

Abstract
Melampomagnolide B (MMB, 3) is a parthenolide (PTL, 1) based sesquiterpene lactone that has been used as a template for the synthesis of a plethora of lead anticancer agents owing to its reactive C-10 primary hydroxyl group. Such compounds have been shown to inhibit the IKKβ subunit, preventing phosphorylation of the cytoplasmic IκB inhibitory complex. The present study focuses on the synthesis and in vitro antitumor properties of novel benzyl and phenethyl carbamates of MMB (7a-7k). Screening of these MMB carbamates identified analogs with potent growth inhibition properties against a panel of 60 human cancer cell lines (71% of the molecules screened had GI50 values <2 μM). Two analogs, the benzyl carbamate 7b and the phenethyl carbamate7k, were the most active compounds. Lead compound 7b inhibited cell proliferation in M9 ENL AML cells, and in TMD-231, OV-MD-231 and SUM149 breast cancer cell lines. Interestingly, mechanistic studies showed that 7b did not inhibit p65 phosphorylation in M9 ENL AML and OV-MD-231 cells, but did inhibit phophorylation of both p65 and IκBα in SUM149 cells. 7b also reduced NFκB binding to DNA in both OV-MD-231 and SUM149 cells. Molecular docking studies indicated that 7b and 7k are both predicted to interact with the ubiquitin-like domain (ULD) of the IKKβ subunit. These data suggest that in SUM149 cells, 7b is likely acting as an allosteric inhibitor of IKKβ, whereas in M9 ENL AML and OV-MD-231 cells 7b is able to inhibit an event after IκB/p65/p50 phosphorylation by IKKβ that leads to inhibition of NFκB activation and reduction in NFκB-DNA binding. Analog 7b was by far the most potent compound in either carbamate series, and was considered an important lead compound for further optimization and development as an anticancer agent.
Keywords: Melampomagnolide B, Benzyl and phenethyl amine, Carbamates, Anticancer activity, Antileukemic activity
Graphical Abstract
1. Introduction
Several drug discovery studies have recently focused on the development of the guaianolide,[1] pseudoguaianolide,[2] germacrolide,[3] melampolide,[4] and heliangolide[5, 6] series of sesquiterpene lactone natural products as antileukemic and anticancer agents. Over the past decade, our research group has extensively investigated the development of the sesquiterpene lactone natural product, parthenolide (PTL, 1) and its analogs as potent antileukemic and anticancer agents that target the NFκB pathway through inhibition of the IκBα/p65/p50 kinase complex (IKK).[7–21]
Initially, these investigations focused on preparing various aminoparthenolide analogs in order to improve the potency and drug-like properties of PTL [14, 16] These aminoparthenolides were shown to be water-soluble and with comparable antileukemic activity when compared to PTL (1, Fig. 1).[9, 14, 16] Among these analogs, the dimethylamino adduct of PTL (DMAPT, 2, Fig. 1) had greatly improved oral bioavailability in rodents (~70%) compared to PTL (~2%) and was successfully developed into a first generation clinical candidate.[16] Recently, we have reported on the sesquiterpene lactone melampomagnolide B (MMB; 3, Fig. 1) and its conjugates as new antileukemic sesquiterpenes.[10, 11, 15] The primary hydroxyl group of MMB has become a key functionality for the synthesis of a variety of MMB conjugates, i.e. esters, carbonates, thiocarbonates, carbamates, and triazole analogs, which exhibit much improved anticancer activities compared to the parent compound.
Fig. 1.

Chemical structures of sesquiterpene lactones parthenolide (PTL; 1), dimethylaminoparthenolide (DMAPT; 2), and melampomagnolide B (MMB; 3).
The carbamate functionality possesses both amide and ester hybrid features. The amide resonance in carbamate moieties displays chemical and proteolytic stability and is broadly utilized as a peptide bond surrogate in medicinal chemistry because of its ability to permeate cell membranes.[22] Carbamates can also modulate inter- and intramolecular interactions with target enzymes or receptors through participation in hydrogen bonding of the carbonyl moiety and the NH functional group.[22] Furthermore, the carbamate group has additional opportunities for chemical modification, such as N-substitution to afford N-alkyl or N-aryl derivatives.
Streptavidin pull-down and LC/MS/MS peptide sequencing studies with an MMB-biotin probe[23] afforded target proteins from both the NFκB and glutathione pathways. Both IKKβ and p65 proteins of the NFκB pathway, as well as the modulatory and catalytic subunits of the ligase and glutathione peroxidase enzyme of the glutathione pathway, were identified as potential targets in the mechanism of action of MMB. Our recent studies on the mechanism of action of MMB-triazole derivatives indicate that these compounds may be down-regulating anti-apoptotic genes under NFκB control by binding to critical sites on the IKKβ subunit of the IKK complex, causing a decrease in phosphorylation of IκB and p50/p65 (NFκB), leading to inhibition of cytoplasmic NFκB activation and reduced binding of NFκB to anti-apoptotic gene regulatory regions on DNA (Fig. 2).
Fig. 2.

MMB-mediated anti-cancer mechanism. MMB and its analogs inhibit IKKβ-mediated phosphorylation of IκB and/or p65, resulting in down-regulation of anti-apoptotic gene transcription and sensitization of cancer cells to apoptotic signals.
Recently, we have reported on imidazole and benzimidazole carbamate analogs of MMB as potential clinical candidates for treatment of acute myelogenous leukemia.[24] In the present study we report on the synthesis and in vitro anticancer activities of novel benzyl and phenethyl carbamate derivatives of MMB (7a-7k), which were evaluated against a panel of 60 human cancer cell lines, against the M9-ENL1 leukemic stem cell line, tumorigenic and ovarian metastasis variants of the MDA-MB-231 breast cancer cell line and the inflammatory breast cancer cell line SUM149PT.
2. Results and Discussion
2.1. Chemistry
The general procedure for the synthesis of the benzyl and phenethyl carbamate derivatives of MMB was as follows: MMB (3) was prepared from PTL via previously reported literature procedures.[15, 17] MMB was reacted with carbonylditriazole (4) to yield the MMB-triazole carbamate intermediate 5.[11, 25] This intermediate was reacted with a variety of substituted benzyl and phenethyl amines (6a-6k) to afford the corresponding benzyl and phenethyl MMB carbamates in 88–93% yield (Scheme 1, 7a-7k). Confirmation of the structure and purity of these analogs was obtained from 1H- and 13C-NMR spectrometry, and from high resolution mass spectrometric analysis.
Scheme 1.

Synthesis of benzyl and phenethyl carbamate derivatives of melampomagnolide B (7a-7k)
2.2. Biological activity
2.2.1. In vitro growth inhibition data against a panel of 60 human cancer cell lines
The benzyl and phenethyl MMB carbamate derivatives 7a-7k were evaluated for antitumor activity in a preliminary screen against an NCI panel of 60 human cancer cell lines, which includes nine different subpanels representing leukemia, non-small cell lung, colon, central nervous system, melanoma, ovary, renal, prostate, and breast cancer cell lines, at a concentration of 10 μM utilizing a sulforhodamine B (SRB) assay procedure described by Rubinstein et al.[26] The NCI-60 screening assay measures the growth inhibition of the test compounds by determining percentage cell growth inhibition by optical density (OD) measurements of SRB-derived color, just before exposing the cells to the test compound (ODtzero), and after 48 h exposure to the test compound (ODtest) or the control vehicle (ODctrl).
From the preliminary single dose screening, all compounds which showed 60% or more growth inhibition in at least eight of the cell lines (namely, 7a-7c, 7h, 7i and 7k) were screened in the same way using five different concentrations (100 μM, 10 μM, 1 μM, 0.1 μM and 10 nM) to afford dose-dependent growth inhibition curves. Growth inhibition values (GI50) representing the molar drug concentration resulting in a 50% reduction in net protein increase compared with control cells, were determined and are presented in Table 1. The Total Growth Inhibition (TGI) and Lethal Concentration values (LC50) are also provided in the Supporting Information. The 3,4,5-trimethoxybenzyl (7f) and 3,4-dimethoxyphenethyl (7j) analogs were not potent enough in the single dose screening to be selected for 5-dose screening.
Table 1.
