

Abstract
Idiopathic Parkinson’s disease is the second most common neurodegenerative disease and is estimated to be approximately 30% heritable. Genome wide association studies have revealed numerous loci associated with risk of development of Parkinson’s disease. The majority of genes identified in these studies are expressed in glia at either similar or greater levels than their expression in neurons, suggesting that glia may play a role in Parkinson’s disease pathogenesis. The role of individual glial risk genes in Parkinson’s disease development or progression is unknown, however. We hypothesized that some Parkinson’s disease risk genes exert their effects through glia. We developed a Drosophila model of α-synucleinopathy in which we can independently manipulate gene expression in neurons and glia. Human wild type α-synuclein is expressed in all neurons, and these flies develop the hallmarks of Parkinson’s disease, including motor impairment, death of dopaminergic and other neurons, and α-synuclein aggregation. In these flies, we performed a candidate genetic screen, using RNAi to knockdown 14 well-validated Parkinson’s disease risk genes in glia and measuring the effect on locomotion in order to identify glial modifiers of the α-synuclein phenotype. We identified 4 modifiers: aux, Lrrk, Ric, and Vps13, orthologs of the human genes GAK, LRRK2, RIT2, and VPS13C, respectively. Knockdown of each gene exacerbated neurodegeneration as measured by total and dopaminergic neuron loss. Knockdown of each modifier also increased α-synuclein oligomerization. These results suggest that some Parkinson’s disease risk genes exert their effects in glia and that glia can influence neuronal α-synuclein proteostasis in a non-cell-autonomous fashion. Further, this study provides proof of concept that our novel Drosophila α-synucleinopathy model can be used to study glial modifier genes, paving the way for future large unbiased screens to identify novel glial risk factors that contribute to PD risk and progression.
Keywords: Parkinson’s disease, Glia, GWAS, LRRK2, Autophagy
1. Introduction
Parkinson’s disease is the second most common neurodegenerative disorder and is defined by α-synuclein aggregation and death of dopaminergic neurons. Approximately 5–10% of Parkinson’s disease is monogenic.(Lesage and Brice, 2009) To date, point mutations in 20 genes have been reported as causative of familial Parkinson’s disease, (Kim and Alcalay, 2017; Blauwendraat et al., 2020) although of these 20, only LRRK2 and GBA mutations are relatively common. Beyond familial Parkinson’s disease, so called “idiopathic” Parkinson’s disease is also estimated to be 30% heritable,(Keller et al., 2012) and genome wide association studies (GWAS) have now identified nearly 100 loci associated with risk of idiopathic Parkinson’s disease.(Nalls et al., 2019) GWAS for Parkinson’s disease demonstrate high face validity, in that several of the same genes that cause monogenic forms of Parkinson’s disease when mutated are also GWAS hits, including α-synuclein (SNCA), LRRK2, GBA, and Vps13C, suggesting that common variants in these genes influence risk of “idiopathic” Parkinson’s disease.
There are many challenges in moving from GWAS results to a biological understanding of disease, however. First, GWAS identify variations in allele frequency in single nucleotide polymorphisms (SNPs), which may or may not regulate expression of nearby genes. Thus, there is a need to combine GWAS with expression quantitative trait loci (eQTL) analysis to prioritize potentially causative genes.(Li et al., 2019; Kia et al., 2019; Grenn et al., 2020) Second, GWAS are agnostic to the cell type of origin of disease and may reflect signals coming from diverse neuronal(Nalls et al., 2019) and non-neuronal(Kia et al., 2019; Reynolds et al., 2019) cell types within the brain, as well as cells outside of the brain.(Reynolds et al., 2019; Pierce and Coetzee, 2017; Coetzee et al., 2016; Gagliano et al., 2016) Although computational analyses are invaluable tools in addressing these questions, even the best approaches available today are dependent on the input data, which may be limited. Thus, there is a strong need for mechanistic studies to validate the role of GWAS nominated genes in disease pathogenesis and to determine in which cell types they are exerting their effects.(Pierce et al., 2020)
Such mechanistic studies rely on model organisms, and emerging studies in primary mouse cells as well as human induced pluripotent stem cell (iPSC) derived models support a pathogenic role for some Parkinson’s disease GWAS candidate genes in glia, particularly astrocytes. Eight of the genes implicated in monogenic Parkinson’s disease have a known function in astrocytes,(Booth et al., 2017) including LRRK2 and GBA, which are also GWAS hits. LRRK2 G2019S expressing iPSC-derived astrocytes have downregulated expression of extracellular matrix proteins,(Booth et al., 2019) accelerated ER stress,(Lee et al., 2019) and impaired autophagy leading to non-cell-autonomous neurodegeneration.(di Domenico et al., 2019) iPSC-derived astrocytes carrying different neuropathic or nonneuropathic GBA mutations have varying degrees of astrogliosis and lysosomal dysfunction,(Aflaki et al., 2020) and GBA D409V knockin mouse astrocytes demonstrate lysosomal morphology and functional defects as well as defects in cytokine production, which, interestingly, were normalized by inhibition of LRRK2 kinase activity,(Sanyal et al., 2020) suggesting potential crosstalk between LRRK2 and GBA within astrocytes. These cell culture studies have provided important mechanistic insight regarding the function of LRRK2 and GBA in astrocytes, but little is known about their role in glia in vivo. Additionally, the time-intensive nature of iPSC-based co-culture systems makes a higher throughput approach to glial gene investigation challenging. Finally, little is known about the role of Parkinson’s disease GWAS candidate genes in other glial cells beyond astrocytes.
To address these challenges and investigate the role of Parkinson’s disease GWAS candidates in glia in vivo in a systematic manner, we developed a novel Drosophila Parkinson’s disease model(Ordonez et al., 2018) in which gene expression can be independently manipulated in neurons and glia(Olsen and Feany, 2019) using the (Potter et al., 2010) and UAS-Gal4 expression systems. (Brand and Perrimon, 1993) In this model, human wild type α-synuclein is expressed in all neurons, and flies develop neurodegeneration, loss of dopaminergic neurons, and motor dysfunction. We performed a candidate screen, knocking down a panel of well-validated GWAS nominated genes in glia in order to identify those that enhanced neuronal α-synuclein toxicity. Specifically, we identified Drosophila orthologs of LRRK2, GAK, Vps13C, and RIT2 as glial enhancers. We confirmed that glial knockdown of these genes exacerbated neurodegeneration and loss of dopaminergic neurons. We then investigated the effects of gene knockdown on proteostasis, impairment in which underlies Parkinson’s disease pathology. Interestingly, while all modifiers enhanced neurodegeneration, they had divergent effects on α-synuclein aggregation and impairment of autophagy, suggesting that different Parkinson’s disease risk genes effect different cellular processes in glia and providing evidence for multiple mechanisms of non-cell-autonomous neurodegeneration.
2. Materials and methods
2.1. Drosophila
All fly crosses and aging were performed at 25 °C. All experiments were performed at 10 days post-eclosion unless otherwise noted in the figure legends. All experiments include both male and female flies in which wild type human α-synuclein is expressed in neurons using the pan-neuronal driver neuronal-synaptobrevin (nSyb)-QF2. Control flies include the driver but lack transgenic human α-synuclein. Additionally, flies contain either the pan-glial driver repo-Gal4 or the astrocyte-like glia driver alrm-Gal4 for knockdown of genes of interest.
Transgenic RNAi stocks were obtained from the Bloomington Drosophila Stock Center and include UAS-Vps13 RNAi HMS01715 (Vps13 RNAi #1), UAS-Vps13 RNAi HMS02460 (Vps13 RNAi #2), UAS-aux RNAi HMS01935 (aux RNAi #1), UAS-aux RNAi GL00213 (aux RNAi #2), UAS-Ric RNAi GL01247 (Ric RNAi #1), UAS-Ric RNAi JF02670 (Ric RNAi #2), UAS-fray RNAi HMJ02228 (fray RNAi #1), UAS-fray RNAi HMC04151 (fray RNAi #2), UAS-Syt4 RNAi HMS01934 (Syt4 RNAi #1), and UAS-Syt4 RNAi JF02272 (Syt4 RNAi #2). The UAS-Lrrk RNAi #22139 (Lrrk RNAi #2) transgenic line was obtained from Vienna Drosophila Resource Center. The following Drosophila stocks were kindly provided by the indicated investigators: 1. nSyb-QF2 by Dr. Christopher Potter, 2. alrm-Gal4 by Dr. Marc Freeman, 3. UAS-Lrrk RNAi #1–1–37 (Lrrk RNAi #1) by Dr. Bingwei Lu. Most transgenic strains used in the study were created using w as a selectable marker and thus carry a w mutation on the X chromosome. Transgenic RNAi lines created by the Transgenic RNAi Project (TRiP) at Harvard Medical School were created in a y1 v1 (HMS01935, GL01247, JF02670, HMJ02228, HMS01934, JF02272) or y1 sc v1 (HMS01715, HMS02460, GL00213, HMC04151) genetic background.
Two RNAi lines were used to identify locomotor deficits in the genetic screen, and a single RNAi was used for all subsequent confirmatory studies. The RNAi lines used in the confirmatory studies were UAS-Vps13 RNAi HMS02460, UAS-aux RNAi GL00213, UAS-Lrrk RNAi #1–1–37, and UAS-Ric RNAi JF02670. Control RNAi lines used to confirm phenotype specificity were UAS-mCherry RNAi, UAS-EGFP RNAi, and UAS-Luciferase RNAi from the Transgenic RNAi Project (TRiP).
