

ABSTRACT
During mycoparasitism, a fungus—the host—is parasitized by another fungus—the mycoparasite. The genetic underpinnings of these relationships have been best characterized in ascomycete fungi. However, within basidiomycete fungi, there are rare instances of mushroom-forming species parasitizing the reproductive structures, or sporocarps, of other mushroom-forming species, which have been rarely investigated on a genetic level. One of the most enigmatic of these occurs between Entoloma abortivum and species of Armillaria, where hyphae of E. abortivum are hypothesized to disrupt the development of Armillaria sporocarps, resulting in the formation of carpophoroids. However, it remains unknown whether carpophoroids are the direct result of a mycoparasitic relationship. To address the nature of this unique interaction, we analyzed gene expression of field-collected Armillaria and E. abortivum sporocarps and carpophoroids. Transcripts in the carpophoroids are primarily from E. abortivum, supporting the hypothesis that this species is parasitizing Armillaria. Most notably, we identified differentially upregulated E. abortivum β-trefoil-type lectins in the carpophoroid, which we hypothesize bind to Armillaria cell wall galactomannoproteins, thereby mediating recognition between the mycoparasite and the host. The most differentially upregulated E. abortivum transcripts in the carpophoroid code for oxalate decarboxylases—enzymes that degrade oxalic acid. Oxalic acid is a virulence factor in many plant pathogens, including Armillaria species; however, E. abortivum has evolved a sophisticated strategy to overcome this defense mechanism. The number of gene models and genes that code for carbohydrate-active enzymes in the E. abortivum transcriptome was reduced compared to other closely related species, perhaps as a result of the specialized nature of this interaction.
IMPORTANCE By studying fungi that parasitize other fungi, we can understand the basic biology of these unique interactions. Studies focused on the genetic mechanisms regulating mycoparasitism between host and parasite have thus far concentrated on a single fungal lineage within the Ascomycota. The work presented here expands our understanding of mycoparasitic relationships to the Basidiomycota and represents the first transcriptomic study to our knowledge that examines fungal-fungal relationships in their natural setting. The results presented here suggest that even distantly related mycoparasites utilize similar mechanisms to parasitize their host. Given that species of the mushroom-forming pathogen Armillaria cause plant root-rot diseases in many agroecosystems, an enhanced understanding of this interaction may contribute to better control of these diseases through biocontrol applications.
KEYWORDS: basidiomycetes, metatranscriptomics, mycoparasitism, plant pathogens, transcriptomics
INTRODUCTION
Fungal mycoparasitism is a nutritional strategy where a living fungus—the host—is parasitized by and acts as a nutrient source for another fungus—the mycoparasite. Certain species of fungi in the Hypocreales (Ascomycota) are among the best-studied mycoparasites. Perhaps the best known of these are species of Trichoderma and Clonostachys rosea, which have biocontrol activity against plant-pathogenic species of Botrytis, Fusarium, Pythium, and Rhizoctonia (1, 2). Other fungal mycoparasites in the Hypocreales include Tolypocladium species, many of which are parasites on the reproductive structures, or sporocarps, of species in the genus Elaphomyces (Eurotiales, Ascomycota) (3); Escovopsis weberi, which is a specialized necrotrophic parasite of fungal gardens of attine ants (4, 5); and Hypomyces lactifluorum, which parasitizes the mushrooms of Russula species and transforms them into the iconic “lobster mushroom” (6). Within the Basidiomycota, one well-studied genus of mycoparasites is Tremella, which contain parasites of corticiaceous basidiomycetes (7) and lichen-forming fungi (8).
Less-studied examples of mycoparasitism involve mushroom-forming fungi that parasitize other mushroom-forming fungi. Fewer than 20 reported mushroom species may engage in this type of interaction, making it an incredibly rare phenomenon given the total number of mushroom-forming fungi (9, 10). Examples of this interaction whereby the parasite does not appear to impact the fitness of the host include Volvariella surrecta, which fruits from the pileus of its host, Clitocybe nebularis (11), and Asterophora species, which colonize their Russula or Lactarius host after it dies (10). More commonly, though, mushroom mycoparasites deform host sporocarps and likely prevent the dispersal of their spores. Pseudoboletus parasiticus fruits from the sporocarps of Scleroderma species, which, after infection, are no longer able to mature and disperse their spores (12). Psathyrella epimyces causes the deformation of sporocarp tissue of its host, Coprinus comatus (13). Additionally, of the 10 mushroom species in the genus Squamanita, all are known to be parasites of sporocarps of species in the genera Cystoderma, Galerina, Hebeloma, and Inocybe (10).
One of the most frequently encountered putative mycoparasitic interactions between two mushrooms involves species of Armillaria and Entoloma abortivum (Fig. 1). Entoloma abortivum is often encountered fruiting in soil, humus, or decaying logs in deciduous woods (14), while Armillaria species are facultative necrotrophs that can cause root rot in forest and agronomic systems worldwide (15, 16). Entoloma abortivum was originally described to occur as two morphologies: the mushroom form, which has a pinkish-gray stipe and pileus and pink gills, and then the carpophoroid form, which is generally white and subglobose and does not develop well-formed gills (Fig. 1). Initially, the carpophoroid form was assumed to be E. abortivum sporocarps that were malformed due to parasitism by Armillaria species (14). However, macro- and microscopic studies of carpophoroid collections determined that they are actually Armillaria sporocarps permeated with E. abortivum hyphae (17). Laboratory inoculation experiments showed that E. abortivum interacts with Armillaria sporocarps to disrupt their morphological development (17).
FIG 1.

The components of this fungal interaction in nature. (A) All three components—Armillaria and E. abortivum sporocarps, along with carpophoroids—fruiting synchronously. (B) A closeup photo of all of the components, with an Armillaria sporocarp indicated by the yellow arrow, an E. abortivum sporocarp indicated by the light pink arrow, and a carpophoroid indicated by the magenta arrow. Scale bars in panel A are equal to 10 cm, and those in panel B are equal to 1 cm. Photos courtesy of Ben Lemmond, University of Florida (© 2021 Lemmond; reproduced with permission).
Whether carpophoroids are the result of a mycoparasitic relationship, where E. abortivum serves as the mycoparasite and Armillaria species serve as the host, remains unknown. To address this, we initiated a transcriptome study after we encountered all three components of this system fruiting in close proximity—carpophoroids and individual sporocarps of E. abortivum and Armillaria. For a mycoparasite to be successful, there are several crucial steps to the utilization of a fungal host for nutrition. These steps include (i) sensing the host, (ii) recognition and attachment to host hyphae, (iii) initiating defense responses, and (iv) the eventual demise of the host (18). Previous genomic and transcriptomic studies elucidated the genetic machinery that model mycoparasites utilize during each of these steps (18–20). In this work, we used transcriptomic and metatranscriptomic techniques to analyze the genomic toolbox of E. abortivum and Armillaria during the carpophoroid stage. We show that the gene expression profiles of E. abortivum resemble those of known mycoparasitic species, as well as predict certain genes in both species that facilitate this interaction. Additionally, we used transcriptomic information to determine the species of Armillaria involved in this association.
RESULTS
Transcriptome assemblies of Entoloma abortivum and Armillaria.
To benchmark gene diversity and baseline expression levels of the field-collected mushroom species in our study, we sequenced the sporocarp transcriptomes of E. abortivum and the Armillaria species found in close proximity to the carpophoroids. To date, there are several transcriptomic studies of Armillaria species (21, 22), but none for any Entoloma species. The assembled transcriptome of E. abortivum was just under 120 million bp. There was a total of 43,599 contigs and an N50 value of 3,527; 94.5% of benchmark universal single-copy orthologs (BUSCOs) from the Agaricales were present in the E. abortivum transcriptome. Nine internal transcribed spacer (ITS) sequences were extracted from the transcriptome, accounting for 5.96 × 10−4% to 1.82 × 10−3% of the mapped reads; all ITS sequences were from E. abortivum. Within the contigs, a total of 9,728 unique gene models were recovered in the transcriptome assembly (Fig. 2) with 603 transcripts differentially upregulated in the carpophoroid tissue compared to the sporocarp tissue and 403 transcripts differentially upregulated in the sporocarp tissue compared to carpophoroid tissue (Fig. 3). The transcriptome contained 195 genes that code for carbohydrate-active enzymes (CAZymes). The transcriptome lacks any genes that code for cellobiohydrolases (glycoside hydrolase [GH] 6 and GH7), xylanases (GH10, GH11, and GH30), and auxiliary proteins like polysaccharide monooxygenases (GH61) but does contain nine chitinases (GH18) (Fig. 2). Transcripts detected in the E. abortivum transcriptome that were previously determined to play important roles in mycoparasitic interactions (18–20) include 10 putative secondary metabolite gene clusters, one G-coupled protein receptor (GCPR), 38 ATP-binding cassette (ABC) transporters, and 113 genes from the major facilitator superfamily (MFS) (Fig. 3). The average gene expression (in normalized units of trimmed mean of m-values [TMM]) of the 10 most abundant E. abortivum transcripts in the sporocarp ranged from 4,333 to 17,890. Information about the 10 most abundant transcripts in the sporocarps is available in Table 1.
FIG 2.
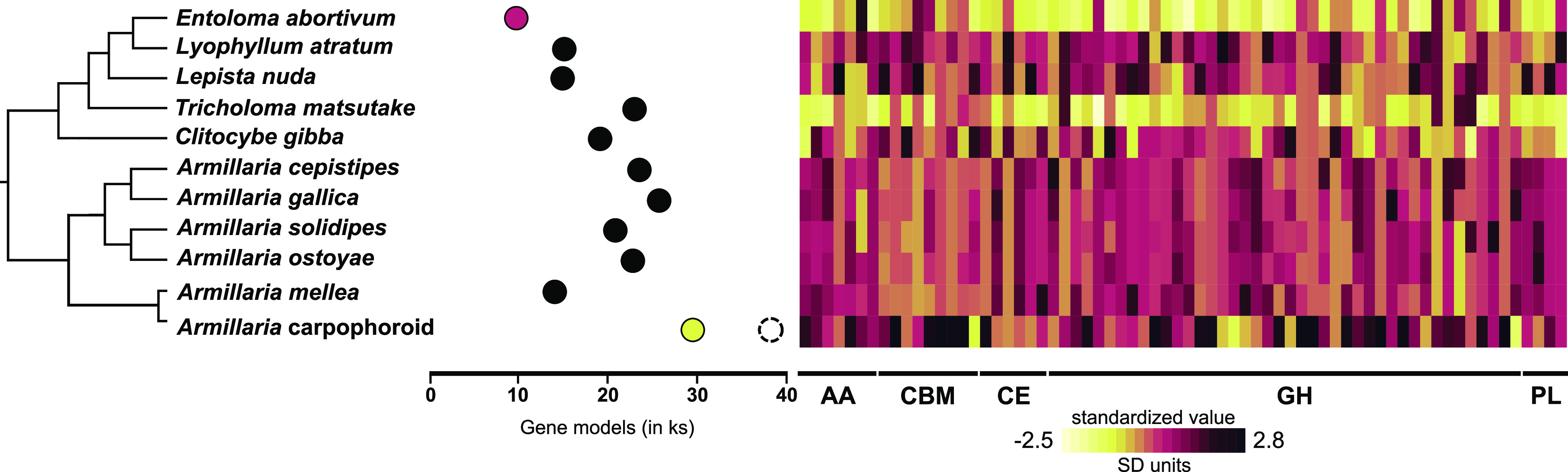
Number of total gene models and gene copy numbers of CAZymes. (Left) A representative phylogeny based off phylogenetic evidence from references 22 and 111. (Middle) Filled-in, colored dots represent the number of fungal gene models in the transcriptomes of Armillaria (yellow) and E. abortivum (pink) generated in this study, along with those of closely related species from previously generated genomic data (filled-in black dots). The dashed circle associated with the Armillaria transcriptome represents the total number of identified gene models (inclusive of additional non-Armillaria genes), showing that nearly one-quarter of the total gene models in the Armillaria transcriptome were from organisms other than the target. (Right) Heat map showing gene copy numbers of plant cell wall-degrading enzymes detected in the transcriptomes of E. abortivum and Armillaria, along with other closely related species. Abbreviations: AA, auxiliary activities; CBM, carbohydrate-binding modules; CE, carbohydrate esterase; GH, glycoside hydrolase; PL, polysaccharide lyase.