Antitumor activity (GI50/μM)a data for the substituted benzyl and phenethyl carbamate derivatives of MMB (7a-c, 7h, 7i and 7k) from the NCI-five dose human cancer cell panel assay
| 1b | 7a | 7b | 7c | 7h | 7i | 7k | |
|---|---|---|---|---|---|---|---|
|
|
|||||||
| Panel/cell line | GI50 | GI50 | GI50 | GI50 | GI50 | GI50 | GI50 |
| Leukemia | |||||||
| CCRF-CEM | 7.94 | 0.47 | 0.26 | 0.48 | 0.34 | 1.98 | 0.27 |
| HL-60(TB) | 5.01 | 1.69 | 0.26 | 1.61 | 1.36 | 2.34 | 1.06 |
| K-562 | 19.9 | 1.57 | 0.37 | 0.82 | 0.61 | 3.08 | 0.53 |
| MOLT-4 | 15.8 | 2.00 | 0.28 | 2.53 | 2.09 | 2.61 | 1.69 |
| RPMI-8226 | 7.94 | 1.8G | 0.26 | 1.77 | 0.91 | 2.85 | 0.62 |
| SR | nd | 0.32 | 0.30 | 0.67 | 0.51 | 2.07 | 0.43 |
| Lung Cancer | |||||||
| A549/ATCC | nd | 2.58 | 2.43 | 3.07 | 7.28 | 13.2 | 2.59 |
| EKVX | nd | 2.06 | 1.29 | 1.98 | 1.78 | 7.45 | 1.54 |
| HOP-62 | nd | 3.06 | 4.81 | 3.69 | 4.39 | 12.2 | 1.99 |
| HOP-92 | 12.5 | 1.42 | 0.24 | 1.49 | 1.84 | 1.99 | 1.28 |
| NCI-H226 | nd | 2.02 | 0.48 | 2.11 | 1.57 | 4.11 | 1.72 |
| NCI-H23 | nd | 1.84 | 0.55 | 1.74 | 1.86 | 4.34 | 1.72 |
| NCI-H322M | nd | 4.72 | nd | nd | nd | 12.6 | nd |
| NCI-H460 | nd | 3.01 | 2.74 | 3.73 | 3.94 | 12.2 | 2.06 |
| NCI-H522 | 5.01 | 0.34 | 0.17 | 0.35 | 1.02 | 1.82 | 1.16 |
| Colon Cancer | |||||||
| COLO 205 | 15.8 | 1.35 | 0.24 | 1.75 | 1.55 | 1.94 | 1.82 |
| HCC-2998 | nd | 1.94 | 0.51 | 1.82 | 1.68 | 2.48 | 1.71 |
| HCT-116 | 10.0 | 0.77 | 0.19 | 0.54 | 0.71 | 1.34 | 0.47 |
| HCT-15 | nd | 0.77 | 0.24 | 1.04 | 1.02 | 1.74 | 1.01 |
| HT29 | nd | 1.20 | 0.26 | 1.22 | 1.14 | 2.15 | 1.32 |
| KM12 | nd | 2.80 | 2.35 | 3.53 | 4.27 | 8.36 | 1.95 |
| SW-620 | 15.8 | 1.12 | 0.22 | 0.76 | 0.52 | 2.51 | 0.71 |
| CNS Cancer | |||||||
| SF-268 | nd | 2.46 | 0.75 | 2.20 | 1.78 | 10.6 | 1.56 |
| SF-295 | nd | 7.78 | 10.8 | 5.02 | 7.26 | 13.5 | 3.81 |
| SF-539 | 19.9 | 1.54 | 0.32 | 1.53 | 1.49 | 1.79 | 1.46 |
| SNB-19 | nd | 5.32 | 0.70 | 1.57 | 1.43 | 8.65 | 1.39 |
| SNB-75 | 50.1 | 1.99 | 0.74 | 2.44 | 1.47 | 4.09 | 1.20 |
| U251 | nd | 3.16 | 0.96 | 2.20 | nd | 11.1 | 1.40 |
| Melanoma | |||||||
| LOX IMVI | 7.94 | 0.54 | 0.27 | 1.43 | 1.34 | 1.38 | 1.18 |
| MALME-3M | 12.5 | 1.86 | 0.20 | 1.89 | 1.84 | 2.44 | 2.09 |
| M14 | nd | 1.64 | 0.19 | 1.10 | 1.29 | 1.98 | 1.26 |
| MDA-MB-435 | nd | 1.99 | 0.42 | 1.70 | 1.58 | 3.53 | 1.58 |
| SK-MEL-2 | nd | 1.66 | 1.18 | 1.71 | 1.73 | 8.97 | 1.58 |
| SK-MEL-28 | nd | 1.47 | 0.27 | 1.52 | 1.35 | 1.99 | 1.47 |
| SK-MEL-5 | nd | 3.48 | 1.20 | 2.86 | 1.84 | 6.23 | 1.71 |
| UACC-257 | nd | 1.54 | 0.32 | 1.48 | 1.53 | 2.49 | 1.63 |
| UACC-62 | nd | 0.66 | nd | nd | nd | 1.25 | nd |
| Ovarian Cancer | |||||||
| IGROV1 | 19.9 | 2.12 | 0.29 | 1.57 | 1.26 | 2.61 | 1.08 |
| OVCAR-3 | 19.9 | 1.01 | 0.28 | 0.87 | 0.99 | 2.19 | 0.98 |
| OVCAR-4 | nd | 1.70 | 0.49 | 1.78 | 1.78 | 3.41 | 1.56 |
| OVCAR-5 | nd | 1.82 | 1.46 | 2.29 | 2.25 | 2.65 | 1.96 |
| OVCAR-8 | nd | 1.60 | 0.49 | 2.45 | 2.26 | 2.77 | 1.49 |
| NCI/ADR-RES | nd | 2.98 | 1.79 | 2.26 | 2.95 | 10.0 | 2.26 |
| Renal Cancer | |||||||
| 786-0 | nd | 1.70 | 0.18 | 1.16 | 1.37 | 1.76 | 1.04 |
| A498 | nd | 2.11 | 0.72 | 3.23 | 2.35 | 4.52 | 2.07 |
| ACHN | nd | 1.29 | 0.27 | 1.27 | 1.12 | 1.72 | 1.32 |
| CAKI-1 | 10.0 | 1.59 | 0.31 | 1.38 | 1.19 | 2.52 | 1.42 |
| RXF 393 | 12.5 | 0.68 | 0.19 | 1.04 | 0.67 | 1.55 | 1.20 |
| TK-10 | nd | 1.66 | 0.35 | 1.79 | 1.61 | 2.73 | 1.73 |
| UO-31 | nd | 1.51 | 0.16 | 1.68 | 1.39 | 1.76 | 1.55 |
| Prostate Cancer | |||||||
| PC-3 | nd | 2.72 | 1.87 | 3.13 | 2.30 | 8.08 | 1.73 |
| DU-145 | nd | 0.72 | 0.37 | 0.69 | 0.89 | 1.92 | 0.77 |
| Breast Cancer | |||||||
| MCF7 | 15.8 | 0.58 | 0.27 | 0.49 | 0.49 | 1.89 | 0.41 |
| MDA-MB-231 | nd | 1.61 | 0.62 | 1.47 | 1.56 | 2.02 | 1.42 |
| HS 578T | nd | 4.12 | 0.79 | 3.40 | 3.43 | 13.8 | 2.41 |
| BT-549 | nd | 1.57 | 0.20 | 1.25 | 1.51 | 2.12 | 1.10 |
| T-47D | nd | 1.85 | 0.34 | 2.01 | 1.61 | 2.47 | 1.38 |
| MDA-MB-468 | nd | 0.58 | 0.24 | 0.66 | 0.85 | 1.55 | 0.84 |
| Average GI50 | 14.96 | 1.92 | 0.86 | 1.80 | 1.82 | 4.4 | 1.44 |
GI50 values <1μM are bolded; nd: not determined;
GI50: concentration of drug resulting in a 50% reduction in net cell growth, as compared to cell numbers on day 0.
5-dose NCI cancer cell screening data for PTL (1).[10]
MMB carbamate derivatives 7a-c, 7h-7i and 7k exhibited greater cytotoxicity in the five dose NCI-60 human cancer cell assay when compared to PTL (Table 1). The 3,5-dimethoxybenzyl analog 7a afforded potent growth inhibition against 71 % of all the cancer cell lines in the panel, with GI50 values ranging from 0.32 to 2.0 μM, and an average GI50 value of 1.9 μM for all the cell lines in the panel. This compound exhibited particularly potent growth inhibition against the SR leukemia and NCI-H522 lung cancer cell lines with GI50 values of 0.32 μM and 0.34 μM, respectively.
The 4-methoxybenzyl analog 7b, exhibited potent growth inhibition against 91 % of the cancer cell lines in the NCI-60 panel, affording GI50 values ranging from 0.16 to 1.9 μM and was by far the most potent of all the carbamate analogs in the series with an average GI50 value of 0.86 μM against all the cancer cell lines in the panel. Compound 7b exhibited particularly potent growth inhibition against the leukemia subpanel cell lines with GI50 values in the range 0.26–0.37 μM, and was also effective against solid tumor cell lines in the renal cancer and breast cancer cell panels with GI50 values of 0.16–0.72 μM and 0.20–0.86 μM, respectively. Against renal cancer cell lines UO-31, 786–0 and RXF-393, 7b afforded GI50 values of 0.16, 0.18 and 0.19 μM, respectively, and against breast cancer cell lines BT-549, MDA-MB-468, and MCF7 7b exhibited GI50 values of 0.20, 0.24, and 0.25 μM, respectively.
The 2-methoxybenzyl analog 7c exhibited potent growth inhibition against 68% of the cancer cell lines in the NCI-60 panel, with GI50 values ranging from 0.35 to 1.98 μM and an average GI50 value of 1.80 μM for all the cell lines in the panel. Compound 7c was most effective in the leukemia subpanel and had a GI50 value of 0.48 μM against the CCRF-CEM cell line; it was less effective than 7b against the solid tumor cell panels, but did exhibit some activity against lung cancer cell line NCI-H522 (GI50=0.35 mM) and breast cancer cell line MCF7 (GI50=0.49 μM).
In the phenethyl carbamate series, the 3-methoxyphenethyl analog 7h exhibited potent growth inhibition against 78% of the cancer cell lines in the NCI-60 panel with GI50 values ranging from 0.34 to 1.86 μM with an average GI50 value of 1.82 μM for all the cell lines in the panel. GI50 values of 0.34 and 0.49 μM were observed against CCRF and MCF7 cell lines, respectively.
The 4-methoxyphenethyl carbamate analog 7i, which was the least potent of the six analogs evaluated in the NCI-60 assay, was active against only 29% of the cancer cell lines in the panel, with GI50 values ranging from 1.25 to 1.99 μM, and an average GI50 value of 4.4 μM for all the cell lines in the panel. Thus, a significant ten-fold reduction in overall anticancer activity is observed for 7i when compared to 7b, as a result of the replacement of the 4-methoxybenzyl moiety for a 4-methoxyphenethyl moiety (Table 1).
The 4-bromophenethyl carbamate analog 7k, exhibited potent growth inhibition against 88% of the cancer cell lines in the NCI-60 panel with GI50 values ranging from 0.27 to 1.99 μM and an average GI50 value of 1.44 μM for all the cell lines in the panel. It was most effective against the leukemia subpanel, but did exhibit some activity against solid tumor cell lines MCF-17 (GI50=0.41 μM) and HCT-15 (colon cancer; GI50=0.47 μM).
