

Abstract
Cell cycle-dependent redox changes can mediate transient covalent modifications of cysteine thiols to modulate the activities of regulatory kinases and phosphatases. Our previously reported finding that protein cysteine oxidation is increased during mitosis relative to other cell cycle phases suggests that redox modifications could play prominent roles in regulating mitotic processes. The Aurora family of kinases and their downstream targets are key components of the cellular machinery that ensures the proper execution of mitosis and the accurate segregation of chromosomes to daughter cells. In this study, X-ray crystal structures of the Aurora A kinase domain delineate redox-sensitive cysteine residues that, upon covalent modification, can allosterically regulate kinase activity and oligomerization state. We showed in both Xenopus laevis egg extracts and mammalian cells that a conserved cysteine residue within the Aurora A activation loop is crucial for Aurora A activation by autophosphorylation. We further showed that covalent disulfide adducts of this residue promote autophosphorylation of the Aurora A kinase domain. These findings reveal a potential mechanistic link between Aurora A activation and changes in the intracellular redox state during mitosis, as well as provide insights into how novel small molecule inhibitors may be developed to target specific subpopulations of Aurora A.
INTRODUCTION
The Aurora kinases are key mitotic kinases conserved in all eukaryotes (1). Three Aurora kinase paralogs are expressed in mammals and share a highly conserved C-terminal Ser/Thr protein kinase domain. Aurora A plays a critical role in regulating centrosome maturation, mitotic entry, and spindle organization as well as correcting improper kinetochore-microtubule attachments near spindle poles (2–4), while Aurora B plays a similar role in destabilizing improper kinetochore-microtubule attachments at the metaphase plate (5, 6). Aurora A is diffusely distributed at low levels in the cytoplasm during interphase but becomes concentrated at centrosomes and spindle microtubules proximal to the spindle poles during late G2 and M phases (7). Aurora B localizes to centromeres in early mitosis and subsequently to the central spindle during anaphase and to the cleavage furrow and midbody during cytokinesis (7). Aurora C shares similar functions and localizations as Aurora B, but its expression is normally restricted to germ cells and is required for spermatogenesis and oocyte development (8).
Because Aurora A and B are expressed in all mitotically active cells and are increased in abundance during late G2 and M phases (7), it is not surprising that the Aurora kinases are overexpressed in multiple cancer types (9). In particular, the gene encoding Aurora A, located within chromosome 20q13, is frequently amplified in breast, colorectal and bladder tumors, and also in ovarian, prostate, neuroblastoma and cervical cancer cell lines (10). Aurora A overexpression is associated with genomic instability and is a poor prognostic marker in patients with head and neck squamous cell carcinomas, ovarian and gastrointestinal tumors, colorectal cancer liver metastases, glioblastomas, and breast carcinomas. These findings have led to intense pharmacological interest in developing small molecule inhibitors of Aurora A. Despite promising results seen in pre-clinical models, none of the ATP-competitive Aurora A inhibitors available to date have shown efficacy in the treatment of cancer patients (11, 12). Whether covalent or allosteric inhibitors might function as better inhibitors is not known, but intriguingly, several type II Aurora A inhibitors that specifically induce an inactive conformation of the kinase domain have been shown to increase survival in a mouse model of N-Myc-driven neuroblastoma by disrupting the ability of Aurora A to bind to and stabilize N-Myc (13, 14) independent of its catalytic activity. Clearly, a better understanding of the mechanisms that control Aurora A kinase localization, activity, structural conformation and interactions with other proteins could lead to more effective development and application of Aurora A inhibitors, both as cancer therapeutics and as research tools.
The regulation of Aurora A activity and function is complex and depends on both the subcellular localization of different pools of Aurora A, and the presence of specific binding partners (15, 16). The best characterized among these interactors are Bora, CEP192 and Targeting protein for Xklp2 (TPX2), which bind directly to Aurora A in a mutually exclusive manner (16). TPX2 localizes Aurora A to spindle microtubules (17), and its binding allosterically activates the Aurora A kinase domain by stabilizing an active conformation of the activation segment (18, 19). Bora binds to both Aurora A and Polo-like kinase 1 (Plk1) specifically in the cytoplasm, followed by Plk1 activation by promoting Aurora A-mediated phosphorylation of Thr210 within the Plk1 activation loop (20, 21). Plk1, in turn, then promotes activation of the cyclin-dependent kinase 1 (Cdk1)/cyclin B complex to trigger mitotic entry (22–24).
An alternative mechanism is involved in regulating Aurora A and Plk1 activity at centrosomes (25, 26). There, CEP192 recruits both Aurora A and Plk1 to centrosomes where Aurora A undergoes activation through autophosphorylation of Thr288 within its activation loop, through a poorly understood process. The activated Aurora A subsequently phosphorylates and activates centrosomal Plk1. This centrosomal Aurora A-Plk1 phosphorylation cascade is required for centrosome maturation and microtubule nucleation, as well as for centrosome separation and bipolar spindle formation (16, 26, 27).
Although Xenopus CEP192 (xCEP192) is required for recruitment of Xenopus Aurora A (xAurora A) to centrosomes, experiments in the Xenopus egg extract system have shown that xAurora A activation/autophosphorylation can also be induced in the absence of centrosomes by forced dimerization through addition of a bivalent antibody to xAurora A (anti-xAurora A) to the egg extracts (25). However, no xAurora A autophosphorylation is observed if the extracts are first depleted of xCEP192 prior to addition of the anti-xAurora A dimerizing antibody. This suggests that in addition to recruiting xAurora A to centrosomes and facilitating dimerization, other factors are required for xAurora A activation. We believe this activation process involves redox modifications of Aurora A itself.
We have previously reported that overall levels of protein thiol oxidation increase as mammalian cells progress through the cell cycle (28). Elegant studies by Rhee, Finkel, Carroll and their colleagues, and others have shown that modification of cysteine residues by thiol oxidation can directly regulate the activity of proteins, including cell cycle regulatory kinases and phosphatases (29–36). Furthermore antioxidant and redox sensor proteins such as peroxiredoxins also contain highly reactive cysteine residues (35, 37). Reactive cysteine residues in all of these proteins can be reversibly oxidized to sulfenic and sulfinic acids (38, 39), or further oxidized irreversibly to sulfonic acid. Cysteine sulfenic acids can further react with other thiols, including other protein cysteine residues and thiol-containing metabolites, to form a range of thiol-disulfide species that in turn can undergo further thiol-disulfide exchange reactions. Through these exchanges, redox signals leading to disulfide bond formation can be transduced directly onto signaling molecules and from sensor proteins to a wide range of target proteins. Links between redox modifications and cell cycle regulation have previously been reported, including the activation of growth factor signaling pathways via the oxidative modifications of cysteine residues that stimulate receptor tyrosine kinases and inhibit protein tyrosine phosphatases (34, 40, 41). Reversible oxidative inhibition of Cdc14B phosphatase to promote CDK1 signaling during mitosis, for example, has also been reported (42). Along these lines, we previously reported that overall protein thiol oxidation is increased during mitosis relative to other cell cycle phases (28), which likely reflects the existence of additional redox-dependent regulatory mechanisms with important roles in mitosis. In a search for new inhibitors of Aurora A, we identified several redox-sensitive cysteine residues within the Aurora A kinase domain. Our data indicate that Cys290 within the activation loop is essential for Aurora A autophosphorylation at Thr288, and that disulfide modifications of Cys290 prime the kinase domain for autophosphorylation upon dimerization.
RESULTS
Conserved redox-sensitive cysteines within the Aurora A kinase domain can be covalently modified to induce an inactive homodimeric conformation
In an effort to identify novel small molecule inhibitors of the Aurora A kinase domain, we created a chimeric construct consisting of a fragment of TPX2 (residues 7 to 20) fused to the N-terminus of the Aurora A kinase domain (residues 116 to 389), hereafter referred to as t-Aurora A. Like other AGC family kinases, the Aurora A kinase domain contains a conserved hydrophobic patch adjacent to helix C, commonly referred to as the PDK1-interacting fragment (PIF) pocket. A C-terminal hydrophobic motif in most AGC kinases docks in cis on to the PIF pocket to stabilize an active conformation of the kinase domain. Although Aurora A does not contain a hydrophobic motif, Tyr8 and Tyr10 within TPX2 functionally serve this purpose in trans (18, 19). Our t-Aurora A construct thus contains a hydrophobic motif in cis, which likely accounts for the improved overall stability relative to constructs of the Aurora A kinase domain alone and allowed us to obtain more ordered crystals with improved diffraction quality. Similar to other Aurora A kinase domain constructs, t-Aurora expressed in bacteria in the absence of phosphatase co-expression or treatment is phosphorylated on both Thr287 and Thr288 within the activation loop. The structure of this autophosphorylated construct was then determined under a variety of chemical modification and buffer conditions, revealing unexpected conformational differences in the kinase domain depending on the modification state of key Cys residues. As expected, the structure of fully reduced and autophosphorylated t-Aurora A showed a monomeric kinase domain adopting the canonical protein kinase fold with an N-terminal lobe mainly consisting of a β-sheet and two α-helices (αB and αC), and a larger predominantly α-helical C-terminal lobe (Fig. 1A). This structure closely resembles the previously determined structure of the Aurora A kinase domain in complex with the N-terminal 43 residues of TPX2 (PDB code 1OL5) (18), with the two structures sharing an rms deviation of 0.64 Å over 264 Cα atoms and show an active conformation with a fully ordered activation segment phosphorylated on Thr288 and Thr287. However, a crystal structure of t-Aurora A obtained with cacodylate as the pH buffering agent, unexpectedly revealed covalent modification of Cys247 and Cys290 with dimethyl arsenic adducts (43), inducing a conformational rearrangement of the kinase domain (Fig. 1A, shown in blue). Furthermore, the structure of this cacodylate-modified form of t-Aurora A showed that the kinase domain dimerized via a displaced activation segment that is swapped between two symmetry-related molecules within the crystal structure (Fig. 1B), with the dimerized kinase domains oriented with their N-terminal lobes pointing in roughly orthogonal directions.
Fig. 1. Crystallization of the Aurora A kinase domain with cacodylate buffer identifies two conserved redox-active cysteines and an activation segment-swapped dimer.
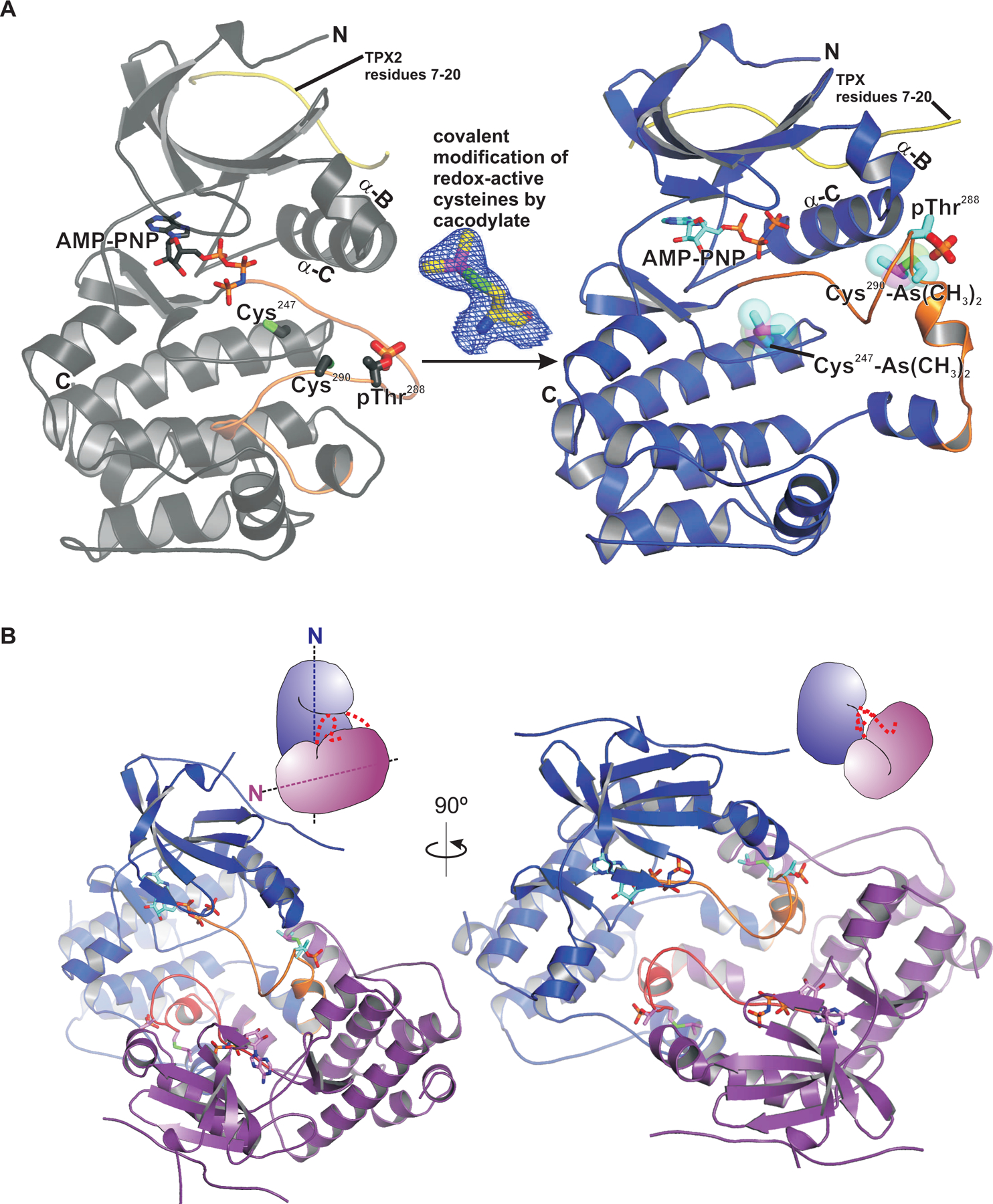
(A) Left, the crystal structure of the Aurora A kinase domain in an active conformation, shown in grey ribbons representation, adopts the canonical kinase domain fold with N-terminal and C-terminal lobes. The ATP binding pocket containing bound AMP-PNP, shown in stick representation (with carbons colored dark grey, oxygens red, and phosphorus orange) is located within the active site cleft in between the two lobes. Side chains of Cys247, Cys290, and phosphorylated Thr288 are also shown in stick representation. Right, the crystal structure of the Aurora A kinase domain obtained with cacodylate buffer, shown in blue ribbons representation, depicting covalent modification of Cys247 and Cys290 (shown in stick representation with cyan carbons, overlayed with transparent space filled rendering) and a large displacement of the activation segment (shown in orange). TPX2 residues 7–20 are shown in yellow. (B) The crystal structure of the activation segment-swapped dimer of the cacodylate-modified Aurora kinase domain. Monomers are colored blue and purple and shown as ribbons representations, with a cartoon indicating the relative orientation of each monomer in the upper right of the structure. The activation segment is orange. Symmetry-related kinase domain monomers within the crystal structure can be seen in a dimeric arrangement with their activation segments exchanged between the monomers at the dimer interface. The monomers are oriented with their N-terminal lobes pointing in near-orthogonal directions, as indicated by the dotted axis lines in the cartoon representation.
