

Abstract
An impaired renal function, including acute and chronic kidney disease and end‐stage renal disease, can be the result of aging, certain disease conditions, the use of some medications, or as a result of smoking. In patients with renal impairment (RI), the pharmacokinetics (PKs) of drugs or drug metabolites may change and result in increased safety risks or decreased efficacy. In order to make specific dose recommendations in the label of drugs for patients with RI, a clinical trial may have to be conducted or, when not feasible, modeling and simulations approaches, such as population PK modeling or physiologically‐based PK modelling may be applied. This tutorial aims to provide an overview of the global regulatory landscape and a practical guidance for successfully designing and conducting clinical RI trials or, alternatively, on applying modeling and simulation tools to come to a dose recommendation for patients with RI in the most efficient manner.
INTRODUCTION
Under normal physiological conditions, the renal excretion clearance of drugs/metabolites is the net result of a three‐step process that includes glomerular filtration, tubular reabsorption, and tubular secretion. Glomerular filtration occurs due to the pressure gradient in the glomerulus whereby solutes and water are filtered out of the blood. 1 The filtrate is collected in the renal tubules, and as the filtrate moves through these tubules, ions, water, and nutrients are actively or passively reabsorbed from the filtrate to the interstitial fluid (tubular reabsorption). In contrast, some toxins and drugs/metabolites are actively transported from the interstitial fluid into the filtrate. Once the filtrate reaches the distal convoluted tubule, most of the urine and solutes have been reabsorbed. Some undesirable products like metabolic waste products, urea, uric acid, and certain drugs/metabolites are excreted by tubular secretion. Glomerular filtration rate (GFR) is the volume of glomerular filtrate formed per minute by the kidneys and is an important indicator of renal function.
An impaired renal function, including acute and chronic kidney disease and end‐stage renal disease (ESRD), can be the result of other conditions, such as hypertension, atherosclerosis, diabetes, or inherited diseases, and may also occur as a result of smoking or the use of some medications. Aging is shown to be associated with significant changes in the structure and function of the kidneys, even in the absence of comorbidities with the diseases mentioned above. 2
In patients with renal impairment (RI), renal clearance, as well as absorption, bioavailability, and plasma protein binding of drugs/drug metabolites can be altered. Additionally, RI is also associated with decreased activity of several hepatic and gastrointestinal drug‐metabolizing enzymes and transporters. As a consequence, the pharmacokinetics (PKs) of drugs or drug metabolites may change and result in the need for an altered dose to avoid a loss of therapeutic effect and/or an increase in safety risks. 3 , 4 It is therefore imperative that the impact of varying degrees of RI on the PKs of a new drug and its metabolite(s) be investigated to be able to define a safe and efficacious dose for these patients. This tutorial aims to provide an overview of the global regulatory landscape and practical guidance for successfully designing and conducting clinical RI trials. On the other hand, RI trials may not always be necessary or feasible to conduct under certain circumstances. In such cases, modeling and simulation methods may be applied as alternate methods, which are also discussed further in this tutorial.
Out of scope of this tutorial is augmented renal clearance in critically ill patients 5 or patients requiring renal replacement therapy (such as dialysis, hemofiltration, hemodiafiltration, and kidney transplantation) 6 although patients with ESRD requiring dialysis are mentioned. Any differences in renal function in the pediatric population are also out of scope. The reader is referred to a recently published tutorial on pediatric studies. 7
GLOBAL REGULATORY LANDSCAPE
On September 4, 2020, the US Food and Drug Administration (FDA) announced the availability of a revised draft guidance on “Pharmacokinetics in Patients with Impaired Renal Function—Study Design, Data Analysis, and Impact on Dosing” 8 which was published to collect additional public comments prior to its finalization. This revised draft of the FDA Guidance 8 and the current European Medicines Agency guideline 9 provide a deeper understanding on the approaches to evaluate the potential impact on the PKs of drugs/new chemical entities in the subjects with RI. They also describe the approaches pharmaceutical companies should follow when developing posology instructions for administering drugs in subjects with varying degree of RI. No specific guidelines by other major regulatory authorities are available. Personal communication with applicants has indicated that Japan (Pharmaceutical and Medicines Devices Agency) and China (National Medical Products Administration) have informally adopted the FDA Guidance while Australia (Therapeutic Goods Administration) has formally adopted the EMA guidelines. A close review of global regulatory documents revealed that, although these guidelines are quite similar, there are subtle differences in agency‐specific requirements around the timing of the study, the methodology of GFR assessment, specific language for biologics, and topically administered drugs. Some of these differences in the agency guidelines have been reviewed previously (Paglialunga et al.). 10 A comparison of the key aspects of the guidelines is provided in Table 1.
TABLE 1.
Comparison of the key guidelines for renal impairment study as per FDA and EMA guidance
| FDA Guidance 8 | EMA Guidance 9 | |
|---|---|---|
| Timing of study | No specific details mentioned | If dosing adjustment is required in patients with impaired renal function, a recommendation is provided to conduct the study in phase III |
| Large molecules/biologics | RI study not necessary for large molecules such as cytokines or cytokine modulators with MW > 69 kDa | Large proteins with MW > 60 kDa, such as monoclonal antibodies are not expected to undergo glomerular filtration, and therefore do not require a full RI study |
| GFR assessment methods | Recommends the use of eGFR assessment from the MDRD or CKD‐EPI methods for renally eliminated drugs. | Recommends the use of absolute GFR and the GFR normalized to a body surface area of 1.73 m2 for renally eliminated drugs |
| ESRD definition | ESRD definition has been modified in the current draft version and now refers to kidney failure (i.e., patients with renal function < 15 ml/min or dialysis patients on non‐dialysis days) | ESRD is defined as subjects with absolute GFR < 15 ml/min and requiring dialysis |
| Topically administered and hepatically eliminated drugs | A dedicated study may not be important for locally acting drugs (i.e., topical products) with limited systemic absorption | Waiver for conducting the RI study for both topically administered as well as hepatically cleared drugs without relevant systemic absorption |
| Data analysis | Model‐based analyses to establish a relationship between renal function (i.e. creatinine clearance CLcr or eGFR) and relevant PK parameters (AUC, C max , CL/F, t 1/2, and renal clearance CLR) | Both graphical and model‐based analysis to establish a relationship between the parameters defining renal elimination capacity and the PK parameters |
Abbreviations: AUC, area under the plasma‐concentration‐time curve; BSA, body surface area; CKD‐EPI, Chronic Kidney Disease Epidemiology Collaboration; CLcr, creatinine clearance; CL/F, apparent clearance; CLR, renal clearance; Cmax, maximum plasma concentration; eGFR, estimated glomerular filtration rate; EMA, European Medicines Agency; ESRD, end‐stage renal disease; FDA, US Food and Drug Administration; GFR, glomerular filtration rate; MDRD, Modification of Diet in Renal Disease; MW, molecular weight; PK, pharmacokinetic; RI, renal impairment; t1/2, terminal half‐life.
ASSESSMENT OF RENAL FUNCTION
Renal function is generally measured by assessing GFR, which is the rate or the volume per unit of time at which renal ultrafiltrate is formed by glomerulus and the normal measure is ~ 120 ml formed per minute. The GFR is a direct measure of renal function; however, measuring this rate is a laborious process. A useful and practical surrogate marker for the GFR is creatinine clearance (CrCL). 11 The CrCL measured over 24 h is used as a measure of renal function. However, this method may be inconvenient for routine measurement during clinical trials as accurately timed urine collection is needed. 12 Exogenous markers, such as iohexol, EDTA, or inulin (preferred in the EMA guideline) can be used to provide an accurate estimation of the GFR 8 , 9 but cannot be routinely applied in clinical studies. Therefore, various mathematical equations can be used to estimate CrCL and GFR. Table 2 describes the classification of RI based on the GFR or CrCL. As per EMA guidance, 9 for renally cleared drugs, the renal elimination capacity is not body size‐adjusted and rather referred to as “absolute GFR” in ml/min. The FDA guidance, 8 suggests individualization of GFR for drug dosing by multiplying the standardized GFR by the individual’s body surface area (BSA) and dividing by 1.73, therefore, now expressed in ml/min as opposed to ml/min/1.73 m2 unit.
TABLE 2.
| Stage | Description | Renal function a , b (ml/min) | GFR d (ml/min) |
|---|---|---|---|
| 1 | Control (normal renal function)7 | ≥90 | ≥90 |
| 2 | Mild | 60–89 | 60 to <90 |
| 3 | Moderate | 30–59 | 30 to <60 |
| 4 | Severe | 15–29 | <30 (dialysis not required) |
| 5 | Kidney failure a , c ; ESRD d | <15 or dialysis patients on non‐dialysis days | <15 requiring dialysis |
Abbreviations: EMA, European Medicines Agency; ESRD, end‐stage renal disease; FDA, US Food and Drug Administration; GFR, glomerular filtration rate.
