

Abstract
Viral entry and egress are important determinants of virus infectivity and pathogenicity. β-coronaviruses, including the COVID-19 virus SARS-CoV-2 and mouse hepatitis virus (MHV), exploit the lysosomal exocytosis pathway for egress. Here, we show that SARS-CoV-2 ORF3a, but not SARS-CoV ORF3a, promotes lysosomal exocytosis. SARS-CoV-2 ORF3a facilitates lysosomal targeting of the BORC-ARL8b complex, which mediates trafficking of lysosomes to the vicinity of the plasma membrane, and exocytosis-related SNARE proteins. The Ca2+ channel TRPML3 is required for SARS-CoV-2 ORF3a-mediated lysosomal exocytosis. Expression of SARS-CoV-2 ORF3a greatly elevates extracellular viral release in cells infected with the coronavirus MHV-A59, which itself lacks ORF3a. In SARS-CoV-2 ORF3a, Ser171 and Trp193 are critical for promoting lysosomal exocytosis and blocking autophagy. When these residues are introduced into SARS-CoV ORF3a, it acquires the ability to promote lysosomal exocytosis and inhibit autophagy. Our results reveal a mechanism by which SARS-CoV-2 interacts with host factors to promote its extracellular egress.
Keywords: COVID-19, SARS-CoV-2, SARS-CoV, ORF3a, lysosomal exocytosis
Graphical abstract
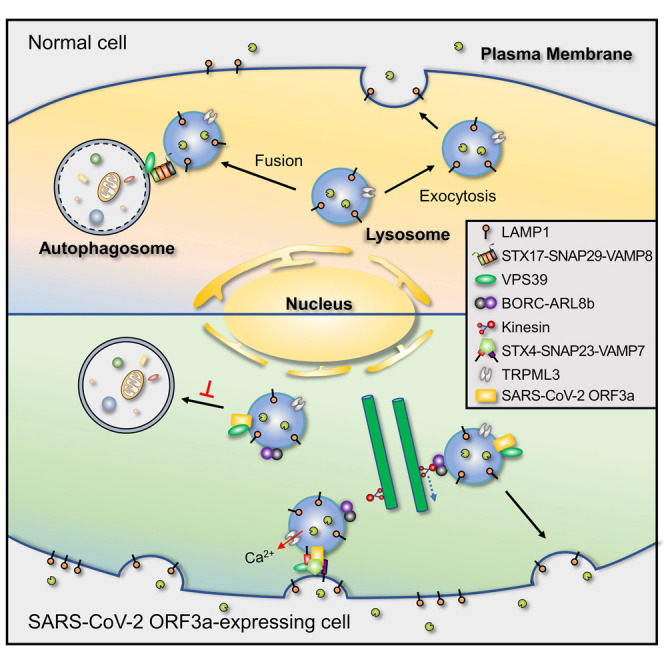
Chen et al. demonstrate that ORF3a of SARS-CoV-2, but not SARS-CoV, promotes lysosomal exocytosis by promoting lysosomal targeting of the BORC-ARL8b complex and exocytosis-related SNARE proteins. The residues at 171 and 193 are key determinants of the differential function of SARS-CoV-2 and SARS-CoV ORF3a in lysosomal exocytosis and autophagy inhibition.
Introduction
β-coronaviruses, including the severe acute respiratory syndrome (SARS) virus SARS-CoV, the COVID-19 virus SARS-CoV-2, and mouse hepatitis virus (MHV), are enveloped, positive-sense, single-stranded RNA viruses (Fung and Liu, 2019; Wu et al., 2020; Zhou et al., 2020). SARS-CoV-2 and SARS-CoV enter host cells via binding of the viral surface spike (S) protein to its receptor ACE2 (angiotensin-converting enzyme 2) on target cells (Hoffmann et al., 2020b; Zhou et al., 2020). The S proteins of SARS-CoV and SARS-CoV-2, which display 87% protein similarity, are composed of S1 and S2 domains and are present as homotrimers (Wrapp et al., 2020). Upon infection, the S protein is proteolytically activated by cellular proteases at the S1/S2 boundary to separate the S1 and S2 domains and subsequently at the S2′ site in the S2 domain (Hoffmann et al., 2020b). S1 contains the receptor-binding domain (RBD) that mediates ACE2 recognition, whereas S2 triggers fusion of the viral and cellular membranes to enable the virus to enter the host cytoplasm (Hartenian et al., 2020). The S protein can be cleaved by the cell surface protease TMPRSS2 to mediate viral/plasma membrane (PM) fusion, or by the lysosomal proteases cathepsin B and L (CTSB/L) when the virus is internalized via endocytosis to trigger viral/endosomal membrane fusion (Hartenian et al., 2020; Hoffmann et al., 2020b; Shang et al., 2020). The extent to which each mechanism is employed depends on the differential expression of TMPRSS2 by different host cell types (Hoffmann et al., 2020b). Compared with SARS-CoV RBD, SARS-CoV-2 RBD exhibits higher ACE2-binding affinity (Shang et al., 2020; Wrapp et al., 2020). The SARS-CoV-2 S protein also contains multiple arginine residues at the S1/S2 cleavage site that can be recognized by the cellular protease furin, enabling pre-cleavage of S proteins during viral packaging in host cells (Hoffmann et al., 2020a; Shang et al., 2020). The differential properties of the S protein contribute to the higher infectivity of SARS-CoV-2 compared with SARS-CoV (Hoffmann et al., 2020b; Peng et al., 2021; Shang et al., 2020). Viral entry has been the focus for developing interventions, such as inhibiting ACE2 binding, inactivating host proteases or blocking membrane fusion (Hartenian et al., 2020; Peng et al., 2021).
After the coronaviral RNA genome is released into the cytoplasm, it is translated into structural proteins and nonstructural proteins (NSPs). NSPs mediate the formation of double-membrane vesicles (DMVs), upon which the viral replication transcription complex (RTC) is anchored (Snijder et al., 2020). The viral particles are assembled and budded into the lumen of the ER and the ER-Golgi intermediate compartment (ERGIC); they then traffic to the trans-Golgi network (TGN) and eventually reach the late endosomes/lysosomes (Fung and Liu, 2019; Ghosh et al., 2020). β-coronaviruses exploit the ARL8b-dependent lysosomal exocytosis pathway for release into the extracellular environment (Ghosh et al., 2020). During lysosomal exocytosis, the BORC-ARL8b complex drives anterograde transport of lysosomes from perinuclear regions to the vicinity of the PM (Pu et al., 2016). Fusion of lysosomes with the PM is mediated by the SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) complex composed of VAMP7, STX4, and SNAP23 and also requires an increase in the intracellular and/or localized Ca2+ level (Rodríguez et al., 1997; Saftig and Klumperman, 2009; Tancini et al., 2020). Very little is known about whether and how coronaviral proteins interact with host factors to control lysosomal exocytosis-mediated viral egress.
The autophagy-lysosome pathway acts as a surveillance system against pathogen invasion (Choi et al., 2018; Deretic et al., 2013; Levine et al., 2011; Wong and Sanyal, 2020). Autophagy involves engulfment of a portion of unselected cytosolic materials or selected cargos (i.e., damaged organelles, protein aggregates, and invading pathogens) in a double-membrane autophagosome (Lamb et al., 2013; Mizushima et al., 2011; Stolz et al., 2014). The autophagosome fuses with endolysosomal vesicles such as late endosomes/lysosomes to form amphisomes, a process known as autophagosome maturation, which eventually leads to the formation of degradative autolysosomes (Zhao and Zhang, 2019; Zhao et al., 2021). Viruses have developed mechanisms to evade autophagic destruction and even to hijack autophagic vacuoles (i.e., autophagosomes, amphisomes, and nondegradative autolysosomes) for their own benefits, for example, by promoting replication and release (Choi et al., 2018; Deretic et al., 2013; Wong and Sanyal, 2020; Zhao et al., 2021). SARS-CoV-2 virus infection blocks autophagy at a step of autophagosome maturation, resulting in accumulation of autophagosomes and amphisomes (Miao et al., 2021). Mechanistically, the late endosomal/lysosomal-localized viral ORF3a protein sequestrates the HOPS complex component VPS39, preventing it from interacting with the autophagosomal SNARE protein STX17. This blocks assembly of the trans-SNARE complex composed of STX17, SNAP29, and VAMP8, which drives fusion of autophagosomes and amphisomes with lysosomes (Miao et al., 2021). ORF3a from the SARS virus SARS-CoV, however, fails to interact with VPS39 or block autophagosome maturation (Miao et al., 2021).
Here, we demonstrated that SARS-CoV-2 ORF3a, but not SARS-CoV ORF3a, promotes lysosomal exocytosis. SARS-CoV-2 ORF3a promotes the lysosomal targeting of the BORC-ARL8b complex and the exocytosis-related SNARE proteins VAMP7 and STX4 and also increases the cytosolic Ca2+ level. Expression of SARS-CoV-2 ORF3a facilitates the egress of MHV-A59 virus, which lacks an ORF3a counterpart. We found that the serine at residue 171 and the tryptophan at residue 193 are essential for SARS-CoV-2 ORF3a to promote lysosomal exocytosis and inhibit autophagy. When these residues are introduced at the corresponding positions in SARS-CoV ORF3a, the mutated protein acquires the ability to promote lysosomal exocytosis and block autophagy. Our study provides mechanistic insights into how SARS-CoV-2 promotes lysosomal exocytosis and also shows that this process is differentially modulated by SARS-CoV-2 and SARS-CoV, which may contribute to the different levels of viral infectivity and pathogenicity.
Results
ORF3a of SARS-CoV-2 promotes lysosomal exocytosis
Lysosomal exocytosis results in PM localization of lysosomal membrane proteins and also the release of lysosomal contents into the extracellular environment (Luzio et al., 2007; Tancini et al., 2020). To determine whether the late endosomal/lysosomal-localized SARS-CoV-2 ORF3a (referred to as ORF3a hereafter unless the viral origin is stated) is involved in lysosomal exocytosis, we examined the cell surface levels of two lysosomal membrane proteins, LAMP1 and LAMP2 in HeLa cells expressing ORF3a. PM was separated from soluble fractions by cell fractionation. Despite the expansion of the lysosome population (Miao et al., 2021), the total levels of LAMP1 and LAMP2 were not evidently changed in ORF3a-expressing cells (Figure 1 A). In the PM fraction derived from cells expressing ORF3a, LAMP1 and LAMP2 were more abundant and the proportion of LAMP1 and LAMP2 on the PM was higher compared with control cells (Figure 1A). Levels of mature cathepsin D (CTSD) and, to a lesser extent, cathepsin B (CTSB) were higher in the culture medium of ORF3a-expressing cells than control cells (Figure 1B).
Figure 1.

ORF3a of SARS-CoV-2 promotes lysosomal exocytosis
(A) Immunoblotting analysis of subcellular fractions from HeLa cells expressing FLAG (Control), SARS-CoV-2 ORF3a-FLAG (ORF3a), and SARS-CoV ORF3a-FLAG (SARS ORF3a). Quantification of LAMP1 and LAMP2 levels is shown. Levels of LAMP1 and LAMP2 were normalized by GAPDH level in cell lysates and by integrin α5 level in the PM fraction. The level in control cells is set to 1.0. HeLa cells were used throughout this study unless otherwise noted.
(B) Immunoblotting analysis of secreted lysosomal cathepsins in the culture medium of cells expressing FLAG (Control) and ORF3a-FLAG (ORF3a). Quantification of mature cathepsin B (CTSB) and cathepsin D (CTSD) (normalized by total protein levels in the medium) is shown. The level in control cells is set to 1.0.
(C–E) Compared with control cells expressing GFP (C), the level of cell membrane-localized LAMP1, detected by phycoerythrin (PE)-conjugated LAMP1 antibody in live cells, is higher in ORF3a-GFP-expressing cells (D). (E) shows quantification of the fluorescence intensity of cell membrane-localized LAMP1 per cell in GFP-expressing cells (n = 33) and ORF3a-GFP-expressing cells (n = 29). The level in control cells is set to 1.0. Data are shown as mean ± SEM ∗∗∗p < 0.001.
