

Abstract
Models that examine genetic differences between populations alongside a genotype–phenotype map can provide insight about phenotypic variation among groups. We generalize a simple model of a completely heritable, additive, selectively neutral quantitative trait to examine the relationship between single-locus genetic differentiation and phenotypic differentiation on quantitative traits. In agreement with similar efforts using different models, we show that the expected degree to which two groups differ on a neutral quantitative trait is not strongly affected by the number of genetic loci that influence the trait: neutral trait differences are expected to have a magnitude comparable to the genetic differences at a single neutral locus. We discuss this result with respect to population differences in disease phenotypes, arguing that although neutral genetic differences between populations can contribute to specific differences between populations in health outcomes, systematic patterns of difference that run in the same direction for many genetically independent health conditions are unlikely to be explained by neutral genetic differentiation.
Keywords: GENETIC DIFFERENTIATION, HEALTH DISPARITIES, POPULATION GENETICS, QUANTITATIVE GENETICS
1. Introduction
Since Lewontin’s (1972) landmark partitioning of human genetic diversity, many studies have supported his claim that allele-frequency differences between geographically defined groups of people are relatively modest (Barbujani et al. 1997; Brown and Armelagos 2001; Rosenberg et al. 2002; Li et al. 2008). The findings of these previous studies have often been reported as estimates of FST, which can be interpreted as the proportion of variance in an allelic indicator variable attributable to allele-frequency differences between populations (Holsinger and Weir 2009). Estimates of worldwide human FST and FST-like quantities have ranged from ~0.05 (e.g., Rosenberg et al. 2002) to ~0.15 (e.g., Barbujani et al. 1997).
Human FST estimates suggest that for phenotypes governed by a single typical genetic locus, population membership is likely to account for a relatively small proportion of the total variance of the trait. However, phenotypes are generally influenced by many loci, not just one. Large sets of loci can contain a great deal of information about population membership and can permit highly accurate ancestry inference, even if each locus has a small FST (Smouse et al. 1982; Bowcock et al. 1994; Mountain and Cavalli-Sforza 1997; Rosenberg et al. 2002; Bamshad et al. 2003; Edwards 2003; Li et al. 2008). Should we expect traits that are influenced by many loci to aggregate information about population membership across loci, leading to differences between populations that are more pronounced than those observed for traits influenced by fewer loci?
This question has been examined in many population-genetic and quantitative-genetic studies, both in theoretical models (Felsenstein 1973, 1986; Chakraborty and Nei 1982; Rogers and Harpending 1983; Lande 1992; Spitze 1993; Lynch and Spitze 1994; Whitlock 1999; Berg and Coop 2014; Edge and Rosenberg 2015) and in empirical applications in humans (Roseman 2004; Roseman and Weaver 2004; Weaver et al. 2007; Relethford 2010) and other organisms (for reviews, see Whitlock 2008; Leinonen et al. 2013), finding that, in the absence of selection, the expected degree to which groups differ on an additive, genetically determined trait does not depend on the number of loci that influence the trait. Put differently, a typical neutral trait conveys roughly the same degree of information about population membership as a single neutral locus, even if the trait is influenced by a large set of loci that would, if considered directly, permit accurate classification by population of origin.
Recently, to facilitate direct comparisons of multilocus genetic classification, single-locus genetic differentiation, and phenotypic differentiation, we developed a model that combines a simple model of multilocus genetic classification with a simple genotype–phenotype map. Our model enables genotype–phenotype comparisons to be performed in a statistical framework that permits exact computation and does not require detailed evolutionary assumptions (Edge and Rosenberg 2015). Our results agreed with those found with other models, highlighting the differences between polygenic phenotypic differentiation and information about population membership at multiple genetic loci.
In our past work (Edge and Rosenberg 2015), we applied strong assumptions about the allele-frequency distribution, and we examined only haploids. Here, we extend our earlier model to allow arbitrary allele-frequency distributions and arbitrary ploidy. Our results provide another way of establishing the result that between-group differentiation on a neutral trait mirrors between-group genetic differentiation at a neutral locus, one that makes minimal evolutionary assumptions. In Section 2, we describe our extended model. In Section 3, we define several measurements of between-group genetic differentiation. In Sections 4 and 5, we describe properties of two statistics that summarize the degree of difference between two populations on a quantitative trait. In Section 6, we introduce two simplifying assumptions that allow us to analyze the problem of inferring an individual’s population of origin using either genetic or phenotypic information. Finally, we discuss the results with respect to the interpretation of population differences in disease phenotypes. Figure 1 provides a conceptual map of the structure of the article.
FIGURE 1.
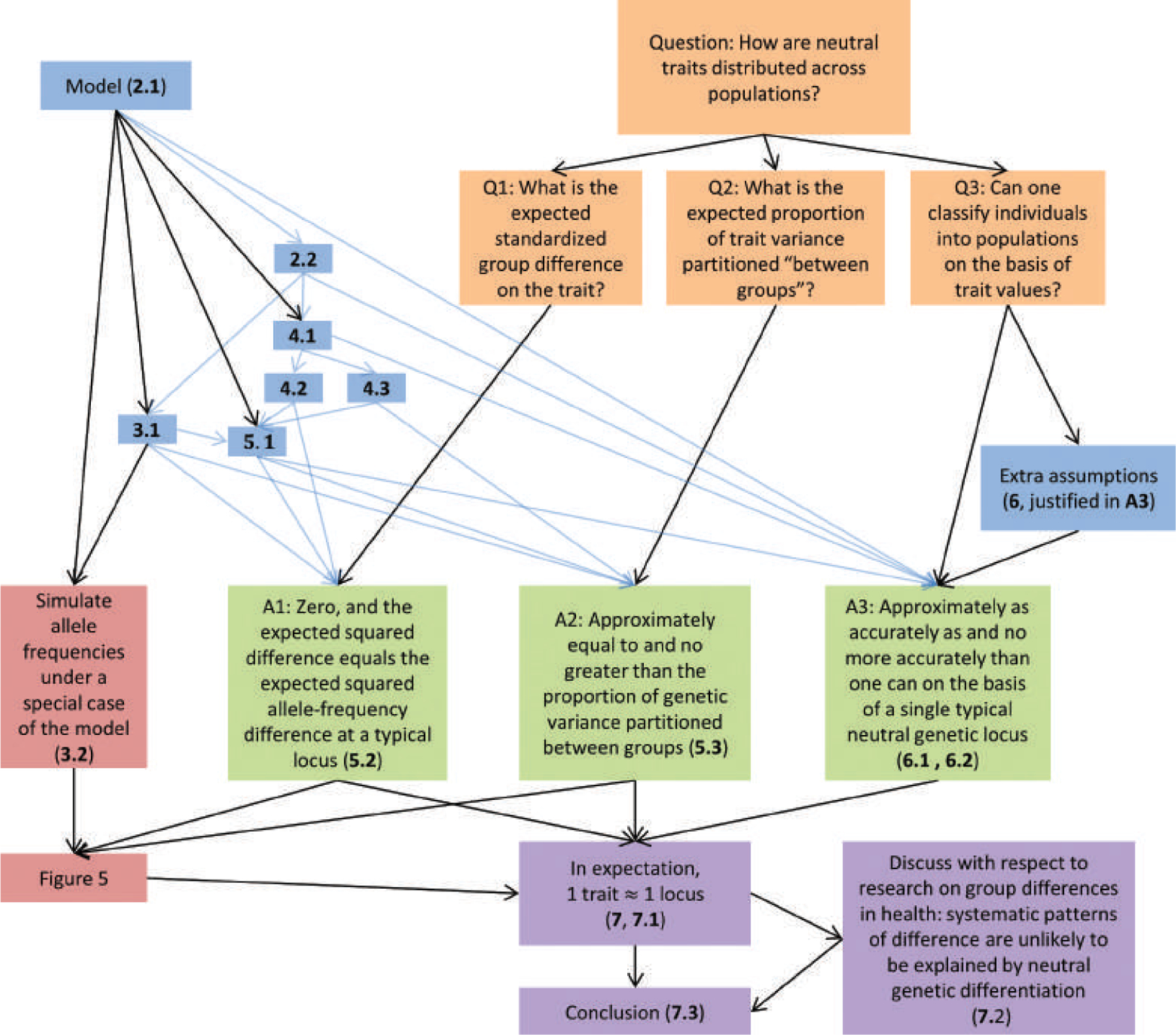
A conceptual map of this article. For boxes that correspond to specific subsections of the article, the subsection number is displayed in bold. Arrows indicate conceptual dependence, with blue arrows indicating that a subsection cites mathematical results obtained in another subsection. Motivating questions are in orange, mathematical machinery is in blue, simulations are in red, mathematical answers are in green, and interpretation of results is in purple.
2. Preliminaries
2.1. Model
Our extended model is parallel to our previously reported model (Edge and Rosenberg 2015) and is similar to models used by Risch et al. (2002), Edwards (2003), and especially Tal (2012) to investigate the problem of classifying individuals into populations using multilocus genetic data. For a summary of our notation, see Table 1.
Table 1.
Summary of Notation
| Symbol | Meaning |
|---|---|
| k | The number of loci that influence a quantitative trait |
| ℓ | The ploidy of the individuals being considered |
| M | An individual’s population membership; takes values A and B |
| L ij | An individual’s allelic type at the jth allele at the ith locus; takes values 0 and 1 |
| p i | The frequency of the 1 allele at locus i in population A (Eq. 1) |
| q i | The frequency of the 1 allele at locus i in population B (Eq. 1) |
| The mean frequency of the 1 allele across loci in population A (Eq. 2) | |
| The mean frequency of the 1 allele across loci in population B (Eq. 2) | |
| The variance across loci in the frequency of the 1 allele in population A (Eq. 3) | |
| The variance across loci in the frequency of the 1 allele in population B (Eq. 3) | |
| V ij | An indicator for whether an individual’s jth allele at the ith locus is a + allele (Eq. 4) |
| T | An individual’s value for a quantitative trait (Eq. 4) |
| X i | An indicator for whether the 0 or the 1 allele is also the + allele at locus i (Eq. 5) |
| S | The number of 1 alleles an individual carries (Eq. 6) |
| δ i | The difference between populations in the frequency of the 1 allele at locus i (Eq. 8) |
| The mean allele-frequency difference between populations, or (Eq. 9) | |
| The mean squared difference between populations in the frequency of the 1 allele (Eq. 10) | |
| The ratio of the mean (across k loci) within-population variance in an allelic indicator variable to the mean (across k loci) total variance in an allelic indicator variable (Eq. 14) | |
| A function of that bears the same relationship to as the square of Cohen’s d does to r2 (Eq. 15) | |
| A generalization of for the sum of ℓ independent allelic indicator variables (Eq. 16) | |
| A function of that bears the same relationship to as the square of Cohen’s d does to r2 (Eq. 17) | |
| U i | A transformation of the Xi: if Xi = 1, then Ui = 1; if Xi = 0, then Ui = −1 (Eq. 18) |
| D T | The standardized difference between populations A and B on the trait (Eqs. 25, 34) |
| The proportion of the total variance in the trait attributable to between-population difference on the trait (Eqs. 26, 27, 41) |
|
| Q ST | A quantitative-trait analogue of FST: if ℓ = 1(haploid organisms), then (Eq. 28) |
| W S | A quantity that equals 1 if an individual is classified into the wrong population on the basis of its value of S, and that equals 0 otherwise (Eq. 55) |
| W T | A quantity that equals 1 if an individual is classified into the wrong population on the basis of its value of T, and that equals 0 otherwise (Eq. 56) |
We consider two populations of equal size, labeled A and B. In each individual, we consider k biallelic genetic loci. Each individual is ℓ-ploid (ℓ ≥ 1), carrying ℓ copies of each locus. At each locus, the allelic type more common in population B than in population A is labeled “1,” and the other allelic type is labeled “0.” Conditional on population membership, all of an individual’s alleles are independent—both alleles at the same locus, as under Hardy-Weinberg equilibrium, and alleles at distinct loci, as under linkage equilibrium.
