

Abstract
Liquid–liquid phase separation (LLPS) contributes to the spatial and functional segregation of molecular processes within the cell nucleus. However, the role played by LLPS in chromatin folding in living cells remains unclear. Here, using stochastic optical reconstruction microscopy (STORM) and Hi-C techniques, we studied the effects of 1,6-hexanediol (1,6-HD)-mediated LLPS disruption/modulation on higher-order chromatin organization in living cells. We found that 1,6-HD treatment caused the enlargement of nucleosome clutches and their more uniform distribution in the nuclear space. At a megabase-scale, chromatin underwent moderate but irreversible perturbations that resulted in the partial mixing of A and B compartments. The removal of 1,6-HD from the culture medium did not allow chromatin to acquire initial configurations, and resulted in more compact repressed chromatin than in untreated cells. 1,6-HD treatment also weakened enhancer-promoter interactions and TAD insulation but did not considerably affect CTCF-dependent loops. Our results suggest that 1,6-HD-sensitive LLPS plays a limited role in chromatin spatial organization by constraining its folding patterns and facilitating compartmentalization at different levels.
INTRODUCTION
The modern concept of hierarchical chromatin folding in the eukaryotic cell nucleus is based on the results of Hi-C analyses (1). Eukaryotic chromosomes are partitioned into semi-independent topologically associating domains (TADs) (2,3), which are typically composed of chromatin loops (4). Low-resolution analyses have demonstrated that active and repressed chromatin are spatially segregated into A and B chromatin compartments, respectively (1), which are comprised of smaller compartmental domains (5). Recent evidence suggests that the basic spatial organization of the genome relies on the interplay between active DNA loop extrusion and the passive spatial segregation of chromatin domains enriched in particular sets of epigenetic marks (such as active and inactive chromatin domains) (5–8). Although the DNA loop extrusion machinery has been properly characterized (9,10), the processes/forces that mediate the spatial segregation of active and inactive chromatin remain poorly understood. Recent studies have disclosed the important role played by liquid–liquid phase separation (LLPS) in the functional compartmentalization of the eukaryotic cell nucleus, particularly in the assembly of various nuclear bodies, such as the nucleolus, splicing speckles, and Cajal bodies (11–15). The proteins that participate in the formation of phase-separated condensates frequently possess intrinsically disordered regions (IDRs) (16–18). These IDRs may mediate weak-affinity and non-specific interactions with multiple target sites that trigger the LLPS (19). IDRs are present in many proteins involved in the assembly of repressive chromatin domains (histone H1, heterochromatin protein one (HP1) and chromobox 2 (CBX2) subunit of mammalian PRC1 complex), which enables these factors to form liquid condensates, both in vitro and in vivo (20–23). On the other hand, the components of the transcriptional machinery, including RNA polymerase II (24), Mediator complex subunits (25), and various transcription factors (26), also possess IDRs and are capable of forming complex phase-separated condensates at enhancers (27,28). The assembly of activating domains at enhancers and the clustering of RNA polymerase II molecules at transcription hubs or factories via phase separation appears to be functionally relevant (28–33). Therefore, the formation of different types of phase-separated chromatin condensates may underlie the segregation of the A and B chromatin compartments (34,35). In addition to the direct LLPS-driven segregation of chromatin domains bearing different epigenetic marks, the spatial clustering of active and repressed chromatin domains may also be mediated by their differential interactions with nuclear bodies, such as nucleoli and nuclear speckles (36). The disruption of nuclear speckles was shown to reduce spatial chromatin interactions within the active compartment (37). Considering that nucleoli and nuclear speckles are LLPS-dependent membrane-free compartments (17), LLPS is likely to contribute to the spatial segregation of A and B chromatin compartments, both directly and indirectly.
To obtain further insights into the possible roles played by LLPS in 3-dimensional (3D) genome organization, here, we studied the effects of 1,6-hexanediol (1,6-HD), an agent known to disrupt liquid-phase condensates (28), on chromatin folding in HeLa cells. Using stochastic optical reconstruction microscopy (STORM) and Hi-C analysis, we demonstrate that the suppression of LLPS leads to the partial decondensation of compact nucleosome clutches, irreversibly changes the internal structures of A and B chromatin compartments, and slightly compromises their spatial segregation. At the level of TADs, 1,6-HD-driven alteration of LLPS changes chromatin compaction and weakens enhancer-promoter loops. Upon the restoration of LLPS after 1,6-HD removal the repressed chromatin does not return to its initial state but instead acquires a novel, more compact configuration.
MATERIALS AND METHODS
Cell culture and treatments
Human HeLa cells were obtained from ATCC. All cells were grown in Dulbecco's modified Eagle's medium (DMEM), supplemented with 10% fetal calf serum. To obtain transiently permeabilized cells, HeLa cells were incubated with 1% Tween 20 in Dulbecco's phosphate-buffered saline (DPBS) for 10 min at room temperature, washed twice with DPBS, and transferred to culture medium. To obtain 1,6-HD-treated cells, transiently-permeabilized HeLa cells were incubated with 5% 1,6-HD (Sigma-Aldrich, #240117) in culture medium at room temperature, washed twice with DPBS, and transferred to culture medium. For recovery experiments, HeLa cells that were treated with 1,6-HD were incubated in culture medium for 1.5 h at 37°C. Treatment with 2,5-hexanediol (2,5-HD; Sigma-Aldrich, #H11904) was performed as described for 1,6-HD. The number of caspase-3/7-positive cells was measured using CellEvent Caspase-3/7 Green Detection Reagent (Invitrogen), according to the manufacturer's instructions.
Measurement of transcriptional activity
For 5-ethyniluridine (EU) incorporation, cells were incubated with 200 μM EU (Jena Bioscience) for 15 min at 37°C. After this incubation, cells were washed three times with PBS and fixed in 100% cold (–20°C) methanol for 10 min before staining. The cells were washed three times with PBS and processed using a Click-iT EU Imaging Kit (Life Technologies), according to the manufacturer's recommendations. The integrated intensities of EU fluorescence were analyzed using CellProfiler software.
Immunofluorescence
For immunostaining, cells were grown on microscope slides. All samples were fixed in 100% cold methanol (−20°C) for 10 min. After washing in PBS, cells were pre-incubated with 1% bovine serum albumin (BSA) in PBS, with 0.05% Tween 20, for 30 min and were then incubated with antibodies (anti-coilin, Abcam, #ab87913; anti-H2B, Active Motif, #61037) in PBS, supplemented with 1% BSA and 0.05% Tween 20, for 1 h at room temperature. After the incubation, cells were washed three times with PBS, supplemented with 0.2% BSA and 0.05% Tween 20. The primary antibodies bound to antigens were visualized using Alexa Fluor 488- or Alexa Fluor 594-conjugated secondary antibodies. The DNA was counterstained with 4,6-diamino-2-phenylindole (DAPI) for 10 min at room temperature. The samples were mounted using Dako fluorescent mounting medium (Life Technologies). The immunostained samples were analyzed using a Zeiss AxioScope A.1 fluorescence microscope (objectives: Zeiss N-Achroplan 40×/0.65 and EC Plan-Neofluar 100×/1.3 oil; camera: Zeiss AxioCam MRm; acquisition software: Zeiss AxioVision Rel. 4.8.2; Jena, Germany). The images were processed using ImageJ software (version 1.44).
Samples for Structured Illumination Microscopy (SIM) were mounted in Dako fluorescent mounting medium (Life Technologies) and examined using a Nikon N-SIM micro-scope (100×/1.49 NA oil immersion objective, 488 and 561 nm diode laser excitation). Image stacks (z-steps of 0.2 μm) were acquired with EMCCD camera (iXon 897, An- dor, effective pixel size 60 nm). Exposure conditions were adjusted to get a typical yield about 5000 max counts (16-bit raw image) while keeping bleaching minimal. Image acquisition, SIM image reconstruction and data alignment were performed using NIS-Elements (Nikon). The identification and analysis of SC35 domains were performed as previously described (38).
Western blot analysis
For chromatin fractionation experiments, cells were permeabilized in RSB buffer containing 10 mM HEPES–NaOH (pH 7.5), 1.5 mM MgCl2, 0.5 mM EDTA, 10 mM KCl, 0.5% NP40, phosphatase, and protease inhibitors. After incubation at 4°C for 10 min, cells were collected by centrifugation at 1000 × g for 5 min. Cells were then incubated in RSB buffer containing 100 mM NaCl. After incubation at 4°C for 10 min, the first soluble fraction (0.1 fraction) was separated by centrifugation at 10 000 × g for 10 min. Cells were then incubated in RSB buffer containing 400 mM NaCl. After incubation at 4°C for 1 h, the second soluble fraction (0.4 fraction) was separated by centrifugation at 8000 × g for 10 min. The insoluble chromatin fraction (pellet) was then sonicated in RSB buffer at 50% amplitude for 30 s with a VirSonic 100 ultrasonic cell disrupter. Immunoblotting was run as described (39). The following antibodies were used: CTCF (Active Motif, 61311; 1:2000), Rad21 (Abcam, ab992; 1:2000), HP1 (Abcam, ab109028, 1:2000), and histone H2B (Active Motif, 39125; 1:2000).
OptoDroplet analysis
HeLa cells were transfected using XFect reagent with the pHR-FUSN-mCh-Cry2WT plasmid (40), which was a gift from Clifford Brangwynne (Addgene plasmid # 101223). OptoDroplet formation was observed using a Nikon Eclipse Ti-E inverted fluorescence microscope equipped with a Nikon Intensilight C-HGFI light source. Acquisitions were performed using the 100× objective, with the TexasRed filter set for the visualization of the mCherry signal and the FITC filter set for Cry2 activation.
STORM sample preparation and image acquisition
To perform immunostaining, the culture medium was aspirated, and the cells were washed with 1× PBS once and fixed in 1% formaldehyde 15 min and then in 100% methanol for 10 min at −20°C. After washing once with 1× PBS, the cells were incubated with H2B antibodies diluted in blocking buffer (1% BSA in 1× PBS with 0.05% Tween 20), at 4°C overnight. The cells were washed three times with 1× PBS, for 5 min per wash, and the Alexa Fluor 647-conjugated secondary antibody in the blocking buffer was added to the sample for 1 h, protected from light. The cells were washed three times with 1× PBS and stored in 1× PBS before imaging. Immediately before imaging, the buffer was replaced with STORM imaging buffer, containing 10% (w/v) glucose (Sigma-Aldrich), 0.56 mg/ml glucose oxidase (Sigma-Aldrich), 0.17 mg/ml catalase (Sigma-Aldrich), and 0.14 M β-mercaptoethanol (Sigma-Aldrich). All imaging experiments were performed using a commercial ONI Nanoimager microscope system. Laser light at 647 nm was used to excite Alexa Fluor 647. The emitted light was collected by an oil immersion 100×, 1.49 NA objective and imaged onto a scientific CMOS (sCMOS) camera. For STORM imaging, 50 000 frames were acquired at an exposure time of 10 ms. The reconstruction of the super-resolution image was performed using NimOS software from ONI.