The above results indicate that the benzyl carbamate conjugate of MMB (7b) is a potent anticancer agent against both hematological and solid human tumor cell lines. Compound 7k was the most potent compound in the phenethyl carbamate series of analogs, but was considerably less potent than 7b.
2.2.2. Inhibition of p65 phosphorylation by 7b in M9-ENL AML cell cultures
We determined the ability of the lead analog, 7b at various concentrations (2.5, 5, and 10 μM), to inhibit NF-κB activation via IKKβ-mediated phophorylation of p65 at SER-536 in M9-ENL AML stem cells, followed by lysis and analysis by immuno blot. We used PTL (1) as a reference compound at the same concentrations. Interestingly, we found that 7b had no inhibitory effect on p65 phophorylation (Fig. 3) in M9-ENL AML cells, which is consistant with the inability of 7b to bind to the KD site of IKKβ in our modeling data, but is inconsistent with the premise that the mechanism of 7b is allosteric inhibition of IKKβ or that 7b somehow blocks the ability of IKKβ to recognise its substrate.
Fig. 3.

(A) relative viability of acute myelogenous leukemia M9-ENL stem cells after treatment with parthenolide (PTL) and 7b. (B) inhibition of p65 phosphorylation by PTL and 7b in leukemia M9-ENL cells.
2.2.3. Compound 7b is a potent inhibitor of NF-κB and cell proliferation in breast cancer cells
Utilizing three additional cancer cell lines: xenograft tumor-derived MDA-MB-231 breast cancer cell line (TMD-231), an ovarian metastasis-derivative of TMD-231 (OV-MD-231) and an inflammatory breast cancer cell line (SUM149PT), we also wanted to determined the ability of lead compound 7b to inhibit cell proliferation and IKKβ phosphorylation of IκBα in solid tumor cells. Cells were treated with various concentrations of 7b over 4–5 days and bromodeoxyuridine incorporation-ELISA assays were performed to measure cell proliferation (Fig. 4A). TMD-231 cells were more sensitive to 7b compared to its OV-MD-231 cells, and SUM149PT cells were even more sensitive to 7b with an IC50 of <1 μM.
Fig. 4.

(A) Compound 7b inhibits proliferation of breast cancer cell lines. The effect of 7b on proliferation of TMD-231, OV-MB-231 and SUM149PT cancer cells. Cells were treated with the indicated concentrations of 7b and cell proliferation was measured by bromodeoxy uridine ELISA assay 4–5 days after treatment. Results represent average values from two experiments with six tachnical replicates. Similar results were obtained in four biological replicates in case of OV-MD-231 and SUM149PT. Experiments were done twice in TMD-231 cells with similar results. (B) Inhibition of TNFα-induced p65 and IκBα phosphorylation by 7b in OV-MD-231 and SUM149PT cancer cell lines.
In studies with the OV-MD-231 breast cancer cell line, as was observed with M9 ENL cells, 7b also failed to act as an inhibitor of p65 phophorylation (Fig. 4B), although in the SUM149PT cell line, an inflammatory breast cancer cell line, 7b at 2 μM did inhibit TNFα-induced phosphorlation of p65 (Fig. 4B). Compound 7b inhibited IκBα phosphorylation in the SUM149PT cell line (Fig. 4B). For unknown reasons, we were unable to detect IκBα in OV-MD-231 cells, raising an intriguing possiblity of ovarian metastasis-associated loss of IκBα.
Treatment of SUM149PT and OV-MD-231 cells with 2 μM 7b for 3 hours demonstrated a significant reduction in NFκB-DNA binding activity in both cell lines (Fig. 5). The effect of 7b was specific to NFκB, since the drug had no effect on DNA binding to SP-1 in either cell line.
Fig. 5.

Inhibition of NFκB-DNA binding in OV-MD-231 and SUM149PT breast cancer cell lines after exposure of cells to 0.5 μM and 2.0 μM of 7b over 3 h. EMSA was performed to measure NFκB and SP-1 DNA binding.
Cytokine-mediated NFκB activation in these breast cancer cell lines involves activation of IKKβ and subsequent phophorylation and degradation of the IκBα inhibitory complex with cytoplasmic release of NFκB. Thus, the inhibitory effect of 7b on NFκB-DNA binding must involve an upstream mechanism(s) that inhibits this degradation of IκBα. In the case of SUM149PT cells, the inhibition of NFκB-DNA binding that results from exposure to 7b is likely due to the ability of this compound to inhibit p65 phosphorylation as well as IκBα phosphorylation (Fig. 4B), although the mechanism of inhibition of p65 phosphorylation by 7b is likely allosteric since this analog binds to the ULD rather than the KD of this kinase (Fig. 6).
Fig. 6.

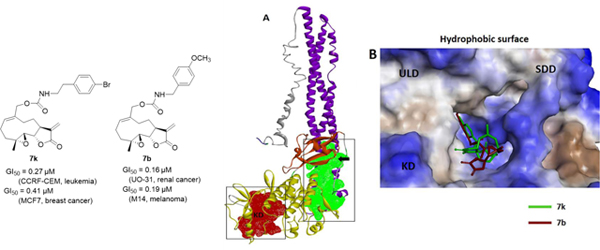
(A) Full length structure of IKKβ showing kinase domain (KD), ubiquitin-like domain (ULD) and scaffold dimerization domain (SDD), and interaction of 7b and 7k with the ULD. (B) predicted Glide score (GScore) for the two molecules, (C&D) predicted binding poses of molecules 7b and 7k, respectively at the ULD. (E) binding affinity estimates from MM-GBSA calculations. (F-G) molecular interactions of 7b and 7k with IKKβ amino acid residues.
The ability of 7b to inhibit NFκB-DNA binding in OV-MD-231 cells is intreging, since 7b does not inhibit p65 phosphorylation in these cells (Fig. 4B), indicating that 7b likely blocks a subsequent post-phosphorylation step that results in reduced binding of NFκB to DNA.
2.3. Molecular Docking and MM/GBSA Binding Affinity Studies
Based on our previous studies, both PTL (1) and MMB (3) inhibit formation/activation of the NFκB transcription factor leading to down-regulation of its down-stream targets [27, 28] and inhibition of nuclear translocation of p65 [29]. We analyzed the interactions of the most potent MMB derivatives 7b and 7k in this present study with a computational model of the IKKβ subunit (Fig. 6).
Monomeric IKKβ has three conserved domains including the kinase domain (KD) or activation domain, the scaffold dimerization domain (SDD), which facilitates dimerization and oligomerization of IKKβ, and the ubiquitin-like domain (ULD) which is important for the catalytic activity of IKKβ[30–32]. Interestingly, molecular modeling studies have determined that PTL and MMB both interact with the KD site of the IKKβ subunit, while the aminoparthenolide, DMAPT, binds at the interface of the KD and SDD sites.33 In the present study we analyzed the interactions of 7b and 7k with the above binding domains on IKKβ to identify the mechanism of action of these lead molecules.
We performed protein-ligand docking studies similar to our previous investigations.[33] We first screened the whole IKKβ subunit for preferential binding cavities for compounds 7b and 7k using autodock-VINA. Results indicated that both 7b and 7k bind specifically to the ubiquitin-like domain (ULD) on the IKKβ subunit (Fig. 6A, 6C and 6D). Further confirmation of these results was obtained when we again performed standard flexible protein-ligand docking using GLIDE (Schrodinger, Inc.), focusing specifically on the ULD. Predicted Glide scores (GScore) and Glide minimum energy docking poses for 7b and 7k are given in Fig. 6B. In addition to autodock-VINA and Glide docking, protein-ligand complexes were minimized and scored for binding affinity (ΔGbinding) with MM/GBSA calculations (Table 2) in implicit solvent conditions. The calculated binding estimates shown in Fig. 6E indicate that both 7b and 7k show the greatest binding affinities for the ULD site on IKKβ.
Table 2.
Calculated binding affinities (ΔGbinding) with MM/GBSA for compounds interacting with IKKβ
| Compound | ΔGbinding (kcal/mol) | Site of Interaction |
|---|---|---|
| 7b | −50.5 | ULD |
| 7k | −42.8 | ULD |
The kinase domain (KD) of the IKKβ subunit extends from amino acids 16 to 307, whereas the ULD extends from amino acids 310 to 394 with an extended polypetide unit from 391 to 410 that connects the ULD to the scaffold dimerization domain (SDD) extending from amino acids 410 to 666. Finally, a NEMO binding domain spans from amino acids 708 to 742. Analyzing these binding residues indicates that both 7b and 7k bind predominantly to the residues that are part of the ULD; specifically, Leu 382, and Ile 322 interact with the benzene ring (π-alkyl bond interaction) of both 7b and 7k; similarly, His 313 of the ULD also interacts with 7b and 7k. Interestingly, binding residue analysis indicated that both 7b and 7k also interact with arginine and proline residues that are part of the amino acid stretch that connects the ULD and SDD. Arg 446 (SDD) is predicted to form a strong π-cation interaction with the phenyl ring of both 7b & 7k. Compound 7b forms a hydrogen bond with Arg 404 and Leu 265. Similarly, 7k forms hydrogen bonds with both Arg 404 and Asn 263. Amino acid residues that are part of the KD, including Asn 263, Asn 264, Leu 266, and Ser 267 also interact with 7b and 7k (Fig. 6F & 6G).
Studies [28, 34, 35] have shown that the ULD is a conserved domain which is absent in the IKKα subunit, and that the role of the ULD is critical for the catalytic activity of IKKβ, since ULD deletion mutants of IKKβ lose their catalytic activity and are potent inhibitors of IκBα/p65/p50 phosphorylation and subsequent nuclear translocation of p65/p50 (NFκB). These studies have also shown that the ULD site is not required for maintenance of the IKK complex architecture, since ULD mutants of IKKβ are still able to form IKK complexes with IKKα and IKKγ (NEMO).
