

Abstract
Core-binding factor α2 (CBFα2; otherwise known as AML1 or PEBP2αB) is a DNA-binding subunit in the family of core-binding factors (CBFs), heterodimeric transcription factors that play pivotal roles in multiple developmental processes in mammals, including hematopoiesis and bone development. The Runt domain in CBFα2 (amino acids 51 to 178) mediates DNA binding and heterodimerization with the non-DNA-binding CBFβ subunit. Both the CBFβ subunit and the DNA-binding protein Ets-1 stimulate DNA binding by the CBFα2 protein. Here we quantify and compare the extent of cooperativity between CBFα2, CBFβ, and Ets-1. We also identify auto-inhibitory sequences within CBFα2 and sequences that modulate its interactions with CBFβ and Ets-1. We show that sequences in the CBFα2 Runt domain and sequences C terminal to amino acid 214 inhibit DNA binding. Sequences C terminal to amino acid 214 also inhibit heterodimerization with the non-DNA-binding CBFβ subunit, particularly heterodimerization off DNA. CBFβ rescinds the intramolecular inhibition of CBFα2, stimulating DNA binding approximately 40-fold. In comparison, Ets-1 stimulates CBFα2 DNA binding 7- to 10-fold. Although the Runt domain alone is sufficient for heterodimerization with CBFβ, sequences N terminal to amino acid 41 and between amino acids 190 and 214 are required for cooperative DNA binding with Ets-1. Cooperative DNA binding with Ets-1 is less pronounced with the CBFα2-CBFβ heterodimer than with CBFα2 alone. These analyses demonstrate that CBFα2 is subject to both negative regulation by intramolecular interactions, and positive regulation by two alternative partnerships.
The complex interplay between transcription factors bound to DNA provides enormous opportunity for regulation of gene expression. Not surprisingly, combinatorial control that utilizes multiple transcription factors is the rule for most eukaryotic enhancers. Recent findings implicate auto-regulation as an integral feature of these protein partnerships. There are regions within proteins that negatively regulate DNA binding or protein-protein interactions, presumably through intramolecular interactions (24). Positive regulation, as mediated by the creation of multiprotein complexes, can inactivate auto-inhibition. The molecular pathways for assembling these multiprotein complexes are beginning to emerge from systems in which both biochemical and structural approaches are aggressively undertaken.
The DNA-binding α subunits of the core-binding factors (CBFs) represent a model system of combinatorial control, as they display auto-inhibition that is rescinded through interactions with two different partner proteins. One partner is CBFβ, a subunit that binds CBFα subunits and stimulates DNA-binding activity without itself binding DNA (56, 85). CBFα subunits also interact with members of the ets family of DNA-binding proteins to form ternary complexes on DNA (19, 33, 41, 78, 86). These different classes of partnerships provide an opportunity to develop a mechanistic model for regulating DNA binding by both intra- and intermolecular interactions.
The CBFs comprise a small family of proteins involved in multiple developmental pathways in vertebrates and invertebrates (75). DNA-binding CBFα subunits in mammals are encoded by three genes (CBFA1, CBFA2 (AML1), and CBFA3), and the non-DNA-binding CBFβ subunit is encoded by the CBFB gene (4, 5, 36, 39, 48, 56, 57, 85). CBFA1 is required for bone development in mammals (34, 60). CBFA2 (AML1) and CBFB are essential for the emergence of definitive hematopoietic progenitors and stem cells in the mammalian embryo (52, 53, 58, 68, 82, 83). The Drosophila CBFA homolog runt functions in three developmental pathways: sex determination, segmentation, and neurogenesis (16, 17, 28, 67). The Drosophila gene lozenge, which also encodes a DNA-binding α subunit, plays a role in developmental pathways involving the eye, antenna, and tarsal claws and in the development of crystal cells, a blood cell lineage (13, 64, 77).
The ets proteins constitute a larger family of transcription factors that share a common DNA-binding domain, termed the ETS domain (25, 71). There are over 50 ets genes identified throughout metazoa, including over 20 paralogs in the human genome. Studies of vertebrate, Caenorhabditis elegans, and Drosophila ets proteins demonstrate roles in cell growth, differentiation, and transformation. For example PointedP2 (PntP2), a proposed ortholog of mammalian Ets-1 and Ets-2, is essential for R7 photoreceptor development in Drosophila and is the nuclear target of phosphorylation in the signal transduction pathway originating from the Sevenless receptor (2, 11, 59). In hematopoiesis, the ets protein PU.1 is required for B-cell and macrophage development (42, 70). Ets-1 is required for natural killer cell development (6), while both Ets-1 and Fli-1 are required for maintaining normal numbers of T cells (9, 44, 50). Both ets and CBF genes (FLI1, ERG, TEL, CBFA2, and CBFB) are frequent targets of chromosomal translocations in human leukemias (63); thus, dysregulation of ets or CBF function appears to be an underlying cause of hematopoietic transformation. One translocation, t(12;21), the most frequent chromosomal rearrangement in pediatric acute lymphocytic leukemia (43, 66, 72), actually fuses the ets gene TEL to CBFA2 (23, 65). Other ets and CBF genes (FLI-1, Pu.1, CBFA1, and ets-1) are preferential proviral insertion sites in leukemias and lymphomas induced by retroviruses (7, 49, 76) or oncogenes captured by acutely transforming retroviruses that cause leukemia (35, 54).
Many cell types in vertebrates express multiple ets genes, leading to a requirement for regulatory pathways that can dictate specificity of action of a particular ets protein. A common pathway to such specificity is partnerships with other transcription factors. Two well-characterized examples are the requisite interaction between serum response factor and one of the ets proteins Elk-1 and SAP-1 (14, 38) and the partnership between the ets protein PU.1 and the insulin response factor-related protein Pip (10). Biochemical and genetic analyses suggest that certain ets and CBF proteins also form partnerships. In Drosophila, both PntP2 and Lozenge are required for R7 cell development; PntP2 receives the signal from the Sevenless receptor, while Lozenge is required for the competency of R7 precursor cells to respond to the Sevenless signal (11, 13, 59). In vertebrates, Ets-1, Ets-2, PU.1, and GABP have been implicated as putative partners for the CBF proteins in regulating transcription of genes expressed in T, B, and myeloid cells (18, 19, 33, 41, 62, 78, 86). Ets-1 and CBFα proteins were shown to bind cooperatively to the T-cell receptor α- and β-chain enhancers, and synergistically activate transcription from the T-cell receptor α-chain enhancer in vivo and in vitro (19, 33, 41, 78, 86). The minimal B-cell-specific enhancer from the immunoglobulin μ-chain gene consists of binding sites for PU.1, CBF, and Ets-1 (or a related ets protein) (18). PU.1 and CBFα2 cooperatively activate transcription from the macrophage colony-stimulating factor promoter in myeloid cells (62). The osteopontin gene, which encodes a major noncollagenous bone matrix protein, contains a promoter responsive to both the CBFα1 protein and Ets-1 (69).
In this study, we used rigorous quantitative analyses to approach the issues of building multiprotein complexes. This methodology provides a framework for mechanistic investigations of both intra- and intermolecular regulation, including key insights for analyzing the structural basis of cooperativity between CBFα2 and two of its partners, CBFβ and Ets-1. The CBFα proteins share a 128-amino-acid region of homology, named the Runt domain after the founding member of the CBFα family (31). The Runt domain constitutes the DNA-binding domain of the CBFα proteins and the heterodimerization domain for CBFβ (31, 45, 57). Here we show that the full-length CBFα2 protein exhibits auto-inhibition, and we identify sequences C terminal to the Runt domain of CBFα2 that inhibit both DNA binding and heterodimerization with the CBFβ subunit. The C-terminal inhibitory sequences in CBFα2, however, do not repress binding of the α-β heterodimer to DNA. The second partnership that we characterize is that between CBFα2 and Ets-1. The sequences within CBFα2 that modulate its interaction with Ets-1 map to the N-terminal 214 amino acids, whereas the C-terminal auto-inhibitory sequences in CBFα2 are not required. Finally, we demonstrate that cooperative binding of CBFα2 with Ets-1 is not augmented by the CBFβ subunit. A model that integrates these phenomena is presented.
MATERIALS AND METHODS
Expression of CBFα2(451) and truncated derivatives.
We created a modified pVL1392 baculovirus transfer vector containing a Kozak sequence followed by sequences encoding a hexahistidine (H6) tag, two FLAG epitopes [(FLAG)2], and coding sequences for full-length CBFα2 [CBFα2(451)] (75) or its truncated derivatives. A PCR primer complementary to the H6 codons in the bacterial expression plasmid pQE30 (Qiagen), with a Kozak sequence and a BglII site at the 5′ end (5′-TTAGATCTCCGCCATGGGAGGATCGCATCACCATC-3′ was used in conjunction with a reverse primer (5′-CATTACTGGATCTATCAACAGG-3′) to amplify the H6 tag from pQE30. The PCR product was digested with BglII and BamHI and subcloned into the pBK-CMV vector (Stratagene) between the BglII (converted from a SpeI site) and BamHI sites. Complementary DNA encoding full-length CBFα2(451) (with an in-frame BamHI site preceding the ATG start codon) was subcloned in frame with the H6 tag, between the BamHI site and a KpnI site in the pBK-CMV polylinker. The resulting plasmid was partially digested with BamHI, and complementary oligonucleotides encoding the FLAG epitope (5′-GATCTATGGACTACAAAGACGATGACGATAAGG-3′ and 3′-ATACCTGATGTTTCTGCTACTGCTATTCCCTAG-5′) were subcloned into the BamHI site.