(F) Column scatter charts showing the distances from LAMP1 puncta to the cell membrane. Data are shown as mean ± SEM (n = 411 puncta from 8 cells in control cells, n = 793 puncta from 8 cells in ORF3a-expressing cells). ∗∗∗p < 0.001.
Scale bars, (C and D) 10 μm.
See also Figure S1.
We next performed immunofluorescence staining to directly visualize cell surface-localized LAMP1. In this assay, live cells are incubated with a phycoerythrin (PE)-conjugated human LAMP1 antibody at 4°C for 90 min, followed by fixation and detection (Rodríguez et al., 1997; Medina et al., 2011). The cells are not permeabilized. Control cells contained low levels of cell-surface-localized LAMP1 (Figure 1C), which results from constitutive fusion of lysosomes with the PM and/or trafficking of LAMP1 to the PM via the conventional secretory pathway (Braulke and Bonifacino, 2009). In cells expressing ORF3a, cell-surface-localized LAMP1 was dramatically elevated (Figures 1D and 1E). The signal was abolished if control cells or ORF3a-expressing cells were treated with trypsin to remove surface proteins before incubating with the anti-LAMP1 antibody (Figures S1A–S1D). Cell-surface-localized LAMP1 was clearly distinct from LysoTracker-stained intracellular lysosomes (Figure S1E). An anti-TOMM20 antibody detected abundant mitochondria in permeabilized cells, but not in live cells (Figures S1G and S1H). These results confirm that the live cell LAMP1 staining assay specifically detects cell-surface-localized LAMP1, but not intracellular lysosomes, and thus the live cell staining assay was used in this study. Expressing ORF3a in other cell types such as COS7 cells also led to elevated levels of cell surface LAMP1 (Figures S1F, S1I, and S1J). Brefeldin A (BFA) treatment, which blocks conventional secretory transport (Miller et al., 1992), failed to suppress the elevated cell surface LAMP1 in ORF3a-expressing cells (Figures S1K–S1M). Together, these results provide evidence that expression of ORF3a promotes lysosomal exocytosis.
The BORC-ARL8b complex is essential for ORF3a-mediated lysosomal exocytosis
During lysosomal exocytosis, lysosomes are transported anterogradely from the perinuclear region to the PM (Tancini et al., 2020). The number and percentage of LAMP1-stained lysosomes close to the PM was greater in ORF3a-expressing cells (Figures 1F, S1N, and S1O). The late endosomal/lysosomal-associated multisubunit BORC complex recruits the small Arf-like Ras family GTPase ARL8b to promote ARL8b-dependent coupling to kinesin motors, which drives the movement of lysosomes toward the PM (Pu et al., 2016). In control cells, the BORC component BORCS6 was largely diffuse in the cytosol and formed only a few puncta (Figure 2 A). In ORF3a-expressing cells, BORCS6 formed a large number of puncta that colocalized or closely associated with ORF3a punctate structures (Figures 2B and 2C). In GFP-TRAP assays, BORCS6, but not ARL8b, was evidently co-immunoprecipitated by ORF3a-GFP (Figures 2G and 2N). Depleting ARL8b or the BORC component BORCS3 caused juxtanuclear accumulation of lysosomes (Figures S1P–S1S). BORCS3 knockdown (KD) and ARL8b KD greatly reduced the PM accumulation of LAMP1 in control cells and ORF3a-expressing cells (Figures 2D–2F and S1T–S1X). Thus, ORF3a facilitates lysosomal targeting of the BORC-ARL8b complex for exocytosis.
Figure 2.

The BORC-ARL8b complex and TRPML3 are required for lysosomal exocytosis in ORF3a-expressing cells
(A–C) Compared with control cells expressing mCherry (A), the number of BORCS6-GFP puncta is dramatically increased in ORF3a-mCherry-expressing cells (B). (C) Shows quantification of the number of BORCS6-GFP puncta per cell. Data are shown as mean ± SEM (n = 31 for each bar). ∗∗∗p < 0.001.
(D–F) Compared with control cells transfected with control siRNA (E), the level of cell membrane-localized LAMP1 is dramatically decreased in ORF3a-expressing cells transfected with BORCS3 siRNA (F). (D) Shows quantification of the fluorescence intensity of cell membrane-localized LAMP1 per cell. The level in control siRNA-treated cells is set to 1.0. Data are shown as mean ± SEM (n = 23 for control cells, n = 27 for ARL8b KD cells, and n = 22 for BORCS3 KD cells). ∗∗∗p < 0.001.
(G) In GFP-Trap assays, ORF3a-mCherry is co-immunoprecipitated by BORCS6-GFP. Cell lysates were immunoprecipitated using GFP-Trap and analyzed by immunoblotting with mCherry and GFP antibodies.
(H) The 340/380 nm fluorescence excitation ratio of HeLa cells expressing GFP (control, n = 55) and ORF3a-GFP (ORF3a, n = 49) after loading with fura-2. Data are shown as mean ± SEM ∗∗∗p < 0.001.
(I) The cytosolic free calcium concentration in HeLa cells expressing mCherry (control, n = 45) and ORF3a-mCherry (ORF3a, n = 9). Data are shown as mean ± SEM ∗∗∗p < 0.001.
(J–L) Compared with control cells expressing mCherry (J), the number of LAMP1-GCaMP6f puncta is dramatically increased in ORF3a-mCherry-expressing cells (K). (L) shows quantification of the number of LAMP1-GCaMP6f puncta per cell. Data are shown as mean ± SEM (n = 15 for control cells, n = 21 for ORF3a-expressing cells). ∗∗∗p < 0.001.
(M) The ratio of F/F0 in control cells expressing mCherry (control) and ORF3a-mCherry (ORF3a) after inducing lysosomal Ca2+ release by HBSS.
(N) In GFP-Trap assays, FLAG-TRPML1 and FLAG-TRPML3 but not FLAG-ARL8b are co-precipitated by ORF3a-GFP. Cell lysates were immunoprecipitated using GFP-Trap and analyzed by immunoblotting with FLAG and GFP antibodies.
(O) Shows quantification of ΔF/F0, related to Figure 2M. Data are shown as mean ± SEM (n = 21 for control, n = 25 for ORF3a). ∗∗∗p < 0.001.
(P and Q) Compared with ORF3a-expressing cells transfected with control siRNA (Figure 2E), the level of cell membrane-localized LAMP1 is dramatically decreased in ORF3a-expressing cells transfected with TRPML3 siRNA (Q).
(P) Shows quantification of the fluorescence intensity of cell membrane-localized LAMP1 per cell. The level in control siRNA-treated cells is set to 1.0. Data are shown as mean ± SEM (n = 23, 17, 20, and 21 for bars from left to right). ∗∗∗p < 0.001; n.s., no significant difference. The data for ORF3a-expressing cells transfected with control siRNA are the same as those in Figure 2D.
Scale bars: (A, B, E, F, J, K, and Q) 10 μm.
See also Figures S1 and S2.
ORF3a-mediated lysosomal exocytosis requires TRPML3
Lysosomal exocytosis is induced by an elevation in cytosolic Ca2+ concentration (Rodríguez et al., 1997; Tancini et al., 2020). Compared with control cells, the cytosolic free calcium concentration ([Ca2+]cyt) was increased in ORF3a-expressing cells, rising from ∼135 to ∼220 nM (Figures 2H and 2I). We examined whether localized lysosomal Ca2+ release was affected by ORF3a expression. To monitor the release of Ca2+ from lysosomes, we constructed a lysosome-targeted genetically encoded Ca2+ sensor in which GCaMP6f was linked to the C-terminal cytosolic tail of LAMP1 (LAMP1-GCaMP6f). In control cells, the majority of the LAMP1-GCaMP6f puncta exhibited very weak fluorescence and were largely distinct from LysoTracker-stained acidic lysosomes (Figure S2A). Only a few strongly fluorescent LAMP1-GCaMP6f puncta were detected (Figure 2J). However, in ORF3a-expressing cells, the number of puncta with strong LAMP1-GCaMP6f signal was dramatically increased (Figures 2K and 2L). LAMP1-GCaMP6f was largely colocalized with ORF3a-labeled late endosomes/lysosomes and also with puncta that were weakly stained by LysoTracker (Figures 2K and S2B). HBSS-induced starvation has been shown to trigger the release of Ca2+ from lysosomes (Medina et al., 2015). Compared with control cells, the amplitude of lysosomal Ca2+ released in ORF3a-expressing cells was much larger than in control cells (Figures 2M and 2O).
SARS-CoV-2 ORF3a has been shown to form Ca2+-permeable non-selective cation channels (Kern et al., 2021). Mutating residues located at the top of the channel cavity, Q57E or S58L Q116L, reduces Ca2+ permeability (Kern et al., 2021). We found that expression of ORF3a(Q57E) or ORF3a(S58L Q116L), like wild-type ORF3a, still caused accumulation of a large number of VPS39-positive puncta (Figures S2C–S2G), increased the number of LAMP1-GCaMP6f puncta (Figures S2H–S2J), and enhanced lysosomal exocytosis (Figures S2K–S2M). Expression of ORF3a(Q57E) and ORF3a(S58L Q116L), similar to ORF3a, caused the formation of a few damaged endosomes/lysosomes labeled by galectin 3 (Figures S2N–S2R). We found that the cytosolic Ca2+ concentration was still slightly increased in cells expressing ORF3a(Q57E) or ORF3a(S58L Q116L) (Figure S2S), which is consistent with the notion that these mutants attenuate, but do not abolish, the Ca2+ permeability (Kern et al., 2021). Therefore, lysosomal-localized Ca2+ release in ORF3a(Q57E)- or ORF3a(S58L Q116L)-expressing cells is likely mediated by the partial Ca2+ permeability retained by these ORF3a mutants.
We next examined whether the TRPML channels are involved in ORF3a-mediated lysosomal exocytosis. In control cells, the puncta strongly labeled by the lysosomal Ca2+ channel TRPML1 were localized close to the PM (Figure S2T). In ORF3a-expressing cells, many more lysosomes showed strong TRPML1 signal and were largely colocalized with ORF3a punctate structures (Figures S2U and S2V). ORF3a-GFP co-immunoprecipitated TRPML1 in GFP-Trap assays (Figure 2N), suggesting that ORF3a may facilitate lysosomal targeting of TRPML1. The PM-localized LAMP1 in ORF3a-expressing cells, however, was not evidently affected in TRPML1 knockout cells (Figures 2P and S2W). TRPML3 has been shown to promote expulsion of bacterium-containing neutralized lysosomes (Miao et al., 2015). Simultaneously depleting TRPML3 in ORF3a-expressing cells resulted in localization of LAMP1-GCaMP6f puncta in the proximity of the PM but did not change the number of puncta (Figures S2X–S2A1). Knocking down TRPML3 reduced the level of PM-localized LAMP1 in control cells and also dramatically inhibited the enhanced lysosomal exocytosis in ORF3a-expressing cells (Figures 2P, 2Q, S2B1, and S2C1). ORF3a-GFP also interacted with TRPML3 in GFP-Trap assays (Figure 2N). Therefore, the increased lysosomal Ca2+ release mediated by ORF3a is not sufficient to drive fusion of lysosomes with the PM. TRPML3 is required for the ORF3a-induced enhancement of lysosomal exocytosis.
