

Abstract
The survivin (BIRC5) expression is very low in normal differentiated adult tissues, but it is one of the most widely upregulated genes in tumor cells. The overexpression of survivin in many cancer types has been positively correlated with resistance to chemotherapy, tumor metastasis, and poor patient survival. Survivin is considered to be a cancer specific biomarker and serves as a potential cancer drug target. In this report, we describe the design and syntheses of a series of novel selective survivin inhibitors based on the hydroxyquinoline scaffold from our previously reported lead compound MX-106. The best compound identified in this study is compound 12b. In vitro, 12b inhibited cancer cell proliferation with an average IC50 value of 1.4 μM, using a panel of melanoma, breast, and ovarian cancer cell lines. The metabolic stability of 12b improved over MX-106 by 1.7-fold (88 vs 51 min in human microsomes). Western blot analyses demonstrated that treatments with 12b selectively decreased survivin protein levels, but negligibly affected other closely related members in the IAP family proteins, and strongly induced cancer cell apoptosis. In vivo, compound 12b effectively inhibited melanoma tumor growth when tested using a human A375 melanoma xenograft model. Further evaluation using an aggressive, orthotopic ovarian cancer mouse model showed that 12b was highly efficacious in suppressing both primary tumor growth in ovaries and tumor metastasis to multiple peritoneal organs. Collectively, results in this study strongly suggest that the hydroxyquinoline scaffold, represented by 12b and our earlier lead compound MX-106, has abilities to selectively target survivin and is promising for further preclinical development.
Keywords: BIRC5 (survivin), selective survivin inhibitors, structure-activity relationships, antiproliferative activities, P-glycoprotein overexpression, ovarian tumor metastasis, orthotopic ovarian cancer mouse model
Graphical Abstract
1. Introduction
The apoptotic process is essential for development and tissue homeostasis and provides some measure of protection against errors in DNA replication and other related processes. Cancer cells can hijack and dysregulate this apoptotic process to protect their survival and growth [1]. Apoptosis is regulated in part by a family of proteins called inhibitors of apoptosis proteins (IAPs), contain a conserved 3-dimensional fold BIR domain that binds zinc. Survivin (BIRC5) is unique among the members of the IAP family in that its expression is low or undetectable in normal terminally differentiated cells while it is highly overexpressed in many types of cancer cells, where it supports dysregulated cellular replication [2]. Clinically, the expression of survivin is positively correlated to cancer cell metastasis, tumor invasiveness, chemoresistance, and poor prognosis [3, 4]. The pivotal role played by survivin in multiple signaling pathways in cancer cells including apoptosis inhibition, mitotic control, and cell cycle promotion make it an attractive therapeutic target against cancer [5]. Several compounds have been reported to inhibit survivin expression, and some of them have been proposed as specific inhibitors of survivin [6], with YM155 as the most known survivin inhibitor reported [7]. YM155 inhibits transcription of the survivin gene, but recent reports have suggested that it may actually be an DNA-damaging agent instead of a bona-fide survivin inhibitor [8]. Thus, the exact mechanism of action of YM155 is still debatable [9]. It made to clinical trials but were suspended in phase II due to its high systemic toxicity and relative low ability to degrade survivin [7, 9]. Additional studies indicate that YM155 is a substrate of the drug efflux pump P-glycoprotein (P-gp), consistent with its limited efficacy in drug resistant cells [10].
We have previously reported the identification of compound UC-112 (Figure 1), which selectively decreases survivin protein levels and activates caspases 3/7 and 9 [11]. UC-112 also retains its potency in multidrug-resistant cancer cell lines that overexpress P-gp. A subsequent structure-activity relationship (SAR) study of UC-112 led to discovery of its derivative MX-106 [5-(((4-isopropylbenzyl)oxy)methyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol] (Figure 1). MX-106 suppresses the in vivo growth of melanoma tumors [12]. Mechanistic studies indicated that MX-106 functions by selectively suppressing expression of survivin and induces apoptosis of cancer cells. The results of the SAR showed that the 8-hydroxyquinoline moiety is important for its antiproliferative activities (A and B rings, shown in Figure 1). However, the benzyloxy ether linkage that connects the C ring to the 8-hydroxyquinoline moiety (linker moiety, Figure 1) is metabolically labile group and we hypothesized that we could improve the metabolic stability while maintaining the potency of this scaffold by replacing the ether linker with more stable moieties such as imidazole and triazole rings. We therefore sought to further optimize the MX-106 hydroxyquinoline scaffold by designing several focused sets of new MX-106 analogs (Figure 1). We introduced four major modifications to the MX-106 structure in the present study. First, we modified the flexible ether linker by cyclization or replacing it with other ring systems. Second, we modified the C ring by replacing the phenyl ring with other bioisosteres or by introducing additional substitutions. Third, we modified the D ring with a variety of cyclic amines. Finally, we removed the methylene linker between the B and D rings to obtain a planar head scaffold. This combined approach led to the production of thirty-one new MX-106 analogs, which we evaluated in vitro and in vivo to identify compound 12b as a new lead compound for future further preclinical studies.
Figure 1.
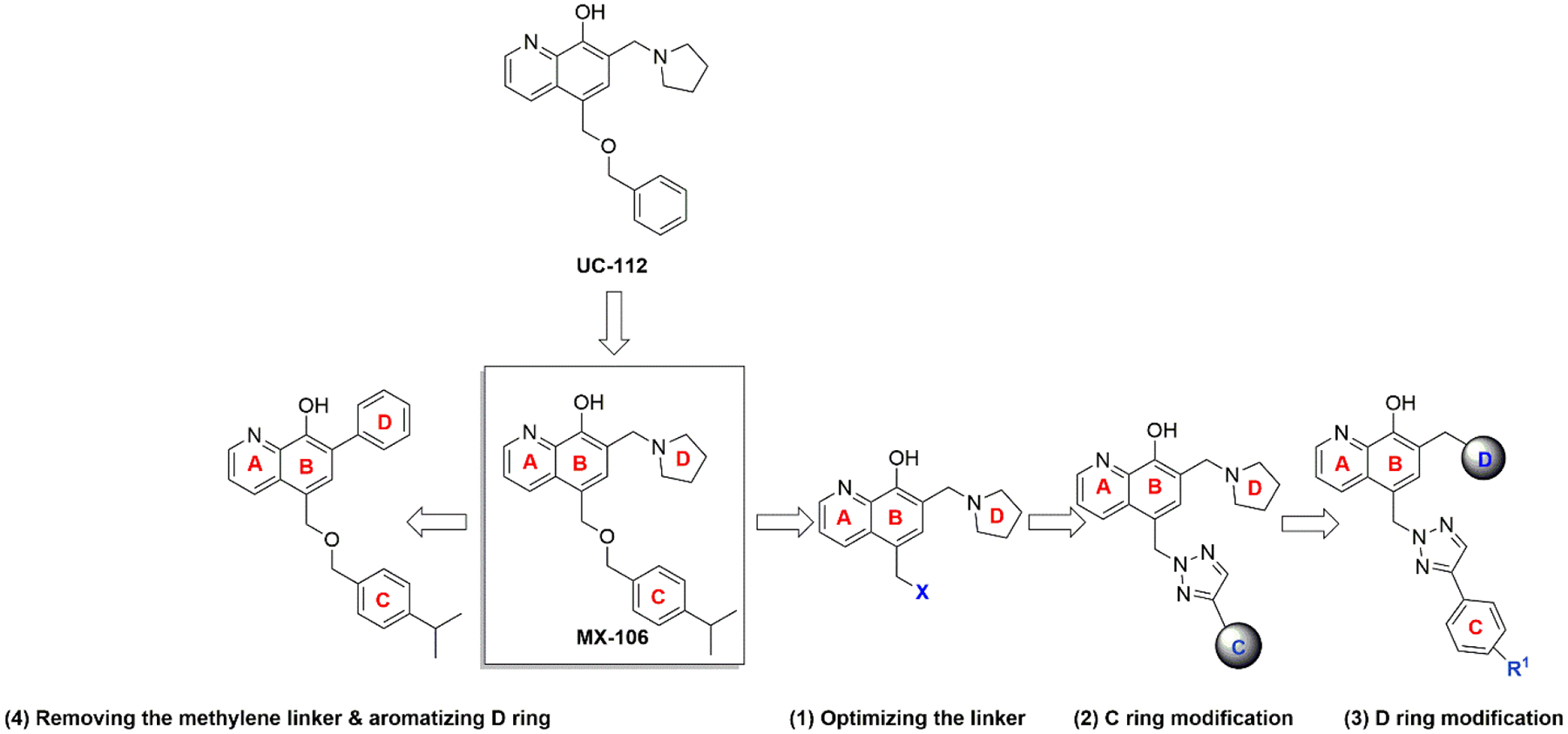
Targeted modifications used to produce new MX-106 analogs.
2. Chemistry
Compounds 4a–4d contained different linkers between the B and C rings relative to the parent compound MX-106 (Scheme 1). They were synthesized by reacting 8-hydroxyquinoline with formaldehyde and catalytic zinc chloride in concentrated hydrochloric acid to generate the salt of compound 2 [12]. Compounds 3a and 3b were synthesized using step b, where phenyl-substituted triazole and imidazole rings reacted with salt 2 in the presence of K2CO3 in DMF to form the desired products. Compounds 3c and 3d were synthesized using steps c and d. In step c, substituted alcohols were reacted directly with salt 2 with heating to form different salts. In step d, these were converted into free bases by adjusting the pH with NaHCO3 solution. Compounds 3a–3d were used in Mannich reactions with paraformaldehyde and pyrrolidine in anhydrous ethanol to form the final compounds 4a–4d.
Scheme 1.
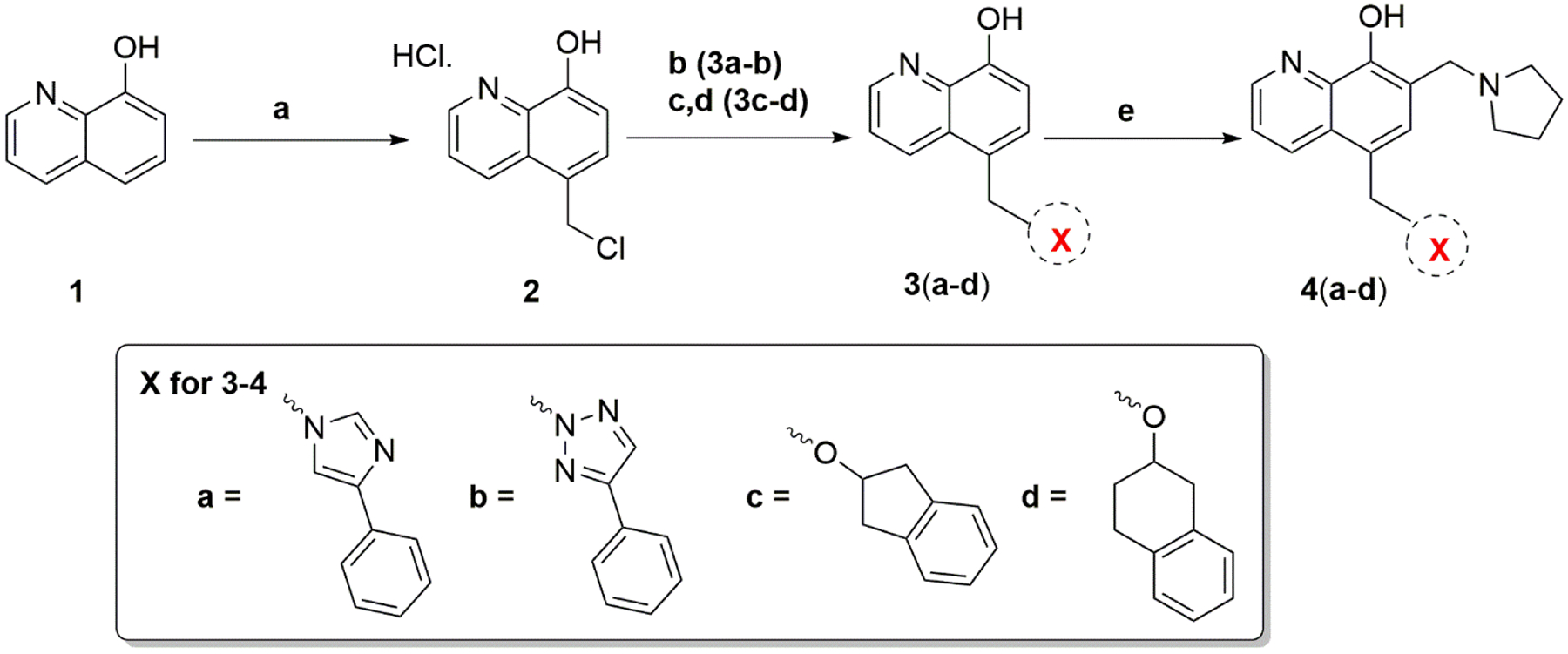
Synthesis of Compounds 4a–4d
Reagents and conditions: (a) Conc. HCl, 37% formaldehyde, ZnCl2, r.t, 24 hr; (b) K2CO3, DMF, r.t, 24 hr; (c) substituted alcohol, 60°C; (d) hexane:ethylacetate (1:1), NaHCO3, H2O, pH 8–10; (e) pyrrolidine, paraformaldehyde, anhydrous EtOH, reflux, 3 hr.
The general synthesis of MX-106 analogs that were substituted in the C ring (compounds 7a–7k and 10) is outlined in Scheme 2. Various terminal alkynes were reacted with azidotrimethylsilane with catalytic amounts of CuI in a mixture of DMF and methanol to prepare different 4-substituted-1-H-triazoles (compounds 5a–5k and 8) [13]. Compounds 5a–5k and 8 were reacted with salt 2 to form intermediate compounds 6a–6k and 9, which were submitted to Mannich reaction with paraformaldehyde and pyrrolidine to form compounds 7a–7k and 10 (Scheme 2).
Scheme 2.
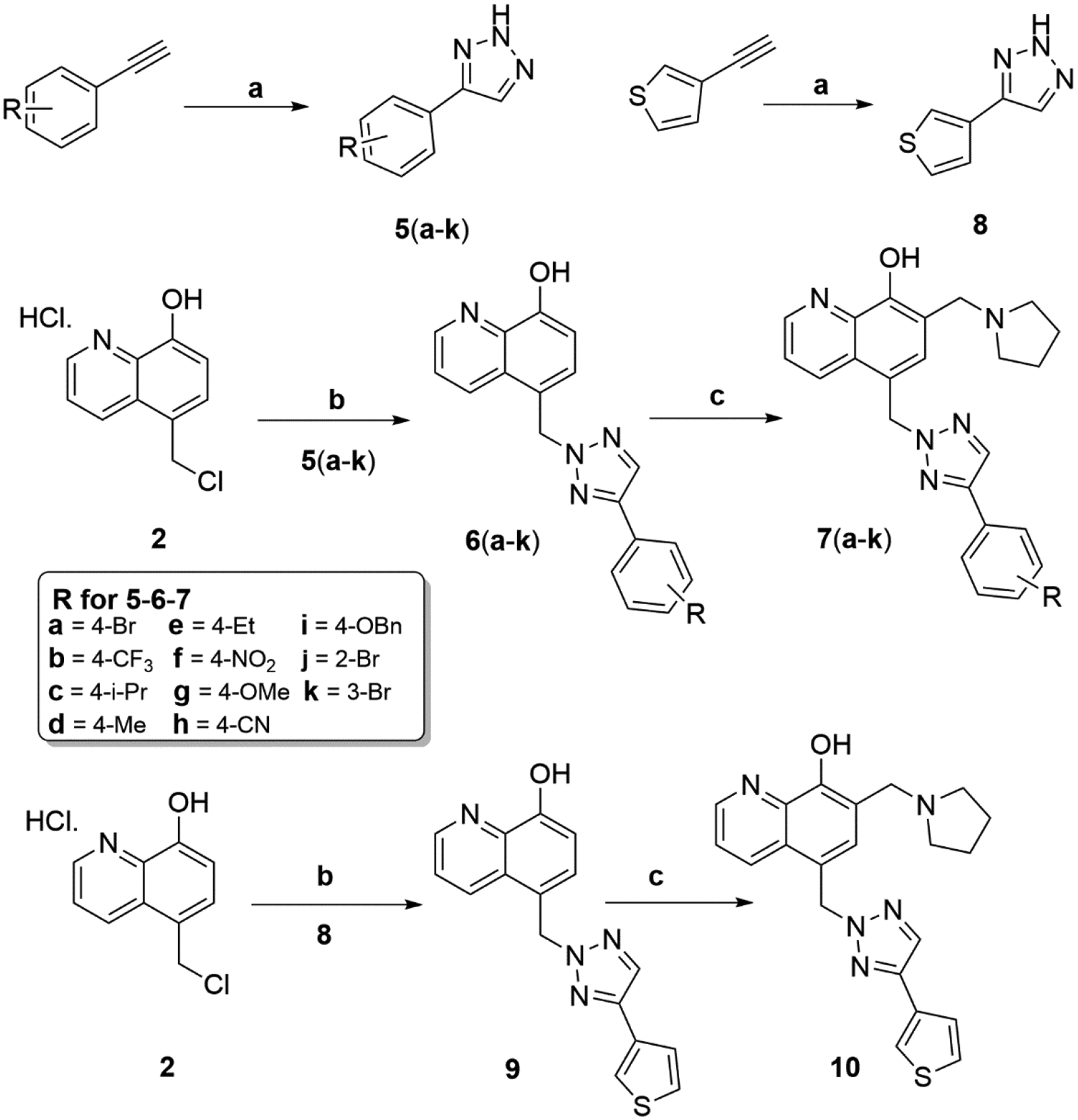
Synthesis of Compounds 7a–7k and 10
Reagents and conditions: (a) azidotrimethylsilane (1.5 equiv), CuI(5 mol%), DMF/MeOH = 5:1, reflux, 12 hr; (b) K2CO3, DMF, r.t, overnight; (c) pyrrolidine, paraformaldehyde, anhydrous EtOH, reflux, 3 hr.
MX-106 analogs that were substituted in the D ring (compounds 12a–12e and 14) are shown in Scheme 3. The intermediate compounds 6a and 6b were reacted with different secondary amines via Mannich reaction to form final compounds 12a–12e. The final compound 14 was obtained by removing the tert-butyloxycarbonyl protecting group (BOC group) from compound 13 using trifluoroacetic acid (TFA) in dichloromethane.
Scheme 3.
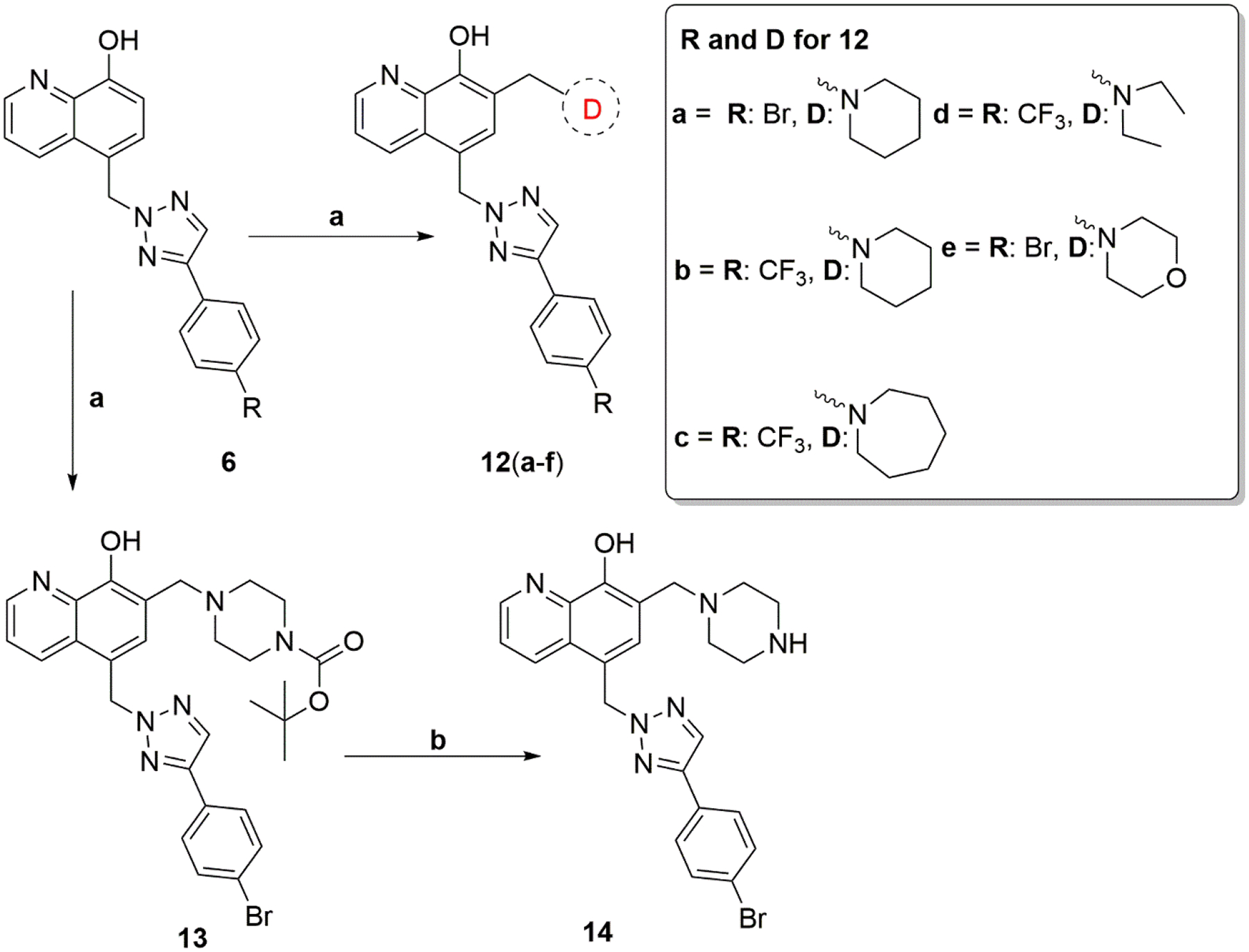
Synthesis of Compounds 12a–12e and 14
Reagents and conditions: (a) amine, paraformaldehyde, anhydrous EtOH, reflux, 3 hr; (b) trifluoroacetic acid (TFA), anhydrous CH2Cl2.
