

ABSTRACT
The ubiquity and long-range transport of the microorganisms inhabiting dust can pose a serious risk to human, animal, and plant health. The well-recognized importance of dust-associated microorganisms contrasts starkly with our limited understanding of the factors determining the variation in the composition of these communities at the global scale. Here, we provide the first insight into the global determinants of dust-associated microorganisms by quantifying the environmental factors shaping bacterial and fungal community composition in 467 outdoor settled dust samples collected from 33 countries and 6 continents. Our results show that the global variation in dust-associated bacterial and fungal community composition was, to some degree, predictable from mean annual precipitation and temperature. Notably, our results show that the fungal genera Alternaria and Aspergillus, which contain many species that can serve as triggers of allergenic disease in humans and as plant pathogens, were more abundant in drier regions. Collectively, these results highlight the key influence of climate on the global distribution of dust-associated microorganisms and provide the baseline information needed to build a more comprehensive understanding of how microbial exposures vary across the globe and in response to climate change.
IMPORTANCE A broad diversity of microorganisms can be found in dust, with some of these microorganisms capable of causing allergenic disease in human via inhalation or affecting plant health by acting as plant pathogens. However, the spatial variation in dust microbiomes and the environmental factors associated with this variation have not been comprehensively assessed at the global scale. Here, we investigated the bacteria and fungi found in outdoor settled dust samples spanning 33 countries and 6 continents. Our results show that dust-associated bacteria and fungi exhibit climate-driven variability in community composition at the global scale. Our results call for the development of strategies to predict the geographic distribution of dust-associated microorganisms and to identify the potential associations between microbial exposures and the health of humans, animals, and plants.
KEYWORDS: aerobiology, allergens, climate change, dust microbiomes
INTRODUCTION
In dust and outdoor air, microorganisms are ubiquitous and diverse, and they can represent a large fraction of aerosolized particles (1). Exposure to these microorganisms can have detrimental effects on human, animal, and plant health (2). For example, allergic asthma, which is often triggered by exposure to airborne microorganisms (3), affects more than 300 million people worldwide, and it is estimated to account for approximately 1,000 deaths per day (4). Moreover, dust-dwelling plant pathogens represent a major threat to agricultural production and food security (5). Furthermore, transoceanic and transcontinental dust events can transport microorganisms thousands of kilometers away from their sources, resulting in pronounced ecological disturbances to distant sink ecosystems (6). Although dust-associated communities of microorganisms have been extensively studied using both cultivation-dependent and cultivation-independent approaches, with these methods yielding important insights into the types of bacteria, fungi, and other taxa that can be found in dust (7–10), few studies have directly investigated how the composition of microorganisms varies across large spatial scales and the environmental factors associated with this variation.
Geographic variability in the composition of dust-associated microorganisms can arise from differential contributions of source environments. Dust settled on external surfaces is considered to be a long-term reservoir of microorganisms that were previously airborne and can reenter the atmosphere through dust resuspension (11). Emissions from a variety of source environments, such as soils, waterbodies, leaf surfaces, humans, and animals, can serve as important contributors to the microbial loading of dust and outdoor air (12). Previous studies have shown that the relative importance of different source environments can vary across seasons, land-use types, and vegetation types (13–16). For example, soil-associated microorganisms are more abundant in the air above forests and agricultural landscapes than in suburban areas (14). In addition, geographic variability in dust-associated microbial community composition can track the changes in environmental attributes. A survey of settled dust collected outside homes across the United States demonstrated that the composition of bacterial and fungal communities varied as a function of climatic conditions and soil properties (17). Likewise, climatic seasonal changes modulate the temporal dynamics of airborne microbial communities, with cold-tolerant and ice-associated microorganisms predominant during snowy seasons (13). However, the majority of previous studies have only focused on studying the environmental drivers of community variability in dust microbiomes at the local or regional scales. Our systematic and comprehensive understanding of dust-associated microorganisms at the global scale remains limited. As we know that the abundances of some microorganisms associated with human diseases (e.g., allergic disorders) can vary across geographic regions (18), a global investigation of the environmental factors shaping the distribution of outdoor dust-associated microorganisms is crucial for understanding the potential impacts of climate change and other global change factors on public health.
Here, we conducted a global survey of outdoor dust settled on external surfaces. The dust samples were collected by swabbing various external surfaces such as window sills, door trim, walls, and fences. A total of 467 dust samples were collected from 33 countries and 6 continents (Fig. 1 and Fig. S1) (19), representing the largest attempt to comprehensively investigate the global distribution of outdoor dust-associated microbial communities. The bacterial and fungal communities in dust samples were characterized by sequencing the V4 region of the 16S rRNA gene and the first internal transcribed spacer region (ITS1) of the rRNA operon, respectively. Because our study was conducted at the global scale, we were primarily interested in understanding how broad-scale environmental factors determine the global variation in dust-associated microbial communities. Specifically, our aims were to (i) identify the key climatic or soil variables predicting the global variation in the community composition of outdoor dust-associated microorganisms and (ii) reveal the associations between climatic or soil variables and the relative abundances of globally prevalent microbial lineages, especially those that are potentially linked to public health.
FIG 1.