2.2. Immunohistochemistry and immunofluorescence
Flies were fixed in formalin and embedded in paraffin. Either 2 or 4 μm serial frontal sections were prepared through the entire fly brain. Slides were processed through xylene, ethanols, and into water. For neuron counts, slides were stained with hematoxylin. For immunohistochemistry, microwave antigen retrieval or pressure cooker antigen retrieval with 10 mM sodium citrate, pH 6.0, was performed. Slides were blocked in 2% milk in PBS with 0.3% triton X-100 for 1 h then incubated with appropriate primary antibody in 2% milk in PBS with 0.3% triton X-100 at room temperature overnight. Primary antibodies used include tyrosine hydroxylase (1:200 to 1:500, mouse, Immunostar), α-synuclein 5G4 (1:50,000, mouse, Millipore), ref. (Kim and Alcalay, 2017) p/p62 (1:5000 rabbit Sarkar et al., n.d.), Atg8a/LC3 (clone EIJ4E, 1:2000, rabbit, Cell Signaling). For immunohistochemistry, slides were incubated in biotin-conjugated secondary antibodies in 2% milk in PBS with 0.3% triton X-100 for 1 h (1:200, Southern Biotech) followed by avidin-biotin-peroxidase complex (Vectastain Elite) in PBS for 1 h. Histochemical detection was performed with diaminobenzidine (ImmPACT DAB, Vector). For immunofluorescence, slides were incubated with fluorophore-conjugated secondary antibodies in 2% milk in PBS with 0.3% triton X-100 for 1 h (1:200, Alexa 488 or Alexa 555, Invitrogen) then mounted with DAPI-containing Fluoromount medium (Southern Biotech). Immunofluorescence microscopy was performed on a Zeiss LSM 800 confocal microscope. Images were processed using Fiji.
2.3. Quantification of total cell counts
Single slice images of the anterior medulla from hematoxylin-stained formalin-fixed parrafin-embedded tissue were captured at 40× magnification using brightfield microscopy. One image (slice) per fly and 6 flies per genotype were used for quantification. The number of nuclei in each tissue section was counted and normalized to the area of the section.
2.4. Quantification of TH+ neuron counts and α-synuclein aggregates
Single slice images of the anterior medulla from immunofluorescence-stained formalin-fixed parrafin-embedded tissue were captured on a confocal microscope at 63× magnification. One image (slice) per fly and 6 flies per genotype were used for quantification. The number of TH+ cells or α-synuclein aggregates in each tissue section was counted and normalized to the area of the section.
2.5. Western blotting
Fly heads were dissected then homogenized in 2× Laemmli buffer, boiled for 10 min, and centrifuged. SDS-PAGE was performed (Lonza) followed by transfer to nitrocellulose membrane (Bio-Rad) and microwave antigen retrieval in PBS. Membranes were blocked in 2% milk in PBS with 0.05% Tween-20 for 1 h, then immunoblotted with appropriate primary antibody in 2% milk in PBS with 0.05% Tween-20 overnight at 4 °C. Primary antibodies used include α-synuclein H3C (1:10,000 to 1:100,000, mouse, Developmental Studies Hybridoma Bank), α-synuclein clone 42 (1:5000 mouse, BD Bioscience), and phospho-serine 129 α-synuclein (1:5000, rabbit, Abcam). Membranes were incubated with appropriate horseradish peroxidase-conjugated secondary antibodies (1:50,000) in 2% milk in PBS with 0.05% Tween-20 for 3 h. Signal was developed with enhanced chemiluminescence (Thermo Scientific).
2.6. Oligomer assay
20 fly heads per genotype were homogenized in 20 ul TNE lysis buffer (10 mm Tris HCl, 150 mM NaCl, 5 mM EGTA, 0.5% nonidet-40) supplemented with HALT protease and phosphatase inhibitor (Roche). The homogenate was briefly spun down to remove debris. The remaining supernatant was ultracentrifuged at 100,000 × g for 1 h at 4C. The supernatant was transferred to a new tube and combined with 2× Laemelli buffer at a 1:1 ratio. SDS-PAGE was then performed as above, except without boiling samples and without microwave antigen retrieval.
2.7. Locomotion assay
Adult flies were aged in vials containing 9–14 flies per vial. At day 10 post-eclosion, flies were transferred to a clean vial (without food) and given 1 min to acclimate to the new vial. The vial was then gently tapped three times to trigger the startle induced locomotion response, then placed on its side for 15 s. The percentage of flies still in motion was then recorded. Differences between genotypes were measured and statistical significance assessed by one-way ANOVA.
2.8. Statistics
All statistical analysis aside was performed using GraphPad Prism version 7.0a.
2.9. Data availability
The data that support the findings of this study are available from the corresponding author upon request.
3. Results
3.1. Parkinson’s disease risk genes are expressed in glia
In the most current Parkinson’s disease GWAS meta-analysis, Nalls et al. identify 90 independent loci associated with risk of Parkinson’s disease.(Nalls et al., 2019) Using publicly available human bulk RNAseq data,(Zhang et al., 2016) we examined expression of the nearest candidate gene for each locus across neurons, fetal astrocytes, mature astrocytes, microglia, oligodendrocytes and endothelial cells (Fig. 1). In some instances, two loci mapped to the same nearest gene. Further, 2 genes were not in the Zhang et al(Zhang et al., 2016) database (GS1–124 K5.11, LINC00693) and an additional 7 genes were in the database but not meaningfully expressed by any of the brain cell populations (CASC16, CD19, CRHR1, HLA-DRB5, SPPL2B, TRIM40, WNT3). Among the remaining 78 genes, only 22 genes (28.5% of the total) were most highly expressed in neurons, and of those, for only 6 genes (SH3GL2, INPP5F, SNCA, ITGA8, PAM, and DLG2) was the neuronal expression greater than 50% of the total expression of that gene. Thus, many genes implicated in Parkinson’s disease risk are expressed predominantly in glia or somewhat ubiquitously throughout the brain. A similar distribution between neuronal versus non-neuronal predominant expression was seen among the genes identified by Nalls et al(Nalls et al., 2019) as likely expression quantitative loci (indicated by diamond symbol in Fig. 1).
Fig. 1.
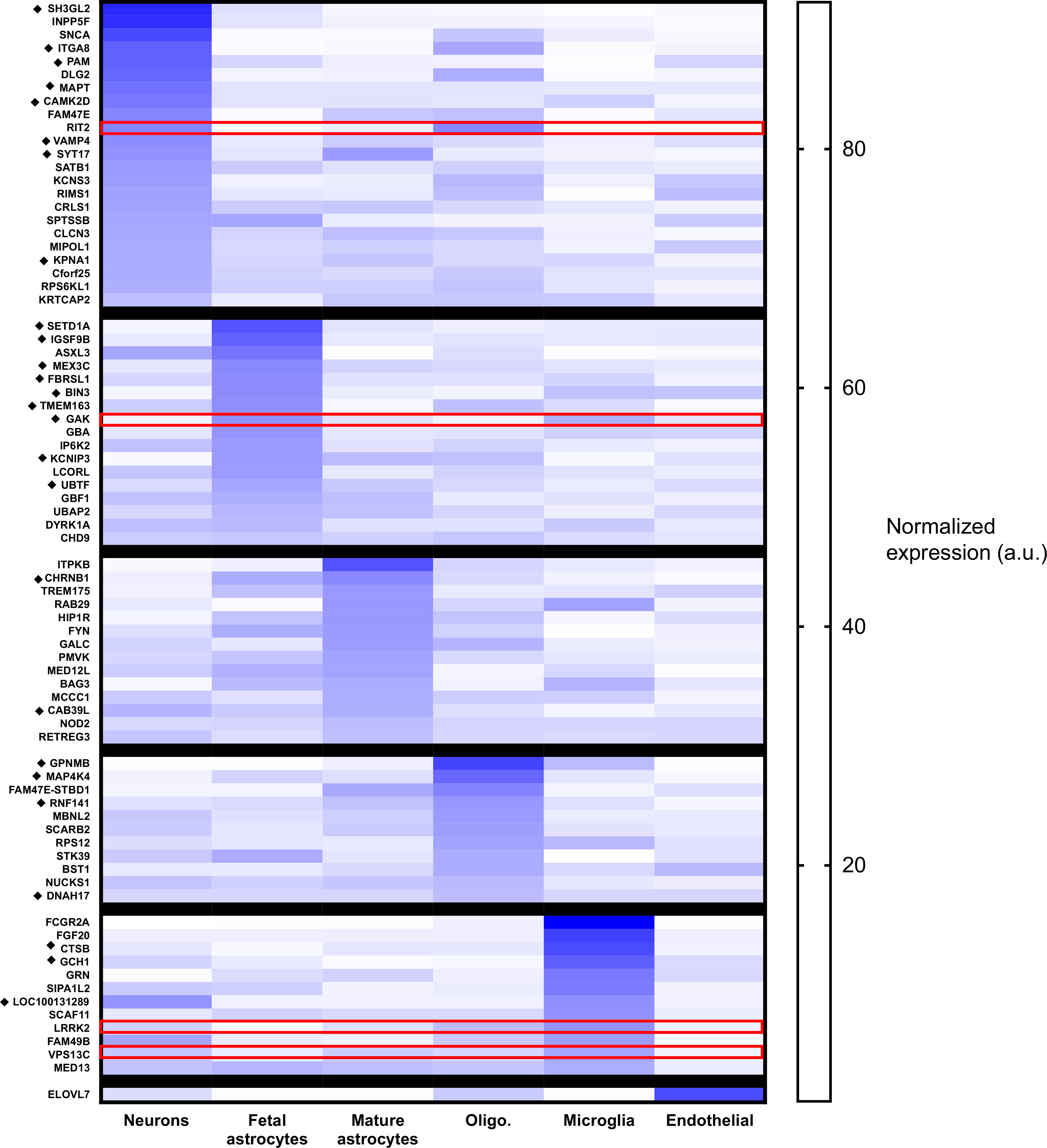
Parkinson’s disease GWAS candidate genes are expressed in glia. The figure demonstrates the relative expression of Parkinson’s disease GWAS candidate genes across glial subtypes using publicly available RNAseq data: Zhang Y, Sloan SA, Clarke LE, et al. Purification and functional characterization of human astrocytes. Neuron. 2016. Oligo. = oligodendrocytes. The diamond symbol (◆) indicates genes identified as likely expression quantitative loci by Nalls et al. Expression pattern for the human orthologs of the Drosophila genes further described in this manuscript are highlighted in red. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.2. A Drosophila model for identifying glial modifiers
We hypothesized that some Parkinson’s disease GWAS candidate genes might exert their effects in glia. To investigate this, we developed a Drosophila model of α-synucleinopathy(Ordonez et al., 2018) in which it is possible to independently manipulate gene expression in glia.(Olsen and Feany, 2019) The flies express human α-synuclein in all neurons, and we performed a candidate screen in order to identify glial enhancers of neuronal α-synuclein toxicity. In glia, we knocked down the Drosophila orthologs of 14 strong GWAS candidate genes (14 genes with genome wide significance,(Nalls et al., 2014) p < 1×−08, Table 1) and measured the effect on locomotion. MAPT and DDRGK1 were not included in the screen due to having expression largely restricted to neurons and lack of available reagents, respectively.