A plasmid containing two consecutive FLAG epitopes in the correct reading frame was identified by DNA sequence analysis. A BglII-KpnI fragment containing the H6(FLAG2-CBFα2(451) coding region was isolated from the pBK-CMV plasmid and subcloned into the corresponding sites in the polylinker of pVL1392. C-terminal truncations in CBFα2(451) were generated by PCR and used to replace C-terminal sequences of H6(FLAG)2-CBFα2(451) in the same pVL1392 plasmid. Subcloning details for the various C-terminal truncations will be provided upon request.
C-terminal H6 tags were introduced onto the truncated CBFα2(1-312) and CBFα2(41-312) proteins by PCR, using an antisense primer complementary to sequences encoding amino acids 306 to 312, preceded by six histidine codons, two stop codons, and a BamHI site, in conjunction with a sense primer complementary to sequences 5′ to a PstI site in CBFα2(451). The PCR product was digested with PstI and BamHI and subcloned into the corresponding sites in pVL1392. Complementary DNA encoding the 5′ end of CBFα2(451) (including 60 bp of 5′ untranslated sequence) was then subcloned into this vector as a NotI (from the polylinker of pBluescript SK+)-PstI fragment. Subcloning details for CBFα2(41-312)-H6 will be provided upon request.
Recombinant baculoviruses (Autographa californica) were produced with a BaculoGold transfection kit (Pharmingen) according to the manufacturer's protocol. Recombinant viruses were used to infect Sf9 cells (600 ml in 1-liter spinner flasks) that were grown to a density of 1.5 × 106 to 2.0 × 106 cells/ml. Cells were collected by centrifugation at 1,000 × g and then resuspended in 50 to 75 ml of serum-free complete medium (EX-400; JRH) supplemented with recombinant virus at a multiplicity of infection of 10. After incubation for 1 h at 27°C, Grace's complete medium (Gibco) was added to bring the final cell density to 1.5 × 106 cells/ml, and the infected cells were cultured at 27°C in spinner flasks for 48 h.
Partial purification of CBFα2(451).
All purification steps were performed at 4°C. Sf9 cells were harvested by centrifugation at 1,000 × g, and crude nuclei were prepared by hypotonic lysis (15). Nuclei were resuspended in 5 packed cell volumes of 6 M guanidine HCl–10 mM sodium phosphate (pH 8.0)–0.1% Triton X-100–10% glycerol (buffer A) and stirred for 1 h. The nuclear debris was pelleted (25,000 × g, 15 min), and the supernatant from 1.5 × 109 Sf9 cells was incubated with 2 to 3 ml of Ni-nitrilotriacetic acid (NTA) resin (Qiagen) for 1 h with continuous agitation. The protein was renatured on the Ni-NTA column by the following batch washes (5 min each, followed by centrifugation at 200 × g for 5 min): two washes with 30 ml of buffer A; three washes with 50 ml of 8 M urea–10 mM sodium phosphate (pH 8.0)–150 mM NaCl–0.1% Triton X-100–10% glycerol (buffer B); three washes with 50 ml of 8 M urea– 10 mM sodium phosphate (pH 7.4)–150 mM NaCl–0.1% Triton X-100–10% glycerol (buffer C); three washes with 50 ml of 1 M urea–10 mM sodium phosphate (pH 7.4)–300 mM NaCl–0.1% Triton X-100–10% glycerol (buffer D); and three washes with 50 ml of 10 mM sodium phosphate (pH 7.4)–300 mM NaCl–0.1% Triton X-100–10% glycerol (buffer E). The resin was then resuspended in 10 ml of 10 mM sodium phosphate (pH 7.4)–150 mM NaCl–0.1% Triton X-100–10% glycerol (buffer F) and poured into a column (5 ml). H6(FLAG)2-CBFα2(451) was eluted from the Ni-NTA resin with 20 ml of buffer F containing 200 mM imidazole. Protein fractions were frozen and stored at −70°C.
Native purification of truncated CBFα2 proteins.
Crude nuclei from infected Sf9 cells were prepared by hypotonic lysis and extracted with 20 ml of 10 mM sodium phosphate (pH 7.8)–500 mM NaCl–10% glycerol (buffer G) for 30 min at 4°C. The nuclei were pelleted (25,000 × g, 20 min), and the supernatant was collected and incubated with 2 ml of Ni-NTA resin for 1 h with continuous agitation. The resin was washed once with 20 ml of 10 mM sodium phosphate (pH 7.8)–500 mM NaCl–0.1% Triton X-100–10% glycerol (buffer H), poured into a column (5 ml), and washed with 20 ml of buffer H plus 15 mM imidazole. H6(FLAG)2-CBFα2 proteins were eluted with 10 ml of 10 mM sodium phosphate–150 mM NaCl–0.1% Triton X-100–10% glycerol (buffer I) plus 200 mM imidazole. Peak fractions from the Ni-NTA column were loaded directly onto an anti-FLAG M2 monoclonal antibody column (1 ml; Sigma), and the flowthrough fraction was readsorbed three times. The column was washed with 50 ml of buffer I, and the H6(FLAG)2-CBFα2 proteins were eluted from the anti-FLAG column with 0.33 mM FLAG peptide in 6 ml of buffer I as instructed by the manufacturer. Protein fractions were frozen and stored at −70°C. The concentrations of CBFα2 active for DNA binding were determined as described previously (12, 29).
CBFβ(187) was purified from bacteria as described previously (26). The activity of the CBFβ(187) protein was assumed to be 100%, based on the consistent quality of nuclear magnetic resonance spectra obtained with 15N-labeled protein (26). The fragment spanning amino acids 41 to 214 of CBFα2, which contains the DNA-binding Runt domain, was purified from bacteria as described elsewhere (B. E. Crute, Y.-Y. Tang, J. J. Kelley III, X. Huang, J. Yan, J. Shi, K. L. Hartman, T. M. Laue, N. A. Speck, and J. H. Bushweller. Submitted for publication). Expression and purification of full-length Ets-1 and Ets-1ΔN280 and determination of their active concentrations were performed as described previously (29, 61).
Synthetic oligonucleotides.
A high-affinity site (81, 84) was used to measure the binding affinity of CBF to DNA. An ets/CBF composite oligonucleotide (SC1/core) derived from the murine leukemia virus (MLV) enhancer was used to measure cooperative DNA binding. SC1/core contains a high-affinity ets site (55) juxtaposed to a core-binding site:
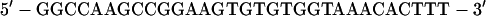 |
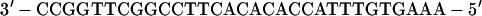 |
The spacing of the native MLV enhancer is retained in SC1/core. The higher affinity of the SC1 site facilitated more accurate quantification.
EMSA.
Equilibrium constants of CBFα2 and Ets-1 were determined by electrophoretic mobility shift assays (EMSA) using conditions described previously (12, 29). When protein titrations were used, the concentrations were in a range that resulted in approximately 0 to 100% binding. For proteins that were added in saturating amounts, the concentrations were at least 10-fold above the KD (equilibrium dissociation constant) of the protein for its specific site (CBFα2 and Ets-1; 2 × 10−8 M), ensuring >90% DNA occupancy. In all assays, the DNA concentrations were at least 10-fold below the estimated KD of either CBFα2 or Ets-1 (10−11 M), ensuring that the total protein [Pt] was an accurate estimate of free protein [P]. For most of the binding reactions, the protein(s) and DNA were added simultaneously and incubated on ice for 20 min. To measure the apparent affinity of CBFα2 in the presence of Ets-1, CBFα2 and DNA were preincubated for 20 min on ice. Saturating amounts of Ets-1 were added following the incubation, and all of the reactions were incubated for an additional 20 min. In most cases, DNA and protein-DNA complexes were resolved on 6% native polyacrylamide gels. Eight percent acrylamide gels were used for measuring cooperative DNA binding with CBFα2 fragments smaller than CBFα2(1-331) and for measuring the KD of CBFβ for CBFα2-DNA complexes. Following electrophoresis, the gels were dried and the radioactivity was quantified by the volume integration of individual bands by phosphorimaging (Molecular Dynamics ImageQuant).
Measurement of KD.
For assays containing only a single binding species, CBFα2 or Ets-1, KDs were measured as described previously (29). In brief, the fraction of free DNA, [D]/[Dt], was determined by measuring the ratio of the free DNA signal analyzed at each protein concentration to the DNA signal in a control lane containing no protein. The fraction of DNA in complex with protein, [PD]/[Dt], was derived from the relationship [PD]/[Dt] = 1 − [D]/[Dt]. Multiple experiments were performed with the same range of protein concentrations to provide a mean and standard error of each data point. Data were fit to the rearranged mass action equation, [PD]/[Dt] = 1/(1 + KD/[P]), using nonlinear least squares analyses (Kaleidagraph; Synergy Software) to derive KD values with standard error.
To measure the affinity of CBFα2-CBFβ heterodimers for DNA, CBFα2 was titrated onto a fixed amount of DNA (10−13 M) in the presence of 1.3 × 10−5 M CBFβ(187) (>10-fold above the KD of CBFβ for CBFα2 in solution). To determine the fraction of DNA bound as described above, the concentration of the α-β heterodimer as defined by the concentration of CBFα2 was substituted as [P] in the rearranged mass action equation. The KD of CBFβ(187) for CBFα2-DNA complexes was measured as described previously (26, 83).
To measure cooperative DNA binding, the apparent DNA binding affinity of the first protein, P1 was determined in the presence of a second protein, P2. The concentration of P2 was ≥10-fold above the KD of P2 for the DNA site. Competitive binding curves were generated from the equation [PD]/[Dt] = 1/(1 + KD/[P]) with the following assumptions. (i) Disappearance of the binary complex (DNA + P2) was measured; therefore, [Dt] was defined as the binary complex signal in a control lane that contained DNA and only P2. (ii) The binary complex signal (DNA + P2) was used as [D] for reaction mixtures with DNA + P1 + P2. (iii) The fraction of DNA in the ternary complex (DNA + P1 +P2) was defined as [PD]/[Dt], which was derived from 1 − [D]/[Dt].