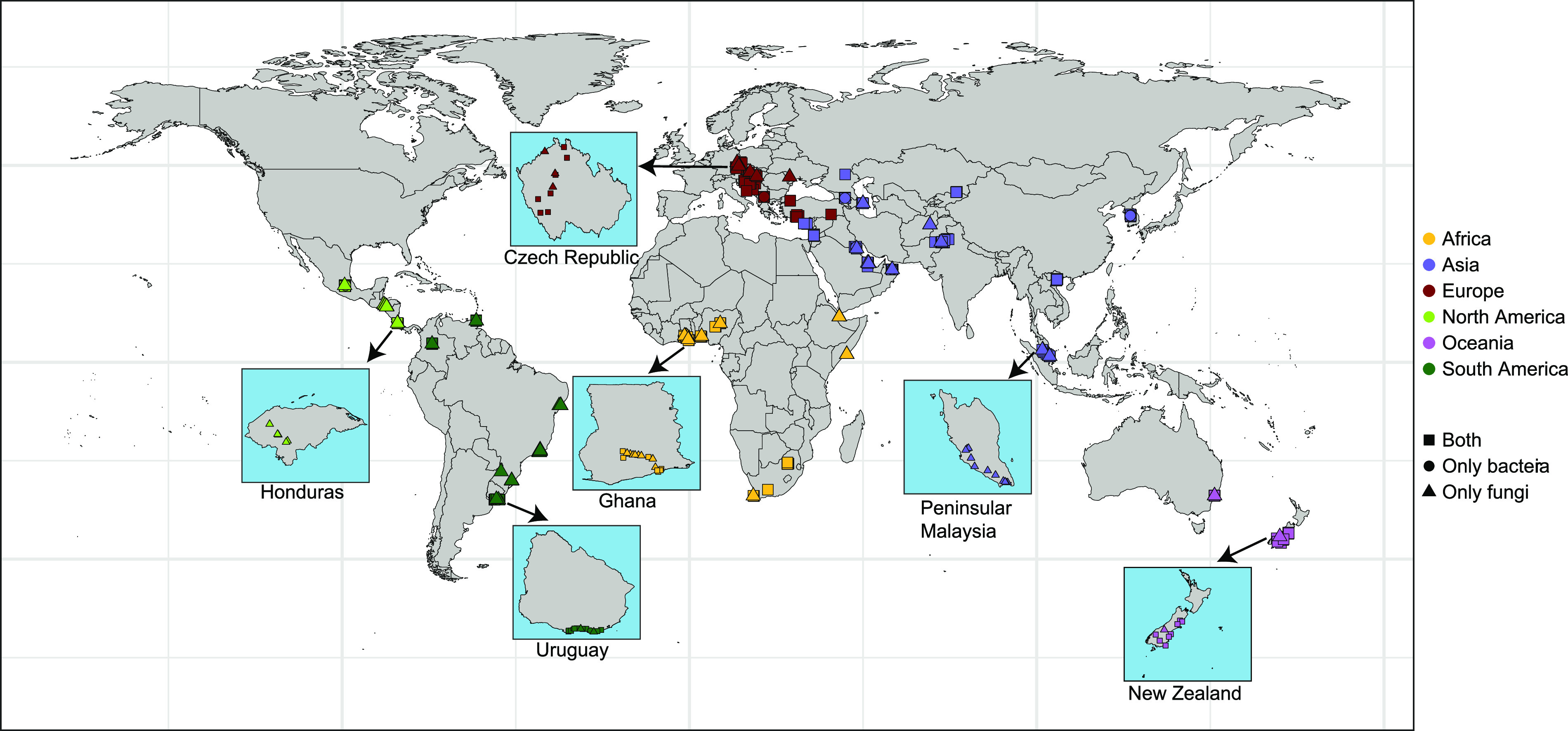
Geographic distribution of dust samples. A total of 467 samples of dust that had accumulated on outdoor surfaces were collected from 33 countries and 6 continents. The squares denote samples having both bacterial and fungal data, the circles denote samples having only bacterial data, and the triangles denote samples having only fungal data. For each continent, a higher resolution map of the sampling locations in one sample country is shown. See Fig. S1 for all the types of external surfaces.
RESULTS
General description of global dust microbial communities.
We obtained a total of 1,813,789 bacterial and 3,861,464 fungal sequences. These sequences were assembled into a total of 13,492 (415 ± 260, mean ± standard deviation [SD]) bacterial and 18,870 (88 ± 51, mean ± SD) fungal phylotypes (Fig. S2 and S3). We found that 42.23% of bacterial phylotypes and 87.10% of fungal phylotypes were restricted to one continent (Fig. S4). Likewise, 96.16% of bacterial phylotypes and all fungal phylotypes were detected in fewer than half of the samples (Fig. S5A and S5B). At the genus level, 91.83% of bacterial genera and 99.10% of fungal genera were found in fewer than half of the samples (Fig. S5C and S5D).
At the class level, the bacterial communities were dominated by Alphaproteobacteria (23.29%), Actinobacteria (16.64%), Bacilli (8.37%), Cytophagia (7.48%), and Gammaproteobacteria (6.13%). The fungal communities were dominated by Dothideomycetes (42.91%), Eurotiomycetes (12.42%), Tremellomycetes (7.74%), Cystobasidiomycetes (6.81%), and Agaricomycetes (6.79%). At the genus level, there were on average 106 bacterial genera (range: 13 to 303) and 47 fungal genera (range: 3 to 153) detected per sample. The top 10 most abundant bacterial genera were Hymenobacter, Paracoccus, Microbispora, Rubellimicrobium, Sphingomonas, Janthinobacterium, Acinetobacter, Chroococcidiopsis, Rubrobacter, and Kaistobacter (Fig. S6A). The dominant fungal genera were Aspergillus, Alternaria, Cladosporium, Symmetrospora, Naganishia, Nigrospora, Aureobasidium, Pseudopithomyces, Knufia, and Toxicocladosporium (Fig. S6B). The relative abundances of these genera were highly variable across the global dust samples (Fig. S6). For example, the relative abundances of the fungal genera Aspergillus and Alternaria in individual samples ranged from 0 to 85.27% and 0 to 68.62%, respectively (Fig. S6B).
Environmental determinants of microbial communities.
Stochastic factors contributed 82.52% and 79.41% of the variation in bacterial and fungal community composition, respectively. Despite the large impact of stochastic factors, the global variation in bacterial and fungal community composition was, to some degree, predictable from environmental factors. The results of variation partitioning analyses showed that environmental factors explained a larger fraction of variation in microbial community composition than geographic distance (Table S1). For bacterial communities, multiple regression on distance matrices (MRM) showed that global variation in community composition was best predicted by mean annual precipitation (5.57%, P = 0.001), and to a lesser extent, mean annual temperature (3.57%, P = 0.001), geographic distance (2.64%, P = 0.001), and precipitation of the driest month (2.43%, P = 0.001). For fungal communities, MRM showed that the strongest driver of community composition was mean annual temperature (7.03%, P = 0.001), followed by mean annual precipitation (5.08%, P = 0.001) and geographic distance (3.96%, P = 0.001). These patterns are evident from the nonmetric multidimensional scaling (NMDS) ordination plots in Fig. 2, which illustrate how dust-associated bacterial and fungal community compositions shift across gradients in mean annual precipitation and temperature.
FIG 2.