Table 1:
Genes tested in Drosophila screen. The human GWAS nominated gene is shown in the left hand column and the Drosophila ortholog(s) on the right.
| Genes tested | |
|---|---|
|
| |
| GWAS Candidate | Drosophila Ortholog |
|
| |
| DGKQ | CG31140 |
| FGF20 | bnl |
| GAK | aux |
| GBA | CG31148, CG31414 |
| GCH1 | Pu |
| INPP5f | CG7956 |
| LRRK2 | Lrrk |
| MCCC1 | CG2118 |
| RIT2 | Ric |
| SCARB2 | Emp |
| STK39 | fray |
| STX1B | Syk1A |
| SYT11 | Syt4 |
| VPS13C | Vps13 |
Six genes were identified as enhancers in the initial screen: aux, fray, Lrrk, Ric, Syt4, and Vps13. Of these, 4 were selected for further analyses based on amplitude of effect and confirmation with a second RNAi line. Syt4 was excluded because there was only modest (though statistically significant) enhancement of the phenotype, not meeting the predetermined threshold of biological significance of 25% decrement in locomotion (Supplemental Fig. 1A). fray was excluded because the second RNAi line resulted in significant toxicity in control flies (Supplemental Fig. 1B). In the case of aux, there was significant toxicity in control flies when the gene was knocked down in all glia using the pan-glial repo driver (data not shown). Therefore, the astrocyte-like glia driver alrm-Gal4 was used for all subsequent studies. Thus, the final experimental conditions were pan-glial knockdown of Lrrk, Ric, and Vps13 (Fig. 2A), and astrocyte-like glia knockdown of aux (Fig. 2B). To confirm that the effect on behavior was specific and not due to genetic background effects, we also expressed multiple control RNAi constructs in glia, finding no significant decrement in locomotion (Supplemental Fig. 1C). Additionally, we validate the knockdown of each gene by qRT-PCR (Supplemental Fig. 2).
Fig. 2.
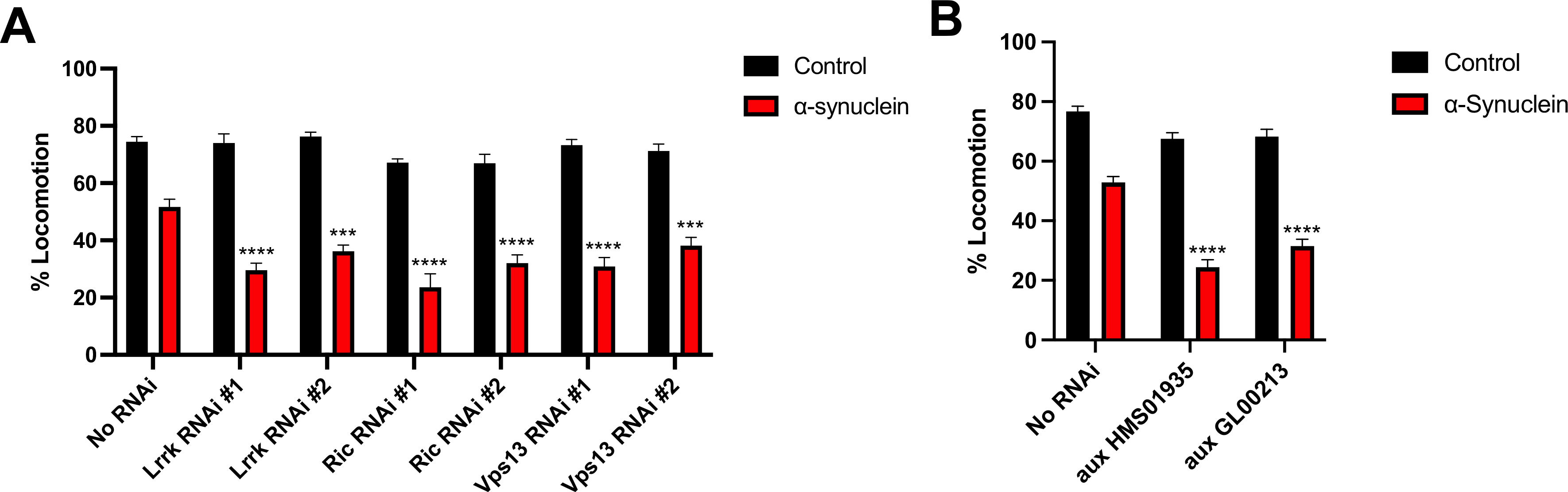
Glial knockdown of aux, Lrrk, Ric, and Vps13 exacerbates the locomotor deficit in α-synuclein expressing flies. A. Lrrk, Ric, and Vps13 were knocked down using the pan-glial driver repo-Gal4. The genotypes of control flies (black) and α-synuclein flies (red) are nSybQF2, repo-Gal4/+; and QUAS-α-Syn, nSybQF2, repo-Gal4/+, respectively. B. aux was knocked down using the astrocyte-like-glia driver alrm-Gal4. The genotypes of control flies (black) and α-synuclein flies (red) are nSybQF2, alrm-Gal4/+; and QUAS-α-Syn, nSybQF2, alrm-Gal4/+, respectively. Locomotion assay was performed 10 days post-eclosion. N = minimum 60 flies per genotype, 6 biological replicates of 9–14 flies/vial. * = p < 0.05, ** = p < 0.01, *** = p < 0.005, *** = p < 0.001. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.3. Knockdown of glial genes enhances neurodegeneration through non-cell-autonomous effects on proteostasis
We next sought to confirm that the knockdown of the glial modifiers enhanced locomotor impairment due to enhanced neurodegeneration, rather than through a symptomatic effect on behavior. We quantified both total numbers of cells (Fig. 3A–B) and the number of dopaminergic neurons (Fig. 3C–D) in the anterior medulla, confirming a decrease in both. The anterior medulla is an easily identified, bilateral anatomical structure with robust pathology and many cell bodies, the vast majority of which are neuronal.(Raji and Potter, 2021)
Fig. 3.