As stated in the FDA guidance.
Estimate of GFR based on an estimation equation and expressed in ml/min. To convert ml/min/1.73 m2 to ml/min multiply by the individual’s body surface area calculated using an appropriate formula and divide by 1.73.
This classification is strictly for the purposes of conducting a dedicated renal impairment study and should not be used for the purposes of classifying kidney disease.
As stated in the EMA guidance.
Methods for GFR estimation and when to use a specific method
Cockcroft‐Gault (CG) calculation of GFR (Equation 1) 13 : this is the traditional method and has been extensively used in the past to estimate the renal function via CrCL estimation. However, creatinine is not only filtered but also secreted via active secretion process. Although this method adjusts for age, weight, and gender differences, it is less accurate than the Modification of Diet in Renal Disease (MDRD) equation (Equation 2) 14 or the Chronic Kidney Disease Epidemiology Collaboration (CKD‐EPI) equation (Equation 3), 15 , 16 which could result in an incorrect estimation of dosage.
Equation 1 (CG):
| (1) |
where, CrCL is creatinine clearance (ml/min) and Scr is serum creatinine (mg/dl).
CG tends to overestimate renal function at lower levels, for instance when a patient is obese or fluid overload exists. As a result, the increase in weight does not reflect an increase in muscle mass. 17 The FDA guidance 8 suggests that for subjects who are overweight or obese, alternative body weight metrics, such as ideal body weight or adjusted body weight, may be used when calculating CrCL. However, alternative methods exist to overcome this disadvantage as well. The MDRD 14 and CKD‐EPI 15 equations are two other common equations for calculating estimated GFR (eGFR) in adults 18 years of age and older. Particularly, the MDRD method has been increasingly applied in dosing recommendations of approved new drug labels. 18
Equation 2 (MDRD):
| (2) |
Factor is 0.742 (female) and 1.210 (African American).
The MDRD equation is adjusted based on body size and avoids the inclusion of weight, the formula is less prone to errors from fluid overload and obesity. There are several variations to the equation for the calculation of eGRF using MDRD. One disadvantage of the MDRD method is that it is not sensitive in estimating the GFR in patients with chronic kidney disease (CKD) with normal SCr level nor in ESRD.
Equation 3 (CKD‐EPI):
| (3) |
Factor is 1.018 (female) and 1.159 (African American); k is 0.7 (female) and 0.9 (male); a is −0.329 (female) and −0.411 (male); min indicates the minimum of SCr/k or 1, and max indicates the maximum of SCr/k or 1.
To estimate GFR accurately in patients with CKD , this equation was proposed. Although kidney disease research initiatives, such as Kidney Disease: Improving Global Outcomes (KDIGO) are promoting eGFR calculation using CKD‐EPI equation, there are no specific recommendations in the regulatory guidances. Using the CKD‐EPI equation for measuring GFR in phase I studies may help in harmonizing CKD staging, population PK (PopPK) analyses, and dosing by estimated renal function. 18 For more information, the reader is referred to a recent paper 19 that found a widespread dosing discordance rate among the CG, MDRD, and CKD‐EPI equations in patients.
WHEN TO CONDUCT A CLINICAL RI STUDY
An RI study aims to study the effect of mild, moderate, and severe RI on the PKs of a drug and its active metabolite(s), to inform a dose recommendation for this population in the drug label. Regulatory guidelines (i.e., FDA 8 and EMA 9 ) provide recommendations on when a study is needed for market registration. Figure 1 summarizes the recommendations for conducting an RI study and enlist the situations when it may not be necessary.
FIGURE 1.

Overview of criteria/conditions when an RI study may or may not be recommended. CL, clearance; FDA, US Food and Drug Administration; EMA, European Medicines Agency; PK, pharmacokinetic; RI, renal impairment
If RI is expected to significantly alter the PKs of the drug or metabolite (i.e., renal excretion is the main route of elimination for the drug or metabolite, as defined by the FDA as the fraction of systemically available drug or active metabolite that is eliminated unchanged in the urine [fe] is 0.3 or greater and by the EMA as renal excretion/metabolism is greater than or equal to one‐third of total elimination), it is recommended to perform a clinical PK study in patients with RI. However, all drugs that are intended for use in patients with RI or drugs with a narrow therapeutic window may likely require PK evaluation for dose recommendation independent of whether this drug is renally excreted or not. 20 , 21 Alternatively, for drugs with a narrow therapeutic window, therapeutic drug monitoring or monitoring of exposure based on clinical markers for efficacy and/or safety may be additionally considered. In contrast, if the maximum tolerated dose and therapeutic window are known through early studies, they may provide a large enough margin for the safety of the approved dose and not require a clinical PK study in patients with RI. 22
It is recognized in the regulatory guidances 8 , 9 that impaired renal function is associated with decreased activity of several hepatic and gastrointestinal drug‐metabolizing enzymes, such as cytochrome p450 enzymes, as well as UGT, NAT2, and transporters. 23 In a recent study, 24 the effect of metabolism of drugs specifically by CYP1A2, CYP2C8, CYP2C9, and CYP2C19, and the transport of drugs specifically by OATP as a function of CKD was assessed. The outcome suggests a decrease in clearance of OATP and CYP2C8 substrates as a function of CKD, although there was some overlap in the substrates, which calls the need for further investigation. In comparison, the effect on the other CYPs was variable and more modest.
The accumulation of uremic toxins in patients with RI or ESRD results in altered transcription and/or direct inhibition of CYP enzymes and transporters. 4 , 23 In addition, plasma protein binding of drugs or drug metabolites can be altered in patients with RI. So even for drugs that are mainly hepatically cleared and have a high protein binding, RI may have a significant impact on the PKs of the drug or drug metabolite as has been previously shown and summarized in literature. 23 , 25
If the PK evaluation in patients with RI shows little or no effect of RI on the PKs of the drug, it may still be required to study the effect of dialysis on the PK in patients with ESRD. 8 Patients with ESRD who need dialysis for therapeutic purposes should be regarded carefully in terms of dose selection, particularly for drugs and active metabolites with extensive renal excretion. 9 Dose reduction in these patients is often recommended to avoid adverse drug reactions. However, a drug or active metabolite can also be removed significantly by dialysis, in which case a dose adjustment would be required to ensure therapeutic efficacy. 9 It is therefore essential to have knowledge of the impact of dialysis on the elimination of drugs and their metabolites. 3 Study design and examples are discussed elsewhere in this tutorial.
Situations when an RI study may not be needed include when the drug is a therapeutic protein with a molecular weight greater than 69 kDa, or gaseous or volatile in nature and excreted by the lungs, or for locally acting drugs (i.e., mainly topically administered drugs), or drugs that do not enter the systemic circulation. 8 , 9 Additionally, supportive (pre)clinical safety and elimination data in the case of a single‐dose administration of a drug may suggest minimal impact in RI and may not require a dedicated clinical study. 8 , 9
CLINICAL RI STUDY DESIGN
The RI study design is primarily intended to compare the drug PKs in subjects with impaired renal function to the subjects with normal renal function. Usually, either a stand‐alone/full study design or a reduced/staged study design is applied. In some cases, a reduced design is followed by a full design.
Considerations for selection of the study design depends on factors, such as the expected effect of impaired renal function on the drug PK (i.e., whether the change in PKs is expected to be clinically relevant based on preclinical studies and/or mechanistic predictions), the expected impact of RI on drug pharmacodynamics (i.e., concentration‐response relationship), therapeutic window, and safety profile of the drug, the predominant route of elimination (renal vs. nonrenal) and the hemodynamic instability in case of patients with severe RI. A decision tree based on these considerations that shows when a full RI study or a reduced RI study should be conducted is shown in Figure 2.
FIGURE 2.
