

Abstract
JAK2 (Janus tyrosine kinase 2) is important for signalling through many cytokine receptors, and a gain-of-function JAK2 mutation in its pseudokinase domain, V617F, has been implicated in Philadelphia chromosome-negative myeloproliferative neoplasms. How this mutation hyperactivates JAK2 is poorly understood. In the present paper we report our findings that the V617F mutation has little effect on the Vmax of JAK2 kinase activity, but lowers the Km value for substrates. Therefore under physiological conditions where the concentration level of substrates is presumably below saturation, JAK2(V617F) exhibits hyperactivation compared with wild-type JAK2. This lower Km of JAK2(V617F) towards substrates requires the JAK2 FERM (4.1/ezrin/radixin/moesin) domain, as deletion of the FERM domain abolished this effect. We also show that, in contrast with its positive role in JAK2(V617F) hyperactivation, the FERM domain in wild-type JAK2 is inhibitory. Deletion or mutations of the FERM domain resulted in increased basal JAK2 kinase activity. The results of the present study provide the biochemical basis for how V617F hyperactivates JAK2, and identifies novel regulating roles of the JAK2 FERM domain to control kinase activity at different activation states.
Keywords: 4.1/ezrin/radixin/moesin (FERM) domain, Janus tyrosine kinase 2 (JAK2), kinase activity, myeloproliferative neoplasm
INTRODUCTION
JAK2 (Janus tyrosine kinase 2) is a member of the Janus family of non-receptor tyrosine kinases and plays a key role in signal transduction of many cytokine receptors, including the haematopoietic cytokine receptors [1]. Upon cytokine stimulation, JAK2 autophosphorylates its activation loop in the kinase domain and becomes active. Activated JAK2 subsequently phosphorylates the cytoplasmic domains of cognate receptors and also signalling proteins such as STAT5 (signal transducer and activator of transcription 5) to turn on downstream signalling pathways, leading to the proliferation and differentiation of haematopoietic cells [1]. Gain-of-function mutations in JAK2 were recently found in the majority of patients with MPNs (myeloproliferative neoplasms), a group of diseases that cause the overproduction of haematopoietic cells [2].
JAK2, like other JAK family members, contains a C-terminal tyrosine kinase domain [JH1 (JAK homology domain 1)], preceded by a pseudokinase domain (JH2) and an N-terminal segment (JH3–JH7). The N-terminal segment of JAKs, consisting of a FERM (4.1/ezrin/radixin/moesin) domain and an atypical SH2 (Src homology 2) domain [1], mediates association with the membrane-proximal region of cytokine receptors [3]. For JAK2, this association also regulates the expression of cytokine receptors on the plasma membrane [4,5]. There is evidence suggesting that the N-terminal segment also regulates JAK kinase activity. For example, mutation of Tyr221 in the FERM domain decreased the catalytic activity of JAK2 [6], whereas mutation of Ser523 increased JAK2 activity [7,8]. Mutations in the FERM domain of JAK1 modulate its kinase activity [9]. Zhou et al. [10] showed that a SCID (severe combined immunodeficiency) patient-derived JAK3 mutant, Y100C, could still associate with the common γ chain, but was inactive [10]. They also showed that the JAK3 FERM domain binds to, and enhances the activity of, the isolated JAK3 kinase domain [10]. In addition, a point mutation in the FERM domain of the Drosophila JAK, Hop, hyperactivates it and causes a leukaemia-like phenotype [11,12].
JAK kinase activity is also critically controlled by the pseudokinase domain, which is predicted to adopt a kinase fold, but lacks residues essential for catalysis [13]. Saharinen and co-workers [14,15] showed that deletion of this domain in JAK2 or JAK3 leads to a marked increase in kinase activity. In addition, fusing the pseudokinase domain to the JAK2 kinase domain reduced its activity (Vmax) without changing the Km for a substrate peptide [16]. Therefore the pseudokinase domain is essential for auto-inhibition. Consistent with this notion, a point mutation in the JAK2 pseudokinase domain, V617F, was identified in the majority of MPN patients, including almost all patients with polycythemia vera, many with essential thrombocythemia or primary myelofibrosis, and some with rarer forms of MPN [2]. This mutation hyperactivates JAK2 and causes constitutive downstream signalling [17,18]. Although it is believed that V617F disrupts the inhibitory interaction between the pseudokinase and kinase domains, mechanisms underlying how this mutation hyperactivates JAK2 remain elusive.
In the present study we used biochemical methods to characterize the underlying mechanisms of how the V617F mutation affects JAK2 kinase activity. Our results show that V617F not only relieves the inhibition posed by the pseudokinase domain towards the kinase domain, it also plays a positive role to lower the Km of the kinase domain towards substrates. We further show that this novel function of V617F is dependent on the JAK2 FERM domain. In addition, our results indicate that the JAK2 FERM domain is inhibitory in wild-type JAK2 to keep JAK2 in an inactive basal state. Therefore the FERM domain has dual roles in regulating JAK2 at different activation states.
EXPERIMENTAL
Plasmid constructs, cell lines and reagents
HA (haemagglutinin)-tagged or GST (glutathione transferase)-tagged wild-type or mutant JAK2s were generated by subcloning cDNAs encoding different truncated fragments of murine JAK2 into the pcDNA3.1 zeo(+) vector (Invitrogen) or the pEBG vector. HA–ΔFERM encodes amino acid residue 390 to the C-terminus, HA–ΔN encodes amino acid residue 500 to the C-terminus, and HA–JH1 encodes amino acid residue 809 to the C-terminus. GFP (green fluorescent protein)-fusion proteins were generated by subcloning cDNAs encoding different JAK2 fragments into the pEGFP vector (Clontech). Mutations were made by the QuikChange® site-directed mutagenesis kit (Stratagene) and verified by sequencing. Antibodies were from the following sources: anti-HA (Covance); anti-JAK2, anti-phospho-JAK2 and anti-GST (Millipore); and anti-Myc (Invitrogen). HA-affinity resin was from Roche Applied Science. Glutathione–Sepharose 4B, HRP (horseradish peroxidase)-coupled secondary antibodies and the ECL (enhanced chemiluminescence) system were from GE Healthcare.