Scheme 4 shows the general synthesis of compounds 19a–19h. 7-Bromo-8-hydroxyquinoline was reacted with formaldehyde and catalytic zinc chloride in concentrated hydrochloric acid at 60°C for 16 hr to produce compound 15. After introducing a chloromethyl group to the 5-position of the hydroxyquinoline ring, compound 15 was reacted directly with 4-isopropylbenzyl alcohol with heating to form a salt that was converted to a free base by adjusting the pH with NaHCO3 solution to form intermediate compound 16. Compound 16 was reacted with 2-(trimethylsilyl)ethoxymethyl chloride (SEMCl) and NaH in THF at RT to produce compound 17. The Suzuki coupling reaction of compound 17 with appropriate aryl boronic acid under Pd (PPh3)4 condition generated the intermediates 18a–18h. The final compounds 19a–19h were obtained by removing the SEMCl protecting group with trifluoroacetic acid (TFA) in dichloromethane.
Scheme 4.
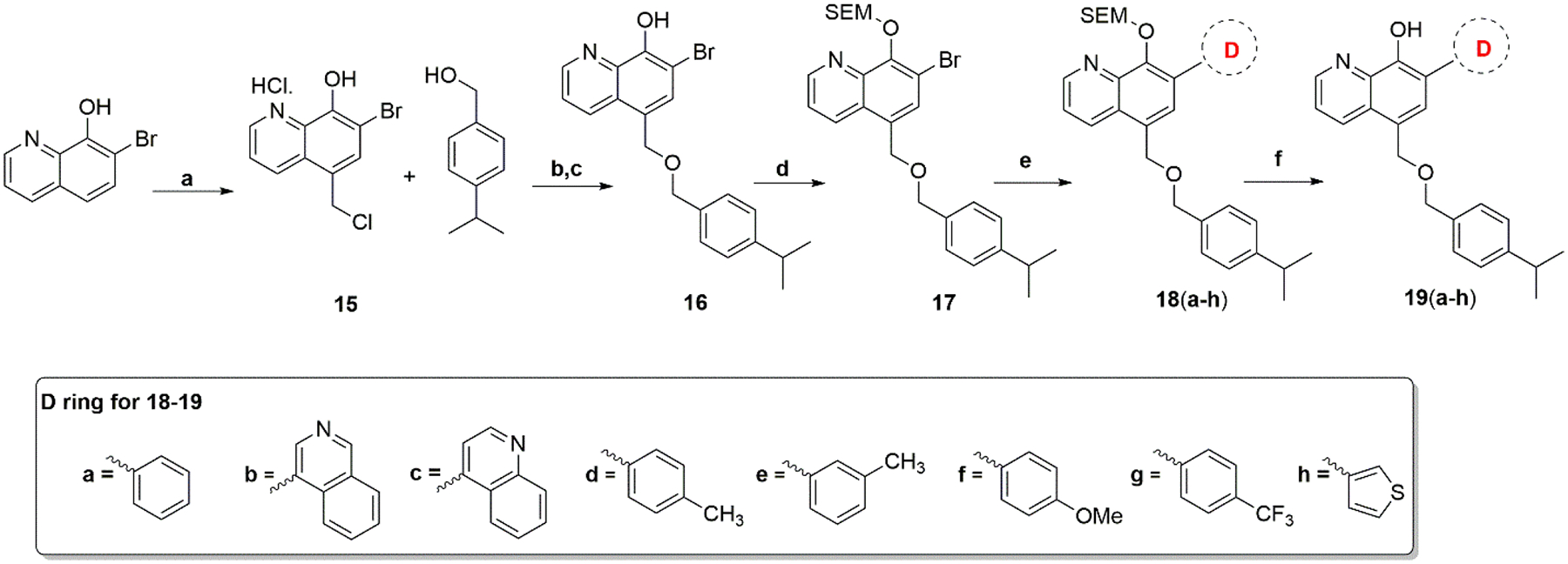
Synthesis of Compounds 19a–19h
Reagents and conditions: (a) Conc. HCl, 37%formaldehyde, ZnCl2, 60°C, 16 hr; (b) 90°C; (c) hexane:ethylacetate (1:1), NaHCO3, H2O, pH 8–10; (d) SEMCl, NaH, THF; (e) Pd(PPh3)4, Na2CO3, dioxane/H2O; (f) trifluoroacetic acid (TFA), CH2Cl2.
3. Results and discussion
3.1. In vitro antiproliferative assay in a panel of melanoma and breast cancer cell lines
All MX-106 analogs were evaluated for their cytotoxicity in a panel of human cancer cell lines including melanoma cancer cell lines A375, RPMI7951, MDA-MB-435 and its multidrug resistant daughter line MDA-MB-435/LCC6MDR1 and breast cancer cell lines MDA-MB-453 and SKBR3. MX-106 was included in the assays as a positive control. These in vitro biological results are summarized in Table 1.
Table 1.
In vitro growth inhibitory effects of MX-106 analogs in a panel of melanoma and breast cancer cell lines (IC50 ± standard error of the mean (SEM) [4], n = 3)
| Compound | A375 | MDA-MB-435 | RPMI7951 | MDA-MB-453 | SKBR3 | MDA-MB-435/LCC6MDRI | Average IC50 | Resistance index (RI)a |
|---|---|---|---|---|---|---|---|---|
| 4a | 2.2±0.2 | 8.9±1.2 | 2.8±0.4 | 3.1±0.3 | 6.9±0.5 | 1.3±0.2 | 4.2 | 0.1 |
| 4b | 2.0±0.3 | 5.0±0.7 | 3.1±0.4 | 2.9±0.4 | 4.0±0.5 | 0.6±0.1 | 2.9 | 0.1 |
| 4c | 1.9±0.3 | 2.2±0.3 | 1.3±0.2 | 2.4±0.3 | 5.9±0.5 | 2.2±0.2 | 2.7 | 1.0 |
| 4d | 1.0±0.2 | 3.3±0.4 | 0.8±0.1 | 1.3±0.2 | 3.9±0.5 | 1.3±0.1 | 1.9 | 0.4 |
| 7a | 0.7±0.1 | 3.1±0.4 | 1.7±0.3 | 3.1±0.4 | 2.7±0.4 | 0.6±0.1 | 2.0 | 0.2 |
| 7b | 0.8±0.1 | 4.1±0.5 | 1.6±0.2 | 3.5±0.5 | 3.2±0.5 | 1.1±0.1 | 2.4 | 0.3 |
| 7c | 1.1±0.2 | 4.6±0.5 | 2.3±0.3 | 5.9±0.7 | 4.9±0.6 | 1.7±0.1 | 3.4 | 0.4 |
| 7d | 0.7±0.1 | 2.0±0.3 | 0.6±0.1 | 2.9±0.5 | 2.9±0.4 | 0.7±0.1 | 1.6 | 0.4 |
| 7e | 0.7±0.1 | 3.2±0.4 | 0.7±0.1 | 3.3±0.5 | 3.3±0.5 | 1.0±0.1 | 2.0 | 0.3 |
| 7f | 0.7±0.1 | 4.1±0.7 | 0.7±0.1 | 4.1±0.6 | 3.6±0.5 | 1.1±0.2 | 2.4 | 0.3 |
| 7g | 0.9±0.1 | 3.9±0.5 | 0.9±0.2 | 2.2±0.4 | 2.8±0.5 | 1.4±0.1 | 2.0 | 0.3 |
| 7h | 1.0±0.1 | 5.9±0.8 | 0.8±0.1 | 4.1±0.7 | 6.0±0.5 | 1.8±0.2 | 3.3 | 0.3 |
| 7i | 0.9±0.1 | 3.2±0.4 | 0.6±0.1 | 1.8±0.2 | 2.7±0.3 | 2.2±0.2 | 1.9 | 0.7 |
| 7j | 0.7±0.1 | 2.4±0.3 | 0.7±0.1 | 3.1±0.4 | 3.0±0.4 | 0.5±0.1 | 1.7 | 0.2 |
| 7k | 0.6±0.1 | 2.6±0.4 | 0.6±0.1 | 2.8±0.5 | 2.7±0.4 | 0.9±0.1 | 1.7 | 0.4 |
| 10 | 1.1±0.2 | 4.8±0.8 | 1.3±0.2 | 2.2±0.4 | 2.7±0.4 | 1.6±0.2 | 2.3 | 0.3 |
| 12a | 1.0±0.1 | 2.2±0.3 | 0.9±0.2 | 1.5±0.3 | 1.9±0.3 | 1.2±0.1 | 1.5 | 0.5 |
| 12b | 0.7±0.1 | 2.6±0.4 | 1.4±0.2 | 2.5±0.3 | 2.4±0.4 | 0.7±0.1 | 1.7 | 0.3 |
| 12c | 0.9±0.1 | 3.1±0.3 | 2.1±0.3 | 6.3±0.7 | 4.2±0.7 | 1.6±0.1 | 3.0 | 0.5 |
| 12d | 1.0±0.1 | 3.8±0.6 | 1.9±0.3 | 6.2±0.7 | 4.0±0.5 | 1.7±0.1 | 3.1 | 0.5 |
| 12e | 1.0±0.2 | 2.4±0.3 | 0.7±0.1 | 2.4±0.3 | 3.0±0.5 | 0.9±0.1 | 1.7 | 0.4 |
| 14 | 0.8±0.1 | 4.7±0.5 | 0.6±0.1 | 2.0±0.3 | 2.4±0.3 | 5.1±0.6 | 2.6 | 1.1 |
| 16 | 1.0±0.2 | 2.1±0.3 | 0.8±0.1 | 1.3±0.3 | 5.7±0.8 | 0.2±0.0 | 1.9 | 0.1 |
| 19a | 0.5±0.1 | 0.5±0.1 | 0.7±0.1 | 0.5±0.1 | 0.8±0.1 | 1.4±0.1 | 0.7 | 2.6 |
| 19b | 0.6±0.1 | 0.6±0.1 | 0.5±0.1 | 1.3±0.1 | 1.5±0.1 | 1.0±0.1 | 0.9 | 1.6 |
| 19c | 0.8±0.1 | 1.2±0.1 | 0.7±0.1 | 1.7±0.1 | 3.1±0.3 | 1.0±0.1 | 1.4 | 0.8 |
| 19d | 0.8±0.1 | 1.8±0.2 | 0.7±0.1 | 0.9±0.1 | 1.6±0.2 | 2.3±0.2 | 1.4 | 1.3 |
| 19e | 0.7±0.1 | 1.8±0.2 | 1.1±0.2 | 1.7±0.2 | 3.3±0.3 | 1.0±0.1 | 1.6 | 0.6 |
| 19f | 0.8±0.1 | 1.9±0.3 | 0.6±0.1 | 0.8±0.1 | 1.3±0.2 | 3.0±0.4 | 1.4 | 1.6 |
| 19g | 0.9±0.1 | 1.9±0.2 | 1.3±0.1 | 4.4±0.6 | 5.7±0.9 | 1.2±0.2 | 2.6 | 0.6 |
| 19h | 0.6±0.1 | 1.1±0.2 | 0.5±0.1 | 0.6±0.1 | 0.8±0.1 | 0.9±0.1 | 0.8 | 0.9 |
| YM-155 (nM) | ndb | 2.4±0.2 | ndb | ndb | ndb | >1μM | ndb | >425 |
| MX-106 | 1.0±0.1 | 3.5±0.4 | 1.1±0.1 | 1.6±0.2 | 2.3±0.2 | 2.1±0.1 | 2.0 | 0.6 |
Resistance Index (IR) is calculated by dividing IC50 values on multidrug-resistant cell line MDA-MB- 435/LCC6MDR1by IC50 values on the matching sensitive parental cell line MDA-MB-435.
Not determined.
Compounds 4a–4d have less conformationally flexible linkers between rings B and C relative to the flexible ether linker in parental MX-106. Compounds 4a–4c showed less potency with an average IC50 values of 4.2, 2.9, and 2.7 μM for compounds 4a, 4b, and 4c respectively as compared to 2.0 μM of MX-106 (unless specified, the IC50 value for each analogue is expressed as the average of results obtained in all six cell lines). Compound 4d showed activity comparable to that of MX-106 in the cell lines tested, with an IC50 value of 1.9 μM. Overall, converting the flexible ether linker in MX-106 to less conformationally flexible linkers do not seem to affect the potency significantly in this scaffold. Next, we kept the trizole ring as a linker and modified the C ring. We synthesized compounds containing several substituted benzene rings in the C ring position by introducing various electron donating groups (methyl, ethyl, isopropyl, methoxy, and benzyloxy) or electron withdrawing groups (bromo, trifluoromethyl, nitro, and cyano) in different positions (para, ortho, and meta) on the phenyl C ring.
We introduced different para-substitutions with electron-withdrawing functional groups, such as −Br (compound 7a), −CF3 (compound 7b), −NO2 (compound 7f), and −CN (compound 7h) to the C phenyl ring. Compound 7a (with a bromo substitution in the para position) exhibited same cytotoxic activity to that of MX-106 (IC50 2.0 μM for 7a vs 2.0 μM for MX-106). But stronger electron withdrawing substitutions resulted in decreased activity (IC50 values are 2.4, 2.4, and 3.3 μM for compounds 7b, 7f, and 7h, respectively).
Next, we shifted the bromo substitution position in this C ring in order to probe its best location. While the para-Br compound 7a was same as MX-106 in potency, 7j (ortho-Br) and 7k (meta-Br) were slightly more active, with IC50 value of 1.7 μM, suggesting that the ortho- and meta-bromine substitutions in the C phenyl ring were slightly favorable for antiproliferative activity. Interestingly, compound 7d, in which an electron donating methyl substitution is introduced in the para position, exhibited improved potency (IC50 1.6 μM vs 2.0 μM for MX-106). But analogs with a larger substitution, 7e (para-CH2CH3), 7g (para-OCH3), 7i (para-benzyloxy), or 7c (para-isopropyl) did not provide further enhancement in potency. Bioisosterically replacing the phenyl ring with a thiophene ring to generate compound 10 resulted in slightly decreased activity (IC50 of 2.3 μM) compared to MX-106.
We continued our modification of MX-106 with the pyrrolidine D ring. We compared the effects of different ring sizes or removed the pyrrolidine ring and replaced it with different cyclic and acyclic amines. Compounds 12a, 12b, and 12e possess larger D rings (12a: para-bromophenyl C ring and piperidine D ring, 12b: para-trifluorophenyl C ring and piperidine D ring, 12e: para-bromophenyl C ring and morpholine D ring). These three analogs exhibited slightly higher cytotoxic activity than the parent compound in all cancer cell lines tested (IC50 values:12a, 1.4 μM; 12b, 1.7 μM; 12e, 1.7 μM; MX-106 2.0 μM). Conversely, compound 14 (with a para-bromophenyl C ring and piperazine D ring) exhibited less cytotoxic activity (IC50 2.6 μM) than did MX-106. The D ring of analog 14 was a piperzine ring rather than the morpholine ring found in 12e, suggesting that perhaps a hydrogen acceptor functional group in D ring is favorable for cytotoxic activity. Compound 12d with diethyl amine, an open ring analog of pyrrolidine, exhibited lower activity (IC50 3.1 μM) than MX-106 in all cell lines tested. In compound 12c, the pyrrolidine ring was replaced by an azepane ring, resulting in decreased activity (IC50 3.0 μM) relative to MX-106. On the whole, a 5-membered or 6-membered D ring is favorable in this hydroxyquinoline scaffold of MX-106.
The parent compound MX-106 contains a methylene linker between the B and D rings. To test the significance of this methylene group for the cytotoxic activity of MX-106, we synthesized several analogs that lacked it. Most of these compounds were more active than the MX-106 parent, suggesting that the methylene linker may not be important for its cytotoxic activity. Analogs 19a (with a phenyl D-ring) and 19h (with thiophene D-ring) demonstrated the best cytotoxic activity in all cancer cell lines tested, with IC50 values of 0.7 and 0.8 μM, respectively. Compounds 19d (para-methylphenyl D ring), 19e (meta-methylphenyl D ring), and 19f (para-methoxyphenyl D ring) exhibited higher cytotoxic activity (IC50 values: 19d, 1.4 μM; 19e, 1.6 μM; 19f, 1.4 μM) than MX-106 in the cancer cell lines tested, although all were lower than that of compound 12a. Interestingly, substituting the phenyl D ring with an electron with drawing group such as para-trifluoromethyl (compound 19g, IC50 2.6 μM) decreased the activity of the compound more than did substituting it with electron donating groups such as in compounds 19d and 19f. Finally, compounds 19b (with 4-isoquinoline D ring) and 19c (with 3-quinoline D ring) exhibited higher potency (IC50 values: 19b, 0.9 μM; 19c, 1.4 μM) than MX-106 in all cancer cell lines tested but they were less cytotoxic than 19a.
3.2. In vitro antiproliferative assay in ovarian cancer cell lines
Survivin is highly expressed in ovarian cancer, where it is negatively correlated with overall patient survival [14]. Knockout (KO) of survivin using CRISPR/Cas9 nickase or pharmacological treatment of ovarian cancer cells with small molecule survivin inhibitor YM155 inhibits the epithelial to mesenchymal transition (EMT), which contributes to ovarian tumor metastasis and chemotherapy drug resistance [15–17]. MX106 effectively overcomes chemoresistance in vitro by inhibiting EMT in ovarian cancer cells. MX106 also suppresses primary tumor growth in mouse ovaries and metastases in multiple peritoneal organs as compared to vehicle-treated control mice in an orthotopic ovarian cancer mouse model [14].Therefore, we selected analogs 7a, 7k, 12a, 12b, and 19a described in this study to be evaluated for their cytotoxicity in the cultured human ovarian cell lines OVCAR3 and OVCAR8. The results of this experimental procedure are summarized in Table 2. Compounds 7a, 7k, 12a, and 12b exhibited average IC50 values of 1.3 μM (7a),1.1 μM (7k, 12a, and 12b) in OVCAR3 and OVCAR8 cells, while that of compound 19a was 2 μM.
Table 2.
In vitro growth inhibitory effects of MX-106 analogs in ovarian cancer cell lines (IC50 ± standard error of the mean (SEM) [1], n = 3)
| Compound | OVCAR3 | OVCAR8 | Average IC50 |
|---|---|---|---|
| 7a | 1.0±0.2 | 1.7±0.2 | 1.3 |
| 7k | 0.8±0.6 | 1.3±0.1 | 1.1 |
| 12a | 0.5±0.0 | 1.7±0.1 | 1.1 |
| 12b | 0.6±0.0 | 1.5±0.1 | 1.1 |
| 19a | 1.3±0.2 | 2.6±1.3 | 2.0 |
3.3. Inhibitory effect of MX-106 analogs against cell lines that overexpress P-gp
P-glycoprotein (P-gp) is a clinically relevant drug transporter that acts as a unidirectional efflux pump. It limits the cellular uptake and intracellular concentration of anticancer drugs, leading to ineffective treatment outcomes in multidrug resistant (MDR) cells [18]. Therefore, drug candidates that possess the ability to overcome or circumvent overexpression of P-gp are expected to retain their therapeutic efficacy even in MDR cancer cells. MDR melanoma cell line MDA-MB-435/LCCMDR1 that overexpresses P-gp and its parental sensitive cancer cell line MDA-MB-435 were used to evaluate the abilities of the newly described analogs to surmount P-gp overexpression. The results are shown in Table 1, where they are reported as a function of the cell line tested. The small molecule survivin inhibitor YM155 was also tested on both the multidrug resistant melanoma cells and their parental cells. The resistance index (RI) was calculated by dividing the IC50 value of the tested compound in resistant MDA-MB-435/LCCMDR1 cells by the IC50 value obtained for the same compound in MDA-MB-435 cells. Therefore, the smaller the RI value, the better the ability of the drug to overcome resistance. Twenty-five of the thirty-one new MX-106 analogs tested exhibited more potent inhibitory effects against the resistant MDA-MB-435/LCCMDR1 cell line that overexpressed P-gp than those observed in the parental drug sensitive MDA-MB-435 cell line. These compounds exhibited RI values that were less than 1, suggesting that they could circumvent drug resistance mediated by P-gp. In contrast, small molecule inhibitor YM155 displayed significantly reduced activity against P-gp-overexpressing MDA-MB-435/LCCMDR1 cells, where it exhibited an IC50 of more than 1 μM comparing to 2.4 nM in the sensitive cell. It is worth noting that compounds 19a, 19b, 19d, and 19f lacking the flexible methylene linker between the B and D rings exhibit poor RI values (RI 2.6, 1.6, 1.3, and 1.6, respectively). In contrast MX-106 (RI 0.6) and the remaining analogs, which retained the methylene linker between the B and D rings, exhibited RI values less than 1 (Table 1). These data suggest that the flexibility of the methylene linker might be beneficial for the ability to overcome P-gp overexpression. Collectively, more than 80% of the new MX-106 analogs we describe here were able to overcome drug-resistance mediated by P-gp overexpression in MDA-MB-435/LCCMDR1 cells.