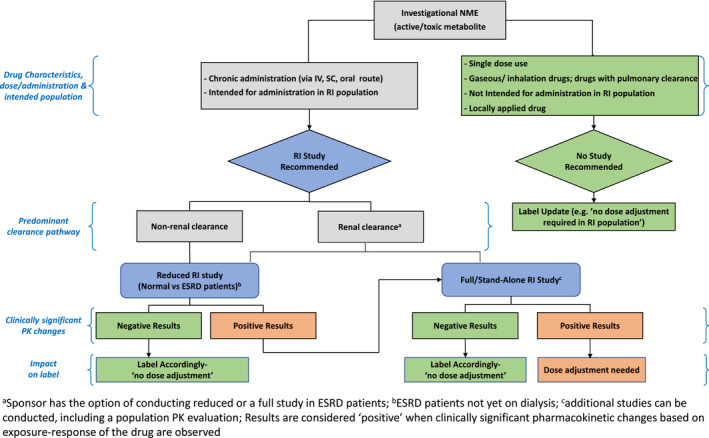
Decision Tree (adapted from Appendix 1 from the FDA guidance 8 ) to assess the need and type of renal impairment study. ESRD, end‐stage renal disease; NME, new molecular entity; PK, pharmacokinetic; RI, renal impairment
Stand‐alone/full study design
The stand‐alone/full study design is a dedicated phase I PK study using subjects with varying degrees of RI. A stand‐alone study design involves comparing the PKs of the drug in patients with varying degrees of RI (mild, moderate, and severe; see Table 2) to the control renal function group. The selection of subjects for a full RI study should ensure adequate representation of each degree of RI and control subjects. A parallel‐group study design is used. The number of subjects enrolled in each group should be adequate to detect the expected differences in the PKs between the groups to enable dosage recommendations for each group. As per EMA guidelines, 9 at least 6–8 subjects per RI group is considered adequate. In contrast to the previous version, the current draft version of the FDA guidance has added some recommendations around sample size selection. Although the FDA guidance 8 does not unequivocally provide a specific number, it provides some details and approaches around selection of adequate sample size. It thereby provides the example, an approach of prospectively targeting 95% confidence interval within 60% and 140% of the geometric mean estimate of relevant PK parameters for the drug in each renal function group with at least 80% power. Consideration should also be given to matching demographic factors, such as age, weight, sex, and ethnicity. Other factors, such as smoking habits and comedication, also require consideration. A 1:1 matching strategy, where individual subjects in each test group are matched with individuals in the control group can be used. A mean matching strategy is sometimes used, especially when the number of patients in each test group is a limiting factor. An example of the application of this study design is in phase I, open‐label, nonrandomized, parallel‐group trial for fulacimstat (BAY1142524). 26 This trial was conducted with the matching of healthy subjects for age, body weight, and gender (male/female) to the groups with RI (N = 36). The RI experimental groups included in the study design were patients with mild, moderate, and severe renal impairment, as classified in Table 2. As a single oral dose of the study drug (i.e., 25 mg immediate‐release tablet fulacimstat) was administered to evaluate the drug PKs in subjects with normal renal function versus patients with mild, moderate, or severe renal impairment. Another example is a study conducted for lesogaberan (AZD 3355), 27 a GABAB agonist drug. One‐to‐one match RI phase I study was conducted with 23 subjects to assess the PKs of the drug in healthy as well as patients with renal impairment (moderate or severe) after a single oral dose of 130 mg lesogaberan.
A single dose study can be used when the drug and its active metabolite(s) show linear PKs and no time‐dependent PKs at the concentrations that would be expected in patients on recommended doses. With single‐dose studies, the same dose is usually given to both the test and control groups. A multiple‐dose study will be more appropriate with nonlinear or time‐dependent PKs. However, with multiple dose studies, careful consideration should be given to patient groups with higher degrees of RI. Lower doses may be warranted in groups with higher degrees of RI to avoid toxicity. Modeling tools to predict doses appropriate in higher degrees of RI may be useful here.
Reduced/staged study design
The reduced/staged study design is an adaptive two‐stage study design often used to evaluate the initial PK effects in subjects at the extremes of renal function, and establish if a full study design is necessary. It may be considered for drugs that are predominately nonrenally eliminated (i.e., for hepatically metabolized and biliary excreted drugs). Under such circumstances, a full study design can be bypassed initially. This study design is also referred to as the “worst‐case scenario” as the PKs are compared between patients with extremes of renal function, meaning normal RI versus patients with severe RI, typically not yet on dialysis. Stage I involves comparing the drug PKs in severely impaired (test group) to the patients with normal renal function (control group). If the results indicate that the RI study has no clinically significant impact on drug PKs, no further study is warranted, and no dosing adjustment required. However, if the results indicate otherwise, a stage II study is warranted. In stage II, the intermediate renal function groups (mild and moderate impairment) should be further compared with patients with normal renal function. Stage II can be achieved by performing a full RI study or by alternative methods, such as a PopPK analysis of data from the phase II/III clinical studies. A description of such PopPK methodology is given later in this tutorial.
Subject inclusion in stage I of the reduced study design should ensure a match between the test group (subjects with ESRD) and the control group (patients with normal renal function). The principles described for dosing in the full study design apply to this study design as well. A reduced RI study was conducted for vismodegib 28 to include subjects with normal renal function (n = 9) and subjects with severe RI (n = 3). This was essentially an RI subgroup study within the indication (i.e., in a small number of patients with locally advanced or metastatic basal cell carcinoma).
Study design for patients with ESRD requiring dialysis
It is recommended by both the FDA 8 and the EMA 9 to investigate the PKs of drugs that are to be used in patients with ESRD requiring dialysis and the impact of the dialysis on the clearance of these drugs. The FDA guidance 8 specifically refers to intermittent hemodialysis (IHD) as this is the most common dialysis method used also briefly refers to the common therapies—intermittent dialytic as well as continuous dialytic therapies used in patients with ESRD in the United States. Dosage adjustments may be warranted if a significant fraction of the drug or its active metabolites are removed during the dialysis process. For drugs that are unlikely to be affected by the dialysis process, such as those with a large molecular weight, those that are tightly bound to plasma proteins, or those that are primarily nonrenally excreted, PK studies during dialysis may be omitted.
The objective of the PK studies in patients with ESRD on dialysis is to determine the extent to which the dialysis process could impact the elimination of the drug both during dialysis as well as in between dialysis sessions. It is therefore necessary to conduct the PK studies under dialysis and non‐dialysis (between dialysis) conditions. There are some recent examples in the literature where this impact on PKs was characterized for various drugs or drug combinations. 29 , 30 , 31 In addition, the FDA guidance 8 describes the need and approach for evaluating the PKs of critical care medications likely to be used in patients on continuous renal replacement therapy (CRRT). The findings from the IHD studies may not be sufficient for patients on CRRT and hence may require a specific study and study design.
The FDA guidance gives recommendations regarding the sample collection and data analysis for the accurate determination of drug clearance during dialysis (IHD, continuous dialysis, or peritoneal dialysis). First, pre‐dialysis blood samples must be collected and during the dialysis process, blood samples from both the arterial and venous sides of the dialyzer must be collected at appropriate intervals. Next, the volume of the entire dialysate should be recorded, and a sample should be used for drug concentration measurement. In addition, the model and make of the dialyzer, blood flow, and dialysate flow during dialysis should be recorded. Then, drug concentrations and, if applicable, active metabolite concentrations in the blood (entering dialyzer) and dialysate must be determined. After calculating the amount of drug retained in the dialysate, the following equation can be used to determine the drug clearance during dialysis (CLD):
Where, t0 is the start of the hemodialysis, and tl is the end of the hemodialysis.
Significant clearance of the drug by the dialysis may warrant dosage adjustments in these patients.
Characterizing the impact of renal function in phase II and phase III trials
The FDA guidance underpins the utility of PopPK analysis as an extremely informative tool to assess impact of renal function on drug exposure quite early in the process of drug development for the population intended for the clinical use of the drug. Given the clinical studies have well characterized PopPK analyses available from phase II and/or phase III clinical trials, accompanied by adequate representation of patients with varying degree of renal function in the dataset, PopPK can be highly recommended to provide appropriate dosing recommendations in labeling for the impaired renal function population. If under certain circumstances, patients with severe RI were not enrolled in sufficient number, a reduced RI design is recommended to cover the impact of RI on drug exposure. In addition, guidance suggests evaluating renal function as an independent predictor of the drug exposure‐response relationship whenever possible. Following are the key considerations for PopPK analysis – sufficient number of patients with representation over the range of renal function, adequate number of samples collection times with sufficient number of samples per patient, accurate dosing records, information of unbound drug concentration as well as active metabolite/parent drug levels (when appropriate). Furthermore, it is recommended to use same measures of estimated renal function, particularly when data is pooled across multiple phase II and/or phase III studies. If exposure‐response analysis is conducted and available from the pooled studies across phase II/III must be used as an independent predictor of response to account for impact of RI.
DATA ANALYSIS IN RI STUDIES
The objectives of the data analysis in RI studies are to determine whether the PKs of drugs differ between patients with normal renal function and those with different degrees of RI and to make dosage recommendations (if warranted) in patients with diminished renal function. The recommendation for dosing is based on the understanding of the relationship between a measure of renal function and relevant PK parameters. Figure 3 illustrates the workflow that can be used to achieve these objectives.