STORM image analysis
Spatial analysis of H2B was performed as previously described (41). Briefly, for each extracted nucleus from a reconstructed STORM image, the bottom 10% of signal intensities were discarded as background. Next, we separated each nucleus into 50 × 50 px squares. For each square, we calculated L- and G-functions, using a custom python code (available at GitHub page: https://github.com/ArtemLuzhin/STORM-microscopy-analysis). As a control, we shuffled the values in each square and then calculated the L- and G-functions for the shuffled squares. The L-function plots were obtained as the ratio between the L-function and the G-function for both raw and shuffled squares.
Hi-C library preparation
Hi-C libraries were prepared as described previously (42), with minor modifications. A total of 5–10 million cells were fixed in 1× PBS containing 2% formaldehyde (Sigma-Aldrich) for 10 min, with occasional mixing. The reaction was quenched with by the addition of 2 M glycine (Sigma-Aldrich), to a final concentration of 125 mM. Cells were pelleted by centrifugation (1000 × g, 10 min, 4°C), resuspended in 50 μl 1× PBS, snap-frozen in liquid nitrogen, and stored at −80°C. Cells were lysed in 1.5 ml isotonic buffer [50 mM Tris–HCl pH 8.0 (Sigma-Aldrich), 150 mM NaCl (Sigma-Aldrich), 0.5% (v/v) NP-40 substitute (Fluka), 1% (v/v) Triton-X100 (Sigma-Aldrich), 1 × Halt™ Protease Inhibitor Cocktail (Thermo Scientific)], on ice for 15 min. Cells were pelleted by centrifugation at 2500 × g for 5 min, resuspended in 100 μl 1× DpnII buffer (New England Biolabs), and pelleted again. The pellet was resuspended in 200 μl 0.3% SDS (Sigma-Aldrich) in 1.1× DpnII buffer (New England Biolabs) and incubated at 37°C for 1 h. Then, 330 μl 1.1× DpnII buffer and 53 μl 20% Triton X-100 were added, and the suspension was incubated at 37°C for 1 h. Next, 600 U DpnII enzyme (New England Biolabs) were added, and the chromatin was digested overnight (14–16 h), at 37°C, with shaking (1,400 rpm). In the morning, 200 U DpnII enzyme was added, and the cells were incubated for 2 h. DpnII was then inactivated by incubation at 65°C for 20 min. The nuclei were harvested for 10 min at 5000 × g, washed with 100 μl 1× NEBuffer 2 (New England Biolabs), and resuspended in 125 μl 1.2× NEBuffer 2. Cohesive DNA ends were biotinylated by adding 25 μl biotin fill-in mixture [0.025 mM dATP (Thermo Scientific), 0.025 mM dGTP (Thermo Scientific), 0.025 mM dTTP (Thermo Scientific), 0.025 mM biotin-14-dCTP (Invitrogen), 0.8 U/μl Klenow enzyme (New England Biolabs)]. The samples were incubated at 37°C for 75 min, with shaking (1400 rpm). Nuclei were pelleted by centrifugation at 3000 × g for 5 min, resuspended in 300 μl 1× T4 DNA ligase buffer (Thermo Scientific), and pelleted again. The pellet was resuspended in 300 μl 1× T4 DNA ligase buffer, and 75 U T4 DNA ligase (Thermo Scientific) was added. Chromatin fragments were ligated at 20°C for 6 h. The cross-links were reversed by overnight incubation at 65°C in the presence of proteinase K (100 μg/ml) (Sigma-Aldrich). After cross-link reversal, the DNA was purified by single phenol-chloroform extraction, followed by ethanol precipitation [glycogen (Thermo Scientific) at a concentration of 20 μg/ml was used as co-precipitator]. After precipitation, the pellets were dissolved in 100 μl 10 mM Tris–HCl, pH 8.0. To remove residual RNA, samples were treated with 50 μg RNase A (Thermo Scientific) for 45 min at 37°C. To remove residual salts and DTT, the DNA was purified using Agencourt AMPure XP beads (Beckman Coulter). Biotinylated nucleotides from the non-ligated DNA ends were removed by incubating the Hi-C libraries (2 μg) in the presence of 6 U T4 DNA polymerase (New England Biolabs) in NEBuffer 2, supplemented with 0.025 mM dATP and 0.025 mM dGTP, at 20°C for 4 h. Next, the DNA was purified using Agencourt AMPure XP beads. The DNA was then dissolved in 500 μl sonication buffer (50 mM Tris–HCl, pH 8.0, 10 mM EDTA, 0.1% SDS) and was sheared to a size of ∼100–1000 bp, using a VirSonic 100 (VerTis). The samples were concentrated (and simultaneously purified), using AMICON Ultra Centrifugal Filter Units (Merck), to a total volume of approximately 50 μl. The DNA ends were repaired by adding 62.5 μl MQ water, 14 μl 10× T4 DNA ligase reaction buffer, 3.5 μl 10 mM dNTP mix (Thermo Scientific), 5 μl 3 U/μl T4 DNA polymerase (New England Biolabs), 5 μl 10 U/μl T4 polynucleotide kinase (New England Biolabs), and 1 μl 5 U/μl Klenow DNA polymerase (New England Biolabs) and incubating at 20°C for 30 min. The DNA was purified with Agencourt AMPure XP beads and eluted with 50 μl 10 mM Tris–HCl (pH 8.0). To perform an A-tailing reaction, the DNA samples were supplemented with 6 μl 10× NEBuffer 2, 1.2 μl 10 mM dATP, 1 μl MQ water and 3.6 μl 5 U/μl Klenow (exo-) (New England Biolabs). The reactions were performed for 30 min at 37°C in a PCR machine, and the enzyme was then heat-inactivated by incubation at 65°C for 20 min. The DNA was purified using Agencourt AMPure XP beads and eluted with 200 μl 10 mM Tris–HCl (pH 8.0). Biotin pulldown of the ligation junctions was performed as described previously, with minor modifications. Briefly, 10 μl MyOne Dynabeads Streptavidin C1 (Invitrogen) beads were used to capture the biotinylated DNA, and the volumes of all buffers were decreased by 4-fold. The washed beads attached to captured ligation junctions were resuspended in 50 μl adapter ligation mixture, composed of 41.5 μl MQ water, 5 μl 10× T4 DNA ligase reaction buffer (Thermo Scientific), 2.5 μl Illumina TruSeq adapters and 1 μl 5 U/μl T4 DNA ligase (Thermo Scientific). Adapter ligation was performed at 22°C, for 2.5 h, and the beads were washed twice with 100 μl TWB [5 mM Tris–HCl, pH 8.0, 0.5 mM EDTA, 1 M NaCl, 0.05% Tween-20 (Sigma-Aldrich)], once with 100 μl 1× binding buffer (10 mM Tris–HCl, pH 8.0, 1 mM EDTA, 2 M NaCl), and once with 100 μl CWB (10 mM Tris–HCl, pH 8.0 and 50 mM NaCl), and then resuspended in 20 μl MQ water. Test PCR reactions containing 4 μl streptavidin-bound Hi-C library were performed to determine the optimal number of PCR cycles necessary to generate sufficient PCR products for sequencing. The PCR reactions were performed using KAPA High Fidelity DNA Polymerase (KAPA) and Illumina PE1.0 and PE2.0 PCR primers (10 pmol each). The temperature profile was 5 min at 98°C, followed by 6, 9, 12, 15 and 18 cycles of 20 s at 98°C, 15 s at 65°C, and 20 s at 72°C. The PCR reactions were separated on a 2% agarose gel supplemented with ethidium bromide, and the number of PCR cycles necessary to obtain a sufficient amount of DNA was determined based on the visual inspection of gels (typically 9–12 cycles). Four preparative PCR reactions were performed for each sample. The PCR mixtures were combined, and the products were purified using Agencourt AMPure XP beads.
Hi-C data processing
Hi-C reads were mapped to the reference human genome hg19 assembly using Bowtie v2.2.3 (43) with the ‘–very-sensitive’ mode and the iterative mapping procedure implemented in hiclib (https://bitbucket.org/mirnylab/hiclib), as described previously (44). The minimal read size was set to 25 bp and the iterative mapping step was increasing by 5 bp until a maximal read length was reached. We then filtered out non-uniquely mapped reads, ‘same fragment’ and ‘dangling end’ reads, PCR duplicates, reads from restriction fragments shorter than 100 bp and longer than 100 kb, and reads from the top 0.5% of restriction fragments with the greatest number of reads. The remaining read pairs were aggregated into genomic bins of different sizes to produce Hi-C contact matrices. To remove low coverage bins and iteratively correct the contact matrices we used cooler v0.7.9 balance function with default parameters (45). As biological replicates in all experiments demonstrated a high correlation (Pearson's r > 0.96), they were pooled together for downstream analyses. Statistics of the Hi-C data processing can be found in Supplementary Table S1.
Pc(s) curves
P c(s) curves were computed using hiclib and the range between 20 kb and 100 Mb was extracted.
Chromatin compartment annotation and analysis
Compartment annotation
Chromatin compartments were annotated using principal component analysis (PCA) implemented in the cooltools v0.3.2 call-compartments function (https://github.com/open2c/cooltools) for 100-kb-resolution contact matrices. Per the established convention, the orientation of the PC1 eigenvector was selected such that it correlated positively with GC content. Consequently, B-compartment bins are those with negative PC1 eigenvector values, and A compartment bins are those with positive values.
Validation of the compartment signal
To validate the annotated compartments, we used ENCODE datasets (46) for the HeLa cell line (Supplementary Table S2). Raw RNA-seq reads from two biological replicates were mapped to the reference human genome hg19 assembly using STAR v2.6.1c (47) with default parameters and merged. Unmapped and low-mapping-quality reads were removed using SAMtools v1.5 (48) with option -q 30. We then calculated transcription levels in 100-kb genomic bins using BEDtools v2.25.0 (49). Processed ChIP-seq signal tracks were downloaded from the UCSC Genome Browser database and aggregated into 100-kb genomic bins using BEDtools v2.25.0. Fold changes of RNA- and ChIP-seq signals in each compartment were then defined as the mean signal in the compartment divided by the mean signal across the entire genome.
Saddle plots
Compartmentalization saddle plots were generated using the cooltools v0.3.2 compute-saddle function using 100-kb observed-over-expected cis and trans matrices, respectively. Prior to the calculation, we clipped 2.5 percentiles of the genome-wide PC1 eigenvector from the top and bottom to exclude extreme values in the analysis. The resulting saddle plots were zoomed into 25 equally sized bins, and a log10 transformation was performed.
Average compartment
Because the currently available tools for the pile-up analysis of the Hi-C maps can only deal with chromatin domains and loops, we developed a tool to compute and visualize the average compartment. This visualization, referred to here as a pentad, is aimed to represent short- and long-range contacts within A and B compartments and contacts between A and B compartments. It consists of several piled-up areas from the observed-over-expected Hi-C matrix that are determined based on the compartment signal provided by the user (Figure 4A). The tool can filter the areas based on their dimensions in genomic bins, the amount of zero-contact pixels, and the distance between the bases that form the area. The areas that pass the filters are extracted from the matrix and rescaled using bilinear interpolation. Rescaled areas of the same type are averaged genome wide and aggregated into a pentad. It is possible to apply the tool to both cis and trans compartment interactions. The tool uses a cool file with a Hi-C contact matrix and a bedGraph file with a compartment signal as the main inputs. The output is the calculated average compartment in text and image formats. The tool, documentation, and examples are available at https://github.com/magnitov/pentads.