3.4. MX-106 analogs retain selective inhibition for survivin among IAPs
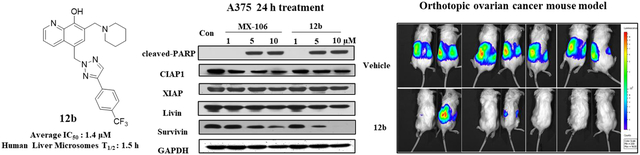
We showed previously that MX-106 selectively downregulated the expression of survivin in cancer cells as compared with other members of the IAP family [12]. To determine whether the new MX-106 analogs maintained the same selective degradation of survivin, we treated A375, a melanoma cell line with compounds 7a, 12b, and 19a and three ovarian cancer cell lines (SKOV3, OVCAR3, and OVCAR8) with compounds 7k and 12b and performed western blotting assay to monitor survivin protein levels (Figures 2 and 3). MX-106 served as positive control. Analogs 7a and 12b selectively suppressed survivin levels in A375 cancer cells in a dose-dependent manner, while the levels of other IAPs were minimally affected (Figures 2A and 2B). Also, compound 19a selectively downregulated survivin in A375 cells, although to a lesser extent than we observed for compounds 7a and 12b (Figure 2C). The western blot result for 19a is not conclusive since 10 μM treatment did not show survivin downregulation unlike 7a and 12b treatments. Consistent with downregulation of survivin, compounds 7a, 12b, and 19a effectively induced apoptosis in cancer cells, as indicated by our detection of elevated levels of an apoptosis, cleaved poly (ADP-ribose) polymerase (cleaved-PARP) (Figure 2A–2C). Similarly, treatment of ovarian cancer cell lines OVCAR8 (Figure 3A), SKOV3 (Figure 3B), and OVCAR3 (Figure 3C) with compounds 7k and 12b reduced the accumulation of survivin protein.
Figure 2.
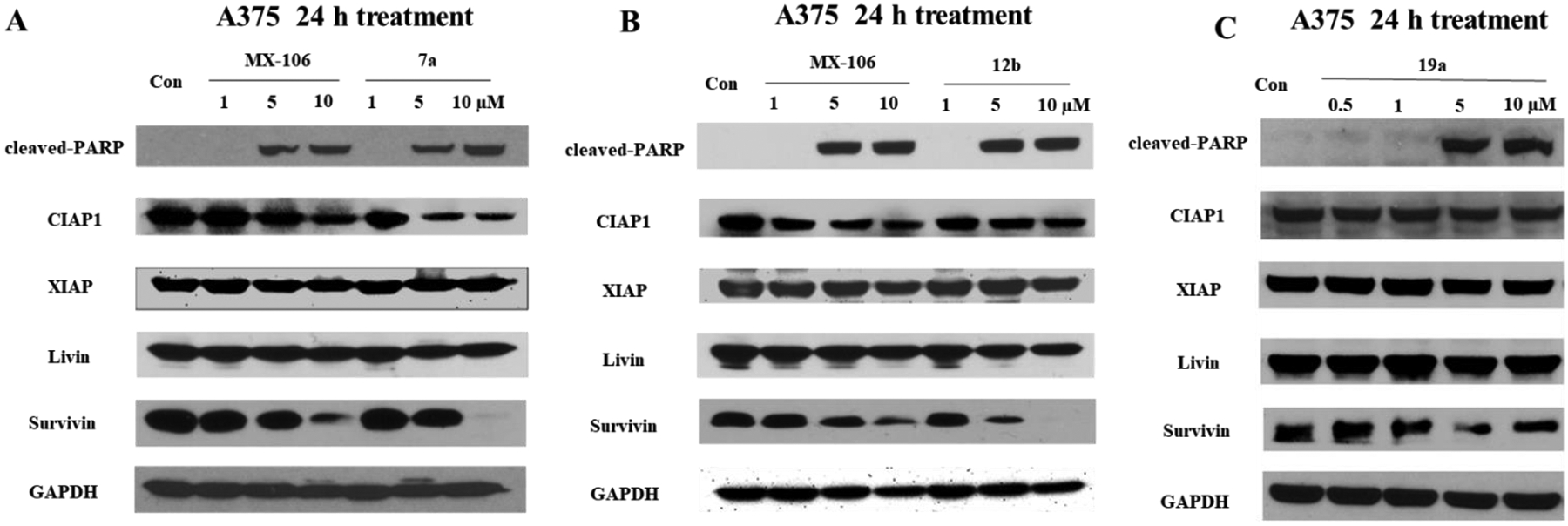
Compounds 7a, 12b, and 19a selectively downregulate survivin in melanoma cancer cells. Western blot analysis of A375 cells treated for 24 h with increasing doses of (A) MX-106 and compound 7a, (B) MX-106 and compound 12b, or (C) compound 19a. Primary antibodies were specific for cleaved apoptosis marker poly(ADP-ribose) polymerase (cleaved-PARP); IAP family members cellular inhibitor of apoptosis protein-1 (CIAP1), X-linked inhibitor of apoptosis protein (XIAP), melanoma inhibitor of apoptosis protein (Livin), and baculoviral inhibitor of apoptosis repeat-containing protein 5 (survivin or BIRC5); and for the internal control glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
Figure 3.
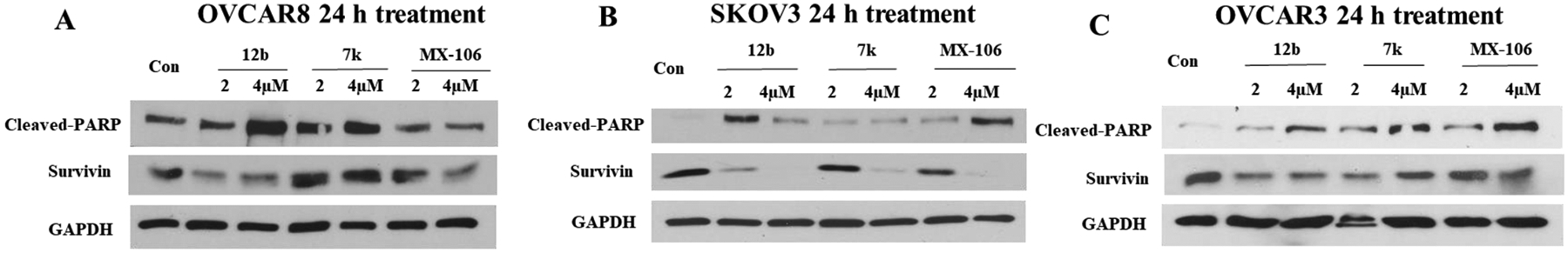
Compounds 7k and 12b downregulate survivin in ovarian cancer cells. Western blot analysis of ovarian cancer cells treated for 24 h with gradient increasing doses of compounds 7k, 12b, and MX-106: (A) OVCAR8 cells, (B) SKOV3 cells and (C) OVCAR3 cells were treated, respectively. Primary antibodies were specific for apoptosis marker cleaved-PARP, IAP survivin, or for the internal control GAPDH as described in the legend to Figure 2.
3.5. Metabolic stability of MX-106 analogs in vitro in liver microsomes
MX-106 has a half-life of 51 minutes with a clearance rate at 136.6 μL/min/mg in human liver microsomes [12]. MX-106 can potentially be deactivated metabolically by the hydrolysis of the ether linker. To improve the metabolic stability of MX-106, we blocked its metabolic labile site and designed new MX-106 analogs described above. We examined the metabolic stabilities of analogs 7a, 7k, 12a, 12b, 19a, and 19b in vitro by measuring their half-lives on incubation with mouse, rat, and human liver microsomes in the presence of an NADPH regenerating system. The results are summarized in Table 3. All tested compounds possessed longer half-lives and lower clearance in comparison to MX-106 in human liver microsomes.
Table 3.
In vitro microsomal stabilities of compounds 7a, 7k, 12a, 12b, 19a, and 19b in liver microsomes from mice, rats, or humans
| Compounds | Metabolic stability in mouse | Metabolic stability in rat | Metabolic stability in human | |||
|---|---|---|---|---|---|---|
| t1/2 (h) | Clint (μL/min/mg) | t1/2 (h) | Clint (μL/min/mg) | t1/2 (h) | Clint (μL/min/mg) | |
| 7a | 0.8 | 27.8 | 1.0 | 22.6 | 1.9 | 12.0 |
| 7k | 1.0 | 24.3 | 0.9 | 24.8 | 2.3 | 10.0 |
| 12a | 0.7 | 35 | 1.3 | 18.0 | 1.3 | 17.5 |
| 12b | 1.1 | 21.6 | 1.6 | 14.2 | 1.5 | 15.7 |
| 19a | >4 | 5.5 | 2.5 | 9.2 | >4 | 3.2 |
| 19b | 1.2 | 19.9 | 0.6 | 36.7 | 1.4 | 16.3 |
| Verapamil | 0.1 | 192.5 | 0.2 | 100.4 | 0.3 | 85.6 |
3.6. Binding kinetics of compound 12b to survivin.
To determine if 12b suppresses survivin expression by directly interacting with the survivin protein, we used a PlexArray HT system (Plexera Bioscience, Woodinville, WA) that employs surface plasmon resonance (SPR) technology to evaluate the interaction and measure its affinity. Biotinylated survivin protein was expressed in E.coli BL21 (DE3) strains (Invitrogen; Thermo Fisher Scientific) and immobilized to a gold-coated sensor chip via avidin-biotin conjugation and compound 12b added at different concentrations for detection of binding in relative resonance units (RU) and determination of affinity of 12b for survivin. Resonance values for 12b increased in a concentration-dependent manner (Figure 4). We used a steady-state fitting model to calculate the equilibrium dissociation constant (KD) of compound 12b binding to survivin at 4.27 μM. Although preliminary and the exact binding site on survivin protein is yet to be determined, this result demonstrates that 12b directly interact with survivin. We are currently working with a CRO (Creative Biostructures, Inc) to optimize the crystallization condition in order to obtain the crystal structures of survivin protein in complexes with this scaffold and will report the results in the future.
Figure 4.
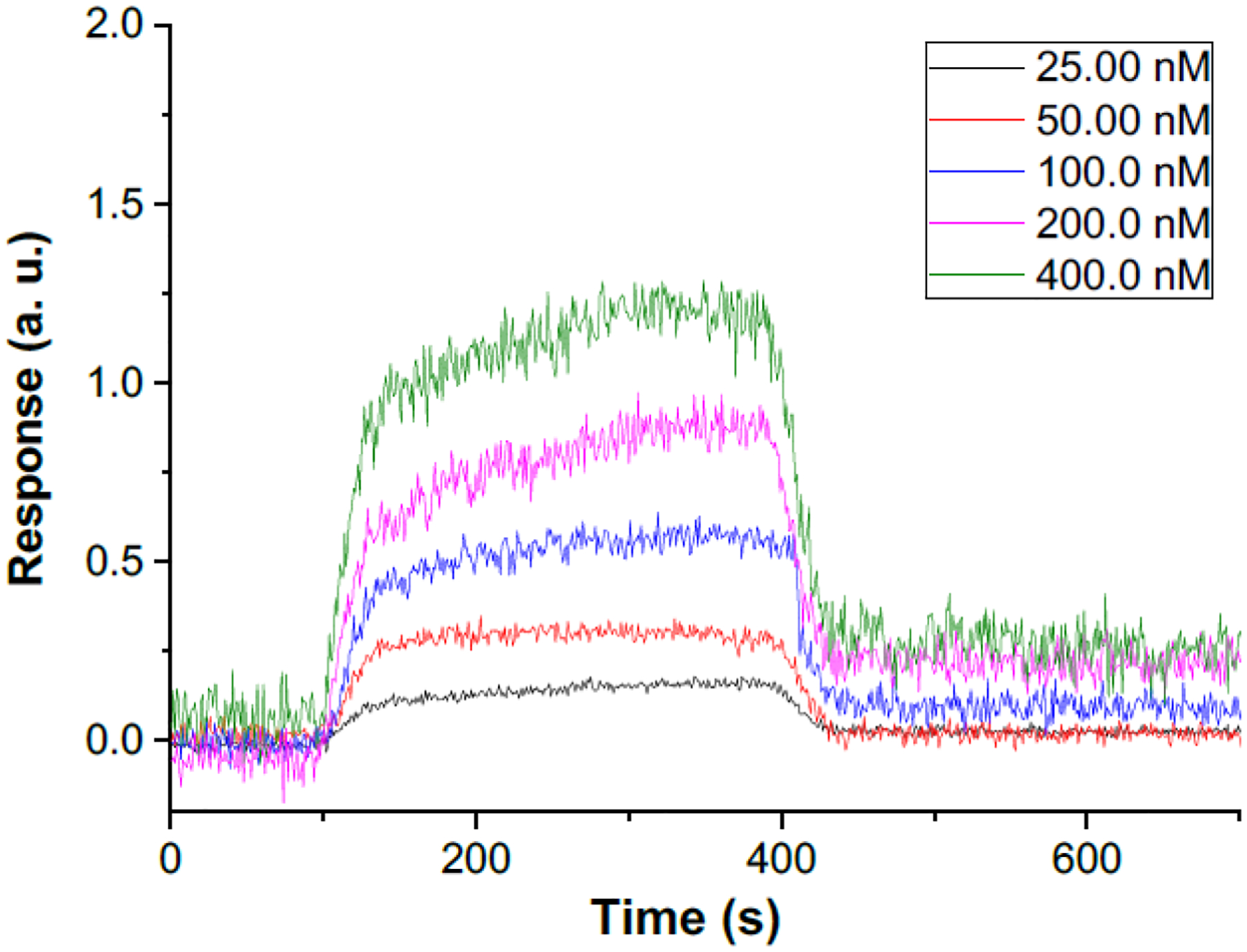
Kinetics of binding survivin protein by compound 12b at 25, 50, 100, 200, 400 nM. Binding of compound 12b to survivin protein was measured using SPR technology. Resonance was measured in arbitrary units at each concentration and plotted as a function of incubation time.
3.7. Molecular modeling of the interaction between survivin and compound 12b
To explain the observed potency of compound 12b we performed a molecular modeling study using the known complex of human survivin-SMAC AVPI, determined by solving the crystal structure for human survivin and the 1–15 peptide from Smac/DIABLO (PDB entry: 3UIH), as a scaffold. The results are shown in Figures 5A and 5B. Compound 12b was predicted to form salt bridge and π-π stacking interactions with the survivin protein BIR domain. The key contacts predicted are as follows: (1) a possible salt bridge interaction between the 8-hydroxyquinoline of 12b and residue Lys79 of survivin (Figure 5B, purple arrow at right); (2) two possible salt bridge interactions between the piperidine of 12b and survivin residues Asp71 and Glu 65 (Figure 5B, purple arrows at top and left); (3) a possible π- π stacking interactions between the 8-hydroxyquinoline and triazole ring of 12b and residue His80 of survivin (Figure 5B, green lines at right); and (4) a π-cation interaction between the piperidine of 12b and residue Trp67 of survivin (Figure 5B, red lines at left).
Figure 5.
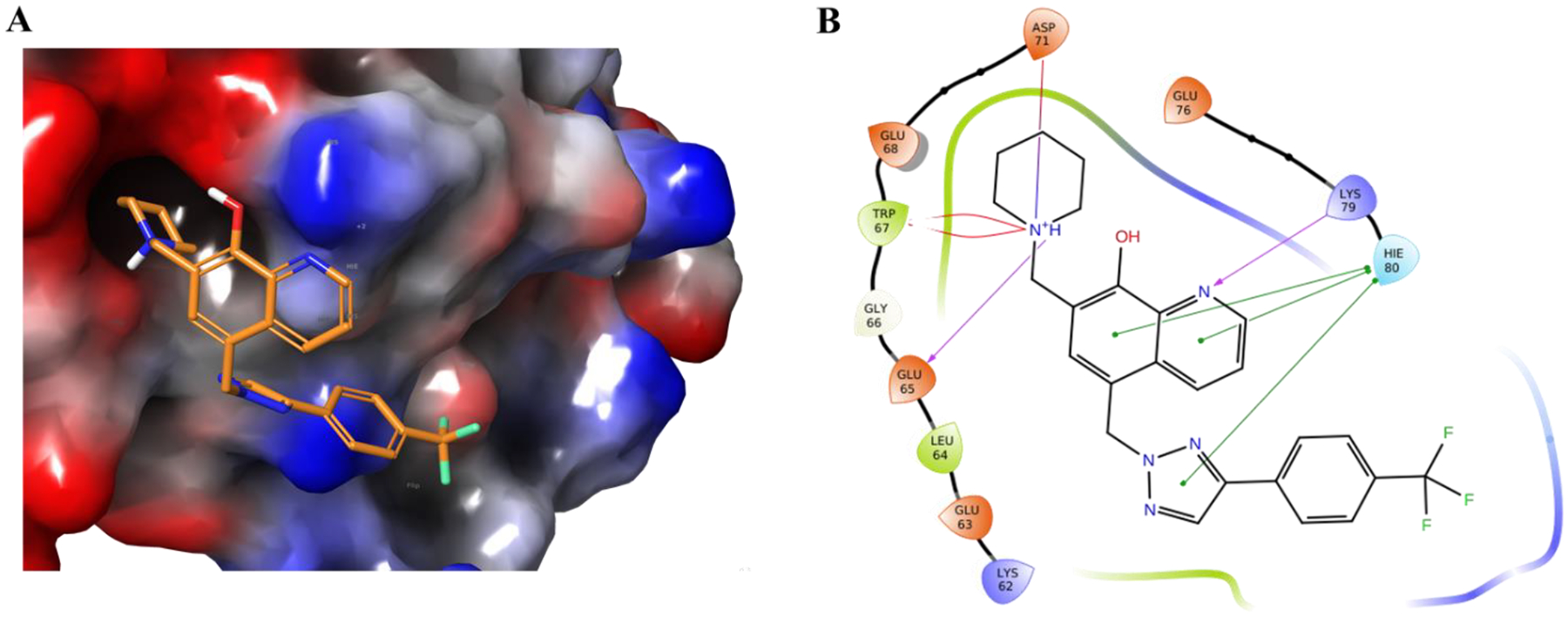
Potential binding of 12b to survivin based on modeling of 12b onto the crystal structure of survivin bound to the Smac/DIABLO AVPI peptide. (A) The best docking pose of 12b binding to survivin (survivin PDB: 3UIH), shown with the surface of the protein color-coded by electron potential (red: electron negative potential; blue: electron positive potential). (B) Types of interactions predicted between 12b and residues in survivin protein are shown in a 2D interaction map. Predicted contacts include: 1) a salt bridge interaction between 8-hydroxyquinoline of 12b and residue Lys79 of survivin (purple arrow at right); (2) two salt bridge interactions between the piperidine of 12b and survivin residues Asp71 and Glu 65 (purple arrows at top and left); (3) a π- π stacking interactions between the 8-hydroxyquinoline and triazole ring of 12b and residue His80 of survivin (green lines at right); and (4) a π-cation interaction between the piperidine of 12b and residue Trp67 of survivin (red lines at left).
3.8. In vivo assessment of antitumor efficacy
3.8.1. Antitumor activity of compound 12b in A375 subcutaneous and OVCAR8 orthotopic xenograft models in mice.
As one of the most potent MX-106 analogs we describe here, compound 12b significantly inhibited the accumulation of survivin protein at a low micromolar concentration in a dose dependent manner and was stable in mouse and human liver microsomes. Therefore, we selected compound 12b for further evaluation of its antitumor efficacy in two xenograft mouse models for cancer. We investigated the effect of treatment with 12b on inhibition of solid tumors in a subcutaneous human melanoma A375 xenograft model in mice. A375 melanoma tumors were implanted in the flank of immunodeficient NOD scid gamma (NSG) mice and allowed to grow for 2 weeks until the average tumor volume reached 100 mm3 prior to treatment. Groups of mice (n=7/group) were treated with vehicle alone or with 20 mg/kg or 40 mg/kg doses of 12b by intraperitoneal (IP) administration 3 days/week for 15 days. Tumor size (volume) and body weight were measured every 2 days.
As shown in Figure 6A, Compound 12b significantly inhibited tumor growth in a dose-dependent manner. All of the mice were stable and there was no significant body weight loss observed, suggesting that doses of 20 mg/kg and 40 mg/kg were equally well tolerated (Figure 6B). At the end of the experiment, the mice were sacrificed, and tumors were removed, weighed, and measured. Compared to the vehicle control, both doses of compound 12b significantly inhibited growth of the tumors, with inhibition of tumor volume by approximately 41% (20 mg/kg) and 66% (40 mg/kg) and reduction of tumor weight by approximately 32% (20 mg/kg) and 55% (40 mg/kg) (Figures 6C–E). These results demonstrated that treatment of mice with compound 12b attenuated the progression of melanoma tumors at a safe dosage.
Figure 6.