The effect of CBFβ on cooperative DNA binding between CBFα2 and Ets-1 was determined by a similar approach. The CBFβ concentration was 2 × 10−5 M, ≥10-fold above its KD for CBFα2. All EMSAs containing either one or two proteins were quantified as described above. To measure the KD of Ets-1 in the presence of CBFα2-CBFβ heterodimer, the disappearance of the DNA signal from the CBFα2-CBFβ-DNA complex was determined and used as [D] to generate binding curves as described above.
RESULTS
Purification of CBFα2 proteins.
The CBFα2 proteins were produced by using a baculovirus expression system and partially purified by His and FLAG tag affinity chromatography (Fig. 1). Full-length CBFα2(451), due to its tight association to the nuclear matrix (32, 87), was obtained from insect cell extracts under denaturing conditions and refolded on the Ni-NTA column (Fig. 1A). Limited quantities of partially purified material were obtained by this method, and no further purification was possible without loss of activity. A series of C-terminal truncations in CBFα2(451) starting at amino acid 331 were engineered (Fig. 2B). These truncated proteins were purified to homogeneity from soluble nuclear extracts by sequential affinity chromatography on Ni-NTA and anti-FLAG antibody columns (Fig. 1A). The concentrations of active full-length and truncated CBFα2 proteins were determined by DNA titrations. Representative examples of the purification and activity determination are shown in Fig. 1.
FIG. 1.

Expression and purification of CBFα2. (A) Coomassie blue-stained sodium dodecyl sulfate-polyacrylamide gel displaying fractions from each step of the purification for CBFα2(451) and two truncated derivatives, CBFα2(1-331) and CBFα2(1-214). Lanes: M, molecular weight markers; NE, unfractionated nuclear extract; Ni-NTA, eluate from the Ni-NTA column; αFLAG, eluate from the anti-FLAG monoclonal antibody column. Arrows indicate expected position of the CBFα2 bands. (B) Activities of CBFα2 proteins quantified by DNA titration in an EMSA. Concentrations (molar) of protein-DNA complex [PD] versus total input DNA [Dt] are plotted.
FIG. 2.

Modulation of CBFα2 DNA binding by C-terminal sequences. (A) Equilibrium DNA binding studies of full-length CBFα2(451) and CBFα2(41-214) were performed by EMSA and used to generate DNA binding curves. Data from at least three experiments provide mean and standard error for each data point. KD values were obtained by curve fitting as described in Materials and Methods. (B) Summary of equilibrium dissociation constants for truncated CBFα2. The black rectangle in the schematic diagram of CBFα2 represents the DNA-binding Runt domain. The gray and stippled boxes represent the H6 and FLAG tags, respectively. Relative affinity was calculated as the ratio of mutant affinity to the affinity of CBFα2(451). (C) Summary of equilibrium dissociation constants for CBFα2 proteins tagged at amino acid 312 with H6 (gray box).
Sequences C terminal to the Runt domain in CBFα2(451) inhibit DNA binding.
Quantitative DNA binding assays detected a significant difference between the affinity of full-length CBFα2(451) and the isolated DNA-binding Runt domain, CBFα2(41-214). Figure 2A presents protein titrations performed on a high-affinity core site. Full-length CBFα2(451) displays a 69-fold-lower affinity for DNA than CBFα2(41-214). Sequences in CBFα2(451) that inhibit DNA binding were mapped by analyzing the affinity of sequentially truncated proteins (Fig. 2B). A C-terminal truncation to amino acid 214 [CBFα2(1-214)] derepressed DNA binding significantly (34-fold). Further truncation from amino acid 214 to 190 had no added effect. All truncated CBFα2 proteins containing additional C-terminal sequences between amino acids 214 and 451 exhibited lower DNA-binding affinity than CBFα2(1-214). However, none of the truncated proteins bound DNA as poorly as CBFα2(451). These results map the C-terminal inhibitory sequences over a large region between amino acids 214 and 451, and they suggest that there are multiple inhibitory elements distributed throughout this large region. Alternatively, the inhibitory sequences are distant from each other in the primary structure but located on a single surface of the folded protein.
Sequences N terminal to the Runt domain modestly affect DNA binding. The affinities of CBFα2(1-312) and CBFα2(41-312) were essentially identical (Fig. 2C), and CBFα2(1-214) and CBFα2(41-214) displayed only a twofold difference in affinity (Fig. 2B). Thus, inhibitory sequences that affect DNA binding appear to be located primarily in the C terminus of the protein, between amino acids 214 and 451.
C-terminal sequences in CBFα2 modulate heterodimerization with CBFβ.
CBFβ increases the affinity of the CBFα subunits for DNA. In quantitative analyses, a sixfold increase in DNA-binding affinity of a Runt domain fragment, CBFα2(41-214), was observed in the presence of the CBFβ subunit (Crute et al., submitted). The auto-inhibition phenomenon raises the question of whether the inhibitory sequences that affect DNA binding also influence binding of the CBFα2-CBFβ heterodimer to DNA or modulate heterodimerization of the CBFα2 and CBFβ subunits. To address these questions, we analyzed DNA binding of inhibited and activated forms of CBFα2 in the presence and absence of CBFβ. CBFα2(1-331) was chosen as the inhibited species, as it is the largest CBFα2 protein fragment that we could purify to homogeneity. The binding properties of CBFα2(1-331) were compared to those of the isolated Runt domain CBFα2(41-214), which represents the uninhibited species. CBFα2(1-214), another uninhibited species, was also analyzed to assess the impact of sequences N terminal to the Runt domain on interactions with CBFβ on and off the DNA.
To facilitate the presentation of these results, we illustrate a simple network of potential interactions between CBFα2, CBFβ, and the DNA as described by four equilibria, with equilibrium dissociation constants K1, K2, K3, and K4 (Fig. 3A). K2 describes CBFα2 binding to DNA in a binary complex. The difference in K2 between the isolated runt domain, CBFα2(41-214), and the C-terminally truncated protein CBFα2(1-214) is twofold, and the K2 values for CBFα2(41-214) and CBFα2(1-331) differ eightfold (Fig. 3B and E). These differences illustrate the autoinhibitory phenomenon of CBFα2 that is mediated primarily by sequences C terminal to amino acid 214.
FIG. 3.

Thermodynamic box describing interactions between CBFα2, CBFβ, and DNA. (A) Schematic diagram of the potential interactions between CBFα2 (α), CBFβ (β), and DNA. The modeled bend in DNA induced by the Runt domain is suggested by both circular permutation analysis and circular dichroism spectroscopy (22; Crute et al., submitted). (B) Equilibrium dissociation constants (K2) of CBFα2(41-214), CBFα2(1-214), and CBFα2(1-331) for DNA. Data from at least three experiments are presented. Standard errors are 1.1 × 10−12 M, 2.1 × 10−12 M, and 7.1 × 10−12 M, respectively. (C) Equilibrium dissociation constants (K4) of CBFα2-CBFβ heterodimers for DNA. Standard errors are 3.9 × 10−13 M for CBFα2(1-214) and 1.8 × 10−13 M for CBFα2(1-331). (D) Equilibrium dissociation constants (K3) of CBFβ for CBFα2-DNA complexes. Data represent at least three experiments. Standard errors are 3.2 × 10−9 M, 1.5 × 10−9 M, and 3.5 × 10−9 M for CBFα2(41-214), CBFα2(1-214), and CBFα2(1-331), respectively. (E) Summary of equilibrium dissociation constants K1, K2, K3, and K4. K4 for CBFα2(41-214) was not determined directly but calculated from K2K3 = K1K4. K1 for CBFα2(41-214) was determined independently (Crute et al., submitted).
The other three equilibria were tested for sensitivity to these same auto-inhibitory sequences. The equilibrium dissociation constant K4 characterizes binding of the CBFα2-CBFβ heterodimer to DNA. This binding affinity was measured by titrating CBFα2 onto a constant, limited amount of DNA (10−13 M) in the presence of a constant, excess amount of CBFβ (1.3 × 10−5 M). These conditions ensured that all the available CBFα2 was in the heterodimeric form. The DNA-binding affinities of all three heterodimeric complexes were approximately equal (Fig. 3C and E), suggesting that neither sequences N terminal to the Runt domain nor the C-terminal inhibitory sequences interfere with binding of the CBFα2-CBFβ heterodimer to DNA.
CBFβ can assemble onto a preformed CBFα2-DNA complex, as represented by K3. To measure K3, a protein titration of CBFβ was performed under conditions in which all CBFα2 was bound to DNA (Fig. 3D). The affinities of CBFβ for the CBFα2(41-214)–DNA and CBFα2(1-214)–DNA complexes are essentially equal, demonstrating that sequences N-terminal to the Runt domain do not affect heterodimerization on DNA, at least in the context of CBFα2 proteins truncated at amino acid 214. In contrast, the affinity of CBFβ for the CBFα2(1-331)–DNA complex is 5.3-fold lower than for the uninhibited CBFα2 proteins. These data suggest that sequences C-terminal to the Runt domain hinder the interaction of CBFβ with CBFα2 when bound to DNA.
Finally, CBFα2 and CBFβ can form heterodimers in the absence of DNA with an equilibrium dissociation constant represented as K1. K1 cannot be directly measured by EMSA; however, the equation K2K3 = K1K4 allows K1 to be calculated. K1 for the uninhibited species, CBFα2(41-214) and CBFα2(1-214), differ from K1 for the inhibited protein CBFα2(1-331) 21-fold, indicating that sequences between amino acids 214 and 331 inhibit CBFα2-CBFβ heterodimerization (Fig. 3E).