Expression of ORF3a promotes the lysosomal targeting of VAMP7 and STX4
During lysosomal exocytosis, fusion of lysosomes with the PM is mediated by the assembly of the trans-SNARE complex composed of VAMP7, STX4, and SNAP23 (Saftig and Klumperman, 2009; Tancini et al., 2020). In control cells, VAMP7-GFP weakly labeled the PM and formed puncta that only partially colocalized with LAMP1-labeled late endosomes/lysosomes (Figures 3 A and S3A). STX4-GFP was also weakly detected on the PM and formed a few puncta (Figure 3D), while SNAP23 was largely localized in the cytosol and the PM (Figure S3E). In ORF3a-expressing cells, the numbers of VAMP7 puncta and STX4 puncta were greatly increased, and they extensively colocalized with ORF3a punctate structures (Figures 3A–3E and S3B). The STX4 puncta were colocalized with VAMP7 in ORF3a-expressing cells (Figures S3C and S3D). Expression of ORF3a also promoted PM localization of SNAP23 and the formation of puncta weakly labeled by SNAP23 that also colocalized with ORF3a (Figures S3E–S3G). The number of VAMP7 puncta in ORF3a-expressing cells was dramatically decreased by STX4 KD (Figures 3F–3H and S3H). VAMP7 KD also reduced the number of STX4 puncta in ORF3a-expressing cells (Figures S3I–S3L), which indicates that STX4 and VAMP7 are involved in each other’s recruitment or stabilization on lysosomes. In GFP-Trap assays, ORF3a-GFP co-immunoprecipitated endogenous STX4 and also VAMP7 (Figures 3J and S3M). After STX4 KD or VAMP7 KD, the level of PM-localized LAMP1 in ORF3a-expressing cells was dramatically decreased (Figures 3I, 3K and 3L). SNAP23 KD did not evidently affect the cell-surface-localized LAMP1 (Figures 3I, S3N, and S3O), which may be because its function is substituted by other PM-localized SNARE proteins such as SNAP25 as reported (Sørensen et al., 2003). In ORF3a-expressing cells with simultaneous knockdown of STX4, the yellow puncta formed by the RFP-GFP-LC3 reporter (unacidified autophagic structures) still accumulated (Figures S3P–S3R), which indicates that the defective autophagosome maturation is not due to reduced availability of intracellular lysosomes in ORF3-expressing cells. Thus, VAMP7 and STX4 are involved in ORF3a-mediated lysosomal exocytosis.
Figure 3.

Enhancement of lysosomal exocytosis by ORF3a expression requires VAMP7 and STX4
(A and B) Compared with control cells expressing FLAG (A), the number of VAMP7-GFP puncta is dramatically increased in ORF3a-FLAG-expressing cells (B).
(C) Quantification of the number of VAMP7-GFP and STX4-GFP puncta per cell. Data are shown as mean ± SEM (n = 26, 31, 28, and 30 for bars from left to right). ∗∗∗p < 0.001.
(D and E) Compared with control cells expressing FLAG (D), the number of STX4-GFP puncta is dramatically increased in ORF3a-FLAG-expressing cells (E).
(F–H) Compared with control cells expressing ORF3a and transfected with control siRNA (F), the number of VAMP7-GFP puncta is dramatically decreased in ORF3a-expressing cells transfected with STX4 siRNA (G). (H) Shows quantification of the number of VAMP7-GFP puncta per cell. Data are shown as mean ± SEM (n = 23 for control cells, n = 30 for STX4 KD cells, and n = 20 for STX6 KD cells). ∗∗∗p < 0.001; n.s., no significant difference.
(I) Quantification of the fluorescence intensity of cell membrane-localized LAMP1 per cell. The level in control siRNA-treated cells is set to 1.0. Data are shown as mean ± SEM (n = 23, 23, 22, 14, and 15 for bars from left to right). ∗∗∗p < 0.001; n.s., no significant difference. The data for ORF3a-expressing cells transfected with control siRNA are the same as those in Figure 2D.
(J) Endogenous STX4 but not STX6 is co-precipitated by ORF3a-GFP in GFP-Trap assays. Cell lysates were immunoprecipitated using GFP-Trap and analyzed by immunoblotting with STX4, FLAG and GFP antibodies.
(K and L) Compared with control cells expressing ORF3a and transfected with control siRNA (Figure 2E), levels of cell membrane-localized LAMP1 are dramatically decreased in ORF3a-expressing cells transfected with STX4 siRNA and VAMP7 siRNA (K and L).
Scale bars: (A, B, D–G, K, and L) 10 μm.
See also Figures S3 and S4.
The expression pattern of FLAG-STX1A, VAMP2-GFP, SNAP25-GFP, GFP-STX18, and VAMP4-GFP remained largely unchanged in ORF3a-expressing cells (Figures S4A–S4K). STX6 is localized in the TGN and forms a few cytosolic punctate structures in control cells (Laufman et al., 2011). In ORF3a-expressing cells, STX6 formed far more puncta (Figures S4L–S4N). However, no interaction was detected between STX6 and ORF3a in GFP-Trap assays (Figure 3J). STX6 KD had no effect on the number of VAMP7 puncta, or on the elevated level of PM-localized LAMP1 in ORF3a-expressing cells (Figures 3H, 3I, and S4O–S4Q).
The enhanced lysosomal exocytosis in ORF3a-expressing cells requires VPS39
Expression of ORF3a results in sequestration of the HOPS component VPS39 on late endosomes/lysosomes (Miao et al., 2021). We determined whether VPS39 contributes to the enhanced lysosomal exocytosis in ORF3a-expressing cells. The level of PM-localized LAMP1 in ORF3a-GFP-expressing cells was dramatically reduced by simultaneously depleting VPS39 (Figures 4A, 4B, and S4R). VPS39 KD also dramatically suppressed the formation of BORCS6, VAMP7, and STX4 puncta in ORF3a-expressing cells (Figures 4C–4G). In GFP-Trap assays, VPS39 co-immunoprecipitated with BORCS6 and STX4 (Figures S4S and S4T). The interaction of ORF3a with BORCS6, VAMP7, and STX4 was decreased after VPS39 KD (Figures 4H, S4U, and S4V), suggesting that VPS39 contributes to the lysosomal targeting and/or stabilization of BORC, VAMP7, and STX4 by ORF3a.
Figure 4.

VPS39 contributes to the enhanced lysosomal exocytosis in ORF3a-expressing cells
(A and B) Compared with control cells expressing ORF3a and transfected with control siRNA (Figure 2E), the level of cell membrane-localized LAMP1 is dramatically decreased in ORF3a-expressing cells transfected with VPS39 siRNA (A). (B) shows quantification of the fluorescence intensity of cell membrane-localized LAMP1 per cell. The level in control siRNA-treated cells is set to 1.0. Data are shown as mean ± SEM n = 23 for VPS39 KD cells. ∗∗∗p < 0.001. The data for ORF3a-expressing cells transfected with control siRNA are the same as those in Figure 2D.
(C–G) Compared with control cells expressing ORF3a and transfected with control siRNA (C, Figures 3F and S3K, respectively), the number of BORCS6-GFP, VAMP7-GFP, and STX4-GFP puncta is dramatically decreased in ORF3a-expressing cells transfected with VPS39 siRNA (D–F). (G) shows quantification of the number of BORCS6-GFP, VAMP7-GFP, and STX4-GFP puncta per cell. The data for VAMP7-GFP and STX4-GFP in control cells are the same as those in Figures 3H and S3J. Data are shown as mean ± SEM (n = 19, 31, 23, 30, 27, and 31 for bars from left to right). ∗∗∗p < 0.001.
(H) The level of ORF3a-mCherry co-precipitated by VAMP7-GFP is decreased after VPS39 KD. Cell lysates were immunoprecipitated using GFP-Trap and analyzed by immunoblotting with mCherry and GFP antibodies. Levels of ORF3a-mCherry in control siRNA- and VPS39 siRNA-treated cells were normalized by VAMP7-GFP level. The level in control siRNA-treated cells is set to 1.0.
Scale bars: (A and C–F) 10 μm.
See also Figure S4.
TFEB has been shown to be predominantly localized in the nucleus in ORF3a-expressing cells via an unknown mechanism (Figures S4W and S4X) (Miao et al., 2021). TFEB overexpression promotes lysosomal exocytosis (Medina et al., 2011). Unlike in ORF3a-expressing cells, the formation of VPS39-GFP, BORCS6-GFP, STX4-GFP, and VAMP7-GFP puncta was not evidently changed in TFEB-overexpressing cells (Figures S5A–S5I). As shown below, a mutant ORF3a causes TFEB nuclear localization but does not enhance lysosomal exocytosis. The nuclear-localized TFEB in ORF3a-expressing cells is not functional, as the mRNA levels of the majority of TFEB targets remain unchanged (Miao et al., 2021). Taken together, our results indicate that ORF3a promotes lysosomal exocytosis independent of nuclear-localized TFEB.
SARS-CoV-2 infection elevates lysosomal exocytosis
We next examined whether SARS-CoV-2 infection also elevates lysosomal exocytosis. In SARS-CoV-2-virus-infected Vero E6 cells or in HeLa cells expressing ACE2, staining with the PE-conjugated anti-LAMP1 antibody showed dramatically elevated cell surface LAMP1 (Figures 5A–5E). In control cells, VAMP7 and STX4 were largely diffuse and formed a very few punctate structures (Figures 5F and 5H). However, a large number of VAMP7 and STX4 puncta were detected in SARS-CoV-2-infected cells (Figures 5G, 5I, and 5J). The numbers of VAMP7 and STX4 puncta were also dramatically increased in SARS-CoV-2-infected Vero E6 cells (Figures 5J–5N). Therefore, as in ORF3a-expressing cells, SARS-CoV-2 virus infection increases the recruitment of STX4 and VAMP7 and also promotes lysosomal exocytosis.
Figure 5.

SARS-CoV-2 virus infection elevates lysosomal exocytosis
(A–E) The level of cell-membrane-localized LAMP1 is dramatically increased in SARS-CoV-2 (SARS2)-infected ACE2-expressing HeLa cells (A and B) and Vero E6 cells (C and D). (E) shows quantification of the fluorescence intensity of cell membrane-localized LAMP1 per cell. Levels in control cells are set to 1.0. Data are shown as mean ± SEM (n = 26, 24, 21, and 22 for bars from left to right). ∗∗p <0.01; ∗∗∗p <0.001.
(F–I) Compared with control cells expressing ACE2 (F and H), the number of VAMP7-GFP puncta and STX4-GFP puncta is dramatically increased in SARS-CoV-2-infected ACE2-expressing HeLa cells (G and I).
(J) Quantification of the number of VAMP7-GFP puncta and STX4-GFP puncta per cell. Data are shown as mean ± SEM (n = 24, 27, 29, 26, 23, 28, 24, and 26 for bars from left to right). ∗∗∗p < 0.001.
(K–N) Compared with control cells (K and M), the number of VAMP7-GFP puncta and STX4-GFP puncta is dramatically increased in SARS2-infected Vero E6 cells (L and N).
(O) Quantification of the fluorescence intensity of cell-membrane-localized LAMP1 per cell in 17Cl-1 cells expressing GFP and ORF3a-GFP. The level in control cells is set to 1.0. Data are shown as mean ± SEM (n = 21 for each bar). ∗∗∗p < 0.001.
(P–R) Plaque assay showing that the vital titer of the supernatant from MHV-A59-infected ORF3a-GFP-expressing cells is higher than that from MHV-A59-infected GFP-expressing cells (control) (P and Q). (R) shows quantification of vital titer at 16 h post-infection. Data are shown as mean ± SEM (n = 3 for each bar). ∗p < 0.05.
Scale bars: (A–D, F–I, and K–N) 10 μm.
See also Figure S5.
ORF3a facilitates MHV-A59 egress
The coronavirus MHV-A59 also exploits lysosomal exocytosis for egress (Ghosh et al., 2020). MHV-A59 does not contain an ORF3a homolog. We investigated whether expression of SARS-CoV-2 ORF3 could facilitate the release of MHV-A59 into the extracellular environment. Expression of ORF3a promotes lysosomal exocytosis in 17Cl-1 cells, which can be effectively infected with MHV-A59 (Figures 5O, S5J, and S5K). Supernatants were collected from MHV-A59-infected control and ORF3a-expressing cells at 16 h post-infection. The plaque assay was then performed to determine the viral titer. We found that more colonies were formed by the supernatant from MHV-59-infected ORF3a-expressing cells than control cells (Figures 5P–5R). At 16 h post-infection, the percentage of cells undergoing apoptotic cell death, judged by propidium iodide (PI) staining, was the same in control and ORF3a-expressing cells (Figure S5L). These results indicate that expression of ORF3a is sufficient to facilitate the extracellular release of MHV-A59.