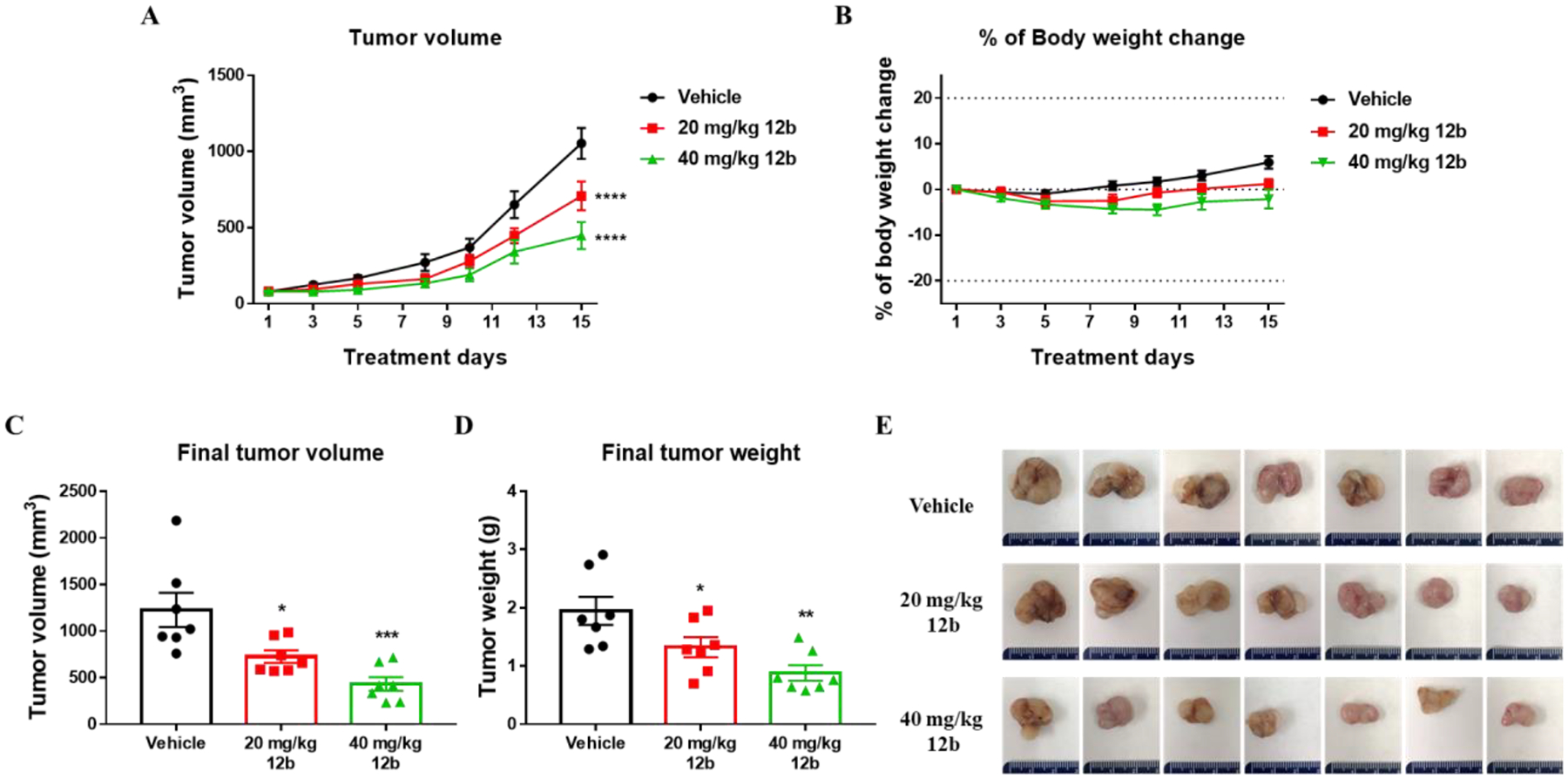
The antitumor efficacy of compound 12b against growth of A375 xenograft tumors in NSG mice. A375 cells were subcutaneously injected into the dorsal right flank of NSG mice. When average tumor volume reached 100 mm3, mice were randomized into 3 groups (n=7/group) and treated with vehicle control or with 20 mg/kg or 40 mg/kg doses of compound 12b. (A) Tumor growth curves with 20 mg/kg and 40 mg/kg compound 12b versus vehicle. Tumor volumes were monitored 3 times/week. Statistical significance was assessed using twoway ANOVA with Dunnett’s multiple comparisons test (****p < 0.0001). (B) Mouse body weight was recorded thrice a week throughout the 15 days treatment period. (C-D) The final volume (C) (one-way ANOVA, *p = 0.0165, ***p = 0.0004) and weight (D) (one-way ANOVA, *p = 0.0450, **p = 0.0015) of inoculated tumors were measured at the end of the experiment. (E) Images of tumors isolated from each treatment group.
3.8.2. Treatment with compound 12b reduced the growth rate and metastasis of ovarian tumors in an orthotopic OVCAR8 ovarian cancer mouse model.
Survivin is overexpressed in ovarian cancer and our lead compound MX106 suppresses the growth of primary ovarian tumors and subsequent metastasis [14]. We utilized an established orthotopic ovarian cancer mouse model to further evaluate the therapeutic potential of compound 12b for treatment of survivin-overexpressing ovarian tumors. This was done by surgically injecting OVCAR8 tumor cells expressing luciferase into the left side of the ovarian bursa (left ovary) of female mice. One week following orthotopic implantation, the mice were separated into 2 groups that were treated with either vehicle or 20 mg/kg compound 12b (3 times/week by IP injection). We began treatment with 4 iterations of the lower dose in case the implantation surgery had weakened the mice, then increased the dose to 40 mg/kg for an additional 4 treatments. The mice were monitored for body weight and bioluminescence imaging after administration of D-luciferin. Mice were sacrificed after 9 days of treatment and all the major organs/tissues were harvested and bioimaged to visualize metastases.
After 2.5 weeks of treatment, mice that had received treatment with compound 12b exhibited a significant reduced total photon flux in tumors relative to that in mice treated with only the vehicle control, (Figures 7A and 7B). Examination of body weight during the treatment period showed no severe weight loss during the course of the 12b treatment (Figure 7C). Bioimaging of the excised uterus and ovaries from each mouse ex vivo is shown in Figure 7D. In each case, the right ovary was normal whereas the left ovary bore the tumor. In the vehicle control group, the tumor-bearing left ovaries exhibited higher luciferase activity than did the normal right ovaries due to increased tumor burden. However, treatment with compound 12b resulted in a remarkable decrease in luciferase activity (photon flux) of the excised ovaries, presumably due to delayed tumor progression (Figure 7D). We confirmed the results of the bioluminescence measurements by visual inspection of the representative uterus images and by measurement of tumor weight (Figure 7E). The tumor masses of the animals treated with compound 12b displayed a very strong decrease in tumor weight compared to the control group (12b: 0.11 ± 0.02; control: 0.18 ± 0.01), as shown in the right side panel of Figure 7E.
Figure 7.
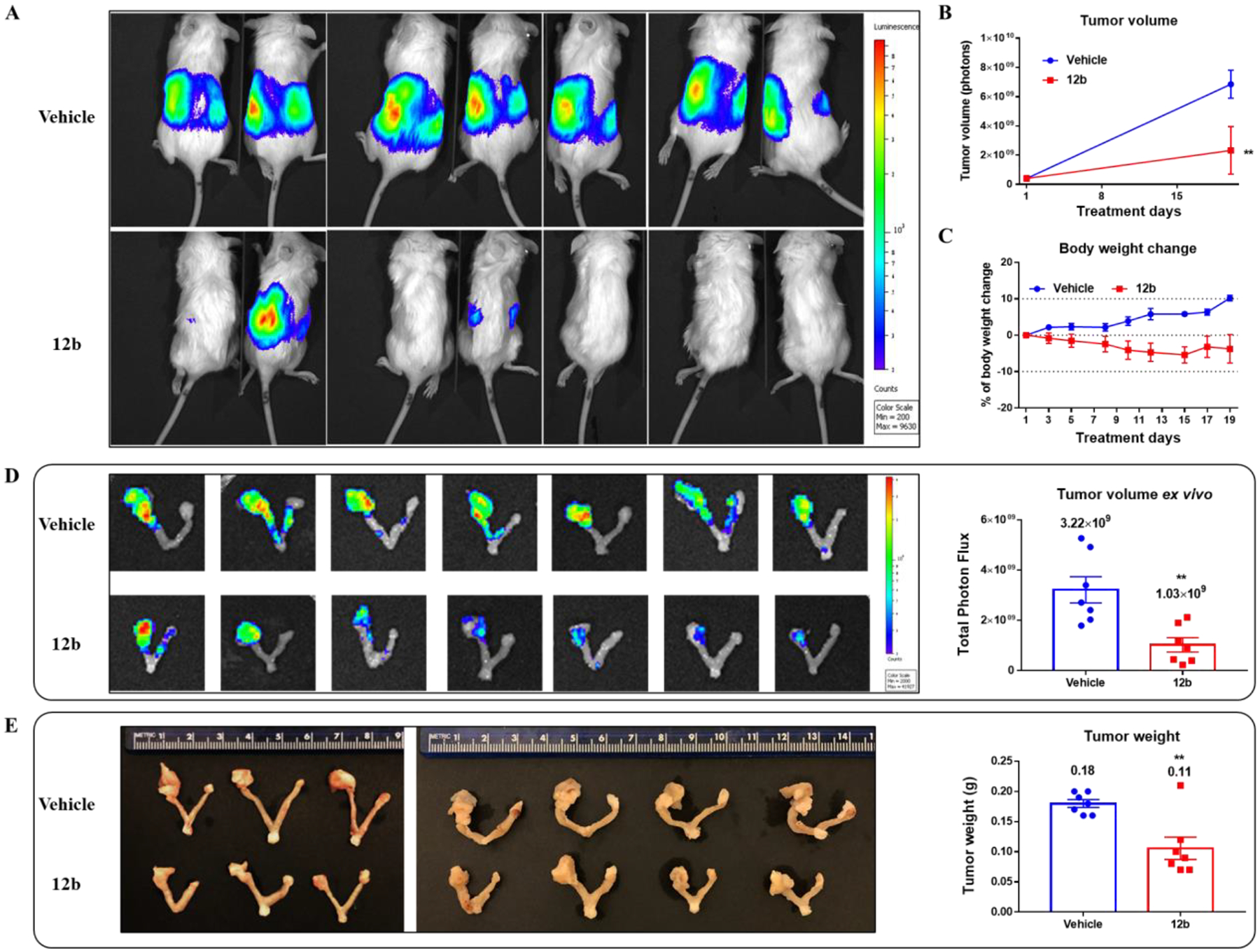
Compound 12b reduced growth of primary tumors in an orthotopic ovarian cancer mouse model. Luciferase-expressing OVCAR8 cells were injected into the left side of the ovarian bursa of 7-week old NSG mice. After one week of tumor progression, mice were randomly separated into 2 groups (vehicle and 12b treatment groups) based on the photon flux in the tumors and mice body weight and drug treatment was initiated. (A) Representative pseudocolor images tracking OVCAR8 xenografts in mice at the experimental endpoint. The tumor photon flux (B) (two-way ANOVA, **p = 0.0068) and mouse body weight (C) were monitored during the treatment. (D) Bioluminescence images of excised ovarian tumor mass ex vivo and scatter plot with a bar showing the final tumor photon flux ± SEM of excised tumors (Student t-test, **p = 0.0031). (E) Pathological images of uterus and ovaries from each mouse and scatter plot with a bar representing the wet weight of ovarian tumor ± SEM (Student t-test, **p = 0.0027).
Since distant metastases often occur as a late complication in ovarian cancer patients, we resected nearby major organs (liver, kidney, spleen, stomach, and intestine) from both groups of mice and imaged them ex vivo to look for bioluminescence signals that represented metastases. This allowed us to determine the effect of 12b in suppressing peritoneal multi-organ metastasis in the orthotopic OVCAR8 ovarian cancer model. The area and total photon flux of metastatic tumors in liver (Figure 8A), kidney (Figure 8B), spleen (Figure 8C) and stomach (Figure 8D) were significantly less in number and smaller in mice treated with compound 12b than in control animals. However, there was no significant inhibition of metastatic intestinal tumors in mice treated with compound 12b (Figure 8E). Taken together, the results of our in vivo studies using the orthotopic ovarian cancer model demonstrate that treatment of mice with compound 12b led to inhibition of tumor growth and visceral metastases in ovarian tumors that overexpressed survivin.
Figure 8.
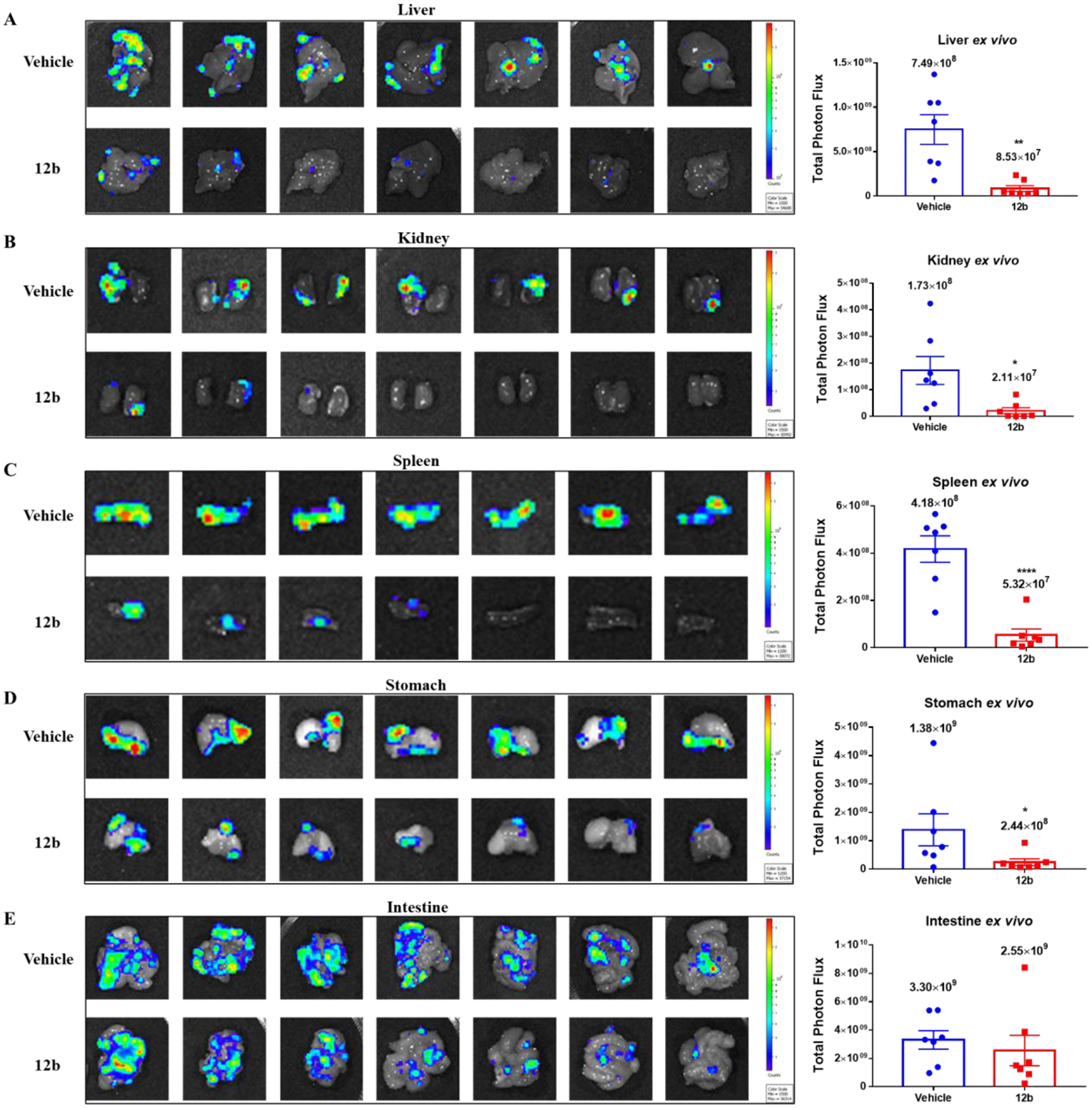
Anti-metastatic effect of compound 12b in the orthotopic OVCAR8 ovarian cancer mouse model. At the endpoint of the study described in Figure 7, all the major organs were harvested from the mice and bioimaged ex vivo to visualize metastases. Images of metastases in (A) liver (Student t-test, **p = 0.0021); (B) kidney (Student t-test, *p = 0.0154); (C) spleen (Student t-test, ****p < 0.0001); (D) stomach (Student t-test, *p = 0.0408); and (E) intestine of each mouse in the vehicle group (top panel) and 12b treatment group (bottom panel) are shown on the left and scatter plots with the bar representing the mean ± SEM of total photon flux for each tissue are shown on the right.
4. Conclusions
We designed and synthesized thirty-one novel MX-106 analogs that optimized the linker, C ring, and D ring moieties of MX-106. Several analogs showed comparable potency to that of MX-106 in killing tumor cells and improved metabolic stability in human liver microsomes. The new MX-106 analogs selectively downregulate the accumulation of survivin protein in a dose-dependent manner. They also exhibited significant abilities to overcome P-gp-mediated drug resistance not observed for small molecule survivin inhibitor YM-155. The in vivo efficacy of compound 12b was determined in melanoma and ovarian cancer xenograft models, where it exhibited significant inhibitory effect of tumor growth. Further optimization of the 8-hydroxyquinoline MX-106 scaffold may generate survivin inhibitors that selective for survivin and even more potent in their anti-tumor activity.
5. Experimental section
5.1. Chemistry
Chemical reagents and solvents were purchased from AK Scientific (Mountain View, CA), Alfa Aesar (Ward Hill, MA), or Sigma-Aldrich Chemical Co. (St. Louis, MO) and were used without further purification. Aluminum-backed uniplates (Analtech, Newark, DE) were used for routine thin layer chromatography (TLC), which was monitored using ultraviolet (UV) light. Silica gel (230–400 mesh; Fisher Scientific, Pittsburgh, PA) was used for flash chromatography. A Bruker Ascend 400 (Billerica, MA) spectrometer was used to obtain NMR spectra. Coupling constants (J) are provided in Hertz (Hz). Chemical shifts are reported as parts per million (ppm) relative to DMSO-d6 or tetramethylsilane (TMS) in CDCl3. Melting points were recorded on a Fisher-Johns melting point apparatus (uncorrected). High resolution mass spectra (HRMS) were collected in positive detection mode on a Waters Xevo G2-S time-of-flight (Tof) instrument equipped with an electron-spray ionization (ESI) source (Milford, MA).
5.1.1. Preparation of 5-(chloromethyl)quinolin-8-ol (2).
A mixture of 5.84 g (40.0 mmol) of 8-hydroxyquinoline, 50 mL of concentrated hydrochloric acid, and 6.4 mL of 37% formaldehyde was treated with 0.6 g of zinc chloride and stirred at room temperature (RT) for 48 hr. The mixture was filtered, washed with copious acetone, and dried to produce compound 2 as a yellow solid (7.2g, 88%). 1H NMR (400 MHz, DMSO-d6) δ 11.77 (s, 1H), 9.11 – 9.07 (m, 2H), 8.03 (dd, J = 8.5, 5.1 Hz, 1H), 7.82 (d, J = 8.0 Hz, 1H), 7.36 (d, J = 8.0 Hz, 1H), 5.32 (s, 2H).
5.1.2. General procedure for the preparation of compounds 3a-3d.
Method A was used to synthesize compounds 3a and 3b. To a solution of compound 3 in DMF (12 mL) was added potassium carbonate (3 equivalents). The suspension was stirred at RT for 30 minutes. Salt of 5-chloromethyl-8-quinolinol hydrochloride (compound 2; 1 equivalent) was added to the suspension. The mixture was stirred overnight at RT. Water was added to the suspension and the mixture was extracted with ethyl acetate (30 mL, 3 times). The combined organic phase was washed with brine, dried over anhydrous Na2SO4 and concentrated to obtain the crude compound, which was purified by flash chromatography (dichloromethane: methanol; 10:1).
Method B was used to synthesize compounds 3c and 3d. Compound 2 was suspended in substituted alcohol 3 (5 equivalents) and the mixture was heated at 70°C for 1 h. The solution was poured into a 200 mL solution of hexane: ethyl acetate (1:1) and filtered, then NaHCO3 solution was added to the filtrate. The mixture was extracted 3 times with dichloromethane (30 mL, 3 times), washed with brine, dried over anhydrous Na2SO4 and evaporated to dryness to yield the desired compounds.
5.1.2.1. 5-((4-phenyl-1H-imidazol-1-yl)methyl)quinolin-8-ol (3a).
Pale brown solid (Yield: 97%). 1H NMR (400 MHz, Chloroform-d) δ 8.83 (dd, J = 4.2, 1.4 Hz, 1H), 8.17 (d, J = 8.5 Hz, 1H), 7.73 – 7.69 (m, 2H), 7.59 (s, 1H), 7.49 (dd, J = 8.6, 4.2 Hz, 1H), 7.42 (d, J = 7.8 Hz, 1H), 7.33 (t, J = 7.6 Hz, 2H), 7.23 – 7.16 (m, 2H), 7.13 (d, J = 1.2 Hz, 1H), 5.49 (s, 2H).
