

Abstract
Rationale:
Diabetic cardiomyopathy is accompanied by increased production of NADH, predominantly through oxidation of fatty acids and consequent increases in oxidative stress. The role of nicotinamide phosphoribosyltransferase (Nampt), the rate-limiting enzyme of the salvage pathway of NAD+ synthesis, in the development of diabetic cardiomyopathy is poorly understood.
Objective:
We investigated the role of endogenous and exogenous Nampt during the development of diabetic cardiomyopathy in response to high fat diet (HFD) consumption and in the context of oxidative stress.
Methods and Results:
HFD consumption upregulated endogenous Nampt, and HFD-induced cardiac diastolic dysfunction, fibrosis, apoptosis and pro-inflammatory signaling were alleviated in transgenic mice with cardiac-specific overexpression of Nampt. The alleviation of diastolic dysfunction observed in these mice was abolished by inhibition of NADP(H) production via NAD kinase (NADK) inhibition. Nampt overexpression decreased the GSSG/GSH ratio, oxidation of thioredoxin 1 (Trx1) targets, dityrosine, and the accumulation of toxic lipids, including ceramides and diglycerides, in the presence of HFD consumption. Nampt overexpression upregulated not only NAD+ but also NADP+ and NADPH in the heart and in cultured cardiomyocytes, which in turn stimulated the GSH and Trx1 systems and alleviated oxidative stress in the heart induced by HFD consumption. In cultured cardiomyocytes, Nampt-induced upregulation of NADPH was abolished in the presence of NADK knockdown, whereas that of NAD+ was not. Nampt overexpression attenuated H2O2-induced oxidative inhibition of Prdx1 and mTOR in an NADK-dependent manner in cultured cardiomyocytes. Nampt overexpression also attenuated H2O2-induced cell death, an effect that was partly abolished by inhibition of NADK, Trx1 or GSH synthesis. In contrast, oxidative stress and the development of diabetic cardiomyopathy in response to HFD consumption were exacerbated in Nampt+/− mice.
Conclusion:
Nampt-mediated production of NAD+ protects against oxidative stress in part through the NADPH-dependent reducing system, thereby alleviating the development of diabetic cardiomyopathy in response to HFD consumption.
Keywords: NAD(H), NADP(H), Nampt, oxidative stress, diabetic cardiomyopathy, Heart Failure, Oxidative Stress, Cell Signaling/Signal Transduction, Obesity, Metabolic Syndrome, Metabolism, Animal Model of Human Disease
Graphical Abstract
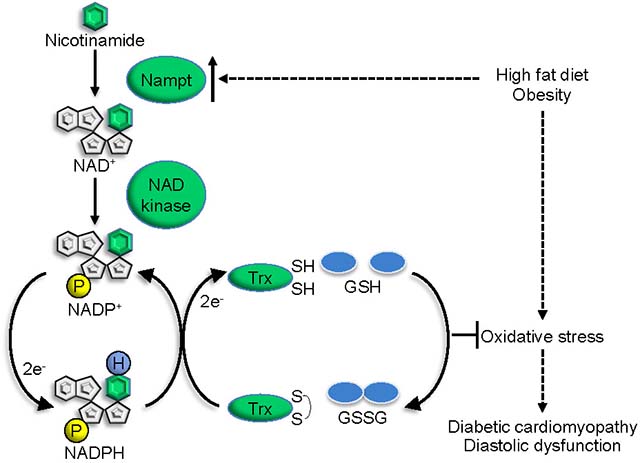
INTRODUCTION
The number of patients with obesity and metabolic syndrome has increased dramatically over the past few decades 1. These patients eventually develop insulin resistance and type II diabetes and more than half of diabetic patients exhibit signs of cardiac dysfunction characterized by cardiac hypertrophy and diastolic dysfunction, termed diabetic cardiomyopathy 2. Although systolic function is often preserved even in the presence of diastolic dysfunction in diabetic cardiomyopathy patients, some patients also develop systolic dysfunction in the long term. Metabolically, the presence of insulin resistance severely attenuates uptake and utilization of glucose for energy; instead, the heart uses fatty acids as its primary energy source 3. Diabetic hearts often exhibit lipotoxicity, where accumulation of toxic lipids, including ceramide and diacylglycerol, induces cardiac dysfunction 4. Diabetic hearts also exhibit increases in oxidative stress and mitochondrial dysfunction. Furthermore, we have shown recently that insufficient activation of mitophagy exacerbates both mitochondrial dysfunction and lipotoxicity 5.
In order to protect against oxidative stress, cells possess antioxidant systems, such as glutathione (GSH), and reducing enzymes, including thioredoxins (Trxs) 6. GSH neutralizes free radicals by donating an electron, which reduces the radicals while oxidizing GSH to form GSSG 7. Trxs reduce proteins with disulfide bonds through thiol disulfide exchange reactions 8. GSH and Trxs also provide electrons to other reducing enzymes, including GSH peroxidases (Gpxs), Glutaredoxins (Grxs), peroxiredoxins (Trx1-dependent peroxidase) and sulfiredoxin 1 (Srxn1), thereby reducing hydrogen peroxide 6. Oxidized GSH and Trxs are recycled to their reduced forms by GSH reductase and Trx reductase, respectively, in the presence of the electron donor, NADPH 9.
NADPH is produced from NADP+ primarily through glucose-6-phosphate dehydrogenase in the pentose phosphate pathway in the cytosol, although it is also produced by NADP-linked isoforms of malic enzyme, isocitrate dehydrogenase and glutamate dehydrogenase in mitochondria 10. NADP+ is generated from NAD+ through NAD kinase (NADK)-mediated phosphorylation 11. Previous work has suggested that the pentose phosphate pathway critically regulates the reduced forms of GSH and Trxs 12, 13. Nicotinamide phosphoribosyltransferase (Nampt), the rate-limiting enzyme in the salvage pathway of NAD+ synthesis, plays a key role in mediating NAD+ synthesis in cardiomyocytes 14. This raises the possibility that Nampt also plays an important role in the production of NADP(H) in the heart. We have shown previously that overexpression of Nampt upregulates NAD+ in the mouse heart 15. However, the extent to which manipulation of NAD+ through Nampt affects NADP(H) production and alleviation of oxidative stress remains unknown.
Thus, we here evaluated the role of Nampt in the regulation of oxidative stress in a mouse model of type II diabetes, the high fat diet (HFD) consumption model. We asked 1) whether Nampt affects the development of diabetic cardiomyopathy, 2) whether Nampt plays an important role in the regulation of NADP(H) and consequent activation of the GSH and Trxs systems, and 3) whether the protective effect of Nampt is mediated through NADP(H) production in the heart.
METHODS
Detailed methods are available in the Online Data Supplement.
Data Availability.
The author declare that all supporting data are available within the article. In addition, any raw data that support the findings of this study are available from the corresponding author upon reasonable request.
Animal Experiments.
Nampt transgenic mice (Tg-Nampt) were generated using the α-myosin heavy chain promoter to achieve cardiac-specific expression of Nampt on an FVB background 15. Non-transgenic mice (NTg) were used as control mice. Systemic Nampt heterozygous knockout mice with a C57BL/6 genetic background were provided by Dr. Yamanaka (Kyoto University, Japan). Numbers of animals used are shown in figure legends. Echocardiographic measurements and PV loop analyses were performed in a blind fashion. No animals were excluded from analyses. All procedures involving animals were performed in accordance with protocols approved by Rutgers Biomedical and Health Sciences.
Diet induced obesity (DIO) mouse model.
Composition of normal diet (ND) and high fat diet (HFD) come from Research Diet Inc. The composition of the diet is shown in Table I in Data Supplement. To induce DIO, 60% HFD or calorie controlled ND was fed to Tg-Nampt or non-transgenic mice. Thionicotinamide, an NADK inhibitor, was intraperitoneally injected into the mice (3 mg/Head, twice a week).
Statistical analysis.
All values in graphs are expressed as the mean ± S.E. Normality was tested with the Shapiro-Wilk normality test. If the data exhibited a normal distribution, pairwise testing was performed with the Student’s t test or multiple group comparisons were performed by 2-way ANOVA, followed by Tukey post-test. If the data failed normality testing or N<6, pairwise testing was performed with the non-parametric Mann-Whitney U test and multiple group comparisons were performed by the non-parametric Kruskal-Wallis test, followed by Dunn’s post-test. Priori power calculations were performed based on data from published studies 5, 16, 17 and pilot experiments. The effect size in this study was 1–5 with an alpha = 0.05 and power = 0.80. Microsoft Excel 2016 was used for Student’s t tests and GraphPad Prism 9 was used for ANOVA and Kruskal-Wallis tests. Precise p-values and N are shown in Table II in Data Supplement. Dot plot was used except where it visually interferes with interpretation (Fig. 1D, 8B and 8F). All quantified Western blot and cell viability data are shown as values relative to control.
Figure 1.
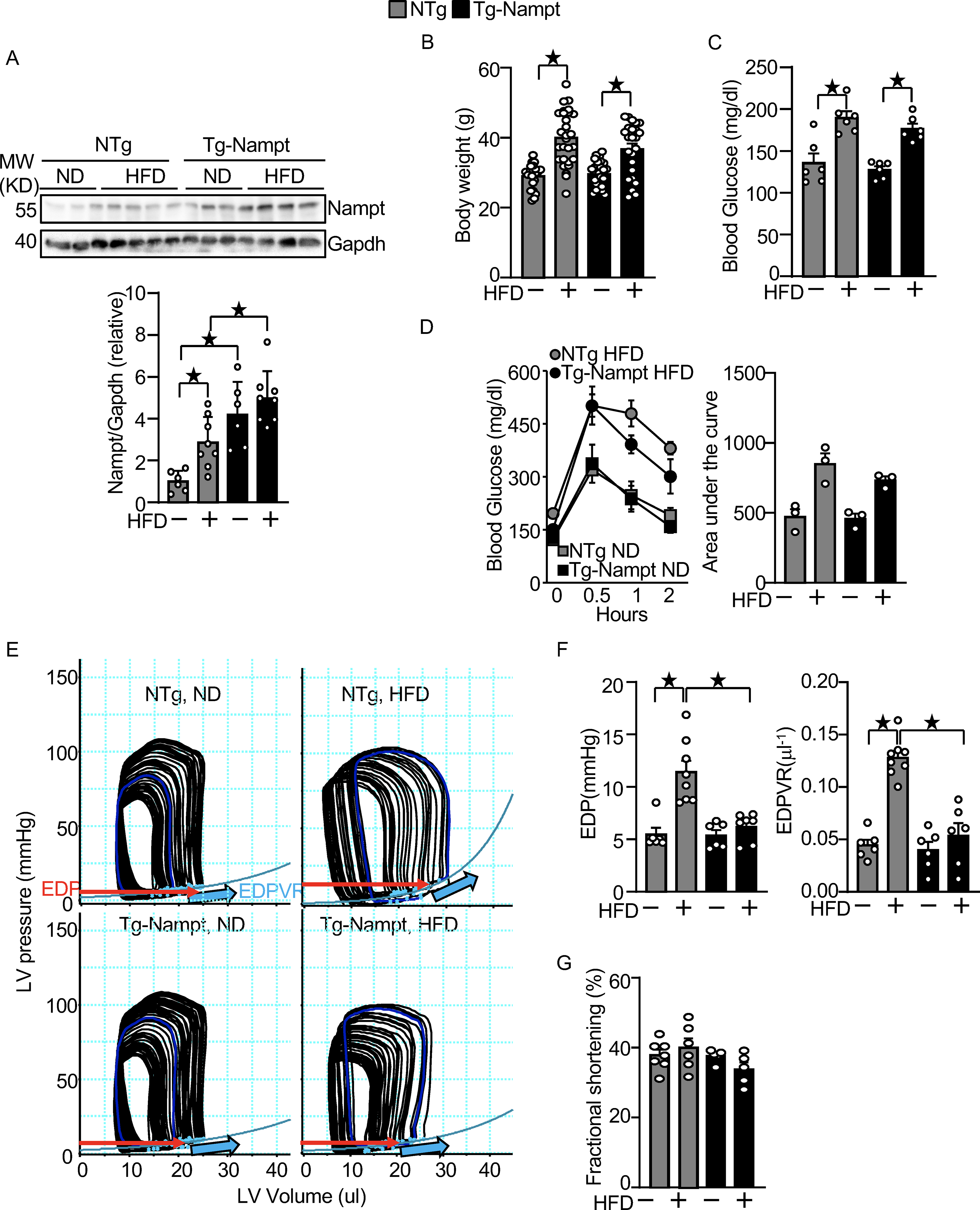
Nampt overexpression ameliorates HFD-induced diastolic dysfunction. (A) HFD consumption upregulates Nampt. Heart lysates were prepared from NTg and Tg-Nampt mice after 3 months of HFD consumption. Western blot analyses were performed with indicated antibodies. (B) Body weights in NTg and Tg-Nampt mice after 3 months of HFD consumption. The mice were fed with HFD beginning at 6–12 weeks of age for 3 months. (C) Blood glucose in NTg and Tg-Nampt mice after 3 months of HFD consumption. (D) Glucose tolerance in NTg and Tg-Nampt mice under ND feeding and 3 months of HFD consumption. (E) Representative PV loop results after 3 months of HFD consumption. (F) Increased EDP and EDPVR were observed after 3 months of HFD consumption in NTg but were normalized in Tg-Nampt mice. (E-F) Mice were anesthetized using pentobarbital (G) LV fractional shortening was preserved in Tg-Nampt mice fed ND and after 3 months of HFD consumption. Statistical significance was determined with ANOVA (A and F (EDPVR)), repeated measures ANOVA (B, C, D (Left) and G) and the Kruskal-Wallis test (D (right) and F (EDP)).
