

ABSTRACT
Acinetobacter baumannii is emerging as a multidrug-resistant (MDR) nosocomial pathogen of increasing threat to human health worldwide. The recent MDR urinary isolate UPAB1 carries the plasmid pAB5, a member of a family of large conjugative plasmids (LCPs). LCPs encode several antibiotic resistance genes and repress the type VI secretion system (T6SS) to enable their dissemination, employing two TetR transcriptional regulators. Furthermore, pAB5 controls the expression of additional chromosomally encoded genes, impacting UPAB1 virulence. Here, we show that a pAB5-encoded H-NS transcriptional regulator represses the synthesis of the exopolysaccharide PNAG and the expression of a previously uncharacterized three-gene cluster that encodes a protein belonging to the CsgG/HfaB family. Members of this protein family are involved in amyloid or polysaccharide formation in other species. Deletion of the CsgG homolog abrogated PNAG production and chaperone-usher pathway (CUP) pilus formation, resulting in a subsequent reduction in biofilm formation. Although this gene cluster is widely distributed in Gram-negative bacteria, it remains largely uninvestigated. Our results illustrate the complex cross-talks that take place between plasmids and the chromosomes of their bacterial host, which in this case can contribute to the pathogenesis of Acinetobacter.
IMPORTANCE The opportunistic human pathogen Acinetobacter baumannii displays the highest reported rates of multidrug resistance among Gram-negative pathogens. Many A. baumannii strains carry large conjugative plasmids like pAB5. In recent years, we have witnessed an increase in knowledge about the regulatory cross-talks between plasmids and bacterial chromosomes. Here, we show that pAB5 controls the composition of the bacterial extracellular matrix, resulting in a drastic reduction in biofilm formation. The association between biofilm formation, virulence, and antibiotic resistance is well documented. Therefore, understanding the factors involved in the regulation of biofilm formation in Acinetobacter has remarkable therapeutic potential.
KEYWORDS: Acinetobacter, biofilm, curli, PNAG, plasmid
INTRODUCTION
Acinetobacter baumannii is regarded as a nosocomial pathogen capable of causing multiple types of infection; however, in the last few years community-acquired infections have become more common. Importantly, A. baumannii is a major threat to global health due to the increasing prevalence of multidrug-resistant (MDR) isolates (1, 2). Genes encoding antibiotic resistance are usually located in chromosomal resistance islands or in plasmids (3, 4). Plasmids are major contributors to horizontal gene transfer, since they facilitate the exchange of genetic material between microorganisms, spreading the MDR phenotype (5–7). For decades, plasmid biology focused on their replication, maintenance, and mobilization, as well as on their contribution to antibiotic resistance and virulence (8). More recently, genomic and transcriptomic analysis has begun to uncover complex and dynamic relationships between plasmids and their host chromosome.
A. baumannii strains harbor different types of plasmids. Regarding MDR, the large conjugative plasmid (LCP) family, which are approximately 150 to 200 kb, are particularly worrisome. pAB3, the LCP carried in the laboratory strain ATCC 17978, isolated in 1951 carries only one cassette conferring resistance to trimethoprim. However, pAB04 or pAB5, LCPs from recent clinical isolates AB04 and UPAB1, contain 12 and 15 antibiotic resistance cassettes, respectively. This increase in the number of antibiotic resistance cassettes illustrates the rapid evolution of these plasmids. LCPs contain three conserved regions: the MDR region, containing various antibiotic resistance cassettes; a region encoding the type IV secretion system conjugative pilus, required for plasmid dissemination via conjugation; and the regulatory region, which contains several transcriptional regulators (9–11). For example, LCPs harbor two TetR regulators that repress the type VI secretion system (T6SS) encoded in A. baumannii chromosome, which allows conjugation and promotes their own dissemination (10).
We have previously shown that besides providing resistance to antibiotics and repressing T6SS, pAB5 can control the expression of additional chromosomally encoded genes, impacting UPAB1 virulence (11). The regulatory activity of pAB5 can be observed by plating cells with or without this plasmid in Congo red-containing plates. The bacterial colonies carrying pAB5 are much lighter, which reflects a clear reduction in Congo red binding. Transcriptomic and proteomic analysis revealed that pAB5 reduced the expression of cell-surface components, including chaperone/usher pathway (CUP) pili, β-1→6-linked poly-N-acetylglucosamine (PNAG), and many additional proteins of unknown functions (11). A bioinformatic analysis of the pAB5 sequence revealed that this LCP encodes at least six genes predicted to function as transcriptional regulators. Here, we explored the cross talk between pAB5 and the chromosome of UPAB1 and identified the transcriptional regulator involved in the regulation of PNAG synthesis. Furthermore, our analysis revealed a novel uncharacterized gene cluster regulated by pAB5, evolutionarily related to curli formation, and whose disruption has a dramatic effect on the surface composition of UPAB1.
RESULTS
The transcriptional regulator H-NS from pAB5 controls UPAB1 phenotype on Congo red binding.
We have recently shown that pAB5 regulates the expression of multiple chromosomally encoded virulence factors in UPAB1 (11). One of the most evident phenotypes controlled by pAB5 is Congo red binding, which has been associated with the presence of either amyloid fibers, such as curli, or polysaccharides in Escherichia coli and A. baumannii species (12, 13). pAB5 encodes at least six putative transcriptional regulators (see Table S1 in the supplemental material). Among these, there are two TetR regulators, TetR1 and TetR2, virtually identical to the ones encoded in plasmid pAB3. These two TetR regulators downregulate the expression of the T6SS machinery, which is employed by multiple Acinetobacter strains to compete with other bacteria (9, 10). pAB5 carries one additional regulator of unknown function, belonging to the TetR family, TetR3. In addition, pAB5 encodes the global regulator H-NS (histone-like nucleoid structuring). H-NS-like proteins have been shown to be implicated in the facilitation of chromosome evolution through their ability to silence transcription, allowing integration of horizontally transferred genes into bacterial chromosomes (14). The H-NS regulator encoded in pAB5 is 100% identical to H-NS regulators encoded in other LCPs, but it is only 50% identical to the UPAB1 chromosomal copy (see Fig. S1). Finally, orthologues of other regulators, such a FrmR, a putative metal/formaldehyde-sensitive transcriptional repressor, and ArsR, a putative repressor belonging to the arsenic-sensitive family transcriptional regulators are also encoded in pAB5. To investigate whether any of these regulators’ controls Congo red binding, we cloned them individually in the pVRL2 expression vector (15). The constructs were transformed into UPAB1p-, a strain cured of pAB5 plasmid (11), and the different strains were plated on Congo red plates. As previously reported, UPAB1 displayed a reduced Congo red binding (white colonies) compared to UPAB1p- (red colonies), which correlated with the quantification of Congo red binding (Fig. 1A and B). From the six regulators expressed in UPAB1p-, only H-NS changed the color of the colonies and diminished Congo red binding (Fig. 1A and B). All other strains expressing the remaining five regulators behaved as UPAB1p-, exhibiting similar levels of Congo red binding. Finally, UPAB1Δhns, a strain carrying pAB5 without hns, showed levels of Congo red binding to comparable to those of strain UPAB1p- (Fig. 1C and D). These results demonstrate that H-NS is solely responsible for the pAB5-dependent repression of Congo red binding.
