

Summary
Monocytic-lineage inflammatory Ly6c+CD103+ dendritic cells (DCs) promote anti-tumor immunity, but these DCs are infrequent in tumors, even upon chemotherapy. Here we examined how targeting pathways that inhibit the differentiation of inflammatory myeloid cells impact anti-tumor immunity. Pharmacologic inhibition of Bruton’s Tyrosine Kinase (BTK) and the tryptophan-degrading enzyme indoleamine 2,3-dioxygenase (IDO), or deletion of Btk or Ido1, allowed robust differentiation of inflammatory Ly6c+CD103+ DCs during chemotherapy, promoting anti-tumor T cell responses and inhibiting tumor growth. Immature Ly6c+c-kit+ precursor cells had epigenetic profiles similar to conventional DC precursors; deletion of Btk or Ido1 promoted differentiation of these cells. Mechanistically, a BTK-IDO axis inhibited a tryptophan-sensitive differentiation pathway driven by GATOR2 and mTORC1, and disruption of the GATOR2 in monocyte-lineage precursors prevented differentiation into inflammatory DCs in vivo. IDO-expressing DCs and monocytic cells were present across a range of human tumors. Thus, a BTK-IDO axis represses differentiation of inflammatory DCs during chemotherapy, with implications for targeted therapies.
eTOC blurb
Dendritic cells (DCs) in tumors are often dysfunctional, failing to effectively cross-present tumor antigens following chemotherapy. Sharma et al. reveal a pathway consisting of the kinase BTK and the tryptophan-depleting enzyme IDO that suppresses activation of monocyte-lineage DCs by inhibiting amino-acid sensitive mTORC1 signaling. Pharmacological blockade of this pathway promotes differentiation of inflammatory DCs and enhances anti-tumor T cells responses.
Graphical Abstract
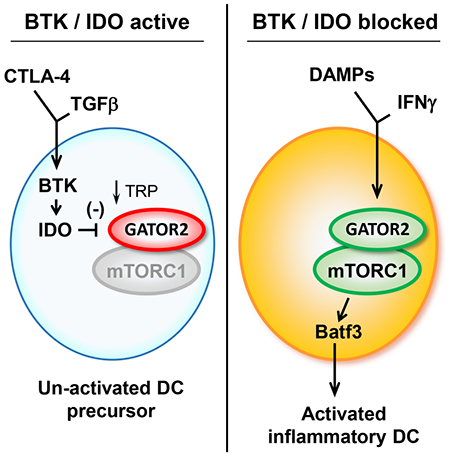
Introduction
In theory, combining immunotherapy with conventional chemotherapy should be a powerful strategy to activate anti-tumor immunity (Salas-Benito et al., 2021). Following chemotherapy, dying cells release a wave of tumor antigens, along with potentially pro-inflammatory signals. In practice, however, the impact of chemo-immunotherapy has been limited, mostly confined to incremental improvement of standard checkpoint-blockade approaches in tumor types that were already checkpoint-responsive (Gandhi et al., 2018).
Insight into this limited responsiveness may lie in understanding the nature of the dendritic cells (DCs) responsible for cross-presenting tumor antigens following chemotherapy (Gardner et al., 2020; Garris and Luke, 2020). Cross-presentation is associated with the conventional type I dendritic cell (cDC1) subset (Böttcher and Reis e Sousa, 2018), but the classical immune-activating form of cDC1 are rare in tumors (Broz et al., 2014). Instead, many tumor-associated DCs appear dysfunctional, or even actively immunosuppressive (Maier et al., 2020; Oh et al., 2020). An inflammation-induced terminal activation step is thought to be critical in order for DCs to coordinate protective immune responses (Bosteels et al., 2020). In tumors, the lack of activated, inflammatory DCs suggests that this key final step may be suppressed or blocked. However, the mechanism of this is unknown.
The regulatory enzyme indoleamine 2,3-dioxygenase (IDO) is part of a pathway that inhibits immune activation and enforces tolerance to apoptotic cells (Ravishankar et al., 2012; Sharma et al., 2015). In mouse models, expression of IDO by tumor-associated DCs activates regulatory T cells and inhibits effector T cell responses (Sharma et al., 2007). IDO also plays a poorly-understood role in immunosuppressive signaling within the DCs themselves (Pallotta et al., 2021). Thus, IDO might contribute to DC dysfunction during chemotherapy (Johnson et al., 2017). However, simply blocking IDO during chemotherapy has only modest effect (Hou et al., 2007), suggesting that some additional pathway still impedes the ability of DCs to present tumor antigens.
Cross-presentation of tumor antigens has traditionally been associated with conventional cDC1 cells (Hildner et al., 2008), but DCs from the monocyte lineage can also play a role during immunotherapy (Domingos-Pereira et al., 2019; Sharma et al., 2018). These inflammatory monocytic DCs differentiate from immature myeloid precursor cells in tumors (Sharma et al., 2018). In tumor-associated myeloid cells, the Bruton’s Tyrosine Kinase (BTK) signaling pathway promotes an immature, immunosuppressive phenotype (Gunderson et al., 2016; Stiff et al., 2016) and inhibits differentiation of immunogenic DCs (Kawakami et al., 2006; Varikuti et al., 2020). Therefore, we asked whether the BTK pathway might cooperate with IDO to suppress DC activation during chemotherapy.
Using mouse tumor models and a combination of pharmacologic and genetic strategies, we found that cell-intrinsic disruption of the BTK and IDO pathways during chemotherapy allowed robust differentiation of inflammatory monocyte-lineage DCs. In a model of cultured human monocytes, BTK acted to stabilize the expression of IDO, while IDO blocked tryptophan-sensitive inflammatory signaling via mTORC1. Single-cell RNA-sequencing databases from human tumors showed IDO-expressing DCs and monocytic cells to be widespread across a range of tumor types. Our findings suggest a BTK-IDO-mTORC1 axis that suppresses the differentiation of inflammatory DCs in tumors, and that may be targetable in human disease with existing pharmacological agents.
Results
Pharmacological inhibition of BTK and IDO promotes differentiation of tumor-associated monocyte-lineage DCs
To test whether monocytic DCs could mediate classical Batf3-dependent cross-presentation (Böttcher and Reis e Sousa, 2018), we asked if committed monocyte-lineage precursor cells (cMoPs) could restore antigen presentation in Batf3-deficient mice (Figure 1A). Adoptive-transfer of wild-type (WT) cMoPs restored cross-presentation of a nominal vaccine antigen as effectively as dendritic-lineage (CDP) precursors (Figure 1B). Both lineages gave rise to Batf3+ DCs after transfer, but the monocyte-lineage cells could be distinguished by their uniform expression of the myeloid marker Ly6c. The resulting CD11c+ DCs co-expressed both monocytic and dendritic markers (Ly6c+CD103+ DCs) (Sharma et al., 2018). This Ly6c+CD103+ population arose spontaneously after vaccination, and was lost in Batf3−/− mice (Figure 1C). Thus, inflammatory monocyte-lineage cells could be mediators of Batf3-dependent cross-presentation.
Figure 1. Pharmacological inhibition of BTK and IDO promotes differentiation of tumor-associated monocyte-lineage DCs.
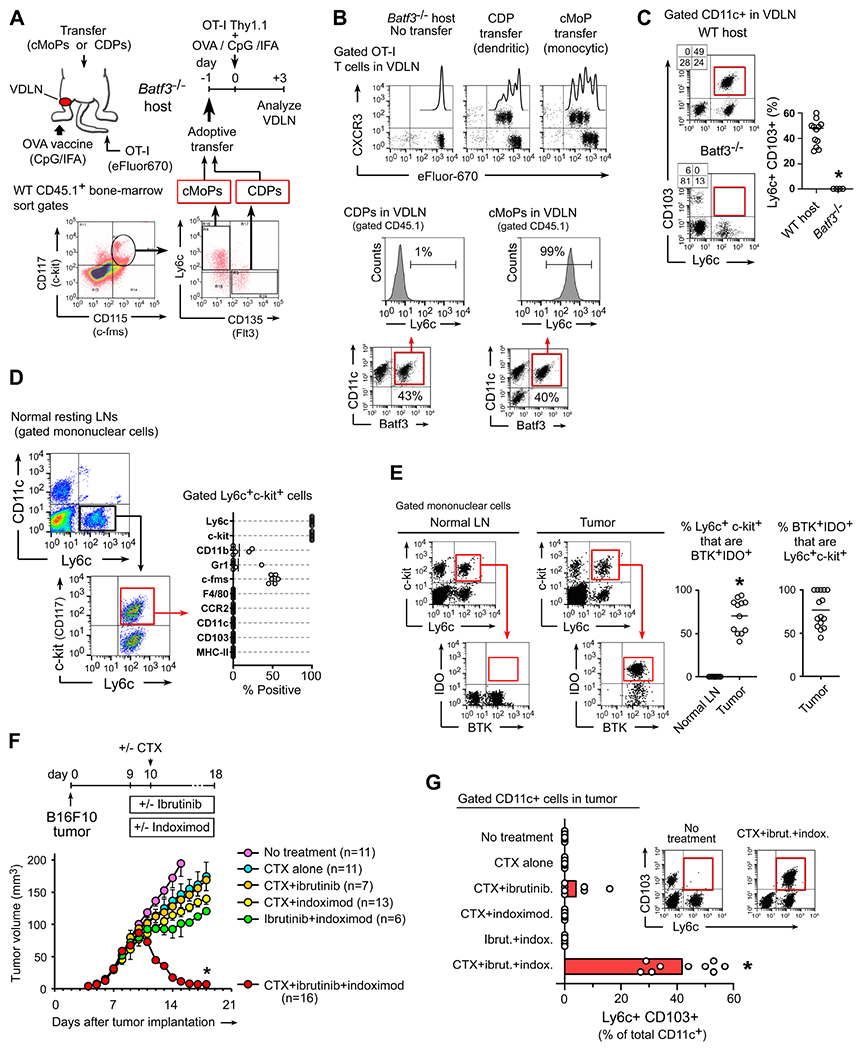
(A-B) Adoptive transfer of monocyte-lineage (cMoP) or dendritic (CDP) precursors into Batf3-deficient hosts, followed by injection of OT-I T cells i.v., and vaccination with ovalbumin protein (OVA) in CpG+incomplete Freund’s adjuvant (IFA), panel A. Flow-cytometry analysis of OT-I proliferation, and phenotype of the transferred precursor cells (B). Representative of 3 independent experiments.
(C) Flow cytometry analysis of endogenous Ly6c+CD103+ DCs (gated CD11c+ cells) in vaccine-draining lymph nodes (VDLNs) following vaccination of WT mice versus Batf3−/− (OVA+CpG+IFA vaccine, no adoptive-transfer). * p<0.001 by t-test.
(D) Flow cytometry phenotyping of immature Ly6c+c-kit+ precursor population in normal resting LNs in C57BL/6 mice.
(E) Flow cytometry analysis of BTK and IDO expression in gated Ly6c+c-kit+ cells from normal LNs, or disaggregated B16F10 melanoma tumors. *p<0.001.
(F-G) B16F10 tumors were treated with combinations of cyclophosphamide (CTX, 150 mg/kg i.p., given on day 10), ibrutinib (16 mg/kg/day i.p.) and indoximod (2 mg/ml in drinking water). Inhibitor drugs were started 1 day before CTX. Raw data pooled from a total of 27 experiments, showing tumor volume (F) and flow cytometry quantitation (G) of Ly6c+CD103+ DCs in tumors. *p<0.001 versus all other groups by ANOVA.
See also Figure S1
The Ly6c+CD103+ DCs emerged so rapidly that we speculated they might arise from cells already present in the lymph nodes (LNs). Bone-marrow cMoPs co-express Ly6c, c-kit and c-fms (Hettinger et al., 2013), and resting LNs revealed a population of cells with similar markers (Figure 1D). These Ly6c+c-kit+ cells did not express CCR2, thus distinguishing them from Ly6c+ circulating monocytes, and they showed no markers of mature DCs. In vitro maturation studies (Figure S1A) showed that the Ly6c+c-kit+ population could rapidly differentiate into Ly6c+CD103+ DCs when co-cultured with activated T cells. No other cells in the LNs could give rise to Ly6c+CD103+ DCs.
Similar Ly6c+c-kit+ precursor cells were present in tumors (Figure S1B). However, we hypothesized that in tumors these immature cells might be prevented from differentiation into inflammatory Ly6c+CD103+ DCs by either BTK or IDO. Analysis of Ly6c+c-kit+ precursors in tumors revealed that many co-expressed both BTK and IDO (Figure 1E). In contrast, Ly6c+c-kit+ cells in normal LNs did not show this co-expression. While B cells, MDSCs or other cells could potentially express BTK or IDO alone, co-expression of both together was less common. Thus, the small population of Ly6c+c-kit+ precursor cells accounted for the majority of BTK+IDO+ cells in tumors.
Based on this, we asked whether differentiation of Ly6c+CD103+ DCs might be enhanced by drugs that targeted the BTK and IDO pathways. These clinical drugs did not have the specificity and selectivity needed to test molecular mechanism (for which we used genetic techniques, below); however, they are already approved or in clinical trials, so they are readily translatable. Mice with B16F10 tumors were treated with ibrutinib (a BTK-inhibitor) and indoximod (an IDO-inhibitor) plus cyclophosphamide (CTX) chemotherapy (Figure 1F; individual traces are shown in Figure S1C). Tumors were allowed to become fully established before receiving chemotherapy on day 10. BTK and IDO inhibitor dugs were started one day prior. CTX alone had little effect on tumor growth; and adding ibrutinib or indoximod individually produced only minor delay. However, ibrutinib and indoximod together were synergistic with chemotherapy, producing rapid tumor regression. Although CTX had little effect by itself, it supplied an indispensable triggering signal, since in the absence of CTX the ibrutinib+indoximod combination by itself produced only minor effect.
The effect on tumor size was paralleled by differentiation of inflammatory Ly6c+CD103+ DCs in tumors (Figure 1G). Only the full combination of CTX + ibrutinib + indoximod elicited consistent differentiation of large numbers of Ly6c+CD103+ DCs. Induction of these DCs was rapid, beginning within 24 hrs of CTX administration (Figure S1D). This was before any change in tumor size, showing that the presence of DCs was not simply an artifact of shrinking tumors.
Pharmacological inhibition of BTK and IDO promotes anti-tumor T cell responses after chemotherapy and reduces tumor growth
The hypothesis predicted that the degree of DC differentiation should correlate with the degree of anti-tumor response. Using the ibrutinib + indoximod combination, titration of the ibrutinib dose produced a range of anti-tumor efficacy (measured as final tumor size), and the induction of Ly6c+CD103+ DCs tracked closely (Figure 2A). Linrodostat is another inhibitor of IDO (Sonpavde et al., 2020). Varying doses of linrodostat in combination with ibrutinib produced different degrees of anti-tumor activity (Figure 2B), and induction of Ly6c+CD103+ DCs was correlated. ArQ531 is a second-generation noncovalent BTK-inhibitor, with greater specificity than ibrutinib (i.e., no binding to ITK) and activity against ibrutinib-resistant disease (Reiff et al., 2018). When Arq531 was combined with chemotherapy, with and without indoximod, the anti-tumor activity of the various combinations again correlated with Ly6c+CD103+ DCs (Figure 2C).
Figure 2. Pharmacological inhibition of BTK and IDO promotes anti-tumor T cell responses after chemotherapy and reduces tumor growth.