Figure 8.
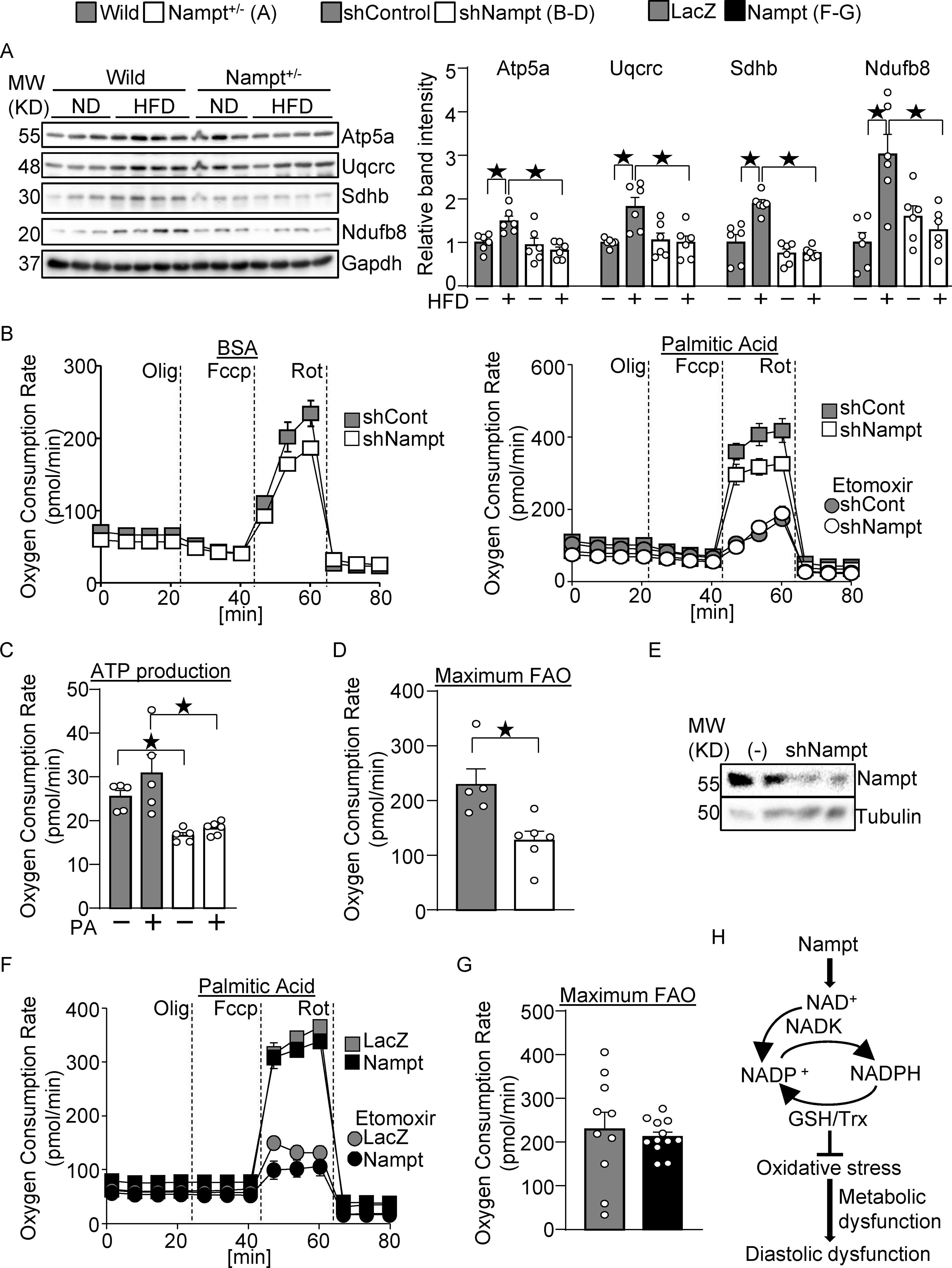
Nampt potentiates metabolic ability. (A) Nampt knockdown suppresses mitochondrial proteins during HFD consumption. Heart lysates were prepared from WT and Nampt+/− mice after 3 months of HFD consumption. Western blot analyses were performed with indicated antibodies. (B-D) Nampt potentiates ATP production and fatty acid oxidation. Cardiomyocytes were transduced with shNampt. Oxygen consumption rate was examined in cardiomyocytes incubated with 100 μmol/L palmitic acid (PA). ATP production coupled oxygen consumption (C) and maximum fatty acid oxidation capacity (D) are shown. (C-D) N=5–6. (E) Western blot showing knockdown of Nampt by adenovirus vector of shNampt. (F-G) Cardiomyocytes were transduced with Ad-Nampt. Oxygen consumption rate was examined in cardiomyocytes incubated with 100 μmol/L palmitic acid (PA). Statistical significance was determined with ANOVA (A), the Kruskal-Wallis test (C), the Mann-Whitney U test (G), and the Student’s t test (G). (H) A schematic representation of the current hypothesis. Nampt promotes NAD production, which promotes NADP production via NADK. Increased NADP(H) confers resistance against oxidative stress, possibly through the GSH and Trx systems, which may in turn attenuate diastolic dysfunction.
RESULTS
Nampt is upregulated in the heart in response to HFD consumption and cardiomyocyte specific upregulation of Nampt protects the heart against the effects of HFD consumption.
We investigated the effect of HFD consumption upon the level of Nampt protein in the mouse heart. HFD consumption for 3 months significantly upregulated Nampt in the heart compared to normal diet (ND) consumption (Fig. 1A). We next investigated whether increasing the level of Nampt in cardiomyocytes protects the heart against the effects of HFD consumption, using transgenic mice with cardiac specific overexpression of Nampt (Tg-Nampt) 15. The level of Nampt was significantly greater in Tg-Nampt than in NTg in both the presence and absence of HFD consumption (Fig. 1A). Non-transgenic (NTg) and Tg-Nampt mice were fed a HFD for 3 months. Body weight and the blood glucose level after 3 months of HFD consumption were not significantly different between NTg and Tg-Nampt mice (Fig. 1B and 1C). Glucose tolerance after 3 months of ND and HFD consumption was also not significantly different between NTg and Tg-Nampt mice (Fig. 1D). Left ventricular (LV) diastolic function was assessed by pressure-volume (PV) loop analyses (Fig. 1E), where diastolic dysfunction is characterized by an increase in the slope of the end-diastolic PV loop relationship (EDPVR) and elevated LV end-diastolic pressure (EDP). HFD consumption significantly increased EDP and EDPVR in NTg mice (Fig. 1E and 1F), indicating that LV diastolic dysfunction is induced by HFD consumption in mice. The increases in EDP and EDPVR in response to HFD consumption were significantly attenuated in Tg-Nampt mice (Fig. 1F). On the other hand, the echocardiographically measured LV fractional shortening (%FS), a measure of LV systolic function, was not significantly affected by 3 months of HFD consumption in either NTg or Tg-Nampt mice (Fig. 1G). Results of the echocardiographic measurements are summarized in Table III in Data Supplement. These results suggest that gain of Nampt function ameliorates diastolic dysfunction induced by HFD consumption.
Gain of Nampt function ameliorates HFD-induced pathological changes relevant to diabetic cardiomyopathy.
Cardiac hypertrophy, fibrosis and inflammation are often observed in diabetic cardiomyopathy hearts 18. HFD consumption induced cardiac hypertrophy, as indicated by increases in LV weight (LVW)/tibia length (TL), in NTg mice, which was attenuated significantly in Tg-Nampt mice (Fig. 2A). Results of the organ weight measurements are summarized in Table IV in Data Supplement. HFD consumption induced increases in cardiomyocyte size in NTg mice but not in Tg-Nampt mice (Fig. 2B). These results suggest that increased expression of Nampt inhibits HFD-induced cardiac hypertrophy. HFD consumption induced significantly more apoptosis in NTg mouse hearts than in Tg-Nampt mouse hearts (Fig. 2C). HFD consumption also significantly induced cardiac fibrosis in NTg mouse hearts but not in Tg-Nampt mouse hearts (Fig. 2D). Fibrosis markers, including collagen 3a1 (Col3a1) and transforming growth factor β1 (TGFβ1), were suppressed in Tg-Nampt hearts in the presence of HFD consumption, further suggesting that Nampt has a protective effect against cardiac fibrosis (Fig. 2E). Since HFD consumption induces inflammation in the heart, we evaluated how Nampt affects inflammatory regulators. HFD consumption significantly activated IκB kinase (IKK) in NTg hearts but not in Tg-Nampt hearts (Fig. 2F). The level of Toll-like receptor 4 (TLR4), a pattern recognition receptor involved in inflammatory responses, was significantly lower in Tg-Nampt hearts than in NTg hearts in both the presence and absence of HFD consumption. HFD consumption, however, did not significantly stimulate other signaling molecules known to be activated by inflammation, including JNK and transforming growth factor-β-activated kinase 1 (TAK1), in either NTg or Tg-Nampt mouse hearts. Taken together, these results suggest that gain of Nampt function ameliorates cardiac hypertrophy, fibrosis and inflammation in response to HFD consumption.
Figure 2.
