

Key Points
Deficiency of EHHADH, a peroxisomal β-oxidation enzyme, causes male-specific kidney hypertrophy and proximal tubular injury in mice.
Our work suggests genetic defects in peroxisomal metabolism may be a cause of CKD.
Our work also indicates that sexual dimorphism in tubular metabolic homeostasis affects susceptibility to kidney disease.
Keywords: chronic kidney disease, androgens, basic science, hypertrophy, kidney, mice, multifunctional protein 1, peroxisomal bifunctional protein, peroxisomes, proximal tubule, sex differences
Visual Abstract
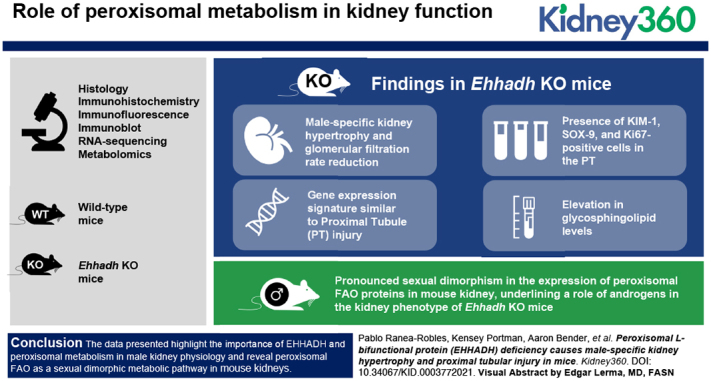
Abstract
Background
Proximal tubular (PT) cells are enriched in mitochondria and peroxisomes. Whereas mitochondrial fatty acid oxidation (FAO) plays an important role in kidney function by supporting the high-energy requirements of PT cells, the role of peroxisomal metabolism remains largely unknown. L-bifunctional protein (EHHADH) catalyzes the second and third step of peroxisomal FAO.
Methods
We studied kidneys of WT and Ehhadh KO mice on a C57BL/6N background using histology, immunohistochemistry, immunofluorescence, immunoblot, RNA-sequencing, and metabolomics. To assess the role of androgens in the kidney phenotype of Ehhadh KO mice, mice underwent orchiectomy.
Results
We observed male-specific kidney hypertrophy and glomerular filtration rate reduction in adult Ehhadh KO mice. Transcriptome analysis unveiled a gene expression signature similar to PT injury in AKI mouse models. This was further illustrated by the presence of kidney injury molecule-1 (KIM-1), SOX-9, and Ki67-positive cells in the PT of male Ehhadh KO kidneys. Male Ehhadh KO kidneys had metabolite changes consistent with peroxisomal dysfunction and an elevation in glycosphingolipid levels. Orchiectomy of Ehhadh KO mice decreased the number of KIM-1–positive cells to WT levels. We revealed a pronounced sexual dimorphism in the expression of peroxisomal FAO proteins in mouse kidney, underlining a role of androgens in the kidney phenotype of Ehhadh KO mice.
Conclusions
Our data highlight the importance of EHHADH and peroxisomal metabolism in male kidney physiology, and reveal peroxisomal FAO as a sexual dimorphic metabolic pathway in mouse kidneys.
Introduction
The kidney uses the mitochondrial fatty acid β-oxidation (FAO) pathway as the predominant energy source (1) to fulfill the high energy requirements of proximal tubule (PT) cells. Dysfunction of mitochondrial FAO has been linked to the development of kidney fibrosis in patients with CKD and mouse models (2,3). PT cells are not only enriched in mitochondria, but also in peroxisomes (4). Peroxisomes have unique metabolic functions that include the β-oxidation of specific carboxylic acids such as very long-chain fatty acids, and the biosynthesis of plasmalogens (5). The importance of peroxisomes in kidney function is highlighted by the presence of renal cysts and/or calcium oxalate stones in patients with Zellweger spectrum disorder and other peroxisomal diseases (6,7). Moreover, peroxisome abundance and function are reduced in several rodent models of kidney injury (8,9). These data indicate that peroxisomes are important for kidney function. The exact roles of peroxisomes in the kidney, however, remain unknown.
Each cycle of peroxisomal FAO consists of four enzymatic steps. The second and third steps are catalyzed by the peroxisomal L- and D-bifunctional proteins (encoded by EHHADH and HSD17B4, respectively). EHHADH is mainly expressed in liver and kidney. We and others have characterized a specific role of EHHADH in the hepatic metabolism of long-chain dicarboxylic acids (10–12), but the role of EHHADH in the metabolic homeostasis of the kidney is unknown.
In this study, we examined the role of EHHADH in the kidney by using a knockout (KO) mouse model (Ehhadh KO mice). Our results demonstrate that the Ehhadh KO mouse is a new model for metabolic kidney injury with enhanced susceptibility in male mice, and underline the role of peroxisomes in kidney physiology.
Materials and Methods
Animal Experiments
All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of the Icahn School of Medicine at Mount Sinai (IACUC-2014–0100) and comply with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (National Institutes of Health Publication 8023, 8th edition, 2011). The generation of Ehhadh KO (Ehhadh−/−) mice has been previously described (13). Wild-type (WT) and Ehhadh KO mice on a pure C57BL/6N background, generated through heterozygous breeding, were fed a regular chow (PicoLab Rodent Diet 20, LabDiet). Mice were housed in rooms with a 12 h light/dark cycle. Mice were euthanized by exposure to CO2 and blood was collected from the inferior vena cava for the preparation of EDTA plasma. Organs were snap frozen in liquid nitrogen and stored at −80°C.
We used 104 mice to generate the data shown in this study (33 male WT mice, 12 female WT mice, 36 male Ehhadh KO mice, and 23 female Ehhadh KO mice). One cohort of 76 mice (22 male WT mice, nine female WT mice, 27 male Ehhadh KO mice, and 20 female Ehhadh KO mice) included the mice used for the generation of the RNA-sequencing (RNA-seq) dataset (four WT mice, average age 7.8 months, four Ehhadh KO mice, average age 8.0 months, all male), kidney mass measurements, histology and immunofluorescence (IF) studies, plasma creatinine and BUN measurements, and immunoblot studies. A second cohort of mice was used to generate the GFR data and consisted of six male WT mice (average age 3.3 months), three female WT mice (average age 5.8 months), four male Ehhadh KO mice (average age 3.7 months), and three female Ehhadh KO mice (average age 6.0 months). A third cohort of mice was used for the orchiectomy experiment and consisted of five WT (average age before orchiectomy 3.2 months) and five Ehhadh KO mice (average age before orchiectomy 3.4 months). The metabolomics dataset was obtained from a fourth cohort that was generated for a previous study (10), consisting of seven WT (males, average age 8.6 months) and five Ehhadh KO (males, average age 8.6 months) mice.
Orchiectomy
To remove the production of gonadal androgens, surgical castration surgery was carried out in 3–4-month-old WT and Ehhadh KO mice under full anesthesia. Sham surgery was performed for control mice. Mice were euthanized 6 weeks after orchiectomy/sham surgery, and kidneys were weighed and collected for further studies.
BUN and Plasma Creatinine
Measurements of BUN and creatinine were performed in mouse plasma. BUN was measured using a quantitative colorimetric QuantiChrom kit (DIUR-100, BioAssay Systems), following the manufacturer’s instructions. Creatinine was analyzed by liquid chromatography–mass spectrometry.
GFR
GFR was measured in conscious mice using clearance kinetics of plasma fluorescein isothiocyanate-inulin after a single bolus injection in the vein tail, as previously described (14). GFR was calculated on the basis of a two-compartment model. GFR was expressed in µl per minute.
Histology, Immunohistochemistry, and IF
Kidneys were collected after CO2 euthanasia, weighed, cut in two transverse halves, and fixed by immersion in 10% formalin (ThermoFisher Scientific) for 24 hours. Next, fixed kidneys were washed in PBS, transferred to 70% EtOH, and embedded in paraffin blocks. Serial transversal sections (4 µm thick) were cut with a microtome. Immunohistochemistry (IHC) and IF studies were carried out using the avidin-peroxidase method and fluorescent antibodies, respectively. For both IHC and IF, we performed antigen retrieval by boiling preparations 20 minutes in 5 mM citrate buffer, pH 6.0. Next, kidney sections were blocked in 0.3% Triton X-100 in 5% normal donkey serum in PBS.
The incubation with primary antibody was performed in 0.1% Triton X-100 in 3% normal donkey serum in PBS, overnight at 4°C, using the following antibodies: anti–KIM-1 (AF1817, RD Systems), anti–SOX-9 (NBP1–8551, Novus Bio), anti-LRP2 (ab76969, Abcam), anti-SGLT1 (ab14685, Abcam), biotinylated-LTL (B-1325, Vector Laboratories), anti-EHHADH (GTX81126, Genetex), anti-Ki67 (MA5–14520, Invitrogen), anti-AQP2 (AQP-002, Alomone labs), anti-CALB (PA5–85669, ThermoFisher Scientific), and anti-SLC34A3 (NPT2c, 20603, BiCell Scientific). The incubation with secondary antibody was performed in PBS for 2 hours at room temperature, using the following secondary antibodies: 705–585–147, 712–545–153, 016–540–084 (Jackson ImmunoResearch), and A21206 (Invitrogen).