It should be noted that the ULD does not appear to play a role in facilitating ubiquitination or ubiquitination-mediated conjugation of IKKβ, and thus is not a likely target for IKKβ ubiquitination or ubiquitin-like conjugation of IKKβ with other proteins.[28] However, Carter et al.[36, 37] have shown that mono ubiquitination of lysine 163 regulates phosphorylation of the activation T loop of IKKβ.
The studies by May et al.[28] have clearly shown that the ULD is a critical domain required for functional activity of IKKβ and that the integrity of the region between Leu 311 and Ala 367 is absolutely critical for the functional activity of IKKβ. This region of the ULD likely interacts with the C-terminal portion of IκBα to facilitate concerted phophorylation at Ser 42 and Ser 36 prior to dissociation of IκBα from the IκBα/p65/p50 complex. ULD deletion results in the ULD mutant IKKβ subunit being no longer being able to recognize and physically interact with its specific substrate, IκBα/p65/p50, resulting in inhibition of phosphorylation of downstream NFκB pathway targets. Xu et al.[34] have hypothesized that KD activity is compromised in the absence of the ULD, since the ULD-SDD region is critical for IKKβ specificity, while the ULD is required for catalytic activity. Thus, the ULD-SDD region of IKKβ directly interacts with the C-terminal portion of IκBα and probably orients IκBα such that its N-terminal cognate phophorylation sites are presented to the KD active site.
Both 7b and 7k show interactions with amino acid residues in the SDD and KD regions, indicating that these molecules are targeting the ULD at a binding area that is at the interface with the KD and SDD.
Computational binding cavity analysis indicates that the IKKβ subunit contains a cavity on the ULD at the interface of the KD, and SDD (Fig 7A, green; and 4B). Studies have shows the importance of activity regulation by the residues present at the interface between these domains of IKKβ. Based on our predictions and previous studies, we propose two possible mechanisms of action for compounds 7b and 7k. One possibility is that binding of 7b and 7k to the ULD may cause allosteric structural changes at the KD site (probably at the T-loop), which would affect downstream phosphorylation and inhibition of the NFκB pathway, or that these compounds may be interacting with IKKβ at the critical Leu 311 to Ala 367 region of the ULD, thereby interfering with the ability of IKKβ to recognize its specific substrate, IκBα/p65/p50, resulting in inhibition of phosphorylation of the downstream elements of the NFκB pathway.
Fig. 7.

(A) Binding cavity prediction indicates two major druggable sites on the IKKβ molecule: one at the KD (red) and one at the ULD at the interface of the KD and SDD (green). (B) ULD binding pocket (at the interface of the KD and SDD) showing molecules 7b (blue) and 7k (crimson red) superimposed on the solid molecular surface.
3. Conclusions
A series of novel substituted benzyl and phenethyl carbamate derivatives of MMB (7a-7k) have been synthesized and evaluated for their anticancer activity against a panel of 60 human cancer cell lines. All the analogs exhibited growth inhibition properties that were improved over MMB. The rank order of compounds based on average GI50 values (Table 1) was 7b>7k>7c>≡7h>7a>71. Within the benzyl carbamate derivatives, the most active compound from the human cancer cell growth inhibition assays was the 4-methoxybenzyl analog, 7b. Moving the methoxy group around the phenyl ring to afford 2-methoxybenzyl and 3-methoxybenzyl analogs resulted in lower growth inhibition, and the presence of additional methoxy groups (i.e. 7a, 7e and 7f) also afforded less effective agents. In the phenethyl carbamate series of analogs, the 4-methoxyphenethyl analog 7i was considerably less active than 7b. The most active phenethyl carbamate derivative was the 4-bromophenethyl analog 7k.
Molecular docking studies with monomeric IKKβ showed that the 4-methoxybenzyl and 4-bromophenethyl carbamate analogs 7b and 7k bind to the ULD site on IKKβ, which represents a potentially new binding site for MMB analogs at the IKKβ subunit. Interestingly, in our modeling data we observed that both 7b and 7k interacted with the ULD site in a similar manner. The aromatic substituent in both compounds interacts with a critical His 313 amino acid residue via different binding modes that help anchor down the aromatic ring in these two compounds at the ULD binding site. In the case of 7b the 4-methoxy moiety interacts with the imidazole ring of His 313 via a hydrogen bonding interaction, whereas with 7k the 4-bromo substituent is involved in a C-Br--π interaction with the imidazole ring of His 313. These compounds are thought to allosterically inhibit IKKβ phosphorylation of its substrate, IκBa/p65/p50, or prevent the ability of IKKβ from recognizing its specific substrate, resulting in inhibition of down-stream targets of the NFκB pathway and leading to down-regulation of anti-apoptotic gene transcription and sensitization of cancer cells to apoptotic signals.
The ULD site on IKKβ constitutes a new therapeutic target for intervention in the NFκB pathway, and analog 7b was considered an important lead compound for further development as an anticancer agent, since it represents a promising new tool for downregulating pro-inflammatory cytokine-mediated activation pathways in cancer cells. One of the main drawbacks of targeting kinase domains is the likelyhood of off-target effects, since many kinase domains share sequence homology and other structural similarities. Targeting a site on the IKKβ complex that is not a kinase site, would likely minimize off-target inhibition of other kinases through active site inhibition.
The inability of compound 7b to inhibit p65 phosphorylation in M9 AML cells and OV-MD-231 cells is interesting, and is consistent with this compound’s inability to bind to the KD site on the IKKβ subunit. However, the fact that 7b is able to inhibit NFκB-DNA binding in the latter cell type suggests that this compound is inhibiting a post-phosphorylation event after IκB/p65/p50 phosphorylation by IKKβ that leads to inhibition of NFκB activation.
It has been shown in several studies that IKKβ-mediated phophorylation of the IκB/p65/p50 complex (induced by TNFα, LPS, and other molecules) does not result directly in disassociation of this inhibitor complex with subsequent activation of NFκB.1–8 Other studies utilizing peptide aldehydes to block subsequent activation of NFκB have shown accumulation of an inducibly phophorylated IκBα complex, but NFκB is not activated, indicating that this inducible phosphorylation of IκBα is not sufficient for dissociation of the IκB/p65/p50 complex; thus, an additional event must be required for post-phosphorylation NFκB activation9–16. Other studies have shown that post-phosphorylation ubiquitination of IκBα in vivo following treatment of cells with inducers of NFκB does not lead to disassociation of the IκB/p65/p50 complex and activation of NFκB. These studies have shown that there is strong evidence suggesting that the proteosome is involved in the degradation of the IκB/p65/p50 complex during NFκB activation, and that NFκB remains associated with the ubiquitinated and phophorylated IκBα subunit during its degradation by the 26S proteosome. These events are summarized in Fig. 8 below.
Fig. 8.

Post-phosphorylation events after IκB/p65/p50 phosphorylation by IKKβ that lead to NFκB activation and DNA binding.
Lead analog 7b interacts with the ULD site on IKKβ, and it has been demonstrated that this site is required for functional activity of this kinase and that a ULD deletion mutant associates with p65, suggesting that this domain is not required to facilitate this interaction, but is in fact necessary for the post phosphorylation disassociation of IKKβ from p65. Most interestingly, a single point mutation at Leu353 of the ULD is sufficient to cause an interaction with p65, suggesting that this is indeed a highly critical residue for this proposed function of the ULD. It is therefore possible that the ULD recruits a protein that is involved in disassembling the IKKβ-IκB/p65/p50 complex following phosphorylation by IKKβ.
Alternatively, a conformational change that requires a functional ULD may be required for IKKβ release, and mutation within the ULD might prevent this dissociation from occurring. Thus, it is possible that compound 7b may be acting in a similar manner to the ULD deletion mutant, and may be either blocking the site that binds the protein involved in disassociation of IKKβ from the IκB/p65/p50 complex, or 7b may be inhibiting the conformational change on the ULD that mediates dissociation of IKKβ from the IκB/p65/p50 complex (Fig 8). Either way, these data suggest that 7b is acting upstream of the proteosomal inhibitor, bortizomib.
It is clear from the Western blot data (Fig. 4B) that in OV-MD-231 cells compound 7b did not inhibit phosphorylation of p65, but did reduce (at about 2 μM) NFκB binding to DNA (Fig. 5). Interestingly, in SUM149PT cells 7b inhibited both phosphorylation of p65 and reduced NFκB binding to DNA, suggesting a different mechanism of action in this inflammatory breast cancer cell line compared to that observed in the OV-MD-231 cell line. These data suggest that in OV-MD-231 cells, compound 7b inhibits NFκB activation at a point that is downstream from IKKβ phosphorylation of p65, perhaps at the point where ubiquitination of the NFκB/p65/p50 inhibitory complex occurs, or at the point where proteosome binding of the ubiquitinated inhibitory complex occurs. Another possibility is that 7b may actually inhibit NFκB binding to DNA. In SUM149PT cells the mechanism of inhibition of 7b appears to be quite different than in OV-MD-231 cells (and perhaps M9-ENL cells); i.e. inhibition of p65 phosphorylation is observed which would support an allosteric mechanism of inhibition of IKKβ-mediated phosphorylation of p65 in this cell line.
These data may have important implications for our understanding of possible mechanisms of inhibition of the NFκB pathway in different cancer cell lines.