Immunoprecipitation and immunoblotting
HEK (human embryonic kidney)-293T cells were transiently transfected with vectors encoding different JAK2 constructs using FuGENE™ 6 transfection reagent (Roche). At 48 h after transfection, cells were lysed with 1% Nonidet P40 lysis buffer [50 mM Tris/HCl (pH 7.4), 150 mM NaCl and 1% Nonidet P40] with protease and phosphatase inhibitors [1 mM sodium orthovanadate, 1 mM sodium fluoride and 1 mM PMSF plus complete protease inhibitor cocktail (Roche)]. Lysates were subjected to immunoprecipitation with HA-affinity resin. The precipitates were eluted with SDS sample buffer, separated by SDS/PAGE, transferred on to nitrocellulose membranes, and probed with anti-phospho-JAK2 or anti-HA antibodies. Bound antibodies were detected by the ECL system after incubation with HRP-coupled secondary antibodies. To examine the interaction between JAK2 and STAT5, HEK-293T cells were co-transfected with vectors encoding STAT5(Y694F)–Myc and HA-tagged JAK2 constructs or vector alone. Lysates were subjected to immunoprecipitation with HA-affinity resin, and the precipitates were separated on SDS/PAGE and probed with anti-Myc antibodies.
GST pulldown assay
HEK-293T cells transiently co-transfected with HA–JH1 and wild-type, or mutant, GST–FERM were lysed in 1% Nonidet P40 lysis buffer with protease inhibitors. The supernatant from these lysates were incubated with glutathione–Sepharose 4B at 4°C for 4 h, and washed three times with lysis buffer. Proteins associated with the resin were eluted with SDS sample buffer, separated on SDS/PAGE, transferred on to nitrocellulose membranes and blotted with antibodies against the HA tag.
In vitro JAK2 kinase assay
HA-tagged JAK2 proteins were immunoprecipitated using HA-affinity resin. Immunoprecipitants were washed three times with lysis buffer, twice with kinase buffer [10 mM Hepes (pH 7.4), 50 mM NaCl, 5 mM MnCl2, 5 mM MgCl2, 1 mM DTT (dithiothreitol), 1 mM sodium orthovanadate, 1 mM sodium fluoride and 1 mM PMSF plus protease inhibitor cocktail] and resuspended in kinase buffer supplemented with 100 μM ATP, 2 μCi [γ-32P]ATP (PerkinElmer), phosphatase inhibitors and protease inhibitors. A peptide derived from STAT5 (AKAADGY694VKPQIKQVV) containing the JAK2 phosphorylation site and a peptide derived from the JAK2 activation loop (VLPQDKEY1007Y1008KVKEPGES) were used as substrates. The reaction mixture was incubated at 30°C. For time course experiments, kinases were incubated with 1 mM substrate at 30°C and reactions were stopped at different time points by adding an equal volume of 20 mM EDTA. An aliquot of the reaction mixture was spotted on to P80 filter paper. After extensive washing with 0.1% phosphoric acid solution, bound radiolabelled phosphate was counted using a scintillation counter. To determine the Km, kinases were incubated with different concentrations of substrates (0, 40, 80, 160, 300 and 600 μM) at 30°C for 30 min, a time within the linear reaction range. Kinase reactions were stopped by the addition of SDS sample buffer. Samples were then boiled at 98°C for 5 min, separated by SDS/PAGE (20% gels) and analysed by Typhoon Trio scanning (GE Healthcare). Data were analysed using the Graphpad Prism 4.0 software. The experiments in the present study have been performed at least three times, and representative results are shown.
RESULTS
JAK2(V617F) has a lower Km value towards peptide substrates than full-length JAK2 or the JAK2 kinase domain in isolation
In order to determine the mechanisms underlying how V617F hyperactivates JAK2, we determined the kinetic parameters for HA-tagged JAK2 and JAK2(V617F) using in vitro kinase assays. For comparison, we also determined the kinetic parameters for the kinase domain (JH1) in isolation. HA–JAK2, HA–JAK2(V617F) or HA–JH1 were transiently expressed in HEK-293T cells. After immunoprecipitation with HA-affinity resin, the precipitants were separated by SDS/PAGE and analysed using HA antibodies or antibodies that recognize activated JAK2 phosphorylated on Tyr1007 and Tyr1008 in the activation loop in JH1. Consistent with the notion that JAK2 is auto-inhibited by the pseudokinase domain in the basal state and that V617F relieves auto-inhibition, both HA–JH1 and HA–JAK2(V617F) had higher activity than HA–JAK2 at similar protein levels (Figure 1A). Immunoprecipitated kinases were then incubated with 1 mM peptide substrate derived from the JAK2 phosphorylation site of STAT5 in the presence of [γ-32P]ATP in in vitro kinase assays. The presence of an equal amount of kinases was estimated based on semi-quantitative immunoblottings of parallel samples. Within the 60 min assay period, all constructs exhibited a linear increase of peptide phosphorylation over time (Figure 1B). Immunoprecipitants from cells expressing kinase-deficient JAK2 had only background levels of activity (results not shown), confirming that the activity measured in our assays was from JAK2. These kinases were then incubated with various peptide concentrations (0–600 μM) for 30 min while keeping the ATP concentration constant. Samples were analysed by SDS/PAGE and quantified by autoradiography. Km and Vmax were deduced from data analysis using Graphpad Prism. HA–JH1 had the highest Vmax among the three constructs [Figure 1C; relative Vmax values, HA–JAK2 = 153, HA–JAK2(V617F) = 182, HA–JH1 = 1159, data normalized to maximal activity of HA–JAK2], and its Km was similar to the Km of HA–JAK2 (Figure 1E; Km values, HA–JAK2 = 306 μM, HA–JH1 = 239 μM). These results are in line with a previous report showing that fusing the pseudokinase domain to the kinase domain lowered its Vmax without changing the Km [16]. Surprisingly, while the Vmax of HA–JAK2(V617F) was slightly higher than that of HA–JAK2 (Figure 1D), HA–JAK2(V617F) had a much lower Km compared with HA–JAK2 or HA–JH1 (Figure 1E; Km for HA–JAK2(V617F) = 67 μM). Therefore, with similar Vmax values between HA–JAK2 and HA–JAK2(V617F), product turnover would be faster for JAK2(V617F) owing to the lower Km at low concentration levels of substrate, consistent with Figure 1(A). In addition, because HA–JAK2(V617F) had a lower Km value than HA–JH1, the V617F mutation does not simply free up the kinase domain from the pseudokinase domain to mimic the isolated kinase domain.