5.1.2.2. 5-((4-phenyl-2H-1,2,3-triazol-2-yl)methyl)quinolin-8-ol (3b).
Pale yellow solid (Yield: 90%) 1H NMR (400 MHz, Chloroform-d) δ 8.80 – 8.77 (m, 1H), 8.63 (dd, J = 8.7, 1.5 Hz, 1H), 7.81 (s, 1H), 7.77 – 7.73 (m, 2H), 7.62 (d, J = 7.8 Hz, 1H), 7.51 (dd, J = 8.6, 4.2 Hz, 1H), 7.41 (t, J = 7.6 Hz, 2H), 7.36 – 7.32 (m, 1H), 7.17 (d, J = 7.8 Hz, 1H), 5.95 (s, 2H).
5.1.2.3. 5-(((2,3-dihydro-1H-inden-2-yl)oxy)methyl)quinolin-8-ol (3c).
White solid (Yield: 70%). 1H NMR (400 MHz, Chloroform-d) δ 8.79 (dd, J = 4.2, 1.6 Hz, 1H), 8.49 (dd, J = 8.5, 1.5 Hz, 1H), 7.49 – 7.42 (m, 2H), 7.21 – 7.18 (m, 2H), 7.17 – 7.13 (m, 2H), 7.10 (d, J = 7.7 Hz, 1H), 4.90 (s, 2H), 4.46 (tt, J = 6.5, 4.9 Hz, 1H), 3.18 (d, J = 6.5 Hz, 1H), 3.14 (d, J = 6.5 Hz, 1H), 3.05 (d, J = 5.0 Hz, 1H), 3.01 (d, J = 5.0 Hz, 1H).
5.1.2.4. 5-(((1,2,3,4-tetrahydronaphthalen-2-yl)oxy)methyl)quinolin-8-ol (3d).
Yellow solid (Yield: 66%). 1H NMR (400 MHz, Chloroform-d) δ 9.01 (s, 1H), 8.87 (dd, J = 4.4, 1.5 Hz, 1H), 8.56 (dd, J = 8.6, 1.5 Hz, 1H), 7.51 (dd, J = 8.5, 4.4 Hz, 1H), 7.46 (d, J = 7.8 Hz, 1H), 7.18 (d, J = 7.8 Hz, 1H), 7.09 (dddd, J = 12.0, 9.2, 5.8, 3.2 Hz, 4H), 4.94 (d, J = 2.0 Hz, 2H), 3.90 (dddd, J = 8.9, 7.7, 4.9, 3.0 Hz, 1H), 3.71 (d, J = 10.9 Hz, 2H), 2.87 (ddt, J = 31.5, 15.3, 7.7 Hz, 2H), 1.88 (dtd, J = 12.7, 8.9, 5.7 Hz, 1H), 1.42 – 1.22 (m, 1H).
5.1.3. General procedure for the preparation of compounds 4a-4d.
An equimolar mixture of compound 3, paraformaldehyde, and pyrrolidine in anhydrous ethanol (20 mL) was refluxed for 3 hr under argon. After cooling, the solvent was evaporated under reduced pressure. The crude compound was purified by flash chromatography (dichloromethane: methanol; 20:1).
5.1.3.1. 5-((4-phenyl-1H-imidazol-1-yl)methyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol (4a).
Yellow solid (Yield: 81%). Mp: 123–125 °C. 1H NMR (400 MHz, Chloroform-d) δ 8.92 (dd, J = 4.1, 1.6 Hz, 1H), 8.12 (dd, J = 8.5, 1.6 Hz, 1H), 7.74 – 7.70 (m, 2H), 7.57 (d, J = 1.4 Hz, 1H), 7.42 (dd, J = 8.6, 4.2 Hz, 1H), 7.34 (t, J = 7.7 Hz, 2H), 7.25 – 7.19 (m, 1H), 7.15 – 7.12 (m, 2H), 5.47 (s, 2H), 4.00 (s, 2H), 2.72 (q, J = 5.2, 4.1 Hz, 4H), 1.89 (p, J = 3.1 Hz, 4H). 13C NMR (101 MHz, Chloroform-d) δ 154.29, 149.04, 142.46, 139.67, 137.31, 133.97, 130.84, 128.60, 128.48, 126.86, 126.22, 124.70, 121.91, 120.06, 118.19, 114.87, 57.55, 53.77, 48.24, 23.66. HRMS: calculated for C24H24N4O [2]+ 385.2023, found 385.2029 (mass error = 1.6 ppm).
5.1.3.2. 5-((4-phenyl-2H-1,2,3-triazol-2-yl)methyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol (4b).
Pale orange solid (Yield: 88%). Mp: 102–103 °C. 1H NMR (400 MHz, Chloroform-d) δ 8.87 (dd, J = 4.2, 1.6 Hz, 1H), 8.57 (dd, J = 8.6, 1.6 Hz, 1H), 7.81 (s, 1H), 7.77 – 7.73 (m, 2H), 7.50 (s, 1H), 7.46 (dd, J = 8.6, 4.2 Hz, 1H), 7.43 – 7.40 (m, 1H), 7.39 (d, J = 1.6 Hz, 1H), 7.37 – 7.31 (m, 1H), 5.93 (s, 2H), 4.11 (s, 2H), 2.84 (br s, 4H), 1.95 – 1.90 (m, 4H). 13C NMR (101 MHz, Chloroform-d) δ 148.71, 147.80, 139.42, 132.19, 131.27, 130.24, 130.14, 128.85, 128.47, 126.99, 125.90, 121.88, 120.45, 56.80, 56.12, 53.61, 23.58. HRMS: calculated for C23H23N5O [9]+ 386.1975, found 386.1976 (mass error = 0.3 ppm).
5.1.3.3. 5-(((2,3-dihydro-1H-inden-2-yl)oxy)methyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol (4c).
Red orange semisolid (Yield: 86%). 1H NMR (400 MHz, Chloroform-d) δ 8.87 – 8.82 (m, 1H), 8.46 (d, J = 8.5 Hz, 1H), 7.59 (s, 1H), 7.46 (dd, J = 8.4, 4.2 Hz, 1H), 7.23 – 7.11 (m, 4H), 4.89 (s, 2H), 4.53 – 4.46 (m, 1H), 4.22 (s, 2H), 3.60 (d, J = 10.7 Hz, 1H), 3.22 (d, J = 6.5 Hz, 1H), 3.18 (d, J = 6.5 Hz, 1H), 3.07 (d, J = 4.8 Hz, 1H), 3.04 – 2.98 (m, 4H), 2.01 – 1.95 (m, 4H). 13C NMR (101 MHz, Chloroform-d) δ 152.73, 148.47, 140.76, 138.99, 133.43, 129.73, 127.67, 126.56, 124.70, 124.41, 122.06, 79.78, 69.12, 53.23, 44.80, 39.30, 24.46, 23.38. HRMS: calculated for C24H26N2O2 [9]+ 375.2067, found 375.2065 (mass error = −0.5 ppm).
5.1.3.4. 7-(pyrrolidin-1-ylmethyl)-5-(((1,2,3,4-tetrahydronaphthalen-2-yl)oxy)methyl)quinolin-8-ol (4d).
Pale orange solid (Yield: 72%). Mp: 72–73 °C. 1H NMR (400 MHz, Chloroform-d) δ 9.88 (s, 1H), 8.86 (d, J = 4.2 Hz, 1H), 8.38 (d, J = 8.5 Hz, 1H), 7.38 (dd, J = 8.7, 4.1 Hz, 1H), 7.30 (s, 1H), 7.09 (th, J = 7.3, 3.6 Hz, 4H), 4.91 (s, 2H), 4.01 (s, 2H), 3.96 – 3.87 (m, 1H), 3.11 (dd, J = 16.3, 4.9 Hz, 1H), 3.00 – 2.78 (m, 3H), 2.77 – 2.72 (m, 4H), 2.13 (dt, J = 13.0, 4.8 Hz, 1H), 1.89 (q, J = 3.1 Hz, 4H), 1.87 – 1.81 (m, 1H). 13C NMR (101 MHz, Chloroform-d) δ 153.34, 148.57, 139.60, 136.02, 134.46, 132.87, 129.42, 128.56, 127.25, 125.91, 125.79, 123.83, 121.32, 117.11, 73.76, 68.32, 57.10, 53.66, 35.48, 28.43, 27.24, 23.59. HRMS: calculated for C25H28N2O2 [M+H]+ 389.2224, found 389.2226 (mass error = 0.5 ppm).
5.1.4. General procedure for the preparation of compounds 5a-5k and 8.
The terminal alkyne and azidotrimethylsilane (1.5 equivalent) were added to a solution of DMF and MeOH (5:1) under argon atmosphere. After adding CuI (5 mol%), the reaction mixture was refluxed for 12 hr and quenched with water (10 mL) [13, 19]. The mixture was extracted with dichloromethane (30 mL.3 times), dried with anhydrous Na2SO4 and concentrated to obtain the crude compounds, which were purified by flash chromatography (hexane: ethyl acetate; 2:1).
5.1.4.1. 4-(4-bromophenyl)-2H-1,2,3-triazole (5a).
White solid (Yield: 96%). 1H NMR (400 MHz, CDCl3) δ 11.78 (s, 1H), 7.97 (s, 1H), 7.71 (d, J = 8.2 Hz, 2H), 7.59 (d, J = 8.1 Hz, 2H).
5.1.4.2. 4-(4-(trifluoromethyl)phenyl)-2H-1,2,3-triazole (5b).
White solid (Yield: 93%). 1H NMR (400 MHz, CDCl3) δ 11.80 (s, 1H), 8.04 (s, 1H), 7.96 (d, J = 8.0 Hz, 2H), 7.72 (d, J = 8.0 Hz, 2H).
5.1.4.3. 4-(4-isopropylphenyl)-2H-1,2,3-triazole (5c).
White solid (Yield: 99%). 1H NMR (400 MHz, Chloroform-d) δ 13.41 (s, 1H), 8.00 (s, 1H), 7.76 (d, J = 7.9 Hz, 2H), 7.32 (d, J = 7.8 Hz, 2H), 2.95 (h, J = 6.9 Hz, 1H), 1.28 (dd, J = 6.9, 1.3 Hz, 6H).
5.1.4.4. 4-(p-tolyl)-2H-1,2,3-triazole (5d).
Pale yellow solid (Yield: 90%). 1H NMR (400 MHz, Chloroform-d) δ 7.95 (s, 1H), 7.75 – 7.68 (m, 2H), 7.28 (d, J = 0.9 Hz, 2H), 2.40 (s, 3H).
5.1.4.5. 4-(4-ethylphenyl)-2H-1,2,3-triazole (5e).
White solid (Yield: 93%) 1H NMR (400 MHz, Chloroform-d) δ 7.96 (s, 1H), 7.76 – 7.72 (m, 2H), 7.31 – 7.28 (m, 2H), 2.70 (q, J = 7.6 Hz, 2H), 1.27 (t, J = 7.6 Hz, 3H).
5.1.4.6. 4-(4-nitrophenyl)-2H-1,2,3-triazole (5f).
Pale brown solid (Yield: 88%) 1H NMR (400 MHz, Chloroform-d) δ 8.37 – 8.29 (m, 2H), 8.09 (s, 1H), 8.03 (d, J = 1.8 Hz, 2H).
5.1.4.7. 4-(4-methoxyphenyl)-2H-1,2,3-triazole (5g).
White solid (Yield: 93%) 1H NMR (400 MHz, CDCl3) δ 7.92 (s, 1H), 7.78 – 7.73 (m, 2H), 7.01 – 6.96 (m, 2H), 3.86 (s, 3H).
5.1.4.8. 4-(2H-1,2,3-triazol-4-yl)benzonitrile (5h).
Pale orange solid (Yield: 73%) 1H NMR (400 MHz, CDCl3) δ 11.88 (s, 1H), 8.05 (s, 1H), 7.98 – 7.94 (m, 2H), 7.77 – 7.73 (m, 2H).
5.1.4.9. 4-(4-(benzyloxy)phenyl)-2H-1,2,3-triazole (5i).
White solid (Yield: 91%) 1H NMR (400 MHz, DMSO-d6) δ 8.18 (s, 1H), 7.78 (d, J = 8.3 Hz, 2H), 7.49 – 7.45 (m, 2H), 7.43 – 7.38 (m, 2H), 7.36 – 7.31 (m, 1H), 7.10 (d, J = 8.4 Hz, 2H), 5.15 (s, 2H).
5.1.4.10. 4-(2-bromophenyl)-2H-1,2,3-triazole (5j).
White solid (Yield: 95%) 1H NMR (400 MHz, DMSO-d6) δ 8.32 (s, 1H), 7.76 (dd, J = 8.0, 1.2 Hz, 3H), 7.49 (td, J = 7.5, 1.3 Hz, 2H), 7.34 (td, J = 7.7, 1.8 Hz, 2H).
5.1.4.11. 4-(3-bromophenyl)-2H-1,2,3-triazole (5k).
White solid (Yield: 94%) 1H NMR (400 MHz, Chloroform-d) δ 8.00 (t, J = 1.8 Hz, 1H), 7.98 (s, 1H), 7.78 – 7.75 (m, 1H), 7.52 (ddd, J = 8.0, 2.0, 1.0 Hz, 1H), 7.33 (t, J = 7.9 Hz, 1H).
2.2.4.12. 4-(thiophen-3-yl)-1H-1,2,3-triazole (8).
White solid (Yield: 95%) 1H NMR (400 MHz, Chloroform-d) δ 7.88 (s, 1H), 7.69 (dd, J = 2.9, 1.3 Hz, 1H), 7.50 (dd, J = 5.1, 1.3 Hz, 1H), 7.43 (dd, J = 5.0, 2.9 Hz, 1H).
5.1.5. General procedure for the preparation of compounds 6a-6k and 9.
These compounds were derived from compounds 5a–5k and 8, respectively, and were synthesized individually in parallel. To a solution of compound 5(a-k) or 8 (5 mmol) in DMF (12 mL) was added potassium carbonate (15 mmol). The suspension was stirred at RT for 30 minutes. Salt of compound 2 (5 mmol) was added to the suspension and the mixture was stirred overnight at RT. Water was added to the suspension and the mixture was extracted 3 times with ethyl acetate (30 mL). The combined organic phase was washed with brine, dried over anhydrous Na2SO4 and concentrated to obtain the crude compounds. The crude compounds were purified by flash chromatography (dichloromethane: ethyl acetate; 20:1).
5.1.5.1. 5-((4-(4-bromophenyl)-2H-1,2,3-triazol-2-yl)methyl)quinolin-8-ol (6a).
White solid (Yield: 90%). 1H NMR (400 MHz, Chloroform-d) δ 8.80 (d, J = 4.2 Hz, 1H), 8.63 (d, J = 8.6 Hz, 1H), 8.40 (s, 1H), 7.79 (s, 1H), 7.62 (d, J = 8.2 Hz, 3H), 7.56 – 7.48 (m, 3H), 7.17 (d, J = 7.8 Hz, 1H), 5.94 (s, 2H).
5.1.5.2. 5-((4-(4-(trifluoromethyl)phenyl)-2H-1,2,3-triazol-2-yl)methyl)quinolin-8-ol (6b).
Pale yellow solid (Yield: 90%). 1H NMR (400 MHz, CDCl3) δ 8.80 (d, J = 4.2 Hz, 1H), 8.64 (d, J = 8.6 Hz, 1H), 7.86 (d, J = 6.3 Hz, 3H), 7.65 (t, J = 7.9 Hz, 3H), 7.53 (dd, J = 8.7, 4.2 Hz, 1H), 7.18 (d, J = 7.8 Hz, 1H), 5.97 (s, 2H).
5.1.5.3. 5-((4-(4-isopropylphenyl)-2H-1,2,3-triazol-2-yl)methyl)quinolin-8-ol (6c).
White solid (Yield: 86%). 1H NMR (400 MHz, Chloroform-d) δ 8.79 (d, J = 4.2 Hz, 1H), 8.62 (d, J = 8.6 Hz, 1H), 8.39 (s, 1H), 7.77 (d, J = 1.3 Hz, 1H), 7.67 (d, J = 7.8 Hz, 2H), 7.60 (d, J = 7.7 Hz, 1H), 7.50 (dd, J = 9.0, 4.2 Hz, 1H), 7.28 (s, 2H), 7.17 (d, J = 7.8 Hz, 1H), 5.94 (s, 2H), 2.92 (p, J = 6.8 Hz, 1H), 1.26 (dd, J = 6.9, 1.2 Hz, 6H).
5.1.5.4. 5-((4-(p-tolyl)-2H-1,2,3-triazol-2-yl)methyl)quinolin-8-ol (6d).
White solid (Yield: 66%) 1H NMR (400 MHz, Chloroform-d) δ 8.78 (dd, J = 4.2, 1.5 Hz, 1H), 8.63 (dd, J = 8.6, 1.5 Hz, 1H), 8.42 (s, 1H), 7.77 (s, 1H), 7.67 – 7.57 (m, 3H), 7.50 (dd, J = 8.6, 4.2 Hz, 1H), 7.23 – 7.15 (m, 3H), 5.94 (s, 2H), 2.37 (s, 3H).
5.1.5.5. 5-((4-(4-ethylphenyl)-2H-1,2,3-triazol-2-yl)methyl)quinolin-8-ol (6e).
Pale brown solid (Yield: 73%) 1H NMR (400 MHz, CDCl3) δ 8.79 (dd, J = 4.2, 1.5 Hz, 1H), 8.63 (dd, J = 8.6, 1.5 Hz, 1H), 7.78 (s, 1H), 7.68 – 7.65 (m, 2H), 7.61 (d, J = 7.8 Hz, 1H), 7.50 (dd, J = 8.6, 4.2 Hz, 1H), 7.24 (d, J = 8.1 Hz, 2H), 7.17 (d, J = 7.8 Hz, 1H), 5.94 (s, 2H), 2.67 (q, J = 7.6 Hz, 2H), 1.24 (t, J = 7.6 Hz, 3H).
5.1.5.6. 5-((4-(4-nitrophenyl)-2H-1,2,3-triazol-2-yl)methyl)quinolin-8-ol (6f).
Pale orange solid (Yield: 59%). 1H NMR (400 MHz, CDCl3) δ 8.81 (dd, J = 4.2, 1.5 Hz, 1H), 8.64 (dd, J = 8.6, 1.5 Hz, 1H), 8.29 – 8.24 (m, 2H), 7.94–7.89 (m, 3H), 7.65 (d, J = 7.8 Hz, 1H), 7.54 (dd, J = 8.6, 4.2 Hz, 1H), 7.19 (d, J = 7.8 Hz, 1H), 5.98 (s, 2H).
5.1.5.7. 5-((4-(4-methoxyphenyl)-2H-1,2,3-triazol-2-yl)methyl)quinolin-8-ol (6g).
White solid (Yield: 76%). 1H NMR (400 MHz, Chloroform-d) δ 8.79 (dd, J = 4.2, 1.5 Hz, 1H), 8.63 (dd, J = 8.6, 1.5 Hz, 1H), 7.73 (s, 1H), 7.69 – 7.65 (m, 2H), 7.61 (d, J = 7.8 Hz, 1H), 7.51 (dd, J = 8.6, 4.2 Hz, 1H), 7.17 (d, J = 7.8 Hz, 1H), 6.96 – 6.91 (m, 2H), 5.93 (s, 2H), 3.83 (s, 3H).
5.1.5.8. 4-(2-((8-hydroxyquinolin-5-yl)methyl)-2H-1,2,3-triazol-4-yl)benzonitrile (6h).
Pale brown solid (Yield: 77%). 1H NMR (400 MHz, CDCl3) δ 8.81 (dd, J = 4.2, 1.5 Hz, 1H), 8.63 (dd, J = 8.6, 1.5 Hz, 1H), 7.88 – 7.83 (m, 3H), 7.71 – 7.67 (m, 2H), 7.64 (d, J = 7.8 Hz, 1H), 7.53 (dd, J = 8.6, 4.2 Hz, 1H), 7.18 (d, J = 7.8 Hz, 1H), 5.97 (s, 2H).
5.1.5.9. 5-((4-(4-(benzyloxy)phenyl)-2H-1,2,3-triazol-2-yl)methyl)quinolin-8-ol (6i).
White solid (Yield: 86%). 1H NMR (400 MHz, Chloroform-d) δ 8.79 (dd, J = 4.2, 1.5 Hz, 1H), 8.63 (dd, J = 8.6, 1.5 Hz, 1H), 7.73 (s, 1H), 7.69 – 7.65 (m, 2H), 7.61 (d, J = 7.8 Hz, 1H), 7.51 (dd, J = 8.6, 4.2 Hz, 1H), 7.44 (d, J = 7.4 Hz, 2H), 7.41 – 7.36 (m, 2H), 7.33 (t, J = 7.1 Hz, 1H), 7.17 (d, J = 7.8 Hz, 1H), 7.03 – 6.98 (m, 2H), 5.93 (s, 2H), 5.09 (s, 2H).
5.1.5.10. 5-((4-(2-bromophenyl)-2H-1,2,3-triazol-2-yl)methyl)quinolin-8-ol (6j).
White solid (Yield: 54%). 1H NMR (400 MHz, Chloroform-d) δ 8.80 (dd, J = 4.2, 1.5 Hz, 1H), 8.66 (dd, J = 8.6, 1.5 Hz, 1H), 8.07 (s, 1H), 7.69 – 7.62 (m, 3H), 7.54 – 7.50 (m, 1H), 7.35 (td, J = 7.6, 1.3 Hz, 1H), 7.22 (dd, J = 7.6, 1.8 Hz, 1H), 7.18 (d, J = 7.8 Hz, 1H), 5.97 (s, 2H).