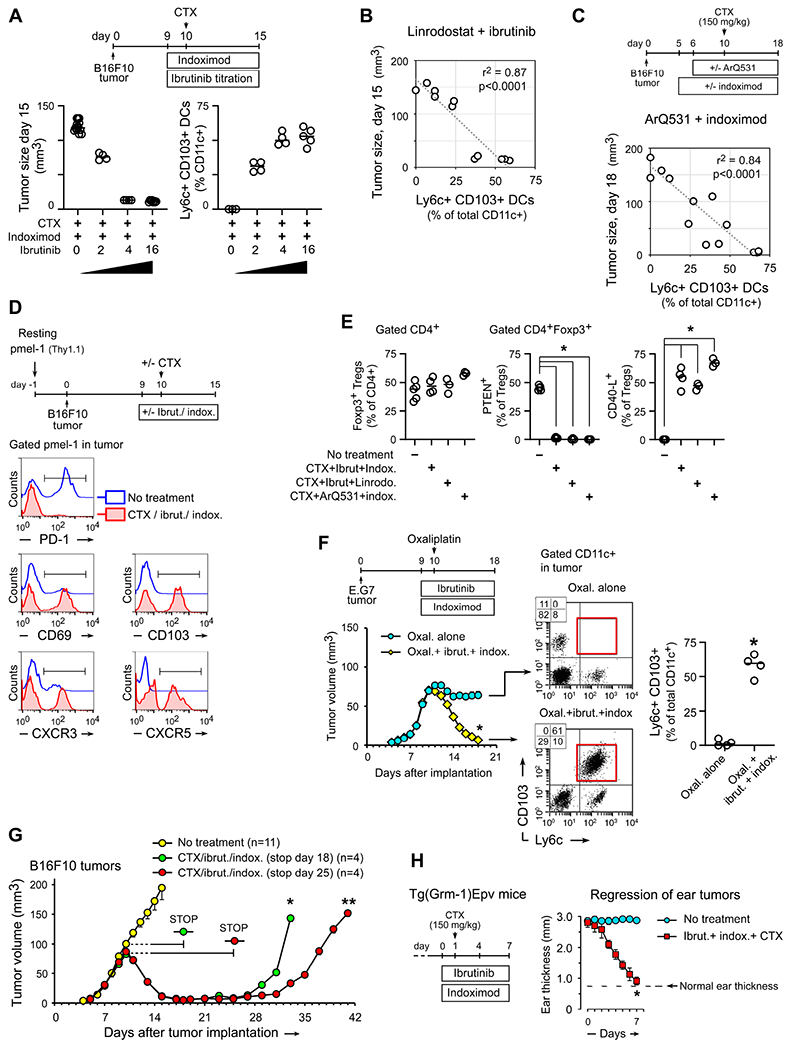
(A-C) Measurement of Ly6c+CD103+ DCs as a pharmacodynamic marker. B16F10 tumors were treated with combinations of CTX, different BTK-inhibitors and different IDO-inhibitors to produce a varying range of antitumor efficacy. Response was measured as final tumor size, and Ly6c+CD103+ DCs were quantitated in tumors by flow cytometry.
(A) Synergy between CTX, indoximod and graded doses of ibrutinib, by tumor size and Ly6c+CD103+ DCs, both measured on day 15.
(B) Linear regression analysis of Ly6c+CD103+ DCs versus tumor response across graded doses of the IDO-inhibitor linrodostat (10-5-2-0 mg/kg/day) plus CTX (given day 10) and ibrutinib (16 mg/kg/day), on a schedule similar to panel A. Regression analysis shows pooled data from all groups, measured on day 15.
(C) Ly6c+CD103+ DCs versus final tumor size (day 18) across different combinations of BTK-inhibitor ArQ531 (50 mg/kg/day i.p.) with CTX (day 10) and indoximod. Regression analysis shows pooled data from all groups, measured on day 18. (Based on preclinical pharmacology, the ArQ531 was started on day 6 in these experiments.)
(D) C57Bl/6 mice were pre-loaded with resting (naïve) TCR-transgenic pmel-1 T cells, then implanted with B16F10 tumors and treated with CTX + ibrutinib + indoximod, or no treatment. Flow cytometry analysis of Thy1.1+ pmel-1 cells in tumors on day 15. Representative of 3 independent experiments.
(E) Flow cytometry analysis showing destabilization of tumor-associated Treg cells (gated CD4+Foxp3+). B16F10 tumors treated with the three regimens as shown, or no treatment. *p<0.001 versus “no-treatment” group by ANOVA.
(F) Growth curves and flow cytometry analysis of E.G7 tumors (EL4-OVA) treated with oxaliplatin chemotherapy, versus oxaliplatin + ibrutinib + indoximod. *p<0.001.
(G) B16F10 tumors treated as in Figure 1F, with ibrutinib+indoximod therapy discontinued on day 18 (day +8 after CTX) or day 25 (day +15 after CTX). *, ** p<0.001 by ANOVA.
(H) Autochthonous melanoma tumors in Tg(Grm1)Epv mice, treated with CTX + ibrutinib + indoximod, versus untreated controls. Tumor response measured as reduction in ear thickness (Sharma et al., 2018); n=6 mice, *p<0.001.
See also Figure S2
To ask whether BTK-IDO blockade allowed activation of effector T cells (van der Leun et al., 2020), mice were pre-loaded with congenically-marked tumor-specific CD8+ T cells (naïve pmel-1) prior to tumor implantation (Figure 2D, S2A). Under these conditions, pmel-1 are rendered unresponsive (anergic) to cognate tumor antigen (Sharma et al., 2018). Without treatment, pmel-1 in tumors became PD-1HI and showed no evidence of activation. In contrast, following BTK-IDO chemo-immunotherapy, pmel-1 cells in tumors showed reduction in PD-1 and increase in multiple activation markers.
Inflammation can also destabilize the immunosuppressive Treg cell phenotype in tumors, manifesting as loss of PTEN phosphatase and expression of pro-inflammatory markers such as CD40-ligand (Sharma et al., 2015). Following BTK-IDO chemo-immunotherapy the number of Treg cells in tumors (expressed as a fraction of infiltrating CD4+ cells) did not change, but there was extensive destabilization of the immunosuppressive phenotype (Figure 2E). This was seen in all of the three drug combinations.
Next we next evaluated a model in which the chemotherapy itself was more intrinsically active, to ask whether BTK-IDO blockade would have any additional effect. EL4-OVA cells (E.G7) treated with oxaliplatin chemotherapy undergo immunogenic cell death (Ghiringhelli et al., 2009). Using this model, oxaliplatin alone caused a significant delay in growth but could not cause objective regression, whereas addition of ibrutinib + indoximod resulted in rapid tumor regression (Figure 2F). Pharmacodynamic analysis showed that oxaliplatin alone was unable to maintain differentiation of intratumoral Ly6c+CD103+ DCs, whereas adding BTK-IDO blockade sustained robust DC differentiation (measured on day +8 after the dose of chemotherapy).
The direct cytolytic effect of a single dose of chemotherapy lasts only a few days, but when IDO and BTK were blocked we saw that tumors continued to shrink for many days. Although the residual tumors were small, intratumoral Ly6c+CD103+ DCs remained strongly induced (see Figure 2F) suggesting that an immune response was on-going. To test this, we used the B16F10 model and discontinued ibrutinib + indoximod therapy at either day +8 or day +15 after chemotherapy (Figure 2G). At either time-point, if ibrutinib + indoximod was stopped, then the tumors progressively regrew. Thus, it appeared that even small tumors were being actively maintained by an on-going anti-tumor response.
Finally, we tested an autochthonous tumor model, since immune responses against such tumors can be difficult to achieve (Ciampricotti et al., 2012). Tg(Grm1)Epv mice progressively develop extensive multi-focal melanomas on skin of tail and ears (Shin et al., 2008). Treatment with CTX + ibrutinib + indoximod caused rapid involution of tumors (Figure 2H), quantitated as the thickness of ear tumors (Sharma et al., 2018). The regressing tumors demonstrated robust differentiation of Ly6c+CD103+ DCs (Figure S2B).
Deletion of Btk or Ido1 replaces pharmacologic inhibition and allows differentiation of inflammatory monocyte-lineage DCs within tumors following chemotherapy
We next tested whether the Ly6c+CD103+ DCs mechanistically drove the response to treatment. Batf3−/− mice lacked the Ly6c+CD103+ DC population during vaccination (cf. Figure 1C), and they also failed to respond to BTK-IDO chemo-immunotherapy (Figure 3A). We asked whether supplying the Ly6c+CD103+ population by adoptive-transfer of wild-type (WT) cMoPs could restore the response to BTK-IDO blockade (Figure 3B). For comparison, we tested transfer of conventional CDP dendritic precursors. Both precursor populations reached the tumor in similar numbers, and both expressed CD11c, Batf3, and CD103. However, only the cMoP-derived DCs expressed Ly6c and produced the Ly6c+CD103+ DCs; and only the cMoPs drove T cell activation and objective tumor regression. CDPs produced only modest growth delay.
Figure 3. Deletion of Btk or Ido1 replaces pharmacologic inhibition and allows differentiation of inflammatory monocyte-lineage DCs within tumors following chemotherapy.
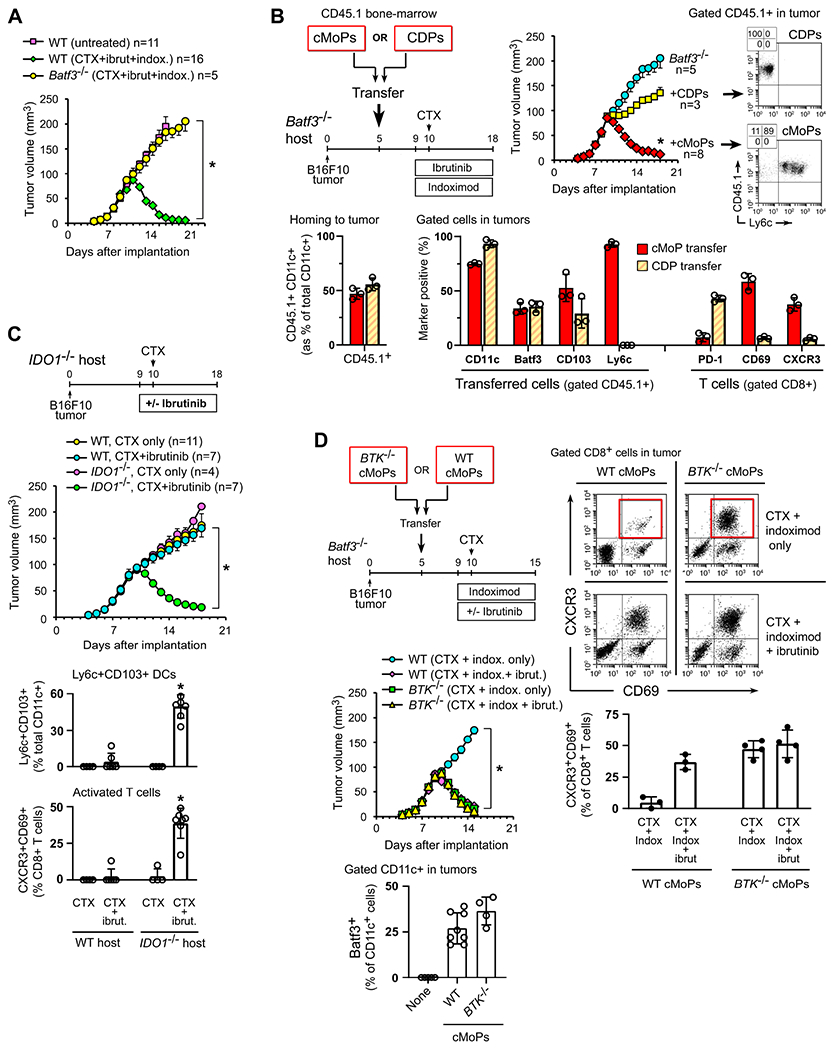
(A) B16F10 tumors implanted in Batf3−/− or WT mice, then treated with CTX + ibrutinib + indoximod, as in Figure 1F. In all panels in the figure, *p<0.001 by ANOVA.
(B) Batf3−/− mice bearing B16F10 tumors received adoptive-transfer of sorted cMoPs or CDPs from CD45.1+ WT bone marrow. Control mice received no transfer. All mice were then treated with CTX + ibrutinib + indoximod. Bar graphs show flow cytometry analysis of homing to tumors (left), and phenotype of transferred cells and endogenous host T cells (right) in tumors at the end of therapy.
(C) Ido1−/− mice or WT C57BL/6 controls were implanted with B16F10 tumors and treated with CTX alone or CTX + ibrutinib doublet. Pooled data from 4 independent experiments.
(D) Batf3−/− mice with established B16F10 tumors received adoptive-transfer of cMoPs from either Btk−/− donors or WT controls. All mice were then treated with CTX + indoximod doublet alone, or CTX + indoximod + ibrutinib. Pooled data from 4 independent experiments. Flow cytometry was performed to measure the ability of each cMoP population to generate Batf3+CD11c+ DCs in tumors (using the CTX + ibrutinib + indoximod regimen), and the phenotype of CD8+ T cells in tumors on day 15.
See also Figure S3
To further test that the inflammatory Ly6c+ DCs elicited by BTK-IDO therapy were originating from the monocytic lineage, we performed lineage-tracing studies using a myelomonocytic Lyz2-YFP reporter strain (Figure S3A). Prior to treatment few CD11c+ cells in tumors expressed Lyz2-YFP, whereas following treatment the majority of CD103+ DCs in tumors now expressed Ly6c and marked with the Lyz2 (LysM) reporter. Similarly, when WT mice were treated with BTK-IDO chemo-immunotherapy, the majority of CD103+ DCs were of the Ly6c+ phenotype (Figure S3B). Thus, for our remaining studies we focused on the cMoP-derived (monocytic) Ly6c+CD103+ DC population.
Re-activation of exhausted T cells in tumors has been reported to require IL-12 from DCs (Garris et al., 2018. Using the Batf3−/− adoptive-transfer model, cMoP cells lacking IL-12p40 failed to rescue anti-tumor activity, compared to IL-12-competent WT cMoPs (Figure S3C). This further supported the hypothesis that the Ly6c+CD103+ DCs were themselves driving the re-activation of anti-tumor immunity.
To test whether the effect of indoximod was on-target, we asked whether ablation of the Ido1 gene could substitute for the IDO-inhibitor drug (Figure 3C). Tumors were implanted in Ido1−/− mice (Baban et al., 2004) or WT controls, then treated with CTX + ibrutinib doublet (without IDO-inhibitor). In WT hosts, the CTX + ibrutinib doublet showed little effect, as expected from Figure 1. However, in Ido1−/− hosts, CTX + ibrutinib alone showed a robust anti-tumor response, accompanied by differentiation of Ly6c+CD103+ DCs and activation of T cells. Thus, genetic ablation of Ido1 functionally replaced the contribution of the IDO-inhibitor drug, implying an on-target effect.
Even in the genetic absence of Ido1, however, the BTK-inhibitor drug was still required, since CTX without ibrutinib had no effect. This suggested that BTK and IDO each made their own contribution to suppression, and removing only one of them was not enough.
It was slightly more complex to test the BTK mechanism, since Btk−/− mice have multiple immune defects (Mangla et al., 2004), so tumor studies directly in these mice would not be informative. Instead, we used the Batf3−/− adoptive-transfer model to ask whether genetic ablation of Btk in the transferred cMoPs would replace the need for BTK-inhibitor drug (Figure 3D). Control Btk-sufficient (WT) cMoPs strictly required the BTK-inhibitor drug and did not respond to CTX + indoximod alone. However, Btk−/− cMoPs responded to CTX + indoximod alone, and adding ibrutinib had no further observable effect. Thus, genetic ablation of Btk in the cMoP population replaced the need for BTK-inhibitor drug, again implying an on-target effect.