In summary, sequences in CBFα2 C terminal to the Runt domain inhibit DNA binding (K2) and heterodimerization with the CBFβ subunit (K1 and K3). Heterodimerization is inhibited both in solution (K1) and on the DNA (K3), but less so on DNA. Finally, DNA binding of the preassembled heterodimer (K4) is not significantly affected by C-terminal inhibitory sequences.
Ets-1 enhances CBFα2 DNA binding.
CBFα2 also functions in association with Ets-1 (33, 86). To compare this partnership to that of the CBFα2-CBFβ heterodimer, quantitative EMSAs were used to investigate DNA binding cooperativity. We chose a composite binding site that contains a high-affinity ets binding site (SC1) (55) juxtaposed to a CBF binding site similar to that found in the Moloney MLV enhancer (74). The spacing between the two sites retains the configuration within the Moloney MLV enhancer. The binding affinity of each protein alone on this engineered composite site, termed SC1/core, was determined by protein titrations with a constant, limited amount of DNA (10−12 M). The KD of CBFα2(1-331) for the SC1/core site was 3.0 × 10−9 M, and the KD of Ets-1 was 8.5 × 10−10 M (Fig. 4A and B).
FIG. 4.

Ets-1 and CBFα2 bind DNA cooperatively. (A) EMSA of equilibrium DNA binding studies of CBFα2(1-331) titrated onto DNA alone or in the presence of Ets-1 (left) or Ets-1 titrated onto DNA alone and in the presence of CBFα2(1-331) (right). (B) Equilibrium DNA binding curves for CBFα2(1-331) (left) and Ets-1 (right); data from panel A. Symbols: ○, binary protein = DNA complexes; ●, ternary complexes. Equilibrium DNA binding curves display [PD/[Dt] as the mean (±standard error) of at least two independent experiments. (C) Thermodynamic box depicting potential interactions between Ets-1, CBFα2, and DNA. Equilibrium dissociation constants were obtained from panels A and B. KD values and standard error were obtained from the curve fit of means as described in Materials and Methods.
We next determined the extent to which Ets-1 enhances CBFα2 DNA binding by measuring the apparent affinity of CBFα2 for the composite element in the presence of Ets-1. The CBFα2 titration was repeated under conditions that predict 90% occupancy of DNA by Ets-1. The apparent DNA-binding affinity of CBFα2(1-331) increased sevenfold in the presence of Ets-1 (Fig. 4A and B). Interestingly, this enhancement was observed only under conditions in which CBFα2 binding was allowed to reach equilibrium prior to addition of Ets-1. The molecular basis of this order-of-addition effect is considered in Discussion.
Thermodynamics dictates that cooperative binding between CBFα2 and Ets-1 will be reciprocal under ideal equilibrium conditions. To test this prediction, a protein titration of Ets-1 was performed under conditions that predict 90% occupancy by CBFα2(1-331). As expected, the presence of CBFα2(1-331) enhanced the apparent DNA-binding affinity of Ets-1 approximately 10-fold (Fig. 4A and B).
Figure 4C illustrates the thermodynamic equilibria describing the reciprocal cooperativity between CBFα2(1-331) and Ets-1. K1, which represents the binding of Ets-1 alone to DNA, is 10-fold higher than K3, the equilibrium dissociation constant for Ets-1 binding to a CBFα2(1-331)–DNA complex. Reciprocally, K2, which describes binding of CBFα2(1-331) to DNA, is sevenfold higher than K4, which represents binding of CBFα2(1-331) to DNA occupied by Ets-1. Note as expected from thermodynamics that K2K3 ≈ K1K4.
The scheme presented in Figure 4C does not include the potential interaction between Ets-1 and CBFα2(1-331) in the absence of DNA. A direct interaction may be excluded under the conditions of our assay only if the binding of Ets-1 to CBFα2(1-331) in solution has a KD at least 10-fold higher than the concentrations of Ets-1 and CBFα2 used to saturate the SC1/core site (2 × 10−8 M). Increasing the concentration of CBFα2(1-331) had no effect on K3 (data not shown), supporting the hypothesis that interactions off DNA do not occur to an appreciable extent at the protein concentrations tested. In addition, no direct interactions between Ets-1 and CBFα2(1-331) could be detected by surface plasmon resonance spectroscopy with a CBFα2 surface and Ets-1 concentrations as high as 10−8 M (see accompanying report [20]). Therefore, we predict that any interaction between Ets-1 and CBFα2(1-331) in solution will have a KD greater than 10−7 M.
Specific regions of CBFα2 are involved in cooperative DNA binding with Ets-1.
Sequences required for cooperative interactions with Ets-1 were mapped by testing deletion mutants of CBFα2. Protein titrations of CBFα2 were performed under saturating conditions for Ets-1 (Fig. 5). Removal of CBFα2 sequences C terminal to amino acid 214 did not affect cooperative binding with Ets-1 (Fig. 5B). Thus, the intramolecular inhibitory sequences in the C terminus of CBFα2(1-331) do not appear to be required for cooperative binding. No cooperative binding was observed between Ets-1 and the isolated CBFα2 Runt domain, CBFα2(41-214) (Fig. 5C). Removal of amino acids 190–214 to create CBFα2(1-190) also disrupted cooperative DNA binding with Ets-1 (Fig. 5D). Again, reciprocal cooperativity was obtained when Ets-1 was titrated onto DNA saturated with the truncated proteins CBFα2(1-331) and CBFα2(1-214) but not with CBFα2(41-214) and CBFα2(1-190) (data not shown). Thus, sequences N terminal to amino acid 41 and between amino acids 190 and 214 in CBFα2 contribute to cooperative binding with Ets-1.
FIG. 5.

Sequences in CBFα2 required for cooperative DNA binding with Ets-1. Equilibrium DNA binding studies were performed by EMSA with truncated CBFα2 proteins in the absence (open circles) or presence (closed circles) of Ets-1 (A to D). Equilibrium DNA binding curves display [PD]/[Dt] as the mean (±standard error) of at least two independent experiments. (E) Summary of equilibrium dissociation constants derived from binding curves in panels A to D. KD values and standard error were obtained from the curve fit of means as described in Materials and Methods.
CBFβ and Ets-1 do not synergistically stimulate CBFα2 DNA binding.
Our findings implicate both CBFβ and Ets-1 as partners for CBFα2. A remaining question is whether these two proteins can work together to enhance CBFα2 DNA binding. To facilitate the visualization of complexes containing all three proteins on DNA, we used an Ets-1 deletion mutant, Ets-1ΔN280, that has a molecular mass of 18 kDa. The accompanying report (20) demonstrates that Ets-1ΔN280 retains all sequences required for cooperative binding with CBFα2(1-331). Protein-DNA complexes containing Ets-1ΔN280 alone, CBFα2 alone, CBFα2-CBFβ, CBFα2–Ets-1ΔN280, and CBFα2–CBFβ–Ets-1ΔN280 can be clearly distinguished by EMSA (Fig. 6A). We titrated the CBFα2(1-331)–CBFβ heterodimer onto DNA alone and onto DNA saturated with Ets-1ΔN280 (Fig. 6A and B). Ets1ΔN280 did not further augment binding of CBFα2-CBFβ to DNA (Fig. 6B and C). In a reciprocal experiment, we titrated Ets-1ΔN280 onto DNA saturated with CBFα2(1-331) in the presence or absence of CBFβ (Fig. 6). The presence of CBFβ did not further augment cooperative DNA binding between CBFα2 and Ets-1ΔN280. In other words, no synergistic activation was observed in the presence of both CBFα2 partner proteins. In an important control, comparable levels of CBFβ (in the absence of CBFα2) did not affect the affinity of Ets-1ΔN280 for DNA (data not shown). We conclude that Ets-1ΔN280 and CBFβ cannot stimulate DNA binding by CBFα2(1-331) in a synergistic or even an additive fashion on this composite site.
FIG. 6.

DNA-binding enhancement by Ets-1 and CBFβ is neither additive nor synergistic. (A) EMSA of equilibrium DNA binding studies of CBFα2(1-331) titrated onto DNA saturated with Ets-1ΔN280 in the presence of CBFβ protein (left) and of Ets-1ΔN280 titrated onto DNA saturated with the CBFα2-CBFβ heterodimer (right). Control lanes to the left of each panel document the position of each of the protein-DNA complexes. (B) Equilibrium DNA binding curves for CBFα2(1-331) (left) and Ets-1ΔN280 (right). The identity of each curve is indicated in panel C. (C) Summary of equilibrium dissociation constants. Relative binding affinities (fold enhancement) compare KD values for multiprotein-DNA complexes to those obtained from DNA binding studies of Ets-1 and CBFα2 in isolation. KD values are presented as the mean (±standard error) of at least two independent experiments.
DISCUSSION
CBFα2-CBFβ partnership.
We quantitatively analyzed DNA binding by CBFα2 and modulation of this activity by intramolecular inhibitory sequences and by two protein partners, CBFβ and Ets-1. CBFα2 DNA binding is inhibited by at least two independent domains. The first domain is the DNA-binding Runt domain itself. The CBFβ subunit stimulates DNA binding by the Runt domain sixfold. We have proposed that the Runt domain assumes an inhibited conformation that is alleviated by association with the CBFβ subunit (Crute et al., submitted). Indeed, circular dichroism spectroscopy reveals that association of the Runt domain and CBFβ, either in solution or on the DNA, is accompanied by a conformational change in one or both proteins (Crute et al., submitted). Our working hypothesis is that CBFβ “locks in” a high-affinity DNA-binding conformation of the Runt domain (Fig. 7A and B). The structural basis for this phenomenon awaits determination of the Runt domain and CBFβ structures, which are under way (8, 21, 27, 51).