When two primary antibodies raised in rabbit were required for double IF, sections were incubated with AffiniPure Fab Fragment Goat Anti-Rabbit IgG (111–007–003) after the first primary antibody incubation to allow its detection with anti-goat secondary antibody. Control slides confirmed the first primary antibody was not detected by the anti-rabbit secondary antibody (data not shown). Nuclei were visualized with a Hoechst stain. For IF studies, autofluorescence was reduced by incubating the slides in 0.3% Sudan Black in 70% EtOH (Electron Microscopy Sciences) for 15 minutes. Microscopy images were taken with a Nikon Eclipse 80i microscope and the NIS-Elements BR 5.20.01 software (Nikon). Images were analyzed with ImageJ (15). The number of Ki67+ and kidney injury molecule 1+ (KIM-1+) cells per mm2 and the number of double SOX-9/KIM-1–positive cells in the kidney were determined by counting positive cells with the Cell Counter plugin in ImageJ (15). The cross-sectional area (in µm2) of 50 random cortical tubules was measured in hematoxylin and eosin–stained sections of WT and Ehhadh KO kidneys using the ROI manager tool of ImageJ (15). The average of three different images per mouse was used for the quantification. The researcher was blinded to the genotype of the sample when analyzing the images.
Immunoblot Analysis
Protein was extracted from frozen mouse kidneys and immunoblot analysis was performed as previously described (10) using the following primary antibodies: anti–KIM-1 (AF1817, RD Systems), anti–SOX-9 (NBP1–8551, Novus Bio), anti-SPTLC2 (51012–2-AP, Proteintech), anti-ABCD3 (PA1–650, Invitrogen), anti-ACOX1 (ab184032, Abcam), anti-EHHADH (GTX81126, Genetex), anti-HSD17B4 (15116–1-AP, Proteintech), anti-ACAA1 (12319–2-AP, Proteintech), anti-SCPx (HPA027135, Atlas Antibodies), anti-CROT (NBP1–85501, Novus Bio), anti-CPT2 (26555–1-AP, Proteintech), anti-MCAD (55210–1-AP, Proteintech), anti–α-tubulin (32–2500, ThermoFisher Scientific), anti-citrate synthase (GTX628143, Genetex), and anti-AMACR (16).
RNA-seq, Differential Gene Expression, Pathway Enrichment
RNA was isolated using QIAzol lysis reagent followed by purification using the RNeasy kit (Qiagen). RNA samples were submitted to the Genomics Core Facility at the Icahn Institute and Department of Genetics and Genomic Sciences for further processing. Briefly, mRNA-focused cDNA libraries were generated using Illumina reagents (polyA capture), and samples were run on an Illumina HiSeq 2500 sequencer to yield a read depth of approximately 56 million 100 nucleotide single-end reads per samples. Reads from fastq files were aligned to the mouse genome mm10 (GRCm38.75) with STAR (release 2.4.0 g1) and summarized to gene- and exon-level counts using featureCounts. Only genes with at least one count per million in at least two samples were considered. Differential gene expression analysis was conducted with the R package limma, as previously described (10). Differentially expressed genes (DEGs) were defined using an adjusted (adj) P value <0.05 with no logFC cutoff. The raw data and the count matrix for all genes can be accessed on the National Center for the Biotechnology Information database (GSE169676).
Pathway enrichment analysis was performed using Fisher’s exact test and P values were adjusted using a Benjamini-Hochberg procedure. Hallmark and BioPlanet Pathways were sourced from Enrichr (17). Input genes included genes up- or downregulated (at adj P<0.05) in Ehhadh KO versus WT kidneys that were converted from mouse to human orthologs using g:Orth from g:Profiler. iRegulon v1.3 was used to predict transcriptional regulators of kidney DEGs (18). Input genes included genes up- or downregulated (at adj P<0.05) in Ehhadh KO versus WT kidneys that were converted from mouse to human orthologs. The option Motif collection 10 K (9713 position weight matrices), putative regulatory region 10 kb centered around transcription start site (10 species), alongside the program default settings for Recovery and transcription factor (TF) prediction options were selected for the analysis.
Genesets associated with murine PT responses to acute injury were curated from a clustering analysis of single cell RNA-seq analysis of kidneys sampled over 12 hours and 2, 14, and 42 days after bilateral ischemia–reperfusion (19). Genesets associated with eight subclusters, namely healthy S1, S2, and S3, repairing PT, injured S1/2, injured S3, severe injured PT, and failed repair PT were curated from Dataset S2 of Kirita et al. (19). Genes found differentially expressed in folic acid nephropathy (FAN) versus control mice in PT cells were curated from Supplementary Table S2 of Dhillon et al. (20). Genes found differentially expressed in PT after unilateral ureteral obstruction (UUO) versus control mice (at adj P<0.01) were curated from Supplemental Table 2 of Wu et al. (21). PT injury associated murine genesets were tested for enrichment in up- or downregulated DEGs (at adj P<0.05) between Ehhadh and WT mice using the Fisher’s exact test and P values were adjusted using a Benjamini-Hochberg procedure.
Genes with sex-specific expression differences in mouse kidney were curated from two sources. One set were the gene differentially expressed (at adj P<0.05) between kidneys of 10-week-old healthy BALB/c male and female mice (n=5 per group) (22). The other set were genes differentially translated (at adj P<0.05) in PT of contralateral kidneys from mice subjected to UUO (n=3 females, n=4 males) (21). DEGs were converted from mouse to human orthologs using g:Orth from g:Profiler. Pathway enrichment analysis was performed using Enrichr (17).
Metabolomics
Global metabolite profiling (mView) from kidney (half, transversal) samples of seven WT males and five Ehhadh KO males was performed by Metabolon, Inc. (Research Triangle Park, NC). To remove protein, to dissociate small molecules bound to protein or trapped in the precipitated protein matrix, and to recover chemically diverse metabolites, proteins were precipitated with methanol under vigorous shaking for 2 minutes (Glen Mills GenoGrinder 2000) followed by centrifugation. The resulting extract was analyzed by two separate reverse phase ultraperformance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) methods with positive ion mode electrospray ionization (ESI), one reverse phase UPLC-MS/MS method with negative ion mode ESI, and one hydrophilic interaction liquid chromatography UPLC-MS/MS method with negative ion mode ESI. The scaled imputed data represent the normalized raw area counts of each metabolite rescaled to set the median equal to 1. Any missing values were imputed with the minimum value. Metabolite pathway enrichment analysis using significantly altered Human Metabolome Database metabolites was performed using MetaboAnalyst platform (23).
Statistical Analysis
Data are displayed as the mean±SD, with individual values shown. Differences were evaluated using unpaired t test with Welch’s correction or two-way ANOVA, as indicated in the figure legends. Significance is indicated in the figures. GraphPad Prism 8 was used to compute statistical values.
Results
EHHADH Deficiency Induces Male-Specific Kidney Hypertrophy without Signs of Severe Pathology
We observed male-specific kidney enlargement in 4–9-month-old Ehhadh KO mice (Figure 1A). Kidney mass and kidney-to-body weight (BW) ratio were higher in male Ehhadh KO mice compared with WT male mice (Figure 1, B and C). No changes in kidney mass were observed in female Ehhadh KO mice (Figure 1, B and C). Histologic analysis revealed that Ehhadh KO mice showed male-specific PT hypertrophy (Figure 1D, 1E, Supplemental Figure 1, A and B), but changes suggestive of kidney damage were not observed (Figure 1D and Supplemental Figure 1A). Female Ehhadh KO mice did not display PT hypertrophy (Supplemental Figure 1, A and B). GFR was decreased in male Ehhadh KO mice when compared with WT mice (Figure 1F), but this was not accompanied by alterations in circulating kidney function markers such as plasma creatinine (Figure 1G) or BUN (Figure 1H). GFR and BUN in female Ehhadh KO mice were similar to WT (Supplemental Figure 1, C and D). In summary, EHHADH deficiency causes male-specific kidney hypertrophy and a decrease in GFR without signs of severe kidney damage.
Figure 1.
