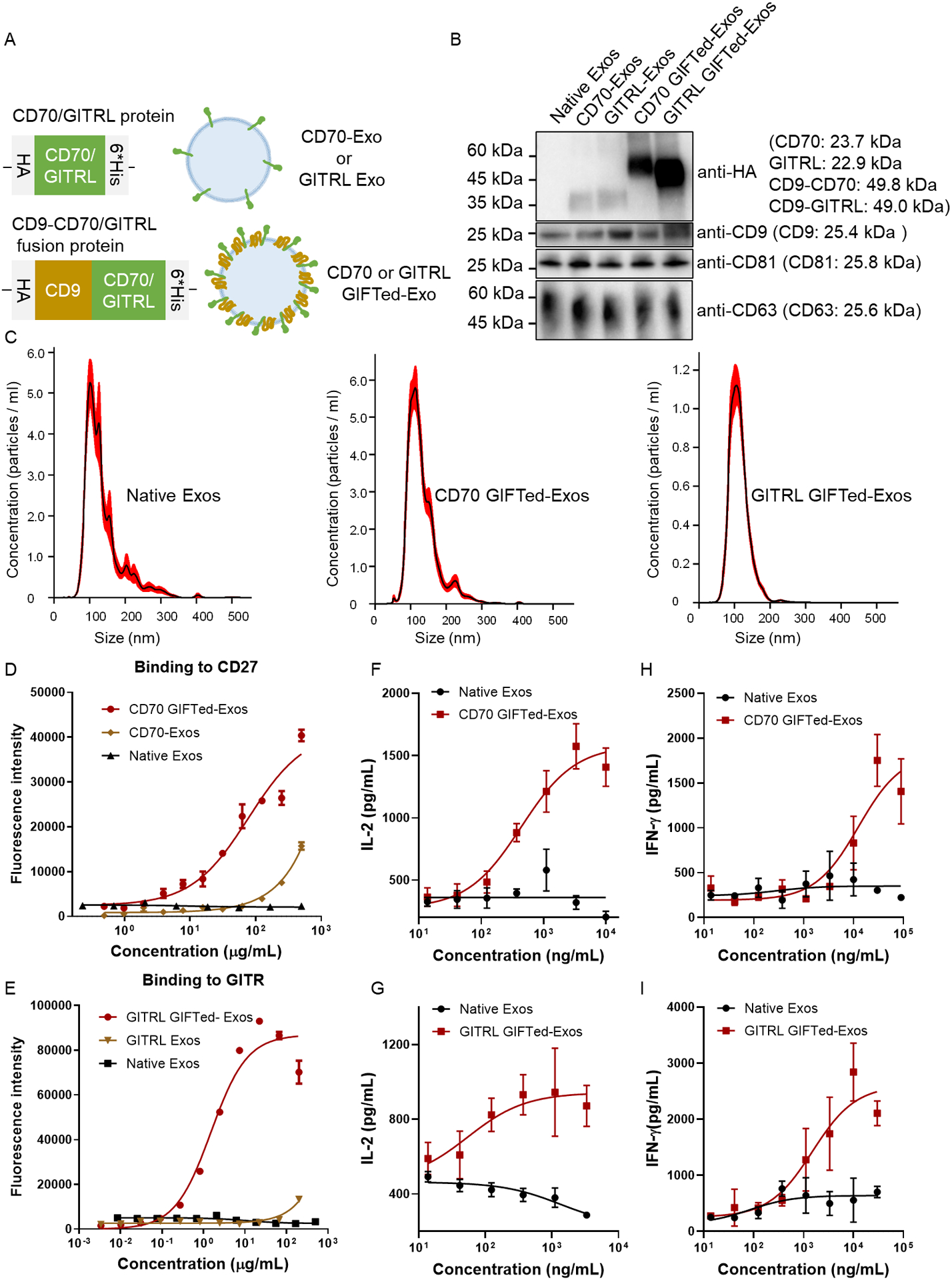
Generation and characterization of CD70 GIFTed-Exos and GITRL GIFTed-Exos. (A) Schematics of the design of CD70- and GITRL-expressing exosomes. (B) Immunoblot analysis of purified exosomes. Theoretical molecular weights of native proteins or fusion proteins are shown. (C) NTA analysis of purified native exosomes and GIFTed-Exos. (D) and (E) ELISA analysis of binding of the GIFTed-Exos to human CD27 (D) or GITR (E). Data are shown as mean ± SD of duplicates. (F)-(I) Dose dependent activation of human T cells by CD70 GIFTed-Exos (F) and (H) and GITRL GIFTed-Exos (G) and (I). Human PBMCs were incubated with pre-coated anti-human CD3 monoclonal antibody in the presence of various concentrations of CD70 GIFTed-Exos or GITRL GIFTed-Exos for 40 hours. The levels of secreted IL-2 (F) and (G) and IFN-γ (H) and (I) were measured by ELISA. Data are shown as mean ± SD of triplicates.
The bindings of displayed CD70 and GITRL on exosomes to their respective receptors, human CD27 and GITR, were then examined by ELISA (Figure 2D and 2E). Unlike CD70-Exos and GITRL-Exos, CD70 GIFTed-Exos and GITRL GIFTed-Exos exhibit much tighter binding to CD27 and GITR, consistent with their higher levels of expression as analyzed by western blots. In comparison, no binding to either receptor was detected for native exosomes. Next, CD70 and GITRL GIFTed-Exos were examined for co-stimulatory activity on T cells through measurements of secreted cytokines interleukin-2 (IL-2) and interferon gamma (IFN-γ). As shown in Figure 2F–I, both CD70 GIFTed-Exos and GITRL GIFTed-Exos dose-dependently induce releases of IL-2 and IFN-r from human peripheral blood mononuclear cells (hPBMCs) pre-activated by an anti-human CD3 monoclonal antibody. By contrast, native exosomes have little stimulatory effects on the release of IL-2 and IFN-γ. These results demonstrate that GIFTed-Exos represent an effective approach for functional display of membrane proteins on exosome surface, allowing generation of engineered exosomes with new and/or improved properties.
Intracellular delivery of fluorescent protein cargos by mCherry GIFTed-Exos.
We next sought to use GIFTed-Exos to deliver soluble proteins into cytoplasm of target cells. Red fluorescence protein mCherry was first chosen as a model protein cargo for incorporation into exosomes. mCherry GIFTed-Exos were generated by placing PhoCl at C-terminus of CD9, followed by the mCherry protein cargo. Two flexible (GGGGS)2 linkers were used to connect these three protein domains. A HA tag was added at N-terminus of CD9-PhoCl-mCherry fusion protein. As a signal peptide, nuclear localization signal (NLS) directs proteins into the nucleus. A NLS sequence and a FLAG tag were attached at C-terminus of the fusion protein (Figure 3A). Immunoblot analysis indicated that the CD9-PhoCl-mCherry fusion protein was successfully expressed in exosomes and underwent 405 nm violet light-mediated cleavage in a time-dependent manner, resulting in the release of mCherry with a short C-terminal 12-residue peptide of PhoCl (cPhoCl-mCherry) into the lumen of mCherry GIFTed-Exos (Figure 3A and B). On the basis of immunoblots, 2 minute-exposure of mCherry GIFTed-Exos to 405 nm violet light could lead to nearly complete detachment of mCherry from the fusion protein.
Figure 3.
