

Abstract
Often referred to as the bridge between innate and adaptive immunity, dendritic cells (DCs) are professional antigen‐presenting cells (APCs) that constitute a unique, yet complex cell system. Among other APCs, DCs display the unique property of inducing protective immune responses against invading microbes, or cancer cells, while safeguarding the proper homeostatic equilibrium of the immune system and maintaining self‐tolerance. Unsurprisingly, DCs play a role in many diseases such as autoimmunity, allergy, infectious disease and cancer. This makes them attractive but challenging targets for therapeutics. Since their initial discovery, research and understanding of DC biology have flourished. We now recognize the presence of multiple subsets of DCs distributed across tissues. Recent studies of phenotype and gene expression at the single cell level have identified heterogeneity even within the same DC type, supporting the idea that DCs have evolved to greatly expand the flexibility of the immune system to react appropriately to a wide range of threats. This review is meant to serve as a quick and robust guide to understand the basic divisions of DC subsets and their role in the immune system. Between mice and humans, there are some differences in how these subsets are identified and function, and we will point out specific distinctions as necessary. Throughout the text, we are using both fundamental and therapeutic lens to describe overlaps and distinctions and what this could mean for future research and therapies.
Keywords: antigen presentation, dendritic cell development, dendritic cell subsets, immune responses, immunotherapy, vaccine approaches
Recent studies of phenotype and gene expression at the single cell level have identified DC heterogeneity even within the same type, supporting the idea that DCs have evolved to greatly expand the flexibility of the immune system to react appropriately to a wide range of threats. This review is meant to serve as a quick and robust guide to understand the basic divisions of DC subsets and their role in the immune system. Throughout the text, we are using both fundamental and therapeutic lens to describe overlaps and distinctions and what this could mean for future research and therapies.
Abbreviations
- AD
atopic dermatitis
- APC
antigen‐presenting cell
- BM
bone marrow
- cDC
conventional DC
- CNS
central nervous system
- DC
dendritic cell
- FDC
follicular DC
- GC
germinal centre
- IFN
type I interferons
- LC
Langerhans’ cell
- MO
monocyte
- MΦ
macrophage
- pDC
plasmacytoid DC
- SCS
subcapsular sinus ; FcR, Fc receptor
- SLE
systemic lupus erythematosus
- Th
T helper
- Treg
T regulatory
- TSLP
thymic stromal lymphopoietin
INTRODUCTION
Ralph Steinman and Zanvil Cohn are credited with the discovery of DCs in 1973 [1]. In recognition of his pivotal findings, Ralph Steinman was awarded several prestigious prizes, including the Nobel Prize in Physiology or Medicine in 2011. Historically, scientists had previously encountered some of the subtypes without appreciating the significance. In 1868, Paul Langerhans had described Langerhans’ cells (LCs) for the first time, although he originally believed they were nerve cells [2], and in 1964, Miller and Nossal observed that within the lymph node follicles, there were specific cells, which kept antigens presented on the surface, only later named follicular DCs (FDCs) [3]. Then, to compare DCs with other APCs and understand the signals that modulate their function, it became fundamental to establish an in vitro cell culture system. In 1994, Sallusto and Lanzavecchia described an in vitro protocol to generate human DCs from blood mononuclear cells in the presence of GM‐CSF and IL‐4 [4] and this methodological advance allowed research in this field to propel and thrive until today.
DCs only make up to 0·1–1% of mononuclear cells but multiple subsets exist and are differentially distributed across the body, blood, skin, organs and lymphoid tissue with CD11c+ DCs also reported in the central nervous system (CNS) [5]. These different DC subsets are equipped with unique features to present antigens and initiate T cell‐mediated responses but also maintain immunological tolerance [6]. Initially, DCs were very simplistically classified into two broad subsets: conventional DCs (cDCs), which primarily function as APCs, and plasmacytoid DCs (pDCs), which are specialized producers of type I interferons (IFNs) that respond to viruses [7]. Subsequently, the cDC subset was further divided into cDC1 and cDC2 (classical DCs) based on the identification of specific surface receptors that perform well across species (DEC‐205, CLEC9a, CD8α, human CD141/BDCA‐3 for cDC1 and CD11b, CD11c and SIRPα/CD172a for CD1c+/BDCA‐1+cDC2) and function (antigen cross‐presentation to CD8 T cells or priming of CD4 T helper (Th) cells, respectively) (Table 1). More recent studies based on an increasing level of resolution of phenotype and gene expression have identified pre‐cDC populations in human blood. See et al. [8] characterized a population containing AXL+ SIGLEC6+ cells as ‘early pre‐DC’ with the ability to develop into cDC1 and cDC2. Villani et al. also identified a CD34+ CD100+ DC precursor in human blood. This cell population expresses lower level of CD123 compared to AXL+ SIGLEC6+ pre‐cDC and appears to develop earlier. In the same report, CD1c+ DCs were shown to express unique markers (e.g. CD1c, CLEC10A, FcεR1A, FcγR2B and CD1d) and yet be distributed across two separate clusters—DC2 with low levels of MHC‐II, and DC3 cells that express CD14 and exhibit a strong inflammatory signature [9].
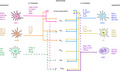
TABLE 1.
Phenotype of human and mouse DC subsets. Summary of the phenotype, pathogen receptor expression profile, key cytokines of human DC subsets and the mouse DC equivalent. Note that mouse equivalent of human AXL+ DCs is missing because they are considered ‘transitional’, and therefore, authors have not defined a specific set of receptors for their identification
Therefore, with the advent of single cell sequencing and high‐dimensional flow cytometry, it is now also possible to elucidate DC origin and development.
DC DEVELOPMENT
Tracing back the origin of DCs is an ever‐evolving field. It was largely accepted that murine DCs stem from a common myeloid progenitor, which becomes a macrophage (MΦ)/DC progenitor that can then split into a common DC progenitor to give rise to the cDC and pDC subsets [10, 11, 12] (Figure 1). Similarly, a common monocyte (MO) intermediate from the macrophage (MΦ)/DC progenitor would differentiate into MO‐derived MΦ or MO‐DC line [13]. However, it has been recently established that also common lymphoid progenitors can generate pDC and cDC1 subsets, perhaps more efficiently than the traditional myeloid lineage [14, 15, 16]. The murine common lymphoid progenitor precursor identified as Ly6Dhi, IL‐7Rα+, CD81+ and CD2hi distinguishes pDC lineage very early as these cells divergence from the cDC lineage [17, 18].
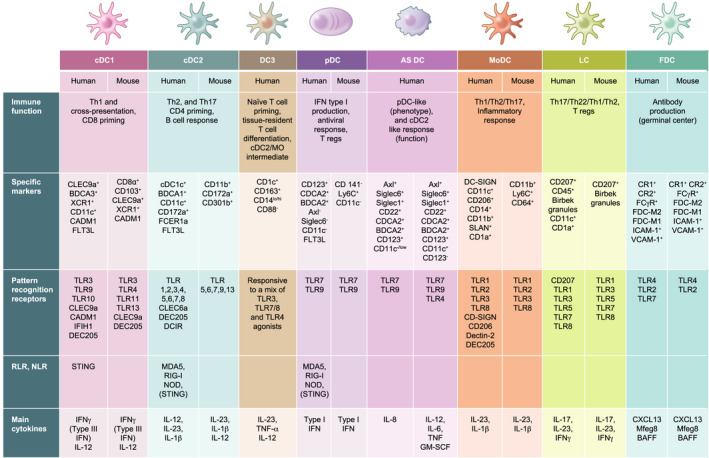
FIGURE 1.
Development of DC lineages. In the BM, a hematopoietic stem cell can differentiate into a common myeloid progenitor or a common lymphoid progenitor. Common myeloid cells become MΦ/DC progenitors and can give rise to pDCs and to a final pre‐DC stage leading to conventional DC1 (cDC1) and cDC2, whereas MOs would give rise to MO‐derived MΦs or moDCs via a DC3 intermediate. Common lymphoid progenitor cells give rise to NK, B and T cells, and also to pDCs and the cDC1 subset. The majority of LCs derive from fetal liver‐derived MOs; however, some LCs can derive from yolk sac progenitors. FDCs are thought to originate from stromal cells of mesenchymal derivation. The illustration depicts also the major transcription factors expressed during the differentiation of the different DC lineages
For FDCs and LCs, the lineage differs from this pathway. Indeed, the majority of LCs derive from the fetal liver‐derived MO lineage [19], and a fraction from yolk sac progenitors [20]. Historically considered representatives of the DC lineage, LCs are now seen more as a specialized subset of tissue‐resident MΦs, considering their cellular development, and tissue residency properties. As discussed in the next section, LCs also display a remarkable profile common to DCs, such as migratory capacity and antigen presentation.