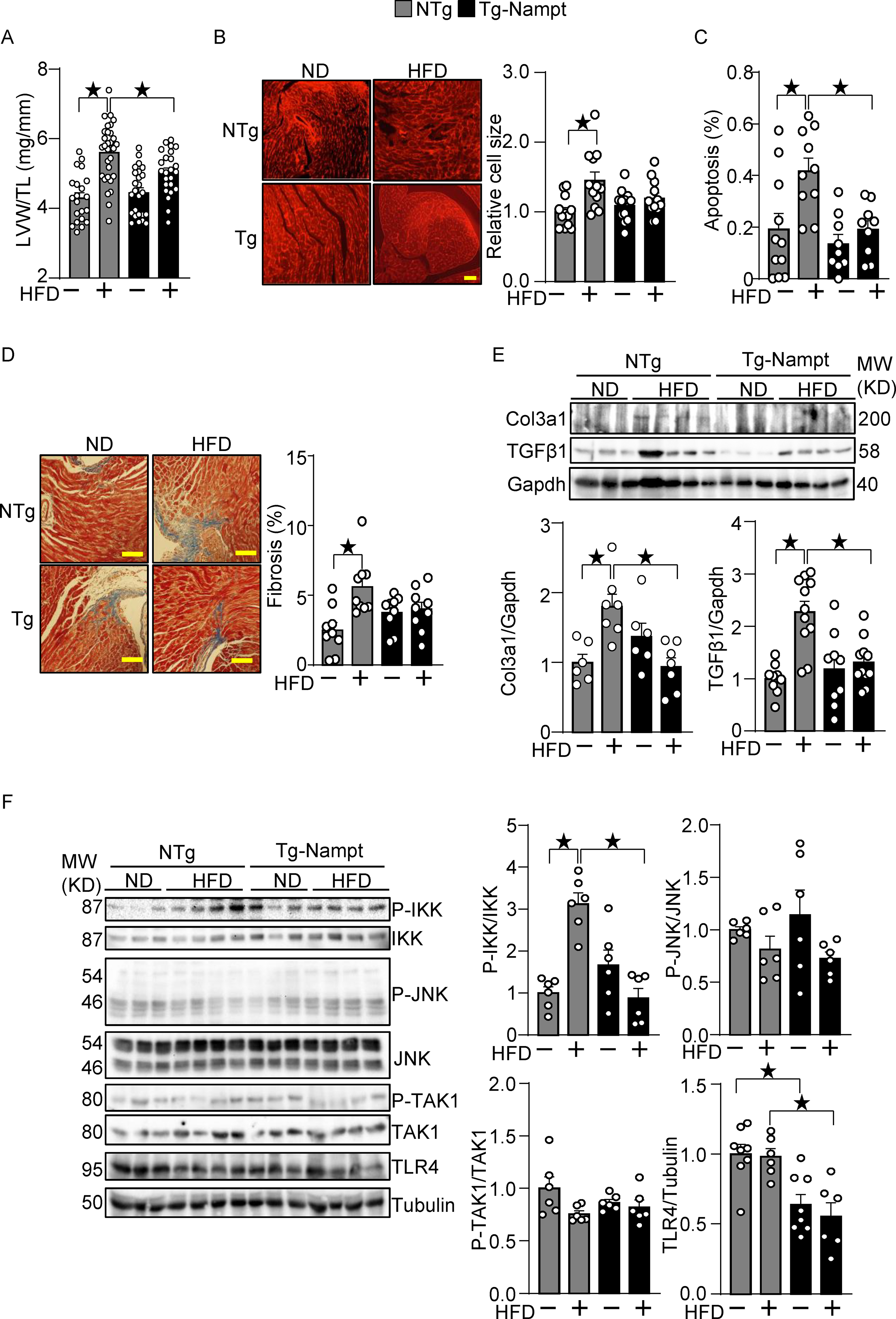
Nampt overexpression ameliorates HFD-induced cardiac pathologies relevant to diastolic dysfunction. (A-B) Nampt ameliorates HFD-induced left ventricular (LV) (A) and cardiomyocyte (B) hypertrophy. (A) LV hypertrophy indicated by LV weight (LVW)/tibial length (TL). (B) Relative cell size was evaluated, using wheat germ agglutinin (WGA) staining. (C) Nampt ameliorates HFD-induced apoptotic cell death. TUNEL-positive cells (apoptotic cells) were quantified as the number of TUNEL-positive/total number of nuclei (%). (D) Nampt ameliorates HFD-induced cardiac fibrosis. Cardiac fibrosis was evaluated using Masson’s trichrome staining. (Left) Representative images from each group are shown. Scale bars, 100 μm. (Right) Quantitative analysis of the fibrotic areas. (E) Nampt inhibits HFD-induced fibrosis markers. (F) Nampt inhibits HFD-induced IKK activation. Heart lysates were prepared from NTg and Tg-Nampt mice after 3 months of HFD feeding. Indicated pro-inflammatory signal regulators were examined with Western blot analyses. Statistical significance was determined with ANOVA (A, B, C, E and F (P-JNK/JNK, P-TAK1/TAK1 and TLR4/Tubulin)) and the Kruskal-Wallis test (D, F (P-IKK/IKK)).
Overexpression of Nampt promotes production of NADP+ and NADPH in the heart.
Nampt plays an essential role in generating NAD+ in the heart. The levels of NAD+, NADH, and NAD++NADH were significantly elevated in Tg-Nampt mouse hearts compared to in NTg mouse hearts in the presence and absence of HFD consumption (Fig. 3A). The NAD+/NADH ratio was not significantly different between NTg and Tg-Nampt mouse hearts. The levels of NADP+, NADPH, and NADP++ NADPH were also all elevated in Tg-Nampt mouse hearts in both the presence and absence of HFD consumption (Fig. 3B). The NADPH/NADP+ ratio was significantly greater in Tg-Nampt mouse hearts than in NTg mouse hearts in the presence of HFD consumption. The levels of NAD+ kinase (NADK), an enzyme that catalyzes NAD+ phosphorylation, and glucose-6-phosphate dehydrogenase (G6PD), the rate limiting enzyme for the pentose phosphate pathway, in the heart did not differ significantly between Tg-Nampt and NTg in either the presence or absence of HFD consumption (Fig. 3C). To test whether NADK mediates Nampt-induced protective effects against diastolic dysfunction, Tg-Nampt mice were treated with an NADK inhibitor, thionicotinamide 19. In order to clarify the role of NADK in the early phase of diastolic heart failure development, we performed PV loop analyses after 1 month of HFD consumption. HFD-induced diastolic dysfunction was inhibited in Tg-Nampt mice, an effect that was abolished in the presence of the NADK inhibitor (Fig. 3D). In addition, heart weight/tibia length was significantly reduced in Tg-Nampt compared to in NTg mice in response to HFD consumption, an effect that was reversed in the presence of the NADK inhibitor (Fig. 3E). As shown in Fig. 3F, we verified that Nampt-induced NADP and NADPH upregulation was partly abolished with the NADK inhibitor. In contrast, Nampt-induced NAD and NADH upregulation was not significantly changed by the NADK inhibitor. These results suggest that Nampt protects against HFD-induced diastolic dysfunction and cardiac hypertrophy partly through NADK-dependent NADP(H) production.
Figure 3.
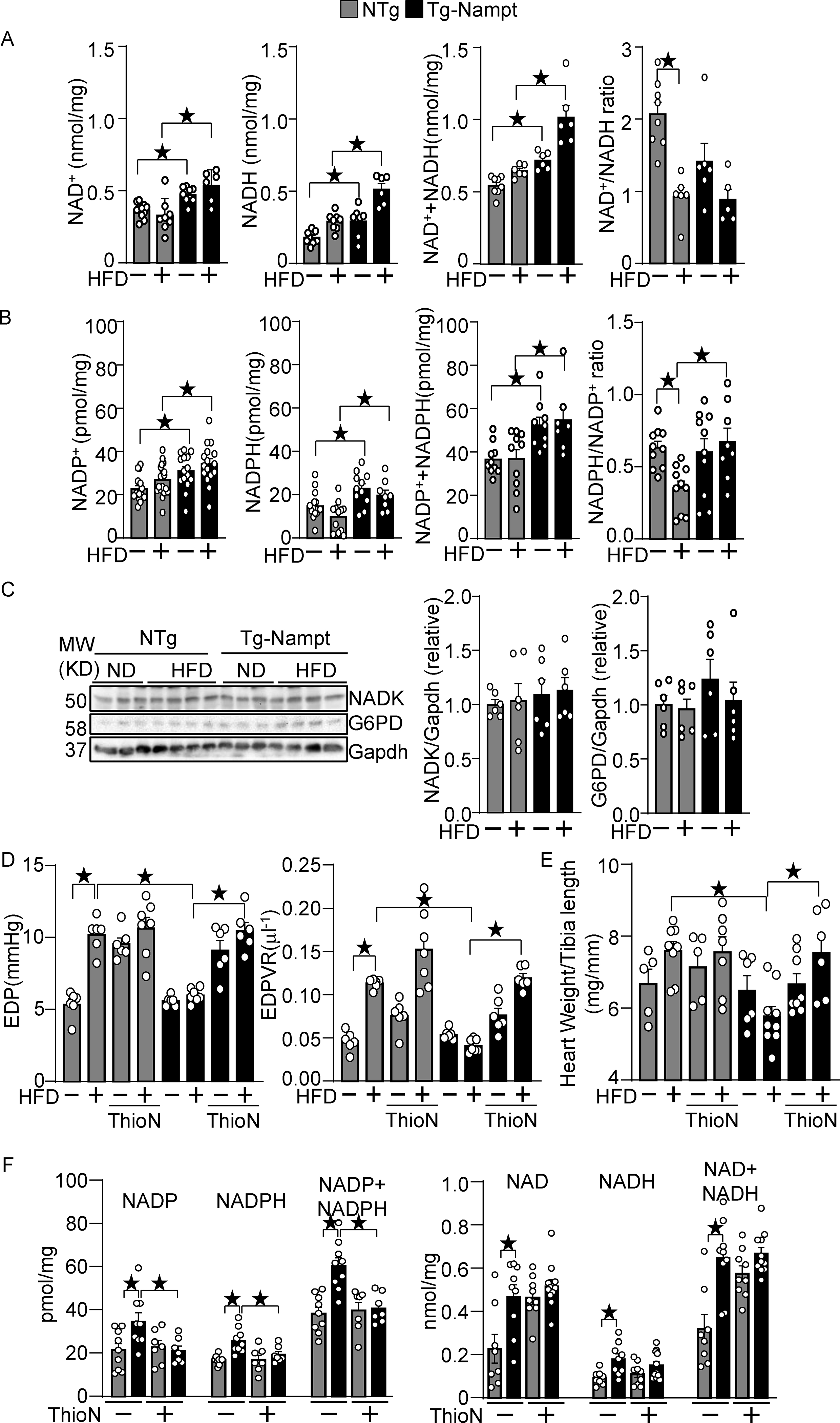
Nampt overexpression upregulates NAD(H) and NADP(H). (A) The levels of NAD+, NADH and total NAD++NADH were examined in Tg-Nampt mice after either normal diet or HFD consumption. (B) The levels of NADP+, NADPH, and total NADP++NADPH and the NADPH/NADP+ ratio were examined in Tg-Nampt mice under HFD feeding conditions. (C) Neither Nampt overexpression nor HFD significantly affects the level of NADK or G6PD (D) Nampt overexpression attenuates HFD-induced diastolic dysfunction in an NADK-dependent manner. Mice were anesthetized with Avertin. (E) Nampt overexpression attenuates HFD-induced cardiac hypertrophy in an NADK-dependent manner. (F) ThioN, an NADK inhibitor, inhibits Nampt-induced NADP(H), but not NAD(H) production. Statistical significance was determined with ANOVA (A-F).
Gain of Nampt function potentiates the GSH and Trx1 systems.