FIG. 7.

Models for interactions between CBFα2, CBFβ, and Ets-1. (A) The Runt domain (RD) is in equilibrium between a high- and low-affinity DNA-binding conformation. (B) Heterodimerization with CBFβ (β) locks the Runt domain into its high-affinity DNA-binding conformation, shifting the DNA-binding equilibrium to the right. (C) C-terminal inhibitory sequences in CBFα2 further shift the equilibrium of the Runt domain toward its low-affinity DNA-binding conformation and mask the CBFβ heterodimerization surface. Association of the C-terminal inhibitory sequences to the Runt domain is destabilized when CBFα2 is bound to DNA. Dissociation of the inhibitory sequences unmasks the CBFβ binding surface on the Runt domain. (D) The high-affinity DNA-binding conformation of the Runt domain is stabilized by the CBFβ subunit. Association of the C-terminal inhibitory sequences to the Runt domain is also directly inhibited by the CBFβ subunit, which masks the interaction site. The DNA-binding affinity of this complex is the same as that of the Runt domain-CBFβ complex in panel B. (E) Binding of CBFα2 to DNA exposes the Ets-1 interaction surface, which includes (but is not restricted to) sequences N terminal to the Runt domain. Tethering of Ets-1 to CBFα2 on the DNA increases the likelihood of a productive binding event, resulting in increased affinity. Ets-1 does not mask the Runt domain surface to which CBFβ and the C-terminal inhibitory domain bind. Conformational changes in the Ets-1 protein itself are not depicted in this diagram (see the accompanying report [20]).
Sequences C terminal to the Runt domain in CBFα2 contain a second intramolecular inhibitory domain that dampens DNA binding (Fig. 7C and 8). Our analysis mapped inhibitory sequences starting between amino acids 214 to 242 and ending somewhere between amino acids 331 and 451. Kanno and colleagues, using less quantitative approaches, also mapped C-terminal inhibitory sequences that affect DNA binding; however, their proposed boundaries lie between amino acids 183 and 291 (32). CBFβ overcomes the effect of the C-terminal inhibitory sequences, causing CBFα2 to bind DNA with the same affinity as truncated proteins lacking C-terminal inhibitory sequences (Fig. 7D). The C-terminal sequences also inhibit heterodimerization with CBFβ both on and, more significantly, off the DNA. A simple model to explain these phenomena is that the inhibitory sequences contact the surface of the Runt domain and both repress DNA binding and mask the heterodimerization surface for CBFβ (Fig. 7C). The association of CBFα2 with DNA may induce a conformational change that partially unmasks the heterodimerization surface for CBFβ on the Runt domain. This would account for the observation that heterodimerization is inhibited to a lesser extent in the presence of DNA. However, the altered conformation of the Runt domain would equilibrate rapidly with the inhibited conformation, causing rapid dissociation from DNA. Once CBFβ heterodimerizes with the Runt domain, the optimal DNA-binding conformation of the Runt domain is stabilized and inhibition by the C-terminal domain is rescinded (Fig. 7D). We speculate that CBFβ counteracts repression mediated by the C-terminal inhibitory sequences in CBFα2 by maintaining an altered conformation of the Runt domain and by occupying the site on the Runt domain to which the C-terminal inhibitory domain associates, preventing its reassociation.
FIG. 8.

Summary of CBFα2(451) functional domains. Shown are boundaries of the DNA-binding and heterodimerization domains as defined by Kagoshima et al. (30). Autoinhibition of both DNA binding and heterodimerization maps to the C-terminal half of the protein. RD, Runt domain.
CBFα2 and CBFβ heterodimerization may provide a key regulatory step for controlling activity in vivo. CBFβ is essential for the embryonic function of CBFα2 in hematopoiesis, as demonstrated by gene disruption experiments (52, 68, 83). Overexpression studies suggest that CBFβ lacks an intrinsic ability to translocate to the nucleus and does so only as an α-β heterodimer (1, 32, 40). Thus, the concentration of active CBFα2-CBFβ heterodimers in the nucleus will be determined, at least in part, by the cytoplasmic concentration of each subunit and by other mechanisms that may affect the affinity of CBFα2 for CBFβ in solution. For example, transcripts from the CBFA2 gene are alternatively spliced (3, 46, 47), yielding multiple CBFα2 isoforms that may have different affinities for CBFβ in solution. C-terminal sequences in the related CBFα1 protein are sensitive to proteolysis in vivo (40), which could also affect affinity for the CBFβ subunit. Chromosomal translocations that create CBFα2 and CBFβ fusion proteins could remove and/or introduce sequences that impact on heterodimerization with the partner protein. For example, the CBFα2 chimeric oncoproteins AML1/ETO and AML1/Evi-1, products of the t(8;21) and t(3;21), respectively, cause CBFβ to accumulate in the nucleus more efficiently than it does in the presence of the wild-type CBFα2 protein (79). Both AML1/ETO and AML1/Evi-1 chimeric proteins lack the intramolecular C-terminal inhibitory sequences in CBFα2. The affinity of CBFα and β subunits in solution could also determine which CBFα subunits are active in cells in which multiple CBFα genes are expressed. For example, recent evidence suggests that the CBFα1 protein has a lower affinity for CBFβ than CBFα2 (80). Concentrations of cytoplasmic CBFβ at or above the KD for CBFα2, but below the KD for CBFα1, will favor the formation of the active CBFα2-CBFβ heterodimer in cells in which both CBFA1 and CBFA2 genes are expressed.
Partnership with Ets-1.
Cooperative DNA binding between CBFα2(1-331) and Ets-1 provides another example whereby auto-inhibition is rescinded through protein-protein interactions. Ets-1 increases the affinity of CBFα2(1-331) for DNA approximately sevenfold. Enhancement of CBFα2(1-331) DNA binding by Ets-1 required preincubating CBFα2(1-331) with DNA prior to addition of Ets-1. This order-of-addition effect strongly suggests a conformational change in CBFα2 or that the DNA is necessary for cooperative DNA binding. The accompanying report (20) demonstrates that cooperative binding between Ets-1 and CBFα2(1-331) also occurs on nicked DNA templates, indicating that cooperativity is unlikely to be mediated by through-DNA effects. Taken together, the data suggest that a DNA-induced conformational change in the CBFα2(1-331) protein is required for cooperative DNA binding with Ets-1 to occur. We hypothesize that this conformational change must precede the entry of Ets-1 into the ternary complex to enable the most stable complex to form.
Ets-1 DNA binding is also regulated by an auto-inhibitory mechanism. In this case, a well-developed structural model of auto-inhibition is available (25, 29, 61, 73). Auto-inhibition requires three inhibitory helices plus a portion of the ETS domain that together form an inhibitory module. The mechanism of inhibition involves a major structural disruption of the inhibitory module that accompanies DNA binding. In the accompanying report (20), quantitative studies demonstrate that the sequences within the inhibitory module of Ets-1 are required for cooperative DNA binding with CBFα2. Furthermore, mutants that are constitutively disrupted and display high affinity do not display cooperativity (20, 33). These data strongly suggest that the role of CBFα2 is to counteract the auto-inhibition of Ets-1 DNA binding by affecting the conformation of the Ets-1 inhibitory module.
Several lines of evidence indicate that Ets-1 mediates its stimulatory effect through sequences on CBFα2 different from those utilized by CBFβ. For example, CBFβ appears to rescind auto-inhibition of CBFα2 mediated by both the Runt domain and the C-terminal inhibitory sequences. In contrast, removal of the C-terminal inhibitory sequences in CBFα2 has no effect on cooperative DNA binding with Ets-1, indicating that Ets-1 does not counteract the C-terminal inhibitory domain. In addition, CBFβ can stimulate DNA binding by Runt domain protein fragments that include amino acids 41 to 214, or even amino acids 59 to 190 (30), where as cooperative binding with Ets-1 requires amino acids 1 to 41 and 190 to 214 (Fig. 8). The sequences flanking the Runt domain that are required for cooperative DNA binding with Ets-1 could form part of the docking site for Ets-1. The order-of-addition experiment suggests that the Ets-1 interaction surface is exposed only when CBFα2 is bound to DNA. The mapping data suggest that the Ets-1 and CBFβ binding sites on CBFα2 are distinct and that the Ets-1 interaction surface on CBFα2 does not overlap with the interface for the C-terminal inhibitory sequences (Fig. 7E).
A recent independent study that also investigated CBFα2 and Ets-1 cooperative DNA binding expands the data presented here. Kim and colleagues reported that a portion of the C-terminal inhibitory sequences in CBFα2 (between amino acids 183 and 292) is required for cooperative DNA binding with Ets-1 (33). Our results document that only the C-terminal sequences between amino acids 190 and 214 are necessary for a sevenfold enhancement of CBFα2 DNA binding by Ets-1 (Fig. 8). Kanno et al. also found that CBFα2(50-292) bound DNA cooperatively with Ets-1 and concluded that sequences N terminal to the Runt domain were not necessary for cooperative DNA binding (33). We, on the other hand, observed cooperative DNA binding with a CBFα2(1-214) but not a CBFα2(41-214) fragment. Taken together, these data suggest that proteins lacking N-terminal sequences require sequences C terminal to amino acid 214 for cooperative binding with Ets-1. To reconcile the data presented herein with those of Kanno et al., we speculate that cooperative DNA binding by Ets-1 and CBFα2 utilizes at least three segments of CBFα2, amino acids 1 to 41, 190 to 214, and 214 to 292, but that any two regions are sufficient.