Immature Ly6c+c-kit+ precursor cells and inflammatory monocytic DCs in tumor-draining lymph nodes share genetic similarities with pre-cDC1 and cDC1
Tumor-draining LNs (TDLNs) are an important site of immune activation during immunotherapy (Yost et al., 2021). In the ibrutinib+indoximod model, adoptive-transfer studies confirmed the presence of Ly6c+CD103+ DCs in TDLNs after therapy (Figure S4A). To ask if Ly6c+c-kit+ precursor cells were epigenetically primed to express genes associated with DCs, immature (CD11cNEG) Ly6c+c-kit+ cells were sorted from TDLNs of untreated B16F10 tumors, and chromatin accessibility measured by ATAC-seq (Figure 4A). Results were compared against analysis of publically-available datasets for mature cDC1 (Brown et al., 2019 and pre-cDC1 precursors from bone-marrow (Durai et al., 2019). Ly6c+c-kit+ cells showed open chromatin at classical cDC1 genes such as Batf3, Itgae, Zbtb46, Flt3, Xcr1, Irf8, Batf3, Zbtb46 and Id2. All of these were shared with conventional pre-cDC1 cells, as were Clec9a, Itgae (CD103), Irf4 and MHC class II (Figure S4B). In addition, however, immature Ly6c+c-kit+ cells also showed open chromatin at monocytic loci such as Ly6c2 (Ly6c), Cebpb, Cx3cr1, Ccr2, Itgam (CD11b), and Csfr1 (CD115, c-fms). This monocytic signature was not shared with the pre-cDC1 or cDC1 cells.
Figure 4. Immature Ly6c+c-kit+ precursor cells and inflammatory monocytic DCs in tumor-draining lymph nodes share genetic similarities with pre-cDC1 and cDC1.
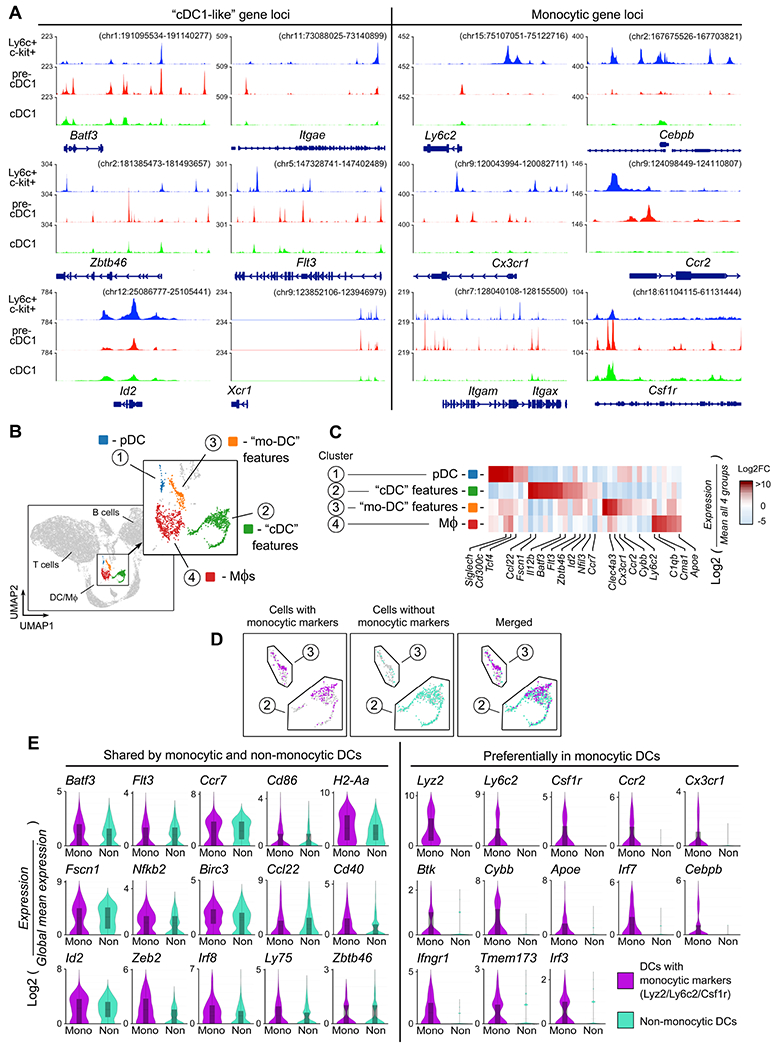
(A) Ly6c+c-kit+ precursor cells (CD11cNEG) were sorted from tumor-draining LNs (TDLNs) of untreated B16F10 tumors (day 14). Chromatin accessibility was assessed by ATAC-seq (A). Tracks are compared against published datasets for mature cDC1 (Brown et al., 2019) and pre-cDC1 (Durai et al., 2019). Three independent experiments were performed with similar results; a representative track is shown.
(B-E) scRNA-seq was performed on total cells from TDLNs of B16F10 tumors, 48 hrs after treatment with CTX + ibrutinib + indoximod. Analysis was performed using Loupe browser. Unsupervised UMAP clustering (B), with heat-map (C) of genes used to assign cluster descriptions. (D) Clusters 2 and 3 were pooled, and individual cells classified as falling in either “monocytic” or “non-monocytic” sub-population based on expression of at least one monocytic-signature gene (Ly6c1, Ly6c2, Csf1r or Lyz2). (E) Expression of selected individual genes by cells classified as either “monocytic-signature” or non-monocytic in pooled Clusters 2+3.
See also Figure S4
While the immature Ly6c+c-kit+ cells showed accessible chromatin at many “cDC1-like” genes, these were not yet expressed at the mRNA level (suppl. Figure S4C). In contrast, many of the monocytic genes were actively expressed as mRNA. Thus, Ly6c+c-kit+ precursor cells were recognizably monocytic in origin, but appeared epigenetically poised for rapid differentiation into a phenotype with features resembling conventional cDC1.
To characterize the mature Ly6c+CD103+ DC population, we performed single-cell RNA sequencing (scRNA-seq). B16F10 tumors were treated with CTX + ibrutinib + indoximod as described in Figure 1F, and cells isolated from TDLNs 48 hrs after the dose of CTX. Two mice were analyzed separately and the biological replicates pooled. Unsupervised clustering was performed by UMAP (Figure 4B), and major immune subsets were assigned based on characteristic markers (Figure S4D). Four clusters were found to contain the DCs and monocyte-macrophage (Mϕ) cells; these were arbitrarily designated Clusters 1-4. Heat-map analysis using a set of curated marker genes (Figure 4C) showed that Cluster 2 expressed genes characteristic of conventional cDCs (Batf3, Flt3, Id2, Nfil3, Fscn1, Ccl22). Cluster 3 expressed some of these same genes, but also showed a prominent monocytic signature (Ly6c2, Cx3cr1, Ccr2, Cybb) suggestive of monocytic DCs. Clusters 1 and 4 were respectively plasmacytoid DCs and Mϕs, and were not analyzed further.
Although Cluster 2 had features of conventional cDCs, we knew that monocyte-lineage cMoPs could give rise to a similar phenotype. The markers that we used to identify such cells by flow cytometry (Ly6c and CD103) proved to be low-abundance transcripts on scRNA-seq, so to identify putative monocyte-lineage cells we employed an “OR” function of Ly6c1, Ly6c2, Csf1r and Lyz2. All cells in Clusters 2 and 3 were pooled, and then each cell was coded as either “monocytic gene signature” if it expressed at least one of these genes, or “non-monocytic” if none were expressed. Cluster 3 contained almost exclusively “monocytic signature” cells by this classification. However, the cDC-like Cluster 2 also contained a large number of cells classified as monocytic-signature (Figure 4D). We therefore pooled Clusters 2 and 3, and compared cells based on monocytic signature vs non-monocytic (Figure 4E). Many of the monocytic-signature and non-monocytic cells shared conventional cDC-like genes such as Flt3, Irf8, Zbtb46, Id2 and Fscn1. However, the monocytic-signature cells also expressed genes such as Ly6c2, Cx3cr1, Ccr2, Cybb, which were not shared with the non-monocytic population. Thus, taken together, the scRNA-seq data were consistent with the epigenetic data, suggesting a population of DCs derived from the monocyte lineage, but with many features overlapping conventional cDC.
BTK and IDO in human monocyte-derived DCs inhibit an inflammatory differentiation pathway driven by GATOR2 and mTORC1
In the mouse models, blocking BTK and IDO together was so synergistic that we suspected the two pathways must interact. To test this, we used an in vitro model of human monocyte-derived “DC-like” cells (MDDCs) (Munn et al., 2002; Munn et al., 2004). These are not authentic tissue DCs, but they can be informative for modeling specific signaling pathways (Balan et al., 2018). Monocytes were cultured in GM-CSF + IL-4 then differentiated to a “DC-like” stage with a standard cocktail of inflammatory cytokines (Jonuleit et al., 1997). The resulting cells expressed the generalized DC marker CD83, but few expressed the more differentiated markers Batf3 or XCR1 (Figure 5A). Completing this final differentiation step required a strong inflammatory signal, which was supplied by adding IFNγ during the last 24 hrs. As an opposing signal we used recombinant TGFβ1. TGFβ is an immunosuppressive pathway in tumors (de Streel and Lucas, 2021), and it activates BTK signaling in DCs (Gujar et al., 2016). Adding TGFβ completely blocked the ability of IFNγ to drive the final DC differentiation step.
Figure 5. BTK and IDO in human monocyte-derived DCs inhibit an inflammatory differentiation pathway driven by GATOR2 and mTORC1.
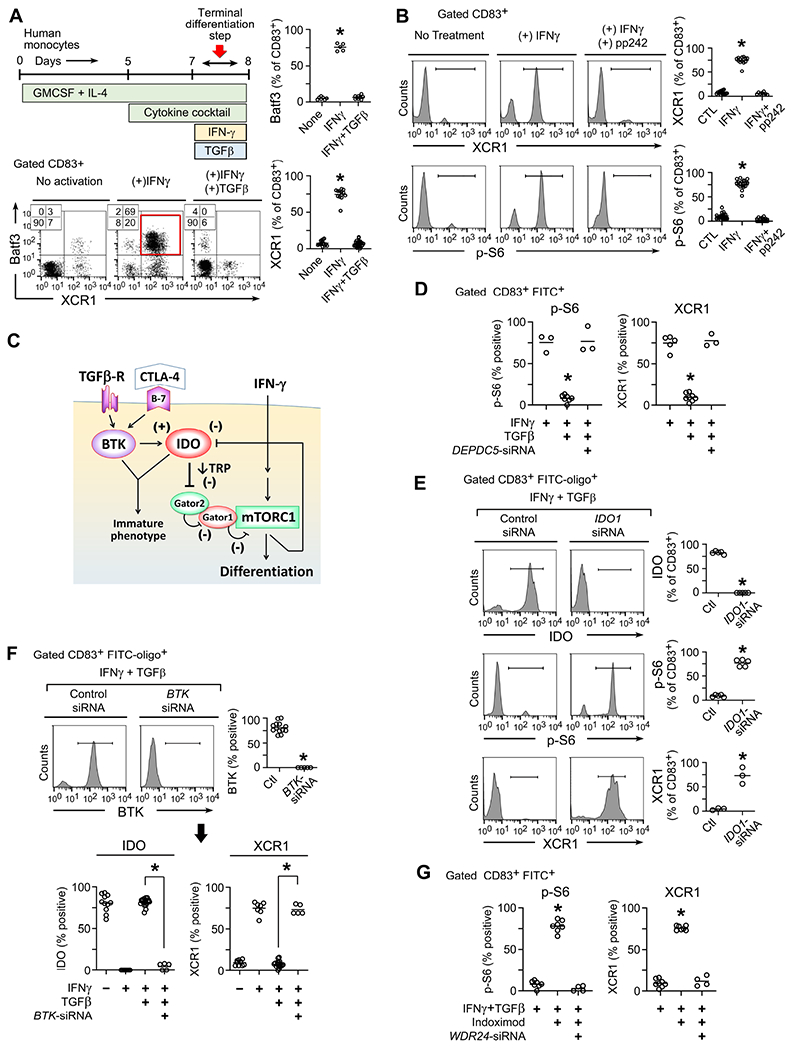
Human monocytes were cultured with growth factors and cytokine cocktail as described in Methods, followed by activation with IFNγ to drive terminal differentiation, with or without inhibitory TGFβ.
(A) In vitro culture schema, and flow cytometry analysis of gated CD83+ cells on day 8 for each treatment group. Representative of at lease 5 independent experiments. *p<0.001 by ANOVA.
(B) Analysis of DC maturation (XCR1 marker) and S6 phosphorylation by flow cytometry in cells treated with IFNγ ± mTOR-inhibitor pp242. At least 6 independent experiments per group.
(C) Proposed signaling model.
(D-G) Silencing of IDO1, BTK or components of the GATOR1-GATOR2 complex using siRNA (or scrambled siRNA control). siRNA was added day 5 along with FITC-labeled tracer oligos to identify transfected cells. Cells were analyzed by flow cytometry on day 8, and transfected cells gated as CD83+ FITC+ .
(D) Effect of silencing of the DEPDC5 subunit of GATOR1. At least 3-5 experiments per group, *p<0.001 vs both other groups.
(E) Effect of silencing IDO1. At least 3-5 experiments per group, *p<0.001 vs both other groups.
(F) Effect of silencing BTK on the stability of IDO. At least 5 experiments per group, *p<0.001.
(G) Effect of disrupting the WDR24 subunit of GATOR2 on the ability of indoximod (200 uM) to restore mTORC1 activity (p-S6) in vitro. At least 4 experiments per group, *p<0.001 vs both other groups.
See also Figure S5
IDO degrades the essential amino acid tryptophan (Munn and Mellor, 2013). In other settings this can inhibit the amino-acid sensitive mTOR pathway (Sharma et al., 2015). As a readout for activity of the mTORC1 complex, we measured phosphorylation of ribosomal S6 protein (Figure 5B). IFNγ caused phosphorylation of S6, which was prevented by the mTOR-inhibitor compound pp242. Blocking mTOR also prevented DC differentiation (XCR1 expression), even in the presence of IFNγ. Thus, we hypothesized that IFNγ might require mTORC1 signaling in order to drive DC differentiation, and this mTOR signal might be blocked by IDO (proposed diagram in Figure 5C)
To test the role of mTORC1 we used siRNA to silence the DEPDC5 subunit of the GATOR1 complex (Figure 5D). GATOR1 normally suppresses mTORC1 (Wolfson and Sabatini, 2017), so disrupting GATOR1 is activating. TGFβ suppressed S6 phosphorylation and XCR1 expression as expected; but disrupting GATOR1 restored mTORC1 activity (p-S6) and allowed DC differentiation (XCR1), despite the presence of TGFβ. Thus, the ability of TGFβ to block DC differentiation appeared to depend on its ability to successfully inhibit mTORC1.
To ask whether the inhibition of mTORC1 occurred via an IDO-dependent mechanism, IDO1 was silenced with siRNA during IFNγ+TGFβ treatment (Figure 5E). (In these experiments we used flow cytometry to follow IDO, but this correlated well with functional enzymatic activity, Figure S5A.) Despite the presence of TGFβ, silencing IDO1 restored mTORC1 activity (p-S6) and DC differentiation (XCR1). Thus, TGFβ required an intact IDO pathway in order to successfully inhibit mTORC1.
In some cell types, IFNγ is an inducer of IDO. In mature MDDCs, however, IFNγ causes rapid loss of IDO, unless expression is maintained by stabilizing signals such as IL-10 or CTLA-4 (Munn et al., 2002; Munn et al., 2004). In our model, IFNγ caused rapid loss of IDO (Figure 5F). Loss of IDO was prevented by TGFβ, and the ability of TGFβ to maintain IDO expression was strictly dependent on BTK, as shown using siRNA silencing. A similar effect was seen with a second IDO-stabilizing signal, CTLA-4-Ig fusion protein (abatacept) (Munn et al., 2004). CTLA-4-Ig maintained strong IDO expression in the face of IFNγ; and, like TGFβ, this was dependent on the BTK pathway (Figure S5B). Thus, the role of BTK appeared to be to allow signals such as TGFβ or CTLA-4-Ig to dominantly stabilize IDO and prevent its loss of expression.