It is well known that the conformation of the activation segment within the C-terminal lobe is a key determinant of whether a kinase domain is in a catalytically active or inactive state (44). In particular, the conserved DFG motif at the N-terminal end of the activation segment is observed in multiple conformations in kinase domain crystal structures, with the DFG-in conformation being required for kinase activity. In the DFG-in conformation, the phenylalanine side chain within the DFG motif points inward and is stacked within a conserved column of buried hydrophobic side chains referred to as the regulatory spine (45, 46). Notably, the activation segment in our t-Aurora A structure with cacodylate-modified cysteine residues, is in an inactive DFG-out conformation, in which the side chain of Phe275 within the DFG motif is flipped outward into the ATP binding pocket (Fig. 2A), resulting in the misplacement of key catalytic residues, including the Mg2+-coordinating Asp274 within the DFG motif, and adenylyl-imidodiphosphate (AMP-PNP, a non-hydrolysable analogue of ATP) bound in a distorted non-catalytic conformation. Superposition of the DFG-out cacodylate-modified form with the fully reduced and active DFG-in t-Aurora A structure shows a steric clash between the dimethyl-arsenic adduct on Cys247 with the side chain of Phe275 in the DFG-in conformation, indicating that covalent modification of the Cys247 side chain would allosterically inhibit Aurora A kinase activity. In addition, an overall widening of the active site cleft and displacement of the side chain of Glu181 on helix αC results in an opening of a small hydrophobic cavity adjacent to the ATP-binding pocket in the cacodylate-modified structure, where it is occupied by an oxidized DTT molecule. This cavity is also observed in the structure of an Aurora A kinase domain bound to a specific 5-aminopyrimidinyl quinazoline inhibitor (PDB code 2C6E), where it is occupied by the benzamide moiety of this type II kinase inhibitor (47) (Fig. 2B). These observations suggest that compounds that selectively modify Cys247 may provide a scaffold for the development of novel covalent non-ATP-competitive Aurora A inhibitors, and further suggests that such inhibitors which re-structure the active site may facilitate the binding of, and thereby work in conjunction with, existing Aurora A inhibitors like the 5-aminopyrimidinyl quinazoline compounds.
Fig. 2. Cacodylate-modification of Cys247 in the Aurora A kinase domain induces an inactive conformation, suggesting that compounds that selectively modify Cys247 may provide novel Aurora A inhibitors.
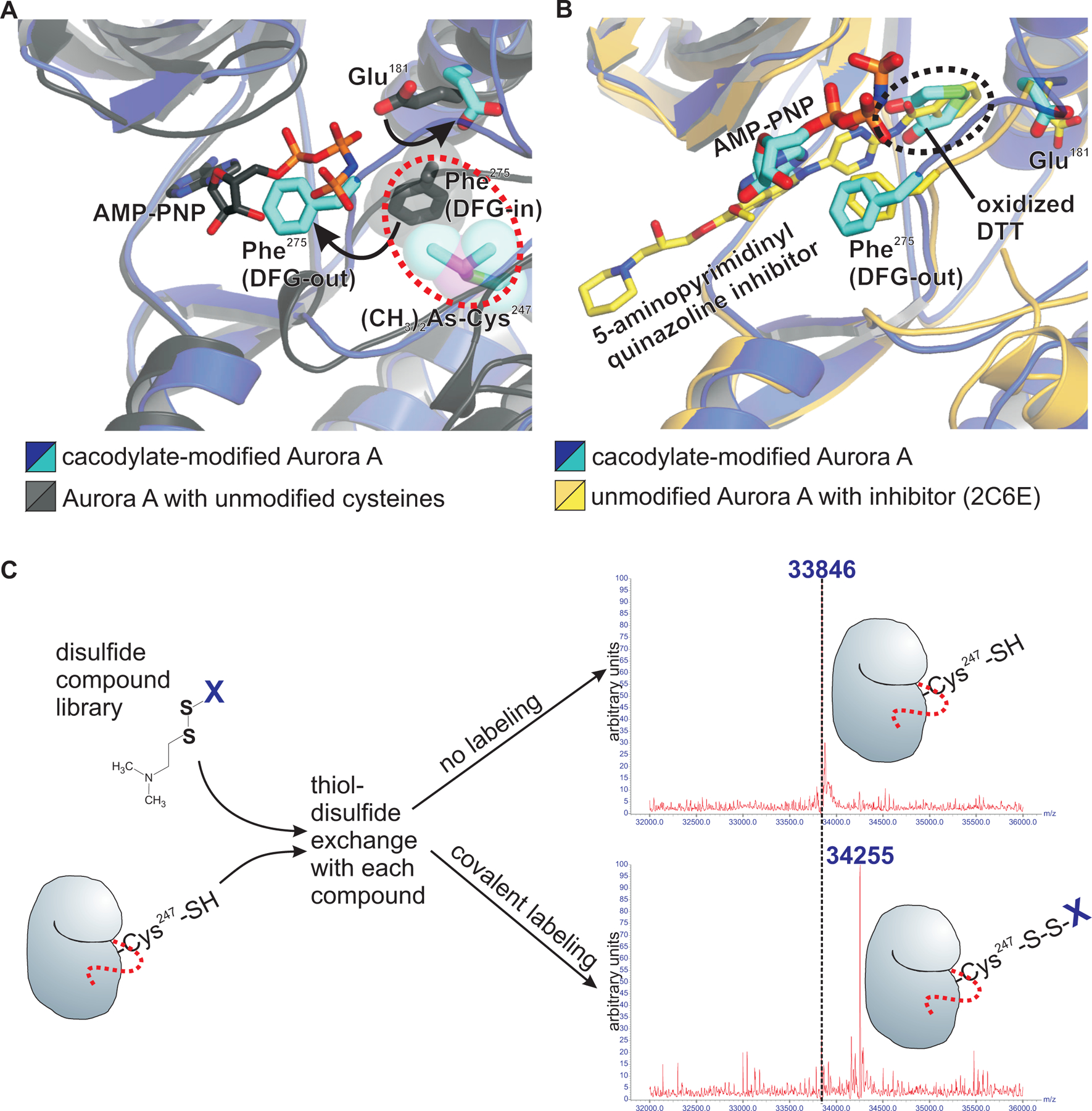
(A) Superposition of the active sites in the cacodylate-modified Aurora A kinase domain (blue and cyan) with the corresponding unmodified structure (grey and dark grey) shows spatial overlap between Phe275 in the active DFG-in conformation (dark grey, in stick representation) and the dimethyl arsenic adduct of the cacodylate-modified Cys247 (cyan and purple in stick representation, overlayed with transparent space filled rendering). AMP-PNP in the active structure (stick representation with dark grey carbons) spatially overlaps with Phe275 in the inactive cacodylate-modified DFG-out structure (cyan), and the side chain of Glu181 is also displaced in the cacodylate-modified structure (cyan) relative to the unmodified structure (dark grey). (B) The displacement of the Glu181 side chain expands the ATP-binding pocket, allowing an oxidized DTT molecule to occupy this space in the cacodylate-modified Aurora A structure. Superposition of the previous structure of the Aurora A kinase domain in complex with a 5-aminopyrimidinyl quinazoline inhibitor (beige and yellow; PDB code 2C6E) shows how this expanded ATP-binding pocket accommodates existing Aurora A-selective inhibitors. (C) Schematic of the experiment in which an Aurora A kinase domain construct containing a single cysteine (Cys247) was used for a mass spectrometry-based high throughput tethering screen of 880 disulfide-containing compounds (listed in data file S1) to identify additional covalent modifiers of Cys247. Each compound was incubated separately with Aurora A kinase domain, to allow thiol-disulfide exchange with the Cys247 side chain thiol in the presence of ß-mercaptoethanol, a non-specific disulfide reducing agent. Stable covalent labeling of Cys247 under these mildly reducing conditions requires additional stabilizing contacts between the particular compound and residues in Aurora A in close proximity to Cys247. These stable disulfide adducts were then detected by an increased total mass of the protein using mass spectrometry (data file S1).
A tethering screen of Cys247-directed disulfide compounds identifies the activation loop cysteine as a site of redox modification that promotes Aurora A autophosphorylation
To identify Cys247-modifying Aurora A inhibitors, we next conducted a high throughput mass spectrometry-based tethering screen (48) of 880 disulfide containing molecules to identify compounds that can stably label Cys247 through thiol-disulfide exchange, with the covalent labeling detectable as an increase in the total mass of the protein (as modelled in Fig. 2C). To selectively target Cys247 in the screen, we used a mutant t-Aurora A construct in which the other two cysteines were mutated (C290A and C319V). The choice of amino acid substitutions for cysteine were chosen based on structural data, with alanine chosen where valine would be expected to cause steric clashes. A number of cysteine-modifying compounds were identified (data file S1), from which a subset was selected, based on diversity of the chemical structures and availability of the compounds, for co-crystallization trials using the wild-type t-Aurora A construct lacking any cysteine mutations. Crystals were obtained with compounds 7–80 and 8–34. Unexpectedly, the resulting structures revealed specific disulfide labeling of Cys290 within the activation loop two residues C-terminal to the site of autophosphorylation on Thr288, and no modification whatsoever of Cys247. Both the 7–80– and 8–34–modified structures were highly similar and showed a symmetric activation segment-swapped dimer, with each of the monomers oriented with their N-terminal lobes pointing in the same direction (Fig. 3, A and B). This is distinct from the dimer configuration observed for cacodylate-modified t-Aurora A, in which the monomers are oriented with their N-terminal lobes pointing in orthogonal directions at roughly 90° relative to each other (Fig. 1B). In particular, the active sites of the 7–80- and 8–34-modified structures revealed an active DFG-in conformation and have the activation segments of each kinase well positioned for trans-phosphorylation. For reference, comparison with the active Akt kinase domain in complex with a GSK3ß substrate peptide (PDB code 1O6L; Fig. 3C) (49) shows that the Thr288 sidechain of each Aurora A monomer in both the 7–80– and 8–34–modified structures is well positioned within the active site of the other monomer for phosphorylation in trans (Fig. 3C). Our structures contrast with a previously published structure of an unphosphorylated Aurora A kinase domain in complex with a TPX2 fragment, containing reduced cysteine residues (PDB code 4C3P) (50). Whereas this unphosphorylated and fully reduced Aurora A-TPX2 complex showed a similar activation segment-swapped dimerization mode as our 7–80– and 8–34–modified structures, key active site residues within our 7–80– and 8–34–modified structures are positioned more favorably for catalysis. The fully reduced Aurora A-TPX2 fragment complex contained a displaced catalytic base (Glul81) in both monomers, and either a misplaced or disordered region of the activation loop containing Thr288 (Fig. 3, D and E). These structural comparisons emphasize the importance of how the 7–80 and 8–34 disulfide adducts on Cys290 stabilize a more trans-phosphorylation–competent conformation within the Aurora A kinase domain dimer than what was previously observed in the fully reduced Aurora A-TPX2 fragment complex. These findings point to a previously unrecognized role of C290 within the activation loop in promoting autophosphorylation of Aurora A.
Fig. 3. Crystal structures of Aurora A kinase domain modified with disulfide adducts on C290 show a catalytically active dimer facilitating autophosphorylation in trans.