Stimulation of CBFα2 DNA binding by CBFβ and Ets-1 together is neither additive nor synergistic, although it is formally possible that these two proteins act cooperatively on other DNA sites. Cooperative DNA binding by Ets-1 and CBFα2 may be biologically significant only in cells in which the CBFβ subunit is present in limiting amounts. A possible example is the precursor cell for the R7 photoreceptor in the Drosophila eye. The effects of a lozenge mutation (lozenge encodes a CBFα protein) are suppressed by overexpression of the Drosophila CBFβ proteins Brother and Big Brother, indicating that the CBFβ proteins are limiting in this developmental context (37). In this situation, cooperative DNA binding by Lozenge and PntP2, an Ets-1 homolog, may contribute to the essential role played by both of these proteins in determining R7 identity (11, 13, 59).
The complexities of the CBFα2-CBFβ and CBFα2–Ets-1 partnerships provide unique insights into the basis of combinatorial control of transcriptional regulation. The rigorous quantification of the phenomena is a critical step in deciphering the molecular mechanisms. Additional mechanistic insights into how Ets-1 and CBFβ modulate DNA binding by CBFα2 will emerge as more structural information on all players becomes available.
ACKNOWLEDGMENTS
We thank Yen-Yee Tang for the CBFα2 Runt domain protein, Barbara Crute for making many of the C-terminal deletions, and John Bushweller for critically reading the manuscript. We are also grateful to Gus Lienhard for his many insightful comments.
N.A.S. is supported by Public Health Service grants RO1 CA58343 and CA75611. B.J.G. acknowledges support from the Public Health Service (grant RO1 GM38663), fellowship support for T.L.G. from NIH training grant CA090602, as well as support to the Huntsman Cancer Institute from grant CA42014.
REFERENCES
- 1.Adja N, Stacy T, Speck N A, Liu P P. The leukemic protein CBFβ-SMMHC sequesters CBFα2 into cytoskeletal filaments and aggregates. Mol Cell Biol. 1998;18:7432–7443. doi: 10.1128/mcb.18.12.7432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Albagli O, Klaes A, Ferreira E, Leprince D, Klambt C. Function of ets genes is conserved between vertebrates and Drosophila. Mech Dev. 1996;59:29–40. doi: 10.1016/0925-4773(96)00568-0. [DOI] [PubMed] [Google Scholar]
- 3.Bae S-C, Ogawa E, Maruyama M, Oka H, Satake M, Shigesada K, Jenkins N A, Gilbert D J, Copeland N G, Ito Y. PEBP2αB/mouse AML1 consists of multiple isoforms that possess differential transactivation potentials. Mol Cell Biol. 1994;14:3242–3252. doi: 10.1128/mcb.14.5.3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bae S-C, Takahashi E, Zhang Y W, Ogawa E, Shigesada K, Namba Y, Satake M, Ito Y. Cloning, mapping and expression of PEBP2αC, a third gene encoding the mammalian Runt domain. Gene. 1995;159:245–248. doi: 10.1016/0378-1119(95)00060-j. [DOI] [PubMed] [Google Scholar]
- 5.Bae S C, Yamaguchi-Iwai Y, Ogawa E, Maruyama M, Inuzuka M, Kagoshima H, Shigesada K, Satake M, Ito Y. Isolation of PEBP2αB cDNA representing the mouse homolog of human acute myeloid leukemia gene, AML1. Oncogene. 1993;8:809–814. [PubMed] [Google Scholar]
- 6.Barton K, Muthusamy N, Fischer C, Ting C N, Walunas T L, Lanier L L, Leiden J M. The Ets-1 transcription factor is required for the development of natural killer cells in mice. Immunity. 1998;9:555–563. doi: 10.1016/s1074-7613(00)80638-x. [DOI] [PubMed] [Google Scholar]
- 7.Ben-David U, Giddens E B, Letwin K, Bernstein A. Erythroleukemia induction by Friend murine leukemia virus: insertional activation of a new member of the ets gene family, Fli-1, closely linked to c-ets-1. Genes Dev. 1991;5:908–918. doi: 10.1101/gad.5.6.908. [DOI] [PubMed] [Google Scholar]
- 8.Berardi M, Sun C, Zehr M, Abildgaard F, Peng J, Speck N A, Bushweller J H. The Ig fold of the core binding factor α Runt domain is a member of a family of structurally and functionally related Ig fold DNA binding domains. Structure. 1999;7:1247–1256. doi: 10.1016/s0969-2126(00)80058-1. [DOI] [PubMed] [Google Scholar]
- 9.Bories J C, Willerford D M, Grevin D, Davidson L, Camus A, Martin P, Stehelin D, Alt F W. Increased T-cell apoptosis and terminal B-cell differentiation induced by inactivation of the Ets-1 proto-oncogene. Nature. 1995;377:635–638. doi: 10.1038/377635a0. [DOI] [PubMed] [Google Scholar]
- 10.Brass A L, Kehrli E, Eisenbeis C F, Storb U, Singh H. Pip, a lymphoid-restricted IRF, contains a regulatory domain that is important for autoinhibition and ternary complex formation with the ets factor PU.1. Genes Dev. 1996;10:2335–2347. doi: 10.1101/gad.10.18.2335. [DOI] [PubMed] [Google Scholar]
- 11.Brunner D, Ducker K, Oellers N, Hafen E, Scholz H, Klambt C. The ETS domain protein pointed-P2 is a target of MAP kinase in the sevenless signal transduction pathway. Nature. 1994;370:386–389. doi: 10.1038/370386a0. [DOI] [PubMed] [Google Scholar]
- 12.Crute B E, Lewis A F, Wu Z, Bushweller J H, Speck N A. Biochemical and biophysical properties of the CBFα2 (AML1) DNA-binding domain. J Biol Chem. 1996;271:26251–26260. doi: 10.1074/jbc.271.42.26251. [DOI] [PubMed] [Google Scholar]
- 13.Daga A, Karlovich C A, Dumstrei K, Banerjee U. Patterning of cells in the Drosophila eye by Lozenge, which shares homologous domains with AML1. Genes Dev. 1996;10:1194–1205. doi: 10.1101/gad.10.10.1194. [DOI] [PubMed] [Google Scholar]
- 14.Dalton S, Treisman R. Characterization of SAP-1, a protein recruited by serum response factor to the c-fos serum response element. Cell. 1992;68:597–612. doi: 10.1016/0092-8674(92)90194-h. [DOI] [PubMed] [Google Scholar]
- 15.Dignam J D, Lebowitz R M, Roeder R G. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duffy J B, Gergen J P. The Drosophila segmentation gene runt acts as a position-specific numerator element necessary for the uniform expression of the sex-determining gene Sex-lethal. Genes Dev. 1991;5:2176–2187. doi: 10.1101/gad.5.12a.2176. [DOI] [PubMed] [Google Scholar]
- 17.Duffy J B, Kania M A, Gergen J P. Expression and function of the Drosophila gene runt in early stages of neural development. Development. 1991;113:1223–1230. doi: 10.1242/dev.113.4.1223. [DOI] [PubMed] [Google Scholar]
- 18.Erman B, Cortes M, Speck N A, Sen R. ETS-core binding factor: a common composite motif in antigen receptor gene enhancers. Mol Cell Biol. 1998;18:1322–1330. doi: 10.1128/mcb.18.3.1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Giese K, Kingsley C, Kirshner J R, Grosschedl R. Assembly and function of a TCRα enhancer complex is dependent on LEF-1-induced DNA binding and multiple protein-protein interactions. Genes Dev. 1995;9:995–1008. doi: 10.1101/gad.9.8.995. [DOI] [PubMed] [Google Scholar]
- 20.Goetz T L, Gu T L, Speck N A, Graves B J. Auto-inhibition of Ets-1 is counteracted by DNA binding cooperativity with core-binding factor α2. Mol Cell Biol. 1999;20:81–90. doi: 10.1128/mcb.20.1.81-90.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goger M, Gupta V, Kim W-Y, Shigesada K, Ito Y, Werner M H. Molecular insights into PEBP2/CBFβ-SMMHC associated acute leukemia revealed from the three-dimensional structure of PEBP2/CBFβ. Nat Struct Biol. 1999;6:620–623. doi: 10.1038/10664. [DOI] [PubMed] [Google Scholar]
- 22.Golling G, Li L-H, Pepling M, Stebbins M, Gergen J P. Drosophila homologues of proto-oncogene product PEBP2/CBFβ regulate the DNA-binding properties of Runt. Mol Cell Biol. 1996;16:932–942. doi: 10.1128/mcb.16.3.932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Golub T R, Barker G F, Bohlander S K, Hiebert S, Ward D C, Bray-Ward P, Morgan E, Raimondi S C, Rowley J D, Gilliland D G. Fusion of the TEL gene on 12p13 to the AML1 gene on 21q22 in acute lymphoblastic leukemia. Proc Natl Acad Sci USA. 1995;92:4917–4921. doi: 10.1073/pnas.92.11.4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Graves B J, Cowley D O, Goetz T L, Petersen J M, Jonsen M D, Gillespie M E. Autoinhibition as a transcriptional regulatory mechanism. Cold Spring Harbor Symp Quant Biol. 1998;63:621–629. doi: 10.1101/sqb.1998.63.621. [DOI] [PubMed] [Google Scholar]
- 25.Graves B J, Peterson J M. Specificity within the ets family of transcription factors. Adv Cancer Res. 1998;75:1–55. doi: 10.1016/s0065-230x(08)60738-1. [DOI] [PubMed] [Google Scholar]