4. Experimental
All reagents, solvents and chemicals utilized in the synthesis of the benzyl and phenethyl carbamates of MMB, were purchased from Oakwood Chemicals and Fisher Scientific. The synthetic reactions were carried out at ambient temperature and the products were purified by flash column chromatography (silica gel; methanol/dichloromethane) to afford pure compounds in 88–93% yield. 1H and 13C NMR spectra were recorded on a Varian 400 MHz spectrometer equipped with a Linux workstation running on vNMRj software. Spectral analyses were carried out in CDCl3 taking the CDCl3 peak as reference standard for both 1H and 13C spectra. Chemical shifts were measured as δ values in parts per million (ppm) and coupling constants (J) were measured in hertz (Hz). HRMS data were recorded on an Agilent 6210 LCTOF instrument operating in multimode. Thin-layer chromatography (TLC) was carried out on pre-coated silica gel glass plates (F 254 Merck). The purity of final compounds was determined to be ≥95% by high pressure liquid chromatographic (HPLC) analysis on a Model 1290A Agilent HPLC-Diode Array unit utilizing an Altima-C18 column (4.6 × 250 mm, 5 μm); a mobile phase of acetonitrile containing 30% Milli Q water at a flow rate of 0.8 mL/min was used. UV detection was at 210 nm.
4.1. General procedure for the synthesis of benzyl and phenethyl carbamate derivatives of MMB (7a-7k):
A mixture of MMB-triazole (5) (1.0 mmol) and an appropriate aryl amine (6a-6j) (1.1 mmol) in dichloromethane (10 mL) was stirred for 4 hrs at ambient temperature. After completion of the reaction (monitored by TLC), the reaction mixture was concentrated under reduced pressure to afford the crude product. The crude product was purified by column chromatography (silica gel, 2% methanol in dichloromethane) to afford the corresponding benzyl and phenethyl carbamate derivatives of MMB (7a-7k).
4.1.1. ((1aR,7aS,10aS,10bS,E)-1a-methyl-8-methylene-9-oxo-1a,2,3,6,7,7a,8,9,10a,10b-decahydrooxireno[2’,3’:9,10]cyclo deca[1,2-b]furan-5-yl)methyl(3,5-dimethoxybenzyl)carbamate (7a).
Yield: 92%, 1H NMR (400 MHz, DMSO-d6): δ 0.87 (t, 1H, J = 13.2 Hz, CH), 1.43 (s, 3H, CH3), 1.59 (m, 2H, CH2), 2.04–2.27 (m, 6H, 3xCH2), 2.79 (m, 1H, CH), 3.00 (m, 1H, CH), 3.67 (s, 6H, 2xOCH3), 4.07 (m, 3H, J = 9.6 Hz, OCH and CH2), 4.35–4.49 (dd, 2H, J = 12.4 Hz, 37.2 Hz, OCH2), 5.54 (d, 1H, J = 2.4 Hz, =CH), 5.99 (d, 1H, J = 2.8 Hz, OCH), 6.32 (m, 3H, Ar-H) 7.69 (bs, 1H, NH) ppm. 13H NMR (100 MHz, CDCl3): δ 17.98, 23.77, 24.45, 25.79, 36.60, 42.60, 45.20, 55.33, 59.91, 63.26, 67.28, 81.04, 99.20, 105.42, 120.28, 130.10, 135.41, 138.72, 140.59, 156.08, 161.07, 169.38 ppm. HRMS calcd. For C25H32NO7 (M+H)+: 458.2173. Found 458.1673.
4.1.2. ((1aR,7aS,10aS,10bS,E)-1a-methyl-8-methylene-9-oxo-1a,2,3,6,7,7a,8,9,10a,10b-decahydrooxireno[2’,3’:9,10]cyclo deca[1,2-b]furan-5-yl)methyl(4-methoxybenzyl)carbamate (7b).
Yield: 93%, 1H NMR (400 MHz, CDCl3): δ 1.07 (t, 1H, J = 13.6 Hz, CH), 1.25 (s, 3H, CH3), 1.64 (m, 2H, CH2), 2.17–2.42 (m, 6H, 3xCH2), 2.84 (d, 1H, J = 5.6 Hz, CH), 3.01 (m, 1H, CH), 3.80 (s, 3H, OCH3), 4.29 (d, 1H, J = 4.8 Hz, OCH), 4.49–4.66 (dd, 2H, J = 12.4 Hz, 37.2 Hz, OCH2), 4.93 (d, 1H, J = 2.4 Hz, =CH), 5.52 (s, 1H, OCH), 5.68 (d, 1H, J = 3.2 Hz, =CH), 6.23 (s, 1H, =CH), 6.86 (d, 2H, J = 8.0 Hz, Ar-H), 7.19 (d, 2H, J = 6.4 Hz, Ar-H) ppm. 13H NMR (100 MHz, CDCl3): δ 18.11, 23.81, 24.66, 25.89, 29.66, 36.65, 42.61, 44.64, 55.37, 60.01, 63.28, 67.30, 81.14, 114.08, 120.39, 128.96, 130.14, 130.40, 156.10, 159.04, 169.50 ppm. HRMS calcd. for C24H30NO6 (M+H)+: 428.2127. Found 428.2125.
4.1.3. ((1aR,7aS,10aS,10bS,E)-1a-methyl-8-methylene-9-oxo-1a,2,3,6,7,7a,8,9,10a,10b-decahydrooxireno[2’,3’:9,10]cyclo deca[1,2-b]furan-5-yl)methyl(2-methoxybenzyl)carbamate (7c).
Yield: 90%, 1H NMR (400 MHz, CDCl3): δ 1.09 (t, 1H, J = 13.6 Hz, CH), 1.25 (s, 3H, CH3), 1.53 (m, 2H, CH2), 2.13–2.40 (m, 6H, 3xCH2), 2.83 (d, 1H, J = 9.6 Hz, CH), 3.01 (m, 1H, CH), 3.83 (s, 3H, OCH3), 4.33 (d, 1H, J = 5.6 Hz, OCH), 4.46–4.61 (dd, 2H, J = 12.4 Hz, 37.2 Hz, OCH2), 5.10 (d, 1H, J = 2.4 Hz, =CH), 5.49 (s, 1H, OCH), 5.66 (d, 1H, J = 3.2 Hz, =CH), 6.19 (s, 1H, =CH), 6.87 (m, 4H, Ar-H) ppm. 13H NMR (100 MHz, CDCl3): δ 17.97, 23.78, 24.63, 25.87, 29.67, 36.62, 41.19, 42.60, 55.29, 59.92, 63.27, 67.28, 81.07, 110.28, 120.23, 120.60, 126.31, 128.79, 129.00, 129.49, 130.19, 135.59, 138.73, 156.04, 157.43 ppm. HRMS calcd. for C24H30NO6 (M+H)+: 428.2114. Found 428.2112.
4.1.4. ((1aR,7aS,10aS,10bS,E)-1a-methyl-8-methylene-9-oxo-1a,2,3,6,7,7a,8,9,10a,10b-decahydrooxireno[2’,3’:9,10]cyclo deca[1,2-b]furan-5-yl)methyl(3-methoxybenzyl)carbamate (7d).
Yield: 89%, 1H NMR (400 MHz, CDCl3): δ 1.08 (t, 1H, J = 13.6 Hz, CH), 1.53 (s, 3H, CH3), 2.17–2.42 (m, 6H, 3xCH2), 2.83–2.86 (d, 1H, J = 9.6 Hz, CH), 2.90 (m, 1H, CH), 3.80 (s, 3H, OCH3), 3.81 (m, 1H, CH), 4.33 (d, 2H, J = 6.0 Hz, CH2), 4.49–4.66 (dd, 2H, J = 12.0 Hz, 44.4 Hz, OCH2), 5.10 (m, 1H, J = 2.4 Hz, =CH), 5.52 (d, 1H, J = 2.8 Hz, OCH), 5.66 (d, 1H, J = 3.2 Hz, =CH), 6.21 (d, 1H, J = 3.6 Hz, =CH), 6.81 (m, 3H, Ar-H),7.23–7.25 (m 1H, Ar-H) ppm. 13H NMR (100 MHz, CDCl3):δ 17.97, 23.76, 24.50, 25.79, 36.60, 42.60, 45.07, 55.22, 59.91, 63.25, 67.28, 81.04, 112.79, 113.26, 119.65, 120.26, 129.77, 130.12, 135.43, 138.74, 139.83, 156.11, 159.87, 169.39 ppm. HRMS calcd. for C24H30NO6 (M+H)+: 428.2114. Found 428.2150.
4.1.5. ((1aR,7aS,10aS,10bS,E)-1a-methyl-8-methylene-9-oxo-1a,2,3,6,7,7a,8,9,10a,10b-decahydrooxireno[2’,3’:9,10]cyclo deca[1,2-b]furan-5-yl)methyl(3,4-dimethoxybenzyl)carbamate (7e).
Yield: 90%, 1H NMR (400 MHz, CDCl3): δ 1.06 (t, 1H, J = 13.6 Hz, CH), 1.51 (s, 3H, CH3), 1.62 (m, 2H, CH2), 2.11–2.39 (m, 6H, 3xCH2), 2.81 (d, 1H, J = 8.8 Hz, CH), 3.01 (m, 1H, CH), 3.81 (s, 3H, OCH3), 3.84 (s, 3H, OCH3), 4.26 (d, 1H, J = 4.8 Hz, OCH), 4.48–4.63 (dd, 2H, J = 12.4 Hz, 37.2 Hz, OCH2), 5.00 (d, 1H, J = 2.4 Hz, =CH), 5.49 (s, 1H, OCH), 5.65 (d, 1H, J = 3.2 Hz, =CH), 6.19 (s, 1H, C2H), 6.79 (m, 3H, =CH and C5H, C6H) ppm. 13H NMR (100 MHz, CDCl3):δ 17.96, 23.77, 24.54, 25.79, 36.61, 42.60, 45.03, 55.89, 55.92, 59.91, 63.24, 67.25, 81.04, 110.98, 111.19, 119.88, 120.19, 130.15, 130.79, 135.43, 138.79, 148.53, 149.11, 156.05, 169.39 ppm. HRMS calcd. For C25H32NO7 (M+H)+: 458.2177. Found 458.2175.
4.1.6. ((1aR,7aS,10aS,10bS,E)-1a-methyl-8-methylene-9-oxo-1a,2,3,6,7,7a,8,9,10a,10b-decahydrooxireno[2’,3’:9,10]cyclo deca[1,2-b]furan-5-yl)methyl(3,4,5-trimethoxybenzyl) carbamate (7f).