FIGURE 3.
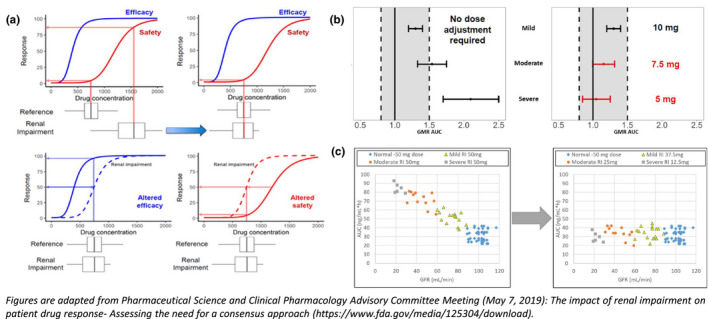
Translation from current to alternative approach: matching exposure‐efficacy/safety relationship between renal impairment and reference groups. AUC, area under the plasma‐concentration‐time curve; GMR, geometric mean ratio
Step 1 Estimation of PK parameters
As with all PK studies, plasma concentration data obtained in the RI studies are used to estimate PK parameters, using either noncompartmental or compartmental analysis. In addition, urinary excretion data may also be analyzed. The key PK parameters estimated in RI studies include apparent clearance (CL/F), renal clearance (CLR), apparent volume of distribution (Vd/F), and elimination terminal half‐life (t 1/2), as well as exposure parameters, such as the area under the plasma‐concentration‐time curve (AUC), maximum plasma concentration (C max), or minimal plasma concentrations (C min), depending on the drug itself and parameters that can be used as markers for efficacy or toxicity.
Step 2 Modeling the effect of different degrees of RI on the PKs of the drug
This step in the data analysis involves the development of mathematical models that can quantitatively relate the change in the specific PK parameter of the drug with the degree of RI as reflected by CrCL or eGFR. Effective models will enable the successful prediction of the relevant PK parameter of the drug, given the renal function status in the patient. Modeling approaches may include linear regression on noncompartmental analysis (NCA) of PK parameters of interest (e.g., CL/F) to determine the relationship between the parameter in healthy subjects and patients with RI. PopPK analysis, using nonlinear mixed effects modeling and covariate analysis can also be used to determine the impact of RI (using a covariate such as CrCL) on the parameter of interest. Models can use the renal function (CrCL or eGFR) as a categorical variable, such as normal, mild, moderate, or severe. Alternatively, regression analysis can be used, where the renal function and PK parameters are continuous variables. The latter approach is the preferred method.
Graphical presentation can be useful and is aimed at describing the relationship between elimination capacity (e.g., the measures of GFR and CrCL) as a continuous variable against PK variables, such as CL/F, AUC, C max, and C min (whenever appropriate).
Model‐based relationships can be assessed to provide a rational quantitative basis for dose adjustment in subjects with RI. The developed models should confidently predict the PK parameters of drug in a defined RI. In most cases, the regression approach is utilized to develop commonly used linear models between GFR and CL/F of the drug. However, other approaches, such as mechanistic modeling, can be used if adequately supported.
The estimated results should include the model predicted PK parameter estimates as well as measures of their precision (i.e., standard error or confidence interval). In addition, the prediction error estimates of drug/active metabolite clearance (such as confidence bounds for the prediction estimates) over a range of define renal function parameters (as CrCL or eGRF).
Step 3 Making dosage recommendations based on PK changes
The objective of this part of the analysis is to use the model developed in the previous step to make recommendations on the dose of the drug that can be used in patients with a specific degree of RI (as indicated by CrCL or eGFR) to achieve drug exposure (e.g., AUC or C max) that is similar to that observed in patients with normal renal function. Typically, a dosage recommendation may include an adjustment in the amount/dose of drug administered or the dosing interval or both. The application of the above model in the simulation of drug exposure with the different recommended doses in different degrees of RI is a useful aid in verifying and supporting the dosage recommendations.
There is a myriad of approaches to dosage selection/recommendation for patients with impaired renal function. One such approach is utilizing PK simulations that project systemic exposures that fall within the 5th and 95th percentiles of those achieved in the reference group. This can serve as boundaries for appropriate dose selection. Another approach is to establish no‐effect boundaries, which represent an interval band within which a change in systemic exposure due to varying degree of RI is deemed not significant enough and therefore warrant no further clinical action or dose modification. An alternate approach could be leveraging the modeling and simulation to determine the renal function threshold below which dosing adjustment is recommended.
Current approach
A generalized dose or dosing interval recommendation for a drug in patients with RI stems from matching the exposures to the reference group (i.e., subjects with normal renal function) enrolled in the clinical trial. As per the current approach, both dose and dosing interval are adjusted to match the plasma concentration/exposure of drug or active metabolite in subjects with RI to the subjects with normal renal function. 32
Limitations of current approach
Although this method serves its utility in making judicious decisions around phase III dose selection or in early dose recommendation for the label (Table 3), this method may have a few disadvantages. The late‐stage clinical trials generally include patients with mild impairment and at times patients with moderate impairment, but may lack subjects with severe RI and thereby any exposure matching to normal function may not represent the clinical experience. Furthermore, these trials also exclude patients with comorbidity and patients taking co‐medications with an intent to maintain a well‐defined patient population with minimum variability. These exclusions could in turn significantly shroud the treatment effect, create an “evidence gap” that limits the utility of these clinical trials in making generalized dosing recommendations in the patients with RI.
TABLE 3.
Example of dose adjustment recommendation based on (a) exposure matching of group mean or (b) matching to point estimates
| Stages of RI | (a) Exposure matching | (b) Matching point estimate | |||
|---|---|---|---|---|---|
| Fold increase in AUC (compared to reference) | Phase III dose evaluated | Labeled dose recommended | GMR for AUC (compared to reference) | Labeled dose recommended | |
| Normal (= reference) | 1× | A mg | A mg | — | A mg |
| Mild impairment | 1.3× a | A mg | A mg | 1.3 | A mg |
| Moderate impairment | 1.5× a | A mg | A mg | 2.5 | A/2 mg |
| Severe impairment | 2× b | Excluded | A/2 mg | 3.8 | A/4 mg |
(a) Compares the mean AUC between renal impairment and reference groups; (b) compares the point estimates (i.e., geometric mean ratio for AUC between groups).
Abbreviations: AUC, area under the plasma‐concentration‐time curve; RI, renal impairment.
<2‐fold of reference AUC.
≥2‐fold reference AUC.
This has recently been the topic of discussion at an FDA Pharmaceutical Science and Clinical Pharmacology Advisory Committee Meeting. 32
Alternative approach
This calls for availability of information on the drug metabolism, disposition, and elimination in the renally impaired subgroups in the early phase drug development. Inclusion of patients with severe RI/ESRD in the late‐stage clinical trials would be more informative; this would enable a robust decision making strategy for dosing recommendation in the RI population via modeling and simulation‐based approaches. Late‐stage trials for various scenarios (such as dose ranging studies, phase III dose selection, registrational trials, etc.) often use sparse PK sampling collection, which can be aptly utilized to assess the impact of RI on drug PK using modeling approaches.
Translation from current to alternative approach: Matching exposure‐efficacy/safety relationship between RI and reference subset population
Specific dosing recommendations as per both current and alternate approaches are based on the overall understanding of the relationship among renal function, drug exposure, and the exposure‐efficacy/safety relationship. 32
For exposure‐matching, we perform a hypothesis testing of whether the exposure‐response relationships for efficacy and safety are similar in the various degrees of RI subgroups (when available) for which the dosing is being derived and the reference exposure group. 8 Traditionally, the “reference group” is defined as the subgroup of patients with normal renal function. However, in certain situations, the definition of reference group appears somewhat ambiguous. The late‐stage clinical trials may/may not include the patients with varying extents of RI. More often, patients with mild to moderate RI are included, that too with/without prospective dose adjustment. Under such circumstances, this definition of reference group appears vague for the purpose of exposure‐matching. It becomes unclear whether reference group should always be the normal renal function subgroup, or instead selecting the patients with most proximal range of renal function and an acceptable benefit‐risk profile selected from the current trial would be more appropriate to unequivocally define the reference group. For drugs with a wide therapeutic range, changes in the drug PK due to renal function may not always result in a dosage adjustment for patients with RI. In such cases, for drugs with wide therapeutic range, subjects with normal function and mild impairment can be considered as reference (Figure 3a). There is still a need to establish a best practice guideline around selection of reference group in RI studies across the pharmaceutical industry. 32
Exposure‐response matching approaches
The documentation of the FDA Advisory Committee 32 includes a summation and explanation of the commonly applied approaches, which are (1) matching to a point estimate whereby the exposure matching is based on deriving doses for RI subgroups based on the geometric mean ratio (GMR) in the AUC; (2) matching the confidence interval of GMR of exposure (i.e. AUC, C max or C min) to predefined “no‐effect boundary,” and (3) matching to the range of exposures observed for the reference group.