Figure 4.
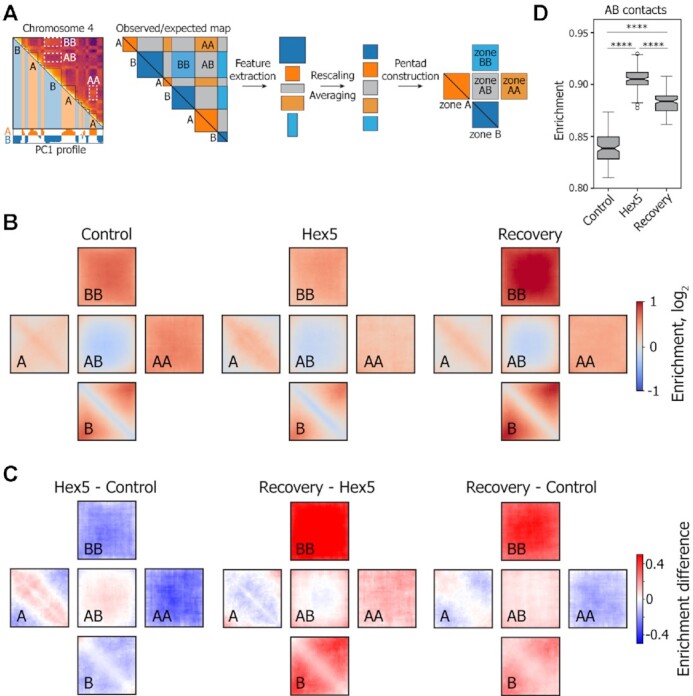
Pentad analysis of A and B compartments upon 1,6-HD treatment. (A) Schematic representation of the principal of pentad analysis. (B) Plots (pentads) showing the observed-over-expected contact frequencies inside the A and B compartments at short (A, B; compartmental domains) and large genomic distances (AA, BB) and between compartments (AB). (C) Pairwise subtractions of Control, Hex5, and Recovery pentads. (D) Boxplots showing the contact frequencies between the A and B compartments. ****P < 0.0001 in a Mann–Whitney U-test.
To follow the distribution and density of contacts within and between A and B compartments, we utilized the described tool in application to 100-kb-resolution contact matrices. We considered the areas that had both dimensions greater than or equal to 3 bins, had a maximum fraction of zeros less than 0.25, and were closer than 0.75 of the chromosome length to the diagonal. Areas passing these filters were extracted from the observed-over-expected matrix and rescaled into 33 × 33 pixel squares using bilinear interpolation. Rescaled areas of the same type were then averaged genome wide using a median value in each pixel and aggregated into one pentad.
Compartment strength
Compartment strength was calculated separately for compartments A and B using the pentads for each chromosome per replicate (N = 46). Compartment strength was defined as the sum of long-range contacts within a compartment (top or right square) divided by the sum of contacts between compartments (middle square) in the pentad. Statistical differences between compartment strengths in A and B were determined by a Mann–Whitney U-test.
AB contacts
The contacts between compartments were approximated from the calculated pentads for each condition (N = 3). We extracted the 11 × 11 pixel square from the middle of the pentad because these contacts represent the ‘true’ interactions between compartments, not biased towards low values from the PC1 eigenvector.
TAD detection and analysis
Domain annotation
In this work, we did not distinguish loop domains generated by loop extrusion (50,51), and compartmental domains arose from compactization of genome regions covered by distinct histone modifications (5). Both types of features are represented by contact domains in Hi-C maps and are referred to as TADs in our analysis. TADs were annotated using the insulation score algorithm (52) implemented in the cooltools v0.3.2 diamond-insulation function for 20-kb-resolution contact matrices. The window size for the insulation score calculation was set to 360 kb. The bins with a boundary strength higher than 0.1 were considered as TAD boundary bins. These boundaries were converted into TADs by continuously joining two neighboring boundaries together. The TAD boundary coordinate was then set to the coordinate between the annotated boundary bin and the adjacent bin that had a lower insulation score. We then removed TADs smaller than 60 kb and those containing a total fraction of zeros greater than 0.5. The resulting number of annotated domains was 4956 in the Control, 4694 in 1,6-hexanediol-treated, and 5010 in Recovery cells.
Assignment to the compartments
The annotated TADs were assigned to compartments A or B if the intersection of the domain with the compartment was greater than 75%. Using this procedure, we assigned 2852 TADs to compartment А and 1576 to compartment В in the Control, 2051 to А and 2142 to В in 1,6-hexanediol-treated, and 3139 to А and 1267 to В in Recovery cells.
Average TAD
The average TAD was calculated using coolpup.py v0.9.5 (53) from 20-kb observed-over-expected contact matrices. TADs of sizes greater than 60 kb and less than 3 Mb were used for pile-up generation. The –rescale and –local options were used, and the rescale size was set to 99 pixels. The density of contacts within the average TAD was calculated as the mean value of the central 33 × 33 pixel square.
Average TAD boundary
The average TAD boundary was calculated using coolpup.py v0.9.5 from 10-kb observed-over-expected contact matrices with a pad size of ±250 kb around the TAD boundaries. The boundary strength was calculated as the mean value of the average intra-TAD interactions (upper-left and bottom-right quarters) divided by the mean value of average inter-TAD interactions (upper-right quarter).
Loop detection and analysis
Loop annotation
Loops were annotated using the CPU version of HiCCUPS (Juicer Tools v1.11.09) (54) for 5-, 10- and 25-kb-resolution contact matrices. For the annotation, we used the recommended parameters for medium-resolution Hi-C maps. Loop annotations at different resolutions were merged as described previously (4). The total number of annotated loops is 2837 in the Control, 2381 in 1,6-HD-treated cells and 5709 in Recovery cells.
Assignment to compartments
The loops were assigned to compartments A or B if both of their anchors belonged to the same compartment.
CTCF-associated loops
To identify CTCF-associated loops, we used the annotated CTCF ChIP-seq peaks. Loops in which both anchors intersected with at least one CTCF peak were considered CTCF associated. Using this procedure, we obtained 1932 loops in the Control (1320 in compartment А and 362 in compartment В), 1585 in 1,6-hexanediol-treated (822 in compartment А and 594 in compartment В), and 3999 in Recovery (3192 in compartment А and 344 in compartment В) cells.
Enhancer-promoter loops
To identify enhancer-promoter loops, we utilized the ENCODE combined chromatin state segmentation for the HeLa cell line. We searched for loops that had a ‘TSS’ state in one loop anchor and either an ‘E’ or ‘WE’ state in the other, as described previously (4) (466 total loops, 392 in compartment A and 36 in compartment B).
Promoter–promoter interactions
Gene pairs with promoter-promoter interactions (loops) detected by ChIA-PET and promoter coordinates detected by CapStarr-seq for the HeLa cell line were obtained from a recent publication (55) (896 total pairs, 819 in compartment A and 30 in compartment B).
Average loop
The average loop was calculated using coolpup.py v0.9.5 on 10-kb observed-over-expected contact matrices with a pad size of ±150 kb around the loop pixel. Loops of sizes 0.1–1.5 Mb were used for the pile-up generation. The loop strength was calculated as the mean value of the central 3 × 3 square pixels. Statistical differences between individual loop strengths were determined by a Mann–Whitney U-test.
ChIP-seq
ChIP-seq was performed with an anti-CTCF antibody (Active Motif, #61311) as described (56,57) for two biological replicates. ChIP samples were prepared for next-generation sequencing using a NEBNext Ultra II DNA library prep kit for Illumina (New England Biolabs). Libraries were sequenced on the Illumina NovaSeq 6000 and resulted in around 40 million 100-bp single-end reads per sample.
Reads were mapped to the reference human genome hg19 assembly using Bowtie v2.2.3 with the ‘–very-sensitive’ mode. Non-uniquely mapped reads, possible PCR and optical duplicates were filtered out using SAMtools v1.5. The bigWig files with the ratio of RPKM normalized ChIP-seq signal to the input were generated using deepTools v3.4.2 bamCompare function (58). ChIP-seq signal regions overlapping with hg19 blacklist regions (59) were discarded. Peaks were called using PePr (60) with a p-value cutoff of 1e–5 and a sliding window size of 100 bp. The peak calling procedure annotated 29 957 peaks in control, 30 287 peaks in 1,6-hexanediol treated, and 31 479 in recovery cells. The pile-ups of the ChIP-seq signal centered at the peak coordinate were generated using deepTools v3.4.2 computeMatrix function with parameters -missingDataAsZero and -skipZeros followed by plotHeatmap function for visualization.
RESULTS
1,6-HD treatment of living cells to study higher-order chromatin organization
To investigate the role played by LLPS in spatial genome organization, we used human HeLa cells that were treated with 1,6-HD. This aliphatic alcohol is predicted to disrupt weak hydrophobic interactions, both in vitro and in vivo, disassembling LLPS-dependent macromolecular condensates (61–63). Molecular condensates depended purely on phase separation driven by electrostatic interactions are expected to be unaffected by 1,6-HD treatment (61). Nevertheless, 1,6-HD is the only available tool to test in vivo the contribution of LLPS in the assembly of macromolecular complexes at this moment (63). 1,6-HD has been used in a number of seminal studies to treat living cells, particularly the studies that described the involvement of LLPS in super-enhancer function and heterochromatin domain formation (21,27,28). Prolonged treatment with 1,6-HD can lead to cellular membrane rupture and cell death (63,64). To minimize 1,6-HD toxicity in vivo and to avoid possible secondary effects associated with such toxicity, we utilized the following precautions: (i) limited 5% 1,6-HD treatment to 15 min; and (ii) applied 1,6-HD to HeLa cells that were first transiently permeabilized with Tween 20 (65). Treatment of living cells with 5% 1,6-HD is widely accepted condition (66–69) that provides disruption of LLPS-driven condensates with minimal cytotoxicity. Using cells transiently permeabilized with Tween 20 additionally lowers 1,6-HD toxicity that in case of intact non-permeabilized cells is related to the cytoplasmic membrane rupture (63,64). Generally, the short-term treatment with Tween 20 to permeabilize eukaryotic cells does not affect cell morphology or metabolic processes, such as transcription and DNA repair foci formation (65,70). Additionally, we have verified that the treatments used in our study did not result in cell death and did not perturb intracellular processes. The treatment of HeLa cells with Tween 20 alone (1%, 10 min) did not lead to the appearance of apoptotic cells and did not inhibit RNA polymerase I and II-dependent transcription (Supplementary Figures S1A and B). The short-term incubation of transiently permeabilized (Tween 20-treated) cells in culture medium containing 1,6-HD (5%, 15 min) induced only moderate levels of apoptosis (Supplementary Figure S1A). Moreover, the number of apoptotic cells did not significantly increase upon the 1.5-h recovery period, during which cells were incubated in Tween 20- and 1,6-HD-free medium (Supplementary Figure S1A). Although transcription was strongly inhibited by 1,6-HD treatment, it was almost fully restored after the recovery period (Supplementary Figure S1B).