Figure 1. JAK2(V617F) has a lower Km towards a STAT5-derived peptide than JAK2 or JAK2 kinase domain.
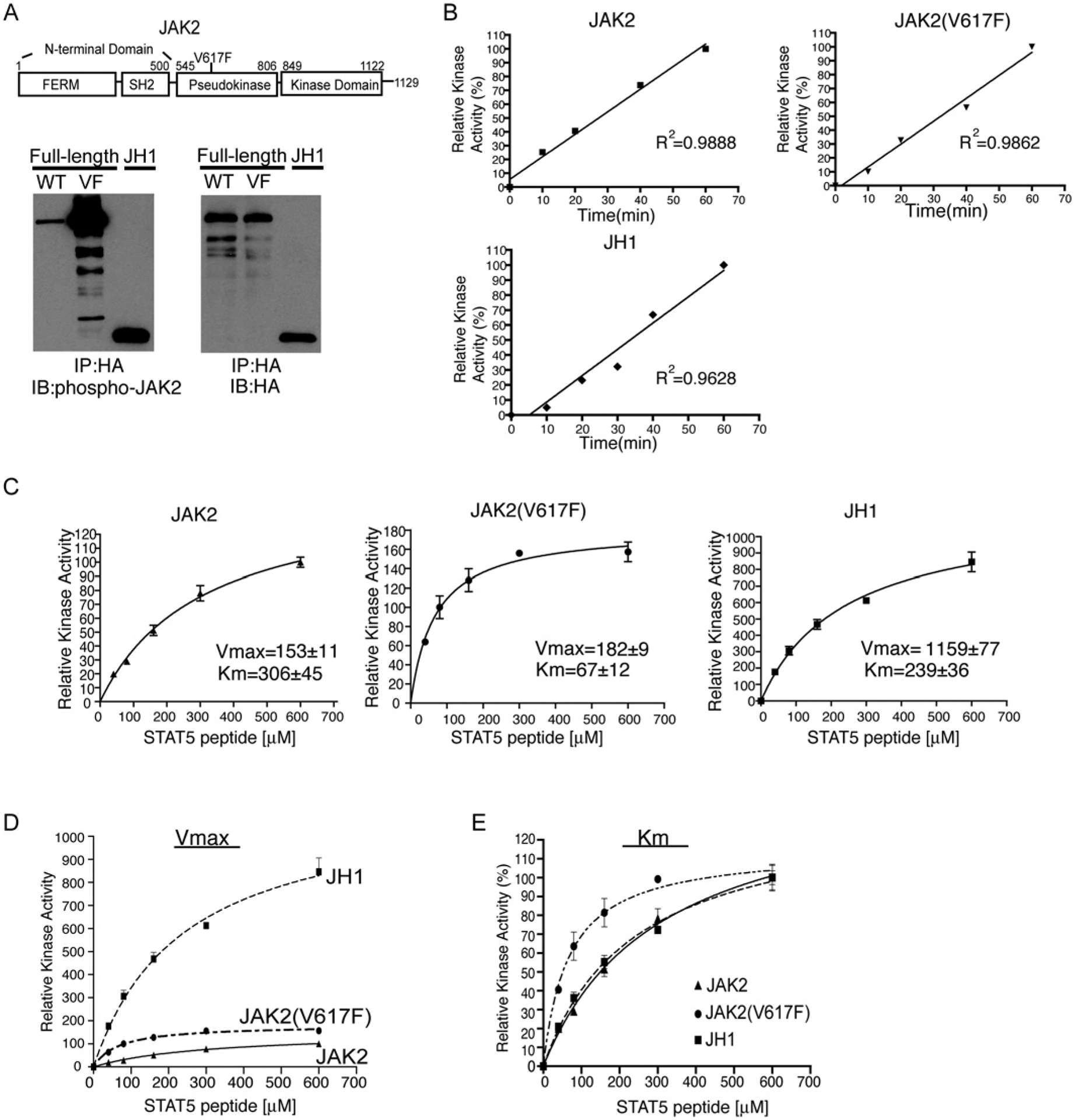
(A) Immunoprecipitated HA–JAK2, HA–JAK2(V617F) or HA–JH1 were immunoblotted with anti-phospho-JAK2 antibodies. The same blot was subsequently probed with antibodies against HA. IB, immunoblot; IP, immunoprecipitate; VF, V617F; WT, wild-type. (B) Kinetic analysis of the catalytic activity of HA–JAK2, HA–JAK2(V617F) and HA–JH1. Immunoprecipitated proteins were subjected to an in vitro kinase assay using a STAT5-derived peptide (1 mM) as a substrate. The reactions were stopped at various time points and the reaction mixtures were spotted on to P80 filter paper and counted with a scintillation counter. (C) Immunoprecipitated proteins were subjected to an in vitro kinase assay with various concentrations of a STAT5-derived peptide substrate. The reaction time was 30 min. The peptides were separated on SDS/PAGE followed by quantification using a PhosphorImager. Vmax and Km for each construct were shown. (D) Comparison of Vmax values for HA–JAK2, HA–JAK2(V617F) and HA–JH1. Data were normalized to maximal wild-type JAK2 activity. (E) Comparison of Km values for HA–JAK2, HA–JAK2(V617F) and HA–JH1. The activity of the kinases was normalized to the maximal activity of each construct.
To corroborate these results, we determined the Km value of these kinases towards another substrate peptide corresponding to the JAK2 activation loop. Consistent with our previous results, HA–JAK2(V617F) had the lowest Km, whereas the Km for HA–JAK2 and HA–JH1 were similar (Figure 2; Km values, HA–JAK2 = 152 μM, HA–JAK2(V617F) = 58 μM, HA–JH1 = 205 μM). Taken together, our results show that with similar Vmax values, JAK2(V617F) has a much lower Km towards substrates compared with JAK2. Therefore, under physiological conditions where the concentration level of substrate is presumably below saturation, JAK2(V617F) exhibits hyperactivation compared with wild-type JAK2. In addition, because the Km value is lower for JAK2(V617F) than JH1, V617F does more than simply free up the kinase domain from the pseudokinase domain to mimic the kinase domain in isolation.