Yield: 91%, 1H NMR (400 MHz, CDCl3): δ 1.06 (t, 1H, J = 12.4 Hz, CH), 1.54 (s, 3H, CH3), 1.63 (m, 2H, CH2), 2.13–2.42 (m, 6H, 3xCH2), 2.84 (d, 1H, J = 9.2 Hz, CH), 2.93 (m, 1H, OCH), 3.82 (s, 3H, OCH3), 3.85 (s, 6H, 2xOCH3), 4.28 (d, 2H, J = 5.6 Hz, CH2), 4.51–4.66 (dd, 2H, J = 12.8 Hz, 49.2 Hz, OCH2), 5.10 (bs, 1H, NH), 5.52 (d, 1H, J = 2.0 Hz, =CH), 5.69 (m, 1H, OCH), 6.21 (d, 1H, J = 2.4 Hz, =CH), 6.50 (s, 2H, Ar-H) ppm. 13H NMR (100 MHz, CDCl3): δ17.96, 23.77, 24.53, 25.76, 36.60, 42.60, 45.49, 56.13, 59.92, 60.83, 67.29, 81.05, 104.63, 120.16, 130.18, 133.18, 135.96, 135.39, 137.35, 138.80, 153.40, 156.09, 169.40 ppm. HRMS calcd. for C26H34NO8 (M+H)+: 488.2321. Found 488.2319.
4.1.7. ((1aR,7aS,10aS,10bS,E)-1a-methyl-8-methylene-9-oxo-1a,2,3,6,7,7a,8,9,10a,10b-decahydrooxireno[2’,3’:9,10]cyclo deca[1,2-b]furan-5-yl)methyl(2-methoxyphenethyl)carbamate (7g).
Yield: 88%, 1H NMR (400 MHz, CDCl3): δ 1.09 (t, 1H, J = 11.6 Hz, CH), 1.54 (s, 3H, CH3), 1.62–1.66 (m, 2H, CH2), 2.14–2.43 (m, 6H, 3xCH2), 2.80–2.90 (m, 2H, CH, NH), 3.39–3.44 (q, 2H, CH2), 3.81 (s, 1H, OCH), 3.82 (s, 3H, OCH3), 3.83 (m, 1H, CH), 4.43–4.62 (dd, 2H, J = 12.4 Hz, 37.2 Hz, OCH2), 4.82 (t, 1H, J = 2.4 Hz, =CH), 5.51 (d, 1H, J = 3.2 Hz, =CH), 5.62 (m, 1H, OCH), 6.21 (d, 1H, J = 3.6 Hz, =CH), 6.85 (dd, 2H, Ar-H), 7.10 (d, 1H, J = 9.6 Hz, Ar-H), 7.20–7.24 (m, 1H, Ar-H) ppm. 13H NMR (100 MHz, CDCl3):δ 17.99, 23.77, 24.50, 25.82, 30.64, 36.65, 41.06, 42.59, 55.26, 59.92, 63.28, 66.98, 81.07, 110.39, 120.27, 120.58, 126.97, 127.93, 129.85, 130.54, 135.62, 138.74, 156.06, 157.53, 169.42 ppm. HRMS calcd. for C25H32NO6 (M+H)+: 442.2257. Found 442.2253.
4.1.8. ((1aR,7aS,10aS,10bS,E)-1a-methyl-8-methylene-9-oxo-1a,2,3,6,7,7a,8,9,10a,10b-decahydrooxireno[2’,3’:9,10]cyclo deca[1,2-b]furan-5-yl)methyl(3-methoxyphenethyl)carbamate (7h).
Yield: 89%, 1H NMR (400 MHz, CDCl3): δ 1.08 (t, 1H, J = 13.2 Hz, CH), 1.52(s, 3H, CH3), 1.56–1.61 (m, 2H, CH2), 2.11–2.42 (m, 6H, 3xCH2), 2.75–2.87 (m, 1H, CH), 3.41–3.46 (m, 2H, CH2), 3.76 (s, 3H, OCH3), 3.81 (t, 1H, J = 9.6 Hz, OCH), 4.42–4.62 (dd, 2H, J = 12.4 Hz, 37.2 Hz, OCH2), 4.65 (d, 1H, J = 2.4 Hz, =CH), 5.51 (d, 1H, J = 2.8 Hz, OCH), 5.61(d, 1H, J = 8.0 Hz, =CH), 6.20 (d, 1H, J = 3.2 Hz, =CH), 6.7 (s, 1H, Ar-H), 6.74 (m, 2H, Ar-H), 7.19 (m, 1H, Ar-H) ppm. 13H NMR (100 MHz, CDCl3):δ 17.97, 23.77, 24.46, 25.79, 35.99, 36.62, 42.00, 42.58, 55.16, 59.92, 63.27, 67.10, 81.06, 111.72, 114.59, 120.23, 121.03, 129.64, 130.04, 135.50, 138.76, 140.13, 155.99, 159.80, 169.40 ppm. HRMS calcd. for C25H32NO6 (M+H)+: 442.2259. Found 442.2257.
4.1.9. ((1aR,7aS,10aS,10bS,E)-1a-methyl-8-methylene-9-oxo-1a,2,3,6,7,7a,8,9,10a,10b-decahydrooxireno[2’,3’:9,10]cyclo deca[1,2-b]furan-5-yl)methyl(4-methoxyphenethyl)carbamate (7i).
Yield: 90%, 1H NMR (400 MHz, CDCl3): δ 1.06 (t, 1H, J = 13.2 Hz, CH), 1.54 (s, 3H, CH3), 1.61 (m, 2H, CH2), 2.13–2.43 (m, 6H, 3xCH2), 2.73–2.90 (m, 3H, CH, NH), 3.40 (d, 2H, J = 6.0 Hz, CH2), 3.79 (s, 3H, OCH3), 3.81 (m, 1H,OCH), 4.43–4.63 (dd, 2H, J = 12.4 Hz, 37.2 Hz, OCH2), 4.69 (m, 1H, =CH), 5.52 (d, 1H, J = 2.8 Hz, =CH), 5.63 (m, 1H, OCH), 6.22 (d, 1H, J = 3.2 Hz, =CH), 6.83 (d, 2H, J = 8.4 Hz, Ar-H), 7.08 (d, J = 8.0 Hz, 2H, Ar-H) ppm. 13H NMR (100 MHz, CDCl3):δ 17.97, 23.78, 24.49, 25.79, 35.08, 36.62, 42.59, 55.26, 59.92, 63.27, 67.09, 81.06, 114.05, 120.23, 129.67, 130.08, 130.46, 135.52, 138.77, 156.00, 158.29, 169.40 ppm. HRMS calcd. for C25H32NO6 (M+H)+: 442.2245. Found 442.2243.
4.1.10. ((1aR,7aS,10aS,10bS,E)-1a-methyl-8-methylene-9-oxo-1a,2,3,6,7,7a,8,9,10a,10b-decahydrooxireno[2’,3’:9,10]cyclo deca[1,2-b]furan-5-yl)methyl(3,4-dimethoxyphenethyl) carbamate (7j).
Yield: 92%, 1H NMR (400 MHz, CDCl3): δ 1.06 (t, 1H, J = 12.8 Hz, CH), 1.54 (s, 3H, CH3), 1.62 (m, 2H, CH2), 2.13–2.44 (m, 7H, 3xCH2, CH), 2.74 (t, 2H, J = 7.2 Hz, CH2), 2.84 (d, 1H, J = 10.0 Hz, CH), 2.91 (m, 1H, CH), 3.40 (q, 2H, CH2), 3.84 (s, 1H, NH), 3.86 (s, 3H,OCH3), 3.87 (s, 3H, OCH3), 4.45–4.63 (dd, 2H, J = 12.4 Hz, 37.2 Hz, OCH2), 4.71 (m, 1H, =CH), 5.52 (d, 1H, J = 3.2 Hz, =CH), 5.64 (m, 1H, OCH), 6.22 (d, 1H, J = 3.6 Hz, =CH), 6.71 (m, 2H, Ar-H), 6.8 (d, J = 8.0 Hz, 1H, Ar-H) ppm. 13H NMR (100 MHz, CDCl3):δ 17.96, 23.77, 24.50, 25.78, 35.60, 36.62, 42.23, 42.59, 55.86, 55.90, 59.92, 63.25, 67.11, 81.06, 111.32, 111.84, 120.18, 120.65, 130.10, 130.96, 135.49, 138.80, 147.71, 149.08, 156.01, 169.40 ppm. HRMS calcd. For C26H34NO7 (M+H)+: 472.2370. Found 472.2368.
4.1.11. ((1aR,7aS,10aS,10bS,E)-1a-methyl-8-methylene-9-oxo-1a,2,3,6,7,7a,8,9,10a,10b-decahydrooxireno[2’,3’:9,10]cyclo deca[1,2-b]furan-5-yl)methyl(4-bromophenethyl)carbamate (7k).
Yield: 89%, 1H NMR (400 MHz, CDCl3): δ 1.10 (t, 1H, J = 13.6 Hz, CH), 1.54 (s, 3H, CH3), 1.59–1.64 (m, 2H, CH2), 2.13–2.44 (m, 6H, 3xCH2), 2.75–2.83 (m, 2H, CH2), 3.40–3.45 (m, 2H, CH2), 3.81 (t, 1H, J = 9.6 Hz, OCH), 4.43–4.63 (dd, 2H, J = 12.4 Hz, 37.2 Hz, OCH2), 4.67 (d, 1H, J = 2.4 Hz, =CH), 5.52 (d, 1H, J = 2.8 Hz, OCH), 5.65 (d, 1H, J = 3.2 Hz, =CH), 6.23 (d, 1H, J = 3.2 Hz, =CH), 7.05 (d, 2H, J = 7.6 Hz, Ar-H), 7.42 (d, 2H, J = 8.0 Hz, Ar-H) ppm. 13H NMR (100 MHz, CDCl3): δ 15.40, 21.2, 21.88, 23.17, 32.92, 34.05, 39.35, 40.01, 57.35, 60.68, 64.54, 78.49, 117.63, 117.49, 127.58, 127.93, 129.11, 132.86, 134.95, 136.20, 153.40, 166.83 ppm. HRMS calcd. for C24H29NO5 (M+H)+:490.1165. Found 490.1163.