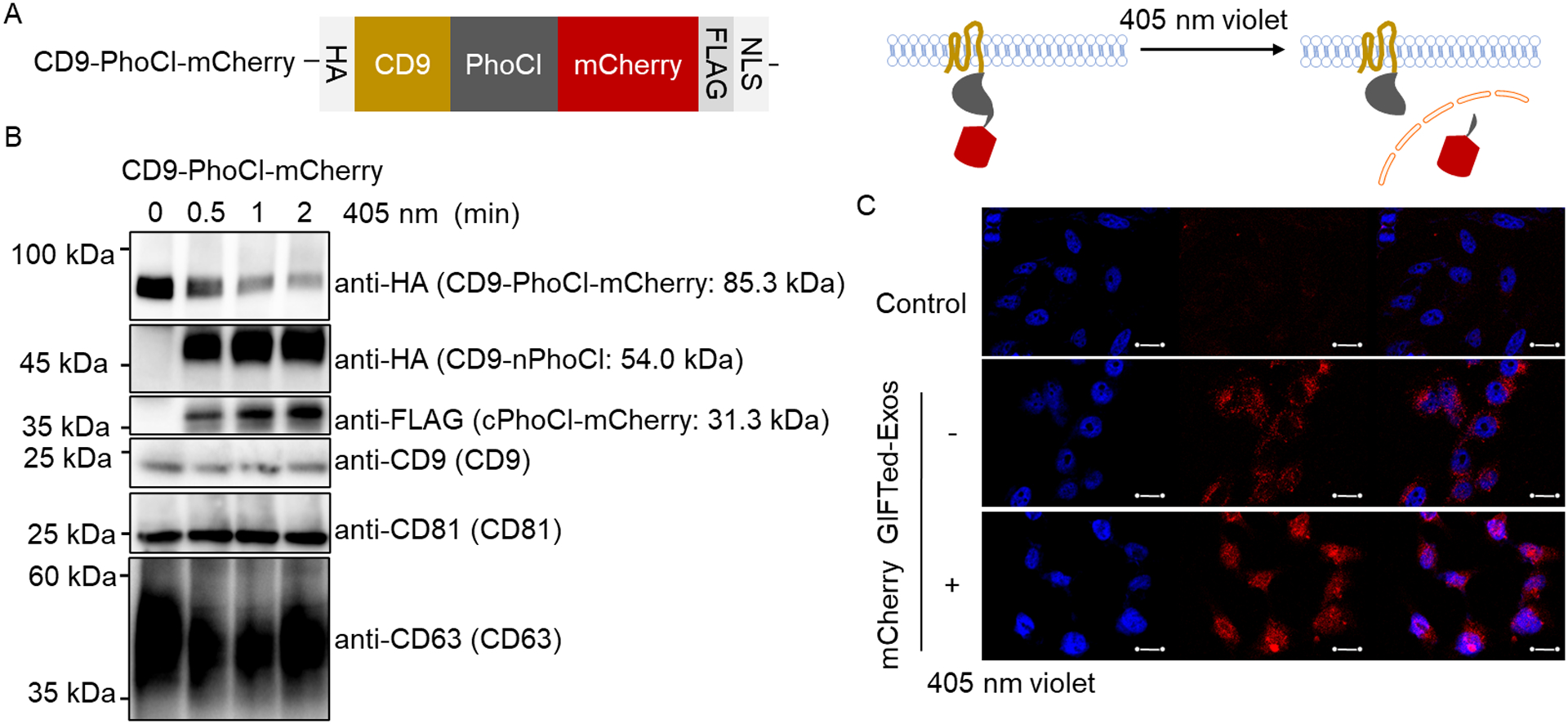
Design, generation and examination of mCherry GIFTed-Exos. (A) Schematic of the design of mCherry GIFTed-Exos. (B) Immunoblot analysis of the mCherry GIFTed-Exos and violet light-induced release of mCherry. Theoretical molecular weights of fusion proteins are shown. (C) Cellular uptake of mCherry GIFTed-Exos. The exosomes (500 μg mL−1) without and with 405 nm violet light irradiation were incubated with HeLa cells for 6 hours at 37°C, followed by washing with PBS, permeabilization, and confocal imaging. Blue: nuclei stained with DAPI; red: mCherry. Scale bars, 20 μm.
Next, cellular uptake of mCherry GIFTed-Exos and intracellular release and distribution of mCherry protein cargo were examined (Figure 3C). Confocal microscopy imaging showed efficient delivery of the mCherry protein cargo into HeLa cells, which is mainly located in the cytosol for cells treated with mCherry GIFTed-Exos without violet light treatment, but translocated into nuclei for cells incubated with mCherry GIFTed-Exos pretreated by violet light, matching the functions of PhoCl and C-terminal NLS in the designed fusion protein. These results support successful loading of the mCherry protein cargo into exosomes, light-controlled release of the mCherry protein cargo, and intracellular delivery of the mCherry protein cargo by GIFTed-Exos.
Inducing cell apoptosis by GIFTed-Exos-mediated delivery of proapoptotic proteins.
The applicability of the GIFTed-Exos-based approach for cytosolic delivery of bioactive proteins was then examined using apoptin, which is also named as chicken anemia virus viral protein 3 (VP3) and reported to possess tumor cell-specific proapoptotic activity [34, 35]. CD9-PhoCl-apoptin fusion construct was generated using the same strategy as described above with a HA tag and a 6×His tag at its N- and C-terminus, respectively (Figure 4A). In addition, the same apoptin expression construct without the fused CD9 and PhoCl (designated as apoptin Exos) was generated for comparison. Immunoblot analysis indicated successful expression of the CD9-PhoCl-Apoptin fusion protein and violet light-dependent release of the apoptin protein cargo from the fusion protein (Figure 4B). In contrast to apoptin Exos, apoptin GIFTed-Exos display significantly increased expression levels of apoptin (Figure S1A), supporting that the CD9-fusion strategy enhances loading of protein cargos into exosomes. Confocal microscopy imaging showed time-dependent uptake of PKH67-labled native exosomes and apoptin GIFTed-Exos in HeLa cells with saturation around 12 hours (Figure S2), consistent with previous reports [36, 37]. No obvious differences in cellular uptake were found between native exosomes and apoptin GIFTed-Exos in HeLa cells (Figure S2).
Figure 4.
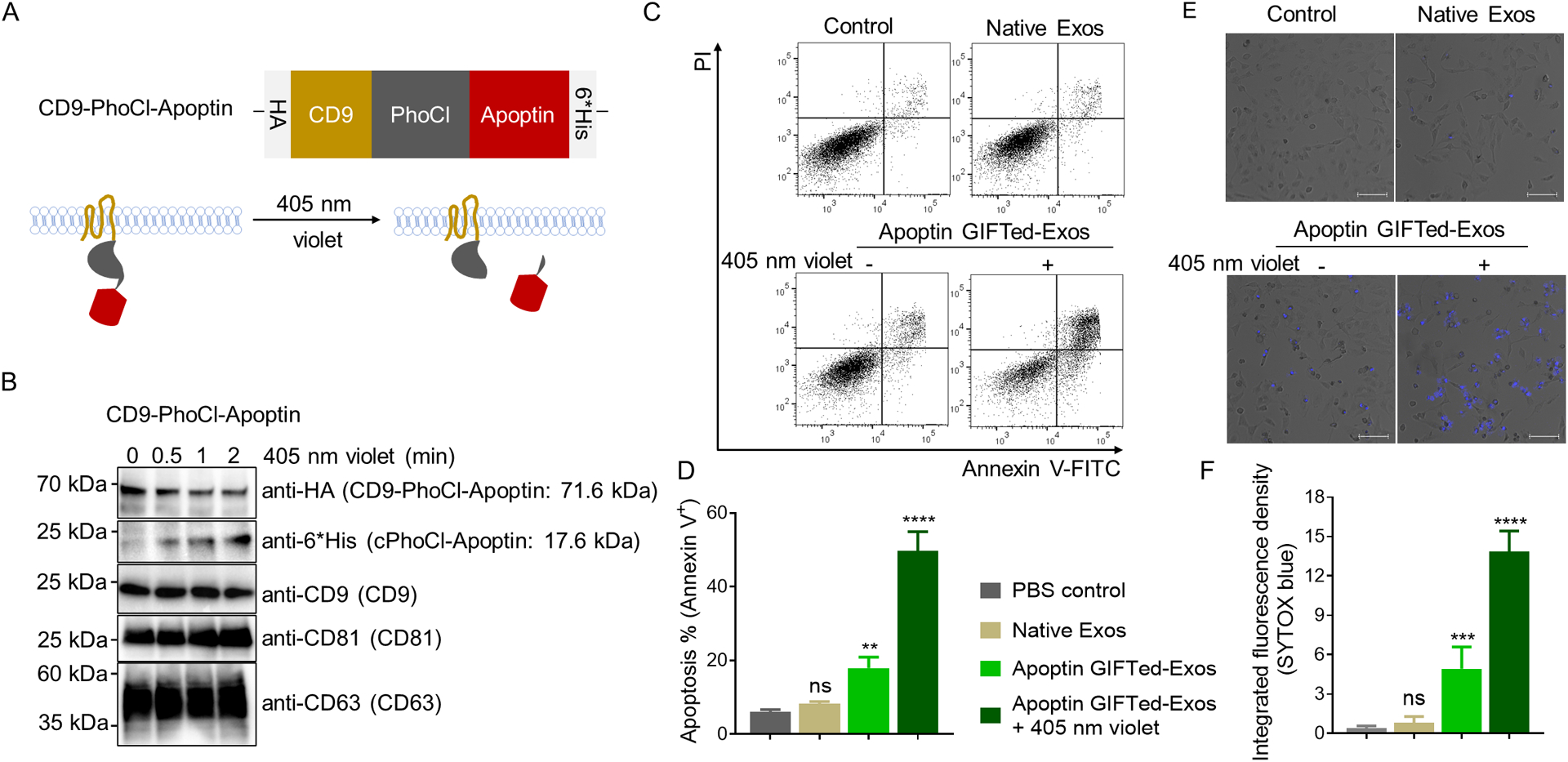
Generation and analysis of apoptin GIFTed-Exos. (A) Schematic of the design of apoptin GIFTed-Exos. (B) Immunoblot analysis of the apoptin GIFTed-Exos and violet light-induced release of apoptin. Theoretical molecular weights of fusion proteins are shown. (C) Representative flow cytometry analysis of cell apoptosis induced by apoptin GIFTed-Exos. HeLa cells were incubated in the absence or presence of native exosomes or apoptin GIFTed-Exos (500 μg mL−1) for 24 hours at 37°C, followed by apoptosis analysis through annexin V-FITC/PI staining. (D) Quantitative representation of the percentages of annexin V-positive apoptotic cells for each group of (C). Data are shown as mean ± SD of triplicates. ns, not significant; ** p < 0.01; **** p < 0.0001. (E) Representative confocal images of cell apoptosis induced by apoptin GIFTed-Exos. Following treatment without and with native or apoptin GIFTed-Exos (500 μg mL−1) for 24 hours at 37°C, HeLa cells were analyzed for apoptosis through SYTOX blue staining. Scale bars: 100 μm (F) Quantitative representation of the percentage of SYTOX blue-positive apoptotic cells for each group of (E) (6 fields of view per region). Data are shown as mean ± SD. ns, not significant; *** p < 0.001; **** p < 0.0001.
