

Summary
Persistent activation of fibroblasts and resistance of myofibroblasts to turnover play important roles in organ-tissue fibrosis development and progression. The mechanism that mediates apoptosis resistance of myofibroblasts is not understood. Here, we report that myofibroblasts express and secrete PKM2. Extracellular PKM2 (EcPKM2) facilitates progression of fibrosis by protecting myofibroblasts from apoptosis. EcPKM2 upregulates arginase-1 expression in myofibroblasts and therefore facilitates proline biosynthesis and subsequent collagen production. EcPKM2 interacts with integrin αvβ3 on myofibroblasts to activate FAK-PI3K signaling axis. Activation of FAK-PI3K by EcPKM2 activates downstream NF-κB survival pathway to prevent myofibroblasts from apoptosis. On the other hand, activation of FAK- PI3K by EcPKM2 suppresses PTEN to subsequently upregulate arginase-1 in myofibroblasts. Our studies uncover an important mechanism for organ fibrosis progression. More importantly, an antibody disrupting the interaction between PKM2 and integrin αvβ3 is effective in reversing fibrosis, suggesting a possible therapeutic strategy and target for treatment of organ fibrosis.
Subject areas: Biological sciences, Molecular biology, Cell biology
Graphical abstract
Highlights
-
•
Extracellular PKM2 (EcPKM2) facilitates organ fibrosis progression
-
•
EcPKM2 promotes apoptosis resistance and collagen production in myofibroblasts
-
•
EcPKM2 activates αvβ3-FAK-PI3K signaling axis
-
•
An antibody disrupting PKM2 and αvβ3 interaction reverses lung and liver fibrosis
Biological sciences; Molecular biology; Cell biology
Introduction
Organ and tissue fibrosis is a typical outcome of persistent inflammatory responses to tissue damages, which leads to constant activation of fibroblasts and apoptosis resistance of these myofibroblasts (Bataller and Brenner, 2005; Elsharkawy et al., 2005; Lim et al., 2011; Moreira, 2007; Weiskirchen et al., 2019; Winau et al., 2008; Yin et al., 2013). High amounts of extracellular matrix (ECM), mostly collagen type I, and matrix metalloproteinase (MMP) inhibitors are secreted by myofibroblasts, which leads to accumulation of collagen fibrils in the organs due to excessive production and decrease in degradation of collagen (Atzori et al., 2009; Puche et al., 2013). In the normal tissue injury repair, attenuation of insults abolishes inflammation response. Upon attenuating inflammation response, myofibroblasts in tissue undergo apoptosis due to the presence of several pro-apoptotic factors, such as TNF-α, FasL, and TRAIL (Habiel et al., 2018; Wallach-Dayan et al., 2015). However, in the case of chronic inflammation, apoptosis of myofibroblasts is prevented. Despite accumulated evidence indicating that interactions of myofibroblasts with ECM and various growth factors and inflammatory cytokines support myofibroblasts resistance to apoptosis (Elsharkawy et al., 2005), the detailed mechanism of this apoptosis resistance is still not clear. Given the critical role of myofibroblasts in organ tissue fibrosis progression, induction of apoptosis of myofibroblasts is an important strategy for developing fibrosis treatment (Elsharkawy et al., 2005). Collagen fibril is excessively produced and deposited during fibrosis progression. Myofibroblasts is the main cell type that is engaged in collagen synthesis and secretion. Gly-x-Pro and Gly-x-Hyp triplet repeats are the main composition of collagen. Collagen contains ∼30% glycine and proline/hydroxyproline respectively. To meet the needs of rapid collagen synthesis and secretion, myofibroblasts must quickly upregulate glycine and proline productions. Regulation of amino acid metabolism in myofibroblasts is not yet fully understood.
Pyruvate kinase is an enzyme that catalyzes the last reaction in glycolysis. There are four isoforms of pyruvate kinases, L/R and M1/M2, which are expressed in different tissue types or under different physiological conditions (Tsutsumi et al., 1988; Yamada et al., 1990). PKM2 can form a homodimer or a homotetramer. The tetramer is active as pyruvate kinase (Elbers et al., 1991; Hacker et al., 1998) while the dimer is a protein kinase (Gao et al., 2012, 2013; Yang et al., 2012). Interestingly, a number of recent studies show that PKM2 is functionally involved in multiple cellular processes in different locations, including metabolism control, transcription regulation, and chromatin packaging (Gao et al., 2012; Luo et al., 2011; Yang et al., 2011). High serum levels of PKM2 have long been observed in patients with various inflammation diseases (Hathurusinghe et al., 2007; Jeffery et al., 2009; Staib et al., 2006), indicating a potential association of EcPKM2 with inflammation responses. PKM2 has been reported to be involved in metabolism adjustment in various immune cells activation during inflammation (Angiari et al., 2020; Damasceno et al., 2020). It is also shown in several laboratories that PKM2 is functionally involved in several non-metabolism cellular processes in various cell types (Dong et al., 2016; Zhang et al., 2019). These studies suggested a potential important functional role of PKM2 in inflammatory responses to tissue injury and cancer progression. Our previous studies demonstrated that EcPKM2 facilitates tumor growth by promoting angiogenesis (Li et al., 2014). During cutaneous wound repair, PKM2 is released by infiltration neutrophils at the wound site. The released PKM2 facilitates wound repair (Zhang et al., 2016). We report here that PKM2 is expressed in and secreted from myofibroblasts. EcPKM2 is present in fibrotic liver/lung tissue and in blood circulation of chronic liver disease (CLD) patients. EcPKM2 facilitates fibrosis progression by protecting myofibroblasts from apoptosis. EcPKM2 also facilitates fibrosis progression by upregulating Arginase-1 (Arg-1) in myofibroblasts, which consequently promotes proline synthesis and subsequent collagen synthesis in and secretion from myofibroblasts. PKM2 acts on myofibroblasts by interacting with integrin αvβ3 and subsequently activating the integrin-FAK-PI3K signaling axis. EcPKM2 activates integrin-FAK-PI3K-NF-κB survival signaling to promote myofibroblast survival. EcPKM2 activates integrin-FAK-PI3K-PTEN signaling to upregulates Arg-1 expression in myofibroblasts. Upregulation of Arg-1 facilitates proline and collagen biosynthesis. Neutralization of EcPKM2 by a PKM2 antibody reverses liver and lung fibrosis, providing a potential therapeutic strategy for organ and tissue fibrosis treatment.
Results
Myofibroblasts express and secret PKM2
We previously reported that EcPKM2 facilitated wound repair and tissue regeneration by promoting angiogenesis (Li et al., 2014; Zhang et al., 2016). Several laboratories observe that PKM2 is expressed in fibroblasts (Forty et al., 2016; Vo et al., 2013). Myofibroblast differentiation plays a vital role in tissue repair and regeneration. We asked whether myofibroblasts express and secrete PKM2. Thus, we examined PKM2 expression and secretion from fibroblasts and myofibroblasts. We employed primary human hepatic stellate cells (hHSC), an immortalized human hepatic stellate cell line (LX2), and primary human lung fibroblasts (NHLF). The cells were activated by TGFβ. Expression and secretion of PKM2 was probed by immunoblot of cell lysate and ELISA of culture media. PKM2 was not expressed or secreted in hHSC and NHLF and expressed in a low level in LX2 without TGFβ treatment. However, PKM2 was expressed and secreted in a high level in and from hHSC, NHLF, and LX2 cells upon TGFβ stimulation (Figures 1A and 1B). Since PKM2 is a pyruvate kinase, we tested pyruvate kinase activity of the secreted PKM2. PKM2 secreted from LX2 cells under the same conditions had about 50% pyruvate kinase activity compared with PKM2 expressed and purified from bacterial E. coli (Figure 1C). We also carried out immuno-histological analyses of tissue samples from liver disease and pulmonary fibrosis patients using an in-house developed rabbit monoclonal antibody against PKM2, IgGPK (Zhang et al., 2016). Interestingly, very high levels of PKM2 were detected in liver fibrosis/cirrhosis (Figures 1D and 1E) and lung fibrosis (Figures 1F and 1G) patient tissues. The EcPKM2 was apparent in the staining. EcPKM2 was also present in most liver tissue samples of hepatitis patients. As control, almost no PKM2 staining was observed in normal liver and lung tissues (Figures 1D–1F). It would be expected that PKM2 should also be detectable in the blood circulation of fibrosis patients if EcPKM2 presents in CLD liver tissues. Thus, we carried out ELISA analyses of serum samples collected from CLD patients and healthy individuals. Clearly, PKM2 was detected in the blood from patients with liver fibrosis/cirrhosis and viral hepatitis, whereas the levels of PKM2 were very low in the blood of healthy individuals (Figure S1A). Altogether, our results indicate that PKM2 is released to extracellular space of CLD and pulmonary fibrosis patients, and PKM2 is expressed and secreted by myofibroblasts.
Figure 1.

Myofibroblasts express and secrete PKM2
(A and B) PKM2 levels in extracts of indicated cells (A) were analyzed by immunoblot (IB:PKM2) and in culture medium (ng/mL) (B) of indicated cells were measured by ELISA. The cells were treated (+) or untreated (−) with TGFβ (10 ng/mL).
(C) Pyruvate kinase activity of PKM2 purified from medium of LX2 cells and rPKM2 expressed and purified from bacterial E. coli were measured using pyruvate kinase kit. Pyruvate kinase activity is presented as relative activity by referencing the activity of rPKM2 as 1. The assay was carried out under the same amounts and same concentration of PKM2.
(D) Representative images of IHC staining of PKM2 in liver disease tissue array. The call-out is enlargement of red square area.
(E) Quantitation of PKM2 staining (PKM2 stain intensity) of liver diseases tissue array. Quantitation was calculated from the analyses of randomly selected four view fields from each spot of the array. The PKM2 staining intensity was calculation of all tissue spots in the array (total 12 normal n = 12, 18 viral hepatitis n = 18, 60 cirrhosis liver n = 60 spots).
(F and G) Representative images (F) and quantitation (G, PKM2 stain intensity) of IHC staining of PKM2 in lung fibrosis tissue spots in lung diseases tissue array (total 4 normal n = 4, 6 pulmonary fibrosis n = 6 spots). In (D) and (F), blue is hematoxylin staining. The call-out is enlargement of black square area. Red arrows indicate extracellular PKM2 in the staining. Error bars in (B), (C), and (G) represent mean ± S.E.M. Scale bars, 100 μm. ∗p<0.05, ∗∗p<0.01, ∗∗∗p<0.001.