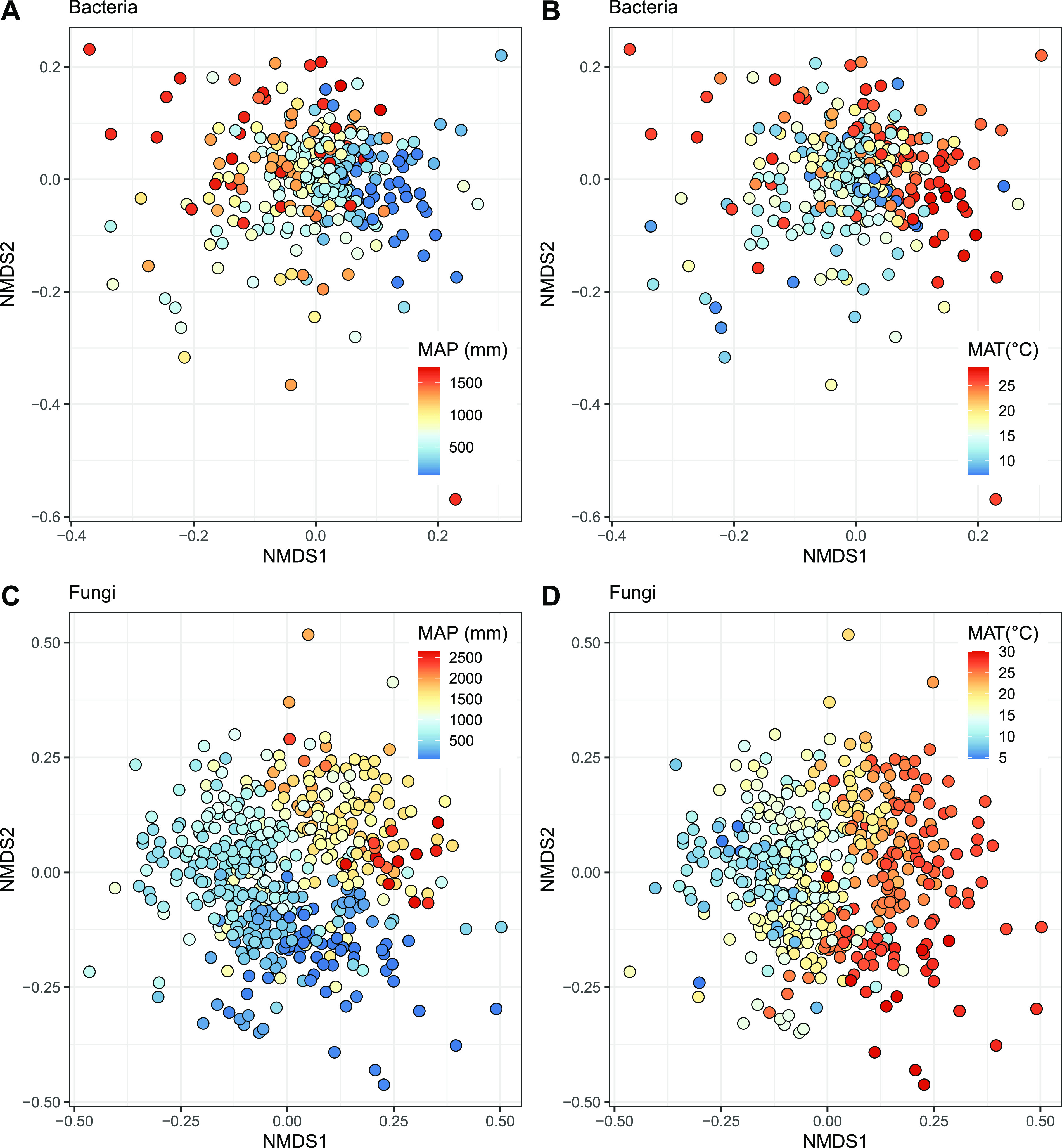
Environmental determinants of microbial community composition. NMDS ordination plots show the changes in bacterial (stress, 0.21) (A and B) and fungal (stress, 0.23) (C and D) community composition along gradients in mean annual precipitation and mean annual temperature (for both, P = 0.001 in MRM models). MAP, mean annual precipitation; MAT, mean annual temperature.
We identified 64 bacterial genera and 32 fungal genera that were prevalent in our global dust samples (Fig. S5C and S5D). The relative abundances of 7 bacterial genera were negatively associated with mean annual temperature (e.g., the cold-climate taxa, Psychrobacter and Polaromonas; Fig. 3A). The relative abundances of Sphingomonas and Hymenobacter were positively associated with precipitation of the driest month, and the relative abundance of Cellulomonas was positively related to soil pH (Fig. 3A). Notably, the relative abundances of two prevalent and dominant fungal genera, Alternaria and Aspergillus, were negatively associated with mean annual precipitation (Fig. 3B and Fig. 4). Both Alternaria and Aspergillus were particularly abundant (representing up to 60 to 80% of ITS sequences) in dust samples collected from drier regions in the Middle East (Fig. 4).
FIG 3.

Environmental determinants of the relative abundances of the prevalent genera of bacteria (A) and fungi (B). Only those genera significantly correlated with at least one environmental variable are shown (semipartial Spearman rank correlation, P < 0.05). The dendrograms show the results of hierarchical cluster analysis, which was conducted based on the statistically significant semipartial Spearman rank correlation coefficients. MAT, mean annual temperature; MTWQ, mean temperature of the wettest quarter; TSM, temperature of the sampling month; MAP, mean annual precipitation; PDM, precipitation of the driest month; PCQ, precipitation of the coldest quarter; PSM, precipitation of the sampling month; soil OC, soil organic carbon; soil BD, soil bulk density.
FIG 4.
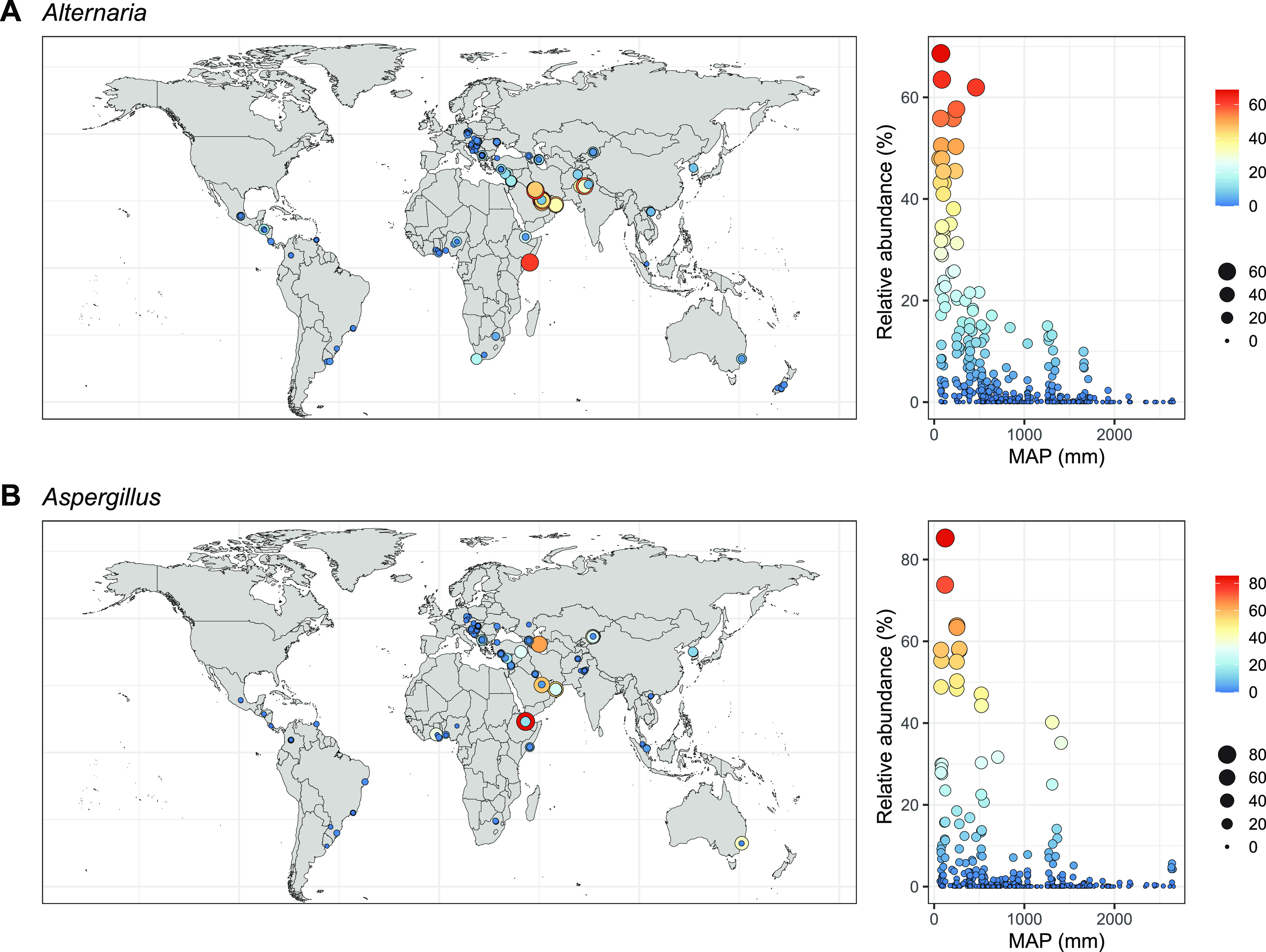
Geographic patterns of the relative abundances of Alternaria (A) and Aspergillus (B). The color of the points indicates the relative abundance, and the point sizes are proportional to the relative abundance. Samples with zero relative abundance of either Alternaria or Aspergillus are omitted from the world map, but they are retained in the scatterplot. The relative abundances of both Alternaria (Spearman’s ρ = −0.42, P < 0.001) and Aspergillus (Spearman’s ρ = −0.26, P < 0.001) were negatively associated with mean annual precipitation (MAP).