Annexin V-FITC/PI-based flow cytometry was then performed to study apoptosis effect induced by apoptin GIFTed-Exos. As shown in Figure 4C, treatment with apoptin GIFTed-Exos pre-exposed to violet light causes a significant increase in cell apoptosis (49.7% ± 5.6%) in HeLa cells relative to those of control and native exosome-treated groups. Apoptin GIFTed-Exos without pre-exposure to violet light could induce less than 20% cell apoptosis, possibly due to inefficient release of apoptin from the exosomes. It was reported that accumulation of apoptin in nuclei of cancer cells is required for induction of apoptosis [38]. In contrast to HeLa cells, no significant cell apoptosis was observed for HEK293 cells in all treatment groups (Figure S3). Consistent with their high levels of apoptin expression, apoptin GIFTed-Exos pre-exposed to violet light induces HeLa cell apoptosis approximately three times higher than that of the apoptin Exos-treated group (16.6% ± 1.3%) (Figure S1B, C). Furthermore, confocal microscopy showed significant apoptosis for HeLa cells incubated with apoptin GIFTed-Exos without and with pre-treatment by violet light, with a higher level for violet light-treated apoptin GIFTed-Exos, and a lack of apoptosis for HEK293 cells treated by all groups, consistent with the flow cytometry results (Figures 4E–F and S3). These results indicate that GIFTed-Exos allow efficient delivery of apoptin for inducing marked apoptosis in cancer cells.
In vitro and in vivo delivery of antioxidant enzymes by GIFTed-Exos.
In addition to mCherry and apoptin, catalase was selected as a model protein cargo for further examining the generality of GIFTed-Exos-based protein delivery approach. By catalyzing decomposition of hydrogen peroxide into water and oxygen, catalase is an important antioxidant enzyme for preventing hepatic ischemia or reperfusion injury by reducing reactive oxygen species (ROS) produced by Kupffer cells and neutrophils infiltrating the organ. CD9-PhoCl-catalase construct was generated using the same approach as described above. A HA tag and a 6×His tag were placed at the N- and C-terminus of the fusion protein, respectively. (Figure 5A). To increase specificity of the resulting catalase GIFTed-Exos for hepatocytes, we co-expressed apolipoprotein A-I (ApoA-I) on the exosome surface (Figure 5A). ApoA-I is a ligand of high density lipoprotein (HDL) scavenger receptor class B type I (SR-BI), which directs transport of cholesterol from cells of the arterial wall to liver and steroidogenic organs [39]. To display ApoA-I on exosome surface, we fused it with the transmembrane domain (TMD) of human platelet-derived growth factor receptor (PDGFR), which has been widely used in displaying peptides and single-chain antibodies on surfaces of mammalian cells [40, 41]. A flexible (GGGGS)3 linker was inserted between these two proteins and a HA tag was added at N-terminus of ApoA-I-PDGFR TMD fusion (Figure 5A).
Figure 5.
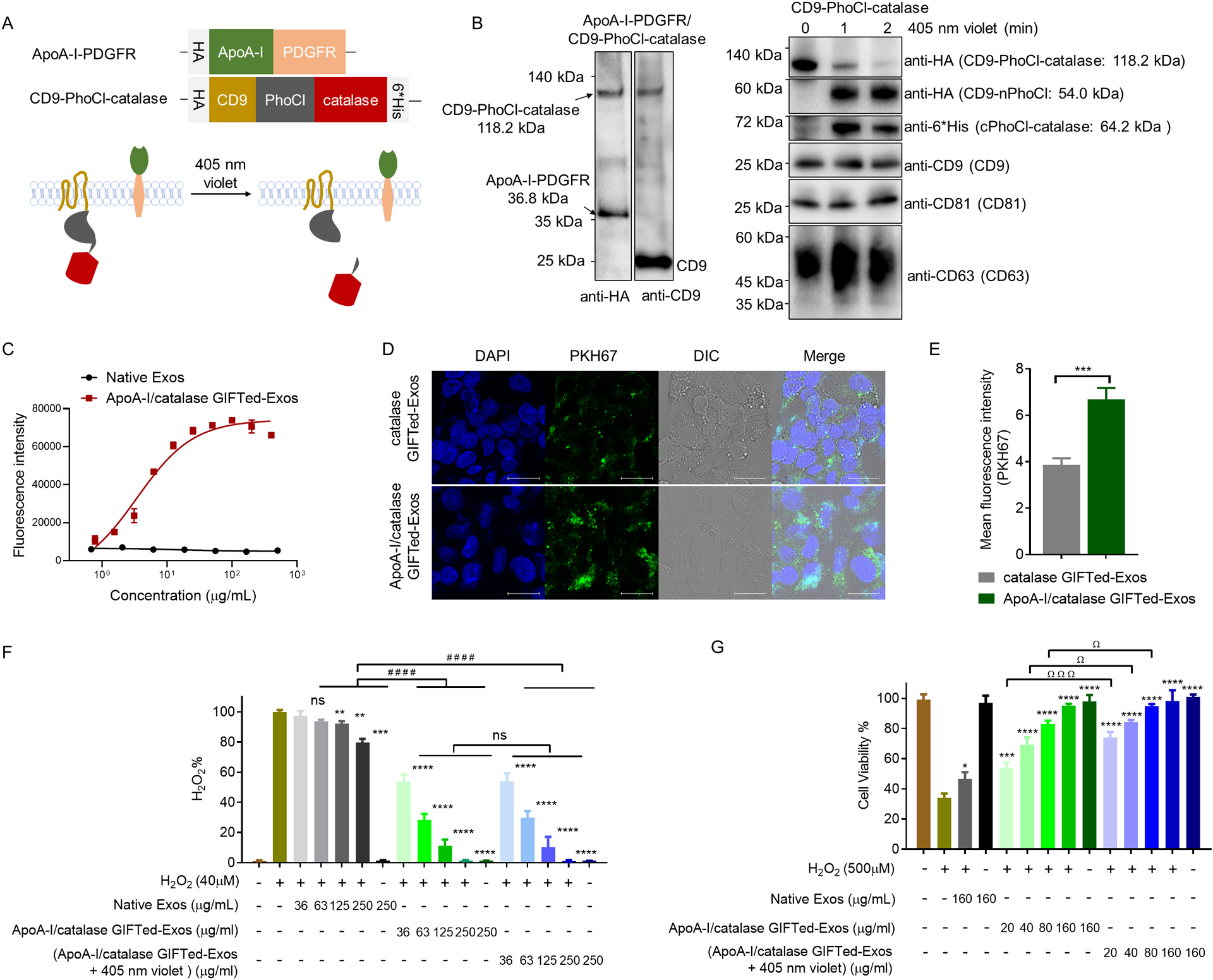
Design, generation, and evaluation of ApoA-I/catalase GIFTed-Exos. (A) Schematic of the design of ApoA-I/catalase GIFTed-Exos. (B) Immunoblot analysis of ApoA-I/catalase GIFTed-Exos and violet light-induced release of catalase. Theoretical molecular weights of fusion proteins are shown. (C) ELISA analysis of binding of ApoA-I/catalase GIFTed-Exos to SR-BI receptor. Data are shown as mean ± SD of duplicates. (D) Representative confocal images of cellular uptake of PKH67-labled GIFTed-Exos. HepG2 cells were treated with catalase GIFTed-Exos or ApoA-I/catalase GIFTed-Exos for 6 hours at 37°C, followed by washing with PBS, permeabilization, and confocal imaging. Blue: nuclei stained with DAPI; green: PKH67-labeled exosomes. Scale bars, 20 μm. (E) Quantitative representation of the fluorescence intensity of PKH67 channel for each group in (D) (6 fields of view per region). Data are shown as mean ± SD. n = 6; ***, p < 0.001. (F) Enzymatic activity of ApoA-I/catalase GIFTed-Exos. The exosomes were exposed to 40 μM of H2O2 for 30 minutes at 37°C. The concentrations of H2O2 were then detected using Amplex red hydrogen peroxide/peroxidase assay kits. (G) ApoA-I/catalase GIFTed-Exos protect HepG2 cells from H2O2-induced cytotoxicity. HepG2 cells were exposed to 500 μM of H2O2 in the absence or presence of native exosomes or ApoA-I/catalase GIFTed-Exos without and with 405 nm violet light treatment for 6 hours at 37°C. Cell viability was measured by MTT assays. Data in (F) and (G) are shown as mean ± SD of triplicates. Symbols indicate relative levels of significance compared with the H2O2 only-treatment group (ns, not significant; *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****,p < 0.0001) or with native exosome-treatment group (#, p < 0.01; ###, p < 0.001; ####, p < 0.0001) or with ApoA-I/catalase GIFTed-Exos-treatment group (ns, not significant; Ω, p < 0.05; ΩΩΩ P < 0.001).