Overall, matching to a point estimate is generally applied to the results of stand‐alone RI studies. An example of the application of this approach is given in Table 3 whereby the GMR for the RI subgroup relative to normal is two and the dose in the RI group is therefore reduced by half relative to the dose for individuals with normal renal function.
Matching the confidence interval of GMR, the “no‐effect boundary” is determined based on the understanding of the dose‐exposure‐response relationships. In the absence of reliable exposure‐response information, a totality of evidence or a conservative standard of bioequivalence principle (0.8–1.25) is invoked to determine the no‐effect boundary. Exposure matching is based on ensuring the 90% confidence interval of the expected AUC with dose adjustment falls within the no‐effect boundary. 8 This approach is further explained in Figure 3b.
When matching to the range of exposures observed for the reference group, the range of exposures observed in the registration trials is considered to have an acceptable benefit/risk profile. Dose adjustment in the RI subgroups is obtained through ensuring that the predicted exposures fall within this range. For example, dosing in patients with RI that result in exposures that fall within the 5th and 95th percentile of those observed in the reference group in clinical trials (Figure 3c).
IMPACT ON LABELING
The FDA 8 and EMA 9 guidances summarizes specifically which information is required to be incorporated in the label. Details of the results of the RI study and its clinical relevance to drug usage in patients with different degrees of RI are presented in the PK subsection of the Clinical Pharmacology section of the drug label (Section 4 in the EU Summary of Product Characteristics and Section 12 in the US Highlights of Prescription). 33 , 34 For drugs with specific dosage recommendations in different subgroups of patients with RI, such information is included in other relevant sections of the label, such as “Use in specific populations,” “Dosage and Administration,” “Warnings and Precautions,” and “Contraindications.” 8 , 33 , 34 One of the pivotal concerns that stems from the current approach of dosing recommendation is that the late‐stage clinical trials exclude patients with severe RI and ESRD. As a consequence, the dosing information in the label is often provided for mild and moderate RI subgroups only. These dosing recommendations more often translate to the labeling language as a statement – “No dose adjustment is needed in mild and moderate RI, however, the impact of severe RI, on safety, efficacy, or drug’s pharmacokinetics is unknown.” Examples of such language can be found in the paper by Xiao et al. 22 Furthermore, for patients with severe RI, the label is often observed to have the language statement as—"Dosing recommendations cannot be provided,” or “Patients with severe RI were not included in phase III studies, and the impact on the pharmacokinetics in this population subgroup is unknown.” 22
APPLICATION OF MODELING AND SIMULATION TOOLS TO ASSESS IMPACT OF RI ON DRUG PKS
Modeling and simulations (M&S) have been used to facilitate regulatory risk/benefit assessments and support drug labeling with increasing frequency. 35 , 36 , 37 , 38 Regulatory authorities, such as the FDA, now support and facilitate the development and application of robust models to enhance the regulatory evaluation process. 39
There may be situations where a dedicated RI study is not feasible, for example, in the event that there are some safety concerns with administering the study drug to healthy individuals, as with some anticancer drugs, a study in patients with RI and healthy matched controls may not be feasible. Another example is when a drug is administered as a long‐acting formulation, as with some psychotropic drugs, which would result in a very long study duration, making it challenging to conduct a study in healthy subjects and patients with RI. During the development of drugs with break‐through therapies, the need for rapid progression of the clinical development may warrant foregoing or deferral of a dedicated RI study. In such cases, M&S tools may be needed to guide dosing recommendations in the target patient population with RI. In the absence of adequate patient numbers for inclusion in a dedicated RI PK study, modeling tools may enable the use of data from various clinical trials during the development of the drug to evaluate the impact of RI on the PKs of the drug. Dose selection for different degrees of RI can also be challenging when designing either reduced or full RI clinical trials. Dose predictions using M&S can be useful here.
Modeling tools based on exposure‐response analysis, PopPK modeling and physiologically‐based pharmacokinetic (PBPK) modeling approaches are frequently used. PopPK analyses are based on models developed using observed clinical data (top‐down approach), whereas PBPK modeling is based on a mechanistic understanding of physiological and biochemical processes (bottom‐up approach), and observed data may be used for verification and enhancement of the modeling (middle out approach). PopPK analyses are well‐established in drug development and numerous examples of its application to drugs in RI are available in the literature. In the early stages of drug development, when limited information is available on the PK of the drug, in vitro data and information generated in preclinical studies can be used with PBPK modeling to predict the PKs of the drug in humans. Such predictions can be used to inform initial dosing in the RI studies as well as the study design. Both PopPK and PBPK modeling can be useful in understanding the drug disposition in patients with RI.
Exposure‐response analysis
Understanding the relationship between drug exposure and response (both efficacy and safety) is critical for identifying the optimal dose to strike a balance between drug efficacy and safety profile. Exposure‐response analysis has been increasingly used as an essential part of model‐based drug development; more often for justification of label dose and in decision making when to conduct an RI study, in early phases of clinical development or as postmarketing study. In a situation when the drug is renally cleared and has exhibited a steep exposure‐safety curve (i.e., a small change in PKs leading to a safety event), an RI study is highly warranted in the early stages of clinical development to guide dose recommendation across varying degrees of RI. On the other hand, if the drug has demonstrated a statistically significant increase in the exposure in patients with RI in a previous analysis (say, PopPK analysis), but this increase in exposure with higher degree of RI does not translate to clinically relevant change in exposure‐safety analysis, an RI study may be deemed unwarranted and no dosing adjustment will be needed for patients with RI.
In totality, the similarity of the exposure‐response relationship is assessed between normal (reference) versus the RI group to make a decision on RI study and dose adjustment. One such example is Crizotinib (Xalkori), a kinase inhibitor. Exposure‐response analysis was conducted to establish correlation between drug exposure (trough concentration) and renal function measure (CrCL), and was presented in the Summary Basis of Approval for Clinical Pharmacology and Biopharmaceutics Review. 40 The regression and categorical analysis indicated that steady state trough concentration (Ctrough) in patients with mild and moderate RI were similar to those in patients with normal renal function.
Population pharmacokinetics modeling in RI studies
PopPK is a well‐established technique used in drug development for more than 3 decades and has been extensively reviewed. 41 , 42 , 43 PopPK analyses can be used to analyze combined sparse sampling data sets from different clinical studies to estimate PK parameters and the impact of specific variables (covariates) on the PK parameters. RI studies usually have limited patient numbers, whereas other clinical trials (efficacy and safety) used throughout the clinical development of the drug may collectively account for rich and informative data on drug behavior in patients. PopPK can be used to analyze data from an RI study together with data from phase I, II, and III studies, to evaluate the impact of a measured covariate, such as CrCL on the specified PK parameter of a drug. A nonlinear mixed effect modeling technique can be used to build a correlation between various covariates (age, weight, gender, and CrCL) and PK parameter. CrCL is generally used as a measure of renal function and may be correlated to the CL/F as a function to drug PKs.
As discussed, positive results observed in an RI study with a reduced study design (normal vs. patients with ESRD) requires the conduct of a full RI study. This can be averted by using PopPK analyses if adequate numbers of patients with mild and moderate RI have been included in the phase II and III clinical trials. PopPK analyses on the collective data from all these studies can be used to estimate PK changes and recommend dosing. Duloxetine is an example where PopPK analyses together with the reduced RI study was used to support the label. 44 This drug is metabolized predominantly hepatically by CYP1A2 and CYP2D6. A reduced RI study showed greater than 100% increase in AUC in the patients with ESRD. PopPK analyses on data from phase II and phase III studies showed no significant difference between patients with normal renal function and those with mild or moderate RI. As a consequence, the label for this drug states that the drug should be avoided in patients with severe RI but refers to the PopPK analysis for the statement on mild to moderate degrees of RI to have no significant effect on duloxetine apparent clearance.
The PopPK analysis for axitinib, a kinase inhibitor, is another example. 45 For this drug, the recruitment of adequate patient numbers for a stand‐alone RI study may have been difficult. A review of the summary basis of approval revealed that a PopPK analysis was performed on data obtained from patients with a range of renal functions to evaluate the impact of RI on drug PK. The FDA accepted the PopPK analysis‐supported evidence and waived the requirement to conduct a dedicated RI study.