DISCUSSION
Associations between climatic factors and the global variation in microbial community composition.
Ecologists have long been intrigued by the widespread distribution and high diversity of microorganisms in outdoor dust (20). However, compared to the vast body of research on the biogeography of microorganisms in habitats such as soil and seawater, microorganisms inhabiting dust represent one of the least-studied microbial components of the biosphere (21). Dust-associated microbial community composition can be homogenized by long-distance atmospheric transport and the complexities of local wind patterns (2). These dispersal mechanisms might lead to nonexistent geographical patterns and pose a daunting challenge to identifying the environmental determinants of microbial community variation. Indeed, some of the taxa that we detected were present on every continent. Our results show that global variation in dust-associated bacterial and fungal community composition was, to some degree, predictable from climatic conditions. These global results are consistent with a continental-scale study that showed how the geographic pattern of microbial community composition in outdoor settled dust was associated with climatic variables (17). However, a large part of the observed variation in microbial community composition was unexplained by the included climatic variables. The unexplained variation could be attributable to unmeasured, but important, factors such as surrounding vegetation composition, land-use type, or wind currents. Moreover, there could be stochastic factors controlling the types of microorganisms found in dust, which is confirmed by our findings that stochastic factors dominantly explained the community variability. For example, the presence of dead insects in one sample might lead to dominance by insect-associated bacteria and fungi (17). Despite the large amount of unexplained variation, we found that mean annual precipitation and temperature were the two most important variables associated with differences in bacterial and fungal community composition across the compiled dust sample set. Overall, our results indicate that dust-associated microorganisms exhibit nonrandom and climate-driven variability in community composition at the global scale.
Climatic conditions may determine the outdoor dust-associated microbial community composition in three interrelated ways. First, climatic conditions may shape the composition of microbial communities in the surrounding source environments. Dust-associated microorganisms can be aerosolized from a myriad of nearby source environments such as soils and waters, with climatic conditions directly or indirectly determining the microbial community composition in these potential source environments. For example, the global soil bacterial and fungal community composition is associated with mean annual precipitation and temperature (22, 23), and the global community patterns of marine bacteria are driven by mean annual temperature (24). Therefore, the selection and filtering effects of climatic conditions on microbial communities in source environments could leave an imprint on dust-associated microbial communities. Second, climatic conditions govern how dust-associated microorganisms are aerosolized and transported from source environments. Emissions from source environments require a reduction in the forces holding the microorganisms to the surfaces of the source environments (i.e., bonding effect [25]). This bonding effect is influenced by the moisture and temperature at the surfaces, which can be related to precipitation and temperature regimes (26). For example, drier and warmer conditions can facilitate the aerosolization and dispersal of soil- and plant-associated microorganisms (27). Therefore, climatic conditions can modulate the relative contributions of different source environments to dust-associated microbial communities. And third, differences in climatic conditions could contribute to the variation in the longer-term preservation of microbial taxa in dust. For example, in drier and warmer areas, spore-forming microorganisms are more likely to be persistent in dust settled on surfaces for a longer time due to their capability to resist desiccation and UV radiation (28).
Associations between climatic factors and the relative abundances of prevalent genera.
For bacteria, our global study identified some of the prevalent genera commonly found in outdoor airborne and dust surveys. For example, it has been shown that the genus Sphingomonas is highly abundant in dust settled on balconies (29), and the genus Hymenobacter is dominant in the atmosphere (30). In our study, both Sphingomonas and Hymenobacter were dominant and prevalent genera, and the relative abundances of both taxa were positively associated with the precipitation of the driest month, indicating that they were more common in regions that were more predictably wet. In addition, the relative abundances of the bacterial genera Psychrobacter and Polaromonas were negatively associated with mean annual temperature. These results are expected because Psychrobacter and Polaromonas are well known to contain many psychrophilic microorganisms commonly found in low-temperature environments (31), and previously, Psychrobacter was found to dominate airborne bacterial communities in winter (13). Furthermore, the relative abundance of the bacterial genus Cellulomonas was positively related to soil pH, which is consistent with a previous study showing that pH is a significant predictor of the continental-scale distribution of Cellulomonas (17).
Our results show that two fungal genera, Alternaria and Aspergillus, were more abundant in drier regions. Both Alternaria and Aspergillus are frequently identified fungi in dust surveys. For example, these genera have been shown to dominate the fungal community in atmospheric particulate matter (32) and in dust settled on patios (8). Consistent with these studies, our global investigation shows that Alternaria and Aspergillus were dominant and ubiquitous taxa in outdoor settled dust. Furthermore, our results show that dry regions in the Middle East were hot spots of Alternaria and Aspergillus. It has been suggested that the spores of Alternaria and Aspergillus require dry weather to become airborne and disseminate (33, 34). Moreover, given that Alternaria and Aspergillus species are frequently found in soils in high abundance (35, 36), it is possible that drier conditions facilitate the aerosolization of soil-associated Alternaria and Aspergillus to the near-surface atmosphere and dust. Indeed, dust storms carry a large proportion of Alternaria and Aspergillus species that originate from arid soils and transport them to downwind terrestrial and aquatic environments (37, 38). The information about the environmental preferences of Alternaria and Aspergillus species and the geographic variation in their relative abundances is noteworthy because these two genera are critical to the health of humans, crops, and livestock (39). Specifically, both fungal genera are well known to contain many species acting as triggers of allergenic disease (e.g., asthma and hay fever) that pose a serious risk to human health and well-being (40). In addition, they contain a myriad of virulent plant pathogens that affect the health and productivity of many economically important crops (e.g., maize, corn, cotton, wheat, and citrus) and plants that are essential food sources for livestock and wildlife animals (41). The high relative abundances of Alternaria and Aspergillus species in dry regions raise significant concerns about the potential impact of climate change on human health and agricultural productivity, especially as aridity and dryland areas are anticipated to increase under projected climate change scenarios (42).