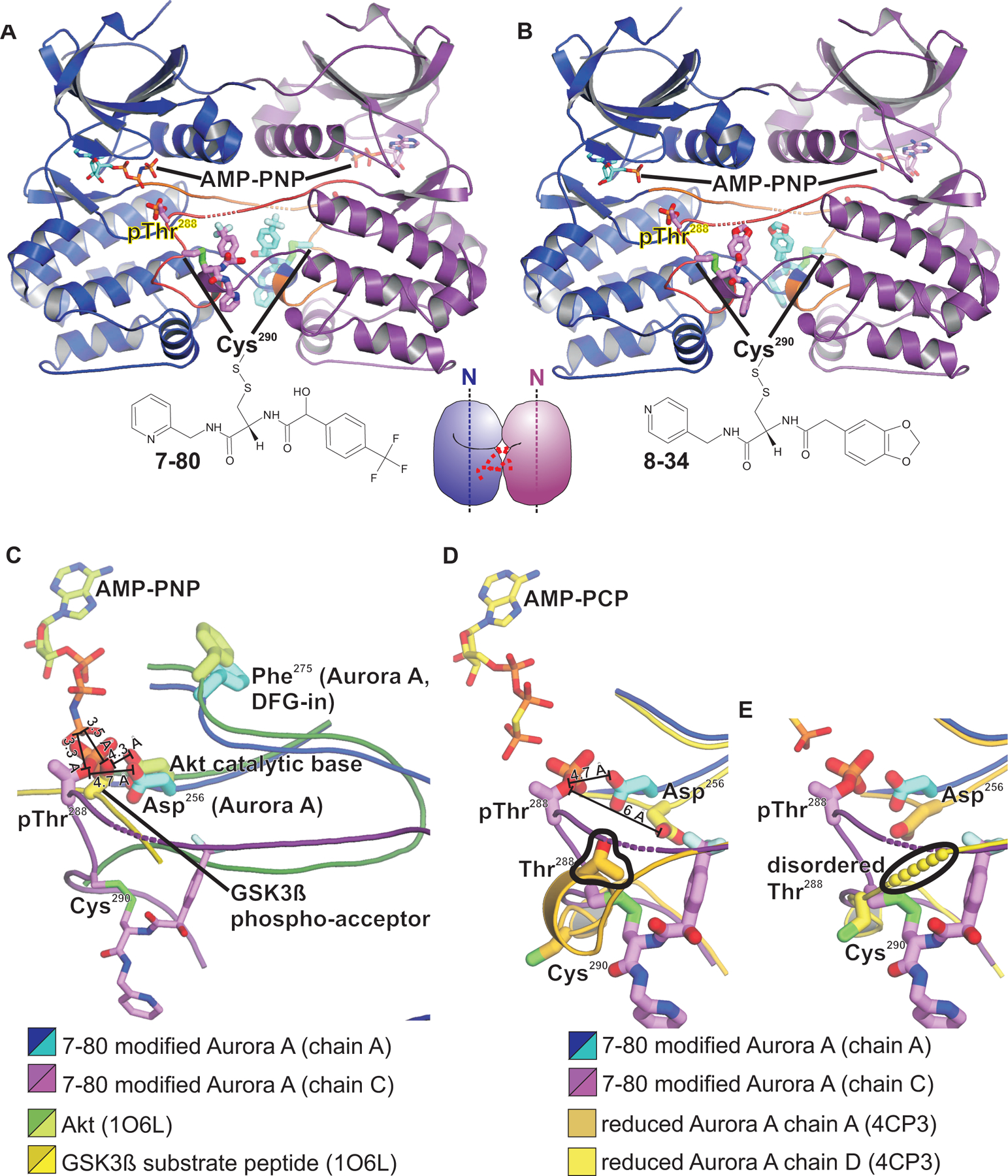
(A and B) Structures of the Aurora A kinase domain modified with compounds 7–80 (A) and 8–34 (B) at Cys290 show an activation segment-swapped dimer, in which the monomers are oriented with their N-terminal lobes pointing in the same direction, as indicated by the dotted axis lines in the cartoon representation in the center bottom. Monomers are colored blue and purple and shown as ribbons representations. The activation segment is orange. This dimer configuration is distinct from the near orthogonal arrangement seen in the cacodylate-modified structure (Fig. 1B). (C) Superposition of the active site regions of the 7–80 modified Aurora A structure with the Akt kinase domain (dark green and chartreuse) in complex with AMP-PNP and a GSK3ß substrate peptide (beige and yellow) from PDB code 1O6L. The active site of one monomer of the 7–80 modified Aurora A is shown in blue and cyan, with the Thr288/Cys290 activation loop from the other monomer shown in purple and magenta. Sulfur atoms of Cys290 are colored bright green. (D and E) Structure of the 7–80-modified structure, superimposed and contrasted with each of the monomers from a structure of a fully reduced and unphosphorylated Aurora A kinase domain in a similar dimer configuration (PDB code 4C3P). Large differences in the positioning of the phopsho acceptor residue (Thr288) and of the catalytic base (Asp256) in the 4C3P structure (emphasized with black outlines) can be seen between the 7–80-modified and the unmodified structures.
Mutation of the activation loop cysteine impairs Aurora A autophosphorylation in Xenopus egg extracts and in mammalian cells
To confirm the importance of the activation loop cysteine in promoting Aurora A autophosphorylation in a more physiologically relevant context, we used the Xenopus egg extract system which, as noted above, represents a convenient tool for dissecting the mechanisms of Aurora A activation. A metaphase-arrested Xenopus egg extract was first depleted of endogenous xAurora A using a bead-immobilized xAurora A-binding fragment of xCEP192. The extract was then supplemented with recombinant wild-type or mutant, FLAG-tagged xAurora A proteins (Fig. 4A). xAurora A activation was induced by addition of demembranated sperm nuclei as a source of centrioles (Fig. 4B) or by addition of a bivalent anti-xAurora A antibody to artificially induce xAurora A dimerization (Fig. 4C), and autophosphorylation was monitored as a function of time by SDS-PAGE followed by immunoblotting. Both centriole-induced clustering and antibody-induced dimerization of endogenous, wild type FLAG-tagged xAurora A resulted in robust autophosphorylation on Thr295 (equivalent to human Thr288) (Fig. 4, A and B). No impairment of Thr295 autophosphorylation was observed for the C254V mutant (equivalent to C247V in human Aurora A), which actually showed stronger Thr295 phosphorylation relative to the wild type. Mutation of the activation loop Cys297 (equivalent to human Cys290), however, completely abolished autophosphorylation to a similar level as that seen with the non-phosphorylatable T295A and the D281A kinase-dead mutants (equivalent to human T288A and D274A, respectively). Similar results were obtained using undepleted Xenopus egg extracts, in which exogenous FLAG-tagged wild-type or mutant xAurora A constructs were selectively dimerized and activated by addition of an anti-FLAG antibody (Fig. 4D). Notably, the loss of autophosphorylation observed upon mutation of the activation loop cysteine occurred despite the fact that Xenopus C297A and the human C290A mutants retain catalytic activity (50–52).
Fig. 4. The activation loop cysteine is crucial for Aurora A activation by autophosphorylation.
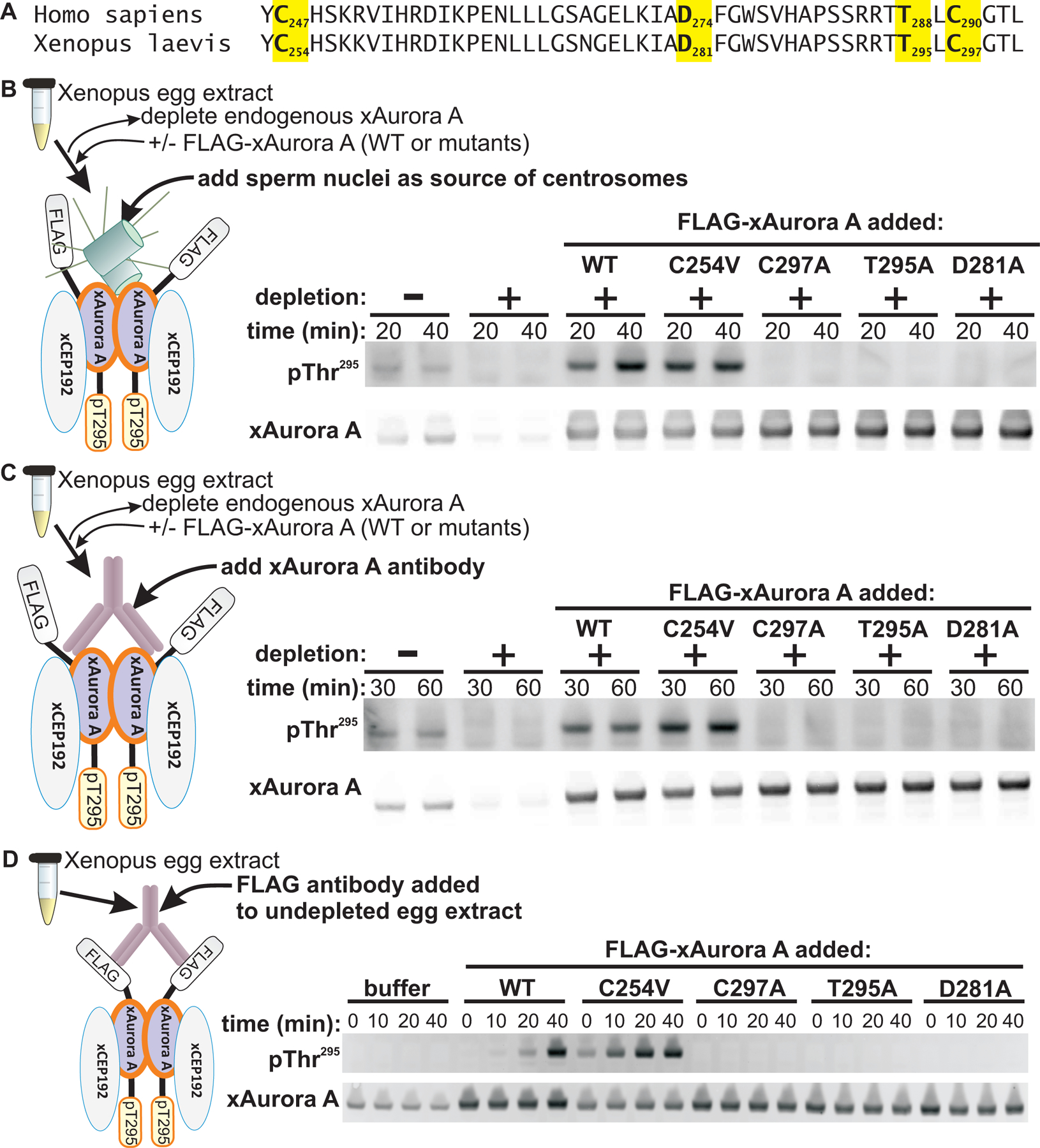
(A) Sequence alignment of human Aurora A (residues 246–293) with X. laevis Aurora A (residues 253–300). The highlighted amino acid residues denote residues mutated in the constructs used in the assays described in the remainder of this figure (B to D). (B to D) Xenopus egg extracts were used for xAurora A activation assays to examine the requirement for the activation loop cysteine (Cys297) of xAurora A in autophosphorylation. Endogenous xAurora A was depleted from Xenopus egg extracts using an immobilized xAurora A-binding fragment of xCEP192. Wild-type and mutant xAurora A constructs were then added to this depleted extract and assayed for activation by addition of either sperm nuclei as a source of centrosomes (B) or by addition of an antibody to xAurora A (C). In (D), xAurora A activation was also assayed using undepleted Xenopus egg extract and selective activation of exogenous wild-type and mutant FLAG-tagged xAurora A constructs using an antibody to FLAG. To the right of each experimental schematic, total and autophosphorylated (pThr295) Aurora A was detected by Western blotting. Blots in (B and D) are representative of 5 independent experiments, and the blots in (C) are representative of 3 independent experiments.
Furthermore, analogous results were obtained in mammalian cells. Stable HeLa cell lines were engineered to express a doxycycline-inducible shRNA to knock down endogenous Aurora A, and shRNA-resistant FLAG-tagged Aurora A constructs driven by a minimal fragment of the native Aurora A promoter (53) (Fig. 5A). Both endogenous and wild-type FLAG-tagged Aurora A showed robust mitotic autophosphorylation on Thr288 (Fig. 5B). Consistent with our findings in the Xenopus egg extract system, mutation of Cys290 to alanine completely eliminated Thr288 phosphorylation, resulting in similar levels of phosphorylation as those observed upon mutation of the phospho-acceptors within the activation loop (T288V, and T287V + T288V), or in the D256N + D274N kinase-dead mutant. In addition, there was also an apparent dominant negative effect of expression of exogenous Aurora A constructs on endogenous Aurora A Thr288 phosphorylation. This likely resulted from exogenous constructs outcompeting endogenous Aurora A for binding to interactors such as CEP192 and TPX2. These interactors are stoichiometrically less abundant than Aurora A (26). Together, these results in the Xenopus egg extract system and in mammalian cells indicate an essential physiological role for Cys290 in Aurora A autophosphorylation on Thr288.
Fig. 5. The activation loop cysteine in Aurora A is critical for autophosphorylation in mammalian cells.