It is recommended that when PopPK analyses are used to support labeling, the following aspects should be carefully considered:
Adequate numbers of individuals within the different degrees of RI should be included to ensure that any PK differences between the groups can be adequately detected and dose adjustments can be predicted where necessary, active metabolites should be analyzed when appropriate, and unbound drug concentrations should be modeled when appropriate.
PBPK modeling to predict drug PK changes in RI
Utilization of PBPK modeling in the prediction of PK differences and potential dosage adjustments associated with RI is on the increase, as evident from the number of submissions to regulatory authorities 35 as well as publications in the literature. 46 , 47 , 48 Details on the general principles and application of PBPK modeling have been described extensively previously. 49 , 50 , 51 , 52 , 53 In generating a virtual RI population in the PBPK software, key differences in specific parameters that differ from the healthy population are incorporated into the mechanistic framework. Some of the fundamental physiological and biochemical parameters that have been quantified include GFR, the fraction of drug unbound in plasma (fu), hematocrit, gastric emptying time, and CYP enzyme activity. The changes in GFR during different states of RI are shown in Table 4. Hyperalbuminuria associated with RI results in reduced albumin in the plasma. Consequently, the fraction of drug bound to albumin is decreased. In addition, anemia is evident in CKD, resulting in reduced hematocrits. 53 In addition, gastric emptying time is prolonged with renal RI. Finally, measured hepatic enzyme expression data in subjects with RI are not available. Rowland‐Yeo et al. 54 extrapolated CYP abundance values using clinical data from patients with mild, moderate, and severe RI. Sayama et al. 55 reported a meta‐analysis of data for 151 drugs in subjects with moderate and severe RI to derive scalars for changes in nonrenal CL via CYP and UGT‐mediated metabolism and other undisclosed mechanisms. Scalars of 0.68 and 0.65 were reported for changes in CYP‐mediated hepatic metabolism for patients with moderate and severe RI relative to healthy subjects, respectively. The values that are applied for albumin concentration, hematocrit, and the gastric emptying time used for patients with moderate and severe RI are summarized in Table 4 in addition to the changes in CYP abundances related to the degree of RI, which can be included in PBPK models of subjects with RI. 54 , 55
TABLE 4.
Changes in the virtual population for RI a
| Control (normal renal function) | Moderate RI (GFR 30 – 59 ml/min/1.73 m2) | Severe RI (GFR <30 ml/min/1.73 m2) | Women | |||
|---|---|---|---|---|---|---|
| Males | Females | Males | Females | Males | ||
| Albumin (g/l) | 50.3 | 49.4 | 41.0 | 39.8 | 37.0 | 31.2 |
| Hematocrit (%) | 43 | 38 | 37.8 | 36.4 | 32.9 | 31.3 |
| Gastric emptying time (h) | 0.4 | 0.55 | 0.65 | |||
| Extrapolated CYP abundance (pmol/mg protein) | ||||||
| CYP1A2 | 52 | 28.4 | 27.4 | |||
| CYP2C8 | 24 | 20 | 13 | |||
| CYP2C9 | 73 | 63 | 29 | |||
| CYP2C19 | 14 | 5.5 | 2.3 | |||
| CYP2D6 | 8.0 | 4.6 | 2.1 | |||
| CYP3A4 | 137 | 95.2 | 87.3 | |||
Abbreviations: GFR, glomerular filtration rate; RI, renal impairment.
Note: All the CYP abundances is referring to the EM group.
Several studies have shown that RI affects both CLR and non‐renal clearance (CLNR) of drugs. 56 , 57 PBPK models for RI therefore consider both CLR and CLNR.
With evidence of changes in CYP enzyme expressions in RI (Table 4), drug clearance by all organs involved in metabolic clearance of the drug is likely to be changed. Hence, drugs that may not have a renal elimination pathway may also show altered clearance during RI.
The kidneys play a crucial role in drug disposition. The elimination may involve metabolites and/or unchanged drug. Factors that influence the GFR, active secretion, or the tubular reabsorption of drug/metabolite impact the extent of elimination via the kidneys. The effects of chronic renal disease and uremia on drug metabolism, transport, and elimination and the potential mechanisms involved in these changes, have been reviewed. 24 , 58 Some of these factors include the ionization or lipophilicity of the molecule, the plasma protein and erythrocyte binding, as well as affinity for transporter proteins in the kidneys. 56 , 57 These factors are used in the PBPK model to calculate the CLR and hence it is evident that the changes in RI that relate to GFR, renal blood flow, plasma protein binding, and blood‐to‐plasma ratio would affect the CLR of the drug.
Information on transporters and CLR during RI (such as potential changes in expression and/or abundance) that is required for the development of reliable PBPK transporter models in RI is limited, resulting in lower confidence in transporter‐related predictions in RI. Transporters significantly influence active secretion by the kidneys. Transporters on the basolateral (between blood and cells) and apical (between the cells and urine) membranes in the proximal tubules facilitate the uptake and efflux, respectively, of substances into the urine and thereby impact the disposition of drugs by the kidneys. Another equation can be used to scale the transporter contribution to clearance (CLint,T), to that relevant to the whole kidney (CLR,T). 46
Using PBPK modeling, Hsu and coworkers 46 were able to define the transporter‐mediated renal secretion and demonstrate that there is likely to be a greater than or equal to 10‐fold reduction in the functional proximal tubule cells per gram kidneys (i.e., the scaling factor PTCPGK) in severe RI, leading to decreased active secretion of their test drugs (oseltamivir carboxylate, cidofovir, and cefuroxime). In another study, seven renally excreted drugs that were substrates of organic anion transporters (OATs) were selected for PBPK modeling and evaluation of predictive performance. 59 It was shown that PBPK models that considered the effects of RI on tubular secretion, hepatic elimination, and inhibition of OATs by uremic solutes (which increase in RI), described the PK of the drugs reasonably in different stages of RI.
PBPK modeling for predictions of the exposure of drugs during different stages of RI is evolving continuously, as more data becomes available to improve the understanding of physiological and biochemical changes in these disease states. PBPK modeling is especially useful in predicting the impact of RI on drugs that are not predominantly excreted by the kidneys. PBPK modeling was used to predict the PK of drugs cleared mainly by CYP enzymes, such as telithromycin, sildenafil, and repaglinide, 60 rivaroxaban, 61 and clarithromycin 62 in RI patients. Another useful application of PBPK modeling is the prediction of drug‐drug interactions in patients with RI, such as those with clarithromycin or metoprolol. 62 A recent report on regulatory submissions states that 4% of all the PBPK submissions between 2008 and 2017 involved PBPK models for RI. 18 The authors highlight the current lack of confidence in the predictive performance of such models due to limited experience and the sparsity of quantitative data on specific changes in different degrees of RI, that enable the construction of reliable models. Studies that contribute to our knowledge of quantitative changes that impact on the PK of drugs during severe RI are warranted.
OTHER CONSIDERATIONS/APPROACHES
Other studies that may be performed or other considerations could also support the decision to perform a clinical PK RI study and/or dose selection for patients with RI. They are summarized here, but the reader is referred to another article for more details and examples. 22
Hepatic impairment study
This could serve as a surrogate for a reduced RI PK study for drugs with minimum renal elimination but nevertheless have a different PK exposure in patients with RI attributed to elevated uremic toxins that affect expression and/or the activity of drug metabolic enzymes and transporters.
Drug‐drug interaction study
Drug‐drug interaction (DDI) data around uremic toxins that cause changes in the expression and/or activity of drug metabolizing enzymes and transporters can be used to assess impact of RI on drug PK in patients. Lack of clinically significant DDI from an enzyme/transporter inhibitor or inducer can be construed as uremic toxic having no impact on the PKs of the drug under study. 22
CONCLUSIONS
Physiological changes observed in patients with RI may have an impact on the overall PK of a drug and/or its metabolite(s). This may result in a loss of efficacy or lead to an increased safety risk. It is therefore important to investigate the impact of varying degrees of RI on the PKs of a new drug and its metabolite(s) and adjust the dosing recommendation in these patients if warranted. Guidelines from regulatory agencies (e.g., the FDA and EMA) as well as ample literature examples are available to help in the design of such an RI study. There are also examples on investigating the effect of dialysis on the PK in patients with ESRD for whom dose recommendation should be carefully evaluated, particularly for drugs that are extensively renally excreted. However, full clinical PK trials in patients with RI may not always be necessary or feasible to conduct. In such cases, M&S methods, such as PopPK or PBPK modeling, may be applied. Whatever approach is taken, the results will be translated into a dose recommendation in the label for patients with RI that is safe and efficacious. This is generally the case for patients with mild and moderate RI but is still lacking for patients with severe RI and patients with ESRD. Therefore, the challenge for the future will be to address these evidentiary and labeling gaps.
