

Abstract
Nonalcoholic fatty liver disease (NAFLD) has been shown to be associated with extrahepatic comorbidities including neuronal inflammation and Alzheimer’s-like pathology. Environmental and genetic factors also act as a second hit to modulate severity and are expected to enhance the NAFLD-linked neuropathology. Using a mouse model of NAFLD, exposed to microcystin subsequent to the onset of fatty liver, we show that the cyanotoxin could significantly increase proinflammatory cytokine expression in the frontal cortex and cause increased expression of Lcn2 and HMGB1. The above effects were NLRP3 inflammasome activation-dependent since the use of NLRP3 knockout mice abrogated the increase in inflammation. NLRP3 was also responsible for decreased expression of the blood-brain barrier (BBB) tight junction proteins Occludin and Claudin 5 suggesting BBB dysfunction was parallel to neuroinflammation following microcystin exposure. An increased circulatory S100B release, a hallmark of astrocyte activation in microcystin exposed NAFLD mice also confirmed BBB integrity loss, but the astrocyte activation observed in vivo was NLRP3 independent suggesting an important role of a secondary S100B mediated crosstalk. Mechanistically, conditioned medium from reactive astrocytes and parallel S100B incubation in neuronal cells caused increased inducible NOS, COX-2, and higher BAX/ Bcl2 protein expression suggesting oxidative stress-mediated neuronal cell apoptosis crucial for neurodegeneration. Taken together, microcystin exacerbated neuronal NAFLD-linked comorbidities leading to cortical inflammation, BBB dysfunction, and neuronal apoptosis.
Keywords: Microcystin-LR, NAFLD, NASH, Lipocalin2, S100B, Blood-brain barrier
Introduction:
Nonalcoholic fatty liver disease (NAFLD) has been characterized by the accumulation of excessive fat (steatosis) in the liver followed by a more progressive phase of inflammation and fibrosis [1]. Most likely the transition of NAFLD to steatohepatitis and fibrosis requires a second or multiple hits [2]. We have shown exclusively that environmental toxins, oxidative stress, and proinflammatory cytokines are part of this multiple hit paradigm [3], [4]. According to CDC, a significant global population are reported to be diagnosed with NAFLD and is considered a major public health burden [5] and comes as no surprise as obesity rises globally. NAFLD more often leads to nonalcoholic steatohepatitis (NASH) characterized by excessive fat accumulation oxidative stress, fibrosis, and hyperinflammatory state that subsequently causes irreversible liver damage [5]. Growing evidence including findings from our laboratory strongly implicated that environmental pollutants worsen liver pathology, particularly in NAFLD and its progressive inflammatory state NASH [6], [3]. Cyanotoxin microcystin (MC) has been recognized as one of such pollutants which exacerbates the risk of NAFLD and its progression to NASH and has been seen as a serious environmental and public health issue in recent years [7]. Recent studies implicated that global climate change and increased temperature promote the proliferation and expansion of cyanobacterial harmful algal bloom (HAB) in the aquatic ecosystem and these harmful cyanobacteria produce toxins as secondary metabolites [8] ([9]). MCs are the most studied among all identified cyanotoxins be as of their potent hepatotoxic activity. MCs are taken up into the liver by organic anion-transporting polypeptides (OATPS), expressed in hepatocytes [10]. In addition, OATPs are also expressed in other organs such as small and large intestines, kidneys, as well as in BBB, the human brain, and primary murine neurons suggesting that the toxicity of MC is not only limited to the liver [11] ([12], [13]). Among approximately 200 different identified congeners, microcystin-LR (MC-LR) is the most studied and is listed as one of the most important algal toxins in the United States [14] and highlighted by Spoof and Catherine.(). MC-LR acts as the protein phosphatase inhibitor, that prevents the catalytic activity of Ser/Thr protein phosphatase 1 (PP1) and protein phosphatase 2A (PP2A) [15]; [16]. PP2A is responsible for maintaining the balance of major cellular functions including cell-cycle regulation, cellular development, tumor suppression, and signal transduction [17]. MC-LR binds to the catalytic subunit of PP2A covalently and diminishes the catalytic activity of PP2A [16]. The decreased catalytic activity of the major dephosphorylating enzyme PP2A alters the normal cellular activities and regulation of signal transductions and even may lead to tumorigenesis. In NAFLD, MC-LR can act as a ‘second hit’ and result in progression to NASH and liver fibrosis as shown by us and others [18], [19], [20]. Over the past few years, our research is exclusively focused on the elucidation of the role of MC-LR on organ pathologies, especially on liver NAFLD/NASH progression. Our studies of MC-LR exposure in NAFLD murine model showed worsening liver pathology with subsequent activation of inflammatory miR-21 and oxidative stress which led to increased fibrosis and heightened chances of progression to NASH [20]. Furthermore, MC-LR-induced oxidative stress led to hepatic insulin resistance and advanced metabolic abnormalities via NLRP3 inflammasome activation and thus increased the risk of diabetes in NAFLD phenotypes [21].
Previous studies have described that increased secretion of adipokines in NAFLD/NASH is associated with several comorbidities associated with NAFLD [22], [23], [24]. Being a potent adipokine, Lipocalin 2 (Lcn2) exerts both acute and chronic hepatic inflammation, insulin resistance, and lipogenesis [25]. Previously we showed that increased circulatory Lcn2 in NASH induces neuroinflammation, blood-brain barrier (BBB) dysfunction [26]. In parallel, studies have implicated that MC-LR can cross the BBB and is associated with neuroinflammation, neuronal apoptosis, and enhanced expression of phospho-Tau protein, a characteristic feature of Alzheimer’s disease (AD) like symptoms [27]. However, the role of MC-LR in worsening neuronal health in underlying NAFLD/NASH remains unclear. As a potent inducer of NAFLD/NASH phenotype and with known effects on neuronal pathology, the role of MC-LR in NAFLD-associated neuroinflammation and BBB dysfunction remains a valid line of inquiry.
With obesity and NAFLD reaching pandemic proportions, the present study investigates the role of MC-LR in neuropathology in underlying NAFLD/NASH. We hypothesized that MC-LR exposure led to an increased abundance of Lcn2 in the circulation which subsequently activates NLRP3 inflammasome to induce neuroinflammation and BBB dysfunction in the murine NAFLD/NASH model. Results showed that MC-LR exposure in NAFLD-associated BBB dysfunction caused astrocyte activation, S100B release, and associated neuronal apoptosis, a hallmark of neurodegeneration thus articulating a mechanistic pathway of NAFLD-linked neuronal comorbidity.
1. Materials and Methods:
Microcystin-LR (MC-LR) was purchased from Cayman Chemical Company (Ann Arbor, MI, USA). Primary antibodies which include anti-matrix metallopeptidase 9 (MMP9), anti-3-nitrotyrosine, anti-β-tubulin were purchased from Abcam (Cambridge, MA, USA), and anti-Interleukin-1β (IL 1β), anti-Interleukin 6 (IL 6) antibodies were purchased from Santacruz Biotechnology (Dallas, TX, USA). Anti-Tumor Necrosis Factor α (TNFα), anti-High mobility group box 1 (HMGB1), anti-Zonula occludens-1 (ZO1), anti-B-cell lymphoma 2 (Bcl2), anti-β-actin antibodies were purchased from ProteinTech (Rosemont, IL, USA) whereas anti-Lipocalin 2 (Lcn2) antibody was obtained from R&D Systems (Minneapolis, MN, USA), anti-Glial fibrillary acidic protein (GFAP) antibody was purchased from Novus Biologicals (Centennial, CO, USA) and anti-Claudin5, anti-S100B, anti-inducible nitric oxide synthase (iNOS) antibodies were bought from Invitrogen (Rockford, IL, USA). Also, anti-Cleaved Caspase3, anti-Bax primary antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA), anti-cyclooxygenase-2 (COX-2) primary antibody was obtained from Abclonal Technology (Woburn, MA, USA). Species-specific biotinylated conjugated secondary antibodies and Streptavidin-HRP (Vectastain Elite ABC kit) were obtained from Vector Laboratories (Burlingame, CA, USA). Fluorescence-conjugated (Alexa Fluor) secondary antibodies, ProLong Gold antifade mounting media with DAPI were bought from Thermo Fisher Scientific (Rockford, IL, USA). Mouse brain tissues were sent to AML laboratories (Baltimore, MD, USA) for Paraffin-embedding and sectioning purposes. All other chemicals used in this project were purchased from Sigma and analytical grade, only if otherwise specified.
2.1. Animals:
Pathogen-free, adult (8 weeks old), male, wild-type (WT) C57BL/6 J mice, and NLRP3 knock-out (NLRP3KO) mice (B6N.129-Nlrp3tm3Hhf/J) with C57BL/6J background were purchased from Jackson Laboratories (Ban Harbor, ME, USA) and used in this study. Mice experiments for this study were carried out by strictly following the National Institutes of Health (NIH) NIH guidelines for human care and use of laboratory animals and local Institutional Animal Care and Use Committee (IACUC) standards. The animal handling procedures for this study were approved by the University of South Carolina, Columbia, SC, United States. All mice were housed at 22–24 °C with a 12 h light/12 h dark cycle upon arrival and had ad libitum access to food and water throughout this study. Every mouse used for this study was sacrificed after completion of the experiment and frontal cortex from individual mouse was collected and fixed in Bouin’s solution (Sigma Aldrich St. Louis, MO, USA). Serum samples were prepared from fresh blood of mice obtained by cardiac puncture method immediately after anesthesia and were kept at −80°C for further analysis.
2.2. Diet-induced NAFLD model and exposure to Microcystin-LR:
The groups used for the study were: WT mice fed with chow diet only (Chow), WT mice fed with methionine choline deficient-high fat diet only (MCD-HFD), WT mice fed with chow diet and then exposed to MC-LR (Chow + MC), WT mice fed with methionine choline deficient-high fat diet and then exposed to MC-LR (MCD-HFD + MC), and another group of NLRP3KO mice fed with methionine choline deficient-high fat diet and then exposed to MC-LR (MCD-HFD+MC/NLRP3KO). Six mice per group (n=6) were randomly allocated to their respective cages. Mice were fed with chow diet or methionine choline deficient-high fat diet (to induce NAFLD in mice) consecutively for 6 weeks and subsequently were exposed to vehicle (PBS) or MC-LR (10 μg/ kg body weight; 5 dosages per week) through the intraperitoneal route for 2 weeks while continuing the same diet. MC-LR dosing was started after the mice had prominent symptoms of NAFLD confirmed by ALT levels, liver histology. The dosage was given each afternoon at the same time to eliminate any bias in the study.