Increased production of NADPH, an electron donor, leads to activation of the glutathione (GSH) and thioredoxin (Trx) systems 12, 13. The level of GSH was significantly higher in Tg-Nampt mouse hearts than in NTg mouse hearts in the presence of HFD consumption (Fig. 4A). There was no significant difference in the level of GSSG between Tg-Nampt and NTg mice with or without HFD consumption (Fig. 4A). The GSH/GSSG+ ratio was significantly greater in Tg-Nampt mouse hearts than in NTg mouse hearts in the presence of HFD consumption. (Fig. 4A). These results suggest that overexpression of Nampt rescues the inactivation of the GSH system during HFD consumption. To investigate whether gain of Nampt function potentiates the Trx system, we examined sulfonation (-SO3H) of Prdx1, a major Trx1 substrate, at catalytic cysteine residues, which is an irreversible oxidative modification that inhibits Prdx1. HFD consumption significantly increased sulfonation of Prdx1 in the heart, but the level of Prdx1 sulfonation in the presence of HFD consumption was significantly lower in Tg-Nampt mouse hearts than in NTg mouse hearts (Fig. 4BC).
Figure 4.
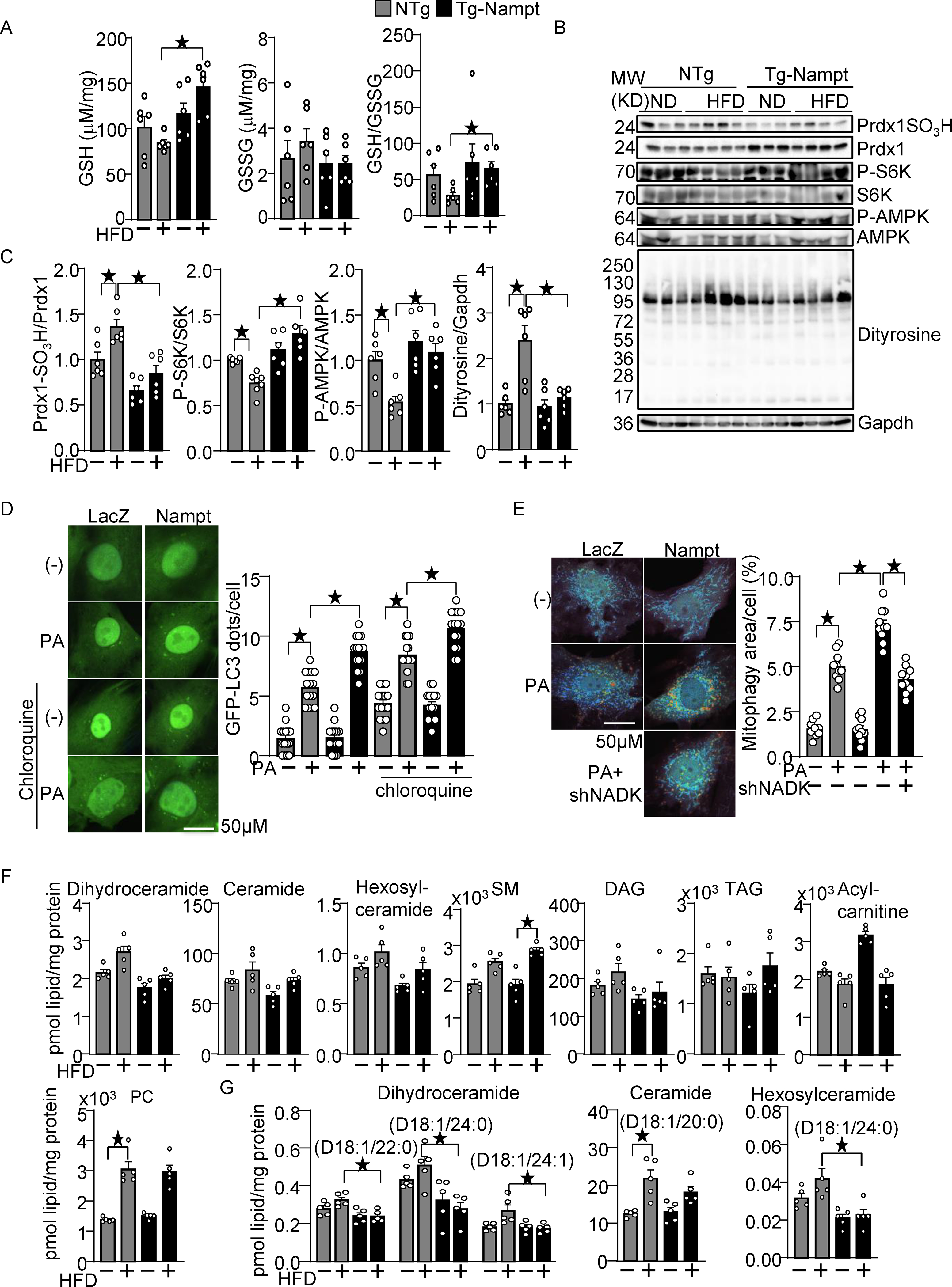
Nampt overexpression attenuates HFD-induced oxidative stress. (A) Nampt overexpression attenuates HFD-induced GSH oxidation. The levels of GSH and GSSG and the GSH/GSSG ratio were examined in Tg-Nampt mice after 3 months of HFD consumption. (B-C) Nampt overexpression attenuates HFD-induced inhibition of Trx1 substrates. Heart lysates were prepared from NTg and Tg-Nampt mice after 3 months of HFD consumption. Western blot analyses were performed with indicated antibodies. (D) Nampt promotes palmitic acid (PA)-induced autophagy. After 2 days of adenovirus transduction of Nampt and GFP-LC3, cardiomyocytes were treated with 100 μM PA and 10 μM chloroquine for 4 hours. GFP-LC3 dots per cell were counted. (E) Nampt promotes PA-induced mitophagy in an NADK-dependent manner. After 3 days of adenovirus transduction of Nampt and Mito-Keima, NADK was knocked down with shNADK. After 3 days, the cardiomyocytes were treated with 100 μmol/L PA for 24 hours. Areas with high 561/457 nm ratios, indicating mitophagy, were measured. (F-G) The effect of Nampt overexpression upon myocardial contents of various lipid species in the presence or absence of HFD consumption. (F) The effect upon the total amount of indicated lipid species. (G) The effect upon several specific subspecies of dihydroceramide, ceramide and hexosylceramide. Statistical significance was determined with ANOVA (A (GSH and GSSG), and C-E) and the Kruskal-Wallis test (A (GSH/GSSG), F and G.
We have shown previously that Trx1 plays an essential role in reducing critical cysteine residues in AMPKα and mTOR, thereby maintaining their activity 20, 21. HFD consumption significantly decreased the level of Thr172 phosphorylated AMPK/total AMPK and Thr389 phosphorylated S6/total S6, indicators of AMPK and mTOR activation respectively, but phosphorylation of AMPK and p70S6K was significantly higher in Tg-Nampt than in NTg in the presence of HFD consumption (Fig. 4BC). These results suggest that gain of Nampt function potentiates the reducing activity of Trx1, thereby rescuing the decreases in the activity of AMPK and mTOR in the presence of HFD consumption. To further investigate whether oxidative stress in the heart is alleviated in Tg-Nampt mice, we evaluated the level of dityrosine, a protein oxidative adduct. The level of dityrosine was elevated in NTg mouse hearts in response to HFD consumption but was significantly lower in Tg-Nampt mouse hearts than in NTg mouse hearts in the presence of HFD consumption (Fig. 4BC).
AMPK promotes autophagy, whereas mTOR inhibits it 22. Since Nampt promotes both AMPK and mTOR activity, we evaluated whether gain of Nampt function promotes or inhibits autophagy and mitophagy. To this end, we expressed GFP-LC3 and Mito-Keima with adenovirus vectors in cultured cardiomyocytes. PA-induced autophagy, indicated by increases in GFP-LC3 dots, was significantly promoted by Nampt overexpression in the presence of chloroquine, a lysosomal inhibitor (Fig. 4D). In addition, PA-induced increases in mitophagy, evidenced by a shift in Mito-Keima excitation to higher wavelengths indicating acidification due to fusion of mitochondria-containing autophagosomes with lysosomes, were significantly enhanced by Nampt. However, this effect was partly inhibited by NADK knockdown with shNADK, suggesting that Nampt potentiates mitophagy in the presence of a fatty acid partly through an NADK dependent mechanism (Fig. 4E).
Myocardial accumulation of toxic lipids, termed lipotoxicity, is an important feature of diabetic cardiomyopathy23. To investigate whether the protective effect of Nampt during HFD consumption is associated with altered accumulation of toxic lipids, we performed lipidomics analyses of ventricular tissues obtained from NTg and Tg-Nampt mice that had been fed either ND or HFD. Using LC-MS, we analyzed myocardial contents of various lipid species (Table V in Data Supplement), including glycerophosphocholines (PCs), sphingomyelins (SMs), triacylglycerols (TAGs), acylcarnitines, ceramides, and diacylglycerols (DAGs). Of note, the amount of toxic lipids24, including ceramides and DAGs, was not significantly different between NTg and Tg-Nampt mice under HFD consumption conditions (Fig. 4F). However, specific lipid species, including dihydroceramide species D18:1/22:0, D18:1/24:0 and D18:1/24:1, ceramide species D18:1/20:0 and hexosylceramide species D18:1/24:0, were significantly lower in Tg-Nampt mice than in NTg mice under HFD consumption conditions (Fig. 4G). These results suggest that a gain of Nampt function downregulates accumulation of toxic lipids. Taken together, these results indicate that Nampt potentiates the GSH and Trx1 systems, autophagy and mitophagy, attenuates oxidative stress, and prevents accumulation of toxic lipid derivatives in the heart in the presence of HFD consumption in vivo.
Nampt promotes NADP(H) production and protects cardiomyocytes against oxidative stress in an NADK-dependent manner.
We next investigated the role of endogenous NADK in mediating the protective effect of Nampt overexpression in cardiomyocytes. Knockdown of NADK with short hairpin RNA (shNADK), either with or without overexpression of Nampt, did not affect the level of NAD+ (Fig. 5A). However, increased production of NADPH in the presence of Nampt overexpression was abolished in the presence of shNADK (Fig. 5B). These results suggest that NADK plays an essential role in mediating the upregulation of NADPH in the presence of Nampt overexpression. Notably, the level of NADP was not significantly altered even in the presence of Nampt overexpression in either the presence or absence of shNADK, suggesting that NADP produced by NADK is instantly converted to NADPH in cultured cardiomyocytes. Taken together, these results suggest that Nampt-induced NAD+ production promotes NADP(H) production in an NADK-dependent manner.
Figure 5.