Despite having a dendritic morphology, FDCs are unrelated to DC and pDC lineages and, as we will see next, they play a unique function as APCs for B cell activation, rather than T cell responses. To date, most scholars accept that FDCs originate from stromal cells of mesenchymal derivation [21, 22, 23] rather than from a bone marrow (BM) precursor [24].
DC differentiation is regulated by environmental cues, and key growth factors are Flt3L, GM‐CSF and M‐CSF [25, 26]. It is proposed that Flt3L is necessary for influencing differentiation of cDC and pDC subsets, but not LC [27, 28]. Overexpression of human Flt3 is sufficient to rescue or enhance DC differentiation potential in Flt3− or Flt3+ hematopoietic progenitors, respectively [29]. It is also known that administration of Flt3L dramatically expands cDC and pDC subsets in mice and in healthy human subjects [25, 30, 31, 32]. Initial data from GM‐CSF‐deficient mice showed marginal impact on DCs and other myeloid subsets [33]. Only in later studies, mice lacking GM‐CSF or its receptor (GM‐CSFR) were shown to have substantially reduced numbers of DCs in skin and gut [34, 35, 36]. It is now widely appreciated that GM‐CSF can induce the differentiation of DCs in vitro and in vivo; however, GM‐CSF is not detectable at steady state in serum and it increases during response to a pathogen and inflammation [37].
M‐CSF, or Csf‐1, regulates the differentiation of MΦ populations [38]. The receptor, Csf1R or CD115, is expressed on MO and MO‐derived MΦ populations, and also on common DC progenitors, but it is gradually lost in some DC lineages and maintained in CD11c+ cDCs and pDCs [39, 40]. These data indicate that the dual action of Flt3L and Csf‐1 determines the differentiation of progenitors into DCs rather than into a MO/ MΦ line. Work from Wang et al. indicated that LCs and microglia are present in Csf‐1‐deficient mice but absent from Csf1R‐deficient mice. They also showed that IL‐34, a ligand of Csf1R secreted by keratinocyte and neurons, is indeed required for the development of both cell types [41].
Studies of human DC progenitors and development pathways have been more challenging than the same studies with murine DCs. Only with an ad hoc cell system, it is possible to identify and characterize human equivalents of MΦ/DC and common DC progenitors, and pre‐DC cells [42, 43, 44]. There is overall a significative degree of conservation for DC developmental pathway between mouse and human. However, the nature of human DC progenitors resulted to be more heterogeneous than anticipated. Within the same population, human progenitor cells undergo distinct developmental pathways while sharing a common transitional phenotype. In human BM and cord blood samples, at least three subpopulations can be identified as early hematopoietic pools with DC potential and sequential progenitor–progeny relationship [43, 44, 45]: one population having granulocyte, monocyte and DC potential; a second population that differentiates into MOs and DCs; and a third population that produces only DCs, which will further produce committed pre‐DCs. These pre‐DCs can be found in the BM, blood and other peripheral lymphoid organs where they will differentiate in cDC1 and cDC2 subsets.
TRANSCRIPTIONAL CONTROL OF DC DEVELOPMENT
Extracellular signals are also responsible for modulating the action of several transcription factors that control DC development and commitment [46, 47, 48] (Figure 1). Mouse genetic models used to compare DC progenitors and differentiated subpopulation revealed that multiple transcription factors interact sequentially to drive DC development and specification of distinct subsets.
The transcription factors Gfi‐1, PU. 1 and STAT3 are active in all DCs and largely responsible for DC commitment and differentiation from a very early stage. Rathinam et al. [49] showed that Gfi1−/− mice have reduced myeloid and lymphoid DCs, whereas skin LC numbers were enhanced. PU.1, encoded by the Sfpi1 gene, has been shown to have disparate roles in haematopoiesis, while Kueh et al. [50] proposed that divergence between the lymphoid and myeloid lineages is determined by either low or high levels of PU.1, respectively. Common progenitors of the myeloid and lymphoid lineages from PU.1−/− mice could not differentiate into DCs [51], suggesting that PU.1 lies upstream of Flt3 and GM‐CSFR and is required for differentiation of steady state DC and MO subsets.
Several models of DC development predict pDCs diverge from cDC lineages at the common DC progenitor stage (Figure 1). In general terms, expression of E2‐2, encoded by the Tcf4 gene, favours pDC differentiation in mice and humans [52], while overexpression of ID family proteins, which bind E2‐2, inhibited development of CD123+ pDCs but did not affect development of myeloid DCs [53]. A balanced action of E2‐2 and ID2 is a mechanism proposed for pDC/cDC divergence. However, the basis for lineage divergence between pDCs and cDCs is not fully explained at present.
Common DC progenitors give rise also to pre‐DC populations, which will lead to two types of cDCs named IRF8+ BATF3−, and IRF4+ cDCs because their differentiation is critically driven by IRF8 and IRF4. These, together with other transcription factors such as KLF4, Notch2, E2.2 and ID2 represent a core genetic signature that differentiates the pDC from DC1 and cDC2 lineages. IRF8‐deficient animals lack spleen‐resident CD8α+ and peripheral CD103+ cDCs, and LCs [54, 55]. Cells isolated from IRF8−/− spleens were unable to produce type I IFN in response to viral stimulation. Very recently, Cytlak et al. reported that pDC, cDC1 and cDC2 cells are strictly dependent on IRF8. These subsets are believed to develop from an IRF8hi progenitor and differential IRF8 expression would impact different lineages in a dose‐dependent manner [56]. During differentiation from pre‐DCs to CD11b+ cDCs, IRF8 is lost and replaced by IRF4 expression. It was suggested that in CD11b+ cDCs, IRF4 serves as the binding partner with PU.1 to turn on expression of CD80, CD86 and CCR7 necessary for antigen presentation [57]. IRF4‐deficient mice have reduced numbers of splenic CD4+ CD11bhi cDCs [58] but no defects in CD8+ cDC development [59, 60].
Interestingly, IRF4+ cDC population presents additional heterogeneity with CD8− CD11b+ cDCs that are dependent on the transcription factors Notch2 and KLF4. Notch2 plays an important role in the maintenance of the splenic CD11b+ cDC compartment. Mice that lack Notch2 in the CD11c+ compartment have a survival disadvantage in CD11b+ splenic cDCs against C. rodentium infection. Functionally, IL‐23 production by Notch2 IRF4+ cDCs is required for effective Th17 responses [61]. Pre‐cDCs express high level of KLF4, which is downregulated in mature splenic cDCs. KLF4 deletion in early hematopoietic progenitors reduced the expression of IRF4 in pre‐cDCs without affecting IRF8+ and IRF4+ cDC differentiation [62, 63]. KLF4 IRF4+ cDCs appear to be required for functional type 2 responses [64]. For practicality, we note that IRF8+ cDCs express XCR1 and IRF4+ cDCs express SIRPα [65, 66]. Alternatively, XCR1+ IRF8+ cDCs are also referred to as cDC1 and SIRPα+ IRF4+ cDCs as cDC2.
The transcriptional network composed by ID2 together with BATF3, and IRF8 is critical for differentiation of cross‐presenting cDCs, also termed ‘BATF3‐IRF8‐ID2‐dependent DCs’ [67]. BATF3 is known for its exclusive role in the development of the CD8α + DC subset. BATF3−/− mice lack CD103+ DCs and have reduced numbers of CD8α+ DCs in the spleen [68]. Importantly, CD8α+ DCs present in BATF3−/− mice have reduced capabilities for cross‐presentation [69]. Nevertheless, compensatory effects of other BATF factors that interact with IRF8 are in place and promote differentiation of CD8α+ DCs [70].
All these observations are emblematic of lineage specification and functionally divergent DC subsets conferred by cytokine‐driven transcription factors.