To test the in vivo LLPS-disrupting properties of 1,6-HD treatment, we applied two different approaches. First, we investigated the influence of 1,6-HD on LLPS using a recently developed optoDroplet system (40). In this system, the intrinsically disordered region of an RNP granule protein FUS is combined with the fluorescent protein mCherry and the light-sensitive oligomerization domain of Arabidopsis thaliana cryptochrome 2 (CRY2) to generate a fusion protein that undergoes LLPS in living cells upon blue light activation (40). HeLa cells, transfected with a plasmid encoding the above-described light-sensitive chimeric protein, were subjected to blue light irradiation in culture medium either with or without 1,6-HD (5%) (Figure 1A). In contrast to Tween 20, 1,6-HD treatment strongly affected the assembly of optoDroplets in HeLa cells (Figure 1A). After the 1.5-h-long incubation of the treated cells in 1,6-HD-free medium, the ability of the transfected cells to assemble optoDroplets in response to blue light irradiation was completely reestablished (Figure 1A). Next, we verified that 1,6-HD not only interfered with the formation of optoDroplets but could also promote their disassembly. For this purpose, we first stimulated optoDroplet formation in HeLa cells and then added to the culture medium 1,6-HD (5%) (Figure 1B). The addition of 1,6-HD stimulated the disassembly of optoDroplets within several seconds (Figure 1B). We applied optoDroplet assay to test also the mode of action of 2,5-hexanediol (2,5-HD), which was considered by some studies as a negative control for 1,6-HD. Notably, 2,5-HD was as effective as 1,6-HD in preventing the assembly of opto-droplets and in promoting their dissolution (Supplementary Figure S1C-D), making it hardly applicable as a negative control for experiments with 1,6-HD.
Figure 1.

1,6-HD compromises LLPS in living human cells. (A) HeLa cells transfected with pHR-FUSN-mCh-Cry2WT were transiently permeabilized and then either mock-treated (control), treated with 1,6-HD (5%, 15 min), or treated with 1,6-HD and allowed to recover for 1.5 h. OptoDroplet formation was monitored as described in (40). Scale bar: 10 μm. (B) HeLa cells transfected with pHR-FUSN-mCh-Cry2WT were light-illuminated to induce optoDroplet formation. Then 1,6-HD (5%) was added to the culture medium; optoDroplet existence was monitored after ten seconds of incubation with the drug. Scale bar: 10 μm. (C) Transiently permeabilized HeLa cells were untreated (control), treated with 1,6-HD (5%, 15 min), or treated with 1,6-HD and allowed to recover for 1.5 h before being stained for coilin (green). Scale bar: 15 μm. (D) Quantification of the samples presented in (C). Percentage of cells containing coilin foci (i.e. Cajal bodies) are shown.
Second, we examined whether the LLPS-dependent membrane-free intranuclear compartments, such as Cajal bodies and splicing speckles, were affected by 1,6-HD treatment. These compartments were chosen because they were reported to be sensitive to 1,6-HD (33,71). The indirect immunofluorescence analysis of coilin, a major proteinaceous component of Cajal bodies, showed that 1,6-HD treatment (5%, 15 min), but not the treatment with Tween 20 alone, disrupted these compartments in living cells (Figures 1C and D). During the 1.5-h-long recovery period in 1,6-HD-free medium, Cajal bodies were fully re-established (Figures 1C and D). Structured illumination microscopy (SIM) of SC35 (SRSF2) combined with the nucleosome clustering analysis (38) also demonstrated that the treatment with 1,6-HD affected the integrity of splicing speckles (Supplementary Figure S1E).
Together, these data examining 1,6-HD toxicity and its mode of action argue for the suitability of 1,6-HD treatments to the study of the role of LLPS in higher-order chromatin organization in eukaryotic cells.
Chromatin domains became more uniformly distributed upon 1,6-HD treatment
First, we analyzed the distribution of histone H2B in HeLa cells permeabilized with Tween 20 and non-treated (Control) or treated with 1,6-HD, using conventional epifluorescence microscopy (Supplementary Figure S1F). Virtually no changes were detected in the distributions of H2B in 1,6-HD-treated cells compared with control cells. To examine the organization of chromatin at a nanoscale resolution, we performed STORM imaging of the core histone H2B in HeLa cells. The cells were first transiently permeabilized with Tween 20 and then either i) not treated (Control), ii) treated with 5% 1,6-HD for 15 min, or iii) treated with 1,6-HD for 15 min and then allowed to recover for 1.5 h in a fresh culture medium that did not contain either Tween 20 or 1,6-HD (Figure 2A). As an additional control, we used HeLa cells that were incubated with the histone deacetylase inhibitor sodium butyrate (SB). Treatment with SB leads to histone hyperacetylation and, thus, can promote global chromatin decondensation (38,41). In agreement with the previously published data (38,41,72), H2B appeared to be clustered into discrete chromatin clutches (Figure 2A). As expected, SB-treated cells showed more distributed H2B/nucleosome signals throughout the nucleus than control cells (Figure 2A), validating the STORM analysis performed.
Figure 2.

Super-resolution microscopy analysis of chromatin organization changes induced by 1,6-HD. (A) Representative STORM images of H2B in human HeLa cells that had been permeabilized with Tween 20 and either untreated (control), treated with 1,6-HD (5%, 15 min), treated with 1,6-HD and then incubated in a drug-free medium for 1.5 h, and non-permeabilized cells treated with sodium butyrate (SB; 5 mM, 14 h). Scale bar: 1 μm. Magnified images from the boxed regions in the image of each nucleus are shown. Scale bar: 0.2 μm. (B) A simplified scheme for L-function analysis. The scheme shows clustered (red circles) or random (blue circles) particles around the origin point (black circle). L-function plots of clustered and random patterns are shown. (C) L-function plots of chromatin using the same conditions as described in (A). The shaded parts of the curves represent 95% confidence intervals (CIs). For each condition, n = 14–23 cells.
To analyze quantitatively the effects of 1,6-HD on chromatin organization in interphase nuclei, we applied the analysis based on the spatial descriptive statistics (41,72). We used the L-function, which quantitatively describes the probability of identifying a molecule (nucleosome) with respect to its neighboring molecules as a function of radial distance (r) (Figure 2B). In control HeLa cells, the L-function for histone H2B shows a narrow sharp peak, suggesting the presence of clustered nucleosome clutches (Figure 2C). Treatment of the cells with SB significantly decreased the peak height of the L-function plot, indicating that SB-mediated histone acetylation decondensed chromatin clutches (Figure 2C). 1,6-HD treatment resulted in reduced levels of H2B clustering and a more uniform distribution of nucleosome clutches throughout the nucleus as compared to Control cells (Figure 2C). In cells that were treated with 1,6-HD and then recovered in a fresh medium, this 1,6-HD-induced trend for uniform distribution of nucleosome clutches became even more prominent (Figure 2C).
In summary, the super-resolution microscopy analysis shows that (i) disruption of weak hydrophobic interactions, particularly those underlying LLPS, in living cells leads to a slight decondensation of nucleosome clutches and increased stochasticity of their distribution throughout the nucleus; and (ii) the chromatin in the cells treated with 1,6-HD and recovered in a fresh medium adopts a distinct conformation as compared to that in both Control and 1,6-HD-treated cells.
Treatment of living cells with 1,6-HD affects chromatin compartment strength
To obtain further insights into the possible contribution of LLPS in 3D genome organization, we performed in situ Hi-C analysis on HeLa cells that were first transiently permeabilized with Tween 20 and then either (i) not treated with 1,6-HD (Control), (ii) treated with 5% 1,6-HD for 15 min (Hex5) or (iii) treated with 1,6-HD for 15 min and then allowed to recover in a fresh culture medium that contained neither Tween 20 nor 1,6-HD for 1.5 h (Recovery). Hi-C analysis was performed in two biological replicates, using the DpnII restriction enzyme. Each Hi-C library was sequenced to ∼150 million paired-end reads per replicate, and 75–98 million unique contacts were obtained after data processing (Supplementary Table S1). Because the biological replicates were highly correlated (Supplementary Figure S2A), we combined them to obtain 158–193 million sequenced ligation junctions per experiment (Supplementary Table S1), which allowed us to construct Hi-C maps at up to 20 kb resolution (Figure 3A, Supplementary Figure S2B). Interestingly, the dependence of contact probability on genomic distance, Pc(s), was virtually the same in the Control and Hex5 samples, but differed in the Recovery sample, which was characterized by decreased spatial interactions over distances shorter than 107 bp and increased spatial interactions at longer distances (Figure 3B and Supplementary Figure S2C). The ratio of cis (intrachromosomal) to trans (interchromosomal) contacts was also similar between the Control and Hex5 samples (Figure 3C, Supplementary Figure S2D) but decreased ∼ 2.5 times in the Recovery sample, indicating an increased intermingling of chromosome territories.
Figure 3.

1,6-HD treatment alters the strength of A and B compartments. (A) Visualization of Control, Hex5, and Recovery contact matrices at a 100-kb resolution. The compartment profiles are shown below the maps. (B) Dependence of contact probability, Pc(s), on genomic distance, s, for Control, Hex5, and Recovery samples. Black lines show the slopes for Pc(s) = s−0.75 and Pc(s) = s−1.5. A magnified view of a section of the graph is presented in the top-right corner of the picture. Note that the Control and Hex5 curves are almost completely merged. (C) Ratio between cis (intrachromosomal) and trans (interchromosomal) contacts in Control, Hex5 and Recovery contact matrices. (D) Heatmaps (saddle plots) showing log10 values of contact enrichments between genomic regions belonging to A and B compartments. (E) Compartment strength in cis in Control, Hex5, and Recovery contact matrices. ****P < 0.0001, **P < 0.01, n.s. – non-significant difference in a Mann–Whitney U-test.