Caveats and conclusions.
To our knowledge, this is one of the more comprehensive studies to document the types of bacteria and fungi found in outdoor settled dust and how their global distributions are associated with climatic conditions. However, it is worth noting that there are important gaps in the geographic coverage of our study. Dust samples from Europe and western Asia were overrepresented, while dust samples from eastern Asia, Africa, North America, South America, and Oceania were scarce. We also want to acknowledge that our analyses did not include local-scale factors such as nearby vegetation, construction activity, and vehicle and pedestrian traffic that could play important roles in shaping the dust microbiomes at finer spatial scales. Future work studying dust-associated microorganisms from understudied geographic regions and incorporating factors operating at local, regional, and continental scales is required to better understand the determinants of dust-associated microbial communities.
We found that the composition of dust-associated bacterial and fungal communities changed predictably along global climatic gradients. Our findings that two putatively pathogenic genera (i.e., Alternaria and Aspergillus) were more abundant in drier regions call for future studies identifying the potential associations between their geographic distributions and disease outbreaks. Our analytical approaches can only be used to assess the relative abundances of microbial taxa, and thus, an important next step is to estimate the absolute abundances of individual taxa by pairing quantitative molecular approaches with sampling methodologies that control for the amount of dust collected. Furthermore, although our study focused on bacterial and fungal communities, viruses also represent a highly diverse group of dust-associated microorganisms that pose a major threat to human health (43). Recent work has also shown that dust acts as a reservoir of antibiotic resistance genes (28). Therefore, future studies that focus on how the environment shapes the geographic distribution and composition of dust-associated viruses and antibiotic-resistant microorganisms/genes will provide a more comprehensive understanding of the impacts of microbial exposures on public health.
MATERIALS AND METHODS
Sample collection.
From November 2016 to February 2017, a total of 467 outdoor settled dust samples were collected from 33 counties and 6 continents (Fig. 1 and Fig. S1) (19). SecurSwab DUO-V DNA collectors (Bode Technology, Lorton, VA, USA) were used to collect outdoor dust settled on various external surfaces such as window sills (150 samples), door trims (46 samples), walls (35 samples), and fences (30 samples) (see Fig. S1 for all types of external surfaces). These external surfaces serve as passive collectors of outdoor dust and aerosols. However, we do not know how long the dust had accumulated on the surfaces, and we cannot exclude the possibility that some microorganisms are growing on the surfaces themselves (44). The SecurSwab DUO-V DNA collectors were rotated slowly and moved back and forth across the surfaces during sampling. The SecurSwab DUO-V DNA collectors were used to prevent sample loss or contamination when the dust samples were transported back to the laboratory. All dust samples were stored desiccated at room temperature and were sent to the University of Colorado for molecular analyses.
Molecular analyses.
DNA from each dust swab was extracted using the Qiagen PowerSoil DNA extraction kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. To characterize the bacterial and archaeal communities, the V4 hypervariable region of the 16S rRNA gene was amplified using the 515-F (GTGCCAGCMGCCGCGGTAA) and 806-R (GGACTACHVGGGTWTCTAAT) primer pair (45). To characterize the fungal communities, the first internal transcribed spacer (ITS1) region of the rRNA operon was amplified using the ITS1-F (CTTGGTCATTTAGAGGAAGTAA) and ITS2 (GCTGCGTTCTTCATCGATGC) primer pair (46). The primers included the appropriate Illumina adapters, with the reverse primers also having an error-correcting 12-bp barcode unique to each sample to permit the multiplexing of samples. PCRs were performed in duplicate in a final mixture volume of 25 μl, including 12.5 μl Promega Hot Start master mix, 10.5 μl PCR-grade water, 1 μl PCR primers (forward and reverse combined at a 10-μM concentration), and 1 μl extracted genomic DNA. The PCR thermocycler conditions for both the 16S rRNA gene and ITS region consisted of 94°C for 3 min, 35 cycles of 94°C for 45 s, 50°C for 1 min, 72°C for 1.5 min, followed by a final elongation step for 10 min at 72°C. Extraction blanks and negative controls without a DNA template were included in each batch of PCRs to check for possible contamination. The PCR products were cleaned and normalized using the SequalPrep 96-well normalization plate kits (Thermo Fisher Scientific, Waltham, MA, USA). The purified PCR products from all samples were pooled in equimolar concentrations and were sequenced on a MiSeq instrument (Illumina, San Diego, CA, USA). The sequencing was conducted at the University of Colorado Next Generation Sequencing Facility.
Sequence processing.
The raw reads were demultiplexed using idemp (https://github.com/yhwu/idemp). Then, the raw reads were processed using DADA2 (47), which can resolve exact biological sequences by assembling reads into error-corrected amplicon sequence variants (hereafter, “phylotypes”). The DADA2 pipeline included quality filtering, modeling of the error rate, dereplication, phylotype inference, merging of the paired-end reads, construction of a phylotype count table, chimera removal, and taxonomy assignment. There were three major differences between the DADA2 pipelines for the 16S and ITS reads. First, the length of the ITS region is highly variable, and thus, Cutadapt (48) was used to remove primer sequences prior to quality filtering. Second, during quality filtering, the 16S reads, but not the ITS reads, were truncated to the same length. This is because some ITS variants might be shorter than the truncation length. Third, taxonomic identities were determined using the RDP classifier (49), trained on the SILVA nonredundant (nr) version 132 database (50) for 16S rRNA phylotypes and the UNITE database (51) for the ITS rRNA phylotypes. Those 16S phylotypes without a bacterial domain assignment and assigned to chloroplast or mitochondrial origin were removed. Those ITS phylotypes without a fungal domain assignment were removed. To remove potential contaminants, phylotypes present in the extraction or PCR blanks were removed. We retained samples with more than 1,000 sequences. The final analyses included 302 samples with both bacterial and fungal data, 11 samples with only bacterial data, and 154 samples with only fungal data. The sequence counts were normalized using a cumulative-sum scaling (52).
Compilation of environmental variables.