Figure 2. JAK2(V617F) has a lower Km value towards a JAK2-derived peptide than JAK2 or JAK2 kinase domain.

Kinetic analysis of HA–JAK2 (A), HA–JAK2(V617F) (B) or HA–JH1 (C) in in vitro kinase assays using a JAK2-derived peptide as the substrate as described in Figure 1. (D) Comparison of Km values between constructs.
The JAK2 FERM domain plays different roles in JAK2 or JAK2(V617F) to regulate kinase activity
Because JAK2(V617F) had a lower Km than JH1, we determined whether other non-catalytic domains in JAK2 play a role in JAK2(V617F) hyperactivation. Two constructs were engineered as depicted in Figure 3(A): HA–ΔFERM contains the SH2, pseudokinase and kinase domains, but lacks the FERM domain, and HA–ΔN contains just the pseudokinase and kinase domains. For each construct, V617F was introduced into the pseudokinase domain. Kinase activities of HA–ΔFERM and HA–ΔN were examined by autophosphorylation. Interestingly, HA–ΔFERM was more active than HA–JAK2 (Figure 3B, lane 1 and lane 4), indicating that the FERM domain contributes to JAK2 auto-inhibition. In contrast, the activity of HA–ΔFERM(V617F) was similar to HA–ΔFERM, and was dramatically reduced compared with HA–JAK2(V617F) (Figure 3B, lane 2 and lane 5). Therefore the FERM domain plays an activating role in JAK2(V617F) hyperactivation. Both HA–ΔN and HA–ΔN(V617F) had similar low kinase activities (Figure 3B, lanes 6 and 7). Because the activity of HA–ΔN and HA–ΔN(V617F) is lower than that of HA–ΔFERM or HA–ΔFERM(V617F), the SH2 domain may participate in the relief of auto-inhibition. These results indicate that the FERM domain exhibits opposite effects towards kinase activity in wild-type JAK2 or JAK2(V617F). Specifically, the FERM domain plays an inhibitory role in keeping wild-type JAK2 in a basal inhibitory state whereas, in JAK2(V617F), the FERM domain is essential for its hyperactivation. Consistent with previous results that the FERM domain is also required for interaction with cognate receptors at the plasma membrane [4], GFP-tagged wild-type JAK2 or JAK2(V617F) localized on the plasma membrane and in the cytoplasm, whereas GFP-tagged ΔFERM localized only in the cytoplasm (Supplementary Figure S1 at http://www.BiochemJ.org/bj/426/bj4260091add.htm).
Figure 3. The JAK2 FERM domain is required for the lower Km value in JAK2(V617F).

(A) Schematic diagram of the various JAK2 constructs. (B) Immunoprecipitated kinases were immunoblotted with anti-phospho-JAK2 or anti-HA antibodies. (C) Immunoprecipitated HA–ΔFERM or HA–ΔFERM(V617F) were subjected to an in vitro kinase assay using a STAT5-derived peptide (1 mM) as a substrate. The reactions were stopped at various time points and the reaction mixtures were spotted on to P80 filter paper and counted with a scintillation counter. (D) Immunoprecipitated proteins were subjected to an in vitro kinase assay with various concentrations of a STAT5-derived peptide substrate. The reaction time was 30 min. The peptides were separated by SDS/PAGE followed by quantification using a PhosphorImager. (E) HA–JAK2, HA–JAK2(V617F), HA–ΔFERM and HA–ΔFERM(V617F) bound STAT5(Y649F)–Myc. Cell lysates expressing STAT5(Y694F)–Myc with HA–JAK2, HA–JAK2(V617F), HA–ΔFERM, HA–ΔFERM(V617F) or vector alone (−) were subjected to immunoprecipitation with HA-affinity resin and probed with anti-Myc antibodies. IB, immunoblot; IP, immunoprecipitate; VF, V617F; WT, wild-type.
To determine whether the FERM domain is required for the lower Km for substrates observed in JAK2(V617F), we determined the Km for HA–ΔFERM and HA–ΔFERM(V617F) by in vitro kinase assays using the STAT5 peptide as a substrate. As shown in Figure 3(D), both HA–ΔFERM and HA–ΔFERM(V617F) had a Km of approx. 200 μM, which is similar to HA–JAK2 and HA–JH1, but is much higher than the Km for HA–JAK2(V617F). These results demonstrate that the FERM domain is required for the lower Km observed in JAK2(V617F) and confirmed the essential role of the JAK2 FERM domain in JAK2(V617F) hyperactivation.
Because a decrease in Km may indicate an increase in substrate affinity, we examined the effect of the V617F mutation on the ability of JAK2 to bind substrate proteins using co-immunoprecipitation assays. Lysates from cells expressing Myc-tagged STAT5(Y694F), a non-phosphorylatable STAT5 mutant, with HA-tagged JAK2, JAK2(V617F), ΔFERM, ΔFERM(V617F) or vector alone were subjected to immunoprecipitation with HA-affinity resin, and the precipitants were probed with anti-Myc antibodies. As shown in Figure 3(E), all JAK2 constructs pulled down STAT5(Y694F) specifically, and no difference in binding was detectable between wild-type JAK2 and JAK2(V617F), or between ΔFERM and ΔFERM(V617F). Because the binding between STAT5 and JAK2 is weak [we estimated that less than 1% of STAT5(Y694F) was pulled down], and is complex and involves multiple domains of both proteins [19,20], the difference in binding affinity between JAK2 and JAK2(V617F) towards STAT5(Y694F) may be below the detection limit in this assay. In addition, the binding between JAK2 or JAK2(V617F) and STAT5(Y694F) may not be the same as the binding between JAK2 or JAK2(V617F) and STAT5. Alternatively, because in addition to the affinity of substrate for enzyme, the value of Km also includes the rate at which the substrate bound to the enzyme is converted into product, differences in Km may not be due to differences in substrate affinity, but in the catalytic steps.
