

Abstract
Brain arteriolosclerosis (B-ASC), characterized by pathologic arteriolar wall thickening, is a common finding at autopsy in aged persons and is associated with cognitive impairment. Hypertension and diabetes are widely recognized as risk factors for B-ASC. Recent research indicates other and more complex risk factors and pathogenetic mechanisms. Here we describe aspects of the unique architecture of brain arterioles, histomorphologic features of B-ASC, relevant neuroimaging findings, epidemiology and association with aging, established genetic risk factors, and the co-occurrence of B-ASC with other neuropathologic conditions such as Alzheimer’s disease and limbic-predominant age-related TDP-43 encephalopathy (LATE). There may also be complex physiologic interactions between metabolic syndrome (e.g. hypertension and inflammation) and brain arteriolar pathology. Although there is no universally applied diagnostic methodology, several classification schemes and neuroimaging techniques are used to diagnose and categorize cerebral small vessel disease pathologies that include B-ASC, microinfarcts, microbleeds, lacunar infarcts, and cerebral amyloid angiopathy (CAA). In clinical-pathologic studies that include consideration of comorbid diseases, B-ASC is independently associated with impairments in global cognition, episodic memory, working memory, and perceptual speed, and has been linked to autonomic dysfunction and motor symptoms including parkinsonism. We conclude by discussing critical knowledge gaps related to B-ASC and suggest that there are probably subcategories of B-ASC that differ in pathogenesis. Observed in over 80% of autopsied individuals beyond 80 years of age, B-ASC is a complex and under-studied contributor to neurologic disability.
Keywords: SVD, arteriosclerosis, cAVU, senescence, neuropathology, neuroimaging
Introduction
Brain arteriolosclerosis (B-ASC), a common subtype of cerebral small vessel disease (cSVD), is associated with substantial neurological morbidity [19, 30, 34, 84, 183, 194]. In the present review we discuss approaches to diagnosis and classification of cSVD pathologic changes including B-ASC. We review the literature on risk factors and biological mechanisms thought to underlie B-ASC, emphasizing the complex anatomy and physiology of arteriolar walls, and discuss relevant clinicopathological studies. We conclude by highlighting critical areas for further investigation.
Background and historical context
Combining the words “arteriole” and the Greek “sklērós”, arteriolosclerosis implies “hardening of the arterioles”. While histopathology provides the gold-standard criteria for determining the presence and severity of B-ASC, prior studies and reference sources have employed differing definitions of arteriolosclerosis. Some are presented in Table 1. A schematic representation of the histologic features of brain arterioles is presented in Fig. 1.
Table 1.
Definitions of Arteriolosclerosis
| Source | Arteriolosclerosis definition |
|---|---|
| Skrobot et. al., 2016 | Hyaline thickening of walls of vessels <150 μm in diameter, not associated with lipid-containing cells replacing the tunica media. Diagnosis requires an absence of intramural inflammation, amyloid or fibrinoid necrosis. |
| Buchman et. al., 2013 | Arteriolosclerosis describes the histologic changes commonly found in the deep penetrating small vessels of the brain in aging, including intimal deterioration, smooth muscle degeneration, and fibrohyalinotic thickening of arterioles with consequent narrowing of the vascular lumen. |
| Grinberg and Thal, 2010 | Concentric hyaline thickening of small arteries (40 – 150 μm in diameter) leading to a concentric stenosis of the vessel lumen. |
| Taber’s Medical Dictionary, June 2020 | A disease of the arterial vessels marked by thickening and loss of elasticity in the arterial walls. It is known colloquially as “hardening of the arteries.” |
| Merriam-Webster Medical Dictionary, June 2020 | Thickening of the intima of arterioles (as of the kidney in hypertension) by hyaline and fatty deposits that reduce the lumen and obstruct blood flow |
Fig. 1. A brain arteriole and constituents of the cerebral arteriolar vascular unit (cAVU).

Brain arterioles are not all alike, but this schematic depiction may orient readers to some cAVU components. Shown here is a relatively thin-walled arteriole--no internal elastic lamina or adventitial nerve fibers are depicted. Inflammatory cells including macrophages and microglia are commonly found in and around the arteriolar wall. Acellular strands of elastin and collagen are interwoven with fibroblast-like cells in the adventitia.
Historically, much of the early descriptions of arteriolosclerosis focused on renal pathology. Following Lobstein’s use of the term “arteriosclerosis” [117], Gull and Sutton applied the term “arterio-capillary fibrosis” [70]. Moritz and Oldt later described how aging and hypertension are associated with renal “arteriolar sclerosis” [145], which evolved to “arteriolosclerosis”[51]. These terms were useful because the histopathologic changes observed in arteriolar vessel walls differ from findings typical of those in arteries. In arteries, lipid laden macrophages may infiltrate the vessel wall, forming a cholesterol-rich (and often calcified) matrix which is termed an atheroma, the pathologic substrate of atherosclerosis [94]. When small atheromas occur in arterioles or narrow arteries (so-called microatheromas [14]), they are distinct from B-ASC. By contrast, B-ASC shows thickened and/or dysmorphic vessel wall changes, with increased arteriolar tortuosity, but no atheroma. B-ASC also lacks amyloid. At least two subtypes of arteriolosclerosis have been recognized: hyperplastic (i.e., vessel wall “onion-skinning”) and hyaline (i.e., glassy-looking acellular mural thickening). However, many arterioles have both histomorphologic patterns [52].
Although early studies of arteriolosclerosis were oriented toward the kidney, vascular pathology in the brain has recently generated intense interest among clinical and translational neuroscientists. The physiology and cellular elements of brain arterioles are unique [195, 211], and as yet incompletely understood. While arteriolosclerosis is commonly present in multiple organs [163], the pathobiology in brain arterioles may have features that are distinct from non-CNS arteriolosclerosis.
Neuropathologic workup: diagnostic and research approaches
B-ASC is typically identified in neuropathologic practice using routine hematoxylin and eosin (H&E) stains (Fig. 2). For diagnostic purposes, a semiquantitative severity scale is commonly used with categories typically corresponding to “none”, “mild”, “moderate”, and “severe” levels of pathologic change (Fig. 3). This type of B-ASC metric was used in Vascular Cognitive Impairment Neuropathology Guidelines (VCING) [183], in the National Alzheimer’s Coordinating Center Neuropathology (NACC NP) Data Set [26, 141], and elsewhere [100, 114, 154, 155].
Fig. 2. Autopsy diagnosis of B-ASC involves staining with hematoxylin and eosin (H&E).

The VCING collaborative group provided a consensus among experts describing criteria for defining B-ASC and for generating parameters that reflect different severity of the pathology. These diagnostic criteria are presented on the right side of the figure. In this study [183], which included evaluation of n=113 brains and sampling from many different brain areas, stained slides of the occipital cortex white matter produced moderately strong inter-rater reliability among different neuropathologists, and a robust association with cognitive impairment, i.e., B-ASC pathology was more often present in cases with dementia than those with normal cognitive status.
Fig. 3. One approach to grading arteriolosclerosis (B-ASC) pathologic severity is on a semiquantitative scale of “none”, “mild”, “moderate”, or “severe” B-ASC.
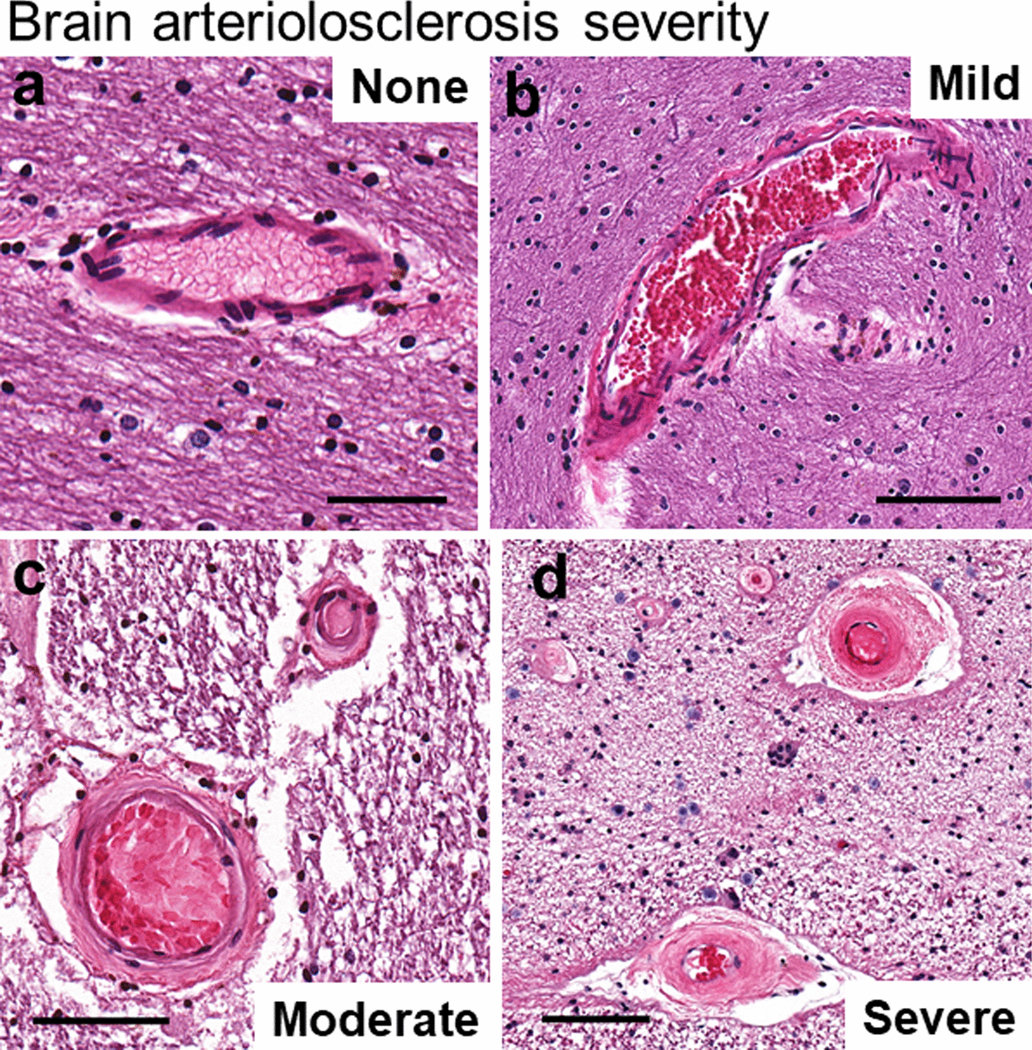
A score of 0 corresponds to no/very minimal changes (a); a score of 1 indicates mild changes (b); a score of 2 is moderately severe B-ASC (c); and a score of 3 indicates severe B-ASC (d). Note that in (d), the arterioles are partly occluded by severe hyperplastic (“onion-skin”-type) arteriolosclerotic changes. Scale bars = (a): 80 μm; (b): 200 μm; (c): 100 μm; and, (d): 100 μm. All of these 4 panels show vascular profiles from occipital cortex white matter of human subjects.
The VCING system described and evaluated a protocol for diagnosing B-ASC neuropathologically (Fig. 2). The NACC data set includes comparable information related to cerebrovascular pathologies (Table 2). However, NACC NP cerebrovascular data have not been assessed for inter-rater reliability among neuropathologists. In the VCING study [183], sampling protocols were also recommended. The inter-rater reliability assessment of the B-ASC severity diagnoses (0–3 scale, Fig. 3) showed “moderate” agreement, whereas most other cSVD subtypes had “high” inter-rater reliability when diagnosed by 14 different neuropathologist raters. This is partly because the criteria for delineating each increment of B-ASC severity (e.g., mild vs. moderate) are highly subjective and investigators may weigh anatomic regions of interest differently. Based on the VCING study, B-ASC evaluation in the occipital cortex white matter provides a diagnostic marker with a combination of acceptable reliability and robust association with cognitive status (Fig. 2).
Table 2.
Small vessel pathologies as noted in Vascular Cognitive Impairment Neuropathology Guidelines (VCING) system and National Alzheimer’s Disease Coordinating Center (NACC) Version 10 Neuropathology data set
| Cerebrovascular Pathology | VCING scoring system[183] | NACC v10 scoring system[26, 141] |
|---|---|---|
| Arteriolosclerosis | 0–3 scale * | 0–3 scale |
| Microinfarcts | 0/1 | Numbers and locations |
| Lacunar infarcts | 0–3 scale | Numbers and locations |
| Large infarcts | 0/1 | Number, size, & location of old large infarcts, also acute and subacute large infarcts |
| Microhemorrhage | 0/1 | Numbers and locations |
| Larger hemorrhage | 0/1 | Gross, subdural or epidural, and epidural hemorrhages noted separately |
| Cerebral amyloid angiopathy (CAA) | Meningeal, cortical, or capillary scored separately | 0–3 overall scale |
| Myelin loss | 0–3 scale | “White matter rarefaction”, 0–3 scale |
| Other | Fibrinoid necrosis (0/1), microaneurysms (0/1), Perivascular space dilation (0–3 scale), Perivascular hemosiderin leakage (0–3 scale) | Any type of aneurysm (0/1), Laminar necrosis (0/1), vascular malformation (0/1), blood vessel mineralization (0/1) |
| Atheroma/atherosclerosis in the Circle of Willis | 0–3 scale | 0–3 scale |
The aged brain may contain a broad spectrum of arteriolar pathologic changes [38, 81, 111, 159, 197, 210] (Fig. 4). Although the VCING system represents a validated neuropathologic method for diagnosing cSVD subtypes (Table 2), there were no recommendations for subclassifying B-ASC. The VCING article also acknowledged that there was some disagreement among the coauthors. A key stipulation of their B-ASC definition was that “[d]iagnosis requires absence of intramural inflammation, amyloid, or fibrinoid necrosis” [183]. Yet it has been noted that “there is clearly morphological overlap with [B-ASC and] the late stage of Fisher’s lipohyalinosis.” [94] Skrobot et al.’s specification that only blood vessels <150 μm in diameter can be affected by B-ASC is debatable, since larger vessels may be affected by the same pathogenetic mechanisms. Some researchers have examined B-ASC in larger vessels (≥300 μm diameter) [43]. As to the lower size limits, cerebral vessels <20μm diameter may have a smooth muscle layer in their walls (Fig. 5c), and thus probably are arterioles. Tissue processing and staining may produce differing effects, and it is challenging to distinguish arterioles from venules in some circumstances. To date, no definition of arterioles based on lumen size is universally accepted.
Fig. 4. Arteriolar walls can show different histomorphologies with aging.
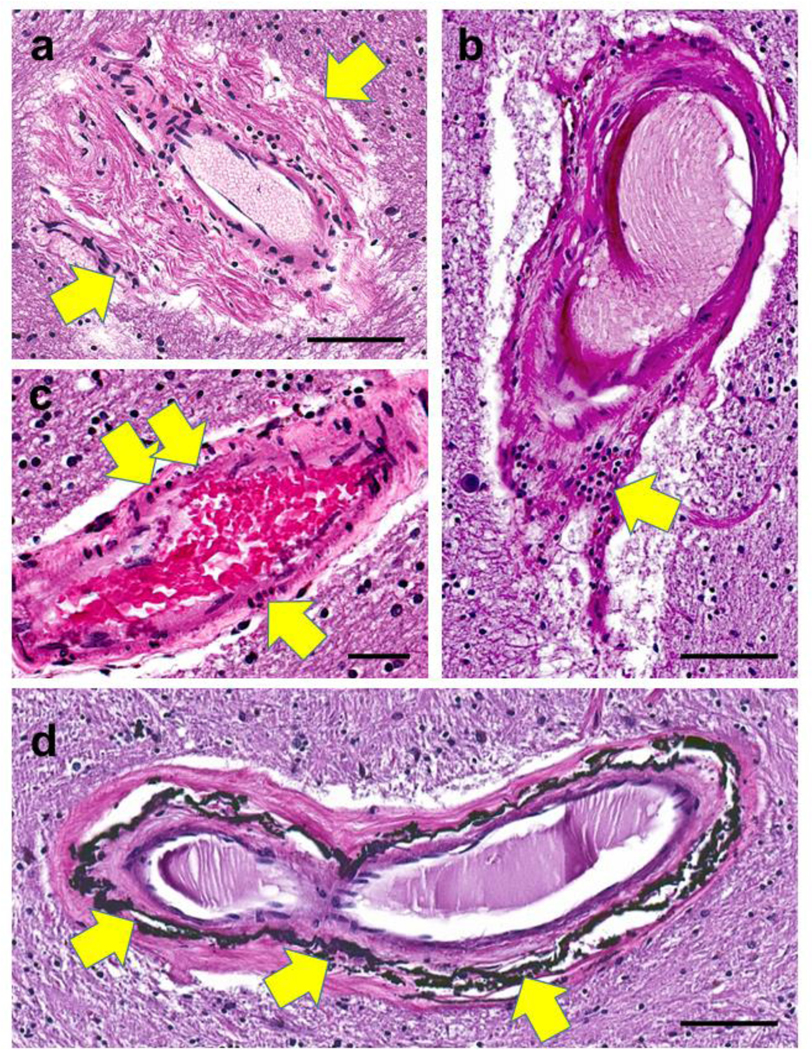
In panel (a), periarteriolar (adventitial) fibrosis is extensive and non-concentric (arrows). Panel (b) shows a collection of lymphocytes (arrow) in portions of the vessel wall. In panel (c), vessel wall changes include pyknotic-appearing smooth muscle cells (arrows). Siderocalcinosis, distinct from B-ASC, has been associated with dementia [194] and is usually seen preferentially in the globus pallidus (arrows in d). Synonyms for this include medial vascular calcification, calcific medial arteriosclerosis, and Monckeberg’s medial sclerosis. Scale bars = (a, b, and d): 100 μm; (c): 60 μm.