The ApoA-I/catalase GIFTed-Exos were generated by co-transfection of ApoA-I-PDGFR TMD and CD9-PhoCl-catalase expression constructs. Immunoblot analysis indicated that both the ApoA-I-PDGFR TMD and CD9-PhoCl-catalase fusions were successfully expressed in exosomes (Left panel in Figure 5B). Violet light exposure triggers cleavage of CD9-PhoCl-catalase fusion protein in exosomes in a time-dependent fashion, releasing human catalase with the C-terminal 12-residue peptide of PhoCl (cPhoCl-catalase) (Figure 5A and right panel in Figure 5B). ELISA analysis showed that ApoA-I/catalase GIFTed-Exos bind tightly to SR-BI in a dose-dependent manner (Figure 5C). This result was further confirmed by flow cytometric analysis using high SR-BI expression HepG2 cells [42] and low SR-BI expression HeLa cells [43], which revealed strong binding of the ApoA-I/catalase GIFTed-Exos to HepG2 cells and little binding to HeLa cells (Figure S4). Confocal microscopy of cellular uptake of fluorescently labeled exosomes indicated that in comparison to catalase GIFTed-Exos, ApoA-I/catalase GIFTed-Exos exhibited higher levels of accumulation in SR-BI-positive HepG2 cells (Figure 5D and E). These results suggest that surface-displayed ApoA-I may increase specificity of catalase GIFTed-Exos for hepatocytes to facilitate liver-specific delivery of antioxidant enzymes.
The enzymatic activity of ApoA-I/catalase GIFTed-Exos was then evaluated (Figure 5F). Fluorescence-based activity assays indicated that ApoA-I/catalase GIFTed-Exos (36–250 μg mL−1) without and with pre-treatment by violet light could efficiently decompose H2O2 in a dose-dependent fashion. In contrast, native exosomes show weak H2O2-degrading activity, which is consistent with previous findings that exosomes from different cell types can carry antioxidant enzymes involved in ROS scavenging [44, 45].
Next, protective effects of GIFTed-Exos-mediated delivery of catalase on H2O2-induced cytotoxicity were evaluated. HepG2 cells were incubated for 6 hours with 500 μM of H2O2 in the absence or presence of various concentrations of native exosomes or ApoA-I/catalase GIFTed-Exos without and with pre-treatment by violet light. In contrast to H2O2-treated cells with more than 60% loss of cell viability (Figures 5G and S5A), cells incubated with ApoA-I/catalase GIFTed-Exos reveal significantly higher levels of viability which correlate with exosome concentrations. Notably, ApoA-I/catalase GIFTed-Exos confer nearly full protection of HepG2 cells from H2O2-induced cytotoxicity at concentrations of 80–160 μg mL−1, whereas 160 μg mL−1 of native exosomes provide modest protection. Compared with ApoA-I/catalase GIFTed-Exos without violet light exposure, ApoA-I/catalase GIFTed-Exos pre-treated by violet light display increased protecting activities, possibly resulting from improved access to the released catalase cargo upon cellular uptake. Moreover, 1-hour pre-incubation of HepG2 cells with ApoA-I/catalase GIFTed-Exos result in greater protection from H2O2-induced cytotoxicity (Figure S5B).
To examine in vivo activity of ApoA-I/catalase GIFTed-Exos, a murine CCl4-induced liver injury model was used, which has been established to screen hepatoprotective or liver treatment drugs [46]. Upon administration, CCl4 is metabolized by hepatic microsomal cytochrome P450 to produce a highly reactive trichloromethyl radical, followed by the generation of hydrogen peroxide [47, 48]. ApoA-I/catalase GIFTed-Exos are expected to accumulate in liver through ApoA-I-mediated binding to the SR-BI receptor on the surface of hepatocytes, and prevent or ameliorate CCl4-induced liver damage by catalyzing the decomposition of hydrogen peroxide. Relative to corn oil-treated group, CCl4-treated group showed dramatically increased plasma ALT (54.3 ± 15.4 vs 1074.4 ± 157.5 U/L; 19.8-fold) and AST (69.9 ± 38.6 vs 886.0 ± 34.4 U/L; 12.7-fold) activities 24 hours post CCl4 administration. Importantly, mice treated with ApoA-I/catalase GIFTed-Exos right after the CCl4 injection showed significantly reduced ALT and AST activities in plasma, supporting that ApoA-I/catalase GIFTed-Exos deliver effective protection against acute liver injury induced by CCl4 (Figure 6). In addition, mice administered with ApoA-I/catalase GIFTed-Exos alone revealed no significant changes in plasma levels of both ALT and AST relative to those of mice receiving PBS or corn oil, suggesting no apparent hepatotoxicity for ApoA-I/catalase GIFTed-Exos. These results demonstrate in vivo therapeutic efficacy of liver-targeted ApoA-I/catalase GIFTed-Exos.
Figure 6.
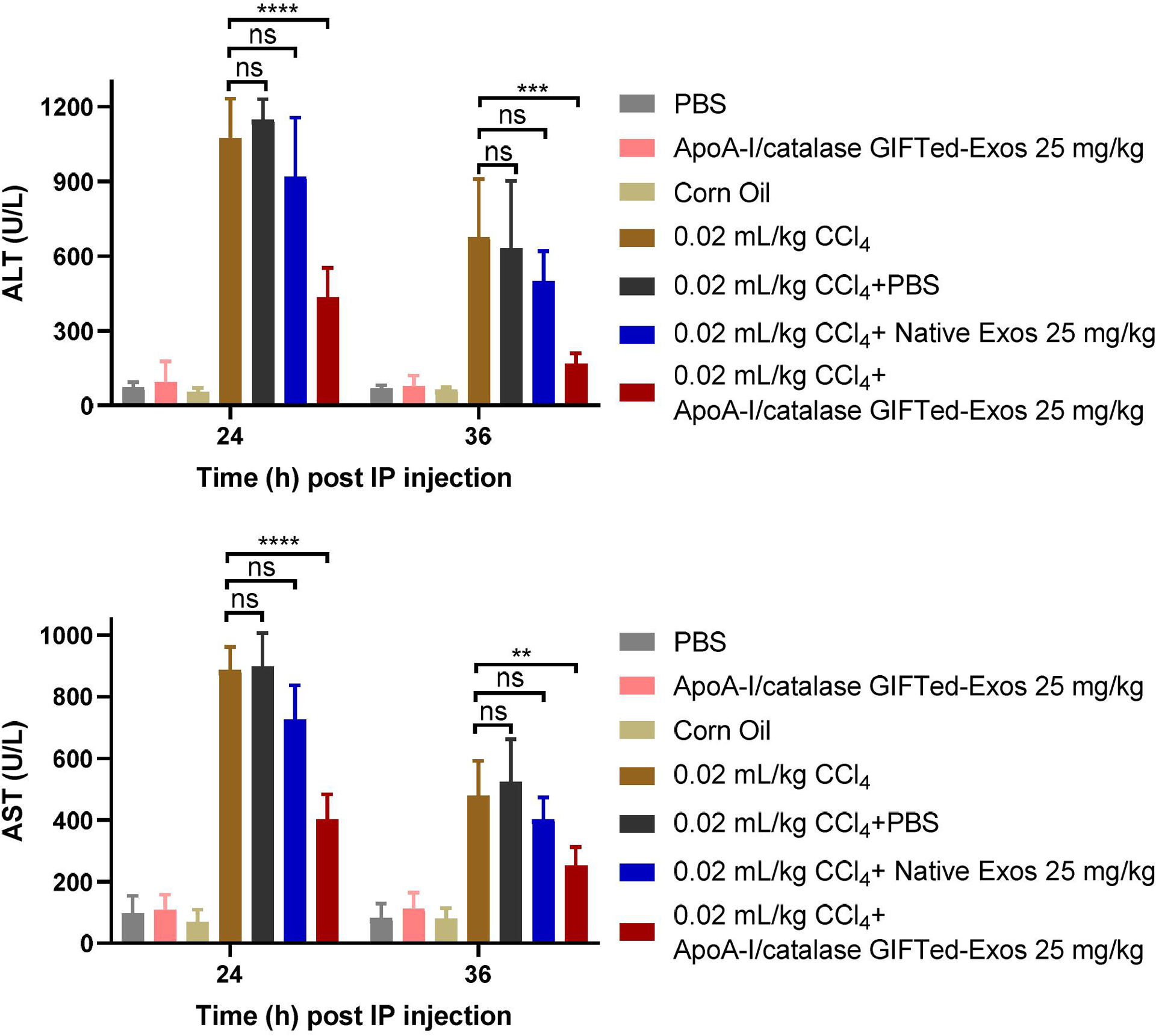
In vivo protective effects of ApoA-I/catalase GIFTed-Exos against CCl4-induced acute liver damage. Mice were intraperitoneally administered with CCl4 (4 mL/kg) dissolved in corn oil (0.5% v/v), followed with immediate intravenous injections of native exosomes or ApoA-I/catalase GIFTed-Exos and two more injections at 6 hours and 12 hours. Enzymatic activities of ALT and AST in plasma were measured 24 hours and 36 hours after CCl4 administration. Value represents mean ± SD (n = 4). ns, not significant; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
Discussion
Through genetic engineering of exosome-associated tetraspanin CD9, we develop a new biocompatible nanoplatform for versatile incorporation of functional proteins of interest. The designed GIFTed-Exos allow to carry considerable amounts of membrane proteins or soluble proteins payloads for potential modulation of target cell functions. Currently, most proteins engineered to exosome surfaces are limited to short peptides, single-chain antibodies, and truncated extracellular domains of transmembrane proteins. And overexpressing exogenous membrane proteins in cells often gives rise to suboptimal expression levels in the isolated exosomes [17, 49, 50]. In comparison, utilizing CD9-based fusions allows to display full-length membrane proteins on exosome surface, which may retain their biological activities. Moreover, fusing with CD9 improves expression levels of target proteins in exosomes. In this study, two type II membrane proteins, CD70 and GITRL, were successfully incorporated into GIFTed-Exos. To expand the utility of this platform, type I membrane proteins and multispanning membrane proteins will be fused to the appropriate terminus of CD9 for creation of GIFTed-Exos with new biological functions. In addition, other exosome-associated proteins may be exploited for generating GIFTed-Exos with potentially improved properties, such as tetraspanins (CD81 and CD63), integrins, and membrane-binding proteins (TSG101 and Alix). The potential impact of the fusion architecture on assembly of the exosomes and the display efficiency of membrane proteins will be further investigated.