Let Lij be an indicator random variable denoting whether the jth allele (1 ≤ j ≤ ℓ) at locus i is the “1” allele, and let M be a random variable that represents an individual’s population membership and that takes values A and B. The conditional probabilities that Lij = 1 are
| (1) |
The pi and qi obey 0 ≤ pi ≤ qi ≤ 1. The constraint qi ≥ pi holds because, by definition, the 1 allele is more common in population B than in population A. We also assume that, for at least one value of i, pi or qi does not equal 0 or 1; and for the limiting results in Section 6, we assume that, as the number of loci k approaches infinity, the number of loci at which pi ∈ (0, 1) and the number of loci at which qi ∈ (0, 1) both approach infinity.
Define and as the means across loci of the allele frequencies pi and qi:
| (2) |
Define and as the variances across loci of the pi and qi, though they are not probabilistic variances because the pi and qi are nonrandom:
| (3) |
We model a completely heritable, selectively neutral, additively determined trait as a function of the k loci described above. Specifically, an individual’s value on the trait—represented by a random variable T—is a weighted sum of the individual’s Lij values. As in our previous work (Edge and Rosenberg 2015, Eq. 9), the weights are determined by labels at each locus, where for each trait we label one allele at each locus—either the 0 allele or the 1 allele—as the “+” allele and the other allele as the “−” allele. T is then equal to the number of + alleles carried by the individual. That is,
| (4) |
where Vij = 1 if the jth allele at the ith locus is a + allele and Vij = 0 otherwise.
Again following our previous work (Edge and Rosenberg 2015, Eq. 7), we assume that whether an allele is more common in population B than in population A (i.e., labeled “1”) is independent of whether it is associated with larger trait values than the other allele (i.e., labeled “+”). This claim amounts to assuming that the alleles at k loci have not been under selection and have reached their current frequencies independently of their effect on the trait. We express this assumption with the random variable Xi, with Xi = 0 if the 0 allele and the + allele are identical at the ith locus and Xi = 1 if the 1 allele and the + allele are identical at the ith locus. Each trait is associated with a set of k values for the Xi, and for each of the k loci,
| (5) |
independently of the Xi for the other loci.
We introduce a statistic for comparison with the trait value T. We summarize the information about population membership available at an individual’s k loci with the genotypic statistic S, the total number of 1 alleles—that is, alleles that are more common in population B than in population A—at k loci:
| (6) |
S is not generally an optimal basis for distinguishing members of population A and B (e.g., Tal 2012)—in principle, we could improve classification by more heavily weighting loci that have a greater allele frequency difference between populations—but we will show that classifications based on S approach perfect accuracy as k increases, as long as .
Figure 2 shows a schematic of our model. The model reduces to the one described in Edge and Rosenberg (2015) if one assumes (a) that pi = p and qi = q for all i, (b) that q = 1 − p, and (c) that the organisms being examined are haploid (ℓ = 1).
FIGURE 2.
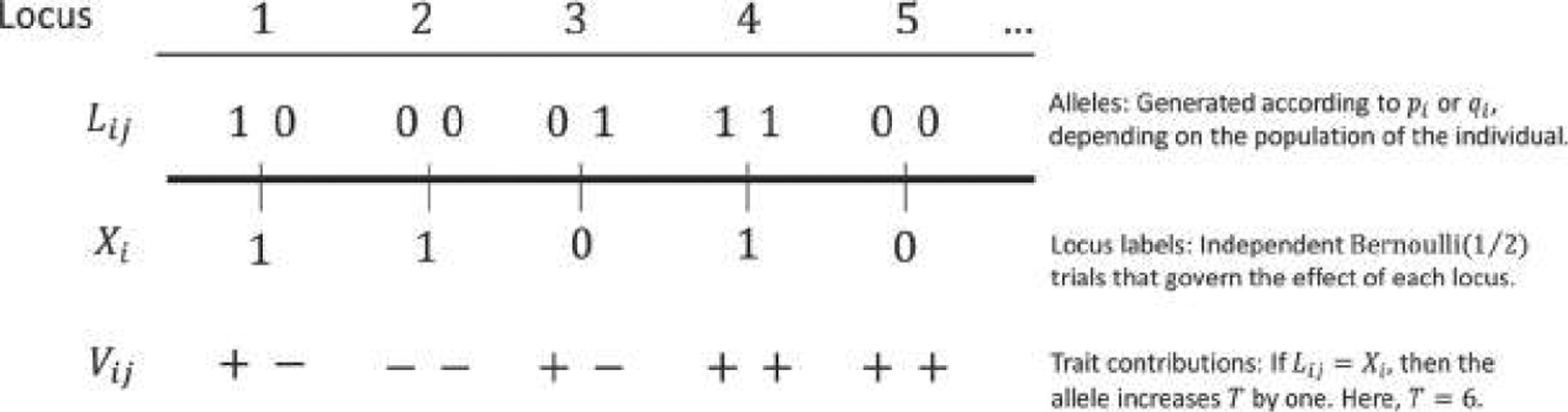
A schematic of our model for generating a quantitative trait. Five loci are shown for a diploid individual. The Lij are the individual’s alleles, which, conditional on population membership, are independent Bernoulli trials with probability at locus i equal either to pi (if the individual is drawn from population A) or to qi (if the individual is drawn from population B). At each locus, the frequency of the 1 allele is at least as large in population B as it is in population A. The Xi are labels indicating which allele at locus i leads to larger values of T; they are independent Bernoulli trials, each with probability ½. If an individual’s jth allele at locus i (Lij) matches the allele that leads to larger values of the trait for that locus (Xi), then Vij takes the value “+”; otherwise, Vij takes the value “−”. T is equal to the sum of + alleles carried by the individual. In the case pictured, T = 6.
2.2. The Poisson Binomial Distribution
Under our model, many relevant quantities have a Poisson binomial distribution, which arises when independent Bernoulli trials with possibly varying success probabilities are summed. By the central limit theorem, the Poisson binomial distribution converges to a normal distribution as the number of terms summed increases without bound, provided that the sum of the variances of the Bernoulli random variables approaches infinity (Deheuvels et al. 1989, Theorem 1.1). If Z is a Poisson binomial random variable with probabilities p1, …, pk, then
| (7) |
where is as in Eq. 2 and is as in Eq. 3 (e.g., Edwards 1960).
3. Genetic Differentiation between Populations at a Single Locus
On the basis of our model, we define several statistics measuring the degree of genetic differentiation between populations at a single typical locus. In Section 5, we use these statistics to compare the degree of genetic differentiation at a typical locus to the expected degree of difference between populations in a neutral trait.
3.1. Single-Locus Differentiation Measures
One summary of the degree of single-locus genetic differentiation between populations is the difference between populations in the frequency of the 1 allele at the locus:
| (8) |
We also define
| (9) |
| (10) |
where , and
| (11) |
where .
The quantity δ (Eq. 8), the difference in population frequencies of one specific allele at a biallelic locus, is closely related to FST for a single locus. Specifically, with two populations and dropping the subscript i, single-locus FST, which we write as , is
| (12) |
(e.g., Weir 1996; Rosenberg et al. 2003, Eq. 8; Holsinger and Weir 2009, Eq. 4). , and δ2 never deviates from by more than (5√5 − 11)/2 ≈ 0.0902 (Rosenberg et al. 2003). can be interpreted in terms of a ratio involving heterozygosity in the subpopulations and in the total population (Nei 1973), or as a ratio of variance components (Holsinger and Weir 2009); we emphasize the latter interpretation. Specifically, if L is an allelic indicator variable representing a single copy of a locus and M denotes population membership, then for a single locus,
| (13) |
The subscript M indicates that the variance in the numerator is taken with respect to group membership. Equation 13 can be verified using the law of total variance, noting that VarM[E(L|M)] = δ2/4 and that EM[Var(L|M)] = [p(1 − p) + q(1 − q)]/2, and comparing with Eq. 12.
To summarize the overall degree of genetic differentiation at a group of k loci, we define an FST measure that summarizes the typical degree of differentiation at a locus chosen from a set of k loci, which we write as . For locus i, Li1 is an allelic indicator variable representing one copy of the locus. To compute , we sum the variance components that appear in Eq. 13 across all k loci and take their ratio:
| (14) |
This ratio is analogous to estimators of FST that involve a ratio of two variance estimates (e.g., Weir and Cockerham 1984, Eq. 10). In our model, however, is known and not estimated because the allele frequencies are known. Though is intended as an index of the degree of genetic differentiation at a single typical locus, is not equal to the mean of the values computed for each locus separately. Rather, it is the ratio of the mean across loci of the between-group variance in allelic type to the mean across loci of the total variance in allelic type.
One interpretation of FST is as the proportion of the variance removed from an indicator variable for one copy of an allele by conditioning on population membership. FST is thus analogous to r2, a measurement of effect size commonly used in meta-analysis, which can be interpreted as the proportion of variance in a dependent variable that is removed by conditioning on an independent variable (Fox 1997: 94). Another commonly used effect-size measurement applicable to differences between two groups is Cohen’s d (Cohen 1988), the difference in group means on a dependent variable divided by the square root of the mean across groups of within-group variances of the independent variable. For equally sized groups, Cohen’s d is related to r2 by d2 = 4r2/(1 − r2) (by inverting Rosenthal 1994, Eqs. 16–24). By analogy, we define another measurement of between-group genetic differentiation across a set of loci:
| (15) |
for a set of loci is not generally equal to the mean across loci of the value that would result by applying Eq. 15 to each locus separately. Rather, like , is a ratio of two means across loci—the mean of the and the mean within-group variance in allelic type.
is a variance partition for allelic indicator variables representing one copy of a locus. At a diploid or polyploid biallelic locus, each copy of the locus provides information about population membership, so more information is available at the locus than is reflected in one copy. We thus define an analogue of for a set of ℓ-ploid loci by partitioning the variance of the sum of the number of 1 alleles at each locus into between-group and within-group components. For a single locus, the between-group variance of the sum is
and by the independence of the allelic copies at a single locus, the within-group variance is
To define , we sum these terms across loci to construct a ratio of the between-group variance to the total variance:
| (16) |
We show in Appendix 1 that with if and only if or . Figure 3 shows the relationship between and for several values of ℓ, illustrating the relative increase in compared with as ℓ increases. Figure 3 also illustrates, as shown in Appendix 1, that is comparable to for close to 0 and comparable to for close to 1.
FIGURE 3.