5.1.5.11. 5-((4-(3-bromophenyl)-2H-1,2,3-triazol-2-yl)methyl)quinolin-8-ol (6k).
White solid (Yield: 62%). 1H NMR (400 MHz, Chloroform-d) δ 8.80 (dd, J = 4.2, 1.5 Hz, 1H), 8.62 (dd, J = 8.6, 1.5 Hz, 1H), 8.43 (s, 1H), 7.91 (t, J = 1.8 Hz, 1H), 7.80 (s, 1H), 7.66 (dt, J = 7.7, 1.2 Hz, 1H), 7.63 (d, J = 7.9 Hz, 1H), 7.53 (dd, J = 8.6, 4.2 Hz, 1H), 7.46 (ddd, J = 8.0, 2.0, 1.0 Hz, 1H), 7.28 (d, J = 7.9 Hz, 1H), 7.18 (d, J = 7.8 Hz, 1H), 5.95 (s, 2H).
5.1.5.12. 5-((4-(thiophen-3-yl)-2H-1,2,3-triazol-2-yl)methyl)quinolin-8-ol (9).
Pale pink solid (Yield: 78%). 1H NMR (400 MHz, Chloroform-d) δ 8.79 (dd, J = 4.2, 1.5 Hz, 1H), 8.61 (dd, J = 8.6, 1.5 Hz, 1H), 7.70 (s, 1H), 7.62 – 7.58 (m, 2H), 7.51 (dd, J = 8.6, 4.2 Hz, 1H), 7.42 (dd, J = 5.0, 1.3 Hz, 1H), 7.37 (dd, J = 5.1, 2.9 Hz, 1H), 7.17 (d, J = 7.8 Hz, 1H), 5.93 (s, 2H).
5.1.6. General procedure for the preparation of compounds 7a-7k and 10.
These compounds were derived from compounds 6a–6k and 9, respectively, and were synthesized individually in parallel. An equimolar mixture of the substrates 6(a–k) or 9, paraformaldehyde, and pyrrolidine in anhydrous ethanol (15 mL) was refluxed for 3 hr under argon. After cooling, the solvent was evaporated under reduced pressure. The crude compound was purified by flash chromatography (dichloromethane: methanol 20: 1) to produce the pure products.
5.1.6.1. 5-((4-(4-bromophenyl)-2H-1,2,3-triazol-2-yl)methyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol (7a).
Pink solid (Yield: 87%). Mp: 146–148 °C. 1H NMR (400 MHz, Chloroform-d) δ 8.87 (d, J = 4.1 Hz, 1H), 8.54 (d, J = 8.5 Hz, 1H), 7.79 (s, 1H), 7.61 (d, J = 8.2 Hz, 2H), 7.51 (d, J = 8.2 Hz, 2H), 7.42 (dd, J = 8.6, 4.1 Hz, 1H), 7.37 (s, 1H), 5.90 (s, 2H), 4.01 (s, 2H), 2.70 (d, J = 5.9 Hz, 4H), 1.87 (q, J = 3.9, 3.4 Hz, 4H). 13C NMR (101 MHz, Chloroform-d) δ 154.43, 148.78, 146.77, 139.63, 131.98, 131.19, 129.77, 129.24,127.43, 126.80, 122.37, 121.63, 119.99, 118.17, 57.65, 56.18, 53.76, 23.65. HRMS: calculated for C23H22BrN5O [M+H]+ 464.1080, found 464.1068 (mass error = −2.6 ppm).
5.1.6.2. 7-(pyrrolidin-1-ylmethyl)-5-((4-(4-(trifluoromethyl)phenyl)-2H-1,2,3-triazol-2-yl)methyl)quinolin-8-ol (7b).
Pale brown solid (Yield: 66%). Mp: 144–145 °C. 1H NMR (400 MHz, Chloroform-d) δ 8.88 (d, J = 4.1 Hz, 1H), 8.56 (d, J = 8.6 Hz, 1H), 7.86 (d, J = 7.6 Hz, 3H), 7.65 (d, J = 8.0 Hz, 2H), 7.47 – 7.38 (m, 2H), 5.93 (s, 2H), 4.03 (s, 2H), 2.73 (d, J = 5.8 Hz, 4H), 1.89 (d, J = 4.9 Hz, 4H). 13C NMR (101 MHz, Chloroform-d) δ 154.46, 148.78, 146.40, 139.61, 133.71, 131.92, 131.62, 130.20 (q, J = 33.3 Hz),, 129.84, 126.80, 126.04, 125.81 (q, J = 3.0 Hz), 124.03 (q, J = 273.7 Hz), 121.64, 119.86, 118.12, 57.59, 56.27, 53.73, 23.63. 19F NMR (376 MHz, Chloroform-d) δ −62.64. HRMS: calculated for C24H22F3N5O [M+H]+ 454.1849, found 454.1839 (mass error = −2.2 ppm).
5.1.6.3. 5-((4-(4-isopropylphenyl)-2H-1,2,3-triazol-2-yl)methyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol (7c).
Pale yellow solid (Yield: 78%). Mp: 106–107 °C. 1H NMR (400 MHz, Chloroform-d) δ 8.87 (d, J = 3.9 Hz, 1H), 8.55 (d, J = 8.6 Hz, 1H), 7.78 (d, J = 1.9 Hz, 1H), 7.71 – 7.64 (m, 2H), 7.44 (s, 2H), 7.27 (d, J = 7.4 Hz, 2H), 5.92 (s, 2H), 4.08 (s, 2H), 2.92 (p, J = 6.9 Hz, 1H), 2.81 (br s, 4H), 1.91 (br s, 4H), 1.26 (dd, J = 7.0, 1.9 Hz, 6H).13C NMR (101 MHz, Chloroform-d) δ 149.39, 147.85, 132.22, 131.14, 127.82, 126.93, 125.92, 56.07, 53.58, 33.96, 23.93, 23.56. HRMS: calculated for C26H29N5O [M+H]+ 428.2445, found 428.2438 (mass error = −1.6 ppm).
5.1.6.4. 7-(pyrrolidin-1-ylmethyl)-5-((4-(p-tolyl)-2H-1,2,3-triazol-2-yl)methyl)quinolin-8-ol (7d).
Pale orange solid (Yield: 74%). Mp: 102–103 °C. 1H NMR (400 MHz, Chloroform-d) δ 10.31 (s, 1H), 8.86 (dd, J = 4.3, 1.6 Hz, 1H), 8.54 (dd, J = 8.6, 1.6 Hz, 1H), 7.78 (s, 1H), 7.66 – 7.61 (m, 2H), 7.39 (dd, J = 8.6, 4.1 Hz, 1H), 7.35 (s, 1H), 7.19 (d, J = 7.9 Hz, 2H), 5.89 (s, 2H), 3.98 (s, 2H), 2.68 (dq, J = 7.6, 4.2, 3.7 Hz, 4H), 2.34 (s, 3H), 1.89 – 1.78 (m, 4H). 13C NMR (101 MHz, Chloroform-d) δ 154.26, 148.69, 147.81, 139.59, 138.30, 132.04, 131.03, 129.61, 129.51, 127.42, 126.78, 125.78, 121.55, 120.26, 118.17, 57.60, 56.01, 53.71, 23.62, 21.30. HRMS: calculated for C24H25N5O [M+H]+ 400.2132, found 400.2141 (mass error = 2.2 ppm).
5.1.6.5. 5-((4-(4-ethylphenyl)-2H-1,2,3-triazol-2-yl)methyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol (7e).
Pale orange solid (Yield: 82%). Mp: 90–92 °C. 1H NMR (400 MHz, Chloroform-d) δ 8.86 (dd, J = 4.2, 1.6 Hz, 1H), 8.54 (dd, J = 8.6, 1.5 Hz, 1H), 7.78 (s, 1H), 7.71 – 7.63 (m, 2H), 7.41 (dd, J = 8.6, 4.1 Hz, 1H), 7.38 (s, 1H), 7.26 – 7.20 (m, 2H), 5.90 (s, 2H), 4.01 (s, 2H), 2.75 – 2.69 (m, 4H), 2.65 (q, J = 7.6 Hz, 2H), 1.93 – 1.82 (m, 4H), 1.23 (t, J = 7.6 Hz, 3H). 13C NMR (101 MHz, Chloroform-d) δ 154.22, 148.70, 147.85, 144.73, 139.56, 132.07, 131.09, 129.71, 128.35, 127.67, 126.83, 125.90, 121.63, 120.32, 117.93, 57.43, 56.05, 53.69, 28.68, 23.62, 15.57. HRMS: calculated for C25H27N5O [M+H]+ 414.2288, found 414.2285 (mass error = −0.7 ppm).
5.1.6.6. 5-((4-(4-nitrophenyl)-2H-1,2,3-triazol-2-yl)methyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol (7f).
Orange solid (Yield: 57%). Mp: 168–170 °C. 1H NMR (400 MHz, Chloroform-d) δ 8.89 (dd, J = 4.2, 1.6 Hz, 1H), 8.56 (dd, J = 8.6, 1.6 Hz, 1H), 8.31 – 8.23 (m, 2H), 7.91 (dd, J = 6.9, 2.0 Hz, 3H), 7.49 – 7.42 (m, 2H), 5.95 (s, 2H), 4.04 (s, 2H), 2.73 (td, J = 5.3, 4.3, 2.3 Hz, 4H), 1.95 – 1.83 (m, 4H). 13C NMR (101 MHz, Chloroform-d) δ 154.56, 148.82, 147.45, 145.64, 139.61, 136.52, 132.09, 131.88, 130.00, 126.78, 126.40, 124.26, 121.70, 119.60, 118.18, 57.60, 56.42, 53.76, 23.65. HRMS: calculated for C23H22N6O3 [M+H]+ 431.1826, found 431.1831 (mass error = 1.2 ppm).
5.1.6.7. 5-((4-(4-methoxyphenyl)-2H-1,2,3-triazol-2-yl)methyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol (7g).
Pink solid (Yield: 77%). Mp: 142–144 °C. 1H NMR (400 MHz, Chloroform-d) δ 8.87 (dd, J = 4.2, 1.6 Hz, 1H), 8.55 (dd, J = 8.6, 1.6 Hz, 1H), 7.74 (s, 1H), 7.69 – 7.65 (m, 2H), 7.41 (dd, J = 8.6, 4.1 Hz, 1H), 7.35 (s, 1H), 6.95 – 6.90 (m, 2H), 5.89 (s, 2H), 4.00 (s, 2H), 3.81 (s, 3H), 2.70 (tt, J = 5.0, 2.4 Hz, 4H), 1.89 – 1.82 (m, 4H). 13C NMR (101 MHz, Chloroform-d) δ 159.78, 154.29, 148.72, 147.64, 139.62, 132.04, 130.70, 129.58, 127.21, 126.80, 122.95, 121.56, 120.28, 118.18, 114.22, 57.69, 56.00, 55.31, 53.74, 23.64. HRMS: calculated for C24H25N5O2 [M+H]+ 416.2081, found 416.2077 (mass error = −1.0 ppm).
5.1.6.8. 4-(2-((8-hydroxy-7-(pyrrolidin-1-ylmethyl)quinolin-5-yl)methyl)-2H-1,2,3-triazol-4-yl)benzonitrile (7h).
White solid (Yield: 68%). Mp: 164–166 °C. 1H NMR (400 MHz, Chloroform-d) δ 8.88 (dd, J = 4.2, 1.6 Hz, 1H), 8.55 (dd, J = 8.6, 1.6 Hz, 1H), 7.90 – 7.81 (m, 3H), 7.72 – 7.64 (m, 2H), 7.49 – 7.40 (m, 2H), 5.94 (s, 2H), 4.04 (s, 2H), 2.74 (q, J = 5.5, 4.7 Hz, 4H), 1.95 – 1.83 (m, 4H).13C NMR (101 MHz, Chloroform-d) δ 154.49, 148.79, 145.97, 139.57, 134.64, 132.68, 131.93, 131.85, 130.02, 126.81, 126.29, 121.72, 119.73, 118.70, 118.02, 111.71, 57.46, 56.35, 53.73, 23.63. HRMS: calculated for C24H22N6O [M+H]+ 411.1928, found 411.1928 (mass error = 0.0 ppm).
5.1.6.9. 5-((4-(4-(benzyloxy)phenyl)-2H-1,2,3-triazol-2-yl)methyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol (7i).
Pale orange (Yield: 68%). Mp: 132–134 °C. 1H NMR (400 MHz, Chloroform-d) δ 8.88 (dd, J = 4.1, 1.6 Hz, 1H), 8.55 (dd, J = 8.6, 1.6 Hz, 1H), 7.76 – 7.64 (m, 3H), 7.47 – 7.29 (m, 7H), 7.06 – 6.97 (m, 2H), 5.91 (s, 2H), 5.10 (s, 2H), 4.03 (s, 2H), 2.74 (br s, 4H), 1.89 (br s, 4H). 13C NMR (101 MHz, Chloroform-d) δ 158.93, 154.28, 148.74, 147.59, 139.59, 136.68, 132.04, 130.73, 129.62, 128.61, 128.04, 127.47, 127.22, 126.82, 123.19, 121.61, 120.29, 117.97, 115.16, 69.99, 57.59, 56.02, 53.71, 23.62. HRMS: calculated for C30H29N5O2 [M+H]+ 492.2394, found 492.2394 (mass error = 0.0 ppm).
5.1.6.10. 5-((4-(2-bromophenyl)-2H-1,2,3-triazol-2-yl)methyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol (7j).
Orange solid (Yield: 59%). Mp: 57–59 °C. 1H NMR (400 MHz, Chloroform-d) δ 8.91 – 8.85 (m, 1H), 8.57 (dd, J = 8.7, 1.6 Hz, 1H), 8.07 (s, 1H), 7.65 (ddd, J = 16.9, 7.9, 1.5 Hz, 2H), 7.43 (dd, J = 8.6, 4.1 Hz, 1H), 7.38 (s, 1H), 7.33 (td, J = 7.5, 1.3 Hz, 1H), 7.18 (td, J = 7.7, 1.8 Hz, 1H), 5.94 (s, 2H), 4.01 (s, 2H), 2.75 – 2.67 (m, 4H), 1.94 – 1.79 (m, 4H). 13C NMR (101 MHz, Chloroform-d) δ 154.42, 148.78, 146.29, 139.64, 134.27, 133.65, 132.07, 131.23, 130.95, 129.76, 129.72, 127.54, 126.83, 121.86, 121.63, 120.06, 118.21, 57.68, 56.15, 53.76, 23.65. HRMS: calculated for C23H22BrN5O [M+H]+ 464.1080, found 464.1082 (mass error = 0.4 ppm).
5.1.6.11. 5-((4-(3-bromophenyl)-2H-1,2,3-triazol-2-yl)methyl)-7-(pyrrolidin-1-ylmethyl)quinolin-8-ol (7k).
Orange solid (Yield: 64%). Mp: 70–72 °C. 1H NMR (400 MHz, Chloroform-d) δ 8.91 – 8.85 (m, 1H), 8.54 (dd, J = 8.6, 1.5 Hz, 1H), 7.92 (t, J = 1.8 Hz, 1H), 7.80 (s, 1H), 7.65 (dt, J = 7.7, 1.4 Hz, 1H), 7.44 (dt, J = 8.7, 3.0 Hz, 2H), 7.39 (s, 1H), 7.25 (t, J = 7.9 Hz, 1H), 5.91 (s, 2H), 4.03 (s, 2H), 2.72 (q, J = 4.6, 3.4 Hz, 4H), 1.95 – 1.82 (m, 4H). 13C NMR (101 MHz, Chloroform-d) δ 154.42, 148.78, 146.39, 139.61, 132.31, 131.96, 131.38, 131.33, 130.39, 129.83, 128.84, 126.81, 124.43, 122.96, 121.69, 119.97, 118.10, 57.58, 56.22, 53.75, 23.65. HRMS: calculated for C23H22BrN5O [M+H]+ 464.1080, found 464.1083 (mass error = 0.6 ppm).
5.1.6.12. 7-(pyrrolidin-1-ylmethyl)-5-((4-(thiophen-3-yl)-2H-1,2,3-triazol-2-yl)methyl)quinolin-8-ol (10).
Orange solid (Yield: 69%). Mp: 57–59 °C. 1H NMR (400 MHz, Chloroform-d) δ 9.14 (s, 1H), 8.84 (d, J = 4.2 Hz, 1H), 8.53 (dd, J = 8.6, 1.5 Hz, 1H), 7.70 (s, 1H), 7.58 (dd, J = 3.0, 1.3 Hz, 1H), 7.49 (s, 1H), 7.46 – 7.37 (m, 2H), 7.35 (dd, J = 5.0, 2.9 Hz, 1H), 5.90 (s, 2H), 4.07 (s, 2H), 2.85 – 2.77 (m, 4H), 1.90 (h, J = 3.0 Hz, 4H). 13C NMR (101 MHz, Chloroform-d) δ 154.03, 148.64, 143.96, 139.35, 132.18, 131.51, 131.37, 130.21, 126.95, 126.43, 125.87, 121.85, 121.61, 120.43, 116.93, 56.56, 55.96, 53.55, 23.54. HRMS: calculated for C21H21N5OS [M+H]+ 392.1540, found 392.1541 (mass error = 0.3 ppm).
5.1.7. General procedure for the preparation of compounds 12a-12e and 13.
The method used to synthesize compounds 12a-12e and 13 was the same used to synthesize compounds 7a-7k and 10.
5.1.7.1. 5-((4-(4-bromophenyl)-2H-1,2,3-triazol-2-yl)methyl)-7-(piperidin-1-ylmethyl)quinolin-8-ol (12a).
White solid (Yield: 74%). Mp: 131–132 °C. 1H NMR (400 MHz, Chloroform-d) δ 9.24 (s, 1H), 8.92 – 8.86 (m, 1H), 8.53 (dd, J = 8.6, 1.6 Hz, 1H), 7.79 (s, 1H), 7.61 (d, J = 8.3 Hz, 2H), 7.52 (d, J = 8.2 Hz, 2H), 7.42 (dd, J = 8.6, 4.2 Hz, 1H), 7.32 (s, 1H), 5.90 (s, 2H), 3.87 (s, 2H), 2.60 (br s, 4H), 1.68 (p, J = 5.6 Hz, 4H), 1.57 – 1.45 (m, 2H). 13C NMR (101 MHz, Chloroform-d) δ 155.02, 148.91, 146.79, 139.76, 131.99, 131.91, 131.19, 129.86, 129.24, 127.44, 126.89, 122.38, 121.62, 119.99, 117.07, 61.49, 56.18, 54.10, 25.80, 23.91. HRMS: calculated for C24H24BrN5O [M+H]+ 478.1237, found 478.1243 (mass error = 1.3 ppm).
5.1.7.2. 7-(piperidin-1-ylmethyl)-5-((4-(4-(trifluoromethyl)phenyl)-2H-1,2,3-triazol-2-yl)methyl)quinolin-8-ol (12b).
Pale yellow solid (Yield: 82%). Mp: 120–121 °C. 1H NMR (400 MHz, Chloroform-d) δ 11.56 (s, 1H), 8.90 (d, J = 4.1 Hz, 1H), 8.55 (d, J = 8.6 Hz, 1H), 7.85 (d, J = 7.2 Hz, 3H), 7.65 (d, J = 7.9 Hz, 2H), 7.43 (dd, J = 8.6, 4.1 Hz, 1H), 7.33 (s, 1H), 5.92 (s, 2H), 3.87 (s, 2H), 2.60 (br s, 4H), 1.67 (q, J = 5.7 Hz, 4H), 1.51 (br s, 2H). 13C NMR (101 MHz, Chloroform-d) δ 155.12, 148.94, 146.42, 139.80, 133.74, 131.85, 131.64, 130.21 (q, J = 32.3 Hz), 129.88, 126.88, 126.07, 125.82 (q, J = 3.0 Hz), 124.04 (q, J = 272.7 Hz), 121.62, 119.85, 117.13, 61.55, 56.28, 54.10, 25.83, 23.92. 19F NMR (376 MHz, Chloroform-d) δ −62.64. HRMS: calculated for C25H24F3N5O [M+H]+ 468.2006, found 468.2010 (mass error = 0.9 ppm). Conclusive structural characterization was obtained from X-ray analysis of single crystals of compound 12b and summarized in Supplementary Tables S1, and S2, and Figure S1.
5.1.7.3. 7-(azepan-1-ylmethyl)-5-((4-(4-(trifluoromethyl)phenyl)-2H-1,2,3-triazol-2-yl)methyl)quinolin-8-ol (12c).