FIG 3.
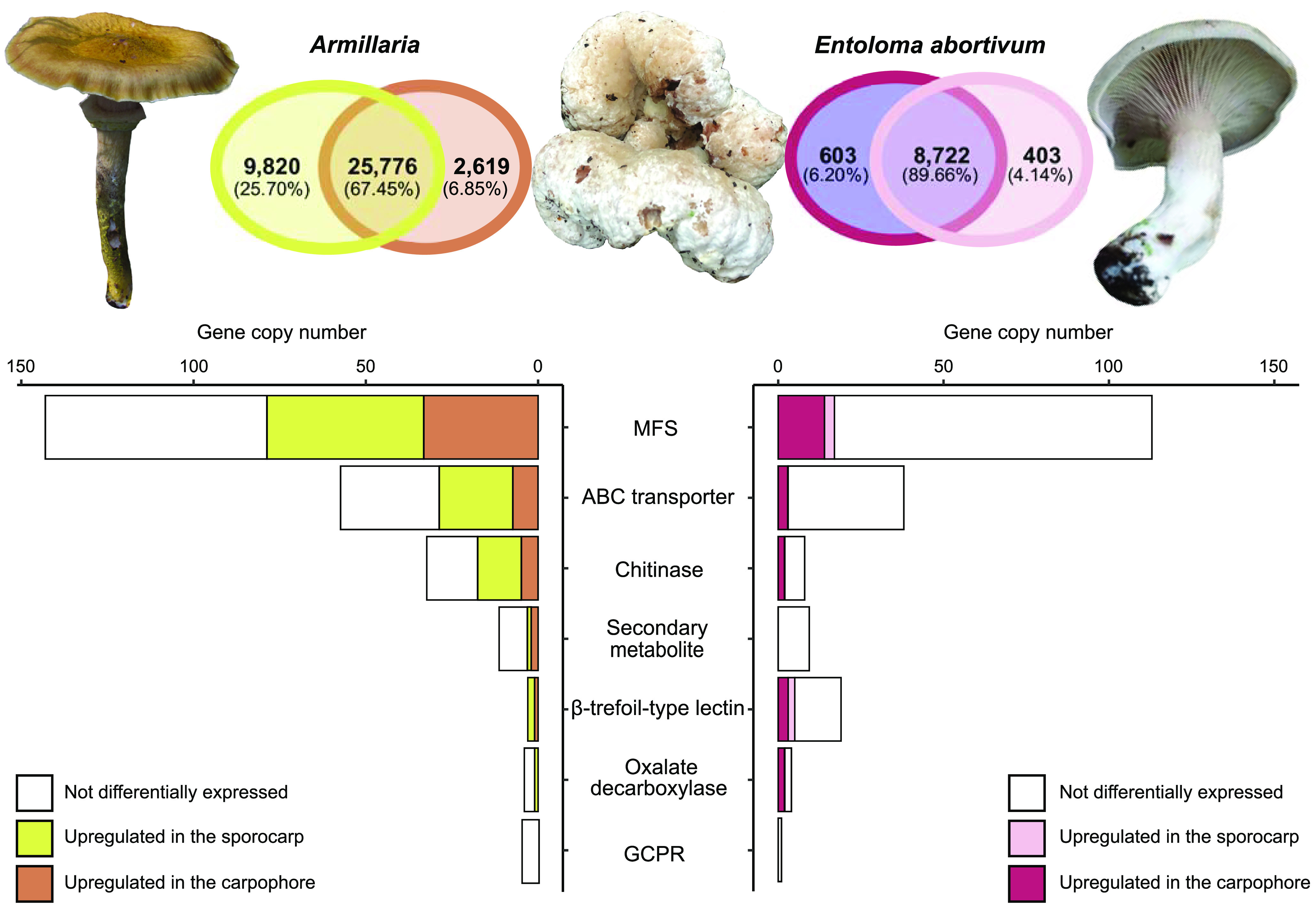
Differentially upregulated transcripts in the sporocarp and carpophoroid. (Top) Venn diagram showing the number of differentially upregulated transcripts between the sporocarp and carpophoroid in Armillaria and E. abortivum. Photo of E. abortivum courtesy of Eva Skific (© 2021 Skific; reproduced with permission). (Bottom) Bar graph showing the number of copies of genes important in mycoparasitic interactions. The white portion of the bar shows the number of these genes detected in the transcriptome but not differentially upregulated, while the darker colors (brown and purple, respectively) show the number of genes that are differentially upregulated by each species in the carpophoroid and the lighter colors (yellow and coral, respectively) show the number of these genes that are differentially upregulated by each species in their respective sporocarp.
TABLE 1.
Ten most abundant E. abortivum transcripts in sporocarps and carpophoroidsa
| Gene | Annotation | Cond. | C (TMM) | S (TMM) | logFC | q value |
|---|---|---|---|---|---|---|
| ENT_DN2762_c0_g3 | Oxalate decarboxylase | C | 68,724 | 238 | −8.5 | 4.5 × 10−317 |
| ENT_DN1212_c0_g1 | Hypothetical protein | C/S | 55,939 | 12,590 | −2.5 | 4.3 × 10−49 |
| ENT_DN3063_c0_g2 | Oxalate decarboxylase | C | 48,157 | 135 | −8.8 | 0 |
| ENT_DN1952_c0_g2 | Acid phosphatase | C | 19,999 | 677 | −5.2 | 1.3 × 10−169 |
| ENT_DN4045_c1_g1 | Hypothetical protein | C | 19,965 | 3,374 | −2.9 | 3.4 × 10−63 |
| ENT_DN5742_c0_g1 | Hypothetical protein | C | 11,229 | 23 | −9.3 | 6.9 × 10−220 |
| ENT_DN4332_c0_g1 | Hypothetical protein | C | 11,038 | 607 | −4.5 | 4.7 × 10−77 |
| ENT_DN1952_c0_g1 | Acid phosphatase | C | 10,766 | 673 | −4.4 | 1.9 × 10−122 |
| ENT_DN2936_c0_g2 | Hypothetical protein | C | 9,212 | 3,686 | NS | |
| ENT_DN3086_c0_g1 | Hypothetical protein | C | 9,076 | 1,563 | −2.9 | 2.2 × 10−62 |
| ENT_DN1375_c0_g1 | Trehalase | S | 1,934 | 17,890 | 2.9 | 6.5 × 10−37 |
| ENT_DN5247_c0_g3 | Hypothetical protein | S | 2,718 | 7,204 | NS | |
| ENT_DN761_c0_g2 | Hypothetical protein | S | 7,405 | 6,918 | NS | |
| ENT_DN521_c0_g2 | Hypothetical protein | S | 6,045 | 6,424 | NS | |
| ENT_DN852_c0_g1 | Hypothetical protein | S | 123 | 5,429 | 5.1 | 2.0 × 10−142 |
| ENT_DN3621_c0_g2 | RNA polymerase | S | 1,977 | 5,138 | NS | |
| ENT_DN6707_c0_g1 | Hypothetical protein | S | 1,654 | 5,027 | NS | |
| ENT_DN2379_c0_g1 | TPR-like protein | S | 4,862 | 5,017 | NS | |
| ENT_DN466_c0_g1 | Auxin efflux carrier | S | 1,205 | 4,333 | NS |
Cond., condition under which each gene was most abundant, referring to either the carpophoroids (C) or sporocarps (S). NS, not significantly differentially upregulated. Negative logFC values are significantly differentially upregulated in the carpophoroid, whereas positive values are significantly differentially upregulated in the sporocarp. Transcripts discussed in the text include two oxalate decarboxylases (ENT_DN2762_c0_g3 and ENT_DN3063_c0_g2), three β-trefoil-type lectins (ENT_DN4359_c0_g1, ENT_DN1877_c0_g1, and ENT_DN4255_c0_g1), three ABC transporters (ENT_DN1537_c0_g1, ENT_DN189_c0_g1, and ENT_DN3860_c0_G1), two chitinases (ENT_DN2096_c0_g1 and ENG_DN4507_c0_g3), and 14 MFS transcripts (ENT_DN409_c0_g1, ENT_DN1954_c0_g1, ENT_DN1998_c0_g1, ENT_DN2070_c0_g1, ENT_DN2744_c0_g1, ENT_DN3292_c0_g1, ENT_DN3474_c0_g1, ENT_DN3861_c0_g1, ENT_DN3943_c0_g1, ENT_DN3943_c0_g2, ENT_DN3981_c0_g1, ENT_DN4751_c0_g2, ENT_DN6588_c0_g1, and ENT_DN6695_c0_g1).
The assembled transcriptome of Armillaria was just over 138 million bp. A total of 63,905 contigs, an N50 value of 2,845, and 97.8% of the BUSCOs representative of the Agaricales were present in the transcriptome. Eleven ITS sequences were extracted from the transcriptome, accounting for 2.77 × 10−5% to 6.26 × 10−4% of the mapped reads; six ITS sequences were from Armillaria, while five were from an uncharacterized species of the yeast Kodamaea. A total of 38,215 unique gene models were recovered, and, after bioinformatic filtering against public Armillaria genomes, 29,936 of these represented orthologs of Armillaria species (Fig. 2). In total, 2,619 transcripts were differentially upregulated in the carpophoroid tissue compared to the sporocarp tissue, whereas 9,820 transcripts were differentially upregulated in the sporocarp tissue compared to the carpophoroid tissue (Fig. 3). The transcriptome contained 580 genes that code for CAZymes, with 34 of those coding for chitinases (Fig. 2). Genes detected in the Armillaria transcriptome that might be important in mycoparasitic interactions include 12 putative secondary metabolite gene clusters, five GCPRs, 59 ABC transporters, and 144 MFS transcripts (Fig. 3). The average TMM (normalized units of trimmed mean of m-values) of the 10 most highly upregulated Armillaria transcripts in the sporocarp ranged from 8,438 to 36,477. The most differentially upregulated transcript was annotated as a cell wall galactomannoprotein (Fig. 4 and 5; Table 2), while, notably, the 14th most differentially upregulated transcript in the sporocarp coded for isocitrate lyase (Table 2; Fig. 5).
FIG 4.

Volcano plots. Each dot represents a transcript plotted according to its log2 fold change and the −log10 of its P value. Transcripts with the highest statistical significance and the largest fold change are represented by dots toward the top of the plot that are far to either the left (carpophoroid) or right (sporocarp) side. All transcripts with a q value of <0.05 are shown. Black dots represent a gene with a nonsignificant logFC (−2 < logFC < 2). (A) Armillaria. (B) E. abortivum. Dots with a black circle around them are annotated according to the abbreviations: OD, oxalate decarboxylase; CWG, cell wall galactomannoprotein.