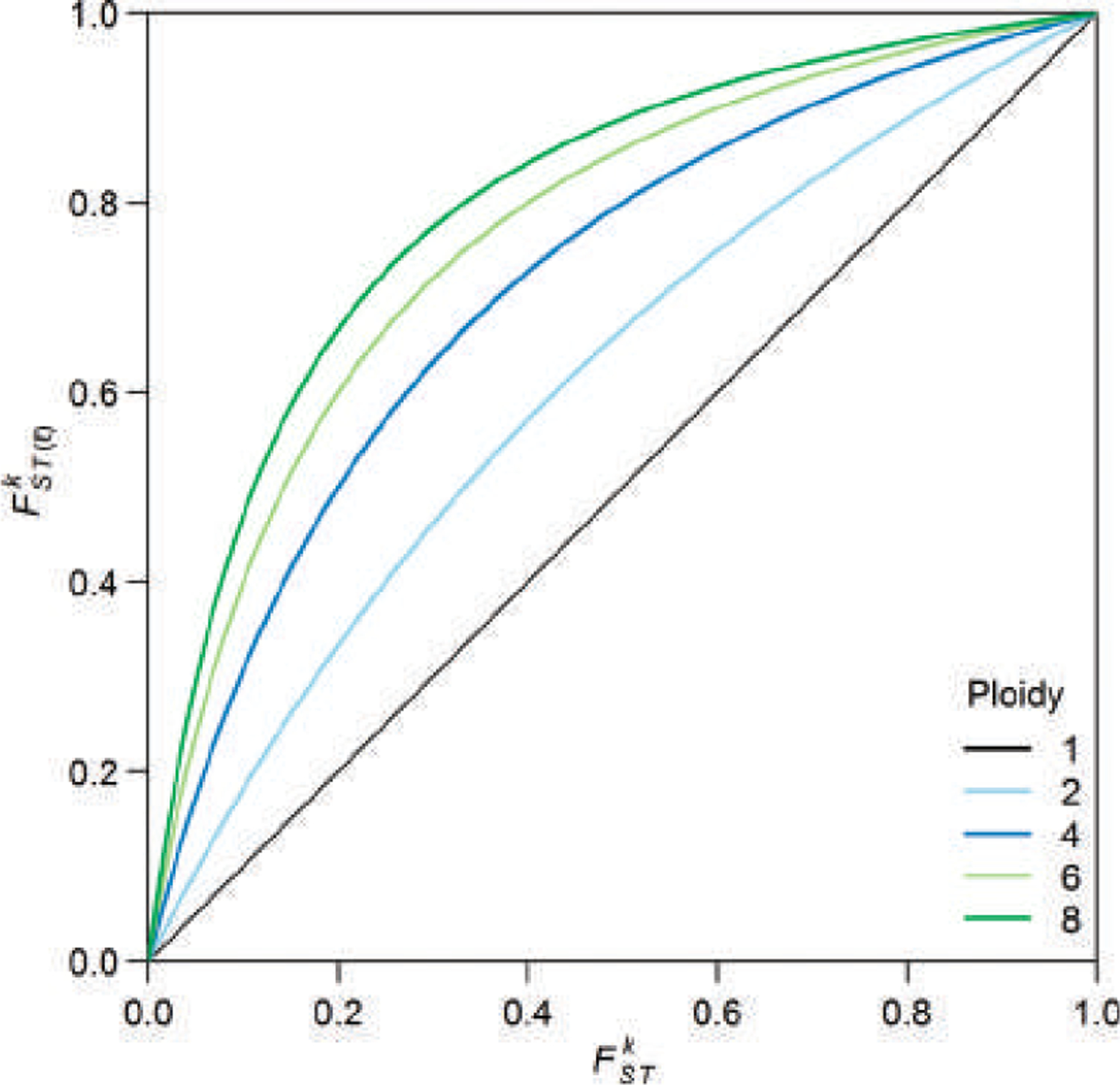
The relationship between (Eq. 14), which partitions the variance of allelic indicator variables representing a single copy of each locus, and (Eq. 16), which partitions the variance of sums of ℓ allelic indicator variables at each locus. Thus, for haploids (ℓ = 1), . For higher ploidy, , with if and only if or (see Appendix 1). The plot is obtained from Eq. A1.4.
Similarly, we can define an analogue of for an ℓ-ploid locus:
| (17) |
Whereas and can be viewed as indices of the amount of information about population membership available in a single copy of a typical locus, and assess the total amount of population membership information at a typical locus, considering all ℓ copies.
3.2. Simulation-Based Allele Frequency Differences
Because some of our results depend on specific characteristics of the pi and qi, we simulated allele frequencies under a model similar to that of Nicholson et al. (2002) to obtain suitable example distributions for the pi and qi (see Figure 4 for a schematic). Specifically, we generated allele frequencies for derived alleles in an ancestral population according to the neutral site frequency spectrum with 2N = 20,000, choosing each allele frequency πi according to P(πi = j/(2N)) ∝ 1/j (Charlesworth and Charlesworth 2010, Eq. B6.6.1). To simulate drift after divergence, we produced postdivergence allele frequencies by adding to each “ancestral” allele frequency πi an independently drawn Normal(0, 0.3πi(1 − πi)) random number, where 0.3 is chosen so that approximates worldwide human FST estimates. Any postdivergence allele frequencies less than 0 or greater than 1 were set to 0 or 1, respectively. After simulating postdivergence frequencies of the derived allele independently in two populations, we assigned the frequencies of either the ancestral or the derived allele in each population to be pi and qi, requiring qi ≥ pi. We generated 106 pairs of allele frequencies (pi, qi) after removing loci at which the same allele fixed in both populations. (Such loci do not contribute to or .) For our simulated allele frequencies, , , , , , , , and .The value of 0.099 is similar to estimates of FST for human populations.
FIGURE 4.
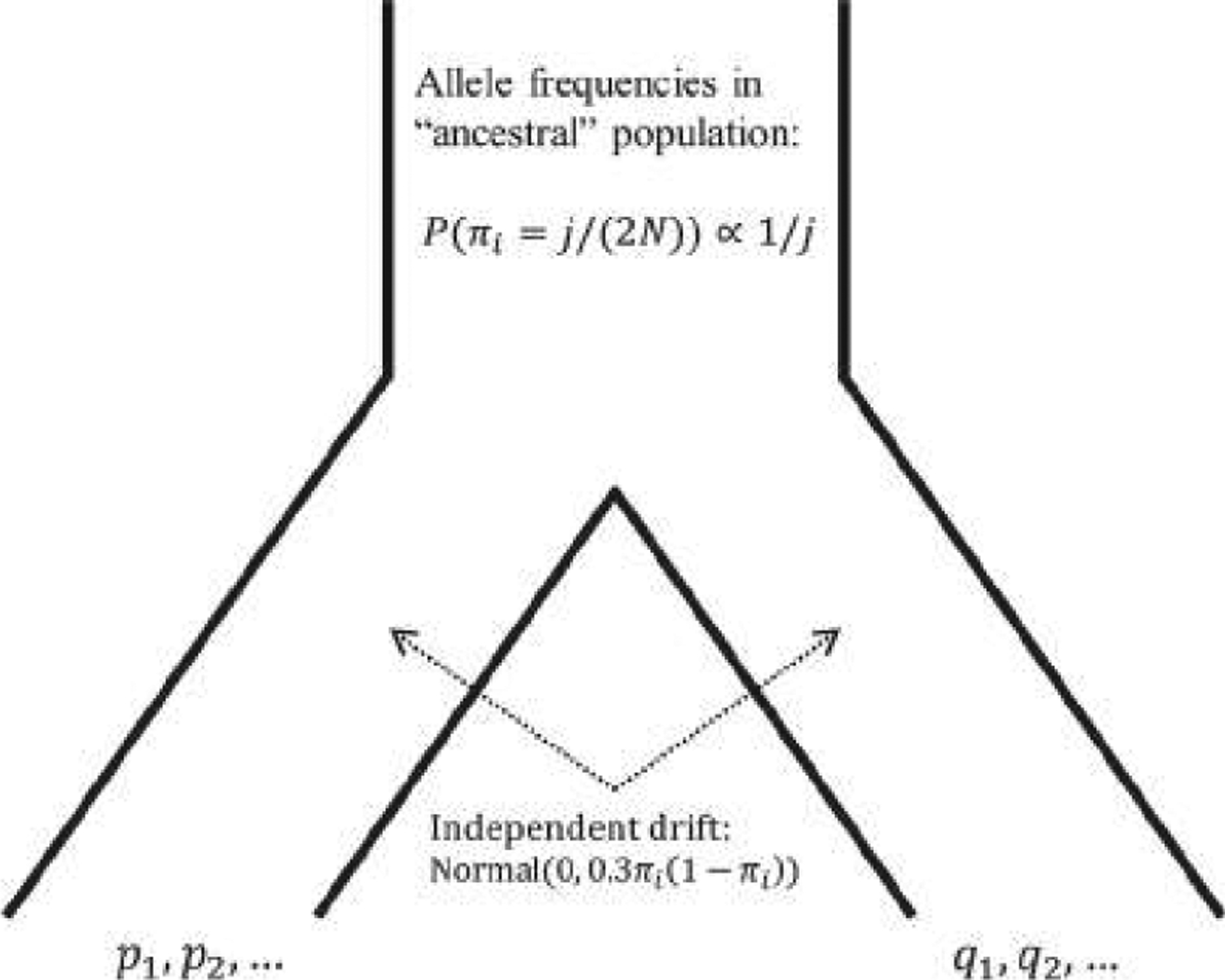
A schematic of the drift model used to simulate allele frequencies (see Section 3.2). Derived allele frequencies in an “ancestral” population are drawn according to the neutral site frequency spectrum. Following a split, the two subpopulations drift independently, with the drift represented by a truncated normal variate with expectation 0. After drift, for each locus i, the allele with greater frequency in population B than in population A is identified, its frequency in population A is labeled pi, and its frequency in population B is labeled qi.
4. Properties of the Trait Value T Conditional on the Labeling Xi
We next consider the distribution and properties of the trait value T in each population. In this section, we condition on the labeling of the alleles at each locus X1, X2, …, Xk. These labels determine, for each locus, whether the 1 or the 0 allele increases an individual’s trait value. In Section 5, we remove this condition and consider the expected behavior of the trait value under random assignment of the labels.
It is convenient to define a transformation of the labels:
| (18) |
If Xi = 1 and the 1 allele is the + allele, then Ui = 1, and if Xi = 0 and the 1 allele is the − allele, then Ui = −1.
4.1. Distribution of T within Each Population Given the Labeling of the Alleles
In either population, conditional on the labeling of the alleles,
Because the Lij are independent Bernoulli random variables with different success probabilities, T has a Poisson binomial distribution in each population. Specifically, within population A, each of the ℓ allelic copies at each locus at which xi = 1 increases T by 1 with probability pi, and each of the ℓ allelic copies at each locus at which xi = 0 increases T by 1 with probability 1 − pi. Within population B, the same statement holds if pi is replaced by qi.
By the properties of the Poisson binomial distribution (Eq. 7), the expectations of T in populations A and B conditional on the labeling are then
| (19) |
The difference in the conditional expectations is then
| (20) |
where, analogously to Eq. 18, ui = 2xi − 1.
By Eq. 20 and the fact that the populations have equal size so that P(M = A) = P(M = B) = ½, the variance across populations of the conditional expectation of T is
| (21) |
By the properties of the Poisson binomial distribution (Eq. 7), the conditional variance of the trait in population A is
the last step follows from the simplification of the variance in Eq. 7. Because this quantity does not depend on {X1, …, Xk}, we can remove the condition on {x1, …, xk}, giving
| (22) |
Similarly, the variance of T in population B is
| (23) |
We use the conditional expectations and variances of T in the two populations to define several measurements of the degree of difference between populations on the trait.