To promote loading of protein cargos into exosomes, other genetical engineering approaches were reported, including direct and indirect additions of ubiquitin tags and blue-light-mediated reversible association of heterogeneous dimers [51–53]. Using CD9-PhoCl-based fusions facilitates enrichment of soluble protein cargos in exosomes. Importantly, the fused cargo proteins could be irreversibly released upon transient exposure to violet light and carry a short 12-residue peptide at N-terminus. These designs prevent concentration-dependent reversible association, avoid extended illumination, and minimize impact of genetically attached tags on the structure and activity of protein cargos, leading to the generation of cargo protein-carrying GIFTed-Exos with potentially excellent loading efficiency and biological activities.
In summary, we generated a novel form of engineered exosome-derived protein carrier GIFTed-Exos that enhance incorporation of both membrane proteins and soluble protein cargos into exosomes and protein payloads release. The generated CD70 and GITRL GIFTed-Exos possess notable T-cell co-stimulatory activities and the mCherry, apoptin, and ApoA-I/catalase GIFT-Exos demonstrate effective loading and intracellular delivery of distinct types of protein cargos. GIFTed-Exos may provide a general approach for generating new exosome-based research and therapeutic tools for modulating functions of target cells and tissues.
Experimental Methods
Materials.
Roswell Park Memorial Institute (RPMI) 1640 medium and Dulbecco’s modified Eagle’s medium (DMEM) were purchased from Corning Inc. (Corning, NY). Fetal bovine serum (FBS), Expi293 expression medium, and Opti-modified Eagle’s medium (Opti-MEM) were purchased from Thermo Fisher Scientific (Waltham, MA). Triton X-100, PKH67 green fluorescent cell linker kit, and carbon tetrachloride (CCl4) were purchased from Sigma-Aldrich (St. Louis, MO). Dead cell apoptosis kit with annexin V FITC and propidium iodide (PI), QuantaBlu fluorogenic peroxidase substrate, Coomassie Plus (Bradford) assay kit, SYTOX Blue, and 4′,6-diamidino-2-phenylindole (DAPI) were purchased from Thermo Fisher Scientific (Waltham, MA).
Cell lines.
Human cervical cancer cell line HeLa, human hepatocellular carcinoma cell line HepG2, and human embryonic kidney 293 cells (HEK293) were obtained from the American Type Culture Collection (ATCC) (Manassas, VA) and maintained in DMEM medium supplemented with 10% FBS at 37°C in 5% CO2. Expi293F cells were purchased from Thermo Fisher Scientific (Waltham, MA) and maintained in Expi293 expression medium with shaking at a speed of 125 rpm min−1 at 37°C in 8% CO2. Human peripheral blood mononuclear cells (PBMCs) were purchased from HemaCare (Van Nuys, CA).
Molecular cloning and expression of GIFTed-Exos in mammalian cells.
All cDNA fragments and PCR primers used in this study are listed in Table S1.
Synthetic genes encoding hemagglutinin (HA)-CD9-CD70-6×His and HA-CD9-GITRL-6×His fragments were purchased from Integrated DNA Technologies, Inc. (Skokie, IL), in which a (GGGGS)3 linker and a SalI restriction enzyme site were inserted between CD9 and CD70/GITRL and a BglII restriction enzyme site was inserted between HA-tag and CD9. A pDisplay vector (Thermo Fisher Scientific, Waltham, MA) without the N-terminal signal peptide and the transmembrane (TM) domain of human platelet-derived growth factor receptor (PDGFR) was used as the backbone vector. The amplified HA-CD9-CD70-6×His and HA-CD9-GITRL-6×His fragments were ligated in-frame using T4 DNA ligase (New England Biolabs, MA) between the EcoRI and NotI restriction enzyme sites in the modified pDisplay vector to generate pDisply-CD9-CD70 and pDisplay-CD9-GITRL constructs, respectively. Constructs for pDisplay-CD70 and pDisplay-GITRL were generated by replacing the CD9-CD70/GITRL fragment with amplified CD70 or GITRL insert between the restriction enzyme sites of BglII and NotI of pDisplay-CD9-CD70 and pDisplay-CD9-GITRL vectors.
A PhoCl-mCherry fragment was amplified from pcDNA-NLS-PhoCl-mCherry vector (Addgene plasmid #87691) [24] with a FLAG-tag and an NLS sequence fused at its C-terminus and an MfeI restriction enzyme site between PhoCl and mCherry regions. pDisplay-CD9-PhoCl-mCherry construct was generated by replacing the CD9-CD70 region with amplified PhoCl-mCherry insert between the restriction enzyme sites of SalI and NotI of pDisplay-CD9-CD70 vector.
A synthetic gene fragment encoding Apoptin-6×His was purchased from Integrated DNA Technologies, Inc. pDisplay-CD9-PhoCl-Apoptin was generated by replacing the mCherry-FLAG-NLS fragment with an amplified Apoptin-6×His insert between the restriction enzyme sites of MfeI and NotI.
A catalase DNA fragment was amplified from pcDNA3-Casp3-myc vector (Addgene plasmid #11813) [54] with a 6×His tag at its C-terminus. pDisplay-CD9-PhoCl-Catalase was generated by replacing the mCherry-FLAG-NLS region with an amplified Catalase-6×His insert between the restriction enzyme sites of MfeI and NotI.
A cDNA fragment encoding mouse ApoA-I was purchased from Dharmacon, Inc (Lafayette, CO). The amplified fragment was inserted between the HA-tag and the TM domain of human PDGFR in pDisplay vector using restriction enzyme sites BglII and SalI.
The generated expression vectors were confirmed by DNA sequencing provided by GENEWIZ (South Plainfield, NJ). Endotoxin-free plasmids for the sequence-verified expression constructs were prepared using ZymoPURE II Plasmid Kits (ZYMO Research, CA), followed by transfection into Expi293F cells using PEI MAX 40K (Polysciences, PA) by following manufacturer’s instructions.
Exosome purification.
Culture media of Expi293F cells transfected with the expression constructs were collected on days 3 and 6 post transfection. Exosomes were isolated through differential centrifugation and ultracentrifugation as previously described with modification [33, 55, 56]. Briefly, cell cultures containing expressed exosomes were centrifuged at 100 ×g for 10 minutes at 4°C to pellet suspension cells. The collected supernatants were then centrifuged at 4000 ×g for 30 minutes to remove dead cells and cell debris, followed by 14,000 ×g for 40 minutes at 4°C to remove large vesicles. Collected supernatants were then centrifuged at 60,000 rpm (371,000 ×g) in a Type 70 Ti rotor (Beckman Instruments, Indianapolis, IN) for 1.5 hours at 4°C. Exosome pellets were washed twice with PBS and resuspended in PBS, followed by filtration with 0.2 μm syringe filters. Protein concentrations of the exosomes were determined by Bradford assays.
Nanoparticle tracking analysis (NTA).
The particle concentration and size distribution of the purified exosomes were determined through NTA using a Nanosight LM10 (Malvern Instruments, U.K.) by following the manufacturer’s instruction. Ten replicates of analysis with 60 seconds for each were performed.
Immunoblot analysis.
Exosome samples (2 μg of protein) were boiled at 95°C for 5 min in NuPAGE LDS sample buffer (Thermo Fisher Scientific, MA). The lysates were then separated by 4–20% ExpressPlus-PAGE gels (GeneScript, Piscataway, NJ), transferred to Immun-Blot PVDF membranes (Bio-Rad Laboratories, Inc, Hercules, CA), and incubated with appropriate primary antibodies (anti-HA (clone: 2–2.2.14), anti-6×His (clone: HIS.H8) and anti-FLAG (FG4R) from Thermo Fisher Scientific, anti-CD63 (clone: H5C6) and anti-CD81 (clone: 5A6) from BioLegend, and anti-CD9 (clone: D8O1A) from Cell Signaling Technology) and secondary antibodies (anti-mouse IgG-HRP (catalog number: 62–6520) or anti-rabbit IgG-HRP (catalog number: 65–6120) from Thermo Fisher Scientific). The immunoblots were developed by additions of SuperSignal West Pico PLUS chemiluminescent substrate (Thermo Fisher Scientific) and imaged with a ChemiDoc Touch Imaging System (Bio-Rad Laboratories, Inc, Hercules, CA).