Ser171 and Trp193 are critical for ORF3a to promote lysosomal exocytosis
SARS-CoV-2 ORF3a and SARS-CoV ORF3a exhibit 72.4% identity (199 out of 275 amino acids are identical). ORF3a contains a short extracellular/luminal-oriented N terminus, three transmembrane regions and a C-terminal cytoplasmic domain (Figure 6 A). SARS-CoV ORF3a, however, fails to induce the formation of VPS39 puncta (Figures 6B–6D) (Miao et al., 2021). We found that expression of SARS-CoV ORF3a had no evident effect on the level of LAMP1 and LAMP2 in the PM fraction (Figure 1A). SARS-CoV ORF3a also failed to elevate the cell surface LAMP1 level (Figures 6G and 6I). Consistent with this, SARS-CoV ORF3a did not induce the accumulation of STX4 and VAMP7 puncta (Figures 6J, S5C1, and S5F1).
Figure 6.

Ser171 and Trp193 are essential for ORF3a to promote lysosomal exocytosis and to block autophagy
(A) Schematic illustration of the domains in ORF3a of SARS-CoV-2 (SARS2) and SARS-CoV (SARS).
(B) Quantification of the number of VPS39-GFP puncta in cells expressing ORF3a-mCherry, ORF3a(42aa–275aa)-mCherry, ORF3a(S171E)-mCherry, SARS ORF3a-mCherry, SARS ORF3a(42aa–274aa)-mCherry, and ORF3a(W193R)-mCherry. Data are shown as mean ± SEM (n = 31, 22, 32, 32, 32, and 25 for bars from left to right). ∗∗∗p < 0.001; n.s., no significant difference.
(C and D) Expression of SARS-CoV-2 ORF3a (D) but not SARS-CoV ORF3a (C) induces the formation of a large number of VPS39 puncta.
(E) Quantification of the number of VPS39-GFP puncta in cells expressing ORF3a-FLAG and a series of ORF3a-FLAG mutants with the indicated regions replaced with the corresponding regions of SARS-CoV ORF3a. Data are shown as mean ± SEM (n = 29 for ORF3a(165–171) chimeric mutants, n = 20 for other bars). ∗∗∗p < 0.001; ∗∗p < 0.01; n.s., no significant difference.
(F) The mutant ORF3a(S171E), in which Ser171 of SARS-CoV-2 ORF3a is replaced by Glu171 of SARS-CoV ORF3a, fails to induce formation of VPS39-GFP puncta.
(G and H) Compared with control cells expressing GFP (Figure 1C), the level of cell-membrane-localized LAMP1 detected by the live cell staining assay is not increased in cells expressing SARS-CoV ORF3a-GFP (G) or SARS-CoV-2 ORF3a(S171E)-GFP (H).
(I) Quantification of the fluorescence intensity of cell-membrane-localized LAMP1 per cell. The level in control cells is set to 1.0. Data are shown as mean ± SEM (n = 33, 23, 21, 28, 29, 23, and 16 for bars from left to right). ∗p < 0.05, ∗∗∗p < 0.001; n.s., no significant difference. The data for control and ORF3a-expressing cells are the same as those in Figure 1E.
(J) Quantification of the number of STX4-GFP and VAMP7-GFP puncta in cells expressing mCherry (control), ORF3a-mCherry, ORF3a(S171E)-mCherry, and SARS-CoV ORF3a-mCherry. Data are shown as mean ± SEM (n = 16, 30, 24, 19, 14, 31, 22, and 19 for bars from left to right). ∗∗∗p < 0.001. The data for ORF3a-expressing cells are the same as those in Figure 3C.
(K) In GFP-Trap assays, FLAG-VPS39 is co-precipitated by ORF3a-GFP and very weakly by ORF3a(W193R)-GFP, but not by ORF3a(S171E)-GFP. Cell lysates were immunoprecipitated using GFP-Trap and analyzed by immunoblotting with FLAG and GFP antibodies.
Scale bars: (C, D, F, G, and H) 10 μm.
See also Figures S5 and S6.
We next determined the region responsible for the differential effect of SARS-CoV-2 ORF3a. SARS-CoV-2 ORF3a(42–275), a truncated protein containing the transmembrane domains and C terminus, retained the ability to induce the formation of a large number of VPS39 puncta, but SARS-CoV ORF3a(42–274) did not (Figures 6B, S5M, and S5N). The differences between the C terminus of SARS-CoV-2 ORF3a and SARS-CoV ORF3a lie in 10 regions. We mutated the amino acids in each region of SARS-CoV-2 ORF3a to those in SARS-CoV ORF3a to generate chimeric proteins. Substituting the amino acids 165 to 171 abolished the formation of VPS39-GFP puncta, substituting the amino acids 190 to 194 reduced the formation of VPS39-GFP puncta, and mutating amino acids in other regions of SARS-CoV-2 ORF3a had no evident effect on the formation of VPS39-GFP puncta (Figures 6E and S5O–S5X). Further mutating serine at 171 of ORF3a to the corresponding glutamic acid in SARS-CoV ORF3a, referred to as ORF3a(S171E), abolished the formation of VPS39-GFP puncta, while mutating 165, 166, and 169 to their counterparts in SARS-CoV ORF3a had no effect (Figures 6B, 6F, and S5Y–S5A1). ORF3a(S171E)-GFP also failed to promote the formation of VAMP7-GFP puncta and STX4-GFP puncta (Figures 6J, and S5E1, and S5H1). Compared with ORF3a-GFP, ORF3a(S171E)-GFP formed fewer punctate structures (Figure S5B1). The elevated PM-localized LAMP1 was also abolished in ORF3a(S171E)-expressing cells (Figures 6H and 6I). The S171E mutation also abolished the ability of ORF3a to increase cytosolic free Ca2+, HBSS-induced lysosomal Ca2+ release, and the number of LAMP1-GCaMP6f puncta (Figures S5I1–S5M1). Expression of the ORF3a(S171E) mutant still led to TFEB nuclear localization (Figure S5N1). Mutating tryptophan at residue 193 to its counterpart arginine in SARS-CoV ORF3a also reduced the number of VPS39 puncta and partially blocked enhanced lysosomal exocytosis (Figures 6B, 6I and S5O1–S5P1). Expression of ORF3a(S171E) failed to cause accumulation of p62 aggregates and LC3 puncta (Figures S6A–S6H). ORF3a(S171E) abolished while ORF3a(W193R) reduced the interaction with VPS39 (Figure 6K). Taken together, these results show that residues 171 and 193 are critical for ORF3a to promote lysosomal exocytosis and inhibit autophagy.
The E171S and R193W mutations endow SARS-CoV ORF3a with the ability to promote lysosomal exocytosis and block autophagy
We investigated which residues are sufficient to endow SARS-CoV ORF3a with the ability to cause the formation of VPS39 and STX4 puncta and to promote lysosomal exocytosis. SARS-CoV ORF3a(E171S) did not promote the formation of VPS39-GFP and STX4-GFP puncta, or PM-localized LAMP1 (Figures 7 A and S6I–S6K). We then mutated amino acids in other regions in the C terminus of SARS-CoV ORF3a(E171S) to those in SARS-CoV-2 ORF3a. The mutant proteins are designated SARS-CoV ORF3a(E171S, X–Y), where X–Y is the range of substituted amino acids. We found that expression of SARS-CoV ORF3a(E171S, 190–194) dramatically induced the formation of VPS39 puncta (Figures 7A and S6P). A few VPS39 puncta were also detected in cells expressing SARS-CoV ORF3a(E171S, 215–220), while other mutants had no effect (Figures 7A and S6L–S6U). Mutating the individual amino acids in the range 190–194 revealed that SARS-CoV ORF3a(E171S R193W), but not SARS-CoV ORF3a(E171S S190T, E171S D192K, or E171S H194E), caused the formation of VPS39 and STX4 puncta that were largely colocalized with LAMP1 (Figures 7A, 7B, and S6V–S6D1). In GFP-Trap assays, SARS-CoV ORF3a(E171S R193W) was co-immunoprecipitated by VPS39 (Figure S7A). Compared with SARS-CoV ORF3a, cells expressing SARS-CoV ORF3a(E171S R193W) contained more LAMP1-GCamP6 fluorescent puncta (Figures 7C–7E). Expression of SARS-CoV ORF3a did not evidently change the cytosolic free Ca2+ (Figures 7F and 7G), while expressing SARS-CoV ORF3a(E171S R193W) caused an increase in cytosolic free Ca2+ as well as lysosomal Ca2+ release (Figures 7F–7H and S7B). Expression of SARS-CoV ORF3a(E171S R193W) also enhanced cell surface-localized LAMP1 (Figures 6I and 7I).
Figure 7.

Mutating E171S and R193W endows SARS-CoV ORF3a with the ability to promote lysosomal exocytosis and block autophagy
(A) Quantification of the number of VPS39-GFP puncta in cells expressing ORF3a-mCherry, SARS ORF3a-mCherry, SARS ORF3a(E171S)-mCherry, SARS ORF3a(E171S R193W)-mCherry, and a series of SARS ORF3a(E171S)-mCherry mutants in which the indicated region of SARS-CoV ORF3a is replaced with that of SARS-CoV-2 ORF3a. Data are shown as mean ± SEM (n = 22 for ORF3a-expressing cells, n = 32 for other bars). ∗∗∗p <0.001; n.s., no significant difference.
(B) A large number of VPS39 puncta are formed in cells expressing SARS-CoV ORF3a(E171S R193W).
(C–E) Compared with control cells expressing SARS ORF3a-mCherry, the number of LAMP1-GCaMP6f puncta is dramatically increased in SARS-CoV ORF3a(E171S R193W)-mCherry-expressing cells (D and E). (C) shows quantification of the number of LAMP1-GCaMP6f puncta per cell. Data are shown as mean ± SEM (n = 21 for SARS ORF3a-expressing cells, n = 19 for SARS-CoV ORF3a(E171S R193W)-expressing cells). ∗∗∗p < 0.001.
(F) The cytosolic free calcium concentration in HeLa cells expressing mCherry (control), SARS ORF3a-mCherry, and SARS ORF3a(E171S R193W)-mCherry. The data for control cells are the same as those in Figure 2I. Data are shown as mean ± SEM (n = 45 for control, n = 14 for SARS ORF3a, n = 25 for SARS-CoV ORF3a(E171S R193W)). ∗∗∗p < 0.001; n.s., no significant difference.
(G) The 340/380 nm fluorescence excitation ratio of HeLa cells expressing GFP (control), SARS ORF3a-GFP, and SARS ORF3a(E171S R193W)-GFP after loading with fura-2. The data for control cells are the same as those in Figure 2H. Data are shown as mean ± SEM (n = 55 for control, n = 68 for SARS ORF3a, and n = 38 for SARS-CoV ORF3a(E171S R193W)). ∗∗∗p < 0.001; n.s., no significant difference.
(H) The ratio of ΔF/F0 in control cells expressing mCherry (control), SARS ORF3a-mCherry, and SARS ORF3a(E171S R193W)-mCherry after inducing lysosomal Ca2+ release by HBSS. (H) shows quantification of ΔF/F0. The data for control cells are the same as those in Figure 2O. Data are shown as mean ± SEM (n = 21 for control, n = 12 for SARS ORF3a, n = 16 for SARS-CoV ORF3a(E171S R193W)). ∗∗∗p < 0.001; n.s., no significant difference.
(I) Compared with cells expressing SARS ORF3a (Figure 6G), the level of cell membrane-localized LAMP1 detected by the live cell staining assay is dramatically increased in cells expressing SARS ORF3a(E171S R193W)-GFP.