FIG 1.

H-NS inhibits Congo red binding. (A and C) UPAB1 and derivatives strain spotted in YESCA Congo red agar plates. (B and D) Quantification of Congo red binding (excitation wavelength at 485 nm and the emission at 612 nm). The values represent the means and standard deviations from five (B) and four (D) independent experiments. A t test was performed by comparison to the wild type (**, P ≤ 0.003; *, P ≤ 0.03; ns, not significant).
Plasmid-encoded H-NS inhibits PNAG production.
It has been proposed that in some A. baumannii strains, Congo red binding is linked to PNAG production (12). To examine whether pAB5 inhibits PNAG production, we used a specific antibody to check for the presence or absence of PNAG. In correlation to Congo red phenotypes, UPAB1 and UPAB1p expressing hns (p-/hns) showed drastically reduced levels of PNAG production, while UPAB1p-, UPAB1p- harboring the empty vector (p-/vec), and UPAB1Δhns strains show similar level of PNAG production (Fig. 2A). The differences observed on the levels of PNAG production were not related to differences on cells loaded to the membrane (Fig. 2B). None of the other putative transcriptional regulators from pAB5, altered PNAG production (see Fig. S2). The locus containing the pgaABCD genes has been shown to be responsible for PNAG production in the clinical isolate A. baumannii S1 (12). UPAB1 harbors a similar gene cluster containing four genes pgaABCD (Fig. 3A). The pgaA-D cluster (including genes pgaA through pgaD) encodes the poly-beta-1,6-N-acetyl-d-glucosamine export porin PgaA; the poly-beta-1,6-N-acetyl-d-glucosamine N-deacetylase PgaB, the poly-beta-1,6-N-acetyl-d-glucosamine synthase PgaC, and the poly-beta-1,6-N-acetyl-d-glucosamine biosynthesis protein PgaD (16). These proteins have an identity ranging from 23% to 55% to the PNAG cluster in E. coli and Yersinia pestis. Our recent transcriptomic data showed that this locus is repressed by pAB5 (11). By RT-PCR, we show that expression of pgaABC was repressed by H-NS (Fig. 3B). Furthermore, the pgaA mutant or the whole pgaA-D cluster mutant abolished Congo red binding (Fig. 4A; see also Fig. S3 in the supplemental material) and PNAG production (Fig. 4B and C). These phenotypes were recovered in the complemented strains, demonstrating that pgaA-D is responsible for PNAG production in UPAB1. The loss of hns on the pAB5 plasmid does not have the same effect on pga mRNA levels as loss of the entire plasmid. This suggest that an additional regulator may be present on pAB5. However, this effect on transcription does not seem enough to produce a phenotypic change. Together, these results show that H-NS downregulates the pgaA-D cluster with the concomitant repression in PNAG production.
FIG 2.
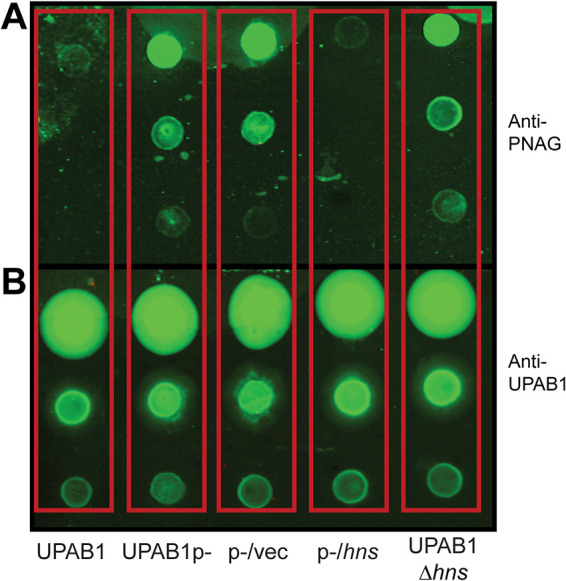
H-NS reduces PNAG production. (A and B) Immunoblotting with the antibody anti-PNAG (A) and the antibody anti-UPAB1 as a loading control (B). Cells were taken from an overnight LB-agar plate at 26°C and adjusted to an OD of 1. After treatment, 5 μl of a 10-fold serial dilution was spotted on nitrocellulose membrane.
FIG 3.
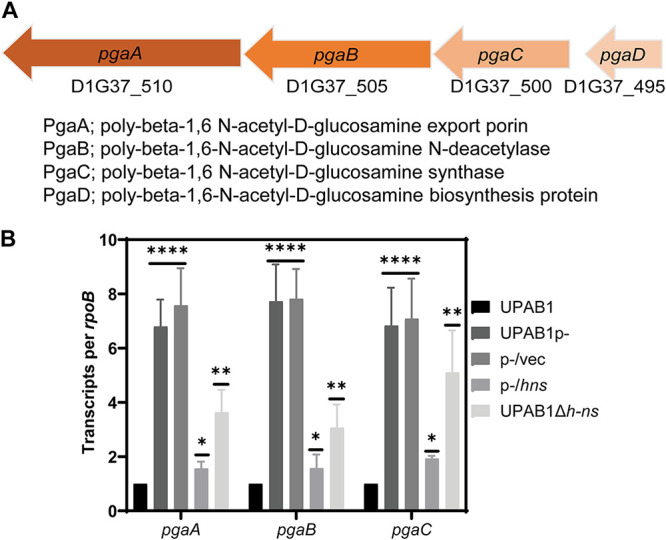
H-NS reduces the expression of pnag cluster. (A) Illustration of the pgaA-D gene cluster. (B) pgaABC expression. Transcripts were measured from cells growing on LB plates at 26°C and adjusted to an OD of 1. The results are shown as rpoB-adjusted transcript values. The values represent the means and standard deviations from four independent experiments. t test results are indicated (****, P ≤ 0.0001; **, P ≤ 0.005; *, P ≤ 0.05).
FIG 4.