EcPKM2 facilitates liver and lung fibrosis progression
Upregulation and secretion of PKM2 in myofibroblasts, and the presence of EcPKM2 in fibrotic liver and lung tissues of CLD and pulmonary fibrosis patients, suggest a potential role of EcPKM2 in organ and tissue fibrosis development and progression. To test the speculation, liver fibrosis was induced by twice weekly i.p. administration of thioacetamide (TAA) and 10% ethanol in drinking water (Hessien et al., 2010). Bacterially expressed recombinant PKM2 (rPKM2), PKM1 (rPKM1), or vehicle was added into the fibrosis induction (mix with TAA) 18 days after the first dose of TAA (Figure 2A). At the end of the experiments, the animals were sacrificed. Body weights, liver weights, and out-surface of the livers were closely examined. Clearly, addition of rPKM2 to the TAA fibrosis induction significantly reduced mouse body weight and increased liver weight (Figures 2B andS1B). Fibrotic features on the liver surface were more apparent with the addition of rPKM2 compared to those with the addition of vehicle to TAA (Figure S1C). Examination of serum markers that typically reflect liver damages suggested that addition of rPKM2 resulted in more damages to the liver (Figure S1D). Sirius red staining of liver sections and hydroxyproline assay demonstrated more and thicker collagen accumulation in the livers of animals that were treated with the addition of rPKM2 to the TAA/alcohol fibrosis induction (Figures 2C, 2D, andS1E). Liver cirrhosis features were identified by dense collagen bands and cross-networks in the livers of animals that were treated with rPKM2 addition (Figure 2D). IHC staining of αSMA indicated that addition of rPKM2 to TAA/alcohol led to increase in αSMA positive myofibroblasts in fibrotic liver (Figures 2E and 2F). These results suggest that addition of rPKM2 into TAA-alcohol exacerbated liver fibrosis progression. We then asked, “Can administration of rPKM2 alone induce liver fibrosis?” To answer this question, a group of mice was only administrated rPKM2 without TAA-alcohol. Clearly, administration of rPKM2 alone did not induce liver fibrosis (Figures S1F–S1I), suggesting that EcPKM2 could not initiate liver fibrosis.
Figure 2.

rPKM2 facilitates fibrosis progression in murine models
(A) Schematic illustration of the schedule of TAA/alcohol liver fibrosis induction and subsequent addition of rPKM2/rPKM1 into TAA. Red arrow indicates addition of rPKM2/rPKM1/buffer to the TAA fibrosis induction.
(B) Liver weight of the animals at endpoint.
(C and D) Representative images (C) and Quantitation (D) of Sirius red staining of liver sections from mice treated with indicated agents. The call-out in (C) is enlarged images showing the Sirius red stain feature.
(E and F) Representative images (E) and Quantitation (F) of IHC staining of αSMA of liver sections from mice treated with indicated agents.
(G) Schematic illustration of the schedule of bleomycin lung fibrosis induction and subsequent addition of rPKM2/rPKM1 into bleomycin. Red arrow indicates addition of rPKM2/rPKM1/vehicle to the bleomycin fibrosis induction.
(H) Lung weight of the animals at endpoint.
(I) Representative images of Sirius red staining (Upper) and IHC staining of αSMA (Bottom) of lung sections from mice treated with indicated agent.
(J and K) Quantitation of Sirius red (J) and αSMA IHC (K) staining of sections of lung from mice treated with indicated agents. Liver tissues from mice treated with indicated agents. In (D), (F), (J) and (K), Quantification was calculated from measurements of 10 mice. Four randomly selected tissue sections per animal and three randomly selected view fields in each section were quantified. The quantity of Sirius red stain is presented as Sirius red+ % of total area, quantity of αSMA IHC is presented as αSMA+ % of total area. Error bars in (D), (F), (H), (J), and (K) represent mean ± S.E.M. Scale bars, 100 μm. ∗p<0.05, ∗∗p<0.01, ∗∗∗p<0.001.ns: statistically no significance.
To test the commonality of the role of EcPKM2 in facilitating organ tissue fibrosis, we analyzed the effects of addition of rPKM2 in the bleomycin-induced lung fibrosis mouse model (Moeller et al., 2008). rPKM2, rPKM1, or vehicle was added into bleomycin 2.5 weeks after the first dose of bleomycin (Figure 2G). Glossy examination of mouse lung indicated that addition of rPKM2 into bleomycin led to more fibrosis features on the lung surface (Figure S2A) and reduced body weight and increased lung weight (Figures 2H andS2B) compare to the groups with addition of rPKM1 and vehicle into bleomycin. Sirius red staining of the lung sections and hydroxyproline assay demonstrated more collagen accumulation in the lungs of animals that were treated with the rPKM2 addition (Figures2I, 2J, andS2C). IHC staining of αSMA indicated that addition of rPKM2 into bleomycin led to increase in αSMA positive myofibroblasts in fibrotic lungs (Figures 2I and 2K). Similarly, administration of rPKM2 alone without bleomycin did not induce lung fibrosis (see Figures 2H–2K).
EcPKM2 facilitates myofibroblasts apoptosis resistance
How does EcPKM2 facilitate liver and lung fibrosis progression? Fibroblasts are activated to myofibroblasts during fibrosis progression. Sustained activation and resistance to apoptosis of myofibroblasts lead to organ tissue fibrosis progression (Bataller and Brenner, 2005; Yin et al., 2013). Since EcPKM2 could not initiate liver/lung fibrosis (see Figures S1F–S1H, 2H–2K), we questioned whether EcPKM2 exerted its effects on myofibroblast turnover. To test this conjecture, we carried out immunofluorescence (IF) staining of αSMA and TUNEL with sections prepared from lungs of mice that were treated with bleomycin with addition of rPKM2 or rPKM1. Evidently, with the addition of rPKM2, significantly more αSMA positive lung myofibroblasts and less TUNEL staining were presented compared to the addition of rPKM1 (Figures 3A and 3B). Less αSMA and TUNEL co-staining was observed in the group with addition of rPKM2 to the bleomycin fibrosis induction (Figure 3C), suggesting less apoptosis of αSMA positive lung myofibroblasts. Similarly, addition of rPKM2 into the TAA/alcohol led to more αSMA and less TUNEL staining (Figure 3D). Less αSMA and TUNEL co-staining was detected in fibrotic liver of the TAA-alcohol fibrosis model with the addition of rPKM2 (Figure 3E). The results of αSMA and TUNEL staining suggest that EcPKM2 may play a role in facilitating myofibroblasts apoptosis resistance. To verify the role of EcPKM2 in myofibroblast apoptosis protection, NHLFs were first activated by TGFβ. Treatment of the TGFβ activated NHLF with FasL + Chlorhexidine (CHX) led to cell apoptosis. Addition of rPKM2, but not rPKM1, into culture medium protected NHLF from the FasL + CHX induced apoptosis as demonstrated by cell viability and Annexin v-FACS apoptosis assays (Figures 3F–3H). Furthermore, addition of rPKM2 into culture medium resulted in a decrease in cleaved PARP and an increase in BCL2 and cIAP1 in the cells (Figure 3I), suggesting that rPKM2 reduced cell apoptosis and increase cell survival signaling. We also analyzed the effects of rPKM2 on hHSC. The hHSC were activated by prolonged culturing and TGFβ treatment as indicated by increased expression of αSMA (Figure S2D). The activated/inactivated hHSC were treated by FasL and TRAIL in the presence and absence of rPKM2 in culture medium. Presence of rPKM2 decreased the activated hHSC from both FasL- and TRAIL-induced apoptosis (Figure 3J). Similarly, rPKM2 also protected LX2 cells from TRAIL induced apoptosis (Figure S2E). We conclude from these experiments that EcPKM2 promotes myofibroblast apoptosis resistance.
Figure 3.

EcPKM2 promotes myofibroblasts apoptosis resistance
(A–C) Representative images of immunofluorescence (IF) staining (A) and quantitation of αSMA and TUNEL staining (B) and αSMA and TUNEL double staining over αSMA staining (%) (C) in sections from lung of mice that were treated by the indicated agents. In (A), Red is IF staining of αSMA. Green is TUNEL staining. Blue is DAPI staining.
(D and E) Quantitation of αSMA and TUNEL staining (D) and αSMA and TUNEL double staining over αSMA staining (%) (E) in sections from liver of mice that were treated by the indicated agents. In (B) and (D), quantity of αSMA is presented as staining intensity pixels per mm2, quantity of TUNEL staining is presented as number of positive nuclei per mm2. In (A)–(E), animals in control group were not treated with any agents.
(F) NHFL cell viability was measured by ATP cell viability kit. Viability of cells without the FasL + CHX treatment (control with addition of vehicle) was set as 100.
(G and H) Apoptosis of NHFL cells was measured by Annexin v-FACS assay, examples of FACS plots (G) and quantitation (H). In (F)–(H), the cells were treated by the indicated agents. Control is the results from cells that were treated with vehicle.
(I) Cellular levels of cleaved PARP (IB:cleaved PARP), full-length PARP (IB:Full-length PARP), BCL2 (IB:BCL2), and cIAP1 (IB:cIAP1) in NHLF that were treated by indicated agents were analyzed by immunoblot. Immunoblot of GAPDH (IB:GAPDH) is a loading control.
(J)Apoptosis of primary human HSCs under treatment of indicated agents were measured by Annexin v-FACS assay. The cells were cultured for 8 days to activate HSC prior to the indicated treatment. Cell apoptosis is presented as relative apoptosis by defining apoptosis of cell treated with buffer (control) as 1. Error bars in (B)–(F), (H), and (J) represent mean ± S.E.M. Scale bars, 100 μm. ∗p<0.05, ∗∗p<0.01, ∗∗∗p<0.001.ns: statistically no significance.