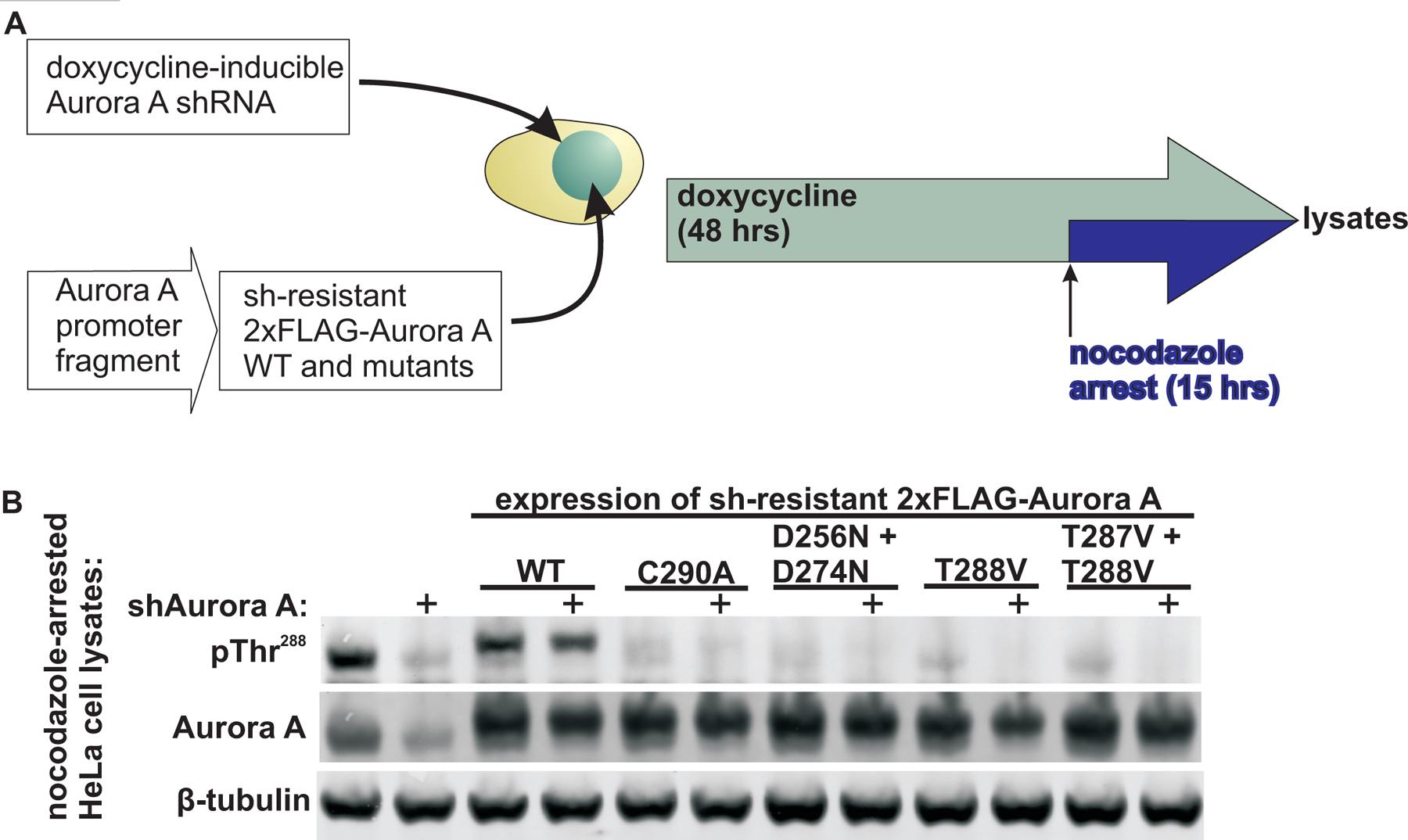
(A) Experimental schematic of the creation of stable HeLa cell lines by incorporating a doxycycline-inducible shRNA against endogenous Aurora A and transfected with or without shRNA-resistant, FLAG-tagged, wild-type and mutant Aurora A constructs driven by a native Aurora A promoter fragment. Following induction of shAurora A, the cells were nocodazole arrested. (B) Western blotting to assess Aurora A autophosphorylation at pThr288 in the nocodazole-arrested lysates described in (A). Total Aurora A and β-tubulin were blotted for reference. Blots are representative of 2 independent experiments.
Oxidative modifications of the activation loop cysteine with coenzyme A (CoAlation) may promote the formation of a disulfide-linked Aurora A kinase domain homodimer
Oxidative modifications of cysteine side chain thiols are increasingly recognized to play important physiological roles in regulating protein structure and function (35, 54). Consistent with this, our structures of 7–80 and 8–34 modified t-Aurora A indicate how disulfide modifications of Cys290 may promote Thr288 autophosphorylation. To further evaluate whether oxidative modifications of Cys290 play a role in Aurora A autophosphorylation, we tested the effect of adding the reducing agent dithiothreitol (DTT) to the Xenopus egg extract-based assay. Given the already strong reducing environment of the cytosol, with concentrations of reduced glutathione in the millimolar range (55), we used a relatively high concentration of DTT in these assays to induce a sufficient redox perturbation. xAurora A autophosphorylation was markedly inhibited by the addition of DTT (Fig. 6A), consistent with the involvement of oxidative modification(s) in xAurora A activation in the context of a cytosolic extract. We cannot, however, completely dismiss the possibility that other redox-sensitive biochemical processes in the Xenopus egg extract may have been disrupted.
Fig. 6. Disulfide modification of Aurora A is involved in its activation by autophosphorylation of its activation loop.
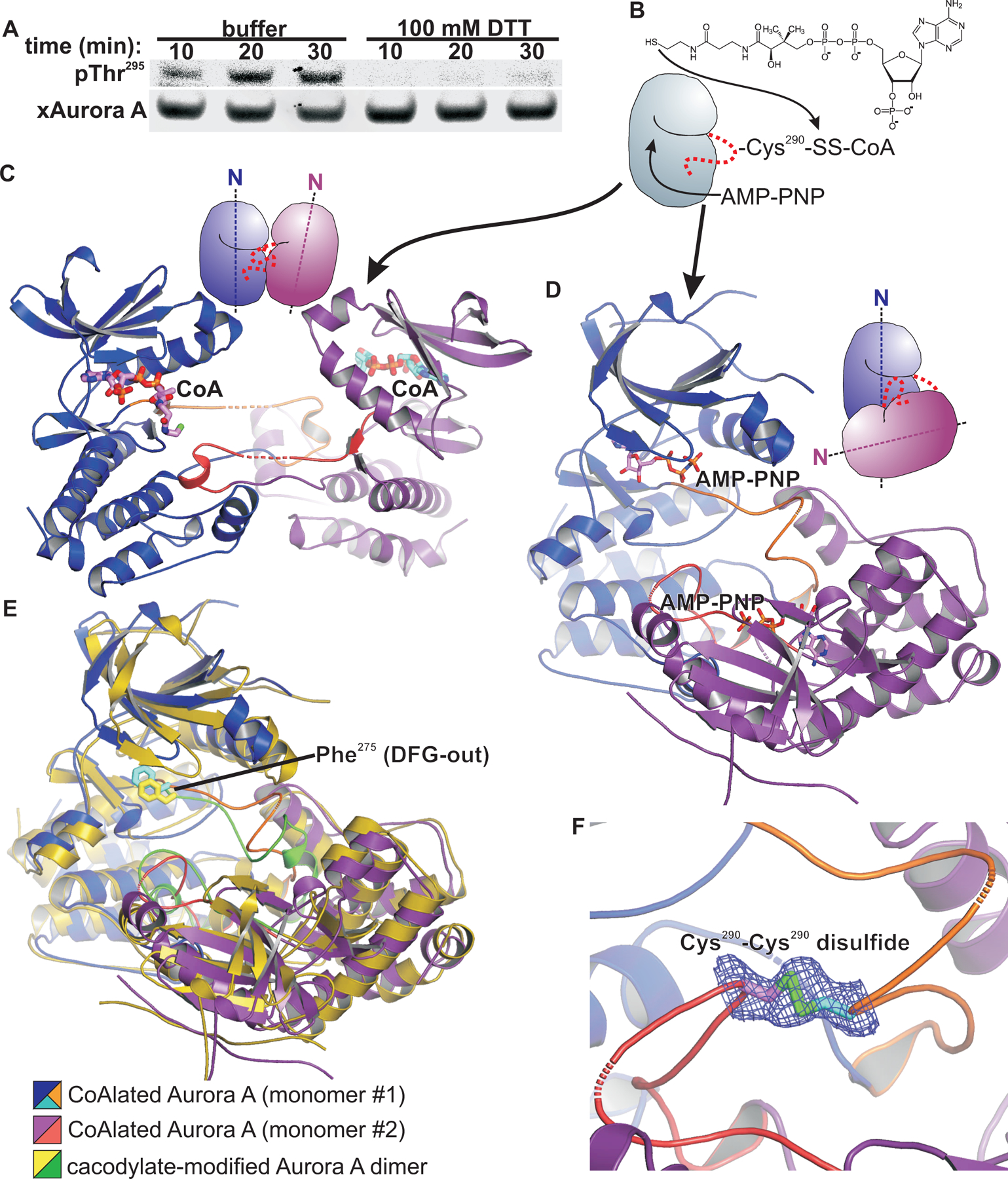
(A) Western blotting for Aurora A autophosphorylation at pThr288 to assess xAurora A activation in Xenopus egg extracts supplemented with demembranated sperm nuclei and exposed to DTT (or buffer, control) for the indicated times. Blots are representative of 4 independent experiments. (B) Schematic for Aurora A kinase domain constructs CoAlated on Cys290 and crystallized in the presence of AMP-PNP. (C) Structure of a wild type Aurora A kinase domain construct CoAlated on Cys290 shows an activation segment-swapped dimer. Monomers are colored blue and purple and shown as ribbons representations, with a cartoon shown above the structure. The activation segments (orange and red) are swapped between the monomers. The CoA adduct of one monomer is bound in the ATP-binding of pocket of the opposing monomer. No electron density was observed for the TPX2 fragment fused to the N-terminus of the Aurora A kinase domain construct used to determine this structure. (D) Structure of a single-cysteine human Aurora A kinase domain construct CoAlated on Cys290 and crystallized in complex with AMP-PNP shows an activation segment-swapped dimer with monomers oriented with their N-terminal lobes pointing in near orthogonal directions (dotted axis lines in the cartoon representation shown in the upper right of the panel), in contrast to the monomer orientation shown in panel C. Monomers are colored blue and purple and shown as ribbons representations. The activation segment is orange. (E) The CoAlated Aurora A kinase domain dimer from (D), with the activation segments colored orange and the DFG phenylalanine in cyan, is shown superimposed on the cacodylate-modified dimer, colored yellow/gold with activation segments colored green. Both structures show a catalytically inactive DFG-out activation segment conformation. (F) A 2mFo-DFc map contoured at 0.5σ shows electron density consistent with a subpopulation of the Aurora A kinase domain molecules in the crystal containing a Cys290-Cys290 symmetric disulfide.
The relevant oxidative modification of Aurora A that occurs in vivo is not known. However, disulfide modification of Aurora A Cys290 by coenzyme A (CoAlation) was recently reported as an inhibitory modification that can occur within mammalian cells under oxidative stress (56, 57). A reported crystal structure of a CoAlated Aurora A kinase domain shows CoA disulfide bonded to the Cys290 thiol with the ADP moiety of CoA inserted into the ATP-binding pocket, accounting for the inhibitory effect of CoAlation on Aurora A kinase activity (57). We hypothesized that, while CoAlated Cys290 is inhibitory in a monomeric Aurora A kinase domain, it might instead promote autophosphorylation in the context of a kinase domain dimer by stabilizing a more catalytically competent conformation, similar to what was observed in our 7–80- and 8–34-modified t-Aurora A structures. To examine this, we CoAlated unphosphorylated wild-type t-Aurora A by thiol-disulfide exchange (Fig. 6B), and crystallized this in the presence of AMP-PNP, with the aim of displacing the ADP moiety of CoA from the ATP-binding pocket. The structure of this wild-type CoAlated t-Aurora A revealed an activation segment-swapped dimer, with the ADP moiety of CoA from one molecule tightly bound in the ATP-binding pocket of the other monomer (Fig. 6C), despite the inclusion of excess AMP-PNP in the buffers during crystallization and subsequent handling and cryoprotection. This was evident by strong electron density for the 3-phosphate group in CoA not present in AMP-PNP. Intriguingly, this dimeric structure shows a more extended arrangement of the swapped activation segments with the kinase domains compared to what was observed in the cacodylate-modified dimer structure (Fig. 1B). Our structure markedly contrasts with the previously reported CoAlated and phosphorylated Aurora A kinase domain structure, which crystallized as a monomer with CoA bound intramolecularly within the ATP-binding pocket (57). In addition, in our wild-type unphosphorylated CoAlated t-Aurora A structure, no electron density was visible for the TPX2 residues fused at the N-terminus, indicating that this segment is now disordered, which contrasts with all of our other t-Aurora A structures determined in this study.
We next prepared a CoAlated and non-phosphorylated form of the C247V + C319V double mutant t-Aurora A for structural analysis. Mass spectrometry data indicated that, in contrast to the wild-type CoAlated construct, only approximately two thirds of the double mutant protein was CoAlated (fig. S1). In contrast to the structure obtained with the wild type unphosphorylated fully CoAlated t-Aurora A, the structure of the C247V + C319V unphosphorylated CoAlated t-Aurora A construct revealed an activation segment swapped dimer in which AMP-PNP was bound at the ATP-binding pocket (Fig. 6D). We did not observe any electron density for CoA. Instead, we observed notable differences in the conformation of the activation segments compared with the cacodylate-modified dimer structure (Figs. 6E and 1B), including weak electron density at the dimer interface consistent with a symmetric disulfide bond between the Cys290 side chains from the two molecules within the dimer (Fig. 6F). Of note, the kinase domain in our CoAlated dimer is also in an inactive DFG-out conformation. Given the incomplete CoAlation of the protein used for crystallization, it is likely that the small amount of t-Aurora A with reduced Cys290 underwent a thiol-disulfide exchange with CoAlated t-Aurora A, resulting in a subpopulation of the dimers in the crystal consisting of disulfide linked monomers. This structure obtained using incompletely CoAlated t-Aurora A therefore suggests that CoAlation may act as a priming modification that facilitates the formation of a symmetric disulfide dimer of the Aurora A kinase domain.