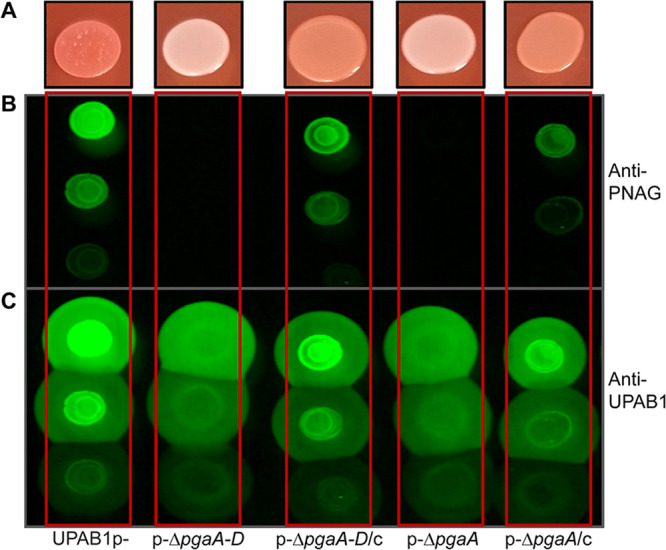
The pgaA-D cluster is involved in PNAG production. (A) Phenotype on YESCA-Congo red agar plates of UPAB1p-, derivative mutant, and complemented strains. PNAG production was measured with the antibodies anti-PNAG (B) and anti-UPAB1 as a loading control (C).
H-NS regulates the expression of a previously unidentified “curli-like” cluster.
Congo-red binding has been routinely employed to monitor Curli amyloid production in E. coli. In this bacterium, curli synthesis involves two operons, csgBAC and csgDEFG. These operons are responsible for curli fiber polymerization, stability, transport, and assembly (13, 17, 18). Particularly, CsgG forms an oligomeric transport complex and is essential for curli assembly. Although curli formation has not been reported in Acinetobacter species, a bioinformatic analysis revealed that a CsgG ortholog (D1G37_12595) is contained within a gene cluster that also comprises two additional genes, D1G37_12600 (“Ab12600”) and D1G37_12605 (“Ab12605”), both encoding putative lipoproteins (Fig. 5A). Our previous transcriptomic and proteomic analysis indicated that these genes are also downregulated by pAB5 (11). We validated these data by RT-PCR and determined that the transcription of csgG and Ab12600 is repressed ∼10-fold by pAB5 or a vector expressing hns (Fig. 5B), indicating that, besides PNAG, H-NS also represses the csgG curli-like cluster in UPAB1.
FIG 5.
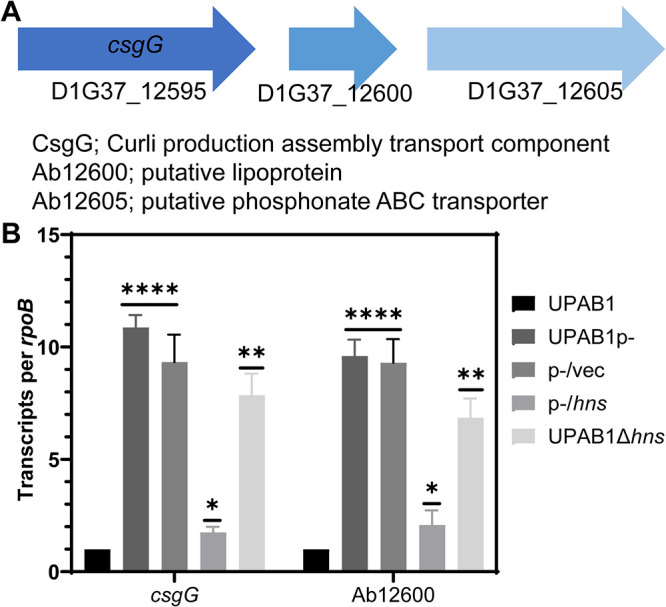
H-NS reduces the expression of curli-like cluster. (A) Illustration of csgG cluster. (B) csgG and Ab12600 expression. Transcripts were measured from cells growing on LB plates at 26°C and adjusted to an OD of 1. The results are shown as rpoB-adjusted transcript values. Values represent means and standard deviations from four independent experiments. t test results are indicated (****, P ≤ 0.0001; **, P ≤ 0.005; *, P ≤ 0.05).
Disruption of CsgG decreases PNAG production, CUP pilus formation, and biofilm.
To explore the role of the CsgG-containing operon in UPAB1, we deleted the csgG and Ab12600 genes in the strains expressing high levels of csgG, i.e., UPAB1p- and UPAB1Δhns. Surprisingly, csgG and Ab12600 mutant strains showed low levels of Congo red binding and reduced PNAG production, similar to the pgaA-D and pgaA mutants (Fig. 6A and B; see also Fig. S3 in the supplemental material). The complementation of the csgG mutant rescued both phenotypes. To further explore the role of the curli-like cluster, we analyzed these cells via scanning electron microscopy (SEM) and transmission electron microscopy (TEM). SEM images showed that UPAB1p- cells were coated with a thick layer of extracellular matrix material (Fig. 7). The csgG mutant displayed drastically reduced attachment to the coverslip and, except for some fibers, lacked most of the extracellular matrix (Fig. 7). This phenotype was partially complemented by expressing the CsgG gene in trans. (Fig. 7). For comparative purposes, we also examined the ΔpgaA-D mutant strain. The ΔcsgG and ΔpgaA-D strains exhibited similar phenotypes, although the reduced binding to the coverslips was less pronounced in the ΔpgaA-D strain (Fig. 7). These phenotypes are not due to growth defects (see Fig. S4), and they correlate with the lower PNAG production in the csgG mutant. Our TEM analysis showed that CsgG, but not PgaA-D deletion, results in an almost total abrogation of CUP pilus formation (Fig. 8). UPAB1 genome harbors two loci encoding putative chaperone-usher pathway (CUP) pili, that we termed CUP1 and CUP2 (11). CUP pilus levels were restored in the complemented cells. Western blot analysis confirmed that deletion of CsgG abrogated CUP pilus expression (see Fig. S5). Moreover, this analysis confirmed that CUP pili are repressed by pAB5, although this repression was independent of H-NS (see Fig. S5). The changes in the extracellular matrix were also reflected in the levels of biofilm formation, since the pgaA-D and csgG mutants produced less biofilm than did wild-type bacteria (Fig. 9). The reduction of biofilm formation was restored in the complemented strains (see Fig. S6). Moreover, cells carrying pAB5, but not pAB5Δhns, displayed lower levels of biofilm formation (Fig. 9). Together, these experiments demonstrate that deletion of CsgG results in reduced production of PNAG and CUP pili, with the subsequent reduction in biofilm formation.
FIG 6.