Fig. 5. Photomicrographs to highlight features of the cAVU with immunohistochemistry.
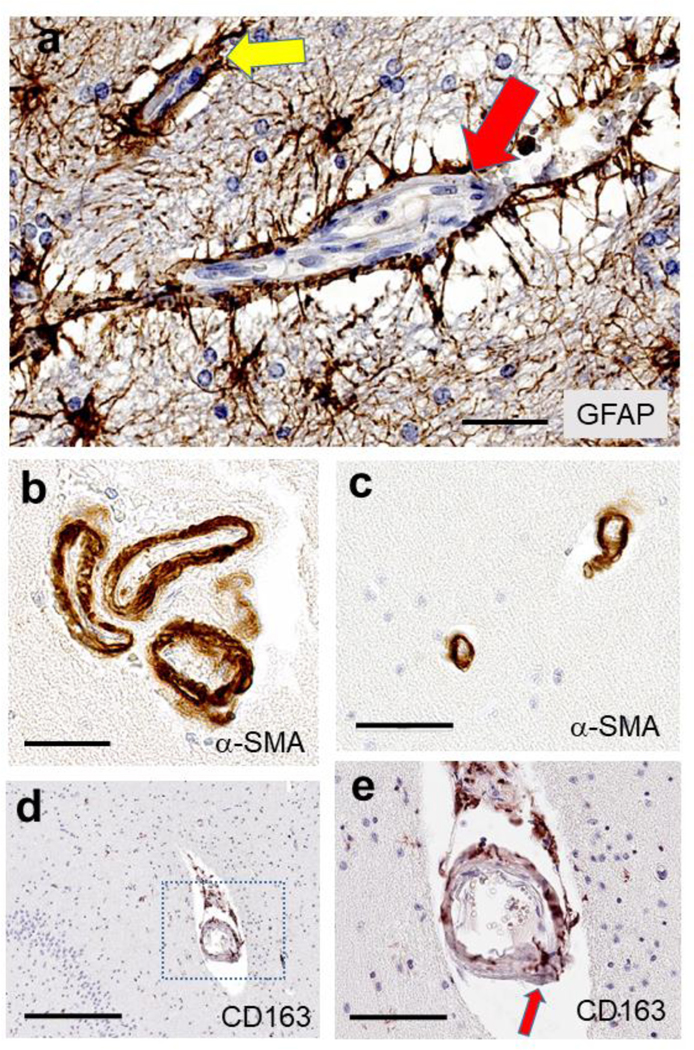
Astrocytes and astrocytic end feet processes (a) are shown immunostained for GFAP, surrounding a capillary (yellow arrow) and a larger arteriole (red arrow). Smooth muscle cells, immunolabeled for α-SMA (panels b, c) in arterioles cut in cross-section, may highlight that multiple arterioles can exist in a single Virchow-Robin space (b), and the vascular profiles may be rather narrow, <20 μm lumen diameter (c). Some cellular constituents have only recently been appreciated, such as the “scavenger microglia” that express CD163 antigen and are arranged in and around small blood vessels, such as this hippocampal arteriole (d, e). In panel (e), a magnified view of the panel (d) inset, a CD163 immunoreactive cell spans the full thickness of the arteriolar wall (red arrow). Scale bars = (a): 60 μm; (b): 50 μm; (c): 60 μm; (d): 300 μm; and, (e): 90 μm.
To grade the severity of B-ASC quantitatively for research purposes (as opposed to the semi-quantitative method described above), several groups have used a “sclerotic index” (SI), which is based on measurements of arteriolar wall thickness and luminal narrowing (SI = 1-[internal diameter/external diameter]) [76, 81, 111]. A similar approach calculated the ratio of the overall vessel radius to the thickness of the vessel medial layer [158, 197]. A third method assessed B-ASC based on the number of different brain areas affected (detected with H&E stains), rather than severity of the pathologic changes in any given region [199]. Importantly, there can be a wide spectrum of B-ASC pathologies in different areas of the same brain, not all necessarily highlighted best by using H&E stains.
As to other stains used in pathologic assessments of B-ASC features such as vessel wall disruption and collagenization, Verhoeff–Van Gieson elastic stain and Masson’s trichrome histochemical stains have been utilized [43, 197]. Immunohistochemical methods have also been used to evaluate B-ASC. Smooth muscle cells should be present in arterioles but not capillaries, whereas pericytes and endothelial cells are present in both arteriole and capillary vessel walls. Hence, α-smooth muscle actin (α-SMA) immunohistochemistry (Fig. 5b,c) was used to mark arterioles for a quantitative assessment of B-ASC in human frontal cortex [154]. The presence of B-ASC has also been inferred from parameters of blood-brain dysfunction, including using antibodies against fibrinogen and IgG, to detect extravasated plasma as evidence of blood-brain barrier leakage [32, 129, 142], which may be an indicator of B-ASC severity in specific circumstances.
Much remains unknown about B-ASC. Experts have previously remarked on the need for a more precise brain-specific terminology [51, 65, 120, 128, 152]. While previous definitions of B-ASC have enabled substantial scientific progress, future diagnostic methods focusing on specific mechanisms may better reflect the complexity of pathologic changes in aged brain arterioles.
Arteriole physiology and the cerebral arteriolar vascular unit
B-ASC affects the cerebral vasculature, which is a unique and complex network of blood vessels. Blood flowing into this network traverses the great cerebral vessels and then flows through pial arteries (usually 200–1,000 μm lumen diameter) located on the cerebral surface [27]. Branches from the pial arteries form penetrating arteries that pass into the brain parenchyma. These descending arteries feed parenchymal arterioles (usually ~20–200 μm lumen diameter) that further subdivide into smaller precapillary arterioles which connect with capillaries (~3–10 μm lumen diameter) [112, 127].
Arterioles serve important physiologic functions in the CNS. The brain is an energy demanding organ that couples synaptic function with fine-tuned vasoregulation. Arterioles are the primary site of vascular resistance, where the greatest changes in blood pressure and flow velocity occur [189]. Smooth muscle and pericyte constriction increases vascular resistance in arterioles, modified by neural input, pH, CO2, and oxygen, as well as paracrine, endocrine, and pharmacologic factors [105, 181].
Encircling the lumens of arterioles are a specialized combination of cells and structural elements (Fig. 1). There is no specific extant term for features of arteriolar walls. We will use a new term, cerebral arteriolar vascular unit (cAVU), which differs from the neurovascular unit (NVU) [46, 83, 95]; the latter term generally but not always [105] refers to capillaries. Some structures (e.g. endothelium) are present in the cAVU as well as in the NVU, and some functions overlap, such as preventing most large molecules from entering brain parenchyma. The terms for histologic layers of the arteriolar wall borrow from the terminology of arteries: intima, media, and adventitia (from the lumen to the periphery). Despite the similarities, the overall structure and functions of brain arterioles are unique. There also is diverse histology even among brain arterioles [112] that “blurs the traditional boundaries of a rigid layered classification.”[127]
Cellular components of the cAVU may include endothelial cells, pericytes, smooth muscle cells, inflammatory cells, fibroblast-like cells, and astrocyte end-feet processes (Fig. 1). The endothelium comprises one cell layer, allowing for exchange of chemicals through the cell membranes. Although commonly associated more with capillaries than arterioles [20], pericytes are found within the basement membrane in narrow arterioles and are vital for modulating cerebral blood flow [25, 220]. Pericytes also function in formation of blood vessels, maintenance of the blood-brain barrier (see below), and regulation of immune cell entry into the brain [139].
Of primary importance for cerebral vasoregulation are the arteriolar smooth muscle cells. These cells contract or relax to modify blood pressure and flow. Arteriolar vessel walls are generally described as having only 1–2 layers of smooth muscle cells [110, 127]. The precapillary sphincter is a microdomain in human cerebral cortex connecting small arterioles and capillaries, dynamically regulating blood flow via smooth muscle cell contraction [69]. Intriguingly, there is significant evolutionary molecular divergence (especially between rodents and humans) in vascular smooth muscle cells [113], underscoring the structural and functional uniqueness of human arterioles.
Astrocytes serve multiple functions and have so-called end feet processes that cover the abluminal (peripheral) aspect of CNS vessels (Fig. 5a). They coordinate the delivery of oxygen and glucose and have inflammation/activation signaling pathways that are in some ways similar to those of leukocytes and microglia [157]. The astrocytic end-feet are enriched with potassium and water channels, which are essential for maintaining appropriate neuronal excitability [23, 24]. They play bidirectional roles in coupling smooth muscle contraction/relaxation to neuronal activity [106], while modulating neuronal activity to adapt to changes in cerebral blood flow and oxygenation [103].
In addition to the long-established components of the cAVU, other cell types have recently been studied. For example, the perivascular microglial “scavenger-type” cells (Figs. 5d,e) are among vascular-tropic inflammatory cells. These cells may be related to neurodegenerative disease pathogenesis and were recently shown to have rich phenotypic variety [107, 208]. In some pathological circumstances, pericytes may adapt a microglia-like phenotype [162]. The arteriolar vessel wall adventitia consists of thin layers of fibroblasts, along with collagen and elastic fibers in healthy brains. By contrast, fibroblasts are not found in capillary walls [211].
The cAVU contains extracellular structures which help demarcate the concentric layers of the vessel wall. Endothelial cells rest on a basal membrane in the intima. An internal elastic lamina may be present in the adluminal (toward the lumen) portion of the media. The internal elastic lamina “is not always found at all levels of the arteriolar tree, but it is present in feed arterioles of … cerebrum.” [127] Peripheral to the astrocytic end-feet, the perivascular space is an incompletely understood functional domain [1, 133, 215].
Precapillary arterioles lead into a highly branched capillary network. The cells and structural elements of the cAVU (arterioles) and capillary NVU are cooperative components of the blood-brain barrier [146, 165]. The blood-brain barrier regulates tightly what can move between the general circulation and the brain, protecting the brain from neurotoxins and pathogens [44]. B-ASC can induce blood-brain barrier leakage, causing plasma protein and other molecules to extravasate into the brain parenchyma [4, 66, 73]. However, multiple subtypes of cSVD can cause this type of injury.
Classifications of cerebral small vessel disease
Neuropathologic criteria-based classification systems provide a framework for evaluating the presence and severity of B-ASC, and a taxonomy to distinguish B-ASC from other cSVD subtypes. Researchers also use these classification systems to perform clinical-pathological correlation studies, to assess associations with genetics and environmental risk factors, to develop experimental models, to help estimate the disease prevalence in various populations, and to identify disease-driving mechanisms that might provide therapeutic targets.
There is no universally agreed-upon method for categorizing cSVD pathologic changes. Different classification methods include histomorphologic (the main method for distinguishing B-ASC from other cSVD), etiologic (e.g., genetic, diabetic, hypertensive), pathologic comorbidity (e.g., amyloid, vasculitis), and radiography-based criteria.
The VCING classification system incorporates neuropathologic criteria for diagnosing a broad range of cSVD (Table 2) [183], built on prior studies and recommendations [8, 45, 47, 49, 143, 172, 184]. Unlike the VCING system, the Newcastle categorization [93, 96] emphasizes common patterns of different pathologies and the corresponding clinical scenarios of those patterns. This approach acknowledges that the different subtypes of cerebrovascular pathology are often seen in combination with each other, with stereotypical clinical manifestations.
It is unlikely that the current versions of the VCING or Newcastle systems provide complete representations of the complexity of cSVD. However, some merging of these different approaches might be useful for identifying both the microscopic lesions and the combinatorial patterns that contribute to clinical diseases. In addition to these specific categorization systems, other cSVD categories include cerebral amyloid angiopathy (CAA), inflammatory, and other subtypes; excellent reviews are available [66, 94, 120, 128, 213, 214].
Neuroimaging
No current neuroimaging modality allows 1-to-1 evaluation of the same phenomena that are observed using neuropathologic methods, and while no neuroimaging method captures the full spectrum of changes seen neuropathologically, the converse is also true. However, these approaches can complement one another in the study of cSVD. Autopsy studies are inherently cross-sectional and limited by sampling and staining methods. By contrast, neuroimaging allows for assessment of whole-brain metrics, multiple analyses of the same brain regions of interest, association with real-time, within-scanner behavioral performance, and longitudinal monitoring of the onset and progression of cerebrovascular changes.
It is important to distinguish between diseases of vessel walls (e.g. B-ASC, CAA) and the secondary parenchymal changes that are often detected with neuroimaging. Specific vessel wall pathologies cannot always be correlated exactly with measures of parenchymal injury. Further, the parenchymal changes that are sometimes described as cSVD can also be caused by extracranial disease processes such as renal disease, microemboli, and other disorders.
Structural magnetic resonance imaging (MRI) is the most widely used and standardized method for cSVD diagnoses and research applications, owing both to its high spatial resolution and its ability to detect a spectrum of cerebrovascular abnormalities without the need for application of extensive post-acquisition image analyses. There is currently no universally accepted method for categorizing cerebral cSVD using MRI but proposed standards are evolving rapidly. Another challenge is that the terminology differs between the neuropathologic and neuroimaging literatures.
White matter hyperintensities (WMH) on T2-weighted MRI are an established, often- studied marker of cerebral cSVD. WMH are readily visible on standard T2-weighted MRI, and their visibility is further enhanced through fluid-attenuation (i.e. MRI FLAIR imaging). Often seen bilaterally in aged persons’ brains, particularly in periventricular or peripheral regions of subcortical white matter (Fig. 6a), WMH have been associated with cognitive impairment, reduced local blood flow, gliosis, and demyelination [12, 57, 115, 153]. B-ASC has been cited as the “main, but not the only” underlying cause of WMH [7, 210].
Fig. 6. Magnetic resonance imaging (MRI) modalities are commonly used in the study of cerebral small vessel disease.

FLAIR imaging (a) showing regions of periventricular white matter hyperintensities (WMHs; arrows) and deep WMHs (orange arrowhead); T1-weighted imaging (b) showing lacunes (arrows); T2-weighted imaging (c) showing expanded perivascular spaces (arrows); and, susceptibility-weighted imaging (d) showing presumed microbleeds (arrows).
WMH is believed to indicate tissue injury, but the specificity, sensitivity, and predictive value of WMH have been debated. Indeed, specific cut-offs and locations for their measurement are not widely agreed upon. Although there are barriers for comparing imaging findings with pathologic changes [64], the study of WMH on brain imaging studies helps motivate further neuropathological investigation [217].
Other markers of cerebral cSVD on MRI include lacunes (Fig. 6b), enlarged perivascular spaces (EPVs; perivascular spaces are also known as Virchow-Robin spaces, Fig. 6c), and cerebral microbleeds (Fig. 6d). Each of these cSVD markers is frequently observed in the basal ganglia and other regions associated with penetrating and tortuous vessels. These may be directly relevant to B-ASC in some contexts. However, these MRI-based markers differ in size, are associated with different vascular events, and are thus best appreciated with different MRI sequences.
Lacunes are ~3–15 mm diameter infarcts visible on T1 or T2 images [216]. Lacunes must be distinguished from EPVs, which tend to be smaller (1–3mm) fluid-filled spaces, rather than infarcts, surrounding perforating arterioles and venules; EPVs are best appreciated on T2 images [216]. Cerebral microbleeds are small signal voids (usually < 5mm) on susceptibility-weighted imaging (SWI), or T2* gradient echo sequences [222]. They may signal the accumulation of hemosiderin (iron) associated with blood and old micro-hemorrhages, but may also be a surrogate for ischemic cSVD rather than necessarily a hemorrhagic diathesis [88]. Notably, some imaging sequences like SWI are associated with blooming effects which artifactually enhance microbleed size on MRI [104, 130].