Dimerization of the Aurora A kinase domain via a symmetric disulfide bond involving the activation loop cysteine residues promotes autophosphorylation
Because the structure of the C247V + C319V t-Aurora A construct was obtained using incompletely CoAlated protein, and corresponded to a mixture of predominantly CoAlated protein with a minor component of the disulfide dimer, we next set out to specifically crystallize the disulfide linked form. To do this we created a C247V + D256N + C319V kinase-dead mutant t-Aurora A construct in which Cys290 was the sole cysteine residue. We then used Ellman’s reagent to generate a reactive disulfide form of this t-Aurora A construct (58), which we reacted with unmodified reduced protein. Thiol-disulfide exchange between these two forms of the protein resulted in a t-Aurora A homodimer disulfide bonded via Cys290, which was then specifically purified by size-exclusion chromatography. The crystal structure of this t-Aurora A disulfide homodimer revealed a dimer configuration very similar to what was observed for our cacodylate-modified and double mutant CoAlated dimer structures (Fig. 7, A and B). However, whereas the cacodylate-modified and CoAlated double mutant dimer structures were catalytically inactive with their activation segments in a DFG-out conformation, this kinase-dead disulfide homodimer structure showed an active DFG-in conformation (Fig. 7C). The Cys290-Cys290 disulfide within the homodimer structure showed strong electron density and is positioned at the center of the dimer interface, similar to what was seen in our CoAlated C247V + C319V dimer structure. As a consequence, the Thr288 residue of each monomer is placed at a midpoint position in between the two active sites and is not optimally positioned for phosphorylation in cis or in trans. However, the fact that the activation segment is seen in multiple distinct conformations in the cacodylate-modified, double mutant CoAlated and Cys290-Cys290 disulfide dimer structures suggests that this dimerization mode shared by these forms of t-Aurora A can accommodate a range of activation segment conformations. We therefore speculate that additional activation segment conformations would be accessible within this dimer that would permit movement of each Thr288 residue into one or both active sites for phosphorylation.
Fig. 7. Crystal structure of a disulfide-linked Aurora A kinase domain homodimer.
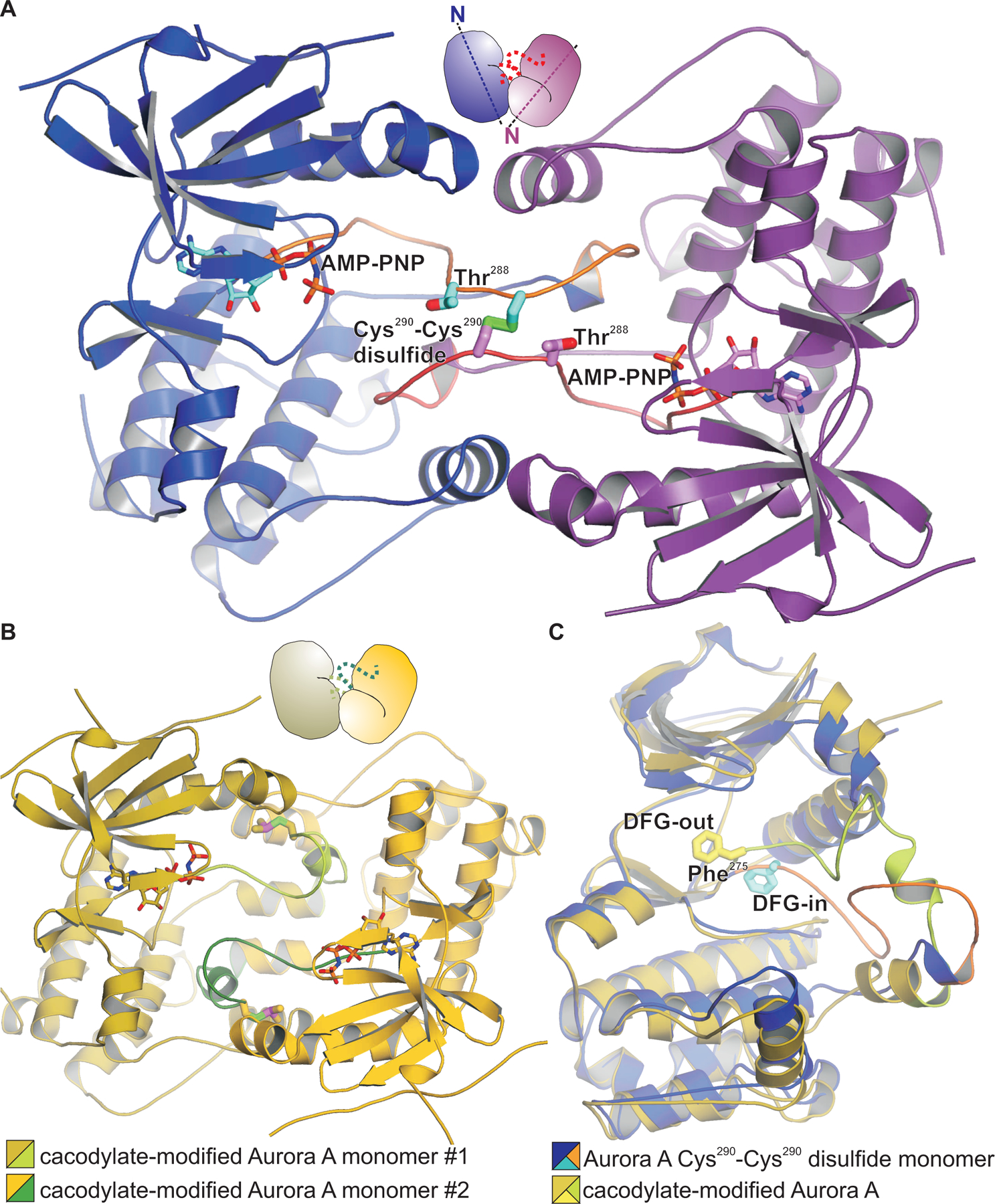
(A) The structure shows an activation segment-swapped dimer with the disulfide bond formed between Cys290 in each monomer (with sulfur atoms colored green) at the center of the dimer interface. Molecules are shown in cartoon representation with the monomers colored blue and magenta, and with a cartoon representation shown above the structure indicating the relative orientations of the monomers. (B) The cacodylate-modified Aurora A kinase domain dimer in an identical orientation as in (A), shows a similar overall dimer configuration but with conformational differences in the activation segments [green in (B) vs orange in (A)]. (C) Monomers of Aurora A from (A) and (B) are shown superimposed. The DFG-in active conformation of a monomer of the Aurora A kinase domain disulfide homodimer (Phe shown in stick representation, colored cyan) contrasts with the inactive DFG-out conformation of the cacodylate-modified kinase domain (colored yellow), and also with the inactive DFG-out conformation of the CoAlated disulfide-linked dimer structure (Fig. 6E).
To directly test this, we examined the ability of the t-Aurora A disulfide dimer to autophosphorylate in vitro. We generated and purified catalytically active disulfide homodimers of wild-type and a C247V + C319V double mutant t-Aurora A constructs. Incubation of these disulfide homodimers with ATP resulted in potent autophosphorylation of the dimer at Thr288 (upper bands), whereas t-Aurora A monomers (lower bands) did not show substantial autophosphorylation at the protein concentrations and incubation times used (Fig. 8A). These findings are consistent with enhanced autophosphorylation occurring intramolecularly within the disulfide homodimer. It is worth noting that we were unable to obtain disulfide dimers in t-Aurora A constructs containing the C290A mutation, even when Cys247 and Cys319 were available, in agreement with a report from Tsuchiya et al (57) that wild type but not C290A Aurora A forms disulfide dimers in vitro upon treatment with H2O2. These findings are consistent with Cys290 being critical for disulfide dimer-enhanced autophosphorylation.
Fig. 8. Disulfide-mediated dimerization of Aurora A promotes autophosphorylation.
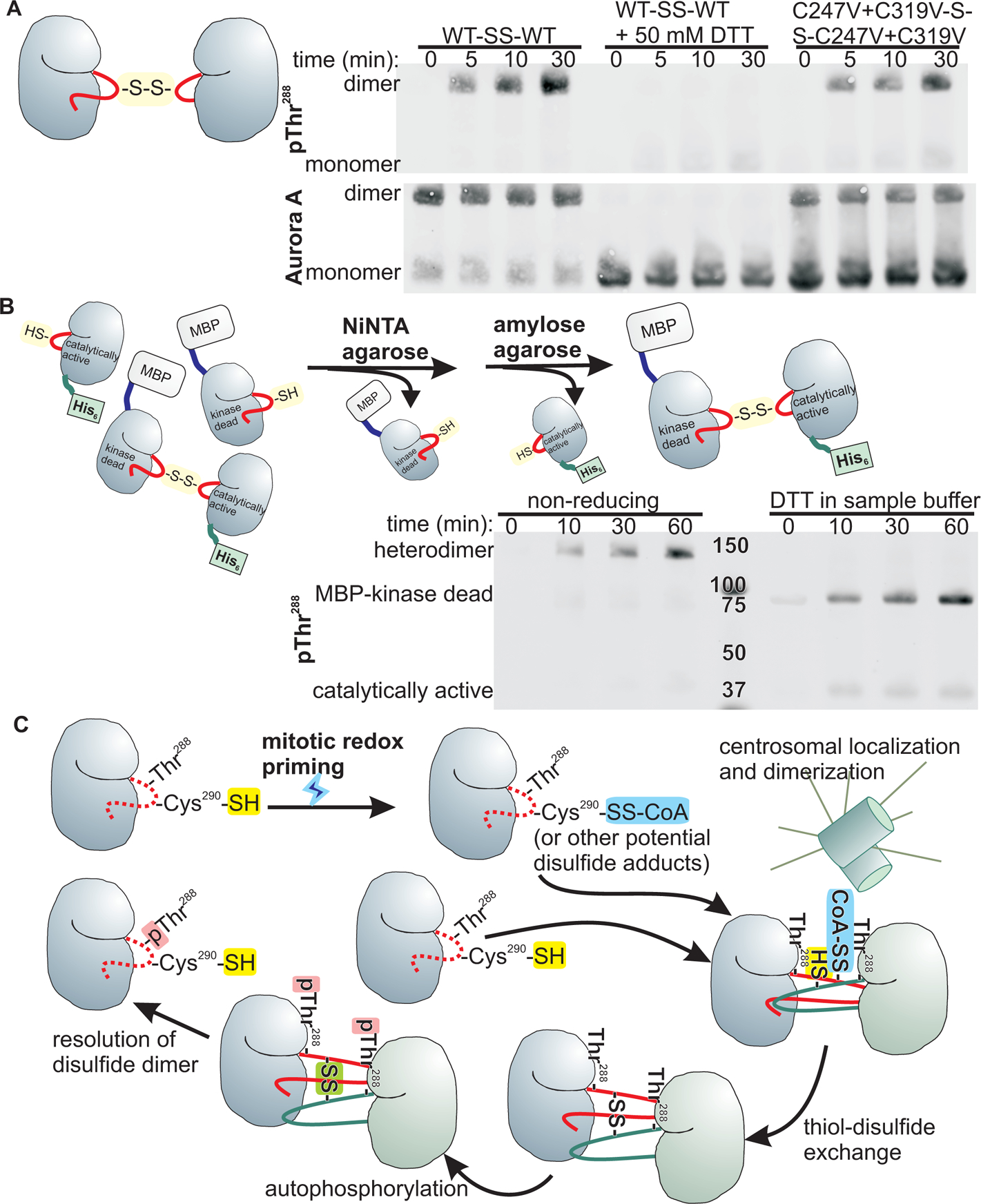
(A) Catalytically active Aurora A kinase domain constructs treated with the disulfide-promoting Ellman’s reagent and incubated with ATP show robust Thr288 autophosphorylation as assayed by Western blotting. Phosphorylation of Thr288 is detected in the bands corresponding to dimers for both the wild type and the C247V + C319V mutant construct. Inclusion of DTT in the kinase assay abrogates Thr288 phosphorylation. Blots are representative of 2 independent experiments. (B) Upper panel, purification scheme for disulfide-linked heterodimers containing an MBP-tagged kinase dead and untagged catalytically active Aurora A kinase domain. Lower panel, following incubation with ATP, the samples were analyzed by SDFS-PAGE under non-reducing or reducing conditions, and probed for autophosphorylation by Western blotting. Blots are representative of 8 independent experiments. (C) Proposed model of redox and dimerization-dependent activation of Aurora A. We posit that increased protein cysteine oxidation during mitosis results in increased levels of disulfide modifications of proteins, such as CoAlation of Aurora A. Clustering of Aurora A molecules upon centrosomal recruitment promotes dimerization and thiol-disulfide exchange between kinase domains to form a disulfide homodimer that facilitates autophosphorylation. Resolution of the disulfide homodimer releases activated (pThr288) Aurora A monomers.
To determine if the phosphorylation occurred in trans or in cis, we heterodimerized a maltose binding protein (MBP)-tagged kinase-dead construct with a His6-tagged catalytically active construct. Assay of this disulfide heterodimer after tandem affinity purification showed potent autophosphorylation as was seen for the disulfide homodimers, and analysis of the autophosphorylated heterodimer following reduction with DTT revealed that phosphorylation occurred on both monomers, but predominantly on the MBP-tagged inactive kinase domain (Fig. 8B), consistent with a strong preference for trans-phosphorylation.
DISCUSSION
In our efforts to discover novel small molecule inhibitors of Aurora A, we unexpectedly obtained a crystal structure of the Aurora A kinase domain in which Cys247 and Cys290 were covalently modified by dimethyl arsenic adducts. These modifications resulted in a large conformational change of the activation segment and dimerization via activation segment exchange between two kinase domain monomers. Both of these cysteine residues are conserved in a number of protein kinases, including members of the CMGC, CAMK and AGC clades of the human kinome tree (59). There is some precedent for these cysteine residues in regulating the activity of other kinases. Redox regulation of AMP-activated protein kinase (AMPK) has been reported to be mediated by intermolecular disulfides involving both of the equivalent cysteine residues within the AMPK kinase domain (60). Reduction of these intermolecular disulfides by thioredoxin1 is required for AMPK activation during energy starvation. Oxidative modifications of the activation loop cysteine within cAMP-dependent protein kinase (PKA) have also been shown to inhibit kinase activity and enhance activation loop dephosphorylation (61–64). Thus, we believe that targeting one or both of these cysteine residues may therefore provide a strategy to develop novel small molecule inhibitors applicable to a number of protein kinases that would offer greater specificity over ATP-competitive inhibitors. In particular, N-alpha-tosyl-L-phenylalanyl chloromethyl ketone (TPCK) has been shown to selectively inhibit AGC kinases by direct covalent modification of activation loop cysteines that are preceded by a phenylalanine residue (65). This raised the possibility that although the TXC motif (consisting of the activation loop cysteine two residues C-terminal to the phospho-acceptor threonine) is conserved in many protein kinases, the immediate surrounding environment of this cysteine residue may provide additional specificity determining features for the development of selective thiol-reactive kinase inhibitors. Furthermore, our findings suggest that cysteine-directed allosteric kinase inhibitors of Aurora A may enhance the potency of type II kinase inhibitors by stabilizing an active site conformation that is more favorable for binding. It is worth noting that the tethering screen reported in this manuscript was initially directed at Cys247 and serendipitously identified compounds that can form disulfide adducts with Cys290 to stabilize a dimeric active conformation of the kinase domain. It would therefore be interesting, in future studies, to perform a tethering screen directed specifically at Cys290 by using a C247V mutant construct.