(J–L) Compared with cells expressing SARS ORF3a-GFP (J), the number of LC3 puncta is dramatically increased in cells expressing SARS-CoV ORF3a(E171S R193W)-GFP (L). (K) shows quantification of the number of LC3 puncta per cell. Data are shown as mean ± SEM (n = 28 for SARS ORF3a-expressing cells; n = 25 for SARS ORF3a(E171S R193W)-expressing cells). ∗∗∗p < 0.001.
(M) The percentage of total LC3 puncta that are RFP+GFP− in cells expressing FLAG vector (control) and SARS ORF3a(E171S R193W)-FLAG after 3 h amino acid starvation. Data are shown as mean ± SEM (n = 28 for control cells, n = 22 for SARS ORF3a(E171S R193W)-expressing cells). ∗∗∗p < 0.001.
(N) A model showing that SARS-CoV-2 ORF3a and SARS-CoV ORF3a(E171S R193W) promote lysosome exocytosis. They facilitate lysosomal targeting of the BORC-ARL8b complex and SNARE proteins. TRPML3 is essential for fusion of lysosomes with the PM in cells expressing ORF3a or SARS-CoV ORF3a(E171S R193W).
Scale bars: (B, D, E, I, J, and L) 10 μm.
See also Figures S6 and S7.
SARS-CoV ORF3a(E171S R193W) also inhibited autophagy, resulting in accumulation of a large number of LC3 puncta and p62 aggregates (Figures 7J–7L, and S7C–S7E). The RFP-GFP-LC3 assay indicated that red-only RFP+GFP− puncta (which indicate acidic compartments because GFP is quenched at low pH) accumulated in control cells under starvation conditions, while in cells expressing SARS-CoV ORF3a(E171S R193W), unacidified LC3 puncta (RFP+GFP+) accumulated under both nutrient-rich and starvation conditions (Figures 7M and S7F–S7I). This further confirms that expression of SARS-CoV ORF3a(E171S R193W) blocks autophagy. Therefore, mutating E171S and R193W is sufficient to endow SARS-CoV ORF3a with the ability to promote lysosomal exocytosis and block autophagy.
Discussion
ORF3a of SARS-CoV-2 promotes lysosomal exocytosis
During the cycle of β-coronavirus infection including SARS-CoV-2 and MHV, newly assembled viral particles traffic to late endosomes/lysosomes and then exploit the lysosomal exocytosis pathway for release (Ghosh et al., 2020). Lysosomal exocytosis is enhanced by β-coronavirus infection (Ghosh et al., 2020), but the underlying mechanism remains largely unknown. Here, we revealed that SARS-CoV-2 ORF3a promotes multiple steps involved in lysosomal exocytosis (Figure 7N). In ORF3a-expressing cells, targeting of the BORC-ARL8b complex to late endosomes/lysosomes is greatly enhanced, suggesting that anterograde lysosome transport is facilitated. VAMP7 and STX4 are also targeted to ORF3a-localized late endosomes/lysosomes at an enhanced level by ORF3a. Depleting BORC, ARL8b, VAMP7, or STX4 abolishes the enhanced lysosomal exocytosis in ORF3a-expressing cells.
Fusion of lysosomes with the PM requires an elevated cytosolic Ca2+ level, which could result from extracellular Ca2+ influx or localized lysosomal Ca2+ release (Rodríguez et al., 1997; Saftig and Klumperman, 2009). TRPML1 has been shown to mediate the localized lysosomal Ca2+ release that is needed for lysosomal exocytosis in response to TFEB expression and at the phagocytic cup during large particle uptake (Medina et al., 2011; Samie et al., 2013). TRPML3 senses lysosome neutralization and triggers Ca2+ efflux to stimulate lysosomal exocytosis such as in expulsion of lysosome-encased uropathogenic E. coli (Miao et al., 2015; Tancini et al., 2020). β-coronavirus infection results in deacidification of lysosomes (Ghosh et al., 2020), which may result from loading with too many viral particles, and/or perturbations in proton pump or ion channel activity by viral proteins. In ORF3a-expressing cells. the LysoTracker fluorescence intensity is weaker in a subpopulation of lysosomes (Ghosh et al., 2020; Miao et al., 2021). We showed here that ORF3a expression elevates cytosolic Ca2+ concentration and localized lysosomal Ca2+ release as shown by an increase in the number of LAMP1-GCaMP6f puncta. ORF3a itself is a Ca2+-permeable channel, whose activity is not affected by pH (Kern et al., 2021). In ORF3a-expressing cells with simultaneous knockdown of TRPML3, a large number of LAMP1-GCaMP6f puncta still accumulate in the vicinity of the PM, but the enhanced lysosomal exocytosis is inhibited. This suggests that TRPML3 is not responsible for localized lysosomal Ca2+ release but is required for triggering the fusion of lysosomes with the PM. TRPML3 is widely distributed, including in the PM, the ER and intracellular compartments (e.g., lysosomes). Thus, TRPML3 can mobilize multiple Ca2+ stores to promote lysosomal exocytosis (Kim et al., 2009). In ORF3a-expressing cells, LAMP1-GCaMP6f puncta are weakly stained by LysoTracker, which suggests that ORF3a-induced lysosome neutralization may activate TRPML3. The lysosomal-localized synaptotagmin VII (SYT7) has also been implicated in promoting fusion of lysosomes with the PM (Tancini et al., 2020). SYT7 labels more puncta in ORF3a-expressing cells and in SARS-CoV-2 infected cells (Figures S7J–S7N). However, SYT7 KD has no evident effect on enhanced lysosomal exocytosis in ORF3a-expressing cells (Figures S7O–S7Q), suggesting that other Syt proteins or Ca2+ sensors may be involved in this process. CQ treatment, which strongly deacidifies lysosomes so that they have no LysoTracker staining signal, facilitates lysosomal exocytosis, but to a lesser extent than ORF3a expression (Figures S7R–S7W). Thus, SARS-CoV-2 ORF3a exploits a mechanism that is at least partly independent of lysosomal alkalinization to enhance lysosomal exocytosis.
Lysosomes/late endosomes exhibit heterogeneity in their size, morphology, and resident enzymes. ORF3a mainly labels late endosomes/lysosomes positive for RAB7 and LAMP1; it only labels a subset of LAMP2-positive lysosomes (Miao et al., 2021). In ORF3a-expressing cells, a certain percentage of accumulated amphisomes are positive for both LC3 and RAB7/LAMP1 (Miao et al., 2021). Virion particles are detected in amphisome-like structures in SARS-CoV-2-infected cells (Miao et al., 2021). TFEB has been shown to induce exocytosis of the arrested autolysosomes in Pompe disease muscle cells (Spampanato et al., 2013). Exocytosis of amphisomes may also be enhanced by ORF3a. VPS39, which is sequestrated on late endosomes/lysosomes by ORF3a (Miao et al., 2021), contributes to recruitment and/or stabilization of BORC, VAMP7, and STX4 on lysosomes and also to enhanced lysosomal exocytosis in ORF3a-expressing cells. Thus, sequestration of VPS39 on late endosomes/lysosomes by ORF3a has the dual ability to block autophagosome maturation and promote lysosomal exocytosis. Although the physiological relevance of ORF3a in SARS-CoV-2 egress has yet to be determined, expression of ORF3a increases the viral titer of supernatants from MHV-A59-infected cells, which supports the notion that ORF3a has an important function in promoting lysosomal exocytosis-mediated viral egress.
The differential function of ORF3a in lysosomal exocytosis may contribute to the higher infectivity and pathogenicity of SARS-CoV-2 compared with SARS-CoV
Unraveling the differential functions of SARS-CoV-2 and SARS-CoV proteins is crucial for us to understand the mechanisms which endow SARS-CoV-2 with much higher infectivity and pathogenicity than SARS-CoV. Previous studies demonstrated that multiple differential properties of the S protein enable SARS-CoV-2 to enter more efficiently into host cells than SARS-CoV (Shang et al., 2020; Wrapp et al., 2020). Compared with the SARS-CoV S protein, SARS-CoV-2 S protein possesses a multibasic domain at the S1/S2 cleavage site that allows pre-cleavage by furin during viral packaging in host cells (Hoffmann et al., 2020a; Shang et al., 2020), its RBD exhibits higher ACE2-binding affinity (Wrapp et al., 2020), and it adopts conformations less susceptible to immune surveillance (Shang et al., 2020).
ORF3a is an accessory protein present only in a subset of coronaviruses. ORF3a of SARS-CoV and SARS-CoV-2 forms a homotetrameric complex and possesses non-selective cation channel activity (Castaño-Rodriguez et al., 2018; Kern et al., 2021; Lu et al., 2006). SARS-CoV ORF3a protein localizes to the PM and Golgi apparatus and also forms a few cytoplasmic punctate structures (Castaño-Rodriguez et al., 2018; Lu et al., 2006; Minakshi and Padhan, 2014). SARS-CoV ORF3a regulates various cellular responses, including apoptosis (Tan et al., 2006), activation of NF-κB and NLRP3 inflammasome (Siu et al., 2019), and production of inflammatory chemokines (Kanzawa et al., 2006). Deletion of ORF3a has no effect on the extracellular release of SARS-CoV from cultured cells (Freundt et al., 2010). In mouse models of SARS-CoV infection, genomic deletion of ORF3a reduces viral titer and virulence (Castaño-Rodriguez et al., 2018). SARS-CoV-2 ORF3a is localized on late endosomes/lysosomes as well as on the PM (Miao et al., 2021). SARS-CoV-2 ORF3a has been shown to contribute to viral pathogenesis in the mouse model of SARS-CoV-2 infection (Silvas et al., 2021). Compared with the parental virus, infection with virus with a deletion of ORF3a results in lower viral titers, faster recovery of lung pathology, and reduced morbidity (Silvas et al., 2021). Our studies revealed that SARS-CoV-2 ORF3a, but not SARS-CoV ORF3a, sequestrates VPS39 on late endosomes/lysosomes, blocks autophagosome maturation (Miao et al., 2021), and promotes lysosomal exocytosis. Ser171 and Trp193 of SARS-CoV-2 ORF3a are critical for these effects. Surprisingly, mutating Glu171 and Arg193 of SARS-CoV ORF3a to the corresponding Ser and Trp in SARS-CoV-2 ORF3a endows it with the ability to promote lysosomal exocytosis and inhibit autophagy activity. Egress is an important determinant of coronaviral infectivity and pathogenesis. The differential function of ORF3a in lysosomal exocytosis-mediated viral release may contribute to the higher infectivity and pathogenicity of SARS-CoV-2 compared with SARS-CoV. Of note, civet SARS ORF3a, an intermediate for bat-to-human spillover, contains Glu171 and Arg193 as in SARS-CoV, while bat RaTG13 and pangolin CoV contain Ser171 and Trp193 as in SARS-CoV-2 (Figure S7X). Our functional analysis of ORF3a supports the hypothesis that pangolin could be a possible intermediate host for SARS-CoV-2 (Lam et al., 2020). Extensive efforts have been focused on blocking SARS-CoV-2 entry as a strategy for COVID-19 treatment (Hartenian et al., 2020; Hoffmann et al., 2020b). Our findings indicate that the mechanisms facilitating viral egress, such as the interaction of ORF3a with host factors, can also be explored for developing new intervention strategies to treat SARS-CoV-2 infection.