Although there are limitations to MRI resolution, newer studies are extending the limit of detection to resolve features related to B-ASC in vivo. Fig. 7 shows results from a novel high-resolution MRI technique [122] which enables the study of vessels of several hundred micrometers in diameter. With this method, which can be accomplished at 3T or ultra-high magnetic field (7T), the vessel wall tortuosity can be readily observed and the decreased vessel number with aging quantified in three dimensions, as well as over time.
Fig. 7. High resolution MRI for the characterization of basal ganglia lenticulostriate arteries using 3T and 7T magnets.

This relatively high-resolution in vivo imaging [122] enables visualization of slender arteries/arterioles. In aging, there is increased tortuosity and decreased number of blood vessels resolved. In the raw images (a), blood is rendered black. Workflow is illustrated in panel (b), beginning with manual vessel segmentation on the raw turbo spin echo variable refocusing flip angles (TSE-VFA) image. The vessel volumes are reconstructed, and a mesh surface is created in preparation for shape analysis. Quantitative measures of vessel length, tortuosity and radius can be calculated. The box plot (c) shows that more vessels were detected in young participants than in older participants and more vessels were detected using 7T VFA-TSE compared to the more conventional 7T time-of-flight magnetic resonance angiography (7T TOF-MRA) [122]. An example comparing an older and younger adult (d) illustrates the smaller number of vessels detected in an older participant (A,C,E) than a younger participant (B, D, F).
A number of other new neuroimaging methods focus more on functional than structural phenomena. For example, one recently developed technique involves cerebral perfusion imaging using pseudo-continuous arterial spin labeling (pCASL), which measures local blood flow without the use of a contrast agent such as gadolinium [209]. This approach permits quantification of blood flow to different brain regions. Unlike the structural techniques described above, many of the newer functional studies require specialized resources that are not readily available at most centers. Despite this limitation, neuroimaging methods have profoundly augmented the study of how cSVD develops over time.
Anatomic distribution
The anatomic distribution of B-ASC is important to consider because vascular pathology may have focal clinical manifestations. One group of investigators report [199] that non-amyloid cSVD appear to affect “first arteries of the basal ganglia and then expands into the peripheral white matter and leptomeningeal arteries, as well as into thalamic and cerebellar white matter vessels” [200], in a stereotypic spatio-temporal pattern. Not all prior studies have found that B-ASC follows a predictable anatomic sequence, however [111]. Anatomic associations with risk factor data, for instance basal ganglia B-ASC related to hypertension, are better established [99, 146]. Neuroimaging studies, by contrast, have emphasized the periventricular and deep white matter changes [59] (Fig. 6).
Other brain regions are also affected by B-ASC in aged persons, with possible clinical manifestations. The amygdala appears vulnerable to B-ASC. The amygdala has an idiosyncratic blood supply [82, 149], with a tendency to show extensive B-ASC in elderly brain, along with venous collagenosis, especially near the inferior horn of the lateral ventricle [94]. In addition to its importance in emotion and behavior, the amygdala serves roles in autonomic nervous system regulation; autonomic dysfunction has been linked to B-ASC [76]. It seems likely that amygdala microangiopathy has other functional consequences, but the correlates of B-ASC in this region have not been extensively studied. We note that the amygdala is also the brain area apparently first affected by TDP-43 proteinopathy in limbic-predominant age-related TDP-43 encephalopathy (LATE) [90, 147], and perivascular TDP-43 proteinopathy can be prominent near small vessels, as discussed below.
Epidemiology and risk factors
The main risk factors generally discussed in association with ASC are hypertension and diabetes [94]. Other comorbidities reported to increase risk of B-ASC include obesity, renal failure, and sleep pattern perturbations [84, 114]. Environmental factors, including air pollution and chemical toxins, may promote B-ASC, and may worsen hypertension and other risk factors, according to both epidemiologic and animal studies [61, 102]. However, in advanced age, risk factors widely associated with B-ASC correlate less robustly with B-ASC pathology than in younger subjects, according to multiple studies, as described below.
Hypertension is a strong risk factor for mortality and morbidity worldwide [67, 136, 221]. Arteriolosclerosis is considered a downstream complication of hypertension, because in hypertensives there is narrowing of the vessel lumens in both arteries and arterioles [99]. Radiographic evidence of hypertension-induced brain injury includes WMHs, small and large infarcts, enlarged perivascular spaces, and cerebral aneurysms [55, 160]. The recent SPRINT-MIND trial found that intensive lowering of blood pressure was associated with a relative decrease of WMH size, and reduced incidence of cognitive impairment [67, 68].
Type II diabetes mellitus (T2DM) is also associated with increased risk for B-ASC, probably via increased exposure of the cAVU to hyperglycemia, inflammatory cytokines, and other factors [28]. Neuropathologic studies establish associations between T2DM and B-ASC as well as with small and large infarcts [3, 153, 172, 188]. Correspondingly, neuroradiography has also showed T2DM-linked increased risk for WMHs, brain infarcts, and blood flow disturbance [57, 196].
Metabolic syndrome (MetS) is a term to describe a common combination of abnormalities in the same individual, including hypertension, T2DM, obesity, and dyslipidemia [109]. Approximately a third of American adults have MetS in some form [177]. MetS has negative impacts on brain function [48, 97], but the mechanism underlying this association requires further study. A number of additional phenomena cluster with MetS, including hyperhomocysteinemia and a generalized pro-inflammatory state [42]. These phenomena may have synergistic deleterious effects on arterioles; for example, hypertension, hyperglycemia, hyperhomocysteinemia, and dyslipidemia may each harm components of the cAVU and promote B-ASC [93, 137, 187].
Trends have been reported for an association of MetS-related conditions (hypertension and T2DM) with more severe B-ASC [18, 84]. Yet these associations were not as strong in persons beyond age 80 years. Arvanitakis et al. [18] and Ighodaro et al. [84] both reported that the association between hypertension and B-ASC was attenuated in advanced age. In another study of aged research participants, neither hypertension nor T2DM was statistically significantly associated with B-ASC [194]. These data indicate that a substantial proportion of elderly persons’ B-ASC may be attributable to causes other than traditional MetS or cardiovascular risk factors. However, these studies and their conclusions need to be considered carefully because of potential confounders such as medicated diseases (a person may have a history of hypertension but have it under control medically), death/survivor bias, racial disparities, differing risk factors in midlife and late life, and comorbid conditions [18, 87, 118].
Table 3 shows analyses of recent data from the NACC NP v10 Data Set. Data were limited to autopsies reported after June 2014 through the March 2020 data freeze, and these volunteers were recruited and evaluated at 32 different U.S. research centers. As reported earlier with a different set of NACC subjects [84], a clinical history of hypertension was associated with increased odds for moderate or severe B-ASC pathology, but the trend was only statistically significant in persons dying before age 80 years. Notably, myocardial infarction was strongly associated with hypertension, T2DM, and hypercholesterolemia (Table 3).
Table 3.
Clinical and pathological correlations in NACC Neuropathology v10 Dataset, stratified by age of death
| Brain arteriolosclerosis (B-ASC) | Association with history of myocardial infarction# (p-value) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| <80 years at death | >=80 years at death | Overall p-value | |||||||
| B-ASC: None or mild, n(%) | B-ASC: Moderate or severe n(%) | p-value* | B-ASC: None or mild n(%) | B-ASC: Moderate or severe n(%) | p-value* | ||||
| Cardiovascular risk factor by clinical history ** | |||||||||
| Diabetes | No | 303 (88.9) | 188 (85.8) | 0.35 | 300 (86.5) | 340 (85.4) | 0.77 | 0.31 | <0.0001 |
| Yes | 38 (11.1) | 31 (14.2) | 47 (13.5) | 58 (14.6) | |||||
| Hypertension | No | 276 (60.5) | 158 (51.6) | 0.018 | 183 (36.2) | 182 (32.2) | 0.19 | 0.0002 | <0.0001 |
| Yes | 180 (39.5) | 148 (48.4) | 322 (63.8) | 383 (67.8) | |||||
| Hypercholesterolemia | No | 149 (44.5) | 86 (39.6) | 0.30 | 126 (36.7) | 135 (34.1) | 0.50 | 0.11 | <0.0001 |
| Yes | 186 (55.5) | 131 (60.4) | 217 (63.3) | 261 (65.9) | |||||
| Body mass index (BMI) >= 30 | No | 215 (81.1) | 138 (73) | 0.053 | 273 (84.3) | 284 (84.5) | 1.0 | 0.32 | 0.75 |
| Yes | 50(18.9) | 51 (27) | 51 (15.7) | 52 (15.5) | |||||
| Cerebrovascular pathology ** | |||||||||
| Cerebral microinfarct(s), >0 | No | 470 (86.9) | 285 (83.3) | 0.17 | 480 (79.2) | 408 (61.4) | <0.0001 | <0.0001 | 0.082 |
| Yes | 71 (13.1) | 57 (16.7) | 126 (20.8) | 257 (38.6) | |||||
| Cerebral microbleed(s), >0 | No | 499 (97.1) | 314(93.5) | 0.018 | 579 (97.1) | 618 (93.6) | 0.0051 | 0.00015 | 0.66 |
| Yes | 15 (2.9) | 22 (6.5) | 17 (2.9) | 42 (6.4) | |||||
| Cerebral large or lacunar mfarct(s), >0 | No | 495 (91.5) | 299 (87.2) | 0.050 | 536 (88.7) | 497 (74.6) | <0.0001 | <0.0001 | 0.50 |
| Yes | 46 (8.5) | 44(12.8) | 68 (11.3) | 169 (25.4) | |||||
| White matter rarefaction (moderate/severe) | No | 405 (84.2) | 168 (53.5) | <0.0001 | 527 (92.8) | 348 (56.4) | <0.0001 | <0.0001 | 0.41 |
| Yes | 76(15.8) | 146 (46.5) | 41(7.2) | 269 (43.6) | |||||
| CAA (moderate/severe) | No | 369 (68.3) | 177 (51) | <0.0001 | 464 (76.6) | 390 (58.7) | <0.0001 | <0.0001 | 0.19 |
| Yes | 171 (31.7) | 170 (49) | 142 (23.4) | 274 (41.3) | |||||
| Neurodegenerative disease pathology ** | |||||||||
| LATE-NC, Stage >=2 | No | 283 (74.5) | 191 (71.8) | 0.51 | 268 (75.9) | 291 (64.2) | 0.00045 | 0.00077 | 0.97 |
| Yes | 97 (25.5) | 75 (28.2) | 85 (24.1) | 162 (35.8) | |||||
| Hippocampal sclerosis, Any | No | 480 (88.2) | 286 (81.9) | 0.012 | 526 (86.8) | 509 (76.3) | <0.0001 | <0.0001 | 0.54 |
| Yes | 64(11.8) | 63 (18.1) | 80 (13.2) | 158 (23.7) | |||||
| Alzheimer’s disease, Intermediate/Severe | No | 121 (22.2) | 64(18.3) | 0.19 | 182 (30) | 133 (19.9) | <0.0001 | 0.00015 | 0.43 |
| Yes | 423 (77.8) | 285 (81.7) | 424 (70) | 534 (80.1) | |||||
| Lewy body disease, Any | No | 289(53.1) | 176 (50.4) | 0.47 | 414(68.3) | 395 (59.2) | 0.00093 | 0.022 | 0.73 |
| Yes | 255 (46.9) | 173 (49.6) | 192 (31.7) | 272 (40.8) | |||||
Chi-square test, 2-tailed
Cardiovascular risk factors are by self-report at last exam; pathological features are autopsy results
Myocardial infarction indicates either remote or recent by clinical history
These data indicate collectively that traditional cardiovascular risk factors are less predictive of B-ASC severity in advanced age than they are predictive of cardiac pathology in the same persons. While the associations between B-ASC and hypertension or T2DM were less strong than might be expected, there are other notable comorbidities robustly associated with B-ASC in aged individuals (Table 3), including cerebrovascular disease and neurodegenerative pathologies [84]. Perhaps the most salient risk factor for B-ASC is aging itself.
Genetics and aging
B-ASC appears to be influenced strongly by genetics and senescence. One clue comes from persons of age <20 years with “progeria” (accelerated senescence phenotype), driven by specific gene mutations, who tend to have substantial B-ASC [78, 174, 185]. B-ASC is also seen with aging in wild-type animal species [72, 180]. Different genetic risk factors affect people within distinct ranges of the aging spectrum, and there has been extensive focus on how aging and genetics influence the vasculature – combining with stress, caloric intake, glycemia/insulinemia, immune factors, and other processes that modulate an individual’s lifespan [95, 140, 198, 207].
Chronologic age is linked consistently to increased prevalence of B-ASC in autopsy series. Smith and colleagues showed that cSVD was evident in ~3% of individuals in their 40s, but ~19% of 70-year-olds [186]. B-ASC increases even more dramatically among persons beyond age 80 years [50, 151, 199]. Fig. 8 depicts how B-ASC pathologic changes evolve in aging. Prior studies agree that over 80% of individuals beyond age 80 had identifiable B-ASC [50, 84, 89]. Further studies are needed in diverse population-based cohorts.
Fig. 8. Brain arteriolosclerosis (B-ASC) pathology by age at death, from the NACC Neuropathology version 10 data set (n=2,284).

B-ASC pathology increases with advanced age. Note that <20% of subjects had moderate or severe B-ASC pathology before age 60, whereas in subjects over 80, >50% had moderate or severe B-ASC pathology, and >80% had some degree of B-ASC.
B-ASC in aged individuals is common, and genetics also appears to have a strong influence. Table 4 presents genetic risk factors of B-ASC identified to date. Recent articles by Marini et al. [125] and Vinters et al. [213] provided overviews of prior studies on the genetic influence on B-ASC associated phenotypes.
Table 4.
Human genes associated with brain arteriolosclerosis and other inherited small vessel disease
| Gene | Chr.* | Known Function of gene product | Associated subtype of small vessel disease | Refs* |
|---|---|---|---|---|
| Mendelian/monogenic | ||||
| NOTCH3 ** | 19p13 | Transcription regulator in vascular smooth muscle cells | WMH*, ischemic stroke, ICH*, CADASIL | [91, 138] |
| COL4A1/A2 ** | 13q34 | Collagen IV, endothelial basement membrane protein | WMH, ischemic stroke, ICH | [92, 169] |
| HTRA1 ** | 10q26 | Serine protease involved in repression of TGF-β | WMH, ischemic stroke, CARASIL | [138, 169] |
| TREX1 | 3p21 | 3’-exonuclease involved in DNA repair | WMH, ischemic stroke | [171, 176] |
| Contributory/polygenic | ||||
| FOXC1/FOXF2 | 6p25 | DNA regulation in eye, brain, heart, and kidney development. | WMH, ischemic stroke | [56] |
| ZCCHC14 | 16q24 | Transcription factor and metal ion-binding protein | WMH, stroke | [202] |
| WDR12/ICA1L | 2q33 | Protein domain-specific binding | WMH, stroke | [124] |
| PMF | 1q22 | Transcription factor involved in mitosis | WMH, ICH | [204, 219] |
| APOE | 19q13 | Apolipoprotein involved in lipid transport and metabolism | B-ASC*, WMH, ischemic stroke | [98, 223] |
| ABCC9 | 12p12 | K+ channel regulators in blood vessels | B-ASC in persons >80y.o. at death | [84] |
| JAZF | 7p15 | Transcription factor involved in transcription repression | B-ASC | [40] |
| TSPAN8 | 12q21 | Cell-membrane protein involved in cell growth regulation | B-ASC | [40] |
| LGR5 | 12q21 | Receptor in Wnt signaling pathway | B-ASC | [40] |
| KCNJ11 | 11p15 | K+ channel regulated by ABCC8andABCC9 | B-ASC | [40] |
| PCSK9 | 1p32 | Protease involved in cholesterol metabolism regulation. | B-ASC | [40] |
| ZFHX3 | 16q22 | Transcription factor associated with atrial fibrillation | B-ASC | [40] |
Abbreviations: Chr.: Chromosomal location; Refs: References; WMH: white matter hyperintensities; ICH: intracerebral hemorrhage seen via radiography; B-ASC: Brain arteriolosclerosis; CADASIL: cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy; CARASIL: Cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy
For NOTCH3, COL4A1/A2, and HTRA1, gene variants also may be contributory in polygenic disease
Rare monogenic forms of cSVD, with Mendelian modes of inheritance, shed light on the genes and biological pathways with potential strong impacts. The most common monogenic cSVD is cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), attributed to NOTCH3 gene mutations [91, 138]. A pathologic hallmark of CADASIL is the presence of granular osmiophilic material adjacent to smooth muscle cells in the walls of arterioles (on electron microscopy), loss of smooth muscle in the tunica media, and adventitial fibrosis [15]. Other aspects of arteriolar pathology in CADASIL were well described by Miao et al. [135].