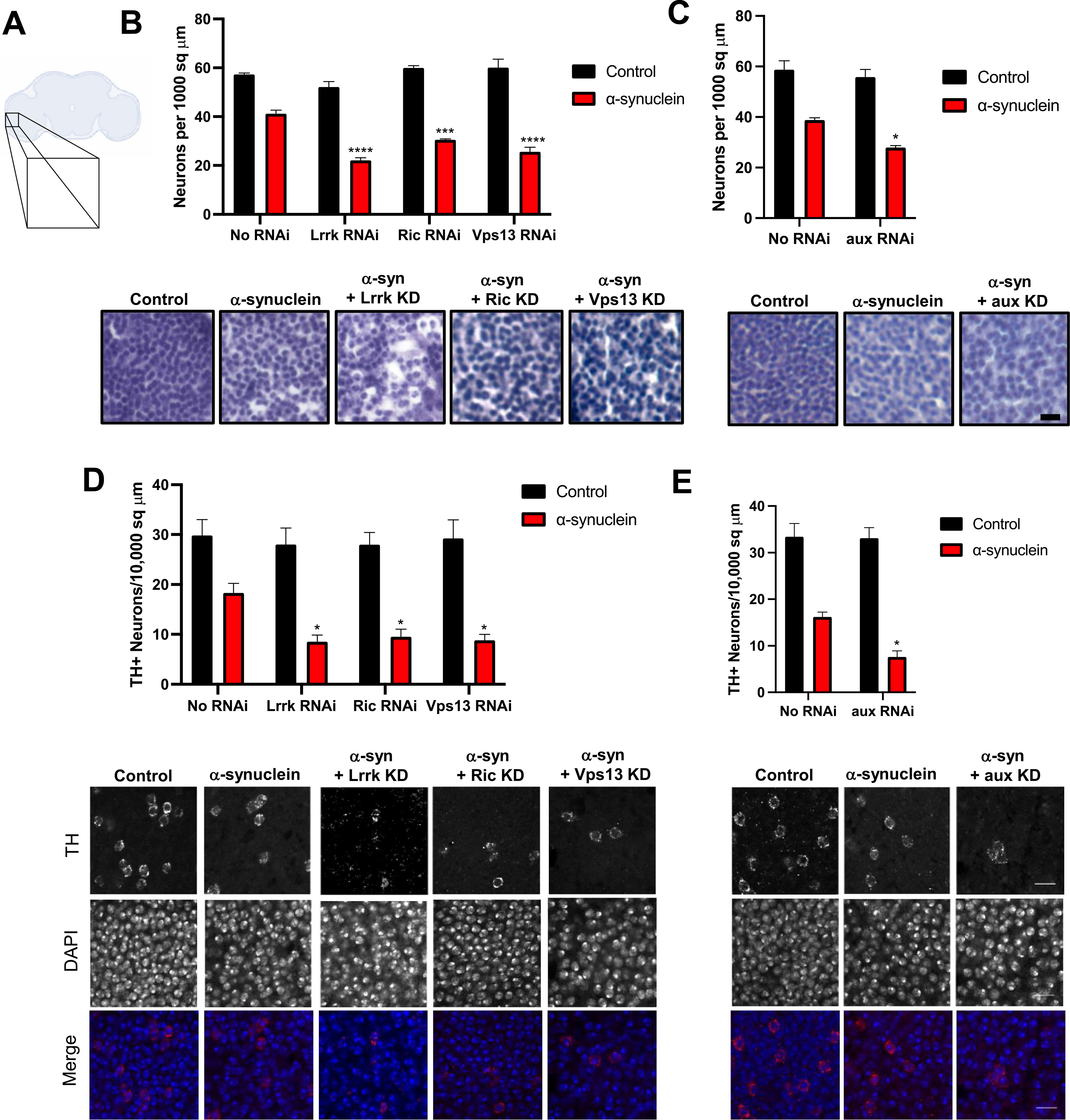
Glial knockdown of aux, Lrrk, Ric, and Vps13 increases α-synuclein induced neurodegeneration. A. schematic of fly brain. Inset corresponds to the medulla. B–C. Total cell counts were measured by staining with hematoxylin. D-E. Immunofluorescence was performed using anti-tyrosine hydroxylase to identify dopaminergic neurons. Nuclei are stained with DAPI. In B and D the genotypes of control flies (black) and α-synuclein flies (red) that lack an RNAi are nSybQF2, repo-Gal4/+ and QUAS-α-Syn, nSybQF2, repo-Gal4/+, respectively. In C and E, the genotypes of control flies (black) and α-synuclein flies (red) that lack an RNAi are nSybQF2, alrm-Gal4/+ and QUAS-α-Syn, nSybQF2, alrm-Gal4/+, respectively. N = 6 flies per genotype. * = p < 0.05, ** = p < 0.01, *** = p < 0.005, *** = p < 0.001. Scale bar = 5 μm. All experiments were performed at 10 days post-eclosion. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
To determine the mechanism of neurodegeneration, we turned to α-synuclein. Interestingly, although knockdown of each glial modifier enhanced neurodegeneration, only Lrrk and Ric knockdown led to an increase in the numbers of large α-synuclein aggregates, whereas Vps13 knockdown did not affect the number of aggregates (Fig. 4A, C), and aux knockdown led to a decrease in aggregates (Fig. 4B, D). To confirm that none of the modifiers altered expression of the α-synuclein transgene, we measured α-synuclein by immunoblotting at day 1 post-eclosion. Additionally, none of the modifiers affected baseline phosphorylation of α-synuclein at serine 129 (Supplemental Fig. 3).
Fig. 4.
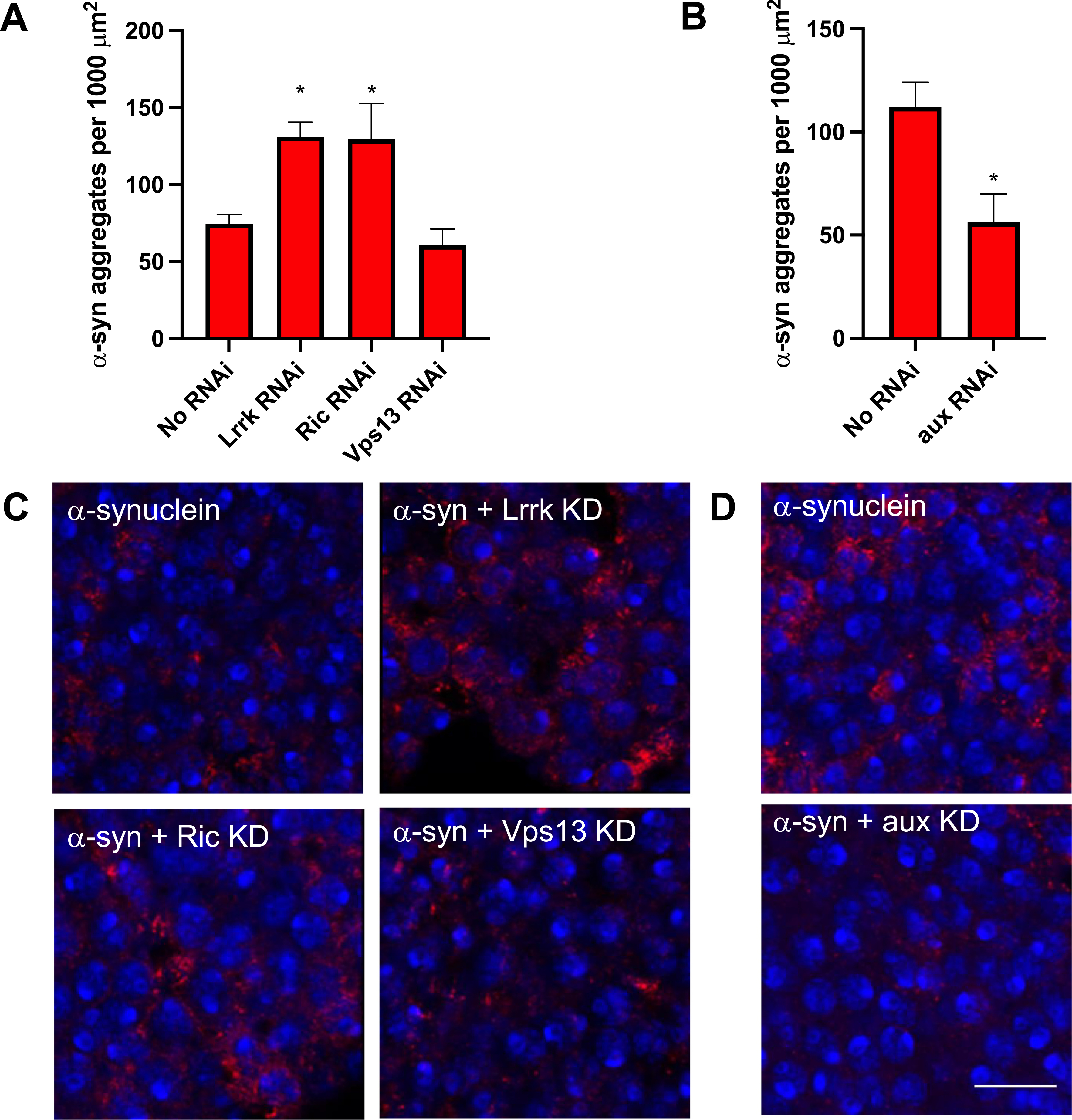
Glial modifiers have divergent effects on α-synuclein aggregation. α-synuclein immunofluorescence was performed using an anti-α-synuclein antibody that preferentially recognizes aggregated α-synuclein. Nuclei are stained with DAPI. In A and C the genotype of α-synuclein flies that lack an RNAi is QUAS-α-Syn, nSybQF2, repo-Gal4/+. In B and D the genotype of α-synuclein flies that lack an RNAi is QUAS-α-Syn, nSybQF2, alrm-Gal4/+. N = 6 flies per genotype. * = p < 0.05. Scale bar = 10 μm. All experiments were performed at 10 days post-eclosion.
There has been extensive debate regarding which molecular species of α-synuclein are pathogenic, with some evidence suggesting that α-synuclein oligomers or fibrils, rather than Lewy bodies or large Lewy body-like inclusions, drive pathology (for recent reviews, see(Bengoa-Vergniory et al., 2017; Alam et al., 2019)). We therefore examined high molecular weight species of α-synuclein by immunoblotting, finding that knockdown of each glial modifier led to an increase in α-synuclein oligomers (Fig. 5). Collectively, these data suggest that manipulation of GWAS candidate genes in glia can enhance neurodegeneration in a non-cell-autonomous manner. Further, they suggest that α-synuclein oligomers, rather than large aggregates, are responsible for this effect.
Fig. 5.