4.2. The Standardized Difference in Trait Means, DT, Given the Labeling of the Alleles
We consider three indices of the degree of difference between populations on the trait—two here, and a third we defer to Section 6. The first is the standardized difference in population means for the trait, DT, which is the difference between population trait means divided by the square root of the mean across populations of within-population trait variances. DT is an instance of the Cohen’s d measure of effect size (Cohen 1988). In this case, conditional on the labeling, DT is
The numerator is given in Eq. 20. By Eqs. 22 and 23 and the fact that the two populations are assumed to be the same size, the square of the denominator is
| (24) |
Therefore, combining Eqs. 20 and 24,
| (25) |
where again, as in Eq. 18, ui = 2xi − 1. In Section 5, we study the distribution of DT across different labelings of the alleles.
4.3. Partitioning the Variance of the Trait Given the Labeling of the Alleles: and QST
A second measure of between-population difference on the trait is the proportion of the trait’s variance attributable to difference between populations. We label this proportion , with
| (26) |
The last step follows from the law of total variance. Conditional on the labeling {X1, …, Xk}, the numerator appears in Eq. 21, and the denominator is the sum of the expressions in Eqs. 21 and 24. Thus, by Eq. 26, conditional on the labeling of the alleles for a given trait,
| (27) |
The proportion is related to QST, which is an analogue of FST developed for quantitative traits. For haploids, QST is the proportion of the heritable variance in a quantitative trait attributable to genetic differences between populations (Whitlock 2008). Because we have assumed that the trait we examine is completely heritable, for haploids. QST is defined so that, like FST, it does not depend on ploidy, which means that for ploidy ℓ > 1 (unless or ). For diploids, again invoking the assumption of perfect heritability of the trait, QST is
(Whitlock 2008), and by analogy, for ℓ-ploid organisms,
| (28) |
Thus, regardless of ploidy ℓ, QST is obtained from the expression in Eq. 27 by setting ℓ to 1. The relationship between QST and is exactly the same as the relationship between and (Figure 3 and Appendix 1); that is, if , then .
5. Properties of DT and across Different Labelings of the Alleles
In this section, we consider properties of the trait value T across different traits, which may have different allelic labels (Xi), so that each trait has its own locus-specific effects for the alleles. Specifically, we consider two indices of the degree of difference between populations on the trait defined in Section 4, the standardized group difference, DT (Section 4.2), and the proportion of trait variance that is attributable to between-group differences, (Section 4.3).
5.1. Properties of
One random variable that appears in expressions for both DT (Eq. 25) and (Eq. 27) is , where Ui is a function of the labels Xi that determine which allele at locus i is the + allele (Eq. 18), taking a value of either −1 or 1 with probability ½ each, and δi is the difference between populations in the frequency of the 1 allele (Eq. 8). We give the relevant moments of here for later reference.
We note first that, for all i and for integers n ≥ 0, the odd and even moments of the Ui obey
| (29) |
| (30) |
Thus, by Eq. 29, for n ∈ {0, 1, 2, …},
| (31) |
The second moment is
| (32) |
by Eq. 30 and because, by the independence of the Ui, E(UiUj) = E(Ui)E(Uj) = 0 for all i ≠ j.
We show in Appendix 2 that the fourth moment is
| (33) |
5.2. The Standardized Group Difference in Trait Means, DT
Removing the condition on the labels in Eq. 25, ui becomes the random variable Ui (Eq. 18), and DT becomes the random variable
| (34) |
By Eq. 29,
| (35) |
Equation 35 reflects the symmetry of the distribution of DT around 0. By Eqs. 32, 34, and 35,
| (36) |
where is as defined in Eq. 10.
(Eq. 36) is one measurement of the typical size of the between-group difference in trait means, irrespective of its direction. For fixed and , is usually larger if k > 1 than if k = 1 because of variation in the allele frequencies: if k = 1, but each may be positive if k > 1, and positive values of each of these terms increase . Nonetheless, does not grow without bound as k increases, and it is equal to one of our indices of between-group genetic differentiation at a single locus, (Eq. 17),
| (37) |
Thus, though increases with higher ploidy, it does not necessarily increase as the number of loci k influencing the trait increases (Figure 5A). Equation 36 reduces to the results we showed for in our previous work (Edge and Rosenberg 2015, Eqs. 37, 38) under the more restrictive assumptions we used there. The correspondences between the main results in this article and the main results in Edge and Rosenberg (2015) are summarized in Table 2.
FIGURE 5.
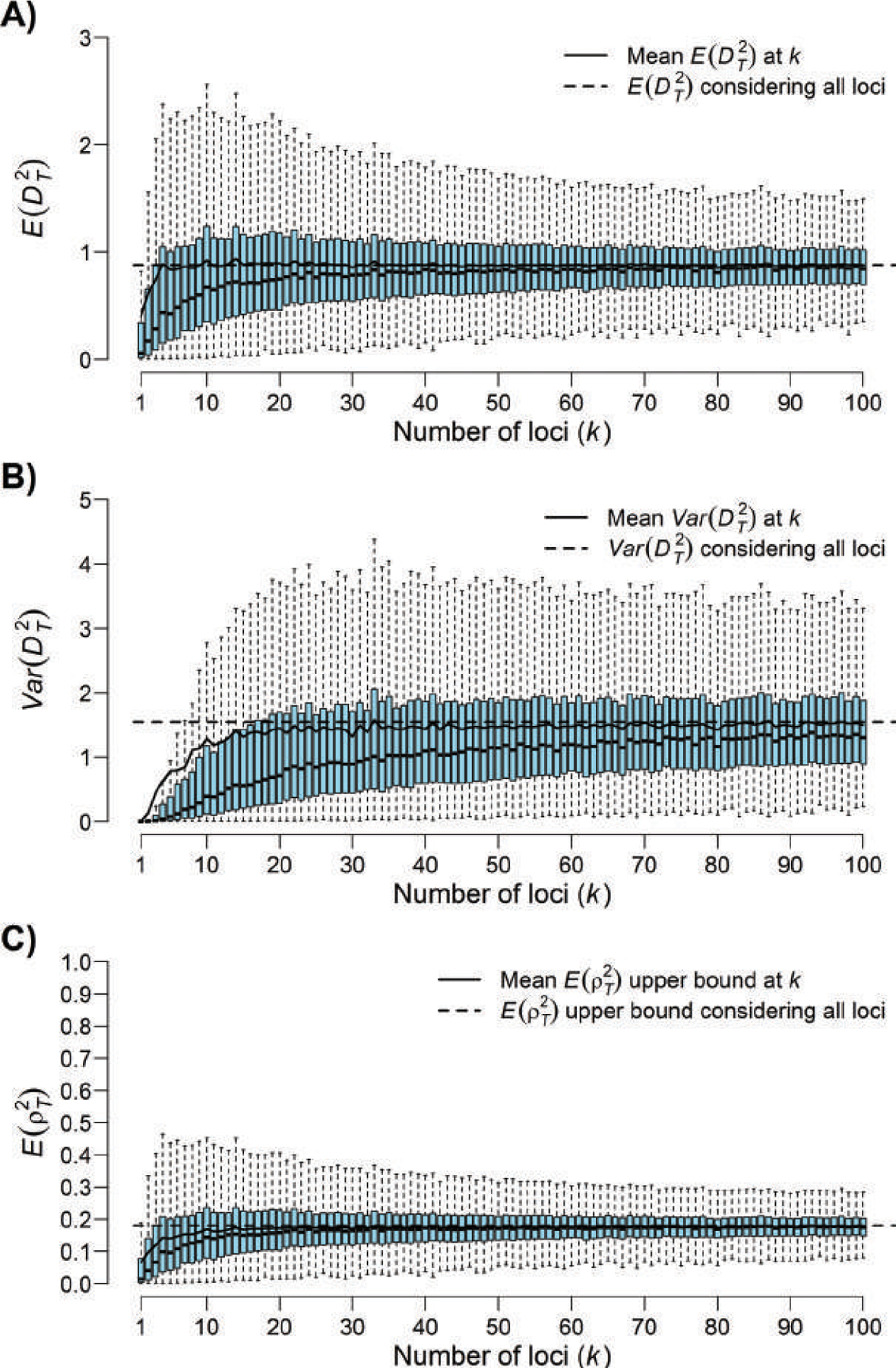
The behavior of expected measures of trait differentiation as the number of randomly selected loci influencing the trait increases. We simulated allele frequencies at 106 neutral loci for a pair of populations that have undergone independent drift since divergence from an ancestral population, with FST ≈ 0.1 (see Section 3.2). For each k ∈ {1, …, 100}, we selected 1,000 size-k random subsets of the 106 pairs of simulated allele frequencies and computed three quantities for each subset, assuming diploidy (ℓ = 2). (A) The expected squared standardized trait difference between groups, (Eq. 36). (B) The variance of the squared standardized trait difference between groups, (Eq. 39). (C) The upper bound on (and approximate value of) the expected proportion of variance in a neutral trait attributable to allele-frequency differences between groups, (Eqs. 43, 44). For each quantity, box plots of the 1,000 values for each k are shown. Boxes represent the middle 50% of data at each k, and whiskers extend 1.5 times the interquartile range beyond the edge of the box or to the most extreme observation, whichever is shorter. Outliers beyond 1.5 times the interquartile range from the edge of the box are not shown. For all three quantities, as k increases, the mean value at k loci (solid line) converges to the value obtained using all loci (dashed line) because larger random sets of loci more precisely reflect the overall degree of between-group differentiation than do smaller sets of loci.
Table 2.
Correspondence between the Main Results in This Article and in Edge and Rosenberg (2015)
| Result | Equation Number | |
|---|---|---|
| This Article | Edge and Rosenberg (2015) | |
| E(DT) = 0 due to symmetry around 0 of the distribution of DT. | 35 | 36 |
| does not increase without bound with the number of loci and is equal to , where DL is an analogue of DT for the allelic count at a single locus. | 36, 37 | 37, 38 |
| FST ≈ QST. | 45, 47, 48 | 42, 43 |
| As the number of loci k increases without bound, the genetic misclassification rate approaches 0. | 55 | 5 |
| The expectation of the approximate trait-based misclassification rate is closely related to the genetic misclassification rate obtained using one locus. | 57 | 47 |
The main results in this article reduce to the main results in Edge and Rosenberg (2015) under the following assumptions: (a) The allele frequencies are the same at each locus, meaning that for all i, for all i, for all i, and . (b) The allele frequencies are symmetric, meaning that . In conjunction with assumption (a), (b) implies that . (c) The organisms are haploid, or in the present article’s notation, ℓ = 1. Assumption (c) implies that (Eqs. 26–28).
In addition to the expectation of , we may wish to know its variance—do traits influenced by many loci vary widely in their level of between-population difference? By Eq. 33,
| (38) |
The required variance, calculated as , is, by Eqs. 36 and 38,
| (39) |
If k = 1, then , and . As k increases, approaches
| (40) |
Equations 39 and 40 indicate that as the number of loci k increases, the variance of does increase, but it asymptotes to a limit that does not depend on k (Figure 5B).