Yield of positive GIFTed-Exos.
The yield of positive exosomes was evaluated through Ni-NTA affinity chromatography by using the GITRL GIFTed-Exo as an example. GITRL GIFTed-Exos were incubated with Ni-NTA beads at 4°C with end-over-end rotation for 2 hours, followed by washing beads twice with PBS buffer and eluting with PBS buffer containing 400 mM imidazole. The yield of positive exosomes was defined as eluted GITRL GIFTed-Exos divided by total GITRL GIFTed-Exos and expressed as a percentage.
Binding of the engineered exosomes to their ligands/receptors as measured by ELISA.
Nighty six-well ELISA plates (Greiner Bio-One, Monroe, NC) were coated with appropriate ligands/receptors (0.4 μg mL−1; human CD27-rFc, human GITR-hFc, and mouse SCARB1-hFc/6×His from Sino Biological Inc., Wayne, PA) or native exosomes at various concentrations overnight at room temperature or 4°C. Non-bound antigens or exosomes were washed away with PBST (PBS with 0.05% Tween-20) for three times. The wells were then blocked with PBS containing 1% BSA for 2 hours at room temperature, followed by washing with PBST. Various concentrations of exosomes (CD70-Exos, GITRL-Exos, CD70 GIFTed-Exos, GITRL GIFTed-Exos, and ApoA-I/Catalase GIFTed-Exos) or corresponding ligands/receptors (0.5 μg mL−1; human CD27-rFc, human GITR-hFc, and mouse SCARB1-hFc/6×His) were added and incubated for 2 hours at room temperature, followed by washing with PBST for three times. For bound engineered exosomes, an anti-HA primary antibody was subsequently added for 2-hour incubation, followed by washing and incubation with an anti-mouse IgG-HRP secondary antibody for 1 hour. For bound ligands/receptors, an anti-rabbit IgG-HRP antibody or anti-human IgG-HRP antibody was added and incubated for 1 hour. QuantaBlu fluorogenic peroxidase substrate (Thermo Fisher Scientific, MA) was then added after washing, and the fluorescence signals were measured using a BioTek Synergy H1 Hybrid Multi-Mode Microplate reader (BioTek, VT).
T-cell co-stimulation assays.
Anti-CD3 monoclonal antibody (mAb) (clone: OKT3, BioLegend, San Diego, CA) (10 μg mL−1) was pre-coated in pre-treated 96-well plates at 37°C for 3 hours. Human PBMCs (1 ×105 per well) incubated in complete RPMI and purified CD70 GIFTed-Exos or GITRL GIFTed-Exos at various concentration were added. As controls, PBMCs were cultured alone with the immobilized anti-CD3 mAb or native exosomes. After 40 hours, cell culture supernatants were collected and assayed for interleukin-2 (IL-2) and interferon gamma (IFN-γ) secretion by ELISA (R&D Systems, Minneapolis, MN). Results are expressed as a mean ± SD from one of at least three separate experiments.
Confocal imaging of cell uptake of exosomes and intracellular distribution of cargo proteins.
HeLa or HepG2 cells were seeded into clear bottoms of 24-well plates (50,000 cells/well) one day before the experiment. HeLa cells were incubated with 500 μg mL−1 mCherry GIFTed-Exos (1.4×1010 particles), mCherry GIFTed-Exos pre-treated with 405 nm violet (405-nm blue laser module (Output Power: 500 mW), Ensfouy, China) for 2 minutes, or native exosomes for 6 hours. HepG2 cells were incubated with 200 μg mL−1 PKH67-labeled catalase GIFTed-Exos or ApoA-I/catalase GIFTed-Exos (5.6×109 particles) for 6 hours at 37°C with 5% CO2. The cells were then gently washed with PBS, fixed with 4% paraformaldehyde (Thermo Fisher Scientific, MA), permeabilized with 0.1% triton X-100 and stained with DAPI. Cells were then washed and mounted on slides in fluromount mounting media (Diagnostic BioSystems Inc., Pleasanton, CA). Images were analyzed with a Leica SP8 confocal laser scanning microscope (Leica Microsystems Inc., Buffalo Grove, IL) and processed using LAS X software (Leica Microsystems Inc., Buffalo Grove, IL).
Confocal imaging of cellular uptake of native exosomes and GIFTed-Exos.
HeLa cells were seeded into clear bottoms of 24-well plates (50,000 cells/well) one day before the experiment. HeLa cells were incubated with 200 μg mL−1 PKH67-labeled native exosomes or apoptin GIFTed-Exos (5.6×109 particles) for 1, 2, 6, 12, or 24 hours at 37°C with 5% CO2. The cells were then gently washed with PBS, fixed with 4% paraformaldehyde (Thermo Fisher Scientific, MA), permeabilized with 0.1% triton X-100 and stained with DAPI. Cells were then washed and mounted on slides in fluromount mounting media (Diagnostic BioSystems Inc., Pleasanton, CA). Images were analyzed with a Zeiss LSM880 confocal microscope with Airyscan (Carl Zeiss AG, Oberkochen, Germany) and processed using LAS X software (Leica Microsystems Inc., Buffalo Grove, IL).
Apoptosis assays.
HeLa or HEK293 cells were incubated with 500 μg mL−1 Apoptin Exos, Apoptin GIFTed-Exos, Apoptin GIFTed-Exos pre-treated with 405 nm violet for 2 minutes, or native exosomes for 24 hours at 37°C with 5% CO2. Cell apoptosis was detected using the Annexin V-FITC/PI apoptosis detection kit (Thermo Fisher Scientific, MA) and SYTOX Blue dead cell stain (Thermo Fisher Scientific, MA) according to the manufacturer’s instructions, followed by analyzing on a flow cytometer or imaging with a Leica SP8 confocal laser scanning microscope (Leica Microsystems Inc., Buffalo Grove, IL), respectively.
Flow cytometric analysis of the binding of ApoA-I/catalase GIFTed-Exos to cells.
SR-BI-high-expression HepG2 cells and SR-BI-low-expression HeLa cells (200,000 cells per tube) were stained with 200 μg mL−1 of PKH67-labled native exosomes or ApoA-I/catalase GIFTed-Exos for 30 minutes at 4°C. Cells were washed three times with PBS containing 2% FBS and resuspended in PBS containing 2% FBS, followed by analysis using a BD Fortessa X20 flow cytometer. Data were processed by FlowJo_V10 software (Tree Star Inc., Ashland, OR).
Enzymatic activity of ApoA-I/catalase GIFTed-Exos.
In vitro enzymatic activity of ApoA-I/catalase GIFTed-Exos was evaluated using hydrogen peroxide (H2O2) decomposition assay with Amplex Red hydrogen peroxide/peroxidase assay Kit (Thermo Fisher Scientific, MA). For this purpose, native exosomes, ApoA-I/catalase GIFTed-Exos, or ApoA-I/catalase GIFTed-Exos pre-treated with 405 nm violet were incubated with 40 μM H2O2 for 30 minutes at 37°C. Following the incubation, the remaining H2O2 was detected according to manufacturer’s instructions. The catalase enzymatic activity was expressed as the percentage of H2O2 decomposition.
Prevention of H2O2-induced cytotoxicity by ApoA-I/catalase GIFTed-Exos.
H2O2-mediated liver cell cytotoxicity was first evaluated by MTT assays. HepG2 cells (1×104 cells per well) were seeded into clear bottoms of 96-well plates and allowed to attach overnight. Then, the cells were exposed to various concentration of H2O2 for 6 hours. Following the treatments, 10 μL MTT (5 mg mL−1) was added into each well. After 3 hours of incubation at 37°C and subsequent additions of 100 μL of lysis buffer (20% SDS in 50% dimethylformamide, pH 4.7), plates were incubated for 2 hours at 37°C and measured for absorbance at 570 nm using a BioTek Synergy H1 Hybrid Multi-Mode Microplate reader (BioTek, Winooski, VT). Cell viability was expressed as a percentage of viable cells in the treated groups compared to the untreated control group.
The protection of HepG2 cells from H2O2-induced cytotoxicity by ApoA-I/catalase GIFTed-Exos was assessed by MTT assays. HepG2 cells (1 × 104 cells per well) were seeded into 96-well plates and allowed to attach overnight. Cells were treated with ApoA-I/catalase GIFTed-Exos or native exosomes in the presence of 500 μM H2O2 for 6 hours. In separate experiments, cells were first pre-incubated with exosomes for 1 hour before exposing to 500 μM H2O2 for 6 hours. Following the treatments, cell viability was determined as described above.
In vivo activity studies of ApoA-I/catalase GIFTed-Exos.
Six to eight-week male BALB/cJ mice were purchased from The Jackson Laboratory (Bar Harbor, ME). All animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Southern California.