Mutations in the JAK2 FERM domain affect catalytic activity
To further test the idea that the FERM domain regulates JAK2 kinase activity, we engineered three mutations at residue Tyr114, Y114A, Y114C and Y114F. Y114C corresponds to the Y100C mutation in the JAK3 FERM domain that was isolated from a SCID patient and was shown to abolish JAK3 kinase activity [10]. Substitution was made on the background of HA–JAK2 or HA–JAK2(V617F), and the kinase activity of the mutants was examined by autophosphorylation. As shown in Figure 4(A), wild-type JAK2 showed a low autophosphorylation level, and Y114A, Y114C or Y114F mutations resulted in slightly increased kinase activities (lanes 2, 3 and 4 compared with lane 1). In contrast, the Y114A and Y114C on the HA–JAK2(V617F) background drastically reduced its kinase activity (Figure 4A, lanes 6 and 7 compared with lane 5), and Y114F had little effect (Figure 4A, lane 8). Consistent with previous results, HA–ΔFERM was more active than HA–JAK2 (Figure 4A, lanes 9 and 10). These results further confirm our previous findings that the FERM domain contributes to auto-inhibition in wild-type JAK2 and is essential for hyperactivation in JAK2(V617F).
Figure 4. Mutations in the JAK2 FERM domain affect JAK2 catalytic activity.
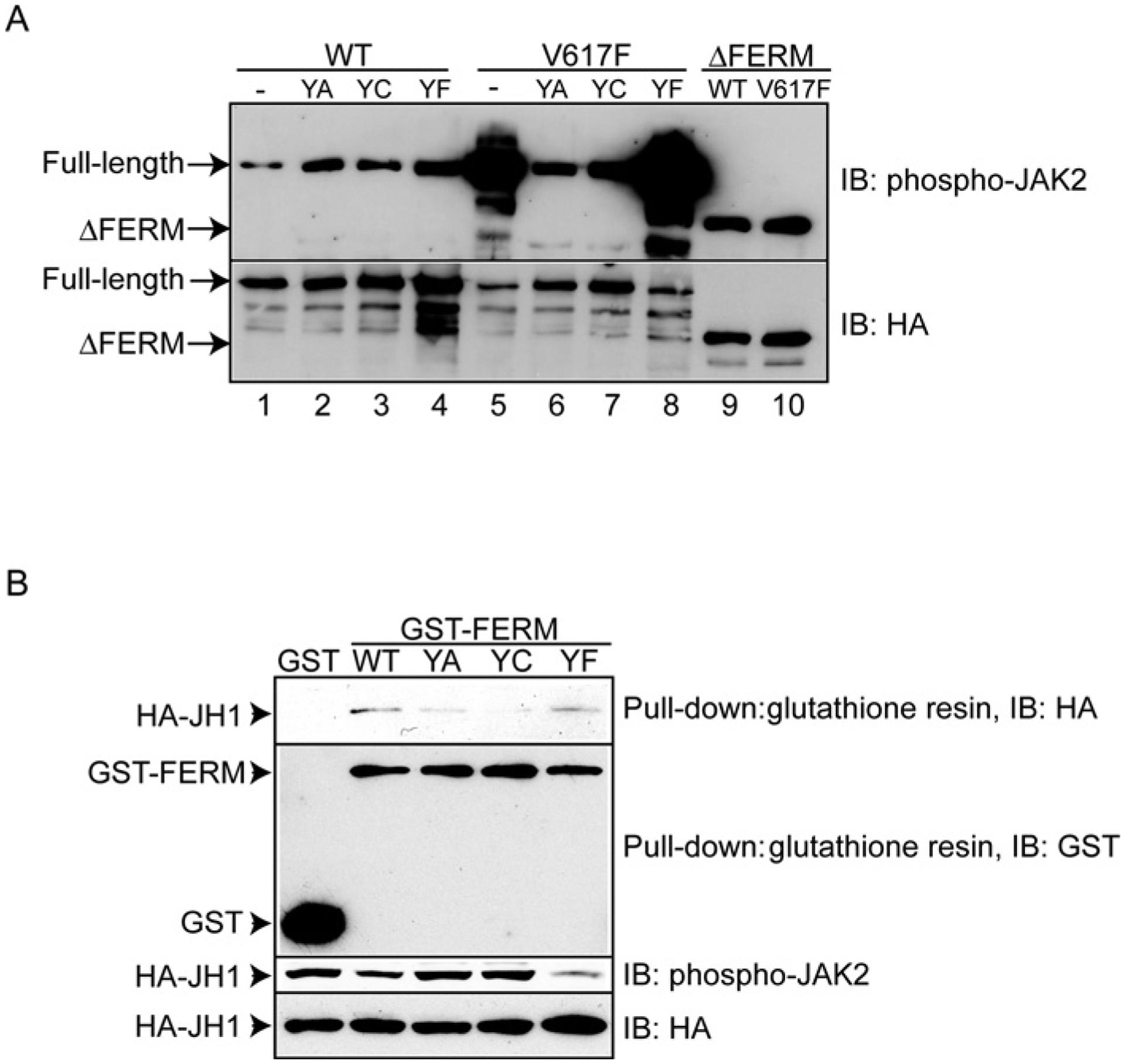
(A) Y114A, Y114C or Y114F mutations were made on the background of HA–JAK2 (lanes 1–4) or HA–JAK2(V617F) (lanes 5–8). Immunoprecipitated kinases from HEK-293T cells were immunoblotted with anti-phospho-JAK2 or anti-HA antibodies. (B) The JAK2 FERM domain interacts with and inhibits the JAK2 kinase domain. Wild-type GST–FERM or GST–FERM with Y114A, Y114C and Y114F were transiently expressed with HA–JH1 in HEK-293T cells. Cell lysates were probed with anti-phospho-JAK2 or anti-HA antibodies (bottom two panels). GST, GST–FERM or GST–FERM with mutations were pulled down by glutathione resin and the precipitants were probed with antibodies against HA or GST (top two panels). IB, immunoblot; WT, wild-type; YA, Y114A; YC, Y114C; YF, Y114F.