Mutations in genes that encode collagen or other vessel-related proteins can also lead to B-ASC. COL4A1 and COL4A2 encode type IV human collagen, which is a key protein in endothelial basement membranes. High-penetrance COL4A1 mutations lead to a variety of rare monogenic brain infarction phenotypes which include severe B-ASC, and more common COL4A1 single nucleotide polymorphisms (SNPs) are also associated with brain cSVD [41, 134]. TREX1 mutations are another interesting cause of degenerative microvascular lesions in the eye, brain, and elsewhere, implicating inflammatory dysfunction at the basement membrane of arterioles and other small vessels [171, 176]. Other genes associated with monogenic B-ASC phenotype include HTRA1 and FOXC1/FOXF2 [108, 125, 213].
In contrast to the rare monogenically inherited conditions, the common, chronic diseases of aging (which presumably include B-ASC) can be influenced by a large number of lowpenetrance risk alleles. Human genetic variants that include chromosomal loci at 16q24, 2q33, 14q22, and 12q24 have been linked to B-ASC-type phenotypes [125]. Specific SNPs were linked via genome-wide association studies (GWAS) to risk for WMH or other radiographic cSVD phenotypes [5, 6, 16, 21, 41, 54, 126, 164, 202, 203]. However, not all of the phenotype-driving genes have been elucidated to date.
Chou and colleagues [39] investigated the association of candidate SNPs with cerebrovascular pathologies including B-ASC. Variants located within or near the following genes were suggestively associated with B-ASC: the diabetes risk genes JAZF, TSPAN8, LGR5, and KCNJ11; the LDL risk gene PCSK9; and, the atrial fibrillation risk gene ZFHX3 [39]. The ===4 isoform of Apolipoprotein E (APOE), the major late-onset AD risk allele, has also been associated with B-ASC severity [223].
Another gene candidate for B-ASC is ABCC9, which encodes SUR2. SUR2 proteins function as K+ channel regulators and can affect the stress response, metabolism, and regulation of blood flow and the cAVU [152]. ABCC9/SUR2is a pericyte marker in mouse brain [211] but is also expressed in vascular smooth muscle cells in humans [152]. ABCC9 mutations in humans are a cause of Cantu syndrome, with a phenotype that often includes tortuous CNS blood vessels [152]. A common ABCC9 genetic polymorphism has been associated with B-ASC severity but only among persons over 80 years of age at death [84], a finding in need of replication. Overall, more work is required on gene variants that are associated with the B-ASC phenotype.
Pathologic mechanisms
We have described “upstream” risk factors that may drive B-ASC. The “downstream” pathologic mechanisms promoted by B-ASC are also important to consider. Harmful mechanisms linked with B-ASC include impaired blood flow [71], mechanical stiffening [148], functional synaptic uncoupling [144], blood-brain barrier leakage [32], synergies with neurodegenerative disease mechanisms [119], impaired angiogenesis [205], vessel wall mineralization [194], failure of “glymphatics” [1, 55, 133, 215], impaired access to growth factors or growth factor receptors [74], release of reactive and oxidizing agents [11, 156], harmful neuroinflammation [4, 175], micro-hemorrhages [191], micro-thrombi [201], vasospasm [62], seizure kindling [86], perturbations of calcium and other electrolytes [167], and various cell type-specific cytopathic changes [73, 168, 206]. Although it is not clear which of these mechanisms are “downstream” of B-ASC, two common diseases with possible synergistic effects with B-ASC are MetS (see above) and LATE.
Whereas it is commonly thought that MetS promotes B-ASC, there is a credible hypothesis that B-ASC works in the other direction, contributing to hypertension and other aspects of MetS [28]. Blood pressure is partly regulated by the brain via hypothalamic and brainstem autonomic signaling [121]. The CNS lacks energy reserves, so maintaining cerebral perfusion is a critical biologic priority. When human cerebral perfusion is impaired, the brain can release paracrine and endocrine factors that augment blood flow and increase systemic blood pressure [218], in a pattern comparable to that of renovascular hypertension [79]. According to this hypothesis, B-ASC and hypertension might promote each other in a deleterious cycle: hypertension leads to blood vessel abnormalities, including more B-ASC, which in turn worsens hypertension, culminating in ischemic brain damage [17, 166]. There may also be additional, analogous feed-forward central regulation (via hormones, adipokines, cytokines, etc.) of hyperglycemia, hyperlipidemia, and inflammatory factors which may maintain cerebral energy supply in advanced age, at the expense of harming the cAVU.
There is also growing evidence that B-ASC is associated with a common neurodegenerative condition referred to as LATE [150]. Among the conditions that may mimic AD clinically, LATE generally affects persons of advanced age (>80 years), with memory loss that can evolve to a dementia syndrome, and is often comorbid with AD (in which case the dementia phenotype is more severe than either AD or LATE alone) [150]. In multiple independent data sets, LATE is more common in persons with moderate-to-severe B-ASC than in those with no or low B-ASC [26, 101, 154, 155]. The papers that reported an association with LATE and B-ASC tended not to find an association between AD and B-ASC. In a recent study by Bourassa et al. [29], brain arteriolar mural cell markers (α-SMA for smooth muscle cells, PDGFRβ and CD13 for pericytes) in parietal cortex were substantially decreased in brains with comorbid TDP-43 proteinopathy. It was previously noted that TDP-43 proteinopathic deposits may be present in the perivascular astrocytic end-feet in frontotemporal lobar degeneration with TDP-43 (FTLD-TDP) as well as in LATE [116, 152]. As shown in Fig. 9, TDP-43 deposits may be detected outside both arterioles and capillaries, particularly the latter. TDP-43 proteinopathy is associated with increased HS pathology [13], which is also linked to cerebral ischemia [152, 212]. These observations raise questions about how LATE and B-ASC interact mechanistically. Microangiopathy, including B-ASC, may contribute to TDP-43 proteinopathy, or vice versa, or there may be a synergistic feed-forward process in which each exacerbates the other. A critical question remains as to which more directly affects neurological function.
Fig. 9. Phospho-TDP-43 immunoreactive proteinopathy can be seen immediately adjacent to small blood vessels in brains with limbic-predominant age-related TDP-43 encephalopathy (LATE).
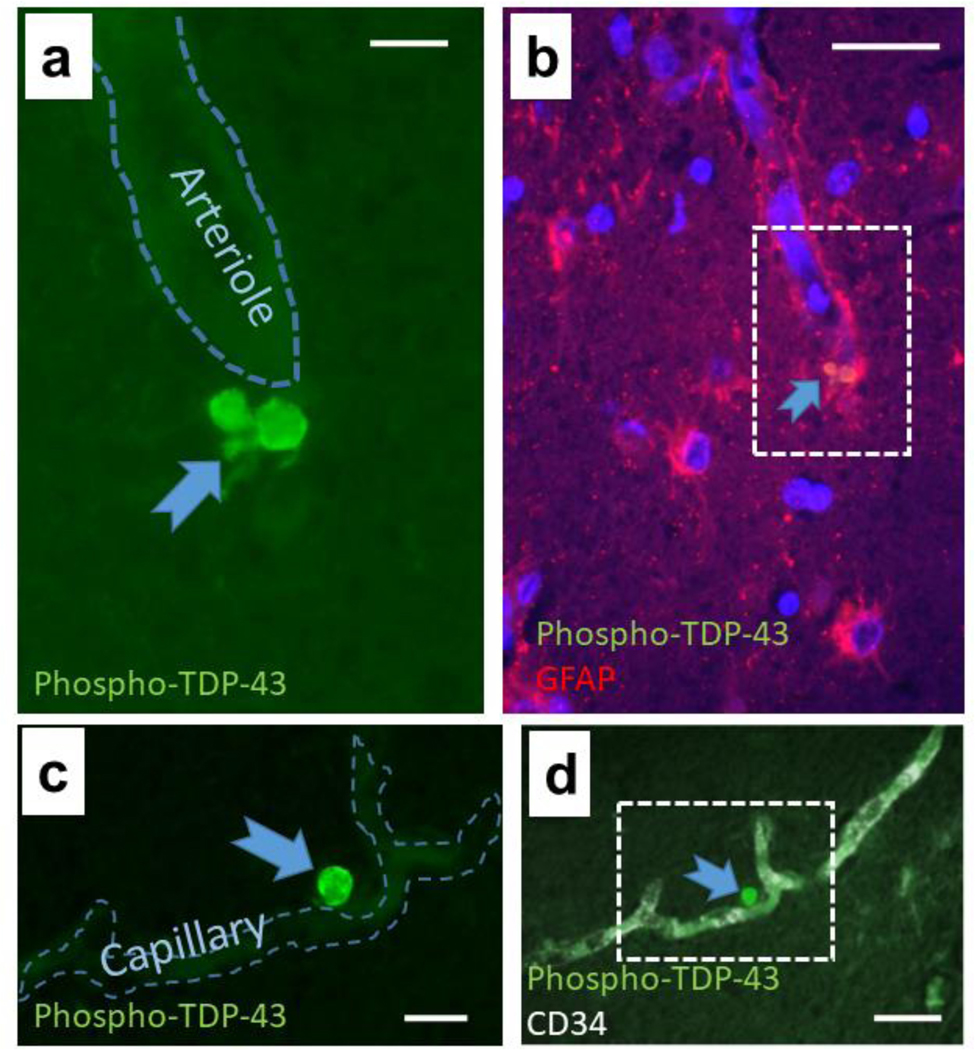
The micrograph shows perivascular TDP-43 proteinopathy near an arteriole (a, b) and a capillary (c, d) in hippocampi of 2 subjects with autopsy-proven LATE. In panel (a), the green filter shows the TDP-43 proteinopathy adjacent to an arteriole. Panel (b) shows lower magnification and the red filter shows GFAP immunoreactivity, revealing the colocalization with GFAP and TDP-43 (arrow). Panel (c) shows a phospho-TDP-43 deposit adjacent to a capillary as indicated by CD34 immunofluorescence at lower magnification (d). The presence of TDP-43 proteinopathy in astrocyte end-feet in FTLD-TDP was shown previously by Lin et al. [116]. LATE neuropathologic change is associated with relatively severe B-ASC [101, 154]. Thus, there may be parallel or synergistic disease-driving mechanisms related to the pathologies that usually are categorized separately as cerebrovascular or neurodegenerative. Scale bars = (a): 15 μm; (b): 40 μm; (c): 20 μm; and, (d): 40 μm.
Clinical-pathological correlation
Multiple studies have supported the hypothesis that the severity of B-ASC is strongly associated with impaired cognition. However, testing this hypothesis is complicated by potential confounders [93, 120, 128]. Many factors can contribute to cognitive impairment, for an individual or a population [100, 123]. Therefore, optimal clinical-pathological correlation studies should employ analytical models that factor in the effects of different neurodegenerative diseases, CAA, lacunar infarcts, microhemorrhages, and microinfarcts, among other co-pathologies.
Some studies have evaluated the clinical impact of B-ASC-associated pathologic findings, rather than B-ASC itself. White matter disruption and astrocytosis attributed to vascular disease were associated with cognitive impairment [53, 131]. Methods that combined B-ASC with other cSVD subtypes also found associations with dementia [184]. Since B-ASC may be partially responsible for microinfarcts and lacunes (B-ASC is indeed strongly associated with those comorbid pathologies, as shown in Table 3), it is a valid approach to focus on those ischemia-related pathologic endpoints [53, 131]. Note that a separate literature exists on the association between WMH and neurological function [9, 214]. Further, B-ASC is a common comorbidity in AD and other neurodegenerative diseases [76, 154]. Although these considerations are germane to B-ASC, the main focus here is on whether autopsy-diagnosed B-ASC pathology is associated with antemortem clinical status.
Clinical-pathologic studies related to B-ASC are listed in Table 5. Most of the relevant prior studies assessed the association between B-ASC and global cognitive status. For example, the VCING study [183] evaluated a sample of 113 autopsies to assess the “likelihood that cerebrovascular disease contributed to a cognitive impairment” of mild or severe degree (dementia). A robust association was found between B-ASC and cognitive impairment [183]. Ighodaro et al. [84] evaluated the NACC dataset (n=2,390) using regression models that adjusted for AD, Lewy body disease, and other cerebrovascular pathologies. In that study, the association between B-ASC severity was tested with final MMSE and CDR Sum of Boxes scores, finding a significant association for both metrics [84]. Two clinical-pathologic series from Brazil also showed positive association between B-ASC and decreased cognitive status [193, 194]. An interesting finding by Suemoto et al. (2019) was that siderocalcinosis in the arterial/arteriolar vessel walls (Fig. 4d) was associated with dementia in participants <80 years of age at death [194].
Table 5.
Clinical-pathological correlations for CNS arteriolosclerosis
| Source | Cohorts and sample sizes | Avg age at death | Main findings |
|---|---|---|---|
| Skobrot et al, 2017[183] | Oxford Brain bank and Newcastle Brain Tissue Resource, n=113 | 83.4yrs | Cognitive impairment (either mild cognitive impairment or dementia) was associated with more severe B-ASC |
| Arvanitakis et al, 2017[19] | Rush University (Chicago, USA) cohorts, n=1,143 | 88.8yrs | Odds of clinically suspected Alzheimer’s type dementia were higher with more severe B-ASC |
| Boyle et al, 2018[30] | Rush University cohorts (Chicago, USA), n=1161 | 89.6yrs | B-ASC accounted for 5.2% of risk for clinical Alzheimer’s type dementia diagnosis |
| Buchman et al, 2011[33] | Rush University cohorts (Chicago, USA), n=418 | 88.5yrs | B-ASC was associated with parkinsonism gait disturbance, same trend for global parkinsonism and tremor |
| Buchman et al, 2017[34] | Rush University cohorts (Chicago, USA), n=163 | 91.2yrs | Spinal arteriolosclerosis was prevalent (24% modsevere) and is associated with parkinsonism |
| Suemoto et al, 2019[194] | Biobank for Aging Studies (BAS) of the University of Sao Paulo, Brazil n=677 <80y.o.; n=412 >80y.o. | <80y.o.: 67.0yrs; >80y.o.: 85.7yrs |
Hyaline B-ASC was associated with cognitive impairment in younger and older groups (effect size trended larger in younger group); siderocalcinosis was associated with cognitive impairment in younger group only |
| Suemoto et al, 2014[193] | The São Paulo Autopsy Service, Brazil, n=1,092 | 74yrs | Hyaline B-ASC severity was associated with cognitive status |
| Ighodaro et al, 2017[84] | NACC data set, n=1008 <80y.o.; n=1,382 >80y.o. | <80y.o.: 78.0yrs; >80y.o.: 88.0yrs |
Moderate and severe B-ASC were associated with global cognition measured by final CDR-SB and MMSE scores. Hypertension was associated with B-ASC in younger but not older group. |
Several relevant studies from the Rush University Religious Orders Study and Memory and Aging Project cohorts further demonstrated that B-ASC is associated with cognitive impairment. In a study of 1,143 individuals, B-ASC was associated with global cognitive status, episodic memory, working memory, and perceptual speed (all p<0.05), with a trend (p=0.052) for visuospatial abilities [19]. Women in this cohort were more likely than men to have severe B-ASC [161], as was the case in the NACC data set [84]. In a separate study, researchers estimated that the attributable risk for AD-type dementia due to B-ASC was 5.2% [30]. Analogous attributable risks in the same sample were 10.8% for all Lewy body diseases, 8.9% for macroscopic infarcts, and 8.1% for CAA [30]. Therefore, in this community-based cohort, the estimated proportion of AD-type dementia risk due to B-ASC was on par with other neuropathologic changes that are known to have a very large public health impact.
Although the overall contribution of B-ASC to cognitive impairment in a cohort can be estimated, it is much more difficult to estimate the degree of cognitive impairment resulting specifically from B-ASC in a given individual, particularly without an autopsy. This has been a challenging problem in the dementia research field. Some of the cognitive impairment attributed to AD-type dementia in clinical studies was almost surely due to B-ASC instead. This phenomenon has noteworthy implications. For example, in studies lacking autopsy confirmation, T2DM was associated repeatedly with increased risk for the clinical diagnosis of Probable AD [37, 170]. However, in studies where autopsy evaluation was part of the study design, the severity of AD pathologic change was not associated with T2DM; rather, the severity of cSVD pathologic change (including B-ASC) was increased in T2DM cases and was associated with antemortem cognitive impairments [3, 153, 172, 188]. Similar phenomena may underlie the observation that AD-type dementia risk is disproportionately high among African-American adults—neuropathologic studies indicate that the relevant disease-driving factor is cSVD pathologies including B-ASC, instead of the amyloid plaques and neurofibrillary tangles of AD [22, 179]. However, there are challenges in studying dementia in African Americans due to historic medical mistreatment, which has led to low willingness to participate in studies that involve brain autopsy [85]. More work is required in this area.