5.3. Properties of and QST
Removing the condition on the labels in Eq. 27, ui becomes the random variable Ui (Eq. 18), and becomes a random variable
| (41) |
To describe the behavior of across different traits, we approximate by replacing in Eq. 41 with its expectation, motivated by a Taylor approximation argument. Making the substitution , we have
| (42) |
Defining μY = E(Y), a first-order Taylor series expansion for g(Y) around Y = μY gives
and taking the expectation gives
By Eq. 32, . Substituting for Y in Eq. 42 gives
| (43) |
The expression on the right side of Eq. 43 is an approximation of , but it is also a strict upper bound on . To see that it is an upper bound, note that is concave in (Eq. 42). Thus, by Jensen’s inequality, which states that if g is a concave function of a random variable X, then E[g(X)] ≤ g[E(X)], we have
| (44) |
As we observed for , if and are fixed, then can take larger values if k > 1 than if k = 1 because increasing or increases the upper bound on . Nonetheless, does not grow without bound as k increases (Figure 5C). Comparing Eqs. 43 and 44 with Eq. 16,
| (45) |
The expected value of is thus approximately equal to, and no greater than, the ratio of the mean across loci of the between-group variance of to the mean across loci of the total variance of , where is a random variable representing the number of 1 alleles carried by an ℓ-ploid individual at locus i.
Because QST is equal to the expression for in Eq. 41 with ℓ set to 1 (Eq. 28), Eqs. 43 and 44 imply that
| (46) |
By Eq. 14, the expression on the right side of Eq. 46 is equal to , so
| (47) |
| (48) |
Equation 47 is consistent with previous work on the relationship between and QST under different models (e.g., Lande 1992; Whitlock 1999), and it provides one justification for the claim that the degree of between-group difference on a neutral trait is approximately equal to the degree of between-group genetic differentiation at a typical locus.
6. Adding Assumptions Inspired by Equal Drift since a Recent Divergence
Having addressed the relationship between neutral genetic and neutral phenotypic differentiation between populations in the context of standardized differences and variance partitioning (Sections 4 and 5), we now consider the accuracy with which individuals can be classified into populations using neutral genetic and phenotypic information. We require two assumptions that will allow us to consider approximate misclassification rates that would arise if we attempted to identify an individual’s population of origin by examining k loci directly or by examining a trait determined additively by those k loci. The case in which these assumptions are met is a restriction of the general case we have been examining. The special case in this section can be viewed as the expectation under a model in which the allele frequencies in populations A and B have experienced equal amounts of drift since a recent divergence (see Appendix 3).
The first assumption is symmetry of average frequency of the 1 allele across loci in populations A and B,
| (49) |
where both and are assumed to be nonzero. By Eq. 9, Eq. 49 implies that
| (50) |
The quantity in Eq. 50 is the value of a locus that has the average frequency of the 1 allele in each population, lending a new interpretation (Eq. 12). The second assumption is that the variance of the allele frequencies has the same value in each population:
| (51) |
6.1. Multilocus Classification
We now consider the problem of identifying the population of an individual of unknown origin. In this subsection, we examine the misclassification rates that arise from an examination of the number of 1 alleles carried by an individual across k loci, S.
Recall that the genotypic statistic S (Eq. 6) is the number of alleles carried by an individual that are more common in population B than in population A. If Xi = 1 for all i, then T = S. Within each population, the Ls are independent Bernoulli random variables with possibly different probabilities. Thus, within each population, S has a Poisson binomial distribution. By the properties of the Poisson binomial distribution (Eq. 7),
| (52) |
where , , , and are as defined in Eqs. 2 and 3. The variances of S within each population are the same as the variances of T within each population (Eqs. 22, 23).
We consider the normal approximation of the misclassification rate obtained if the genotypic statistic S is used for classification. If the assumptions in Eqs. 49 and 51 hold, then the within-population variances of S in the two populations are equal:
| (53) |
Further, when k is large, as a sum of independent Bernoulli variables the sum of whose variances increases without bound, the distribution of S is approximately normal:
| (54) |
(Deheuvels et al. 1989, Theorem 1.1).
Denoting the normal density that approximates the distribution of S in population A by fA(s) and the corresponding normal density for population B by fB(s), then when we observe that S = s, we classify the individual into population A if fA(s) > fB(s) and into population B if fA(s) < fB(s). In this case, fA(s) > fB(s) if and fA(s) < fB(s) if . We ignore the case of , which is negligible for large k. By the assumption in Eq. 49, , and we therefore classify an individual into population A if S < ℓk/2 and into population B if S > ℓk/2.
We represent the event that an individual is misclassified on the basis of S with the random indicator variable WS, which equals 1 if and only if an individual is misclassified on the basis of S and equals 0 otherwise. In population A, the approximate probability of misclassification is
where Φ is the cumulative distribution function of the standard normal distribution. A similar calculation for population B gives the same misclassification rate. Thus, in both populations, the approximate misclassification probability obtained using S is
| (55) |
As k increases, with , , and held constant, the argument to the cumulative distribution function in Eq. 55 approaches infinity, and the value of the cumulative distribution function approaches 1. Thus, as the number of loci increases, the misclassification probability obtained when using the genotypic statistic S, P(WS = 1), approaches 0.
6.2. Trait-Based Classification
Next we consider the approximate misclassification rate obtained on the basis of an individual’s trait value. We represent the event that an individual is misclassified on the basis of T with the random indicator variable WT, which equals 1 if an individual is misclassified on the basis of its trait value and equals 0 otherwise. Using an argument similar to the one used to justify Eq. 55 (detailed in Appendix 4), the approximate trait-based misclassification rate, conditional on X1, …, Xk, is
| (56) |
where Φ is the cumulative distribution function of the standard normal distribution.
To understand how the misclassification rate is expected to behave across different labelings of the loci, we consider the expectation of the normal approximation of the misclassification rate obtained using the trait value T, P(WT = 1). Removing the condition on the allelic labeling in Eq. 56 and rearranging gives
where Ui is as defined in Eq. 18. Taking the expectation of both sides gives
Noticing that Φ is concave for positive values of its argument gives, by Jensen’s inequality,
Because , and because the square root is a concave function, Jensen’s inequality gives
Then
Because (Eq. 32),
We then have an approximate lower bound on the expected probability of misclassification on the basis of T:
| (57) |
If , then Eq. 57 produces the same result as Eq. 55 with k = 1. The expectation of the approximate misclassification probability on the basis of the trait is therefore, if , greater than or equal to the approximate misclassification probability obtained using a single locus.
For fixed , , and , as k increases, the argument to the cumulative distribution function in Eq. 57 does not approach infinity. The value of the cumulative distribution function approaches an asymptotic value obtained as the increasingly many loci converge on large-k values of and . Thus, as the number of loci considered increases, the misclassification probability obtained when using the trait value T, P(WT = 1) does not approach 0.
7. Discussion
We have extended a model of multilocus allele frequency differences and polygenic trait differences between groups to accommodate more general allele-frequency distributions and arbitrary ploidy. Our results recapitulate our original conclusion (Edge and Rosenberg 2015): a single neutral trait provides approximately the same amount of information about population membership as does a single neutral genetic locus. This general claim is reflected in three specific ways of asking about the relative magnitude of population-membership information in genotypes and in phenotypes: using the standardized trait difference between groups (DT, Eq. 37), using the between-group variance in the trait ( and DST, Eqs. 45, 47), and using the misclassification rate obtained when attempting to classify individuals into groups by their trait values (P(WT = 1), Eq. 57). We also provide two main updates to our previous work. First, under our model, within-population variation in allele frequency across loci tends to increase the degree of expected difference between groups on a polygenic trait (Eq. 36). Second, the degree of information about population membership is greater for a diploid (or polyploid) than for a haploid locus (Figure 3), and it is also correspondingly greater for a trait in a diploid (or polyploid) organism than in a haploid (Eq. 37).
What accounts for the difference between the ancestry information of multiple genetic loci and that of a trait governed by those same loci? When examining multiple genetic loci, information about population membership can be cumulated from multiple sites—for example, by counting in an individual the alleles that are more common in population A than in population B. In contrast, although an individual’s trait value implicitly encodes information about its genotype at many loci, random genetic drift prevents the trait from accumulating information about population membership. In our model, for every locus at which the allele at higher frequency in population A is associated with larger trait values, there is likely to be another locus at which the “A-like” allele is associated with smaller trait values. The cumulative effect of this locus-by-locus shuffling of the choice of population associated with the higher trait value is that a single neutral trait is, in expectation, approximately as informative about population membership as a single neutral locus.
7.1. Models of Genotypic and Phenotypic Differentiation
Our results accord with those of previous efforts to address similar questions with different models (Felsenstein 1973, 1986; Rogers and Harpending 1983; Lande 1992; Spitze 1993; Lynch and Spitze 1994; Whitlock 1999; Berg and Coop 2014), which have repeatedly found that group or population differences in neutral, completely heritable traits mirror neutral genetic differentiation. Previous examinations of trait differentiation have often proceeded by relating assumptions about quantitative traits to models of evolutionary change in allele frequencies, such as a Wright–Fisher model (Felsenstein 1973), an island migration model (Lande 1992), or a coalescent framework (Whitlock 1999). The use of such evolutionary models can suggest connections with other areas of evolutionary genetics and can also provide insights with considerable generality; for example, Whitlock’s (1999) results hold for coalescent models with arbitrary population structure.
In contrast with some previous models of phenotypic diversity, our models here and in our previous work (Edge and Rosenberg 2015) are more similar to the genetic classification models of Risch et al. (2002), Edwards (2003), and Tal (2012) in that we directly consider allele frequencies, using simple probabilistic arguments and minimal evolutionary assumptions. This approach complements earlier evolutionary work on the relationship of genetic and phenotypic differentiation in at least two ways. First, our model allows for computations with quantities that are of interest in epidemiological and biomedical studies but that do not necessarily arise naturally under evolutionary models, such as Cohen’s d (equal to our DT for a completely heritable trait) and the effect size r2 (equal to our for a completely heritable trait). Second, the fact that our model makes only minimal evolutionary assumptions shows that similar results obtained under evolutionary models are robust in that they are also produced via a substantially different modeling approach.
7.2. Interpreting Group Differences in Phenotype
How can this work aid in the interpretation of phenotypic differences between human groups? Consider health outcomes, an important set of phenotypes for which genetic and phenotypic differentiation across populations have been of interest.
Among people in the United States, for example, well-established differences exist between socially defined racial groups in the incidence of many health conditions, including heart disease (e.g., Lloyd-Jones et al. 2010), various cancers (e.g., Ward et al. 2004; Siegel et al. 2013), and diabetes (e.g., LaVeist et al. 2009), with African Americans having worse health outcomes than European Americans across many domains (reviewed in Dressler et al. 2005; Adler and Rehkopf 2008). Such phenotypic differences between pairs of groups arise from a combination of interacting factors, which can be viewed as modifications of a baseline prediction made on the basis of neutral genetic differences. Evolutionary genetics can then contribute to understanding group differences in health outcomes by providing models that predict the degree to which phenotypic differences between human groups are likely to be based in neutral genetic differences. Such models do not necessarily explain the source of any particular phenotypic difference, but they do provide an idea of what patterns of difference are expected.
Using population-genetic models to build intuition about phenotypic differences between human groups does not require that group classifications are reducible to, caused by, or primarily based in genetic differences between populations. Rather, population-genetic models of neutral genetic variation are applicable to sets of groups that are correlated with some degree of genetic population structure and that thus distinguish groups that differ in allele frequency at sites across the genome. Although the relationship between population-genetic groupings and socially defined groupings is complex (e.g., Kittles and Weiss 2003; Bamshad et al. 2004; Kitcher 2007; Hunley et al. 2016), we can gain some intuition about the possible sources of a group difference in phenotype for socially defined groups by comparing its size to the associated degree of between-group differentiation at a typical genetic locus.