In the coming years, we anticipate to see a shift in the renal function assessment methods in clinical practice, which would drive utility toward the use of MDRD and CKD‐EPI methods for accurate assessment of GFR in clinical setting. Furthermore, we also expect to see more acceptance in the clinical community toward use of 24‐h urine collection for the healthy match subject, in the cases where MDRD or CG assessment methods fails to accurately capture GFR.
In addition, recently, the FDA Pharmaceutical Science and Clinical Pharmacology Advisory Committee Meeting has put forth the best practices considerations for translating PK information into dose individualization instructions. By far, this document most eloquently outlines the current and alternate paradigm/approaches along with delicately covering the niche area of translation from current to alternate approach. Furthermore, this shares an elaborate discussion centered around identifying the key issues in current practices, such as enrollment of subjects with RI in the early phases (phase II and III) of clinical drug development, choice of appropriate reference group, and proposing high‐level mitigation strategies to address these issues. Last, the ultimate utility of this information lies in the translation recommendation via the use of exposure‐response relationship to build a better correlation between efficacy/safety and drug exposure in the patients with varying degree of RI. We anticipate more information will surface in this field in the coming years and perhaps an updated position on the FDA draft guidance will cover some of these most relevant best practices discussed here. Undoubtedly, these recent advancements in this field will emphasize the need for all the stakeholders across industry, regulatory agencies, and Clinical Research Organisations to develop the study protocols and design encompassing these newly proposed alternate approaches in an attempt to evaluate dosing regimen for inclusion in labeling with the full range of renal functions.
CONFLICT OF INTEREST
The authors declared no competing interests for this work.
AUTHOR CONTRIBUTIONS
P.R., M.C. and P.M. equally contributed to the writing of the manuscript. P.R. supervised the project.
REFERENCES
- 1. Murray I, Histology PMA, Histology, kidney and glomerulus. [Updated May 5, 2020]. In: StatPearls [Internet]. Treasure Island, FL: StatPearls Publishing; 2020. https://www.ncbi.nlm.nih.gov/books/NBK554544/. Accessed August 27, 2020 [Google Scholar]
- 2. Hommos MS, Glassock RJ, Rule AD. Structural and functional changes in human kidneys with healthy aging. J Am Soc Nephrol. 2017;28(10):2838‐2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Velenosi TJ, Urquhart BL. Pharmacokinetic considerations in chronic kidney disease and patients requiring dialysis. Expert Opin Drug Metab Toxicol. 2014;10(8):1131‐1143. [DOI] [PubMed] [Google Scholar]
- 4. Miners JO, Yang X, Knights KM, Zhang L. The role of the kidney in drug elimination: transport, metabolism, and the impact of kidney disease on drug clearance. Clin Pharmacol Ther. 2017;102(3):436‐449. [DOI] [PubMed] [Google Scholar]
- 5. Mahmoud SH, Shen C. Augmented renal clearance in critical illness: an important consideration in drug dosing. Pharmaceutics. 2017;9(3):36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ricci Z, Romagnoli S, Ronco CR. Renal replacement therapy. F1000Research. 2016;5:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shakhnovich V, Hornik CP, Kearns GL, Weigel J, Abdel‐Rahman SM. How to conduct clinical trials in children: a tutorial. Clin Transl Sci. 2019;12(3):218‐230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Food and Drug Administration, Center for Drug Evaluation and Research . Pharmacokinetics in patients with impaired renal function ‐ study design, data analysis, and impact on dosing. https://www.fda.gov/media/78573/download Cited (2020). Accessed November 4, 2020.
- 9. European Medicines Agency . Guideline on the evaluation of the pharmacokinetics of medicinal products in patients with decreased renal function. https://www.ema.europa.eu/en/documents/scientific‐guideline/guideline‐evaluation‐pharmacokinetics‐medicinal‐products‐patients‐decreased‐renal‐function_en.pdf Cited (2015). Accessed August 27, 2020.
- 10. Paglialunga S, Offman E, Ichhpurani N, Marbury TC, Morimoto BH. Update and trends on pharmacokinetic studies in patients with impaired renal function: practical insight into application of the FDA and EMA guidelines. Expert Rev Clin Pharmacol. 2017;10(3):273‐283. [DOI] [PubMed] [Google Scholar]
- 11. Nankivell BJ. Creatinine clearance and the assessment of renal function. Australian Prescriber. 2001;24(1):15‐17. [Google Scholar]
- 12. Fallahzadeh MK, Singh N. The 24 hour urine creatinine clearance for prediction of glomerular filtration rate in liver cirrhosis patients: have we considered all elements? Hepat Mon. 2013;13(7):e13398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16(1):31‐41. [DOI] [PubMed] [Google Scholar]
- 14. Levey AS, Coresh J, Greene T et al. Using standardized serum creatinine values in the modification of diet in renal disease study equation for estimating glomerular filtration rate. Ann Intern Med. 2006;145(4):247‐254. [DOI] [PubMed] [Google Scholar]
- 15. Levey AS, Stevens LA, Schmid CH, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009;150(9):604‐612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Michels WM, Grootendorst DC, Verduijn M, Elliott EG, Dekker FW, Krediet RT. Performance of the Cockcroft‐Gault, MDRD, and new CKD‐EPI formulas in relation to GFR, age, and body size. Clin J Am Soc Nephrol. 2010;5(6):1003‐1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Traynor J, Mactier R, Geddes CG, Jonathan GF. How to measure renal function in clinical practice. BMJ. 2006;333(7571):733‐737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Crass RL, Pai MP. Estimating renal function in drug development: time to take the fork in the road. J Clin Pharmacol. 2019;59(2):159‐167. [DOI] [PubMed] [Google Scholar]
- 19. McConachie SM, Shammout L, Martirosov DM. Clearance confusion: an exploratory analysis of inpatient dosing discordances between renal estimating equations. Ann Pharmacother. 2020;54(11):1102‐1108. [DOI] [PubMed] [Google Scholar]
- 20. Huang SM, Temple R, Xiao S, Zhang L, Lesko LJ. When to conduct a renal impairment study: US Food and Drug Administration perspective. Clin Pharmacol Ther. 2009;86(5):475‐479. [DOI] [PubMed] [Google Scholar]
- 21. Ibrahim S, Honig P, Huang SM, Gillespie W, Lesko LJ, Williams RL. Clinical pharmacology studies in patients with renal impairment: past experience and regulatory perspectives. J Clin Pharmacol. 2000;40(1):31‐38. [DOI] [PubMed] [Google Scholar]
- 22. Xiao JJ, Chen JS, Lum BL, Graham RA. A survey of renal impairment pharmacokinetic studies for new oncology drug approvals in the USA from 2010 to early 2015: a focus on development strategies and future directions. Anticancer Drugs. 2017;28(7):677‐701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nolin TD, Naud J, Leblond FA, Pichette V. Emerging evidence of the impact of kidney disease on drug metabolism and transport. Clin Pharmacol Ther. 2008;83(6):898‐903. [DOI] [PubMed] [Google Scholar]
- 24. Tan ML, Yoshida K, Zhao P et al. Effect of chronic kidney disease on nonrenal elimination pathways: a systematic assessment of CYP1A2, CYP2C8, CYP2C9, CYP2C19, and OATP. Clin Pharmacol Ther. 2018;103(5):854‐867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fujita K, Matsumoto N, Ishida H et al. Decreased disposition of anticancer drugs predominantly eliminated via the liver in patients with renal failure. Curr Drug Metab. 2019;20(5):361‐376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. ClinicalTrials.gov: Investigation of pharmacokinetics, safety, and tolerability of a single oral 25 mg BAY 1142524 IR tablet dose in male and female subjects with renal impairment and in age‐, gender‐, and weight‐matched healthy subjects in a single Center, non‐controlled, open‐label, observational design. ClinicalTrials.gov Identifier: NCT03402438.
- 27. ClinicalTrials.gov: A Phase I, open label, non‐randomized, parallel group, pharmacokinetic study in subjects with normal renal function, moderate or severe renal impairment receiving a single dose of oral 130 mg AZD3355. ClinicalTrials.gov Identifier: NCT00863161
- 28. Food and Drug Administration, Center for Drug Evaluation and Research . Division Director Summary Review NDA #203388. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/203388Orig1s000SumR.pdf. Cited (2012). Accessed August 27, 2020.