We compiled a set of climatic and soil variables from public databases using the geographic coordinates of the dust samples. Specifically, we compiled 19 climatic variables and the temperature and precipitation of the sampling months from the WorldClim database (53). We compiled four soil variables from the Harmonized World Soil database (54) to represent the soil physical and chemical properties. A list of these environmental variables is provided in Table S2.
Identification of prevalent genera.
To identify the genera that were widespread in our global dust samples (we identified prevalent genera instead of phylotypes because there were too few globally prevalent phylotypes; see Fig. S5A and S5B), we selected the bacterial genera that occurred in more than 50% of the samples (55) and the fungal genera that occurred in more than 30% of the samples (35). We focused on these more widespread genera, given their ubiquity and because it would have been difficult to model the distributions of taxa that occurred in small numbers of samples.
Statistical analyses.
Statistical analyses were implemented in R (56). The contribution of stochastic factors to microbial community variability was estimated following reference 57. A total of 10 multicollinearity-free variables were retained (variance inflation factor, <5). These environmental variables included seven climatic variables (mean annual temperature, mean annual precipitation, mean temperature of the wettest quarter, temperature of the sampling month, precipitation of the driest month, precipitation of the coldest quarter, and precipitation of the sampling month) and three soil variables (pH, organic carbon, and bulk density). The best subset of environmental variables accounting for the variation in community dissimilarity (Bray-Curtis metric) was selected by the bioenv procedure within the vegan package (58). MRM was used to fit the microbial community dissimilarity as a function of environmental variables and geographic distance (59). The relative importance of explanatory variables was estimated using the relaimpo package (60). Variation partitioning was used to decompose the variation in the microbial community composition into fractions explained uniquely and jointly by environmental factors and geographic distance. The associations between the relative abundances of the prevalent genera and environmental variables were examined using semipartial Spearman rank correlation (61), which allowed us to examine the relationships between the relative abundances of specific genera and particular environmental variables while controlling for other environmental variables. Statistically significant correlation coefficients (P < 0.05) were used to group the prevalent genera using hierarchical cluster analysis (the “complete” method).
Data availability.
The data supporting the conclusions of this article are available in Figshare (https://doi.org/10.6084/m9.figshare.14213519).
ACKNOWLEDGMENTS
This research was supported by the US Army Research Office and the Defense Forensic Science Center and was accomplished under cooperative agreement W911NF‐16‐2‐0195. Y.C. is supported by the USDA-NIFA (2017-68005-26867) Sustainable Bioeconomy for Arid Lands Center.
N.F., R.R.D., and S.A.F. conceived and designed the study. M.J.G. conducted laboratory analyses and sequencing. Y.C. performed statistical analyses and wrote the manuscript with substantial contributions from N.F. and A.B. All authors contributed to the writing of the manuscript and assisted with revisions.
We declare no conflicts of interest.
Footnotes
Supplemental material is available online only.
Contributor Information
Yongjian Chen, Email: chenyj@email.arizona.edu.
Jeffrey A. Gralnick, University of Minnesota
REFERENCES
- 1.Després VR, Huffman JA, Burrows SM, Hoose C, Safatov AS, Buryak G, Fröhlich-Nowoisky J, Elbert W, Andreae MO, Pöschl U, Jaenicke R. 2012. Primary biological aerosol particles in the atmosphere: a review. Tellus B Chem Phys Meteorol 64:15598. doi: 10.3402/tellusb.v64i0.15598. [DOI] [Google Scholar]
- 2.Griffin DW. 2007. Atmospheric movement of microorganisms in clouds of desert dust and implications for human health. Clin Microbiol Rev 20:459–477. doi: 10.1128/CMR.00039-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Douwes J, Thorne P, Pearce N, Heederik D. 2003. Bioaerosol health effects and exposure assessment: progress and prospects. Ann Occup Hyg 47:187–200. doi: 10.1093/annhyg/meg032. [DOI] [PubMed] [Google Scholar]
- 4.Global Asthma Network. 2018. The global asthma report 2018. Global Asthma Network, Auckland, New Zealand. [Google Scholar]
- 5.Dietzel K, Valle D, Fierer N, U’Ren JM, Barberán A. 2019. Geographical distribution of fungal plant pathogens in dust across the United States. Front Ecol Evol 7:304. doi: 10.3389/fevo.2019.00304. [DOI] [Google Scholar]
- 6.Kellogg CA, Griffin DW. 2006. Aerobiology and the global transport of desert dust. Trends Ecol Evol 21:638–644. doi: 10.1016/j.tree.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 7.Yassin MF, Almouqatea S. 2010. Assessment of airborne bacteria and fungi in an indoor and outdoor environment. Int J Environ Sci Technol 7:535–544. doi: 10.1007/BF03326162. [DOI] [Google Scholar]
- 8.Adams RI, Miletto M, Taylor JW, Bruns TD. 2013. Dispersal in microbes: fungi in indoor air are dominated by outdoor air and show dispersal limitation at short distances. ISME J 7:1262–1273. doi: 10.1038/ismej.2013.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Al Salameen F, Habibi N, Uddin S, Al Mataqi K, Kumar V, Al Doaij B, Al Amad S, Al Ali E, Shirshikhar F. 2020. Spatio-temporal variations in bacterial and fungal community associated with dust aerosol in Kuwait. PLoS One 15:e0241283. doi: 10.1371/journal.pone.0241283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aalismail NA, Ngugi DK, Díaz-Rúa R, Alam I, Cusack M, Duarte CM. 2019. Functional metagenomic analysis of dust-associated microbiomes above the Red Sea. Sci Rep 9:13741. doi: 10.1038/s41598-019-50194-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prussin AJ, II, Marr LC. 2015. Sources of airborne microorganisms in the built environment. Microbiome 3:78. doi: 10.1186/s40168-015-0144-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ruiz-Gil T, Acuña JJ, Fujiyoshi S, Tanaka D, Noda J, Maruyama F, Jorquera MA. 2020. Airborne bacterial communities of outdoor environments and their associated influencing factors. Environ Int 145:106156. doi: 10.1016/j.envint.2020.106156. [DOI] [PubMed] [Google Scholar]