Two possible causes of group phenotypic differences that are larger or smaller than the degree of between-group differentiation at a typical selectively neutral locus are natural selection and environmental difference. If a trait has been under selection in two populations in a manner that leads to divergence—for example, if the trait is advantageous in one population and disadvantageous or neutral in the other—then the between-group difference on the trait will typically exceed the average between-group genetic difference. An example relevant to health differences between socially defined racial groups is skin pigmentation: lighter skin has been positively selected among populations at higher latitudes (Jablonski and Chaplin 2000; Relethford 2002; Berg and Coop 2014), but it is also a risk factor for skin cancer (Lin and Fisher 2007). In turn, European Americans, most of whose recent ancestors generally lived at high latitude, have substantially higher rates of skin cancer than do African Americans (Halder and Bridgeman-Shah 1995), a larger fraction of whose recent ancestors generally lived at lower latitudes. In contrast, many other health-related traits are likely associated with similar reproductive fitness wherever they occur. Such conditions would be expected to experience convergent selection, which would lead to smaller between-group trait differences than might be predicted from neutral genetic diversity. Thus, one explanation for larger-than-expected phenotypic differences among groups is divergent selection, and one explanation for smaller-than-expected phenotypic differences among groups is convergent selection. This reasoning is the basis for the use of comparisons between QST and FST to test hypotheses about phenotypic evolution, a productive approach for model organisms that can be raised in a “common garden” situation (Whitlock 2008; Leinonen et al. 2013).
In humans, assessing whether selection has magnified health differences beyond the neutral prediction is difficult. In some specific cases, divergent selection is regarded as an important driver of group difference in disease burden—including, for example, sickle-cell disease, which occurs at higher rates in malarial regions of Africa and the Mediterranean (e.g., Piel et al. 2010). In many other cases of phenotypic difference, hypotheses of divergent selection—sometimes paired with gene–environment interaction—have also been proposed (Knowler et al. 1983; Meindl 1987; Zlotogora et al. 1988; Wilson and Grim 1991; Bindon and Baker 1997). Many such hypotheses have been criticized individually (Curtin 1992; Risch et al. 2003), and concern has been raised that selective explanations for differences in health outcomes often assume a degree of endurance and importance unwarranted by the evidence (Kaufman and Hall 2003). As new cases connecting selection pressures to molecular evidence of adaptation emerge (e.g., Fumagalli et al. 2015), the empirical basis for assessing the role of divergent selection in explaining differences in health outcomes will expand.
Much recent discussion has focused on an alternative approach to examining worldwide consequences of selection, considering demographic factors that influence the strength of selection against deleterious mutations in different populations (Lohmueller 2014; Henn et al. 2015). Recent studies have not found pronounced difference between groups of primarily European and African descent in the overall frequency of putatively deleterious alleles (Fu et al. 2014; Simons et al. 2014; Do et al. 2015), but populations differ in how these alleles are distributed among individuals, with Europeans carrying more genotypes homozygous for putatively deleterious alleles compared with Africans (Lohmueller et al. 2008; Fu et al. 2014; Do et al. 2015). Though these studies do not identify differences between populations in the influence of experienced selective pressures, their results suggest that a systematic difference across populations in the outcomes of selection on disease phenotypes, if it exists at all, would likely tilt toward a greater disease burden in non-Africans.
Finally, environmental differences between groups are important sources of between-group trait differences. Environmental differences and associated differences in the effect of gene–environment interactions can act in concert with or in opposition to any genetic differences that influence a trait, leading to between-group trait differences that are larger or smaller than would be expected on the basis of neutral genetic differences alone (Pujol et al. 2008). In the United States, the environments of people of different socially defined races differ in myriad factors that could contribute to differences in health outcomes (Williams and Jackson 2005), including socioeconomic status (Adler and Newman 2002), education (Non et al. 2012), residential segregation (Williams and Collins 2001), discrimination (Williams and Mohammed 2009), targeting by the criminal justice system (Iguchi et al. 2005), access to medical care (Mayberry et al. 2000), and doctor–patient communication (Ashton et al. 2003).
We can examine an environmental hypothesis about group differences in health outcomes in relation to the predicted pattern of differences across outcomes for our neutral model of two groups. Under the neutral model, each group has an equal chance of having a larger trait value for each trait. Thus, each population would have a larger mean value for roughly half the traits on which the groups differed, with each trait independent if there are no genetic correlations. An environmental explanation of differences in health outcomes might hold that social differences, such as differences in access to health care, are likely to cause a pattern in which differences between groups run in the same direction across many diseases and causes of mortality. Under this reasoning, the fact that African Americans suffer more than do European Americans from a wide variety of diseases is more consistent with environmental sources of phenotypic difference than with a neutral-genetic explanation. As an example, Wong et al. (2002) tabulated racial differences in causes of death in 36 categories over a 9-year period. After adjusting for age, sex, and years of education, they estimated that black Americans lost more life years than white Americans on average in 28 of those categories. Informally, assuming under our neutral model that no genetic correlation exists between phenotypic outcomes and that for each outcome the larger value has equal probability of occurring in either group, the binomial probability that one population would have a larger trait value than the other on at least 28 of 36 independent phenotypes is only 0.001. A single systematic environmental effect that simultaneously inflates many nongenetic risk factors in African Americans, on the other hand, can provide a simple explanation for such skewed outcomes.
7.3. Conclusions
Our model provides a general framework for describing the relevance of single-locus genetic diversity partitioning for predictions about the sources of phenotypic differences between groups. For neutral, heritable traits, group differences in phenotype will be random in direction and will reflect the degree of genetic difference at a single locus—modest in size for humans, but likely not zero—regardless of how many loci influence the trait. Such neutral differences are a baseline on top of which selection and environmental influences act (Figure 6). In the case of health-related differences between socially defined races in the United States, the occurrence of genetic differentiation as measured by FST suggests that neutral genetic differences are likely to exist for many heritable health outcomes that are not under selection. Such genetically based differences may run in the opposite direction of the apparent phenotypic difference between groups, and typical values of human FST suggest that they will likely be modest in size on average. Nonetheless, their existence supports the view that genetic research designs that capitalize on group differences (e.g., Winkler et al. 2010; Zaitlen et al. 2014) can be informative about genetic architecture or the genetic variants that influence phenotypes (Rosenberg et al. 2010; Teo et al. 2010). At the same time, any patterns of difference in which one group suffers more than others from the majority of many genetically independent diseases are unlikely to be explained by neutral genetic variation. For humans, our model supports the view that coordinated group differences across a preponderance of independent health-related traits suggest an important role for systematic differences in environmental risk factors.
FIGURE 6.
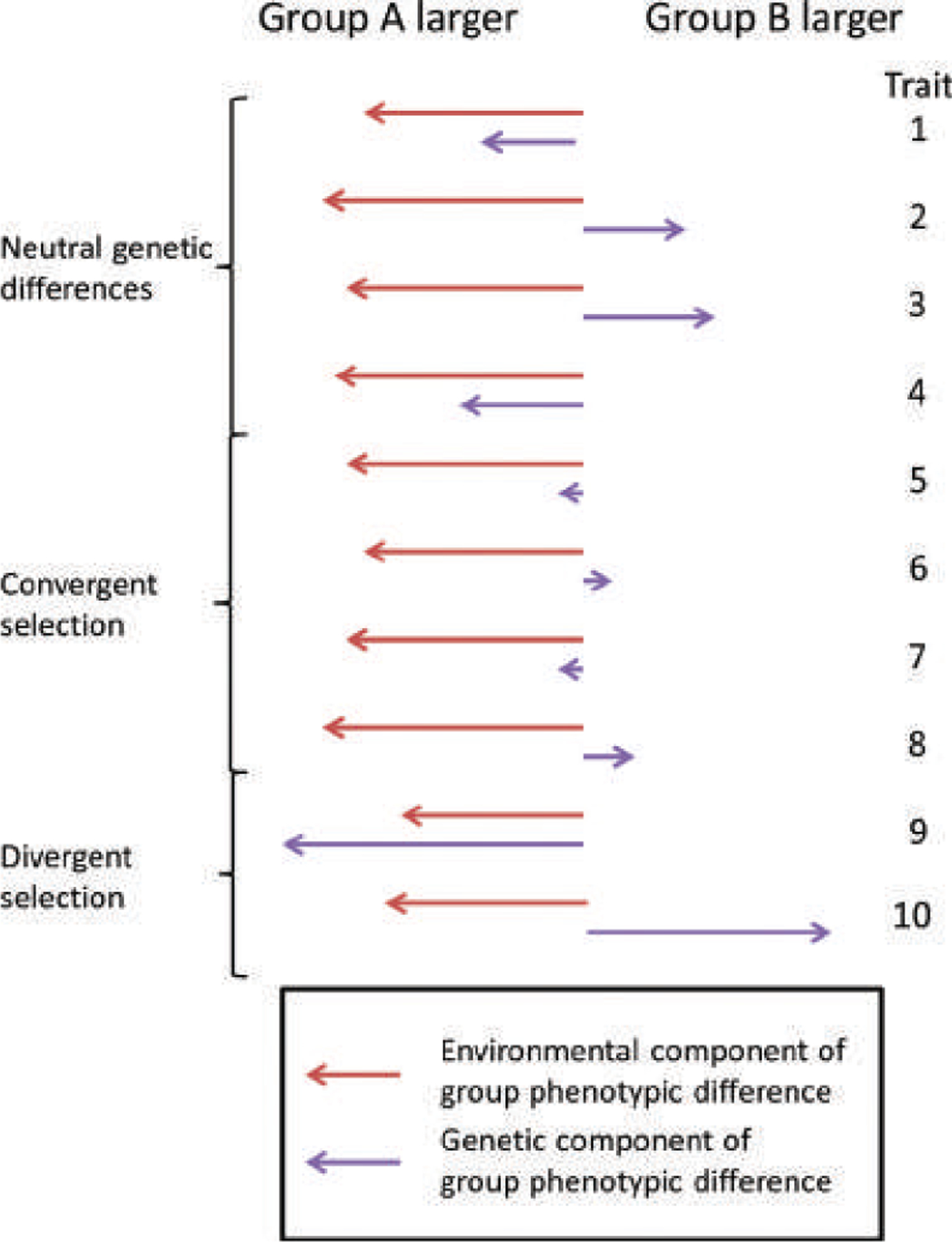
A schematic for thinking about health differences between socially defined racial groups in the United States. Groups such as African Americans face environmental differences likely to lead to worse health outcomes across a range of different diseases (red arrows). For health phenotypes that have been selectively neutral, genetic differences between groups will be random in direction and comparable in size to genetic differences at a single locus—modest for humans, but usually not zero, depending on the groups being considered (purple arrows in traits 1–4). For health phenotypes under convergent selection, such as those that lead to reduced reproductive success in most or all human environments, genetic differences will be random in direction and smaller than for neutral phenotypes (purple arrows in traits 5–8). Some health outcomes, such as skin cancer and sickle-cell disease, differ between groups in part because of divergent selection. (These differences do not necessarily coincide neatly with socially relevant racial divisions.) For such phenotypes, the genetic component of a group difference in phenotype can be large (purple arrows in traits 9 and 10). Not considered in this simple diagram are gene–environment interactions, which often are especially important in cases of divergent selection. For example, in the case of skin cancer, the degree to which genetic variants that lead to darker skin are protective depends on sun exposure.