Activation segment exchange has been observed in a number of protein kinase crystal structures, including those of Aurora A (50), and provides a mechanism for kinase domains to homodimerize and autophosphorylate in trans (66). This dimerization is known to be required, though not sufficient, for activation during cell cycle progression (25). Interestingly, our dimeric Aurora A kinase domain structures determined in this study show two distinct modes of dimerization; comparison of these dimeric structures revealed differences in both the DFG-in/out status and the positioning of Thr288, indicating that differences in the dimerization interface and the nature of the cysteine thiol modifications can regulate the positioning of both the DFG motif and the Thr288 phospho-acceptor. These differences in activation segment conformations likely underlie how the relative orientations of the kinase domain monomers differ between the dimers, since the swapped activation segments are central to the dimer interface. Our data indicate that the Aurora A kinase domain can dimerize in both inactive and catalytically competent conformations, with specific thiol modifications of the activation loop required to induce an active dimeric configuration for efficient autophosphorylation. These thiol modifications may therefore help to explain the previously published finding that dimerization alone is required but insufficient for xAurora A activation in Xenopus egg extracts (25). The catalytic activity exhibited by disulfide-linked homodimers of the Aurora A kinase domain raises questions as to whether these disulfide-linked Aurora A dimers can directly phosphorylate the downstream substrate Plk1, or whether Aurora A and Plk1 can form a specific disulfide-linked heterodimer via their activation loop cysteines to promote Plk1 activation. These questions, along with kinetic studies of Aurora A dimerization rates, warrant further investigation in future studies.
Our results have also led us to provide an alternative interpretation for the previously reported non-covalent “long-lived” Aurora A kinase domain dimer that was proposed by Zorba et al. to form in concentrated protein stock solutions and, due to a slow dissociation rate, to contribute to a faster initial rate of autophosphorylation upon dilution and addition of ATP (50). The impaired autophosphorylation seen for the C290A mutant Aurora A kinase domain under these conditions was proposed to be due to the inability of the C290A mutant to form this non-covalent long-lived dimer. In light of our finding that the Aurora A kinase domain can form a Cys290-Cys290 disulfide homodimer that potently autophosphorylates, this previously proposed long-lived dimer may alternatively be interpreted as the disulfide homodimer, which is more likely to be formed in more concentrated protein stock solutions, and the apparent slow dissociation rate may be due to a slow reduction of the disulfide homodimer under the assay conditions. Additionally, the impaired autophosphorylation of the C290A mutant reported in these assays can alternatively be attributed to the inability of the C290A mutant to form the disulfide homodimer.
Electron density for the TPX2 fragment fused at the N-terminus of the t-Aurora A constructs used for our structural studies is clearly visible in the 7–80-, 8–34- and cacodylate-modified dimers, and in the disulfide-linked homodimers, suggesting that these dimers are compatible with binding to interactors such as TPX2 and CEP192. In contrast, no density was visible for the CoAlated unphosphorylated wild type t-Aurora A structure with CoA bound at the ATP-binding pocket, suggesting that TPX2 and CoA binding may be mutually exclusive (Fig. 6C). A common feature among the activation segment-swapped dimer structures determined in this study is the covalent modification of the activation loop Cys290, suggesting that covalent modifications of the activation loop such as CoAlation promotes the displacement of the activation segment, particularly in the unphosphorylated state, to facilitate activation segment swapping and dimerization. Subsequent binding of Aurora A interactors such as TPX2 or CEP192 may then promote displacement of CoA from the active site to allow ATP binding and autophosphorylation.
We propose a model in which a transient redox priming modification of Aurora A during mitosis, when combined with localization and clustering of Aurora A at centrosomes, leads to activation by autophosphorylation of Thr288 within its activation loop (Fig. 8C). We and others have shown that overall levels of ROS and protein thiol oxidation increase during mitosis (28), reflecting a more oxidative cytoplasmic environment that would favor an increased fraction of Aurora A molecules being transiently modified by disulfide adducts, such as CoA. Clustering of Aurora A molecules upon their recruitment at centrosomes creates a high local concentration of kinase domains that would promote their dimerization via activation segment exchange. In this model, thiol-disulfide exchange between an Aurora A kinase molecule CoAlated (or otherwise modified) on Cys290 with a second Aurora A kinase molecule with an unmodified, reduced Cys290 residue would result in the formation of a disulfide homodimer that facilitates autophosphorylation on Thr288. Subsequent resolution of the dimer by reduction of the disulfide bond releases activated Aurora A kinase monomers that can then phosphorylate downstream targets.
This model is further supported by our findings here that mutation of Cys290 (or its equivalent) is sufficient to prevent Aurora A autophosphorylation and that high levels of DTT are sufficient to suppress Aurora A activation in Xenopus egg extracts, as well as by reports from other laboratories that treatment of mammalian cells in culture with oxidizing agents results in increased Aurora A Thr288 phosphorylation (57, 67). Although this model is strongly supported by structural, biochemical, and cell biological data, a number of features of the model remain unresolved. Whether CoA is truly the critical thiol-reactive compound that initiates centrosomal thiol-disulfide exchange in vivo is not known with certainty, nor is it clear what role, if any, CEP192 might play in this process. Whether Aurora A activation at other subcellular locations involves a similar activation mechanism is also unclear, though our data suggests that this activation mechanism is likely to be broadly conserved. Further studies will be needed to identify the physiologically relevant oxidatively modified Aurora A species that are expected to form as cells progress through the cell cycle, as well as the initial oxidative events that trigger the formation of such species. Nonetheless, the redox and dimerization-dependent mechanism of Aurora A activation that we propose provides a means to time Aurora A autophosphorylation to coincide with other mitotic processes, and to localize this Thr288 phosphorylation only to sites where Aurora A is recruited and concentrated. In addition, these findings identify Cys290 as a target site for the development of new covalent inhibitors of Aurora A that function in an allosteric manner.
MATERIALS AND METHODS
Protein expression and purification
Recombinant t-Aurora A protein constructs were expressed from pET28a-based plasmids in Escherichia coli (E. coli) Rosetta 2 (Novagen), containing an N-terminal His6-maltose-binding protein (MBP) purification tag. Unphosphorylated active constructs were produced by phosphatase co-expression using an N-terminal His6-λ-phosphatase-MBP tag. For large scale preparations, the protein was purified in tandem by nickel metal affinity chromatography, followed by amylose affinity chromatography (New England Biolabs), cleavage of the N-terminal purification tags by tobacco etch virus (TEV) protease cleavage, removal of the purification tags by cation exchange chromatography (SP-sepharose), and a final gel filtration step. The protein was then concentrated and frozen in aliquots in liquid nitrogen and stored at −80°C. Recombinant FLAG-tagged wild type and mutant Xenopus Aurora A proteins were produced using TNT SP6 High-Yield Wheat Germ Protein Expression System (Promega) as described previously (25, 26).
Protein crystallization and data collection
Crystals were grown by hanging drop vapor diffusion, by mixing 1 µL of 10 mg/mL protein stock with 1 µL of precipitant solution and equilibrating over a reservoir of precipitant solution at room temperature. Crystals typically appeared within a few days and grew to full size within a couple of weeks. CoAlated t-Aurora A C247V + C319V protein crystallized in tetragonal and hexagonal crystal forms under identical conditions. We focused our analyses on the tetragonal crystal form as it demonstrated substantially better diffraction. For data collection, crystals were harvested in stabilization buffer and cryoprotected by stepwise transfer into cryoprotectant buffer. The composition of the protein, reservoir, stabilization and cryoprotectant buffers are listed in table S1. Cryoprotected crystals were flash frozen in a nitrogen gas stream at 100 K, for immediate data collection on a home source, or stored in liquid nitrogen for data collection at the Northeastern Collaborative Access Team beamlines at Argonne National Laboratory. Home data were collected on a Rigaku rotating anode source outfitted with Yale mirror optics with either an Raxis IV detector or a Saturn 944 CCD detector, or on a Bruker Microstar source with multi-layer optics with MAR345 detector. All data were processed using HKL2000 (68) and the CCP4 software suite (69).
Structure determination and refinement
All structures of t-Aurora A were determined by molecular replacement. The native unmodified wild type structure was determined with AMoRe (70) using the structure of Aurora A residues 122–403 bound to TPX2 residues 1–43 (PDB code 1OL5) (18) as the search model. The cacodylate-modified structure was determined first by using the C-terminal lobe of the Aurora A kinase domain (residues 213 to 388 from 1OL5) as an initial search model with AMoRe. The position and orientation of the N-terminal lobe (residues 123 to 212) were then determined with MolRep (71) using the C-terminal lobe as a partial solution. The 7–80-modified structure was determined with PHASER (72) using the native unmodified t-Aurora A structure as a search model. Refinement and manual fitting of the 8–34-modified structure utilized the 7–80-modified structure as an initial model. The disulfide homodimer structure was determined using PHASER with 1OL5 as the search model. The CoAlated unphosphorylated wild-type structure was determined with PHASER using our native unmodified structure as the search model. The CoAlated unphosphorylated C247V + C317V disulfide-liked homodimer structure was determined with PHASER using the monomer asymmetric unit of the disulfide homodimer structure as the search model. Models were manually fitted using XtalView (73), MIFit (74) and Coot (75), and refined using PHENIX (76). Rosetta was used to improve the initial model geometry for the native unmodified wild type t-Aurora A and the CoAlated unphosphorylated wild-type structures (77). Structure figures were prepared using PyMOL (78) and XtalView (73). KinaseTool was used for analysis of the conformation of the DFG motif within t-Aurora A crystal structures (79). The structures and X-ray data have been deposited in the Protein Data Bank (PDB). Accession codes are listed in table S2.
Tethering screen for Aurora A-cysteine reactive compounds
A single cysteine-containing t-Aurora A construct containing C290A and C319V mutations was produced and purified in an unphosphorylated form in E. coli Rosetta 2 with an N-terminal purification tag consisting of a His6-tag, λ-phosphatase, glutathione S-transferase (GST), MBP, Streptococcus B1 domain (Gb1) and TEV cleavage site, as previously described (80). The purification tag was removed by TEV cleavage. This construct was subsequently found to contain an additional A241T mutation that was not expected to substantially affect the structure of the kinase domain. The tethering screen was performed as previously described (48), using a protein concentration of 4 µM, and buffer containing 10 mM Tris pH 8, 0.5 M NaCl, 1 mM adenosine, and 200 µM ß-mercaptoethanol. The disulfide-containing monophore compounds were screened at a concentration of 100 µM each. Mass changes due to covalent labeling of the protein were detected on a Waters LCT-Premier LC/ESI-MS.
Experiments in Xenopus egg extracts
Crude M-phase cytostatic-factor-arrested Xenopus egg extract was prepared as described previously (81), and supplemented with 15 µM nocodazole for all xAurora A activation assays. Endogenous xAurora was depleted from the extracts using a GST-tagged xCEP192 fragment (residues 521–757) immobilized on AminoLink Coupling Resin (Thermo Scientific) as described previously (26). xAurora A activation was initiated by the addition of demembranated sperm nuclei to a final concentration of 20000 to 40000 nuclei per µL, by addition of soluble rabbit xAurora A antibody to a final concentration of 100 ng/µL, or by addition of mouse FLAG M2 antibody (Sigma F3165) to a final concentration of 100–250 ng/µL. Assays were performed at 22°C by incubation of the reactions in a water bath or a PCR thermocycler, and aliquots were taken at the indicated time points and stopped with non-reducing SDS sample buffer. Total xAurora A protein and pThr295 were detected by Western blotting with a rabbit polyclonal xAurora A antibody and a rabbit monoclonal phospho-Aurora A [human pT288 / Xenopus pT295) antibody (D13A11, Cell Signaling Technology), respectively], and IRDye fluorescent secondary antibodies (LI-COR Biosciences). For reactions activated with demembranated sperm nuclei or soluble rabbit xAurora A antibody, pThr295 was detected by enhanced chemiluminescence using horse radish peroxidase conjugated to protein A.
Cell culture experiments
Mammalian cells lines were maintained in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum and antibiotics in a humidified incubator at 37°C with 5% CO2. Lentiviruses were produced in HEK293T cells, by calcium phosphate co-transfection (CalPhos mammalian transfection kit, Clontech Laboratories) of packaging vectors (pCMV-VSV-G envelope and pCMV-dR8.2 dvpr) together with either pTRIPZ-V2THS_12364 (doxycycline inducible human Aurora A shRNA, Dharmacon) or a pHAGE2-based lentiviral construct (82) expressing an shRNA-resistant 2X FLAG-tagged Aurora A (wild type or mutant) driven by a native Aurora A promoter fragment consisting of −367 to +356 base pairs around the transcriptional start site (53). Virus-containing media were filtered through 0.45 µm filters and used to transduce HeLa cells. HeLa cells stably transduced with inducible shAurora A were selected with puromycin (2 µg/mL). Doubly transduced HeLa cells were produced by subsequent transduction of these cells with pHAGE2 expressing shRNA-resistant 2X FLAG-tagged Aurora A-IRES-GFP followed by selection with blasticidin (25 µg/mL) and sorting for GFP-positive cells. Knockdown of endogenous Aurora A in transduced HeLa cell lines was induced by treatment with doxycycline (2 µg/mL) for 48 hours. The cells were then arrested by addition of nocodazole (10 µM), harvested 15 hours later and stored as frozen cell pellets. Lysates were produced by sonication of the cell pellets in PBS supplemented with 1% SDS, 1 mM NaF, 1 mM Na3VO4, 10 mM ß-glycerol phosphate, and 10 mM EDTA. Total and phospho-Aurora A in the lysates were detected by Western blotting, using rabbit monoclonal α-Aurora A antibody (1G4, Cell Signaling Technology), and α-phospho-Aurora A (pT288) antibody (D13A11, Cell Signaling Technology), respectively, and IRDye fluorescent secondary antibodies.