We note that the transcription factors associated with human progenitors and their development are still little defined when compared to the extensive information acquired for murine DCs. However, human genetic observations are very helpful to point out the significance of some of these transcriptional regulators during human DC development. Namely, heterozygous mutations of GATA2 (a zinc finger transcription factor involved in the homeostasis of hematopoietic stem cells) cause DC, MO, B and NK lymphoid deficiency, combined with mononuclear cell deficiency and absence of blood and interstitial tissue DCs [71]. IRF8 mutations are reported in humans [72]. These defects appear to block the transition of early progenitors to the MΦ/DC progenitor stage, and as a result, MOs and DCs fail to develop in these patients. These subjects show susceptibility to Mycobacteria, including the Bacillus Calmette–Guérin vaccine strain. A patient with the IRF8 K108E mutation manifested complete loss of blood myeloid DCs, pDCs, and MOs, while another patient with the IRF8 T80A mutation lacked CD1c+ DCs [72]. Worthy to note, these human phenotypes are different from the phenotype of IRF8‐deficient mice discussed above. In spite of discordant phenotypes, these observations support a critical role of IRF8 for keeping DC homeostasis in both mice and humans.
IDENTIFICATION OF DC SUBSETS
Recent advances in multiparametric flow cytometry and single cell RNA technologies revealed the great heterogeneity of DCs, even within the same subset. Nonetheless, these sophisticated methods still support the initial classification of DC populations defined by surface receptor expression (Figure 2).
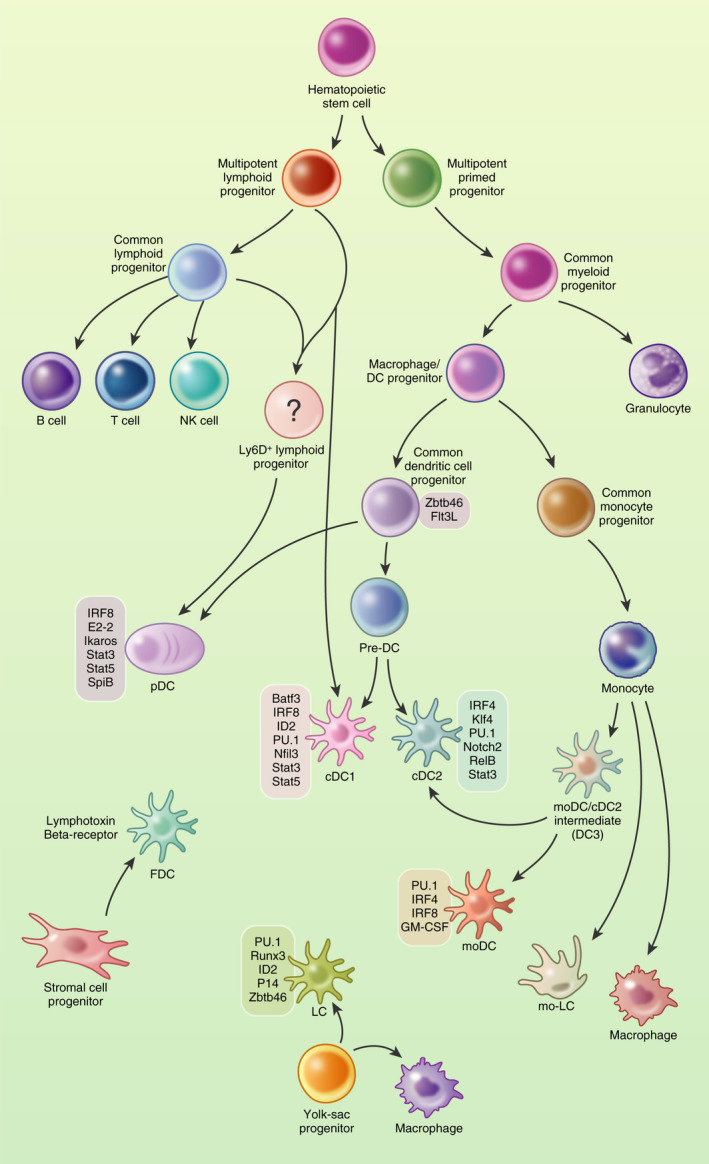
FIGURE 2.
Schematic of human DC subsets, their markers, key cytokines and corresponding T cell function. Dashed lines represent weaker associations or places where more research is needed to further define the role of the subset in the given response
Classical cell markers for murine and human cDC1s are CADM1 [73], CLEC9a (DNGR‐1), BDCA‐3/CD141, XCR1 and CD11c [74]. The markers CD103 and CD8α are specific for murine models, although it should be noted that cDC1s in human spleen may express CD8α while resident murine cDC1s express CD8α, but migratory cDC1s (CD103+) are lacking [75, 76]. Murine cDC1 cells express langerin (CD207) in the spleen and have higher expression of TLR4 and TLR9, whereas human cDC1s do not express langerin and tend to have higher expression levels of TLR3 for detecting double‐stranded RNA [77] (Table 1). The cDC2 subtype in humans is distinguished by the expression of CD1c, CD11c and SIRPα/CD172a and can be additionally screened for receptors like CD11b, Dectin‐1 (CLEC7a), Dectin‐2 (CLEC6a) and DEC‐205. These same receptors are expressed by other subtypes, although expression levels may vary (Figure 2, Table 1). The murine cDC2 equivalent is defined by CD11b, CD172a and CD11c, with markers like CD206 indicating whether the DC is migratory or resident [78, 79]. Their functional flexibility may be further explained by their wide range of surface markers and TLR receptors. Human cDC2s express nearly every TLR except TLR9 [8].
Several groups have independently demonstrated that cDC2s are very heterogeneous. Specifically they include CD5+ DC2s and CD5− CD163+/− CD14+/ ‐ DC3s, the latter with a pro‐inflammatory profile that correlated with systemic lupus erythematosus (SLE) progression in patients [80] and expansion of tissue‐resident memory T cell in primary breast cancer [81] (Table 1).
The human pDC subset is characterized by CD123/IL3RA expression, BDCA‐2/CLEC4c/ CD303, Siglec6 and DCIR/CLEC4a. Murine pDCs can be marked by B220, CD11c, SiglecH, and CD317 [7, 82, 83] although they may also display detectable levels of TLR5 and are particularly enriched in TLR7 and TLR9 [84]. Two groups independently reported the presence of a unique, novel DC subset that shares traditional markers of pDCs and cDC2s. This subset was identified as AXL+/Siglec6+ and when removed from the bulk pDC population, the remaining pDC induced very low levels of T cell proliferation in response to TLR stimulation [8, 9] (Figure 2, Table 1).
Observations of a new cell type in adult mice, with similarities between pDC and cDC2, were recently made by Leylek et al. using high‐dimension mass‐cytometry and transcriptomic analysis. The authors named this murine homolog of human AXL+ cells as ‘transitional DCs’ and demonstrated that they efficiently activate T cells. Like murine pDCs, these transitional DCs are characterized by CX3CR1 expression and are recruited to the site of infection, however they are inefficient producers of IFN‐I [85]. In accordance with their transitional phenotype, the authors could not identify specific markers that would unambiguously distinguish them, indicating that more research on this newly defined subset is needed to determine to what extent these cells play in pDC plasticity. Contrary to this model of pDC plasticity, Abbas et al. demonstrated with a comprehensive analysis that during MCMV infection, pDCs undergo different and sequential activation states, and that the same pDC can secrete IFN‐I and promote T cell activation but sequentially in time and in different microenvironments [86].
Sometimes referred to as inflammatory DCs, moDCs are marked by the expression of CD11c, CD16, CD14, CD11b, CD172α, and CD209 (DC‐SIGN), although the expression of CD14 and CD16 may vary depending on the MO subset [87]. Murine moDCs can be marked by Ly6C, CD11b and CD115 [88], and like cDC2s, moDCs express a wide range of TLRs, and produce multiple cytokines [89] (Figure 2, Table 1).
Unlike the other subsets, FDCs do not express their own MHC‐II but may obtain small amounts from exosomes [90, 91] and can be marked by expression of CR1/CD35, CR2/CD21, FcγRIIb/CD32 and FcεR2/CD23 [92, 93, 94]. Expression of LTβR, TNFR1, VCAM, BP‐3 and ICAM is shared with fibroblast reticular cells and marginal reticular cells, and while these receptors are important for function and development, they are not specific markers for FDCs [94, 95] (Figure 2, Table 1).