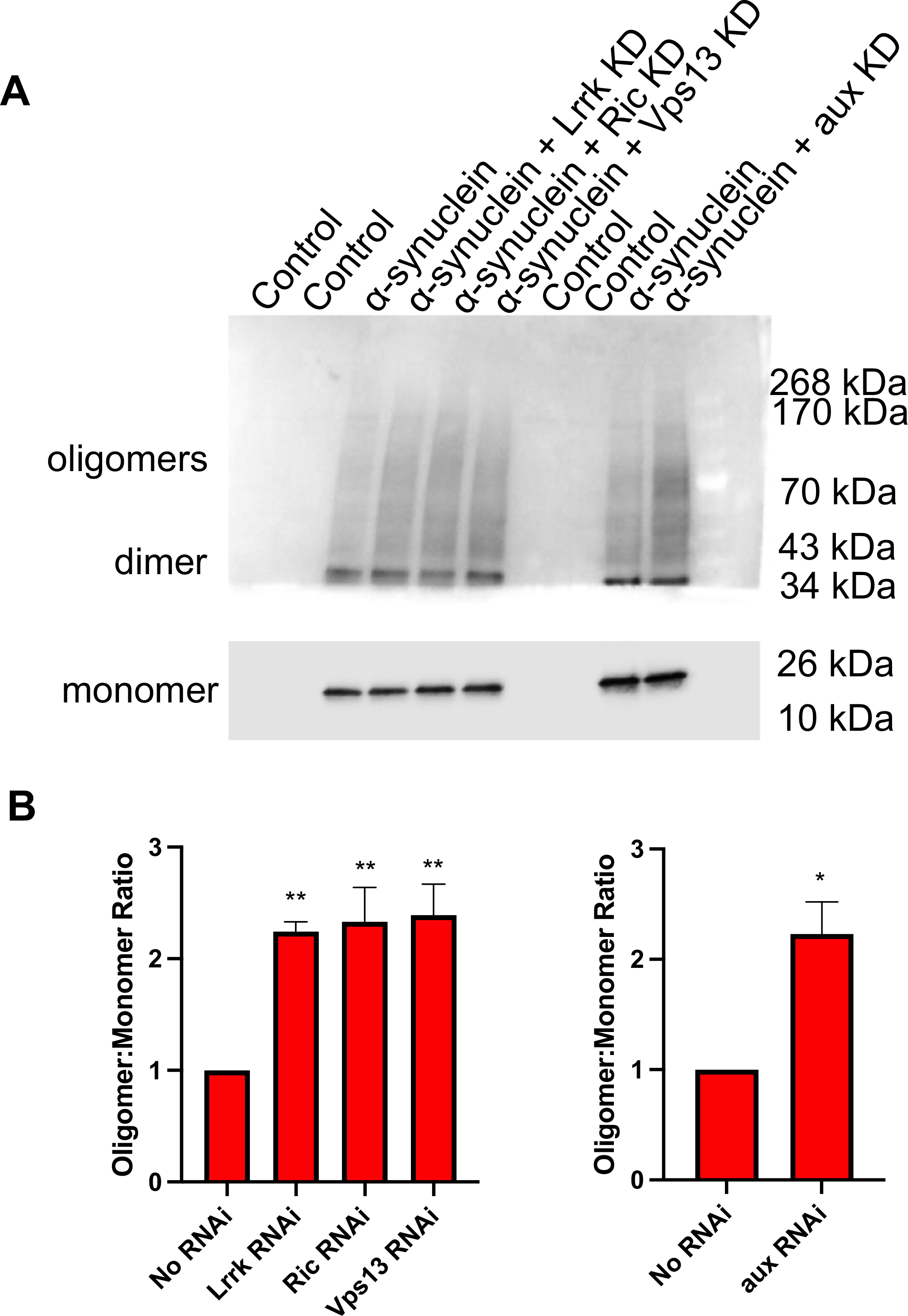
Glial modifiers increase α-synuclein oligomerization. A. Representative immunoblot using. This immunoblot was cut prior to application of the primary antibody. The top portion demonstrating oligomers and dimers was exposed for 1 min and the bottom portion demonstrating the α-synuclein monomers was exposed for 6 s. B. Quantification from 4 independent experiments. The genotype of control or α-synuclein flies that lack an RNAi is nSybQF2, repo-Gal4/+ or QUAS-α-Syn, nSybQF2, repo-Gal4/+, respectively, in the left half of the immunoblot and left panel of B. The genotype of control or α-synuclein flies that lack an RNAi is nSybQF2, alrm-Gal4/+ or QUAS-α-Syn, nSybQF2, alrm-Gal4/+, respectively, in the right half of the immunoblot and the right panel of B. All experiments were performed at 10 days post-eclosion using homogenized fly heads.
3.4. Different glial modifiers affect neuronal proteostasis through different mechanisms
To further explore the effects of glial modifiers on proteostasis, we examined the lysosomal-autophagy system. As we have previously demonstrated,(Sarkar et al., n.d.) autophagic flux is impaired in the α-synuclein-expressing flies, leading to accumulation of autophagosomes. Autophagic flux and autophagosomes can be measured in Drosophila by immunostaining for ref. (Kim and Alcalay, 2017)p and Atg8a, Drosophila orthologs of p62 and LC3, respectively. LC3 is involved in phagophore and autophagosome formation as well as selection of targets for degradation, and p62 mediates degradation of ubiquitinated proteins by binding to ubiquitin and LC3. Glial Lrrk knockdown led to a further increase in Atg8a (LC3) and ref. (Kim and Alcalay, 2017)p (p62) puncta beyond that induced by neuronal α-synuclein alone. In contrast, knockdown of Vps13 and Ric had no effect on these markers, and knockdown of aux decreased them (Fig. 6), suggesting divergent effects of the modifiers on autophagy. The increase or decrease in Atg8a (LC3) and ref. (Kim and Alcalay, 2017) p (p62) puncta seen with glial Lrrk knockdown and glial aux knockdown, respectively, appears to be occurring in neurons, as the puncta co-localize with α-synuclein (Supplemental Fig. 4). We have previously demonstrated that knockdown of Lrrk directly in neurons exacerbates the impairment in autophagy induced by α-synuclein,(Sarkar et al., n.d.) suggesting that inhibiting Lrrk expression causes similar downstream cell biological effects in neurons regardless of whether the inhibition occurs in neurons or glia.
Fig. 6.
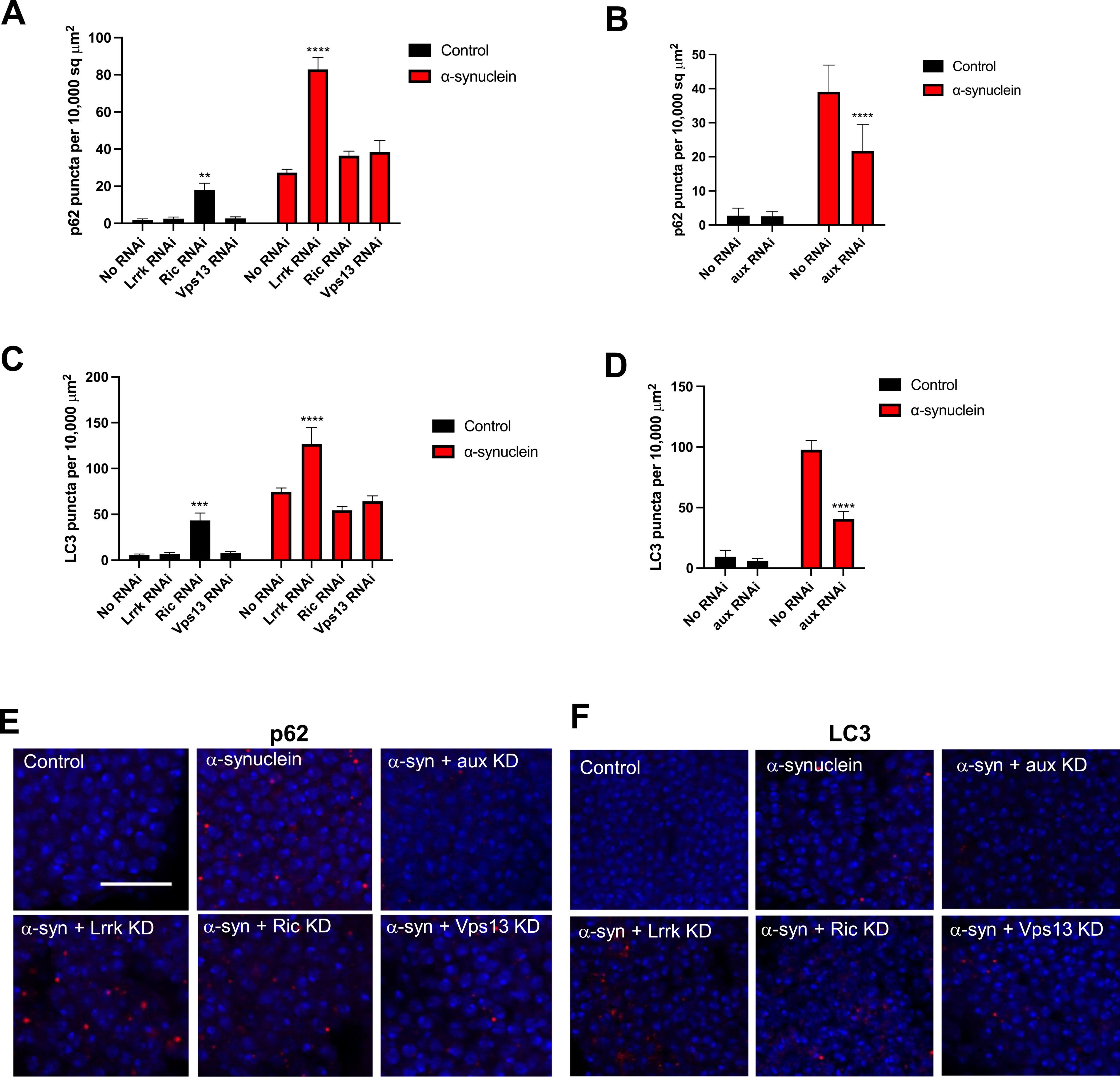
Glial Lrrk knockdown impairs autophagy. p62 (A, C, E) and LC3 (B, D, F) were measured by immunofluorescence. The antibodies used are ref. (Kim and Alcalay, 2017)p/p62, Atg8a/LC3. In A, C, E the genotype of α-synuclein flies that lack an RNAi is QUAS-α-Syn, nSybQF2, repo-Gal4/+. In B, D, F the genotype of α-synuclein flies that lack an RNAi is QUAS-α-Syn, nSybQF2, alrm-Gal4/+. Nuclei are stained with DAPI. Scale bar = 10 μm. All experiments were performed at 10 days post-eclosion.
In summary, we identified 4 Parkinson’s disease GWAS candidate genes that enhance neuronal α-synuclein induced neurodegeneration in a non-cell-autonomous manner when knocked down in glia. Despite having divergent effects on the accumulation of α-synuclein aggregates, knockdown of each modifier resulted in increased α-synuclein oligomerization, supporting a role for α-synuclein oligomers as drivers of neurotoxicity and suggesting that glia regulate neuronal proteostasis. Further, in the case of Lrrk knockdown, we identify impaired autophagy as a potential upstream mechanism, consistent with the known role of LRRK2 (Giaime et al., 2017; Gómez-Suaga et al., 2012; Orenstein et al., 2013; Plowey et al., 2008; Tong et al., 2012) and with our prior work on neuronal Lrrk (Sarkar et al., n.d). Interestingly, knockdown of the other modifiers did not similarly cause an increase in autophagosomes, suggesting that different glial modifiers act through different, specific, mechanisms, rather than simply due to non-specific glial injury or toxicity.