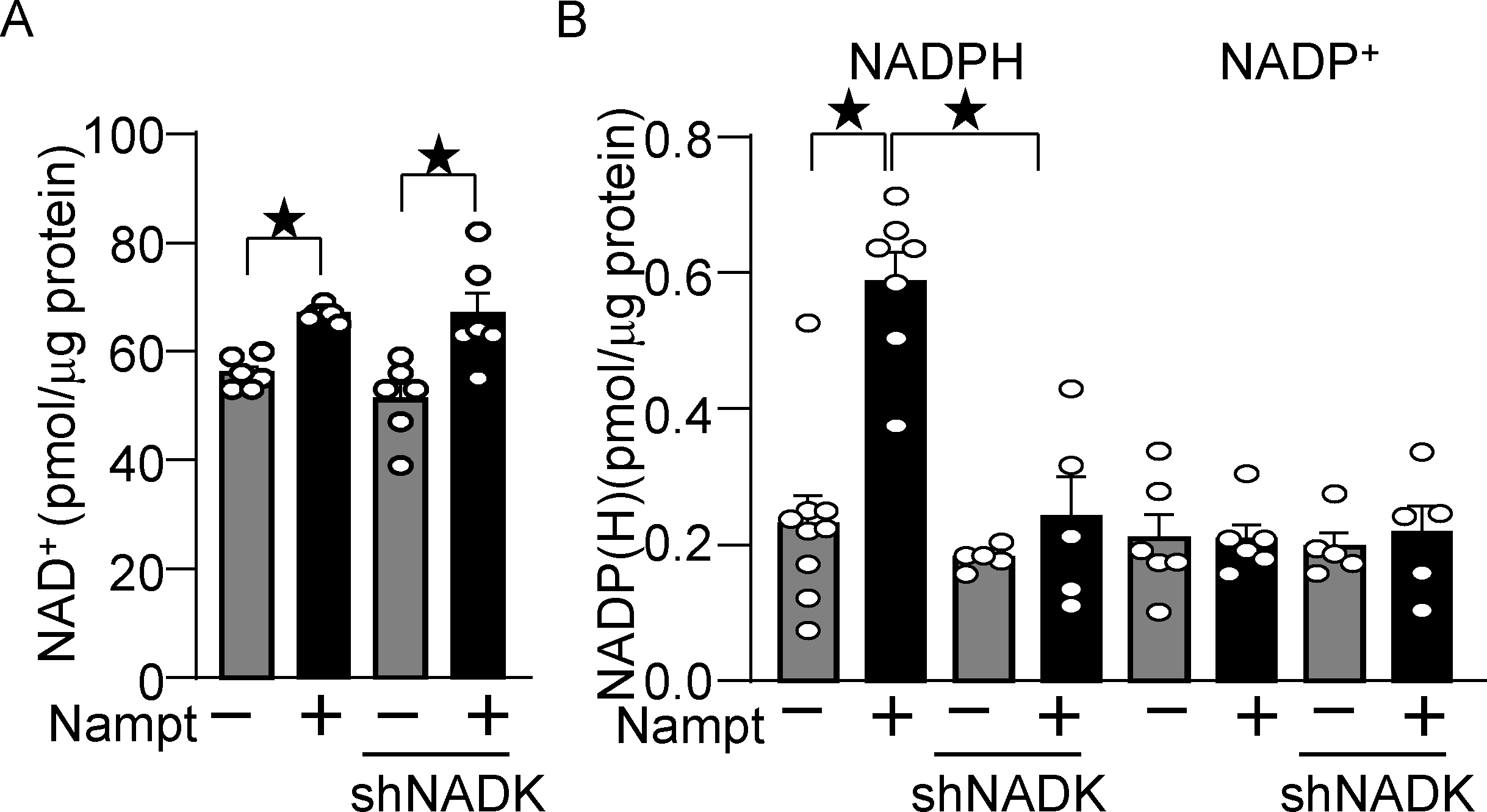
Nampt overexpression upregulates NADP(H) in an NADK-dependent manner. (A) Nampt upregulates NAD+, which is not affected by NADK knockdown. Statistical significance was determined with ANOVA. (B) Nampt upregulates NADPH in an NADK-dependent manner. After 1 day of adenovirus vector (Nampt) transduction, shNADK was transfected into cardiomyocytes. After 2 days of adenovirus transduction, the levels of NAD+, NADP+ and NADPH were examined. Statistical significance was determined with ANOVA (A and B (NADP)) and the Kruskal-Wallis test (B (NADPH)).
We next investigated whether Nampt protects against oxidative stress through NADPH production. As shown in Fig. 6A, treatment of cardiomyocytes with H2O2 induced Prdx1 sulfonation, which was inhibited in the presence of Nampt overexpression. The suppression of H2O2-induced sulfonation of Prdx1 was reversed in the presence NADK knockdown (Fig. 6A), suggesting that NADK and the consequent production of NADP+ and NADPH play an important role in mediating the protective effect of Nampt against Prdx1 sulfonation. We have shown previously that AMPK and mTOR are oxidized and form intermolecular disulfide bonds in the presence of oxidative stress 20, 21. H2O2 treatment induced band shifts of AMPKα and mTOR to higher molecular weights under non-reducing conditions, which were inhibited in the presence of Nampt overexpression in cardiomyocytes. Nampt-induced alleviation of AMPK and mTOR oxidation was reversed in the presence of NADK knockdown (Fig. 6B). H2O2 treatment inhibited phosphorylation of mTOR, an effect that was alleviated in the presence of Nampt overexpression in cardiomyocytes. The protective effect of Nampt in the presence of oxidative stress was reversed in the presence of NADK knockdown (Fig. 6C). Phosphorylation of S6K, an mTOR substrate, in the presence of oxidative stress was higher in the presence of Nampt overexpression, an effect that was abolished with NADK knockdown (Fig. 6C). As shown in Fig. 6D, H2O2 treatment induced death in cardiomyocytes, which was attenuated in the presence of Nampt overexpression. Nampt-induced suppression of H2O2-induced cell death was reversed in the presence of NADK knockdown. The protective effects of Nampt were also abolished in the presence of buthionine sulfoximine (BSO), an inhibitor of GSH synthesis, or downregulation of Trx1. These results suggest that Nampt confers resistance against oxidative stress to cardiomyocytes through NADK-dependent mechanisms and consequent activation of NADPH-dependent antioxidant systems, including the GSH and Trx1 systems. Besides the NADK-dependent mechanism, sirtuins, NAD+-dependent enzymes, may mediate the protective effect of Nampt against oxidative stress. To test this possibility, we knocked down sirtuins, including Sirt1 and Sirt3. Knockdown of Sirt1 abolished the protective effect of Nampt, whereas that of Sirt3 promoted the protective effect (Fig. 6E). Thus, Sirt1 mediates, whereas Sirt3 inhibits, the protective effect of Nampt. Knockdown of Trx1, Sirt1 and Sirt3 proteins was verified (Fig. 6F). Interestingly, knockdown of Sirt1 downregulated Trx1, whereas that of Sirt3 upregulated Trx1. Conversely, knockdown of Trx1 upregulated Sirt1, most likely due to compensatory mechanisms. Thus, Trx1 may also mediate Sirt1- and loss of Sirt3-induced protective effects. Taken together, both the NADK-dependent anti-oxidant system and Sirt1 plays a crucial role in Nampt-induced stress resistance.
Figure 6.
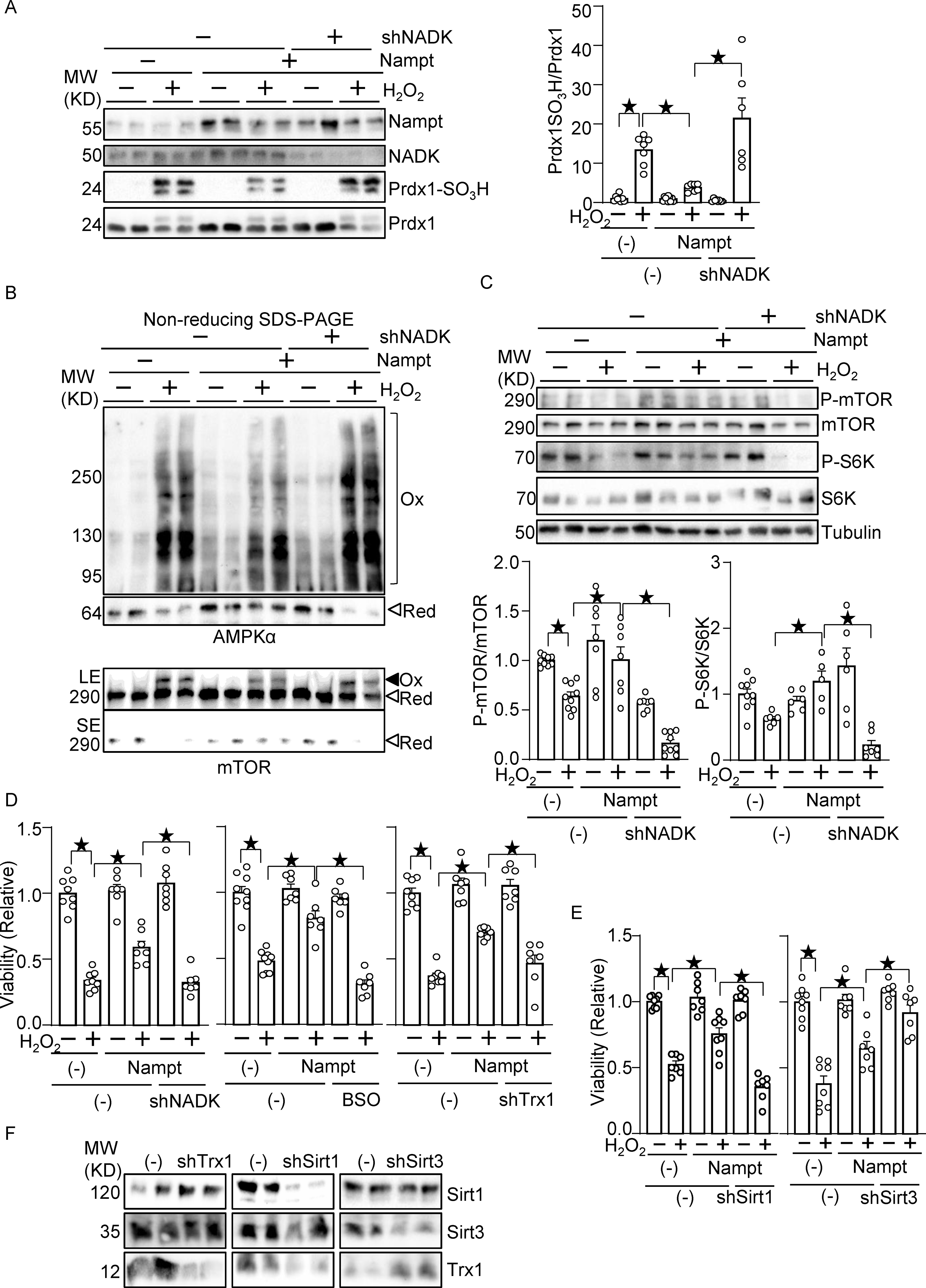
Nampt alleviates oxidative stress in an NADK-dependent manner. (A) Nampt prevents H2O2-induced Prdx1 oxidation in an NADK-dependent manner. (B) Nampt prevents H2O2-induced intermolecular disulfide bond formation in AMPKα and mTOR in an NADK-dependent manner (Ox: oxidized, Red: reduced, LE: long exposure, SE: short exposure). (C) Nampt prevents H2O2-induced mTOR inhibition in an NADK-dependent manner. After 1 day of adenovirus vector (Nampt) transduction, shNADK was transfected into cardiomyocytes. After 1 or 2 days of transfection, cells were treated with 100 μM H2O2 for 30 minutes. Western blot analyses were performed following SDS-PAGE under reducing (A and C) and non-reducing (B) conditions using indicated antibodies. (D) Nampt prevents H2O2-induced myocyte cell death, an effect that is partly abolished by inhibition of NADK, GSH synthesis or Trx1. (Left) After adenovirus transduction and shRNA transfection as described above, cells were treated with 100 μmol/L H2O2 for 6 hours. Cell viability was assessed by trypan blue dye exclusion. (Middle) Cells were treated with buthionine sulfoximine (BSO, 100 μmol/L), a GSH synthesis inhibitor, for 16 hours prior to H2O2 treatment. (Right) Adenovirus vector harboring shTrx1 was transduced together with that for Nampt. (E) Sirt1, but not Sirt3, mediates the protective effects of Nampt against H2O2-induced myocyte cell death. After adenovirus transduction and shRNA transfection, cells were treated with 100 μmol/L H2O2 for 6 hours. Cell viability was assessed by trypan blue dye exclusion. (F) Western blot showing knockdown of Trx1, Sirt1 and Sirt3. Statistical significance was determined with ANOVA (A-E).
Nampt is upregulated in the heart in response to HFD consumption, thereby maintaining the cellular redox status and protecting against the development of diastolic dysfunction.