LCs are specialized cells of the skin and mucosal tissues. They express low levels of MHC class II, CD1a, CD24, intermediate levels of CD11c, CD207 (langerin), the epithelial cell adhesion molecule (EpCAM), SIRPα. To note that human LCs are negative for the lineage markers (CD14, CD16, CD3, CD56 and CD19) and MΦ marker F4/80, while murine LCs are CD11b+ F4/80+ and lack CX3CR1 expression [73, 96] (Figure 2, Table 1). Human LCs isolated from skin expressed mRNA for TLR1, TLR2, TLR3, TLR5, TLR6 and TLR10, and they are extremely flexible in function.
MULTI‐FACETED IN VIVO DC FUNCTION AND ORGANIZATION IN LYMPHOID TISSUE
We might wonder why does the immune system allow for this redundancy? One answer to this question lays in the location, abundance and organization of the different DC subsets in the lymphoid tissue (Figure 3).
FIGURE 3.
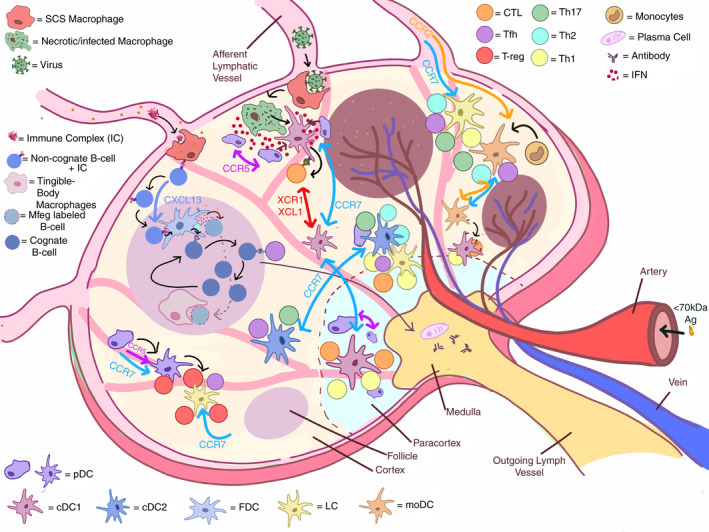
Schematic of DC organization in the lymph node. The majority of the cDC1 subset is advantageously positioned in the paracortex. The remaining cDC1s are found in the interfollicular zone. cDC2 can be found in the interfollicular zone, whereas a small population occupies the T cell zone. The pDC subset is found ubiquitously across the lymph node, in both the T cell and the interfollicular zone. cDC1, cDC2, pDC, LC and moDC populations all respond to CCR7, the most relevant chemotactic signal, which is typically generated by a gradient of CCL21/CCL19 ligand cytokines secreted by stromal cells, T cell zone reticular cells and fibroblast reticular cells. To note that moDCs are recruited to sites of inflammation via CCR2. During viral infection, pDCs migrated either to the site of CD8+ T cell priming via CCR5, or to subcapsular infected MΦs via CXCR3. pDCs were also found to respond to CCL3 and CCL4 chemokines produced in the context of productive XCR1+ cDC1 and CD8+ T cell interactions. B cells and FDCs find themselves in a peculiar loop as FDCs need B cells to mature and B cells need interaction from FDCs to survive and generate antibodies. FDCs secrete Mfge8 important for antigen uptake. IC larger than 70 kDa can enter the lymph node via afferent lymphatic vessels, whereas those smaller can directly enter the conduit network. The large ICs will be met by SCS MΦs, who will pass them to non‐cognate B cells, which will relay to the FDCs for uptake, recycling and prolonged epitope presentation
cDC1s promote Th1 immunity and are capable of cross‐presentation of exogenous antigens to CD8+ T cells, and therefore, cDC1 are particularly geared to help fight tumours and viral threats. Alcántara‐Hernández et al. estimated that cDC1s are <10% in blood, tonsil and skin, but are nearly a fifth of all DCs in the spleen.[6] Studies have shown that cDC1s can bind necrotic cells via CLEC9a when near the subcapsular sinus (SCS) MΦs [97, 98]. Van Dinther et al. [99] showed that CLEC9a on cDC1s enhanced CD8+ T cell cross‐priming of antigens targeted to CD169+ MΦs. Mohapatra et al. similarly showed how CD8α+ DCs captured cell‐associated antigens from both live and apoptotic tumour cells, whereas CD169+ MΦs picked up antigens mostly from apoptotic cells [99, 100]. These data together suggested that cell interactions between cDC1s and MΦs and cross‐presenting CD8α+ DCs are all mechanisms in place to maximize a rather inefficient event of antigen cross‐presentation (Figure 3).
The cDC2 subset is the most abundant of all DC subsets representing approximately half of the DC population in the blood, spleen and skin, but only about 15% of DCs in the tonsils [6, 101]. The majority of cDC2s can be found in the interfollicular zone, whereas a small population occupies the T cell zone [102]. Human cDC2s are capable of coordinating Th1 and Th17 responses, and cross‐presenting exogenous antigens to CD8+ T cells, whereas this function is not well defined in mice [103, 104]. Recently, Bosteels et al. [105] demonstrated that during inflammation, cDC2s acquire an inflammatory phenotype, sharing a transcription profile and function with cDC1s and moDCs, that optimally boost CD4 and CD8 T cell immunity via Fc receptors (FcR).
The pDC subset is estimated to represent about a third of DCs in the blood, about a fifth in the spleen, absent in skin and nearly 80% of DCs in tonsils [6]. The presence of rare pDCs in healthy skin is debatable; however, pDCs have been found in various skin diseases, suggesting an active mechanism of pDC recruitment under pathological conditions. pDCs are found ubiquitously across the lymph node, in both the T cell and the interfollicular zone [106]. These cells are known for promptly producing large amounts of type I IFN upon viral challenge and a study conducted by Brewitz et al. [107] observed that upon injecting modified vaccinia virus Ankara in the footpad of mice, the cDCs interacting with OT‐I T cells were surrounded by pDCs, highlighting the critical role of those IFN‐I producing cells for cDC functionality. The ability of pDCs to produce abundant type I IFN quickly helps to drive maturation the XCR1+ DCs, promoting antigen cross‐presentation. More recently, Fu et al. [108] demonstrated that pDCs enabled cDC1 cells to cross‐prime CD8+ T cells by transferring antigens to cDCs in the form of pDC‐derived exosomes.
LCs are capable of orchestrating a Th response or T regulatory (Treg) response [109, 110]. Due to their location in the skin, they are often the first DC subtype sensing the external environment, which is consistent with their functional plasticity. Seré et al. [111] showed that after UV light exposure, two waves of LC recruitment were observed one wave of short‐lived LCs, involving MOs and the second wave of steady state BM precursors. In response to an invading pathogen in skin, dermal DCs and LCs migrate to the lymph node to recruit and prime T cells. These effector T cells secrete cytokines to differentiate MOs in moDCs for a second wave of response; however, it remains unclear whether these MO‐derived LC‐like cells replace the yolk sac LCs temporarily or are long lived [111]. Likewise, moDCs appear at sites of inflammation, and can induce multiple responses including Th1, Th2 and Th17 responses [112, 113]. moDCs are also able to transfer their MHC‐I complex to other DC subsets in both mice and humans and can cross‐present to CD8+ T cells.
FDCs are found strictly in the follicles and germinal centres (GCs) in the spleen and lymph nodes and secrete CXCL13, a known chemoattractant to B and follicular Th cells [114], as well BAFF, which is critical to B cell survival and affinity maturation [115]. Ablation of FDCs leads to complete disappearance of GCs [89]. Recently, TLR7 activation on FDCs was found to be mediated by antigen uptake and resulted in sustained levels of IFNα secretion [116]. A unique function of FDCs is the presentation of immune complexes (ICs), which involves a step where the subcapsular MΦs take the incoming ICs and pass them through non‐cognate B cells, to FDCs [117] (Figure 3). Heesters et al. described how FDCs take ICs into recycling endosomal compartments preventing antigen degradation and presenting it for long periods of time, even weeks [117, 118]. Thus, FDCs have evolved to provide a large variety of antigenic epitopes on their surface for B cell antibody responses.