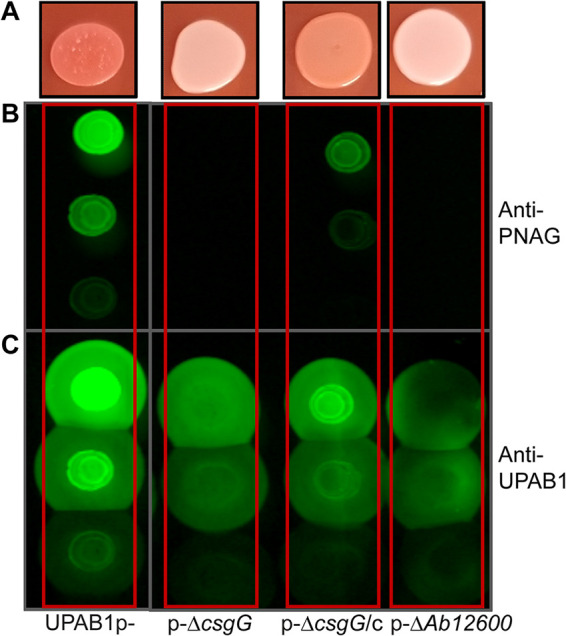
A curli-like cluster is also involved in PNAG production. (A) Phenotype on YESCA-Congo red agar plates of UPAB1p-, derivative mutant, and complemented strains. PNAG production was measured with the antibodies anti-PNAG (B) and anti-UPAB1 as a loading control (C).
FIG 7.
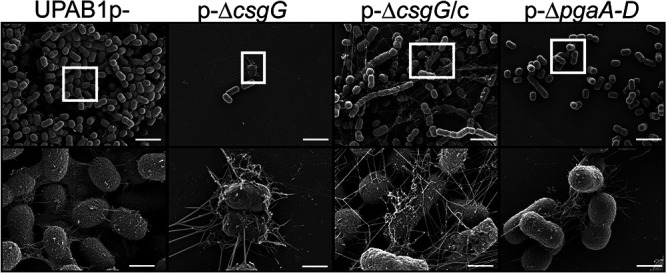
CsgG and PNAG are involved in extracellular matrix production. SEM analysis of UPAB1p-, p-ΔcsgG, p-ΔcsgG/c, and p-ΔpgaA-D (as a control) was performed. Cells were grown in YESCA media with 4% DMSO in 24-well plates with glass coverslips. After overnight growth, the glass coverslips were removed, washed (150 mM cacodylate buffer with 2 mM CaCl2), fixed (2.5% glutaraldehyde, 2% paraformaldehyde, and 0.2% tannic acid in 150 mM cacodylate buffer [pH 7.4] with 2 mM CaCl2), and treated for observation. The bottom panel is a magnification of the white square in the top panel. The scale bars indicate 1 μm for the top panel and 200 nm for the bottom panel.
FIG 8.
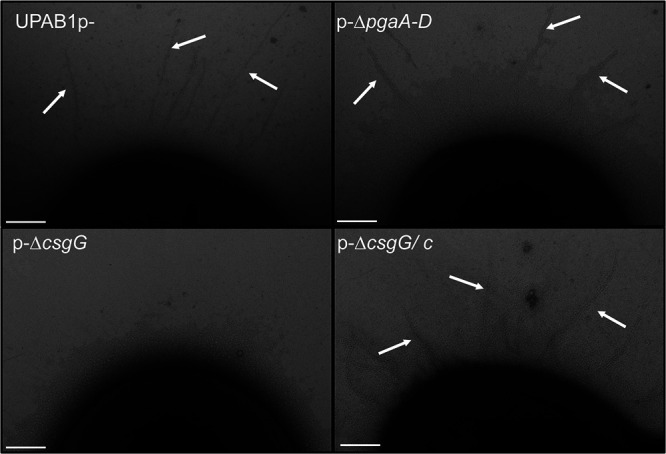
CsgG is involved in CUP pilus formation. TEM images show CUP pili. The pilus structures were absent in the p-ΔcsgG strain and restored in the complemented strain (p-ΔcsgG/c). The cells grew for 48 h in YESCA media supplemented with 4% DMSO with shaking at 26°C. Scale bar, 100 nm.
FIG 9.
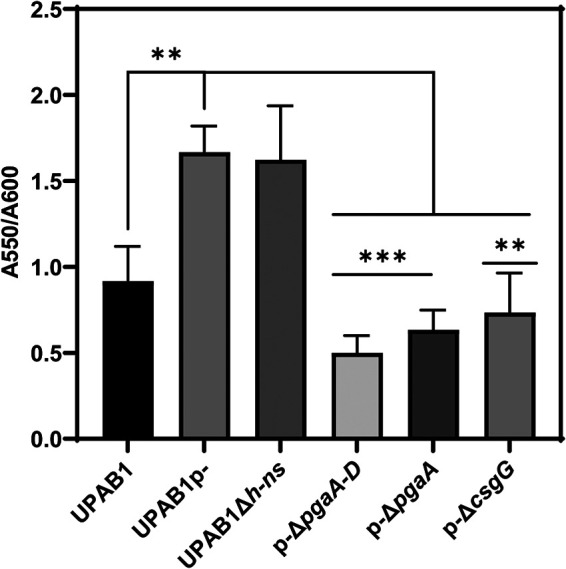
pAB5 reduces biofilm formation. Cells were grown for 8 h on LB broth at 37°C under static conditions. Biofilm formation was measured by crystal violet staining and normalized to the growth. The values represent the means and standard deviations from three independent experiments. A t test was performed in comparison to the pAB5- strain (***, P ≤ 0.0005; **, P ≤ 0.005).
The csgG cluster is widely distributed in Gram-negative bacteria.
Our results show that CsgG is implicated in different phenotypes in UPAB1. A bioinformatics analysis revealed that this cluster is widespread among Gram-negative bacteria (Fig. 10). In some bacterial species, such as Neisseria meningitidis or Vibrio harveyi, the locus contains additional genes predicted to be cotranscribed (19, 20). Recently, the crystal structure of GNA1162 from N. meningitidis, a homologue to D1G37_12605, has been solved (21). GNA1162 exhibited structural similarities to TolB, and the authors of that study speculate that this protein may act as an accessory protein to an unidentified transport machinery. Although this cluster is present in a very large number of bacteria, its roles remain unknown, and further studies are needed to identify the target of this cluster and its importance in virulence.
FIG 10.

Distribution of csgG cluster in Gram-negative bacteria. The csgG cluster is indicated in blue.