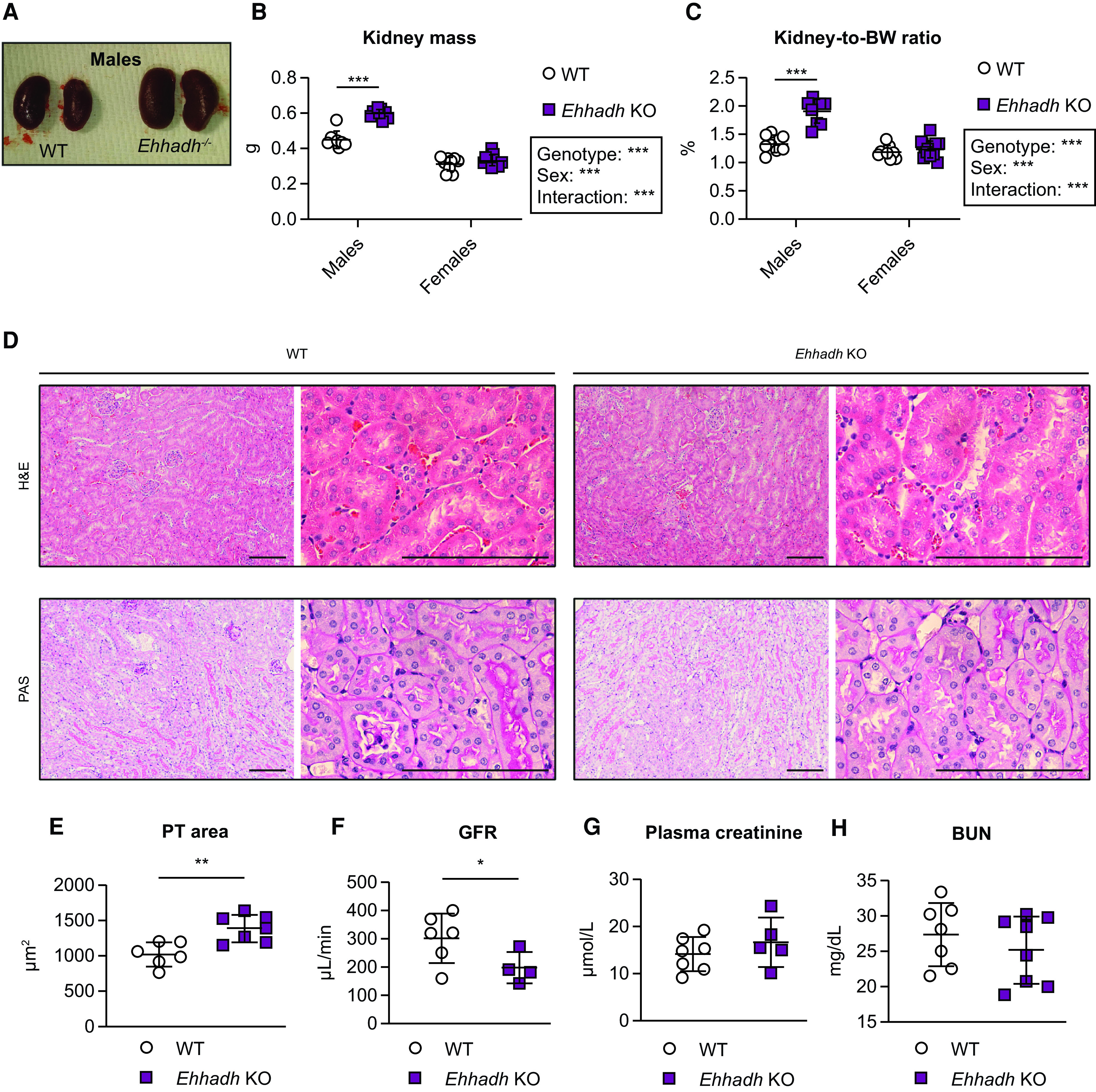
EHHADH deficiency induces male-specific kidney hypertrophy without signs of severe pathology. (A) Representative images of WT and Ehhadh KO kidneys from male mice. Measurement of (B) kidney weight (combined weight of both kidneys per animal) and (C) kidney-to-body weight (BW) ratio in 4–9-month-old male WT (n=8), male Ehhadh KO (n=9), female WT (n=9), and female Ehhadh KO mice (n=12). (D) Representative images of hematoxylin and eosin and periodic acid–Schiff staining of kidney sections from male WT and Ehhadh KO mice. Scale bars = 100 µm. (E) Morphometric analysis of the cross-sectional tubule areas in WT (n=6) and Ehhadh KO (n=7) male mice. (F) GFR in WT (n=6) and Ehhadh KO (n=4) male mice. (G) Plasma creatinine levels (μmol/L) in WT (n=5) and Ehhadh KO (n=7) male mice. (H) BUN levels (mg/dl) in WT (n=7) and Ehhadh KO (n=8) mice male. Data are presented as mean±SD with individual values plotted. ***P<0.001, **P<0.01, *P<0.05, by two-way ANOVA (B and C), or unpaired t test with Welch's correction (E and F). EHHADH, L-bifunctional protein; WT, wild-type; KO, knockout.
Transcriptional Activation of PT Kidney Injury Signatures in Male Ehhadh KO Kidneys
We performed RNA-seq analysis on whole kidneys from male adult WT and Ehhadh KO mice (n=4). We identified 1475 significant DEGs (at adj P<0.05), of which 806 genes were up- and 669 genes were downregulated (Figure 2A and Supplemental Table 1A). Among the top upregulated genes by fold-change, we identified KIM-1 (encoded by Havcr1) and the transcription factor SOX-9 (Sox9), which are considered markers of PT injury (24) and PT regeneration (25,26), respectively (Figure 2A and Supplemental Table 1A).
Figure 2.
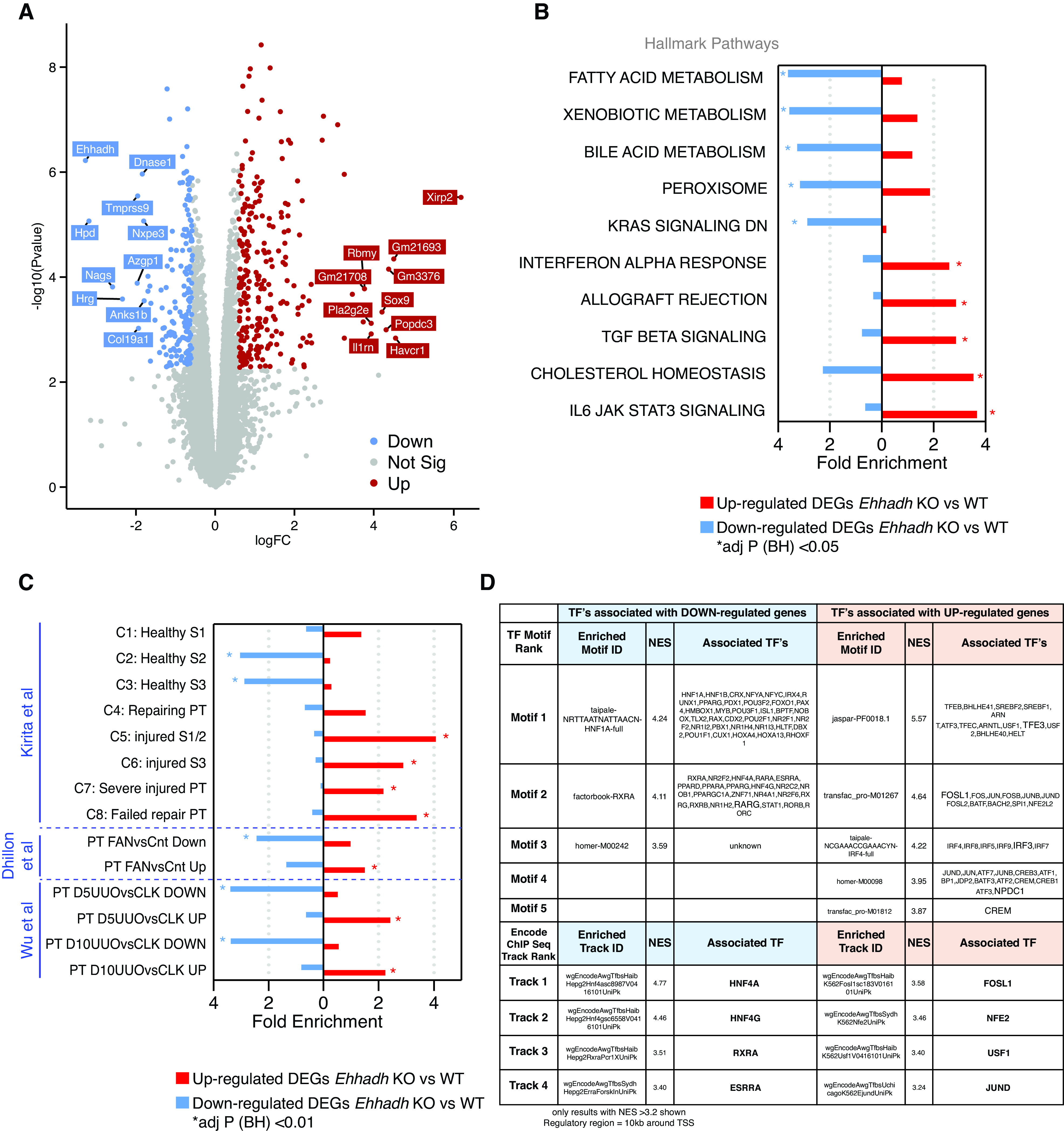
Transcriptomics of male Ehhadh KO kidneys. (A) Volcano plot with significantly differentially expressed genes (DEGs) highlighted. Decreased genes (down) are highlighted in blue, increased genes (up) are highlighted in red. Gene names indicate the top 10 significant up and down genes by logFC using an adjusted (adj) P<0.05. Genes that were not statistically significant are represented in gray. (B) Top 10 pathways (by fold enrichment) after pathway enrichment analysis of genes either significantly up- or downregulated in Ehhadh KO mice versus WT according to the Hallmark database. Values represent the fold enrichment and significance is indicated with an asterisk (*) (adj P<0.05). Full table of results are in Supplemental Table 1, B and C. (C) Results of enrichment analysis of genes either up- or downregulated in Ehhadh KO mice versus WT, in gene sets curated from three independent murine proximal tubule (PT) AKI models (see Methods). Values represent the fold enrichment and significance is indicated with an asterisk (*) (adj P<0.01). Full table of results are in Supplemental Table 1D. (D) Predicted transcriptional regulators as identified by Iregulon through Motif or Encode ChIP-seq enrichment analysis for the genes either up- or downregulated in Ehhadh KO mice versus WT kidney samples. Only results with normalized enrichment score (NES) >3.2 are shown. The full table of results is in Supplemental Table 1E. The bolded transcription factor (TF) represents the most likely TF associated with the enriched motif. Human orthologs of the murine DEGs (at adj P<0.05) were the input for (B), (C), and (D). BH, Benjamini-Hochberg.
Pathway enrichment analysis of the downregulated DEGs using the Hallmarks database highlighted “Fatty acid metabolism,” “Bile acid metabolism,” and “Peroxisome,” among other pathways (Figure 2B, Supplemental Figure 2A, Supplemental Table 1B). Pathway enrichment analysis of the upregulated DEGs highlighted inflammatory pathways, such as “Interferon alpha response,” “TGF beta signaling,” and “IL6 JAK STAT3 signaling.” Similar pathways were found enriched using the Bioplanet database (Supplemental Figure 2B, Supplemental Table 1C). These changes resemble reported transcriptional changes in kidneys after AKI (2,19–21), suggesting that EHHADH deficiency causes kidney injury in male mice.