FIG 5.

Genes in both Armillaria and E. abortivum that are putatively important in the recognition and defense responses during this mycoparasitic interaction. (A) An Armillaria cell wall galactomannan that is differentially upregulated in the sporocarp; (B) three E. abortivum β-trefoil-type lectins that are differentially upregulated in the carpophoroid; (C) an Armillaria transcript that codes for isocitrate lyase that is differentially upregulated in its sporocarps; (D) two E. abortivum transcripts that code for oxalate decarboxylases that are differentially upregulated in the carpophoroids. All results are shown as means ± standard errors of the means. TMM, normalized units of trimmed mean of m-values.
TABLE 2.
Ten most abundant Armillaria transcripts in sporocarps and carpophoroidsa
| Gene | Annotation | Cond. | C (TMM) | S (TMM) | logFC | q value |
|---|---|---|---|---|---|---|
| ARM_DN1755_c2_g1 | Cell wall galactomannoprotein | S | 0 | 36,479 | 13.5 | 4.0 × 10−182 |
| ARM_DN3840_c0_g1 | Serine carboxypeptidase | S | 12 | 29,472 | 6.7 | 7.9 × 10−100 |
| ARM_DN22943_c1_g1 | Rab geranylgeranyltransferase | S | 21 | 18,649 | 5.1 | 3.8 × 10−89 |
| ARM_DN1737_c0_g1 | LysM-domain-containing protein | S | 0 | 14,557 | 15.9 | 3.0 × 10−159 |
| ARM_DN1699_c3_g1 | Hypothetical protein | S | 9 | 12,848 | 5.8 | 1.6 × 10−94 |
| ARM_DN1314_c0_g1 | Chondroitin AC/alginate lyase | S | 102 | 11,436 | 2.2 | 3.7 × 10−25 |
| ARM_DN20980_c0_g1 | Glycopeptide | S | 0 | 10,449 | 15.4 | 3.4 × 10−139 |
| ARM_DN8135_c0_g1 | Rasp f 7 allergen | S | 0 | 9,014 | 11.5 | 1.8 × 10−58 |
| ARM_DN4971_c0_g2 | Aldehyde dehydrogenase | S | 103 | 8,688 | NS | |
| ARM_DN1205_c0_g1 | Hypothetical protein | S/C | 9,049 | 8,348 | −4.8 | 2.1 × 10−74 |
| ARM_DN5170_c0_g1 | Hypothetical protein | C | 2,086 | 377 | −7.1 | 6.9 × 10−150 |
| ARM_DN23207_c0_g2 | Senescence associated | C | 1,711 | 2,482 | −4.1 | 6.6 × 10−61 |
| ARM_DN996_c0_g1 | Senescence associated | C | 1,423 | 1,090 | −5.0 | 3.0 × 10−73 |
| ARM_DN409_c1_g1 | Elongation factor 1-alpha | C | 769 | 1,154 | −4.1 | 4.0 × 10−155 |
| ARM_DN1893_c0_g1 | Hypothetical protein | C | 722 | 58 | −8.3 | 5.3 × 10−109 |
| ARM_DN693_c0_g1 | Hypothetical protein | C | 520 | 840 | −3.9 | 1.1 × 10−48 |
| ARM_DN1222_c0_g1 | CYS3-cystathionine gamma-lyase | C | 485 | 717 | −4.1 | 5.5 × 10−27 |
| ARM_DN1146_c0_g4 | Heat shock protein 70 | C | 431 | 2,361 | −2.2 | 1.7 × 10−54 |
| ARM_DN3800_c0_g1 | ATP synthase F1 | C | 300 | 4,686 | NS |
Cond., condition under which each gene was most abundant, referring to either the carpophoroids (C) or sporocarps (S). NS, not significantly differentially upregulated. Negative logFC values are significantly differentially upregulated in the carpophoroid, whereas positive values are significantly differentially upregulated in the sporocarp. Transcripts discussed in the text were an isocitrate lyase (ARM_DN1_c7_g1), two putative senescence-associated proteins (ARM_DN23207_c0_g2 and ARM_DN996_c0_g1), and a heat shock protein (ARM_DN1146_c0_g4).
Metatranscriptomic analysis of combined fungal hyphae in carpophoroid tissue.
We sequenced the metatranscriptome of the mixed tissue in the carpophoroids that are typically found when E. abortivum and species of Armillaria are found in proximity. In the carpophoroid tissue, significantly more transcriptomic reads from E. abortivum were identified than from Armillaria sp. (t[8] = 16.6, P = 1.77 × 10−7, n = 9 replicates per species) (Fig. 6). The average number of E. abortivum mapped reads in the carpophoroid tissue was 2,613,988, while the average number of Armillaria mapped reads was 74,880 (Fig. 6).
FIG 6.

Boxplots of number of carpophoroid reads that mapped to Armillaria and E. abortivum when MAPQ (MAPping Quality) was 30. Individual data points are indicated for each species with an open circle. The continuous line within each box represents the mean number of mapped reads. Species labeled with different letters (a to b) have a statistically significant (P < 0.05) different number of mapped reads in the carpophoroid.
The average TMM of the top 10 most abundant E. abortivum transcripts in the carpophoroid ranged from 9,075 to 68,720 (Table 1), while the average TMM of the top 10 most abundant Armillaria transcripts in the carpophoroid ranged from 300 to 9,049 (Table 2). The first and third most highly upregulated E. abortivum transcripts in the carpophoroid tissue code for two oxalate decarboxylases, both of which were differentially upregulated in the carpophoroid tissue compared to the sporocarp (Fig. 3 to 5 and Table 1). There were other differentially upregulated E. abortivum transcripts in the carpophoroid that were not as highly upregulated that may also play a role in mycoparasitism. These include three β-trefoil-type lectins, three ABC transporters, two chitinases, and 14 MFS transcripts (Fig. 3; Table 1). We were unable to detect any transcripts belonging to secondary metabolite gene clusters that were differentially upregulated in the carpophoroids (Fig. 3). Two Armillaria transcripts coding for putative senescence-associated proteins, as well as a heat shock protein associated with cell death, were differentially upregulated in the carpophoroid compared to the sporocarp (Table 2).
Phylogenetic placement of Armillaria reads.
Phylogenomic analysis of 100 Armillaria BUSCOs generated in this study, in conjunction with previously published Armillaria genomes, shows a strongly supported sister relationship with an Armillaria mellea specimen from France (100% bootstrap support [BS]) (Fig. 7A). Phylogenetic analysis of all ITS sequences characterized as A. mellea from GenBank shows that this specimen is conspecific with specimens from eastern North America (Fig. 7B).
FIG 7.

Phylogenetic placement of the Armillaria species used in this analysis. (A) Maximum likelihood phylogeny of Armillaria species and Guyanagaster necrorhizus generated from the analysis of 100 random BUSCOs. The outgroup taxon is Cylindrobasidium torrendii. Each node is fully supported with 100% bootstrap support. (B) Maximum likelihood phylogeny of Armillaria mellea phylogenetic tree generated from the analysis of the ITS region. Branches with 70% or more bootstrap support are thickened. Outgroup taxa include A. gemina, A. luteobubalina, A. puiggarii, and A. sinapina. The Armillaria specimen analyzed during this study is in bold and yellow in both phylogenies.
DISCUSSION
The formation of carpophoroids associated with species of E. abortivum has traditionally been thought to be the result of an Armillaria species attacking and parasitizing Entoloma sporocarps (14), hypothesized on the basis that Armillaria species are widespread generalist forest pathogens that have a broad range of host plants (15). However, subsequent studies suggested the opposite: the production of carpophoroids is the result of E. abortivum disrupting the development of Armillaria sporocarps (17). Here, we employed RNA sequencing and differential gene expression analysis on field-collected fungal tissue of each of the three components of this association to better understand the mechanistic basis of this interaction. We determined that E. abortivum reads in the metatranscriptome of the carpophoroid tissue—which can be interpreted as a measure of living tissue—are almost 35 times more abundant than A. mellea reads (Fig. 6). This finding suggests that carpophoroids are structures that result from E. abortivum parasitizing, and eventually killing, its Armillaria host under natural conditions.
Fungal-fungal necrotrophic mycoparasitic interactions are multistage processes that are best studied in model species, such as those in the genera Trichoderma, Coniothyrium, Clonostachys, and Tolypocladium (20). Genomic and transcriptomic studies of necrotrophic mycoparasites show a convergence of significant genetic mechanisms at each stage (20). The E. abortivum genes that are differentially upregulated in the carpophoroid tissue are largely consistent with other examples of necrotrophic mycoparasites in the Ascomycota. In the carpophoroid tissues we analyzed, E. abortivum appears to employ much of its energy on recognition and defense responses (Fig. 5). Inversely, the Armillaria sporocarps we analyzed illuminate possible mechanisms by which these two species recognize one another and how Armillaria responds to parasitism (Fig. 5).
The genetics of the Entoloma-Armillaria mycoparasitic interaction.
A crucial step in a successful mycoparasite’s life history is the ability to sense its host. Genes involved in the recognition of the fungal prey include those that code for GPCRs (18, 20). However, we did not find any of these genes that were differentially upregulated by E. abortivum in the carpophoroid tissue (Fig. 3). Given the significantly lower number of Armillaria reads in the carpophoroid tissue compared to E. abortivum, we presume that these carpophoroids are relatively advanced in age, and expression of the genes used for sensing the presence of the host is no longer necessary.
We also identified three E. abortivum transcripts that code for β-trefoil-type lectins—proteins that bind to galactose units of sugar chains (23)—that were differentially upregulated in the carpophoroid tissue (Fig. 3 and 5). In well-studied mycoparasitic interactions, the recognition, attachment, and coiling around a fungal substrate are mediated by lectins expressed by at least one of the fungal partners (24–26). More specifically, basidiomycete β-trefoil-type lectins have previously been shown to play a role in the recognition of nonself glycans (27). Interestingly, the most abundant and differentially upregulated transcript produced in the Armillaria sporocarps, the substrate to which E. abortivum hyphae attach, codes for a cell wall galactomannoprotein (Fig. 4 and 5). These proteins belong to a group of glycans which consist of a mannose backbone with galactose side chains and are known to make up a major part of the cell wall of some fungal species (28). This particular galactomannoprotein appears to be specific to Armillaria species. Watling (14) commented on the highly specific nature of this interaction and that it has been documented only occurring between E. abortivum and Armillaria species. One possible mediator of the specificity of this interaction could be the galactose sugars on the mannose protein (known thus far only from Armillaria species) that are the means by which E. abortivum β-trefoil-type lectins recognize and attach to the Armillaria host. However, more genome sequencing of other Agaricales species is needed to determine whether this protein is truly specific to species in the genus Armillaria.