2.3. Cell Culture:
2.3.1. Mouse primary astrocyte cell culture:
Primary mouse astrocytes C8-D1A [Astrocyte type I clone] (ATCC CRL-2541) were purchased from ATCC (Manassas, VA, USA). The cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Catalog number: 11995065, Thermo Fisher Scientific, Rockford, IL, USA) and supplemented with 10% fetal bovine serum (FBS) (Catalog number: F-0500-D, Atlas Biologicals, Fort Collins, CO, USA). The cells were plated in 12- and 6-well tissue culture plates and growth were allowed until 70% confluency. The cells were sera starved using a 1% fetal bovine serum FBS in DMEM for 18 h and then exposed with mouse recombinant Lcn2 (100 ng/mL) or MC-LR (20 μM) or both for 24 h. To inhibit NLRP3 inflammasome, cells were treated with CRID3 (TOCRIS Life Sc, Minneapolis, MN, USA) at a concentration of 7 µM or with a RAGE antagonist FPS-ZM1 at 230 nM for 3 h prior to treatment.
Cell supernatants were collected for ELISA and proteins were extracted by RIPA buffer.
2.3.2. Mouse neuronal cell culture:
Mouse neuroblastoma cell line Neuro-2a (N2a) (ATCC® CCL-131™) was purchased from ATCC (Manassas, VA, USA). These N2a cells were maintained on Eagle’s Minimum Essential Medium (EMEM) (ATCC® 30–2003™) supplemented with 10% FBS, 100 U/mL Penicillin, 100 µg/mL Streptomycin (Gibco, NY, USA) and incubated in a humidified 5% CO2 incubator at 37°C.
Cells were plated on a 6 well plate with a seeding density of 0.3×106 cells/well and were grown till the cells reached almost 70% confluency. Then, the N2a cells were serum-starved for 18 h using EMEM supplemented with 1% FBS. Following serum starvation, the cells were then exposed with mouse recombinant S100B (Sino-Biological, Chesterbrook, PA, USA) or with supernatant collected from astrocyte cells, treated with Lcn2 and MC-LR. For inhibition experiments for RAGE receptor, cells were exposed with 230 nM FPS-ZM1 (Sigma-Millipore, St. Louis, MO, USA) for 3 h prior to treatment.
2.3.3. Mouse brain endothelial cell culture:
Mouse brain endothelial cells were purchased from Cell Biologics Inc (Chicago, IL, USA). Cells were grown and maintained according to the manufacturer’s instructions. Cells were treated with Lcn2 (100 ng/mL) or MC-LR (20 μM) or both for 24 h. To inhibit the NLRP3 inflammasome, cells were treated with CRID3 (TOCRIS Life Sc, Minneapolis, MN, USA) in a concentration of 7 µM for 3 h prior to treatment. Cell supernatants were collected for ELISA and proteins were extracted for Western blot analysis.
2.4. Laboratory analysis:
2.4.1. Immunohistochemistry:
Paraffin-embedded, 5 μm thick brain tissue sections were deparaffinized using our standard laboratory procedure. Briefly, brain tissues were immersed successively in 100% xylene, 1:1 solution of xylene and ethanol, 100% ethanol, 95% ethanol, 70% ethanol, 50% ethanol, deionized water for 3 min each. Epitope retrieval of the deparaffinized sections was performed using the epitope retrieval solution and steamer (IHC-World, Woodstock, MD, USA) for 45 min. To block the endogenous peroxidases, 3% H2O2 solution was used on the tissue sections for 15 min. The brain sections were blocked with 5% goat serum, followed by overnight incubation with primary antibodies of IL 1β, IL 6, TNFα, and MMP9 in recommended dilutions overnight at 4°C. After washing with 1X PBS-T20 (PBS + 0.05% Tween 20) 3 times, species-specific biotinylated conjugated secondary antibodies and streptavidin-conjugated with horseradish peroxidase (HRP) were diluted in recommended dilutions and applied on the sections. Finally, 3,3’ Diaminobenzidine (Sigma-Aldrich, St. Louis, MO, USA) was used as a chromogenic substrate and the sections were counterstained with Mayer’s hematoxylin (Sigma-Aldrich, St. Louis, MO, USA). Mounting of the sections was done by using Aqua-Mount (Lerner Laboratories, Kalamazoo, MI, USA). Tissue sections were observed using an Olympus BX63 microscope (Olympus, USA) under a × 20 objective. CellSens Software from Olympus America (Centre Valley, PA, USA) was used for morphometry analysis of images.
2.4.2. Immunofluorescence staining and microscopy:
Paraffin-embedded, 5 μm thick brain tissue sections were subjected to deparaffinization using standard laboratory protocol. The tissue sections were permeabilized using PBS-T100 (PBS + 0.1% Triton X-100) solution for 1 h, followed by serum blocking using 5% goat serum following epitope retrieval. The brain sections were incubated with primary antibodies which include anti-HMGB1, anti-Lcn2, anti-Claudin5, anti-CD31, anti-ZO1, anti-GFAP, anti-S100B, anti-3-nitrotyrosine, anti-β-tubulin and anti-Cleaved Caspase 3 at recommended dilutions for overnight at 4°C. Species-specific secondary antibodies conjugated with Alexa Fluor (633-red and 488-green) (Invitrogen) were used at recommended dilutions. The sections were mounted with ProLong™ Diamond Antifade Mountant 4’,6-diamidino-2-phenylindole (DAPI) (Life Technologies, Carlsbad, CA, USA). Sections were observed under the BX63 Olympus fluorescence microscope using × 40 and × 60 objective lenses. CellSens Software from Olympus America (Centre Valley, PA, USA) was used for morphometry analysis of images.
2.4.3. Western blot:
Protein samples from the brain tissues were first extracted using 1X RIPA-lysis buffer supplemented with protease and phosphatase inhibitors and then quantified by the BCA assay kit (Thermo Fisher Scientific, Rockford, IL, USA). Approximately, 50 μg of tissue lysate were resolved using SDS-PAGE, protein bands were transferred to nitrocellulose membrane using pre-cut nitrocellulose/filter paper sandwiches (Bio-Rad Laboratories, Hercules, CA, USA) and Trans-Blot Turbo transfer system (Bio-Rad). Ponceau S staining was performed to ensure proper transfer of proteins onto the nitrocellulose membrane and blocking was done using 5% bovine serum albumin (BSA) for 1 h. Primary antibodies against iNOS, COX-2, Bax, Bcl2, ZO-1, Claudin5, β-Actin were probed at recommended dilutions overnight at 4°C. Species-specific HRP-conjugated secondary antibodies were used to tag primary antibodies and Pierce-enhanced chemiluminescence (ECL) Western Blotting Substrate (Thermo Fisher Scientific, Rockford, IL, USA) was applied for immunoreactivity detection. Image of the blot was captured by using G: BoxChemi XX6 (Syngene imaging systems) and densitometry analysis of the blot images was performed using Image J software.
2.4.4. Enzyme-linked immunosorbent assay (ELISA):
Quantification of Lcn2 and S100B was performed by mouse-specific Lcn2 and S100B ELISA kits from R&D Systems (Minneapolis, MN, USA) and Abclonal Technology (Woburn, MA, USA) respectively using sera collected from the Chow, Chow+MC, MCD-HFD, MCD-HFD+MC, and MCD-HFD+MC/NLRP3KO mice groups. IL 1β ELISA was performed with the collected supernatants from mouse primary brain endothelial cells. IL 1β ELISA kit was purchased from ProteinTech (Rosemont, IL, USA). All the ELISAs were performed following the manufacturer’s protocol.
2.5. Statistical analyses:
All in vivo and in vitro experiments were repeated 3 times. Statistical analysis was performed by unpaired, paired t-test and analysis of variance (ANOVA) followed by Bonferroni post-hoc correction for intergroup comparisons. For all statistical analyses, a p-value < 0.05 was considered statistically significant.
2. Results:
3.1. MC-LR exposure exacerbates neuroinflammation in NAFLD/NASH:
Previous studies including our research findings have demonstrated that NAFLD/NASH exhibited neuroinflammation [26]. To study the role of MC-induced worsening of neuroinflammation, we performed immunohistochemistry to study the tissue-based protein expression of proinflammatory cytokines IL 1β, IL 6, and TNFα in CHOW, CHOW+MC, HCD-HFD, and MCD-HFD+MC mouse groups. Immunoreactivities of proinflammatory cytokines were measured in the hippocampus and cerebral cortex of the brain tissue sections, displayed in Figures 1A and 1B. Results showed a 1.5-fold increased immunoreactivity of IL 6, and TNFα in MC-LR exposure in the CHOW diet group (p<0.01, n=6). However, no significant difference was observed in IL 1β expression on MC-LR exposure in the CHOW diet group. Notably, MC-LR exposure in the MCD-HFD mouse group showed 16-fold increased immunoreactivity of IL 1β (p<0.01, n=6), and 2-fold increased TNFα expression (p<0.05, n=6). Moreover, results showed a 5-fold increased immunoreactivity of IL 1β (p<0.01, n=6) and 1.5-fold higher immunoreactivity of both TNFα and IL 6 (p<0.05, n=6) in MCD-HFD+MC when compared to MCD-HFD alone. The above results suggested an increased neuroinflammatory response following MC-LR exposure in underlying NAFLD/NASH pathology.
Fig. 1.