- 26.Huang X, Crute B E, Sun C, Tang Y-Y, Kelley III J J, Lewis A F, Hartman K L, Laue T M, Speck N A, Bushweller J H. Overexpression, purification, and biophysical characterization of the heterodimerization domain of the core-binding factor β subunit. J Biol Chem. 1998;273:2480–2487. doi: 10.1074/jbc.273.4.2480. [DOI] [PubMed] [Google Scholar]
- 27.Huang X, Peng J W, Speck N A, Bushweller J H. Core binding factor β revealed: solution structure and map of the CBFα binding site. Nat Struct Biol. 1999;6:624–627. doi: 10.1038/10670. [DOI] [PubMed] [Google Scholar]
- 28.Ingham P, Gergen P. Interactions between the pair-rule genes runt, hairy, even-skipped and fushi tarazu and the establishment of periodic pattern in the Drosophila embryo. Development. 1988;104(Suppl.):51–60. [Google Scholar]
- 29.Jonsen M D, Petersen J M, Xu Q-P, Graves B J. Characterization of cooperative function of inhibitory sequences in Ets-1. Mol Cell Biol. 1996;16:2065–2073. doi: 10.1128/mcb.16.5.2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kagoshima H, Akamatsu Y, Ito Y, Shigesada K. Functional dissection of the α and β subunits of the transcription factor PEBP2 and the redox susceptibility of its DNA binding activity. J Biol Chem. 1996;271:33074–33082. doi: 10.1074/jbc.271.51.33074. [DOI] [PubMed] [Google Scholar]
- 31.Kagoshima H, Shigesada K, Satake M, Ito Y, Miyoshi H, Ohki M, Pepling M, Gergen J P. The Runt-domain identifies a new family of heteromeric DNA-binding transcriptional regulatory proteins. Trends Genet. 1993;9:338–341. doi: 10.1016/0168-9525(93)90026-e. [DOI] [PubMed] [Google Scholar]
- 32.Kanno T, Kanno Y, Chen L-F, Ogawa E, Kim W-Y, Ito Y. Intrinsic transcriptional activation-inhibition domains of the polyomavirus enhancer binding protein 2/core binding factor α subunit revealed in the presence of the β subunit. Mol Cell Biol. 1998;18:2444–2454. doi: 10.1128/mcb.18.5.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim W-Y, Sieweke M, Ogawa E, Wee H-J, Englmeier U, Graf T, Ito Y. Mutual activation of Ets-1 and AML1 DNA binding by direct interaction of their autoinhibitory domains. EMBO J. 1999;18:1609–1620. doi: 10.1093/emboj/18.6.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, Shimizu Y, Bronson R T, Gao Y-H, Inada M, Sato M, Okamoto R, Kitamura Y, Yoshiki S, Kishimoto T. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89:755–764. doi: 10.1016/s0092-8674(00)80258-5. [DOI] [PubMed] [Google Scholar]
- 35.Leprince D, Gegonne A, Coll J, de Taisne C, Schneeberger A, Lagrou C, Stehelin D. A putative second cell-derived oncogene of the avian leukaemia retrovirus E26. Nature. 1983;306:395–397. doi: 10.1038/306395a0. [DOI] [PubMed] [Google Scholar]
- 36.Levanon D, Negreanu V, Bernstein Y, Bar-Am I, Avivi L, Groner Y. AML1, AML2, and AML3, the human members of the runt domain gene-family: cDNA structure, expression, and chromosomal localization. Genomics. 1994;23:425–432. doi: 10.1006/geno.1994.1519. [DOI] [PubMed] [Google Scholar]
- 37.Li L H, Gergen J P. Differential interactions between Brother proteins and Runt domain proteins in the Drosophila embryo and eye. Development. 1999;126:3313–3322. doi: 10.1242/dev.126.15.3313. [DOI] [PubMed] [Google Scholar]
- 38.Ling Y, Lakey J H, Roberts C E, Sharrocks A D. Molecular characterization of the B-box protein-protein interaction motif of the ETS-domain transcription factor Elk-1. EMBO J. 1997;16:2431–2440. doi: 10.1093/emboj/16.9.2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu P, Tarle S A, Hajra A, Claxton D F, Marlton P, Freedman M, Siciliano M J, Collins F S. Fusion between transcription factor CBFβ/PEBP2β and a myosin heavy chain in acute myeloid leukemia. Science. 1993;261:1041–1044. doi: 10.1126/science.8351518. [DOI] [PubMed] [Google Scholar]
- 40.Lu J, Maruyama M, Satake M, Bae S-C, Ogawa E, Kagoshima H, Shigesada K, Ito Y. Subcellular localization of the α and β subunits of acute myeloid leukemia-linked transcription factor PEBP2/CBF. Mol Cell Biol. 1995;15:1651–1661. doi: 10.1128/mcb.15.3.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mayall T P, Sheridan P L, Montminy M R, Jones K A. Distinct roles for P-CREB and LEF-1 in TCR enhancer assembly and activation on chromatin templates in vitro. Genes Dev. 1997;11:887–899. doi: 10.1101/gad.11.7.887. [DOI] [PubMed] [Google Scholar]
- 42.McKercher S R, Torbett B E, Anderson K L, Henkel G W, Vestal D J, Baribault H, Klemsz M, Feeney A J, Wu G E, Paige C J, Maki R A. Targeted disruption of the PU.1 gene results in multiple hematopoietic abnormalities. EMBO J. 1996;15:5647–5658. [PMC free article] [PubMed] [Google Scholar]
- 43.McLean T W, Ringold S, Neuberg D, Stegmaier K, Tantravahi R, Ritz J, Koeffler H P, Takeuchi S, Janssen J W, Seriu T, Bartram C R, Sallan S E, Gilliland D G, Golub T R. TEL/AML-1 dimerizes and is associated with a favorable outcome in childhood acute lymphoblastic leukemia. Blood. 1996;88:4252–4258. [PubMed] [Google Scholar]
- 44.Melet F, Motro B, Rossi D J, Zhang L, Bernstein A. Generation of a novel Fli-1 protein by gene targeting leads to a defect in thymus development and a delay in Friend virus-induced erythroleukemia. Mol Cell Biol. 1996;16:2708–2718. doi: 10.1128/mcb.16.6.2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meyers S, Downing J R, Hiebert S W. Identification of AML-1 and the (8;21) translocation protein (AML-1/ETO) as sequence-specific DNA-binding proteins: the runt homology domain is required for DNA binding and protein-protein interactions. Mol Cell Biol. 1993;13:6336–6345. doi: 10.1128/mcb.13.10.6336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Meyers S, Lenny N, Hiebert S W. The t(8;21) fusion protein interferes with AML1-1B-dependent transcriptional activation. Mol Cell Biol. 1995;15:1974–1982. doi: 10.1128/mcb.15.4.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miyoshi H, Ohira M, Shimizu K, Mitani K, Hirai H, Imai T, Yokoyama K, Soeda E, Ohki M. Alternative splicing and genomic structure of the AML1 gene involved in acute myeloid leukemia. Nucleic Acids Res. 1995;23:2762–2769. doi: 10.1093/nar/23.14.2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miyoshi H, Shimizu K, Kozu T, Maseki N, Kaneko Y, Ohki M. t(8;21) breakpoints on chromosome 21 in acute myeloid leukemia are clustered within a limited region of a single gene, AML1. Proc Natl Acad Sci USA. 1991;88:10431–10434. doi: 10.1073/pnas.88.23.10431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moreau-Gachelin F, Tavitian A, Tambourin P. Spi-1 is a putative oncogene in virally induced murine erythroleukaemias. Nature. 1988;331:277–280. doi: 10.1038/331277a0. [DOI] [PubMed] [Google Scholar]
- 50.Muthusamy N, Barton K, Leiden J M. Defective activation and survival of T cells lacking the Ets-1 transcription factor. Nature. 1995;377:639–642. doi: 10.1038/377639a0. [DOI] [PubMed] [Google Scholar]
- 51.Nagata T, Gupta V, Sorce D, Kim W-Y, Sali A, Chait B T, Shigesada K, Ito Y, Werner M H. Immunoglobulin motif DNA-binding and heterodimerization for the PEBP2/CBF Runt-domain. Nat Struct Biol. 1999;6:615–619. doi: 10.1038/10658. [DOI] [PubMed] [Google Scholar]
- 52.Niki M, Okada H, Takano H, Kuno J, Tani K, Hibino H, Asano S, Ito Y, Satake M, Noda T. Hematopoiesis in the fetal liver is impaired by targeted mutagenesis of a gene encoding a non-DNA binding subunit of the transcription factor, polyomavirus enhancer binding protein 2/core binding factor. Proc Natl Acad Sci USA. 1997;94:5697–5702. doi: 10.1073/pnas.94.11.5697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.North T E, Gu T-L, Stacy T, Wang Q, Howard L, Binder M, Marín-Padilla M, Speck N A. Cbfa2 is required for the formation of intra-aortic hematopoietic clusters. Development. 1999;126:2563–2575. doi: 10.1242/dev.126.11.2563. [DOI] [PubMed] [Google Scholar]
- 54.Nunn M F, Seeberg P H, Moscovici C, Duesberg P H. Tripartite structure of the avian erythroblastosis virus E26 transforming gene. Nature. 1983;306:391–395. doi: 10.1038/306391a0. [DOI] [PubMed] [Google Scholar]
- 55.Nye J A, Petersen J M, Gunther C V, Jonsen M D, Graves B J. Interaction of murine Ets-1 with GGA-binding sites establishes the ETS domain as a new DNA-binding motif. Genes Dev. 1992;6:975–990. doi: 10.1101/gad.6.6.975. [DOI] [PubMed] [Google Scholar]
- 56.Ogawa E, Inuzuka M, Maruyama M, Satake M, Naito-Fujimoto M, Ito Y, Shigesada K. Molecular cloning and characterization of PEBP2β, the heterodimeric partner of a novel Drosophila runt-related DNA binding protein PEBP2α. Virology. 1993;194:314–331. doi: 10.1006/viro.1993.1262. [DOI] [PubMed] [Google Scholar]