DISCUSSION
We have previously shown that the Acinetobacter LCPs play a key role in the dissemination of MDR and influences the pathogenesis of this bacterium by controlling the expression of chromosomally encoded virulence factors. Here, we show that pAB5, the LCP from UPAB1, encodes an H-NS transcriptional regulator that inhibits biofilm formation by repressing the expression of PNAG. In addition, we found that pAB5-encoded H-NS represses the expression of a three-gene cluster that contains a homolog of CsgG, a protein involved in curli assembly. We found that disruption of CsgG dramatically reduced PNAG production, CUP pilus assembly, and consequently biofilm formation. Bioinformatic analysis revealed that the “curli-like cluster” is widely distributed among Gram-negative bacteria without any attributed function.
The cross-regulatory pathways between plasmids and the bacterial chromosomes have been recently reviewed (22). Plasmid-encoded transcriptional regulators can modulate the expression of genes involved in many different processes, including motility, glycogen synthesis, adherence, and quorum sensing, among others (22). H-NS-like proteins are encoded in plasmids in Shigella flexneri and Salmonella enterica. However, in these species, plasmid-encoded H-NS appear to regulate only genes horizontally acquired that maintain the energetic cost of their expression at a lower level, without affecting expression of other chromosomal genes (22). We have previously shown that repressing chromosomally encoded T6SS enable the dissemination of LCPs via conjugation (10). By downregulating PNAG, CsgG, and CUP pili, pAB5 represses biofilm formation in UPAB1. In monospecies biofilms, bacteria are surrounded by their kin, which limits the dissemination capacity of the plasmids. It is tempting to speculate that promoting the planktonic lifestyle of the host increases the chances of dissemination of pAB5 to other bacterial hosts. However, further work is required to understand the physiological implication of this process.
Our discovery that pAB5-encoded H-NS represses biofilm formation in UPAB1 is preceded by similar findings in E. coli and Salmonella (23–28). However, in these species H-NS is encoded in the chromosome. In E. coli and Salmonella, the CsgG protein is part of a multiprotein complex responsible for curli fiber formation, and H-NS regulates its expression by repressing csgD, a key regulator for curli synthesis. In these species, H-NS is part of an intricate regulatory network that integrates diverse environmental conditions and ultimately controls curli biogenesis. CsgG is part of a predicted operon together with two putative lipoproteins of unknown function that is widely distributed in Gram-negative bacteria. Acinetobacter spp. do not encode a complete curli biosynthetic machinery, and curli fibers have not been reported. In Caulobacter, the CsgG ortholog, named HfaB is part of a cluster containing HfaABD, where HfaA has properties of amyloid proteins (CsgA). The HfaABD complex is critical for anchoring holdfast, a polysaccharide made of N-acetyl-d-glucosamine (NAG), and other sugars to the Caulobacter cell surface. It has been proposed that holdfast is attached to HfaA by an unknown mechanism (29–31). We hypothesize that a similar function anchoring PNAG to the cell surface is accomplished by a multimeric complex formed by CsgG and the two lipoproteins in the operon. Further work is necessary to determine the exact role of this gene cluster in the assembly of the extracellular matrix in these species.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Bacterial strains, plasmids, and oligonucleotides used in this study are listed in the supplemental material. Unless otherwise noted, all strains were grown in lysogeny broth (LB) broth at 37°C with shaking (200 rpm). For strain constructions, we used gentamicin (15 or 20 μg ml−1), kanamycin (7.5 or 50 μg ml−1), zeocin (50 μg ml−1 with low-salt LB), and hygromycin (300 μg ml−1).
Construction of A. baumannii mutants and complement strains and pVRL2 constructs.
Plasmids and oligonucleotides used in this study are listed in Tables S2 and S3, respectively, in the supplemental material. The constructs for generating deletions in hns, pgaA-D, pgaA, csgG, and D1G37_126000 were made by substituting the gene with an antibiotic (kanamycin or zeocin) cassette, as described previously (32). Selection of mutants was carried out using the proper antibiotic. To make unmarked strains, electrocompetent mutants were transformed with pAT03 to remove the FRT-flanked antibiotic cassette. Transformants were plated on LB-agar plates containing 2 mM IPTG plus hygromycin to express FLP recombinase. All strains were verified by antibiotic resistance, PCR amplification, and gene sequencing. To generate genetic complementation, genes of interest were cloned into the pUC18T-miniTn7T-Gm (zeo) vector and introduced to UPAB1p- strains via four-parental mating methods, as described previously (33, 34). Briefly, 100 μl of stationary cultures was normalized to an optical density at 600 nm (OD600) of 2.0 of each recipient strain, and HB101(pRK2013), EC100D(pTNS2), and EC100D containing the pUC18T-miniTn7T constructs were added to 600 μl of warm LB. Each suspension was washed twice by centrifugation at 7,000 × g, followed by resuspension of the bacterial pellet in 1 ml of warm LB. On the final wash, the bacterial pellet was resuspended in 25 μl of LB, and the suspension was spotted on a prewarmed LB agar plate (or low-salt LB agar plate), followed by incubation overnight at 37°C. The bacteria were scraped from the plate, resuspended in 1 ml of LB, vortexed, and serial dilutions were plated on LB-agar plates supplemented with chloramphenicol to select against E. coli strains and gentamicin or zeocin to select for A. baumannii strains that had received the mini-Tn7 constructs. Correct insertion of the constructs was verified by PCR amplification and sequencing. The expression of different regulators of the pAB5 plasmid as p/h-ns, p/tetR1, and others were made using the arabinose-inducible pVRL2 vector (15). Constructs were generated by restriction enzyme cloning using the HindIII and PstI sites. All the constructs were introduced to UPAB1p- strains by electroporation, and transformants were selected on gentamicin.
Congo red plate and Congo red binding quantification.
The red versus white color of cells was investigated using YESCA agar media (35) supplemented with 50 μg/ml Congo red. To quantify the Congo red binding for bacteria prestained on the YESCA Congo red plates (36), cells were recovered from YESCA Congo red plates after incubation at 26°C for 48 h. The cells were washed twice in 50 mM potassium phosphate buffer by centrifugation at 16,000 × g for 2 min and resuspended in 1 ml of 50 mM potassium phosphate buffer, and the OD was adjusted to 1. Then, 100 μl of each sample was loaded onto a 96-well opaque plate, and the fluorescence of Congo red was measured using the plate reader (BioTek microplate spectrophotometer) with an excitation wavelength at 485 nm and emission at 612 nm. Buffer was used as the blank.
Reverse transcription-PCR.