EcPKM2 upregulates Arg-1 in myofibroblasts to promote proline synthesis
A hallmark of organ tissue fibrosis is synthesis and secretion of excess amounts of collagen by myofibroblasts. We asked whether EcPKM2 affected collagen synthesis and secretion in myofibroblasts. Immunoblot analyses demonstrated that rPKM2 increased collagen in NHLF and LX2 upon TGFβ stimulation. The collagen increase was not observed with addition of rPKM2 without TGFβ (Figures 4A and 4B). Measurement of collagen in the culture medium showed that addition of rPKM2 into cell culture increased collagen secretion (Figures 4C and 4D). However, RT-PCR analyses revealed that rPKM2 did not upregulate the collagen 1A1 mRNA in NHLF and LX2 (Figures 4E and 4F), indicating that rPKM2 likely promoted collagen production after mRNA synthesis in the cells. Due to unique amino acid composition (∼30% Gly and Pro) in collagen, myofibroblasts must rapidly turn on glycine and proline biosynthesis for rapid collagen production. We analyzed amino acid abundance in NHLF, hHSC, and LX2 upon TGFβ with the addition of rPKM2 or rPKM1. An increase in proline and a decrease in arginine in the cells were evident upon addition of rPKM2 into the culture medium (Figures 4G, 4H, S3A, and S3B). Proline is mainly synthesized in cells by Arginine – ornithine, catalyzing by arginase, and subsequent to proline or polyamine (Wu and Morris, 1998). We therefore quantified ornithine in NHLF, hHSC, and LX2 upon the TGFβ and rPKM2 treatment. Clearly, rPKM2 increased ornithine (Figures S3C–S3E). Decrease in arginine and increase in ornithine and proline suggest that rPKM2 might regulate the arginine metabolism enzyme arginase-1 (Arg-1). Immunoblot, RT-PCR, and arginase activity assays demonstrated that addition of rPKM2 into the cell culture medium increased Arg-1 both in protein and mRNA levels (Figures 4A, 4B, 4I, and 4J) as well as arginase activity (Figures S3F and S3G). Furthermore, Arg-1 inhibitor and L-arginine deficiency medium abrogated the effects of rPKM2 in upregulation of collagen production (Figures S3H and S3I). Thus, our results suggest that EcPKM2 facilitates collagen synthesis and secretion by upregulating Arg-1 in myofibroblasts, thereby increasing proline production from arginine. To verify that EcPKM2 upregulates Arg-1 in myofibroblasts, we probed the expression of Arg-1 in the liver and lung of the fibrosis mice, with the addition of rPKM2 or rPKM1 to the fibrosis induction. Evidently, addition of rPKM2 into bleomycin increased Arg-1 and αSMA staining and Arg-1/αSMA co-staining in the fibrotic lungs (Figures 4K–4M). Arg-1 staining levels are very high in normal healthy and fibrotic livers, indicating Arg-1 expression in liver cells other than myofibroblasts. Although, no significant increase in Arg-1 staining with addition of rPKM2 into the TAA fibrosis induction as compared to addition of rPKM1 was observed, an increase in Arg-1/αSMA co-staining (Figures S3J–S3L) suggested increase in Arg-1 expression in myofibroblasts (activated HSC) in the fibrotic liver by addition of rPKM2. Therefore, our results support that EcPKM2 upregulates Arg-1 in myofibroblasts during fibrosis progression to facilitate proline and subsequent collagen synthesis and secretion.
Figure 4.

EcPKM2 facilitates collagen synthesis in myofibroblasts by upregulating Arg-1
(A andB) Cellular levels of collagen 1A1 (IB:Col1a1) and Arg-1 (IB:Arg1) in NHLF (A) and LX2 (B) cells were analyzed by immunoblot. The cells were treated by the indicated agents. Bottom panels are immunoblot analyses of cellular levels of collagen 1a1 in NHLF (A) and LX2 (B) cells without TGFβ treatment. Immunoblot of Actin (IB:Actin) is a loading control.
(C and D) Representative pictures of secreted collagen (with Sirius Red) in culture medium that spung down at bottom of centrifuge tube (C) and quantification of the collagen in culture media (D). The cells were treated by the indicated agents. The quantity of precipitated collagen is presented as relative collagen secretion by reference the control cells as 1.
(E and F) Cellular levels of collagen 1A1 mRNA in NHLF (E) and LX2 (F) cells were analyzed by qRT-PCR. The cells were treated by the indicated agents. The quantity of collagen 1A1 mRNA is presented as Relative Col1A1 mRNA level by reference the control cells as 1. In (A)–(F), the control is the cells with no treatment.
(G and H) Relative abundance of selected amino acids in NHLF (G) and LX2 (H) cells were analyzed by HPLC-MS. The cells were treated by TGFβ + rPKM1 (black bars) or TGFβ + rPKM2 (gray bars). The amino acid levels in the cells that were treated with TGFβ + rPKM1 are set as reference 1.
(I and J) Cellular levels of Arg-1 mRNA in NHLF (I) and LX2 (J) cells were analyzed by qRT-PCR. The cells were treated by the indicated agents. The quantity of Arg-1 mRNA is presented as Relative ARG1 mRNA level by reference in the Arg-1 mRNA level in cells that were treated with TGFβ + rPKM1 as 1.
(K–M) Representative images (K) and quantitation of immunofluorescence (IF) staining of αSMA and Arg1 staining (L) and αSMA and Arg1 double staining over αSMA staining (%) (M) in sections from lung of mice that were treated by the indicated agents. In (K), Green is IF staining of αSMA. Red is Arg1 staining. Blue is DAPI staining. In (L), quantities of αSMA and Arg1 are presented as staining intensity pixels per mm2. Error bars in (D)–(J), (L), and (M) represent mean ± S.E.M. Scale bars, 100 μm. ∗p<0.05, ∗∗p<0.01, ∗∗∗p<0.001.ns: statistically no significance.
EcPKM2 interacts with and activates integrin αvβ3
An open question is: What is the cell surface receptor(s) that mediates the effects of EcPKM2 on myofibroblasts? We carried out chemical crosslinking with His-tagged both rPKM2 and rPKM1 and LX2 cells activated by TGFβ using glutaraldehyde as the crosslinker. Denatured cell lysates were prepared from the crosslinks of rPKM2/rPKM1 and LX2 cells. The crosslinked proteins were separated by His-tag pull-down. Immunoblot of PKM2/PKM1 revealed the rPKM2/rPKM1 crosslinked proteins (Figure 5A). Clearly, no overlap in rPKM2/rPKM1 crosslinked protein was observed (Figure 5A). Bands corresponding to the rPKM2 crosslinked proteins were sliced out and digested by trypsin, and subsequently analyzed by MALDI-tof/tof. The MS-analyses showed that rPKM2 crosslinked to integrin αv and β3 (Figure S4A). Interaction of PKM2 with integrin αvβ3 was verified by co-immunoprecipitation of rPKM2 with β3 in cell extracts prepared from LX2 and NHLF activated by TGFβ (Figures 5B andS4B). To characterize the PKM2 and integrin αvβ3 interaction, we carried out in vitro SPR binding analyses using different recombinant integrin pairs and rPKM2. Evidently, rPKM2 interacted with recombinant integrin αvβ3 with an estimated kd of 5.1 nM and 1:1 binding stoichiometry. In contrast, rPKM2 interacted with other three integrin pairsα1β1, αvβ5, and αIIbβ3 with much lower affinities (Figure 5C). The rPKM2 and integrin αvβ3 interaction was further verified by cell attachment assay using culture plate coated with rPKM2. CHO cells do not express αvβ3 (Stupack et al., 2001; Tani et al., 2003). As a result, the cells did not attach strongly to the plates. We then exogenously expressed αvβ3 in CHO cells (Figure S4C) and performed the same attachment analyses. Evidently, the αvβ3 expressing CHO cells attached to the rPKM2 coated plates (Figure 5D). As a control, the CHO cells expressing integrin αIIbβ3, αvβ6, and αvβ1 (Figures S4C and S4D) did not attach or weakly attached to the plates (Figure 5D). LX2 cells express high levels of αvβ3 (Figure S4E). The cells attached strongly to the rPKM2 coated plates. However, the attachment was completely blocked by IgGPK and LM609, an antibody that recognizes integrin αvβ3 and blocks integrin activation (Figure 5E). Moreover, knockdown of integrin β3 (Figure S4F) abolished the attachment of LX2 to rPKM2 (Figure 5F). Results of our crosslinking, co-precipitation, in vitro SPR binding, and cell attachment assays suggest that EcPKM2 specifically interacted with integrin αvβ3 on myofibroblasts. To test if EcPKM2 and integrin αvβ3 interaction had any functional relevance, we examined LX2 cell migration on vitronectin in the presence of rPKM2 and rPKM1. Evidently, cell migration on vitronectin was significantly increased by addition of rPKM2 but not rPKM1. The increases in migration were diminished by addition of IgGPK and LM609 (Figure 5G). As a control, addition of IgGPK did not affect LX2 cell migration if rPKM2 was not added to the culture medium (Figure S4G). It is well documented that integrin activation often leads to activation of FAK and integrin clustering (Cluzel et al., 2005). Consistently, an increase in activation of FAK was observed in LX2 and NHLF cells upon addition of rPKM2 to the culture medium (Figures 5H andS5A). The activation of FAK was diminished by the antibody IgGPK (Figure 5H). Integrin clustering on LX2 cells was clearly visible by immunefluorescence (IF) staining of integrin αvβ3 upon addition of rPKM2 to the culture medium. As a control, the integrin clustering was not induced by rPKM1 (Figure 5I). We conclude from our experiments that EcPKM2 interacts with and activates integrin αvβ3 signaling on myofibroblasts.
Figure 5.

EcPKM2 interacts with integrin αvβ3 and activates the integrin signaling
(A) Crosslinking of His-rPKM2 (right) or His-rPKM1 (left) with LX2 cell surface proteins was revealed by immunoblot of PKM2/PKM1 (PKM2+CL, PKM1+CL). The rPKM2 or rPKM1 was crosslinked to LX2 cells. The denatured extracts were prepared from the crosslinked cells. His-PKM1 or His-rPKM2 was pulled down from the extracts. The precipitations were analyzed by immunoblot (Lane PKM2+CL or PKM1+CL). Lane PKM2/PKM1 is the immunoblot analyses of extracts without crosslinking. Arrows indicated the crosslinked bands that were sliced out and subjected to MALDI/tof-tof analyses.
(B) Co-immunoprecipitation of integrin β3 with PKM2 (IP:IgGPK) in extracts of LX2 cells with TGFβ treatment was examined by immunoblot of integrin β3 (IB: Integrin β3). Immunoblot of PKM2 (IB:PKM2) indicates amounts of PKM2 that are precipitated down. Immunoprecipitation using IgG purified from pre-immune serum (IP:IgGCon) is a control. Immunoblot of integrin β3 (IB: Integrin β3) in whole cell lysate (WCL) is a control indicating equal amounts of cell extracts used in the IPs.
(C) (Left) The representative binding curves of binding of PKM2 to integrins αvβ3, α1β1, and αIIbβ3 that were monitored by SPR with integrin that were immobilized on the Biacore chip.(Right) The deduced disassociation constants (kd) and binding stoichiometry of PKM2 to integrins αvβ3,α1β1, αvβ5, and αIIbβ3 from the SPR binding analyses. nd means not determined.
(D) Attachment of CHO cells to rPKM2 coated plates. Integrin αvβ3, αIIbβ3, αvβ1, αvβ6, and empty vector (Veh) were exogenously expressed in the cells. The attachment is presented as total number of cells attached to the plate per view field.