- 13.Bowers RM, McCubbin IB, Hallar AG, Fierer N. 2012. Seasonal variability in airborne bacterial communities at a high-elevation site. Atmos Environ 50:41–49. doi: 10.1016/j.atmosenv.2012.01.005. [DOI] [Google Scholar]
- 14.Bowers RM, McLetchie S, Knight R, Fierer N. 2011. Spatial variability in airborne bacterial communities across land-use types and their relationship to the bacterial communities of potential source environments. ISME J 5:601–612. doi: 10.1038/ismej.2010.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bowers RM, Sullivan AP, Costello EK, Collett JL, Jr, Knight R, Fierer N. 2011. Sources of bacteria in outdoor air across cities in the midwestern United States. Appl Environ Microbiol 77:6350–6356. doi: 10.1128/AEM.05498-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lymperopoulou DS, Adams RI, Lindow SE. 2016. Contribution of vegetation to the microbial composition of nearby outdoor air. Appl Environ Microbiol 82:3822–3833. doi: 10.1128/AEM.00610-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barberán A, Ladau J, Leff JW, Pollard KS, Menninger HL, Dunn RR, Fierer N. 2015. Continental-scale distributions of dust-associated bacteria and fungi. Proc Natl Acad Sci USA 112:5756–5761. doi: 10.1073/pnas.1420815112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith KF, Guégan J-F. 2010. Changing geographic distributions of human pathogens. Annu Rev Ecol Evol Syst 41:231–250. doi: 10.1146/annurev-ecolsys-102209-144634. [DOI] [Google Scholar]
- 19.Grantham NS, Reich BJ, Laber EB, Pacifici K, Dunn RR, Fierer N, Gebert M, Allwood JS, Faith SA. 2020. Global forensic geolocation with deep neural networks. J R Stat Soc Ser C Appl Stat 69:909–929. doi: 10.1111/rssc.12427. [DOI] [Google Scholar]
- 20.Darwin C. 1846. An account of the fine dust which often falls on vessels in the Atlantic Ocean. Q J Geol Soc 2:26–30. doi: 10.1144/GSL.JGS.1846.002.01-02.09. [DOI] [Google Scholar]
- 21.Thompson LR, Sanders JG, McDonald D, Amir A, Ladau J, Locey KJ, Prill RJ, Tripathi A, Gibbons SM, Ackermann G, Navas-Molina JA, Janssen S, Kopylova E, Vázquez-Baeza Y, González A, Morton JT, Mirarab S, Xu ZZ, Jiang L, Haroon MF, Kanbar J, Zhu Q, Song SJ, Kosciolek T, Bokulich NA, Lefler J, Brislawn CJ, Humphrey G, Owens SM, Hampton-Marcell J, Berg-Lyons D, McKenzie V, Fierer N, Fuhrman JA, Clauset A, Stevens RL, Shade A, Pollard KS, Goodwin KD, Jansson JK, Gilbert JA, Knight R, Earth Microbiome Project Consortium . 2017. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature 551:457–463. doi: 10.1038/nature24621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bahram M, Hildebrand F, Forslund SK, Anderson JL, Soudzilovskaia NA, Bodegom PM, Bengtsson-Palme J, Anslan S, Coelho LP, Harend H, Huerta-Cepas J, Medema MH, Maltz MR, Mundra S, Olsson PA, Pent M, Põlme S, Sunagawa S, Ryberg M, Tedersoo L, Bork P. 2018. Structure and function of the global topsoil microbiome. Nature 560:233–237. doi: 10.1038/s41586-018-0386-6. [DOI] [PubMed] [Google Scholar]
- 23.Větrovský T, Kohout P, Kopecký M, Machac A, Man M, Bahnmann BD, Brabcová V, Choi J, Meszárošová L, Human ZR, Lepinay C, Lladó S, López-Mondéjar R, Martinović T, Mašínová T, Morais D, Navrátilová D, Odriozola I, Štursová M, Švec K, Tláskal V, Urbanová M, Wan J, Žifčáková L, Howe A, Ladau J, Peay KG, Storch D, Wild J, Baldrian P. 2019. A meta-analysis of global fungal distribution reveals climate-driven patterns. Nat Commun 10:5142. doi: 10.1038/s41467-019-13164-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sunagawa S, Coelho LP, Chaffron S, Kultima JR, Labadie K, Salazar G, Djahanschiri B, Zeller G, Mende DR, Alberti A, Cornejo-Castillo FM, Costea PI, Cruaud C, D’Ovidio F, Engelen S, Ferrera I, Gasol JM, Guidi L, Hildebrand F, Kokoszka F, Lepoivre C, Lima-Mendez G, Poulain J, Poulos BT, Royo-Llonch M, Sarmento H, Vieira-Silva S, Dimier C, Picheral M, Searson S, Kandels-Lewis S, Bowler C, de Vargas C, Gorsky G, Grimsley N, Hingamp P, Iudicone D, Jaillon O, Not F, Ogata H, Pesant S, Speich S, Stemmann L, Sullivan MB, Weissenbach J, Wincker P, Karsenti E, Raes J, Acinas SG, Bork P, Tara Oceans coordinators . 2015. Structure and function of the global ocean microbiome. Science 348:1261359. doi: 10.1126/science.1261359. [DOI] [PubMed] [Google Scholar]
- 25.Tuson HH, Weibel DB. 2013. Bacteria–surface interactions. Soft Matter 9:4368–4380. doi: 10.1039/C3SM27705D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jones AM, Harrison RM. 2004. The effects of meteorological factors on atmospheric bioaerosol concentrations—a review. Sci Total Environ 326:151–180. doi: 10.1016/j.scitotenv.2003.11.021. [DOI] [PubMed] [Google Scholar]
- 27.Brodie EL, DeSantis TZ, Parker JPM, Zubietta IX, Piceno YM, Andersen GL. 2007. Urban aerosols harbor diverse and dynamic bacterial populations. Proc Natl Acad Sci USA 104:299–304. doi: 10.1073/pnas.0608255104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou H, Wang X, Li Z, Kuang Y, Mao D, Luo Y. 2018. Occurrence and distribution of urban dust-associated bacterial antibiotic resistance in Northern China. Environ Sci Technol Lett 5:50–55. doi: 10.1021/acs.estlett.7b00571. [DOI] [Google Scholar]
- 29.Adams RI, Miletto M, Lindow SE, Taylor JW, Bruns TD. 2014. Airborne bacterial communities in residences: similarities and differences with fungi. PLoS One 9:e91283. doi: 10.1371/journal.pone.0091283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tignat-Perrier R, Dommergue A, Thollot A, Keuschnig C, Magand O, Vogel TM, Larose C. 2019. Global airborne microbial communities controlled by surrounding landscapes and wind conditions. Sci Rep 9:14441. doi: 10.1038/s41598-019-51073-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gawor J, Grzesiak J, Sasin-Kurowska J, Borsuk P, Gromadka R, Górniak D, Świątecki A, Aleksandrzak-Piekarczyk T, Zdanowski MK. 2016. Evidence of adaptation, niche separation and microevolution within the genus Polaromonas on Arctic and Antarctic glacial surfaces. Extremophiles 20:403–413. doi: 10.1007/s00792-016-0831-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yan D, Zhang T, Su J, Zhao L-L, Wang H, Fang X-M, Zhang Y-Q, Liu H-Y, Yu L-Y. 2016. Diversity and composition of airborne fungal community associated with particulate matters in Beijing during haze and non-haze days. Front Microbiol 7:487. doi: 10.3389/fmicb.2016.00487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oliveira M, Ribeiro H, Delgado JL, Abreu I. 2009. Seasonal and intradiurnal variation of allergenic fungal spores in urban and rural areas of the North of Portugal. Aerobiologia 25:85–98. doi: 10.1007/s10453-009-9112-z. [DOI] [Google Scholar]