In order to investigate the role of endogenous Nampt in the heart during HFD consumption, we used systemic Nampt heterozygous knockout (Nampt+/−) mice. Due to embryonic lethality 25, Nampt homozygous knockout mice could not be used for this investigation. As reported previously, Nampt+/− mice showed a lean phenotype under ND feeding conditions 16, but the body weight of Nampt+/− mice did not differ significantly from that of wild type (WT) mice after 3 months of HFD consumption (Fig. 7A). The blood glucose levels of Nampt+/− mice and WT mice also did not differ significantly after 3 months of HFD consumption (Fig. 7B). As expected, upregulation of Nampt in response to HFD consumption was abolished in Nampt+/− mice (Fig. 7C). HFD consumption for 3 months induced diastolic dysfunction in WT mice, as indicated by significant elevations in EDP and the slope of the EDPVR, which was exacerbated in Nampt+/− mice (Fig. 7D). Cardiac systolic function, evaluated via echocardiographically measured %FS, was not significantly different between WT and Nampt+/− mice, with or without HFD consumption (Table VI in Data Supplement). Data obtained by echocardiography are summarized in Table VI in Data Supplement. These results suggest that endogenous Nampt plays an important role in mediating diastolic dysfunction in the heart in response to HFD consumption. HFD consumption significantly increased the LVW/TL ratio in both WT and Nampt+/− mice but LVW/TL did not differ significantly between Nampt+/− and WT mice (Table VII in Data Supplement). Results of organ weight measurements are summarized in Table VII in Data Supplement.
Figure 7.
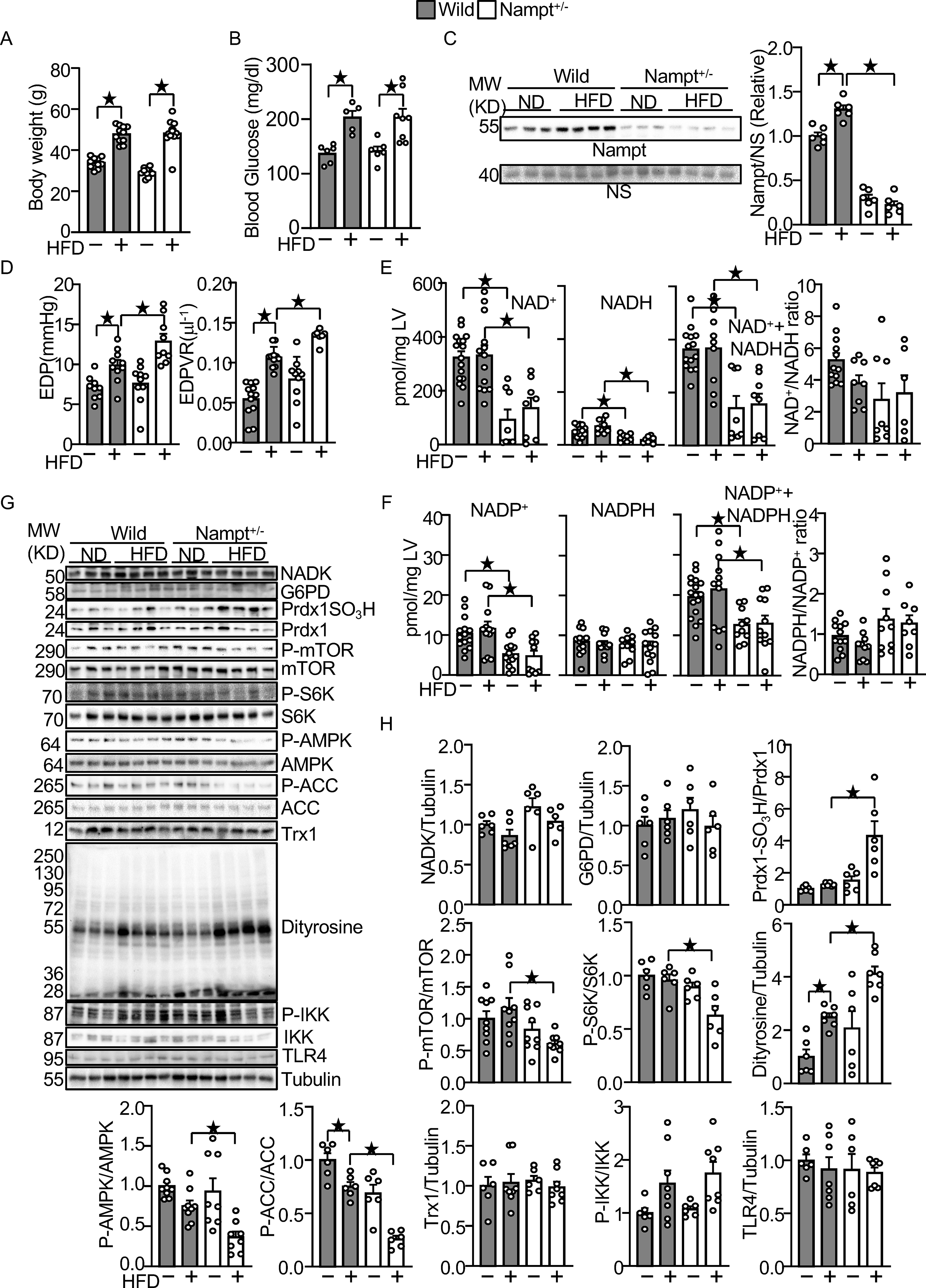
Endogenous Nampt prevents HFD-induced diastolic dysfunction and oxidative stress.
(A) Body weights in wild type and Nampt+/− mice under HFD feeding conditions. (B) Blood glucose in wild type and Nampt+/− mice under HFD feeding conditions (C) HFD-induced Nampt expression is inhibited in Nampt+/− mice. NS: Non-specific band. (D) HFD-induced diastolic dysfunction is exacerbated in Nampt+/− mice. (E) Endogenous Nampt maintains NAD(H). (F) Endogenous Nampt maintains NADP. (G) Neither Nampt knockdown nor HFD significantly affects the level of NADK and G6PD. (H) Nampt knockdown promotes HFD-induced inhibition of Trx1 substrates. Heart lysates were prepared from WT and Nampt+/− mice after 3 months of HFD consumption. Western blot analyses were performed with indicated antibodies. Statistical significance was determined with ANOVA (C, E (NADH), F (NADPH, NADP+NADPH, NADPH/NADP), G, H (NADK/Tubulin, G6PD/Tubulin, PrdxSO3H/Prdx1, P-mTOR/mTOR. P-S6K/S6K, P-AMPKα/AMPKα, P-ACC/ACC, Dityrosine/Tubulin and P-IKK/IKK), repeated measures ANOVA (A, B) and the Kruskal-Wallis test (D, E (NAD, NAD+NADH, NAD/NADH), F (NADP) and H (Trx1/Tubulin and TLR4/Tubulin).
The levels of NAD+, NADH, and NAD++NADH were significantly lower in Nampt+/− mice after both ND and HFD consumption (Fig. 7E). The NAD+/NADH ratio was not significantly different between Nampt+/− and WT mice in either the presence or absence of HFD consumption. The levels of NADP+ and NADP++NADPH were also significantly lower in Nampt+/− mice after both ND and HFD consumption (Fig. 7F). However, the level of NADPH and the NADPH/NADP+ ratio did not differ significantly between Nampt+/− and WT mice in either the presence or absence of HFD consumption (Fig. 7F). These results suggest that endogenous Nampt plays an important role in mediating NAD+ and NADP+ production in the heart in both the presence and absence of HFD consumption. Protein levels of NADK and G6PD in the heart did not differ significantly between Nampt+/− and WT mice in the presence or absence of HFD consumption. (Fig. 7GH). We also evaluated how the haploinsufficiency of Nampt affects downstream effectors of Trx1 and GSH during HFD consumption. The level of sulfonated Prdx1 was significantly higher in Nampt+/− mice than in WT mice in the presence of HFD consumption (Fig. 7GH). The activity of mTOR, as indicated by phosphorylation of mTOR and S6K, was significantly lower in Nampt+/− mice than in WT mice in the presence of HFD consumption (Fig. 7GH). The activity of AMPK, as indicated by phosphorylation of AMPK and ACC, a substrate of AMPK, was also significantly lower in Nampt+/− mice than in WT mice in the presence of HFD consumption (Fig. 7GH). The levels of sulfonated Prdx1 and dityrosine were significantly increased in Nampt+/− mice (Fig. 7GH). In contrast, the levels of Trx1, phosphorylated-IKK and TLR4 were not significantly changed (Fig. 7GH). These results suggest that endogenous Nampt plays an essential role in maintaining cardiac diastolic function and redox homeostasis by maintaining the activity of the Trx1 system in the presence of HFD consumption.
Nampt potentiates metabolic ability.
To investigate the role of endogenous Nampt in metabolic ability, we examined expression of mitochondrial proteins, and triglyceride and ATP contents. The expression of mitochondrial proteins, including Atp5a, Uqcrc, Sdhb, and Ndufb8, was significantly lower in Nampt+/− mice than in WT mice in the presence of HFD consumption (Fig. 8A). To investigate whether endogenous Nampt promotes fatty acid oxidation, oxygen consumption rate was examined in cardiomyocytes transduced with shNampt adenovirus vector and treated with PA (Fig. 8B). Knockdown of Nampt inhibited ATP-production coupled oxygen consumption in both the presence and absence of PA, suggesting that Nampt maintains ATP production (Fig. 8C). Maximum fatty acid oxidation ability, characterized by the etomoxir-, a fatty acid oxidation inhibitor, sensitive oxygen consumption rate, was inhibited by Nampt knockdown (Fig. 8D). Knockdown of Nampt was verified in cultured cardiomyocytes (Fig. 8E). In contrast, maximum fatty acid oxidation ability was not significantly changed in Nampt-overexpressing cardiomyocytes (Fig. 8F–G). Thus, Nampt is essential for maintenance of fatty acid oxidation, but gain of Nampt function does not significantly affect it. Taken together, these results suggest that endogenous Nampt plays a role in maintaining cardiac energy homeostasis, including expression of mitochondrial proteins, ATP production and fatty acid oxidation.
DISCUSSION
Our results suggest that Nampt, the rate limiting enzyme in the NAD+ salvage pathway, inhibits the development of diastolic dysfunction, hypertrophy and fibrosis in the heart in response to HFD consumption. Endogenous Nampt is upregulated in the heart during HFD consumption and downregulation of endogenous Nampt exacerbates cardiomyopathy, indicating that upregulation of Nampt in response to HFD consumption is an adaptive mechanism to prevent the development of diabetic cardiomyopathy. Nampt increases NADPH in an NADK-dependent manner, thereby stimulating the GSH and Trx1 systems and alleviating oxidative stress (Fig. 8H). Importantly, gain of Nampt function inhibits HFD-induced diastolic dysfunction, whereas the protective effect is abolished with NADK inhibition. Since increased oxidative stress is a major driving factor for the development of diabetic cardiomyopathy 26, we propose that Nampt protects the heart against diabetic cardiomyopathy in part by alleviating oxidative stress through an increased supply of NADPH to the GSH and Trx1 systems. The metabolic rigidity in diabetic hearts, characterized by increased fatty acid uptake and mitochondrial oxidation, leads to decreases in the NAD+/NADH ratio, due to increased production of NADH and increased production of acetyl-CoA. This, in turn, leads to, suppression of sirtuins, and increases in protein acetylation, which potentially lead to mitochondrial dysfunction in the heart 27.