(E) Attachment of LX2 cells to BSA, rPKM1, and rPKM2 coated plates (Left), and to rPKM2 coated plates with addition of indicated IgG antibodies, anti-PKM2 antibody (IgGPK), integrin αvβ3 (LM609), and pre-bleeding serum IgG (IgGCon), (Right). The attachment is presented as Relative attachment by defining the cells attached to rPKM2 coated plate as 100.
(F) Attachment of LX2 cells to rPKM1 and rPKM2 coated plates. Integrin β3 was knocked down (KD−β3). The attachment is presented as Relative attachment by defining the cells attached to rPKM2 coated plate with knockdown using scrambled siRNA (Scramble) as 100.
(G) Migration of LX2 cells treated with TGFβ in the presence of rPKM2 and rPKM1 (Left) and with addition of antibodies IgGPK, LM609, or IgGCon (Right) were analyzed. The migration was assayed in plats with vitronectin (VN) coating. The cell migrations are presented as relative migration by defining the migration of controls, without rPKM2/rPKM1 (left) or without antibody treatment (right), as 100 (Control).
(H) Activation of FAK was analyzed by immunoblot of cell extracts of LX2 cells that were seeded on ECM coated culture plate with addition of the indicated proteins (5 μM) for 60 min using antibody against phosphorylated FAK (IB:p-FAK397). Immunoblot of extracts using antibody against FAK (IB:FAK) is a control to indicate the cellular levels FAK and loading control.
(I) Representative images of IF staining of integrin αvβ3 (red) of LX2 cells treated with indicated agents. Error bars in (D)–(F), and (G) represent mean ± S.E.M. Scale bars, 100 μm. ∗p<0.05, ∗∗p<0.01, ∗∗∗p<0.001.ns: statistically no significance.
EcPKM2 activates integrin-FAK-PI3K signaling axis in myofibroblasts
Our next question is how EcPKM2 protects myofibroblasts from apoptosis, and how EcPKM2 upregulates Arg-1 in myofibroblasts. Interaction of EcPKM2 with integrin αvβ3 activates FAK (see Figures 5H andS5A) and it is well established that activation of integrin FAK signaling leads to activation of downstream PI3K (Wegener and Campbell, 2008). Thus, we reasoned whether the effect of EcPKM2 on apoptosis protection is mediated through activation of PI3K. We examined activation of PI3K in NHFL and LX2 upon TGFβ and rPKM2 treatments. Immunoblot and PI3K activity assay demonstrated that the phosphorylated PI3K (phosphor-PI3K) and PI3K activity increased in the cells (Figure 6A, 6B, S5A, and S5B). Furthermore, activation of PI3K was abrogated by an FAK inhibitor (2.5 μM) and the antibody LM609 (Figures6B andS5B). Knockdown of integrin β3 and antibody LM609 also decreased phosphor-PI3K in TGFβ and rPKM2 treated NHLF and LX2 cells (Figures S5C and S5D). If apoptosis protection by EcPKM2 is mediated by PI3K activation, the effects would be abolished by PI3K inhibitor. As expected, the effects of rPKM2 on apoptosis protection of NHFL and LX2 cells were inhibited by a commercially available PI3K inhibitor (20 μg/mL) (Figures 6C andS5E). A well-known downstream target of activated PI3K is activation of NF-κB survival signaling (Dan et al., 2008). Clearly, NF-κB was highly activated in NHFL and LX2 cells in the presence of 50 nM rPKM2, and activation of NF-κB was abolished in the presence of the antibody LM609 or the inhibitors to PI3K or FAK (Figures 6D andS5F). The results indicate that the effects of EcPKM2 in protecting activated HSCs and myofibroblast from apoptosis are mediated via activation of NF-κB survival by FAK-PI3K signaling axis.
Figure 6.

EcPKM2 Activates FAK-PI3K signaling axis
(A) Cellular levels of phosphor-PI3K (IB:p-PI3K), PI3K (IB:PI3K), and phosphor-ERK (IB:p-ERK) in LX2 cells were analyzed by immunoblot. The cells were treated by TGFβ plus rPKM1 or rPKM2.
(B) Activation of PI3K in LX2 cells in the presence of indicated agents was measured by ELISA PI3K kit. The PI3K activities are presented as relative activity by defining the activities of PI3K in buffer treated cells as 1 (Control).
(C)Apoptosis of LX2 cells under treatment of indicated agents were measured by apoptosis kit. Cell apoptosis is presented as Relative apoptosis by defining apoptosis of cell treated with buffer (control) as 1.
(D) NF-kB activity in LX2 cells in the presence of indicated agents was measured using transcription assay kit. The NF-kB activity is present as Relative NF-kB activity by defining the measured NF-kB activity in buffer treated LX2 cells as 1.
(E) Cellular levels of PTEN (IB:PTEN) in LX2 cells treated with indicated agents were analyzed by immunoblot.
(F) Cellular levels of PTEN (IB:PTEN), collagen 1A1 (IB:collagen1a1), and Arg-1 (IB:Arg1) in LX2 cells were analyzed by immunoblot. The cells were treated with TGFβ and rPKM2. PTEN was exogenously expressed in the cells using adenoviral vector (Ad-PTEN). Ad-Null is empty viral vector.
(G) Cellular levels of Arg-1 mRNA in LX2 cells were analyzed by qRT-PCR.
(H) Secreted collagen in culture medium of LX-2 cells were analyzed by Sirius red assay (as in Figures 4C and 4D). The cells were treated with TGFβ and rPKM2. PTEN was exogenously expressed in the cells using adenoviral vector. Immunoblots of GAPDH (IB:GAPDH) in (A) and (F) and Actin (IB:Actin) in (E) are loading controls. Error bars in (B)–(D), (G), and (H) represent mean ± S.E.M. ∗p<0.05, ∗∗p<0.01, ∗∗∗p<0.001. ns: statistically no significance.
It is well established that PI3K – PTEN pathway regulates Arg-1 expression in macrophages (Carracedo and Pandolfi, 2008; Chalhoub and Baker, 2009). Reduction of PTEN in fibroblasts inhibits myofibroblast differentiation and αSMA expression and collagen synthesis (Liu et al., 2013; White et al., 2006). We reasoned whether EcPKM2 regulated Arg-1 expression and subsequent collagen expression in myofibroblasts by the same pathway via the FAK-PI3K signaling axis. Downregulation of PTEN plays an important role in myofibroblast differentiation. However, the role of PTEN in myofibroblast differentiation is TGF-β-independent (White et al., 2006). Thus, we probed the PTEN levels and activity in the rPKM2 treated myofibroblasts. Clearly, TGFβ had an effect on reduction of PTEN in NHLF and LX2. Addition of rPKM2, but not rPKM1, into culture medium in addition to the TGF-β treatment further reduced PTEN in the cells (Figures S5G and6E), suggesting that EcPKM2 might play a role in downregulation of PTEN in myofibroblasts during fibrosis progression, which subsequently controlled Arg-1 expression. Consistent with this notion, knockdown of integrin β3 and the antibody LM609 reduced Arg-1 in NHLF and LX2 cells that were treated with TGFβ and rPKM2 (see Figures S5C and S5D). To further test this conjecture, PTEN was exogenously expressed in NHLF and LX2 cells (Figures 6F andS5H). The cells were treated by TGFβ and rPKM2. Immunoblot and RT-PCR analyses demonstrated reduction in Arg-1 levels in the TGFβ and rPKM2 treated cells (Figures 6F, 6G, S5H, and S5I). Consistently, exogenous expression of PTEN abolished the effects of rPKM2 in increase of collagen synthesis and secretion in LX2 and NHLF cells (Figures 6H andS5J).
An antibody disrupting the PKM2 and integrin αvβ3 interaction reverses organ fibrosis
If EcPKM2 facilitates organ tissue fibrosis progression by protecting activated HSC from turnover and promoting proline synthesis and subsequent collagen production, it would be expected that an antibody that disrupts the PKM2-integrin interaction would abolish the effects of apoptosis protection and Arg-1 expression. This would suggest a novel strategy and target for organ-tissue fibrosis treatment. Thus, we employed a pair of rabbit monoclonal PKM2 antibodies (Figure S6A); one is active in disrupting the PKM2-integrin interaction (16-2), and the other (21-3) is not (Figure S6B). Liver fibrosis was induced by treatment with TAA-alcohol. At the end of fibrosis induction, purified antibodies from both hybridoma lines were administered at 4 mg/kg i.p. for 10 doses (Figure 7A). At the end of the treatment, animals were sacrificed. Body weight, liver weight, and outer surface of livers were examined. Antibody 16-2 treatment led to an increase in body weight and a decrease in liver weight compared to those of IgG and 21-3 treated groups (Figures 7B andS6C). Fibrosis features on the liver surface of 16-2 treated group were clearly reduced compared to those of IgG and 21-3 treated groups (Figure S6D). Examination of serum markers suggested a recovery from liver damages by 16-2 treatment (Figure S6E). Sirius red staining of liver sections revealed a decrease in collagen content and density, indicating resolve of collagen accumulation in fibrotic liver by the antibody 16-2 treatment (Figures 7C and 7D). IHC staining of αSMA indicated reduction in αSMA positive HSCs in fibrotic liver by the antibody 16-2 (Figures 7E and 7F).
Figure 7.

Antibody against PKM2 reversed organ/tissue fibrosis
(A) Schematic illustration the schedule of TAA/alcohol liver fibrosis induction and subsequent PKM2 antibody treatments.
(B) Liver weight of the animals at endpoint of the treatment.
(C–F) Representative images of Sirius red staining (C) and IHC staining of αSMA (E) of liver tissues from mice treated with indicated agents. The call-out in (C) is enlarged images showing the Sirius red staining feature. Quantitation of Sirius red staining (D) and IHC staining of αSMA (F).
(G) Schematic illustration of the schedule of bleomycin lung fibrosis induction and subsequent PKM2 antibody treatments.
(H) Lung weight of the animals at endpoint of the treatment.
(I–K) Representative images of Sirius red staining (I, Upper) and IHC staining of αSMA (I, Bottom) of lung sections from mice treated with indicated agents. Quantitation of Sirius red staining (J) and IHC staining of αSMA (K). In (D), (F), (J), and (K), quantification was calculated from measurements of 10 mice. Four randomly selected tissue sections per animal and three randomly selected view fields in each section were quantified. The quantity of Sirius red staining is presented as Sirius red+ % of total areas. The quantity of αSMA staining is presented as αSMA+ % of total areas.