4. Discussion
We and others have previously demonstrated neuronal functions for some Parkinson’s disease GWAS candidate genes, including SNCA, LRRK2, MAPT, and GBA (Bardai et al., 2018a,b; Blauwendraat et al., 2020; Sarkar et al., 2020; Tran et al., 2020). Here, we suggest that 4 well-validated Parkinson’s disease GWAS candidate genes (LRRK2, GAK, VPS13C, RIT2) may influence Parkinson’s disease pathogenesis at least partially by acting through glia, specifically by influencing glial control of neuronal proteostasis. Proteostasis refers to multiple coordinated homeostatic mechanisms by which cells maintain a healthy and functional proteome. It includes processes that control protein synthesis and folding, disaggregation, and degradation.(Labbadia and Morimoto, 2015) These processes are widely disturbed across the neurodegenerative proteinopathies,(Klaips et al., 2018) likely in part due to a complex feed-forward cycle in which aggregation-prone proteins such as α-synuclein induce proteostatic dysfunction, and this dysfunction in turn exacerbates the pathogenic cascade of α-synuclein misfolding, oligomerization, fibrilization, and aggregation.(Han et al., 2020) The individual steps of this cascade may be controlled by different proteostatic processes, and work in cell culture suggests that different species of α-synuclein may be differentially degraded by the proteosome, macroautophagy, and chaperone mediated autophagy.(Xilouri et al., 2008; Vogiatzi et al., 2008; Gao et al., 2019; Stefanis et al., 2019) Similarly, Prasad et al. found in Drosophila that soluble α-synuclein oligomers were removed by the proteosome,(Prasad et al., 2019) whereas insoluble fibrils and aggregates were removed by autophagy, suggesting that different mechanisms are also at play in vivo. Here, we find that knockdown of all 4 glial modifiers led to an increase in α-synuclein oligomers, with varying effects on large Lewy-body-like inclusions (Figs. 4, 5, 7). Given that different proteostatic systems (the proteosome, macro-autophagy, and chaperone-mediated autophagy) are responsible for clearance of different α-synuclein species in neurons, it is possible that individual glial modifiers influence these systems to varying extents.
Fig. 7.
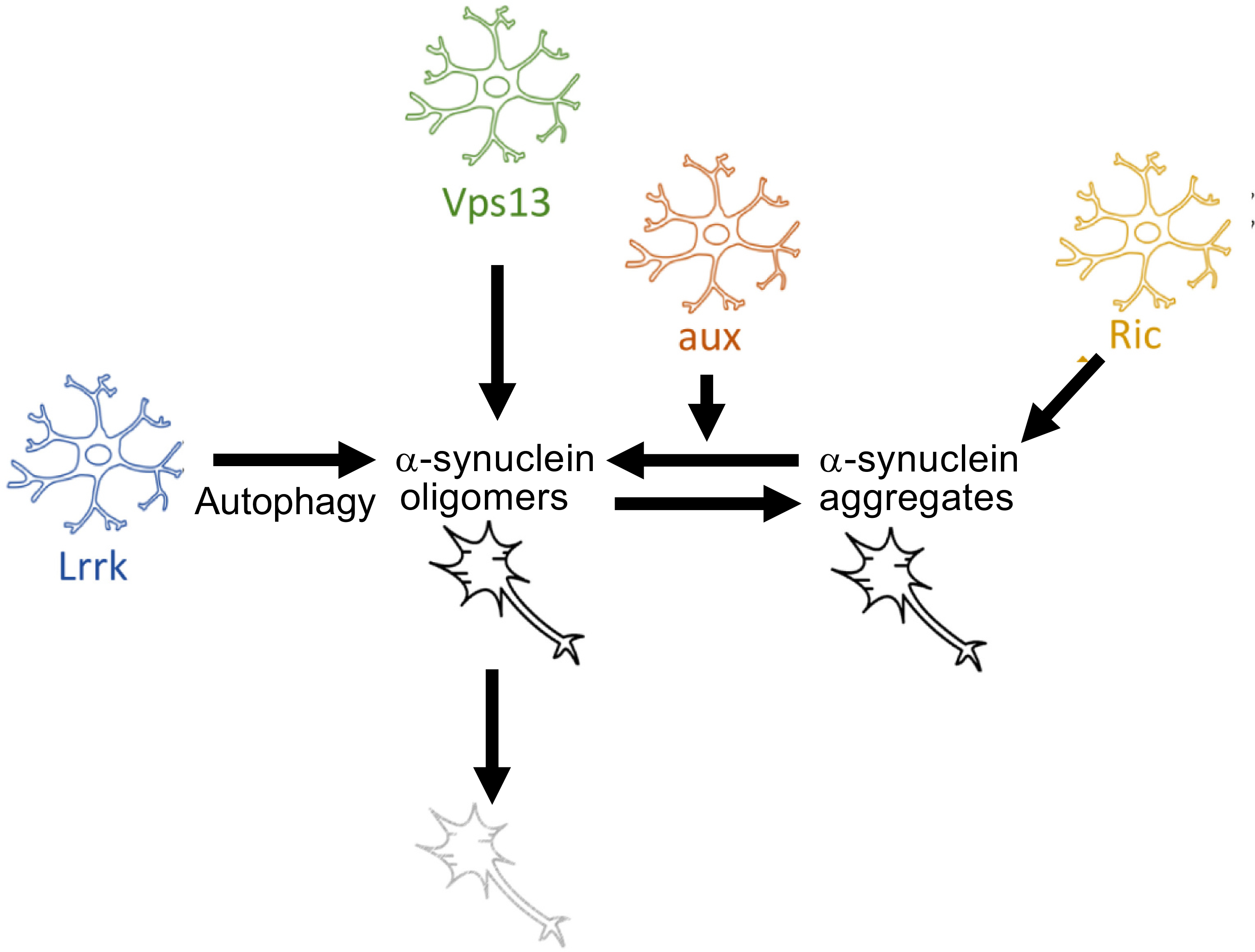
Proposed model. All glial modifiers act by increasing α-synuclein oligomerization but have divergent upstream mechanisms. Glial Lrrk knockdown impairs neuronal autophagy. Glial aux knockdown may favor formation of oligomers as opposed to aggregates. The precise mechanism by which glial Ric or Vps13 knockdown increases α-synuclein oligomerization is unknown.
Non-cell-autonomous control of proteostasis is not well understood. Emerging evidence suggests that glia can alter neuronal autophagy in a variety of diseases,(Kulkarni et al., 2018; Alirezaei et al., 2008; Madill et al., 2017) including in the A53T α-synuclein mouse model of Parkinson’s disease.(Gan et al., 2012) This is consistent with our findings for glial Lrrk knockdown, which led to an increase in markers of autophagosomes and impaired autophagic flux (Fig. 6, Fig. 7). Our finding that the other glial modifiers increase α-synuclein oligomers without seemingly impairing neuronal autophagy suggests that different modifiers can exert their effects on proteostasis through different upstream mechanisms. Beyond macro-autophagy, there are numerous other cellular processes that affect α-synuclein toxicity, including proteosome function, interactions with chaperones(Burmann et al., 2020) that prevent misfolding or induce chaperone-mediated autophagy, phosphorylation(Zhao et al., 2020; Pinho et al., 2019) and other(Ho et al., 2021) post-translational modifications of α-synuclein, and subcellular localization(Pinho et al., 2019) of α-synuclein. Whether glia can indirectly affect these processes is unknown. Interestingly, one recent study suggests an additional mechanism of non-cell-autonomous control of α-synuclein proteostasis. That is, neuronal α-synuclein aggregation can activate the “immunoproteosome,” an inducible form of the proteosome that is assembled in response to inflammatory stimuli,(Driscoll et al., 1993; Seifert et al., 2010) in both neurons and glia.(Ugras et al., 2018) It is therefore possible that changes in proteosome composition or flux in either neurons or glia underlie the effect of other modifiers, particularly in light of the proposed role of the proteosome in clearing oligomers within neurons, as discussed above. (Xilouri et al., 2008; Vogiatzi et al., 2008; Gao et al., 2019; Stefanis et al., 2019; Prasad et al., 2019)
There has been much scientific debate regarding which α-synuclein molecular species are responsible for neurotoxicity.(Lansbury and Lashuel, 2006; Lashuel et al., 2013) Here we find that α-synuclein oligomers, rather than the large Lewy-body-like inclusions, were influenced by glial gene manipulation, and the increase in these species was correlated with increased neurodegeneration. This result is in line with prior work(Chen et al., 2009) using our original Drosophila α-synucleinopathy model,(Feany and Bender, 2000) as well as with studies in yeast, where α-synuclein toxicity occurs in the absence of Lewy-body-like inclusions.(Outeiro and Lindquist, 2003; Volles and Lansbury, 2007) α-synuclein mis-folding and oligomerization may be influenced by phosphorylation and other post-translational modifications, as has been shown in some though not all studies.(Oueslati, 2016) While we did not observe a change in baseline serine 129 phosphorylation of α-synuclein in this study, a more comprehensive analysis of post-translational modifications is beyond the scope of this manuscript. Further, a growing body of evidence suggests that not all pathologic α-synuclein conformations are created equal; different α-synuclein strains may result in different α-synucleinopathies(Shahnawaz et al., 2020) or even different prognosis within one disease.(Lau et al., 2020) Whether glia have any non-cell-autonomous influence on post-translational modifications of α-synuclein or differential α-synuclein strain formation in neurons is unknown, though it is interesting that in multiple system atrophy, defined pathologically by glial cytoplasmic α-synuclein inclusions, oligodendrocytes may be responsible for generating a more pathogenic strain.(Peng et al., 2018) Emerging methods for detecting oligomers in human tissue should shed further light on this critical issue.(Bengoa-Vergniory et al., 2017)
In addition to highlighting the importance of non-cell-autonomous control of proteostasis in Parkinson’s disease pathogenesis, our results have several important implications for the design of future studies. First, they underline the critical need to include investigation of glia in models of Parkinson’s disease, as glia may directly contribute to both risk and progression of disease. Second, they suggest that specific glial genes affect Parkinson’s disease pathogenesis by influencing specific pathways (e.g. Lrrk knockdown exacerbating neuronal autophagy impairment, Fig. 7) rather than by a non-specific injury or “reactive” process. Indeed, there may be innumerable different specific reactive states of glia, including both neuroprotective and detrimental states. (Smith et al., 2020; Yun et al., 2018) This additionally has implications for a future of personalized medicine, as different patients may have different disease drivers, both genetically and cellularly (Bandres-Ciga et al., 2020). Our model can serve as an experimental paradigm for further validation of GWAS candidate genes in glia as well as for dissecting these complex glial-neuronal interactions in vivo. This represents an essential step in getting from GWAS to an increased understanding of pathogenesis and eventually to creation of novel disease-modifying therapies.