Visual inspection of the Hi-C heat maps revealed that the plaid pattern became less pronounced in Hex5 heat map as compared with the Control or Recovery heat maps (Figure 3A), suggesting that the disruption of LLPS by 1,6-HD compromised chromatin compartmentalization. In the Recovery, the plaid pattern became even more prominent as compared to both Control and Hex5, indicating strong compartmentalization. To systematically analyze the effects of 1,6-HD treatment on the A/B compartments, we performed independent principal component analyses (PCA) (1) for the Control, Hex5, and Recovery contact matrices and used publicly available profiles of epigenetic markers (Supplementary Table S2) to validate compartment segmentation (Supplementary Figure S3A). The values of the principal component 1 (PC1) demonstrated a good correlation between the Control, Hex5, and Recovery samples (Spearman's correlation, ρ = 0.82–0.92; Supplementary Figure S3B). Only approximately 14.5% of the genomic bins switched their compartment states between the Hex5 and Control samples (Supplementary Figure S3C). We therefore concluded that 1,6-HD treatment did not substantially change the A/B-compartment profile genome wide. We next separately analyzed the compartment strength in the Control, Hex5, and Recovery matrices for cis-interactions. Of note, for all samples, interactions in cis were more prominent in the B compartment, whereas interactions in trans occurred preferentially within the A compartment (Figure 3D), in line with previous observations (73,74). The strength of the A compartment decreased upon treatment with 1,6-HD and did not change further in the Recovery sample. The strength of the B compartment also reduced in the Hex5 sample, but in the Recovery sample, this strength increased to a level exceeding that of the Control sample (Figure 3E). To consider separately the contribution of transient cell permeabilization with Tween 20 to the changes in higher-order chromatin organization observed in the Recovery experiment, we performed an additional in situ Hi-C analysis on HeLa cells either (i) treated with 1% Tween 20 for 10 min, or (ii) treated with Tween 20 for 10 min and then incubated for 1 h 45 min in a fresh culture medium that did not contain Tween 20. Hi-C analysis was performed in two biological replicates (Supplementary Figure S4A); sequencing statistics are shown in Supplementary Table S1. In these experiments, we did not observe a decrease of the cis/trans ratio (Supplementary Figure S4B), considerable changes of a plaid pattern in Hi-C maps (Supplementary Figure S4C), or significant differences in genome coverage with compartments (Supplementary Figure S4D) after prolonged (1 h 45 min) incubation in a fresh medium of cells that had been permeabilized with Tween 20. Moreover, after the recovery period, we did not observe a substantial increase of B compartment strength as compared to cells analyzed immediately after permeabilization (Supplementary Figure S4E). We therefore concluded that the above-discussed changes of the 3D genome observed upon recovery after 1,6-HD treatment could be attributed solely to the effects of 1,6-HD treatment.
Taken together, our observations suggest that 1,6-HD-mediated LLPS suppression leads to a partial intermixing of chromatin compartments, manifested in a smoothing of the plaid pattern in the Hex5 Hi-C map and a decrease in compartment strength. The different behaviors of the A and B compartments upon LLPS restoration after 1,6-HD removal may indicate that distinct molecular mechanisms are responsible for the generation and/or maintenance of each compartment.
1,6-HD treatment irreversibly modifies the internal structure of compartments and compromises their spatial segregation
To gain a more comprehensive picture of the intra- and intercompartment interactions that occur at various genomic distances, we developed a pentad analysis. In this approach, we divided the entire Hi-C map for each chromosome into five characteristic fields, which formed a pentad that was averaged genome wide (Figure 4A). The A and B types lie on the map diagonal and represent interactions within continuous genomic fragments constituting either A or B compartmental domains. Analyses of these zones allowed us to track contact frequencies at short distances inside compartmental domains. Zones AA, BB, and AB do not lean on the map diagonal and represent long-range interactions both inside the compartments (AA and BB) and between them (AB). The pentads clearly showed that the distributions of spatial contacts within zones A and B were strikingly different (Figure 4B). For all samples, the observed contact frequency at the map diagonal was higher than expected for the A compartment and lower than expected for the B compartment, which was enriched with more distant interactions. This result reflects the more compact chromatin state in the B compartment.
Comparisons between the Control and Hex5 pentads showed that the above-described total decrease in the A compartment strength observed for the Hex5 sample (Figure 3D and E) was entirely mediated by the loss of contacts at long distances (zone AA), whereas short-range interactions (near the diagonal within zone A) were enriched in 1,6-HD-treated cells (Figure 4C). In contrast, the overall decrease in the B compartment strength observed in the Hex5 sample (Figure 3D and E) was mediated by the depletion of spatial contacts at both short- and long-range distances. These changes in the intracompartmental contact profiles following 1,6-HD treatment were accompanied by increased interactions between compartments, indicating the weakening of their spatial segregation (Figure 4C and D) that was also observed as smoothing of the plaid pattern in Hex5 Hi-C maps (Figure 3A). Strikingly, removing 1,6-HD from the culture medium (Recovery) did not restore the initial contact frequencies, either inside compartments or between them. In the Recovery sample, compared with the Control sample, the B compartment became much more compact, both at short- and long-range distances, whereas the number of contacts inside the entire A compartment did not attain the control level. The quantitation of contact frequencies demonstrated a slight decrease in AB contacts following 1,6-HD removal. However, the level of AB contacts still exceeded that observed in the Control (Figure 4D). Notably, pentad analysis of Tween 20-permeabilized cells and cells incubated in a fresh culture medium after Tween 20 removal showed only modest changes in contact frequency within compartments, and the changes in B compartment strength occurred in the opposite direction as compared to those observed in Recovery sample (Supplementary Figure S4F).
Taken together, these results suggested that 1,6-HD treatment removed some constrains on large-scale chromatin folding, which resulted in perturbations that could not be easily reversed but allowed for the alternative refolding of chromatin fiber during the Recovery experiment. Previously, Amat et al. (75) reported hyperosmotic shock-induced changes in chromatin compartments that closely resembled the changes observed in our study, as demonstrated by the analysis of their data using the pentad algorithm (Supplementary Figure S5). However, hyperosmotic shock-induced changes were fully reversed upon the return of cells to normal conditions.
1,6-HD treatment reversibly changes TAD compaction and weakens enhancer-promoter loops
We next analyzed the effects of 1,6-HD treatment on chromatin folding, at the scale of TADs and loops (Supplementary Figure S6A, B). In the Hex5 sample, compared with the Control sample, TADs became more compact in the A compartment and a little less compact in the B compartment (Figure 5A). In contrast to the contact frequencies in the compartments, the contact frequencies inside TADs almost fully reverted to the Control level in the Recovery sample. As revealed by the analysis of contact frequency between adjacent TADs, 1,6-HD treatment resulted in a remarkable decrease in TAD insulation in the A but not in the B compartment (Figure 5B, C). Interestingly, similar to the B compartment strength (Figure 3E), TAD insulation in A compartment did not revert to the Control level in the Recovery sample, but became more prominent. This result is in agreement with the observed reduction in contact frequencies at genomic distances of 0.1–1 Mb (Figure 3B) and the decreased number of cis contacts in the Recovery (Figure 3C). Notably, we did not observe any changes in TAD insulation in the experiments with Tween-permeabilized cells (Supplementary Figure S6C, D). Because mammalian TADs are formed by CTCF/cohesin-mediated loop extrusion (50,51,76), we considered a possibility that the observed decrease in TAD insulation could be triggered by a depletion of the chromatin-bound fraction of CTCF and cohesin. To test this supposition, we performed chromatin immunoprecipitation with anti-CTCF antibodies, followed by DNA sequencing (ChIP-seq; Supplementary Figure S6E). The CTCF binding profiles in the Control, Hex5, and Recovery samples were virtually identical (Figure 5D, E, Supplementary Figure S6F). Accordingly, the strength of CTCF-mediated loops only slightly decreased upon 1,6-HD treatment (Figure 5F, G, Supplementary Figure S6G). Additionally, as revealed by Western blot analysis of chromatin fractions, neither CTCF nor cohesin (Rad21 subunit) were depleted from the fraction of proteins strongly associated with chromatin upon 1,6-HD treatment (Supplementary Figure S6H).
Figure 5.

1,6-HD treatment reversibly changes TAD compaction, does not affect CTCF binding, and compromises enhancer-promoter interactions. (A) Average TADs for the Control, Hex5, and Recovery samples. Number in the upper-left corner shows the enrichment of contacts inside the TAD square over the background. (B) Average TAD boundary for the Control, Hex5, and Recovery samples. Number in the upper-right corner shows the boundary strength calculated as the mean value of the average intra-TAD interactions (upper-left and bottom-right quarters) divided by the mean value of average inter-TAD interactions (upper-right quarter). (C) Averaged insulation score profile around TAD boundaries (±0.3 Mb). (D) Examples of CTCF-binding profiles in the Control, Hex5, and Recovery samples. The dark-blue rectangles below the profiles schematically show gene positions. (E) Heatmaps of CTCF ChIP-seq signal centered at CTCF peak position (±1 kb). (F) Average CTCF-mediated loop. Number in the upper-left corner shows the enrichment of contacts inside the loop pixel over the background. (G) Related to (F): boxplots showing the enrichment of observed signal over expected in the central pixel of the corresponding average loop plot. ****P < 0.0001, **P < 0.01 in a Mann–Whitney U-test. (H) Average enhancer- (E–P) and promoter–promoter (P–P) loops. Number in the upper-left corner shows the enrichment of contacts inside the loop pixel over the background. (I) Related to (H): boxplots showing the enrichment of observed signal over expected in the central pixel of the average CTCF loop plot. ****P < 0.0001, ***P < 0.001, **P < 0.01, n.s. – non-significant difference in a Mann–Whitney U-test. (J) Representative examples of weakened E–P loops following 1,6-HD treatment. Loop pixels are highlighted with a circle and arrow. E, enhancer region, P, promoter region. H3K4me3, H3K27ac, chromatin state (ChromHMM), and gene profiles are shown according to the UCSC genome browser. The resolution of the Hi-C maps is 20 kb.
Taken together, these observations suggest that 1,6-HD treatment did not affect the cohesin and CTCF binding across the genome, allowing chromatin fibers to adopt their initial compaction profiles inside TADs after 1,6-HD removal. The decrease of TAD insulation upon 1,6-HD treatment suggests, therefore, that LLPS along with loop extrusion contributes to the TAD compaction.
To study the effects of 1,6-HD treatment on the spatial communication among genome regulatory elements, we identified enhancer- (E–P) and promoter–promoter (P–P) loops in the Control Hi-C matrix using chromatin state annotation (77). In total, we annotated 466 E–P loops (392 in compartment A and 36 in compartment B) and 896 P–P loops (819 in compartment A and 30 in compartment B). Previously, E–P communications have been reported to occur within phase-separated multi-molecular assemblies (27,28). However, whether 1,6-HD-mediated LLPS suppression can affect preformed interactions that were potentially stabilized by other mechanisms, such as specific protein–protein interactions between transcription factors, remains unclear (78). Our analysis demonstrated that 1,6-HD treatment significantly reduced the strength of E–P loops (P = 9.3 × 10−9, Mann–Whitney U-test; Figure 5H–J) in both A and B compartments (Supplementary Figure S6I); in the Recovery sample, E–P loops reverted to the Control level. In contrast, the overall strength of P–P loops, a major part of which also belonged to the A compartment, remained practically unaffected upon 1,6-HD treatment (P = 0.11, Mann–Whitney U-test; Figure 5H, I). Only loops located in compartment B and that constituted a minor fraction of P–P loops (3.3%) were weakened in Hex5 and returned to a nearly Control level in Recovery (Supplementary Figure S6J). We suppose that the decrease of E–P and P–P loop strength in the B compartment in the Hex5 sample might be a result of the general decrease in contact density within the B compartment at short genomic distances (Figure 4C).