- 34.Antón SF, de la Cruz DR, Sánchez JS, Sánchez Reyes E. 2019. Analysis of the airborne fungal spores present in the atmosphere of Salamanca (MW Spain): a preliminary survey. Aerobiologia 35:447–462. doi: 10.1007/s10453-019-09569-z. [DOI] [Google Scholar]
- 35.Egidi E, Delgado-Baquerizo M, Plett JM, Wang J, Eldridge DJ, Bardgett RD, Maestre FT, Singh BK. 2019. A few Ascomycota taxa dominate soil fungal communities worldwide. Nat Commun 10:2369. doi: 10.1038/s41467-019-10373-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Makhalanyane TP, Valverde A, Gunnigle E, Frossard A, Ramond J-B, Cowan DA. 2015. Microbial ecology of hot desert edaphic systems. FEMS Microbiol Rev 39:203–221. doi: 10.1093/femsre/fuu011. [DOI] [PubMed] [Google Scholar]
- 37.Goudie AS. 2014. Desert dust and human health disorders. Environ Int 63:101–113. doi: 10.1016/j.envint.2013.10.011. [DOI] [PubMed] [Google Scholar]
- 38.Behzad H, Mineta K, Gojobori T. 2018. Global ramifications of dust and sandstorm microbiota. Genome Biol Evol 10:1970–1987. doi: 10.1093/gbe/evy134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fisher MC, Henk DA, Briggs CJ, Brownstein JS, Madoff LC, McCraw SL, Gurr SJ. 2012. Emerging fungal threats to animal, plant and ecosystem health. Nature 484:186–194. doi: 10.1038/nature10947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Salo PM, Arbes SJ, Jr, Sever M, Jaramillo R, Cohn RD, London SJ, Zeldin DC. 2006. Exposure to Alternaria alternata in US homes is associated with asthma symptoms. J Allergy Clin Immunol 118:892–898. doi: 10.1016/j.jaci.2006.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Esch RE, Hartsell CJ, Crenshaw R, Jacobson RS. 2001. Common allergenic pollens, fungi, animals, and arthropods. Clin Rev Allergy Immunol 21:261–292. doi: 10.1385/CRIAI:21:2-3:261. [DOI] [PubMed] [Google Scholar]
- 42.Berdugo M, Delgado-Baquerizo M, Soliveres S, Hernández-Clemente R, Zhao Y, Gaitán JJ, Gross N, Saiz H, Maire V, Lehman A, Rillig MC, Solé RV, Maestre FT. 2020. Global ecosystem thresholds driven by aridity. Science 367:787–790. doi: 10.1126/science.aay5958. [DOI] [PubMed] [Google Scholar]
- 43.Asadi S, Gaaloul Ben Hnia N, Barre RS, Wexler AS, Ristenpart WD, Bouvier NM. 2020. Influenza A virus is transmissible via aerosolized fomites. Nat Commun 11:4062. doi: 10.1038/s41467-020-17888-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Caldwell DE, Brannan DK, Morris ME, Betlach MR. 1981. Quantitation of microbial growth on surfaces. Microb Ecol 7:1–11. doi: 10.1007/BF02010473. [DOI] [PubMed] [Google Scholar]
- 45.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, Gormley N, Gilbert JA, Smith G, Knight R. 2012. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6:1621–1624. doi: 10.1038/ismej.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schoch CL, Seifert KA, Huhndorf S, Robert V, Spouge JL, Levesque CA, Chen W, Fungal Barcoding Consortium . 2012. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for fungi. Proc Natl Acad Sci USA 109:6241–6246. doi: 10.1073/pnas.1117018109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. 2016. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods 13:581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17:10–12. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 49.Wang Q, Garrity GM, Tiedje JM, Cole JR. 2007. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. 2013. The SILVA ribosomal RNA gene database project: improved data processing and Web-based tools. Nucleic Acids Res 41:D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Abarenkov K, Henrik Nilsson R, Larsson K-H, Alexander IJ, Eberhardt U, Erland S, Høiland K, Kjøller R, Larsson E, Pennanen T, Sen R, Taylor AFS, Tedersoo L, Ursing BM, Vrålstad T, Liimatainen K, Peintner U, Kõljalg U. 2010. The UNITE database for molecular identification of fungi—recent updates and future perspectives. New Phytol 186:281–285. doi: 10.1111/j.1469-8137.2009.03160.x. [DOI] [PubMed] [Google Scholar]
- 52.Paulson JN, Stine OC, Bravo HC, Pop M. 2013. Differential abundance analysis for microbial marker-gene surveys. Nat Methods 10:1200–1202. doi: 10.1038/nmeth.2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hijmans RJ, Cameron SE, Parra JL, Jones PG, Jarvis A. 2005. Very high resolution interpolated climate surfaces for global land areas. Int J Climatol 25:1965–1978. doi: 10.1002/joc.1276. [DOI] [Google Scholar]
- 54.Wieder WR, Boehnert J, Bonan GB. 2014. Evaluating soil biogeochemistry parameterizations in Earth system models with observations. Global Biogeochem Cycles 28:211–222. doi: 10.1002/2013GB004665. [DOI] [Google Scholar]
- 55.Delgado-Baquerizo M, Oliverio AM, Brewer TE, Benavent-González A, Eldridge DJ, Bardgett RD, Maestre FT, Singh BK, Fierer N. 2018. A global atlas of the dominant bacteria found in soil. Science 359:320–325. doi: 10.1126/science.aap9516. [DOI] [PubMed] [Google Scholar]
- 56.R Core Team. 2020. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. http://www.R-project.org/. [Google Scholar]
- 57.Ning D, Deng Y, Tiedje JM, Zhou J. 2019. A general framework for quantitatively assessing ecological stochasticity. Proc Natl Acad Sci USA 116:16892–16898. doi: 10.1073/pnas.1904623116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, Minchin PR, O’Hara RB, Simpson GL, Solymos P, Stevens MHH, Szoecs E, Wagner H. 2020. vegan: community ecology package. R package version 2.5–7. https://cran.r-project.org/package=vegan.
- 59.Goslee SC, Urban DL. 2007. The ecodist package for dissimilarity-based analysis of ecological data. J Stat Softw 22:1–19. doi: 10.18637/jss.v022.i07. [DOI] [Google Scholar]
- 60.Grömping U. 2006. Relative importance for linear regression in R : the package relaimpo. J Stat Softw 17:1–27. doi: 10.18637/jss.v017.i01. [DOI] [Google Scholar]
- 61.Kim S. 2015. ppcor: an R package for a fast calculation to semi-partial correlation coefficients. Commun Stat Appl Methods 22:665–674. doi: 10.5351/CSAM.2015.22.6.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material. Download SPECTRUM01447-21_Supp_1_seq4.pdf, PDF file, 3.3 MB (3.4MB, pdf)
Data Availability Statement
The data supporting the conclusions of this article are available in Figshare (https://doi.org/10.6084/m9.figshare.14213519).