- 57.Ogawa E, Maruyama M, Kagoshima H, Inuzuka M, Lu J, Satake M, Shigesada K, Ito Y. PEBP2/PEA2 represents a new family of transcription factor homologous to the products of the Drosophila runt and the human AML1 gene. Proc Natl Acad Sci USA. 1993;90:6859–6863. doi: 10.1073/pnas.90.14.6859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Okuda T, van Deursen J, Hiebert S W, Grosveld G, Downing J R. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell. 1996;84:321–330. doi: 10.1016/s0092-8674(00)80986-1. [DOI] [PubMed] [Google Scholar]
- 59.O'Neill E M, Rebay I, Tjian R, Rubin G M. The activities of two Ets-related transcription factors required for Drosophila eye development are modulated by the Ras/MAPK pathway. Cell. 1994;78:137–147. doi: 10.1016/0092-8674(94)90580-0. [DOI] [PubMed] [Google Scholar]
- 60.Otto F, Thornell A P, Crompton T, Denzel A, Gilmour K C, Rosewell I R, Stamp G W H, Beddington R S P, Mundlos S, Olsen B R, Selby P B, Owen M J. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997;89:765–772. doi: 10.1016/s0092-8674(00)80259-7. [DOI] [PubMed] [Google Scholar]
- 61.Petersen J M, Skalicky J J, Donaldson L W, McIntosh L P, Alber T, Graves B J. Modulation of transcription factor Ets-1 DNA binding: DNA-induced unfolding of an alpha helix. Science. 1995;269:1866–1869. doi: 10.1126/science.7569926. [DOI] [PubMed] [Google Scholar]
- 62.Petrovick M S, Hiebert S W, Friedman A D, Hetherington C J, Tenen T G, Zhang D-E. Multiple functional domains of AML1: PU.1 and C/EBPα synergize with different regions of AML1. Mol Cell Biol. 1998;18:3915–3925. doi: 10.1128/mcb.18.7.3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rabbitts T H. Chromosomal translocations in human cancer. Nature. 1994;372:143–149. doi: 10.1038/372143a0. [DOI] [PubMed] [Google Scholar]
- 64.Rizki T M, Rizki R M, Bellotti R. Genetics of a Drosophila phenoloxidase. Mol Gen Genet. 1985;201:7–13. doi: 10.1007/BF00397978. [DOI] [PubMed] [Google Scholar]
- 65.Romana S P, Mauchauffe M, Le Coniat M, Chumakow I, Le Paslier D, Berger R, Bernard O A. The t(12;21) of acute lymphoblastic leukemia results in a tel-AML1 gene fusion. Blood. 1995;85:3662–3670. [PubMed] [Google Scholar]
- 66.Romana S P, Poirel H, Leconiat M, Flexor M-A, Mauchauffé M, Jonveaux P, Macintyre E A, Berger R, Bernard O A. High frequency of t(12;21) in childhood B-lineage acute lymphoblastic leukemia. Blood. 1995;86:4263–4269. [PubMed] [Google Scholar]
- 67.Sanchez L, Nothiger R. Sex determination and dosage compensation in Drosophila melanogaster: production of male clones in XX females. EMBO J. 1983;2:485–491. doi: 10.1002/j.1460-2075.1983.tb01451.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sasaki K, Yagi H, Bronson R T, Tominaga K, Matsunashi T, Deguchi K, Tani Y, Kishimoto T, Komori T. Absence of fetal liver hematopoiesis in transcriptional co-activator, core binding factor β (Cbfb) deficient mice. Proc Natl Acad Sci USA. 1996;93:12359–12363. doi: 10.1073/pnas.93.22.12359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sato M, Morii E, Komori T, Kawahata H, Sugimoto M, Terai K, Shimizu H, Yasui Y, Ogihara H, Yasui N, Ochi T, Kitamura Y, Ito Y, Nomura S. Transcriptional regulation of osteopontin gene in vivo by PEBP2αA/CBFA1 and ETS1 in the skeletal tissues. Oncogene. 1998;17:1517–1525. doi: 10.1038/sj.onc.1202064. [DOI] [PubMed] [Google Scholar]
- 70.Scott E W, Simon M C, Anastasi J, Singh H. Requirement of transcription factor PU.1 in the development of multiple hematopoietic lineages. Science. 1994;265:1573–1577. doi: 10.1126/science.8079170. [DOI] [PubMed] [Google Scholar]
- 71.Sharrocks A D, Brown A L, Ling Y, Yates P R. The Ets-domain transcription factor family. Internat J Biochem. 1998;29:1371–1387. doi: 10.1016/s1357-2725(97)00086-1. [DOI] [PubMed] [Google Scholar]
- 72.Shurtleff S A, Buijs A, Behm F G, Rubnitz J E, Raimondi S C, Hancock M L, Chan G C-F, Pui C-H, Grosveld G, Downing J R. TEL/AML1 fusion resulting from a cryptic t(12;21) is the most common genetic lesion in pediatric ALL and defines a subgroup of patients with an excellent prognosis. Leukemia. 1995;9:1985–1989. [PubMed] [Google Scholar]
- 73.Skalicky J J, Donaldson L W, Petersen J M, Graves B J, McIntosh L P. Structural coupling of the inhibitory regions flanking the ETS domain of murine Ets-1. Protein Sci. 1996;5:296–309. doi: 10.1002/pro.5560050214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Speck N A, Baltimore D. Six distinct nuclear factors interact with the 75-base-pair repeat of the Moloney murine leukemia virus enhancer. Mol Cell Biol. 1987;7:1101–1110. doi: 10.1128/mcb.7.3.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Speck N A, Stacy T. A new transcription factor family associated with human leukemias. Crit Rev Eukaryotic Gene Expr. 1995;5:337–364. doi: 10.1615/critreveukargeneexpr.v5.i3-4.60. [DOI] [PubMed] [Google Scholar]
- 76.Stewart M, Terry A, Hu M, O'Hara M, Blyth K, Baxter E, Cameron E, Onions D E, Neil J C. Proviral insertions induce the expression of bone-specific isoforms of PEBP2αA (CBFA1): evidence for a new myc collaborating oncogene. Proc Natl Acad Sci USA. 1997;94:8646–8651. doi: 10.1073/pnas.94.16.8646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stocker F R, Gendre N. Peripheral and central nervous system effects of lz3, a Drosophila mutant lacking basiconic antennal sensilla. Dev Biol. 1988;127:12–27. doi: 10.1016/0012-1606(88)90184-4. [DOI] [PubMed] [Google Scholar]
- 78.Sun W, Graves B J, Speck N A. Transactivation of the Moloney murine leukemia virus and T-cell receptor β-chain enhancers by cbf and ets requires intact binding sites for both proteins. J Virol. 1995;69:4941–4949. doi: 10.1128/jvi.69.8.4941-4949.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tanaka K, Tanaka T, Kurokawa M, Imai Y, Ogawa S, Mitani K, Yazaki Y, Hirai H. The AML/ETO(MTG8) and AML1/Evi-1 leukemia-associated chimeric oncoproteins accumulate PEBP2β(CBFβ) in the nucleus more efficiently than wild-type AML1. Blood. 1998;91:1688–1699. [PubMed] [Google Scholar]
- 80.Thirunavukkarasu K, Mahajan M, McLarren K W, Stifani S, Karsenty G. Two domains unique to osteoblast-specific transcription factor Osf2/Cbfa1 contribute to its transactivation function and its inability to heterodimerize with Cbfβ. Mol Cell Biol. 1998;18:4197–4208. doi: 10.1128/mcb.18.7.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Thornell A, Hallberg B, Grundstrom T. Binding of SL3-3 enhancer factor 1 transcriptional activators to viral and chromosomal enhancer sequences. J Virol. 1991;65:42–50. doi: 10.1128/jvi.65.1.42-50.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang Q, Stacy T, Binder M, Marín-Padilla M, Sharpe A H, Speck N A. Disruption of the Cbfa2 gene causes necrosis and hemorrhaging in the central nervous system and blocks definitive hematopoiesis. Proc Natl Acad Sci USA. 1996;93:3444–3449. doi: 10.1073/pnas.93.8.3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang Q, Stacy T, Miller J D, Lewis A F, Huang X, Bories J-C, Bushweller J H, Alt F W, Binder M, Marín-Padilla M, Sharpe A, Speck N A. The CBFβ subunit is essential for CBFα2 (AML1) function in vivo. Cell. 1996;87:697–708. doi: 10.1016/s0092-8674(00)81389-6. [DOI] [PubMed] [Google Scholar]
- 84.Wang S, Speck N A. Purification of core-binding factor, a protein that binds the conserved core site in murine leukemia virus enhancers. Mol Cell Biol. 1992;12:89–102. doi: 10.1128/mcb.12.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang S, Wang Q, Crute B E, Melnikova I N, Keller S R, Speck N A. Cloning and characterization of subunits of the T-cell receptor and murine leukemia virus enhancer core-binding factor. Mol Cell Biol. 1993;13:3324–3339. doi: 10.1128/mcb.13.6.3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wotton D, Ghysdael J, Wang S, Speck N A, Owen M J. Cooperative binding of Ets-1 and core binding factor to DNA. Mol Cell Biol. 1994;14:840–850. doi: 10.1128/mcb.14.1.840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zeng C, Van Wignen A J, Stein J L, Meyers S, Sun W, Shopland L, Lawrence J B, Penman S, Lian J B, Stein G S, Hiebert S W. Identification of a nuclear matrix targeting signal in the leukemia and bone-related AML/CBF-α transcription factors. Proc Natl Acad Sci USA. 1997;94:6746–6751. doi: 10.1073/pnas.94.13.6746. [DOI] [PMC free article] [PubMed] [Google Scholar]