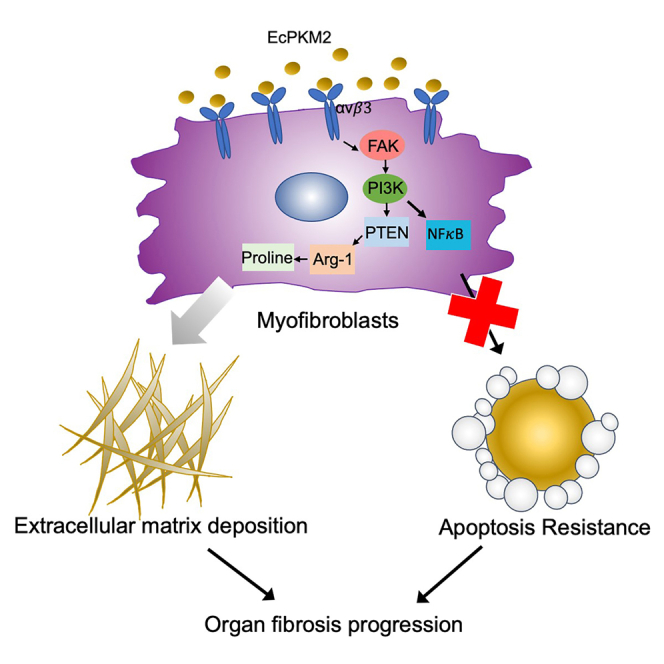
(L) Schematic illustration of mechanism of action of EcPKM2 in organ/tissue fibrosis progression. Error bars in (D), (F), (H), (J), and (K) represent mean ± S.E.M. Scale bars, 100 μm. ∗p<0.05, ∗∗p<0.01, ∗∗∗p<0.001.ns: statistically no significance.
The same anti-PKM2 antibody from clone 16-2 was tested with bleomycin induced lung fibrosis mouse model (Figure 7G). The antibody treatment led to an increase in body weight and a decrease in lung weight compared to those of IgG treated groups (Figures 7H andS6F). Fibrosis features on the lung surface of the antibody treated group were clearly reduced compared to those of IgG treated groups (Figure S6G). Sirius red staining and hydroxyproline assay of lung tissue sections and extracts revealed a substantial decrease in collagen contents and density, indicating reduction of collagen accumulation in fibrotic lung by the antibody treatment (Figures7I and 7J). IHC staining of αSMA indicated reduction of αSMA positive myofibrobalsts in fibrotic lung by the antibody (Figures 7I and 7K). From studies of both liver and lung fibrosis models, we conclude that antibody that disrupts the PKM2 and integrin αvβ3 interaction can potentially be an effective anti-fibrotic agent.
EcPKM2 protects myofibroblasts from apoptosis induction. We therefore analyzed whether the PKM2 neutralization antibody would specifically relieve myofibroblast apoptosis protection in fibrotic liver and lung. Evidently, with treatment of the antibody, significantly less activated lung myofibroblasts and more TUNEL staining were observed compared to the IgG treatment (Figures S7A and S7B). More αSMA and TUNEL co-staining was observed in the sections of fibrotic lungs from the mice that were induced fibrosis by bleomycin and subsequently treated with the antibody, suggesting more apoptosis of αSMA positive lung myofibroblasts (Figure S7C). Similarly, the PKM2 antibody led to less αSMA and more TUNEL staining. More αSMA and TUNEL co-staining was detected in fibrotic liver of the TAA-alcohol fibrosis model with the antibody treatment (Figures S7D and S7E). EcPKM2 upregulates Arg-1 in myofibroblasts to facilitate collagen production. We reasoned that the PKM2 neutralization antibody would abolish the effects of EcPKM2 in regulation of Arg-1 in myofibroblasts in fibrotic liver and lung. Treatment of bleomycin-induced lung fibrosis mice with the anti-PKM2 antibody decreased Arg-1 and αSMA staining and Arg-1/αSMA co-staining in the sections from fibrotic lungs (Figures S8A–S8C). Similarly, treatment of TAA-alcohol-induced liver fibrosis mice with the anti-PKM2 antibody also decreased the Arg-1/αSMA co-staining in the sections from fibrotic liver (Figures S8D–S8F). Significantly decreased Arg-1/αSMA co-staining suggests a decrease in Arg-1 expression in myofibroblasts in the fibrotic lung and liver.
Discussion
Our study revealed an unexpected role of EcPKM2 in facilitating organ tissue fibrosis progression by promoting myofibroblasts apoptosis resistance and upregulating Arg-1 expression via activating integrin αvβ3. Along with our previous observations that EcPKM2 facilitates tumor growth (Li et al., 2014) and cutaneous wound healing (Zhang et al., 2016) by promoting angiogenesis, we propose a theme that EcPKM2 may bridge the early inflammatory response to a later proliferation phase (fibrogenesis/angiogenesis) during tissue injury repair. During cutaneous wound repair, PKM2 is released from infiltrating neutrophils. The released PKM2 promotes angiogenesis and myofibroblasts differentiation to facilitate wound repair (Zhang et al., 2016). In the case of chronic wound and inflammation, PKM2 is upregulated in myofibroblasts which secrete high amounts of PKM2. The released EcPKM2 acts on the myofibroblasts in autocrine to protect them from turnover and to promote Arg-1 expression in the myofibroblasts, which in turn facilitates proline biosynthesis from arginine and subsequent collagen production. In the case of cancers, cancer cells and cancer-associated fibroblasts (CAF) secrete PKM2. Our studies demonstrated that EcPKM2 facilitates tumor angiogenesis (Li et al., 2014), similarly to in the case of cutaneous wound repair. It is intriguing to analyze whether and how EcPKM2 acts on CAF in tumors. EcPKM2 promotes regeneration in wound repair. This functionality may be hijacked by cancers to facilitate cancer progression. EcPKM2 exert its action by interacting with integrin αvβ3 and activating the integrin signaling. Activation of integrin signaling by EcPKM2 triggers activation of FAK-PI3K signaling axis. This signaling axis upregulates NF-κB to turn on myofibroblast survival. This signaling axis also suppresses PTEN to upregulate Arg-1, which promotes proline and collagen synthesis (Figure 7L). Quiescent fibroblasts and HSCs do not express or express low levels of integrin αvβ3. Activation of fibroblasts and HSC by TGFβ upregulates integrin αvβ3 expression (Henderson et al., 2013) (Figures S9A and S9B). Consistently, IHC staining of integrin β3 in sections from liver fibrosis and pulmonary fibrosis patients reveals that the integrin is not expressed in normal healthy liver and lung tissues, while the integrin is highly expressed in liver and pulmonary fibrosis disease tissues (Figures S9C–S9F). The observations explain why EcPKM2 does not initial fibrosis induction and by itself does not induce myofibroblast differentiation due to no integrin αvβ3, the EcPKM2 target, expression in quiescent fibroblasts and stellate cells. EcPKM2 is an auxiliary factor in promoting fibrosis progression in the case of chronic conditions. It is well documented that cancer cells secrete PKM2. PKM2 in blood circulation has been used as a prognosis marker (Kumar et al., 2007). We showed that neutrophils release PKM2 by degranulation in early cutaneous wound healing (Zhang et al., 2016). Here, we demonstrate that myofibroblasts express and secrete PKM2 in fibrotic organs and tissues. It is intriguing whether any other cell types express and secrete PKM2.
A number of previous studies show that activation of several pairs of integrin facilitates fibrosis progression, including αvβ6, αvβ8, and αvβ1. It is believed that the effects of these integrin pairs on fibrosis progression are mediated via activation of latent TGFβ (Henderson et al., 2013; Henderson and Sheppard, 2013; Munger et al., 1999; Reed et al., 2015). Thus, we asked whether EcPKM2 acted by a similar mechanism in facilitating fibrosis progression. We tested the effects of a TGFβ neutralizing antibody and a TGFβR inhibitor on the activity of EcPKM2. Clearly, the function of EcPKM2 was TGFβ signaling independent (Figure S9G). Furthermore, TGFβR was not activated in LX2 cells upon rPKM2 treatment (Figure S9H). We would conclude that the function of EcPKM2 in promoting liver and lung fibrosis progression is not mediated via activation of latent TGFβ. It is reported that integrin β3 KO mice are not protected from CCl4 induced liver fibrosis (Henderson et al., 2013). The observation seems not consistent with our model that EcPKM2 acts via interacting/activating integrin αvβ3 in promoting organ/tissue fibrosis progression. However, it is known that compensatory responses frequently occur in KO models when a protein of interest is deleted during development (Yang and Hynes, 1996; Yang et al., 1996). It was believed that integrin αvβ3 in angiogenic endothelial cells play an important role in angiogenesis (Brooks et al., 1994). However, the observation that integrin β3 KO mice demonstrate enhanced angiogenesis questions the function role(s) of the integrin in angiogenesis (Reynolds et al., 2002). Using an integrin β3 signal deficiency mutant knock-in model, Mahabeleshwar, G.H. et al. show that integrin β3 signaling is essential for pathological angiogenesis. Presence of β3 signal deficiency mutant may prevent developmental compensatory responses in the β3 KO model. It is plausible that the function(s) of integrin β3 in tissue fibrosis, as in angiogenesis, is compensated during development in the β3 KO mice (Mahabeleshwar et al., 2006). Functional roles of intracellular and subcellular localization of PKM1/PKM2 in regulation of metabolism adjustment and other cellular processes in various cell types have been extensively characterized (Dayton et al., 2016). It is also well documented that PKM2 is released into extracellular space in various pathological conditions (Hathurusinghe et al., 2007; Jeffery et al., 2009; Staib et al., 2006). However, the functional significance of extracellular PKM2 is less well studied. Wang, C. et al. report that secreted PKM2 interacts with integrin β1 on cancer cells to promote cancer metastasis. The binding affinity kd of rPKM2 to integrin β1 is measured at around the micromolar scale in the report (Wang et al., 2020). Our SPR binding measurement also showed a weak affinity of rPKM2 to integrin α1β1 with an estimated kd in μM range (consistent with the report of Wang C. et al.), almost 10,000 folds weaker than that of rPKM2 to integrin αvβ3 (see Figure 5C). Cell attachment assays demonstrated that integrin αvβ6 and αvβ1 expressing CHO cells did not attach or only weakly attached to rPKM2 coated plates (see Figure 5D), indicating that PKM2 did not interact or weakly interacted with these integrin pairs.