The GATOR2 complex is sensitive to levels of key amino acids (Wolfson and Sabatini, 2017). One way in which IDO might block mTORC1 activity was by creating an intracellular “tryptophan-insufficiency” condition. Consistent with this possibility, addition of 10-fold excess tryptophan in the medium overcame the ability of TGFβ to inhibit mTORC1 activity (p-S6) and restored DC maturation (XCR1) (Figure S5C). Based on this, we asked whether restoring the “sufficiency” signal from GATOR2 was the functional molecular target when cells were treated with an IDO-inhibitor drug. Cells were treated with IFNγ+TGFβ, and indoximod was added to block IDO (Figure 5G). Indoximod restored mTORC1 activity (p-S6) and DC differentiation (XCR1). Disrupting the WDR24 subunit of GATOR2 entirely reversed the effect of indoximod, abrogating S6 phosphorylation and preventing DC differentiation. Taken together, these studies suggested that the mechanistic target of BTK-IDO blockade in vivo might therefore be to restore the “sufficiency” signal delivered by GATOR2.
Disrupting the GATOR2-mediated sufficiency signal in monocyte-lineage precursors prevents differentiation into inflammatory DCs in vivo
To test this, we first asked whether the GATOR2 signal was required for physiologic differentiation of Ly6c+CD103+ DCs during a normal vaccination. Using the Batf3−/− vaccine model from Figure 1A, we disrupted the GATOR2 complex in the donor cMoPs by treating with siRNA against mouse Wdr24 prior to transfer (Figure 6A) Control cMoPs received irrelevant siRNA. Following adoptive transfer and vaccination, the control cMoPs expressed high levels of mTORC1 activity (p-S6); acquired expression of CD103 and Batf3; and supported cross-presentation to OT-I. Disrupting the GATOR2 complex abrogated mTORC1 activity, blocked DC differentiation, and prevented cross-presentation.
Figure 6. Disrupting the GATOR2-mediated sufficiency signal in monocyte-lineage precursors prevents differentiation into inflammatory DCs in vivo.
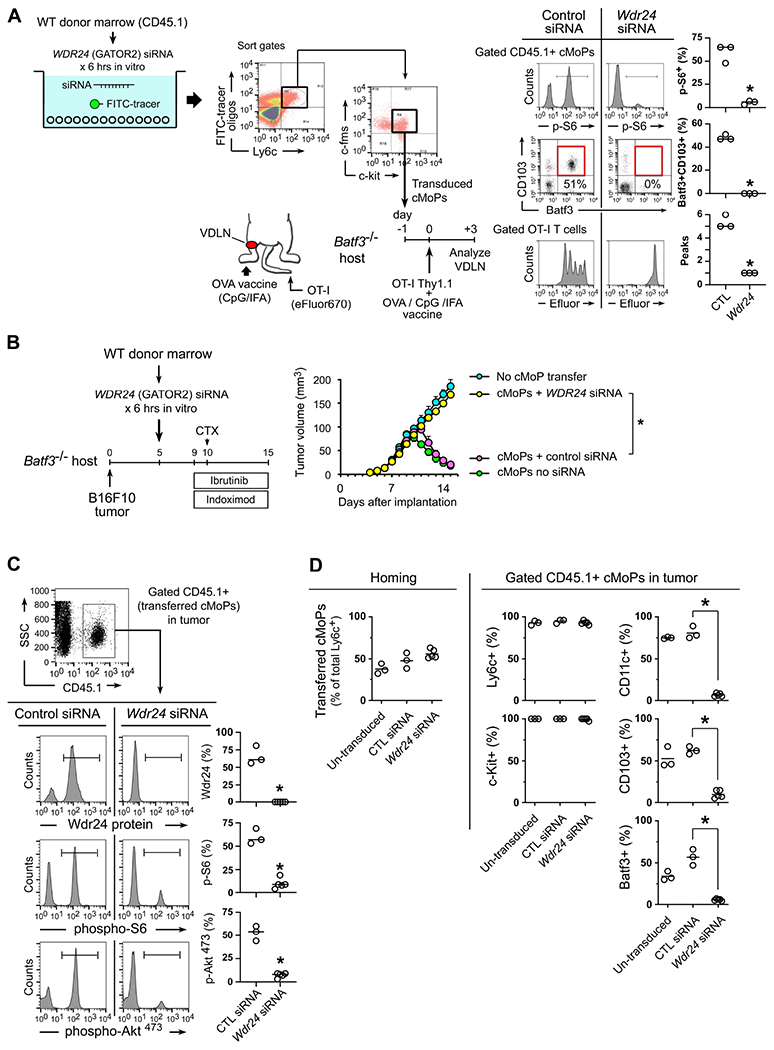
(A-D) In vitro transduction of wild-type CD45.1+ mouse bulk bone marrow cells, using siRNA against the Wdr24 subunit of GATOR2 (or scrambled control siRNA), plus a FITC-oligo tracer. FITC+ (transduced) cMoPs cells (Ly6c+ c-fms+ c-kit+) were then sorted and transferred into Batf3−/− recipients for vaccination or tumor studies.
(A) Rescue of vaccine response by WT cMoPs with either control siRNA or Wdr24 silencing.
(B-D) Rescue of anti-tumor response to CTX + ibrutinib + indoximod therapy in Batf3−/− mice by transfer of WT cMoPs, with or without siRNA silencing of Wdr24. All figures represent pooled data from 3-5 independent experiments.
(B) Growth curves. *p<0.001 by ANOVA. Additional pooled controls (Batf3−/− with no cMoP transfer; and WT cMoPs with no siRNA transduction) are included for comparison.
(C) Wdr24 protein and markers of mTOR activity in tumors on day 15 (+5 days after CTX).
(D) Homing of cMoPs to tumors (left, showing transferred cells as a fraction of total Ly6c+ cells in tumor) for un-transduced cMoPs vs Wdr24-silenced and control siRNA. Phenotypic markers (right) on gated CD45.1+ transferred cMoPs at the end of treatment (day 15). p<0.001 by ANOVA.
In the Batf3−/− B16F10 tumor model, cMoPs with a disrupted GATOR2 complex (treated with Wdr24 siRNA) were unable to rescue response to BTK-IDO chemo-immunotherapy (Figure 6B). Control cMoPs with irrelevant siRNA rescued anti-tumor activity as expected. When transferred cMoPs were analyzed in tumors (Figure 6C), silencing Wdr24 efficiently reduced expression of WDR24 protein. Consistent with the proposed role of GATOR2, lack of WDR24 greatly reduced mTORC1 activity (p-S6), as well as the tightly-coupled phosphorylation Akt-Ser473 (a marker for mTORC2 activity). Phenotyping of the transferred cells (Figure 6D) showed that silencing Wdr24 did not prevent their homing to the tumor, nor expression of Ly6c or c-kit; however, in the absence of a functional GATOR2 complex, BTK-IDO chemo-immunotherapy lost all ability to promote DC differentiation (CD11c, CD103 and Batf3). Taken together, these data were consistent with the hypothesis that the mechanistic target of BTK-IDO blockade was to restore the permissive signal from GATOR2 to mTORC1.
Identification of IDO-expressing DCs and monocytic cells across a range of human tumors
Human DCs show many basic similarities to mice (Zilionis et al., 2019) but can have substantial differences. We adopted an unbiased approach, asking whether there were cells in human tumors that co-expressed BTK and IDO. Biopsies from malignant melanomas (Figure 7A-7C) showed a distinct population of BTK+IDO+ dual-positive cells (green and red pseudocolors, forming yellow when co-expressed), often displaying a dendritic-like morphology. However, the distribution of these cells was patchy and localized, making analysis by histology non-quantitative. To better quantitate and characterize these cells, we analyzed published databases of single-cell RNA-sequencing from a range of human tumors.
Figure 7. Identification of IDO-expressing DCs and monocytic cells across a range of human tumors.
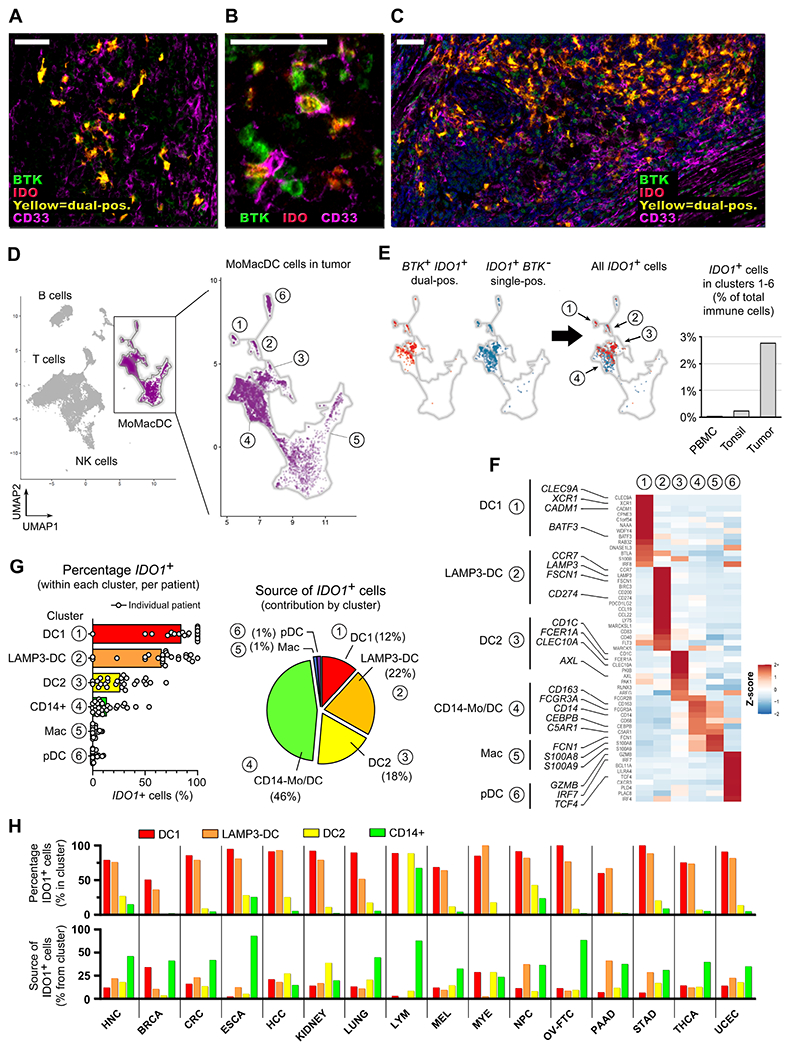
(A-C) Multi-spectral immunofluorescence images of human melanoma biopsies stained for BTK (green pseudocolor), IDO (red) and CD33 (magenta). Dual-positive BTK+IDO+ cells show as yellow. Morphology of the BTK+IDO+ population is representative of over 20 melanoma biopsies. Scale bars are 50 μm.
(D-G) UMAP analysis was performed on scRNA-seq data from ref. (Cillo et al., 2020), comprising pooled populations of tumor-infiltrating immune cells, control tonsil, and peripheral blood mononuclear cells (PBMC).
(D) Outline shows the monocyte-Mϕ-DC (MoMacDC) region, with clusters arbitrarily numbered 1-6.
(E) UMAP analysis of gated tumor-infiltrating immune cells, showing cluster distribution of IDO-expressing cells. BTK+IDO1+ dual-positive cells (red) and single-positive IDO1+BTK − cells (blue), with merged distribution (right). Bar graph compares total IDO1+ cells in the MoMacDC region from tumor versus tonsil and PBMC, as a percentage of total immune cells (T, B and all other cells). See also Figure S6B.
(F) Heatmap analysis of the clusters in panel D, showing Z-score (normalized gene expression relative to the total of tumor infiltrating cells) for a curated list of genes used to assign identification of each cluster.
(G) Percentage of total IDO1+ cells in Clusters 1-6 in tumor-infiltrating cells. Data-points represent values for individual patients (n=26), bars represent mean of pooled samples. Pie chart shows the contribution from each Cluster 1-6 to the total IDO1+ population (pooled samples).
(H) UMAP analysis of sc-RNAseq data for 15 additional tumor types from ref. (Cheng et al., 2021) (see also Figure S7). Analysis of total IDO1+ cells in the myeloid population (which corresponds to the MoMacDC region in panel D). Cells were classed as either DC1, LAMP3+ cDC, or DC2, using the authors’ assignment in the original reference; then CD14+ cells were identified in the remaining cells. The corresponding data from panel G are included as the first tumor type (HNC), for comparison. The lower bar graph shows the contribution of each cluster to the total IDO1+ population (corresponding to the pie-chart of panel G). Abbreviations: HNC, head and neck; BRCA, breast; CRC, colorectal; ESCA, esophageal; HCC, hepatocellular; LYM, lymphoma; MEL, melanoma; MYE, myeloma; NPC, nasopharyngeal; OV-FTC, ovarian; PAAD, pancreatic; STAD, gastric; THCA, thyroid; UCEC, endometrial.
We began with a large dataset from head-and-neck tumors (Cillo et al., 2020). We first used the characteristic BTK+IDO1+ dual-positive phenotype as a hallmark population to locate the cells of interest on UMAP cluster analysis. The BTK+IDO1+ population was restricted to small number of clusters falling in the monocyte-Mϕ-DC (MoMacDC) region (Figure 7D, 7E). Overall, 99% of the BTK+IDO1+ dual-positive cells fell in the MoMacDC region, so the remaining analysis focused on MoMacDC cells. Clusters were arbitrarily numbered 1-6 and assigned an identity based on a curated list of marker genes (Villar and Segura, 2020; Zilionis et al., 2019) (Figure 7F). The BTK+IDO1+ cells were prominent in Cluster 1 (conventional cDC1); Cluster 2 (corresponding to the “activated DCs” of ref. (Zilionis et al., 2019) or the “LAMP3+” DCs of ref. (Cheng et al., 2021)); Cluster 3 (conventional DC2); and Cluster 4 (containing a mix of CD14+ cells, see below).
While the hallmark BTK+IDO1+ phenotype was distinctive, we had seen in the mice that expression of both these genes could vary with local conditions (e.g., not present in Ly6c+c-kit+ cells from resting LNs, but induced by tumors). Therefore, we asked whether cells expressing only IDO alone, or only BTK alone, might be similar to the hallmark double-positive cells. UMAP dimensional reduction is well suited for detecting such self-similarity within populations (Becht et al., 2019). Single-positive IDO1+BTK− cells essentially overlapped the BTK+IDO1+ double-positive cells on UMAP analysis (Figure 7E), suggesting that these two populations appeared closely similar to the clustering algorithm. This same tight co-clustering of all IDO1+ cells, with or without BTK expression, was observed in each dataset of 15 additional tumor types analyzed (Figure S7). In contrast, single-positive BTK+IDO− cells were randomly distributed throughout the MoMacDC space, and did not appear related to the IDO1+ cells (Figure S6A). Thus, for our analysis we treated all IDO1+ DCs together as a superset of the hallmark BTK+IDO1+ population.
While tumors contained many IDO1+ DCs in Clusters 1-4, in normal tonsil and peripheral blood (PBMC) such IDO1+ cells were rare (Figures 7E, S6B). In tumors, the percentage of IDO1+ DCs was highest in Cluster 1 (DC1) and Cluster 2 (LAMP3-DC) (Figure 7G). The dataset contained 26 individual patients, shown as separate dots in the figure, and most patients had a high proportion of IDO1+ cells in these two clusters. Within Cluster 3 (DC2 cells) fewer cells expressed IDO, and these clustered adjacent to Cluster 4. Cluster 4 was a mixed population characterized by CD14 expression (Figure S6C). This included CD163+ cells with markers consistent with monocytic DCs (C5AR1, also known as CD88), plus other cells consistent with the recently-described population of CD14+ inflammatory conventional DCs (Bourdely et al., 2020; Dutertre et al., 2019). These two populations show substantial phenotypic overlap (Villar and Segura, 2020), so we considered them together. Although Cluster 4 had a relatively low percentage of cells expressed IDO1, it was a much larger cluster; thus, overall, the CD14+ region was the largest single contributor to the total IDO1+ DCs in this dataset (Figure 7G).