We then compared the DEGs identified in Ehhadh KO male kidneys with expression signatures from PT cells of three different AKI mouse models; ischemia-reperfusion injury (19), FAN (20), and UUO (21) (Supplemental Table 1D). Genes upregulated or downregulated in the male Ehhadh KO kidneys generally changed in the same direction in the FAN and UUO models (Figure 2C). A recent report of Kirita et al. identified different cell states in the PT of mice subjected to ischemia-reperfusion injury, including a distinct proinflammatory and profibrotic PT cell state that fails to repair (19). Our enrichment analysis revealed a significant enrichment of downregulated DEGs from Ehhadh KO kidneys in the healthy S2 and healthy S3 PT cell states, and an enrichment of upregulated DEGs in the injured S1/2, the injured S3, the severe injured PT, and the failed repair PT cell states (Figure 2C).
Next, we performed cis-regulatory sequence analysis to uncover the transcriptional regulatory network underlying the transcriptional response of Ehhadh KO male mouse kidneys (18) (Supplemental Table 1E). We identified enriched TF motifs in the down-regulated DEGs of Ehhadh KO kidneys that mapped to known regulators of PT differentiation such as HNF1A, HNF4A, RXRA, and ESRRA (19,20,27) (Figure 2D). In the upregulated DEGs, we found enriched TF motifs that mapped to regulators of lysosomal biogenesis (TFEB/TFE3), and TFs whose activation is associated with PT cell injury (FOSL1) (19), among others (Figure 2D). In summary, these data show the transcriptional response of male mouse kidney to EHHADH deficiency is very similar to the transcriptional signature present in AKI mouse models and is characteristic for PT cell injury.
EHHADH Deficiency Activates the PT Injury Response in Male Mice
To validate the results of the RNA-seq, we performed an IHC assessment of KIM-1 in kidney. In contrast to the rare appearance or absence of KIM-1+ cells in male WT kidneys and female WT and Ehhadh KO kidneys, we identified many KIM-1+ cells in male Ehhadh KO kidneys (Figure 3, A and B). KIM-1+ cells were scattered among tubules in Ehhadh KO male kidneys with a prominent cytoplasmic pattern, combined with some tubular cells that showed the typical apical location of KIM-1 that is present in AKI (28). We further validated the increase in KIM-1 protein in male Ehhadh KO kidneys by immunoblotting (Supplemental Figure 3A).
Figure 3.
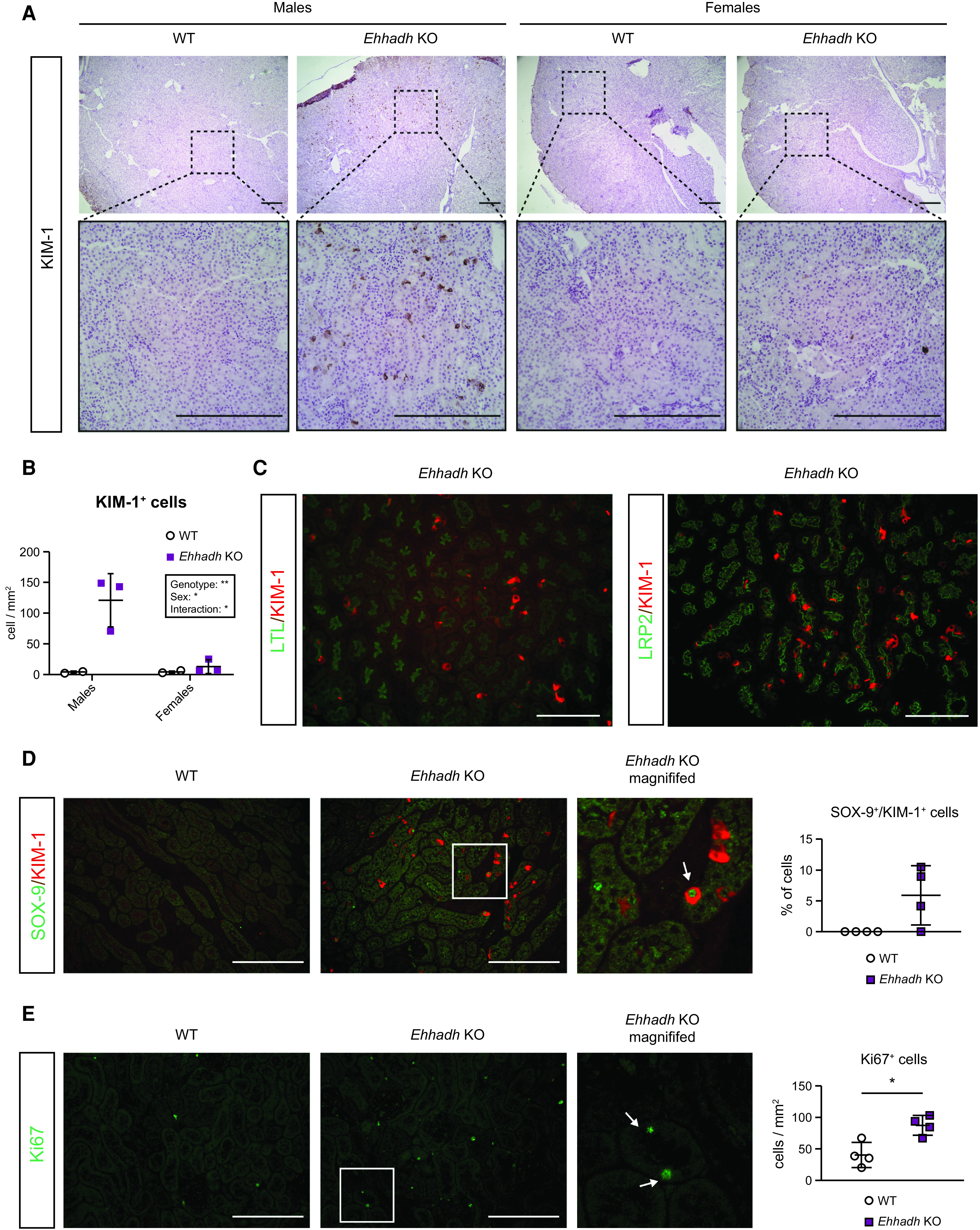
EHHADH deficiency activates the proximal tubule injury response in male mice. (A) Representative images of kidney injury molecule-1 (KIM-1) immunohistochemistry (IHC) in the cortical area of WT and Ehhadh KO mouse kidneys (n=3–4 per sex and genotype). Upper panels: 4× objective. Lower panels: 20× objective. (B) Quantification of KIM-1+ cells per mm2 in WT and Ehhadh KO male and female mice. (C) Representative images of double immunofluorescence (IF) of KIM-1 with pantubular markers (LTL and LRP2) in the kidneys of Ehhadh KO male mice. There was no KIM-1 signal in male WT mice, or female WT and Ehhadh KO mice (not shown). (D) Representative images of double IF of KIM-1 (red) and SOX-9 (green) in WT and Ehhadh KO male mice (n=4). Inset shows expanded region of the Ehhadh KO section. The white arrow points to a double positive Sox9+/Kim-1+ cell. Graph shows quantification of double positive SOX-9+/KIM-1+ cells in WT and Ehhadh KO mice. (E) Representative images of Ki67 IF (green) in WT and Ehhadh KO male mice (n=4). Inset shows expanded region of the Ehhadh KO section. White arrows point to Ki67+ cells. Graph shows quantification of Ki67+ cells per mm2 in WT and Ehhadh KO mice. Data are presented as mean±SD with individual values plotted (D) and (E). Statistical significance was tested using unpaired t test with Welch’s correction (D) and (E). *P<0.05. Scale bar = 250 µm (A), 100 µm (C)–(E).
Using double IF, we localized the KIM-1+ cells in the PT of Ehhadh KO male kidneys using the pantubular markers LTL and Megalin (LRP2) (Figure 3C). Most of the KIM-1+ cells colocalized with SGLT1 (SLC5A1), a marker for the S2/S3 PT segments, whereas a small number of KIM-1+ cells colocalized with the S1 marker NPT2c (SLC34A3) (Supplemental Figure 3, B and C). The localization of the majority of KIM-1+ cells in the S2/S3 segments is consistent with the expression pattern of EHHADH in mouse kidney, which is higher in S2/S3 than in S1 (29) (Supplemental Figure 3, D and E). We did not find colocalization between KIM-1 and markers for distal tubule cells (CALB) or principal cells (AQP2) (Supplemental Figure 3, F and G).
Double IF against KIM-1 and the transcription factor SOX-9 showed the presence of KIM-1+ and SOX-9+ cells in Ehhadh KO male kidneys (Figure 3D). Among the KIM-1+ cells, 5.9±4.8% were also SOX-9+ (Figure 3D), reflecting a tubular cell population with an ongoing injury/repair response (26). Increased SOX-9 protein levels were validated in male Ehhadh KO renal homogenates (Supplemental Figure 3A). We also found an increase in Ki67+ cells in male Ehhadh KO kidneys when compared with WT kidneys (Figure 3E), which is consistent with the RNA-seq data (Supplemental Table 1). Ki67 labels cycling cells, thus showing an increase in the number of proliferating epithelial cells in Ehhadh KO male kidneys. These results show that EHHADH deficiency causes a male-specific PT injury characterized by scattered KIM-1+ cells and the detection of regenerating SOX-9+ and Ki67+ cells.
Metabolic Profiling Unveils Peroxisomal Dysfunction and an Increase in Glycosphingolipid Levels in Male Ehhadh KO kidneys
To assess the effect of EHHADH deficiency on kidney metabolism in a nonbiased way, we performed metabolite profiling in kidneys from WT and Ehhadh KO male mice (Supplemental Table 2). Within the mouse kidneys, 838 known metabolites were detected and quantified. We found 190 metabolites that were significantly altered in the Ehhadh KO kidneys compared with WT kidneys (P<0.05), of which 100 metabolites were increased and 90 metabolites were decreased. Increased metabolites in Ehhadh KO kidneys include fatty acids and their conjugates, notably those that are markers of peroxisomal dysfunction, such as tetracosahexaenoic acid (C24:6n-3) (30), pipecolate (31), and very long-chain acylcarnitines, such as C26-carnitine and C26:1-carnitine (32).