EcPKM2 facilitates liver and lung fibrosis progression via a novel mechanism of action by activation of integrin αvβ3. It is intriguing that an antibody against PKM2 that disrupts the PKM2-integrin αvβ3 interaction is very effective in reversing liver and lung fibrosis, indicating a potential anti-fibrotic strategy and target. Our study with the PKM2 antibody provided the proof of principle evidence for targeting EcPKM2 as a treatment of liver and lung fibrosis.
Limitations of the study
Our data demonstrate that EcPKM2 facilitates progression of liver and lung fibrosis. In fact, sustained activation of stellate cells and fibroblasts and apoptosis resistance of these activated myofibroblasts are common features of many human organ and tissue fibrosis diseases associated with chronic inflammation. Upregulation of biosynthesis of glycine/proline and subsequent collagen by myofibroblasts is a common feature of fibrosis of all organs. Thus, further study is needed to understand whether EcPKM2 plays a similar role in fibrosis progression of other organs and tissue. Cancer associated fibroblasts (CAF) share many common characteristics with myofibroblasts. It is well established that EcPKM2 presents in cancer of all types. Additional work can be done to elucidate whether EcPKM2 plays a similar role promoting survival of CAF and facilitating collagen synthesis and secretion in CAF, thus facilitates cancer progression. Release of EcPKM2 and elevation in integrin αvβ3 expression are closely associated with a number of pathological conditions, particularly various inflammation-associated chronic diseases and cancers. It is plausible that targeted disruption of PKM2 and integrin αvβ3 interaction may be beneficial for treatment of these human diseases. Besides, our data suggest that EcPKM2 interacts with integrin αvβ3, but how they bind with each other is not clear. In addition, integrin αvβ3 can be expressed by various cell types, but whether EcPKM2 has an effect on other integrin αvβ3 -expressing cells requires further investigations.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Arginase 1 | Santa Cruz Biotechnology | Cat#sc-20150 RRID: AB_2058955 |
| Integrin αVβ3 (LM609) | Millipore | Cat#MAB1976 RRID: AB_2296419 |
| Col1a1 | Santa Cruz Biotechnology | Cat#sc-293182 RRID: AB_2797597 |
| PKM2 | Cell Signaling Technology | Cat#4053 RRID: AB_1904096 |
| Chemicals, peptides, and recombinant proteins | ||
| FAK inhibitor 14 | Sigma Aldrich | SML0837 |
| LY294002 | Sigma Aldrich | L9908 |
| Fas ligand | Sigma Aldrich | SRP3036 |
| Cycloheximide | Sigma Aldrich | 01810 |
| TGF-β1 | PeproTech | 100-21C |
| Bleomycin Sulfate | BioTang | RB003 |
| Experimental models: Cell lines | ||
| Normal Human Lung Fibroblast (NHLF) | Lonza | CC-2512 |
| LX-2 Human Hepatic Stellate Cell Line | Millipore | SCC064 |
| Human Hepatic Stellate Cells | Zen-Bio | HP-F-S |
| Other | ||
| ATP EnerCountCell Growth Assay Kit | Codex Biosolutions | CB-80551 |
| Annexin V Apoptosis Detection Kit FITC | Thermo Fisher | 88-8005-74 |
| NFkB p65 Transcription Factor Assay Kit | Millipore | 70-520 |
| Picro Sirus red staining kit | IHC world | IW-3012 |
| NovaUltra H&E Stain Kit | IHC World | IW-3100 |
| Purified PI3K Class I Lipid Kinases | Promega | V1721 |
| ADP-Glo™Kinase Assay with Lipid Kinase Substrates | Promega | V1781 |
| TdT In Situ Apoptosis Detection Kit-Fluorescein | R&D | 4812-30-K |
| Proline Assay Kit | Mybiosource | MBS8305379 |
| Ornithine Assay Kit | Abcam | ab252903 |
| Arginine Assay Kit | Abcam | ab252892 |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Zhi-Ren Liu (zliu8@gsu.edu).
Materials availability
The study did not generate any unique reagents.
Experimental model and subject details
Patient tissue analyses were carried out under the guidelines of NIH. All tissue samples are de-identified. It falls under IRB exemption 4. Samples were sectioned and analyzed by different staining. Liver and lung disease tissue arrays were purchased from US Biomax Inc. The study was approved by Institutional Review Board (IRB).
Liver fibrosis/cirrhosis and lung fibrosis induction and treatments
All animal experiments were carried out in accordance with the guidelines of, and was approved by, IACUC of Georgia State University. Liver fibrosis: 7-week-old male Balb/c mice were treated with appropriate agents (TAA 100 mg/kg for 12 days followed by 200 mg/kg i.p. injection.10% alcohol was added into animal drinking water) for the indicated time periods (as shown in figure). At the end of the treatments, animals were sacrificed. Balb/c mice were used because intensive liver fibrosis was developed with the mouse stain in our hands. Lung fibrosis: 7-week old male C57BL/6J mice were administrated 25mg/kg bleomycin twice a week for 5 weeks and treated with appropriate agents for the indicated time period. Organs and blood samples were collected for subsequent analyses. Statistical analyses were done in comparison to the control group.
Method details
Reagents, antibodies, cell lines, expression cDNAs, and protein expression/purifications
Commercial primary cells, cell lines, reagents, antibodies, and PCR primers are listed in Table S1. Rabbit monoclonal antibodies (16-2 and 21-3) against PKM2 was raised using recombinant PKM2 expressed/purified from E. coli.as an antigen. IgG were purified from cell culture of hybridoma over a protein G column.
The cDNAs that encode human PKM2 and PKM1 were purchased from Addgenes. The cDNAs were subcloned into bacterial expression vector pET-32a. The recombinant proteins were purified from bacterial lysates by a two-column procedure as in previous reports (Gao et al., 2012; Li et al., 2014). The cDNAs for integrins αv, αIIb, β1, β6, and β3 were purchased from Addgene. The cDNAs were subcloned into a mammalian expression vector pCDNA3.1 for exogenous expression of different integrin pairs in cultured cells. Viral vector for expression of PTEN were purchased from Vector Biolabs.
Chemical crosslinking using glutaraldehyde as crosslinkers
His-rPKM2 or His-rPKM1 (1 μM) was incubated with LX2 cells (5 x 104 cells in 1 ml) in culture dish. The cells were activated by TGFβ 5 nM. The freshly prepared glutaraldehyde (1 mM) was added into the cell culture to final concentration of 50 μM. After 1 h incubation on ice, the crosslinking was stopped by adding in 200 μM Tris-HCl pH = 7.4 buffer. The His-rPKM2-crosslinked or His-rPKM1-crosslinked cells were collected by centrifugation. After extensive washes, cell lysate were prepared from the crosslinked cells using a membrane protein extraction kit. The crosslinked proteins were pulled down by Ni-NTA beads. After elution from the Ni-NTA beads, precipitates were dialysed against HEPES buffer pH = 6.9. A small portion of the recovered PKM2 or PKM1 crosslinks was analyzed by immunoblot using anti-PKM2/PKM1 antibody to identify the rPKM2/rPKM1 crosslinked protein. The rest of His-tag pull-down were separated by SDS-PAGE and stained by gel-code blue. The band corresponding to the rPKM2 crosslinks was sliced out and digested by trypsin. The digestion mixture was subsequently analyzed by MALDI-tof/tof to identify the crosslinked proteins.
Tissue section and cell staining
Sirius red
Sirius Red staining was carried out using NovaUltraTM Sirius Red Stain Kit from IHC WORLD following the instruction of vendor.
IHC and IF
IHC and IF staining procedures were similar to those of previous reports (Turaga et al., 2016) (Zhang et al., 2016).
Staining quantitation
Quantitation of Sirius red, IHC, and IF staining (including patient tissues and tissues from animal experiments) was carried out using ImageJ. Quantifications are positive stain areas in each view field or fold change by comparing with the controls unless otherwise specified in the figures and legends. All quantitation results were means of randomly selected 3 view fields per section, 5 sections per animal, and 6–10 mice per experimental group, unless otherwise specified in the figures and legends.
Apoptosis assay, PI3K and NFκB transcription activity assay, SPR binding, binding affinity from SPR, cell attachment, cell migration assays were performed by the procedures similar to our previous reports (Turaga et al., 2016; Zhang et al., 2016).
SPR
The commercial recombinant integrins α1β1, αvβ3, and αIIbβ3 were diluted to a concentration of 100 μg ml-1 and immobilized on to the CM5 chip with response units around 200. Then rPKM2 bindings were carried out at various concentrations with the running buffer of 1x HBSS 10 mM HEPES, 150 mM NaCl, 5 mM MgCl2, pH7.4. The flow rate was 50 μl min-1, and the binding time was 12 min. Kds and binding stoichiometry were deduced by calculation via binding curves of various concentrations.
Cell attachment assays
The cells were cultured overnight under standard conditions. Next day, different cells (with appropriate cell numbers) were transferred to a new plate with wells that were coated with different proteins (indicated in the figures) with fresh medium with the addition of appropriate agents (indicated in the figures). The cells were further cultured for 35 min and washed three times gently. The attached cells were either directly counted or lysed. The cell lysates were then measured to determine the amounts of attached cells. In all attachment assays, the attachment is presented as total number of cells attached to the plate per microscopic view field (calculated from average of three view fields) or as relative attachment by comparing to the attached cells of a reference.
PI3K and NFκB activities and transcription activity assays
PI3K activity was measured by ADP-GloTM Lipid Kinase Systems from Promega by following vendor’s instruction. Briefly, cells were activated with TGF-β or rPKM2 for two days, and cell lysates were prepared using RIPA buffer. PI3K in cell lysates were pulled-down by PI3K antibody and incubated with ATP and lipid substrate included in the kit. PI3K activity was measured by monitoring increase in ADP levels. Activity of NFκB was measured by NFκB p50/p65 EZ-TFA transcription factor assay kit. The flanked DNA binding consensus sequence for NFκB was coated on the plate. After incubation with cell extracts, activated NFκB was retained on the plate. The bound NFκB transcription factors p50 and p65 were analyzed using specific antibodies against p50 and p65. For assays with FAK inhibitor FI14 and PI3K inhibitor, the FAK inhibitor FI14 was used in 2.5 μM and the PI3KIN was used at 20 μg/ml (Cabrita et al., 2011; Min et al., 2018).
Apoptosis
Cells were cultured overnight. The cells were shifted to fresh medium next morning. Appropriate agents were added into the medium to indicated final concentrations. After 24 h incubation, cell apoptosis was determined by cell counting or by TUNEL kit stain using untreated cells as a reference. The apoptosis measurements were repeated five times in each experiment.