B-ASC has also been associated with motor [34, 35, 77] and autonomic [76] impairments. For example, Buchman and colleagues analyzed motor assessments in 850 participants with postmortem cSVD workup [35]. When the global motor scores of individuals with and without arteriolosclerosis were compared, the average global motor score was lower in individuals with more severe B-ASC. In a separate study, Buchman and colleagues evaluated autopsy data from 165 elderly adults [34] and found that “spinal arteriolosclerosis is common and may contribute to the severity of spinal white matter pallor and parkinsonism in older adults.” [34] In this cohort, spinal ASC explained substantial variability in parkinsonism as defined by the Unified Parkinson’s Disease Rating Scale [34, 63]. These non-cognitive findings underscore the widespread effects of B-ASC and the complex inter-relationship between vascular disease and clinical phenotypes.
Possible future directions: differentiating B-ASC subtypes
Diagnostic classification may evolve to recognize clinically meaningful subtypes of B-ASC. It is possible that some, but not other, mechanisms that cause what we currently refer to as B-ASC may be targetable by specific therapeutic or disease-prevention strategies in the future. We highlight four broad categories of phenomena that could form the bases of classifiable B-ASC subtypes: (1) the influence of different cellular subcomponents of the cAVU; (2) interactions with comorbid CNS vascular and/or neurodegenerative diseases; (3) B-ASC caused by systemic diseases including endocrine, inflammatory, and coagulopathic conditions; and, (4) the biology of human aging.
There may be contributions to vasculopathy from multiple different cellular and extracellular elements. Potential cell-specific influences of astrocytes [192], pericytes [95], endothelial cells [73], smooth muscle cells [58, 80], microglia [75], non-cellular and shared micro-domains [2, 182] have been hypothesized [173]. In the future, it may become possible to categorize B-ASC according to the cell type or micro-domain that is primarily responsible for dysfunction in a given vascular segment. We have suggested a new term that may help in focusing efforts to understand the pathobiology of B-ASC, namely the cAVU, comprising the components within the arteriolar portions of the cerebral vasculature.
In addition to the B-ASC related dysfunction attributable to specific cAVU domains, another important area which merits further attention is understanding how B-ASC contributes to, or is affected by, comorbid brain conditions including neurodegenerative “misfoldingopathies” and various cerebrovascular diseases. As shown in Table 3, the associations between B-ASC and comorbid brain conditions are strong in advanced age. Extensive research and prior review articles have focused on the associations between cSVD and AD [31, 93, 137, 187]. Moreover, B-ASC was substantially more severe in both the white matter and neocortex in many different dementias compared to age-matched controls [76]. It has also been shown that in patients with one type of cSVD, other subtypes of cSVD are often present (e.g., CAA and microinfarcts, along with B-ASC) [31].
B-ASC is also affected by systemic medical conditions. There are many ways that non-CNS organ systems may influence the physiology of brain arterioles. The complex influences of MetS are discussed above. Many other systemic perturbations including auto-immunity, coagulopathy, cardiopulmonary disease, and renal dysfunction are common in aged persons and may affect cells of the cAVU. Drugs and surgical (including anesthetic) interventions used to treat the various aging-related diseases may also influence brain vascular health in ways that are not now fully known.
Finally, biological senescence itself may be a critical factor affecting arteriolar walls in aging brains. The literature on the interactions between biological aging and the vasculature, although extensive [95, 140, 198, 207], is incomplete with respect to B-ASC. Related areas of research have included age-related protein modifications (e.g., carbamylation, citrullination, and deamidation) [60], effects of oxidative stress [190], insulin signaling abnormalities [178, 224], mitochondrial dysfunction [10, 36], and dysregulated inflammation and autophagy [132]. Arguably, to study each of these topics in isolation is a reductionist approach to an organismwide senescence program with a genetic component, which includes progression of B-ASC at the final stage of the human aging spectrum. Investigating these pathways more comprehensively, and further addressing the biologic complexity of B-ASC, may help researchers to tailor strategies designed to preserve neurologic function in aging.
Supplementary Material
Acknowledgment
We are very grateful to the research volunteers, their families, and clinicians, as well as the other researchers who made this work possible. This study was also supported by California Dept of Public Health grant A20–2947-5001 and NIH grants P30 AG028383, R01 AG057187, R01 AG039621,R01 AG055449,K24 AG053435, R56 AG057191, R01 HD064993, U54 NS100717, U01 AG016976, RF1 NS118584, and S10 OD023573. Additional support came from Medical Research Council (MRC, G0500247) and previous Newcastle Centre for Brain Ageing and Vitality (BBSRC, EPSRC, ESRC and MRC, LLHW), and Alzheimer’s Research (ARUK). The Newcastle Brain Tissue Resource is funded in part by a grant from the UK MRC (G0400074), by the Newcastle NIHR Biomedical Research Centre in Ageing and Age Related Diseases award to the Newcastle upon Tyne Hospitals NHS Foundation Trust, and by a grant from the Alzheimer’s Society and ARUK as part of the Brains for Dementia Research Project. See Supplemental Acknowledgment for additional acknowledgments.
Supplemental Acknowledgement
NACC data are contributed by the NIA-funded ADRCs: P30 AG019610 (PI Eric Reiman, MD), P30 AG013846 (PI Neil Kowall, MD), P30 AG062428–01 (PI James Leverenz, MD) P50 AG008702 (PI Scott Small, MD), P50 AG025688 (PI Allan Levey, MD, PhD), P50 AG047266 (PI Todd Golde, MD, PhD), P30 AG010133 (PI Andrew Saykin, PsyD), P50 AG005146 (PI Marilyn Albert, PhD), P30 AG062421–01 (PI Bradley Hyman, MD, PhD), P30 AG062422–01 (PI Ronald Petersen, MD, PhD), P50 AG005138 (PI Mary Sano, PhD), P30 AG008051 (PI Thomas Wisniewski, MD), P30 AG013854 (PI Robert Vassar, PhD), P30 AG008017 (PI Jeffrey Kaye, MD), P30 AG010161 (PI David Bennett, MD), P50 AG047366 (PI Victor Henderson, MD, MS), P30 AG010129 (PI Charles DeCarli, MD), P50 AG016573 (PI Frank LaFerla, PhD), P30 AG062429–01(PI James Brewer, MD, PhD), P50 AG023501 (PI Bruce Miller, MD), P30 AG035982 (PI Russell Swerdlow, MD), P30 AG028383 (PI Linda Van Eldik, PhD), P30 AG053760 (PI Henry Paulson, MD, PhD), P30 AG010124 (PI John Trojanowski, MD, PhD), P50 AG005133 (PI Oscar Lopez, MD), P50 AG005142 (PI Helena Chui, MD), P30 AG012300 (PI Roger Rosenberg, MD), P30 AG049638 (PI Suzanne Craft, PhD), P50 AG005136 (PI Thomas Grabowski, MD), P30 AG06271501 (PI Sanjay Asthana, MD, FRCP), P50 AG005681 (PI John Morris, MD), P50 AG047270 (PI Stephen Strittmatter, MD, PhD).
Abbreviations (gene and protein names not listed):
- AD
Alzheimer’s disease
- B-ASC
brain arteriolosclerosis
- CAA
cerebral amyloid angiopathy
- CADASIL
cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy
- cAVU
cerebral arteriolar vascular unit
- cSVD
cerebral small vessel disease
- EPV
enlarged perivascular space
- FLAIR
fluid-attenuated inversion recovery
- FTLD-TDP
frontotemporal lobar degeneration with TDP-43 proteinopathy
- H&E
hematoxylin and eosin
- HS
hippocampal sclerosis
- LATE
limbic-predominant age-related TDP-43 encephalopathy
- MetS
metabolic syndrome
- NACC NP
National Alzheimer’s Coordinating Center Neuropathology
- NVU
neurovascular unit
- pCASL
pseudo-continuous arterial spin labeling
- SI
sclerotic index
- SNP
single nucleotide polymorphism
- SWI
susceptibility weighted imaging
- T2DM
type II diabetes mellitus
- VCING
vascular cognitive impairment neuropathology guidelines
- WMH
white matter hyperintensity
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Contributor Information
Brittney L. Blevins, Dept of Neuroscience, U. Kentucky, Lexington, KY, 40536
Harry V. Vinters, Depts. of Pathology & Laboratory Medicine & Neurology, David Geffen SOM at UCLA & Ronald Reagan UCLA Medical Center, Los Angeles,CA 90095-1732
Seth Love, U. of Bristol and Southmead Hospital, Bristol, BS10 5NB, UK.
Donna M. Wilcock, Sanders-Brown Center on Aging and Dept of Neuroscience, U.Kentucky, Lexington, KY, 40536
Lea T. Grinberg, Department of Neurology and Pathology, UCSF, San Francisco, CA, USA.; Global Brain Health Institute, UCSF, San Francisco, CA, USA; and, LIM-22, Department of Pathology, University of Sao Paulo Medical School, Sao Paulo, Brazil
Julie A. Schneider, Depts of Neurology and Pathology, Rush University Medical Center, Chicago, IL 60612
Rajesh N. Kalaria, Translational and Clinical Research Institute, Newcastle University, Campus for Ageing and Vitality, Newcastle upon Tyne NE4 5PL, UK
Yuriko Katsumata, Sanders-Brown Center on Aging and Dept of Biostatistics, U.Kentucky, Lexington, KY, 40536.
Brian T. Gold, Sanders-Brown Center on Aging and Dept of Neuroscience, U.Kentucky, Lexington, KY, 40536
Danny J. J. Wang, Laboratory of FMRI Technology (LOFT), USC Mark & Mary Stevens Neuroimaging and Informatics Institute, Keck School of Medicine, University of Southern California
Samantha J. Ma, Laboratory of FMRI Technology (LOFT), USC Mark & Mary Stevens Neuroimaging and Informatics Institute, Keck School of Medicine, University of Southern California
Lincoln M. P. Shade, Sanders-Brown Center on Aging and Dept of Biostatistics, U.Kentucky, Lexington, KY, 40536
David W. Fardo, Sanders-Brown Center on Aging and Dept of Biostatistics, U.Kentucky, Lexington, KY, 40536
Anika M. S. Hartz, Sanders-Brown Center on Aging and Dept of Pharmacology and Nutritional Sciences, U.Kentucky, Lexington, KY, 40536
Gregory A. Jicha, Sanders-Brown Center on Aging and Dept of Neurology, U.Kentucky, Lexington, KY, 40536
Karin B. Nelson, National Institutes of Health, Bethesda, MD
Shino D. Magaki, Dept. of Pathology & Laboratory Medicine David Geffen SOM at UCLA & Ronald Reagan UCLA Medical Center, Los Angeles,CA 90095-1732
Frederick A. Schmitt, Sanders-Brown Center on Aging and Dept of Neurology, U.Kentucky, Lexington, KY, 40536
Merilee A. Teylan, Dept of Epidemiology, U. Washington, Seattle, WA 98105
Eseosa T. Ighodaro, Dept of Neurology, Mayo Clinic, Rochester, MN 55905
Panhavuth Phe, Sanders-Brown Center on Aging, U.Kentucky, Lexington, KY, 40536.
Erin L. Abner, Sanders-Brown Center on Aging and Dept of Epidemiology, U.Kentucky, Lexington, KY, 40536
Matthew D. Cykowski, Departments of Pathology and Genomic Medicine and Neurology, Houston Methodist Hospital, Houston, TX 77030
Linda J. Van Eldik, Sanders-Brown Center on Aging and Dept of Neuroscience, U.Kentucky, Lexington, KY, 40536
Peter T. Nelson, Sanders-Brown Center on Aging and Dept of Pathology, U.Kentucky, Lexington, KY, 40536
Literature Cited
- [1].Abbott NJ, Pizzo ME, Preston JE, Janigro D, Thorne RG (2018) The role of brain barriers in fluid movement in the CNS: is there a ‘glymphatic’ system? Acta Neuropathol 135: 387–407. 10.1007/s00401-018-1812-4 [DOI] [PubMed] [Google Scholar]
- [2].Abbott NJ, Revest PA, Romero IA (1992) Astrocyte-endothelial interaction: physiology and pathology. Neuropathol Appl Neurobiol 18: 424–433. 10.1111/j.13652990.1992.tb00808.x [DOI] [PubMed] [Google Scholar]
- [3].Abner EL, Nelson PT, Kryscio RJ, et al. (2016) Diabetes is associated with cerebrovascular but not Alzheimer neuropathology. Alzheimers Dement ePub Jan 23, Available on PubMed. 10.1016/j.jalz.2015.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Abrahamson EE, Ikonomovic MD (2020) Brain injury-induced dysfunction of the blood brain barrier as a risk for dementia. Exp Neurol 328: 113257. 10.1016/j.expneurol.2020.113257 [DOI] [PubMed] [Google Scholar]
- [5].Adib-Samii P, Devan W, Traylor M, et al. (2015) Genetic architecture of white matter hyperintensities differs in hypertensive and nonhypertensive ischemic stroke. Stroke 46: 348–353. 10.1161/STROKEAHA.114.006849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Adib-Samii P, Rost N, Traylor M, et al. (2013) 17q25 Locus is associated with white matter hyperintensity volume in ischemic stroke, but not with lacunar stroke status. Stroke 44: 1609–1615. 10.1161/STROKEAHA.113.679936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Al-Janabi OM, Brown CA, Bahrani AA, et al. (2018) Distinct white matter changes associated with cerebrospinal fluid Amyloid-beta1–42 and hypertension. J Alzheimers Dis 66: 1095–1104. 10.3233/JAD-180663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Alafuzoff I, Gelpi E, Al-Sarraj S, et al. (2012) The need to unify neuropathological assessments of vascular alterations in the ageing brain: multicentre survey by the BrainNet Europe consortium. Exp Gerontol 47: 825–833. 10.1016/j.exger.2012.06.001 [DOI] [PubMed] [Google Scholar]
- [9].Alber J, Alladi S, Bae HJ, et al. (2019) White matter hyperintensities in vascular contributions to cognitive impairment and dementia (VCID): Knowledge gaps and opportunities. Alzheimers Dement (N Y) 5: 107–117. 10.1016/j.trci.2019.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Aliev G, Priyadarshini M, Reddy VP, et al. (2014) Oxidative stress mediated mitochondrial and vascular lesions as markers in the pathogenesis of Alzheimer disease. Curr Med Chem 21: 2208–2217. 10.2174/0929867321666131227161303 [DOI] [PubMed] [Google Scholar]
- [11].Aliev G, Smith MA, Seyidov D, et al. (2002) The role of oxidative stress in the pathophysiology of cerebrovascular lesions in Alzheimer’s disease. Brain Pathol 12: 21–35. 10.1111/j.1750-3639.2002.tb00419.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Altamura C, Scrascia F, Quattrocchi CC, et al. (2016) Regional MRI diffusion, white-matter hyperintensities, and cognitive function in Alzheimer’s disease and vascular dementia. J Clin Neurol 12: 201–208. 10.3988/jcn.2016.12.2.201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Amador-Ortiz C, Lin WL, Ahmed Z, et al. (2007) TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann Neurol 61: 435–445. 10.1002/ana.21154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Arboix A, Blanco-Rojas L, Marti-Vilalta JL (2014) Advancements in understanding the mechanisms of symptomatic lacunar ischemic stroke: translation of knowledge to prevention strategies. Expert Rev Neurother 14: 261–276. 10.1586/14737175.2014.884926 [DOI] [PubMed] [Google Scholar]
- [15].Arima K, Yanagawa S, Ito N, Ikeda S (2003) Cerebral arterial pathology of CADASIL and CARASIL (Maeda syndrome). Neuropathology 23: 327–334. 10.1046/j.1440-1789.2003.00519.x [DOI] [PubMed] [Google Scholar]