Next, we performed pathway enrichment analysis. We unveiled a significant enrichment of “Sphingolipid metabolism” among the KEGG pathways (Figure 4A). Indeed, the top seven metabolites increased in Ehhadh KO kidneys were glucosylceramides and lactosylceramides, which are all sphingolipids (Figure 4B and Supplemental Table 2). In the RNA-seq data, we noted an increase in the expression of Sptlc2 (serine palmitoyltransferase, long-chain base subunit 2), the enzyme that initiates de novo sphingolipid biosynthesis. We also found a male-specific increase in the protein level of SPTLC2 in kidney homogenates from Ehhadh KO mice (Figure 4C). Other metabolites that were altered in Ehhadh KO mice included increased monoacylglycerols and polyamines, and decreased deoxyribonucleosides, deoxyribonucleotides, glutarylcarnitine and adipoylcarnitine (Supplemental Table 2). These results suggest that EHHADH deficiency leads to a profound metabolic remodeling in male mouse kidney with a prominent increase in glycosphingolipid levels.
Figure 4.

Metabolite profiling in Ehhadh KO male kidneys. (A) Pathway enrichmentanalysis using significantly altered Human Metabolome Database metabolites. Scatterplot represents unadj P values from integrated enrichment analysis and impact values from pathway topology analysis. The node color is on the basis of the P values and the node radius represents the pathway impact values. The only significantly altered KEGG pathway using Fisher’s exact t test (adj P<0.05) was “Sphingolipid metabolism”. (B) Glucosyl- and lactosyl-ceramides levels in WT (n=7) and Ehhadh KO (n=5) male mice. The scaled imputed data (Scaled Imp Data) represent the normalized raw area counts of each metabolite rescaled to set the median equal to 1. Any missing values are imputed with the minimum value. (C) Immunoblots of SPTLC2 and the loading control citrate synthase in WT (n=3 per sex) and Ehhadh KO (n=4 per sex) mice, and the corresponding quantification. Data are presented as mean±SD with individual values plotted (B) and (C). Statistical significance was tested using unpaired t test with Welch’s correction (B) or two-way ANOVA with “Genotype” and “Sex” as the two factors, followed by Tukey's multiple comparisons test (C). *P<0.05; **P<0.01; ***P<0.001.
The Kidney Phenotype Caused by EHHADH Deficiency in Mice Is Androgen-dependent
We hypothesized that androgens mediate the male-specific kidney phenotype in Ehhadh KO mice. To test this, we first studied the progression of kidney-to-BW ratio in WT and Ehhadh KO male mice, from 2 to 52 weeks of age. We found the kidney-to-BW ratio began to increase after 10 weeks of age (Supplemental Figure 4A), the time when mice reach sexual maturity and plasma testosterone levels peak (33). To determine if androgens are directly mediating the kidney phenotype caused by EHHADH deficiency, we performed orchiectomy in adult WT and Ehhadh KO mice and studied the kidneys 6 weeks after the operation. Orchiectomy decreased kidney-to-BW ratio in WT and Ehhadh KO mice (Supplemental Figure 4B). Sham-operated Ehhadh KO mice had many KIM-1+ cells in the renal cortex (Figure 5, A and B), similar to the male Ehhadh KO mice displayed in Figure 3B. In contrast, orchiectomized Ehhadh KO mice resembled the WT mice with KIM-1+ cells rarely detected or absent (Figure 5, A and B). We conclude that androgens mediate PT injury in Ehhadh KO mice.
Figure 5.

The kidney phenotype caused by EHHADH deficiency in mice is androgen dependent. (A) Representative images of KIM-1 IF in sham-operated and orchiectomized WT and Ehhadh KO male mice. (B) Quantification of KIM-1+ cells per mm2 in sham-operated and orchiectomized WT and Ehhadh KO male mice. Statistical significance was tested using two-way ANOVA with “Genotype” and “Orchiectomy” as the two factors (B). *P<0.05. Scale bars = 100 µm.
Peroxisomal FAO Proteins Are Sexually Dimorphic in Mouse Kidney
Several reports have shown significant gene expression differences between sexes in the adult mouse kidney (21,22,34). We performed pathway enrichment analysis on two available datasets, one comparing DEGs between male and female healthy kidneys of BALB/c mice (22), and the other one a PT-specific translational profile comparing male versus female PT cells from mouse kidneys (21). We revealed a significant enrichment in genes encoding peroxisomal proteins in both datasets, among other pathways that included fatty acid metabolic pathways (Supplemental Figure 4, A and B and Supplemental Table 3). We then analyzed the levels of peroxisomal FAO proteins in healthy male and female C57BL/6N mouse kidneys. Acyl-CoA oxidase 1, sterol carrier protein x (SCPx, encoded by Scp2), peroxisomal carnitine O-octanoyltransferase, and α-methylacyl-CoA racemase (AMACR) protein levels were higher in male kidneys (Figure 6A). Notably, EHHADH was the only peroxisomal FAO protein whose levels were higher in female kidneys (Figure 6A). No change was found for ATP-binding cassette subfamily D member 3, D-bifunctional enzyme (DBP), or 3-ketoacyl-CoA thiolase, peroxisomal. To determine if the sexual dimorphism was specific for peroxisomal FAO proteins, we also measured protein levels of two mitochondrial FAO proteins, carnitine palmitoyltransferase 2 (CPT2) and medium-chain acyl-CoA dehydrogenase (MCAD). We found increased levels of MCAD in female kidneys, and no change in the levels of CPT2 (Figure 6A). These results show that most of the peroxisomal FAO proteins show pronounced sexual dimorphism in mouse kidney.
Figure 6.
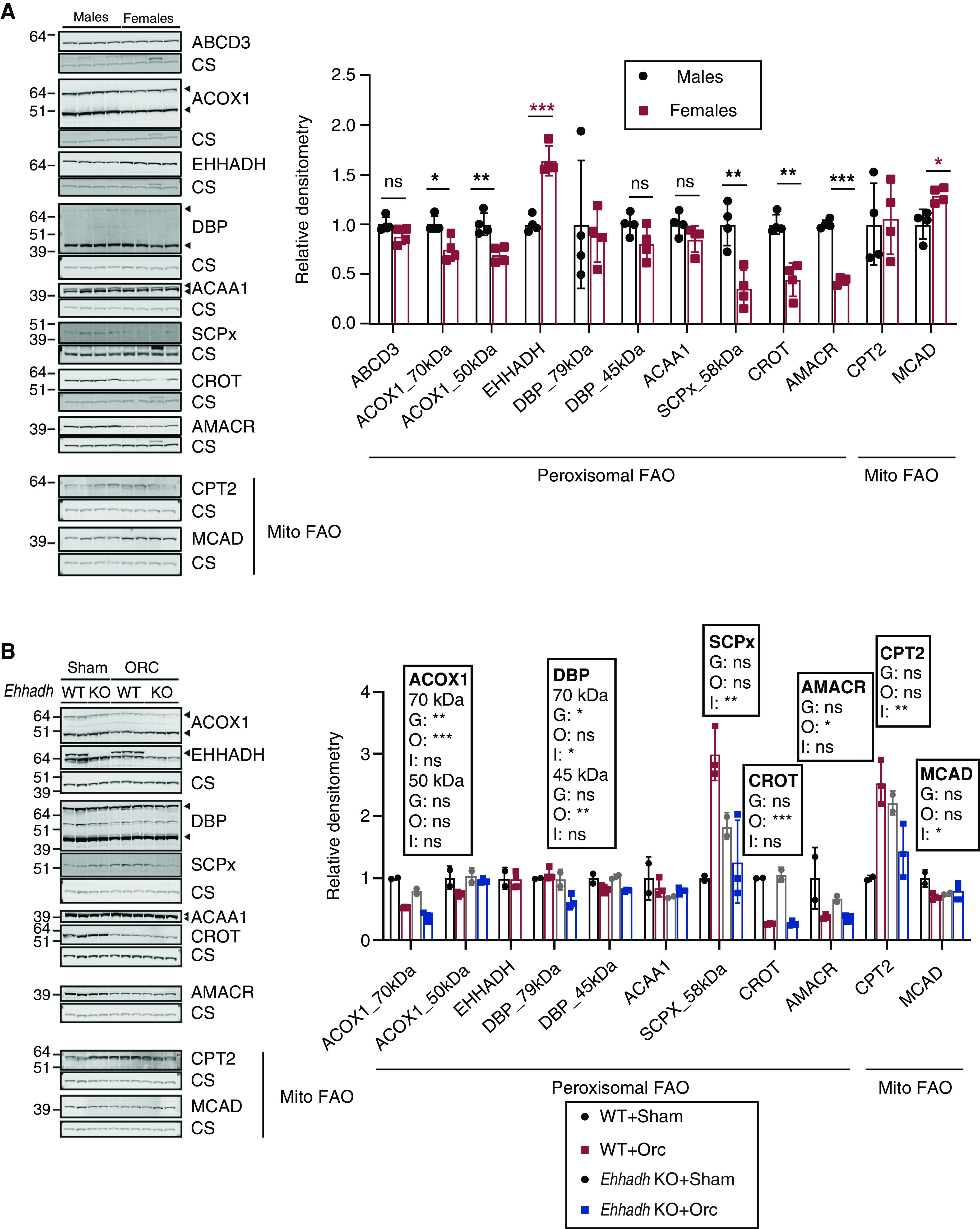
Sexually dimorphic expression of proteins involved in peroxisomal fatty acid oxidation (FAO) in mouse kidneys. (A) Immunoblots of peroxisomal and mitochondrial (Mito) FAO proteins with corresponding loading control (citrate synthase) in male and female WT mice (n=4 per sex), and the corresponding quantification. Black asterisks are shown when protein levels were significantly higher in males. Red asterisks are shown when protein levels were significantly higher in females. (B) Immunoblots of peroxisomal and Mito FAO proteins in sham-operated (n=2 per genotype) and orchiectomized (n=3 per genotype) WT and Ehhadh KO male mice. Data are presented as mean±SD with individual values plotted. Statistical significance was tested using unpaired t test with Welch’s correction (A) or two-way ANOVA with “Genotype” (G) and “Orchiectomy” (O) as the two factors, (“Interaction”: I) (B). *P<0.05; **P<0.01; ***P<0.001.