Across all clusters, one notable attribute of the IDO1+ cells was that they expressed the characteristic genes of each cluster that they occupied (Figure S6D). Thus, the IDO1+ DCs were not a separate hematopoietic lineage of DCs, but rather represented a condition or “state” within each DC subset. In many cases the IDO1+ state comprised the majority of cells in that subset.
Analogous results were seen across sc-RNAseq datasets for 15 additional tumor types from ref. (Cheng et al., 2021) (Figures 7H, S7). For comparison, the colors are the same as shown in Figure 7G (which is included as the first tumor type, labeled HNC). Most tumor types had at least 50% IDO1+ cells in the conventional cDC1 and mature LAMP3-DC clusters, often with substantial contribution from the CD14+ population. Thus, overall, the high proportion of IDO1+ cells among tumor-infiltrating DCs was a feature shared across an array of human tumors.
Discussion
Here we describe a metabolic checkpoint in tumor-associated DCs created by BTK and IDO. These are both well-studied genes, but the role that we describe is distinct because the two pathways function together to form an intracellular signal-transduction system. The cell-intrinsic nature of this signaling was shown by the fact that loss of BTK in just the cMoP population (while intact in all other cells) was sufficient to replace the need for systemic BTK-inhibitor drug. Conversely, silencing the IDO-responsive GATOR2 complex in cMoPs alone was sufficient to ablate the whole downstream cascade of immune activation during ibrutinib+indoximod therapy. Thus, the joint BTK-IDO pathway functioned as a cell-intrinsic checkpoint in the DCs, and hence as a target for therapy.
The actual regulatory signal was delivered by IDO, via a “low-tryptophan” condition sensed by the GATOR2 complex. Metabolic signals, including the level of key amino acids, are emerging as important control elements in immune cells (Leone and Powell, 2020). mTORC1 serves as an integration point for many such metabolic signals (Liu and Sabatini, 2020). The role that we propose for amino-acid sensing does not rule out additional downstream effects of IDO such as kynurenine production. However, our in vivo models suggest that restoring the amino-acid sufficiency signal from GATOR2 was a required target of BTK-IDO immunotherapy.
While IDO regulated mTORC1 the stability of IDO itself was controlled by BTK. IDO is labile (Munn et al., 2002; Munn et al., 2004) and pro-inflammatory IFNγ caused rapid loss of IDO expression. In other systems, this active elimination of IDO has been tied to the SOCS3 pathway (Orabona et al., 2008). Whether SOCS3 plays a similar role in our system remains to be determined, but the elimination of IDO was functionally important because the DCs could not differentiate as long as IDO was active. The role of BTK was to dominantly maintain expression of IDO, despite the presence of inflammatory signals such as IFNγ. This stabilizing signal via BTK could be driven by TGFβ or CTLA-4; and other reports suggest that IL-10 and Tim-3 can also drive BTK (Gujar et al., 2016). Thus, we propose that BTK serves to integrate multiple upstream tolerogenic signals in the tumor microenvironment, allowing them to dominantly maintain expression of IDO and thereby block differentiation of inflammatory DCs.
In mice, we focused on DCs arising from the monocytic lineage. This was not because conventional cDCs or plasmacytoid DCs are unimportant, but because monocytic cells were empirically found to be the most effective at initiating the response to BTK-IDO immunotherapy. Our models focused on this key initiation step, from cold, resting tumor to activated immune response. Once this transition occurred, however, it is quite possible that additional populations (cDCs, pDCs and activated Mϕs) might also then contribute. The current study did not address these downstream mechanisms.
The finding that the inflammatory DCs in mice were of monocytic origin does not imply that all inflammatory DCs in human tumors must therefore be monocytic. Even in the mice, the monocytic precursors rapidly converged on a phenotype similar to conventional cDC1. In human tumors, we found that over half of IDO1-expressing cells were conventional DCs, including cDC1, cDC2 and LAMP3+ cDCs. The balance expressed CD14, suggestive of a monocytic origin. Even here, however, conventional DCs can also acquire CD14 expression in response to inflammation (Bourdely et al., 2020; Dutertre et al., 2019; Villar and Segura, 2020). Thus, in humans, the DCs population expressing IDO appeared to be a mix of both conventional and monocytic DCs.
In all of these DCs, we propose that the BTK-IDO checkpoint may act to repress functional immunogenic activation. Increasing evidence suggests that, although DCs in tumors may appear phenotypically “mature”, they are functionally profoundly impaired, and may be actively immunosuppressive (Maier et al., 2020; Oh et al., 2020). We speculate that the inhibitory signal delivered by IDO may be one mechanism contributing to this dysfunction, by preventing the transition to an activated, inflammatory phenotype.
The close mechanistic link between the BTK and IDO pathways has implications for clinical therapy. In our in vitro model, where we artificially supplied a strong inflammatory signal, blocking either IDO or BTK individually was enough to destabilize the checkpoint. In vivo, however, under the more realistic conditions of a “cold”, immunosuppressive tumor, our mouse studies showed that IDO and BTK each made their own individual contribution, and blocking both was required to initiate a full response. This may help explain why clinical immunotherapy trials targeting BTK alone (Hong et al., 2019; Overman et al., 2020) or IDO alone (Long et al., 2019) have been disappointing, even when combined with PD-1/L1 blockade. That said, patients treated with indoximod can show molecular evidence of robust attempted immune activation in those tumors that respond (Zakharia et al., 2021). Blocking the additional upstream BTK signal may provide synergy, and allow greater response. BTK-inhibitor drugs are already approved for other uses, and can be readily repurposed. Clinical trials combining BTK and IDO blockade are planned to open soon, and will provide a direct test of this hypothesis.
Limitations of the study
Our study leaves open several conceptual questions. The most basic is why BTK and IDO seem to play such an important role in anti-tumor immunity. We speculate that this relates to the normal physiologic role played by IDO in tolerance to apoptotic cells (Ravishankar et al., 2012), but this will require further investigation. Mechanistically, we did not establish how the inflammatory DCs that emerge after BTK-IDO blockade function to re-activate tumor-associated T cells, and if these reactivated T cells formally mediate the anti-tumor response. Finally, the evidence for our proposed “low-tryptophan” signal rests on in vitro tryptophan-supplementation experiments. IDO can have other effects besides tryptophan depletion (e.g., kynurenine production), so further studies are required to test the link between IDO and mTORC1 in vivo. The human and mouse systems were not identical. In human tumors, the cells expressing IDO were not just immature precursors, but were recognizably DCs. It will require on-treatment biopsies during immunotherapy to establish whether blocking the BTK-IDO pathway allows activation of these IDO+ DCs into an inflammatory, immunogenic state. Many of our in vivo studies relied on implantable mouse tumor models. These can be affected by variability in the cell lines, experimental technique, and differences in inbred mouse strains; as well as significant effects on anti-tumor immunity of differing gut flora between colonies (Mandal et al., 2020). We attempted to use multiple reinforcing models and readouts; ultimately, however, the definitive test of the hypothesis will come from our planned clinical trials combining ibrutinib and indoximod together with chemotherapy.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, David H. Munn (dmunn@augusta.edu).
Materials Availability
This study did not generate new unique reagents
Data and Code Availability
The scRNA-seq, bulk RNA-seq and ATAC-seq datasets are deposited in the Genome Expression Omnibus (GEO) under Accession Number GSE180039 and are publicly available. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals.
Animal studies were approved by the Institutional Animal Care and Use Committee, Augusta University. Mice were obtained from Jackson Laboratories or bred in-house, and maintained at 2-5 mice per cage in a Specific Pathogen-Free environment with enrichment. For experiments, mice were used between 3-6 months of age. During the 2-3 week experimental period (tumor implantation and immunotherapy), mice were housed individually in an ABSL2 facility. Animals of both sexes were randomly assigned, and the results shown reflect pooled results with both genders.
Human materials.
Human peripheral blood mononuclear (PBMC) samples for in vitro experiments were obtained by apheresis of normal volunteers and enriched by counter-flow centrifugal elutriation as previously described (Munn et al., 2004), following informed consent under a protocol approved by the Institutional Review Board of Augusta University. Samples were anonymized. Immunofluorescence histology studies were performed on existing, anonymized tumor samples obtained from the Georgia Cancer Center Human Tumor BioBank; or from banked samples at the University of Iowa or Emory University, under IRB-approved protocols.
Tumor cell lines.
B16F10 and E.G7 (EL4-OVA) cell lines were obtained from ATCC, certified mycoplasma-free. Cells were cultured for a single passage and re-frozen in multiple aliquots. Replicate aliquots (all at the same early passage) were thawed, used for a single experiment and discarded. Melanoma lines were monitored and photographed for melanin production in vivo.
METHODS DETAILS
Reagents
Indoximod (1-methyl-D-tryptophan, clinical grade) was obtained from Lumos Pharma (NewLink Genetics) and was dissolved as described (Sharma et al., 2013). Ibrutinib (cat. # HY-10997) and linrodostat (cat. # HY-101560) were purchased from MedChemExpress. The mTOR-inhibitor pp242 (from Selleckchem #S2218) was dissolved in DMSO and used in vitro at 100 nM final concentration (1:20,000 dilution of DMSO). Oxaliplatin and cyclophosphamide were from Sigma.
Mouse strains
The following were obtained from Jackson Laboratories and bred in our colony:
OT-I mice (CD8+, recognizing the SIINFEKL peptide of ovalbumin on H2Kb )
B6.PL-Thy1a/CyJ (Thy1.1 congenic)
B6.SJL-Ptprca Pepcb/BoyJ (CD45.1 congenic)
pmel-1 mice (B6.Cg-Thy1a/CyTg(TcraTcrb)8Rest/J) recognizing a peptide from human gp100
Batf3−/− mice (B6.129S(C)-Batf3tm1Kmm/J) (Hildner et al., 2008)
IDO1−/− mice (B6.129-Ido1tm1Alm/J) (Baban et al., 2004)
BTK−/− mice (B6;129S-Btktm1Wk/J (Stock No: 002536)
IL12p40−/− (B6.129S1-Il12btm1Jm/J (Stock No. 002693)
Control strains: Controls for all Jackson Laboratories mutant strains were wild-type C57BL/6J mice from Jackson.
For other strains, OT-I mice were crossed with Thy1.1 mice and used in the F1 generation as double-heterozygotes. Tg(Grm1)Epv mice (Pollock et al., 2003) were the generous gift of S. Chen, Rutgers University, and was monitored weekly for development of autochthonous melanoma. Lyz2-cre-YFP reporter mice were produced as previously described (Sharma et al., 2018) by crossing Lyz2-cre mice with Rosa26-STOP-flox-YFP reporter mice (B6.129X1Gt(ROSA)26Sortm1(EYFP)Cos/J, strain 006148, Jax), which have a Loxp-flanked STOP site upstream of YFP driven by the ROSA26 promoter.
Analysis of published single-cell RNA-seq datasets
Published scRNA dataset for head and neck squamous cell carcinoma (HNSCC) (Cillo et al., 2020) was downloaded and analyzed (UMAP dimensionality reduction) using BBrowser version 2.4.3 software for analysis of public scRNA-seq datasets (BioTuring). To define monocytes, macrophages and dendritic cells we used authors’ definitions of clusters and cell type annotations or as described in the figures. Annotated expression data and UMAP coordinates were exported and analyzed using Seurat v3 package (Butler et al., 2018) installed on R 3.5.2 (R_Core_Team, 2018). Scaled Z-score for gene expression was calculated using ScaleData() function in Seurat v3 package. To calculate percentage of cells expressing a particular gene within a population, we considered cell as positive if expression was above 0.
Published datasets of tumor-infiltrating myeloid cells across 15 human cancer types were downloaded from http://panmyeloid.cancer-pku.cn, from reference (Cheng et al., 2021). UMAP coordinates, cluster designations and cell expression values were used for visualization using ggplot2 R package.
cMoP and CDP adoptive transfer
For sorting of bone-marrow precursors (cMoPs and CDPs), tibias were flushed, filtered through 40 um mesh, stained briefly on ice then sorted into ice-cold medium using low-shear fluidics and a large-aperture nozzle (Mo-Flo cell sorter). Cells were sorted for CDPs (CD115+ CD117+ Ly6cNEG CD135+) or cMoPs (CD115+ CD117+ Ly6c+ CD135NEG), then 1×105 cells injected i.v. via tail vein. To preserve function, no more than 3 hrs was allowed to elapse between initial harvesting and final injection.
Vaccination
Whole-protein chicken ovalbumin was from Sigma (catalog #A-5503). Human hgp10025-33 (KVPRNQDWL) peptide was synthesized by Southern Biotechnology from the published sequences (Hogquist et al., 1994; Overwijk et al., 2003). CpG-1826 (phosphorothioate oligo 5′-TCCATGACGTTCCTGAGCTT-3′) was synthesized from the published sequence (Chu et al., 1997) by Tri-link Biotechnologies. Vaccines were prepared with 25 ug peptide and 50 ug CpG-1826 in incomplete Freund’s adjuvant (IFA, Sigma F-5506) and administered in the hind-limb footpad. For VDLNs, draining popliteal LNs were removed on day 3-4.
T cell adoptive transfer
OT-I or pmel-1 spleen cells were enriched by negative selection using magnetic beads (mouse naive CD8 isolation kit, #130-096-543, Miltenyi Biotech), or by MoFlo cell sorting using a large-aperture low-shear nozzle. Staining for sorting was performed on ice with short incubation times to keep the cells viable but unactivated. Mice received 2×106 enriched CD8+ cells via tail-vein. To track proliferation of transferred T cells, in some experiments enriched CD8+ cells were labelled with the cell proliferation dye eFluor 670 (Thermo-Fisher, cat. #65-0840 ) according to the manufacturer’s protocol.
Mouse myeloid-cell maturation cultures
Resting lymph nodes were disaggregated by passing once through a 40 um mesh, then stained briefly on ice and flow-sorted into ice-cold medium using low-shear fluidics and a large nozzle (Mo-Flo cell sorter). Co-cultures were performed in V-bottom wells (Nunc 249952 V96), using RPMI-1640 medium with 10% fetal bovine serum, as described (Sharma et al., 2013). As an inflammatory stimulus for DC maturation, CD8+ effector cells (5×104) were flow-sorted from spleens of either OT-I or pmel-1 mice (both performed equivalently), and added to co-cultures along with 100 nM cognate peptide (SIINFEKL or hgp100). All cultures received a feeder layer of 1×105 T cell-depleted B6 spleen cells (CD4−CD8−) plus sorted CD4+ T cells to provide help. All cells other than the myeloid cells of interest were from CD45.1+ or Thy1.1+ congenic donors, so that they could be excluded from analysis.