CCl4 dissolved in corn oil (0.5% v/v) was administered to the peritoneal cavity of mice at a dose of 4 mL/kg body weight to induce acute liver damage [46, 57]. Mice in the control group were treated with same volume of corn oil. To examine the hepatoprotective effect of exosomes, ApoA-I/catalase GIFTed-Exos pre-exposed to 405 nm violet for 2 minutes (25 mg/kg) were injected intravenously into the tail vein of mice immediately and 6 hours and 12 hours after CCl4 administration. Mice in the treatment control groups were administered with same dose of native exosomes or PBS at the same time points. In addition, mice injected with same dose of ApoA-I/catalase GIFTed-Exos or PBS alone were used to evaluate systemic toxicity of ApoA-I/catalase GIFTed-Exos. At 24 hours and 36 hours post CCl4 administration, blood samples were collected from the tail vein and plasma was separated by centrifugation for 10 minutes at 10,000 ×g.
The alanine aminotransferase (ALT) and aspartate Aminotransferase (AST) activities in plasma were assayed. Briefly, collected mouse plasma (5 μL) or a series of dilutions of standard solution (sodium pyruvate) was added to wells of clear 96-well plates, followed by additions of 25 μL of ALT substrate solution (0.2 M alanine, 2 mM 2-oxoglutarate, pH 7.4) or 25 μL AST substrate solution (0.1 M aspartic acid, 2 mM 2-oxoglutarate, pH 7.4) and incubation at 37°C for 20 minutes. Next, 50 μL of 2, 4-dinitrophenylhydrazine (2,4-DNPH) (solution of 1 mM in 1 M HCl) was added and incubated at room temperature for 20 minutes. Lastly, 0.5 M sodium hydroxide was added, and the absorbance was measured at 510 nm using a BioTek Synergy H1 Hybrid Multi-Mode Microplate reader (BioTek, VT). Amounts of pyruvate generated were calculated from the standard. The ALT/AST activity was reported as nmole/min/mL = unit/L, where one milliunit (mU) of ALT/AST is defined as the amount of enzyme that can generate one nmole of pyruvate per minute at 37°C.
Statistical analysis.
Two-tailed unpaired t tests were performed for comparison between two groups. One-way ANOVA with Tukey post-hoc tests were carried out for comparing multiple groups. A p < 0.05 was considered statistically significant. Significance of finding was defined as: ns = not significant, p > 0.05; *, p < 0.05; **, p < 0.01; ***, p < 0.001 and ****, p < 0.0001. Data are shown as mean ± SD. All statistical analyses were calculated using GraphPad Prism (GraphPad Software, La Jolla, CA).
Supplementary Material
Acknowledgments
This work was supported in part by STOP CANCER Research Career Development Award (to Y.Z.), Department of Defense CDMRP Career Development Award W81XWH-19-1-0272 (to Y.Z.), California Breast Cancer Research Program Innovative Development and Exploratory Award 25IB-0080 (to Y.Z.), Tobacco Related-Disease Research Program New Investigator Award T30KT1021 (to Y.Z.), National Institute of Biomedical Imaging and Bioengineering (NIBIB) of the National Institutes of Health (NIH) grant R01EB031830 (to Y. Z.), NIH grant P30CA014089 to the USC Norris Comprehensive Cancer Center, and NIH grant P30DK048522 to the USC Research Center for Liver Diseases.
Footnotes
Competing interests
The authors declare no competing interests.
Data Availability
The raw/processed data required to reproduce these findings are available from the authors upon request.
References
- [1].Kalluri R, LeBleu VS, The biology, function, and biomedical applications of exosomes, Science 367(6478) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Pegtel DM, Gould SJ, Exosomes, Annual Review of Biochemistry 88(1) (2019) 487–514. [DOI] [PubMed] [Google Scholar]
- [3].Johnsen KB, Gudbergsson JM, Skov MN, Pilgaard L, Moos T, Duroux M, A comprehensive overview of exosomes as drug delivery vehicles - endogenous nanocarriers for targeted cancer therapy, Biochim. Biophys. Acta 1846(1) (2014) 75–87. [DOI] [PubMed] [Google Scholar]
- [4].Prada I, Meldolesi J, Binding and Fusion of Extracellular Vesicles to the Plasma Membrane of Their Cell Targets, Int J Mol Sci 17(8) (2016) 1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Tian T, Zhu Y-L, Hu F-H, Wang Y-Y, Huang N-P, Xiao Z-D, Dynamics of exosome internalization and trafficking, Journal of Cellular Physiology 228(7) (2013) 1487–1495. [DOI] [PubMed] [Google Scholar]
- [6].Raposo G, Stoorvogel W, Extracellular vesicles: Exosomes, microvesicles, and friends, Journal of Cell Biology 200(4) (2013) 373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Colombo M, Raposo G, Thery C, Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles, Annu. Rev. Cell Dev. Biol 30 (2014) 255–89. [DOI] [PubMed] [Google Scholar]
- [8].van den Boorn JG, Schlee M, Coch C, Hartmann G, SiRNA delivery with exosome nanoparticles, Nat. Biotechnol 29(4) (2011) 325–6. [DOI] [PubMed] [Google Scholar]
- [9].Li J, Wang Y, Liang R, An X, Wang K, Shen G, Tu Y, Zhu J, Tao J, Recent advances in targeted nanoparticles drug delivery to melanoma, Nanomedicine : nanotechnology, biology, and medicine 11(3) (2015) 769–94. [DOI] [PubMed] [Google Scholar]
- [10].Torchilin VP, Multifunctional, stimuli-sensitive nanoparticulate systems for drug delivery, Nature reviews. Drug discovery 13(11) (2014) 813–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJ, Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes, Nat. Biotechnol 29(4) (2011) 341–5. [DOI] [PubMed] [Google Scholar]
- [12].Saeedi S, Israel S, Nagy C, Turecki G, The emerging role of exosomes in mental disorders, Translational Psychiatry 9(1) (2019) 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Robbins PD, Morelli AE, Regulation of immune responses by extracellular vesicles, Nature Reviews Immunology 14(3) (2014) 195–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Yan Y, Jiang W, Tan Y, Zou S, Zhang H, Mao F, Gong A, Qian H, Xu W, hucMSC Exosome-Derived GPX1 Is Required for the Recovery of Hepatic Oxidant Injury, Mol Ther 25(2) (2017) 465–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Rani S, Ritter T, The Exosome - A Naturally Secreted Nanoparticle and its Application to Wound Healing, Adv Mater 28(27) (2016) 5542–52. [DOI] [PubMed] [Google Scholar]
- [16].Zhu Q, Ling X, Yang Y, Zhang J, Li Q, Niu X, Hu G, Chen B, Li H, Wang Y, Deng Z, Embryonic Stem Cells-Derived Exosomes Endowed with Targeting Properties as Chemotherapeutics Delivery Vehicles for Glioblastoma Therapy, Advanced Science 6(6) (2019) 1801899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kamerkar S, LeBleu VS, Sugimoto H, Yang S, Ruivo CF, Melo SA, Lee JJ, Kalluri R, Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer, Nature 546(7659) (2017) 498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zhuang X, Xiang X, Grizzle W, Sun D, Zhang S, Axtell RC, Ju S, Mu J, Zhang L, Steinman L, Miller D, Zhang HG, Treatment of brain inflammatory diseases by delivering exosome encapsulated anti-inflammatory drugs from the nasal region to the brain, Molecular therapy : the journal of the American Society of Gene Therapy 19(10) (2011) 1769–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].El-Andaloussi S, Lee Y, Lakhal-Littleton S, Li J, Seow Y, Gardiner C, Alvarez-Erviti L, Sargent IL, Wood MJA, Exosome-mediated delivery of siRNA in vitro and in vivo, Nature Protocols 7(12) (2012) 2112–2126. [DOI] [PubMed] [Google Scholar]
- [20].Kim MS, Haney MJ, Zhao Y, Mahajan V, Deygen I, Klyachko NL, Inskoe E, Piroyan A, Sokolsky M, Okolie O, Hingtgen SD, Kabanov AV, Batrakova EV, Development of exosome-encapsulated paclitaxel to overcome MDR in cancer cells, Nanomedicine 12(3) (2016) 655–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Fuhrmann G, Serio A, Mazo M, Nair R, Stevens MM, Active loading into extracellular vesicles significantly improves the cellular uptake and photodynamic effect of porphyrins, Journal of Controlled Release 205 (2015) 35–44. [DOI] [PubMed] [Google Scholar]
- [22].Sato YT, Umezaki K, Sawada S, Mukai S.-a., Sasaki Y, Harada N, Shiku H, Akiyoshi K, Engineering hybrid exosomes by membrane fusion with liposomes, Sci Rep 6 (2016) 21933–21933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Donoso-Quezada J, Ayala-Mar S, González-Valdez J, State-of-the-art exosome loading and functionalization techniques for enhanced therapeutics: a review, Critical Reviews in Biotechnology 40(6) (2020) 804–820. [DOI] [PubMed] [Google Scholar]