TEAMWORK FOR HUMORAL AND CELL‐MEDIATED IMMUNE RESPONSES
We appreciate that when it comes to immune responses to different pathogens, the specific role and function of DC subsets are critical. The balance of Th2 versus Th1 immunity relies greatly on the activity of the cDC1 subset. Jongbloed et al. [119] demonstrated that upon challenge with human CMV and poly I:C, human cDC1s were more effective than the cDC2 counterparts at producing IL‐12 and Th1 cytokines. Genetic deletion of XCR1+ DCs in Baft3−/− in vivo decreased CD8+ T cell response but had no effect on CD4+ T cell activation upon LPS +OVA stimulation [102]. Of note, Ferris et al. [120] proposed that cDC1s prime and are licensed by CD4+ T cells to induce anti‐tumour immunity, showing that MHC‐II expression and CD40 signalling on cDC1 cells are needed for CD4+ T cell priming in response to cell‐associated antigens, and for rejection of fibrosarcoma.
The route of antigen exposure also determines the involvement of a specific DC subset and not another. Via intranasal injection, for example, cDC1s and cDC2s could obtain Ag comparably, but when Ag was injected subcutaneously, only migrating CD11b+ cDC2s took up the antigen efficiently [121]. Deletion of the cDC2 subset or impairing of their migration reduced Tfh cell responses to OVA and influenza virus [121]. Interestingly, during allergic airway Th2 responses, thymic stromal lymphopoietin (TSLP) enhances cDC2‐mediated activation of allergen‐specific CD4+ T cells, while cytokines secreted by Th2 cells reduce TSLP‐mediated CCR7 induction on cDC2s, thus inhibiting migration of cDC2s to the draining lymph nodes [122]. This results in the retention of cDC2s in situ and further exacerbation of local inflammation. Similarly, moDCs appear at sites of inflammation, and can induce multiple responses including Th1, Th2 and Th17 responses [112, 113, 123]. By transfer of their MHC‐I complex to other DC subsets, mouse and human moDCs can cross‐present to CD8+ T cells [124]. Studies in Flt3l−/− mice, lacking all cDCs, revealed that moDCs were sufficient to induce Th2 cell‐mediated immunity but only when high doses of house dust mite were given [125].
The type of invading pathogen determines the type of response mediated by DCs. For example, Staphylococcus aureus and Corynebacterium bovis, are commonly observed in atopic dermatitis (AD). When experimentally inoculated in mice to reproduce the dysbiosis seen in AD patients, S. aureus prominently drove the eczema phenotype, while C. bovis induced a robust Th2 response. In this model, Langerhans’ cells were shown to mediate Th17 immune responses specifically against S. aureus inoculation [126]. Recently, a CD11c+ DC subset residing in the lower layer of the epidermis, and resembling cDC2s, was identified in humans, showing better capability at transferring HIV to CD4+ T cells [127]. In another study [128], it was shown that skin CD103+ DCs are capable of inducing CXCR5+ Tfh cells; however, they are significantly less efficient than skin LCs in inducing GC B cells and IgG1 titres. Both dermal cDC1s and LCs have demonstrated efficient antigen cross‐presentation [129]. Some of these findings have been challenged because, at the time of study, it was not known that dermal cDC1s also express langerin. Indeed, when keratinocytes were induced to express OVA antigens, the dermal cDC1s were surprisingly effective in cross‐presenting keratinocyte‐derived antigen, even in the absence of LCs [130]. It is unknown whether the heterogeneity of LC populations can help explain some of the discrepancies in recent data.
In conclusion, while some subsets are more potent inducers of a certain response and take charge in certain settings, they appear to work as a team. In the event that the antigen is not accessible to cDC1s, T cell immunity can still be carried out by cDC2s, moDCs, or LCs, and as we will see in the next paragraph, in the absence of pDCs, LCs can construct immune tolerance.
IMMUNE TOLERANCE—HOW TO MAKE IT OR BREAK IT
DCs have evolved to identify tissue damage or invading pathogens to promptly mediate protective immune responses. However, DCs are also essential to maintain immune tolerance to self and contribute to tissue homeostasis. Early on, it was shown that when dying TAP−/− cells loaded with OVA were given with immature DCs, they induced an antigen‐specific tolerance with an initial burst of OT‐I CD8+ T cells followed by their deletion [131]. The ability of immature DCs to continuously phagocytize apoptotic cells and present antigenic peptides without undergoing maturation is key to maintaining self‐tolerance.
Lutz et al. have very recently revised some of the historical assumptions about steady state and tolerance. The authors discussed that there is little evidence for a tolerogenic function in vivo of immature DCs. Contrary, tolerogenic DCs undergo molecular changes—upregulation of MHC and costimulatory molecules, CCR7, RelB, and IL‐12p40 secretion, that differ quantitatively, and not qualitatively, from immunogenic DCs. These molecular changes together with different transcriptional activities (NF‐κB (RelA/p50) and c‐Rel for immunogenic DCs) better define the tolerogenic or immunogenic activation status of DCs [132].
Regarding specific DC subsets, finer details on the function of pDC in maintaining immune tolerance have been investigated only in recent years. One study found that upon pDC activation via CpG, there was a heterogeneity of responses depending if the CpG structure was multimeric or monomeric, influencing whether IFNα was secreted or pDC maturation was induced, respectively [133]. The ability of pDCs to reverse Treg anergy requires cell contact and is partially CD86 dependent and IL‐2 independent [134].
The LC subset has been long studied for their estranged abilities of inducing tolerance despite initial antigen presentation to CD8+ T cells [135]. Strandt et al. [136] showed that in the steady state, LCs were capable of stimulating a CTL response to OVA but upon re‐challenge the authors observed induction of Tregs and resistance to immunization. In addition, they reported that when LCs were activated with anti‐CD40 and the TLR3 agonist poly I:C, a memory CTL response was efficiently developed. Similarly human LCs, when co‐cultured with T cells, expanded a Treg subset that inhibited proliferation of T effector memory cells in a mixed lymphocyte reaction [137]. This same study also showed that in the presence of Candida albicans, LCs induced both Treg and T effector memory cells in an antigen dose‐dependent manner. At low dose of antigen, Treg function dominated, while at high dose T effector memory cells proliferated vigorously.
It is evident how pDCs and LCs complement each other in their role of maintaining tolerance; while in physiological conditions LCs are absent in blood, lymphoid tissue and other organs, pDCs are absent in the skin in the steady state but abundant elsewhere.
THERAPEUTIC APPLICATIONS
DCs are attractive therapeutic tools to manipulate adaptive T cell‐driven immune responses in patients or healthy subjects for vaccine purposes. Most of the clinical trials (e.g. NCT00576537, NCT04335890, NCT00833781, NCT01734564, NCT04078269) testing DC‐based vaccines were conducted with ex vivo moDCs matured with the standard cocktail of TNF‐α, IL‐1β, IL‐6 and PGE2. Unfortunately, this approach presents limitations due to the laborious ex vivo cell manipulation required and, more importantly, the risk of infection to patients. Immunization with FLt3L mobilized antigen‐loaded DCs was already long ago showed to induce CD8+ cytotoxic T cells that recognized tumour cells [58]. In 2015, Anandasabapathy et al. [32] found that treatment of healthy volunteers with Flt3L expanded and mobilized specific subsets of DCs such as CD34+ precursors, myeloid and pDC subsets up to 20‐fold in vivo. Phase I/II clinical trials are being conducted to assess how well Flt3L works in combination with other adjuvants or radiation therapy in treating patients with advanced malignancies.
Currently, efforts have shifted into targeting the DC receptors to initiate an immune response [138]. CDX‐1401 is a vaccine composed of a monoclonal antibody to DEC‐205 fused with tumour antigen NY‐ESO‐1 in combination with resiquimod (TLR7/8 agonist) and Hiltonol (TLR3). Data from a clinical study showed humoral and cell‐mediated immunity and the authors also reported tumour regression in patients who received a combination with immune‐checkpoint inhibitors [139]. In 2020, Bhardwaj et al. [140] evaluated the ability of Flt3L to enhance responses to CDX‐1401 in a phase II trial (NCT02129075). The clinical study reported significant T cell responses to NY‐ESO after only one vaccination in patients receiving Flt3L.
To overcome traditional allergen immunotherapy, Sirvent et al. [141] used DC targeting in mice and human studies and observed that glutaraldehyde‐polymerized allergoids conjugated to non‐oxidized mannan generated blocking antibodies to the native allergen and induced Tregs specifically via PD‐L1.