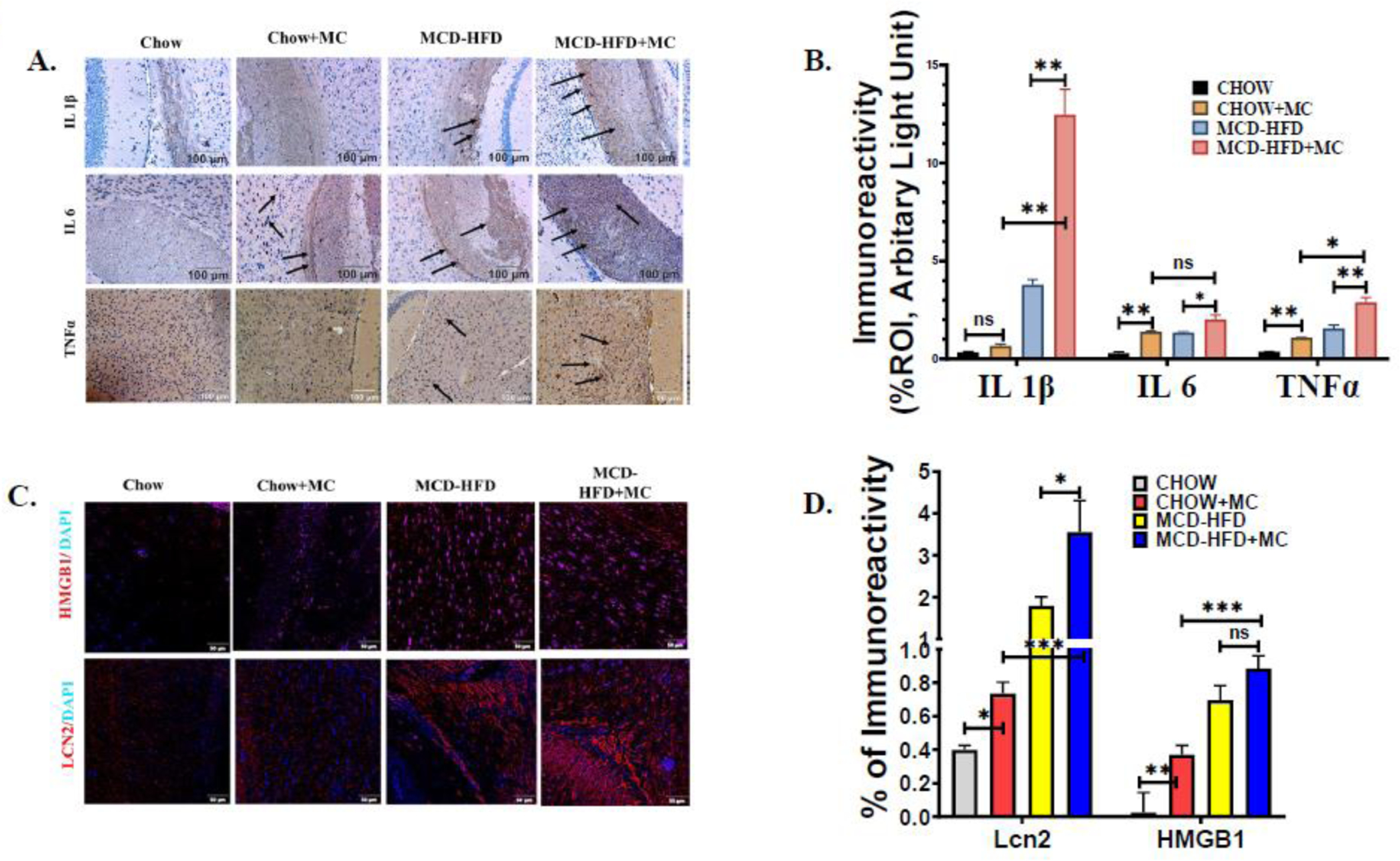
MC-LR exposure results in increased expression of Proinflammatory cytokines, HMGB1, and Lipocalin 2 in brain sections of NAFLD/NASH mice. The in vivo experimental groups include WT Chow diet-fed mice (Chow), WT chow diet-fed mice exposed to MC-LR (Chow+MC), WT mice fed with methionine choline-deficient and high-fat diet (MCD-HFD), and another WT methionine choline-deficient and a high-fat diet-fed group of mice exposed to MC-LR (MCD-HFD+MC). (A.) Immunoreactivity of IL 1β, IL 6, and TNFα expression as depicted by immunohistochemistry images in brain sections from Chow, Chow+MC, MCD-HFD, and MCD-HFD+MC mice groups. All images were taken at × 20 magnification (Scale: 100 μm) and immunoreactivity was indicated by black arrows. (B.) Morphometric analysis of IL 1β, IL 6, and TNFα immunoreactivity [mean data plotted on y-axis was measured as % positive immunoreactive area (% ROI) in arbitrary light units from three different microscopic fields] in Chow, Chow+MC, MCD-HFD, and MCD-HFD+MC mice groups (*p< 0.05, **p< 0.01, ns=non-significant). (C.) Immunoreactivity of HMGB1 (red) and Lcn2 (red) in brain sections counterstained with DAPI from Chow, Chow+MC, MCD-HFD, and MCD-HFD+MC mice groups as shown by immunofluorescence microscopy. All images were taken at × 40 magnification (Scale: 50 μm) (D.) Morphometric analysis of HMGB1, and Lcn2 immunoreactivity [mean data plotted on y-axis was measured as % positive immunoreactive area (% ROI) in arbitrary light units from three different microscopic fields] in Chow, Chow+MC, MCD-HFD, and MCD-HFD+MC mice groups (*p< 0.05, **p< 0.01, ***p< 0.001, ns=non-significant). All statistical significance was tested by performing unpaired t-test between the groups (*p< 0.05, **p< 0.01, ***p< 0.001, ns=non-significant), followed by Bonferroni Dunn Post hoc corrections.
Following our observation of increased neuroinflammation in MC-LR exposure in NAFLD/NASH we were further interested to study the protein expression of Lcn2 and HMGB1 in the brain tissue sections of the above mouse groups. In a previous study, we found that Lcn2 induced neuroinflammation in the murine NASH model by overproduction of HMGB1, a Damage-associated molecular pattern (DAMP) [26]. Following the same argument, immunoreactivity of both Lcn2 and HMGB1 was performed. Lcn2 and HMGB1 immunoreactivity was depicted as red fluorescence on the brain tissue sections, and the nucleus was depicted as blue (DAPI) color (Fig 1C and 1D). Results showed that there was a 2-fold higher immunoreactivity of Lcn2 (p<0.05, n=6) in the CHOW+MC group compared to the control CHOW group. Notably, results showed an increased reactivity of Lcn2 in the brain tissue section MCD-HFD+MC mouse group when compared to CHOW+MC (5-fold, p<0.001) and MCD-HFD groups (2-fold, p<0.05). Our results on HMGB1 immunoreactivity strongly correlated with Lcn2 immunoreactivity in the different mouse groups. Results also showed that MC-LR exposure significantly increased the expression of HMGB1 in the CHOW diet group. Moreover, we observed a 2-fold increase in HMGB1 expression in the MCD-HFD+MC mouse group when compared to the CHOW+MC group while the levels were not significant between MCD+HFD and MC+MCD+HFD group (Fig. 1D) The following results clearly indicated that MC-LR exposure induced Lcn2 to exacerbate neuroinflammation in NAFLD/NASH murine model.
3.2. Elevated neuroinflammation in MC-LR exposure in underlying NAFLD/NASH is mediated by NLRP3 inflammasome activation:
Our previous report suggested that increased Lcn2 and HMGB1 in brain tissue of NAFLD/NASH mouse model induced neuroinflammation via NLRP3 inflammasome activation [26]. To study the effect of NLRP3 inflammasome on neuroinflammation on MC-LR-induced exacerbation of NAFLD/NASH pathology, a NLRP3KO mouse group was used. Immunohistochemistry of IL 1β, IL 6, and TNFα was performed with brain tissue sections as described previously. Results showed a subsequent decrease of proinflammatory cytokines IL 1β and TNFα in the MCD-HFD+MC/ NLRP3KO mouse group when compared to the MCD-HFD+MC group (Fig 2A and 2B). Notably, IL 1β expression was decreased around 2.5 folds in the NLRP3KO compared to its WT phenotype. However, no significant change was observed in IL 6 expression in the NLRP3KO group in comparison with its WT phenotype. Furthermore, we studied the expression of Lcn2 and HMGB1 by immunofluorescence microscopy as described previously. Notably, both Lcn2 and HMGB1 expression were significantly decreased in NLRP3KO phenotypes compared to the WT group. More than 2-fold decreased immunoreactivity of Lcn2 was observed in the NLRP3KO mouse group (Fig 2C and 2D). Interestingly, NLRP3KO mice also had decreased serum Lcn2 when compared to its wild type of phenotype fed with MCD-HFD-MC. Taken together, the above results demonstrated that MC-LR exposure in NAFLD/ NASH abrogated brain pathology by inducing excessive neuroinflammation mediated by an increased circulatory level and protein overexpression of Lcn2, followed by NLRP3 inflammasome activation.
Fig. 2.