- [16].Armstrong NJ, Mather KA, Sargurupremraj M, et al. (2020) Common genetic variation indicates separate causes for periventricular and deep white matter hyperintensities. Stroke STROKEAHA119027544. 10.1161/STROKEAHA.119.027544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Arnold AC, Gallagher PE, Diz DI (2013) Brain renin-angiotensin system in the nexus of hypertension and aging. Hypertens Res 36: 5–13. 10.1038/hr.2012.161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Arvanitakis Z, Capuano AW, Lamar M, et al. (2018) Late-life blood pressure association with cerebrovascular and Alzheimer disease pathology. Neurology 91: e517–e525. 10.1212/WNL.0000000000005951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Arvanitakis Z, Capuano AW, Leurgans SE, Bennett DA, Schneider JA (2016) Relation of cerebral vessel disease to Alzheimer’s disease dementia and cognitive function in elderly people: a cross-sectional study. Lancet Neurol 15: 934–943. 10.1016/S1474-4422(16)30029-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Attwell D, Mishra A, Hall CN, O’Farrell FM, Dalkara T (2016) What is a pericyte? J Cereb Blood Flow Metab 36: 451–455. 10.1177/0271678X15610340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Baik I, Seo HS, Yoon D, Kim SH, Shin C (2015) Associations of sleep apnea, NRG1 polymorphisms, alcohol consumption, and cerebral white matter hyperintensities: analysis with genome-wide association data. Sleep 38: 1137–1143. 10.5665/sleep.4830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Barnes LL, Leurgans S, Aggarwal NT, et al. (2015) Mixed pathology is more likely in black than white decedents with Alzheimer dementia. Neurology 85: 528–534. 10.1212/WNL.0000000000001834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Beckner ME (2020) A roadmap for potassium buffering/dispersion via the glial network of the CNS. Neurochem Int 136: 104727. 10.1016/j.neuint.2020.104727 [DOI] [PubMed] [Google Scholar]
- [24].Bellot-Saez A, Kekesi O, Morley JW, Buskila Y (2017) Astrocytic modulation of neuronal excitability through K(+) spatial buffering. Neurosci Biobehav Rev 77: 87–97. 10.1016/j.neubiorev.2017.03.002 [DOI] [PubMed] [Google Scholar]
- [25].Bergers G, Song S (2005) The role of pericytes in blood-vessel formation and maintenance. Neuro Oncol 7: 452–464. 10.1215/S1152851705000232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Besser LM, Kukull WA, Teylan MA, et al. (2018) The revised national alzheimer’s coordinating center’s neuropathology form-available data and new analyses. J Neuropathol Exp Neurol 77: 717–726. 10.1093/jnen/nly049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Bevan JA, Dodge J, Walters CL, Wellman T, Bevan RD (1999) As human pial arteries (internal diameter 200–1000 microm) get smaller, their wall thickness and capacity to develop tension relative to their diameter increase. Life Sci 65: 1153–1161. 10.1016/s0024-3205(99)00349-5 [DOI] [PubMed] [Google Scholar]
- [28].Borshchev YY, Uspensky YP, Galagudza MM (2019) Pathogenetic pathways of cognitive dysfunction and dementia in metabolic syndrome. Life Sci 237: 116932. 10.1016/j.lfs.2019.116932 [DOI] [PubMed] [Google Scholar]
- [29].Bourassa P, Tremblay C, Schneider JA, Bennett DA, Calon F (2020) Brain mural cell loss in the parietal cortex in Alzheimer’s disease correlates with cognitive decline and TDP-43 pathology. Neuropathol Appl Neurobiol 46(5): 458–477. 10.1111/nan.12599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Boyle PA, Yu L, Leurgans SE, et al. (2019) Attributable risk of Alzheimer’s dementia attributed to age-related neuropathologies. Ann Neurol 85: 114–124. 10.1002/ana.25380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Brenowitz WD, Nelson PT, Besser LM, Heller KB, Kukull WA (2015) Cerebral amyloid angiopathy and its co-occurrence with Alzheimer’s disease and other cerebrovascular neuropathologic changes. Neurobiol Aging 36(10): 2702–2708. 10.1016/j.neurobiolaging.2015.06.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Bridges LR, Andoh J, Lawrence AJ, et al. (2014) Blood-brain barrier dysfunction and cerebral small vessel disease (arteriolosclerosis) in brains of older people. J Neuropathol Exp Neurol 73: 1026–1033. 10.1097/NEN.0000000000000124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Buchman AS, Leurgans SE, Nag S, Bennett DA, Schneider JA (2011) Cerebrovascular disease pathology and parkinsonian signs in old age. Stroke 42: 3183–3189. 10.1161/STROKEAHA.111.623462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Buchman AS, Leurgans SE, Nag S, et al. (2017) Spinal arteriolosclerosis is common in older adults and associated with parkinsonism. Stroke 48: 2792–2798. 10.1161/STROKEAHA.117.017643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Buchman AS, Wilson RS, Shulman JM, et al. (2016) Parkinsonism in older adults and Its association with adverse health outcomes and neuropathology. J Gerontol A Biol Sci Med Sci 71: 549–556. 10.1093/gerona/glv153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Camici GG, Savarese G, Akhmedov A, Luscher TF (2015) Molecular mechanism of endothelial and vascular aging: implications for cardiovascular disease. Eur Heart J 36: 3392–3403. 10.1093/eurheartj/ehv587 [DOI] [PubMed] [Google Scholar]
- [37].Cechetto DF, Hachinski V, Whitehead SN (2008) Vascular risk factors and Alzheimer’s disease. Expert Rev Neurother 8: 743–750. 10.1586/14737175.8.5.743 [DOI] [PubMed] [Google Scholar]
- [38].Cervos-Navarro J, Gertz HJ, Frydl V (1987) Cerebral blood vessel changes in old people. Mech Ageing Dev 39: 223–231. 10.1016/0047-6374(87)90062-5 [DOI] [PubMed] [Google Scholar]
- [39].Chou SH, Shulman JM, Keenan BT, et al. (2013) Genetic susceptibility for ischemic infarction and arteriolosclerosis based on neuropathologic evaluations. Cerebrovasc Dis 36: 181–188. 10.1159/000352054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Chou SY, Shulman JM, Keenan BT, et al. (2013) Genetic susceptibility for ischemic infarction and arteriolosclerosis based on neuropathologic evaluations. Cerebrovasc Dis 36: 181–188. 10.1159/000352054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Chung J, Marini S, Pera J, et al. (2019) Genome-wide association study of cerebral small vessel disease reveals established and novel loci. Brain 142: 3176–3189. 10.1093/brain/awz233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Craft S (2006) Insulin resistance syndrome and Alzheimer disease: pathophysiologic mechanisms and therapeutic implications. Alzheimer Dis Assoc Disord 20: 298–301. 10.1097/01.wad.0000213866.86934.7e [DOI] [PubMed] [Google Scholar]
- [43].Craggs LJ, Yamamoto Y, Deramecourt V, Kalaria RN (2014) Microvascular pathology and morphometrics of sporadic and hereditary small vessel diseases of the brain. Brain Pathol 24: 495–509. 10.1111/bpa.12177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Daneman R, Prat A (2015) The blood-brain barrier. Cold Spring Harb Perspect Biol 7: a020412. 10.1101/cshperspect.a020412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Deramecourt V, Slade JY, Oakley AE, et al. (2012) Staging and natural history of cerebrovascular pathology in dementia. Neurology 78: 1043–1050. 10.1212/WNL.0b013e31824e8e7f [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Dirnagl U (2012) Pathobiology of injury after stroke: the neurovascular unit and beyond. Ann N Y Acad Sci 1268: 21–25. 10.1111/j.1749-6632.2012.06691.x [DOI] [PubMed] [Google Scholar]
- [47].Esiri MM, Wilcock GK, Morris JH (1997) Neuropathological assessment of the lesions of significance in vascular dementia. J Neurol Neurosurg Psychiatry 63: 749–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Feinkohl I, Janke J, Hadzidiakos D, et al. (2019) Associations of the metabolic syndrome and its components with cognitive impairment in older adults. BMC Geriatr 19: 77. 10.1186/s12877-019-1073-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Ferrer I (2010) Cognitive impairment of vascular origin: neuropathology of cognitive impairment of vascular origin. J Neurol Sci 299: 139–149. 10.1016/j.jns.2010.08.039 [DOI] [PubMed] [Google Scholar]
- [50].Ferrer I, Bella R, Serrano MT, Marti E, Guionnet N (1990) Arteriolosclerotic leucoencephalopathy in the elderly and its relation to white matter lesions in Binswanger’s disease, multi-infarct encephalopathy and Alzheimer’s disease. J Neurol Sci 98: 37–50. 10.1016/0022-510x(90)90180-u [DOI] [PubMed] [Google Scholar]
- [51].Fishbein GA, Fishbein MC (2009) Arteriosclerosis: rethinking the current classification. Arch Pathol Lab Med 133: 1309–1316. 10.1043/1543-2165-133.8.1309 [DOI] [PubMed] [Google Scholar]
- [52].Fishbein MC, Fishbein GA (2015) Arteriosclerosis: facts and fancy. Cardiovasc Pathol 24: 335–342. 10.1016/j.carpath.2015.07.007 [DOI] [PubMed] [Google Scholar]
- [53].Flanagan M, Larson EB, Latimer CS, et al. (2016) Clinical-pathologic correlations in vascular cognitive impairment and dementia. Biochim Biophys Acta 1862: 945–951. 10.1016/j.bbadis.2015.08.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Fornage M, Debette S, Bis JC, et al. (2011) Genome-wide association studies of cerebral white matter lesion burden: the CHARGE consortium. Ann Neurol 69: 928–939. 10.1002/ana.22403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Francis F, Ballerini L, Wardlaw JM (2019) Perivascular spaces and their associations with risk factors, clinical disorders and neuroimaging features: A systematic review and meta-analysis. Int J Stroke 14: 359–371. 10.1177/1747493019830321 [DOI] [PubMed] [Google Scholar]
- [56].French CR, Seshadri S, Destefano AL, et al. (2014) Mutation of FOXC1 and PITX2 induces cerebral small-vessel disease. J Clin Invest 124: 4877–4881. 10.1172/JCI75109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Frey BM, Petersen M, Mayer C, et al. (2019) Characterization of white matter hyperintensities in large-scale MRI-studies. Front Neurol 10: 238. 10.3389/fneur.2019.00238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Frosen J, Joutel A (2018) Smooth muscle cells of intracranial vessels: from development to disease. Cardiovasc Res 114: 501–512. 10.1093/cvr/cvy002 [DOI] [PubMed] [Google Scholar]
- [59].Furie KL, Smith EE (2007) Metabolic syndrome: a target for preventing leukoaraiosis and age-related dementia? Neurology 69: 951–952. 10.1212/01.wnl.0000271094.43789.0e [DOI] [PubMed] [Google Scholar]
- [60].Gallart-Palau X, Tan LM, Serra A, et al. (2019) Degenerative protein modifications in the aging vasculature and central nervous system: A problem shared is not always halved. Ageing Res Rev 53: 100909. 10.1016/j.arr.2019.100909 [DOI] [PubMed] [Google Scholar]
- [61].Gangwar RS, Bevan GH, Palanivel R, Das L, Rajagopalan S (2020) Oxidative stress pathways of air pollution mediated toxicity: Recent insights. Redox Biol 101545. 10.1016/j.redox.2020.101545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Garland P, Morton MJ, Haskins W, et al. (2020) Haemoglobin causes neuronal damage in vivo which is preventable by haptoglobin. Brain Commun 2: fcz053. 10.1093/braincomms/fcz053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Goetz CG, Tilley BC, Shaftman SR, et al. (2008) Movement Disorder Society-sponsored revision of the Unified Parkinson’s Disease Rating Scale (MDS-UPDRS): scale presentation and clinimetric testing results. Mov Disord 23: 2129–2170. 10.1002/mds.22340 [DOI] [PubMed] [Google Scholar]
- [64].Grinberg LT, Amaro E Jr., Teipel S, et al. (2008) Assessment of factors that confound MRI and neuropathological correlation of human postmortem brain tissue. Cell Tissue Bank 9: 195–203. 10.1007/s10561-008-9080-5 [DOI] [PubMed] [Google Scholar]
- [65].Grinberg LT, Heinsen H (2010) Toward a pathological definition of vascular dementia. J Neurol Sci 299: 136–138. 10.1016/j.jns.2010.08.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Grinberg LT, Thal DR (2010) Vascular pathology in the aged human brain. Acta Neuropathol 119: 277–290. 10.1007/s00401-010-0652-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Group SMIftSR, Nasrallah IM, Pajewski NM, et al. (2019) Association of intensive vs standard blood pressure control with cerebral white matter lesions. JAMA 322: 524–534. 10.1001/jama.2019.10551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Group SMIftSR, Williamson JD, Pajewski NM, et al. (2019) Effect of intensive vs standard blood pressure control on probable dementia: A randomized clinical trial. JAMA 321: 553–561. 10.1001/jama.2018.21442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Grubb S, Cai C, Hald BO, et al. (2020) Precapillary sphincters maintain perfusion in the cerebral cortex. Nat Commun 11: 395. 10.1038/s41467-020-14330-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Gull WW, Sutton HG (1872) On the pathology of the morbid state commonly called chronic Bright’s Disease with contracted kidney, (“Arterio-capillary fibrosis.”). Med Chir Trans 55: 273–330 271. 10.1177/095952877205500116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Gurol ME, Biessels GJ, Polimeni JR (2020) Advanced Neuroimaging to Unravel Mechanisms of Cerebral Small Vessel Diseases. Stroke 51: 29–37. 10.1161/STROKEAHA.119.024149 [DOI] [PubMed] [Google Scholar]
- [72].Hainsworth AH, Allan SM, Boltze J, et al. (2017) Translational models for vascular cognitive impairment: a review including larger species. BMC Med 15: 16. 10.1186/s12916-017-0793-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Hainsworth AH, Oommen AT, Bridges LR (2015) Endothelial cells and human cerebral small vessel disease. Brain Pathol 25: 44–50. 10.1111/bpa.12224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Harris R, Miners JS, Allen S, Love S (2018) VEGFR1 and VEGFR2 in Alzheimer’s Disease. J Alzheimers Dis 61: 741–752. 10.3233/JAD-170745 [DOI] [PubMed] [Google Scholar]
- [75].Haruwaka K, Ikegami A, Tachibana Y, et al. (2019) Dual microglia effects on blood brain barrier permeability induced by systemic inflammation. Nat Commun 10: 5816. 10.1038/s41467-019-13812-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Hase Y, Polvikoski TM, Firbank MJ, et al. (2020) Small vessel disease pathological changes in neurodegenerative and vascular dementias concomitant with autonomic dysfunction. Brain Pathol 30: 191–202. 10.1111/bpa.12769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Hatate J, Miwa K, Matsumoto M, et al. (2016) Association between cerebral small vessel diseases and mild parkinsonian signs in the elderly with vascular risk factors. Parkinsonism Relat Disord 26: 29–34. 10.1016/j.parkreldis.2016.02.011 [DOI] [PubMed] [Google Scholar]
- [78].Hayashi M, Miwa-Saito N, Tanuma N, Kubota M (2012) Brain vascular changes in Cockayne syndrome. Neuropathology 32: 113–117. 10.1111/j.14401789.2011.01241.x [DOI] [PubMed] [Google Scholar]
- [79].Herrmann SM, Textor SC (2019) Renovascular Hypertension. Endocrinol Metab Clin North Am 48: 765–778. 10.1016/j.ecl.2019.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Hill RA, Tong L, Yuan P, et al. (2015) Regional blood flow in the normal and ischemic brain Is controlled by arteriolar smooth muscle cell contractility and not by capillary pericytes. Neuron 87: 95–110. 10.1016/j.neuron.2015.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Huang YH, Zhang WW, Lin L, et al. (2010) Could changes in arterioles impede the perivascular drainage of interstitial fluid from the cerebral white matter in leukoaraiosis? Neuropathol Appl Neurobiol 36: 237–247. 10.1111/j.13652990.2009.01049.x [DOI] [PubMed] [Google Scholar]
- [82].Huther G, Dorfl J, Van der Loos H, Jeanmonod D (1998) Microanatomic and vascular aspects of the temporomesial region. Neurosurgery 43: 1118–1136. [DOI] [PubMed] [Google Scholar]
- [83].Iadecola C (2017) The neurovascular unit coming of age: A journey through neurovascular coupling in health and disease. Neuron 96: 17–42. 10.1016/j.neuron.2017.07.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Ighodaro ET, Abner EL, Fardo DW, et al. (2017) Risk factors and global cognitive status related to brain arteriolosclerosis in elderly individuals. J Cereb Blood Flow Metab 37: 201–216. 10.1177/0271678X15621574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Ighodaro ET, Nelson PT, Kukull WA, et al. (2017) Challenges and considerations related to studying dementia in Blacks/African Americans. J Alzheimers Dis 60: 1–10. 10.3233/JAD-170242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Imfeld P, Bodmer M, Schuerch M, Jick SS, Meier CR (2013) Seizures in patients with Alzheimer’s disease or vascular dementia: a population-based nested case-control analysis. Epilepsia 54: 700–707. 10.1111/epi.12045 [DOI] [PubMed] [Google Scholar]
- [87].Iulita MF, Noriega de la Colina A, Girouard H (2018) Arterial stiffness, cognitive impairment and dementia: confounding factor or real risk? J Neurochem 144: 527–548. 10.1111/jnc.14235 [DOI] [PubMed] [Google Scholar]