If the goal is to orchestrate a Th1 type immunity, targeting cDC1s through the XCR1 or CLEC9a receptor seems an attractive option [142]. The recent results of Oba et al. [143] demonstrate that in situ induction and activation of cDC1s by Flt3L and TLR3/CD40 stimulation facilitates priming, expansion and infiltration of tumour‐specific T cells in poorly infiltrated tumours and renders those tumours responsive to anti‐PD‐L1 therapy. Also note that murine pDCs express CLEC9a, so it is therefore possible to observe tolerogenic effects when the CLEC9a is targeted in the absence of adjuvant [144]. Kato et al. [145] more recently showed that targeting antigens to CLEC9a enhances early B cell activation and Ab responses. This occurs independently of T cell help and would facilitate B cell migration to the T‐B cell zone in the lymph node, where priming of T cell‐dependent humoral responses is taking place. These new data suggest that follicle‐associated B cells are activated by native antigen displayed via CLEC9a on cDC1s.
Targeting cDC1s via the XCR1‐XCL1 axis with chimeric fusion vaccines may be another great way to enhance a CD8+ T cell response [146, 147]. XCL1‐HA DNA vaccine protected mice against a lethal challenge with influenza virus [148], while Hartung et al. [149] showed that vaccination with XCR1‐OVA or XCL1‐OVA fusion monoclonal antibody prevented the outgrowth of OVA‐expressing tumours. In comparison with CLEC9a, XCR1 is also expressed in a subset of skin cDC1s, while CLEC9a protein expression is almost undetectable in the same compartment [6]. As a consequence, in one study anti‐XCR1 monoclonal antibody was captured more efficiently by human skin cDC1s when compared to anti‐CLEC9a [6].
Targeting antigens through BDCA‐2 on pDCs were shown to promote a tolerogenic effect [150], and it is currently being studied in clinical trials as a functional antagonist in SLE and related diseases [151]. pDCs may not be the only suitable therapeutic target for SLE because evidence has shown that TLR7 activation is critical for FDC antigen uptake and IFNα secretion [116]. This is considered important for self‐reactivity, as TLR7−/− mice had a significant reduction in autoimmunity to self‐antigens. Moreover, lack of Mfge8 results in a deficiency for clearing apoptotic cells in the GC [152, 153], ultimately contributing to the release of self‐antigens which play a pathogenic role in SLE [154]. Their role in long term antigen presentation, B cell survival, and IFN production makes FDCs a great potential target for therapeutics in autoimmune disease.
Research targeting CD1a has been limited in the past due to the lack of receptor expression by murine LCs though it is abundantly expressed by human skin LCs [155]. In an effort to study the effect of CD1a in LC function, Kim et al. used transgenic mice to induce CD1a expression on mouse LCs. They showed convincing evidence for the receptor's role in inducing Th17 responses in models of poison ivy and psoriatic inflammation [155]. In addition, the authors reported that that blocking CD1a reduced inflammation significantly, illustrating this receptor's potential in therapeutics for inflammatory and allergic skin disease.
Finally, to choose the best molecular target, it is important to understand the biology of the DC receptor and the specific cell subset that is ‘marked’ by this receptor. Also, DC‐based vaccines require adjuvants to reduce their tolerogenic profile, and to enhance DC activation and vaccine potency. Exploring the research data on adjuvants to preferentially modulate the activity of certain DC subsets is critical to advance our understanding of antigen presentation in disease states.
CONCLUSIONS
DC subsets are heterogenous distinguished by surface receptors, TLR subtype and cytokine expression, and their ability to coordinate adaptive responses. While DC subsets can be simplified for practicality by clear‐cut functions ‐cDC1s directed towards Th1 and cross‐presentation, cDC2s and moDCs for Th2/Th17, pDCs for IFN secretion, LCs for Th2/Th17/Treg, and FDCs for B cell antigen presentation, these strict definitions do not accurately reflect the multi‐faceted biology of each subset. With the growing interest in using DCs as therapeutic targets, it is critical that the conversation about a given subset accounts for the functional plasticity observed. The consequences of reducing each subset can be seen in cases like DEC‐205 or CLEC9a targeting, where opposing responses of either inflammation or tolerance are common across the literature. This is not to dismiss the unique qualities of a given subset. Distinctions between the subsets, like XCR1 expression on cDC1, BDCA‐2 expression on pDC or the location of the subsets and other characteristics discussed throughout the review, should all be considered as viable ways to specific targeting. We also expect that future DC targeting studies will yield research tools and high‐quality molecules that will add to the definition of DC heterogeneity and will offer innovative translational opportunities for DC approaches to medicine.
CONFLICT OF INTEREST
L.B is an employee of Regeneron Pharmaceuticals, Inc. H.M.G has declared that no conflict of interest exists.
AUTHOR CONTRIBUTIONS
L.B conceptualized the manuscript. H.M.G and L.B conducted literature search and review. H.M.G and L.B. drafted the manuscript. H.M.G prepared the figures. L.B revised manuscript and figures. H.M.G and L.B approved the final version for submission.
ACKNOWLEDGEMENTS
We would like to thank Dr. Stephen Jaspers, in the Target Information Group at Regeneron Pharmaceuticals, Inc., for support, valid discussions and final manuscript editing. We are grateful to Mrs. Samantha Intriligator for her kind assistance with Regeneron library services. We thank Dr. Svetlana Mojsov in the Laboratory of Molecular Immunology at the Rockefeller University, Dr. Arielle Glatman Zaretsky and Dr. Matthew A. Sleeman in the Immunology and Inflammation Department at Regeneron, for critical reading of this manuscript and helpful suggestions. We note that part of this work, consisting in literature review and graphical illustrations, was conducted while Heather M. Giza worked as a Summer Intern at Regeneron.
Giza HM, Bozzacco L. Unboxing dendritic cells: Tales of multi‐faceted biology and function. Immunology. 2021;164:433–449. 10.1111/imm.13394
Funding information
Regeneron Pharmaceuticals, Inc.
REFERENCES
- 1. Steinman RM, Cohn ZA. Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J Exp Med. 1973;137:1142–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. van Spriel AB, de Jong EC. Dendritic cell science: more than 40 years of history. J Leukoc Biol. 2013;93:33–8. [DOI] [PubMed] [Google Scholar]
- 3. Miller JJ 3rd, Nossal GJ. Antigens in immunity. Vi. The phagocytic reticulum of lymph node follicles. J Exp Med. 1964;120:1075–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sallusto F, Lanzavecchia A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony‐stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J Exp Med. 1994;179:1109–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Prodinger C, Bunse J, Kruger M, Schiefenhovel F, Brandt C, Laman JD, et al. CD11c‐expressing cells reside in the juxtavascular parenchyma and extend processes into the glia limitans of the mouse nervous system. Acta Neuropathol. 2011;121:445–58. [DOI] [PubMed] [Google Scholar]
- 6. Alcantara‐Hernandez M, Leylek R, Wagar LE, Engleman EG, Keler T, Marinkovich MP, et al. High‐dimensional phenotypic mapping of human dendritic cells reveals interindividual variation and tissue specialization. Immunity. 2017;47:1037–50.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Asselin‐Paturel C, Trinchieri G. Production of type I interferons: plasmacytoid dendritic cells and beyond. J Exp Med. 2005;202:461–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. See P, Dutertre CA, Chen J, Günther P, McGovern N, Irac SE, et al. Mapping the human DC lineage through the integration of high‐dimensional techniques. Science (New York, N.Y.). 2017;356:eaag3009. 10.1126/science.aag3009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Villani AC, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, et al. Single‐cell RNA‐seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science. 2017;356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Traver D, Akashi K, Manz M, Merad M, Miyamoto T, Engleman EG, et al. Development of CD8alpha‐positive dendritic cells from a common myeloid progenitor. Science. 2000;290:2152–4. [DOI] [PubMed] [Google Scholar]