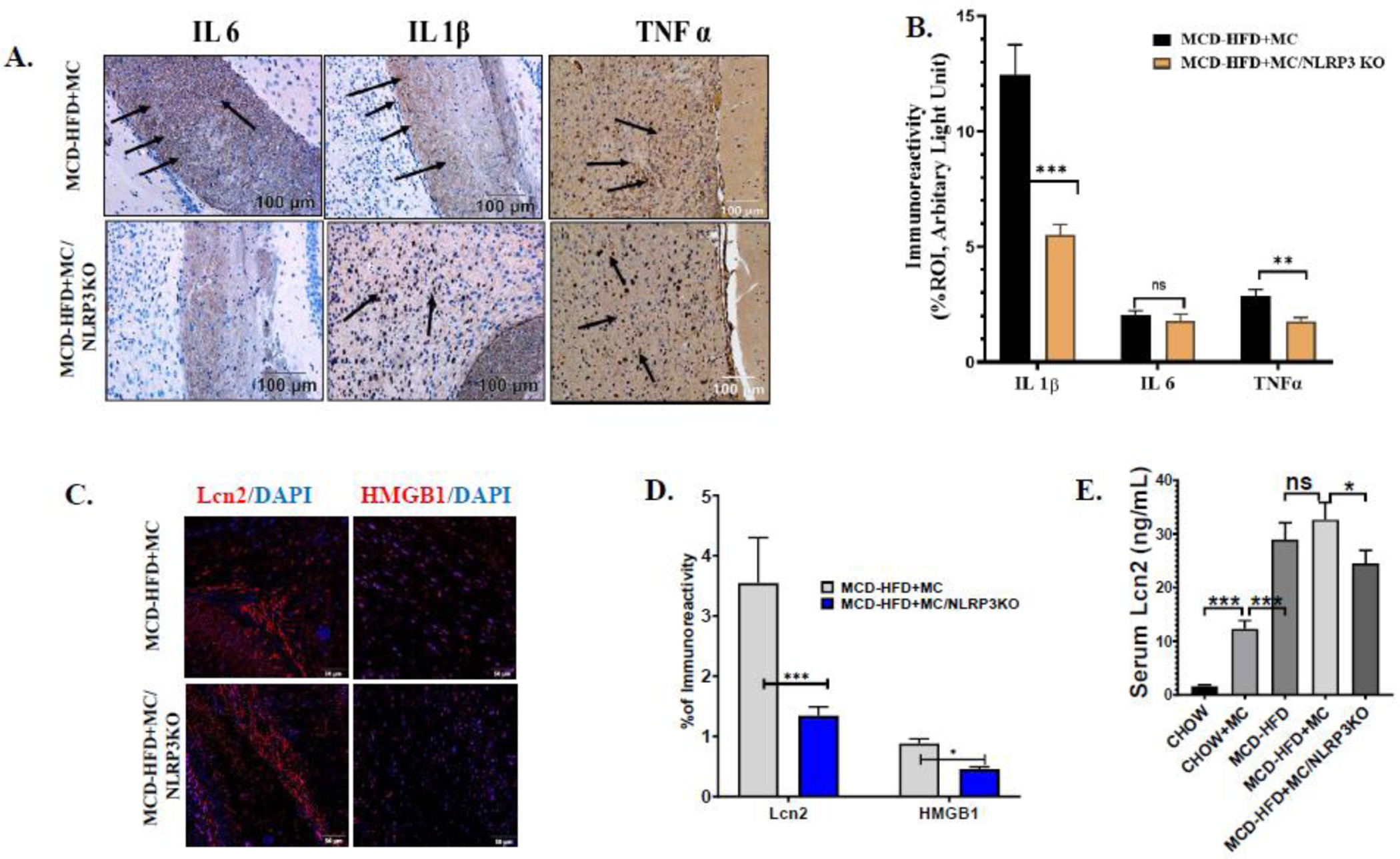
Pathological signs of MC-LR toxicity on WT, NAFLD/NASH mice brain are attenuated in NLRP3KO, NAFLD/NASH mice. (A.) Immunoreactivity of IL 1β, IL 6, and TNFα expression as depicted by immunohistochemistry images in brain sections of methionine choline-deficient and high-fat diet-fed, MC-LR exposed WT mice (MCD-HFD+MC) and NLRP3KO mice (MCD-HFD+MC/NLRP3KO). All images were taken at × 20 magnification (Scale: 100 μm) and immunoreactivity was indicated by black arrows. (B.) Morphometric analysis of IL 1β, IL 6, and TNFα immunoreactivity [mean data plotted on y-axis was measured as % positive immunoreactive area (% ROI) in arbitrary light units from three different microscopic fields] in MCD-HFD+MC and MCD-HFD+MC/NLRP3KO mice groups (**p< 0.01, ***p< 0.001, ns=non-significant). (C.) Immunoreactivity of Lcn2 (red) and HMGB1 (red) in brain sections counterstained with DAPI from MCD-HFD+MC and MCD-HFD+MC/NLRP3KO mice groups as shown by immunofluorescence microscopy. All images were taken at × 40 magnification (Scale: 50 μm) (D.) Morphometric analysis of HMGB1, and Lcn2 immunoreactivity [mean data plotted on y-axis was measured as % positive immunoreactive area (% ROI) in arbitrary light units from three different microscopic fields] in MCD-HFD+MC and MCD-HFD+MC/NLRP3KO mice groups (*p< 0.05, ***p< 0.001). (E.) Lcn2 level (ng/mL) was quantified by ELISA using serum from Chow, Chow+MC, MCD-HFD, MCD-HFD+MC, and MCD-HFD+MC/NLRP3KO mice groups and plotted as a bar graph (**p< 0.01, ***p< 0.001, ns=non-significant). All statistical significance was tested by performing unpaired t-test between the groups (*p< 0.05, **p< 0.01, ***p< 0.001, ns=non-significant), followed by Bonferroni Dunn Post hoc corrections.
3.3. MC-LR induced BBB dysfunction by NLRP3 inflammasome activation in NAFLD/NASH:
Previous studies have shown that increased expression of IL 1β and TNFα and concomitant downregulation of tight junction proteins increase BBB permeability [26]. Brain tissue sections from the mentioned mouse groups were stained with a specific endothelial cell marker CD31, co-stained with either Claudin 5 or with ZO1. The expression of the tight junction protein was confirmed by colocalization analysis (yellow dots). Results showed that there was a 2.5-fold decreased expression of Claudin 5 and a 3-fold decreased expression of ZO1 in the brain tissue sections, on CD31 positive cells (endothelial cells) of MCD-HFD+MC mouse groups when compared to the CHOW control group (Fig 3A–3D). These results indicated that it was highly likely that there was a disruption of tight junction architecture on brain endothelial cells on MC-LR exposure. Strikingly, MC-LR exposure in the MCD-HFD mouse group showed the least immunoreactivity of both Claudin 5 and ZO1. Claudin 5 expression was decreased 10-fold, whereas ZO1 expression was decreased 2-fold in the MCD-HFD+MC group compared to the CHOW+MC mouse group (Fig. 3A–3D). The above observations clearly suggest a likelihood of an exacerbation of BBB dysfunction in NAFLD/NASH phenotype following MC-LR exposure. Furthermore, we studied MMP9 expression by immunohistochemistry. MMP9 has been shown to degrade tight junction proteins Claudin 5, Occludin, and ZO1 in cultured brain endothelial cells and an animal model of focal cerebral ischemia [28]. Results showed that there was an increased MMP 9 expression following MC-LR exposure in the CHOW diet group and the MCD-HFD group. Notably, a 15-fold higher MMP9 expression was observed in the MCD-HFD+MC mouse group when compared to the CHOW+MC group (Fig 3B and D). The above results suggested that MC-LR exposure exacerbated BBB dysfunction in NAFLD/NASH. Next, we investigated the role of NLRP3 inflammasome on exacerbation of BBB dysfunction by MC-LR in the NAFLD/NASH mouse model. We have shown previously that NLRP3 inflammasome activation may play an important role in BBB pathology via overexpression of proinflammatory cytokines IL 1β and IL 6 in NAFLD/NASH. Interestingly, results showed that TJ proteins Claudin 5 and ZO1 expression were restored in the endothelial cells of BBB followed by a significant decrease in MMP 9 expression in brain sections of NLRP3KO mouse exposed with MC-LR and MCD-HFD diet. Claudin 5 expression increased 15-fold, whereas a 2-fold increase in expression of ZO1 was observed in MCD-HFD+MC/NLRP3KO mouse group. The results suggested that MC-LR exacerbates BBB dysfunction in NAFLD/NASH, and it is mediated primarily via the NLRP3 inflammasome.
Fig. 3.
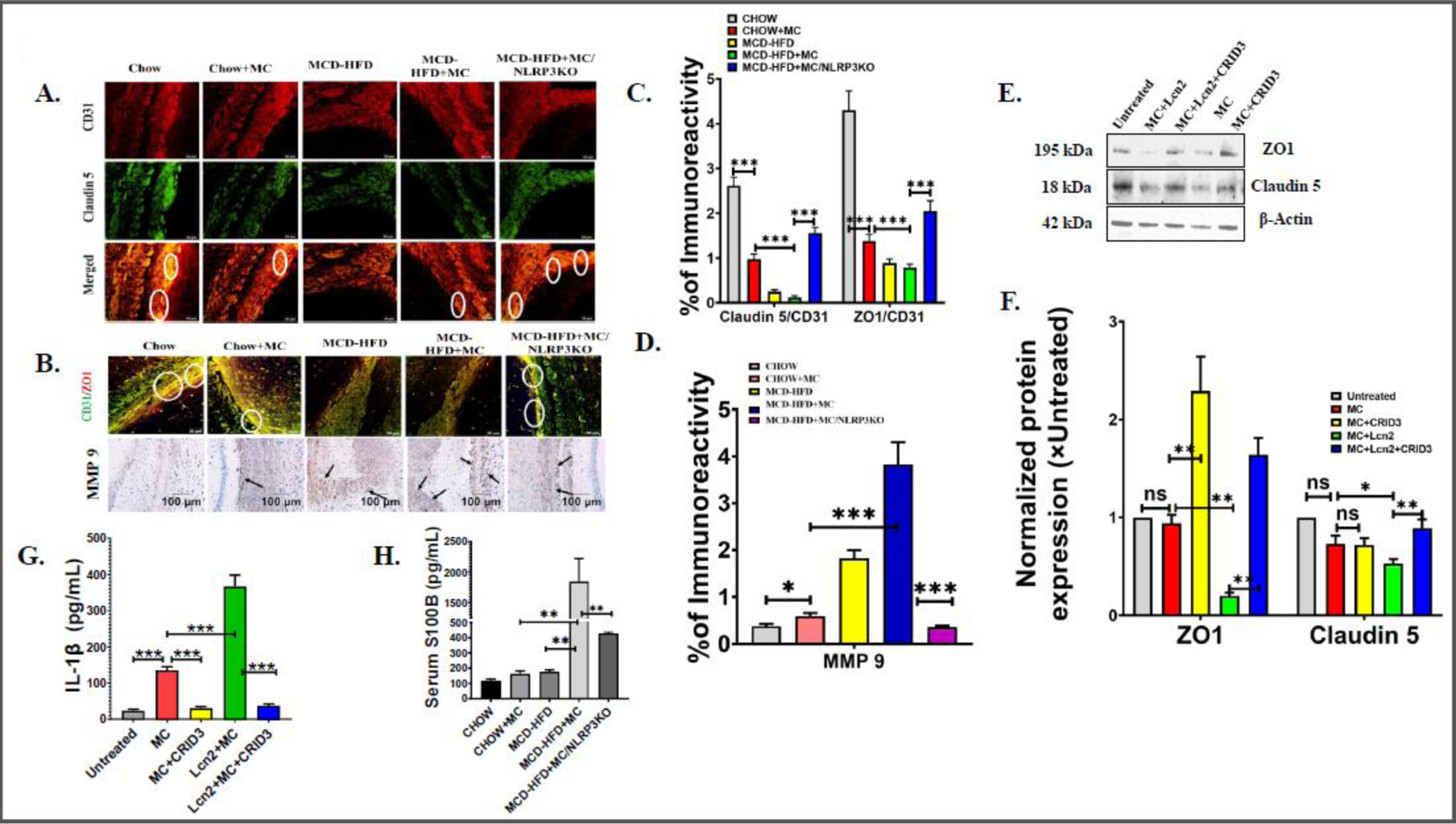
(A.) Colocalization events of CD31 (red) and Claudin5 (green) immunoreactivity in brain sections from Chow, Chow+MC, MCD-HFD, MCD-HFD+MC, and MCD-HFD+MC/NLRP3KO mice groups as shown by immunofluorescence microscopy. Colocalization of CD31 and Claudin5 was represented by yellow dots and highlighted by white circles. All images were taken at × 40 magnification (Scale: 50 μm). (B.) Colocalization events of CD31 (green) and ZO1 (red) and Immunoreactivity of MMP9 expression in Chow, Chow+MC, MCD-HFD, MCD-HFD+MC, and MCD-HFD+MC/NLRP3KO mice groups as depicted by immunofluorescence microscopy and immunohistochemistry, respectively. Colocalization of CD31 and ZO1 was represented by yellow dots and highlighted by white circles whereas MMP9 immunoreactivity was indicated by black arrows. Morphometric analysis of (C.) Claudin5/CD31 and ZO1/CD31 colocalization events, (D.) MMP9 immunoreactivity [mean data plotted on y-axis was measured as % positive