ACKNOWLEDGMENTS
We thank the organizers for including us in this special issue, Jeff Long and Alan Rogers for stimulating discussion, and National Institutes of Health grant R01 HG005855, National Science Foundation grant DBI-1458059, and the Stanford Center for Computational, Evolutionary, and Human Genomics for support.
Appendix 1: and
In this appendix, we derive some results about the relationship between (Eq. 14), which summarizes the information about population membership available in one copy of a typical locus chosen from within a set of k loci, and (Eq. 16), which summarizes the corresponding population membership information available in ℓ independent copies of a typical locus chosen from within the set of k loci.
For convenience, define v as
| (A1.1) |
where , , , and are as defined in Eqs. 2 and 3. Then by Eq. 14,
| (A1.2) |
and by Eq. 16,
| (A1.3) |
where ℓ is the number of copies of the locus being considered and is defined in Eq. 10. Because v, , and ℓ are all nonnegative, and . Equations A1.2 and A1.3 also imply that if and only if .
Noting that
we then have, for ,
Consequently, for ,
| (A1.4) |
Because and if , Eq. A1.4 implies that
with if ℓ =1 or if either or , but with . In other words, if ℓ > 1, for very small but nonzero , but for near 1, The relationship between and is plotted in Figure 3.
Appendix 2: The Fourth Moment of
In this appendix, we show that the fourth moment of is equal to the expression in Eq. 33.
By the independence of the Ui and Eqs. 29 and 30,
| (A2.1) |
To simplify the sum in Eq. A2.1, notice that , so
| (A2.2) |
where is as defined in Eq. 11. Plugging the expression for from Eq. A2.2 into Eq. A2.1 gives
| (A2.3) |
which proves the statement in Eq. 33.
Appendix 3: Allele Frequencies in a Drift Model
In this appendix, we show that the assumptions in Eqs. 49 and 51 are the expectations of allele frequencies under a population-genetic model in which the two populations experience equal degrees of drift since a recent divergence. The model we use for drift is similar to some models used in previous work (Nicholson et al. 2002; Falush et al. 2003). Note that, although the allele frequencies and pi and qi are treated as fixed quantities in the main text, they are treated as random variables in this appendix.
A.3.1. Drift Model
Let π1, π2, …, πk, represent the frequencies of one of two alleles at each of k loci in a predivergence population. The πi may be outcomes of a random process with arbitrary distribution. After a divergence event, the predivergence population splits into two populations—populations A and B—that undergo drift. The amount of drift at each locus is represented by a set of continuous random variables, αA1, αA2, …, αAk for population A and αB1, αB2, …, αBk for population B. For all i, conditional on πi, the αAi and αBi are independent, and E(αAi) = E(αBi) = 0. Further, if the two subpopulations have experienced equal amounts of drift, then conditional on πi, the αAi and αBi are identically distributed. The postdrift allele frequencies in population A are π1 + αA1, π2 + αA2, …, πk + αAk, and in population B they are π1 + αB1, π2 + αB2, …, πk + αBk.
At each locus, define the 1 allele as the allele that is more frequent in population B than in population A. If the allele frequencies are the same in both populations at a locus, then the 1 allele at that locus is chosen randomly, with probability ½ for each allele. The frequency of the 1 allele at locus i is pi in population A and qi in population B.
A.3.2. Proposition 1
Proposition 1:
If populations A and B have experienced equal amounts of drift since divergence, then E(pi) + E(qi) = 1.
Proof:
Under the drift model outlined above,
if αAi < αBi, then pi = πi + αAi and qi = πi + αBi;
if αAi = αBi, then pi = qi = τi, where τi is either πi + αAi or 1 − πi − αAi with probability ½ of each possibility; and
if αAi > αBi, then pi = 1 − πi − αAi and qi = 1 − πi − αBi.
If the two subpopulations have experienced equal amounts of drift since divergence, then conditional on πi, the drift variables αAi and αBi are independent and identically distributed. Thus, P(αAi < αBi) = P(αAi > αBi), and therefore
Conditional on πi = w, we have
Similarly,
And so, conditional on πi = w,
Because E(pi + qi|πi = w) = 1 for all w, the unconditional expectation is
This completes the proof of Proposition 1.
Thus, taking the mean across loci, , and the assumption in Eq. 49 characterizes the expectations of allele frequencies under the model of equal drift since a recent divergence.
A.3.3. Proposition 2
Proposition 2:
If populations A and B have experienced equal amounts of drift since divergence, then .
Proof:
Define a random indicator variable Gi with the property
This property implies that
because pi ≤ qi. By the law of total variance,
Under the assumption of equal drift since divergence, P(Gi = 0) = P(Gi = 1) = ½. Thus, Var(pi|πi = w) is equal to the sum of
and
A parallel calculation for qi reveals that Var(qi| πi = w) is equal to the sum of two equivalent terms, meaning that for all i, Var(pi| πi = w) = Var(qi| πi = w). Because this claim holds for all i and because πi is by definition the same for populations A and B,
Finally, by Eq. 3, is the biased sample variance of the pi and is the biased sample variance of the qi. Applying Bessel’s correction to the biased sample variance, the expectations of and under the drift model are, for k loci,
and because Var(pi) = Var(qi),
Thus, the assumption in Eq. 51 represents the expectation for properties of the variance of allele frequencies across loci under a model of equal drift since a recent divergence. This completes the proof of Proposition 2.
Appendix 4: The Approximate Trait-Based Misclassification Rate, Conditional on the Labeling of the Alleles
In this appendix, we justify Eq. 56 using an argument similar to the one used to justify Eq. 55. We assume the conditions that apply in Section 6, stated in Eqs. 49 and 51.
WT is an indicator variable that equals 1 if an individual is misclassified on the basis of its value for T. Conditional on the allelic labels X1, …, Xk, T has a Poisson binomial distribution (see Section 4.1), which, for large k, is well approximated by a normal distribution (Deheuvels et al. 1989, Theorem 1.1). By the conditional expectations and variances in Eqs. 19, 22, and 23 and the assumptions in Eqs. 49 and 51, the large-k distributions of T in the two populations are approximately
conditional on the labeling of the alleles, X1, …, Xk.
Denoting the normal density that approximates the distribution of T in population A by fA.T(t) and the corresponding normal density for population B by fB.T(t), then after observing that T = t, we classify the individual into population A if fA.T(t) > fB.T(t) and into population B if fA.T(t) < fB.T(t). Because the variances of the two limiting normal distributions are equal, the relationship of the densities depends only on whether the observed trait value t is closer to its expectation in population A or population B. That is, defining
we have
Consider an individual drawn from population A, who will be misclassified if fA.T(t) < fB.T(t). If μA.T < μB.T, then
whereas if μA.T > μB.T, then
(We defer for a moment the case μA.T = μB.T.) Thus, the approximate probability of misclassifying an individual from population A on the basis of its trait value is, if μA.T < μB.T,
| (A4.1) |
where the last step follows from Eq. 20. Similarly, if μA.T > μB.T, then
| (A4.2) |
The expressions in Eqs. A4.1 and A4.2 are equal. Further, the expression they provide also applies to the case of μA.T = μB.T. If μA.T = μB.T, then P(WT = 1) = ½ because the trait has the same distribution in each population. The expression in Eqs. A4.1 and A4.2 applies because if μA.T = μB.T, then by Eq. 20, , and Φ(0) = ½ as required. A similar set of calculations for population B gives the same expression, so we remove the condition on population membership, arriving at the statement in Eq. 56:
LITERATURE CITED
- Adler NE, and Newman K. 2002. Socioeconomic disparities in health: Pathways and policies. Health Aff. (Millwood) 21:60–76. [DOI] [PubMed] [Google Scholar]
- Adler NE, and Rehkopf DH. 2008. US disparities in health: Descriptions, causes, and mechanisms. Annu. Rev. Public Health 29:235–252. [DOI] [PubMed] [Google Scholar]
- Ashton CM, Haidet P, Paterniti DA et al. 2003. Racial and ethnic disparities in the use of health services: Bias, preferences, or poor communication? J. Gen. Intern. Med 18:146–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamshad M, Wooding S, Salisbury BA et al. 2004. Deconstructing the relationship between genetics and race. Nat. Rev. Genet 5:598–609. [DOI] [PubMed] [Google Scholar]
- Bamshad MJ, Wooding S, Watkins WS et al. 2003. Human population genetic structure and inference of group membership. Am. J. Hum. Genet 72:578–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbujani G, Magagni A, Minch E et al. 1997. An apportionment of human DNA diversity. Proc. Natl. Acad. Sci. USA 94:4,516–4,519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg JJ, and Coop G. 2014. A population genetic signal of polygenic adaptation. PLoS Genet 10:e1004412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindon JR, and Baker PT. 1997. Bergmann’s rule and the thrifty genotype. Am. J. Phys. Anthropol 104:201–210. [DOI] [PubMed] [Google Scholar]
- Bowcock AM, Ruiz-Linares A, Tomfohrde J et al. 1994. High resolution of human evolutionary trees with polymorphic microsatellites. Nature 368:455–457. [DOI] [PubMed] [Google Scholar]
- Brown RA, and Armelagos GJ. 2001. Apportionment of racial diversity: A review. Evol. Anthropol 10:34–40. [Google Scholar]
- Chakraborty R, and Nei M. 1982. Genetic differentiation of quantitative characters between populations or species: I. Mutation and random genetic drift. Genet. Res 39:303–314. [Google Scholar]
- Charlesworth B, and Charlesworth D. 2010. Elements of Evolutionary Genetics Greenwood Village, CO: Roberts. [Google Scholar]
- Cohen J 1988. Statistical Power Analysis for the Behavioral Sciences 2nd ed. Hillsdale, NJ: Erlbaum. [Google Scholar]
- Curtin PD 1992. The slavery hypothesis for hypertension among African Americans: The historical evidence. Am. J. Public Health 82:1,681–1,686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deheuvels P, Puri ML, and Ralescu SS. 1989. Asymptotic expansions for sums of nonidentically distributed Bernoulli random variables. J. Multivar. Anal 28:282–303. [Google Scholar]
- Do R, Balick D, Li H et al. 2015. No evidence that selection has been less effective at removing deleterious mutations in Europeans than in Africans. Nat. Genet 47:126–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dressler WW, Oths KS, and Gravlee CC. 2005. Race and ethnicity in public health research: Models to explain health disparities. Annu. Rev. Anthropol 34:231–252. [Google Scholar]
- Edge MD, and Rosenberg NA. 2015. Implications of the apportionment of human genetic diversity for the apportionment of human phenotypic diversity. Stud. Hist. Philos. Biol. Biomed. Sci 52:32–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards AWF 1960. The meaning of binomial distribution. Nature 186:1,074. [DOI] [PubMed] [Google Scholar]
- Edwards AWF 2003. Human genetic diversity: Lewontin’s fallacy. Bioessays 25:798–801. [DOI] [PubMed] [Google Scholar]