- 11. Manz MG, Traver D, Miyamoto T, Weissman IL, Akashi K. Dendritic cell potentials of early lymphoid and myeloid progenitors. Blood. 2001;97:3333–41. [DOI] [PubMed] [Google Scholar]
- 12. Chicha L, Jarrossay D, Manz MG. Clonal type I interferon‐producing and dendritic cell precursors are contained in both human lymphoid and myeloid progenitor populations. J Exp Med. 2004;200:1519–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fogg DK, Sibon C, Miled C, Jung S, Aucouturier P, Littman DR, et al. A clonogenic bone marrow progenitor specific for macrophages and dendritic cells. Science. 2006;311:83–7. [DOI] [PubMed] [Google Scholar]
- 14. Shigematsu H, Reizis B, Iwasaki H, Mizuno S, Hu D, Traver D, et al. Plasmacytoid dendritic cells activate lymphoid‐specific genetic programs irrespective of their cellular origin. Immunity. 2004;21:43–53. [DOI] [PubMed] [Google Scholar]
- 15. Corcoran L, Ferrero I, Vremec D, Lucas K, Waithman J, O'Keeffe M, et al. The lymphoid past of mouse plasmacytoid cells and thymic dendritic cells. J Immunol. 2003;170:4926–32. [DOI] [PubMed] [Google Scholar]
- 16. Pelayo R, Welner R, Perry SS, Huang J, Baba Y, Yokota T, et al. Lymphoid progenitors and primary routes to becoming cells of the immune system. Curr Opin Immunol. 2005;17:100–7. [DOI] [PubMed] [Google Scholar]
- 17. Dress RJ, Dutertre CA, Giladi A, Schlitzer A, Low I, Shadan NB, et al. Plasmacytoid dendritic cells develop from Ly6D(+) lymphoid progenitors distinct from the myeloid lineage. Nat Immunol. 2019;20:852–64. [DOI] [PubMed] [Google Scholar]
- 18. Rodrigues PF, Alberti‐Servera L, Eremin A, Grajales‐Reyes GE, Ivanek R, Tussiwand R. Distinct progenitor lineages contribute to the heterogeneity of plasmacytoid dendritic cells. Nat Immunol. 2018;19:711–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hoeffel G, Wang Y, Greter M, See P, Teo P, Malleret B, et al. Adult Langerhans cells derive predominantly from embryonic fetal liver monocytes with a minor contribution of yolk sac‐derived macrophages. J Exp Med. 2012;209:1167–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schulz C, Gomez Perdiguero E, Chorro L, Szabo‐Rogers H, Cagnard N, Kierdorf K, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. 2012;336:86–90. [DOI] [PubMed] [Google Scholar]
- 21. Bofill M, Akbar AN, Amlot PL. Follicular dendritic cells share a membrane‐bound protein with fibroblasts. J Pathol. 2000;191:217–26. [DOI] [PubMed] [Google Scholar]
- 22. Lindhout E, van Eijk M, van Pel M, Lindeman J, Dinant HJ, de Groot C. Fibroblast‐like synoviocytes from rheumatoid arthritis patients have intrinsic properties of follicular dendritic cells. J Immunol. 1999;162:5949–56. [PubMed] [Google Scholar]
- 23. Munoz‐Fernandez R, Blanco FJ, Frecha C, Martin F, Kimatrai M, Abadia‐Molina AC, et al. Follicular dendritic cells are related to bone marrow stromal cell progenitors and to myofibroblasts. J Immunol. 2006;177:280–9. [DOI] [PubMed] [Google Scholar]
- 24. Aguzzi A, Kranich J, Krautler NJ. Follicular dendritic cells: origin, phenotype, and function in health and disease. Trends Immunol. 2014;35:105–13. [DOI] [PubMed] [Google Scholar]
- 25. Naik SH, Proietto AI, Wilson NS, Dakic A, Schnorrer P, Fuchsberger M, et al. Cutting edge: generation of splenic CD8+ and CD8‐ dendritic cell equivalents in Fms‐like tyrosine kinase 3 ligand bone marrow cultures. J Immunol. 2005;174:6592–7. [DOI] [PubMed] [Google Scholar]
- 26. Waskow C, Liu K, Darrasse‐Jeze G, Guermonprez P, Ginhoux F, Merad M, et al. The receptor tyrosine kinase Flt3 is required for dendritic cell development in peripheral lymphoid tissues. Nat Immunol. 2008;9:676–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Merad M, Manz MG. Dendritic cell homeostasis. Blood. 2009;113:3418–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chorro L, Sarde A, Li M, Woollard KJ, Chambon P, Malissen B, et al. Langerhans cell (LC) proliferation mediates neonatal development, homeostasis, and inflammation‐associated expansion of the epidermal LC network. J Exp Med. 2009;206:3089–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Onai N, Obata‐Onai A, Tussiwand R, Lanzavecchia A, Manz MG. Activation of the Flt3 signal transduction cascade rescues and enhances type I interferon‐producing and dendritic cell development. J Exp Med. 2006;203:227–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Maraskovsky E, Brasel K, Teepe M, Roux ER, Lyman SD, Shortman K, et al. Dramatic increase in the numbers of functionally mature dendritic cells in Flt3 ligand‐treated mice: multiple dendritic cell subpopulations identified. J Exp Med. 1996;184:1953–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Maraskovsky E, Daro E, Roux E, Teepe M, Maliszewski CR, Hoek J, et al. In vivo generation of human dendritic cell subsets by Flt3 ligand. Blood. 2000;96:878–84. [PubMed] [Google Scholar]
- 32. Anandasabapathy N, Breton G, Hurley A, Caskey M, Trumpfheller C, Sarma P, et al. Efficacy and safety of CDX‐301, recombinant human Flt3L, at expanding dendritic cells and hematopoietic stem cells in healthy human volunteers. Bone Marrow Transplant. 2015;50:924–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vremec D, Lieschke GJ, Dunn AR, Robb L, Metcalf D, Shortman K. The influence of granulocyte/macrophage colony‐stimulating factor on dendritic cell levels in mouse lymphoid organs. Eur J Immunol. 1997;27:40–4. [DOI] [PubMed] [Google Scholar]
- 34. Kingston D, Schmid MA, Onai N, Obata‐Onai A, Baumjohann D, Manz MG. The concerted action of GM‐CSF and Flt3‐ligand on in vivo dendritic cell homeostasis. Blood. 2009;114:835–43. [DOI] [PubMed] [Google Scholar]
- 35. Bogunovic M, Ginhoux F, Helft J, Shang L, Hashimoto D, Greter M, et al. Origin of the lamina propria dendritic cell network. Immunity. 2009;31:513–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. King IL, Kroenke MA, Segal BM. GM‐CSF‐dependent, CD103+ dermal dendritic cells play a critical role in Th effector cell differentiation after subcutaneous immunization. J Exp Med. 2010;207:953–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhan Y, Lieschke GJ, Grail D, Dunn AR, Cheers C. Essential roles for granulocyte‐macrophage colony‐stimulating factor (GM‐CSF) and G‐CSF in the sustained hematopoietic response of Listeria monocytogenes‐infected mice. Blood. 1998;91:863–9. [PubMed] [Google Scholar]
- 38. Hume DA, Pavli P, Donahue RE, Fidler IJ. The effect of human recombinant macrophage colony‐stimulating factor (CSF‐1) on the murine mononuclear phagocyte system in vivo. J Immunol. 1988;141:3405–9. [PubMed] [Google Scholar]
- 39. Onai N, Obata‐Onai A, Schmid MA, Ohteki T, Jarrossay D, Manz MG. Identification of clonogenic common Flt3+M‐CSFR+ plasmacytoid and conventional dendritic cell progenitors in mouse bone marrow. Nat Immunol. 2007;8:1207–16. [DOI] [PubMed] [Google Scholar]
- 40. Fancke B, Suter M, Hochrein H, O'Keeffe M. M‐CSF: a novel plasmacytoid and conventional dendritic cell poietin. Blood. 2008;111:150–9. [DOI] [PubMed] [Google Scholar]
- 41. Wang Y, Szretter KJ, Vermi W, Gilfillan S, Rossini C, Cella M, et al. IL‐34 is a tissue‐restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat Immunol. 2012;13:753–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lee J, Breton G, Aljoufi A, Zhou YJ, Puhr S, Nussenzweig MC, et al. Clonal analysis of human dendritic cell progenitor using a stromal cell culture. J Immunol Methods. 2015;425:21–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lee J, Breton G, Oliveira TY, Zhou YJ, Aljoufi A, Puhr S, et al. Restricted dendritic cell and monocyte progenitors in human cord blood and bone marrow. J Exp Med. 2015;212:385–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Breton G, Lee J, Zhou YJ, Schreiber JJ, Keler T, Puhr S, et al. Circulating precursors of human CD1c+ and CD141+ dendritic cells. J Exp Med. 2015;212:401–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Manz MG, Miyamoto T, Akashi K, Weissman IL. Prospective isolation of human clonogenic common myeloid progenitors. Proc Natl Acad Sci USA. 2002;99:11872–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Haniffa M, Collin M, Ginhoux F. Ontogeny and functional specialization of dendritic cells in human and mouse. Adv Immunol. 2013;120:1–49. [DOI] [PubMed] [Google Scholar]