immunoreactive area (% ROI) in arbitrary light units from three different microscopic fields] in Chow, Chow+MC, MCD-HFD, MCD-HFD+MC, and MCD-HFD+MC/NLRP3KO mice groups (*p< 0.05, ***p< 0.001). (E.) Western blot analysis of ZO1, Claudin5 protein expression levels in the mouse primary brain endothelial cell lysates. Lanes 1–5 represent untreated cells, MC+Lcn2, MC+Lcn2+CRID3, MC, and MC+CRID3 groups of cells, respectively. (F.) Band quantification of ZO1, and Claudin5 immunoblot normalized against β-actin (*p< 0.05, **p< 0.01, ns=non-significant). (G.) IL −1β level (pg/mL) was detected by ELISA in supernatants obtained from untreated cells, MC, MC+CRID3, Lcn2+MC, and MC+Lcn2+CRID3 groups of mouse primary brain endothelial cells and displayed by bar graph. (***p< 0.001) (H.) S100B level (pg/mL) was quantified by ELISA using serum from Chow, Chow+MC, MCD-HFD, MCD-HFD+MC, and MCD-HFD+MC/NLRP3KO mice groups and plotted as a bar graph (**p< 0.01). All statistical significance was tested by performing unpaired t-test between the groups (*p< 0.05, **p< 0.01, ***p< 0.001, ns=non-significant), followed by Bonferroni Dunn Post hoc corrections.
Extending our findings on TJ protein expression in the murine model, we studied the expression of TJ proteins (Claudin 5 and ZO1) on mouse primary endothelial cells following MC-LR exposure to further explain the mechanism. Cells were incubated with or without mouse recombinant Lcn2 (100ng/mL) for at least 18 hrs to mimic the in vivo NAFLD/NASH condition, followed by MC-LR exposure (20 uM) in the presence or absence of CRID3, a NLRP3 inflammasome signaling inhibitor. Claudin 5 and ZO1 expression were studied by immunoblot, and IL 1β expression was studied by ELISA with the supernatants from all treatment groups including untreated control. Notably, both Claudin 5 and ZO1 expression were significantly decreased when the cell was co-exposed with MC and Lcn2, compared to untreated and MC exposed cells, and expression was restored on NLRP3 inhibition by CRID3 (Fig 3E and F). However, no change in expression of Claudin 5, ZO1 was observed when cells were exposed with MC-LR only thereby suggesting MC-LR is ineffective in TJ proteins disruption alone but acts synergistically with Lcn2 in exacerbating TJ proteins protein expression. Most interestingly, upon inhibition by CRID3 both ZO1 and Claudin 5 expression were restored and suggesting an improvement of BBB dysfunction via increased TJ protein expression crucial to maintain BBB integrity. Results showed more than 5-fold higher expression of ZO1 and 1.5-fold higher Claudin 5 expression in the MC+Lcn2+CRID3 group compared to the MC+Lcn2 group (Fig 3E and F). We also studied IL 1β secretion in the supernatants of the above groups. Results showed a 5-fold higher IL 1β concentration compared to the untreated group of cells (p<0.001, n=3). Moreover, supernatant from the Lcn2+MC group of cells showed a more than 10-fold increase in IL 1β concentration when compared to untreated control. Upon inhibition with CRID3, IL 1β concentration was decreased 10-fold when compared to the Lcn2+MC group. The above IL 1β ELISA levels in brain endothelial cell culture supernatants suggested that the Lcn2 and MC-LR acted synergistically in IL 1β protein overexpression and release and is likely mediated by the NLRP3 inflammasome. In lieu of the above findings of exacerbation of BBB dysfunction in the murine model and cultured brain endothelial cells, we next studied the circulatory S100B levels to confirm BBB dysfunction in MC-LR exposure in NAFLD/NASH. The study of serum S100B be is based on the rationale that increased S100B in the circulation is believed to be a hallmark for extensive BBB disruption [29]. Serum S100B results showed a significantly elevated S100B level in the MCD-HFD+MC group when compared to all other mouse groups (Fig 3H) and strongly correlated with excessive BBB damage in the MC-LR exposure group in underlying NAFLD.
3.4. MC-LR exposure induces S100B expression independent of NLRP3 inflammasome activation in murine NAFLD/NASH model:
Following observation of increased circulatory S100B levels in the MCD-HFD+MC group, we were keen to study S100B expression in the brain. S100B has been observed to be released by reactive astrocytes in models of brain injury and exerts both beneficial and detrimental effects on the brain, an effect depending on concentrations in the local microenvironment [30]. We studied S100B protein expression along with glial fibrillary acidic protein (GFAP), a biomarker for reactive astrocytes in the brain tissue sections by immunofluorescence analysis. Dual fluorescence labeling was used to stain GFAP (green) and S100B (red) and co-localization was confirmed by yellow dots. Results showed, significantly increased immunoreactivity of GFAP-S100B in the MCD-HFD+MC mouse group, compared to both CHOW+MC and MCD-HFD groups (Fig 4A and 4B). Results also showed a significantly higher expression of GFAP (green fluorescence) in the MCD+HFD+MC group when compared to either CHOW+MC or only Chow groups, suggesting the presence of increased reactive astrocytes population in the brain of underlying NAFLD/NASH with MC-LR exposure group. However, S100B expression was not decreased in the NLRP3KO group that had NAFLD and was exposed to MC compared to the MCD-HFD+MC group. The following observation suggested that the NLRP3 inflammasome is not responsible for the regulation of reactive astrocytes and S100B abundance on MC-LR exposure underlying NAFLD/NASH.
Fig. 4.