Disulfide modifications of t-Aurora A at C290
t-Aurora A modified with compounds 7–80 and 8–34 were produced by incubating wild type t-Aurora A in 10 mM Tris pH 8, 0.5 M NaCl and 1 mM adenosine with 2 mM final concentration of either 7–80 or 8–34 (and 2% DMSO) at 4°C for 3 days. The modified protein was then used for crystallization trials.
To produce t-Aurora A disulfide-linked homodimers, His6-λ-phosphatase-MBP-tagged t-Aurora A wild type and mutant constructs were first batch purified using NiNTA-agarose beads (Qiagen), followed by TEV cleavage and subsequent removal of the N-terminal purification tags by incubation with amylose-agarose beads. The samples were further purified by gel filtration under reducing conditions (10 mM HEPES pH 7, 0.5 M NaCl, 1 mM adenosine, 2 mM DTT), and subsequently exchanged into non-reducing buffer (10 mM HEPES pH 7, 0.5 M NaCl, 1 mM adenosine) by ultrafiltration (Amicon Ultra-4 10 kDa MWCO). Each construct was then split into two samples. One sample was reacted with 10 mM of the sulfhydryl-reactive compound 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB) overnight at 4°C to form the t-Aurora A-C290–2-nitro-5-thiobenzoate adduct. Unreacted DTNB was subsequently removed by buffer exchange via ultrafiltration. The DTNB-treated and untreated samples were then mixed together in equimolar amounts to allow dimerization via thiol-disulfide exchange for 2 hours at room temperature and overnight at 4°C.
To produce the active-inactive t-Aurora A disulfide heterodimer, the catalytically active construct was expressed with a TEV-cleavable N-terminal λ-phosphatase-MBP tag and a non-cleavable C-terminal His6 tag, while the inactive kinase dead (D256N)) construct was expressed with an N-terminal MBP-tag. Both constructs contained C247V and C319V mutations. The catalytically active construct was first purified by NiNTA chromatography, followed by TEV cleavage, flowed through amylose-agarose resin, and further purified by gel filtration. The protein was then reacted with 10 mM DTNB at 4°C for 3 days and subsequently buffer exchanged into 10 mM Tris pH 8, 0.5 M NaCl, 1 mM adenosine by ultrafiltration. The kinase dead construct was purified on amylose-agarose beads, eluted with 1 M methyl-α-D-glucopyranoside, treated with GST-tagged λ-phosphatase (overnight at 4°C), flowed through GSH-agarose resin, and buffer exchanged into 10 mM HEPES pH 7, 0.5 M NaCl, 1 mM adenosine by ultrafiltration. The catalytically active and kinase-dead constructs were then mixed together and incubated at room temperature for ~6 hours. The resulting disulfide heterodimer was then isolated by tandem affinity purification using NiNTA-agarose beads, followed by amylose-agarose beads.
To obtain the crystal structure of the t-Aurora A disulfide homodimer, a His6-MBP-tagged C247V + D256N + C319V triple mutant was expressed and purified as described above. The protein was buffer exchanged into 10 mM HEPES pH 7, 0.5 M NaCl, and 1 mM adenosine, and an aliquot (at ~37 mg/mL) was incubated with 30 mM DTNB at 4°C overnight. Excess DTNB was removed by buffer exchange by ultrafiltration. The disulfide homodimer was formed by mixing the DTNB-modified and unmodified protein together in equimolar amounts followed by incubation at 4°C overnight. The dimer was then purified using two rounds of gel filtration.
For production of CoAlated t-Aurora A constructs, the proteins were expressed as fusion constructs with an N-terminal His6-λ-phosphatase-MBP tag and purified as described above. Reduced coenzyme A (Sigma) was reacted with DTNB at 1:1 stoichiometry (~50 mM final concentration) overnight at room temperature, and the resulting coenzyme A-thionitrobenzoate (CoA-TNB) product was then reacted with C247V + C319V t-Aurora A protein in 10 mM Tris pH 8, 0.5 M NaCl, and 10 mM MgCl2 at 4°C for 3 days. AMP-PNP was added to a final concentration of 5 mM prior to crystallization trials.
Aurora A autophosphorylation assays
t-Aurora A autophosphorylation reactions were carried out in 10 mM HEPES pH 7 and 0.5 M NaCl at room temperature with enzyme concentrations of ~400 nM wild type disulfide homodimer, ~130 nM C247V + C319V double mutant disulfide homodimer and ~33 nM active-inactive disulfide heterodimer. Reactions were initiated by addition of 2 mM ATP and 4 mM MgCl2 and incubated at 25°C for the indicated periods of time. Total and phospho-Aurora A were detected by Western blotting, using a mouse monoclonal Aurora A antibody (35C1, Sigma), a rabbit monoclonal phospho-Aurora A (pT288) antibody (D13A11, Cell Signaling Technology), and IRDye fluorescent secondary antibodies as described above.
Supplementary Material
Figure S1. Mass spectrum of t-Aurora A C247V + C319V construct consistent with ~2/3 of the CoAlated protein.
Table S1: Crystallization and cryoprotection conditions for t-Aurora A constructs.
Table S2: Crystallographic data collection and refinement statistics.
Data file S1: Data from tethering screen.
Acknowledgements:
The authors thank Robert A. Grant, Gregory J. Dodge, and Barbara Imperiali (Massachusetts Institute of Technology) for assistance with shipping of the crystals and for help with the synchrotron data collection. We thank Michael J. Eck and John Genova (Dana-Farber Cancer Institute) for use of their X-ray facilities and for assistance with data collection, Johannes C. Walter (Harvard Medical School) for providing laboratory facilities and advice, and Bashar Alhoch (California Institute of Technology) for assisting with the preparation of some of the Xenopus egg extract. We thank Angela Koehler (Massachusetts Institute of Technology, Broad Institute of Harvard and MIT) and Timothy Lewis (Broad Institute of Harvard and MIT) for initiating the Aurora A inhibitor studies that led up to this work, and Hiroyuki Hayakawa and Karl Munger (Brigham and Women’s Hospital, and Harvard Medical School) for providing a human Aurora A cDNA construct. The authors would also like to thank Jesse Patterson, Bert van de Kooij, Brian Joughin, and Pau Creixell (Yaffe laboratory, MIT) for helpful discussions.
Funding:
This work was supported by NIH grants R01-ES015339, R35-ES028374, R01-GM104047, and R21-ES020466 to M.B.Y., the Charles and Marjorie Holloway Foundation, and the MIT Center for Precision Cancer Medicine. This work was supported in part by the Koch Institute Support (core) Grant P30-CA14051 from the National Cancer Institute. We thank the Koch Institute’s Robert A. Swanson (1969) Biotechnology Center for technical support, specifically Glenn Paradis, Michael Jennings, Michele Griffin and Mervelina Saturno-Condon (Flow Cytometry), and Richard Cook, Alla Leshinsky, and Richard P. Schiavoni (Biopolymers and proteomics). Support for this research was provided by a core center grant P30-ES002109 from the National Institute of Environmental Health Sciences, National Institutes of Health. D. C.L. was also supported by a Merck-MIT Biology postdoctoral fellowship. Work from the Wells lab was supported by R01 CA191018. A.K. and W.G.D were supported by NIH grant GM043974. This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P30 GM124165). The Eiger 16M detector on 24-ID-E beam line is funded by a NIH-ORIP HEI grant (S10OD021527). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02–06CH11357.
Footnotes
Competing interests: The authors declare that they have no competing interests.
Data and materials availability:
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. The structures and X-ray data have been deposited to the Protein Data Bank (PDB); accession codes are listed in table S2. The raw mass spectrometry data is deposited in the MassIVE repository as dataset MSV000085641 (ftp://massive.ucsd.edu/MSV000085641/).
REFERENCES AND NOTES
- 1.Willems E, Dedobbeleer M, Digregorio M, Lombard A, Lumapat PN, Rogister B, The functional diversity of Aurora kinases: A comprehensive review. Cell Div 13 (2018), pp. 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Magnaghi-Jaulin L, Eot-Houllier G, Gallaud E, Giet R, Aurora A protein kinase: To the centrosome and beyond. Biomolecules 9 (2019), pp. 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ye AA, Deretic J, Hoel CM, Hinman AW, Cimini D, Welburn JP, Maresca TJ, Aurora A Kinase Contributes to a Pole-Based Error Correction Pathway. Curr. Biol 25, 1842–1851 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chmátal L, Yang K, Schultz RM, Lampson MA, Spatial Regulation of Kinetochore Microtubule Attachments by Destabilization at Spindle Poles in Meiosis i. Curr. Biol 25, 1835–1841 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu D, Vader G, Vromans MJM, Lampson MA, Lens SMA, Sensing chromosome bi-orientation by spatial separation of Aurora B kinase from kinetochore substrates. Science (80-.) 323, 1350–1353 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krenn V, Musacchio A, The Aurora B kinase in chromosome bi-orientation and spindle checkpoint signaling. Front. Oncol 5 (2015), 10.3389/fonc.2015.00225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bischoff JR, Anderson L, Zhu Y, Mossie K, Ng L, Souza B, Schryver B, Flanagan P, Clairvoyant F, Ginther C, Chan CSM, Novotny M, Slamon DJ, Plowman GD, A homologue of Drosophila aurora kinase is oncogenic and amplified in human colorectal cancers. EMBO J 17, 3052–3065 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quartuccio SM, Schindler K, Functions of Aurora kinase C in meiosis and cancer. Front. Cell Dev. Biol 3 (2015), 10.3389/fcell.2015.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin Y-S, Su L-J, Yu C-TR, Wong F-H, Yeh H-H, Chen S-L, Wu J-C, Lin W-J, Shiue Y-L, Liu H-S, Hsu S-L, Lai J-M, Huang C-YF, Gene expression profiles of the Aurora family kinases. Gene Expr 13, 15–26 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.D’Assoro AB, Haddad T, Galanis E, Aurora-A kinase as a promising therapeutic target in cancer. Front. Oncol 5, 1–8 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Borisa AC, Bhatt HG, A comprehensive review on Aurora kinase: Small molecule inhibitors and clinical trial studies. Eur. J. Med. Chem 140, 1–19 (2017). [DOI] [PubMed] [Google Scholar]
- 12.O’Connor OA, Ozcan M, Jacobsen ED, Roncero JM, Trotman J, Demeter J, Masszi T, Pereira J, Ramchandren R, Beaven A, Caballero D, Horwitz SM, Lennard A, Turgut M, Hamerschlak N, d’Amore FA, Foss F, Kim WS, Leonard JP, Zinzani PL, Chiattone CS, Hsi ED, Trümper L, Liu H, Sheldon-Waniga E, Dansky Ullmann C, Venkatakrishnan K, Jane Leonard E, Shustov AR, Randomized phase III study of alisertib or investigator’s choice (selected single agent) in patients with relapsed or refractory peripheral T-cell lymphoma¨. J. Clin. Oncol 37, 613–623 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brockmann M, Poon E, Berry T, Carstensen A, Deubzer HE, Rycak L, Jamin Y, Thway K, Robinson SP, Roels F, Witt O, Fischer M, Chesler L, Eilers M, Small Molecule Inhibitors of Aurora-A Induce Proteasomal Degradation of N-Myc in Childhood Neuroblastoma. Cancer Cell 24, 75–89 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Richards MW, Burgess SG, Poon E, Carstensen A, Eilers M, Chesler L, Bayliss R, Structural basis of N-Myc binding by Aurora-A and its destabilization by kinase inhibitors. Proc. Natl. Acad. Sci. U. S. A 113, 13726–13731 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Asteriti IA, De Mattia F, Guarguaglini G, Cross-talk between AURKA and Plk1 in mitotic entry and spindle assembly. Front. Oncol 5, 1–9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Joukov V, De Nicolo A, Aurora-PLK1 cascades as key signaling modules in the regulation of mitosis. Sci. Signal 11, 1–26 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Kufer TA, Silljé HHW, Körner R, Gruss OJ, Meraldi P, Nigg EA, Human TPX2 is required for targeting Aurora-A kinase to the spindle. J. Cell Biol 158, 617–623 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bayliss R, Sardon T, Vernos I, Conti E, Structural basis of Aurora-A activation by TPX2 at the mitotic spindle. Mol. Cell 12, 851–862 (2003). [DOI] [PubMed] [Google Scholar]
- 19.Cyphers S, Ruff EF, Behr JM, Chodera JD, Levinson NM, A water-mediated allosteric network governs activation of Aurora kinase A. Nat. Chem. Biol 13, 402–408 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Macůrek L, Lindqvist A, Lim D, Lampson MA, Klompmaker R, Freire R, Clouin C, Taylor SS, Yaffe MB, Medema RH, Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature 455, 119–123 (2008). [DOI] [PubMed] [Google Scholar]
- 21.Seki A, Coppinger JA, Jang CY, Yates JR, Fang G, Bora and the kinase Aurora A cooperatively activate the kinase Plk1 and control mitotic entry. Science (80-.) 320, 1655–1658 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Elia AEH, Cantley LC, Yaffe MB, Proteomic screen finds pSer/pThr-binding domain localizing Plk1 to mitotic substrates. Science (80-.) 299, 1228–1231 (2003). [DOI] [PubMed] [Google Scholar]