During mycoparasitic interactions, the fungal host responds by mounting its own defense, and a successful mycoparasite must be able to cope with this counterattack (18, 20). Oxalic acid (OA) is a virulence factor employed by some plant pathogens, including species of Armillaria, to compromise the defense responses of the host plant by creating an acidic environment (29, 30). One differentially upregulated Armillaria transcript in the sporocarps codes for isocitrate lyase (Fig. 5), which is involved in OA biosynthesis in other fungal pathogens (31, 32), and we suspect that Armillaria sporocarps may utilize OA to defend against the initial infection by E. abortivum. However, two of the most abundant and differentially upregulated E. abortivum transcripts in the carpophoroid tissue code for oxalate decarboxylases—enzymes responsible for the degradation of OA (Fig. 3, 4, and 5D; Table 1). In at least one known well-studied mycoparasitic interaction, OA is secreted by the fungal host, Sclerotinia sclerotiorum, in reaction to penetration by its mycoparasite, Coniothyrium minitans. The acidic environment created by the secreted OA inhibits conidial germination and suppresses mycelial growth of C. minitans (33). However, C. minitans nullifies the growth-suppressing effects of OA or OA-mediated low pH by degrading the OA (34, 35), an enzymatic process largely mediated by oxalate decarboxylase. Because of this, oxalate decarboxylase plays an imperative role in mycoparasitism as OA degradation is vital for infection of the fungal host (36). Given the abundance of these genes produced by E. abortivum in the carpophoroid, we suspect a similar scenario in this mycoparasitic interaction. Subsequent downregulation of isocitrate lyase by Armillaria in the carpophoroids suggests that its genetic defense responses to infection were likely over at the time of our sampling.
Other ways that mycoparasites cope with the counterattack launched by their host include actively excreting host-secreted toxins. Here, we hypothesize that active extrusion of toxins secreted by the host occurs in the E. abortivum carpophoroid tissue via membrane transporters in the ABC superfamily (37–39). Three ABC transporters were differentially upregulated in the carpophoroid tissue (Fig. 3). Another group of genes that were differentially upregulated by E. abortivum in the carpophoroid belong to the major facilitator superfamily (MFS) transporters (Fig. 3). In C. rosea, there was selection for genes in this family that were related to drug resistance and the transport of secondary metabolites, small organic compounds, and carbohydrates (40). Their importance to mycoparasitism in C. rosea is predicted to invoke efflux-mediated protection against exogenous or endogenous secondary metabolites and nutrient uptake (40). MFS transporters have also been shown to be induced in other mycoparasitic species (19, 41), but their exact biological roles have not been investigated.
In mycoparasitism, the final death of the host often results from the synergistic actions of cell wall-hydrolytic enzymes and antifungal secondary metabolites (18, 20). No secondary metabolite gene clusters identified in the E. abortivum transcriptome were differentially upregulated in the carpophoroid tissue (Fig. 3). In some mycoparasitic relationships, the secretion of secondary metabolites occurs early in the interaction, including in Escovopsis weberi, which secretes toxic compounds that kill the leafcutter ant garden before contact (5). In culture experiments between Armillaria isolates and E. abortivum, the growth of Armillaria was severely inhibited by the presence of E. abortivum (42). This suggests that E. abortivum may potentially secrete a toxic compound early in the interaction that inhibits the growth of Armillaria. Given that significantly more of the living tissue in the carpophoroids belonged to E. abortivum (Fig. 6), it is possible that much of the Armillaria tissue was killed preceding the full development of the carpophoroid.
Additionally, we hypothesize that the upregulated β-trefoil-type lectin in E. abortivum that may be important in hyphal recognition may also be cytotoxic toward Armillaria. This type of lectin has sequence homology, as well as putative structural similarity, to the B-subunit of ricin, a toxic protein from the castor bean Ricinus communis (43). An array of β-trefoil-type lectins have been characterized from the sporocarps of the mushroom-forming species Clitocybe nebularis (44), Coprinus cinerea (27), Macrolepiota procera (45), and Boletus edulis (46). Besides being important for nonself recognition, these same mushroom lectins also exhibit entomotoxic activity (47) as well as nematoxic activity (27, 45, 48). Taken together, it is possible that the E. abortivum β-trefoil-type lectins may also function as toxins toward Armillaria. While the E. abortivum transcripts coding for these lectins are not in the highest abundance in the carpophoroid tissue (Fig. 5), this could be because most of the Armillaria sporocarp tissue is already dead and the potential lethal effects produced by them are no longer necessary.
Chitin is an essential polymer in fungal cell walls (49) and is an important target during mycoparasitic attack (20). Indicative of the importance of chitinases in mycoparasitic interactions, members of the genus Trichoderma, as well as Tolypocladium ophioglossoides and Escovopsis weberi, have an increased number of genes coding for them (19, 50–53). Nine fungal chitinases were detected in the transcriptome of E. abortivum, which is fewer than the 13, 19, 20, and 29 detected in the closely related species Tricholoma matsutake (54), Clitocybe gibba (55), Lyophyllum atratum (56), and Lepista nuda (55), respectively (Fig. 2). Only two of those E. abortivum chitinases were differentially upregulated in the carpophoroid tissue (Fig. 3) and were not abundant in comparison to other genes, suggesting minimal significance at this stage in carpophoroid development. One possibility for this difference in abundance could be the result of the putatively acidic pH in the carpophoroid that we infer based on the high gene expression of oxalate decarboxylases. In C. minitans, chitinase activity is positively correlated with ambient pH ranging from 3 to 8 (57), so it is possible that chitinase activity in E. abortivum will increase after a neutral pH is restored. Another possibility for the low abundance is that most of the Armillaria host tissue was already broken down, reducing the need for chitinase activity.
Some of the putatively mycoparasitism-related genes outlined above were also differentially upregulated by Armillaria in the carpophoroid tissue. These include genes that code for MFS, ABC transporters, chitinases, and secondary metabolite gene clusters (Fig. 3). This suggests that Armillaria may be using many of the same genetic mechanisms to defend itself against parasitism by E. abortivum. Additionally, the degree of expression changes—in both the number of differentially upregulated transcripts and the log fold change (logFC)—between the sporocarp and carpophoroid is much greater in Armillaria than in E. abortivum (Fig. 3 and 4), which could reflect an increase in the level of defense from Armillaria. However, this defense is apparently not enough to overcome the parasitic adaptations of E. abortivum.
Gene and CAZyme content of E. abortivum.
The number of predicted gene models in the transcriptome of E. abortivum was 9,728, which is markedly fewer than the number of gene models in the genomes of its closest sequenced relatives (Fig. 2, middle panel). Additionally, relative to other closely related mushroom species, E. abortivum also exhibits a strong reduction in several gene families encoding CAZymes (Fig. 2, right panel) and contains no cellobiohydrolases, xylanases, or polysaccharide monooxygenases. This finding is consistent with what has been observed in obligate mycoparasites and animal pathogens, which also have reduced CAZyme repertoires compared to fungi that utilize other nutritional strategies, hypothesized to be the result of their highly specialized interactions with their hosts (50, 58, 59). Therefore, it is possible that E. abortivum retained only the CAZymes and accessory genes necessary to interact with Armillaria species. While a genome sequence of E. abortivum will be necessary to confirm this reduction, the BUSCO analysis verified that the E. abortivum transcriptome contains nearly 95% of the core set of eukaryotic genes, which suggests that our transcriptome is relatively complete, and it is unlikely that entire gene families are underrepresented.
Another possibility for the reduction in CAZymes could be explained by a broader nutritional strategy employed by Entoloma species, some of which form ectomycorrhiza-like structures on host plant species (60–62). Ectomycorrhizal species have a marked reduction in CAZymes in comparison to their saprotrophic ancestors (63), which we also observe with Tricholoma matsutake (Fig. 2, right panel). However, microscopic analyses of Entoloma ectomycorrhiza-like structures suggest that some species destroy root meristems and young root cells, suggestive of a more parasitic relationship (60, 61). One explanation is that Entoloma species, in general, are parasites of true ectomycorrhizae. More broadly, other species within the Entolomataceae are suspected mycoparasites, as they have been reported growing in close association with the sporocarps of other species (reviewed in reference 17), including Entoloma parasiticum (=Claudopus parasiticus) and Clitopilus fasciculatus (64, 65). This explanation would also add credence to the evidence that Entolomataceae species are difficult to culture and are slow growing (62). Additional research utilizing genomes and laboratory studies to understand the nutritional strategy employed by this lineage will inform us as to whether fungal parasitism in this group is more common than it is currently understood to be. Lastly, it should be noted that Entoloma species, and E. abortivum in particular, form sclerotia in culture and presumably in soil (14, 17). These resting structures are perhaps a dormancy mechanism in the soil to survive seasonality when host tissue is unavailable. Because the timing and presence of E. abortivum fruitings have been hard to predict, we were not able to observe or acquire a transcriptome for the sclerotia in the field.
Gene content and identity of Armillaria species in this interaction.
The number of predicted gene models in the transcriptome of this Armillaria species was 38,215 (Fig. 2, middle panel). This is substantially greater than all other sequenced Armillaria species, which range from 14,473 to 25,704 gene models (22, 66). However, when we looked for gene models in our transcriptome that belong to known Armillaria species, this reduced the total number of gene models to 29,936 (Fig. 2, middle panel). The excess gene models in the Armillaria transcriptome, compared to reference genomes, likely represent duplicated gene models with splice variants, a common artifact of transcriptome sequencing (67). Additionally, nearly one-quarter of the gene models in the Armillaria sporocarps were from organisms other than Armillaria, including the yeast Kodamaea, highlighting the fact that field-collected sporocarps are not composed of tissue from a single organism. However, none of these contaminating organisms had genes that were both abundant and differentially upregulated that we predicted to play a role in this interaction.
Phylogenomic analysis of the Armillaria transcripts generated in this study suggests that the specific Armillaria species parasitized in this relationship is sister to an A. mellea specimen collected from western Europe (Fig. 7A). An ITS-based phylogenetic analysis shows the Armillaria specimen collected in this study is conspecific with other A. mellea collections from eastern North America (Fig. 7B). Before now, observations of A. mellea sensu stricto fruiting in proximity to carpophoroids (17) hinted that it may be a host to E. abortivum. Here, though, we show for the first time using genomic data that A. mellea sensu stricto can definitively serve as a host for E. abortivum. However, our hypothesis is that this interaction does not appear specific to just A. mellea sensu stricto as Armillaria gallica, Armillaria ostoyae, Armillaria jezoensis, Armillaria sp. Nag. E, and Desarmillaria tabescens have been previously confirmed as hosts in this interaction as well (17, 42, 68). However, a broad geographic sampling of carpophoroids using molecular markers or genomic information could address this question more thoroughly. Interestingly, Armillaria species parasitized by E. abortivum appear to be only those present in eastern North America and eastern Asia (17, 42, 68).
Conclusions.
Data from this study support the hypothesis that E. abortivum is a mycoparasite of Armillaria sporocarps. Three β-trefoil-type lectins are differentially upregulated by E. abortivum in the carpophoroid tissue, and we propose that these lectins mediate recognition with Armillaria sporocarps through binding to an Armillaria-specific galactomannoprotein. We hypothesize that by using oxalate decarboxylase, E. abortivum is likely defending against the secretion of OA by Armillaria. These strategies employed by E. abortivum for recognition and defense are similar to mechanisms utilized by other mycoparasites, suggesting that even distantly related mycoparasites utilize similar genetic mechanisms to mediate mycoparasitic interactions. One weakness of this study is that we were limited to the carpophoroid life stage that was available at the point of collection, which led us to speculate about what is occurring during other stages of mycoparasitism (i.e., sensing the host, initiating the interaction, and killing and consuming the host). Therefore, future studies using culture methods, isotopic analysis, and metatranscriptomics of naturally collected carpophoroids at different life stages (i.e., younger and older carpophoroid specimens) will be necessary to completely tease apart the putative mycoparasitic strategies employed by E. abortivum and the defense responses by the Armillaria host. Finally, given that Armillaria species are pathogens in both natural and agronomic systems, a better understanding of this interaction may lead to the development of biocontrol methods for the control of Armillaria root rot.