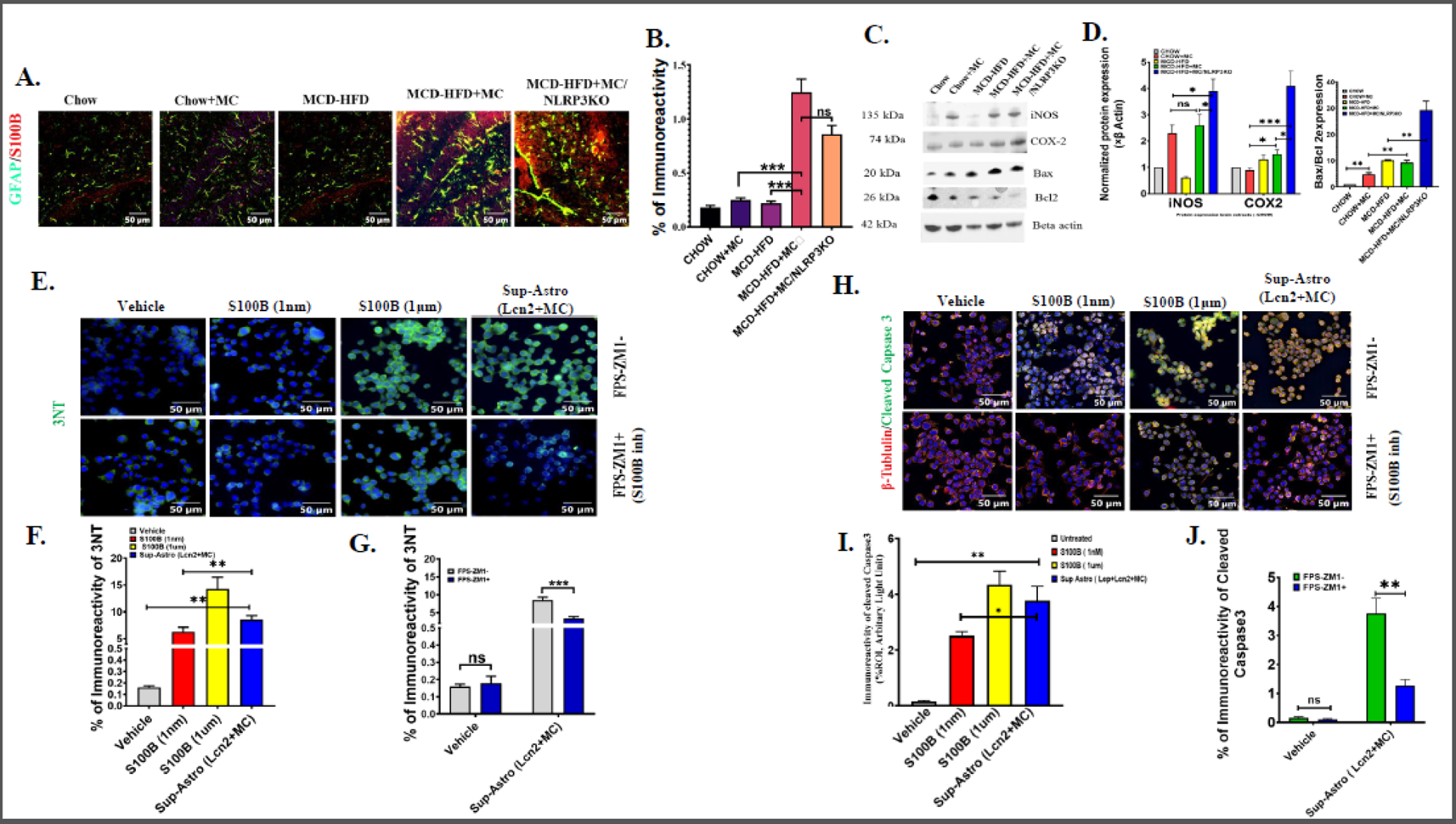
(A.) Colocalization events of GFAP (green) and S100B (red) immunoreactivity in brain sections from Chow, Chow+MC, MCD-HFD, MCD-HFD+MC, and MCD-HFD+MC/NLRP3KO mice groups as shown by immunofluorescence microscopy. Colocalization of GFAP and S100B was represented by yellow dots. All images were taken at × 40 magnification (Scale: 50 μm). (B.) Morphometric analysis of GFAP/S100B colocalization events [mean data plotted on y-axis was measured as % positive immunoreactive area (% ROI) in arbitrary light units from three different microscopic fields] in Chow, Chow+MC, MCD-HFD, MCD-HFD+MC, and MCD-HFD+MC/NLRP3KO mice groups (***p< 0.001, ns=non-significant). (C.) Western blot analysis of iNOS, COX-2, Bax, Bcl2 protein expression levels in Chow, Chow+MC, MCD-HFD, MCD-HFD+MC, and MCD-HFD+MC/NLRP3KO mice groups. Lanes 1–5 represent Chow, Chow+MC, MCD-HFD, MCD-HFD+MC, and MCD-HFD+MC/NLRP3KO mice groups, respectively. (D.) Band quantification of iNOS, COX-2 immunoblots normalized against β-actin and Bax immunoblot normalized against Bcl2 (*p< 0.05, **p< 0.01 ***p< 0.001, ns=non-significant). (E.) Immunoreactivity of 3-nitrotyrosine (3NT) (green) counterstained with DAPI in Neuro-2a cells exposed without or with FPS-ZM1 (S100B inhibitor) and treated with Vehicle, 1nM S100B, 1 μM S100B, and Astrocyte cell culture supernatant (MC+Lcn2) separately. All images were taken at × 40 magnification (Scale: 50 μm). (F.) Morphometric analysis of 3NT immunoreactivity [mean data plotted on y-axis was measured as % positive immunoreactive area (% ROI) in arbitrary light units from three different microscopic fields] in Neuro-2a cells treated with Vehicle, 1nM S100B, 1 μM S100B, and Astrocyte culture Sup (MC+Lcn2) separately (**p< 0.01, ns=non-significant) (G.) Comparative analysis of 3NT immunoreactivity, in Neuro-2a cells, exposed with Astroctye culture Sup (MC+Lcn2) or with Vehicle (Control) in presence of S100B inhibitor FPS-ZM1, or in its absence (***p< 0.001, ns=non-significant). (H.) Colocalization events of β-tubulin (red) and Cleaved Caspase3 (green) immunoreactivity counterstained with DAPI in Neuro-2a cells exposed without or with FPS-ZM1 and treated with vehicle (Control), 1nM S100B, 1 μM S100B, and Cell culture Sup (MC+Lcn2) separately. All images were taken at × 40 magnification (Scale: 50 μm). (I.) Morphometric analysis of β-tubulin and Cleaved Caspase3 colocalization events [mean data plotted on y-axis was measured as % positive immunoreactive area (% ROI) in arbitrary light units from three different microscopic fields] in Neuro-2a cells treated with vehicle (Control), 1nM S100B, 1 μM S100B, and Astrocyte culture Sup (MC +Lcn2) separately (*p< 0.05, **p< 0.01, ***p<0.001, ns=non-significant). All statistical significance was tested by performing unpaired t-test between the groups (*p< 0.05, **p< 0.01, ***p< 0.001, ns=non-significant), followed by Bonferroni Dunn Post hoc corrections. (J) Comparative analysis of Cleaved caspas3 and β-tubulin colocalization in Neuro-2a cells, exposed with Astroctye culture Sup (MC+Lcn2) or with Vehicle (Control) in presence of S100B inhibitor FPS-ZM1, or in its absence (**p< 0.01, ns=non-significant).
Next, we studied the protein expression of COX-2, iNOS, and apoptosis marker Bax and Bcl2 with the brain protein extracts. The above proteins have been reported to be induced by increased S100B levels as has been previously reported [31]. Notably, results showed that iNOS protein expression was significantly increased following MC-LR exposure on mice with CHOW diet as well as in NAFLD/NASH mouse group (Fig 4C), however, no significant difference was observed in the iNOS levels in groups CHOW+MC and MCD-HFD+MC group. Subsequently, results showed significantly elevated expression of COX-2 in the MCD-HFD+MC group when compared to CHOW +MC group. Interestingly, MCD-HFD+MC/NLRP3KO mouse also showed significantly higher expression of both iNOS and COX-2 proteins expression compared to the MCD-HFD+MC mouse group suggesting NLRP3 might not affect the levels of the above proteins in the brain tissue. Since neuronal apoptosis is observed in degeneration, we studied apoptosis by analyzing the relative expression of pro-apoptotic Bax protein and anti-apoptotic Bcl2 proteins. Results showed that MC-LR exposure in the normal chow diet group resulted in a 5-fold increased ratio of Bax/Bcl2 when compared to the CHOW control diet group. Interestingly, Bax/Bcl2 ratio was largely unaltered between MC exposed or unexposed mice that were fed with high fat diet+MCD). However, NLRP3 knock-out mice that were exposed to MCD-HFD had a significant increase in Bax/Bcl2 ratio suggesting higher neuronal apoptosis triggered by the absence of NLRP3 and is consistent with our previous findings that S100B associated increased iNOS, COX-2, and subsequent apoptosis were independent of NLRP3.
To advance our understanding of the association of higher S100B levels in the brain tissue following MC exposure, we resorted to understand the mechanism by using an in vitro model. Primary mouse astrocyte cells were primed with Lcn2 to mimic the condition likely prevalent in underlying NAFLD/NASH, followed by treatment with 20 µm MC-LR. Expression of S100B was quantified by performing ELISA from the culture supernatants of the different treatment groups. Results showed cells, primed with Lcn2 followed by MC-LR exposure induced significantly higher extracellular S100B secretion when compared to untreated cells. Most importantly, inhibition by CRID3, a NLRP3 inhibitor failed to decrease elevated S100B expression (Supplementary Fig. 1). The above results support our findings in the in vivo study that showed increased apoptosis in the NLRP3 KO mice group.
Culture supernatant of astrocytes from the above experimental group MC-LR+Lcn2 was concentrated and treated (conditioned medium treatment) in mouse neuronal cells Neuro-2a for 24 h. 500 µL volume of the concentrated supernatant (119.5 pM S100B) was used for treatment in Neuro-2a cells in the presence or absence of a RAGE antagonist 230 nM FPS-ZM1 to show the direct involvement of S100B since the above directs its action via the RAGE receptor. 3-nitrotyrosine and Cleaved Caspase 3 expressions were studied by immunofluorescence to confirm oxidative stress and apoptosis in Neuro-2a cells. Mouse recombinant S100B was used as a positive control for this study. Results showed that supernatant of astrocyte cells (conditioned medium) significantly increased Cleaved Caspase 3 and 3-nitrotyrosine expression in Neuro-2a cells (Fig 4E and H). Inhibition of RAGE receptor with FPS-ZM1 showed a 3-fold decreased expression of Cleaved Caspase 3 and a 2-fold decreased expression of 3NT, respectively. The results suggested that MC-LR via its actions on the release of S100B from the astrocytes plays a pivotal role in neuronal cell apoptosis and increased oxidative stress.
Discussion:
NAFLD or its progressive inflammatory counterpart NASH has been shown to induce ectopic manifestations that include neuroinflammation and AD-like pathology[26] [32]. In the current study, we show that environmental MC-LR exposure in an underlying NAFLD aggravated neuronal pathology by causing heightened neuroinflammation and blood-brain barrier dysfunction primarily through a NLRP3 dependent process. Further, a MC-LR exposure could cause astrocyte activation and neuronal apoptosis via S100B that was NLRP3 independent thus pointing to a two-pronged mechanism from the single MC-LR trigger. Previous studies have shown that MC-LR can cross BBB and induce neurotoxicity by inducing structural, functional, and behavioral changes [10]. Histopathological changes and oxidative stress in the hippocampus were observed by Li et al in MC-LR exposed rat model [33]. A dose-dependent exposure of MC-LR in nematodes has been shown to cause increased neuronal loss due to Tau protein aggregation and apoptosis and can lead to behavioral changes in animals [34]. In a recent study, Wang et al showed that MC-LR induced BBB dysfunction by impairing tight junction proteins in mouse brain and cultured brain endothelial cells [35]. Our current study supports the findings by Wang et al. Notably, we observed an extensive degradation of tight junction proteins (TJ) Claudin 5, and ZO1 in both in vivo and in vitro studies on MC-LR exposure in underlying NAFLD/NASH suggesting the role of this environmental cyanotoxin in exacerbation of brain pathology. TJ proteins especially Claudin 5 are expressed on brain microvascular endothelial cells and maintain the integrity of BBB [36]. Disruption and decreased expression of Claudin 5 increase the permeability and cause BBB dysfunction. It is to be mentioned though that we reported an increased Lcn2 in the circulation in a similar NASH model previously that was associated with neuroinflammation [26]. We found a similar increase of Lcn2 in the present study that was increased over and above the levels found in our previous NASH study. The increased levels of Lcn2 following MC-LR exposure in the serum certainly correlated with increased neuroinflammation in the present study. However, mechanistic and clear evidence of the systemic Lcn2 involvement is lacking since we did not use liver-specific knockout mice to study its specific role. To overcome the lack of specific evidence for the role of Lcn2 in the in vivo mouse model, we chose to use a cell-based model to provide mechanistic insights into the BBB-TJ protein expression. Our results of a decrease in Claudin5 and Occludin following Lcn2 incubation with MC in brain endothelial cells and a corresponding increase of the protein expression following the use of CRID3, a NLRP3 inhibitor shows that Lcn2 mediation of BBB TJ protein expression is NLRP3 dependent.