Next, we used kidney samples from the adult mice that underwent orchiectomy (Figure 5) to investigate if androgens play a role in regulating the expression of peroxisomal FAO proteins. We found that orchiectomy decreased the levels of acyl-CoA oxidase 1, peroxisomal carnitine O-octanoyltransferase, and AMACR in the kidneys of WT mice (Figure 6B). The levels of these three peroxisomal proteins were higher in the kidneys of male WT mice compared with female mice (Figure 6A), suggesting that androgens modulate their expression. DBP protein levels were also decreased after orchiectomy but only significantly in the kidneys of Ehhadh KO mice (Figure 6B). We did not find any effect of orchiectomy in the levels of EHHADH and 3-ketoacyl-CoA thiolase (Figure 6B). SCPx protein levels showed a differential response to orchiectomy, increasing after orchiectomy in WT kidneys but decreasing in Ehhadh KO kidneys (Figure 6B). CPT2 protein levels followed a pattern that was similar to SCPx (Figure 6B), whereas MCAD protein levels tended to decrease after orchiectomy only in WT kidneys (Figure 6B). Altogether, these data provide additional evidence of sexual dimorphism in the expression of peroxisomal FAO proteins in mouse kidney.
Discussion
In 1954, Rhodin discovered peroxisomes in the PT cells of mouse kidneys, and named them microbodies (4). Although peroxisomes are highly abundant in the kidney, their function in this organ remains largely unknown. We report previously unnoticed male-specific kidney hypertrophy and proximal tubular injury in Ehhadh KO mice. Our work establishes an important function of EHHADH and the peroxisome in male PT metabolic homeostasis.
The renal phenotype of Ehhadh KO mice is associated with a transcriptional signature that shares many features with models of PT injury after AKI (19–21), and is characterized by a downregulation of fatty acid metabolic processes and an upregulation of inflammatory pathways. Notably, PT hypertrophy and injury develop spontaneously in Ehhadh KO mice in a male-specific fashion. Despite the larger size of male Ehhadh KO kidneys, GFR was decreased. Furthermore, the PT of Ehhadh KO male mice displayed many KIM-1, SOX-9, and Ki67-positive cells. Therefore, we postulate that the Ehhadh KO mouse constitutes a model for male-specific metabolic PT injury. Our data suggest a reduction in EHHADH activity and peroxisomal FAO may contribute to the development of kidney disease.
In keeping with the notion of a prominent role for peroxisomes in kidney function are observations that peroxisomal abundance and function are decreased in several models of kidney disease (2,8,9). Patients with peroxisomal disorders such as the Zellweger spectrum disorder and DBP deficiency (7) are prone to develop kidney pathology in the form of renal cysts and/or calcium oxalate stones (6). However, underlying mechanisms that link peroxisomal dysfunction with kidney damage other than defective glyoxylate metabolism have not been identified yet. Mouse models with defects in peroxisome biogenesis factors (peroxins) show a more severe renal phenotype when subjected to kidney injury (35,36). However, peroxin deficiencies impair all peroxisomal functions, hindering the study of the specific role of peroxisomal FAO in kidney physiology. A dominant negative mutation in the EHHADH gene was found in patients with an inherited form of renal Fanconi’s syndrome, but the pathophysiological cause was the disruption in mitochondrial oxidative phosphorylation caused by the mistargeting of the mutant EHHADH protein to mitochondria (37). This is the first study reporting the renal consequences of a single peroxisomal enzyme deficiency in a mouse model.
Our metabolomics analysis provides some insight into the mechanisms underlying the PT injury in Ehhadh KO mice. Among the metabolites that accumulated in male Ehhadh KO mouse kidneys were very long-chain acylcarnitines (C26- and C26:1-carnitine), pipecolate, and tetracosahexaenoic acid (C24:6n-3). Glutarylcarnitine and adipoylcarnitine were decreased. The direction of change in these metabolites is consistent with defective peroxisomal metabolism (30–32). Our data suggest EHHADH deficiency induces a rewiring of cellular metabolism that results in the accumulation of glucosyl- and lactosylceramides in the kidney, with many of these species containing at least one very long-chain acyl chain (C22 or longer). The accumulation of these complex sphingolipids has been associated with an increase in kidney size mediated by androgens (38,39), but a link with peroxisomes has not been established.
The primary metabolic cause of the male-specific renal phenotype of Ehhadh KO mice remains unknown. We speculate that EHHADH deficiency impairs the degradation of an intrinsic metabolite that is toxic to the PT epithelium, although we cannot exclude an extra-renal source with high EHHADH expression such as the liver. We envision two possible models to explain the male-specificity (Supplemental Figure 6). In the first model (A), such a toxic metabolite is produced by one of the CYP enzymes whose expression is known to be sexually dimorphic (21,22,34). Dicarboxylic acids and eicosanoids are likely candidate metabolites because both undergo several cycles of peroxisomal β-oxidation after initial ω-oxidation by CYP enzymes (10,12,40–45). Unfortunately, our metabolomics analysis did not reveal a clear candidate toxic molecule, but the detection may have been obscured by the ongoing disease process. Future studies should focus on differences between the metabolome of male and female kidneys focusing on lipid substrates of CYP enzymes.
In the alternative model (B), the toxic insult caused by EHHADH deficiency may occur in both males and females, but only progresses to PT injury in males due to higher androgen levels. We found that the relative kidney enlargement was first detectable around 10 weeks of age in male Ehhadh KO mice, right after plasma androgen levels peak in mice (33). Moreover, castration reversed kidney enlargement and PT injury in male Ehhadh KO mice. We also found a male-specific increase in SPTLC2 protein levels in renal homogenates. SPTLC2 catalyzes the first and rate-limiting step of sphingolipid biosynthesis, suggesting the metabolic rewiring caused by EHHADH deficiency is also restricted to male animals.
Sex-specific differential susceptibility is reproduced in other rodent models of kidney disease. Androgens, particularly testosterone, are known to induce renal hypertrophy in rodents (46) and to increase the susceptibility of mice to develop kidney injury (47). In humans, the available evidence suggests that male sex is associated with a more rapid rate of disease progression and a worse renal outcome in patients with CKD (48,49). Moreover, a recent study found that genetically predicted levels of testosterone in men were associated with higher risk of CKD and worse kidney function (50).
In conclusion, we show that deficiency of a single peroxisomal FAO enzyme, EHHADH, causes kidney hypertrophy, GFR decrease, and PT injury in male mice. The results of our study suggest that EHHADH and peroxisomal metabolism play an important role in metabolic homeostasis of the PT. Altogether, this study underlines the role of peroxisomes and EHHADH in the mouse kidney and provides evidence for a sexual dimorphic pathophysiologic mechanism within the kidney.
Disclosures
J.C. He reports having consultancy agreements with and having an ownership interest in Renalytix AI; reports receiving research funding from Shangpharma Innovation; reports receiving honoraria from AstraZeneca ($3400); and reports being a scientific advisor or member of the Editorial Board for the American Journal of Physiology, Diabetes, Journal of the American Society of Nephrology, and Kidney International, board member of Chinese American Society of Nephrology and International Chinese Society of Nephrology, Associate Editor for Kidney Disease, and Section Editor for Nephron. K. Lee reports having an ownership interest in Rila Therapeutics Inc. All remaining authors have nothing to disclose.
Funding
This work was supported by the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health under award number R01DK113172.
Acknowledgments
We thank Dr. Sacha Ferdinandusse (Amsterdam University Medical Center, the Netherlands) for providing the AMACR antibodies, and acknowledge the help of the shared resource facilities at the Icahn School of Medicine at Mount Sinai (Biorepository and Pathology, Colony Management, Mouse Genetics and Gene Targeting, Scientific Computing and the Genomics Core). Parts of this manuscript were previously posted to preprint server bioRxiv as https://doi.org/10.1101/2021.03.14.435187. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author Contributions
C. Argmann, J. He, S. Houten, K. Lee, and P. Ranea-Robles conceptualized the study; C. Argmann, A. Bender, S. Houten, K. Portman, and P. Ranea-Robles were responsible for data curation; C. Argmann, A. Bender, S. Houten, K. Portman, and P. Ranea-Robles were responsible for formal analysis; C. Argmann, A. Bender, J. He, S. Houten, K. Lee, D. Mulholland, and P. Ranea-Robles were responsible for investigation; C. Argmann, A. Bender, J. He, S. Houten, K. Lee, D. Mulholland, K. Portman, and P. Ranea-Robles were responsible for the methodology; C. Argmann, S. Houten, and P. Ranea-Robles were responsible for visualization; S. Houten and P. Ranea-Robles wrote the original draft; C. Argmann, J. He, S. Houten, K. Lee, D. Mulholland, and P. Ranea-Robles reviewed and edited the manuscript; C. Argmann, J. He, K. Lee, and D. Mulholland were responsible for the resources; C. Argmann and S. Houten were responsible for the validation; S. Houten was responsible for the funding acquisition, project administration, and resources, and provided supervision; all authors revised the paper and approved the final version of the manuscript.
Supplemental Material
This article contains Supplemental material online at http://kidney360.asnjournals.org/lookup/suppl/doi:10.34067/KID.0005102020/-/DCSupplemental.