DISCUSSION
Recent evidence suggests that LLPS may contribute to the spatial organization of the eukaryotic genome, particularly to the assembly of the inactive chromatin compartment (21–23) and to the mediation of E–P contacts (30). However, most observations related to the role played by LLPS in chromatin folding have either been indirect or based on in vitro experiments (20,21,24,79–81). At the same time, it has been reported that condensed chromatin behaves as a solid rather than a liquid both in vitro and in vivo (82,83). Here, we used the LLPS compromising agent, 1,6-HD (28), to directly address the role played by LLPS in the shaping of the 3D genome in living cells. We found that under conditions that were sufficient to affect Cajal bodies and splicing speckles and that prevented optoDroplet formation, LLPS suppression did not lead to substantial changes in chromatin folding. However, our results suggest that LLPS partially contributes to chromatin compaction at both the local (nucleosome clutches) and whole-chromosome (A/B compartments) levels. As revealed by STORM analysis, 1,6-HD-mediated compromising of LLPS in living cells resulted in a more uniform distribution of nucleosome clutches over the nuclear space. This result agrees with recently published data showing that short, artificial nucleosome arrays can form liquid phase-separated droplets, both under physiological conditions in vitro and when injected into the cell nucleus (79). However, 1,6-HD treatment did not destroy the clutches, suggesting the participation of other factors, such as electrostatic interactions of nucleosomes (82) and macromolecule crowding, in the formation of nucleosome assemblies (34). It should be noted that our observations do not agree with the results of Maeshima’ group, which showed that 1,6-HD treatment results in chromatin condensation (84). However, these authors worked with living cells and did not use permeabilization. The effects observed in their study could, therefore, be attributed to both membrane rupture and liquid condensate disruption by 1,6-HD, whereas we specifically analyzed the effect of the liquid condensate disruption.
Although the megabase-scale chromatin compartmentalization was generally retained in 1,6-HD-treated nuclei, moderate compartment mixing, a decrease of the strength of both A and B compartments, and changes in both intra- and inter-TAD interactions occurred as a result of 1,6-HD treatment. Three important observations are particularly worth commenting upon.
First, the strength of the A compartment decreased at long distances but increased at short distances after 1,6-HD treatment. This finding is potentially related to the special features of transcriptionally active chromatin, which may form distinct liquid condensates due to the recruitment of multi-bromodomain proteins (79). Within these condensates, which also contain various components of the transcription machinery and nascent RNAs, associations of remote promoters within transcription factories and enhancer-promoter interactions result in the generation of a network of long-distance contacts (85). The disruption of liquid condensates associated with active chromatin could, therefore, explain the decrease of long-distance contacts within the A compartment and the establishment of short-distance interactions that could reflect a partial compaction of loci active in Control cells and which is transcriptionally repressed upon 1,6-HD treatment. In line with this supposition, we observed a weakening of E–P interactions upon 1,6-HD treatment and their reestablishment after 1,6-HD removal. Interestingly, the association of promoters in transcription factories manifested in P–P loops was not affected by 1,6-HD. Although transcription factories have attracted significant attention (reviewed in (86,87)), their nature remains obscure. It has been reported that transcription factories remain stable in the absence of transcription (88). Although a contribution of LLPS in the assembly of transcription factories cannot be excluded, our data suggest that transcription factories are also stabilized by forces distinct from LLPS.
The second important observation is that the 1,6-HD treatment did not affect CTCF and cohesin abundance on DNA but resulted in a remarkable decrease in TAD insulation, specifically in the A compartment. This observation suggests that LLPS contributes to the compaction of TADs initially formed by the loop extrusion mechanism (50,51,76). Cohesin-driven extrusion generates numerous transient contacts between chromatin fiber fragments upon passage of the extrusion complex. Juxtoposing of transcriptionally active loci bound by IDR-containing proteins (such as active enhancers and promoters occupied with Bromodomain transcriprion factors and RNA polymerase II) might potentially facilitate the formation of liquid condensates via the increasing of a local concentration of factors liable to establish weak non-specific interactions. 1,6-HD-mediated suppression of LLPS results in at least partial disassembly of intra-TAD condensates (manifested, in particular, in a weakening of E–P loops, see above) that leads to the loss of contacts between corresponding loci and, consequently, to the reversible decrease in TAD packaging. However, because the LLPS suppression does not impact CTCF and cohesin binding, preformed CTCF/cohesin-mediated loops (and, perhaps, ongoing loop extrusion) maintain overall TAD integrity.
The third noticeable observation is that the removal of 1,6-HD from the culture medium did not allow chromatin to adopt its initial configuration: the strength of the A compartment did not increase to the Control level, whereas the B compartment became more densely packed as compared to Control cells. These results suggest that the compromising of LLPS releases certain constraints imposed on chromatin folding by a variety of biological processes. We assume that a complex of mechanisms governing chromatin compartmentalization could be considered as a ‘check and balance system’ constraining chromatin in a biologically relevant configuration (Figure 6). Compromising of the system's elements (in this case, 1,6-HD-mediated LLPS disruption) with its subsequent restoration does not provide a rapid reversion to a normal state because the entire system has been disbalanced. Rather, during the Recovery experiment, chromatin adopts a novel (apparently, thermodynamically favorable but not relevant biologically) configuration.
Figure 6.
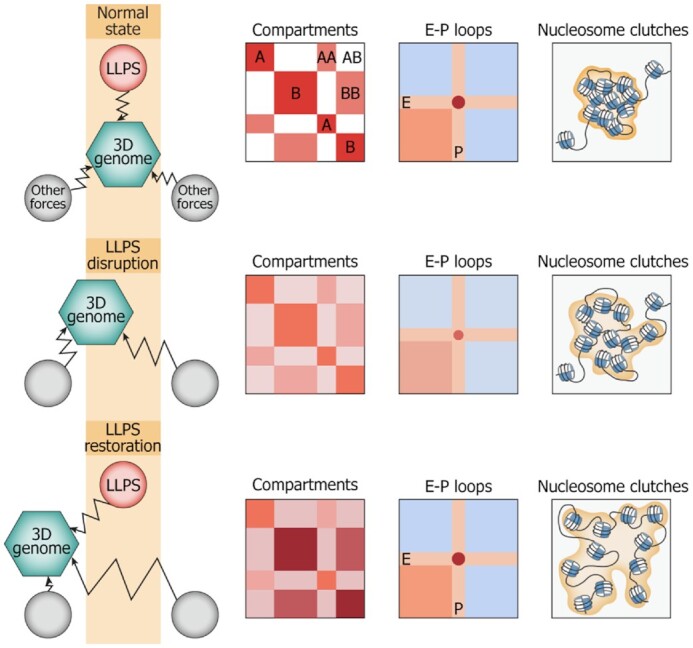
Schematic summarizing all of the obtained results. LLPS is a part of a ‘check-and-balances system’ of different forces shaping the 3D genome. LLPS disruption affects the system, resulting in the partial compromise of nucleosome clutch assemblies, decrease in compartment strength, and enhancer-promoter communication. LLPS restoration in such an unbalanced system does not allow chromatin to adopt its initial configuration but results in a new configuration.
It has been proposed that heterochromatin initially formed by nucleosome interactions (82) serves as a scaffold for the assembly of an inactive-type liquid condensate via the attraction of HP1 and other architectural proteins (89,90). Assuming that ‘active’ condensates containing RNA-polymerase, mediator and multi-bromodomain proteins, and ‘repressive condensates’ containing HP1 are not compatible with each other, it is easy to see why their disruption results in a partial mixing of compartments. However, due to the fact that the observed mixing is far from prominent, we suggest that chromatin compartmentalization is driven and also maintained (and, perhaps, predominantly) by other features of active and repressed chromatin, including the inability of interactions between acetylated nucleosomes of active loci and non-acetylated nucleosomes of repressed regions (42). Furthermore, the overall nuclear architecture might facilitate a spatial segregation of A and B compartments. For example, repressed loci are positioned at the nuclear lamina and in the perinucleolar space, whereas active chromatin tends to be located towards the nuclear interior (91). Because chromatin attachment to the nuclear lamina is supported by specific protein-protein interactions (92), LLPS disruption might affect the internal structure of lamina-associated domains (decondensation of B compartments observed in 1,6-HD-treated cells) but cannot trigger their repositioning inside the nucleus, with a subsequent substantial intermingling with the active portion of the genome. Thus, the spatial segregation of active and repressed chromatin represents a stable feature of the 3D genome organization that persists under various influences such as LLPS disruption, hyperosmotic shock (75), or heat shock (93).
In conclusion, our results demonstrated that 1,6-HD-sensitive LLPS is not the primary force supporting local and global spatial genome organization but rather contributes to the fine-tuning of the 3D genome. The possibility that LLPS may be more important for 3D genome assembly under certain conditions (for example, after mitosis) and the relationships between LLPS and the other chromatin-shaping factors deserve further consideration.
DATA AVAILABILITY
The accession numbers of the published data used and re-analyzed in this work are indicated in Supplementary Table S2. Raw sequencing reads for the Hi-C and ChIP-seq libraries as well as processed Hi-C matrices, annotated compartments, TADs and loops, ChIP-seq signals and peaks are available in the GEO repository under accession number GSE138543. The code of the pentad generation and analysis with examples and documentation is provided at https://github.com/magnitov/pentads.
Supplementary Material
ACKNOWLEDGEMENTS
This work was performed using the equipment of IGB RAS facilities, supported by the Ministry of Science and Higher Education of the Russian Federation. SIM and STORM experiments were performed using the equipment purchased within the framework of Lomonosov Moscow State University development program. This research has been supported by the Interdisciplinary Scientific and Educational School of Moscow University ‘Molecular Technologies of the Living Systems and Synthetic Biology’.
Contributor Information
Sergey V Ulianov, Institute of Gene Biology Russian Academy of Science, 119334 Moscow, Russia; Faculty of Biology, Lomonosov Moscow State University, 119992 Moscow, Russia.
Artem K Velichko, Institute of Gene Biology Russian Academy of Science, 119334 Moscow, Russia; Center for Precision Genome Editing and Genetic Technologies for Biomedicine, Institute of Gene Biology Russian Academy of Sciences, 119334 Moscow, Russia; Institute for Translational Medicine and Biotechnology, Sechenov First Moscow State Medical University, 119991 Moscow, Russia.
Mikhail D Magnitov, Institute of Gene Biology Russian Academy of Science, 119334 Moscow, Russia; Center for Precision Genome Editing and Genetic Technologies for Biomedicine, Institute of Gene Biology Russian Academy of Sciences, 119334 Moscow, Russia; Department of Biological and Medical Physics, Moscow Institute of Physics and Technology (National Research University), 141701 Dolgoprudny, Russia.
Artem V Luzhin, Institute of Gene Biology Russian Academy of Science, 119334 Moscow, Russia; Center for Precision Genome Editing and Genetic Technologies for Biomedicine, Institute of Gene Biology Russian Academy of Sciences, 119334 Moscow, Russia.
Arkadiy K Golov, Institute of Gene Biology Russian Academy of Science, 119334 Moscow, Russia.
Natalia Ovsyannikova, A.N. Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119992 Moscow, Russia.
Igor I Kireev, A.N. Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119992 Moscow, Russia; V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology, and Perinatology, 117997 Moscow, Russia.
Alexey S Gavrikov, Shemyakin−Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, 117997 Moscow, Russia.