Flow cytometry staining and antibodies
Flow cytometry was performed as described previously (Sharma et al., 2018). The conjugated antibodies used for flow cytometry are listed in the Key Resources Table. For staining of mouse lymph nodes, cells were prepared by rapidly passing the whole node through a 40 um mesh, then stained using short incubation times (10 min on ice), as described (Sharma et al., 2013). Tumors were disaggregated by treating for 1 hr with collagenase, DNAse and hyaluronidase in RPMI 1640 medium, as described (Sharma et al., 2015). Mouse CD11c was detected with clone HL3, to minimize cross-reactivity with macrophages. Since the available antibodies for human XCR1 have not been validated for in vitro diagnostic use, two different clones were used for confirmation (clone RTK2758 from BioLegend, and clone 1097A from R&D systems); both gave identical staining results, so the marker appeared informative.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies against mouse antigens | ||
| CD4 (mouse) (clone RM4-5) | BD Biosciences | Cat# 553051 |
| CD8a (mouse) (clone 53-6.7) | BD Biosciences | Cat# 553036 |
| CD11c (clone HL3) | BD Biosciences | Cat# 561119 |
| Ly6c (mouse) (clone AL-21) | BD Biosciences | Cat# 560592 |
| Ly6c (mouse) (clone HK1.4) | eBioscience | Cat# 45-5932 |
| granzyme B (mouse) (clone NGZB) | eBioscience | Cat# 12-8898 |
| PD1 (mouse) (Clone: J43) | eBioscience | Cat# 12-9985 |
| CD103 (mouse) (M290) | BD Biosciences | Cat# 557495 |
| CD69 (mouse) (clone H1.2F3) | eBioscience | Cat# 11-0691 |
| Batf3 (mouse and human) (clone 841702) | R&D Systems | Cat# FAB7437R |
| CD117 (clone 104D2) | eBioscience | Cat# 11-1178 |
| CD115 (clone 12-3A3-1B10) | eBioscience | Cat# 12-1159 |
| C135 (mouse) (clone A2F10.1) | eBioscience | Cat# 12-1351 |
| CXCR3 (mouse) (clone CXCR3-173) | eBioscience | Cat# 12-1831 |
| CXCR5 (mouse) (clone 614641) | R&D Systems | Cat# FAB6198A |
| MHC-II (mouse) (clone M5/114.15.2) | eBioscience | Cat# 12-5321 |
| CCR2 (mouse) (clone 475301) | R&D Systems | Cat# FAB5538P |
| Ly-6G/Gr1 (mouse) cloneRB6-8C5) | eBioscience | Cat# 12-5931 |
| F4/80 (mouse) (clone BM8) | eBioscience | Cat# 12-4801 |
| CD11b (mouse, human) (clone M1/70) | BD Biosciences | Cat# 557396 |
| Foxp3 (mouse) (clone FJK-16 s) | eBioscience | Cat# 11-5773 |
| PTEN (mouse and human) (clone A2B1) | BD Biosciences | Cat# 560002 |
| CD40L (mouse) (clone MR1 | BD Biosciences | Cat# 553658 |
| IDO (mouse) (clone mIDO-48) | eBioscience | Cat# 50-9473 |
| BTK (mouse) (clone) | Santa Cruz Biotech. | Cat# sc-81735 |
| anti-CTLA4 blocking antibody (clone 9D9) | BioXcell | Cat# BE0164 |
| anti-PD-1 blocking antibody (clone J43) | BioXcell | Cat# BP0033-2 |
| anti-PDL2 blocking antibody (clone TY25) | BioXcell | Cat# BE0112 |
| anti-PD-L1 blocking antibody (clone MIH7) | Laboratory of Dr. Miyuki Azuma | (Tsushima et al., 2003) |
| Antibodies against human antigens | ||
| Batf3 (mouse and human) (clone 841702) | R&D Systems | Cat# FAB7437R |
| CD33 (human) (clone SP266) | Abcam | Cat# AB199432 |
| CD8 (human) (clone C8/144B) | Sigma-Aldrich | Cat# 108M-94 |
| phospho-p53 (human) (clone S15) | R&D Systems | Cat# AF1043 |
| phospho-S6 (human, mouse) (clone D57.2.2E) | Cell Signaling | Cat# 5316S |
| phospho-Akt-473 (human, mouse) (clone D9E) | Cell Signaling | Cat# 4075S |
| CD83 (human) (clone HB15e) | BD Biosciences | Cat# 550634 |
| IRF5 (human) (cloneEPR17067) | Abcam | Cat# AB181553 |
| BTK (human) (clone D3H5) | Cell Signaling | Cat# 8547 |
| IDO (human) (clone 998743) | R&D Systems | Cat# MAB603101 |
| XCR1 (human) (clone RTK2758) | BioLegend | Cat# 372603 |
| XCR1 (human) (clone 1097A) | R&D Systems | Cat# FAB8571N |
| Biological Samples | ||
| Healthy human peripheral blood mononuclear cells (PBMC) | Augusta University | N/A |
| Anonymized FFPE tumor samples | Augusta University GCC Biorepository | N/A |
| Anonymized FFPE tumor samples | University of Iowa Melanoma Bank | N/A |
| Anonymized FFPE pediatric tumor samples | Emory University tumor bank | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Human hgp10025-33 (KVPRNQDWL) peptide | SouthernBiotech | custom, based on (Overwijk et al., 2003) |
| VO-OHpic | Biovision | Cat# 1801-5 |
| Incomplete Freund’s adjuvant | Sigma | Cat# F-5506 |
| human IL-1a Recombinant Protein | R&D Systems | Cat# 200-LA |
| human IL-4 Recombinant Protein | R&D Systems | Cat# 204-IL |
| human TNFa Recombinant Protein | R&D Systems | Cat# 210-TA |
| human IL-1b Recombinant Protein | BD Biosciences | Cat# 554602 |
| human IL-6 Recombinant Protein | R&D Systems | Cat# 206-IL |
| human GM-CSF Recombinant Protein | R&D Systems | Cat# 215-GM |
| human IFN-g Recombinant Protein | R&D Systems | Cat# 285-IF |
| human TGF-b1 Recombinant Protein | R&D Systems | Cat# 7754-BH |
| human CTLA-4 Ig (human):Fc(human)(rec.) | Chimerigen Lab. | Cat# CHI-HF-210A4 |
| Anonymized FFPE tumor samples | University of Iowa Melanoma Bank | N/A |
| Prostaglandin E2 | Sigma | Cat# P0409 |
| Whole OVA protein | Sigma | Cat# A-5503 |
| MACH 2 Mouse HRP-Polymer | Biocare Medica | Cat# MHRP520 |
| MACH 2 Rabbit HRP-Polymer | Biocare Medica | Cat# RHRP520 |
| AR6 buffer | Perkin Elmer | Cat# AR6001KT |
| AR9 buffer | Akoya Biosciences | Cat# AR9001KT |
| Background Sniper | Biocare Medical | Cat# BS966L |
| Indoximod (1 methyl-D-tryptophan, clinical grade) | Lumos Pharma (NewLink Genetics) | N/A |
| Ibrutinib | MedChemExpress | Cat# HY-10997 |
| Linrodostat | MedChemExpress | Cat# HY-101560 |
| PP242 (mTOR-inhibitor) | Selleckchem | Cat# S2218 |
| Oxaliplatin | Sigma | Cat# O9512 |
| Cyclophosphamide (CTX) | Sandoz | Cat# NDC0781-3233-94 |
| ArQ531 | ArQule Inc. | ARQ531 v.02 |
| 2-OH-propyl-b-cyclodextrin | Sigma | Cat# PHR1440 |
| CpG1826 (phosphorothioate oligo 5TCCATGACGTTCCTGAGCTT3) | Tri-link Biotechnologies | (Chu et al., 1997) |
| Critical Commercial Assays | ||
| Fixation-permeabilization kit | eBioscience | Cat# 00-5523 |
| mouse naive CD8 isolation kit | Miltenyi Biotech | Cat# 130-096-543 |
| Agencourt AMPure XP | Beckan Coulter | Cat# A63880 |
| DNA Clean & Concentrator-5 | Zymo Research | Cat# D4013 |
| Nextera DNA Sample Preparation kit | Illumina | Cat# FC-121-1031 |
| Nextera index kit | Illumina | Cat# FC-121-1011 |
| Q5 Hot Start High-Fidelity 2X Master Mix | New England Biolabs | Cat# M0494L |
| siRNA Reagent Kit | Santa Cruz Biotech. | Cat# sc-45064 |
| Opal seven-color IHC Kit | PerkinElmer / Akoya Biosciences | Cat# NEL703001KT |
| Kynurenine ELISA kit | IBLAmerica | Cat# IB89190 |
| eBioscience Cell Proliferation Dye eFluor 670 | Thermo Fisher Scientific | Cat# 65-0840 |
| Deposited Data | ||
| bulk RNAseq dataset | This paper | GEO: GSE180039 (GSE180033) |
| sc-RNAseq dataset | This paper | GEO: GSE180039 (GSE180038) |
| ATAC-seq dataset | This paper | GEO: GSE180039 (GSE180026) |
| Experimental Models: Cell Lines | ||
| B16F10 | ATCC | CRL-6475 |
| E.G7-OVA | ATCC | CRL-2113 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | Jackson Laboratory | JAX:000664 |
| Mouse: pmel1 : B6.CgThy1a/CyTg(TcraTcrb)8Rest/J | Jackson Laboratory | JAX:005023 |
| Mouse: Tg(Grm1)Epv | S. Chen, Rutgers Univ. | (Pollock et al., 2003) |
| Mouse: IDO1−/−: B6.129-Ido1tm1Alm/J | Jackson Laboratory | JAX:005867 |
| Mouse: Lyz2-cre-YFP | bred in house from Jax strains | (Sharma et al., 2018) |
| Mouse: IL-12p40 −/− : B6.129S1-Il12btm1Jm/J | Jackson Laboratory | JAX:002693 |
| Mouse: BTK −/− : B6;129S-Btktm1Wk/J | Jackson Laboratory | JAX:002536 |
| Mouse: CD45.1: B6.SJL-Ptprca Pepcb/BoyJ | Jackson Laboratory | JAX:002014 |
| Mouse: Batf3−/− : B6.129S(C)-Batf3tm1Kmm/J | Jackson Laboratory | JAX:013755 |
| Mouse: OT-I : C57BL/6-Tg(TcraTcrb)1100Mjb/J | Jackson Laboratory | JAX:003831 |
| Mouse: Thy1.1: B6.PL-Thy1a/CyJ | Jackson Laboratory | JAX:000406 |
| siRNA pools | ||
| siRNA pool: Human BTK | Santa Cruz Biotech. | Cat# sc-29841 |
| siRNA pool: Human IDO1 | Santa Cruz Biotech. | Cat# sc-45939 |
| siRNA pool: Human WDR24 | Santa Cruz Biotech. | Cat# sc-93306 |
| siRNA pool: Human DEPDC5 | Santa Cruz Biotech. | Cat# sc-77132 |
| siRNA pool: Human scrambled control siRNA | Santa Cruz Biotech. | Cat# sc-44230 |
| siRNA pool: Mouse Wdr24 | Santa Cruz Biotech. | Cat# sc-155268 |
| siRNA pool: Mouse scrambled control siRNA | Santa Cruz Biotech. | Cat# sc-37007 |
| FITC-labeled tracer oligonucleotides | Santa Cruz Biotech. | Cat# sc-36869 |
| Recombinant DNA | ||
| SMARTvector Inducible Non-targeting Control (mCMV/TurboRFP) Lentiviral particles | GE Healthcare Dharmacon, Inc | Item # VSC6571 |
| SMARTvector Inducible Mouse IL-1a (mCMV/TurboRFP) Lentiviral particles | GE Healthcare Dharmacon, Inc | Item # V3SM7671-231534641 |
| Software and Algorithms | ||
| Prism 7 | GraphPad | graphpad.com/scientific-software/prism/ |
| R Project for Statistical Computing v3.5.1 | (R_Core_Team, 2018) | http://www.r-project.org/ |
| BBrowser version 2.4.3 | BioTuring | https://bioturing.com/ |
| Seurat v3.0 | (Butler et al., 2018) | https://satijalab.org/seurat/ |
| Galaxy | https://usegalaxy.org | |
| Integrated genomics viewer (IGV) | (Robinson et al., 2011) | http://software.broadinstitute.org/software/igv/ |
| Fiji/ImageJ software | open source | https://imagej.net/Fiji |
| Other | ||
Intracellular antigens (other than phospho-specific staining) were detected using fixation-permeabilization reagent and matching perm-wash buffer from eBioscience (Cat. #00-5523), with blocking using 5% normal donkey serum, then acquired immediately after staining. For phospho-specific staining, cells were washed in PBS, fixed with 2% paraformaldehyde for 10 min at 37°C, pre-chilled for 1 min, then permeabilized by slow addition of ice-cold methanol to a final concentration of 90%. Cells were then incubated on ice for 30 min, washed with 1% FCS in PBS, blocked with the same solution for 10 min at room temperature, then for 1 h at room temperature and washed. Cells were acquired immediately after staining.
Detection of kynurenine by ELISA
Detection of kynurenine in samples was performed using competitive ELISA kit (IBL America Cat# IB89190) according to manufacturer’s instructions. In brief, 10ul of samples and standards were mixed with 250ul of acylation buffer and 25ul of Acylation Reagent and incubated for 90min at 37°C. 20ul of acylation reaction was used for overnight incubation with Kynurenine Antiserum in Kynurenine Microtiter Strips. Next day, wells were washed and incubated for 30 min. with 100 ul of goat anti-rabbit conjugated with perodixase. After washing, wells were incubated for 25 min with 100 ul of substrate followed by 100ul of Stop solution. Absorbance at 450nm was read using microplate reader.
ATAC-seq analysis
TDLNs were used rather than tumors in order to minimize artifacts introduced by enzymatic disaggregation; the Ly6c+c-kit+ precursor cells were similar in both locations. Cells were isolated by rapid mechanical disaggregation of TDLNs from established (day 14) untreated B16F10 tumors. LNs were pooled and stained for CD11c, Ly6c and c-kit, then the CD11cNEGLy6c+c-kit+ precursor population was sorted and used for ATAC-seq. Three independent samples were isolated in 3 separate experiments. For comparison, ATAC-seq data for “pre-cDC1” samples (identified as LinNEGCD117intCD135+CD226+) were downloaded from ref. (Durai et al., 2019); and data for cDC1 were downloaded from ref. (Brown et al., 2019).
The ATAC-seq libraries were constructed according to the Omni-ATAC-seq protocol as described (Corces et al., 2017). For each sample, 50,000 nuclei prepared from FACS-sorted Ly6c+c-kit+ precursor cells were incubated with 2.5 μL Nextera Tn5 transposase (Illumina) in 50μL 1X transposition reaction mixture in a thermocycler at 37 °C for 1hr. The transposition reaction mixtures were purified with a DNA Clean and Concentrator kit (Zymo Research), and amplified for 11-13 cycles using NEBNext High-Fidelity 2X Master Mix (New England Biolabs) and Nextera Index primers (Illumina). The amplified libraries were size-selected by two rounds of AMPure beads purification (0.5X and 1.2X). ATAC-seq libraries are prepared in triplicates and sequenced on a Hi-Seq instrument in a pair-end 150 cycle run. 60-100 million read pairs were obtained for each replicate. Bioinformatic analysis of ATAC-seq data was carried out in Galaxy (https://usegalaxy.org). Raw reads generated in this study in fastq format and those downloaded from two published ATAC-seq datasets for pre-cDC1 (GSE132240) (Durai et al., 2019) and cDC1 (GSE130201) (Brown et al., 2019) were first examined by FastQC v0.11.8 for quality control purpose. Adaptor and quality trimming of the raw sequencing reads was performed using Trim Glore! v0.6.3. Trimmed reads were aligned to mouse genome mm10 using Bowtie2 v2.3.4.1. Reads aligned to the mitochondria genome were filtered using samtools v1.8. PCR duplicates were removed using MarkDuplicates from Picard v2.18.2. The aligned reads overlapping with ENCODE blacklist regions (version 2) were removed using samtools. Before peak calling, reads mapped on the “+” strand were shifted by 4bp, and on the “−” strand by 5bp using a custom script. ATAC-seq peak calling was performed using MACS2 v2.1.1.20160309 with the following command “-p 0.01 --nomodel --shift --100 --extsize 200 -B --SPMR --keep-dup all --call -summits”. BigWig files were generated by bedGraphtoBigwig and visualized in Integrated genome viewer (IGV) v2.8.3. The bigwig track plot for individual locus was generated using karyoploteR v1.16 in R v3.6.3.