Limitations of the study
There are several limitations to our study: an elevation in cytosolic Ca2+ concentration and/or localized lysosomal Ca2+ release has been previously shown to promote lysosomal exocytosis (Rodríguez et al., 1997; Tancini et al., 2020). The mechanism underlying the additional requirement for TRPML3 in the ORF3a-induced enhancement of lysosomal exocytosis has not been investigated. In addition, the location (e.g., the PM, the ER, or lysosomes) where TRPML3 acts to mobilize Ca2+ should be explored in the future. Sequestration of VPS39 on late endosomes/lysosomes is required for the enhanced lysosomal exocytosis in ORF3a-expressing cells. VPS39 is a subunit of the HOPS complex and also forms a subcomplex with VPS11 or VPS41. The interactions of VPS39 with different subunits of the HOPS complex is differentially affected by ORF3a expression (Miao et al., 2021). Our study does not determine whether VPS39 acts in a functional HOPS complex or in a subcomplex to regulate lysosomal exocytosis in ORF3a-expressing cells. We found that ORF3a overexpression facilitates MHV-A59 egress. Future studies will be required to investigate the role of ORF3a in lysosomal exocytosis-mediated egress of SARS-CoV-2.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-LC3 (clone 4E12) | MBL | Cat# M152-3; RRID: AB_1279144 |
| Rabbit polyclonal anti-p62 | MBL | Cat# PM045; RRID: AB_1279301 |
| Rat monoclonal anti-LAMP1 (clone 1D4B) | BD Biosciences | Cat# 553792; RRID : AB_2134499 |
| Mouse monoclonal anti-LAMP1 | BD Biosciences | Cat# 555798; RRID: AB_396132 |
| Rabbit polyclonal anti-Flag | Cell Signaling Technology | Cat# 14793S; RRID: AB_2572291 _ |
| Mouse polyclonal anti-Flag (clone M2) | Sigma-Aldrich | Cat# F1804; RRID: AB_262044 |
| Mouse PE-conjugated anti-LAMP1 | BioLegend | Cat# 328608; RRID: AB_1186040_ |
| Rabbit polyclonal anti-LAMP1 | Abcam | Cat# ab24170; RRID: AB_775978 |
| Rabbit polyclonal anti-LAMP2 | Sigma | Cat# PRS3627; RRID: AB_1846223 |
| Mouse monoclonal anti-Actin (clone 7D2 C10) | Proteintech | Cat# 60008-1-Ig; RRID: AB_2289225 |
| Mouse monoclonal anti-GFP (clone 7.1 and 13.1) | Roche | Cat# 11814460001; RRID: AB_390913 |
| Rabbit monoclonal anti-VAMP7 (clone D4D5J) | Cell Signaling Technology | Cat# 14811S; RRID: AB_2798625 |
| Rabbit polyclonal anti-GAPDH | Biodragon | Cat# B1034; N/A |
| Rabbit polyclonal anti-Integrin α5 | Cell Signaling Technology | Cat# 4705; RRID: AB_2233962 |
| Mouse monoclonal anti-STX6 (clone 3D10) | Abcam | Cat# ab12370; RRID: AB 2196497 |
| Rabbit monoclonal anti-STX4 (clone E6W7B) | Cell Signaling Technology | Cat# 67657S; N/A |
| Rabbit polyclonal anti-VPS39 | Proteintech | Cat# 16219-1-AP; RRID: AB_11043179 |
| Rabbit polyclonal anti-mCherry | Proteintech | Cat# 26765-1-AP; RRID: AB_2876881 |
| Goat polyclonal anti-CTSB | R and D Systems | Cat# AF953; RRID: AB_355738 |
| Goat polyclonal anti-CTSD | Santa Cruz | Cat# sc-6486, RRID: AB_637896 |
| Mouse monoclonal anti-SARS-CoV2 Nucleocapsid | Sino Biological | Cat# 40143-MM05; RRID: AB_2827977 |
| Bacterial and Virus Strains | ||
| E. coli BL21-CodonPlus (DE3) | Agilent | Cat#280230 |
| E. coli Strain OP50 | Caenorhabditis Genetics Center | Cat#OP50; RRID: WB-STRAIN:OP50 |
| SARS-CoV-2 (hCoV-19/China/CAS-B001/2020) | Institute of Microbiology, Chinese Academy of Sciences | GISAID No. EPI_ISL_514256-7 |
| MHV A59 | Laboratory of Dr. Hongyu Deng (IBP) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Protease inhibitor | Biotool | Cat# B14003 |
| LysoTracker Deep Red | Thermo Fisher Scientific | Cat# L12492 |
| LysoTracker Green | Thermo Fisher Scientific | Cat# L7526 |
| Torin 1 | Cell Signaling Technology | Cat# 14379 |
| Brefeldin A | Beyotime | Cat# S1536 |
| Chloroquine diphosphate salt (CQ) | Sigma | Cat# C6628 |
| Critical Commercial Assays | ||
| RevertAid First Strand cDNA Synthesis Kit | Thermo Fisher Scientific | Cat# K1621 |
| Cell Fractionation kit | Invent Biotechnologies | Cat# SM-005 |
| Deposited Data | ||
| Original Data | This paper, Mendeley Data | N/A |
| Experimental Models: Cell Lines | ||
| Vero E6 cells | Dr. Yan Li (Institute of Microbiology, CAS) | N/A |
| 17Cl-1 cells | Laboratory of Dr. Hongyu Deng (IBP, CAS) | N/A |
| HeLa cells | ATCC | CCL-2 |
| HEK293T cells | ATCC | CRL-11268 |
| Oligonucleotides | ||
| Primers used for qRT-PCR experiments, see Table S1 | This paper | N/A |
| siRNA sequence, see Table S1 | This paper | N/A |
| Recombinant DNA | ||
| Plasmid: SARS2 ORF3a-FLAG | Laboratory of Dr. Peihui Wang (Shandong University) | N/A |
| Plasmid: SARS ORF3a-FLAG | Laboratory of Dr. Peihui Wang (Shandong University) | N/A |
| Plasmid: SARS2 ORF3a-GFP | Miao et al., 2021 | N/A |
| Plasmid: SARS ORF3a-GFP | Miao et al., 2021 | N/A |
| Plasmid: SARS2 ORF3a-mCherry | This paper | N/A |
| Plasmid: SARS ORF3a-mCherry | This paper | N/A |
| Plasmid: BORCS6-GFP | This paper | N/A |
| Plasmid: STX4-GFP | This paper | N/A |
| Plasmid: VAMP7-GFP | This paper | N/A |
| Plasmid: VAMP2-GFP | This paper | N/A |
| Plasmid: SNAP23-GFP | This paper | N/A |
| Plasmid: Flag-TRPML1 | This paper | N/A |
| Plasmid: Flag-STX1A | This paper | N/A |
| Plasmid: VPS39-GFP | Miao et al., 2021 | N/A |
| Plasmid: SYT7-GFP | This paper | N/A |
| Plasmid: RFP-GFP-LC3 | Kimura et al., 2007 | Addgene Plasmid #21074 |
| Software and Algorithms | ||
| ImageJ | NIH | https://imagej.en.softonic.com/ |
| GraphPad Prism | GraphPad Software | https://www.graphpad.com/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Hong Zhang (hongzhang@ibp.ac.cn).
Materials availability
Plasmids and other reagents generated in this study will be available upon request from the Lead Contact with a completed Materials Transfer Agreement.
Experimental model and subject details
Cell lines
HeLa cells and COS7 cells were obtained from ATCC. Mouse fibroblast 17Cl-1 cells were kindly gifted by Dr. Hongyu Deng’s lab (IBP, CAS). Vero E6 cells were kindly gifted by Dr. Yan Li (Institute of Microbiology, CAS). The HeLa cell line with stable TFEB-GFP expression was kindly gifted by Dr. Richard Youle’s lab (NINDS). All of the cell lines were maintained in DMEM (SH30022.01B, Hyclone) with 10% FBS (SH30084.03, Hyclone) and 50 mg/ml penicillin-streptomycin at 37 °C and 5% CO2. For amino acid and HBSS starvation, cells were incubated with DMEM without amino acids (SH4007.01, Hyclone) or HBSS (14025-092, Gibco) for the indicated time. For Brefeldin A (BFA) treatment, cells were incubated with 5 μg/mL BFA (S1536, Beyotime) at 37 °C for 6 h. For chloroquine diphosphate salt (CQ) treatment, cells were incubated with 100 mg/ml CQ (C6628, Sigma) at 37 °C for 4 h. For Torin 1 (14379, Cell Signaling Technology) treatment, cells were incubated with 1 μM Torin 1 at 37 °C for 4 h. For LysoTracker staining, samples were incubated with LysoTracker (L7528, Life Technologies) at 37 °C for 30 min according to the manufacturer’s instructions.
Method details
Plasmids
Genes encoding SARS-CoV-2 ORF3a and SARS-CoV ORF3a were amplified by PCR and cloned into pcDNA6B-FLAG, pEGFP-N1 and pECherry-N1 vector. Coding sequences for BORCS6, STX4, VAMP7, VAMP2, SNAP23, STX1A, VPS39, TRPML1 were amplified by PCR from human cDNA libraries and inserted into pEGFP-N1 or pFLAG-CMV-2 vector. SYT7-GFP was generated by inserting the mouse SYT7 cDNA into pEGFP-N1 vector. All of the SARS-CoV-2 ORF3a mutants and SARS-CoV ORF3a mutants were constructed by mutating the indicated sites of specific plasmids and confirmed by sequencing.
Transfection and siRNA in cell lines
DNA transfections were performed with Lipofectamine 2000 (Life Technologies, 12566014) for 24 h and siRNAs were transfected with Lipofectamine RNAiMAX (13778150, Invitrogen) for 72 h. Double-stranded siRNAs were purchased from GenePharma. Knockdown efficiencies of all siRNAs were verified by Western blot or Real-Time PCR. The siRNA sequences are listed in Table S1.
SARS-CoV-2 virus infection
HeLa cells were cultured on coverslips in 24-well cell culture plates and transfected with human ACE2 and indicated plasmids. 12 h later, cells were infected with 1105 TCID50/ml SARS-CoV-2 virus (hCoV-19/China/CAS-B001/2020, GISAID No. EPI_ISL_514256-7) for 1 hour. After 3 washes with PBS, cells were incubated in culture medium for 24 h at 37 °C with 5% CO2, fixed with 4% PFA for 24 h at room temperature, and then subjected to normal procedures for immunostaining analysis. All of the experiments with SARS-CoV-2 virus were performed inside biosafety cabinets in the biosafety level 3 facility at the Institute of Microbiology, Chinese Academy of Sciences. SARS-CoV-2 virus was propagated in Vero E6 cells and the virus titer was determined by cytopathic efficiency (CPE) assays.
MHV-A59 virus infection
MHV-A59 was propagated by infecting 17Cl-1 cells at a multiplicity of infection (MOI) of 0.05. To infect 17Cl-1 cells, the viral inoculum in DMEM was incubated with cells for 1 h with occasional swirling. The inoculum was then removed and replaced with fresh DMEM plus 10% fetal bovine serum. The titer of MHV-A59 was determined by plaque assay on monolayer 17Cl-1 cells. All titrations were performed in duplicate by incubating cells with serially diluted cell culture supernatants for 36 h. Plaques were counted and calculated as plaque forming units (PFU) per milliliter of supernatant. To determine the effect of SARS-CoV-2 ORF3a on MHV-A59 production, 17Cl-1 cells were transfected with pEGFP-C1 vector or pEGFP-ORF3a plasmid. At 24 h post-transfection, cells were infected with MHV-A59 at a MOI of 3. Supernatants were collected at 16 h post-infection and virus production was quantified by plaque assay.
For propidium iodide (PI) staining, 17Cl-1 cells were cultured on glass-bottom dishes (801001, NEST) and transfected with the indicated plasmids. At 24 h post-transfection, cells were infected with MHV-A59 at a MOI of 3. After 16 h infection, cells were treated with 10 μg/ml propidium iodide (ST511, Beyotime) and 1 μg/ml Hoechst 33342 (C1022, Beyotime).
Cytosolic free calcium ([Ca2+]cyt) imaging
Ratiometric single cell [Ca2+]cyt imaging was performed on an IX-81 microscope (Olympus)-based system as described previously (Zheng et al., 2013; Zhou et al., 2014). HeLa cells were incubated in DMEM containing 2 μM Fura-2 AM at 37 °C for 30 minutes. Transfected cells were identified by the presence of fused or co-expressed eGFP. Semrock filters (BrightLine single-band multi-exciter filter set, optimized for Fura-2) were used to minimize contamination of Fura-2 fluorescence by bleed-through of eGFP fluorescence. Data were acquired with Metafluor software (Universal Imaging) and analyzed with Origin2020b software (OriginLab) and are expressed as means ± S.E.M..