Cells were taken from LB plates after an overnight growth at 26°C, normalized to an OD of 1, and treated with RNAprotect. RNA purification was prepared using the Quick RNA fungal/bacterial miniprep (Zymo Research) by following the manufacturer’s instructions with some modifications in the DNA digestion step as follows. Contaminating DNA was removed using the Turbo DNA-free kit (Invitrogen) by following the manufacturer’s instructions. For reverse transcription-PCR (RT-PCR), cDNA was prepared from 1 μg of RNA using a high-capacity RNA-to-cDNA kit (Applied Biosystems), according to the manufacturer’s protocol. Real-time quantitative PCR (qPCR) was performed using Power SYBR green PCR master mix reagents (Applied Biosystems) on a ViiA7 real-time PCR system (Applied Biosystems) according to the manufacturer’s suggested protocol. In all cases, a no-template control was run with no detectable transcripts.
Biofilm formation.
Cells were grown overnight in 5 ml of LB broth (or YESCA), and then cultures were diluted to an OD600 of 0.01 in LB broth (or YESCA). Cultures were deposited in 96-well plates and incubated at 37°C for 8 or 24 h without shaking. Cultures were removed to read the absorbance at 600 nm. Then, the plates were washed three times with water, stained with 0.1% crystal violet (wt/vol), and quantified at 550 nm after solubilization with 30% acetic acid.
Detection of PNAG production.
Immunoblotting to detect PNAG production was performed as described previously (12) with some modifications. Cells grown overnight on LB, diluted to an OD of 1, spotted onto YESCA plates, and incubated for 48 h at 26°C. Cells were scraped from plate and normalized to an OD of 1, pelleted, and resuspended in 300 μl of 0.5 M EDTA (pH 8.0). The cells were incubated for 5 min at 98°C and centrifuged at 9,000 × g for 5 min. After centrifugation, the supernatants were diluted 1:3 in Tris-buffered saline (TBS) and incubated first with 100 μl of proteinase K (20 mg/ml) for 60 min at 65°C and then for 30 min at 80°C (to inactivate the protease). The preparations were serially diluted 10-fold in TBS, and 5 μl were spotted on nitrocellulose membrane; the membrane was then allowed to dry completely. Next, the membrane was blocked, followed by incubation with an anti-PNAG antibody (kindly provided by Gerald B. Pier, Harvard Medical School) and an anti-human IgG (IRDye 800 CW) antibody (LI-COR Biosciences, Lincoln, NE), and visualized with an Odyssey CLx imaging system (LI-COR Biosciences). Next, the membrane was incubated with anti-UPAB1 primary antibody (Ref), followed by an incubation with anti-rabbit IgG (IRDye 800 CW) antibody (LI-COR Biosciences).
Scanning electron microscopy.
Overnight cultures on YESCA media were diluted in YESCA plus 4% dimethyl sulfoxide (DMSO) (37) to an OD600 of 0.02 in 24 well-plate containing glass coverslips and incubated at 26°C for 24 h with shaking. Next, the medium was removed, and the 24 well-plate was washed with 0.15 M cacodylate buffer. The cells were fixed overnight at room temperature on a shaker using the fixative solution (2.5% glutaraldehyde, 2% paraformaldehyde, and 0.2% tannic acid in 0.15 M cacodylate buffer [pH 7.4] with 2 mM calcium chloride). Postfixation, the coverslips were rinsed in 0.15 M cacodylate buffer three times for 10 min each time, followed by a secondary fixation in 1% OsO4 in 0.15 M cacodylate buffer for 45 min in the dark. The coverslips were then rinsed three times in ultrapure water for 10 min each time and dehydrated in a graded ethanol series (10, 30, 50, 70, 90, and 100%, ×2) for 10 min each step. Once dehydrated, the samples were loaded into a critical point drier (EM CPD 300; Leica, Vienna, Austria), which was set to perform 12 CO2 exchanges at the slowest speed. Once dried, coverslips were mounted on aluminum stubs with carbon adhesive tabs and coated with 10 nm of carbon and 6 nm of iridium (ACE 600; Leica). SEM images were acquired on a FE-SEM (Zeiss Merlin, Oberkochen, Germany) at 1.5 kV and 0.1 nA.
Transmission electron microscopy and CUP pilus detection.
Overnight cultures on YESCA media were diluted in YESCA plus 4% DMSO (37) to an OD600 of 0.02, followed by incubation at 26°C for 48 h with shaking. Next, cultures were washed with phosphate-buffered saline (PBS) and used for TEM and Western blotting. For negative staining and analysis by TEM, bacterial samples were fixed with 1% glutaraldehyde (Ted Pella, Inc., Redding, CA) and allowed to absorb onto freshly glow discharged Formvar/carbon-coated copper grids for 10 min. Grids were then washed in dH2O and stained with 1% aqueous uranyl acetate (Ted Pella, Inc.) for 1 min. Excess liquid was gently wicked off, and grids were allowed to air dry. Samples were viewed on a JEOL 1200EX transmission electron microscope (JEOL USA, Peabody, MA) equipped with an AMT 8-megapixel digital camera (Advanced Microscopy Techniques, Woburn, MA). For cup pilus detection, cells were resuspended in Laemmli buffer to a final OD of 10. Samples were loaded onto 15% SDS-PAGE gel for separation, transferred to a nitrocellulose membrane, and probed with polyclonal rabbit anti CupA (1:1,000) and monoclonal mouse anti-RNA polymerase (1:3,000; BioLegend, San Diego, CA). Western blots were then probed with IRDye-conjugated anti-mouse and anti-rabbit secondary antibodies (both at 1:15,000; LI-COR Biosciences) and visualized with an Odyssey CLx imaging system (LI-COR Biosciences).
Generation of polyclonal rabbit sera against CupA.