We next examined whether the JAK2 FERM domain can associate with the kinase domain. As shown in Figure 4(B), GST–FERM bound specifically to the JAK2 kinase domain (HA–JH1) in pulldown assays (top panel) and inhibited JH1 kinase activity (third panel). GST–FERM with Y114A or Y114C had a reproducible, albeit modest, decrease in binding to the kinase domain, and they failed to inhibit the kinase domain activity. These results are consistent with results showing that Y114A and Y114C increased basal JAK2 activity on the full-length JAK2 background (Figure 4A). Although Y114F increased basal JAK2 activity in full-length JAK2 (Figure 4A), GST–FERM with the Y114F mutation bound to the kinase domain normally and inhibited the kinase domain similar to the wild-type FERM domain in trans. We reason that Y114F may have additional effects in the full-length background, which was eliminated when the FERM and kinase domains were expressed in isolation. For example, it may affect the SH2 domain, which may regulate auto-inhibition. Our results suggest that the JAK2 FERM domain plays dual roles in maintaining proper catalytic activity at different JAK2 activation states.
DISCUSSION
Tyrosine kinases are essential for the regulation of cell proliferation and differentiation, and their activities are tightly controlled. The V617F mutation in the JAK2 pseudokinase domain is the major molecular lesion in patients with BCR/Abl (breakpoint cluster region/Abelson)-negative MPN, underlining the importance of regulatory mechanisms controlling this kinase. The mechanisms of how the V617F mutation hyperactivates JAK2 are not completely understood. In the present study we show that the FERM domain is necessary for the V617F mutation to hyperactivate JAK2, and it does so by lowering the Km of the kinase domain towards substrates. In addition, we show that, in wild-type JAK2, the FERM domain helps to keep the kinase in a basal inactive state. Therefore our results suggest that the FERM domain plays different regulatory roles in wild-type JAK2 compared with JAK2(V617F) to control JAK2 kinase activity at different activation states.
The JAK2 pseudokinase domain is essential for auto-inhibition. Deletion of this domain resulted in JAK2 activation, and a construct lacking the JAK2 kinase domain inhibited the isolated kinase domain in trans [14]. Fusing the wild-type pseudokinase domain to the kinase domain reduced its Vmax without changing the Km for a substrate peptide [16]. Therefore it is believed that the inhibitory interaction between the pseudokinase and kinase domains keeps the kinase domain in an inactive state, and this inhibition is disrupted by V617F, thus hyperactivating JAK2. It was proposed that Val617 directly interacts with the kinase activation loop to keep it in an inactive conformation [21], and that V617F disrupts this critical interaction and allows the activation loop to break free for transphosphorylation/activation [18]. We show in the present study that, consistent with previous findings, the Km values of JAK2 and the isolated JAK2 kinase domain (JH1) are similar. Interestingly, the V617F mutation had little effect on the Vmax of JAK2 kinase activity, but dramatically lowered the Km value for substrates. JAK2(V617F) had a 5-fold lower Km value for two different substrate peptides compared with JAK2 and JH1. Therefore our results suggest that, besides relieving the inhibitory interaction between the pseudokinase and kinase domains, the V617F mutation hyperactivates JAK2 through a novel mechanism that lowers the Km of the kinase domain towards substrates. One hypothesis for this mechanism is that V617F increases the substrate binding affinity of the kinase domain. We did not detect any difference in binding between JAK2 and JAK2(V617F) towards STAT5(Y694F) in co-immunoprecipitation assays. We reason that because the binding between STAT5 and JAK2 is weak and was shown to involve multiple domains of both proteins [19,20], the difference in binding affinity between JAK2 or JAK2(V617F) and STAT5(Y694F) may be below the detection limit in this assay. In addition, the binding between JAK2 or JAK2(V617F) and STAT5(Y694F) may not be the same as the binding between JAK2 or JAK2(V617F) and STAT5. Alternatively, the V617F mutation may affect the catalytic steps instead of substrate affinity to lower the Km.
The Km value for substrates was lower for JAK2(V617F) than JH1, thus we determined whether other JAK2 domains may be involved. Our results show that the Km value for ΔFERM(V617F) was similar to ΔFERM and full-length JAK2, but was much higher than JAK2(V617F). In addition, mutations of Tyr114 in the JAK2 FERM domain drastically reduced the kinase activity of JAK2(V617F). Therefore the lower Km in JAK2(V617F) requires the FERM domain.
Contrary to a positive role in hyperactivating kinase activity in JAK2(V617F), the FERM domain plays an inhibitory role in wild-type JAK2 to maintain it in an inactive basal state. Deletion of the FERM domain in ΔFERM or mutations of Tyr114 herein resulted in higher kinase activity. We also showed that the FERM domain interacts with the isolated kinase domain and inhibits kinase activity in trans. Y114A and Y114C mutations in the FERM domain decreased its binding to the kinase domain and abolished its ability to inhibit the kinase domain activity. Consistent with the notion that the FERM domain has opposite functions in JAK2 compared with JAK2(V617F), the same mutations (Y114A and Y114C) that drastically reduced JAK2(V617F) activity increased activity in the wild-type JAK2 context. Similar observations that FERM domains can regulate kinase activity have been reported in other JAKs [6–12] and in the focal adhesion kinase, which also contains a FERM domain [22]. Therefore the FERM domain in JAKs serves two essential functions: one to interact with cognate receptors and the other to regulate kinase activity. Moreover, the FERM domain plays different roles in regulating JAK2 at different activation states.
We envision a model summarizing our results in Figure 5. In this model, both the FERM domain and the pseudokinase domain (JH2) participate in inhibiting the kinase domain (JH1) in the basal state. For simplicity, we have drawn the model so that the FERM domain and the pseudokinase domain interact with different sites on the kinase domain: the pseudokinase domain interacts with the activation loop, and the FERM domain interacts with another site. This model is consistent with a model put forth by Funakoshi-Tago et al. [23], suggesting that the FERM domain interacts with the pseudokinase and kinase domains, thus preventing the inappropriate activation of JAK2 in the basal state. In the presence of the V617F mutation, the inhibition from the pseudokinase domain is released. Moreover, the FERM domain in JAK2(V617F) interacts with the kinase domain to shape its active site for a lower Km towards substrates. In contrast, a construct harbouring V617F, but lacking the FERM domain, has a similar Km to the isolated kinase domain or wild-type JAK2. Further verification of this model will require additional studies and a detailed structure of the full-length JAK2 protein.