MATERIALS AND METHODS
Sample collection, preparation, and sequencing.
Sporocarps of Armillaria sp., E. abortivum, and the mixed-tissue carpophoroids were observed fruiting in proximity to one another on 18 September 2015 within the Baker Woodlot and Rajendra Neotropical Migrant Bird Sanctuary, Michigan State University, East Lansing, MI (42°42′56.4″ N, 84°28′34.4″ W) (collection accession no. JRH 2446). Entire sporocarps were collected, immediately flash frozen in liquid nitrogen, and subsequently stored at −80°C. At the time of processing, three biological replicates of each of the three tissue types (Armillaria sp. sporocarp, E. abortivum sporocarp, and carpophoroid) were individually ground in liquid N2. Total RNA was then extracted from the ground tissue using the Qiagen RNeasy kit (Qiagen Inc., Hilden, Germany) according to the manufacturer’s protocol. RNA concentration and quality for each of the samples were assessed on a DeNovix DS-11 FX spectrophotometer (DeNovix Inc., Wilmington, DE, USA) and then shipped directly to the University of Minnesota’s Genomics Center (https://genomics.umn.edu). Three technical replicates were sequenced for each biological replicate. Transcriptomic and metatranscriptomic libraries were constructed with the TruSeq standard total RNA library preparation kit with Ribo-Zero ribosomal reduction following the protocol developed by Schuierer et al. (69). Nucleotide sequencing was performed on the Illumina HiSeq 2500 system (Illumina Inc., San Diego, CA, USA), and paired-end RNA sequence reads of 51 bp were generated for further analysis.
De novo transcriptome assembly, transcript abundance estimation, and gene expression analysis.
The quality of the raw reads was assessed using FastQC version 0.11.9 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc). The range of the number of reads for each condition is as follows: E. abortivum sporocarps ranged from 10,584,302 to 14,473,328, Armillaria sporocarps ranged from 11,712,320 to 12,431,979, and the carpophoroids (containing reads from both organisms) ranged from 9,146,682 to 12,852,086. Sequencing adaptors were trimmed, and PhiX contaminants were filtered for each sample using BBDuk (https://jgi.doe.gov/data-and-tools/bbtools/bb-tools-user-guide/bbduk-guide/). Prior to transcriptome assembly, k-mer hash sizes were estimated with khmer (70). De novo assemblies were constructed independently for both Armillaria sp. and E. abortivum with Trinity version 2.11.0 (71) using the trimmed reads generated from the respective sporocarp reads. Assembly statistics for both transcriptomes were generated with QUAST version 5 (72), and transcriptome completeness was assessed by determining the percentage of sequenced BUSCOs in each (73).
The results of the de novo transcriptome assemblies were used as references to perform sample-specific expression analysis. The trimmed sporocarp reads from each of the nine replicates were mapped against their respective reference transcriptomes using Bowtie 2 (74) followed by calculation of abundance estimates using RSEM (75). The trimmed carpophoroid reads were also subsequently mapped, following the same protocol as described above, to both the Armillaria sp. and E. abortivum transcriptomes. Because of the close phylogenetic relatedness between these two species, and to filter out poorly aligned reads, we retained only mapped reads for all samples that had a MAPQ (MAPping Quality) value of 30 and above, which is equivalent to reads that have a 99.9% chance of hitting the correct match. The R package edgeR (76) and “trimmed mean of m-value” (TMM) normalization (77) were used to determine differentially upregulated transcripts between (i) Armillaria sporocarps and carpophoroids and (ii) E. abortivum sporocarps and carpophoroids. Transcripts were considered differentially upregulated if they had a logFC of two or greater and a false-discovery rate (FDR)-adjusted P value, or q value, of <0.05. All statistical analyses for the packages listed above were conducted using R version 4.0.3 (http://www.r-project.org/).
We used SAMtools (78) to determine the number of reads from the carpophoroids that mapped to our reference transcriptomes of E. abortivum and our particular Armillaria species. To understand whether the number of reads that mapped to the carpophoroids differed significantly between each fungal species, we performed an F test of equality of variances and then a two-tailed t test assuming unequal variance with a P value of <0.05 denoting significance.
Sporocarp transcriptome and carpophoroid metatranscriptome annotation.
We annotated the Armillaria sp. and E. abortivum transcriptomes using Trinotate version 3.2 (79). Briefly, the transcripts were translated to coding protein sequences using TransDecoder version 5.5.0 (http://transdecoder.github.io) following identification of the longest open reading frames. To identify the most likely homologous sequence data, we used blastx on the transcripts and blastp on the predicted protein sequences (80). Using the predicted protein sequences, we also ran an HMMER (81) search against the PFAM database (82) to identify conserved domains that might be suggestive of function. We also compared these results to currently curated annotation databases such as Gene Ontology (GO) (83) and Kyoto Encyclopedia of Genes and Genomes (KEGG) (84–86). We used dbCAN2 (87) to annotate the CAZymes present in both species and compared their CAZy content to other closely related Agaricales species (22, 54–56), along with antiSMASH version 5.0 (88) to identify transcripts that belong to secondary metabolite gene clusters for both Armillaria and E. abortivum.
Finally, because this work was based off de novo transcriptome assemblies, there was the possibility that transcripts from contaminating organisms, including insects, yeasts, and other fungi, were also present and might be confounding the results. We approached this issue multiple ways. First, we extracted all internal transcribed spacer (ITS) regions from both transcriptomes, including the regions 18S, ITS1, 5.8S, ITS2, and 28S, using ITSx (89), and identified the origin of those sequences. To verify the number of mapped reads that represent rRNA, we also calculated the proportion of reads that mapped to the transcripts that contained the ITS sequences to the total number of mapped reads. Second, because high-quality genome assemblies of Armillaria exist (22, 66), we used Exonerate (90) to determine how many of the gene models in our Armillaria transcriptome are orthologs of A. gallica—the Armillaria species with the largest number of known gene models. Finally, all genes that we report on as important to this interaction were manually verified to be from the target organism—Armillaria or E. abortivum—using the NCBI blastx tool (80) against the nonredundant protein sequences database and confirming a hit from the Physalacriaceae or Entolomataceae/Lyophyllaceae, respectively.
Phylogenetic analysis of Armillaria transcripts.
In order to identify the specific species of Armillaria associated in this relationship, we identified BUSCOs (73) from the transcriptome of our Armillaria sporocarps along with other Armillaria and Physalacriaceae species with previously sequenced genomes (22, 66, 91, 92). We randomly selected 100 BUSCOs to reconstruct a phylogenomic tree from the six Armillaria specimens (22, 66), Guyanagaster necrorhizus (92), and Cylindrobasidium torrendii (91), which served as the outgroup. Protein-coding sequences were aligned using MAFFT version 7 (93), and noninformative sites and nonaligning regions were trimmed with Gblocks (94). The 100 BUSCOs were concatenated into a supermatrix with 64,436 sites. This supermatrix was used to infer a species tree and branch support using RAxML-NG (95), using a partitioned WAG+G model, where each data partition represented an individual BUSCO.
To expand on the phylogenomic analysis above, we used the representative Armillaria ITS sequence obtained from ITSx, and given the close relationship of our Armillaria species to A. mellea, we pulled all A. mellea ITS sequences from GenBank that included associated location metadata (96–110) (see Table S1 in the supplemental material). These sequences were aligned using MAFFT version 7 (93), with refinements to the alignment performed manually. RAxML-NG (95) was used to reconstruct this phylogeny. Taxa used to root this phylogeny included Armillaria gemina, Armillaria sinapina, Armillaria puiggarii, and Armillaria luteobubalina—all members of the sister lineage to A. mellea fide (16).
Location and GenBank accession numbers of Armillaria mellea sequences used in the phylogenetic analysis. Download Table S1, DOCX file, 0.02 MB (24.2KB, docx) .
Copyright © 2021 Koch and Herr.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Data and code availability.
The raw reads and assembled transcriptomes generated during this study have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO series accession number GSE183699 or under the NCBI BioProject accession no. PRJNA761704. All other associated data, including analysis code and information on voucher collections, are available at https://github.com/HerrLab/Koch_Arma-Ento_2021.
Supplementary Material
ACKNOWLEDGMENTS
We give special thanks to Ben Lemmond and Eva Skific for providing photos of this system and to Greg Bonito at Michigan State University for generously storing sporocarp and carpophoroid tissue until we were able to transport the tissue to our laboratory. We also thank three anonymous reviewers who provided feedback and comments on the initial submission of the manuscript.
This work was completed using the Holland Computing Center of the University of Nebraska, which receives support from the Nebraska Research Initiative. This research was directly supported by startup funding from the University of Nebraska Agricultural Research Division and the University of Nebraska Office of Research and Economic Development. Additionally, J.R.H. acknowledges funding from the U.S. National Air and Space Administration (grant 80NSSC17K0737), the U.S. National Science Foundation (EPSCoR grant 1557417), and the U.S. National Institute of Justice (grant 2017-IJ-CX-0025), all of which indirectly supported this research through the support of research in his laboratory. Funding agencies had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
On behalf of all authors, the corresponding author states that there is no conflict of interest.
J.R.H. initiated the work and sampled field collections; R.A.K. extracted RNA from all the tissue samples and performed the laboratory work; R.A.K. and J.R.H. processed the experimental data, analyzed the data, designed the figures, and drafted the manuscript.
Contributor Information
Joshua R. Herr, Email: jherr@unl.edu.
Jeffrey Blanchard, University of Massachusetts Amherst.
Todd Osmundson, University of Wisconsin-La Crosse.
Dan Lindner, US Forest Service.