- Falush D, Stephens M, and Pritchard JK. 2003. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 164:1,567–1,587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J 1973. Maximum-likelihood estimation of evolutionary trees from continuous characters. Am. J. Hum. Genet 25:471–492. [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J 1986. Population differences in quantitative characters and gene frequencies: A comment on papers by Lewontin and Rogers. Am. Nat 127:731–732. [Google Scholar]
- Fox J 1997. Applied Regression Analysis, Linear Models, and Related Methods Thousand Oaks, CA: Sage. [Google Scholar]
- Fu W, Gittelman RM, Bamshad MJ et al. 2014. Characteristics of neutral and deleterious protein-coding variation among individuals and populations. Am. J. Hum. Genet 95:421–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli M, Moltke I, Grarup N et al. 2015. Greenlandic Inuit show genetic signatures of diet and climate adaptation. Science 349:1,343–1,347. [DOI] [PubMed] [Google Scholar]
- Halder RM, and Bridgeman‐Shah S. 1995. Skin cancer in African Americans. Cancer 75:667–673. [DOI] [PubMed] [Google Scholar]
- Henn BM, Botigué LR, Bustamante CD et al. 2015. Estimating the mutation load in human genomes. Nat. Rev. Genet 16:333–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holsinger KE, and Weir BS. 2009. Genetics in geographically structured populations: Defining, estimating and interpreting FST. Nat. Rev. Genet 10:639–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunley KL, Cabana GS, and Long JC. 2016. The apportionment of human diversity revisited. Am. J. Phys. Anthropol, 160:561–569. [DOI] [PubMed] [Google Scholar]
- Iguchi MY, Bell J, Ramchand RN et al. 2005. How criminal system racial disparities may translate into health disparities. J. Health Care Poor Underserved 16:48–56. [DOI] [PubMed] [Google Scholar]
- Jablonski NG, and Chaplin G. 2000. The evolution of human skin coloration. J. Hum. Evol 39:57–106. [DOI] [PubMed] [Google Scholar]
- Kaufman JS, and Hall SA. 2003. The slavery hypertension hypothesis: Dissemination and appeal of a modern race theory. Epidemiology 14:111–118. [DOI] [PubMed] [Google Scholar]
- Kitcher P 2007. Does “race” have a future? Philos. Public Aff 35:293–317. [Google Scholar]
- Kittles RA, and Weiss KM. 2003. Race, ancestry, and genes: Implications for defining disease risk. Annu. Rev. Genomics Hum. Genet 4:33–67. [DOI] [PubMed] [Google Scholar]
- Knowler WC, Pettitt DJ, Bennett PH et al. 1983. Diabetes mellitus in the Pima Indians: Genetic and evolutionary considerations. Am. J. Phys. Anthropol 62:107–114. [DOI] [PubMed] [Google Scholar]
- Lande R 1992. Neutral theory of quantitative genetic variance in an island model with local extinction and colonization. Evolution 46:381–389. [DOI] [PubMed] [Google Scholar]
- LaVeist TA, Thorpe RJ Jr., Galarraga JE et al. 2009. Environmental and socio-economic factors as contributors to racial disparities in diabetes prevalence. J. Gen. Intern. Med 24:1,144–1,148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinonen T, McCairns RJS, O’Hara RB et al. 2013. QST-FST comparisons: Evolutionary and ecological insights from genomic heterogeneity. Nat. Rev. Genet 14:179–190. [DOI] [PubMed] [Google Scholar]
- Lewontin RC 1972. The apportionment of human diversity. In Evolutionary Biology, vol. 6, Dobzhansky T, Hecht MK, and Steere WC, eds. New York: Appleton-Century-Crofts, 381–398. [Google Scholar]
- Li JZ, Absher DM, Tang H et al. 2008. Worldwide human relationships inferred from genome-wide patterns of variation. Science 319:1,100–1,104. [DOI] [PubMed] [Google Scholar]
- Lin JY, and Fisher DE. 2007. Melanocyte biology and skin pigmentation. Nature 445:843–850. [DOI] [PubMed] [Google Scholar]
- Lloyd-Jones D, Adams RJ, Brown TM et al. 2010. Heart disease and stroke statistics—2010 update: A report from the American Heart Association. Circulation 121:e46–e215. [DOI] [PubMed] [Google Scholar]
- Lohmueller KE 2014. The distribution of deleterious genetic variation in human populations. Curr. Opin. Genet. Dev 29:139–146. [DOI] [PubMed] [Google Scholar]
- Lohmueller KE, Indap AR, Schmidt S et al. 2008. Proportionally more deleterious genetic variation in European than in African populations. Nature 451:994–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, and Spitze K 1994. Evolutionary genetics of Daphnia. In Ecological Genetics, Real L, ed. Princeton, NJ: Princeton University Press, 109–128. [Google Scholar]
- Mayberry RM, Mili F, and Ofili E. 2000. Racial and ethnic differences in access to medical care. Med. Care Res. Rev 57(suppl. 1):108–145. [DOI] [PubMed] [Google Scholar]
- Meindl RS 1987. Hypothesis: A selective advantage for cystic fibrosis heterozygotes. Am. J. Phys. Anthropol 74:39–45. [DOI] [PubMed] [Google Scholar]
- Mountain JL, and Cavalli-Sforza LL. 1997. Multilocus genotypes, a tree of individuals, and human evolutionary history. Am. J. Hum. Genet 61:705–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M 1973. Analysis of gene diversity in subdivided populations. Proc. Natl. Acad. Sci. USA 70:3,321–3,323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson G, Smith AV, Jónsson F et al. 2002. Assessing population differentiation and isolation from single-nucleotide polymorphism data. J. R. Stat. Soc. Series B Stat. Methodol 64:695–715. [Google Scholar]
- Non AL, Gravlee CC, and Mulligan CJ. 2012. Education, genetic ancestry, and blood pressure in African Americans and whites. Am. J. Public Health 102:1,559–1,565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piel FB, Patil AP, Howes RE et al. 2010. Global distribution of the sickle cell gene and geographical confirmation of the malaria hypothesis. Nat. Commun 1:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pujol B, Wilson AJ, Ross RIC et al. 2008. Are QST-FST comparisons for natural populations meaningful? Mol. Ecol 17:4,782–4,785. [DOI] [PubMed] [Google Scholar]
- Relethford JH 2002. Apportionment of global human genetic diversity based on craniometrics and skin color. Am. J. Phys. Anthropol 118:393–398. [DOI] [PubMed] [Google Scholar]
- Relethford JH 2010. Population‐specific deviations of global human craniometric variation from a neutral model. Am. J. Phys. Anthropol 142:105–111. [DOI] [PubMed] [Google Scholar]
- Risch N, Burchard E, Ziv E et al. 2002. Categorization of humans in biomedical research: Genes, race and disease. Genome Biol 3:comment2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risch N, Tang H, Katzenstein H et al. 2003. Geographic distribution of disease mutations in the Ashkenazi Jewish population supports genetic drift over selection. Am. J. Hum. Genet 72:812–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers AR, and Harpending HC. 1983. Population structure and quantitative characters. Genetics 105: 985–1,002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roseman CC 2004. Detecting interregionally diversifying natural selection on modern human cranial form by using matched molecular and morphometric data. Proc. Natl. Acad. Sci. USA 101:12,824–12,829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roseman CC, and Weaver TD. 2004. Multivariate apportionment of global human craniometric diversity. Am. J. Phys. Anthropol 125:257–263. [DOI] [PubMed] [Google Scholar]
- Rosenberg NA, Huang L, Jewett EM et al. 2010. Genome-wide association studies in diverse populations. Nat. Rev. Genet 11:356–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg NA, Li LM, Ward R et al. 2003. Informativeness of genetic markers for inference of ancestry. Am. J. Hum. Genet 73:1,402–1,422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg NA, Pritchard JK, Weber JL et al. 2002. Genetic structure of human populations. Science 298:2,381–2,385. [DOI] [PubMed] [Google Scholar]
- Rosenthal R 1994. Parametric measures of effect size. In The Handbook of Research Synthesis, Cooper H and Hedges LV, eds. New York: Russell Sage Foundation, 231–244. [Google Scholar]
- Siegel R, Naishadham D, and Jemal A. 2013. Cancer statistics, 2013. CA Cancer J. Clin 63:11–30. [DOI] [PubMed] [Google Scholar]
- Simons YB, Turchin MC, Pritchard JK et al. 2014. The deleterious mutation load is insensitive to recent population history. Nat. Genet 46:220–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smouse PE, Spielman RS, and Park MH. 1982. Multiple-locus allocation of individuals to groups as a function of the genetic variation within and differences among human populations. Am. Nat 119:445–460. [Google Scholar]
- Spitze K 1993. Population structure in Daphnia obtusa: Quantitative genetic and allozymic variation. Genetics 135:367–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tal O 2012. The cumulative effect of genetic markers on classification performance: Insights from simple models. J. Theor. Biol 293:206–218. [DOI] [PubMed] [Google Scholar]
- Teo YY, Small KS, and Kwiatkowski DP. 2010. Methodological challenges of genome-wide association analysis in Africa. Nat. Rev. Genet 11:149–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward E, Jemal A, Cokkinides V et al. 2004. Cancer disparities by race/ethnicity and socioeconomic status. CA Cancer J. Clin 54:78–93. [DOI] [PubMed] [Google Scholar]
- Weaver TD, Roseman CC, and Stringer CB. 2007. Were Neandertal and modern human cranial differences produced by natural selection or genetic drift? J. Hum. Evol 53:135–145. [DOI] [PubMed] [Google Scholar]
- Weir BS 1996. Genetic Data Analysis II Sunderland, MA: Sinauer. [Google Scholar]
- Weir BS, and Cockerham CC. 1984. Estimating F-statistics for the analysis of population structure. Evolution 38:1,358–1,370. [DOI] [PubMed] [Google Scholar]
- Whitlock MC 1999. Neutral additive genetic variance in a metapopulation. Genet. Res 74:215–221. [DOI] [PubMed] [Google Scholar]
- Whitlock MC 2008. Evolutionary inference from QST. Mol. Ecol 17:1,885–1,896. [DOI] [PubMed] [Google Scholar]
- Williams DR, and Collins C. 2001. Racial residential segregation: A fundamental cause of racial disparities in health. Public Health Rep 116:404–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DR, and Jackson PB. 2005. Social sources of racial disparities in health. Health Aff. (Millwood) 24:325–334. [DOI] [PubMed] [Google Scholar]
- Williams DR, and Mohammed SA. 2009. Discrimination and racial disparities in health: Evidence and needed research. J. Behav. Med 32:20–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson TW, and Grim CE. 1991. Biohistory of slavery and blood pressure differences in blacks today: A hypothesis. Hypertension 17:I122–I128. [DOI] [PubMed] [Google Scholar]
- Winkler CA, Nelson GW, and Smith MW. 2010. Admixture mapping comes of age. Annu. Rev. Genomics. Hum. Genet 11:65–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong MD, Shapiro MF, Boscardin WJ et al. 2002. Contribution of major diseases to disparities in mortality. New Engl. J. Med 347:1,585–1,592. [DOI] [PubMed] [Google Scholar]
- Zaitlen N, Pasaniuc B, Sankararaman S et al. 2014. Leveraging population admixture to characterize the heritability of complex traits. Nat. Genet 46:1,356–1,362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlotogora J, Zeigler M, and Bach G. 1988. Selection in favor of lysosomal storage disorders? Am. J. Hum. Genet 42:271–273. [PMC free article] [PubMed] [Google Scholar]