EHHADH deficiency does not cause morphological or functional changes in female mouse kidneys. Download Supplemental Figure 1, PDF file, 1.5 MB (1.5MB, pdf)
Transcriptomics of male Ehhadh KO kidneys. Download Supplemental Figure 2, PDF file, 1.5 MB (1.5MB, pdf)
EHHADH deficiency activates the proximal tubule injury response in male mice. Download Supplemental Figure 3, PDF file, 1.5 MB (1.5MB, pdf)
The kidney phenotype caused by EHHADH deficiency in mice isandrogen-dependent. Download Supplemental Figure 4, PDF file, 1.5 MB (1.5MB, pdf)
Pathway enrichment analysis of sexually dimorphic DEGs in mouse kidneys. Download Supplemental Figure 5, PDF file, 1.5 MB (1.5MB, pdf)
Proposed working models to explain how EHHADH deficiency causes male-specific PT injury. Download Supplemental Figure 6, PDF file, 1.5 MB (1.5MB, pdf)
Ehhadh KO kidney RNA-seq signature. Download Supplemental Table 1, XLSX file, 1.9 MB (1.9MB, xlsx)
Untargeted metabolomics dataset comparing WT and Ehhadh KO male kidneys. Download Supplemental Table 2, XLSX file, 571 KB (570.8KB, xlsx)
Pathway enrichment analysis of two published datasets containing sexually dimorphic genes in mouse kidneys. Download Supplemental Table 3, XLSX file, 84 KB (83.7KB, xlsx)
References
- 1.Nieth H, Schollmeyer P: Substrate-utilization of the human kidney. Nature 209: 1244–1245, 1966. 10.1038/2091244a0 [DOI] [PubMed] [Google Scholar]
- 2.Kang HM, Ahn SH, Choi P, Ko Y-A, Han SH, Chinga F, Park ASD, Tao J, Sharma K, Pullman J, Bottinger EP, Goldberg IJ, Susztak K: Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med 21: 37–46, 2015. 10.1038/nm.3762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Afshinnia F, Rajendiran TM, Soni T, Byun J, Wernisch S, Sas KM, Hawkins J, Bellovich K, Gipson D, Michailidis G, Pennathur S; Michigan Kidney Translational Core CPROBE Investigator Group: Impaired B-oxidation and altered complex lipid fatty acid partitioning with advancing CKD. J Am Soc Nephrol 29: 295–306, 2018. 10.1681/ASN.2017030350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rhodin JAG: Correlation of ultrastructural organization and function in normal and experimentally changed proximal convoluted tubule cells of the mouse kidney. Doctoral Thesis., Karolinska Institutet, Stockholm, Aktiebolaget Godvil, 1954
- 5.Wanders RJA, Waterham HR: Biochemistry of mammalian peroxisomes revisited. Annu Rev Biochem 75: 295–332, 2006. 10.1146/annurev.biochem.74.082803.133329 [DOI] [PubMed] [Google Scholar]
- 6.van Woerden CS, Groothoff JW, Wijburg FA, Duran M, Wanders RJA, Barth PG, Poll-The BT: High incidence of hyperoxaluria in generalized peroxisomal disorders. Mol Genet Metab 88: 346–350, 2006. 10.1016/j.ymgme.2006.03.004 [DOI] [PubMed] [Google Scholar]
- 7.Huyghe S, Mannaerts GP, Baes M, Van Veldhoven PP: Peroxisomal multifunctional protein-2: the enzyme, the patients and the knockout mouse model. Biochim Biophys Acta 1761: 973–994, 2006. 10.1016/j.bbalip.2006.04.006 [DOI] [PubMed] [Google Scholar]
- 8.Gulati S, Singh AK, Irazu C, Orak J, Rajagopalan PR, Fitts CT, Singh I: Ischemia-reperfusion injury: biochemical alterations in peroxisomes of rat kidney. Arch Biochem Biophys 295: 90–100, 1992. 10.1016/0003-9861(92)90492-F [DOI] [PubMed] [Google Scholar]
- 9.Kalakeche R, Hato T, Rhodes G, Dunn KW, El-Achkar TM, Plotkin Z, Sandoval RM, Dagher PC: Endotoxin uptake by S1 proximal tubular segment causes oxidative stress in the downstream S2 segment. J Am Soc Nephrol 22: 1505–1516, 2011. 10.1681/ASN.2011020203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ranea-Robles P, Violante S, Argmann C, Dodatko T, Bhattacharya D, Chen H, Yu C, Friedman SL, Puchowicz M, Houten SM: Murine deficiency of peroxisomal L-bifunctional protein (EHHADH) causes medium-chain 3-hydroxydicarboxylic aciduria and perturbs hepatic cholesterol homeostasis. Cell Mol Life Sci 78: 5631–5646, 2021. 10.1007/s00018-021-03869-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nguyen SD, Baes M, Van Veldhoven PP: Degradation of very long chain dicarboxylic polyunsaturated fatty acids in mouse hepatocytes, a peroxisomal process. Biochim Biophys Acta 1781: 400–405, 2008. 10.1016/j.bbalip.2008.06.004 [DOI] [PubMed] [Google Scholar]
- 12.Ferdinandusse S, Denis S, Van Roermund CWT, Wanders RJA, Dacremont G: Identification of the peroxisomal β-oxidation enzymes involved in the degradation of long-chain dicarboxylic acids. J Lipid Res 45: 1104–1111, 2004. 10.1194/jlr.M300512-JLR200 [DOI] [PubMed] [Google Scholar]
- 13.Qi C, Zhu Y, Pan J, Usuda N, Maeda N, Yeldandi AV, Rao MS, Hashimoto T, Reddy JK: Absence of spontaneous peroxisome proliferation in enoyl-CoA Hydratase/L-3-hydroxyacyl-CoA dehydrogenase-deficient mouse liver. Further support for the role of fatty acyl CoA oxidase in PPARalpha ligand metabolism. J Biol Chem 274: 15775–15780, 1999. 10.1074/jbc.274.22.15775 [DOI] [PubMed] [Google Scholar]
- 14.Qi Z, Whitt I, Mehta A, Jin J, Zhao M, Harris RC, Fogo AB, Breyer MD: Serial determination of glomerular filtration rate in conscious mice using FITC-inulin clearance. Am J Physiol Ren Physiol 286: F590–F596, 2004 [DOI] [PubMed] [Google Scholar]
- 15.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A: Fiji: an open-source platform for biological-image analysis. Nat Methods 9: 676–682, 2012. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ferdinandusse S, Denis S, IJlst L, Dacremont G, Waterham HR, Wanders RJA: Subcellular localization and physiological role of α-methylacyl-CoA racemase. J Lipid Res 41: 1890–1896, 2000. 10.1016/S0022-2275(20)31983-0 [DOI] [PubMed] [Google Scholar]
- 17.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma’ayan A: Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14: 128, 2013. 10.1186/1471-2105-14-128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Janky R, Verfaillie A, Imrichová H, Van de Sande B, Standaert L, Christiaens V, Hulselmans G, Herten K, Naval Sanchez M, Potier D, Svetlichnyy D, Kalender Atak Z, Fiers M, Marine JC, Aerts S: iRegulon: From a gene list to a gene regulatory network using large motif and track collections. PLOS Comput Biol 10: e1003731, 2014. 10.1371/journal.pcbi.1003731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kirita Y, Wu H, Uchimura K, Wilson PC, Humphreys BD: Cell profiling of mouse acute kidney injury reveals conserved cellular responses to injury. Proc Natl Acad Sci U S A 117: 15874–15883, 2020. 10.1073/pnas.2005477117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dhillon P, Park J, Hurtado Del Pozo C, Li L, Doke T, Huang S, Zhao J, Kang HM, Shrestra R, Balzer MS, Chatterjee S, Prado P, Han SY, Liu H, Sheng X, Dierickx P, Batmanov K, Romero JP, Prósper F, Li M, Pei L, Kim J, Montserrat N, Susztak K: The nuclear receptor ESRRA protects from kidney disease by coupling metabolism and differentiation. Cell Metab 33: 379–394.e8, 2021. 10.1016/j.cmet.2020.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu H, Lai CF, Chang-Panesso M, Humphreys BD: Proximal tubule translational profiling during kidney fibrosis reveals proinflammatory and long noncoding RNA expression patterns with sexual dimorphism. J Am Soc Nephrol 31: 23–38, 2020. 10.1681/ASN.2019040337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Si H, Banga RS, Kapitsinou P, Ramaiah M, Lawrence J, Kambhampati G, Gruenwald A, Bottinger E, Glicklich D, Tellis V, Greenstein S, Thomas DB, Pullman J, Fazzari M, Susztak K: Human and murine kidneys show gender- and species-specific gene expression differences in response to injury. PLoS One 4: e4802, 2009. 10.1371/journal.pone.0004802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chong J, Soufan O, Li C, Caraus I, Li S, Bourque G, Wishart DS, Xia J: MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res 46[W1]: W486–W494, 2018. 10.1093/nar/gky310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ichimura T, Bonventre JV, Bailly V, Wei H, Hession CA, Cate RL, Sanicola M: Kidney injury molecule-1 (KIM-1), a putative epithelial cell adhesion molecule containing a novel immunoglobulin domain, is up-regulated in renal cells after injury. J Biol Chem 273: 4135–4142, 1998. 10.1074/jbc.273.7.4135 [DOI] [PubMed] [Google Scholar]