Amino acids isolation and mass spectrometry analysis
1x106 cells or 100 mg tissues were used for amino acid analysis with LC-MS. Cells were washed with 0.9% NaCl, scraped off and centrifuged at 1000 rpm, 4°C for 10 min, after which the pellets were saved for the next step. 100 mg of tissues were weighed and homogenized in liquid nitrogen. The tissue dry powder was collected for the next step. 50% pre-cold Methonal was added for fixation. Then chloroform was added to lyse the cells at 4°C for 30 min on the rotator. The lysates were centrifuged at 14000 rpm, 4°C for 10 min and the top aqueous layer was used as samples for LC-MS analysis with a Sciex API3200 esi-triple quadrupole mass spectrometer coupled with an Agilent 1200 HPLC. 10 μl of each standard and sample was injected into the system. Amino Acid Standard H (Thermo Fisher Cat. # 20088) was used as standard. The MS parameters used are as follows: Ion source (IS) voltage, 5400 v, ion source temperature 450C, collision energy 5v. Analyst 1.5.1 was used for data analysis.
RNA isolation and RT-qPCR analysis
The total RNA of cells was isolated using the RNeasy Plus mini kit (Qiagen) according to the manufacturer’s instructions. For RT-qPCR, 0.5 μg of total RNA was reverse-transcribed to cDNA using Maxima First Strand cDNA Synthesis Kit (Thermo Fisher K1641). Real-time qPCR was then performed using 2 μl of cDNA using Luna Universal qPCR Master Mix (NEB M3003) with SYBR® Green as the fluorescent dye and the 7500 Fast Real-Time PCR System (Life Technologies). The primers were used at a final concentration of 250 nM. β-actin was used as the reference gene. The relative transcript abundance of target genes compared with the reference gene was expressed in the cycle threshold (ΔCt) as ΔCt = Ct(target) – Ct(reference). The relative difference in transcript levels of the treated group compared with the control group was expressed as ΔΔCt = ΔCt(treated) –ΔCt(control). The transcript levels are represented as relative fold changes.
Measurement of collagen in cell culture media
The collagen contents in the cell culture medium were determined with Sirius Red (IHC world), (adopting from (Chen et al., 2013; Kliment et al., 2011)). Equal volumes of culture medium were added to 1 ml of Sirius Red dye, followed by 1 h incubation at room temperature in a shaker. After centrifugation at 12,000 rpm for 10 min, the pellets of collagen-dye complex were washed with acidified water (5% acetic acid) twice. Then 250 μl 0.1 M NaOH was added and incubated for 5 min. The contents of tubes were transferred to a 96-well plate and read at 540 nm on an MD plate reader (Molecular Dynamics). Collagen contents were calculated using a standard curve with type 1 collagen (Sigma).
Quantification and statistical analyses
All analyses were performed using GraphPad Prism8. Comparisons were made by two-tailed Student’s t-test when comparing two experimental groups. For two-group comparison of unequal variance, two-tailed Welch’s t-test was used. For a comparison between more than two groups, the statistics analyses were performed using one-way ANOVA with Tukey’s test. P values <0.05 are regarded as statistically significant. In all figures and tables, NS means p > 0.05 and statistically insignificant, ∗ means p < 0.05, ∗∗ means P < 0.01, and ∗∗∗ means P < 0.001.
Acknowledgments
We thank Ravi Chakra Turaga, Malvika Sharma, and Ganesh Satyanarayana for excellent suggestions for our studies. This work is supported in part by research grants fromNational Institutes of Health (CA175112, CA118113, CA178730) andGeorgia Cancer Coalition to ZR. Liu.
Author contributions
ZR Liu conceptualized, planned, and coordinated the study. ZR Liu wrote the paper. H. Han, Y. Zhang, and G. Peng conducted most of the experiments, data analyses, and participated in paper writing, L. Li, Y. Yuan prepared recombinant rPKM2/rPKM1 and integrin αvβ3 expression and purification. J. Yang helped SPR and other in vitro binding studies. Y. Xu helped in data statistical analyses. All authors discussed the results and commented on the paper.
Declaration of interests
The authors declare no competing conflict of interests.
Published: October 22, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103165.
Supplementalinformation
. Cell lines, reagents, antibodies, PCR primers, and kits used in the study. Related to STAR Methods
Data and code availability
-
•
This published article includes all datasets generated or analyzed during this study.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Angiari S., Runtsch M.C., Sutton C.E., Palsson-McDermott E.M., Kelly B., Rana N., Kane H., Papadopoulou G., Pearce E.L., Mills K.H.G. Pharmacological activation of pyruvate kinase M2 inhibits CD4(+) T cell pathogenicity and suppresses autoimmunity. Cell Metab. 2020;31:391–405.e398. doi: 10.1016/j.cmet.2019.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atzori L., Poli G., Perra A. Hepatic stellate cell: a star cell in the liver. Int. J. Biochem. CellBiol. 2009;41:1639–1642. doi: 10.1016/j.biocel.2009.03.001. [DOI] [PubMed] [Google Scholar]
- Bataller R., Brenner D.A. Liver fibrosis. J. Clin. Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks P.C., Clark R.A., Cheresh D.A. Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science. 1994;264:569–571. doi: 10.1126/science.7512751. [DOI] [PubMed] [Google Scholar]
- Cabrita M.A., Jones L.M., Quizi J.L., Sabourin L.A., McKay B.C., Addison C.L. Focal adhesion kinase inhibitors are potent anti-angiogenic agents. Mol. Oncol. 2011;5:517–526. doi: 10.1016/j.molonc.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carracedo A., Pandolfi P.P. The PTEN-PI3K pathway: of feedbacks and cross-talks. Oncogene. 2008;27:5527–5541. doi: 10.1038/onc.2008.247. [DOI] [PubMed] [Google Scholar]
- Chalhoub N., Baker S.J. PTEN and the PI3-kinase pathway in cancer. Annu. Rev. Pathol. 2009;4:127–150. doi: 10.1146/annurev.pathol.4.110807.092311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C., Yang S., Zhang M., Zhang Z., Zhang B., Han D., Ma J., Wang X., Hong J., Guo Y. In vitro Sirius Red collagen assay measures the pattern shift from soluble to deposited collagen. Adv. Exp. Med. Biol. 2013;765:47–53. doi: 10.1007/978-1-4614-4989-8_7. [DOI] [PubMed] [Google Scholar]
- Cluzel C., Saltel F., Lussi J., Paulhe F., Imhof B.A., Wehrle-Haller B. The mechanisms and dynamics of (alpha)v(beta)3 integrin clustering in living cells. J. Cell Biol. 2005;171:383–392. doi: 10.1083/jcb.200503017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damasceno L.E.A., Prado D.S., Veras F.P., Fonseca M.M., Toller-Kawahisa J.E., Rosa M.H., Publio G.A., Martins T.V., Ramalho F.S., Waisman A. PKM2 promotes Th17 cell differentiation and autoimmune inflammation by fine-tuning STAT3 activation. J. Exp. Med. 2020;217:e20190613. doi: 10.1084/jem.20190613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dan H.C., Cooper M.J., Cogswell P.C., Duncan J.A., Ting J.P., Baldwin A.S. Akt-dependent regulation of NF-κB is controlled by mTOR and Raptor in association with IKK. Genes Dev. 2008;22:1490–1500. doi: 10.1101/gad.1662308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayton T.L., Jacks T., Vander Heiden M.G. PKM2, cancer metabolism, and the road ahead. EMBO Rep. 2016;17:1721–1730. doi: 10.15252/embr.201643300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong G., Mao Q., Xia W., Xu Y., Wang J., Xu L., Jiang F. PKM2 and cancer: the function of PKM2 beyond glycolysis. Oncol. Lett. 2016;11:1980–1986. doi: 10.3892/ol.2016.4168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbers J.R., van Unnik J.A., Rijksen G., van Oirschot B.A., Roholl P.J., Oosting J., Staal G.E. Pyruvate kinase activity and isozyme composition in normal fibrous tissue and fibroblastic proliferations. Cancer. 1991;67:2552–2559. doi: 10.1002/1097-0142(19910515)67:10<2552::aid-cncr2820671027>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- Elsharkawy A.M., Oakley F., Mann D.A. The role and regulation of hepatic stellate cell apoptosis in reversal of liver fibrosis. Apoptosis. 2005;10:927–939. doi: 10.1007/s10495-005-1055-4. [DOI] [PubMed] [Google Scholar]
- Forty E., Mercer P., Anastrasiou D., Chambers R. Targetingpyruvate kinase M2 isoform inhibits proliferation of human lung fibroblasts. Eur. Respir. J. 2016;48:PA3893. [Google Scholar]
- Gao X., Wang H., Yang J.J., Chen J., Jie J., Li L., Zhang Y., Liu Z.R. Reciprocal regulation of protein kinase and pyruvate kinase activities of pyruvate kinase m2 by growth signals. J. Biol. Chem. 2013;288:15971–15979. doi: 10.1074/jbc.M112.448753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X., Wang H., Yang J.J., Liu X., Liu Z.R. Pyruvate kinase m2 regulates gene transcription by acting as a protein kinase. Mol.Cell. 2012;45:598–609. doi: 10.1016/j.molcel.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habiel D.M., Moreira A.P., Ismailoglu U.B., Dunleavy M.P., Cavassani K.A., van Rooijen N., Coelho A.L., Hogaboam C.M. TRAIL-dependent resolution of pulmonary fibrosis. Mediators Inflamm. 2018;2018:7934362. doi: 10.1155/2018/7934362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacker H.J., Steinberg P., Bannasch P. Pyruvate kinase isoenzyme shift from L-type to M2-type is a late event in hepatocarcinogenesis induced in rats by a choline-deficient/DL-ethionine-supplemented diet. Carcinogenesis. 1998;19:99–107. doi: 10.1093/carcin/19.1.99. [DOI] [PubMed] [Google Scholar]
- Hathurusinghe H.R., Goonetilleke K.S., Siriwardena A.K. Current status of tumor M2 pyruvate kinase (tumor M2-PK) as a biomarker of gastrointestinal malignancy. Ann. Surg. Oncol. 2007;14:2714–2720. doi: 10.1245/s10434-007-9481-x. [DOI] [PubMed] [Google Scholar]