Bulk RNA-seq analysis
Total RNA was isolated from the sorted CD11cNEGLy6c+c-kit+ precursor cells using TRIzol (Invitrogen) and subjected to RNA-seq analysis. The RNA-seq libraries were constructed using 8 ng of total RNA with SMARTer Stranded Total RNA-seq kit v2-Pico Input library preparation kit (TaKaRa Bio). The libraries were sequenced using Illumina NextSeq500 sequencer. 40-60 millions of single-end 75bp sequencing reads were obtained for each sample, respectively. The raw reads of fastq files were QC-checked and trimmed as described above. Trimmed reads were aligned to mm10 reference genome using STAR v2.75 and raw counts were captured using featurecounts v1.6.4. Raw RNA-seq data for cDC1 (GSE130201) (Brown et al., 2019) was downed and analysis in the same way. The differential expression analysis between CD11cNEGLy6c+c-kit+ cells and cDC1 cells (Brown et al., 2019) was carried out using DESeq2 v2.11. Heatmap was generated using ComplexHeatmap v2.7 in R v3.6.3.
scRNA-seq on mouse lymph nodes
LNs were used for scRNA-seq to avoid prolonged enzymatic disaggregation. B16F10 tumors were implanted bilaterally in the upper anterior thigh. On day 9, mice were begun on oral indoximod (2 mg/ml in drinking water) and daily ibrutinib (350 ug i.p). On day 10, mice received cyclophosphamide 150 mg/kg i.p. Two days later, tumor-draining inguinal LNs were harvested, mechanically disaggregated, passed through 40 um mesh strainer, and washed 4 times with PBS + 1% BSA. scRNA-seq libraries were generated using Chromium Next GEM Single Cell 3’ Reagent kit v3.1 (10x Genomics). Individual libraries were prepared for two independent biologic replicates, approximately 8,000 cells on average per sample (total 16,000 cells each group). Cells were captured using the Chromium Controller (10x Genomics). The scRNA-seq libraries were sequenced using Illumina NextSeq500 sequencer with a high output kit under the following sequencing protocol: 28 bp (Read 1), 8 bp (indexing Run), and 91 bp (RNA Read 2) having Q30 bases in RNA read greater than 86% to collect approximately 30 – 44K mean reads per cell, at the range of 1,466-1,677 median genes per cell. The raw reads in fastq format were processed using 10x Genomics Cell Ranger (5.0.0) analysis pipeline via STAR v2.5.0 alignment against murine mm10 reference genome. The Cell Ranger outputs containing gene-by-cell differential expression data among samples were imported into Loupe Browser (10x Genomics) for further analysis in UMAP plots as shown in Figure 4.
Human monocyte-derived DC cultures
Monocytic cells were enriched from normal volunteer donors by counter-flow elutriation as described (Munn et al., 2004) and frozen in 10% DMSO. Cells were cultured for 8 days as previously described (Munn et al., 2002; Munn et al., 2004) using serum-free X-vivo15 medium (BioWhittaker, Walkersville, MD) plus recombinant human GM-CSF (50ng/ml, R&D Systems cat. #215-GM) and IL-4 (50 ng/ml, R&D Systems cat. #204-IL). Culture media with cytokines was refreshed on day 3. On day 5, a cocktail of cytokines was added comprising TNFα (1100 U/ml, R&D Systems cat. # 210-TA), IL1β (2000 U/ml, BD Biosciences cat. # 554602), IL6 (1000 U/ml, R&D Systems cat. #206-IL) and PGE2 (1 ug/ml, Sigma cat# P0409). On day 7, recombinant human IFNγ (30ng/ml, R&D Systems cat. #285-IF), with or without TGFβ (20ng/ml, R&D Systems cat. #7754-BH) or recombinant CTLA-4-Ig fusion protein (10 ug/ml, Chimerigen), was added as the terminal differentiation step. Cultures were harvested 24 hrs later.
siRNA transduction of human cells
For transduction, gentle conditions were chosen using Santa Cruz siRNA Reagent Kit #sc-45064 for cell permeabilization. Times and concentrations were optimized for gentle conditions, with a goal of ~50-60% transduction efficiency. The transduction medium also contained FITC-labeled tracer oligonucleotides (sc-36869), so that cells that were successfully transduced became FITC-labeled. These FITC+ cells were gated for the final analysis. The presence of non-transduced cells did not affect the outcome, and provided a useful internal control for specificity of the siRNA silencing. Commercial siRNA pools were obtained from Santa Cruz Biotechnology against the following human mRNA: BTK (Santa Cruz #sc-29841). IDO1 (Santa Cruz #sc-45939). WDR24 (Santa Cruz #sc-93306). DEPDC5 (Santa Cruz #sc-77132), plus scrambled control siRNA (Santa Cruz #sc-44230). Transduction was performed on day 5 (at the time of cocktail addition). After 6 hrs at 37°C, most of the transfection medium was gently aspirated and replaced by culture medium with fresh cytokines, and cultures continued.
siRNA transduction of mouse bone marrow cells
Bulk bone-marrow cells were harvested from CD45.1 congenic donors and transduced in vitro for 6 hrs using siRNA Reagent Kit (Santa Cruz #sc-45064). Cultures received either siRNA against mouse Wdr24 (Cat #sc-155268) or control irrelevant siRNA (Cat #sc-37007). The transduction medium also contained FITC-labeled tracer oligonucleotides (sc-36869), so that all cells that were successfully transduced became fluorescent.
Preliminary validation studies confirmed that the cMoP progenitors were efficiently transfected with this reagent system. After 6 hrs, the marrow was washed, stained, and sorted for cMoP cells (CD115+CD117+Ly6c+) that were also FITC+ (transduced). Sorted cells were then injected i.v. into Batf3−/− recipients (1 x 105 per mouse).
Immunohistochemistry on human tissues
Formalin-fixed paraffin-embedded tissues were cut in 4μm sections and deparaffinized in xylene and graded ethanol. For each antibody the following steps were performed between each incubation in primary: Epitope retrieval using either AR6 or AR9 buffer (Perkin Elmer / Akoya Biosciences, Cat# AR6001KT or AR9001KT respectively) by boiling sections in a microwave for 20 minutes, with a 20 minute cool down; followed by blocking for 10 min using Background Sniper (Biocare Medical, Cat # BS966L). Sections were incubated for 1 hr in the following primary antibodies diluted in Background Sniper, with the listed detection reagents and antigen retrieval: CD33 (ca. t# AB199432, clone SP266, Abcam) using Opal 520 and AR6; BTK (cat. # D3H5, Cell Signaling) using Opal 650 and AR6; IDO (cat. # MAB603101, clone 998743, R&D Systems) using Opal 690 and AR6. For each antibody sections were incubated in either MACH 2 Mouse HRP-Polymer or MACH 2 Rabbit HRP-Polymer (Biocare Medical, Cat# MHRP520 or RHRP520 respectively) for 10 mins. Fluorescence staining was performed with Opal seven-color IHC Kit (Cat# NEL703001KT, PerkinElmer / Akoya Biosciences) and DAPI nuclear counterstain. Microwave Retrieval was performed after each antibody staining was complete. Stained slides were mounted in Prolong Gold mounting media and scanned using the PerkinElmer Vectra 3.0 imaging system and Vectra 3.0.5 software. Multispectral images were unmixed using Inform Advanced Image Analysis software (InForm 2.4, PerkinElmer), then pseudocolors assigned using Fiji/ImageJ software.
In vivo tumor studies
Tumor cell lines were maintained as described in the “Experimental Models” section. Tumors were implanted in a shallow subcutaneous location in syngeneic C57Bl/6 mice as previously described (Sharma et al., 2013), using 1×105 cells for B16F10 melanoma and 1×106 cells for E.G7 T-cell lymphoma cells. Tumors were implanted bilaterally on both flanks of each mouse to confirm reproducible implantation technique (replicate tumor diameters in a single mouse were typically within ±15%, and the average of the two tumors was used). Tumor volume was calculated from orthogonal diameters using the formula V=L×W2×π/6. Mice received approved euthanasia if any tumors reached a size of 300 mm2 (product of orthogonal diameters), or at the planned end-date of the experiment; death was never used a planned endpoint.
Chemotherapy was given on day 10 (or as specified in the figure) using either cyclophosphamide (150 mg/kg i.p × 1 dose) or oxaliplatin (5 mg/kg i.p × 1 dose). BTK- and IDO-inhibitor drugs were started 1 day prior to chemotherapy unless otherwise specified. Ibrutinib (MedChemExpress, cat. # HY-10997) was dissolved in 10% DMSO and 0.4% 2-OH-propyl-β-cyclodextrin (HP-β-CD) (Sigma cat. # PHR1440) in phosphate-buffered saline, and administered at 16 mg/kg i.p. daily or as stated in the figure. Indoximod (1-methyl-D-tryptophan) was supplied as clinical-grade material by NewLink Genetics Inc. (Lumos). It was dissolved in a small amount of 0.1N NaOH (1/20th of the final volume) as described (Munn et al., 2005) then restored to pH 7 with HCl and diluted in drinking water to a concentration of 2 mg/ml. The indoximod preparation was flavored with a small amount of aspartame and administered continuously in the drinking water bottle. Arq531 was supplied by Arqule Inc. (now a wholly-owned subsidiary of Merck & Co., Inc) and given at 50 mg/kg as a daily i.p. injection. Linrodostat (MedChemExpress, cat. # HY-101560) was dissolved in 10% DMSO and 0.4% HP-β-CD in phosphate-buffered saline, and administered in graded doses at 10-5-2-0 mg/kg/day i.p. daily.
QUANTIFICATION AND STATISTICAL ANALYSIS
Reproducibility and rigor
Experiments were designed to minimize sources of variability and achieve high reproducibility. For the transplantable tumor studies, we identified that a major source of variability was the length of time from cultured-cell harvesting to implantation. This was minimized by limiting the number of mice injected in one experiment to less than eight (von Kortzfleisch et al., 2020). When combined with stringent implantation technique, this produced reproducible tumor growth within and across repeated experiments. This allowed us to pool results of multiple small experiments (each with its own internal controls) performed over a span of many months, across different litters, with different colonies of mice, all while maintaining acceptable standard deviations. As quality-control for implantation technique, each mouse was implanted with identical bilateral flank tumors, and the mean of the two tumors reported.
For adoptive-transfer experiments, the main source of variability was the time required for FACS sorting (reproducibility declined markedly with sort times beyond 45 minutes). Therefore, our strategy was again to perform repeated small experiments (≤ 4 sorted populations), with suitable internal controls, and pool identical replicate experiments until the desired group size was accumulated (von Kortzfleisch et al., 2020).
Minimizing the number of vertebrate animals
One motive for achieving low variability was to minimize the number of vertebrate animals required for statistically robust and meaningful results. To this end, the experiments were framed to achieve large, unambiguous effect size compared to controls. Group sizes were then accumulated to achieve adequate statistical power.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 7.01 software. For two groups, means were compared by t-test. For multiple treatment groups, analysis was by one-way ANOVA with Tukey’s correction for multiple comparisons. Tumor-growth studies were treated as end-point analyses, and groups compared by t-test (2 groups) or ANOVA at the end of the measurement period. For all scatter-plots, the data shown were pooled from at least 3 independent experiments, and the number of experiments are indicated in the legends. All error bars in figures show mean ± standard deviation. A P value of <0.01 was considered significant.
Power calculation and sample size
For power calculation of mouse tumor experiments, we assumed a standard deviation of ±20% for tumor measurements. Sample size calculation was based on the ability to distinguish 150 ± 30 mm3 (control) vs 30 mm3 (experimental) with p<0.001 and power = 90%. For this, a minimum of 3 mice/group was required.
For each figure, the values for a given treatment group were pooled from at least 2 independent experiments (typically 3-6 experiments) performed at different times. The raw tumor measurements, taken from the same day in each experiment, were pooled, then averaged across experiments. No normalization or transformation was applied. For each treatment group, every experiment containing that group was pooled and reported in the average; no data were censored or discarded.
Supplementary Material
Highlights.
Certain DC precursors in mouse tumors co-express BTK and IDO
The BTK-IDO pathway inhibits mTORC1 signaling and suppresses DC differentiation
Blocking BTK and IDO during chemotherapy synergistically enhances immune activation
DCs expressing BTK and IDO are present in a variety of human tumors
Acknowledgements
The authors thank Anita Sharma, Joyce Wilson and Gabriela Pacholczyk for expert technical assistance; Jeanene Pihkala and the Cancer Center Flow Cytometry Core for cell-sorting; Eiko Kitamura and the Cancer Center Integrated Genomics Core for single-cell sequencing; and Dr. S. Chen, Rutgers University, for the gift of Tg(Grm1)Epv mice. Supported by NIH R01CA103320 and R01CA211229 to D.H.M.; R50CA232983 to M.D.S.; R01HL56067 and R01HL11879 to B.R.B.; and support from the Press-On Foundation and the Beloco Foundation to D.H.M. and T.S.J.
Declaration of Interests
Y.Z. has received clinical trial support from NewLink Genetics (now Lumos Pharma).
S. E. was an employee of ArQule, Inc. (now a wholly-owned subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA), which holds the rights to ArQ531.
E.K. was an employee of NewLink Genetics (now Lumos Pharma).
T.L.M. receives consulting income from FLX Therapeutics.
B.R.B. holds intellectual property interests in the therapeutic use of IDO inhibitors; receives remuneration as an advisor to Magenta Therapeutics and BlueRock Therapeutics; receives research funding from BlueRock Therapeutics, Rheos Medicines, and Equilibre Biopharmaceuticals Corp.
T.S.J. has received clinical trial funding from NewLink Genetics (now Lumos Pharma).
D.H.M. (corresponding author) holds patents and intellectual property interests in the therapeutic use of IDO inhibitors; and has received consulting income and research support from NewLink Genetics (now Lumos Pharma).