Gain of Nampt function reduced the accumulation of some subspecies of dihydroceramide, ceramide and hexosylceramide (Fig. 4G). Conversely, loss of Nampt function limited the myocardial capacity for fatty acid oxidation and mitochondrial respiration, in association with downregulation of enzymes that are involved in mitochondrial energy metabolism (Fig. 8A–D). Thus, endogenous Nampt maintains fatty acid oxidation partly through mitochondrial respiration, which presumably prevents accumulation of toxic lipids in the heart. Since maintaining the NAD(H) level is essential for proper mitochondrial function, our data suggest that Nampt determines mitochondrial respiration capacity via NAD+ production. In addition, maintenance of AMPK and mTOR function via the NADP(H)-Trx1 pathway may also play a role in maintenance of mitochondria function.
Increased oxidative stress also activates nuclear poly (ADP-ribose) polymerase (PARP), a DNA repair enzyme that consumes cellular NAD+ 28. Previous studies have shown that diabetic cardiomyopathy is accompanied by decreases in NAD+/NADH, and restoring the level of NAD+/NADH has been proposed as an effective treatment for diabetic cardiomyopathy 27. Here we show that HFD consumption is accompanied by increases in endogenous Nampt and that exogenous Nampt expression alleviates the development of diabetic cardiomyopathy in response to HFD consumption. These results suggest that upregulation of Nampt prevents the pathogenesis of diabetic cardiomyopathy. Our results provide a basis for the development of a treatment for diabetic cardiomyopathy based on stimulation of NAD+ synthesis through stimulation of Nampt, such as with a small molecule activator of Nampt 29.
Although NADP+ is produced from NAD+ in the presence of NADK, how NAD+ is supplied has not been clearly shown in the heart and cardiomyocytes therein. Using genetically altered mouse models, our study clearly shows that NAD+ produced through the salvage pathway, including Nampt, couples to the production of NADP(H). Downregulation of endogenous Nampt in the presence of HFD consumption was accompanied by decreases in NAD+ and NADP+, whereas the rescue of Nampt by transgenic overexpression restored the levels of NAD+, NADP+ and NADPH, suggesting that endogenous Nampt is a critical regulator of NADP(H) during diabetic cardiomyopathy. Since NADPH is a major donor of electrons for the GSH and Trx systems, which in turn inhibit protein oxidation directly or reduce the level of H2O2 through reduction of GSH peroxidases and peroxiredoxins, our study indicates that Nampt is a critical regulator of the redox status in the heart in the presence of HFD consumption. Nampt alleviates H2O2-induced Prdx1 oxidation and mTOR inhibition in cardiomyocytes through activation of Trx1, whereas these protective effects of Nampt were inhibited in the presence of NADK knockdown. These results suggest that the NADP(H) pathway significantly mediates the protective effects of Nampt under stress conditions.
NAD+ acts as a major substrate of sirtuins and supports sirtuins’ deacetylase activity 30. We have shown previously that sirtuins, including Sirt1, Sirt3 and Sirt5, are upregulated in Tg-Nampt mice, accompanied by decreases in overall protein acetylation 16. We have also shown that the protective effects of NAD+ replenishment in the heart during ischemia and reperfusion are mediated primarily through Sirt1 31. Interestingly, Sirt1 is inhibited by direct carbonyl modification in response to oxidative stress 32. Thus, Nampt may activate Sirt1 not only through NAD+ production but also by preventing Sirt1 oxidation due to a reductive redox environment. In addition, Sirt1 inhibits HFD-induced diastolic dysfunction 33. Thus, Nampt may inhibit diastolic dysfunction partly through Sirt1 activation. The current study shows that the protective effect of Nampt against H2O2-induced cell death is abolished in the presence of Sirt1 knockdown but promoted in the presence of Sirt3 knockdown (Fig. 6E). Sirt1 protects the heart against oxidative stress, possibly through transcription of reducing enzymes such as Trx1 34. Indeed, Sirt1 knockdown downregulated Trx1 (Fig. 6F). Currently, how loss of Sirt3 potentiates Nampt function remains to be elucidated. However, downregulation of Sirt3 in mitochondria may increase NAD+, since Sirt3 is a major consumer of NAD+ in mitochondria. In addition, we observed that Sirt3 knockdown upregulated Trx1 (Fig. 6F). Thus, Sirt3 knockdown may potentiate both NADP(H) production and the Trx1 system, thereby promoting Nampt-induced protective effects as a negative feedback mechanism to offset the potentially harmful effect of Sirt3 downregulation. Further investigation is required to clarify the role of sirtuins in mediating the protective effect of Nampt in response to HFD consumption.
We have shown recently that pressure overload-induced failing heart phenotypes are exacerbated in Tg-Nampt mice partly through Sirt1 16. The molecular mechanisms through which overexpression of Nampt in the heart differentially affects cardiac function in pressure overload and HFD consumption remain to be elucidated. In addition to acting as the rate-limiting enzyme in the NAD+ salvage pathway, Nampt can also be secreted, and its extracellular form, called eNampt, acts as a cytokine 25. Phosphorylation of IKK is elevated in Tg-Nampt mice during pressure overload 16. Interestingly, although HFD consumption activates IKK in the heart, IKK was completely inhibited in Tg-Nampt mice. Thus, how Nampt affects the inflammatory signaling mechanism appears context-dependent and may determine the overall effect of Nampt overexpression in the heart. Further investigation is required to address this issue. In addition, the role of eNampt during HFD consumption remains to be elucidated.
Gain of Nampt function prevents HFD-induced cardiac hypertrophy (Fig. 2A and 2B). Importantly, the Trx1 and GSH systems possess anti-hypertrophic effects. In fact, we have shown previously that Trx1 inhibits cardiac hypertrophy in part through miR-98 35. The heart weight/tibia length ratio (HW/TL), an index of cardiac hypertrophy, after 1 month of HFD consumption was reduced in Tg-Nampt compared to in NTg mice, whereas the decrease in the HW/TL was abolished in the presence of an NADK inhibitor (Fig. 3E). Thus, Nampt may inhibit HFD-induced cardiac hypertrophy partly through NADK-dependent mechanisms such as Trx1 and GSH.
Our in vivo Nampt loss-of-function experiments were conducted with systemic Nampt+/− mice. HFD-induced exacerbation of cardiac dysfunction is at least in part mediated through the loss of Nampt function in cardiomyocytes since cardiomyocyte-specific gain of Nampt function exhibited opposite effects in the heart in the presence of HFD consumption. The loss of Nampt function may negatively affect insulin secretion in pancreatic β-cells 25. However, we did not observe any significant difference in glucose levels between Nampt+/− and WT mice in response to HFD consumption. Further investigation with cardiac-specific Nampt knockout mice would confirm the role of Nampt in cardiomyocytes during HFD consumption.
In summary, Nampt protects the heart against the development of diabetic cardiomyopathy in response to HFD consumption. Therapeutic interventions to increase NADP(H) through activation of Nampt in cardiomyocytes may be effective in treating diastolic dysfunction, hypertrophy and fibrosis in diabetic patients.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Diabetic cardiomyopathy is often accompanied by decreases in NAD+/NADH and increases in oxidative stress.
Nicotinamide phosphoribosyltransferase (Nampt) is a rate-limiting enzyme of NAD+ synthesis.
NADP is converted to NADPH, an electron donor for glutathione (GSH) and thioredoxin (Trx), and is generated from NAD+ in the presence of NAD kinase (NADK).
What New Information Does This Article Contribute?
Nampt is necessary and sufficient to protect the heart against the development of diabetic cardiomyopathy in response to high fat diet (HFD) consumption.
The protective effect of Nampt in the heart in the presence of HFD consumption is mediated in part through NADK-dependent-mechanisms and stimulation of GSH and Trx1.
Consumption of a western diet and obesity promote insulin resistance and the development of diastolic dysfunction and fibrosis in the heart, termed diabetic cardiomyopathy. Diabetic cardiomyopathy is accompanied by decreases in electron acceptors/donors (NAD+/NADH) and increases in oxidative stress. We here demonstrate that Nampt, a rate-limiting enzyme in the NAD+ salvage pathway, plays an essential role in protecting the heart against the development of diabetic cardiomyopathy in response to high fat diet consumption in mice. Nampt not only increases NAD+/NADH but also activates NADPH-dependent anti-oxidant systems, including GSH and the Trx system, in the diabetic heart, which in turn alleviates oxidative stress and improves mitochondrial function. The protective effect of Nampt is inhibited when NADK is inhibited, suggesting that NADK plays an important role in mediating the protective effect of Nampt through production of NADP. Intervention to promote NAD+ not only activates NAD+-dependent enzymes, including sirtuins, but also activates anti-oxidants, and, thus, could be effective in alleviating cardiac dysfunction in diabetic patients.
ACKNOWLEDGEMENTS
We thank Daniela Zablocki for critical reading of the manuscript.
SOURCES OF FUNDING
This work was supported in part by the American Heart Association (AHA) Grant in Aid 17GRNT33440031 (SO), Transformational Project Award 19TPA34850170 (SO), New Jersey Health Foundation research grants PC56-16 and PC80-17 (SO), Foundation Leducq Transatlantic Networks 15CVD04 (JS), and US Public Health Service Grants HL067724, HL091469, HL138720, HL112330, and AG23039 (JS).
Nonstandard Abbreviations and Acronyms:
- BSO
buthionine sulfoximine
- NAD+
nicotinamide adenine dinucleotide
- NADP+
nicotinamide adenine dinucleotide phosphate
- HFD
high fat diet
- Nampt
nicotinamide phosphoribosyltransferase
- NADK
NAD kinase
- G6PD
Glucose-6-phosphate dehydrogenase
- GSH
Glutathione
- PV
Pressure-volume
- EDP
End-diastolic pressure
- EDPVR
EDP volume relationship
- LV
Left ventricular
- JNK
c-Jun N-terminal kinase
- TAK1
Transforming growth factor β-activated kinase 1
- TL
tibial length
- TLR4
Toll like receptor 4
- Trx1
Thioredoxin 1
- Prdx1
Peroxiredoxin 1
Footnotes
DISCLOSURES
None.
SUPPLEMENTAL MATERIALS
Contributor Information
Shin-ichi Oka, Department of Cell Biology and Molecular Medicine, Rutgers New Jersey Medical School, Newark..
Jaemin Byun, Department of Cell Biology and Molecular Medicine, Rutgers New Jersey Medical School, Newark..
Chun-yang Huang, Department of Cell Biology and Molecular Medicine, Rutgers New Jersey Medical School, Newark.; Division of Cardiovascular Surgery, Department of Surgery, Taipei Veterans General Hospital, Taiwan Institute of Clinical Medicine, School of Medicine National Yang-Ming University, Taipei, Taiwan.
Nobushige Imai, Department of Cell Biology and Molecular Medicine, Rutgers New Jersey Medical School, Newark..
Guersom Ralda, Department of Cell Biology and Molecular Medicine, Rutgers New Jersey Medical School, Newark..
Peiyong Zhai, Department of Cell Biology and Molecular Medicine, Rutgers New Jersey Medical School, Newark..
Xiaoyong Xu, Department of Cell Biology and Molecular Medicine, Rutgers New Jersey Medical School, Newark.; Department of Cardiology, Ningbo Medical Center Lihuili Hospital, Zhejiang, China
Sanchita Kashyap, Department of Cell Biology and Molecular Medicine, Rutgers New Jersey Medical School, Newark..
Junco S. Warren, Nora Eccles Harrison Cardiovascular Research and Training Institute, University of Utah, Salt Lake City
John Alan Maschek, Metabolomics, Proteomics, and Mass Spectrometry Cores, University of Utah, Salt Lake City; Department of Nutrition and Integrative Physiology, University of Utah, Salt Lake City.
Trevor S. Tippetts, Department of Nutrition and Integrative Physiology and the Diabetes and Metabolism Research Center, University of Utah, Salt Lake City
Mingming Tong, Department of Cell Biology and Molecular Medicine, Rutgers New Jersey Medical School, Newark..