Measurement of starvation-induced lysosome Ca2+ release and data analysis
Time-lapse imaging was performed on an inverted confocal microscope Zeiss LSM880 microscope with a 63×, 1.3 NA oil-immersion objective. The axial resolution was set to 1.0 μm. Ex=488 nm, Em=490∼660 nm. Images were acquired at 512×512 pixels, 1 s/frame, 30 frames/min.
HeLa cells were transfected with the lysosome-targeted genetically encoded Ca2+ sensor LAMP1-GCaMP6f and plated in Mattek glass-bottomed dishes 24 h before imaging and measurement. After 24 h, cells were firstly placed in 1 ml of complete DMEM medium for 1 minute to obtain resting F 0. Then the medium was carefully changed from DMEM to HBSS, and imaging was continued for 10 minutes to obtain starvation-induced lysosome Ca2+ release (Medina et al., 2015).
The time-lapse LSM files generated from the Zeiss LSM 880 were analyzed using custom-developed programs written in interactive data language (IDL Research Systems). Cell motion artefacts and background fluorescence changes were corrected by image processing. For parametric quantification, we measured the amplitude (ΔF/F0, dimensionless) of each cell (Lu et al., 2019).
Cytosolic Ca2+ measurement (Fluo4-AM)
Cells were seeded on Mattek glass-bottomed dishes and cultured with DMEM medium. For Ca2+ measurement, the cells were loaded with 5 μM Fluo4-AM (Molecular Probes) for 20 min at 37 °C in Tyrode solution: 137 mM NaCl, 20 mM HEPES (Sigma), 10 mM glucose (Sigma), 1.2 mM MgCl2⋅6H2O (Sigma), 1.2 mM NaH2PO4⋅2H2O (Sigma), and 5.4 mM KCl (Sigma), pH 7.35. Cells were then rinsed twice with Tyrode solution and mounted on the inverted stage of a confocal scope (Zeiss LSM 880). Fluorescence excitation was performed using a 488 nm laser, and detection filters were set at 493–622 nm. Images were acquired at 1 s/frame, every 2 s. Cells were scanned for 20–30 s to obtain resting [Ca2+]cyt (Fresting). 50 μM BAPTA-AM was added to chelate cytosolic Ca2+ to measure the minimum fluorescence level (F min). Then, for measurement of the maximal level (F max), the solution was replaced with 10 mM Ca2+, 5 μM thapsigargin, 12 μM A23187 (Sigma), 3 μM FCCP (Sigma), and 20 mM 2-DG (Sigma) in Tyrode solution. The stable value was Fmax. Finally, [Ca2+]cyt was calibrated using the equation [Ca2+] = K d × (F–F min)/(F max–F). K d was assumed to be 1000 nM for Fluo4 in intact cells (Sun et al., 2019; Li et al., 2019). Images were analyzed using Interactive Data Language (IDL, Research Systems) software.
Immunostaining assays
For immunostaining, cells were seeded on coverslips and cultured in 24-well cell culture plates. After transfection with the indicated plasmids, cells were washed 3 times with PBS and fixed with 4% paraformaldehyde for 20 min. Then, cells were permeabilized with 100 μg/ml digitonin (D141, Sigma) for 10 min at room temperature with gentle shaking and blocked with 5% goat serum for 60 min. After incubation with the indicated primary antibodies (diluted in 5% goat serum) for 1.5 h at room temperature or overnight at 4 °C, cells were washed with PBS and stained with fluorescently-labeled secondary antibodies for 1 h at room temperature. After 3 washes with PBS, the coverslips were mounted with DAPI in 50% glycerol. Confocal images were acquired by a Zeiss LSM 880 Meta plus Zeiss Axiovert zoom. The following antibodies were used in this study: LC3 antibody (M152-3, MBL), p62 antibody (PM045, MBL), LAMP1 antibody (553792, BD Biosciences), Flag antibody (F1804, Sigma), Flag antibody (14793S, Cell Signaling Technology).
Immunofluorescence staining of PM-localized LAMP1
Cells were cultured on coverslips in 24-well cell culture plates. For live-cell staining, cells were incubated with PE-conjugated human LAMP1 antibody (328608, BioLegend) in culture medium at 4 °C for 90 min according to the manufacturer’s instructions. After gently washed 3 times by PBS, cells were immediately fixed with 4% paraformaldehyde for 20 min, then stained with fluorescently-labeled secondary antibodies for 2 h at room temperature to amplify the signal. For 17Cl-1 cells, cells were stained with a mouse LAMP1 antibody (560948, BD Biosciences) for 90 min in culture medium at 4 °C. Cells were fixed and then stained with fluorescently-labeled secondary antibodies for 2 h at room temperature.
For trypsin treatment, cells were transfected with indicated plasmids for 24 h. After 3 washes by PBS, cells were treated by trypsin (25200072, Gibco) for 10 min, plated on coverslips coated with 1% poly-lysine for 10 min, then subjected to the PE-anti-LAMP1 staining protocol for live cells as described above.
For LysoTracker Green staining, cells were incubated with LysoTracker Green (L7526, Life Technologies) at 37°C for 30 min according to the manufacturer’s instructions. The cells were then used for live cell staining as described above.
Immunoblotting assays
After the indicated operations, cells were washed 3 times by PBS and lysed with lysis buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100) supplemented with protease inhibitor cocktail for 15 min on ice. After centrifuging (13,000 rpm) for 15 min at 4 °C, cell lysates were mixed with SDS loading buffer and boiled for 10 min. For analysis of CTSB and CTSD in culture medium, equal numbers of HeLa cells were seeded into 24-well cell culture plates with an equal volume of culture medium and transfected by the indicated plasmids. After 24 h culturing at 37 °C, culture medium was mixed with SDS loading buffer and boiled for 10 min.
Samples were subjected to SDS-PAGE electrophoresis and transferred onto 0.45 μm PVDF membranes. Signals on PVDF membranes were detected using the indicated primary and secondary antibodies. Total proteins in culture medium were detected by Ponceau S (P0022, Beyotime) staining. Antibodies used in immunoblotting assays are listed in Key Resources Table.
Co-immunoprecipitation assays
After transfection with the indicated plasmids and washing with PBS buffer, HeLa cells were lysed in lysis buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100 and protease inhibitor cocktail) on ice for 20 min and centrifuged at 13,000 rpm for 15 min at 4 °C. The supernatant was immunoprecipitated by GFP-Trap agarose beads (GNA-20-400, V-nanoab) for 1 h at 4 °C with rotation. After 5 washes with lysis buffer, agarose beads were collected by centrifugation (3000 rpm, 1 min) and boiled with SDS loading buffer for 10 min. Samples were subjected to immunoblotting assays.
Subcellular Fractionation
Plasma membranes were isolated by using a cell fractionation kit (SM-005, Invent Biotechnologies) according to the manufacturer’s protocol. The purity of the different fractions was estimated by immunoblotting analysis of diagnostic proteins.
Quantitative real-time PCR (qRT-PCR)
Total RNA was isolated using Trizol reagent (15596018, Life Technologies). cDNA libraries were constructed with a RevertAid First Strand cDNA Synthesis Kit (K1622, Thermo Scientific) according to manufacturer’s instructions. RT-PCR was performed using specific primers and UltraSYBR Mixture (High ROX) (CW2602M, CWBIO) on a QuantStudio 7 Flex (Applied Biosystems) instrument. mRNA levels were normalized by GAPDH mRNA levels. The RT primers are listed in Table S1.
Generation of TRPML1 knockout cells by CRISPR/Cas9
The sgRNA targeting human TRPML1 (5’-TGCCAGCGGTACTACCACCG-3′) was designed using the CRISPR design tool (http://chopchop.cbu.uib.no/) and cloned into a vector (pX458) containing human codon-optimized Cas9, RNA components and EGFP (Addgene, 48138). The plasmid was transfected into HeLa cells using Lipofectamine 2000 (11668019, Invitrogen) for 24 h. Then GFP-positive cells were single-cell sorted into 96-well plates.
DNA was extracted from single cell clones and sequenced to analyze CRISPR targeting. Sequences were analyzed by CRISPR-ID software (Dehairs et al., 2016) and decoded manually. The following primers were used for sequencing:
Forward: 5’-GATGACACCTTCGCAGCCTA-3’
Reverse: 5’-AGACAAGGTCTTCCTACGTGG-3’.
Quantification and statistical analysis
All of the experiments were repeated at least 3 times on separate days with separate, independent populations. Graph plots and P-values were generated using GraphPad Prism 5 software. Density of immunoblot bands was quantified using Image J software (NIH Image). Image J was used to quantify the immunofluorescence intensity of the images. Statistical comparisons were made using the unpaired Student’s t test and are shown as mean ± S.E.M.. ∗, P<0.05; ∗∗, P<0.01; ∗∗∗, P<0.001 and n.s. represents "no significant difference". Sample size (with n representing the number of cells) and statistical significance (P value) are reported in the figure legends.
Acknowledgments
We are grateful to Dr. Isabel Hanson for editing work. We thank Dr. Yuhai Bi (Institute of Microbiology, CAS) for the SARS-CoV-2 virus and the staff of the Biosafety Level 3 Laboratory (Institute of Microbiology, CAS) for their help with live virus experiments. This work was supported by the following grants to H.Z.: National Natural Science Foundation of China (92054301 and 31790403), Chinese Ministry of Science and Technology (2017YFA0503401), and Key Research Program of Frontier Sciences, CAS (grant QYZDY-SSW-SMC006).
Author contributions
H.Z. and H.D. designed the experiments. D.C., Q.Z., and M.J. performed all mammalian tissue culture experiments. L.S. conducted the MHV-A59 experiments. Y.L. did the SARS-CoV-2 virus infection experiments. H.Z. wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: October 10, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.devcel.2021.10.006.