Supplementary Material
Acknowledgements
The HC3 α-synuclein antibody was provided by the Developmental Studies Hybridoma Bank, created by the NICHD of the NIH and maintained at The University of Iowa, Department of Biology, Iowa City, IA 52242. Drosophila stocks obtained from the Bloomington Drosophila Stock Center (NIH P40OD018537) were used in this study. We thank the Transgenic RNAi Project (TRiP) at the Harvard Medical School (NIH-NIGMS R01GM084947) for making transgenic RNAi stocks. Kit Tuen and Sabrina Clemens provided excellent technical assistance.
Funding information
This work was funded by 1R21 NS0105151 (MBF), 1R01 NS098821 (MBF), 5 K08- K08NS109344-03 (ALO), and W81XWH-18-1-0395 (ALO).
Abbreviations:
- GBA
Glucosylceramidase beta
- GWAS
Genome wide association study
- LRRK2
Leucine-rich repeat kinase 2
- SNCA
α-synuclein
- Vps13C
Vacuolar protein sorting 13 homolog C
Footnotes
Declaration of Competing Interest
The authors have no competing financial interests.
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.nbd.2021.105482.
References
- Aflaki E, Stubblefield BK, McGlinchey RP, McMahon B, Ory DS, Sidransky E, 2020. A characterization of Gaucher iPS-derived astrocytes: potential implications for Parkinson’s disease. Neurobiol. Dis. 134, 104647. 10.1016/j.nbd.2019.104647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam P, Bousset L, Melki R, Otzen DE, 2019. α-Synuclein oligomers and fibrils: a spectrum of species, a spectrum of toxicities. J. Neurochem. 150 (5), 522–534. 10.1111/jnc.14808. [DOI] [PubMed] [Google Scholar]
- Alirezaei M, Kiosses WB, Flynn CT, Brady NR, Fox HS, 2008. Disruption of neuronal autophagy by infected microglia results in neurodegeneration. PLoS One 3 (8), e2906. 10.1371/journal.pone.0002906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandres-Ciga S, Diez-Fairen M, Kim JJ, Singleton AB, 2020. Genetics of Parkinson’s disease: an introspection of its journey towards precision medicine. Neurobiol. Dis. 137, 104782. 10.1016/j.nbd.2020.104782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardai FH, Ordonez DG, Bailey RM, Hamm M, Lewis J, Feany MB, 2018a. Lrrk promotes tau neurotoxicity through dysregulation of actin and mitochondrial dynamics. PLoS Biol. 16 (12), e2006265 10.1371/journal.pbio.2006265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardai FH, Wang L, Mutreja Y, Yenjerla M, Gamblin TC, Feany MB, 2018b. A conserved cytoskeletal signaling Cascade mediates neurotoxicity of FTDP-17 tau mutations in vivo. J. Neurosci. 38 (1), 108–119. 10.1523/JNEUROSCI.1550-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengoa-Vergniory N, Roberts RF, Wade-Martins R, Alegre-Abarrategui J, 2017. Alpha-synuclein oligomers: a new hope. Acta Neuropathol (Berl). 134 (6), 819–838. 10.1007/s00401-017-1755-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blauwendraat C, Nalls MA, Singleton AB, 2020. The genetic architecture of Parkinson’s disease. Lancet Neurol. 19 (2), 170–178. 10.1016/S1474-4422(19)30287-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth HDE, Hirst WD, Wade-Martins R, 2017. The role of astrocyte dysfunction in Parkinson’s disease pathogenesis. Trends Neurosci. 40 (6), 358–370. 10.1016/j.tins.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth HDE, Wessely F, Connor-Robson N, et al. , 2019. RNA sequencing reveals MMP2 and TGFB1 downregulation in LRRK2 G2019S Parkinson’s iPSC-derived astrocytes. Neurobiol. Dis. 129, 56–66. 10.1016/j.nbd.2019.05.006. [DOI] [PubMed] [Google Scholar]
- Brand AH, Perrimon N, 1993. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Dev Camb Engl. 118 (2), 401–415. [DOI] [PubMed] [Google Scholar]
- Burmann BM, Gerez JA, Matečko-Burmann I, et al. , 2020. Regulation of α-synuclein by chaperones in mammalian cells. Nature. 577 (7788), 127–132. 10.1038/s41586-019-1808-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Periquet M, Wang X, et al. , 2009. Tyrosine and serine phosphorylation of alpha-synuclein have opposing effects on neurotoxicity and soluble oligomer formation. J. Clin. Invest. 119 (11), 3257–3265. 10.1172/JCI39088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coetzee SG, Pierce S, Brundin P, Brundin L, Hazelett DJ, Coetzee GA, 2016. Enrichment of risk SNPs in regulatory regions implicate diverse tissues in Parkinson’s disease etiology. Sci. Rep. 6, 30509. 10.1038/srep30509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- di Domenico A, Carola G, Calatayud C, et al. , 2019. Patient-specific iPSC-derived astrocytes contribute to non-cell-autonomous neurodegeneration in Parkinson’s disease. Stem Cell Rep. 12 (2), 213–229. 10.1016/j.stemcr.2018.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll J, Brown MG, Finley D, Monaco JJ, 1993. MHC-linked LMP gene products specifically alter peptidase activities of the proteasome. Nature. 365 (6443), 262–264. 10.1038/365262a0. [DOI] [PubMed] [Google Scholar]
- Feany MB, Bender WW, 2000. A Drosophila model of Parkinson’s disease. Nature. 404 (6776), 394–398. 10.1038/35006074. [DOI] [PubMed] [Google Scholar]
- Gagliano SA, Pouget JG, Hardy J, et al. , 2016. Genomics implicates adaptive and innate immunity in Alzheimer’s and Parkinson’s diseases. Ann Clin Transl Neurol. 3 (12), 924–933. 10.1002/acn3.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan L, Vargas MR, Johnson DA, Johnson JA, 2012. Astrocyte-specific overexpression of Nrf2 delays motor pathology and synuclein aggregation throughout the CNS in the alpha-synuclein mutant (A53T) mouse model. J. Neurosci. 32 (49), 17775–17787. 10.1523/JNEUROSCI.3049-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Perera G, Bhadbhade M, Halliday GM, Dzamko N, 2019. Autophagy activation promotes clearance of α-synuclein inclusions in fibril-seeded human neural cells. J. Biol. Chem. 294 (39), 14241–14256. 10.1074/jbc.RA119.008733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaime E, Tong Y, Wagner LK, Yuan Y, Huang G, Shen J, 2017. Age-Dependent Dopaminergic Neurodegeneration and Impairment of the Autophagy-Lysosomal Pathway in LRRK-Deficient Mice. Neuron 96 (4), 796–807 e6. 10.1016/j.neuron.2017.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Suaga P, Luzón-Toro B, Churamani D, et al. , 2012. Leucine-rich repeat kinase 2 regulates autophagy through a calcium-dependent pathway involving NAADP. Hum. Mol. Genet. 21 (3), 511–525. 10.1093/hmg/ddr481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grenn FP, Kim JJ, Makarious MB, et al. , 2020. The Parkinson’s disease genome-wide association study locus browser. Mov Disord Off J Mov Disord Soc. 35 (11), 2056–2067. 10.1002/mds.28197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han D, Zheng W, Wang X, Chen Z, 2020. Proteostasis of α-Synuclein and its role in the pathogenesis of Parkinson’s disease. Front. Cell. Neurosci. 14, 45. 10.3389/fncel.2020.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho GPH, Ramalingam N, Imberdis T, Wilkie EC, Dettmer U, Selkoe DJ, 2021. Upregulation of cellular Palmitoylation mitigates α-Synuclein accumulation and neurotoxicity. Mov Disord Off J Mov Disord Soc. 36 (2), 348–359. 10.1002/mds.28346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller MF, Saad M, Bras J, et al. , 2012. Using genome-wide complex trait analysis to quantify “missing heritability” in Parkinson’s disease. Hum. Mol. Genet. 21 (22), 4996–5009. 10.1093/hmg/dds335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kia DM, Zhang D, Guelfi S, Manzoni C, Hubbard L, United Kingdom Brain Expression Consortium (UKBEC), International Parkinson’s Disease Genomics Consortium (IPDGC), Reynolds RH, Botia J, Ryten M, Ferrari R, Lews PA, Williams N, Trabzuni T, Hardy J, Wood N, 2019. Integration of eQTL and Parkinson’s disease GWAS data implicates 11 disease genes. Published online May 5. [Google Scholar]
- Kim CY, Alcalay RN, 2017. Genetic forms of Parkinson’s disease. Semin. Neurol. 37 (2), 135–146. 10.1055/s-0037-1601567. [DOI] [PubMed] [Google Scholar]
- Klaips CL, Jayaraj GG, Hartl FU, 2018. Pathways of cellular proteostasis in aging and disease. J. Cell Biol. 217 (1), 51–63. 10.1083/jcb.201709072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni A, Chen J, Maday S, 2018. Neuronal autophagy and intercellular regulation of homeostasis in the brain. Curr. Opin. Neurobiol. 51, 29–36. 10.1016/j.conb.2018.02.008. [DOI] [PubMed] [Google Scholar]
- Labbadia J, Morimoto RI, 2015. The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 84, 435–464. 10.1146/annurev-biochem-060614-033955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansbury PT, Lashuel HA, 2006. A century-old debate on protein aggregation and neurodegeneration enters the clinic. Nature. 443 (7113), 774–779. 10.1038/nature05290. [DOI] [PubMed] [Google Scholar]