Sundararajan Venkatesh, Department of Microbiology, Biochemistry, and Molecular Genetics, Rutgers New Jersey Medical School, Newark.; Department of Nutrition and Integrative Physiology and the Diabetes and Metabolism Research Center, University of Utah, Salt Lake City
Yoshiyuki Ikeda, Department of Cell Biology and Molecular Medicine, Rutgers New Jersey Medical School, Newark..
Wataru Mizushima, Department of Cell Biology and Molecular Medicine, Rutgers New Jersey Medical School, Newark..
Toshihide Kashihara, Department of Cell Biology and Molecular Medicine, Rutgers New Jersey Medical School, Newark..
Junichi Sadoshima, Department of Cell Biology and Molecular Medicine, Rutgers New Jersey Medical School, Newark..
REFERENCES
- 1.Engin A The Definition and Prevalence of Obesity and Metabolic Syndrome. Adv Exp Med Biol. 2017;960:1–17. [DOI] [PubMed] [Google Scholar]
- 2.Paolillo S, Marsico F, Prastaro M, Renga F, Esposito L, De Martino F, Di Napoli P, Esposito I, Ambrosio A, Ianniruberto M, Mennella R, Paolillo R and Gargiulo P. Diabetic Cardiomyopathy: Definition, Diagnosis, and Therapeutic Implications. Heart Fail Clin. 2019;15:341–347. [DOI] [PubMed] [Google Scholar]
- 3.Koutroumpakis E, Jozwik B, Aguilar D and Taegtmeyer H. Strategies of Unloading the Failing Heart from Metabolic Stress. Am J Med. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schulze PC, Drosatos K and Goldberg IJ. Lipid Use and Misuse by the Heart. Circ Res. 2016;118:1736–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tong M, Saito T, Zhai P, Oka SI, Mizushima W, Nakamura M, Ikeda S, Shirakabe A and Sadoshima J. Mitophagy Is Essential for Maintaining Cardiac Function During High Fat Diet-Induced Diabetic Cardiomyopathy. Circ Res. 2019;124:1360–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oka S, Hsu CP and Sadoshima J. Regulation of cell survival and death by pyridine nucleotides. Circ Res. 2012;111:611–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bajic VP, Van Neste C, Obradovic M, Zafirovic S, Radak D, Bajic VB, Essack M and Isenovic ER. Glutathione “Redox Homeostasis” and Its Relation to Cardiovascular Disease. Oxid Med Cell Longev. 2019;2019:5028181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nagarajan N, Oka S and Sadoshima J. Modulation of signaling mechanisms in the heart by thioredoxin 1. Free Radic Biol Med. 2017;109:125–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller CG, Holmgren A, Arner ESJ and Schmidt EE. NADPH-dependent and -independent disulfide reductase systems. Free Radic Biol Med. 2018;127:248–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xie HB, Cammarato A, Rajasekaran NS, Zhang H, Suggs JA, Lin HC, Bernstein SI, Benjamin IJ and Golic KG. The NADPH metabolic network regulates human alphaB-crystallin cardiomyopathy and reductive stress in Drosophila melanogaster. PLoS Genet. 2013;9:e1003544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoxhaj G, Ben-Sahra I, Lockwood SE, Timson RC, Byles V, Henning GT, Gao P, Selfors LM, Asara JM and Manning BD. Direct stimulation of NADP(+) synthesis through Akt-mediated phosphorylation of NAD kinase. Science. 2019;363:1088–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rajasekaran NS, Connell P, Christians ES, Yan LJ, Taylor RP, Orosz A, Zhang XQ, Stevenson TJ, Peshock RM, Leopold JA, Barry WH, Loscalzo J, Odelberg SJ and Benjamin IJ. Human alpha B-crystallin mutation causes oxido-reductive stress and protein aggregation cardiomyopathy in mice. Cell. 2007;130:427–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Y, Lee JH, Paull TT, Gehrke S, D’Alessandro A, Dou Q, Gladyshev VN, Schroeder EA, Steyl SK, Christian BE and Shadel GS. Mitochondrial redox sensing by the kinase ATM maintains cellular antioxidant capacity. Sci Signal. 2018;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hsu CP, Hariharan N, Alcendor RR, Oka S and Sadoshima J. Nicotinamide phosphoribosyltransferase regulates cell survival through autophagy in cardiomyocytes. Autophagy. 2009;5:1229–31. [DOI] [PubMed] [Google Scholar]
- 15.Hsu CP, Oka S, Shao D, Hariharan N and Sadoshima J. Nicotinamide phosphoribosyltransferase regulates cell survival through NAD+ synthesis in cardiac myocytes. Circ Res. 2009;105:481–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Byun J, Oka SI, Imai N, Huang CY, Ralda G, Zhai P, Ikeda Y, Ikeda S and Sadoshima J. Both gain and loss of Nampt function promote pressure overload-induced heart failure. Am J Physiol Heart Circ Physiol. 2019;317:H711–H725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nakamura M, Liu T, Husain S, Zhai P, Warren JS, Hsu CP, Matsuda T, Phiel CJ, J.E. C, Tian B, Li H and Sadoshima J. Glucogen synthase kinase-3a promotes fatty acid uptake and lipotoxic cardiomyopathy. Cell Metab. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Glezeva N and Baugh JA. Role of inflammation in the pathogenesis of heart failure with preserved ejection fraction and its potential as a therapeutic target. Heart failure reviews. 2014;19:681–94. [DOI] [PubMed] [Google Scholar]
- 19.Tedeschi PM, Lin H, Gounder M, Kerrigan JE, Abali EE, Scotto K and Bertino JR. Suppression of Cytosolic NADPH Pool by Thionicotinamide Increases Oxidative Stress and Synergizes with Chemotherapy. Mol Pharmacol. 2015;88:720–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oka SI, Hirata T, Suzuki W, Naito D, Chen Y, Chin A, Yaginuma H, Saito T, Nagarajan N, Zhai P, Bhat S, Schesing K, Shao D, Hirabayashi Y, Yodoi J, Sciarretta S and Sadoshima J. Thioredoxin-1 maintains mechanistic target of rapamycin (mTOR) function during oxidative stress in cardiomyocytes. J Biol Chem. 2017;292:18988–19000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shao D, Oka S, Liu T, Zhai P, Ago T, Sciarretta S, Li H and Sadoshima J. A Redox-Dependent Mechanism for Regulation of AMPK Activation by Thioredoxin1 during Energy Starvation. Cell Metab. 2014;19:232–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim J, Kundu M, Viollet B and Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakamura M and Sadoshima J. Cardiomyopathy in obesity, insulin resistance and diabetes. J Physiol. 2020;598:2977–2993. [DOI] [PubMed] [Google Scholar]
- 24.Itani SI, Ruderman NB, Schmieder F and Boden G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes. 2002;51:2005–11. [DOI] [PubMed] [Google Scholar]
- 25.Revollo JR, Korner A, Mills KF, Satoh A, Wang T, Garten A, Dasgupta B, Sasaki Y, Wolberger C, Townsend RR, Milbrandt J, Kiess W and Imai S. Nampt/PBEF/Visfatin regulates insulin secretion in beta cells as a systemic NAD biosynthetic enzyme. Cell Metab. 2007;6:363–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sharma K and Kass DA. Heart failure with preserved ejection fraction: mechanisms, clinical features, and therapies. Circ Res. 2014;115:79–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berthiaume JM, Kurdys JG, Muntean DM and Rosca MG. Mitochondrial NAD(+)/NADH Redox State and Diabetic Cardiomyopathy. Antioxid Redox Signal. 2019;30:375–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pacher P and Szabo C. Role of poly(ADP-ribose) polymerase-1 activation in the pathogenesis of diabetic complications: endothelial dysfunction, as a common underlying theme. Antioxid Redox Signal. 2005;7:1568–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gardell SJ, Hopf M, Khan A, Dispagna M, Hampton Sessions E, Falter R, Kapoor N, Brooks J, Culver J, Petucci C, Ma CT, Cohen SE, Tanaka J, Burgos ES, Hirschi JS, Smith SR, Sergienko E and Pinkerton AB. Boosting NAD(+) with a small molecule that activates NAMPT. Nature communications. 2019;10:3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Imai S [The sirtuin family : regulators that connect metabolism, aging, and longevity]. Clin Calcium. 2013;23:29–38. [PubMed] [Google Scholar]
- 31.Yamamoto T, Byun J, Zhai P, Ikeda Y, Oka S and Sadoshima J. Nicotinamide mononucleotide, an intermediate of NAD+ synthesis, protects the heart from ischemia and reperfusion. PLoS One. 2014;9:e98972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Caito S, Rajendrasozhan S, Cook S, Chung S, Yao H, Friedman AE, Brookes PS and Rahman I. SIRT1 is a redox-sensitive deacetylase that is post-translationally modified by oxidants and carbonyl stress. FASEB J. 2010;24:3145–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yamamoto T, Endo J, Kataoka M, Matsuhashi T, Katsumata Y, Shirakawa K, Yoshida N, Isobe S, Moriyama H, Goto S, Yamashita K, Ohto-Nakanishi T, Nakanishi H, Shimanaka Y, Kono N, Shinmura K, Arai H, Fukuda K and Sano M. Sirt1 counteracts decrease in membrane phospholipid unsaturation and diastolic dysfunction during saturated fatty acid overload. Journal of molecular and cellular cardiology. 2019;133:1–11. [DOI] [PubMed] [Google Scholar]
- 34.Hsu CP, Zhai P, Yamamoto T, Maejima Y, Matsushima S, Hariharan N, Shao D, Takagi H, Oka S and Sadoshima J. Silent information regulator 1 protects the heart from ischemia/reperfusion. Circulation. 2010;122:2170–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang Y, Ago T, Zhai P, Abdellatif M and Sadoshima J. Thioredoxin 1 negatively regulates angiotensin II-induced cardiac hypertrophy through upregulation of miR-98/let-7. Circ Res. 2011;108:305–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oka S, Alcendor R, Zhai P, Park JY, Shao D, Cho J, Yamamoto T, Tian B and Sadoshima J. PPARalpha-Sirt1 complex mediates cardiac hypertrophy and failure through suppression of the ERR transcriptional pathway. Cell Metab. 2011;14:598–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matyash V, Liebisch G, Kurzchalia TV, Shevchenko A and Schwudke D. Lipid extraction by methyl-tert-butyl ether for high-throughput lipidomics. Journal of lipid research. 2008;49:1137–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xia J, Sinelnikov IV, Han B and Wishart DS. MetaboAnalyst 3.0--making metabolomics more meaningful. Nucleic Acids Res. 2015;43:W251–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oka SI, Chin A, Park JY, Ikeda S, Mizushima W, Ralda G, Zhai P, Tong M, Byun J, Tang F, Einaga Y, Huang CY, Kashihara T, Zhao M, Nah J, Tian B, Hirabayashi Y, Yodoi J and Sadoshima J. Thioredoxin-1 maintains mitochondrial function via mechanistic target of rapamycin signalling in the heart. Cardiovasc Res. 2020;116:1742–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oka SI, Chin A, Park JY, Ikeda S, Mizushima W, Ralda G, Zhai P, Tong M, Byun J, Tang F, Einaga Y, Huang CY, Kashihara T, Zhao M, Nah J, Tian B, Hirabayashi Y, Yodoi J and Sadoshima J. Thioredoxin-1 maintains mitochondrial function via mTOR signaling in the heart. Cardiovasc Res. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Byun J, Oka SI, Imai N, Huang CY, Ralda G, Zhai P, Ikeda Y, Ikeda S and Sadoshima J. Both gain and loss of Nampt function promote pressure-overload-induced heart failure. Am J Physiol Heart Circ Physiol. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The author declare that all supporting data are available within the article. In addition, any raw data that support the findings of this study are available from the corresponding author upon reasonable request.