- 29. Scoville BA, Segal JH, Salama NN et al. Single dose oral ranolazine pharmacokinetics in patients receiving maintenance hemodialysis. Ren Fail. 2019;41(1):118‐125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Leuppi‐Taegtmeyer A, Duthaler U, Hammann F, et al. Pharmacokinetics of oxycodone/naloxone and its metabolites in patients with end‐stage renal disease during and between haemodialysis sessions. Nephrol Dial Transplant. 2019;34(4):692‐702. [DOI] [PubMed] [Google Scholar]
- 31. Kosloski MP, Zhao W, Marbury TC et al. Effects of renal impairment and hemodialysis on the pharmacokinetics and safety of the glecaprevir and pibrentasvir combination in hepatitis C virus‐negative subjects. Antimicrob Agents Chemother. 2018;62(3):e01990‐e2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Food and Drug Administration . Pharmaceutical Science and Clinical Pharmacology Advisory Committee Meeting May 7, 2019. https://www.fda.gov/advisory‐committees/advisory‐committee‐calendar/may‐7‐2019‐meeting‐pharmaceutical‐science‐and‐clinical‐pharmacology‐advisory‐committee‐meeting. Accessed August 27, 2020.
- 33. European Medicines Agency . A guideline on summary of product characteristics. https://ec.europa.eu/health/sites/health/files/files/eudralex/vol‐2/c/smpc_guideline_rev2_en.pdf. Cited (2009). Accessed August 27, 2020.
- 34. Food and Drug Administration, Center for Drug Evaluation and Research . Clinical pharmacology section of labeling for human prescription drug and biological products. https://www.fda.gov/media/74346/download. Cited (2016). Accessed August 27, 2020.
- 35. Grimstein M, Yang Y, Zang X, et al. Physiologically based pharmacokinetic modelling in regulatory science. J Pharm Sci. 2019;108:21‐25. [DOI] [PubMed] [Google Scholar]
- 36. Huang SM, Abernethy DR, Wang Y, Zhou P, Zineh I. The utility of modelling and simulation in drug development and regulatory review. J Pharm Sci. 2013;102:2912‐2923. [DOI] [PubMed] [Google Scholar]
- 37. Zineh I, Abernethy D, Hop C, et al. Improving the tools of clinical pharmacology goals for 2017 and beyond. Clin Pharmacol Ther. 2017;101:22‐24. [DOI] [PubMed] [Google Scholar]
- 38. Guo Y, Chu X, Parrot NJ, et al. Advancing predictions of tissue and intracellular drug concentrations using in vitro, imaging and PBPK modelling approaches. Clin Pharmacol Ther. 2018;104:865‐889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. US Food and Drug Administration . PDUFA reauthorization performance goals and procedures fiscal years 2018 through 2022 (PDUFA IV). https://www.fda.gov/media/99140/download. Accessed August 27, 2020.
- 40. Food and Drug Administration, Center for Drug Evaluation and Research . Clinical Pharmacology & Biopharmaceutics Review NDA #202570. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/202570Orig1s000ClinPharmR.pdf. Cited (2011). Accessed August 27, 2020.
- 41. Bonate PL, Strougo A, Desai A, et al. Guidelines for the quality control of population pharmacokinetic‐pharmacodynamic analyses: an industry perspective. AAPS J. 2012;14(4):749‐758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hu C, Zhang J, Zhou H. Confirmatory analysis for phase III population pharmacokinetics. Pharm Stat. 2011;10(1):14‐26. [DOI] [PubMed] [Google Scholar]
- 43. Duan JZ. Applications of population pharmacokinetics in current drug labelling. J Clin Pharm Ther. 2007;32(1):57‐79. [DOI] [PubMed] [Google Scholar]
- 44. Cymbalta® Prescribing information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/021427s051lbl.pdf. Accessed August 27, 2020.
- 45. Food and Drug Administration, Center for Drug Evaluation and Research . Clinical Pharmacology & Biopharmaceutics Review NDA #202324. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/202324Orig1s000ClinPharmR.pdf. Cited (2012). Accessed August 27, 2020.
- 46. Hsu V, De LT, Zhang P et al. Towards quantitation of the effects of renal impairment and probenecid inhibition on kidney uptake and efflux transporters, using physiologically based pharmacokinetic modelling and simulations. Clin Pharmacokinet. 2014;53:283‐293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li G, Wang K, Chen R, Zhao HR, Zheng QS. Simulation of the pharmacokinetics of bisoprolol in healthy adults and patients with impaired renal function using whole‐body physiologically based pharmacokinetic modeling. Acta Pharmacol Sin. 2012;33:1359‐1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. De Sousa Mendes M, Chetty M. Are standard doses of renally‐excreted antiretrovirals in older patients appropriate: A PBPK Study comparing exposures in the elderly population with those in renal impairment. Drugs R D. 2019;19(4):339‐350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jones HM, Chen Y, Gibson C et al. Physiologically based pharmacokinetic modeling in drug discovery and development: a pharmaceutical industry perspective. Clin Pharmacol Ther. 2015;97:247‐262. [DOI] [PubMed] [Google Scholar]
- 50. Zhao P, Rowland M, Huang S‐M. Best practice in the use of physiologically based pharmacokinetic modeling and simulation to address clinical pharmacology regulatory questions. Clin Pharmacol Ther. 2012;92:17‐20. [DOI] [PubMed] [Google Scholar]
- 51. Rostami‐Hodjegan A, Tucker GT. Simulation and prediction of in vivo drug metabolism in human populations from in vitro data. Nat Rev Drug Discov. 2007;6:140‐148. [DOI] [PubMed] [Google Scholar]
- 52. Jamei M, Dickinson GL, Rostami‐Hodjegan A. A framework for assessing inter‐individual variability in pharmacokinetics using virtual human populations and integrating general knowledge of physical chemistry, biology, anatomy, physiology and genetics: a tale of ‘bottom‐up’ vs ‘top‐down’ recognition of covariates. Drug Metab Pharmacokinet. 2009;24:53‐75. [DOI] [PubMed] [Google Scholar]
- 53. Jones H, Rowland‐Yeo K. Basic concepts in physiologically based pharmacokinetic modelling in drug discovery and development. CPT Pharmacomet Sys Pharmacol. 2013;2:e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rowland‐Yeo K, Aarabi M, Jamei M, Rostami‐Hodjegan A. Modeling and predicting drug pharmacokinetics in patients with renal impairment. Expert Rev Clin Pharmacol. 2011;4:261‐274. [DOI] [PubMed] [Google Scholar]
- 55. Sayama H, Takubo H, Komura H, Kogayu M, Iwaki M. Application of a physiologically based pharmacokinetic model informed by a top‐down approach for the prediction of pharmacokinetics in chronic kidney disease patients. AAPS J. 2014;16(5):1018‐1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhang Y, Zhang L, Abraham S, et al. Assessment of the impact of renal impairment on systemic exposure of new molecular entities: evaluation of recent new drug applications. Clin Pharmacol Ther. 2009;85:305‐311. [DOI] [PubMed] [Google Scholar]
- 57. Zhang L, Xu N, Xiao S et al. Regulatory perspectives on designing pharmacokinetic studies and optimizing labeling recommendations for patients with chronic kidney disease. J Clin Pharmacol. 2012;52:79S‐90S. [DOI] [PubMed] [Google Scholar]
- 58. Yeung CK, Shen DD, Thummel KE, Himmelfarb J. Effects of chronic kidney disease and uremia on hepatic drug metabolism and transport. Kidney Int. 2014;85(3):522‐528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hsueh CH, Hsu V, Zhao P, et al. PBPK modeling of the effect of reduced kidney function on the pharmacokinetics of drugs excreted renally by organic anion transporters. Clin Pharmacol Ther. 2018;103:485‐492. [DOI] [PubMed] [Google Scholar]
- 60. Zhao P, Vieira M, Grillo JA, et al. Evaluation of exposure change of non‐renally eliminated drugs in patients with chronic kidney disease using physiologically based pharmacokinetic modeling and simulation. J Clin Pharmacol. 2012;52(suppl 1):91S‐108S. [DOI] [PubMed] [Google Scholar]
- 61. Grillo JA, Zhao P, Bullock J, et al. Utility of a physiologically based pharmacokinetic (PBPK) modeling approach to quantitatively predict a complex drug‐drug‐disease interaction scenario for rivaroxaban during the drug review process: implications for clinical practice. Biopharm Drug Dispos. 2012;33:99‐110. [DOI] [PubMed] [Google Scholar]
- 62. Tortorici MA, Cutler DL, Hazra A et al. Emerging areas of research in the assessment of pharmacokinetics in patients with chronic kidney disease. J Clin Pharmacol. 2015;55(3):241‐250. [DOI] [PubMed] [Google Scholar]