Pale yellow solid (Yield: 69%). Mp: 64–66 °C. 1H NMR (400 MHz, Chloroform-d) δ 8.91 (dd, J = 4.1, 1.6 Hz, 1H), 8.56 (dd, J = 8.6, 1.6 Hz, 1H), 7.90 – 7.83 (m, 3H), 7.66 (d, J = 8.2 Hz, 2H), 7.44 (dd, J = 8.6, 4.1 Hz, 1H), 7.32 (s, 1H), 5.93 (s, 2H), 4.01 (s, 2H), 2.81 (t, J = 5.7 Hz, 4H), 1.76 (dd, J = 7.6, 4.0 Hz, 4H), 1.67 (q, J = 3.2 Hz, 4H). 13C NMR (101 MHz, Chloroform-d) δ 155.65, 148.97, 146.44, 139.90, 133.75, 131.86, 131.66, 130.24 (q, J = 32.3 Hz),, 129.71, 127.00, 126.09, 125.84 (q, J = 4.0 Hz), 124.04 (q, J = 272.7 Hz), 121.65, 119.75, 117.72, 61.90, 56.30, 55.67, 27.65, 26.59. 19F NMR (376 MHz, Chloroform-d) δ −62.64. HRMS: calculated for C26H26F3N5O [M+H]+ 482.2162, found 482.2159 (mass error = −0.6 ppm).
5.1.7.4. 7-((diethylamino)methyl)-5-((4-(4-(trifluoromethyl)phenyl)-2H-1,2,3-triazol-2-yl)methyl)quinolin-8-ol (12d).
Pale orange solid (Yield: 69%). Mp: 116–117 °C. 1H NMR (400 MHz, Chloroform-d) δ 8.90 (dd, J = 4.2, 1.6 Hz, 1H), 8.55 (dd, J = 8.7, 1.6 Hz, 1H), 7.86 (d, J = 6.5 Hz, 3H), 7.66 (d, J = 8.2 Hz, 2H), 7.43 (dd, J = 8.5, 4.1 Hz, 1H), 7.35 (s, 1H), 5.93 (s, 2H), 3.98 (s, 2H), 2.72 (q, J = 7.2 Hz, 4H), 1.17 (t, J = 7.2 Hz, 6H). 13C NMR (101 MHz, Chloroform-d) δ 155.37, 148.91, 146.41, 139.82, 133.72, 131.85, 131.64, 130.21 (q, J = 33.3 Hz), 129.76, 126.84, 126.06, 125.81 (q, J = 4.0 Hz), 124.01 (q, J = 273.7 Hz), 121.61, 119.75, 117.49, 56.52, 56.28, 46.65, 11.33.19F NMR (376 MHz, Chloroform-d) δ −62.64. HRMS: calculated for C24H24F3N5O [M+H]+ 456.2006, found 456.2010 (mass error = 0.9 ppm).
5.1.7.5. 5-((4-(4-bromophenyl)-2H-1,2,3-triazol-2-yl)methyl)-7-(morpholinomethyl)quinolin-8-ol (12e).
White solid (Yield: 66%). Mp: 142–144 °C. 1H NMR (400 MHz, Chloroform-d) δ 8.87 (dd, J = 4.2, 1.6 Hz, 1H), 8.56 (dd, J = 8.6, 1.6 Hz, 1H), 7.79 (s, 1H), 7.63 – 7.58 (m, 2H), 7.51 (d, J = 8.4 Hz, 2H), 7.45 (d, J = 12.6 Hz, 2H), 5.91 (s, 2H), 3.88 (s, 2H), 3.77 (t, J = 4.6 Hz, 4H), 2.63 (d, J = 9.1 Hz, 4H). 13C NMR (101 MHz, Chloroform-d) δ 153.53, 148.79, 146.85, 139.35, 132.14, 132.00, 131.22, 130.47, 129.18, 127.41, 126.83, 122.43, 121.89, 120.60, 116.88, 66.86, 59.94, 56.09, 53.20. HRMS: calculated for C23H22BrN5O2 [M+H]+ 480.1030, found 480.1039 (mass error = 1.9 ppm).
5.1.7.6. tert-butyl 4-((5-((4-(4-bromophenyl)-2H-1,2,3-triazol-2-yl)methyl)-8 hydroxyquinolin-7-yl)methyl)piperazine-1-carboxylate (13).
Brown solid (Yield: 86%).1H NMR (400 MHz, Chloroform-d) δ 8.86 (dd, J = 4.1, 1.5 Hz, 1H), 8.56 (dd, J = 8.6, 1.6 Hz, 1H), 7.79 (s, 1H), 7.62 – 7.58 (m, 2H), 7.52 – 7.43 (m, 4H), 5.91 (s, 2H), 3.88 (s, 2H), 3.51 (t, J = 5.0 Hz, 4H), 2.57 (t, J = 5.1 Hz, 4H), 1.46 (d, J = 1.5 Hz, 9H).
5.1.7.8. Preparation of 5-((4-(4-bromophenyl)-2H-1,2,3-triazol-2-yl)methyl)-7-(piperazin-1-ylmethyl)quinolin-8-ol (14).
To a solution of tert-butyl 4-((5-((4-(4-bromophenyl)-2H-1,2,3-triazol-2-yl)methyl)-8 hydroxyquinolin-7-yl)methyl)piperazine-1-carboxylate (compound 13) (100 mg, 0.2 mmol) in 10 mL of CH2Cl2 was added TFA (2 mL) at 0 °C. The mixture was stirred at RT for 1 h. Upon completion, the mixture was poured into NaHCO3 solution and extracted three times with CH2Cl2 (30 mL). The combined organic layer was washed with brine, dried over Na2SO4, and evaporated to dryness. The crude product was purified by column chromatography (dichloromethane: methanol 20: 1) to yield target compound 14 as a pale green solid (Yield: 90%). Mp: 212–213 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.87 (dd, J = 4.2, 1.4 Hz, 1H), 8.64 (dd, J = 8.6, 1.5 Hz, 1H), 8.28 (s, 1H), 7.73 (d, J = 8.3 Hz, 2H), 7.63 (dd, J = 8.2, 4.0 Hz, 4H), 6.07 (s, 2H), 3.75 (s, 2H), 3.05 (t, J = 5.2 Hz, 4H), 2.63 (br s, 4H).13C NMR (101 MHz, Chloroform-d) δ 153.53, 148.79, 146.85, 139.35, 132.14, 132.00, 131.22, 130.47, 129.18, 127.41, 126.83, 122.43, 121.89, 120.60, 116.88, 66.86, 59.94, 56.09, 53.20. HRMS: calculated for C23H23BrN6O [M+H]+ 479.1189, found 479.1179 (mass error = −2.1 ppm).
5.1.8. Preparation of 7-bromo-5-(chloromethyl)quinolin-8-ol (15).
A mixture of 3 g (13.38 mmol) of 7-bromo-8-hydroxyquinoline, 20 mL of concentrated hydrochloric acid, and 6 mL of 37% formaldehyde was treated with 0.6 g of zinc chloride and stirred at 60°C for 12 hr. The mixture was filtered, washed with copious acetone, and dried to produce compound 15 as a white solid (1.67g, Yield: 44.5%). 1H NMR (400 MHz, DMSO-d6) δ 8.94 (dd, J = 4.3, 1.5 Hz, 1H), 8.63 – 8.59 (m, 1H), 7.73 – 7.70 (m, 1H), 7.69 (s, 1H), 4.86 (s, 2H).
5.1.9. Preparation of 7-bromo-5-(((4-isopropylbenzyl)oxy)methyl)quinolin-8-ol (16).
7-Bromo-5-chloromethyl-8-quinolinol hydrochloride, compound 15 (1.5 g, 5 mmol) was suspended in 4-isopropyl benzyl alcohol (25 mmol) and the mixture was heated at 90°C for 2 h, during which time the solution became completely homogeneous. The solution was poured into 200 mL solution of hexane: ethyl acetate (1:1) and filtered, then NaHCO3 solution was added to the filtrate. The mixture was extracted by dichloromethane, washed with brine, dried over anhydrous Na2SO4 and evaporated to dryness to yield the desired compound as white solid (Yield: 76%). Mp: 167–168 °C. 1H NMR (400 MHz, Chloroform-d) δ 8.80 (dd, J = 4.3, 1.5 Hz, 1H), 8.45 (dd, J = 8.5, 1.6 Hz, 1H), 7.61 (s, 1H), 7.50 (dd, J = 8.5, 4.2 Hz, 1H), 7.26 – 7.20 (m, 4H), 4.84 (s, 2H), 4.53 (s, 2H), 2.91 (h, J = 6.9 Hz, 1H), 1.25 (d, J = 6.9 Hz, 6H). 13C NMR (101 MHz, Chloroform-d) δ 149.81, 148.69, 148.53, 138.73, 135.07, 133.84, 131.88, 128.14, 126.58, 126.37, 125.62, 122.07, 102.84, 72.03, 69.36, 33.89, 24.02. HRMS: calculated for C20H20BrNO2 [M+H]+ 386.0750, found 386.0754 (mass error = 1.0 ppm).
5.1.10. Preparation of 7-bromo-5-(((4-isopropylbenzyl)oxy)methyl)-8 ((2(trimethylsilyl)ethoxy)methoxy)quinoline (17).
To a solution of compound 16 in THF was added sodium hydride (3 equivalents) at 0°C under inert conditions and stirred at RT for 30 minutes. SEMCl (2 equivalents) was added dropwise to the reaction mixture and the solution was stirred for 3 hr. The reaction was quenched with water and extracted 3 times with ethyl acetate (15 mL). The combined organic phase was washed by brine and water and dried over Na2SO4, then concentrated under reduced pressure to yield a crude product. The crude material was purified by column chromatography (hexane: ethyl acetate; 2:1) to obtain the pure compound as yellow oily liquid (Yield: 88%). 1H NMR (400 MHz, Chloroform-d) δ 8.87 (dd, J = 4.2, 1.6 Hz, 1H), 8.37 (dd, J = 8.6, 1.7 Hz, 1H), 7.66 (s, 1H), 7.39 (dd, J = 8.5, 4.1 Hz, 1H), 7.26 – 7.18 (m, 4H), 5.67 (s, 2H), 4.83 (s, 2H), 4.52 (s, 2H), 4.07 – 4.01 (m, 2H), 2.88 (p, J = 6.9 Hz, 1H), 1.22 (d, J = 6.9 Hz, 6H), 1.00 – 0.90 (m, 2H), 0.00 (s, 9H).
5.1.11. General procedure for the preparation of compounds 18a-18h.
To a solution of compound 17 (1 g, 5.4 mmol) and different aryl boronic acid or aryl boronic acid pinacol ester reagents (0.83 g, 5.4 mmol) in 1,4-dioxane/H2O (45 mL, v/v=2/1) was added Pd (PPh3)4 and Na2CO3 (1.1 g, 10.8 mmol). The reaction mixture was refluxed for 4 h under argon. The solvent was removed, 20 mL of water was added, and the mixture was extracted with dichloromethane (15 mL.3 times).The combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure to yield a crude product, which was used for the next step without purification.
5.1.12. General procedure for the preparation of compounds 19a-19h.
To a solution of 18(a-h) (100 mg) in 10 mL of CH2Cl2 was added TFA (2 mL) at 0°C. The mixtures were stirred at RT for 1 h. After completion of the reaction, the mixture was poured into NaHCO3 solution, and extracted 3 times with dichloromethane (10 mL). The combined organic layer was washed with brine, dried over Na2SO4, and concentrated to dryness. The crude products were purified by column chromatography (hexane/ethyl acetate 2:1) to produce the target compounds.
5.1.12.1. 5-(((4-isopropylbenzyl)oxy)methyl)-7-phenylquinolin-8-ol (19a).
Yellow solid (Yield: 57%). Mp: 112–113 °C. 1H NMR (400 MHz, Chloroform-d) δ 8.78 (dd, J = 4.3, 1.6 Hz, 1H), 8.45 (dd, J = 8.4, 1.6 Hz, 1H), 7.82 – 7.78 (m, 2H), 7.58 (s, 1H), 7.48 (t, J = 7.7 Hz, 2H), 7.42 (dd, J = 8.5, 4.2 Hz, 1H), 7.37 – 7.33 (m, 1H), 7.28 – 7.17 (m, 4H), 4.88 (s, 2H), 4.53 (s, 2H), 2.89 (h, J = 6.9 Hz, 1H), 1.23 (d, J = 6.9 Hz, 6H). 13C NMR (101 MHz, Chloroform-d) δ 148.98, 148.59, 147.97, 139.07, 137.55, 135.45, 133.58, 130.65, 129.38, 128.48, 128.21, 127.32, 126.62, 126.59, 124.31, 121.81, 121.55, 71.93, 70.22, 33.95, 24.12. HRMS: calculated for C26H25NO2 [M+H]+ 384.1958, found 384.1944 (mass error = −3.6 ppm).
5.1.12.2. 5-(((4-isopropylbenzyl)oxy)methyl)-7-(isoquinolin-4-yl)quinolin-8-ol (19b).
Pale green solid (Yield: 55%). Mp: 64–65 °C. 1H NMR (400 MHz, Chloroform-d) δ 9.32 (s, 1H), 8.87 (dd, J = 4.3, 1.5 Hz, 1H), 8.63 (s, 1H), 8.57 (dd, J = 8.5, 1.5 Hz, 1H), 8.09 – 8.05 (m, 1H), 7.78 – 7.73 (m, 1H), 7.65 (qd, J = 7.0, 3.4 Hz, 2H), 7.57 (dd, J = 8.5, 4.2 Hz, 1H), 7.52 (s, 1H), 7.28 (d, J = 7.9 Hz, 2H), 7.21 (d, J = 8.0 Hz, 2H), 4.93 (s, 2H), 4.59 (s, 2H), 2.90 (h, J = 6.8 Hz, 1H), 1.23 (d, J = 7.0 Hz, 6H). 13C NMR (101 MHz, Chloroform-d) δ 152.39, 150.12, 148.65, 148.24, 143.90, 138.64, 135.21, 133.75, 131.57, 130.41, 128.17, 127.95, 127.24, 126.57, 125.44, 124.43, 122.28, 117.25, 72.12, 69.98, 33.88, 24.02. HRMS: calculated for C29H26N2O2 [M+H]+ 435.2067, found 435.2064 (mass error = −0.7 ppm).
5.1.12.3. 5’-(((4-isopropylbenzyl)oxy)methyl)-[4,7’-biquinolin]-8’-ol (19c).
Pale orange solid (Yield: 53%). Mp: 59–61 °C 1H NMR (400 MHz, Chloroform-d) δ 9.02 (d, J = 4.5 Hz, 1H), 8.86 (dd, J = 4.2, 1.5 Hz, 1H), 8.55 (dd, J = 8.5, 1.6 Hz, 1H), 8.24 – 8.20 (m, 1H), 7.79 (dd, J = 8.4, 1.4 Hz, 1H), 7.73 (ddd, J = 8.4, 6.8, 1.4 Hz, 1H), 7.56 (dd, J = 8.5, 4.2 Hz, 1H), 7.53 – 7.50 (m, 1H), 7.49 – 7.46 (m, 2H), 7.28 (d, J = 8.2 Hz, 2H), 7.22 – 7.18 (m, 2H), 4.91 (s, 2H), 4.59 (s, 2H), 2.89 (h, J = 6.9 Hz, 1H), 1.23 (d, J = 6.9 Hz, 6H). 13C NMR (101 MHz, Chloroform-d) δ 149.80, 149.68, 148.69, 148.39, 148.34, 144.75, 138.66, 135.17, 133.77, 130.67, 129.67, 129.52, 128.18, 127.40, 126.91, 126.61, 126.59, 126.38, 124.53, 122.52, 118.08, 72.21, 69.91, 33.89, 24.04. HRMS: calculated for C29H26N2O2 [M+H]+ 435.2067, found 435.2084 (mass error = 3.9 ppm).
5.1.12.4. 5-(((4-isopropylbenzyl)oxy)methyl)-7-(p-tolyl)quinolin-8-ol (19d).
White solid (Yield: 53%). Mp: 85–87 °C. 1H NMR (400 MHz, CDCl3) δ 8.80 (dd, J = 4.3, 1.6 Hz, 1H), 8.48 (dd, J = 8.5, 1.6 Hz, 1H), 7.72 – 7.67 (m, 2H), 7.59 (s, 1H), 7.45 (dd, J = 8.5, 4.2 Hz, 1H), 7.32 – 7.28 (m,2H), 7.25 (s, 2H), 7.20 (d, J = 8.2 Hz, 2H), 4.90 (s, 2H), 4.53 (s, 2H), 2.90 (hept, J = 6.9 Hz, 1H), 2.41 (s, 3H), 1.24 (d, J = 7.0 Hz, 6H). 13C NMR (101 MHz, Chloroform-d) δ 148.68, 148.51, 147.68, 138.83, 137.03, 135.34, 134.45, 133.74, 130.68, 129.12, 128.11, 126.50, 126.45, 124.18, 121.81, 121.55, 71.80, 70.13, 33.86, 24.01, 21.26. HRMS: calculated for C27H27NO2 [M+H]+ 398.2115, found 398.2114 (mass error = −0.3 ppm).
5.1.12.5. 5-(((4-isopropylbenzyl)oxy)methyl)-7-(m-tolyl)quinolin-8-ol (19e).
Pale yellow solid (Yield: 56%). Mp: 72–74 °C. 1H NMR (400 MHz, Chloroform-d) δ 8.82 (dd, J = 4.3, 1.5 Hz, 1H), 8.49 (dd, J = 8.4, 1.5 Hz, 1H), 7.62 – 7.56 (m, 3H), 7.47 (dd, J = 8.5, 4.2 Hz, 1H), 7.39 (t, J = 7.8 Hz, 1H), 7.27 (d, J = 8.1 Hz, 2H), 7.20 (dd, J = 8.4, 6.8 Hz, 3H), 4.92 (s, 2H), 4.54 (s, 2H), 2.90 (h, J = 6.9 Hz, 1H), 2.45 (s, 3H), 1.24 (d, J = 6.9 Hz, 6H). 13C NMR (101 MHz, Chloroform-d) δ 148.87, 148.55, 147.89, 139.02, 137.97, 137.40, 135.38, 133.55, 130.76, 129.96, 128.32, 128.15, 128.07, 126.53, 126.42, 124.14, 121.72, 121.65, 71.82, 70.19, 33.89, 24.04, 21.65. HRMS: calculated for C27H27NO2 [M+H]+ 398.2115, found 398.2113 (mass error = −0.5 ppm).
5.1.12.6. 5-(((4-isopropylbenzyl)oxy)methyl)-7-(4-methoxyphenyl)quinolin-8-ol (19f).
Pale yellow solid (Yield: 58%). Mp: 107–108 °C. 1H NMR (400 MHz, Chloroform-d) δ 9.43 (s, 1H), 8.92 (dd, J = 4.9, 1.5 Hz, 1H), 8.84 (dd, J = 8.5, 1.5 Hz, 1H), 7.70 (d, J = 8.9 Hz, 3H), 7.66 (dd, J = 8.5, 4.8 Hz, 1H), 7.24 (q, J = 8.2 Hz, 4H), 7.05 – 7.00 (m, 2H), 4.94 (s, 2H), 4.56 (s, 2H), 3.87 (s, 3H), 2.91 (p, J = 6.9 Hz, 1H), 1.24 (d, J = 6.9 Hz, 6H). 13C NMR (101 MHz, Chloroform-d) δ 159.48, 148.84, 146.39, 144.75, 139.25, 135.40, 134.85, 132.49, 130.77, 128.74, 128.18, 127.41, 126.62, 125.71, 120.73, 114.05, 72.21, 69.72, 55.38, 33.87, 24.00. HRMS: calculated for C27H27NO3 [M+H]+ 414.2064, found 414.2060 (mass error = −1.0 ppm).
5.1.12.7. 5-(((4-isopropylbenzyl)oxy)methyl)-7-(4-(trifluoromethyl)phenyl)quinolin-8-ol (19g).
Pale brown solid (Yield: 56%). Mp: 99–100 °C. 1H NMR (400 MHz, Chloroform-d) δ 8.83 (dd, J = 4.3, 1.5 Hz, 1H), 8.48 (dd, J = 8.5, 1.6 Hz, 1H), 7.94 – 7.90 (m, 2H), 7.73 (d, J = 8.2 Hz, 2H), 7.57 (s, 1H), 7.50 (dd, J = 8.5, 4.2 Hz, 1H), 7.27 (d, J = 8.1 Hz, 2H), 7.22 (d, J = 8.2 Hz, 2H), 4.91 (s, 2H), 4.57 (s, 2H), 2.90 (h, J = 6.9 Hz, 1H), 1.24 (d, J = 6.9 Hz, 6H). 13C NMR (101 MHz, Chloroform-d) δ 149.31, 148.66, 148.16, 141.11, 138.93, 135.25, 133.61, 129.83, 129.56, 129.09 (q, J = 32.3 Hz), 128.15, 126.95, 126.58, 125.31 (q, J = 4.0 Hz), 124.68, 124.33(q, J = 272.7 Hz), 122.23, 119.89, 72.07, 70.02, 33.90, 24.04. 19F NMR (376 MHz, Chloroform-d) δ −62.45. HRMS: calculated for C27H24F3NO2 [M+H]+ 452.1832, found 452.1831 (mass error = −0.2ppm).
5.1.12.8. 5-(((4-isopropylbenzyl)oxy)methyl)-7-(thiophen-3-yl)quinolin-8-ol (19h).