REFERENCES
- 1.Zeilinger S, Omann M. 2007. Trichoderma biocontrol: signal transduction pathways involved in host sensing and mycoparasitism. Gene Regul Syst Bio 1:227–234. doi: 10.4137/grsb.s397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sun Z-B, Li S-D, Ren Q, Xu J-L, Lu X, Sun M-H. 2020. Biology and applications of Clonostachys rosea. J Appl Microbiol 129:486–495. doi: 10.1111/jam.14625. [DOI] [PubMed] [Google Scholar]
- 3.Quandt CA, Kepler RM, Gams W, Araújo JPM, Ban S, Evans HC, Hughes D, Humber R, Hywel-Jones N, Li Z, Luangsa-Ard JJ, Rehner SA, Sanjuan T, Sato H, Shrestha B, Sung G-H, Yao Y-J, Zare R, Spatafora JW. 2014. Phylogenetic-based nomenclatural proposals for Ophiocordycipitaceae (Hypocreales) with new combinations in Tolypocladium. IMA Fungus 5:121–134. doi: 10.5598/imafungus.2014.05.01.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Currie CR, Mueller UG, Malloch D. 1999. The agricultural pathology of ant fungus gardens. Proc Natl Acad Sci USA 96:7998–8002. doi: 10.1073/pnas.96.14.7998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reynolds HT, Currie CR. 2004. Pathogenicity of Escovopsis weberi: the parasite of the attine ant-microbe symbiosis directly consumes the ant-cultivated fungus. Mycologia 96:955–959. doi: 10.2307/3762079. [DOI] [PubMed] [Google Scholar]
- 6.Rogerson CT, Samuels GJ. 1994. Agaricicolous species of Hypomyces. Mycologia 86:839–866. doi: 10.2307/3760597. [DOI] [Google Scholar]
- 7.Zugmaier W, Bauer R, Oberwinkler F. 1994. Mycoparasitism of some Tremella species. Mycologia 86:49–56. doi: 10.2307/3760718. [DOI] [Google Scholar]
- 8.Zamora JC, Millanes AM, Wedin M, Rico VJ, Pérez-Ortega S. 2016. Understanding lichenicolous heterobasidiomycetes: new taxa and reproductive innovations in Tremella s.l. Mycologia 108:381–396. doi: 10.3852/15-090. [DOI] [PubMed] [Google Scholar]
- 9.Redhead SA, Ammirati JF, Walker GR, Norvell LL, Puccio MB. 1994. Squamanita contortipes, the Rosetta Stone of a mycoparasitic agaric genus. Can J Bot 72:1814–1828. [Google Scholar]
- 10.Griffith GW, Piotr Gajda K, Detheridge AP, Douglas B, Bingham J, Turner A, Bowmaker V, Evans DA, McAdoo WG, Dentinger BTM. 2019. Strangler unmasked: parasitism of Cystoderma amianthinum by Squamanita paradoxa and S. pearsonii. Fungal Ecol 39:131–141. doi: 10.1016/j.funeco.2018.11.012. [DOI] [Google Scholar]
- 11.Weber R, Webster J. 1996. Volvariella surrecta: an uncommon mycoparasite. Mycologist 10:160. doi: 10.1016/S0269-915X(96)80008-5. [DOI] [Google Scholar]
- 12.Both EE. 2006. Personal encounters with the parasitic bolete. Field Mycol 7:1–7. [Google Scholar]
- 13.Buller A. 1924. Researches on fungi. Longmans, London, United Kingdom. [Google Scholar]
- 14.Watling R. 1974. Dimorphism in Entoloma abortivum. Bull Soc Linn Lyon Num Spec 43:449–470. [Google Scholar]
- 15.Baumgartner K, Coetzee MPA, Hoffmeister D. 2011. Secrets of the subterranean pathosystem of Armillaria. Mol Plant Pathol 12:515–534. doi: 10.1111/j.1364-3703.2010.00693.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koch RA, Wilson AW, Séné O, Henkel TW, Aime MC. 2017. Resolved phylogeny and biogeography of the root pathogen Armillaria and its gasteroid relative, Guyanagaster. BMC Evol Biol 17:33. doi: 10.1186/s12862-017-0877-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lindner Czederpiltz DL, Volk TJ, Burdsall HH, Jr.. 2001. Field observations and inoculation experiments to determine the nature of the carpophoroids associated with Entoloma abortivum and Armillaria. Mycologia 93:841–851. doi: 10.2307/3761750. [DOI] [Google Scholar]
- 18.Druzhinina IS, Seidl-Seiboth V, Herrera-Estrella A, Horwitz BA, Kenerley CM, Monte E, Mukherjee PK, Zeilinger S, Grigoriev IV, Kubicek CP. 2011. Trichoderma: the genomics of opportunistic success. Nat Rev Microbiol 9:749–759. doi: 10.1038/nrmicro2637. [DOI] [PubMed] [Google Scholar]
- 19.Quandt CA, Di Y, Elser J, Jaiswal P, Spatafora JW. 2016. Differential expression of genes involved in host recognition, attachment, and degradation in the mycoparasite Tolypocladium ophioglossoides. G3 (Bethesda) 6:731–741. doi: 10.1534/g3.116.027045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karlsson M, Atanasova L, Jensen DF, Zeilinger S. 2017. Necrotrophic mycoparasites and their genomes. Microbiol Spectr 5:FUNK-0016–2016. doi: 10.1128/microbiolspec.FUNK-0016-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ross‐Davis AL, Stewart JE, Hanna JW, Kim MS, Knaus BJ, Cronn R, Rai H, Richardson BA, McDonald GI, Klopfenstein NB. 2013. Transcriptome of an Armillaria root disease pathogen reveals candidate genes involved in host substrate utilization at the host–pathogen interface. For Pathol 43:468–477. doi: 10.1111/efp.12056. [DOI] [Google Scholar]
- 22.Sipos G, Prasanna AN, Walter MC, O’Connor E, Bálint B, Krizsán K, Kiss B, Hess J, Varga T, Slot J, Riley R, Bóka B, Rigling D, Barry K, Lee J, Mihaltcheva S, LaButti K, Lipzen A, Waldron R, Moloney NM, Sperisen C, Kredics L, Vágvölgyi C, Patrignani A, Fitzpatrick D, Nagy I, Doyle S, Anderson JB, Grigoriev IV, Güldener U, Münsterkötter M, Nagy LG. 2017. Genome expansion and lineage-specific genetic innovations in the forest pathogenic fungi Armillaria. Nat Ecol Evol 1:1931–1941. doi: 10.1038/s41559-017-0347-8. [DOI] [PubMed] [Google Scholar]
- 23.Dodd RB, Drickamer K. 2001. Lectin-like proteins in model organisms: implications for evolution of carbohydrate-binding activity. Glycobiology 11:71R–79R. doi: 10.1093/glycob/11.5.71r. [DOI] [PubMed] [Google Scholar]
- 24.Inbar J, Chet I. 1992. Biomimics of fungal cell-cell recognition by use of lectin-coated nylon fibers. J Bacteriol 174:1055–1059. doi: 10.1128/jb.174.3.1055-1059.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Inbar J, Chet I. 1996. The role of lectins in recognition and adhesion of the mycoparasitic fungus Trichoderma spp. to its host. Adv Exp Med Biol 408:229–231. doi: 10.1007/978-1-4613-0415-9_27. [DOI] [PubMed] [Google Scholar]
- 26.Neethling D, Nevalainen H. 1996. Mycoparasitic species of Trichoderma produce lectins. Can J Microbiol 42:141–146. doi: 10.1139/m96-022. [DOI] [PubMed] [Google Scholar]
- 27.Schubert M, Bleuler-Martinez S, Butschi A, Wälti MA, Egloff P, Stutz K, Yan S, Collot M, Mallet J-M, Wilson IBH, Hengartner MO, Aebi M, Allain FH-T, Künzler M. 2012. Plasticity of the β-trefoil protein fold in the recognition and control of invertebrate predators and parasites by a fungal defense system. PLoS Pathog 8:e1002706. doi: 10.1371/journal.ppat.1002706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Latgé JP, Kobayashi H, Debeaupuis JP, Diaquin M, Sarfati J, Wieruszeski JM, Parra E, Bouchara JP, Fournet B. 1994. Chemical and immunological characterization of the extracellular galactomannan of Aspergillus fumigatus. Infect Immun 62:5424–5433. doi: 10.1128/iai.62.12.5424-5433.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cessna SG, Sears VE, Dickman MB, Low PS. 2000. Oxalic acid, a pathogenicity factor for Sclerotinia sclerotiorum, suppresses the oxidative burst of the host plant. Plant Cell 12:2191–2200. doi: 10.1105/tpc.12.11.2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dumas MT, Boyonoski N, Jeng RS. 1989. Oxalic acid production by species of Armillaria in the boreal mixedwood forest, p 593–601. In Proceedings of the 7th International Conference on Root and Butt Rots. Forestry Canada, Pacific Forestry Centre, Victoria, BC, Canada. [Google Scholar]
- 31.Munir E, Yoon JJ, Tokimatsu T, Hattori T, Shimada M. 2001. A physiological role for oxalic acid biosynthesis in the wood-rotting basidiomycete Fomitopsis palustris. Proc Natl Acad Sci USA 98:11126–11130. doi: 10.1073/pnas.191389598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Y, Wang Y. 2020. Oxalic acid metabolism contributes to full virulence and pycnidial development in the poplar canker fungus Cytospora chrysosperma. Phytopathology 110:1319–1325. doi: 10.1094/PHYTO-10-19-0381-R. [DOI] [PubMed] [Google Scholar]
- 33.Wei SJ, Li GQ, Jiang DH, Wang DB. 2004. Effect of oxalic acid on spore germination and mycelial growth of the mycoparasite Coniothyrium minitans. Acta Phytopathol Sin 34:199–203. [Google Scholar]
- 34.Ren L, Li GQ, Han YC, Jiang DH, Huang HC. 2007. Degradation of oxalic acid by Coniothyrium minitans and its effects on production and activity of β-1,3-glucanases of this mycoparasite. Biol Control 43:1–11. doi: 10.1016/j.biocontrol.2007.06.006. [DOI] [Google Scholar]
- 35.Ren L, Li GQ, Jiang DH. 2010. Characterization of some culture factors affecting oxalate degradation by the mycoparasite Coniothyrium minitans. J Appl Microbiol 108:173–180. doi: 10.1111/j.1365-2672.2009.04415.x. [DOI] [PubMed] [Google Scholar]
- 36.Zeng L-M, Zhang J, Han Y-C, Yang L, Wu M-D, Jiang D-H, Chen W, Li G-Q. 2014. Degradation of oxalic acid by the mycoparasite Coniothyrium minitans plays an important role in interacting with Sclerotinia sclerotiorum. Environ Microbiol 16:2591–2610. doi: 10.1111/1462-2920.12409. [DOI] [PubMed] [Google Scholar]
- 37.Dubey M, Jensen DF, Karlsson M. 2016. The ABC transporter ABCG29 is involved in H2O2 tolerance and biocontrol traits in the fungus Clonostachys rosea. Mol Genet Genomics 291:677–686. doi: 10.1007/s00438-015-1139-y. [DOI] [PubMed] [Google Scholar]
- 38.Ruocco M, Lanzuise S, Vinale F, Marra R, Turrà L, Woo S, Lorito M. 2009. Identification of a new biocontrol gene in Trichoderma atroviride: the role of an ABC transporter membrane pump in the interaction with different plant-pathogenic fungi. Mol Plant Microbe Interact 22:291–301. doi: 10.1094/MPMI-22-3-0291. [DOI] [PubMed] [Google Scholar]
- 39.Zhao H, Zhou T, Xie J, Cheng J, Chen T, Jiang D, Fu Y. 2020. Mycoparasitism illuminated by genome and transcriptome sequencing of Coniothyrium minitans, an important biocontrol fungus of the plant pathogen Sclerotinia sclerotiorum. Microb Genom 6:e000345. doi: 10.1099/mgen.0.000345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nygren K, Dubey M, Zapparata A, Iqbal M, Tzelepis GD, Brandström Durling M, Funck Jensen D, Karlsson M. 2018. The mycoparasitic fungus Clonostachys rosea responds with both common and specific gene expression during interspecific interactions with fungal prey. Evol Appl 11:931–949. doi: 10.1111/eva.12609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Atanasova L, Le Crom S, Gruber S, Coulpier F, Seidl-Seiboth V, Kubick CP, Druzhinina IS. 2013. Comparative transcriptomics reveals different strategies of Trichoderma mycoparasitism. BMC Genomics 14:121. doi: 10.1186/1471-2164-14-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cha JY, Igarashi T. 1996. Biological species of Armillaria and their mycoparasitic associations with Rhodophyllus abortivus in Hokkaido. Mycoscience 37:25–30. doi: 10.1007/BF02461452. [DOI] [Google Scholar]