Alexander S Mishin, Shemyakin−Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, 117997 Moscow, Russia.
Azat K Garaev, Institute of Gene Biology Russian Academy of Science, 119334 Moscow, Russia.
Alexander V Tyakht, Institute of Gene Biology Russian Academy of Science, 119334 Moscow, Russia; Center for Precision Genome Editing and Genetic Technologies for Biomedicine, Institute of Gene Biology Russian Academy of Sciences, 119334 Moscow, Russia.
Alexey A Gavrilov, Institute of Gene Biology Russian Academy of Science, 119334 Moscow, Russia; Center for Precision Genome Editing and Genetic Technologies for Biomedicine, Institute of Gene Biology Russian Academy of Sciences, 119334 Moscow, Russia.
Omar L Kantidze, Institute of Gene Biology Russian Academy of Science, 119334 Moscow, Russia.
Sergey V Razin, Institute of Gene Biology Russian Academy of Science, 119334 Moscow, Russia.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Russian Science Foundation [19-74-10009]; Ministry of Science and Higher Education of the Russian Federation [075-15-2019-1661]. The open access publication charge for this paper has been waived by Oxford University Press – NAR Editorial Board members are entitled to one free paper per year in recognition of their work on behalf of the journal.
Conflict of interest statement. None declared.
REFERENCES
- 1. Lieberman-Aiden E., van Berkum N.L., Williams L., Imakaev M., Ragoczy T., Telling A., Amit I., Lajoie B.R., Sabo P.J., Dorschner M.O.et al.. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009; 326:289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dixon J.R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., Hu M., Liu J.S., Ren B.. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012; 485:376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nora E.P., Lajoie B.R., Schulz E.G., Giorgetti L., Okamoto I., Servant N., Piolot T., van Berkum N.L., Meisig J., Sedat J.et al.. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 2012; 485:381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rao S.S., Huntley M.H., Durand N.C., Stamenova E.K., Bochkov I.D., Robinson J.T., Sanborn A.L., Machol I., Omer A.D., Lander E.S.et al.. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014; 159:1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rowley M.J., Nichols M.H., Lyu X., Ando-Kuri M., Rivera I.S.M., Hermetz K., Wang P., Ruan Y., Corces V.G.. Evolutionarily conserved principles predict 3D chromatin organization. Mol. Cell. 2017; 67:837–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rowley M.J., Corces V.G.. Organizational principles of 3D genome architecture. Nat. Rev. Genet. 2018; 19:789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schwarzer W., Abdennur N., Goloborodko A., Pekowska A., Fudenberg G., Loe-Mie Y., Fonseca N.A., Huber W., Haering C.H., Mirny L.et al.. Two independent modes of chromatin organization revealed by cohesin removal. Nature. 2017; 551:51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nuebler J., Fudenberg G., Imakaev M., Abdennur N., Mirny L.A.. Chromatin organization by an interplay of loop extrusion and compartmental segregation. Proc. Natl Acad. Sci. U.S.A. 2018; 115:E6697–E6706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fudenberg G., Abdennur N., Imakaev M., Goloborodko A., Mirny L.A.. Emerging evidence of chromosome folding by loop extrusion. Cold Spring Harb. Symp. Quant. Biol. 2017; 82:45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Davidson I.F., Bauer B., Goetz D., Tang W., Wutz G., Peters J.M.. DNA loop extrusion by human cohesin. Science. 2019; 366:1338–1345. [DOI] [PubMed] [Google Scholar]
- 11. Courchaine E.M., Lu A., Neugebauer K.M.. Droplet organelles. EMBO J. 2016; 35:1603–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Banani S.F., Lee H.O., Hyman A.A., Rosen M.K.. Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017; 18:285–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Boeynaems S., Alberti S., Fawzi N.L., Mittag T., Polymenidou M., Rousseau F., Schymkowitz J., Shorter J., Wolozin B., Van Den Bosch L.et al.. Protein phase separation: a new phase in cell biology. Trends Cell Biol. 2018; 28:420–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gomes E., Shorter J.. The molecular language of membraneless organelles. J. Biol. Chem. 2019; 294:7115–7127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Strom A.R., Brangwynne C.P.. The liquid nucleome - phase transitions in the nucleus at a glance. J. Cell Sci. 2019; 132:jcs235093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Meng F., Na I., Kurgan L., Uversky V.N.. Compartmentalization and functionality of nuclear disorder: intrinsic disorder and protein-protein interactions in intra-nuclear compartments. Int. J. Mol. Sci. 2015; 17:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Uversky V.N. Intrinsically disordered proteins in overcrowded milieu: membrane-less organelles, phase separation, and intrinsic disorder. Curr. Opin. Struct. Biol. 2017; 44:18–30. [DOI] [PubMed] [Google Scholar]
- 18. Darling A.L., Liu Y., Oldfield C.J., Uversky V.N.. Intrinsically disordered proteome of human membrane-less organelles. Proteomics. 2018; 18:e1700193. [DOI] [PubMed] [Google Scholar]
- 19. Uversky V.N. Protein intrinsic disorder-based liquid–liquid phase transitions in biological systems: complex coacervates and membrane-less organelles. Adv. Colloid Interface Sci. 2017; 239:97–114. [DOI] [PubMed] [Google Scholar]
- 20. Strom A.R., Emelyanov A.V., Mir M., Fyodorov D.V., Darzacq X., Karpen G.H.. Phase separation drives heterochromatin domain formation. Nature. 2017; 547:241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Larson A.G., Elnatan D., Keenen M.M., Trnka M.J., Johnston J.B., Burlingame A.L., Agard D.A., Redding S., Narlikar G.J.. Liquid droplet formation by HP1alpha suggests a role for phase separation in heterochromatin. Nature. 2017; 547:236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Turner A.L., Watson M., Wilkins O.G., Cato L., Travers A., Thomas J.O., Stott K.. Highly disordered histone H1-DNA model complexes and their condensates. Proc. Natl Acad. Sci. U.S.A. 2018; 115:11964–11969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tatavosian R., Kent S., Brown K., Yao T., Duc H.N., Huynh T.N., Zhen C.Y., Ma B., Wang H., Ren X.. Nuclear condensates of the Polycomb protein chromobox 2 (CBX2) assemble through phase separation. J. Biol. Chem. 2019; 294:1451–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Boehning M., Dugast-Darzacq C., Rankovic M., Hansen A.S., Yu T., Marie-Nelly H., McSwiggen D.T., Kokic G., Dailey G.M., Cramer P.et al.. RNA polymerase II clustering through carboxy-terminal domain phase separation. Nat. Struct. Mol. Biol. 2018; 25:833–840. [DOI] [PubMed] [Google Scholar]
- 25. Nagulapalli M., Maji S., Dwivedi N., Dahiya P., Thakur J.K.. Evolution of disorder in Mediator complex and its functional relevance. Nucleic Acids Res. 2016; 44:1591–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Boija A., Klein I.A., Sabari B.R., Dall’Agnese A., Coffey E.L., Zamudio A.V., Li C.H., Shrinivas K., Manteiga J.C., Hannett N.M.et al.. Transcription factors activate genes through the phase-separation capacity of their activation domains. Cell. 2018; 175:1842–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cho W.K., Spille J.H., Hecht M., Lee C., Li C., Grube V., Cisse II.. Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science. 2018; 361:412–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sabari B.R., Dall’Agnese A., Boija A., Klein I.A., Coffey E.L., Shrinivas K., Abraham B.J., Hannett N.M., Zamudio A.V., Manteiga J.C.et al.. Coactivator condensation at super-enhancers links phase separation and gene control. Science. 2018; 361:eaar3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hnisz D., Shrinivas K., Young R.A., Chakraborty A.K., Sharp P.A.. A phase separation model for transcriptional control. Cell. 2017; 169:13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hahn S. Phase separation, protein disorder, and enhancer function. Cell. 2018; 175:1723–1725. [DOI] [PubMed] [Google Scholar]
- 31. Gurumurthy A., Shen Y., Gunn E.M., Bungert J.. Phase separation and transcription regulation: are super-enhancers and locus control regions primary sites of transcription complex assembly. Bioessays. 2019; 41:e1800164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nair S.J., Yang L., Meluzzi D., Oh S., Yang F., Friedman M.J., Wang S., Suter T., Alshareedah I., Gamliel A.et al.. Phase separation of ligand-activated enhancers licenses cooperative chromosomal enhancer assembly. Nat. Struct. Mol. Biol. 2019; 26:193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Guo Y.E., Manteiga J.C., Henninger J.E., Sabari B.R., Dall’Agnese A., Hannett N.M., Spille J.H., Afeyan L.K., Zamudio A.V., Shrinivas K.et al.. Pol II phosphorylation regulates a switch between transcriptional and splicing condensates. Nature. 2019; 572:543–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kantidze O.L., Razin S.V.. Weak interactions in higher-order chromatin organization. Nucleic Acids Res. 2020; 48:4614–4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Razin S.V., Gavrilov A.A.. The role of liquid-liquid phase separation in the compartmentalization of cell nucleus and spatial genome organization. Biochemistry (Mosc.). 2020; 85:643–650. [DOI] [PubMed] [Google Scholar]
- 36. Quinodoz S.A., Ollikainen N., Tabak B., Palla A., Schmidt J.M., Detmar E., Lai M.M., Shishkin A.A., Bhat P., Takei Y.et al.. Higher-order inter-chromosomal hubs shape 3D genome organization in the nucleus. Cell. 2018; 174:744–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hu S., Lv P., Yan Z., Wen B.. Disruption of nuclear speckles reduces chromatin interactions in active compartments. Epigenet. Chromatin. 2019; 12:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ricci M.A., Manzo C., Garcia-Parajo M.F., Lakadamyali M., Cosma M.P.. Chromatin fibers are formed by heterogeneous groups of nucleosomes in vivo. Cell. 2015; 160:1145–1158. [DOI] [PubMed] [Google Scholar]
- 39. Garcia H., Miecznikowski J.C., Safina A., Commane M., Ruusulehto A., Kilpinen S., Leach R.W., Attwood K., Li Y., Degan S.et al.. Facilitates chromatin transcription complex is an “accelerator” of tumor transformation and potential marker and target of aggressive cancers. Cell Rep. 2013; 4:159–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shin Y., Berry J., Pannucci N., Haataja M.P., Toettcher J.E., Brangwynne C.P.. Spatiotemporal control of intracellular phase transitions using light-activated optodroplets. Cell. 2017; 168:159–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nozaki T., Imai R., Tanbo M., Nagashima R., Tamura S., Tani T., Joti Y., Tomita M., Hibino K., Kanemaki M.T.et al.. Dynamic organization of chromatin domains revealed by super-resolution live-cell imaging. Mol. Cell. 2017; 67:282–293. [DOI] [PubMed] [Google Scholar]