- Henderson N.C., Arnold T.D., Katamura Y., Giacomini M.M., Rodriguez J.D., McCarty J.H., Pellicoro A., Raschperger E., Betsholtz C., Ruminski P.G. Targeting of alphav integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat. Med. 2013;19:1617–1624. doi: 10.1038/nm.3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson N.C., Sheppard D. Integrin-mediated regulation of TGFbeta in fibrosis. Biochim.Biophys.Acta. 2013;1832:891–896. doi: 10.1016/j.bbadis.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hessien M.H., El-Sharkawi I.M., El-Barbary A.A., El-Beltagy D.M., Snyder N. Non-invasive index of liver fibrosis induced by alcohol, thioacetamide and Schistosomal infection in mice. BMC Gastroenterol. 2010;10:53. doi: 10.1186/1471-230X-10-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffery J., Lewis S.J., Ayling R.M. Fecal dimeric M2-pyruvate kinase (tumor M2-PK) in the differential diagnosis of functional and organic bowel disorders. Inflamm.Bowel Dis. 2009;15:1630–1634. doi: 10.1002/ibd.20946. [DOI] [PubMed] [Google Scholar]
- Kliment C.R., Englert J.M., Crum L.P., Oury T.D. A novel method for accurate collagen and biochemical assessment of pulmonary tissue utilizing one animal. Int. J. Clin. Exp. Pathol. 2011;4:349–355. [PMC free article] [PubMed] [Google Scholar]
- Kumar Y., Tapuria N., Kirmani N., Davidson B.R. Tumour M2-pyruvate kinase: a gastrointestinal cancer marker. Eur. J. Gastroenterol.Hepatol. 2007;19:265–276. doi: 10.1097/MEG.0b013e3280102f78. [DOI] [PubMed] [Google Scholar]
- Li L., Zhang Y., Qiao J., Yang J.J., Liu Z.R. Pyruvate kinase M2 in blood circulation facilitates tumor growth by promoting angiogenesis. J. Biol. Chem. 2014;289:25812–25821. doi: 10.1074/jbc.M114.576934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim M.P., Devi L.A., Rozenfeld R. Cannabidiol causes activated hepatic stellate cell death through a mechanism of endoplasmic reticulum stress-induced apoptosis. Cell Death Dis. 2011;2:e170. doi: 10.1038/cddis.2011.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S., Parapuram S.K., Leask A. Fibrosis caused by loss of PTEN expression in mouse fibroblasts is crucially dependent on CCN2. Arthritis Rheum. 2013;65:2940–2944. doi: 10.1002/art.38121. [DOI] [PubMed] [Google Scholar]
- Luo W., Hu H., Chang R., Zhong J., Knabel M., O'Meally R., Cole R.N., Pandey A., Semenza G.L. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell. 2011;145:732–744. doi: 10.1016/j.cell.2011.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahabeleshwar G.H., Feng W., Phillips D.R., Byzova T.V. Integrin signaling is critical for pathological angiogenesis. J. Exp. Med. 2006;203:2495–2507. doi: 10.1084/jem.20060807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min S.Y., Ha D.S., Ha T.S. Puromycin aminonucleoside triggers apoptosis in podocytes by inducing endoplasmic reticulum stress. Kidney Res. Clin.Pract. 2018;37:210–221. doi: 10.23876/j.krcp.2018.37.3.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeller A., Ask K., Warburton D., Gauldie J., Kolb M. The bleomycin animal model: a useful tool to investigate treatment options for idiopathic pulmonary fibrosis? Int. J. Biochem.Cell Biol. 2008;40:362–382. doi: 10.1016/j.biocel.2007.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira R.K. Hepatic stellate cells and liver fibrosis. Arch. Pathol.Lab. Med. 2007;131:1728–1734. doi: 10.5858/2007-131-1728-HSCALF. [DOI] [PubMed] [Google Scholar]
- Munger J.S., Huang X., Kawakatsu H., Griffiths M.J., Dalton S.L., Wu J., Pittet J.F., Kaminski N., Garat C., Matthay M.A. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96:319–328. doi: 10.1016/s0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- Puche J.E., Saiman Y., Friedman S.L. Hepatic stellate cells and liver fibrosis. Compr. Physiol. 2013;3:1473–1492. doi: 10.1002/cphy.c120035. [DOI] [PubMed] [Google Scholar]
- Reed N.I., Jo H., Chen C., Tsujino K., Arnold T.D., DeGrado W.F., Sheppard D. The alphavbeta1 integrin plays a critical in vivo role in tissue fibrosis. Sci.Transl.Med. 2015;7:288ra279. doi: 10.1126/scitranslmed.aaa5094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds L.E., Wyder L., Lively J.C., Taverna D., Robinson S.D., Huang X., Sheppard D., Hynes R.O., Hodivala-Dilke K.M. Enhanced pathological angiogenesis in mice lacking beta3 integrin or beta3 and beta5 integrins. Nat. Med. 2002;8:27–34. doi: 10.1038/nm0102-27. [DOI] [PubMed] [Google Scholar]
- Staib P., Hoffmann M., Schinkothe T. Plasma levels of tumor M2-pyruvate kinase should not be used as a tumor marker for hematological malignancies and solid tumors. Clin.Chem.Lab.Med. CCLM/FESCC. 2006;44:28–31. doi: 10.1515/CCLM.2006.006. [DOI] [PubMed] [Google Scholar]
- Stupack D.G., Puente X.S., Boutsaboualoy S., Storgard C.M., Cheresh D.A. Apoptosis of adherent cells by recruitment of caspase-8 to unligated integrins. J.Cell Biol. 2001;155:459–470. doi: 10.1083/jcb.200106070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tani N., Higashiyama S., Kawaguchi N., Madarame J., Ota I., Ito Y., Ohoka Y., Shiosaka S., Takada Y., Matsuura N. Expression level of integrin alpha 5 on tumour cells affects the rate of metastasis to the kidney. Br.J.Cancer. 2003;88:327–333. doi: 10.1038/sj.bjc.6600710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsumi H., Tani K., Fujii H., Miwa S. Expression of L- and M-type pyruvate kinase in human tissues. Genomics. 1988;2:86–89. doi: 10.1016/0888-7543(88)90112-7. [DOI] [PubMed] [Google Scholar]
- Turaga R.C., Yin L., Yang J.J., Lee H., Ivanov I., Yan C., Yang H., Grossniklaus H.E., Wang S., Ma C. Rational design of a protein that binds integrin alphavbeta3 outside the ligand binding site. Nat. Commun. 2016;7:11675. doi: 10.1038/ncomms11675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vo A.H., Swaroop A., Liu Y., Norris Z.G., Shavit J.A. Loss of fibrinogen in zebrafish results in symptoms consistent with human hypofibrinogenemia. PLoS One. 2013;8:e74682. doi: 10.1371/journal.pone.0074682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallach-Dayan S.B., Elkayam L., Golan-Gerstl R., Konikov J., Zisman P., Dayan M.R., Arish N., Breuer R. Cutting edge: FasL(+) immune cells promote resolution of fibrosis. J. Autoimmun. 2015;59:67–76. doi: 10.1016/j.jaut.2015.02.006. [DOI] [PubMed] [Google Scholar]
- Wang C., Zhang S., Liu J., Tian Y., Ma B., Xu S., Fu Y., Luo Y. Secreted pyruvate kinase M2 promotes lung cancer metastasis through activating the integrin Beta1/FAK signaling pathway. Cell Rep. 2020;30:1780–1797.e6. doi: 10.1016/j.celrep.2020.01.037. [DOI] [PubMed] [Google Scholar]
- Wegener K.L., Campbell I.D. Transmembrane and cytoplasmic domains in integrin activation and protein-protein interactions (review) Mol.Membr.Biol. 2008;25:376–387. doi: 10.1080/09687680802269886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiskirchen R., Weiskirchen S., Tacke F. Organ and tissue fibrosis: molecular signals, cellular mechanisms and translational implications. Mol. Aspects Med. 2019;65:2–15. doi: 10.1016/j.mam.2018.06.003. [DOI] [PubMed] [Google Scholar]
- White E.S., Atrasz R.G., Hu B., Phan S.H., Stambolic V., Mak T.W., Hogaboam C.M., Flaherty K.R., Martinez F.J., Kontos C.D. Negative regulation of myofibroblast differentiation by PTEN (phosphatase and tensin homolog deleted on chromosome 10) Am. J. Respir. Crit. Care Med. 2006;173:112–121. doi: 10.1164/rccm.200507-1058OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winau F., Quack C., Darmoise A., Kaufmann S.H. Starring stellate cells in liver immunology. Curr.Opin.Immunol. 2008;20:68–74. doi: 10.1016/j.coi.2007.10.006. [DOI] [PubMed] [Google Scholar]
- Wu G., Morris S.M., Jr. Arginine metabolism: nitric oxide and beyond. Biochem. J. 1998;336:1–17. doi: 10.1042/bj3360001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K., Noguchi T., Matsuda T., Takenaka M., Monaci P., Nicosia A., Tanaka T. Identification and characterization of hepatocyte-specific regulatory regions of the rat pyruvate kinase L gene.The synergistic effect of multiple elements. J. Biol. Chem. 1990;265:19885–19891. [PubMed] [Google Scholar]
- Yang J.T., Hynes R.O. Fibronectin receptor functions in embryonic cells deficient in alpha 5 beta 1 integrin can be replaced by alpha V integrins. Mol. Biol.Cell. 1996;7:1737–1748. doi: 10.1091/mbc.7.11.1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.T., Rando T.A., Mohler W.A., Rayburn H., Blau H.M., Hynes R.O. Genetic analysis of alpha 4 integrin functions in the development of mouse skeletal muscle. J. Cell Biol. 1996;135:829–835. doi: 10.1083/jcb.135.3.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W., Xia Y., Hawke D., Li X., Liang J., Xing D., Aldape K., Hunter T., Alfred Yung W.K., Lu Z. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell. 2012;150:685–696. doi: 10.1016/j.cell.2012.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W., Xia Y., Ji H., Zheng Y., Liang J., Huang W., Gao X., Aldape K., Lu Z. Nuclear PKM2 regulates beta-catenin transactivation upon EGFR activation. Nature. 2011;480:118–122. doi: 10.1038/nature10598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin C., Evason K.J., Asahina K., Stainier D.Y. Hepatic stellate cells in liver development, regeneration, and cancer. J. Clin. Invest. 2013;123:1902–1910. doi: 10.1172/JCI66369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Li L., Liu Y., Liu Z.R. PKM2 released by neutrophils at wound site facilitates early wound healing by promoting angiogenesis. Wound Repair Regen. 2016;24:328–336. doi: 10.1111/wrr.12411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z., Deng X., Liu Y., Liu Y., Sun L., Chen F. PKM2, function and expression and regulation. Cell Biosci. 2019;9:52. doi: 10.1186/s13578-019-0317-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
. Cell lines, reagents, antibodies, PCR primers, and kits used in the study. Related to STAR Methods
Data Availability Statement
-
•
This published article includes all datasets generated or analyzed during this study.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.