It has been shown that IL 1β and TNFα also play active roles in neuroinflammation and subsequent BBB dysfunction. In our present study, we showed elevated expression of proinflammatory cytokines IL 1β, IL 6, and TNF α in brain tissue sections of the NAFLD/NASH murine model when exposed to MC-LR. Furthermore, IL 1β expression was found to be significantly high compared to other overexpressed proinflammatory markers such as TNFα and IL 6. IL 1β is considered a major proinflammatory mediator in the brain that elicits rapid and robust neuroinflammation in vivo by activating microglia and astrocytes [37]. Parenchymal expression of IL 1β increases expression of proinflammatory cytokines such as IL 6 and TNFα, chemokines, cyclooxygenase 2 (COX-2), and matrix metalloproteases [38]. IL 1β also increases BBB permeability by suppressing astrocytic sonic hedgehog production and increased chemokine secretion that leads to exacerbated BBB dysfunction [39]. MMPs, especially MMP9 has been reported to play an important role in BBB impairment by degradation of tight junction proteins Claudin 1, Occludin, and ZO1 [40]. Previous studies have described the role of proinflammatory cytokines especially IL 1β and TNFα in MMP9 overexpression [41], [42]. In parallel, proinflammatory cytokine TNFα also has been shown to increase MMP 9 transcription by AP1 and NF-kB activation. Interestingly, our results in vivo showed a robust increase of MMP9 parallel to increases in IL 1β and TNFα suggesting that a plethora of proinflammatory stimuli arising from a NLRP3-Lcn2 axis may have played a significant role in exacerbating the MCLR-induced neuroinflammation and BBB dysfunction. This is also supported by the fact that IL 1β levels in brain endothelial cell culture supernatants decreased significantly following co-incubation with the NLRP3 inhibitor CRID3.
The significant increase in BBB-TJ proteins in NLRP3 KO mice administered MC-LR had parallel decreases in serum S100B levels. Clinical and basic research studies have demonstrated that increased serum S100B level is correlated with BBB disruption and suggested to be a good prognostic indicator for BBB injury [43]. The primary source of S100B is thought to be the reactive astrocytes and it is restricted to brain parenchyma in normal physiological conditions. S100B possesses both beneficial and detrimental effects on neuropathology depending on its concentration in the microenvironment. Reports also suggested that S100B binding to its RAGE receptors expressed on different brain cell types in nanomolar concentration promotes neuronal survival and astrocytic proliferation, whereas the micromolar concentration of S100B has been shown to be detrimental as it induces oxidative stress, neuronal cell apoptosis, and proinflammatory responses and that has been correlated with several neuropathology such as AD and Down syndrome [44], [45], [46], [47]. Astrocyte activation as shown by increased S100B protein was evident in MC-LR exposed NAFLD mice that was not NLRP3 dependent. Our results also showed that MC-LR exposure in NAFLD/NASH triggered robust increased levels of COX-2, and iNOS that are known to be associated with oxidative injury in the neurons following MC-LR exposure underlying NAFLD/NASH. Further, to show the causal link between increased astrocyte activation-led S100B release in causing neuronal apoptosis and associated pathology, we used a neuronal cell culture model where conditioned medium from MC-LR-Lcn2 stimulated Astrocyte culture was used in addition to a S100B stimulation. Supernatants from MC-LR- Lcn2- treated astrocytes showed maximum S100B release compared to all corresponding treatment groups and hence was used for treatment in neuronal cells hoping that S100B might be the prime driver for neuronal cell death. Our results of increased oxidative stress and apoptosis in the cultured neurons that used cultured supernatants of MC-LR-Lcn2 containing approximately 120 pM of S100B, showed that increased release of S100 B, presence of Lcn2 and MC-LR in the culture supernatants can all act synergistically to induce cell death in the neurons. However, the use of S100B inhibitor in restricting cell death in the same neurons suggested that S100B may be the principal mediator in the cellular process. Further, though traces of Lcn2 and MC-LR effects may not be ruled out, the effects of such a mechanism might also depend on the residual half-life and effectiveness of MC-LR in the culture supernatant. The independent effects of either Lcn2 or MC-LR in neuronal cells irrespective of the crosstalk should be probed further to ensure the molecular effects of this toxin after they are transported across the BBB. The above results not only showed the role of S100B and MC-LR but also demonstrated a crosstalk between activated astrocytes and neurons in causing a damaged neuronal pathology. This mechanism bears relevance to environmental factors such as HAB toxins to not only advance NAFLD pathology but their ability to induce neuronal comorbidity.
In summary, we show that an environmental cyanotoxin exposure can advance NAFLD pathology not only by acting on the liver but can have longstanding neuronal inflammation and degenerative pathology. To our knowledge, this is the first mechanistic demonstration of a long-term ectopic consequence of microcystin exposure in a translatable model though the results in large human cohorts comprising of NAFLD patients would need to be verified. Further, the significance of cyanotoxin exposure and its effect on human health needs to be taken on a serious footing as more incidents of harmful algal blooms are discovered and we find ourselves being reminded that along with HABs, NAFLD is also now a global pandemic ([48], [49], [50], https://doi.org/10.1186/s12302–019-0212–2).
Supplementary Material
Acknowledgment:
The authors gratefully acknowledge the technical services at the IRF, University of South Carolina School of Medicine, and AML Labs (Baltimore MD). We also thank the Instrumentation resource facility (IRF) at the University of South Carolina for equipment usage and consulting services.
Funding:
This study was supported by NIH grants P20GM103641–06, P01ES028942–01, and P01AT003961 to Saurabh Chatterjee (Project PI).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest:
The authors declare that there is no conflict of financial or competing interests.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Loomba R, Friedman SL and Shulman GI (2021) Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 184:2537–2564. [DOI] [PubMed] [Google Scholar]
- 2.Tilg H and Moschen AR (2010) Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology 52:1836–46. [DOI] [PubMed] [Google Scholar]
- 3.Seth RK, Kumar A, Das S, Kadiiska MB, Michelotti G, Diehl AM and Chatterjee S (2013) Environmental toxin-linked nonalcoholic steatohepatitis and hepatic metabolic reprogramming in obese mice. Toxicol Sci 134:291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Das S, Alhasson F, Dattaroy D, Pourhoseini S, Seth RK, Nagarkatti M, Nagarkatti PS, Michelotti GA, Diehl AM, Kalyanaraman B and Chatterjee S (2015) NADPH Oxidase-Derived Peroxynitrite Drives Inflammation in Mice and Human Nonalcoholic Steatohepatitis via TLR4-Lipid Raft Recruitment. Am J Pathol 185:1944–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L and Wymer M (2016) Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 64:73–84. [DOI] [PubMed] [Google Scholar]
- 6.Wahlang B, Beier JI, Clair HB, Bellis-Jones HJ, Falkner KC, McClain CJ and Cave MC (2013) Toxicant-associated steatohepatitis. Toxicol Pathol 41:343–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dittmann E and Wiegand C (2006) Cyanobacterial toxins--occurrence, biosynthesis and impact on human affairs. Mol Nutr Food Res 50:7–17. [DOI] [PubMed] [Google Scholar]
- 8.Newcombe G, Chorus I, Falconer I and Lin TF (2012) Cyanobacteria: impacts of climate change on occurrence, toxicity and water quality management. Water Res 46:1347–8. [DOI] [PubMed] [Google Scholar]
- 9.Huisman J, Codd GA, Paerl HW, Ibelings BW, Verspagen JMH and Visser PM (2018) Cyanobacterial blooms. Nat Rev Microbiol 16:471–483. [DOI] [PubMed] [Google Scholar]
- 10.Fischer WJ, Altheimer S, Cattori V, Meier PJ, Dietrich DR and Hagenbuch B (2005) Organic anion transporting polypeptides expressed in liver and brain mediate uptake of microcystin. Toxicol Appl Pharmacol 203:257–63. [DOI] [PubMed] [Google Scholar]
- 11.Zegura B, Gajski G, Straser A, Garaj-Vrhovac V and Filipic M (2011) Microcystin-LR induced DNA damage in human peripheral blood lymphocytes. Mutat Res 726:116–22. [DOI] [PubMed] [Google Scholar]
- 12.Feurstein D, Holst K, Fischer A and Dietrich DR (2009) Oatp-associated uptake and toxicity of microcystins in primary murine whole brain cells. Toxicol Appl Pharmacol 234:247–55. [DOI] [PubMed] [Google Scholar]
- 13.Feurstein D, Kleinteich J, Heussner AH, Stemmer K and Dietrich DR (2010) Investigation of microcystin congener-dependent uptake into primary murine neurons. Environ Health Perspect 118:1370–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Greer B, Meneely JP and Elliott CT (2018) Uptake and accumulation of Microcystin-LR based on exposure through drinking water: An animal model assessing the human health risk. Sci Rep 8:4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.MacKintosh C, Beattie KA, Klumpp S, Cohen P and Codd GA (1990) Cyanobacterial microcystin-LR is a potent and specific inhibitor of protein phosphatases 1 and 2A from both mammals and higher plants. FEBS Lett 264:187–92. [DOI] [PubMed] [Google Scholar]