The UPAB1 gene cupA was cloned into pET28a+ with a 10-histidine tag using the primers SB cupA bamh1 F and SB cupA hind3 R, creating pET28-CupA10His, and electroporated into E. coli DH5α. pET28-CupA10His was confirmed by sequencing. E. coli Rosetta 2 cells were used for CupA purification. Then, 1 liter of LB was inoculated from an overnight culture of Rosetta 2/pET28-CupA10His at an OD600 of 0.05. The culture was grown to an OD600 of ∼0.5 before induction with 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside). The cultures were grown for an additional 4 h. Cells were harvested at 12,000 × g for 20 min. The cells were washed with cold PBS and resuspended in binding buffer supplemented with protease inhibitor (300 mM NaCl, 10 mM imidazole, 30 mM Tris-HCl [pH 8.0]). The cells were lysed with a cell disruptor using three rounds at 35 kpsi (Constant System, Ltd., Kennesaw, GA). Cell lysates were centrifuged at 20,000 × g (or 11,000 rpm) for 20 min to collect inclusion bodies. Pellet was resuspended in binding buffer and centrifuged as described before twice. The pellet was then resuspended in binding buffer containing urea and incubated for 3 h at 4°C with continuous stirring (6 M urea, 300 mM NaCl, 10 mM imidazole, 30 mM Tris-HCl [pH 8.0]). The lysates were then centrifuged at 35,000 × g for 20 min, and the supernatant was filtered using a 0.45-μm-pore size filter. Next, cell lysates were passed over a nickel-NTA agarose column (Gold Bio, St. Louis, MO) equilibrated with 10 column volumes of binding buffer. The load fraction is the total cell lysate. The flowthrough was collected as what passed through the column and did not bind the nickel-NTA resin. The column was washed first with 15 column volumes of washing buffer (5 M urea, 20 mM imidazole, 300 mM NaCl, 30 mM Tris-HCl [pH 8.0]) and second with 10 column volumes of washing buffer (4 M urea, 20 mM imidazole, 300 mM NaCl, 30 mM Tris-HCl [pH 8.0]). Proteins were eluted using elution buffer (2 M urea, 250 mM imidazole, 300 mM NaCl, 30 mM Tris-HCl [pH 8.0]). Elution fractions were analyzed by SDS-PAGE analysis and Coomassie blue staining. The polyacrylamide gel band corresponding to CupA-His was sent to Antibody Research Corporation (St. Louis, MO) for peptide extraction and development of rabbit-derived polyclonal antibodies.
Growth assays.
Bacteria were cultured overnight in YESCA liquid medium at 26°C under shaking conditions. Cultures were washed with PBS and diluted to an OD600 of 0.01 in 150 μl of YESCA liquid medium in 96-well plates, followed by incubation at 26°C under shaking conditions. The OD600 values were measured every 30 min for 16 h via a BioTek microplate spectrophotometer. Three separate experiments were performed with four wells per experiment for each strain.
ACKNOWLEDGMENTS
We acknowledge the assistance of Wandy Beatty with transmission electron microscopy studies at the Molecular Microbiology Department Imaging Facility at the Washington University School of Medicine, and we acknowledge the assistance of Sanja Sviben and James Fitzpatrick at the Washington University Center for Cellular Imaging (WUCCI) in the scanning electron microscopy studies (WUCCI is supported by Washington University School of Medicine, The Children’s Discovery Institute of University, St. Louis Children’s Hospital [CDI-CORE-2015-505 and CDI-CORE-2019-813], and the Foundation for Barnes-Jewish Hospital [grant 3770]). We also thank Gerald B. Pier for sharing the anti-PNAG antibody.
This study was supported by grants from the National Institute of Allergy and Infectious Diseases (grants R01AI144120 and R01AI125363).
Footnotes
Supplemental material is available online only.
Contributor Information
Saida Benomar, Email: sbenomar@wustl.edu.
Mario F. Feldman, Email: mariofeldman@wustl.edu.
Yves V. Brun, Université de Montréal
REFERENCES
- 1.Giammanco A, Calà C, Fasciana T, Dowzicky MJ. 2017. Global assessment of the activity of tigecycline against multidrug-resistant gram-negative pathogens between 2004 and 2014 as part of the Tigecycline Evaluation and Surveillance Trial. mSphere 2:e00310-16. 10.1128/mSphere.00310-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rolain JM, Diene SM, Kempf M, Gimenez G, Robert C, Raoult D. 2013. Real-time sequencing to decipher the molecular mechanism of resistance of a clinical pan-drug-resistant Acinetobacter baumannii isolate from Marseille, France. Antimicrob Agents Chemother 57:592–596. 10.1128/AAC.01314-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blackwell GA, Hamidian M, Hall RM. 2016. IncM Plasmid R1215 is the source of chromosomally located regions containing multiple antibiotic resistance genes in the globally disseminated Acinetobacter baumannii GC1 and GC2 clones. mSphere 1:e00117-16. 10.1128/mSphere.00117-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wright MS, Iovleva A, Jacobs MR, Bonomo RA, Adams MD. 2016. Genome dynamics of multidrug-resistant Acinetobacter baumannii during infection and treatment. Genome Med 8:26. 10.1186/s13073-016-0279-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Norman A, Hansen LH, Sørensen SJ. 2009. Conjugative plasmids: vessels of the communal gene pool. Philos Trans R Soc Lond B Biol Sci 364:2275–2289. 10.1098/rstb.2009.0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Luque A, Paytubi S, Sánchez-Montejo J, Gibert M, Balsalobre C, Madrid C. 2019. Crosstalk between bacterial conjugation and motility is mediated by plasmid-borne regulators. Environ Microbiol Rep 11:708–717. 10.1111/1758-2229.12784. [DOI] [PubMed] [Google Scholar]
- 7.Smalla K, Jechalke S, Top EM. 2015. Plasmid detection, characterization, and ecology. Microbiol Spectr 10.1128/microbiolspec.PLAS-0038-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Orlek A, Stoesser N, Anjum MF, Doumith M, Ellington MJ, Peto T, Crook D, Woodford N, Sarah Walker A, Phan H, Sheppard AE. 2017. Plasmid classification in an era of whole-genome sequencing: application in studies of antibiotic resistance epidemiology. Front Microbiol 8:182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weber BS, Ly PM, Irwin JN, Pukatzki S, Feldman MF. 2015. A multidrug resistance plasmid contains the molecular switch for type VI secretion in Acinetobacter baumannii. Proc Natl Acad Sci USA 112:9442–9447. 10.1073/pnas.1502966112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Di Venanzio G, Moon KH, Weber BS, Lopez J, Ly PM, Potter RF, Dantas G, Feldman MF. 2019. Multidrug-resistant plasmids repress chromosomally encoded T6SS to enable their dissemination. Proc Natl Acad Sci USA 116:1378–1383. 10.1073/pnas.1812557116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Di Venanzio G, Flores-Mireles AL, Calix JJ, Haurat MF, Scott NE, Palmer LD, Potter RF, Hibbing ME, Friedman L, Wang B, Dantas G, Skaar EP, Hultgren SJ, Feldman MF. 2019. Urinary tract colonization is enhanced by a plasmid that regulates uropathogenic Acinetobacter baumannii chromosomal genes. Nat Commun 10:2763. 10.1038/s41467-019-10706-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choi AHK, Slamti L, Avci FY, Pier GB, Maira-Litrán T. 2009. The pgaABCD locus of Acinetobacter baumannii encodes the production of poly-beta-1-6-N-acetylglucosamine, which is critical for biofilm formation. J Bacteriol 191:5953–5963. 10.1128/JB.00647-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barnhart MM, Chapman MR. 2006. Curli biogenesis and function. Annu Rev Microbiol 60:131–147. 10.1146/annurev.micro.60.080805.142106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doyle M, Fookes M, Ivens A, Mangan MW, Wain J, Dorman CJ. 2007. An H-NS-like stealth protein aids horizontal DNA transmission in bacteria. Science 315:251–252. 10.1126/science.1137550. [DOI] [PubMed] [Google Scholar]