The other authors declare no competing interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baban B, Chandler P, McCool D, Marshall B, Munn DH, and Mellor AL, (2004). Indoleamine 2,3-dioxygenase expression is restricted to fetal trophoblast giant cells during murine gestation and is maternal genome specific. J. Reprod. Immunol 61, 67–77. [DOI] [PubMed] [Google Scholar]
- Balan S, Arnold-Schrauf C, Abbas A, Couespel N, Savoret J, Imperatore F, Villani AC, Vu Manh TP, Bhardwaj N, and Dalod M, (2018). Large-Scale Human Dendritic Cell Differentiation Revealing Notch-Dependent Lineage Bifurcation and Heterogeneity. Cell Reports 24, 1902–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becht E, McInnes L, Healy J, Dutertre C-A, Kwok IWH, Ng LG, Ginhoux F, and Newell EW, (2019). Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol 37, 38–44. [DOI] [PubMed] [Google Scholar]
- Bosteels C, Neyt K, Vanheerswynghels M, van Helden MJ, Sichien D, Debeuf N, De Prijck S, Bosteels V, Vandamme N, Martens L, et al. (2020). Inflammatory Type 2 cDCs Acquire Features of cDC1s and Macrophages to Orchestrate Immunity to Respiratory Virus Infection. Immunity 52, 1039–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böttcher JP, and Reis e Sousa C, (2018). The Role of Type 1 Conventional Dendritic Cells in Cancer Immunity. Trends Cancer 4, 784–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourdely P, Anselmi G, Vaivode K, Ramos RN, Missolo-Koussou Y, Hidalgo S, Tosselo J, Nunez N, Richer W, Vincent-Salomon A, et al. (2020). Transcriptional and Functional Analysis of CD1c(+) Human Dendritic Cells Identifies a CD163(+) Subset Priming CD8(+)CD103(+) T Cells. Immunity 53, 335–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CC, Gudjonson H, Pritykin Y, Deep D, Lavallee VP, Mendoza A, Fromme R, Mazutis L, Ariyan C, Leslie C, et al. (2019). Transcriptional Basis of Mouse and Human Dendritic Cell Heterogeneity. Cell 179, 846–863 e824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, Barczak A, Rosenblum MD, Daud A, Barber DL, et al. (2014). Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell 26, 638–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R, (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S, Li Z, Gao R, Xing B, Gao Y, Yang Y, Qin S, Zhang L, Ouyang H, Du P, et al. (2021). A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell 184, 792–809. [DOI] [PubMed] [Google Scholar]
- Chu RS, Targoni OS, Krieg AM, Lehmann PV, and Harding CV, (1997). CpG oligodeoxynucleotides act as adjuvants that switch on T helper 1 (Th1) immunity. J. Exp. Med 186, 1623–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciampricotti M, Hau CS, Doornebal CW, Jonkers J, and de Visser KE, (2012). Chemotherapy response of spontaneous mammary tumors is independent of the adaptive immune system. Nat. Med 18, 344–346; author reply 346. [DOI] [PubMed] [Google Scholar]
- Cillo AR, Kurten CHL, Tabib T, Qi Z, Onkar S, Wang T, Liu A, Duvvuri U, Kim S, Soose RJ, et al. (2020). Immune Landscape of Viral- and Carcinogen-Driven Head and Neck Cancer. Immunity 52, 183–199 e189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, and Wu B, (2017). An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods 14, 959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Streel G, and Lucas S, (2021). Targeting immunosuppression by TGF-beta1 for cancer immunotherapy. Biochem Pharmacol 192, 114697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingos-Pereira S, Sathiyanadan K, La Rosa S, Polak L, Chevalier MF, Martel P, Hojeij R, Derre L, Haefliger JA, Jichlinski P, and Nardelli-Haefliger D, (2019). Intravesical Ty21a Vaccine Promotes Dendritic Cells and T Cell-Mediated Tumor Regression in the MB49 Bladder Cancer Model. Cancer. Immunol. Res 7, 621–629. [DOI] [PubMed] [Google Scholar]
- Durai V, Bagadia P, Granja JM, Satpathy AT, Kulkarni DH, Davidson JT, Wu R, Patel SJ, Iwata A, and Liu T-T, (2019). Cryptic activation of an Irf8 enhancer governs cDC1 fate specification. Nat. Immunol 20, 1161–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutertre C-A, Becht E, Irac SE, Khalilnezhad A, Narang V, Khalilnezhad S, Ng PY, van den Hoogen LL, Leong JY, and Lee B, (2019). Single-Cell Analysis of Human Mononuclear Phagocytes Reveals Subset-Defining Markers and Identifies Circulating Inflammatory Dendritic Cells. Immunity 51, 573–589. [DOI] [PubMed] [Google Scholar]
- Gandhi L, Rodriguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, Domine M, Clingan P, Hochmair MJ, Powell SF, et al. (2018). Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N. Engl. J. Med 378, 2078–2092. [DOI] [PubMed] [Google Scholar]
- Gardner A, de Mingo Pulido A, and Ruffell B, (2020). Dendritic Cells and Their Role in Immunotherapy. Front. Immunol 11, 924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garris CS, Arlauckas SP, Kohler RH, Trefny MP, Garren S, Piot C, Engblom C, Pfirschke C, Siwicki M, Gungabeesoon J, et al. (2018). Successful Anti-PD-1 Cancer Immunotherapy Requires T Cell-Dendritic Cell Crosstalk Involving the Cytokines IFN-gamma and IL-12. Immunity 49, 1148–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garris CS, and Luke JJ, (2020). Dendritic Cells, the T-cell-inflamed Tumor Microenvironment, and Immunotherapy Treatment Response. Clin. Cancer Res 26, 3901–3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, Vermaelen K, Panaretakis T, Mignot G, Ullrich E, et al. (2009). Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat. Med 15, 1170–1178. [DOI] [PubMed] [Google Scholar]
- Gujar R, Maurya N, Yadav V, Gupta M, Arora S, Khatri N, and Sen P, (2016). c-Src Suppresses Dendritic Cell Antitumor Activity via T Cell Ig and Mucin Protein-3 Receptor. J. Immunol 197, 1650–1662. [DOI] [PubMed] [Google Scholar]
- Gunderson AJ, Kaneda MM, Tsujikawa T, Nguyen AV, Affara NI, Ruffell B, Gorjestani S, Liudahl SM, Truitt M, Olson P, et al. (2016). Bruton Tyrosine Kinase-Dependent Immune Cell Cross-talk Drives Pancreas Cancer. Cancer Discov. 6, 270–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hettinger J, Richards DM, Hansson J, Barra MM, Joschko AC, Krijgsveld J, and Feuerer M, (2013). Origin of monocytes and macrophages in a committed progenitor. Nat. Immunol 14, 821–830. [DOI] [PubMed] [Google Scholar]
- Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, et al. (2008). Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 322, 1097–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, and Carbone FR, (1994). T cell receptor antagonist peptides induce positive selection. Cell 76, 17–27. [DOI] [PubMed] [Google Scholar]
- Hong D, Rasco D, Veeder M, Luke JJ, Chandler J, Balmanoukian A, George TJ, Munster P, Berlin JD, Gutierrez M, et al. (2019). A Phase 1b/2 Study of the Bruton Tyrosine Kinase Inhibitor Ibrutinib and the PD-L1 Inhibitor Durvalumab in Patients with Pretreated Solid Tumors. Oncology 97, 102–111. [DOI] [PubMed] [Google Scholar]
- Hou DY, Muller AJ, Sharma MD, Duhadaway JB, Banerjee T, Johnson M, Mellor AL, Prendergast GC, and Munn DH, (2007). Inhibition of IDO in dendritic cells by stereoisomers of 1-methyl-tryptophan correlates with anti-tumor responses. Cancer Res. 67, 792–801. [DOI] [PubMed] [Google Scholar]
- Johnson TS, McGaha T, and Munn DH, (2017). Chemo-Immunotherapy: Role of Indoleamine 2,3-Dioxygenase in Defining Immunogenic Versus Tolerogenic Cell Death in the Tumor Microenvironment. Adv. Exp. Med. Biol 1036, 91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonuleit H, Kuhn U, Muller G, Steinbrink K, Paragnik L, Schmitt E, Knop J, and Enk AH, (1997). Pro-inflammatory cytokines and prostaglandins induce maturation of potent immunostimulatory dendritic cells under fetal calf serum-free conditions. Eur. J. Immunol 27, 3135–3142. [DOI] [PubMed] [Google Scholar]
- Kawakami Y, Inagaki N, Salek-Ardakani S, Kitaura J, Tanaka H, Nagao K, Kawakami Y, Xiao W, Nagai H, Croft M, and Kawakami T, (2006). Regulation of dendritic cell maturation and function by Bruton’s tyrosine kinase via IL-10 and Stat3. Proc Natl Acad Sci U S A 103, 153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leone RD, and Powell JD, (2020). Metabolism of immune cells in cancer. Nat. Rev. Cancer 20, 516–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu GY, and Sabatini DM, (2020). mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol 21, 183–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long GV, Dummer R, Hamid O, Gajewski TF, Caglevic C, Dalle S, Arance A, Carlino MS, Grob JJ, Kim TM, et al. (2019). Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): a phase 3, randomised, double-blind study. Lancet Oncol. 20, 1083–1097. [DOI] [PubMed] [Google Scholar]
- Maier B, Leader AM, Chen ST, Tung N, Chang C, LeBerichel J, Chudnovskiy A, Maskey S, Walker L, Finnigan JP, et al. (2020). A conserved dendritic-cell regulatory program limits antitumour immunity. Nature 580, 257–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal RK, Denny JE, Waide ML, Li Q, Bhutiani N, Anderson CD, Baby BV, Jala VR, Egilmez NK, and Schmidt NW, (2020). Temporospatial shifts within commercial laboratory mouse gut microbiota impact experimental reproducibility. BMC Biology 18, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangla A, Khare A, Vineeth V, Panday NN, Mukhopadhyay A, Ravindran B, Bal V, George A, and Rath S, (2004). Pleiotropic consequences of Bruton tyrosine kinase deficiency in myeloid lineages lead to poor inflammatory responses. Blood 104, 1191–1197. [DOI] [PubMed] [Google Scholar]
- Munn DH, and Mellor AL, (2013). Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol. 34, 137–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, and Mellor AL, (2005). GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity 22, 633–642. [DOI] [PubMed] [Google Scholar]
- Munn DH, Sharma MD, Lee JR, Jhaver KG, Johnson TS, Keskin DB, Marshall B, Chandler P, Antonia SJ, Burgess R, et al. (2002). Potential regulatory function of human dendritic cells expressing indoleamine 2,3-dioxygenase. Science 297, 1867–1870. [DOI] [PubMed] [Google Scholar]
- Munn DH, Sharma MD, and Mellor AL, (2004). Ligation of B7-1/B7-2 by human CD4+ T cells triggers indoleamine 2,3-dioxygenase activity in dendritic cells. J. Immunol 172, 4100–4110. [DOI] [PubMed] [Google Scholar]
- Oh SA, Wu D-C, Cheung J, Navarro A, Xiong H, Cubas R, Totpal K, Chiu H, Wu Y, Comps-Agrar L, et al. (2020). PD-L1 expression by dendritic cells is a key regulator of T-cell immunity in cancer. Nature Cancer 1, 681–691. [DOI] [PubMed] [Google Scholar]
- Orabona C, Pallotta MT, Volpi C, Fallarino F, Vacca C, Bianchi R, Belladonna ML, Fioretti MC, Grohmann U, and Puccetti P, (2008). SOCS3 drives proteasomal degradation of indoleamine 2,3-dioxygenase (IDO) and antagonizes IDO-dependent tolerogenesis. Proc. Natl. Acad. Sci. USA 105, 20828–20833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overman M, Javle M, Davis RE, Vats P, Kumar-Sinha C, Xiao L, Mettu NB, Parra ER, Benson AB, Lopez CD, et al. (2020). Randomized phase II study of the Bruton tyrosine kinase inhibitor acalabrutinib, alone or with pembrolizumab in patients with advanced pancreatic cancer. J. Immunother. Cancer (JITC) 8, e000587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overwijk WW, Theoret MR, Finkelstein SE, Surman DR, de Jong LA, Vyth-Dreese FA, Dellemijn TA, Antony PA, Spiess PJ, Palmer DC, et al. (2003). Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J. Exp. Med 198, 569–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallotta MT, Rossini S, Suvieri C, Coletti A, Orabona C, Macchiarulo A, Volpi C, and Grohmann U, (2021). Indoleamine 2,3-dioxygenase 1 (IDO1): an up-to-date overview of an eclectic immunoregulatory enzyme. FEBS J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollock PM, Cohen-Solal K, Sood R, Namkoong J, Martino JJ, Koganti A, Zhu H, Robbins C, Makalowska I, Shin SS, et al. (2003). Melanoma mouse model implicates metabotropic glutamate signaling in melanocytic neoplasia. Nat. Genet 34, 108–112. [DOI] [PubMed] [Google Scholar]
- R_Core_Team (2018). R: A language and environment for statistical computing. (Vienna, R Foundation for Statistical Computing; ). [Google Scholar]
- Ravishankar B, Liu H, Shinde R, Chandler P, Baban B, Tanaka M, Munn DH, Mellor AL, Karlsson MC, and McGaha TL, (2012). Tolerance to apoptotic cells is regulated by indoleamine 2,3-dioxygenase. Proc. Natl. Acad. Sci. USA 109, 3909–3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiff SD, Mantel R, Smith LL, Greene JT, Muhowski EM, Fabian CA, Goettl VM, Tran M, Harrington BK, Rogers KA, et al. (2018). The BTK Inhibitor ARQ 531 Targets Ibrutinib-Resistant CLL and Richter Transformation. Cancer Discov. 8, 1300–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salas-Benito D, Perez-Gracia JL, Ponz-Sarvise M, Rodriguez-Ruiz ME, Martinez-Forero I, Castanon E, Lopez-Picazo JM, Sanmamed MF, and Melero I, (2021). Paradigms on Immunotherapy Combinations with Chemotherapy. Cancer Discov. 11, 1353–1367. [DOI] [PubMed] [Google Scholar]
- Sharma MD, Baban B, Chandler P, Hou DY, Singh N, Yagita H, Azuma M, Blazar BR, Mellor AL, and Munn DH, (2007). Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J. Clin. Invest 117, 2570–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma MD, Huang L, Choi JH, Lee EJ, Wilson JM, Lemos H, Pan F, Blazar BR, Pardoll DM, Mellor AL, et al. (2013). An inherently bifunctional subset of Foxp3 T helper cells is controlled by the transcription factor Eos. Immunity 38, 998–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma MD, Rodriguez PC, Koehn BH, Baban B, Cui Y, Guo G, Shimoda M, Pacholczyk R, Shi H, Lee EJ, et al. (2018). Activation of p53 in Immature Myeloid Precursor Cells Controls Differentiation into Ly6c(+)CD103(+) Monocytic Antigen-Presenting Cells in Tumors. Immunity 48, 91–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma MD, Shinde R, McGaha T, Huang L, Holmgaard RB, Wolchok JD, Mautino MR, Celis E, Sharpe A, Francisco LM, et al. (2015). The PTEN pathway in Tregs is a critical driver of the suppressive tumor microenvironment. Science Advances 1, e1500845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin SS, Namkoong J, Wall BA, Gleason R, Lee HJ, and Chen S, (2008). Oncogenic activities of metabotropic glutamate receptor 1 (Grm1) in melanocyte transformation. Pigment Cell Mel. Res 21, 368–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonpavde G, Necchi A, Gupta S, Steinberg GD, Gschwend JE, Van Der Heijden MS, Garzon N, Ibrahim M, Raybold B, and Liaw D, (2020). ENERGIZE: a Phase III study of neoadjuvant chemotherapy alone or with nivolumab with/without linrodostat mesylate for muscle-invasive bladder cancer. Future Oncology 16, 4359–4368. [DOI] [PubMed] [Google Scholar]
- Stiff A, Trikha P, Wesolowski R, Kendra K, Hsu V, Uppati S, McMichael E, Duggan M, Campbell A, Keller K, et al. (2016). Myeloid-Derived Suppressor Cells Express Bruton’s Tyrosine Kinase and Can Be Depleted in Tumor-Bearing Hosts by Ibrutinib Treatment. Cancer Res. 76, 2125–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Leun AM, Thommen DS, and Schumacher TN, (2020). CD8(+) T cell states in human cancer: insights from single-cell analysis. Nat. Rev. Cancer 20, 218–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varikuti S, Singh B, Volpedo G, Ahirwar DK, Jha BK, Saljoughian N, Viana AG, Verma C, Hamza O, Halsey G, et al. (2020). Ibrutinib treatment inhibits breast cancer progression and metastasis by inducing conversion of myeloid-derived suppressor cells to dendritic cells. Br J Cancer 122, 1005–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villar J, and Segura E, (2020). Decoding the Heterogeneity of Human Dendritic Cell Subsets. Trends Immunol. 41, 1062–1071. [DOI] [PubMed] [Google Scholar]
- von Kortzfleisch VT, Karp NA, Palme R, Kaiser S, Sachser N, and Richter SH, (2020). Improving reproducibility in animal research by splitting the study population into several ‘mini-experiments’. Sci. Rep 10, 16579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfson RL, and Sabatini DM, (2017). The Dawn of the Age of Amino Acid Sensors for the mTORC1 Pathway. Cell Metab. 26, 301–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yost KE, Chang HY, and Satpathy AT, (2021). Recruiting T cells in cancer immunotherapy. Science 372, 130–131. [DOI] [PubMed] [Google Scholar]
- Zakharia Y, McWilliams RR, Rixe O, Drabick J, Shaheen MF, Grossmann KF, Kolhe R, Pacholczyk R, Sadek R, Tennant LL, et al. (2021). Phase II trial of the IDO pathway inhibitor indoximod plus pembrolizumab for the treatment of patients with advanced melanoma. J. Immunother. Cancer 9, e002057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilionis R, Engblom C, Pfirschke C, Savova V, Zemmour D, Saatcioglu HD, Krishnan I, Maroni G, Meyerovitz CV, Kerwin CM, et al. (2019). Single-Cell Transcriptomics of Human and Mouse Lung Cancers Reveals Conserved Myeloid Populations across Individuals and Species. Immunity 50, 1317–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The scRNA-seq, bulk RNA-seq and ATAC-seq datasets are deposited in the Genome Expression Omnibus (GEO) under Accession Number GSE180039 and are publicly available. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.