Supplemental information
Data and code availability
-
•
All data reported in this paper are available from the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Braulke T., Bonifacino J.S. Sorting of lysosomal proteins. Biochim. Biophys. Acta. 2009;1793:605–614. doi: 10.1016/j.bbamcr.2008.10.016. [DOI] [PubMed] [Google Scholar]
- Castaño-Rodriguez C., Honrubia J.M., Gutiérrez-Álvarez J., DeDiego M.L., Nieto-Torres J.L., Jimenez-Guardeño J.M., Regla-Nava J.A., Fernandez-Delgado R., Verdia-Báguena C., Queralt-Martín M., et al. Role of severe acute respiratory syndrome coronavirus Viroporins E, 3a, and 8a in Replication and pathogenesis. mBio. 2018;9 doi: 10.1128/mBio.02325-17. e02325–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehairs J., Talebi A., Cherifi Y., Swinnen J.V. CRISP-ID: decoding CRISPR mediated indels by Sanger sequencing. Sci. Rep. 2016;6:28973. doi: 10.1038/srep28973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deretic V., Saitoh T., Akira S. Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 2013;13:722–737. doi: 10.1038/nri3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freundt E.C., Yu L., Goldsmith C.S., Welsh S., Cheng A., Yount B., Liu W., Frieman M.B., Buchholz U.J., Screaton G.R., et al. The open reading frame 3a protein of severe acute respiratory syndrome- associated coronavirus promotes membrane rearrangement and cell death. J. Virol. 2010;84:1097–1109. doi: 10.1128/JVI.01662-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung T.S., Liu D.X. Human coronavirus: host-pathogen interaction. Annu. Rev. Microbiol. 2019;73:529–557. doi: 10.1146/annurev-micro-020518-115759. [DOI] [PubMed] [Google Scholar]
- Ghosh S., Dellibovi-Ragheb T.A., Kerviel A., Pak E., Qiu Q., Fisher M., Takvorian P.M., Bleck C., Hsu V.W., Fehr A.R., et al. β-coronaviruses use lysosomes for Egress instead of the biosynthetic secretory pathway. Cell. 2020;183:1520–1535.e14. doi: 10.1016/j.cell.2020.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartenian E., Nandakumar D., Lari A., Ly M., Tucker J.M., Glaunsinger B.A. The molecular virology of coronaviruses. J. Biol. Chem. 2020;295:12910–12934. doi: 10.1074/jbc.REV120.013930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M., Kleine-Weber H., Pöhlmann S. A multibasic cleavage site in the spike protein of SARS-CoV-2 is essential for infection of human lung cells. Mol. Cell. 2020;78:779–784. doi: 10.1016/j.molcel.2020.04.022. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.H., Nitsche A., et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–280.e8. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanzawa N., Nishigaki K., Hayashi T., Ishii Y., Furukawa S., Niiro A., Yasui F., Kohara M., Morita K., Matsushima K., et al. Augmentation of chemokine production by severe acute respiratory syndrome coronavirus 3a/X1 and 7a/X4 proteins through NF-kappaB activation. FEBS Lett. 2006;580:6807–6812. doi: 10.1016/j.febslet.2006.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern D.M., Sorum B., Mali S.S., Hoel C.M., Sridharan S., Remis J.P., Toso D.B., Kotecha A., Bautista D.M., Brohawn S.G. Cryo-EM structure of SARS-CoV-2 ORF3a in lipid nanodiscs. Nat. Struct. Mol. Biol. 2021;28:573–582. doi: 10.1038/s41594-021-00619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.J., Soyombo A.A., Tjon-Kon-Sang S., So I., Muallem S. The Ca(2+) channel TRPML3 regulates membrane trafficking and autophagy. Traffic. 2009;10:1157–1167. doi: 10.1111/j.1600-0854.2009.00924.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam T.T., Jia N., Zhang Y.W., Shum M.H., Jiang J.F., Zhu H.C., Tong Y.G., Shi Y.X., Ni X.B., Liao Y.-S., et al. Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature. 2020;583:282–285. doi: 10.1038/s41586-020-2169-0. [DOI] [PubMed] [Google Scholar]
- Lamb C.A., Yoshimori T., Tooze S.A. The autophagosome: origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 2013;14:759–774. doi: 10.1038/nrm3696. [DOI] [PubMed] [Google Scholar]
- Laufman O., Hong W., Lev S. The COG complex interacts directly with syntaxin 6 and positively regulates endosome-to-TGN retrograde transport. J. Cell Biol. 2011;194:459–472. doi: 10.1083/jcb.201102045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B., Mizushima N., Virgin H.W. Autophagy in immunity and inflammation. Nature. 2011;469:323–335. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.W., Liu C.Z., Li Y.H., Zheng Q.X., Xu Y.J., Liu B.B., Sun W.J., Li Y., Ji S.H., Liu M.W., et al. TMCO1-mediated Ca2+ leak underlies osteoblast functions via CaMKII signaling. Nat. Commun. 2019;10:1589. doi: 10.1038/s41467-019-09653-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu F., Sun J., Zheng Q., Li J., Hu Y., Yu P., He H., Zhao Y., Wang X., Yang S., et al. Imaging elemental events of store-operated Ca2+ entry in invading cancer cells with plasmalemmal targeted sensors. J. Cell Sci. 2019;132:jcs224923. doi: 10.1242/jcs.224923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W., Zheng B.J., Xu K., Schwarz W., Du L.Y., Wong C.K.L., Chen J.D., Duan S.M., Deubel V., Sun B. Severe acute respiratory syndrome-associated coronavirus 3a protein forms an ion channel and modulates virus release. Proc. Natl. Acad. Sci. USA. 2006;103:12540–12545. doi: 10.1073/pnas.0605402103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzio J.P., Pryor P.R., Bright N.A. Lysosomes: fusion and function. Nat. Rev. Mol. Cell Biol. 2007;8:622–632. doi: 10.1038/nrm2217. [DOI] [PubMed] [Google Scholar]
- Medina D.L., Di Paola S., Peluso I., Armani A., De Stefani D., Venditti R., Montefusco S., Scotto-Rosato A., Prezioso C., Forrester A., et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015;17:288–299. doi: 10.1038/ncb3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina D.L., Fraldi A., Bouche V., Annunziata F., Mansueto G., Spampanato C., Puri C., Pignata A., Martina J.A., Sardiello M., et al. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev. Cell. 2011;21:421–430. doi: 10.1016/j.devcel.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao G.Y., Zhao H.Y., Li Y., Ji M.M., Chen Y., Shi Y., Bi Y.H., Wang P.H., Zhang H. ORF3a of the COVID-19 virus SARS-CoV-2 blocks HOPS complex-mediated assembly of the SNARE complex required for autolysosome formation. Dev. Cell. 2021;56:427–442.e5. doi: 10.1016/j.devcel.2020.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao Y.X., Li G.J., Zhang X.L., Xu H.X., Abraham S.N. A TRP channel senses lysosome neutralization by pathogens to trigger their expulsion. Cell. 2015;161:1306–1319. doi: 10.1016/j.cell.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller S.G., Carnell L., Moore H.H. Post-Golgi membrane traffic: brefeldin A inhibits export from distal Golgi compartments to the cell surface but not recycling. J. Cell Biol. 1992;118:267–283. doi: 10.1083/jcb.118.2.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minakshi R., Padhan K. The YXXΦ set motif within the severe acute respiratory syndrome coronavirus (SARS-CoV) 3a protein is crucial for its intracellular transport. Virol. J. 2014;11:75. doi: 10.1186/1743-422X-11-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N., Yoshimori T., Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011;27:107–132. doi: 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- Peng R., Wu L.A., Wang Q., Qi J., Gao G.F. Cell entry by SARS-CoV-2. Trends Biochem. Sci. 2021;46:848–860. doi: 10.1016/j.tibs.2021.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu J., Guardia C.M., Keren-Kaplan T., Bonifacino J.S. Mechanisms and functions of lysosome positioning. J. Cell Sci. 2016;129:4329–4339. doi: 10.1242/jcs.196287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez A., Webster P., Ortego J., Andrews N.W. Lysosomes behave as Ca2+-regulated exocytic vesicles in fibroblasts and epithelial cells. J. Cell Biol. 1997;137:93–104. doi: 10.1083/jcb.137.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saftig P., Klumperman J. Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat. Rev. Mol. Cell Biol. 2009;10:623–635. doi: 10.1038/nrm2745. [DOI] [PubMed] [Google Scholar]
- Samie M., Wang X., Zhang X., Goschka A., Li X., Cheng X., Gregg E., Azar M., Zhuo Y., Garrity A.G., et al. A TRP channel in the lysosome regulates large particle phagocytosis via focal exocytosis. Dev. Cell. 2013;26:511–524. doi: 10.1016/j.devcel.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang J., Wan Y.S., Luo C.M., Ye G., Geng Q.B., Auerbach A., Li F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA. 2020;117:11727–11734. doi: 10.1073/pnas.2003138117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvas J.A., Vasquez D.M., Park J.G., Chiem K., Allué-Guardia A., Garcia-Vilanova A., Platt R.N., Miorin L., Kehrer T., Cupic A., et al. Contribution of SARS-CoV-2 accessory proteins to viral pathogenicity in K18 human ACE2 transgenic mice. J. Virol. 2021;95:e0040221. doi: 10.1128/JVI.00402-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu K.L., Yuen K.S., Castaño-Rodriguez C., Ye Z.W., Yeung M.L., Fung S.Y., Yuan S.F., Chan C.P., Yuen K.-Y., Enjuanes L., Jin D.-Y. Severe acute respiratory syndrome coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J. 2019;33:8865–8877. doi: 10.1096/fj.201802418R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder E.J., Limpens R.W.A.L., de Wilde A.H., de Jong A.W.M., Zevenhoven-Dobbe J.C., Maier H.J., Faas F.F.G.A., Koster A.J., Bárcena M. A unifying structural and functional model of the coronavirus replication organelle: tracking down RNA synthesis. PLoS Biol. 2020;18:e3000715. doi: 10.1371/journal.pbio.3000715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sørensen J.B., Nagy G., Varoqueaux F., Nehring R.B., Brose N., Wilson M.C., Neher E. Differential control of the releasable vesicle pools by SNAP-25 splice variants and SNAP-23. Cell. 2003;114:75–86. doi: 10.1016/s0092-8674(03)00477-x. [DOI] [PubMed] [Google Scholar]
- Spampanato C., Feeney E., Li L., Cardone M., Lim J.A., Annunziata F., Zare H., Polishchuk R., Puertollano R., Parenti G., et al. Transcription factor EB (TFEB) is a new therapeutic target for Pompe disease. EMBO Mol. Med. 2013;5:691–706. doi: 10.1002/emmm.201202176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolz A., Ernst A., Dikic I. Cargo recognition and trafficking in selective autophagy. Nat. Cell Biol. 2014;16:495–501. doi: 10.1038/ncb2979. [DOI] [PubMed] [Google Scholar]
- Sun C.W., Shui B., Zhao W., Liu H., Li W.W., Lee J.C., Doran R., Lee F.K., Sun T., Shen Q.S., et al. Central role of IP3R2-mediated Ca2+ oscillation in self-renewal of liver cancer stem cells elucidated by high-signal ER sensor. Cell Death Dis. 2019;10:396. doi: 10.1038/s41419-019-1613-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Y.J., Lim S.G., Hong W.J. Understanding the accessory viral proteins unique to the severe acute respiratory syndrome (SARS) coronavirus. Antiviral Res. 2006;72:78–88. doi: 10.1016/j.antiviral.2006.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tancini B., Buratta S., Delo F., Sagini K., Chiaradia E., Pellegrino R.M., Emiliani C., Urbanelli L. Lysosomal exocytosis: the extracellular role of an intracellular organelle. Membranes. 2020;10:406. doi: 10.3390/membranes10120406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong H.H., Sanyal S. Manipulation of autophagy by (+) RNA viruses. Semin. Cell Dev. Biol. 2020;101:3–11. doi: 10.1016/j.semcdb.2019.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrapp D., Wang N., Corbett K.S., Goldsmith J.A., Hsieh C.L., Abiona O., Graham B.S., McLellan J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367:1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F., Zhao S., Yu B., Chen Y.-M., Wang W., Song Z.-G., Hu Y., Tao Z.W., Tian J.-H., Pei Y.-Y., et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.G., Codogno P., Zhang H. Machinery, regulation and pathophysiological implications of autophagosome maturation. Nat. Rev. Mol. Cell Biol. 2021 doi: 10.1038/s41580-021-00392-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.G., Zhang H. Autophagosome maturation: an epic journey from the ER to lysosomes. J. Cell Biol. 2019;218:757–770. doi: 10.1083/jcb.201810099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H.Y., Zhou M.H., Hu C.L., Kuo E., Peng X., Hu J.J., Kuo L., Zhang S.L. Differential roles of the C and N Termini of Orai1 protein in interacting with stromal interaction Molecule 1 (STIM1) for Ca2+ release-activated Ca2+ (CRAC) channel activation. J. Biol. Chem. 2013;288:11263–11272. doi: 10.1074/jbc.M113.450254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M.H., Zheng H.Y., Si H.J., Jin Y.X., Peng J.M., He L., Zhou Y.B., Muñoz-Garay C., Zawieja D.C., Kuo L., et al. Stromal interaction Molecule 1 (STIM1) and Orai1 mediate histamine-evoked calcium entry and nuclear factor of activated T-cells (NFAT) signaling in human umbilical vein endothelial cells. J. Biol. Chem. 2014;289:29446–29456. doi: 10.1074/jbc.M114.578492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P., Yang X.-L., Wang X.-G., Hu B., Zhang L., Zhang W., Si H.-R., Zhu Y., Li B., Huang C.-L., et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper are available from the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.