Pale yellow solid (Yield: 55%). Mp: 126–127 °C. 1H NMR (400 MHz, CDCl3) δ 8.78 (dd, J = 4.2, 1.5 Hz, 1H), 8.43 (dd, J = 8.5, 1.6 Hz, 1H), 7.96 (dd, J = 3.0, 1.3 Hz, 1H), 7.73 – 7.70 (m, 2H), 7.45 – 7.39 (m, 2H), 7.26 (d, J = 8.2 Hz, 2H), 7.21 (d, J = 8.2 Hz, 2H), 4.89 (s, 2H), 4.53 (s, 2H), 2.90 (h, J = 6.9 Hz, 1H), 1.24 (d, J = 6.9 Hz, 6H). 13C NMR (101 MHz, Chloroform-d) δ 148.52, 148.50, 147.86, 139.18, 137.44, 135.31, 133.42, 129.17, 128.12, 127.68, 126.50, 126.21, 125.03, 124.11, 123.70, 121.55, 116.24, 71.78, 70.09, 33.85, 24.01. HRMS: calculated for C24H23NO2S [M+H]+ 390.1522, found 390.1528 (mass error = 1.5 ppm).
5.2. Cell culture and reagents.
Two human melanoma (A375 and RPMI7951) and two human breast (SKBR3 and MDA-MB-453) cell lines were purchased from ATCC. The P-gp overexpressed MDA-MB-435/LCC6MDR1 cell line and its parental MDA-MB-435 melanoma cell line were gifts from Dr. Robert Clarke at Georgetown University[20]. All these cell lines were grown in DMEM (Mediatech, Inc) supplemented with 10% FBS (Atlanta Biologicals) and 1% antibiotic-antimycotic solution (Sigma-Aldrich) at 37°C in a humidified incubator containing 5% carbon dioxide (CO2). Two ovarian cancer cell lines, OVCAR3 and OVCAR8 were obtained from ATCC and cultured in RPMI 1640 supplemented with 10% FBS (Hyclone; Logan, UT), 100 U/ml penicillin, and 100 μg/ml streptomycin (Invitrogen; Carlsbad, CA) at 37°C in 5% CO2. Cell lines were authenticated using Short Tandem Repeat (STR) analysis by ATCC and tested negative for mycoplasma contamination using luciferase assay (Lonza, Allendale, NJ).
5.3. Cytotoxicity assay.
5.3.1. Melanoma and breast cancer cell lines.
The human melanoma (A375, RPMI7951, MDA-MB-435, and MDA-MB-435/LCC6MDR1) and human breast (SKBR3 and MDA-MB-453) cell lines were plated into 96-well plates at a density of 3,500 to 5,000 cells per well, depending on the proliferation rate of each cell line. After overnight incubation, cells were treated with the test compounds (concentration range: 3 nM-100 μM) for 72 hr. Each compound treatment was carried out in four replicates. At the endpoint, the MTS reagent (Promega) was added to the cells, they were incubated in the dark 37°C for 1.5 hr, and cell viability was determined by the absorbance a wavelength of 490 nm using a plate reader (BioTek Instruments Inc., Winooski, VT). IC50 values were normalized against untreated cells and calculated using GraphPad Prism 7 software using nonlinear regression.
5.3.2. Ovarian cancer cell lines.
OVCAR3 and OVCAR8 cells (3,000 per well) were seeded into 96-well plates. The culture medium was changed, and cells were treated with compounds 7a, 7k, 12a, 12b, or 19a at 10 concentrations ranging from 3 nM to 100 μM or with a DMSO vehicle control in three replicates. Cells were incubated at 37°C for 72 hr. After treatment, MTS reagent (Promega, Madison, WI) was added and the cells were incubated at 37°C for 1 h. We subsequently measured the absorbance at a wavelength of 490 nm using a plate reader (BioTek Instruments). IC50 values were calculated by nonlinear regression analysis using OriginLab dats analysis and graphing software.
5.4. Liver microsome stability assay [21, 22].
This assay showed the metabolic liability of drug discovery compounds in liver microsomes. Degradation of the compounds in human, mouse, and rat liver microsomes was determined in multiple time points to monitor the rate of disappearance of the parent compound during incubation. Pooled human liver microsomes were obtained from XenoTech (Lenexa, KS), while pooled mouse (CD-1) and rat (Sprague Dawley) liver microsomes were purchased from Gibco (Ottawa, Ontario). NADPH regenerating agent solutions A and B were purchased from BD Gentest (Woburn, MA). Ninety-six-well analytical plates were obtained from Corning Incorporated (Acton, MA), while ninety-six-well deep well plates were purchased from Midsci (St. Louis, MO).
Preparation of samples for determination of stability in microsomes was modified from the methods described by Di et al [23, 24]. We used incubation times of 0, 15, 30, 60, 120, and 240 min. Test compounds and system control (verapamil) were prepared as 10 mM stock solutions in DMSO. Concentrated mouse or human liver microsomes (20 mg/mL protein concentration) and 0.5 M EDTA were diluted into 0.1 M potassium phosphate buffer (pH7.4) and mixed well prior to and after addition of each compound solution. 90 μL each of these solutions were transferred to triplicate wells on each of six 96-well plates (one for each time point). For the time 0 plate, a 3-fold (v/v) excess of cold acetonitrile containing 4 μg/ml warfarin as an internal standard was added to each well, along with NADPH regenerating solutions A and B, which were mixed together in PBS, pH7.4. The samples were prepared for analysis without further incubation. For the other five time-point plates, NADPH regenerating agent was added to each well to initiate the reaction, the plate was incubated at 37° C for the indicated time, and reactions were quenched with a 3-fold volume excess of cold acetonitrile containing the internal standard. The final concentration of each component in this reaction was as follows: 1 mM EDTA, 0.5 mg/mL liver microsome, 10 μM test compound, 1.3 mM NADPH A, and 0.4 U/mL NADPH B (Glucose-6-phosphate dehydrogeneous). Plates were sealed, mixed at 600 rpm for 10 min after quenching, and centrifuged at 4,000 rpm for 20 min to remove debris. The supernatants were transferred to the appropriate plates for analysis by UPLC–MS. Microsomal half-life was determined for each compound (and the control) and the metabolic stability of each compound was calculated from the half-life by least-squares fit of the multiple time points, based on first-order kinetics.
5.5. Western blotting.
5.5.1. A375
A375 cells were seeded into 10-cm dishes at a density of 2 × 106 cells/plate and allowed to attach overnight. The medium was replaced with either fresh complete DMEM medium or DMEM containing the desired concentration (1, 5 and 10 μM) of MX-106, or compounds 7a, 12b, or 19a. After 24 hr of incubation, cells were washed with ice-cold PBS, lysed with RIPA buffer (Thermo Fisher Scientific) containing Halt protease and phosphatase inhibitor (Thermo Fisher Scientific), and centrifuged at 13,000 rpm at 4°C for 15 min. The Bradford assay method was used to quantify the protein in each sample. 50 μg of each samples were subjected to western blot analysis as described [25]. Briefly, the protein samples were separated via SDS-PAGE, transferred to PVDF membranes, blocked with 5% nonfat milk for 1 h at RT followed by overnight incubation at 4°C with primary antibodies, which included cleaved-PARP (#9185), CIAP1 (#7065), XIAP (#2045), Livin (#5471), Survivin (#2808) and GAPDH (HRP Conjugate) (#3683), all purchased from Cell Signaling Technology, Inc. Primary antibodies were detected with HRP-conjugated secondary antibodies (#7074, Cell Signaling Technology, Inc). and immunoreactive bands were visualized on film using enhanced chemiluminescent substrate (ECL, Thermo Fisher Scientific).
5.5.2. Ovarian cancer cell lines.
Ovarian cancer cells were treated with vehicle, or compounds 7k (2 and 4 μM), 12b (2, 4 μM), or MX-106 (2, 4 μM) for 24 hr in serum-free RPMI 1640 medium and collected in RIPA buffer (Thermo Scientific; Rockford, IL) containing 1% Halt Proteinase Inhibitor Cocktail (Thermo Scientific; Rockford, IL). Protein was quantitated using a Bradford assay as above. An equal amount of protein (100 μg/lane) was loaded onto 10% SDS-PAGE gels and transferred onto PVDF membranes. The membranes were blocked with 5% nonfat milk for 1 hr and incubated with primary antibodies against GAPDH (Santa Cruz Biotechnology; St. Louis, MO), MCL1 and Survivin (both from Cell Signaling Technology, Inc.). Primary antibodies were detected with HRP-conjugated secondary antibodies (#7074, Cell Signaling Technology, Inc). and immunoreactive bands were visualized on film using enhanced chemiluminescent substrate (ECL, Thermo Fisher Scientific).
5.6. Surface plasmon resonance (SPR) for to measure binding affinity of 12b for survivin.
The binding affinity of survivin was analyzed using SPR technology using a PlexAray HT system equipped with a bare gold-coated (thickness 47 nm) PlexArray® Nanocapture® Sensor Chip (Plexera Bioscience, Woodinville, WA). Biodo bioprinting was used to manually print recombinant biotinylated survivin protein (in water) at various concentrations onto the sensor chip at 40% humidity via avidin-biotin conjugation. Each spot contained 0.2 μL of 12b solution and each concentration was printed in triplicate. The chip was incubated overnight at 4°C in 80% humidity, then rinsed for 10 min with 10× phosphate buffered saline with Tween 20 detergent (PBST), 10 min with 1× PBST, and twice for 10 min each in deionized water. The chip was blocked at RT overnight with 5% (w/v) non-fat milk in MilliQ water, and washed for 10 min in 10× PBST, 10 min in 1× PBST, and twice for 10 min each in deionized water, then dried under a stream of nitrogen. SPR measurements were performed using the PlexAray HT system (Plexera Bioscience, Seattle, WA, US). Collimated light of wavelength 660 nm passes through a coupling prism, reflects off the gold surface of the chip, is received by an integrated CCD camera. A nonpulsatile piston pump was used to inject samples and buffers into a 30 μL flowcell mounted on the coupling prim. All measurements were performed at RT (25°C). Each measurement cycle involved 1) washing with running buffer (PBST) at a constant rate (2 μL/s) to obtain a stable baseline, 2) injection of the 12b solution (5 μL/s for binding), 3) washing of the surface with PBST for 300 sec (2 μL/s), and 4) regeneration with 0.5% (v/v) H3PO4 for 300 sec (2 μL/s). Changes in signal after binding and washing were recorded as the assay values (in AU). We analyzed selected protein-grafted regions in the SPR images and the average reflectivity variations of the selected areas were plotted as a function of time. Real-time binding signals were collected and analyzed using the PlexArray® Analysis Software Package (Plexera Bioscience). Kinetic analysis was performed using BIAevaluation 4.1 software (Biacore, Inc., a division of Cytiva, Uppsala, Sweden).
5.7. Molecular modeling.
Molecular docking studies of 12b with survivin were conducted in Schrodinger Molecular Modeling Suite 2014 (Schrodinger Inc., Portland, OR) following previously described procedures [11, 12]. Compound 12b was prepared to generate various conformations, then docked into the AVPI SMAC binding motif in a human survivin crystal structure (Protein Data Bank entry: 3UIH). Docking to minimize the energy of potential ligand binding poses was performed prior to calculation of the molecular dynamics. Results were visualized using the Maestro interface of the Schrodinger software.
5.8. In vivo efficacies of compound 12b on A375 subcutaneous melanoma and OVCAR8 orthotopic ovary models in mice.
All animal studies were performed under the guidelines of NIH Principles of Laboratory Animal Care and protocols approved by the UTHSC Institutional Animal Care and Use Committee (IACUC) (protocol #17-056 for melanoma xenograft model and protocol #19-0117 for ovary xenograft model). Animals were housed under a 12:12 hr light/dark cycle at 20–26°C with 30–70% humidity.
5.8.1. A375 melanoma model.
6–7-week-old male humanized immunodeficient NOD scid gamma (NSG) mice (Jackson Laboratory) were used for implantation of A375 melanoma tumors. 2.5 × 106 logarithmically growing A375 cells in 50 μL of phenol red-free, FBS-free DMEM medium were mixed with 50 μL Matrigel and subcutaneously injected into the dorsal right flank of each NSG mouse. The tumors were allowed to grow for 2 weeks until the average tumor volume reached 100 mm3. The mice were randomly assigned into 3 groups (n = 7/group): vehicle (5% DMSO, 20% PEG300, 5% Tween 80, 70% saline), 20 mg/kg compound 12b treatment group and 40 mg/kg compound 12b treatment group. Vehicle or 12b treatments were administrated intraperitoneally (IP) 3 times/week for 15 consecutive days. During the treatment, tumor volume and body weight were recorded 3 times/week; tumor volume was calculated using the formula V = (L × W2)/2, where L and W are the length and width of the tumor. All mice were sacrificed at the conclusion of the treatment and tumors were collected, weighed, and photographed.
5.8.2. Ovarian cancer model.
The therapeutic experiment was further conducted in an orthotopic ovarian cancer mouse model. 5 × 105 OVCAR8 cells were transduced with the lentiviral vector pEF1a-Luc2 to label them with luciferase and surgically injected into left side of the ovarian bursa of 7-week-old female (NSG) mice (Jackson Laboratory) under a dissecting microscope. After one week, the mice were subjected to bioluminescence imaging using the IVIS In Vivo Imaging System (PerkinElmer XMRS) and Living Image software after IP administration of the substrate D-luciferin. Based on the total flux (photons/s), calculated by the Living Image software from luminescence and body weight, mice were randomized into two groups (n=7/group) and subjected to IP administration of vehicle or vehicle-compound 12b. The 12b treatment was initiated at a dose of 20 mg/kg thrice per week. After 4 treatments, the dose was increased to 40 mg/kg for an additional 4 treatments. Body weight were recorded 3 times/week throughout the course of the experiment. Mice were euthanized after 19 days of treatment and all the major organs/tissues were harvested and bioimaged to visualize metastases specific to that organ.
Supplementary Material
Highlights.
The hydroxyquinoline scaffold, represented by 12b and MX-106, has abilities to selectively target survivin without affecting other closely related members in the IAP family proteins.
12b inhibited melanoma tumor growth in human A375 melanoma xenograft model.
12b effectively suppressed primary tumor growth in ovaries and tumor metastasis in an aggressive orthotopic ovarian cancer mouse model.
Acknowledgments
This work was supported by NIH grant 1R01CA193609 to WL. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank Dr. Lei Yang and Dr. Yong Li at St. Jude Children’s Research Hospital for the metabolic stability evaluation of compounds 7a, 7k, 12a, 12b, 19a, and 19b.
Abbreviations
- ESI
electron-spray ionization
- HPLC
high performance liquid chromatography
- IAP
inhibitor of apoptosis proteins
- ppm
parts per million
- Pgp
P-glycoprotein
- SAR
structure-activity relationships
- TLC
thin layer chromatography
- TMS
tetramethylsilane
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Altieri DC, Survivin, cancer networks and pathway-directed drug discovery, Nat Rev Cancer, 8 (2008) 61–70. [DOI] [PubMed] [Google Scholar]
- [2].Deveraux QL, Reed JC, IAP family proteins--suppressors of apoptosis, Genes Dev, 13 (1999) 239–252. [DOI] [PubMed] [Google Scholar]
- [3].Nomura T, Yamasaki M, Nomura Y, Mimata H, Expression of the inhibitors of apoptosis proteins in cisplatin-resistant prostate cancer cells, Oncol Rep, 14 (2005) 993–997. [PubMed] [Google Scholar]
- [4].Tirro E, Consoli ML, Massimino M, Manzella L, Frasca F, Sciacca L, Vicari L, Stassi G, Messina L, Messina A, Vigneri P, Altered expression of c-IAP1, survivin, and Smac contributes to chemotherapy resistance in thyroid cancer cells, Cancer Res, 66 (2006) 4263–4272. [DOI] [PubMed] [Google Scholar]
- [5].Ryan BM, O’Donovan N, Duffy MJ, Survivin: a new target for anti-cancer therapy, Cancer Treat Rev, 35 (2009) 553–562. [DOI] [PubMed] [Google Scholar]
- [6].Roy K, Singh N, Kanwar RK, Kanwar JR, Survivin Modulators: An Updated Patent Review (2011–2015), Recent Pat Anticancer Drug Discov, 11 (2016) 152–169. [DOI] [PubMed] [Google Scholar]
- [7].Nakahara T, Kita A, Yamanaka K, Mori M, Amino N, Takeuchi M, Tominaga F, Hatakeyama S, Kinoyama I, Matsuhisa A, Kudoh M, Sasamata M, YM155, a novel small-molecule survivin suppressant, induces regression of established human hormone-refractory prostate tumor xenografts, Cancer Res, 67 (2007) 8014–8021. [DOI] [PubMed] [Google Scholar]
- [8].Hong M, Ren MQ, Silva J, Paul A, Wilson WD, Schroeder C, Weinberger P, Janik J, Hao Z, YM155 inhibits topoisomerase function, Anticancer Drugs, 28 (2017) 142–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Rauch A, Hennig D, Schafer C, Wirth M, Marx C, Heinzel T, Schneider G, Kramer OH, Survivin and YM155: how faithful is the liaison?, Biochim Biophys Acta, 1845 (2014) 202–220. [DOI] [PubMed] [Google Scholar]
- [10].Iwai M, Minematsu T, Li Q, Iwatsubo T, Usui T, Utility of P-glycoprotein and organic cation transporter 1 double-transfected LLC-PK1 cells for studying the interaction of YM155 monobromide, novel small-molecule survivin suppressant, with P-glycoprotein, Drug Metab Dispos, 39 (2011) 2314–2320. [DOI] [PubMed] [Google Scholar]
- [11].Wang J, Li W, Discovery of novel second mitochondria-derived activator of caspase mimetics as selective inhibitor of apoptosis protein inhibitors, J Pharmacol Exp Ther, 349 (2014) 319–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Xiao M, Wang J, Lin Z, Lu Y, Li Z, White SW, Miller DD, Li W, Design, Synthesis and Structure-Activity Relationship Studies of Novel Survivin Inhibitors with Potent Anti-Proliferative Properties, PLoS One, 10 (2015) e0129807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Oh S, Shin WS, Ham J, Lee S, Acid-catalyzed synthesis of 10-substituted triazolyl artemisinins and their growth inhibitory activity against various cancer cells, Bioorg Med Chem Lett, 20 (2010) 4112–4115. [DOI] [PubMed] [Google Scholar]
- [14].Zhao G, Wang Q, Wu Z, Tian X, Yan H, Wang B, Dong P, Watari H, Pfeffer LM, Guo Y, Li W, Yue J, Ovarian Primary and Metastatic Tumors Suppressed by Survivin Knockout or a Novel Survivin Inhibitor, Mol Cancer Ther, 18 (2019) 2233–2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zhao G, Wang Q, Gu Q, Qiang W, Wei JJ, Dong P, Watari H, Li W, Yue J, Lentiviral CRISPR/Cas9 nickase vector mediated BIRC5 editing inhibits epithelial to mesenchymal transition in ovarian cancer cells, Oncotarget, 8 (2017) 94666–94680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ahmed N, Abubaker K, Findlay J, Quinn M, Epithelial mesenchymal transition and cancer stem cell-like phenotypes facilitate chemoresistance in recurrent ovarian cancer, Curr Cancer Drug Targets, 10 (2010) 268–278. [DOI] [PubMed] [Google Scholar]
- [17].Yoshida S, Furukawa N, Haruta S, Tanase Y, Kanayama S, Noguchi T, Sakata M, Yamada Y, Oi H, Kobayashi H, Expression profiles of genes involved in poor prognosis of epithelial ovarian carcinoma: a review, Int J Gynecol Cancer, 19 (2009) 992–997. [DOI] [PubMed] [Google Scholar]
- [18].Amin ML, P-glycoprotein Inhibition for Optimal Drug Delivery, Drug Target Insights, 7 (2013) 27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Jin T, Kamijo S, Yamamoto Y, Copper-Catalyzed Synthesis of N-Unsubstituted 1,2,3-Triazoles from Nonactivated Terminal Alkynes, 2004 (2004) 3789–3791. [Google Scholar]
- [20].Rae JM, Ramus SJ, Waltham M, Armes JE, Campbell IG, Clarke R, Barndt RJ, Johnson MD, Thompson EW, Common origins of MDA-MB-435 cells from various sources with those shown to have melonoma properties, Clinical & Experimental Metastasis, 21 (2004) 543–552. [DOI] [PubMed] [Google Scholar]
- [21].Rakesh, Bruhn D, Madhura DB, Maddox M, Lee RB, Trivedi A, Yang L, Scherman MS, Gilliland JC, Gruppo V, McNeil MR, Lenaerts AJ, Meibohm B, Lee RE, Antitubercular nitrofuran isoxazolines with improved pharmacokinetic properties, Bioorg Med Chem, 20 (2012) 6063–6072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Mahindroo N, Connelly MC, Punchihewa C, Yang L, Yan B, Fujii N, Amide conjugates of ketoprofen and indole as inhibitors of Gli1-mediated transcription in the Hedgehog pathway, Bioorg Med Chem, 18 (2010) 4801–4811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Di L, Kerns EH, Li SQ, Petusky SL, High throughput microsomal stability assay for insoluble compounds, Int J Pharm, 317 (2006) 54–60. [DOI] [PubMed] [Google Scholar]
- [24].Di L, Kerns EH, Ma XJ, Huang Y, Carter GT, Applications of high throughput microsomal stability assay in drug discovery, Comb Chem High Throughput Screen, 11 (2008) 469–476. [DOI] [PubMed] [Google Scholar]
- [25].Deng S, Krutilina RI, Wang Q, Lin Z, Parke DN, Playa HC, Chen H, Miller DD, Seagroves TN, Li W, An Orally Available Tubulin Inhibitor, VERU-111, Suppresses Triple-Negative Breast Cancer Tumor Growth and Metastasis and Bypasses Taxane Resistance, Mol Cancer Ther, 19 (2020) 348–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.