- 43.Tregear JW, Roberts LM. 1992. The lectin gene family of Ricinus communis: cloning of a functional ricin gene and three lectin pseudogenes. Plant Mol Biol 18:515–525. doi: 10.1007/BF00040667. [DOI] [PubMed] [Google Scholar]
- 44.Pohleven J, Obermajer N, Sabotič J, Anžlovar S, Sepčić K, Kos J, Kralj B, Štrukelj B, Brzin J. 2009. Purification, characterization and cloning of a ricin B-like lectin from mushroom Clitocybe nebularis with antiproliferative activity against human leukemic T cells. Biochim Biophys Acta 1790:173–181. doi: 10.1016/j.bbagen.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Žurga S, Pohleven J, Renko M, Bleuler-Martínez S, Sosnowski P, Turk D, Künzler M, Kos J, Sabotič J. 2014. A novel beta-trefoil lectin from the parasol mushroom (Macrolepiota procera) is nematotoxic. FEBS J 281:3489–3506. doi: 10.1111/febs.12875. [DOI] [PubMed] [Google Scholar]
- 46.Bovi M, Cenci L, Perduca M, Capaldi S, Carrizo ME, Civiero L, Chiarelli LR, Galliano M, Monaco HL. 2013. BEL beta-trefoil: a novel lectin with antineoplastic properties in king bolete (Boletus edulis) mushrooms. Glycobiology 23:578–592. doi: 10.1093/glycob/cws164. [DOI] [PubMed] [Google Scholar]
- 47.Sabotič J, Ohm RA, Künzler M. 2016. Entomotoxic and nematotoxic lectins and protease inhibitors from fungal fruiting bodies. Appl Microbiol Biotechnol 100:91–111. doi: 10.1007/s00253-015-7075-2. [DOI] [PubMed] [Google Scholar]
- 48.Pohleven J, Renko M, Magister S, Smith DF, Künzler M, Strukelj B, Turk D, Kos J, Sabotič J. 2012. Bivalent carbohydrate binding is required for biological activity of Clitocybe nebularis lectin (CNL), the N,N’-diacetyllactosediamine (GalNAcbeta1-4GlcNAc, LacdiNAc)-specific lectin from basidiomycete C. nebularis. J Biol Chem 287:10602–10612. doi: 10.1074/jbc.M111.317263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Latgé JP. 2007. The cell wall: a carbohydrate armour for the fungal cell. Mol Microbiol 66:279–290. doi: 10.1111/j.1365-2958.2007.05872.x. [DOI] [PubMed] [Google Scholar]
- 50.de Man TJ, Stajich JE, Kubicek CP, Teiling C, Chenthamara K, Atanasova L, Druzhinina IS, Levenkova N, Birnbaum SS, Barribeau SM, Bozick BA, Suen G, Currie CR, Gerardo NM. 2016. Small genome of the fungus Escovopsis weberi, a specialized disease agent of ant agriculture. Proc Natl Acad Sci USA 113:3567–3572. doi: 10.1073/pnas.1518501113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kubicek CP, Herrera-Estrella A, Seidl-Seiboth V, Martinez DA, Druzhinina IS, Thon M, Zeilinger S, Casas-Flores S, Horwitz BA, Mukherjee PK, Mukherjee M, Kredics L, Alcaraz LD, Aerts A, Antal Z, Atanasova L, Cervantes-Badillo MG, Challacombe J, Chertkov O, McCluskey K, Coulpier F, Deshpande N, von Döhren H, Ebbole DJ, Esquivel-Naranjo EU, Fekete E, Flipphi M, Glaser F, Gómez-Rodríguez EY, Gruber S, Han C, Henrissat B, Hermosa R, Hernández-Oñate M, Karaffa L, Kosti I, Le Crom S, Lindquist E, Lucas S, Lübeck M, Lübeck PS, Margeot A, Metz B, Misra M, Nevalainen H, Omann M, Packer N, Perrone G, Uresti-Rivera EE, Salamov A, Schmoll M, Seiboth B, Shapiro H, Sukno S, Tamayo-Ramos JA, Tisch D, Wiest A, Wilkinson HH, Zhang M, Coutinho PM, Kenerley CM, Monte E, Baker SE, Grigoriev IV. 2011. Comparative genome sequence analysis underscores mycoparasitism as the ancestral life style of Trichoderma. Genome Biol 12:R40. doi: 10.1186/gb-2011-12-4-r40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ihrmark K, Asmail N, Ubhayasekera W, Melin P, Stenlid J, Karlsson M. 2010. Comparative molecular evolution of Trichoderma chitinases in response to mycoparasitic interactions. Evol Bioinform Online 6:1–26. doi: 10.4137/ebo.s4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xie BB, Qin QL, Shi M, Chen LL, Shu YL, Luo Y, Wang XW, Rong JC, Gong ZT, Li D, Sun CY, Liu GM, Dong XW, Pang XH, Huang F, Liu W, Chen XL, Zhou BC, Zhang YZ, Song ZY. 2014. Comparative genomics provides insights into evolution of Trichoderma nutrition style. Genome Biol Evol 6:379–390. doi: 10.1093/gbe/evu018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miyauchi S, Kiss E, Kuo A, Drula E, Kohler A, Sánchez-García M, Morin E, Andreopoulos B, Barry KW, Bonito G, Buée M, Carver A, Chen C, Cichocki N, Clum A, Culley D, Crous PW, Fauchery L, Girlanda M, Hayes RD, Kéri Z, LaButti K, Lipzen A, Lombard V, Magnuson J, Maillard F, Murat C, Nolan M, Ohm RA, Pangilinan J, Pereira MF, Perotto S, Peter M, Pfister S, Riley R, Sitrit Y, Stielow JB, Szöllősi G, Žifčáková L, Štursová M, Spatafora JW, Tedersoo L, Vaario LM, Yamada A, Yan M, Wang P, Xu J, Bruns T, Baldrian P, Vilgalys R, Dunand C, Henrissat B, Grigoriev IV, Hibbett D, Nagy LG, Martin FM. 2020. Large-scale genome sequencing of mycorrhizal fungi provides insights into the early evolution of symbiotic traits. Nat Commun 11:5125. doi: 10.1038/s41467-020-18795-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ruiz-Dueñas FJ, Barrasa JM, Sánchez-García M, Camarero S, Miyauchi S, Serrano A, Linde D, Babiker R, Drula E, Ayuso-Fernández I, Pacheco R, Padilla G, Ferreira P, Barriuso J, Kellner H, Castanera R, Alfaro M, Ramirez L, Pisabarro AG, Riley R, Kuo A, Andreopoulos W, LaButti K, Pangilinan J, Tritt A, Lipzen A, He G, Yan M, Ng V, Grigoriev IV, Cullen D, Martin F, Rosso MN, Henrissat B, Hibbett D, Martínez AT. 2021. Genomic analysis enlightens Agaricales lifestyle evolution and increasing peroxidase diversity. Mol Biol Evol 38:1428–1446. doi: 10.1093/molbev/msaa301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Steindorff AS, Carver A, Calhoun S, Stillman K, Liu H, Lipzen A, He G, Yan M, Pangilinan J, LaButti K, Ng V, Bruns TD, Grigoriev IV. 2021. Comparative genomics of pyrophilous fungi reveals a link between fire events and developmental genes. Environ Microbiol 23:99–109. doi: 10.1111/1462-2920.15273. [DOI] [PubMed] [Google Scholar]
- 57.Han Y-C, Li G-Q, Yang L, Jiang D-H. 2011. Molecular cloning, characterization and expression analysis of a pacC homolog in the mycoparasite Coniothyrium minitans. World J Microbiol Biotechnol 27:381–391. doi: 10.1007/s11274-010-0469-5. [DOI] [Google Scholar]
- 58.Ahrendt SR, Quandt CA, Ciobanu D, Clum A, Salamov A, Andreopoulos B, Cheng J-F, Woyke T, Pelin A, Henrissat B, Reynolds NK, Benny GL, Smith ME, James TY, Grigoriev IV. 2018. Leveraging single-cell genomics to expand the fungal tree of life. Nat Microbiol 3:1417–1428. doi: 10.1038/s41564-018-0261-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Palmer JM, Drees KP, Foster JT, Lindner DL. 2018. Extreme sensitivity to ultraviolet light in the fungal pathogen causing white-nose syndrome of bats. Nat Commun 9:35. doi: 10.1038/s41467-017-02441-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Agerer R, Waller K. 1993. Mycorrhizae of Entoloma saepium: parasitism or symbiosis? Mycorrhiza 3:145–154. doi: 10.1007/BF00203608. [DOI] [Google Scholar]
- 61.Kobayashi H, Hatano K. 2001. A morphological study of the mycorrhiza of Entoloma clypeatum f. hybridum on Rosa multiflora. Mycoscience 42:83–90. doi: 10.1007/BF02463979. [DOI] [Google Scholar]
- 62.Shishikura M, Takemura Y, Sotome K, Maekawa N, Nakagiri A, Endo N. 2021. Four mycelial strains of Entoloma clypeatum species complex form ectomycorrhiza-like roots with Pyrus betulifolia seedlings in vitro, and one develops fruiting bodies 2 months after inoculation. Mycorrhiza 31:31–42. doi: 10.1007/s00572-020-00994-4. [DOI] [PubMed] [Google Scholar]
- 63.Kohler A, Kuo A, Nagy LG, Morin E, Barry KW, Buscot F, Canbäck B, Choi C, Cichocki N, Clum A, Colpaert J, Copeland A, Costa MD, Doré J, Floudas D, Gay G, Girlanda M, Henrissat B, Herrmann S, Hess J, Högberg N, Johansson T, Khouja H-R, LaButti K, Lahrmann U, Levasseur A, Lindquist EA, Lipzen A, Marmeisse R, Martino E, Murat C, Ngan CY, Nehls U, Plett JM, Pringle A, Ohm RA, Perotto S, Peter M, Riley R, Rineau F, Ruytinx J, Salamov A, Shah F, Sun H, Tarkka M, Tritt A, Veneault-Fourrey C, Zuccaro A, Mycorrhizal Genomics Initiative Consortium, Tunlid A, Grigoriev IV, Hibbett DS, Martin F. 2015. Convergent losses of decay mechanisms and rapid turnover of symbiosis genes in mycorrhizal mutualists. Nat Genet 47:410–415. doi: 10.1038/ng.3223. [DOI] [PubMed] [Google Scholar]
- 64.Noordeloos ME. 1987. Entoloma (Agaricales) in Europe (Nova Hedwigia Beihefte). Lubrecht & Cramer Ltd, Port Jervis, NY. [Google Scholar]
- 65.Noordeloos ME. 1993. Studies in Clitopilus (Basidiomycetes, Agaricales) in Europe. Persoonia 15:241–248. [Google Scholar]
- 66.Collins C, Keane TM, Turner DJ, O’Keeffe G, Fitzpatrick DA, Doyle S. 2013. Genomic and proteomic dissection of the ubiquitous plant pathogen, Armillaria mellea: toward a new infection model system. J Proteome Res 12:2552–2570. doi: 10.1021/pr301131t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Strickler SR, Bombarely A, Mueller LA. 2012. Designing a transcriptome next-generation sequencing project for a nonmodel plant species. Am J Bot 99:257–266. doi: 10.3732/ajb.1100292. [DOI] [PubMed] [Google Scholar]
- 68.Fukuda M, Nakashima E, Hayashi K, Nagasawa E. 2003. Identification of the biological species of Armillaria associated with Wynnea and Entoloma abortivum using PCR-RFLP analysis of the intergenic region (IGR) of ribosomal DNA. Mycol Res 107:1435–1441. doi: 10.1017/s0953756203008633. [DOI] [PubMed] [Google Scholar]