- Lashuel HA, Overk CR, Oueslati A, Masliah E, 2013. The many faces of α-synuclein: from structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 14 (1), 38–48. 10.1038/nrn3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau A, So RWL, Lau HHC, et al. , 2020. α-Synuclein strains target distinct brain regions and cell types. Nat. Neurosci. 23 (1), 21–31. 10.1038/s41593-019-0541-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Han J-H, Kim H, Park SM, Joe E-H, Jou I, 2019. Parkinson’s disease-associated LRRK2-G2019S mutant acts through regulation of SERCA activity to control ER stress in astrocytes. Acta Neuropathol Commun. 7 (1), 68. 10.1186/s40478-019-0716-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesage S, Brice A, 2009. Parkinson’s disease: from monogenic forms to genetic susceptibility factors. Hum. Mol. Genet. 18 (R1), R48–R59. 10.1093/hmg/ddp012. [DOI] [PubMed] [Google Scholar]
- Li YI, Wong G, Humphrey J, Raj T, 2019. Prioritizing Parkinson’s disease genes using population-scale transcriptomic data. Nat. Commun. 10 (1), 994. 10.1038/s41467-019-08912-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madill M, McDonagh K, Ma J, et al. , 2017. Amyotrophic lateral sclerosis patient iPSC-derived astrocytes impair autophagy via non-cell autonomous mechanisms. Mol Brain. 10 (1), 22. 10.1186/s13041-017-0300-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalls MA, Saad M, Noyce AJ, et al. , 2014. Genetic comorbidities in Parkinson’s disease. Hum. Mol. Genet. 23 (3), 831–841. 10.1093/hmg/ddt465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalls MA, Blauwendraat C, Vallerga CL, et al. , 2019. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 18 (12), 1091–1102. 10.1016/S1474-4422(19)30320-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen AL, Feany MB, 2019. Glial α-synuclein promotes neurodegeneration characterized by a distinct transcriptional program in vivo. Glia. 67 (10), 1933–1957. 10.1002/glia.23671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordonez DG, Lee MK, Feany MB, 2018. α-synuclein Induces mitochondrial dysfunction through spectrin and the actin cytoskeleton. Neuron 97 (1), 108–124 e6. 10.1016/j.neuron.2017.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orenstein SJ, Kuo S-H, Tasset I, et al. , 2013. Interplay of LRRK2 with chaperone-mediated autophagy. Nat. Neurosci. 16 (4), 394–406. 10.1038/nn.3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oueslati A, 2016. Implication of alpha-Synuclein phosphorylation at S129 in Synucleinopathies: what have we learned in the last decade? J. Parkinsons Dis. 6 (1), 39–51. 10.3233/JPD-160779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Outeiro TF, Lindquist S, 2003. Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science. 302 (5651), 1772–1775. 10.1126/science.1090439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng C, Gathagan RJ, Covell DJ, et al. , 2018. Cellular milieu imparts distinct pathological α-synuclein strains in α-synucleinopathies. Nature. 557 (7706), 558–563. 10.1038/s41586-018-0104-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce S, Coetzee GA, 2017. Parkinson’s disease-associated genetic variation is linked to quantitative expression of inflammatory genes. PLoS One 12 (4), e0175882. 10.1371/journal.pone.0175882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce SE, Booms A, Prahl J, van der Schans EJC, Tyson T, Coetzee GA, 2020. Post-GWAS knowledge gap: the how, where, and when. NPJ Park Dis. 6, 23. 10.1038/s41531-020-00125-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinho R, Paiva I, Jercic KG, et al. , 2019. Nuclear localization and phosphorylation modulate pathological effects of alpha-synuclein. Hum. Mol. Genet. 28 (1), 31–50. 10.1093/hmg/ddy326. [DOI] [PubMed] [Google Scholar]
- Plowey ED, Cherra SJ, Liu Y-J, Chu CT, 2008. Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J. Neurochem. 105 (3), 1048–1056. 10.1111/j.1471-4159.2008.05217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter CJ, Tasic B, Russler EV, Liang L, Luo L, 2010. The Q system: a repressible binary system for transgene expression, lineage tracing, and mosaic analysis. Cell. 141 (3), 536–548. 10.1016/j.cell.2010.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad V, Wasser Y, Hans F, et al. , 2019. Monitoring α-synuclein multimerization in vivo. FASEB J Off Publ Fed Am Soc Exp Biol. 33 (2), 2116–2131. 10.1096/fj.201800148RRR. [DOI] [PubMed] [Google Scholar]
- Raji JI, Potter CJ, 2021. The number of neurons in Drosophila and mosquito brains. PLoS One 16 (5), e0250381. 10.1371/journal.pone.0250381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds RH, Botía J, Nalls MA, et al. , 2019. Moving beyond neurons: the role of cell type-specific gene regulation in Parkinson’s disease heritability. NPJ Park Dis. 5, 6. 10.1038/s41531-019-0076-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanyal A, DeAndrade MP, Novis HS, et al. , 2020. Lysosome and inflammatory defects in GBA1-mutant astrocytes are normalized by LRRK2 inhibition. Mov Disord Off J Mov Disord Soc. 35 (5), 760–773. 10.1002/mds.27994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S, Murphy MA, Dammer EB, et al. , 2020. Comparative proteomic analysis highlights metabolic dysfunction in α-synucleinopathy. NPJ Park Dis. 6 (1), 40. 10.1038/s41531-020-00143-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S, Olsen AL, Syngecka K, Lohr K, Feany MB. α-synuclein impairs autophagosome maturation through abnormal actin stabilization. PLoS Genet.. Published online 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert U, Bialy LP, Ebstein F, et al. , 2010. Immunoproteasomes preserve protein homeostasis upon interferon-induced oxidative stress. Cell. 142 (4), 613–624. 10.1016/j.cell.2010.07.036. [DOI] [PubMed] [Google Scholar]
- Shahnawaz M, Mukherjee A, Pritzkow S, et al. , 2020. Discriminating α-synuclein strains in Parkinson’s disease and multiple system atrophy. Nature. 578 (7794), 273–277. 10.1038/s41586-020-1984-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith HL, Freeman OJ, Butcher AJ, et al. , 2020. Astrocyte unfolded protein response induces a specific reactivity state that causes non-cell-autonomous neuronal degeneration. Neuron 105 (5), 855–866 e5. 10.1016/j.neuron.2019.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanis L, Emmanouilidou E, Pantazopoulou M, Kirik D, Vekrellis K, Tofaris GK, 2019. How is alpha-synuclein cleared from the cell? J. Neurochem. 150 (5), 577–590. 10.1111/jnc.14704. [DOI] [PubMed] [Google Scholar]
- Tong Y, Giaime E, Yamaguchi H, et al. , 2012. Loss of leucine-rich repeat kinase 2 causes age-dependent bi-phasic alterations of the autophagy pathway. Mol. Neurodegener. 7, 2. 10.1186/1750-1326-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran J, Anastacio H, Bardy C, 2020. Genetic predispositions of Parkinson’s disease revealed in patient-derived brain cells. NPJ Park Dis. 6, 8. 10.1038/s41531-020-0110-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugras S, Daniels MJ, Fazelinia H, et al. , 2018. Induction of the immunoproteasome subunit Lmp7 links Proteostasis and immunity in α-Synuclein aggregation disorders. EBioMedicine. 31, 307–319. 10.1016/j.ebiom.2018.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogiatzi T, Xilouri M, Vekrellis K, Stefanis L, 2008. Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J. Biol. Chem. 283 (35), 23542–23556. 10.1074/jbc.M801992200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volles MJ, Lansbury PT, 2007. Relationships between the sequence of alpha-synuclein and its membrane affinity, fibrillization propensity, and yeast toxicity. J. Mol. Biol. 366 (5), 1510–1522. 10.1016/j.jmb.2006.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xilouri M, Vogiatzi T, Vekrellis K, Stefanis L, 2008. Alpha-synuclein degradation by autophagic pathways: a potential key to Parkinson’s disease pathogenesis. Autophagy. 4 (7), 917–919. 10.4161/auto.6685. [DOI] [PubMed] [Google Scholar]
- Yun SP, Kam T-I, Panicker N, et al. , 2018. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat. Med. 24 (7), 931–938. 10.1038/s41591-018-0051-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Sloan SA, Clarke LE, et al. , 2016. Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron. 89 (1), 37–53. 10.1016/j.neuron.2015.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao K, Lim Y-J, Liu Z, et al. , 2020. Parkinson’s disease-related phosphorylation at Tyr39 rearranges α-synuclein amyloid fibril structure revealed by cryo-EM. Proc. Natl. Acad. Sci. U. S. A. 117 (33), 20305–20315. 10.1073/pnas.1922741117. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon request.