Figure 5. A model of intramolecular interactions in JAK2 and JAK2(V617F).
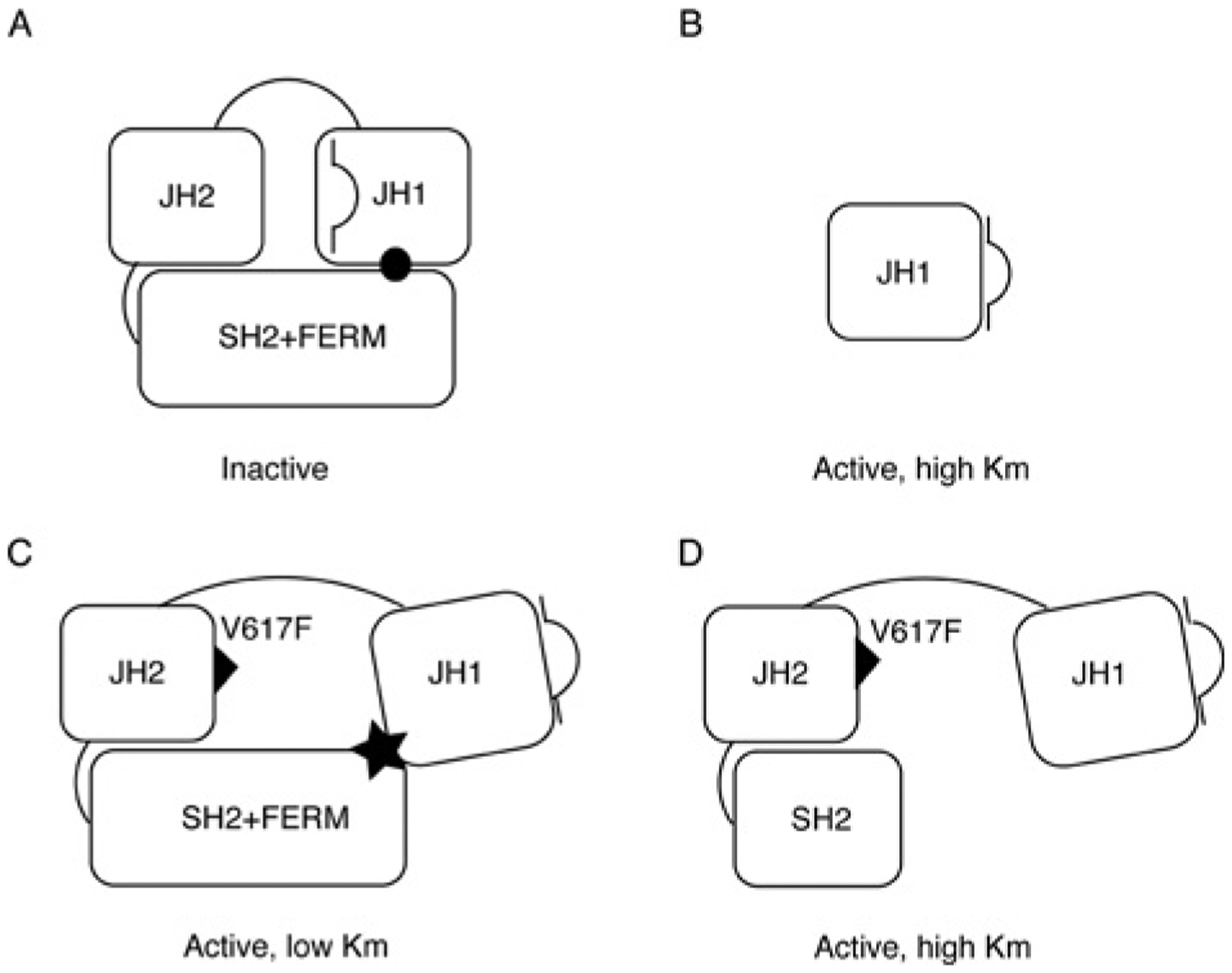
(A) Wild-type JAK2 in the basal state is auto-inhibited by intramolecular interactions. The interaction between JH2 and JH1 keeps the activation loop folded inside the ATP-binding pocket, and the interaction between the FERM domain and JH1 is inhibitory (indicated by ●). (B) JH1 in isolation is active, but has a high Km. (C) In JAK2(V617F), the inhibitory JH1–JH2 interaction is disrupted so that the activation loop is released, and the FERM domain participates in stabilizing the JH1 domain in a conformation that has a lower Km compared with JH1 alone (indicated by a filled star). (D) In the absence of the FERM domain, although the inhibitory JH1–JH2 interaction is disrupted by V617F, JH1 cannot adopt the conformation with the lower Km towards the substrate.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Yu Zhou (University of California, San Francisco, CA, U.S.A.), Dr Qiuxing Jiang (University of Texas Southwestern Medical Center, Dallas, TX, U.S.A.), Dr Nai-wen Chi (University of California, San Diego, CA, U.S.A.) and Dr Xuewu Zhang (University of Texas Southwestern Medical Center, Dallas, TX, U.S.A.) for reagents and helpful discussions, and Hongyun Dong for technical assistance.
FUNDING
This work was supported by the National Institutes of Health [grant number R01 HL089966].