- 69.Schuierer S, Carbone W, Knehr J, Petitjean V, Fernandez A, Sultan M, Roma G. 2017. A comprehensive assessment of RNA-seq protocols for degraded and low-quantity samples. BMC Genomics 18:442. doi: 10.1186/s12864-017-3827-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Crusoe MR, Alameldin HF, Awad S, Boucher E, Caldwell A, Cartwright R, Charbonneau A, Constantinides B, Edvenson G, Fay S, Fenton J, Fenzl T, Fish J, Garcia-Gutierrez L, Garland P, Gluck J, González I, Guermond S, Guo J, Gupta A, Herr JR, Howe A, Hyer A, Härpfer A, Irber L, Kidd R, Lin D, Lippi J, Mansour T, McA’Nulty P, McDonald E, Mizzi J, Murray KD, Nahum JR, Nanlohy K, Nederbragt AJ, Ortiz-Zuazaga H, Ory J, Pell J, Pepe-Ranney C, Russ ZN, Schwarz E, Scott C, Seaman J, Sievert S, Simpson J, Skennerton CT, Spencer J, Srinivasan R, Standage D, Stapleton JA, Steinman SR, Stein J, Taylor B, Trimble W, Wiencko HL, Wright M, Wyss B, Zhang Q, Zyme E, Brown CT. 2015. The khmer software package: enabling efficient nucleotide sequence analysis. F1000Res 4:900. doi: 10.12688/f1000research.6924.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A. 2011. Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat Biotechnol 29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mikheenko A, Prjibelski A, Saveliev V, Antipov D, Gurevich A. 2018. Versatile genome assembly evaluation with QUAST-LG. Bioinformatics 34:i142–i150. doi: 10.1093/bioinformatics/bty266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Seppey M, Manni M, Zdobnov EM. 2019. BUSCO: assessing genome assembly and annotation completeness. Methods Mol Biol 1962:227–245. doi: 10.1007/978-1-4939-9173-0_14. [DOI] [PubMed] [Google Scholar]
- 74.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li B, Dewey CN. 2011. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Robinson MD, McCarthy DJ, Smyth GK. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Robinson MD, Oshlack A. 2010. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol 11:R25. doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup . 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bryant DM, Johnson K, DiTommaso T, Tickle T, Brian Couger M, Payzin-Dogru D, Lee TJ, Leigh ND, Kuo T-H, Davis FG, Bateman J, Bryant S, Guzikowski AR, Tsai SL, Coyne S, Ye WW, Freeman RM, Jr, Peshkin L, Tabin CJ, Regev A, Haas BJ, Whited JL. 2017. A tissue-mapped azolotl de novo transcriptome enables identification of limb regeneration factors. Cell Rep 18:762–P776. doi: 10.1016/j.celrep.2016.12.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. 2009. BLAST+: architecture and application. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wheeler TJ, Eddy SR. 2013. nhmmer: DNA homology search with profile HMMs. Bioinformatics 29:2487–2489. doi: 10.1093/bioinformatics/btt403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, Sonnhammer ELL, Tate J, Punta M. 2014. Pfam: the protein families database. Nucleic Acids Res 42:D222–D230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. 2000. Gene Ontology: tool for the unification of biology. Nat Genet 25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kanehisa M, Goto S. 2000. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kanehisa M. 2019. Toward understanding the origin and evolution of cellular organisms. Protein Sci 28:1947–1951. doi: 10.1002/pro.3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kanehisa M, Furumichi M, Sato Y, Ishiguro-Watanabe M, Tanabe M. 2021. KEGG: integrating viruses and cellular organisms. Nucleic Acids Res 49:D545–D551. doi: 10.1093/nar/gkaa970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang H, Yohe T, Huang L, Entwistle S, Wu P, Yang Z, Busk PK, Xu Y, Yin Y. 2018. dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res 46:W95–W101. doi: 10.1093/nar/gky418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Blin K, Shaw S, Steinke K, Villebro R, Ziemert N, Yup Lee S, Medema MH, Weber T. 2019. antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res 47:W81–W87. doi: 10.1093/nar/gkz310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bengtsson-Palme J, Ryberg M, Hartmann M, Branco S, Wang Z, Godhe A, De Wit P, Sánchez-García M, Ebersberger I, de Sousa F, Amend A, Jumpponen A, Unterseher M, Kristiansson E, Abarenkov K, Bertrand YJK, Sanli K, Eriksson KM, Vik U, Veldre V, Nilsson RH. 2013. Improved software detection and extraction of ITS1 and ITS2 from ribosomal ITS sequences of fungi and other eukaryotes for analysis of environmental sequencing data. Methods Ecol Evol 4:914–919. doi: 10.1111/2041-210X.12073. [DOI] [Google Scholar]
- 90.Slater GSC, Birney E. 2005. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6:31. doi: 10.1186/1471-2105-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Floudas D, Held BW, Riley R, Nagy LG, Koehler G, Ransdell AS, Younus H, Chow J, Chiniquy J, Lipzen A, Tritt A, Sun H, Haridas S, LaButti K, Ohm RA, Kües U, Blanchette RA, Grigoriev IV, Minto RE, Hibbett DS. 2015. Evolution of novel wood decay mechanisms in Agaricales revealed by the genome sequences of Fistulina hepatica and Cylindrobasidium torrendii. Fungal Genet Biol 76:78–92. doi: 10.1016/j.fgb.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Koch RA, Yoon GM, Aryal UK, Lail K, Amirebrahimi M, LaButti K, Lipzen A, Riley R, Barry K, Henrissat B, Grigoriev IV, Herr JR, Aime MC. 2021. Symbiotic nitrogen fixation in the reproductive structures of a basidiomycete fungus. Curr Biol 31:3905–3914. doi: 10.1016/j.cub.2021.06.033. [DOI] [PubMed] [Google Scholar]
- 93.Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Castresana J. 2000. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 17:540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- 95.Kozlov AM, Darriba D, Flouri T, Morel B, Stamatakis A. 2019. RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 35:4453–4455. doi: 10.1093/bioinformatics/btz305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Guo T, Wang HC, Xue WQ, Zhao J, Yang ZL. 2016. Phylogenetic analyses of Armillaria reveal at least 15 phylogenetic lineages in China, seven of which are associated with cultivated Gastrodia elata. PLoS One 11:e0154794. doi: 10.1371/journal.pone.0154794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Coetzee MPA, Wingfield BD, Harrington TC, Dalevi D, Coutinho TA, Wingfield MJ. 2000. Geographical diversity of Armillaria mellea s. s. based on phylogenetic analysis. Mycologia 92:105–113. doi: 10.2307/3761454. [DOI] [Google Scholar]
- 98.Coetzee MPA, Wingfield BD, Bloomer P, Wingfield MJ. 2005. Phylogenetic analyses of DNA sequences reveal species partitions amongst isolates of Armillaria from Africa. Mycol Res 109:1223–1234. doi: 10.1017/s095375620500393x. [DOI] [PubMed] [Google Scholar]
- 99.Schneider-Maunoury L, Deveau A, Moreno M, Todesco F, Belmondo S, Murat C, Courty P-E, Jąkalski M, Selosse M-A. 2020. Two ectomycorrhizal truffles, Tuber melanosporum and T. aestivum, endophytically colonise roots of non-ectomycorrhizal plants in natural environments. New Phytol 225:2542–2556. doi: 10.1111/nph.16321. [DOI] [PubMed] [Google Scholar]
- 100.Coetzee MPA, Bloomer P, Wingfield MJ, Wingfield BD. 2011. Paleogene radiation of a plant pathogenic mushroom. PLoS One 6:e28545. doi: 10.1371/journal.pone.0028545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Osmundson TW, Robert VA, Schoch CL, Baker LJ, Smith A, Robich G, Mizzan L, Garbelotto MM. 2013. Filling gaps in biodiversity knowledge for macrofungi: contributions and assessment of an herbarium collection DNA barcode sequencing project. PLoS One 8:e62419. doi: 10.1371/journal.pone.0062419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kasper-Pakosz R, Pietras M, Łuczaj Ł. 2016. Wild and native plants and mushrooms sold in the open-air markets of south-eastern Poland. J Ethnobiol Ethnomed 12:45. doi: 10.1186/s13002-016-0117-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hughes KW, Petersen RH, Lodge DJ, Bergemann SE, Baumgartner K, Tulloss RE, Lickey E, Cifuentes J. 2013. Evolutionary consequences of putative intra- and interspecific hybridization in agaric fungi. Mycologia 105:1577–1594. doi: 10.3852/13-041. [DOI] [PubMed] [Google Scholar]
- 104.Potyralska A, Schmidt O, Moreth U, Łakomy P, Siwecki R. 2002. rDNA-ITS sequence of Armillaria species and a specific primer for A. mellea. For Genet 9:119–124. [Google Scholar]
- 105.Ortega A, Moreno G, Platas G, Pelaez F. 2008. A new Armillaria mellea variant from tops of coastal sand dunes in southern Spain. Mycotaxon 104:349–364. [Google Scholar]
- 106.Vu D, Groenewald M, de Vries M, Gehrmann T, Stielow B, Eberhardt U, Al-Hatmi A, Groenewald JZ, Cardinali G, Houbraken J, Boekhout T, Crous PW, Robert V, Verkley GJM. 2019. Large-scale generation and analysis of filamentous fungal DNA barcodes boosts coverage for kingdom fungi and reveals thresholds for fungal species and higher taxon delimitation. Stud Mycol 92:135–154. doi: 10.1016/j.simyco.2018.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Binder M, Hibbett DS, Wang Z, Farnham WF. 2006. Evolutionary relationships of Mycaureola dilseae (Agaricales), a basidiomycete pathogen of a subtidal rhodophyte. Am J Bot 93:547–556. doi: 10.3732/ajb.93.4.547. [DOI] [PubMed] [Google Scholar]
- 108.Haelewaters D, Dirks AC, Kappler LA, Mitchell JK, Quijada L, Vandegrift R, Buyck B, Pfister DH. 2018. A preliminary checklist of fungi at the Boston Harbor Islands. Northeast Nat 25:45–76. doi: 10.1656/045.025.s904. [DOI] [Google Scholar]
- 109.Kim M-S, Klopfenstein NB, Hanna JW, McDonald GI. 2006. Characterization of North American Armillaria species: genetic relationships determined by ribosomal DNA sequences and AFLP markers. For Pathol 36:145–164. doi: 10.1111/j.1439-0329.2006.00441.x. [DOI] [Google Scholar]
- 110.Schnabel G, Ash JS, Bryson PK. 2005. Identification and characterization of Armillaria tabescens from the southeastern United States. Mycol Res 109:1208–1222. doi: 10.1017/s0953756205003916. [DOI] [PubMed] [Google Scholar]
- 111.Dentinger BTM, Gaya E, O’Brien H, Suz LM, Lachlan R, Díaz-Valderrama JR, Koch RA, Aime MC. 2016. Tales from the crypt: genome mining from fungarium specimens improves resolution of the mushroom tree of life. Biol J Linn Soc 117:11–32. doi: 10.1111/bij.12553. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Location and GenBank accession numbers of Armillaria mellea sequences used in the phylogenetic analysis. Download Table S1, DOCX file, 0.02 MB (24.2KB, docx) .
Copyright © 2021 Koch and Herr.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Data Availability Statement
The raw reads and assembled transcriptomes generated during this study have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO series accession number GSE183699 or under the NCBI BioProject accession no. PRJNA761704. All other associated data, including analysis code and information on voucher collections, are available at https://github.com/HerrLab/Koch_Arma-Ento_2021.