- 15.Lucidi M, Runci F, Rampioni G, Frangipani E, Leoni L, Visca P. 2018. New shuttle vectors for gene cloning and expression in multidrug-resistant Acinetobacter species. Antimicrob Agents Chemother 62:e02480-17. 10.1128/AAC.02480-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Whitney JC, Howell PL. 2013. Synthase-dependent exopolysaccharide secretion in Gram-negative bacteria. Trends Microbiol 21:63–72. 10.1016/j.tim.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Evans ML, Chapman MR. 2014. Curli biogenesis: order out of disorder. Biochim Biophys Acta 1843:1551–1558. 10.1016/j.bbamcr.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Van Gerven N, Klein RD, Hultgren SJ, Remaut H. 2015. Bacterial amyloid formation: structural insights into curli biogenesis. Trends Microbiol Elsevier, Ltd, New York, NY. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vella P, Rudraraju RS, Lundbäck T, Axelsson H, Almqvist H, Vallin M, Schneider G, Schnell R. 2021. A FabG inhibitor targeting an allosteric binding site inhibits several orthologs from Gram-negative ESKAPE pathogens. Bioorg Med Chem 30:115898. 10.1016/j.bmc.2020.115898. [DOI] [PubMed] [Google Scholar]
- 20.Ge B, McDermott PF, White DG, Meng J. 2005. Role of efflux pumps and topoisomerase mutations in fluoroquinolone resistance in Campylobacter jejuni and Campylobacter coli. Antimicrob Agents Chemother 49:3347–3354. 10.1128/AAC.49.8.3347-3354.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cai X, Lu J, Wu Z, Yang C, Xu H, Lin Z, Shen Y. 2013. Structure of Neisseria meningitidis lipoprotein GNA1162. Acta Crystallogr Sect F Struct Biol Cryst Commun 69:362–368. 10.1107/S1744309113004417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vial L, Hommais F. 2020. Plasmid-chromosome cross-talks. Environ Microbiol 22:540–556. 10.1111/1462-2920.14880. [DOI] [PubMed] [Google Scholar]
- 23.RöMling U, RöMling R, Bian Z, Hammar M, Sierralta WD, Normark S. 1998. Curli fibers are highly conserved between Salmonella Typhimurium and Escherichia coli with respect to operon structure and regulation. J Bacteriol 180:722–731. 10.1128/JB.180.3.722-731.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arnqvist A, Olsen A, Normark S. 1994. Sigma S-dependent growth-phase induction of the csgBA promoter in Escherichia coli can be achieved in vivo by sigma 70 in the absence of the nucleoid-associated protein H-NS. Mol Microbiol 13:1021–1032. 10.1111/j.1365-2958.1994.tb00493.x. [DOI] [PubMed] [Google Scholar]
- 25.Gerstel U, Park C, Römling U. 2003. Complex regulation of csgD promoter activity by global regulatory proteins. Mol Microbiol 49:639–654. 10.1046/j.1365-2958.2003.03594.x. [DOI] [PubMed] [Google Scholar]
- 26.Olsén A, Arnqvist A, Hammar M, Sukupolvi S, Normark S. 1993. The RpoS Sigma factor relieves H-NS-mediated transcriptional repression of csgA, the subunit gene of fibronectin-binding curii in Escherichia coliMolecular. Mol Microbiol 7:523–536. 10.1111/j.1365-2958.1993.tb01143.x. [DOI] [PubMed] [Google Scholar]
- 27.Desai SK, Winardhi RS, Periasamy S, Dykas MM, Jie Y, Kenney LJ. 2016. The horizontally-acquired response regulator SsrB drives a Salmonella lifestyle switch by relieving biofilm silencing. eLife 5:e10747. 10.7554/eLife.10747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Desai SK, Kenney LJ. 2019. Switching lifestyles is an in vivo adaptive strategy of bacterial pathogens. Front Cell Infect Microbiol 9:421. 10.3389/fcimb.2019.00421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hardy GG, Allen RC, Toh E, Long M, Brown PJB, Cole-Tobian JL, Brun YV. 2010. A localized multimeric anchor attaches the Caulobacter holdfast to the cell pole. Mol Microbiol 76:409–427. 10.1111/j.1365-2958.2010.07106.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hardy GG, Toh E, Berne C, Brun YV. 2018. Mutations in sugar-nucleotide synthesis genes restore holdfast polysaccharide anchoring to Caulobacter crescentus holdfast anchor mutants. J Bacteriol 200:e00597-17. 10.1128/JB.00597-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sulkowski NI, Hardy GG, Brun YV, Bharat TAM. 2019. A multiprotein complex anchors adhesive holdfast at the outer membrane of Caulobacter crescentus. J Bacteriol 201:112–131. 10.1128/JB.00112-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tucker AT, Nowicki EM, Boll JM, Knauf GA, Burdis NC, Stephen Trent M, Davies BW. 2014. Defining gene-phenotype relationships in Acinetobacter baumannii through one-step chromosomal gene inactivation. mBio 5:e01313-14. 10.1128/mBio.01313-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kumar A, Dalton C, Cortez-Cordova J, Schweizer HP. 2010. Mini-Tn7 vectors as genetic tools for single copy gene cloning in Acinetobacter baumannii. J Microbiol Methods 82:296–300. 10.1016/j.mimet.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 34.Ducas-Mowchun K, Malaka P, Silva D, Crisostomo L, Fernando DM, Chao T-C, Pelka P, Schweizer HP, Kumar A. 2019. Next generation of Tn7-based single-copy insertion elements for use in multi- and pan-drug-resistant strains of Acinetobacter baumannii. Appl Environ Microbiol 85:e00066-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nenninger AA, Robinson LS, Hultgren SJ. 2009. Localized and efficient curli nucleation requires the chaperone-like amyloid assembly protein CsgF. Proc Natl Acad Sci USA 106:900–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou Y, Blanco LP, Smith DR, Chapman MR. 2012. Bacterial amyloids. Methods Mol Biol 849:303–320. 10.1007/978-1-61779-551-0_21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lim JY, May JM, Cegelski L. 2012. Dimethyl sulfoxide and ethanol elicit increased amyloid biogenesis and amyloid-integrated biofilm formation in Escherichia coli. Appl Environ Microbiol 78:3369–3378. 10.1128/AEM.07743-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 to S6 and Tables S1 to S4. Download JB.00277-21-s0001.pdf, PDF file, 0.9 MB (961.3KB, pdf)