- [88].Janaway BM, Simpson JE, Hoggard N, et al. (2014) Brain haemosiderin in older people: pathological evidence for an ischaemic origin of magnetic resonance imaging (MRI) microbleeds. Neuropathol Appl Neurobiol 40: 258–269. 10.1111/nan.12062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Jellinger KA, Attems J (2010) Prevalence of dementia disorders in the oldest-old: an autopsy study. Acta Neuropathol 119: 421–433. 10.1007/s00401-0100654-5 [DOI] [PubMed] [Google Scholar]
- [90].Josephs KA, Murray ME, Whitwell JL, et al. (2014) Staging TDP-43 pathology in Alzheimer’s disease. Acta Neuropathol 127: 441–450. 10.1007/s00401013-1211-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Joutel A, Corpechot C, Ducros A, et al. (1996) Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature 383: 707–710. 10.1038/383707a0 [DOI] [PubMed] [Google Scholar]
- [92].Joutel A, Haddad I, Ratelade J, Nelson MT (2016) Perturbations of the cerebrovascular matrisome: A convergent mechanism in small vessel disease of the brain? J Cereb Blood Flow Metab 36: 143–157. 10.1038/jcbfm.2015.62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Kalaria RN (2016) Neuropathological diagnosis of vascular cognitive impairment and vascular dementia with implications for Alzheimer’s disease. Acta Neuropathol 131: 659–685. 10.1007/s00401-016-1571-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Kalaria RN, Ferrer I, Love S, Vascular disease, hypoxia, and related conditions. In: Love S, Perry A, Ironside JW, Budka H. (Eds.), Greenfield’s Neuropathology, CRC Press, 2015. [Google Scholar]
- [95].Kalaria RN, Hase Y (2019) Neurovascular ageing and age-related diseases. Subcell Biochem 91: 477–499. 10.1007/978-981-13-3681-2_17 [DOI] [PubMed] [Google Scholar]
- [96].Kalaria RN, Kenny RA, Ballard CG, et al. (2004) Towards defining the neuropathological substrates of vascular dementia. J Neurol Sci 226: 75–80. 10.1016/j.jns.2004.09.019 [DOI] [PubMed] [Google Scholar]
- [97].Kalmijn S, Foley D, White L, et al. (2000) Metabolic cardiovascular syndrome and risk of dementia in Japanese-American elderly men. The Honolulu-Asia aging study. Arterioscler Thromb Vasc Biol 20: 2255–2260. [DOI] [PubMed] [Google Scholar]
- [98].Kamp JA, Moursel LG, Haan J, et al. (2014) Amyloid beta in hereditary cerebral hemorrhage with amyloidosis-Dutch type. Rev Neurosci 25: 641–651. 10.1515/revneuro-2014-0008 [DOI] [PubMed] [Google Scholar]
- [99].Kang CK, Park CA, Lee H, et al. (2009) Hypertension correlates with lenticulostriate arteries visualized by 7T magnetic resonance angiography. Hypertension 54: 1050–1056. 10.1161/HYPERTENSIONAHA.109.140350 [DOI] [PubMed] [Google Scholar]
- [100].Kapasi A, DeCarli C, Schneider JA (2017) Impact of multiple pathologies on the threshold for clinically overt dementia. Acta Neuropathol 134: 171–186. 10.1007/s00401-017-1717-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Katsumata Y, Fardo DW, Kukull WA, Nelson PT (2018) Dichotomous scoring of TDP-43 proteinopathy from specific brain regions in 27 academic research centers: associations with Alzheimer’s disease and cerebrovascular disease pathologies. Acta Neuropathol Commun 6: 142. 10.1186/s40478-018-0641-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Kim JB, Prunicki M, Haddad F, et al. (2020) Cumulative lifetime burden ofcardiovascular disease from early exposure to air pollution. J Am Heart Assoc 9: e014944. 10.1161/JAHA.119.014944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Kim KJ, Ramiro Diaz J, Iddings JA, Filosa JA (2016) Vasculo-neuronal coupling: retrograde vascular communication to brain neurons. J Neurosci 36: 12624–12639. 10.1523/JNEUROSCI.1300-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Kim SH, Shin DW, Yun JM, et al. (2017) Kidney dysfunction and cerebral microbleeds in neurologically healthy adults. PLoS One 12: e0172210. 10.1371/journal.pone.0172210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Kisler K, Nelson AR, Montagne A, Zlokovic BV (2017) Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat Rev Neurosci 18: 419–434. 10.1038/nrn.2017.48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Koehler RC, Roman RJ, Harder DR (2009) Astrocytes and the regulation of cerebral blood flow. Trends Neurosci 32: 160–169. 10.1016/j.tins.2008.11.005 [DOI] [PubMed] [Google Scholar]
- [107].Koizumi T, Kerkhofs D, Mizuno T, Steinbusch HWM, Foulquier S (2019) Vessel-associated immune cells in cerebrovascular diseases: From perivascular macrophages to vessel-associated microglia. Front Neurosci 13: 1291. 10.3389/fnins.2019.01291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Kolar GR, Kothari PH, Khanlou N, et al. (2014) Neuropathology and genetics of cerebroretinal vasculopathies. Brain Pathol 24: 510–518. 10.1111/bpa.12178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Lam DW, LeRoith D, Metabolic Syndrome. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, Dungan K, Grossman A, Hershman JM, Kaltsas G, Koch C, Kopp P, Korbonits M, McLachlan R, Morley JE, New M, Perreault L, Purnell J, Rebar R, Singer F, Trence DL, Vinik A, Wilson DP (Eds.), Endotext, South Dartmouth (MA), 2000. [Google Scholar]
- [110].Lammie GA (2002) Hypertensive cerebral small vessel disease and stroke. Brain Pathol 12: 358–370. 10.1111/j.1750-3639.2002.tb00450.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Lammie GA, Brannan F, Slattery J, Warlow C (1997) Nonhypertensive cerebral small-vessel disease. An autopsy study. Stroke 28: 2222–2229. 10.1161/01.str.28.11.2222 [DOI] [PubMed] [Google Scholar]
- [112].Lee RM (1995) Morphology of cerebral arteries. Pharmacol Ther 66: 149–173. 10.1016/0163-7258(94)00071-a [DOI] [PubMed] [Google Scholar]
- [113].Lee SJ, Blanchett-Anderson S, Keep SG, Gasche MB, Wang MM (2020) Tripartite factors leading to molecular divergence between human and murine smooth muscle. PLoS One 15: e0227672. 10.1371/journal.pone.0227672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Lim AS, Yu L, Schneider JA, Bennett DA, Buchman AS (2016) Sleep fragmentation, cerebral arteriolosclerosis, and brain infarct pathology in community-dwelling older people. Stroke 47: 516–518. 10.1161/STROKEAHA.115.011608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Lin Q, Huang WQ, Tzeng CM (2015) Genetic associations of leukoaraiosis indicate pathophysiological mechanisms in white matter lesions etiology. Rev Neurosci 26: 343–358. 10.1515/revneuro-2014-0082 [DOI] [PubMed] [Google Scholar]
- [116].Lin WL, Castanedes-Casey M, Dickson DW (2009) Transactivation response DNA-binding protein 43 microvasculopathy in frontotemporal degeneration and familial Lewy body disease. J Neuropathol Exp Neurol 68: 1167–1176. 10.1097/NEN.0b013e3181baacec [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Lobstein JF (1833) Traite d’anatomie pathologique. Levrault. [Google Scholar]
- [118].Long DL, Howard G, Long DM, et al. (2019) An investigation of selection bias in estimating racial disparity in stroke risk factors. Am J Epidemiol 188: 587–597. 10.1093/aje/kwy253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Love S, Miners JS (2016) Cerebrovascular disease in ageing and Alzheimer’s disease. Acta Neuropathol 131: 645–658. 10.1007/s00401-015-1522-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Love S, Miners JS (2017) Small vessel disease, neurovascular regulation and cognitive impairment: post-mortem studies reveal a complex relationship, still poorly understood. Clin Sci (Lond) 131: 1579–1589. 10.1042/CS20170148 [DOI] [PubMed] [Google Scholar]
- [121].Lozic M, Sarenac O, Murphy D, Japundzic-Zigon N (2018) Vasopressin, central autonomic control and blood pressure regulation. Curr Hypertens Rep 20: 11. 10.1007/s11906-018-0811-0 [DOI] [PubMed] [Google Scholar]
- [122].Ma SJ, Sarabi MS, Yan L, et al. (2019) Characterization of lenticulostriate arteries with high resolution black-blood T1-weighted turbo spin echo with variable flip angles at 3 and 7Tesla. Neuroimage 199: 184–193. 10.1016/j.neuroimage.2019.05.065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Magaki S, Yong WH, Khanlou N, Tung S, Vinters HV (2014) Comorbidity in dementia: update of an ongoing autopsy study. J Am Geriatr Soc 62: 1722–1728. 10.1111/jgs.12977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Malik R, Chauhan G, Traylor M, et al. (2018) Multiancestry genome-wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat Genet 50: 524–537. 10.1038/s41588-018-0058-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Marini S, Anderson CD, Rosand J (2020) Genetics of cerebral small vessel disease. Stroke 51: 12–20. 10.1161/STROKEAHA.119.024151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Marini S, Georgakis MK, Chung J, et al. (2020) Genetic overlap and causal inferences between kidney function and cerebrovascular disease. Neurology. 10.1212/WNL.0000000000009642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Martinez-Lemus LA (2012) The dynamic structure of arterioles. Basic Clin Pharmacol Toxicol 110: 5–11. 10.1111/j.1742-7843.2011.00813.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].McAleese KE, Alafuzoff I, Charidimou A, et al. (2016) Post-mortem assessment in vascular dementia: advances and aspirations. BMC Med 14: 129. 10.1186/s12916-016-0676-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].McAleese KE, Graham S, Dey M, et al. (2019) Extravascular fibrinogen in the white matter of Alzheimer’s disease and normal aged brains: implications for fibrinogen as a biomarker for Alzheimer’s disease. Brain Pathol 29: 414–424. 10.1111/bpa.12685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].McAuley G, Schrag M, Barnes S, et al. (2011) Iron quantification of microbleeds in postmortem brain. Magn Reson Med 65: 1592–1601. 10.1002/mrm.22745 [DOI] [PubMed] [Google Scholar]
- [131].McNeal DW, Brandner DD, Gong X, et al. (2016) Unbiased stereological analysis of reactive astrogliosis to estimate age-associated cerebral white matter injury. J Neuropathol Exp Neurol 75: 539–554. 10.1093/jnen/nlw032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Meng N, Mu X, Lv X, et al. (2019) Autophagy represses fascaplysin-induced apoptosiss and angiogenesis inhibition via ROS and p8 in vascular endothelia cells. Biomed Pharmacother 114: 108866. 10.1016/j.biopha.2019.108866 [DOI] [PubMed] [Google Scholar]
- [133].Mestre H, Mori Y, Nedergaard M (2020) The brain’s glymphatic system: current controversies. Trends Neurosci 43 (7): 458–466. 10.1016/j.tins.2020.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Meuwissen ME, Halley DJ, Smit LS, et al. (2015) The expanding phenotype of COL4A1 and COL4A2 mutations: clinical data on 13 newly identified families and a review of the literature. Genet Med 17: 843–853. 10.1038/gim.2014.210 [DOI] [PubMed] [Google Scholar]
- [135].Miao Q, Paloneva T, Tuominen S, et al. (2004) Fibrosis and stenosis of the long penetrating cerebral arteries: the cause of the white matter pathology in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Brain Pathol 14: 358–364. 10.1111/j.1750-3639.2004.tb00078.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Mills KT, Stefanescu A, He J (2020) The global epidemiology of hypertension. Nat Rev Nephrol 16: 223–237. 10.1038/s41581-019-0244-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Miners JS, Palmer JC, Love S (2016) Pathophysiology of hypoperfusion of the precuneus in early Alzheimer’s disease. Brain Pathol 26: 533–541. 10.1111/bpa.12331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Mishra A, Chauhan G, Violleau MH, et al. (2019) Association of variants in HTRA1 and NOTCH3 with MRI-defined extremes of cerebral small vessel disease in older subjects. Brain 142: 1009–1023. 10.1093/brain/awz024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Mishra A, Reynolds JP, Chen Y, et al. (2016) Astrocytes mediate neurovascular signaling to capillary pericytes but not to arterioles. Nat Neurosci 19: 1619–1627. 10.1038/nn.4428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Mistriotis P, Andreadis ST (2017) Vascular aging: Molecular mechanisms and potential treatments for vascular rejuvenation. Ageing Res Rev 37: 94–116. 10.1016/j.arr.2017.05.006 [DOI] [PubMed] [Google Scholar]
- [141].Mock C, Teylan M, Beecham G, et al. (2020) The utility of the National Alzheimer’s Coordinating Center’s Database for the rapid assessment of evolving neuropathologic conditions. Alzheimer Dis Assoc Disord 34: 105–111. 10.1097/WAD.0000000000000380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Montagne A, Nation DA, Sagare AP, et al. (2020) APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline. Nature 581: 71–76. 10.1038/s41586-020-2247-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Montine TJ, Phelps CH, Beach TG, et al. (2012) National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol 123: 1–11. 10.1007/s00401-0110910-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Moretti R, Caruso P (2020) Small vessel disease-related dementia: An invalid neurovascular coupling? Int J Mol Sci 21. 10.3390/ijms21031095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Moritz AR, Oldt MR (1937) Arteriolar sclerosis in hypertensive and non-hypertensive individuals. Am J Pathol 13: 679–728 677. [PMC free article] [PubMed] [Google Scholar]
- [146].Nag S, Kilty DW (1997) Cerebrovascular changes in chronic hypertension. Protective effects of enalapril in rats. Stroke 28: 1028–1034. 10.1161/01.str.28.5.1028 [DOI] [PubMed] [Google Scholar]
- [147].Nag S, Yu L, Boyle PA, et al. (2018) TDP-43 pathology in anterior temporal pole cortex in aging and Alzheimer’s disease. Acta Neuropathol Commun 6: 33. 10.1186/s40478-018-0531-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Nakano T, Munakata A, Shimaura N, Asano K, Ohkuma H (2012) Augmentation index is related to white matter lesions. Hypertens Res 35: 729–732. 10.1038/hr.2012.24 [DOI] [PubMed] [Google Scholar]
- [149].Nelson PT, Abner EL, Patel E, et al. (2018) The amygdala as a locus of pathologic misfolding in neurodegenerative diseases. J Neuropathol Exp Neurol 77: 2–20. 10.1093/jnen/nlx099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Nelson PT, Dickson DW, Trojanowski JQ, et al. (2019) Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain. 10.1093/brain/awz099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Nelson PT, Head E, Schmitt FA, et al. (2011) Alzheimer’s disease is not “brain aging”: neuropathological, genetic, and epidemiological human studies. Acta Neuropathol 121: 571–587. 10.1007/s00401-011-0826-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Nelson PT, Jicha GA, Wang WX, et al. (2015) ABCC9/SUR2 in the brain: Implications for hippocampal sclerosis of aging and a potential therapeutic target. Ageing Res Rev 24: 111–125. 10.1016/j.arr.2015.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Nelson PT, Smith CD, Abner EA, et al. (2009) Human cerebral neuropathology of Type 2 diabetes mellitus. Biochim Biophys Acta 1792: 454–469. 10.1016/j.bbadis.2008.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Neltner JH, Abner EL, Baker S, et al. (2014) Arteriolosclerosis that affects multiple brain regions is linked to hippocampal sclerosis of ageing. Brain 137: 255–267. 10.1093/brain/awt318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Neltner JH, Abner EL, Jicha GA, et al. (2016) Brain pathologies in extreme old age. Neurobiol Aging 37: 1–11. 10.1016/j.neurobiolaging.2015.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Niiya Y, Abumiya T, Shichinohe H, et al. (2006) Susceptibility of brain microvascular endothelial cells to advanced glycation end products-induced tissue factor upregulation is associated with intracellular reactive oxygen species. Brain Res 1108: 179–187. 10.1016/j.brainres.2006.06.038 [DOI] [PubMed] [Google Scholar]