4.2. Methodology for 60 human cancer cell screening assay
The anti-proliferative activity of the benzyl and phenethyl MMB carbamates (7a-7k) was evaluated at the National Cancer Institute (NCI) under the developmental therapeutics program (DTP) utilizing a literature procedure.[26] All human cancer cells were grown in RPMI 1640 medium containing 5% fetal bovine serum and 2 mM L-glutamine. The tumor cells were inoculated into 96-well microtiter plates in 100 μL at plating densities ranging from 5,000 to 40,000 cells per well, depending on the doubling time of individual cell lines. After completion of cell inoculation, the microtiter plates were incubated at 37 °C, 5 % CO2, 95 % air and 100 % relative humidity for 24 hours prior to the addition test compounds. After 24 h of incubation, two microtiter plates of each tumor cell line were fixed in situ with TCA. The optical density reading at this point represents a measurement of the cell population for each cell line at the time of drug addition (Tz). The optical density reading (ODtzero) was measured and represents the cell population for each tumor cell line at the time of compound addition. The benzyl and phenethyl MMB carbamates (7a-7k) were solubilized in DMSO at 400-fold desired final maximum test concentration, and stored at −80 °C. An aliquot part of the frozen concentrate was thawed and diluted to 10−4 M concentration with medium containing 50 μg/ml gentamicin along with another control sample of just DMSO. Different benzyl and phenethyl MMB carbamates aliquots of 100 microliters were added to the appropriate microtiter wells containing 100 μL of medium, to achieve 10−5 M and 0 M (control) final drug concentrations. After the addition of benzyl and phenethyl MMB carbamates the microtiter plates were then incubated for 48 hrs at 37 °C and 100% humidity.
Assays were terminated for adherent cells by the addition of cold TCA. Cells were fixed in situ by the addition of 50 μL of cold 50 % (w/v) TCA followed by incubation for 60 minutes at 4 °C. After discarding the supernatant, the microtiter plates were washed five times with water and air dried. Then, sulforhodamine B (SRB) solution (100 microliters) at 0.4 % (w/v) in 1 % acetic acid was added to each well, and plates were incubated for another 10 minutes at room temperature. The unbound dye was removed by washing five times with 1 % acetic acid and the plates were air-dried after SRB staining. Bound SRB stain was subsequently solubilized with 10 mM trizma base, and the absorbance read on an automated plate reader at a wavelength of 515 nm. By using the seven absorbance measurements [time zero, (Tz), control growth, (C), and test growth in the presence of drug at the five concentration levels (Ti)], the percentage growth was calculated at each of the drug concentrations levels. The benzyl and phenethyl MMB carbamate’s growth inhibitory or cytotoxicity effects were calculated by determining percentage cell growth (PG) inhibition. Optical density (OD), before exposing the cells to the test compound (ODtzero), was measured for SRB-derived color, and after 48hrs, exposure to the test compound (ODtest) or the control vehicle (ODctrl) was also measured. The NCI-60 cell panel methodology can be found on the NCI database at http://dtp.nci.nih.gov/branches/btb/ivclsp.html.
4.3. Inhibition of p65 phosphorylation by 7b in M9 ENL1 AML cells
Primary AML cells were exposed to variable concentrations of PTL or 7b (2.5, 5.0, and 10 μM) for 6h. Cell lysates were obtained and diluted in 5× SDS–PAGE sample buffer (10% w/v SDS, 10 mM DDT, 20% glycerol, 0.2 M Tris–HCl, pH 6.8, 0.05% w/v bromophenol blue), and run on 8–10% SDS–PAGE gels. Protein gels were then transferred to PVDF membrane and blocked with 5% milk in 0.1% TBST (20 mM Tris–HCl pH 7.5, 137 mM NaCl, 0.1% Tween 20), followed by incubation with antibodies against p-p65(ser-536) (Cell Signaling). All analyses were conducted in triplicate.
4.4. Inhibition of breast cancer cell proliferation and NFκB DNA binding by compound 7b in OV-MD-231 and SUM149PT breast cancer cells
4.4.1. Cell proliferation assays
500–1000 TMD-231, OV-MD-231 or SUM149 cells were plated in 96-well plates and treatment with 7b was initiated a day after plating. Each treatment condition contained six wells. After 4–5 days of treatment, bromodeoxyuridine incorporation-ELISA was performed as per manufacturer’s instructions (Millipore; cat # QIA58–1000TEST). Results presented are from technical replicates with six technical replicates of two experiments. Similar results were obtained with 2–4 biological replicates.
4.4.2. Electrophoretic mobility shift assay (EMSA)
EMSAs on whole cell lysates from OV-MD-231 or SUM149PT cells using 32P-radiolabelled probes with NF-κB and SP-1 (as a control) binding sites was performed as described previously[38]. Oligonucleotides with NFκB and SP-1 binding sites were purchased from Promega (NFκB, Cat# E329A; Oct-1, Cat# E324B).
4.5. Molecular modeling
The amino acid sequence for IKKβ was obtained from the Uniprot database (http://www.uniprot.org/). For full length three-dimensional structure prediction, the amino acid sequence of IKKβ was submitted to the I-TASSER server (http://zhanglab.ccmb.med.umich.edu/I-TASSER/). I-TASSER predicts protein structure utilizing the fold recognition and ab initio approach. The structure from I-TASSER was then energy-minimized in a cubic box containing water (SPC) and solvent (NaCl) for 5,000 steps using GROMACS simulation package. Energy minimized structures were then used for docking studies.
4.6. Protein-ligand docking.
Protein ligand docking was performed in two steps; 1) First molecules were screened against the entire IKKβ molecule using Autodock-VINA running on a 32 core Linux server with Raccoon2 interface (Windows 7). Ligands were converted to autodock format (.pdbqt) using OpenBabel before the docking search. Grid box definition was performed in Raccoon interface and set to cover the entire molecule. 2) Second stage docking calculations were set up with Schrodinger 2017–4. Protein preparation was made prior to the docking using default protocol from protein preparation wizard with OPLS3 forcefield. Similarly, ligands (7b and 7k) were prepared and minimized at gas-phase with OPLS3 forcefield using using LigPrep module. Docking calculations were carried out using Glide software in standard precision (SP) mode with default parameters for grid and pose generation. Protein-ligand viewed and interacting residues were identified using Discovery Studio (Biovia).
4.7. Binding affinity calculations with MM/GBSA
Docking poses from Glide results were rescored with the MM/GBSA approach from Prime module in Schrodinger suite.[39] MM/GBSA calculations were performed with variable dielectric solvent model VSGB 2.0.
Supplementary Material
Highlights.
Synthesis of a novel series of sesquiterpene lactone analogs
Analogs evaluated for anti-cancer activity against 60 human cancer cell lines
Lead compound inhibited cell proliferation and reduced NFκB-DNA binding in breast cancer cells
Active compounds interact with the ubiquitin-like domain (ULD) of the IKKβ subunit
These analogs are predicted to inhibit down-stream targets of the NFκB pathway
Acknowledgements
We are grateful to the NIH (grants CA158275 and AG012411), an Arkansas Research Alliance Scholar Award to PAC, and an Inglewood Scholars Program Award to MB. We would also like to acknowledge the NCI drug-screening program for support of this study.