Abbreviations used:
- ECL
enhanced chemiluminescence
- FERM
4.1/ezrin/radixin/moesin
- GFP
green fluorescent protein
- GST
glutathione transferase
- HA
haemagglutinin
- HEK
human embryonic kidney
- HRP
horseradish peroxidase
- JAK
Janus tyrosine kinase
- JH1
JAK homology domain 1
- MPN
myeloproliferative neoplasm
- SCID
severe combined immunodeficiency
- SH2
Src homology 2
- STAT
signal transducer and activator of transcription
REFERENCES
- 1.Yamaoka K, Saharinen P, Pesu M, Holt VE III, Silvennoinen O and O’Shea JJ (2004) The Janus kinases (Jaks). Genome Biol. 5, 253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Levine RL, Pardanani A, Tefferi A and Gilliland DG (2007) Role of JAK2 in the pathogenesis and therapy of myeloproliferative disorders. Nat. Rev. Cancer 7, 673–683 [DOI] [PubMed] [Google Scholar]
- 3.Haan C, Kreis S, Margue C and Behrmann I (2006) Jaks and cytokine receptors: an intimate relationship. Biochem. Pharmacol 72, 1538–1546 [DOI] [PubMed] [Google Scholar]
- 4.Huang LJ, Constantinescu SN and Lodish HF (2001) The N-terminal domain of Janus kinase 2 is required for Golgi processing and cell surface expression of erythropoietin receptor. Mol. Cell 8, 1327–1338 [DOI] [PubMed] [Google Scholar]
- 5.Tong W, Sulahian R, Gross AW, Hendon N, Lodish HF and Huang LJ (2006) The membrane-proximal region of the thrombopoietin receptor confers its high surface expression by JAK2-dependent and -independent mechanisms. J. Biol. Chem 281, 38930–38940 [DOI] [PubMed] [Google Scholar]
- 6.Argetsinger LS, Kouadio JL, Steen H, Stensballe A, Jensen ON and Carter-Su C (2004) Autophosphorylation of JAK2 on tyrosines 221 and 570 regulates its activity. Mol. Cell. Biol 24, 4955–4967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ishida-Takahashi R, Rosario F, Gong Y, Kopp K, Stancheva Z, Chen X, Feener EP and Myers MG Jr (2006) Phosphorylation of Jak2 on Ser523 inhibits Jak2-dependent leptin receptor signaling. Mol. Cell. Biol 26, 4063–4073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mazurkiewicz-Munoz AM, Argetsinger LS, Kouadio JL, Stensballe A, Jensen ON, Cline JM and Carter-Su C (2006) Phosphorylation of JAK2 at serine 523: a negative regulator of JAK2 that is stimulated by growth hormone and epidermal growth factor. Mol. Cell. Biol 26, 4052–4062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haan S, Margue C, Engrand A, Rolvering C, Schmitz-Van de Leur H, Heinrich PC, Behrmann I and Haan C (2008) Dual role of the Jak1 FERM and kinase domains in cytokine receptor binding and in stimulation-dependent Jak activation. J. Immunol 180, 998–1007 [DOI] [PubMed] [Google Scholar]
- 10.Zhou YJ, Chen M, Cusack NA, Kimmel LH, Magnuson KS, Boyd JG, Lin W, Roberts JL, Lengi A, Buckley RH et al. (2001) Unexpected effects of FERM domain mutations on catalytic activity of Jak3: structural implication for Janus kinases. Mol. Cell 8, 959–969 [DOI] [PubMed] [Google Scholar]
- 11.Luo H, Hanratty WP and Dearolf CR (1995) An amino acid substitution in the Drosophila hopTum-l Jak kinase causes leukemia-like hematopoietic defects. EMBO J. 14, 1412–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harrison DA, Binari R, Nahreini TS, Gilman M and Perrimon N (1995) Activation of a Drosophila Janus kinase (JAK) causes hematopoietic neoplasia and developmental defects. EMBO J. 14, 2857–2865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boudeau J, Miranda-Saavedra D, Barton GJ and Alessi DR (2006) Emerging roles of pseudokinases. Trends Cell Biol. 16, 443–452 [DOI] [PubMed] [Google Scholar]
- 14.Saharinen P, Takaluoma K and Silvennoinen O (2000) Regulation of the Jak2 tyrosine kinase by its pseudokinase domain. Mol. Cell. Biol 20, 3387–3395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saharinen P and Silvennoinen O (2002) The pseudokinase domain is required for suppression of basal activity of Jak2 and Jak3 tyrosine kinases and for cytokine-inducible activation of signal transduction. J. Biol. Chem 277, 47954–47963 [DOI] [PubMed] [Google Scholar]
- 16.Saharinen P, Vihinen M and Silvennoinen O (2003) Autoinhibition of Jak2 tyrosine kinase is dependent on specific regions in its pseudokinase domain. Mol. Biol. Cell 14, 1448–1459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu X, Levine R, Tong W, Wernig G, Pikman Y, Zarnegar S, Gilliland DG and Lodish H (2005) Expression of a homodimeric type I cytokine receptor is required for JAK2V617F-mediated transformation. Proc. Natl. Acad. Sci. U.S.A 102, 18962–18967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu X, Huang LJ and Lodish HF (2008) Dimerization by a cytokine receptor is necessary for constitutive activation of JAK2V617F. J. Biol. Chem 283, 5258–5266 [DOI] [PubMed] [Google Scholar]
- 19.Barahmand-Pour F, Meinke A, Groner B and Decker T (1998) Jak2-Stat5 interactions analyzed in yeast. J. Biol. Chem 273, 12567–12575 [DOI] [PubMed] [Google Scholar]
- 20.Fujitani Y, Hibi M, Fukada T, Takahashi-Tezuka M, Yoshida H, Yamaguchi T, Sugiyama K, Yamanaka Y, Nakajima K and Hirano T (1997) An alternative pathway for STAT activation that is mediated by the direct interaction between JAK and STAT. Oncogene 14, 751–761 [DOI] [PubMed] [Google Scholar]
- 21.Lindauer K, Loerting T, Liedl KR and Kroemer RT (2001) Prediction of the structure of human Janus kinase 2 (JAK2) comprising the two carboxy-terminal domains reveals a mechanism for autoregulation. Protein Eng. 14, 27–37 [DOI] [PubMed] [Google Scholar]
- 22.Lietha D, Cai X, Ceccarelli DF, Li Y, Schaller MD and Eck MJ (2007) Structural basis for the autoinhibition of focal adhesion kinase. Cell 129, 1177–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Funakoshi-Tago M, Pelletier S, Moritake H, Parganas E and Ihle JN (2008) Jak2 FERM domain interaction with the erythropoietin receptor regulates Jak2 kinase activity. Mol. Cell. Biol 28, 1792–1801 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.