- 42. Ulianov S.V., Khrameeva E.E., Gavrilov A.A., Flyamer I.M., Kos P., Mikhaleva E.A., Penin A.A., Logacheva M.D., Imakaev M.V., Chertovich A.et al.. Active chromatin and transcription play a key role in chromosome partitioning into topologically associating domains. Genome Res. 2016; 26:70–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Langmead B., Salzberg S.L.. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Imakaev M., Fudenberg G., McCord R.P., Naumova N., Goloborodko A., Lajoie B.R., Dekker J., Mirny L.A.. Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat. Methods. 2012; 9:999–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Abdennur N., Mirny L.. Cooler: scalable storage for Hi-C data and other genomically-labeled arrays. Bioinformatics. 2019; 36:311–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Davis C.A., Hitz B.C., Sloan C.A., Chan E.T., Davidson J.M., Gabdank I., Hilton J.A., Jain K., Baymuradov U.K., Narayanan A.K.et al.. The encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res. 2018; 46:D794–D801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.Genome Project Data Processing, S. . The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sanborn A.L., Rao S.S., Huang S.C., Durand N.C., Huntley M.H., Jewett A.I., Bochkov I.D., Chinnappan D., Cutkosky A., Li J.et al.. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc. Natl Acad. Sci. U.S.A. 2015; 112:E6456–E6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fudenberg G., Imakaev M., Lu C., Goloborodko A., Abdennur N., Mirny L.A.. Formation of chromosomal domains by loop extrusion. Cell Rep. 2016; 15:2038–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Crane E., Bian Q., McCord R.P., Lajoie B.R., Wheeler B.S., Ralston E.J., Uzawa S., Dekker J., Meyer B.J.. Condensin-driven remodelling of X chromosome topology during dosage compensation. Nature. 2015; 523:240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Flyamer I.M., Illingworth R.S., Bickmore W.A.. Coolpup.py: versatile pile-up analysis of Hi-C data. Bioinformatics. 2020; 36:2980–2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Durand N.C., Shamim M.S., Machol I., Rao S.S., Huntley M.H., Lander E.S., Aiden E.L.. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 2016; 3:95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dao L.T.M., Galindo-Albarran A.O., Castro-Mondragon J.A., Andrieu-Soler C., Medina-Rivera A., Souaid C., Charbonnier G., Griffon A., Vanhille L., Stephen T.et al.. Genome-wide characterization of mammalian promoters with distal enhancer functions. Nat. Genet. 2017; 49:1073–1081. [DOI] [PubMed] [Google Scholar]
- 56. Arrigoni L., Richter A.S., Betancourt E., Bruder K., Diehl S., Manke T., Bonisch U.. Standardizing chromatin research: a simple and universal method for ChIP-seq. Nucleic Acids Res. 2016; 44:e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pena-Hernandez R., Marques M., Hilmi K., Zhao T., Saad A., Alaoui-Jamali M.A., del Rincon S.V., Ashworth T., Roy A.L., Emerson B.M.et al.. Genome-wide targeting of the epigenetic regulatory protein CTCF to gene promoters by the transcription factor TFII-I. Proc. Natl Acad. Sci. U.S.A. 2015; 112:E677–E686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ramirez F., Ryan D.P., Gruning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dundar F., Manke T.. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016; 44:W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Amemiya H.M., Kundaje A., Boyle A.P.. The ENCODE blacklist: identification of problematic regions of the genome. Sci. Rep. 2019; 9:9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhang Y., Lin Y.H., Johnson T.D., Rozek L.S., Sartor M.A.. PePr: a peak-calling prioritization pipeline to identify consistent or differential peaks from replicated ChIP-Seq data. Bioinformatics. 2014; 30:2568–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lesne A., Baudement M.O., Rebouissou C., Forne T.. Exploring mammalian genome within phase-separated nuclear bodies: experimental methods and implications for gene expression. Genes. 2019; 10:1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Alberti S., Gladfelter A., Mittag T.. Considerations and challenges in studying liquid-liquid phase separation and biomolecular condensates. Cell. 2019; 176:419–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kroschwald S., Maharana S., Simon A.. Hexanediol: a chemical probe to investigate the material properties of membrane-less compartments. Matters. 2017; doi:10.19185/matters.201702000010. [Google Scholar]
- 64. Ming Y., Chen X., Xu Y., Wu Y., Wang C., Zhang T., Mao R., Fan Y.. Targeting liquid–liquid phase separation in pancreatic cancer. Transl. Cancer Res. 2019; 8:96–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Michelini F., Rossiello F., d’Adda di Fagagna F., Francia S.. RNase A treatment and reconstitution with DNA damage response RNA in living cells as a tool to study the role of non-coding RNA in the formation of DNA damage response foci. Nat. Protoc. 2019; 14:1489–1508. [DOI] [PubMed] [Google Scholar]
- 66. Molliex A., Temirov J., Lee J., Coughlin M., Kanagaraj A.P., Kim H.J., Mittag T., Taylor J.P.. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell. 2015; 163:123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Oshidari R., Huang R., Medghalchi M., Tse E.Y.W., Ashgriz N., Lee H.O., Wyatt H., Mekhail K.. DNA repair by Rad52 liquid droplets. Nat. Commun. 2020; 11:695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yamazaki T., Souquere S., Chujo T., Kobelke S., Chong Y.S., Fox A.H., Bond C.S., Nakagawa S., Pierron G., Hirose T.. Functional domains of NEAT1 architectural lncRNA induce paraspeckle assembly through phase separation. Mol. Cell. 2018; 70:1038–1053. [DOI] [PubMed] [Google Scholar]
- 69. Zhang S., Hinde E., Parkyn Schneider M., Jans D.A., Bogoyevitch M.A.. Nuclear bodies formed by polyQ-ataxin-1 protein are liquid RNA/protein droplets with tunable dynamics. Sci. Rep. 2020; 10:1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Francia S., Michelini F., Saxena A., Tang D., de Hoon M., Anelli V., Mione M., Carninci P., d’Adda di Fagagna F.. Site-specific DICER and DROSHA RNA products control the DNA-damage response. Nature. 2012; 488:231–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lin Y., Mori E., Kato M., Xiang S., Wu L., Kwon I., McKnight S.L.. Toxic PR poly-dipeptides encoded by the C9orf72 repeat expansion target LC domain polymers. Cell. 2016; 167:789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Xu J., Ma H., Jin J., Uttam S., Fu R., Huang Y., Liu Y.. Super-resolution imaging of higher-order chromatin structures at different epigenomic states in single mammalian cells. Cell Rep. 2018; 24:873–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Su J.H., Zheng P., Kinrot S.S., Bintu B., Zhuang X.. Genome-scale imaging of the 3D organization and transcriptional activity of chromatin. Cell. 2020; 182:1641–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Tan L., Xing D., Chang C.H., Li H., Xie X.S.. Three-dimensional genome structures of single diploid human cells. Science. 2018; 361:924–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Amat R., Bottcher R., Le Dily F., Vidal E., Quilez J., Cuartero Y., Beato M., de Nadal E., Posas F.. Rapid reversible changes in compartments and local chromatin organization revealed by hyperosmotic shock. Genome Res. 2019; 29:18–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Mirny L.A., Imakaev M., Abdennur N.. Two major mechanisms of chromosome organization. Curr. Opin. Cell Biol. 2019; 58:142–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ernst J., Kellis M.. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat. Biotechnol. 2010; 28:817–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Deng W., Rupon J.W., Krivega I., Breda L., Motta I., Jahn K.S., Reik A., Gregory P.D., Rivella S., Dean A.et al.. Reactivation of developmentally silenced globin genes by forced chromatin looping. Cell. 2014; 158:849–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Gibson B.A., Doolittle L.K., Schneider M.W.G., Jensen L.E., Gamarra N., Henry L., Gerlich D.W., Redding S., Rosen M.K.. Organization of chromatin by intrinsic and regulated phase separation. Cell. 2019; 179:470–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Wang L., Gao Y., Zheng X., Liu C., Dong S., Li R., Zhang G., Wei Y., Qu H., Li Y.et al.. Histone modifications regulate chromatin compartmentalization by contributing to a phase separation mechanism. Mol. Cell. 2019; 76:646–659. [DOI] [PubMed] [Google Scholar]
- 81. Plys A.J., Davis C.P., Kim J., Rizki G., Keenen M.M., Marr S.K., Kingston R.E.. Phase separation of Polycomb-repressive complex 1 is governed by a charged disordered region of CBX2. Genes Dev. 2019; 33:799–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Strickfaden H., Tolsma T.O., Sharma A., Underhill D.A., Hansen J.C., Hendzel M.J.. Condensed chromatin behaves like a solid on the mesoscale in vitro and in living cells. Cell. 2020; 183:1772–1784. [DOI] [PubMed] [Google Scholar]
- 83. Stephens A.D., Liu P.Z., Kandula V., Chen H., Almassalha L.M., Herman C., Backman V., O’Halloran T., Adam S.A., Goldman R.D.et al.. Physicochemical mechanotransduction alters nuclear shape and mechanics via heterochromatin formation. Mol. Biol. Cell. 2019; 30:2320–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Itoh Y., Iida S., Tamura S., Nagashima R., Shiraki K., Goto T., Hibino K., Ide S., Maeshima K.. 1,6-hexanediol rapidly immobilizes and condenses chromatin in living human cells. Life Sci. Alliance. 2021; 4:e202001005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Razin S.V., Ulianov S.V.. Divide and rule: phase separation in eukaryotic genome functioning. Cells. 2020; 9:2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Razin S.V., Gavrilov A.A., Pichugin A., Lipinski M., Iarovaia O.V., Vassetzky Y.S.. Transcription factories in the context of the nuclear and genome organization. Nucleic Acids Res. 2011; 39:9085–9092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Cook P.R., Marenduzzo D.. Transcription-driven genome organization: a model for chromosome structure and the regulation of gene expression tested through simulations. Nucleic Acids Res. 2018; 46:9895–9906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Mitchell J.A., Fraser P.. Transcription factories are nuclear subcompartments that remain in the absence of transcription. Genes Dev. 2008; 22:20–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Sanulli S., Gross J.D., Narlikar G.J.. Biophysical properties of HP1-mediated heterochromatin. Cold Spring Harb. Symp. Quant. Biol. 2019; 84:217–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Sanulli S., Geeta J.N.. Liquid-like interactions in heterochromatin: implications for mechanism and regulation. Curr. Opin. Cell Biol. 2020; 64:90–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Buchwalter A., Kaneshiro J.M., Hetzer M.W.. Coaching from the sidelines: the nuclear periphery in genome regulation. Nat. Rev. Genet. 2019; 20:39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Shevelyov Y.Y., Ulianov S.V.. The nuclear lamina as an organizer of chromosome architecture. Cells. 2019; 8:136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Ray J., Munn P.R., Vihervaara A., Lewis J.J., Ozer A., Danko C.G., Lis J.T.. Chromatin conformation remains stable upon extensive transcriptional changes driven by heat shock. Proc. Natl Acad. Sci. U.S.A. 2019; 116:19431–19439. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession numbers of the published data used and re-analyzed in this work are indicated in Supplementary Table S2. Raw sequencing reads for the Hi-C and ChIP-seq libraries as well as processed Hi-C matrices, annotated compartments, TADs and loops, ChIP-seq signals and peaks are available in the GEO repository under accession number GSE138543. The code of the pentad generation and analysis with examples and documentation is provided at https://github.com/magnitov/pentads.