- [157].Norris CM, Kadish I, Blalock EM, et al. (2005) Calcineurin triggers reactive/inflammatory processes in astrocytes and is upregulated in aging and Alzheimer’s models. J Neurosci 25: 4649–4658. 10.1523/JNEUROSCI.0365-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Okeda R (2003) Pathological changes in the cerebral medullary arteries of five autopsy cases of malignant nephrosclerosis: observation by morphometry and reconstruction of serial sections. Neuropathology 23: 153–160. 10.1046/j.14401789.2003.00492.x [DOI] [PubMed] [Google Scholar]
- [159].Okeda R, Murayama S, Sawabe M, Kuroiwa T (2004) Pathology of the cerebral artery in Binswanger’s disease in the aged: observation by serial sections and morphometry of the cerebral arteries. Neuropathology 24: 21–29. 10.1111/j.14401789.2003.00534.x [DOI] [PubMed] [Google Scholar]
- [160].Ovbiagele B, Saver JL (2006) Cerebral white matter hyperintensities on MRI: Current concepts and therapeutic implications. Cerebrovasc Dis 22: 83–90. 10.1159/000093235 [DOI] [PubMed] [Google Scholar]
- [161].Oveisgharan S, Arvanitakis Z, Yu L, et al. (2018) Sex differences in Alzheimer’s disease and common neuropathologies of aging. Acta Neuropathol 136: 887–900. 10.1007/s00401-018-1920-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Ozen I, Deierborg T, Miharada K, et al. (2014) Brain pericytes acquire a microglial phenotype after stroke. Acta Neuropathol 128: 381–396. 10.1007/s00401014-1295-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Pasi M, Rocha E, Samore W, et al. (2020) Premature vascular disease in young adult stroke: a pathology-based case series. J Neurol 267: 1063–1069. 10.1007/s00415-019-09623-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Paternoster L, Chen W, Sudlow CL (2009) Genetic determinants of white matter hyperintensities on brain scans: a systematic assessment of 19 candidate gene polymorphisms in 46 studies in 19,000 subjects. Stroke 40: 2020–2026. 10.1161/STROKEAHA.108.542050 [DOI] [PubMed] [Google Scholar]
- [165].Petito CK, Levy DE (1980) The importance of cerebral arterioles in alterations of the blood-brain barrier. Lab Invest 43: 262–268. [PubMed] [Google Scholar]
- [166].Popa-Wagner A, Buga AM, Popescu B, Muresanu D (2015) Vascular cognitive impairment, dementia, aging and energy demand. A vicious cycle. J Neural Transm (Vienna) 122 Suppl 1: S47–54. 10.1007/s00702-013-1129-3 [DOI] [PubMed] [Google Scholar]
- [167].Popescu BO, Toescu EC, Popescu LM, et al. (2009) Blood-brain barrier alterations in ageing and dementia. J Neurol Sci 283: 99–106. 10.1016/j.jns.2009.02.321 [DOI] [PubMed] [Google Scholar]
- [168].Price BR, Norris CM, Sompol P, Wilcock DM (2018) An emerging role of astrocytes in vascular contributions to cognitive impairment and dementia. J Neurochem 144: 644–650. 10.1111/jnc.14273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Rannikmae K, Sivakumaran V, Millar H, et al. (2017) COL4A2 is associated with lacunar ischemic stroke and deep ICH: Meta-analyses among 21,500 cases and 40,600 controls. Neurology 89: 1829–1839. 10.1212/WNL.0000000000004560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Reitz C, Patel B, Tang MX, et al. (2007) Relation between vascular risk factors and neuropsychological test performance among elderly persons with Alzheimer’s disease. J Neurol Sci 257: 194–201. 10.1016/j.jns.2007.01.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Richards A, van den Maagdenberg AM, Jen JC, et al. (2007) C-terminal truncations in human 3’−5’ DNA exonuclease TREX1 cause autosomal dominant retinal vasculopathy with cerebral leukodystrophy. Nat Genet 39: 1068–1070. 10.1038/ng2082 [DOI] [PubMed] [Google Scholar]
- [172].Richardson K, Stephan BC, Ince PG, et al. (2012) The neuropathology of vascular disease in the Medical Research Council Cognitive Function and Ageing Study (MRC CFAS). Curr Alzheimer Res 9: 687–696. [DOI] [PubMed] [Google Scholar]
- [173].Rosenberg GA (2017) Extracellular matrix inflammation in vascular cognitive impairment and dementia. Clin Sci (Lond) 131: 425–437. 10.1042/CS20160604 [DOI] [PubMed] [Google Scholar]
- [174].Rosman NP, Anselm I, Bhadelia RA (2001) Progressive intracranial vascular disease with strokes and seizures in a boy with progeria. J Child Neurol 16: 212–215. 10.1177/088307380101600309 [DOI] [PubMed] [Google Scholar]
- [175].Rouhl RP, Damoiseaux JG, Lodder J, et al. (2012) Vascular inflammation in cerebral small vessel disease. Neurobiol Aging 33: 1800–1806. 10.1016/j.neurobiolaging.2011.04.008 [DOI] [PubMed] [Google Scholar]
- [176].Saito R, Nozaki H, Kato T, et al. (2019) Retinal vasculopathy with cerebral leukodystrophy: Clinicopathologic features of an autopsied patient with a heterozygous TREX 1 mutation. J Neuropathol Exp Neurol 78: 181–186. 10.1093/jnen/nly115 [DOI] [PubMed] [Google Scholar]
- [177].Saklayen MG (2018) The global epidemic of the metabolic syndrome. Curr Hypertens Rep 20: 12. 10.1007/s11906-018-0812-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Sartorius T, Peter A, Heni M, et al. (2015) The brain response to peripheral insulin declines with age: a contribution of the blood-brain barrier? PLoS One 10: e0126804. 10.1371/journal.pone.0126804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Schlesinger D, Grinberg LT, Alba JG, et al. (2013) African ancestry protects against Alzheimer’s disease-related neuropathology. Mol Psychiatry 18: 79–85. 10.1038/mp.2011.136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Shindo A, Ishikawa H, Ii Y, Niwa A, Tomimoto H (2020) Clinical features and experimental models of cerebral small vessel disease. Front Aging Neurosci 12: 109. 10.3389/fnagi.2020.00109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Silverman A, Petersen NH, Physiology, Cerebral Autoregulation. StatPearls, Treasure Island (FL), 2020. [PubMed] [Google Scholar]
- [182].Simard M, Arcuino G, Takano T, Liu QS, Nedergaard M (2003) Signaling at the gliovascular interface. J Neurosci 23: 9254–9262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Skrobot OA, Attems J, Esiri M, et al. (2016) Vascular cognitive impairment neuropathology guidelines (VCING): the contribution of cerebrovascular pathology to cognitive impairment. Brain 139: 2957–2969. 10.1093/brain/aww214 [DOI] [PubMed] [Google Scholar]
- [184].Smallwood A, Oulhaj A, Joachim C, et al. (2012) Cerebral subcortical small vessel disease and its relation to cognition in elderly subjects: a pathological study in the Oxford Project to Investigate Memory and Ageing (OPTIMA) cohort. Neuropathol Appl Neurobiol 38: 337–343. 10.1111/j.1365-2990.2011.01221.x [DOI] [PubMed] [Google Scholar]
- [185].Smith AS, Wiznitzer M, Karaman BA, Horwitz SJ, Lanzieri CF (1993) MRA detection of vascular occlusion in a child with progeria. AJNR Am J Neuroradiol 14: 441–443. [PMC free article] [PubMed] [Google Scholar]
- [186].Smith EE, O’Donnell M, Dagenais G, et al. (2015) Early cerebral small vessel disease and brain volume, cognition, and gait. Ann Neurol 77: 251–261. 10.1002/ana.24320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Snyder HM, Corriveau RA, Craft S, et al. (2015) Vascular contributions to cognitive impairment and dementia including Alzheimer’s disease. Alzheimers Dement 11: 710717. 10.1016/j.jalz.2014.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Sonnen JA, Larson EB, Haneuse S, et al. (2009) Neuropathology in the adult changes in thought study: a review. J Alzheimers Dis 18: 703–711. 10.3233/JAD-2009-1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Spence JD (2019) Blood pressure gradients in the brain: Their importance to understanding pathogenesis of cerebral small vessel disease. Brain Sci 9. 10.3390/brainsci9020021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Staiculescu MC, Foote C, Meininger GA, Martinez-Lemus LA (2014) The role of reactive oxygen species in microvascular remodeling. Int J Mol Sci 15: 23792–23835. 10.3390/ijms151223792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Sudduth TL, Powell DK, Smith CD, Greenstein A, Wilcock DM (2013) Induction of hyperhomocysteinemia models vascular dementia by induction of cerebral microhemorrhages and neuroinflammation. J Cereb Blood Flow Metab 33: 708–715. 10.1038/jcbfm.2013.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Sudduth TL, Weekman EM, Price BR, et al. (2017) Time-course of glial changes in the hyperhomocysteinemia model of vascular cognitive impairment and dementia (VCID). Neuroscience 341: 42–51. 10.1016/j.neuroscience.2016.11.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Suemoto CK, Ferretti-Rebustini RE, Rodriguez RD, et al. (2017) Neuropathological diagnoses and clinical correlates in older adults in Brazil: A cross-sectional study. PLoS Med 14: e1002267. 10.1371/journal.pmed.1002267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Suemoto CK, Leite REP, Ferretti-Rebustini REL, et al. (2019) Neuropathological lesions in the very old: results from a large Brazilian autopsy study. Brain Pathol 29: 771–781. 10.1111/bpa.12719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Sweeney MD, Ayyadurai S, Zlokovic BV (2016) Pericytes of the neurovascular unit: key functions and signaling pathways. Nat Neurosci 19: 771–783. 10.1038/nn.4288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Tamura Y, Araki A (2015) Diabetes mellitus and white matter hyperintensity. Geriatr Gerontol Int 15 Suppl 1: 34–42. 10.1111/ggi.12666 [DOI] [PubMed] [Google Scholar]
- [197].Tanoi Y, Okeda R, Budka H (2000) Binswanger’s encephalopathy: serial sections and morphometry of the cerebral arteries. Acta Neuropathol 100: 347–355. 10.1007/s004010000203 [DOI] [PubMed] [Google Scholar]
- [198].Tesauro M, Mauriello A, Rovella V, et al. (2017) Arterial ageing: from endothelial dysfunction to vascular calcification. J Intern Med 281: 471–482. 10.1111/joim.12605 [DOI] [PubMed] [Google Scholar]
- [199].Thal DR, Ghebremedhin E, Orantes M, Wiestler OD (2003) Vascular pathology in Alzheimer disease: correlation of cerebral amyloid angiopathy and arteriosclerosis/lipohyalinosis with cognitive decline. J Neuropathol Exp Neurol 62: 1287–1301. [DOI] [PubMed] [Google Scholar]
- [200].Thal DR, Grinberg LT, Attems J (2012) Vascular dementia: different forms of vessel disorders contribute to the development of dementia in the elderly brain. Exp Gerontol 47: 816–824. 10.1016/j.exger.2012.05.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Tomimoto H, Akiguchi I, Wakita H, et al. (1999) Coagulation activation in patients with Binswanger disease. Arch Neurol 56: 1104–1108. 10.1001/archneur.56.9.1104 [DOI] [PubMed] [Google Scholar]
- [202].Traylor M, Malik R, Nalls MA, et al. (2017) Genetic variation at 16q24.2 is associated with small vessel stroke. Ann Neurol 81: 383–394. 10.1002/ana.24840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Traylor M, Tozer DJ, Croall ID, et al. (2019) Genetic variation in PLEKHG1 is associated with white matter hyperintensities (n = 11,226). Neurology 92: e749–e757. 10.1212/WNL.0000000000006952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Traylor M, Zhang CR, Adib-Samii P, et al. (2016) Genome-wide meta-analysis of cerebral white matter hyperintensities in patients with stroke. Neurology 86: 146–153. 10.1212/WNL.0000000000002263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Uemura MT, Ihara M, Maki T, et al. (2018) Pericyte-derived bone morphogenetic protein 4 underlies white matter damage after chronic hypoperfusion. Brain Pathol 28: 521–535. 10.1111/bpa.12523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Uemura MT, Maki T, Ihara M, Lee VMY, Trojanowski JQ (2020) Brain microvascular pericytes in vascular cognitive impairment and dementia. Front Aging Neurosci 12: 80. 10.3389/fnagi.2020.00080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Ungvari Z, Tarantini S, Donato AJ, Galvan V, Csiszar A (2018) Mechanisms of vascular aging. Circ Res 123: 849–867. 10.1161/CIRCRESAHA.118.311378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Utz SG, See P, Mildenberger W, et al. (2020) Early fate defines microglia and non-parenchymal brain macrophage development. Cell 181: 557–573 e518. 10.1016/j.cell.2020.03.021 [DOI] [PubMed] [Google Scholar]
- [209].van Osch MJ, Teeuwisse WM, Chen Z, et al. (2018) Advances in arterial spin labelling MRI methods for measuring perfusion and collateral flow. J Cereb Blood Flow Metab 38: 1461–1480. 10.1177/0271678X17713434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].van Swieten JC, van den Hout JH, van Ketel BA, et al. (1991) Periventricular lesions in the white matter on magnetic resonance imaging in the elderly. A morphometric correlation with arteriolosclerosis and dilated perivascular spaces. Brain 114 ( Pt 2): 761–774. 10.1093/brain/114.2.761 [DOI] [PubMed] [Google Scholar]
- [211].Vanlandewijck M, He L, Mae MA, et al. (2018) A molecular atlas of cell types and zonation in the brain vasculature. Nature 554: 475–480. 10.1038/nature25739 [DOI] [PubMed] [Google Scholar]
- [212].Vinters HV, Ellis WG, Zarow C, et al. (2000) Neuropathologic substrates of ischemic vascular dementia. J Neuropathol Exp Neurol 59: 931–945. [DOI] [PubMed] [Google Scholar]
- [213].Vinters HV, Zarow C, Borys E, et al. (2018) Review: Vascular dementia: clinicopathologic and genetic considerations. Neuropathol Appl Neurobiol 44: 247–266. 10.1111/nan.12472 [DOI] [PubMed] [Google Scholar]
- [214].Wallin A, Roman GC, Esiri M, et al. (2018) Update on vascular cognitive impairment Associated with subcortical small-vessel disease. J Alzheimers Dis 62: 1417–1441. 10.3233/JAD-170803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Wardlaw JM, Benveniste H, Nedergaard M, et al. (2020) Perivascular spaces in the brain: anatomy, physiology and pathology. Nat Rev Neurol 16: 137–153. 10.1038/s41582-020-0312-z [DOI] [PubMed] [Google Scholar]
- [216].Wardlaw JM, Smith EE, Biessels GJ, et al. (2013) Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol 12: 822–838. 10.1016/S1474-4422(13)70124-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Wardlaw JM, Valdes Hernandez MC, Munoz-Maniega S (2015) What are white matter hyperintensities made of? Relevance to vascular cognitive impairment. J Am Heart Assoc 4: 001140. 10.1161/JAHA.114.001140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Warnert EA, Rodrigues JC, Burchell AE, et al. (2016) Is high blood pressure aelf-protection for the brain? Circ Res 119: e140–e151. 10.1161/CIRCRESAHA.116.309493 [DOI] [PubMed] [Google Scholar]
- [219].Woo D, Falcone GJ, Devan WJ, et al. (2014) Meta-analysis of genome-wide association studies identifies 1q22 as a susceptibility locus for intracerebral hemorrhage. Am J Hum Genet 94: 511–521. 10.1016/j.ajhg.2014.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Yamazaki T, Mukouyama YS (2018) Tissue specific origin, development, and pathological perspectives of pericytes. Front Cardiovasc Med 5: 78. 10.3389/fcvm.2018.00078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Yang F, Qian D, Hu D, et al. (2016) Prevalence, awareness, treatment, and control of hypertension in the older population: results from the multiple national studies on ageing. J Am Soc Hypertens 10: 140–148. 10.1016/j.jash.2015.11.016 [DOI] [PubMed] [Google Scholar]
- [222].Yates PA, Villemagne VL, Ellis KA, et al. (2014) Cerebral microbleeds: a review of clinical, genetic, and neuroimaging associations. Front Neurol 4: 205. 10.3389/fneur.2013.00205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Yip AG, McKee AC, Green RC, et al. (2005) APOE, vascular pathology, and the AD brain. Neurology 65: 259–265. 10.1212/01.wnl.0000168863.49053.4d [DOI] [PubMed] [Google Scholar]
- [224].Zeng Y, Zhang L, Hu Z (2016) Cerebral insulin, insulin signaling pathway, and brain angiogenesis. Neurol Sci 37: 9–16. 10.1007/s10072-015-2386-8 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.