- [24].Zhang W, Lohman AW, Zhuravlova Y, Lu X, Wiens MD, Hoi H, Yaganoglu S, Mohr MA, Kitova EN, Klassen JS, Pantazis P, Thompson RJ, Campbell RE, Optogenetic control with a photocleavable protein, PhoCl, Nature Methods 14(4) (2017) 391–394. [DOI] [PubMed] [Google Scholar]
- [25].Keller AM, Xiao Y, Peperzak V, Naik SH, Borst J, Costimulatory ligand CD70 allows induction of CD8+ T-cell immunity by immature dendritic cells in a vaccination setting, Blood 113(21) (2009) 5167–5175. [DOI] [PubMed] [Google Scholar]
- [26].Keller AM, Groothuis TA, Veraar EAM, Marsman M, de Buy Wenniger LM, Janssen H, Neefjes J, Borst J, Costimulatory ligand CD70 is delivered to the immunological synapse by shared intracellular trafficking with MHC class II molecules, Proceedings of the National Academy of Sciences 104(14) (2007) 5989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kobata T, Jacquot S, Kozlowski S, Agematsu K, Schlossman SF, Morimoto C, CD27-CD70 interactions regulate B-cell activation by T cells, Proceedings of the National Academy of Sciences 92(24) (1995) 11249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].van de Ven K, Borst J, Targeting the T-cell co-stimulatory CD27/CD70 pathway in cancer immunotherapy: rationale and potential, Immunotherapy 7(6) (2015) 655–67. [DOI] [PubMed] [Google Scholar]
- [29].Rowley TF, Al-Shamkhani A, Stimulation by Soluble CD70 Promotes Strong Primary and Secondary CD8+ Cytotoxic T Cell Responses In Vivo, The Journal of Immunology 172(10) (2004) 6039. [DOI] [PubMed] [Google Scholar]
- [30].Stephens GL, McHugh RS, Whitters MJ, Young DA, Luxenberg D, Carreno BM, Collins M, Shevach EM, Engagement of Glucocorticoid-Induced TNFR Family-Related Receptor on Effector T Cells by its Ligand Mediates Resistance to Suppression by CD4+CD25+ T Cells, The Journal of Immunology 173(8) (2004) 5008. [DOI] [PubMed] [Google Scholar]
- [31].Cohen AD, Diab A, Perales MA, Wolchok JD, Rizzuto G, Merghoub T, Huggins D, Liu C, Turk MJ, Restifo NP, Sakaguchi S, Houghton AN, Agonist anti-GITR antibody enhances vaccine-induced CD8(+) T-cell responses and tumor immunity, Cancer Res 66(9) (2006) 4904–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Hu P, Arias RS, Sadun RE, Nien Y-C, Zhang N, Sabzevari H, Lutsiak MEC, Khawli LA, Epstein AL, Construction and Preclinical Characterization of Fc-mGITRL for the Immunotherapy of Cancer, Clinical Cancer Research 14(2) (2008) 579. [DOI] [PubMed] [Google Scholar]
- [33].Cheng Q, Shi X, Zhang Y, Reprogramming Exosomes for Immunotherapy, Methods Mol. Biol 2097 (2020) 197–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Danen-Van Oorschot AA, Fischer DF, Grimbergen JM, Klein B, Zhuang S, Falkenburg JH, Backendorf C, Quax PH, Van der Eb AJ, Noteborn MH, Apoptin induces apoptosis in human transformed and malignant cells but not in normal cells, Proc Natl Acad Sci U S A 94(11) (1997) 5843–5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Guelen L, Paterson H, Gäken J, Meyers M, Farzaneh F, Tavassoli M, TAT-apoptin is efficiently delivered and induces apoptosis in cancer cells, Oncogene 23(5) (2004) 1153–1165. [DOI] [PubMed] [Google Scholar]
- [36].Franzen CA, Simms PE, Van Huis AF, Foreman KE, Kuo PC, Gupta GN, Characterization of Uptake and Internalization of Exosomes by Bladder Cancer Cells, BioMed Research International 2014 (2014) 619829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Xu L, Faruqu FN, Liam-or R, Abu Abed O, Li D, Venner K, Errington RJ, Summers H, Wang JT-W, Al-Jamal KT, Design of experiment (DoE)-driven in vitro and in vivo uptake studies of exosomes for pancreatic cancer delivery enabled by copper-free click chemistry-based labelling, Journal of Extracellular Vesicles 9(1) (2020) 1779458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Poon IK, Oro C, Dias MM, Zhang J, Jans DA, Apoptin nuclear accumulation is modulated by a CRM1-recognized nuclear export signal that is active in normal but not in tumor cells, Cancer Res 65(16) (2005) 7059–64. [DOI] [PubMed] [Google Scholar]
- [39].Kim SI, Shin D, Choi TH, Lee JC, Cheon GJ, Kim KY, Park M, Kim M, Systemic and specific delivery of small interfering RNAs to the liver mediated by apolipoprotein A-I, Molecular therapy : the journal of the American Society of Gene Therapy 15(6) (2007) 1145–52. [DOI] [PubMed] [Google Scholar]
- [40].Ho M, Nagata S, Pastan I, Isolation of anti-CD22 Fv with high affinity by Fv display on human cells, Proc. Natl. Acad. Sci. U.S.A 103(25) (2006) 9637–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Beerli RR, Bauer M, Buser RB, Gwerder M, Muntwiler S, Maurer P, Saudan P, Bachmann MF, Isolation of human monoclonal antibodies by mammalian cell display, Proc. Natl. Acad. Sci. U.S.A 105(38) (2008) 14336–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Rhainds D, Bourgeois P, Bourret G, Huard K, Falstrault L, Brissette L, Localization and regulation of SR-BI in membrane rafts of HepG2 cells, J Cell Sci 117(Pt 15) (2004) 3095–105. [DOI] [PubMed] [Google Scholar]
- [43].Yang X-P, Amar MJ, Vaisman B, Bocharov AV, Vishnyakova TG, Freeman LA, Kurlander RJ, Patterson AP, Becker LC, Remaley AT, Scavenger receptor-BI is a receptor for lipoprotein(a), J Lipid Res 54(9) (2013) 2450–2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Bodega G, Alique M, Puebla L, Carracedo J, Ramirez RM, Microvesicles: ROS scavengers and ROS producers, Journal of extracellular vesicles 8(1) (2019) 1626654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Bonafede R, Brandi J, Manfredi M, Scambi I, Schiaffino L, Merigo F, Turano E, Bonetti B, Marengo E, Cecconi D, Mariotti R, The Anti-Apoptotic Effect of ASC-Exosomes in an In Vitro ALS Model and Their Proteomic Analysis, Cells 8(9) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Scholten D, Trebicka J, Liedtke C, Weiskirchen R, The carbon tetrachloride model in mice, Lab Anim 49(1 Suppl) (2015) 4–11. [DOI] [PubMed] [Google Scholar]
- [47].Nishikawa M, Hashida M, Takakura Y, Catalase delivery for inhibiting ROS-mediated tissue injury and tumor metastasis, Adv Drug Deliv Rev 61(4) (2009) 319–26. [DOI] [PubMed] [Google Scholar]
- [48].Wang D-H, Ishii K, Zhen L-X, Taketa K, Enhanced liver injury in acatalasemic mice following exposure to carbon tetrachloride, Archives of Toxicology 70(3) (1996) 189–194. [DOI] [PubMed] [Google Scholar]
- [49].Yuan Z, Kolluri KK, Gowers KHC, Janes SM, TRAIL delivery by MSC-derived extracellular vesicles is an effective anticancer therapy, Journal of extracellular vesicles 6(1) (2017) 1265291–1265291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Yang Y, Hong Y, Nam G-H, Chung JH, Koh E, Kim I-S, Virus-Mimetic Fusogenic Exosomes for Direct Delivery of Integral Membrane Proteins to Target Cell Membranes, Advanced Materials 29(13) (2017) 1605604. [DOI] [PubMed] [Google Scholar]
- [51].Cheng Y, Schorey JS, Targeting soluble proteins to exosomes using a ubiquitin tag, Biotechnol Bioeng 113(6) (2016) 1315–24. [DOI] [PubMed] [Google Scholar]
- [52].Sterzenbach U, Putz U, Low LH, Silke J, Tan SS, Howitt J, Engineered Exosomes as Vehicles for Biologically Active Proteins, Mol Ther 25(6) (2017) 1269–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Yim N, Ryu SW, Choi K, Lee KR, Lee S, Choi H, Kim J, Shaker MR, Sun W, Park JH, Kim D, Heo WD, Choi C, Exosome engineering for efficient intracellular delivery of soluble proteins using optically reversible protein-protein interaction module, Nat Commun 7 (2016) 12277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Stennicke HR, Salvesen GS, Biochemical characteristics of caspases-3, -6, -7, and -8, J Biol Chem 272(41) (1997) 25719–23. [DOI] [PubMed] [Google Scholar]
- [55].Cheng Q, Shi X, Han M, Smbatyan G, Lenz HJ, Zhang Y, Reprogramming Exosomes as Nanoscale Controllers of Cellular Immunity, J Am Chem Soc 140(48) (2018) 16413–16417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Shi X, Cheng Q, Zhang Y, Reprogramming extracellular vesicles with engineered proteins, Methods 177 (2020) 95–102. [DOI] [PubMed] [Google Scholar]
- [57].Ma SF, Nishikawa M, Katsumi H, Yamashita F, Hashida M, Liver targeting of catalase by cationization for prevention of acute liver failure in mice, J Control Release 110(2) (2006) 273–282. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw/processed data required to reproduce these findings are available from the authors upon request.