- 23.Elia AEH, Rellos P, Haire LF, Chao JW, Ivins FJ, Hoepker K, Mohammad D, Cantley LC, Smerdon SJ, Yaffe MB, The molecular basis for phosphodependent substrate targeting and regulation of Plks by the Polo-box domain. Cell 115, 83–95 (2003). [DOI] [PubMed] [Google Scholar]
- 24.Lens SMA, Voest EE, Medema RH, Shared and separate functions of polo-like kinases and aurora kinases in cancer. Nat. Rev. Cancer 10, 825–841 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Joukov V, De Nicolo A, Rodriguez A, Walter JC, Livingston DM, Centrosomal protein of 192 kDa (Cep192) promotes centrosome-driven spindle assembly by engaging in organelle-specific Aurora A activation. Proc. Natl. Acad. Sci. U. S. A 107, 21022–21027 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Joukov V, Walter JC, De Nicolo A, The Cep192-Organized Aurora A-Plk1 Cascade Is Essential for Centrosome Cycle and Bipolar Spindle Assembly. Mol. Cell 55, 578–591 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meng L, Park J-E, Kim T-S, Lee EH, Park S-Y, Zhou M, Bang JK, Lee KS, Bimodal Interaction of Mammalian Polo-Like Kinase 1 and a Centrosomal Scaffold, Cep192, in the Regulation of Bipolar Spindle Formation. Mol. Cell. Biol 35, 2626–2640 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Patterson JC, Joughin BA, van de Kooij B, Lim DC, Lauffenburger DA, Yaffe MB, ROS and Oxidative Stress Are Elevated in Mitosis during Asynchronous Cell Cycle Progression and Are Exacerbated by Mitotic Arrest. Cell Syst 8, 163–167.e2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Denu JM, Tanner KG, Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: Evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry 37, 5633–5642 (1998). [DOI] [PubMed] [Google Scholar]
- 30.Lee SR, Kwon KS, Kirn SR, Rhee SG, Reversible inactivation of protein tyrosine phosphatase IB in A431 cells stimulated with epidermal growth factor. FASEB J 12, 15366–15372 (1998). [DOI] [PubMed] [Google Scholar]
- 31.Lee S-R, Yang K-S, Kwon J, Lee C, Jeong W, Rhee SG, Reversible Inactivation of the Tumor Suppressor PTEN by H2O2. J. Biol. Chem 277, 20336–20342 (2002). [DOI] [PubMed] [Google Scholar]
- 32.Savitsky PA, Finkel T, Redox regulation of Cdc25C. J. Biol. Chem 277, 20535–20540 (2002). [DOI] [PubMed] [Google Scholar]
- 33.Paulsen CE, Truong TH, Garcia FJ, Homann A, Gupta V, Leonard SE, Carroll KS, Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nat. Chem. Biol 8, 57–64 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Truong TH, Carroll KS, Redox regulation of protein kinases. Crit. Rev. Biochem. Mol. Biol 48, 332–56 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paulsen CE, Carroll KS, Orchestrating Redox Signaling Networks through Regulatory Cysteine Switches. ACS Chem. Biol 5, 47–62 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Finkel T, Signal transduction by reactive oxygen species. J. Cell Biol 194, 7–15 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Winterbourn CC, Biological production, detection, and fate of hydrogen peroxide. Antioxidants Redox Signal 29, 541–551 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Biteau B, Labarre J, Toledano MB, ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature 425, 980–984 (2003). [DOI] [PubMed] [Google Scholar]
- 39.Woo HA, Chae HZ, Hwang SC, Yang KS, Kang SW, Kim K, Rhee SG, Reversing the inactivation of peroxiredoxins caused by cysteine sulfinic acid formation. Science (80-.) 300, 653–656 (2003). [DOI] [PubMed] [Google Scholar]
- 40.Schmid E, El Benna J, Galter D, Klein G, Dröge W, Redox priming of the insulin receptor β-chain associated with altered tyrosine kinase activity and insulin responsiveness in the absence of tyrosine autophosphorylation. FASEB J 12, 863–870 (1998). [DOI] [PubMed] [Google Scholar]
- 41.Corcoran A, Cotter TG, Redox regulation of protein kinases. FEBS J 280, 1944–1965 (2013). [DOI] [PubMed] [Google Scholar]
- 42.Lim JM, Lee KS, Woo HA, Kang D, Rhee SG, Control of the pericentrosomal H2O2 level by peroxiredoxin I is critical for mitotic progression. J. Cell Biol 210, 23–33 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maignan S, Guilloteau JP, Zhou-Liu Q, Clément-Mella C, Mikol V, Crystal structures of the catalytic domain of HIV-1 integrase free and complexed with its metal cofactor: High level of similarity of the active site with other viral integrases. J. Mol. Biol 282, 359–368 (1998). [DOI] [PubMed] [Google Scholar]
- 44.Nolen B, Taylor S, Ghosh G, Regulation of protein kinases: Controlling activity through activation segment conformation. Mol. Cell 15, 661–675 (2004). [DOI] [PubMed] [Google Scholar]
- 45.Kornev AP, Haste NM, Taylor SS, Ten Eyck LF, Surface comparison of active and inactive protein kinases identifies a conserved activation mechanism. Proc. Natl. Acad. Sci. U. S. A 103, 17783–17788 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kornev AP, Taylor SS, Ten Eyck LF, A helix scaffold for the assembly of active protein kinases. Proc. Natl. Acad. Sci. U. S. A 105, 14377–14382 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Heron NM, Anderson M, Blowers DP, Breed J, Eden JM, Green S, Hill GB, Johnson T, Jung FH, McMiken HHJ, Mortlock AA, Pannifer AD, Pauptit RA, Pink J, Roberts NJ, Rowsell S, SAR and inhibitor complex structure determination of a novel class of potent and specific Aurora kinase inhibitors. Bioorganic Med. Chem. Lett 16, 1320–1323 (2006). [DOI] [PubMed] [Google Scholar]
- 48.Erlanson DA, Braisted AC, Raphael DR, Randal M, Stroud RM, Gordon EM, Wells JA, Site-directed ligand discovery. Proc. Natl. Acad. Sci. U. S. A 97, 9367–9372 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang J, Cron P, Good VM, Thompson V, Hemmings BA, Barford D, Crystal structure of an activated Akt/Protein Kinase B ternary complex with GSK3-peptide and AMP-PNP. Nat. Struct. Biol 9, 940–944 (2002). [DOI] [PubMed] [Google Scholar]
- 50.Zorba A, Buosi V, Kutter S, Kern N, Pontiggia F, Cho YJ, Kern D, Molecular mechanism of Aurora A kinase autophosphorylation and its allosteric activation by TPX2. Elife 2014, 1–24 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rowan FC, Richards M, Bibby RA, Thompson A, Bayliss R, Blagg J, Insights into aurora-A kinase activation using unnatural amino acids incorporated by chemical modification. ACS Chem. Biol 8, 2184–2191 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lim DC, Yaffe MB, “Unpublished.”
- 53.Tanaka M, Ueda A, Kanamori H, Ideguchi H, Yang J, Kitajima S, Ishigatsubo Y, Cell-cycle-dependent regulation of human aurora A transcription is mediated by periodic repression of E4TF1. J. Biol. Chem 277, 10719–10726 (2002). [DOI] [PubMed] [Google Scholar]
- 54.Holmström KM, Finkel T, Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol 15, 411–21 (2014). [DOI] [PubMed] [Google Scholar]
- 55.Deponte M, Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. Biochim. Biophys. Acta - Gen. Subj 1830, 3217–3266 (2013). [DOI] [PubMed] [Google Scholar]
- 56.Gout I, Coenzyme A, protein CoAlation and redox regulation in mammalian cells. Biochem. Soc. Trans 46, 721–728 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tsuchiya Y, Byrne DP, Burgess SG, Bormann J, Bakovic J, Huang Y, Zhyvoloup A, Peak-chew S, Tran T, Bellany F, Eyers CE, Carroll J, Skehel M, Bayliss R, Eyers PA, Gout I, Baković J, Huang Y, Zhyvoloup A, Kun Yu BY, Peak-chew S, Tran T, Bellany F, Tabor AB, Chan AE, Guruprasad L, Garifulin O, Filonenko V, Vonderach M, Ferries S, Eyers CE, Carroll J, Skehel M, Bayliss R, Eyers PA, Gout I, Covalent Aurora A regulation by the metabolic integrator coenzyme A. Redox Biol 28, 101318 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wells JA, Yount RG, Reaction of 5, 5ʹ-dithiobis(2-nitrobenzoic acid) with myosin subfragment one: Evidence for formation of a single protein disulfide with trapping of metal nucleotide at the active site. Biochemistry 19, 1711–1717 (1980). [DOI] [PubMed] [Google Scholar]
- 59.Manning G, Whyte DB, Martinez R, Hunter T, p38 Kinases. Science (80-.) 298, 1912–1934 (2002). [DOI] [PubMed] [Google Scholar]
- 60.Shao D, Oka SI, Liu T, Zhai P, Ago T, Sciarretta S, Li H, Sadoshima J, A redox-dependent mechanism for regulation of AMPK activation by thioredoxin1 during energy starvation. Cell Metab 19, 232–245 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.De Piña MZ, Vázquez-Meza H, Pardo JP, Rendón JL, Villalobos-Molina R, Riveros-Rosas H, Piña E, Signaling the signal, cyclic AMP-dependent protein kinase inhibition by insulin-formed H2O2 and reactivation by thioredoxin. J. Biol. Chem 283, 12373–12386 (2008). [DOI] [PubMed] [Google Scholar]
- 62.Humphries KM, Juliano C, Taylor SS, Regulation of cAMP-dependent protein kinase activity by glutathionylation. J. Biol. Chem 277, 43505–11 (2002). [DOI] [PubMed] [Google Scholar]
- 63.Humphries KM, Deal MS, Taylor SS, Enhanced dephosphorylation of cAMP-dependent protein kinase by oxidation and thiol modification. J. Biol. Chem 280, 2750–8 (2005). [DOI] [PubMed] [Google Scholar]
- 64.First EA, Taylor SS, Subunit interaction sites between the regulatory and catalytic subunits of cAMP-dependent protein kinase. Identification of a specific interchain disulfide bond. J. Biol. Chem 263, 5176–5181 (1988). [PubMed] [Google Scholar]
- 65.Anjum R, Pae E, Blenis J, Ballif BA, TPCK inhibits AGC kinases by direct activation loop adduction at phenylalanine-directed cysteine residues. Febs Lett 586, 3471–3476 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Oliver AW, Knapp S, Pearl LH, Activation segment exchange: a common mechanism of kinase autophosphorylation? Trends Biochem. Sci 32, 351–356 (2007). [DOI] [PubMed] [Google Scholar]
- 67.Wang GF, Dong Q, Bai Y, Yuan J, Xu Q, Cao C, Liu X, Oxidative stress induces mitotic arrest by inhibiting Aurora A-involved mitotic spindle formation. Free Radic. Biol. Med 103, 177–187 (2017). [DOI] [PubMed] [Google Scholar]
- 68.Otwinowski Z, Minor W, Processing of X-Ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol 276, 307–326 (1997). [DOI] [PubMed] [Google Scholar]
- 69.Collaborative Computational Project Number 4, The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr 50, 760–763 (1994). [DOI] [PubMed] [Google Scholar]
- 70.Navaza J, AMoRe: an automated package for molecular replacement. Acta Crystallogr. Sect. A 50, 157–163 (1994). [Google Scholar]
- 71.Vagin A, Teplyakov A, MOLREP: An Automated Program for Molecular Replacement. J. Appl. Crystallogr 30, 1022–1025 (1997). [Google Scholar]
- 72.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ, Phaser crystallographic software. J. Appl. Crystallogr 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McRee DE, XtalView/Xfit - A versatile program for manipulating atomic coordinates and electron density. J. Struct. Biol 125, 156–165 (1999). [DOI] [PubMed] [Google Scholar]
- 74.MIFit (2010).
- 75.Emsley P, Lohkamp B, Scott WG, Cowtan K, Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH, PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dimaio F, Echols N, Headd JJ, Terwilliger TC, Adams PD, Baker D, Improved low-resolution crystallographic refinement with Phenix and Rosetta. Nat. Methods 10, 1102–4 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.DeLano, The PyMOL Molecular Graphics System (2002), (available at http://www.pymol.org).
- 79.Modi V, Dunbrack RL, Defining a new nomenclature for the structures of active and inactive kinases. Proc. Natl. Acad. Sci. U. S. A 116, 6818–6827 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lim D, Gold DA, Julien L, Rosowski EE, Niedelman W, Yaffe MB, Saeij JPJ, Structure of the toxoplasma gondii ROP18 kinase domain reveals a second ligand binding pocket required for acute virulence. J. Biol. Chem 288, 34968–34980 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Murray AW, in Methods in Cell Biology Volume 36 (1991), pp. 581–605. [PubMed] [Google Scholar]
- 82.Naldini L, Blömer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D, In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science (80-. ) 272, 263–267 (1996). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Mass spectrum of t-Aurora A C247V + C319V construct consistent with ~2/3 of the CoAlated protein.
Table S1: Crystallization and cryoprotection conditions for t-Aurora A constructs.
Table S2: Crystallographic data collection and refinement statistics.
Data file S1: Data from tethering screen.
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. The structures and X-ray data have been deposited to the Protein Data Bank (PDB); accession codes are listed in table S2. The raw mass spectrometry data is deposited in the MassIVE repository as dataset MSV000085641 (ftp://massive.ucsd.edu/MSV000085641/).