- 16.Xing Y, Xu Y, Chen Y, Jeffrey PD, Chao Y, Lin Z, Li Z, Strack S, Stock JB and Shi Y (2006) Structure of protein phosphatase 2A core enzyme bound to tumor-inducing toxins. Cell 127:341–53. [DOI] [PubMed] [Google Scholar]
- 17.Reynhout S and Janssens V (2019) Physiologic functions of PP2A: Lessons from genetically modified mice. Biochim Biophys Acta Mol Cell Res 1866:31–50. [DOI] [PubMed] [Google Scholar]
- 18.Sarkar S, Kimono D, Albadrani M, Seth RK, Busbee P, Alghetaa H, Porter DE, Scott GI, Brooks B, Nagarkatti M, Nagarkatti P and Chatterjee S (2019) Environmental microcystin targets the microbiome and increases the risk of intestinal inflammatory pathology via NOX2 in underlying murine model of Nonalcoholic Fatty Liver Disease. Sci Rep 9:8742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao Y, Yan Y, Xie L, Wang L, He Y, Wan X and Xue Q (2020) Long-term environmental exposure to microcystins increases the risk of nonalcoholic fatty liver disease in humans: A combined fisher-based investigation and murine model study. Environ Int 138:105648. [DOI] [PubMed] [Google Scholar]
- 20.Albadrani M, Seth RK, Sarkar S, Kimono D, Mondal A, Bose D, Porter DE, Scott GI, Brooks B, Raychoudhury S, Nagarkatti M, Nagarkatti P, Jule Y, Diehl AM and Chatterjee S (2019) Exogenous PP2A inhibitor exacerbates the progression of nonalcoholic fatty liver disease via NOX2-dependent activation of miR21. Am J Physiol Gastrointest Liver Physiol 317:G408–G428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Al-Badrani M, Saha P, Mondal A, Seth RK, Sarkar S, Kimono D, Bose D, Porter DE, Scott GI, Brooks B, Raychoudhury S, Nagarkatti M, Nagarkatti P and Chatterjee S (2020) Early microcystin-LR exposure-linked inflammasome activation in mice causes development of fatty liver disease and insulin resistance. Environ Toxicol Pharmacol 80:103457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zelber-Sagi S, Lotan R, Shlomai A, Webb M, Harrari G, Buch A, Nitzan Kaluski D, Halpern Z and Oren R (2012) Predictors for incidence and remission of NAFLD in the general population during a seven-year prospective follow-up. J Hepatol 56:1145–1151. [DOI] [PubMed] [Google Scholar]
- 23.Meier EM, Pohl R, Rein-Fischboeck L, Schacherer D, Eisinger K, Wiest R, Krautbauer S and Buechler C (2016) Circulating lipocalin 2 is neither related to liver steatosis in patients with non-alcoholic fatty liver disease nor to residual liver function in cirrhosis. Cytokine 85:45–50. [DOI] [PubMed] [Google Scholar]
- 24.Das S, Kumar A, Seth RK, Tokar EJ, Kadiiska MB, Waalkes MP, Mason RP and Chatterjee S (2013) Proinflammatory adipokine leptin mediates disinfection byproduct bromodichloromethane-induced early steatohepatitic injury in obesity. Toxicol Appl Pharmacol 269:297–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ye D, Yang K, Zang S, Lin Z, Chau HT, Wang Y, Zhang J, Shi J, Xu A, Lin S and Wang Y (2016) Lipocalin-2 mediates non-alcoholic steatohepatitis by promoting neutrophil-macrophage crosstalk via the induction of CXCR2. J Hepatol 65:988–997. [DOI] [PubMed] [Google Scholar]
- 26.Mondal A, Bose D, Saha P, Sarkar S, Seth R, Kimono D, Albadrani M, Nagarkatti M, Nagarkatti P and Chatterjee S (2020) Lipocalin 2 induces neuroinflammation and blood-brain barrier dysfunction through liver-brain axis in murine model of nonalcoholic steatohepatitis. J Neuroinflammation 17:201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li XB, Zhang X, Ju J, Li Y, Yin L and Pu Y (2014) Alterations in neurobehaviors and inflammation in hippocampus of rats induced by oral administration of microcystin-LR. Environ Sci Pollut Res Int 21:12419–25. [DOI] [PubMed] [Google Scholar]
- 28.Chen F, Ohashi N, Li W, Eckman C and Nguyen JH (2009) Disruptions of occludin and claudin-5 in brain endothelial cells in vitro and in brains of mice with acute liver failure. Hepatology 50:1914–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Greene C, Hanley N and Campbell M (2020) Blood-brain barrier associated tight junction disruption is a hallmark feature of major psychiatric disorders. Transl Psychiatry 10:373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Villarreal A, Seoane R, Gonzalez Torres A, Rosciszewski G, Angelo MF, Rossi A, Barker PA and Ramos AJ (2014) S100B protein activates a RAGE-dependent autocrine loop in astrocytes: implications for its role in the propagation of reactive gliosis. J Neurochem 131:190–205. [DOI] [PubMed] [Google Scholar]
- 31.Esposito G, Cirillo C, Sarnelli G, De Filippis D, D’Armiento FP, Rocco A, Nardone G, Petruzzelli R, Grosso M, Izzo P, Iuvone T and Cuomo R (2007) Enteric glial-derived S100B protein stimulates nitric oxide production in celiac disease. Gastroenterology 133:918–25. [DOI] [PubMed] [Google Scholar]
- 32.Bosoi CR, Vandal M, Tournissac M, Leclerc M, Fanet H, Mitchell PL, Verreault M, Trottier J, Virgili J, Tremblay C, Lippman HR, Bajaj JS, Barbier O, Marette A and Calon F (2021) High-Fat Diet Modulates Hepatic Amyloid beta and Cerebrosterol Metabolism in the Triple Transgenic Mouse Model of Alzheimer’s Disease. Hepatol Commun 5:446–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li G, Yan W, Cai F, Li C, Chen N and Wang J (2014) Spatial learning and memory impairment and pathological change in rats induced by acute exposure to microcystin-LR. Environ Toxicol 29:261–8. [DOI] [PubMed] [Google Scholar]
- 34.Ju J, Ruan Q, Li X, Liu R, Li Y, Pu Y, Yin L and Wang D (2013) Neurotoxicological evaluation of microcystin-LR exposure at environmental relevant concentrations on nematode Caenorhabditis elegans. Environ Sci Pollut Res Int 20:1823–30. [DOI] [PubMed] [Google Scholar]
- 35.Wang J, Zhang C, Zhu J, Ding J, Chen Y and Han X (2019) Blood-brain barrier disruption and inflammation reaction in mice after chronic exposure to Microcystin-LR. Sci Total Environ 689:662–678. [DOI] [PubMed] [Google Scholar]
- 36.Stamatovic SM, Keep RF and Andjelkovic AV (2011) Tracing the endocytosis of claudin-5 in brain endothelial cells. Methods Mol Biol 762:303–20. [DOI] [PubMed] [Google Scholar]
- 37.Shaftel SS, Kyrkanides S, Olschowka JA, Miller JN, Johnson RE and O’Banion MK (2007) Sustained hippocampal IL-1 beta overexpression mediates chronic neuroinflammation and ameliorates Alzheimer plaque pathology. J Clin Invest 117:1595–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shaftel SS, Griffin WS and O’Banion MK (2008) The role of interleukin-1 in neuroinflammation and Alzheimer disease: an evolving perspective. J Neuroinflammation 5:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Y, Jin S, Sonobe Y, Cheng Y, Horiuchi H, Parajuli B, Kawanokuchi J, Mizuno T, Takeuchi H and Suzumura A (2014) Interleukin-1beta induces blood-brain barrier disruption by downregulating Sonic hedgehog in astrocytes. PLoS One 9:e110024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jiang X, Andjelkovic AV, Zhu L, Yang T, Bennett MVL, Chen J, Keep RF and Shi Y (2018) Blood-brain barrier dysfunction and recovery after ischemic stroke. Prog Neurobiol 163–164:144–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee WJ, Shin CY, Yoo BK, Ryu JR, Choi EY, Cheong JH, Ryu JH and Ko KH (2003) Induction of matrix metalloproteinase-9 (MMP-9) in lipopolysaccharide-stimulated primary astrocytes is mediated by extracellular signal-regulated protein kinase 1/2 (Erk1/2). Glia 41:15–24. [DOI] [PubMed] [Google Scholar]
- 42.Yokoo T and Kitamura M (1996) Dual regulation of IL-1 beta-mediated matrix metalloproteinase-9 expression in mesangial cells by NF-kappa B and AP-1. Am J Physiol 270:F123–30. [DOI] [PubMed] [Google Scholar]
- 43.Rothermundt M, Ponath G, Glaser T, Hetzel G and Arolt V (2004) S100B serum levels and long-term improvement of negative symptoms in patients with schizophrenia. Neuropsychopharmacology 29:1004–11. [DOI] [PubMed] [Google Scholar]
- 44.Huttunen HJ, Kuja-Panula J, Sorci G, Agneletti AL, Donato R and Rauvala H (2000) Coregulation of neurite outgrowth and cell survival by amphoterin and S100 proteins through receptor for advanced glycation end products (RAGE) activation. J Biol Chem 275:40096–105. [DOI] [PubMed] [Google Scholar]
- 45.Griffin WS, Sheng JG, Royston MC, Gentleman SM, McKenzie JE, Graham DI, Roberts GW and Mrak RE (1998) Glial-neuronal interactions in Alzheimer’s disease: the potential role of a ‘cytokine cycle’ in disease progression. Brain Pathol 8:65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bianchi R, Giambanco I and Donato R (2010) S100B/RAGE-dependent activation of microglia via NF-kappaB and AP-1 Co-regulation of COX-2 expression by S100B, IL-1beta and TNF-alpha. Neurobiol Aging 31:665–77. [DOI] [PubMed] [Google Scholar]
- 47.Mori T, Asano T and Town T (2010) Targeting S100B in Cerebral Ischemia and in Alzheimer’s Disease. Cardiovasc Psychiatry Neurol 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Buratti FM, Manganelli M, Vichi S, Stefanelli M, Scardala S, Testai E and Funari E (2017) Cyanotoxins: producing organisms, occurrence, toxicity, mechanism of action and human health toxicological risk evaluation. Arch Toxicol 91:1049–1130. [DOI] [PubMed] [Google Scholar]
- 49.Svircev Z, Drobac D, Tokodi N, Mijovic B, Codd GA and Meriluoto J (2017) Toxicology of microcystins with reference to cases of human intoxications and epidemiological investigations of exposures to cyanobacteria and cyanotoxins. Arch Toxicol 91:621–650. [DOI] [PubMed] [Google Scholar]
- 50.Svircev Z, Lalic D, Bojadzija Savic G, Tokodi N, Drobac Backovic D, Chen L, Meriluoto J and Codd GA (2019) Global geographical and historical overview of cyanotoxin distribution and cyanobacterial poisonings. Arch Toxicol 93:2429–2481. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.