- 25.Kang HM, Huang S, Reidy K, Han SH, Chinga F, Susztak K: Sox9-positive progenitor cells play a key role in renal tubule epithelial regeneration in mice. Cell Rep 14: 861–871, 2016. 10.1016/j.celrep.2015.12.071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kumar S, Liu J, Pang P, Krautzberger AM, Reginensi A, Akiyama H, Schedl A, Humphreys BD, McMahon AP: Sox9 activation highlights a cellular pathway of renal repair in the acutely injured mammalian kidney. Cell Rep 12: 1325–1338, 2015. 10.1016/j.celrep.2015.07.034 [DOI] [PubMed] [Google Scholar]
- 27.Marable SS, Chung E, Park JS: Hnf4a is required for the development of cdh6-expressing progenitors into proximal tubules in the mouse kidney. J Am Soc Nephrol 31: 2543–2558, 2020. 10.1681/ASN.2020020184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ichimura T, Hung CC, Yang SA, Stevens JL, Bonventre JV: Kidney injury molecule-1: A tissue and urinary biomarker for nephrotoxicant-induced renal injury. Am J Physiol Ren Physiol 286: F552–F563, 2004. 10.1152/ajprenal.00285.2002 [DOI] [PubMed] [Google Scholar]
- 29.Limbutara K, Chou CL, Knepper MA: Quantitative proteomics of all 14 renal tubule segments in rat. J Am Soc Nephrol 31: 1255–1266, 2020. 10.1681/ASN.2020010071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ferdinandusse S, Denis S, Mooijer PA, Zhang Z, Reddy JK, Spector AA, Wanders RJ: Identification of the peroxisomal beta-oxidation enzymes involved in the biosynthesis of docosahexaenoic acid. J Lipid Res 42: 1987–1995, 2001. 10.1016/S0022-2275(20)31527-3 [DOI] [PubMed] [Google Scholar]
- 31.Mihalik SJ, Moser HW, Watkins PA, Danks DM, Poulos A, Rhead WJ: Peroxisomal L-pipecolic acid oxidation is deficient in liver from Zellweger syndrome patients. Pediatr Res 25: 548–552, 1989. 10.1203/00006450-198905000-00024 [DOI] [PubMed] [Google Scholar]
- 32.Klouwer FCC, Ferdinandusse S, van Lenthe H, Kulik W, Wanders RJA, Poll-The BT, Waterham HR, Vaz FM: Evaluation of C26:0-lysophosphatidylcholine and C26:0-carnitine as diagnostic markers for Zellweger spectrum disorders. J Inherit Metab Dis 40: 875–881, 2017. 10.1007/s10545-017-0064-0 [DOI] [PubMed] [Google Scholar]
- 33.Bell MR: Comparing postnatal development of gonadal hormones and associated social behaviors in rats, mice, and humans. Endocrinology 159: 2596–2613, 2018. 10.1210/en.2018-00220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rinn JL, Rozowsky JS, Laurenzi IJ, Petersen PH, Zou K, Zhong W, Gerstein M, Snyder M: Major molecular differences between mammalian sexes are involved in drug metabolism and renal function. Dev Cell 6: 791–800, 2004. 10.1016/j.devcel.2004.05.005 [DOI] [PubMed] [Google Scholar]
- 35.Weng H, Ji X, Endo K, Iwai N: Pex11a deficiency is associated with a reduced abundance of functional peroxisomes and aggravated renal interstitial lesions. Hypertension 64: 1054–1060, 2014. 10.1161/HYPERTENSIONAHA.114.04094 [DOI] [PubMed] [Google Scholar]
- 36.Maxwell M, Bjorkman J, Nguyen T, Sharp P, Finnie J, Paterson C, Tonks I, Paton BC, Kay GF, Crane DI: Pex13 inactivation in the mouse disrupts peroxisome biogenesis and leads to a Zellweger syndrome phenotype. Mol Cell Biol 23: 5947–5957, 2003. 10.1128/MCB.23.16.5947-5957.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Klootwijk ED, Reichold M, Helip-Wooley A, Tolaymat A, Broeker C, Robinette SL, Reinders J, Peindl D, Renner K, Eberhart K, Assmann N, Oefner PJ, Dettmer K, Sterner C, Schroeder J, Zorger N, Witzgall R, Reinhold SW, Stanescu HC, Bockenhauer D, Jaureguiberry G, Courtneidge H, Hall AM, Wijeyesekera AD, Holmes E, Nicholson JK, O’Brien K, Bernardini I, Krasnewich DM, Arcos-Burgos M, Izumi Y, Nonoguchi H, Jia Y, Reddy JK, Ilyas M, Unwin RJ, Gahl WA, Warth R, Kleta R: Mistargeting of peroxisomal EHHADH and inherited renal Fanconi’s syndrome. N Engl J Med 370: 129–138, 2014. 10.1056/NEJMoa1307581 [DOI] [PubMed] [Google Scholar]
- 38.Koenig H, Goldstone A, Blume G, Lu CY: Testosterone-mediated sexual dimorphism of mitochondria and lysosomes in mouse kidney proximal tubules. Science 209: 1023–1026, 1980. 10.1126/science.7403864 [DOI] [PubMed] [Google Scholar]
- 39.Hay JB, Gray GM: The effect of testosterone on the glycosphingolipid composition of mouse kidney. Biochim Biophys Acta Lipids Lipid Metab 202: 566–568, 1970. 10.1016/0005-2760(70)90132-3 [DOI] [PubMed] [Google Scholar]
- 40.Tomoko S, Reiko N, Tadayoshi O, Kunihiko S: Metabolism of prostaglandins D2 and F2α in primary cultures of rat hepatocytes. Biochim Biophys Acta Lipids Lipid Metab 879: 330–338, 1986. 10.1016/0005-2760(86)90222-5 [DOI] [PubMed] [Google Scholar]
- 41.Diczfalusy U, Kase BF, Alexson SEH, Björkhem I: Metabolism of prostaglandin F2 α in Zellweger syndrome. Peroxisomal β-oxidation is a major importance for in vivo degradation of prostaglandins in humans. J Clin Invest 88: 978–984, 1991. 10.1172/JCI115401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mayatepek E, Lehmann WD, Fauler J, Tsikas D, Frölich JC, Schutgens RBH, Wanders RJA, Keppler D: Impaired degradation of leukotrienes in patients with peroxisome deficiency disorders. J Clin Invest 91: 881–888, 1993. 10.1172/JCI116309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.de Waart DR, Koomen GCM, Wanders RJA: Studies on the urinary excretion of thromboxane B2 in Zellweger patients and control subjects: evidence for a major role for peroxisomes in the β-oxidative chain-shortening of thromboxane B2. Biochim Biophys Acta 1226: 44–48, 1994. 10.1016/0925-4439(94)90057-4 [DOI] [PubMed] [Google Scholar]
- 44.Dirkx R, Meyhi E, Asselberghs S, Reddy J, Baes M, Van Veldhoven PP: β-oxidation in hepatocyte cultures from mice with peroxisomal gene knockouts. Biochem Biophys Res Commun 357: 718–723, 2007. 10.1016/j.bbrc.2007.03.198 [DOI] [PubMed] [Google Scholar]
- 45.Ferdinandusse S, Meissner T, Wanders RJA, Mayatepek E: Identification of the peroxisomal β-oxidation enzymes involved in the degradation of leukotrienes. Biochem Biophys Res Commun 293: 269–273, 2002. 10.1016/S0006-291X(02)00214-0 [DOI] [PubMed] [Google Scholar]
- 46.Selye H: The effect of testosterone on the kidney. J Urol 42: 637–641, 1939. 10.1016/S0022-5347(17)71560-1 [DOI] [Google Scholar]
- 47.Park KM, Kim JI, Ahn Y, Bonventre AJ, Bonventre JV: Testosterone is responsible for enhanced susceptibility of males to ischemic renal injury. J Biol Chem 279: 52282–52292, 2004. 10.1074/jbc.M407629200 [DOI] [PubMed] [Google Scholar]
- 48.Neugarten J, Golestaneh L: Influence of sex on the progression of chronic kidney disease. Mayo Clin Proc 94: 1339–1356, 2019. 10.1016/j.mayocp.2018.12.024 [DOI] [PubMed] [Google Scholar]
- 49.Neugarten J, Acharya A, Silbiger SR: Effect of gender on the progression of nondiabetic renal disease: A meta-analysis. J Am Soc Nephrol 11: 319–329, 2000. 10.1681/ASN.V112319 [DOI] [PubMed] [Google Scholar]
- 50.Zhao JV, Schooling CM: The role of testosterone in chronic kidney disease and kidney function in men and women: A bi-directional Mendelian randomization study in the UK Biobank. BMC Med 18: 122, 2020. 10.1186/s12916-020-01594-x [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
EHHADH deficiency does not cause morphological or functional changes in female mouse kidneys. Download Supplemental Figure 1, PDF file, 1.5 MB (1.5MB, pdf)
Transcriptomics of male Ehhadh KO kidneys. Download Supplemental Figure 2, PDF file, 1.5 MB (1.5MB, pdf)
EHHADH deficiency activates the proximal tubule injury response in male mice. Download Supplemental Figure 3, PDF file, 1.5 MB (1.5MB, pdf)
The kidney phenotype caused by EHHADH deficiency in mice isandrogen-dependent. Download Supplemental Figure 4, PDF file, 1.5 MB (1.5MB, pdf)
Pathway enrichment analysis of sexually dimorphic DEGs in mouse kidneys. Download Supplemental Figure 5, PDF file, 1.5 MB (1.5MB, pdf)
Proposed working models to explain how EHHADH deficiency causes male-specific PT injury. Download Supplemental Figure 6, PDF file, 1.5 MB (1.5MB, pdf)
Ehhadh KO kidney RNA-seq signature. Download Supplemental Table 1, XLSX file, 1.9 MB (1.9MB, xlsx)
Untargeted metabolomics dataset comparing WT and Ehhadh KO male kidneys. Download Supplemental Table 2, XLSX file, 571 KB (570.8KB, xlsx)
Pathway enrichment analysis of two published datasets containing sexually dimorphic genes in mouse kidneys. Download Supplemental Table 3, XLSX file, 84 KB (83.7KB, xlsx)