Footnotes
Appendix A. Supplementary data
1H and 13C NMR spectra, and HRMS data can be found in the Supporting Information
Conflict of interest statement
The authors declare the following competing financial interest(s): The University of Arkansas for Medical Sciences (UAMS) holds patents on the molecules described in this study. A potential royalty stream to NRP, JP, CTJ, and PAC may occur consistent with UAMS policy.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Zhang Q, Lu Y, Ding Y, Zhai J, Ji Q, Ma W, Yang M, Fan H, Long J, Tong Z, Shi Y, Jia Y, Han B, Zhang W, Qiu C, Ma X, Li Q, Shi Q, Zhang H, Li D, Zhang J, Lin J, Li LY, Gao Y, Chen Y, Guaianolide sesquiterpene lactones, a source to discover agents that selectively inhibit acute myelogenous leukemia stem and progenitor cells, J. Med. Chem, 55 (2012) 8757–8769. [DOI] [PubMed] [Google Scholar]
- [2].Pettit GR, Cragg GM, Antineoplastic agents. 32. The pseudoguaianolide helenalin, Experientia, 29 (1973) 781. [DOI] [PubMed] [Google Scholar]
- [3].Kupchan SM, Fujita T, Maruyama M, Britton RW, The isolation and structural elucidation of eupaserrin and deacetyleupaserrin, new antileukemic sesquiterpene lactones from Eupatorium semiserratum, J. Org. Chem, 38 (1973) 1260–1264. [DOI] [PubMed] [Google Scholar]
- [4].Ma G, Khan SI, Benavides G, Schuhly W, Fischer NH, Khan IA, Pasco DS, Inhibition of NF-kappaB-mediated transcription and induction of apoptosis by melampolides and repandolides, Cancer Chemoth. Pharmacol, 60 (2007) 35–43. [DOI] [PubMed] [Google Scholar]
- [5].Shen YC, Jang JY, Khalil AT, Chiang LC, New gemacranolides from Eupatorium hualienense, Chem. Biodivers, 2 (2005) 244–252. [DOI] [PubMed] [Google Scholar]
- [6].Kretschmer N, Blunder M, Kunert O, Rechberger GN, Bauer R, Schuehly W, Cytotoxic furanogermacranolides from the flowers of Helianthus angustifolius, Planta Med., 77 (2011) 1912–1915. [DOI] [PubMed] [Google Scholar]
- [7].Guzman ML, Rossi RM, Neelakantan S, Li X, Corbett CA, Hassane DC, Becker MW, Bennett JM, Sullivan E, Lachowicz JL, Vaughan A, Sweeney CJ, Matthews W, Carroll M, Liesveld JL, Crooks PA, Jordan CT, An orally bioavailable parthenolide analog selectively eradicates acute myelogenous leukemia stem and progenitor cells, Blood, 110 (2007) 4427–4435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Holcomb BK, Yip-Schneider MT, Waters JA, Beane JD, Crooks PA, Schmidt CM, Dimethylamino parthenolide enhances the inhibitory effects of gemcitabine in human pancreatic cancer cells, J. Gastrointest. Surg, 16 (2012) 1333–1340. [DOI] [PubMed] [Google Scholar]
- [9].Janganati V, Penthala NR, Cragle CE, MacNicol AM, Crooks PA, Heterocyclic aminoparthenolide derivatives modulate G(2)-M cell cycle progression during Xenopus oocyte maturation, Bioorg. Med. Chem. Lett, 24 (2014) 1963–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Janganati V, Penthala NR, Madadi NR, Chen Z, Crooks PA, Anti-cancer activity of carbamate derivatives of melampomagnolide B, Bioorg Med. Chem. Lett, 24 (2014) 3499–3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Janganati V, Ponder J, Jordan CT, Borrelli MJ, Penthala NR, Crooks PA, Dimers of Melampomagnolide B Exhibit Potent Anticancer Activity against Hematological and Solid Tumor Cells, J. Med. Chem, 58 (2015) 8896–8906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Karmakar A, Xu Y, Mustafa T, Kannarpady G, Bratton SM, Radominska-Pandya A, Crooks PA, Biris AS, Nanodelivery of Parthenolide Using Functionalized Nanographene Enhances its Anticancer Activity, RSC Adv., 5 (2015) 2411–2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Nakshatri H, Appaiah HN, Anjanappa M, Gilley D, Tanaka H, Badve S, Crooks PA, Mathews W, Sweeney C, Bhat-Nakshatri P, NF-kappaB-dependent and -independent epigenetic modulation using the novel anti-cancer agent DMAPT, Cell Death. Dis, 6 (2015) e1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Nasim S, Crooks PA, Antileukemic activity of aminoparthenolide analogs, Bioorg. Med. Chem. Lett, 18 (2008) 3870–3873. [DOI] [PubMed] [Google Scholar]
- [15].Nasim S, Pei S, Hagen FK, Jordan CT, Crooks PA, Melampomagnolide B: a new antileukemic sesquiterpene, Bioorg. Med. Chem, 19 (2011) 1515–1519. [DOI] [PubMed] [Google Scholar]
- [16].Neelakantan S, Nasim S, Guzman ML, Jordan CT, Crooks PA, Aminoparthenolides as novel anti-leukemic agents: Discovery of the NF-kappaB inhibitor, DMAPT (LC-1), Bioorg. Med. Chem. Lett, 19 (2009) 4346–4349. [DOI] [PubMed] [Google Scholar]
- [17].Penthala NR, Bommagani S, Janganati V, MacNicol KB, Cragle CE, Madadi NR, Hardy LL, MacNicol AM, Crooks PA, Heck products of parthenolide and melampomagnolide-B as anticancer modulators that modify cell cycle progression, Eur. J. Med. Chem, 85 (2014) 517–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Sun Y, St Clair DK, Xu Y, Crooks PA, St Clair WH, A NADPH oxidase-dependent redox signaling pathway mediates the selective radiosensitization effect of parthenolide in prostate cancer cells, Cancer Res., 70 (2010) 2880–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Yip-Schneider MT, Wu H, Hruban RH, Lowy AM, Crooks PA, Schmidt CM, Efficacy of dimethylaminoparthenolide and sulindac in combination with gemcitabine in a genetically engineered mouse model of pancreatic cancer, Pancreas, 42 (2013) 160–167. [DOI] [PubMed] [Google Scholar]
- [20].Yip-Schneider MT, Wu H, Stantz K, Agaram N, Crooks PA, Schmidt CM, Dimethylaminoparthenolide and gemcitabine: a survival study using a genetically engineered mouse model of pancreatic cancer, BMC Cancer, 13 (2013) 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zong H, Sen S, Zhang G, Mu C, Albayati ZF, Gorenstein DG, Liu X, Ferrari M, Crooks PA, Roboz GJ, Shen H, Guzman ML, In vivo targeting of leukemia stem cells by directing parthenolide-loaded nanoparticles to the bone marrow niche, Leukemia, 30, (2015) 1582–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ghosh AK, Brindisi M, Organic carbamates in drug design and medicinal chemistry, J. Med. Chem, 58 (2015) 2895–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Pei S, Minhajuddin M, Callahan KP, Balys M, Ashton JM, Neering SJ, Lagadinou ED, Corbett C, Ye H, Liesveld JL, O’Dwyer KM, Li Z, Shi L, Greninger P, Settleman J, Benes C, Hagen FK, Munger J, Crooks PA, Becker MW, Jordan CT, Targeting aberrant glutathione metabolism to eradicate human acute myelogenous leukemia cells, J. Biol. Chem, 288 (2013) 33542–33558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Albayati ZA, Janganati V, Chen Z, Ponder J, Breen PJ, Jordan CT, Crooks PA, Identification of a melampomagnolide B analog as a potential lead molecule for treatment of acute myelogenous leukemia, Bioorg Med Chem, 25 (2017) 1235–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Tsunokawa Y, Iwasaki S, Okuda S, A new oxygenating method using 1-alkoxycarbonyl-1,2,4-triazoles and hydrogen peroxide relative reactivity of O-alkylperoxycarbonic acids, Chem. Pharm. Bull, 31 (1983) 4578–4581. [Google Scholar]
- [26].Rubinstein LV, Shoemaker RH, Paull KD, Simon RM, Tosini S, Skehan P, Scudiero DA, Monks A, Boyd MR, Comparison of in vitro anticancer-drug-screening data generated with a tetrazolium assay versus a protein assay against a diverse panel of human tumor cell lines, J. Natl. Cancer Inst, 82 (1990) 1113–1118. [DOI] [PubMed] [Google Scholar]
- [27].Polley S, Huang DB, Hauenstein AV, Fusco AJ, Zhong X, Vu D, Schrofelbauer B, Kim Y, Hoffmann A, Verma IM, Ghosh G, Huxford T, A structural basis for IkappaB kinase 2 activation via oligomerization-dependent trans auto-phosphorylation, PLoS Biol, 11 (2013) e1001581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].May MJ, Larsen SE, Shim JH, Madge LA, Ghosh S, A novel ubiquitin-like domain in IkappaB kinase beta is required for functional activity of the kinase, J. Biol. Chem, 279 (2004) 45528–45539. [DOI] [PubMed] [Google Scholar]
- [29].Hehner SP, Heinrich M, Bork PM, Vogt M, Ratter F, Lehmann V, Schulze-Osthoff K, Droge W, Schmitz ML, Sesquiterpene lactones specifically inhibit activation of NF-kappa B by preventing the degradation of I kappa B-alpha and I kappa B-beta, J. Biol. Chem, 273 (1998) 1288–1297. [DOI] [PubMed] [Google Scholar]
- [30].Adli M, Merkhofer E, Cogswell P, Baldwin AS, IKKalpha and IKKbeta each function to regulate NF-kappaB activation in the TNF-induced/canonical pathway, PLoS One, 5 (2010) e9428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Tang ED, Inohara N, Wang CY, Nunez G, Guan KL, Roles for homotypic interactions and transautophosphorylation in IkappaB kinase beta IKKbeta) activation [corrected], J. Biol. Chem, 278 (2003) 38566–38570. [DOI] [PubMed] [Google Scholar]
- [32].Israel A, The IKK complex, a central regulator of NF-kappaB activation, Cold Spring Harb. Perspect Biol, 2 (2010) a000158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Janganati V, Ponder J, Balasubramaniam M, Bhat-Nakshatri P, Bar EE, Nakshatri H, Jordan CT, Crooks PA, MMB triazole analogs are potent NF-κB inhibitors and anti-cancer agents against both hematological and solid tumor cells, Eur. J. Med. Chem, 157 (2018) 562–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Xu G, Lo YC, Li Q, Napolitano G, Wu X, Jiang X, Dreano M, Karin M, Wu H, Crystal structure of inhibitor of kappaB kinase beta, Nature, 472 (2011) 325–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zhang Y, Otero JE, Abu-Amer Y, Ubiquitin-like domain of IKKbeta regulates osteoclastogenesis and osteolysis, Calcif. Tissue Int, 93 (2013) 78–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Carter RS, Pennington KN, Ungurait BJ, Arrate P, Ballard DW, Signal-induced ubiquitination of I kappaB Kinase-beta, J. Biol. Chem, 278 (2003) 48903–48906. [DOI] [PubMed] [Google Scholar]
- [37].Carter RS, Pennington KN, Arrate P, Oltz EM, Ballard DW, Site-specific monoubiquitination of IkappaB kinase IKKbeta regulates its phosphorylation and persistent activation, J. Biol. Chem, 280 (2005) 43272–43279. [DOI] [PubMed] [Google Scholar]
- [38].Bhat-Nakshatri P, Newton TR, Goulet R, Nakshatri H, NF-κB activation and interleukin 6 production in fibroblasts by estrogen receptor-negative breast cancer cell-derived interleukin 1α, Proceedings of the National Academy of Sciences, 95 (1998) 6971–6976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Li J, Abel R, Zhu K, Cao Y, Zhao S, Friesner RA, The VSGB 2.0 model: a next generation energy model for high resolution protein structure modeling, Proteins, 79 (2011) 2794–2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.