- 47. Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. 2013;31:563–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Murphy TL, Grajales‐Reyes GE, Wu X, Tussiwand R, Briseno CG, Iwata A, et al. Transcriptional control of dendritic cell development. Annu Rev Immunol. 2016;34:93–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rathinam C, Geffers R, Yucel R, Buer J, Welte K, Moroy T, et al. The transcriptional repressor Gfi1 controls STAT3‐dependent dendritic cell development and function. Immunity. 2005;22: 717–28. [DOI] [PubMed] [Google Scholar]
- 50. Kueh HY, Champhekar A, Nutt SL, Elowitz MB, Rothenberg EV. Positive feedback between PU.1 and the cell cycle controls myeloid differentiation. Science. 2013;341:670–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Carotta S, Dakic A, D'Amico A, Pang SH, Greig KT, Nutt SL, et al. The transcription factor PU.1 controls dendritic cell development and Flt3 cytokine receptor expression in a dose‐dependent manner. Immunity. 2010;32:628–41. [DOI] [PubMed] [Google Scholar]
- 52. Cisse B, Caton ML, Lehner M, Maeda T, Scheu S, Locksley R, et al. Transcription factor E2–2 is an essential and specific regulator of plasmacytoid dendritic cell development. Cell. 2008;135:37–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Spits H, Couwenberg F, Bakker AQ, Weijer K, Uittenbogaart CH. Id2 and Id3 inhibit development of CD34(+) stem cells into predendritic cell (pre‐DC)2 but not into pre‐DC1. Evidence for a lymphoid origin of pre‐DC2. J Exp Med. 2000;192:1775–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schiavoni G, Mattei F, Sestili P, Borghi P, Venditti M, Morse HC 3rd, et al. ICSBP is essential for the development of mouse type I interferon‐producing cells and for the generation and activation of CD8alpha(+) dendritic cells. J Exp Med. 2002;196:1415–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Schiavoni G, Mattei F, Borghi P, Sestili P, Venditti M, Morse HC 3rd, et al. ICSBP is critically involved in the normal development and trafficking of Langerhans cells and dermal dendritic cells. Blood. 2004;103:2221–8. [DOI] [PubMed] [Google Scholar]
- 56. Cytlak U, Resteu A, Pagan S, Green K, Milne P, Maisuria S, et al. Differential IRF8 transcription factor requirement defines two pathways of dendritic cell development in humans. Immunity. 2020;53:353–70.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Vander Lugt B, Khan AA, Hackney JA, Agrawal S, Lesch J, Zhou M, et al. Transcriptional programming of dendritic cells for enhanced MHC class II antigen presentation. Nat Immunol. 2014;15:161–7. [DOI] [PubMed] [Google Scholar]
- 58. Fong L, Hou Y, Rivas A, Benike C, Yuen A, Fisher GA, et al. Altered peptide ligand vaccination with Flt3 ligand expanded dendritic cells for tumor immunotherapy. Proc Natl Acad Sci USA. 2001;98:8809–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Suzuki S, Honma K, Matsuyama T, Suzuki K, Toriyama K, Akitoyo I, et al. Critical roles of interferon regulatory factor 4 in CD11bhighCD8alpha‐ dendritic cell development. Proc Natl Acad Sci USA. 2004;101:8981–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Esashi E, Wang YH, Perng O, Qin XF, Liu YJ, Watowich SS. The signal transducer STAT5 inhibits plasmacytoid dendritic cell development by suppressing transcription factor IRF8. Immunity. 2008;28:509–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Satpathy AT, Briseno CG, Lee JS, Ng D, Manieri NA, Kc W, et al. Notch2‐dependent classical dendritic cells orchestrate intestinal immunity to attaching‐and‐effacing bacterial pathogens. Nat Immunol. 2013;14:937–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Grajales‐Reyes GE, Iwata A, Albring J, Wu X, Tussiwand R, Kc W, et al. Batf3 maintains autoactivation of Irf8 for commitment of a CD8alpha(+) conventional DC clonogenic progenitor. Nat Immunol. 2015;16:708–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Schlitzer A, Sivakamasundari V, Chen J, Sumatoh HR, Schreuder J, Lum J, et al. Identification of cDC1‐ and cDC2‐committed DC progenitors reveals early lineage priming at the common DC progenitor stage in the bone marrow. Nat Immunol. 2015;16:718–28. [DOI] [PubMed] [Google Scholar]
- 64. Tussiwand R, Everts B, Grajales‐Reyes GE, Kretzer NM, Iwata A, Bagaitkar J, et al. Klf4 expression in conventional dendritic cells is required for T helper 2 cell responses. Immunity. 2015;42:916–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bagadia P, Huang X, Liu TT, Durai V, Grajales‐Reyes GE, Nitschke M, et al. An Nfil3‐Zeb2‐Id2 pathway imposes Irf8 enhancer switching during cDC1 development. Nat Immunol. 2019;20:1174–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Bajana S, Turner S, Paul J, Ainsua‐Enrich E, Kovats S. IRF4 and IRF8 act in CD11c+ cells to regulate terminal differentiation of lung tissue dendritic cells. J Immunol. 2016;196:1666–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hashimoto D, Miller J, Merad M. Dendritic cell and macrophage heterogeneity in vivo. Immunity. 2011;35:323–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Edelson BT, Bradstreet TR, Kc W, Hildner K, Herzog JW, Sim J, et al. Batf3‐dependent CD11b(low/‐) peripheral dendritic cells are GM‐CSF‐independent and are not required for Th cell priming after subcutaneous immunization. PLoS One. 2011;6:e25660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science. 2008;322:1097–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Tussiwand R, Lee WL, Murphy TL, Mashayekhi M, Kc W, Albring JC, et al. Compensatory dendritic cell development mediated by BATF‐IRF interactions. Nature. 2012;490:502–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Dickinson RE, Griffin H, Bigley V, Reynard LN, Hussain R, Haniffa M, et al. Exome sequencing identifies GATA‐2 mutation as the cause of dendritic cell, monocyte, B and NK lymphoid deficiency. Blood. 2011;118:2656–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hambleton S, Salem S, Bustamante J, Bigley V, Boisson‐Dupuis S, Azevedo J, et al. IRF8 mutations and human dendritic‐cell immunodeficiency. N Engl J Med. 2011;365:127–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Guilliams M, Dutertre CA, Scott CL, McGovern N, Sichien D, Chakarov S, et al. Unsupervised high‐dimensional analysis aligns dendritic cells across tissues and species. Immunity. 2016;45:669–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. van der Aa E, van Montfoort N, Woltman AM. BDCA3(+)CLEC9A(+) human dendritic cell function and development. Semin Cell Dev Biol. 2015;41:39–48. [DOI] [PubMed] [Google Scholar]
- 75. Vremec D, Zorbas M, Scollay R, Saunders DJ, Ardavin CF, Wu L, et al. The surface phenotype of dendritic cells purified from mouse thymus and spleen: investigation of the CD8 expression by a subpopulation of dendritic cells. J Exp Med. 1992;176:47–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Annacker O, Coombes JL, Malmstrom V, Uhlig HH, Bourne T, Johansson‐Lindbom B, et al. Essential role for CD103 in the T cell‐mediated regulation of experimental colitis. J Exp Med. 2005;202:1051–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Haniffa M, Shin A, Bigley V, McGovern N, Teo P, See P, et al. Human tissues contain CD141hi cross‐presenting dendritic cells with functional homology to mouse CD103+ nonlymphoid dendritic cells. Immunity. 2012;37:60–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. McKenzie EJ, Taylor PR, Stillion RJ, Lucas AD, Harris J, Gordon S, et al. Mannose receptor expression and function define a new population of murine dendritic cells. J Immunol. 2007;178:4975–83. [DOI] [PubMed] [Google Scholar]
- 79. Segura E, Valladeau‐Guilemond J, Donnadieu MH, Sastre‐Garau X, Soumelis V, Amigorena S. Characterization of resident and migratory dendritic cells in human lymph nodes. J Exp Med. 2012;209:653–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Dutertre CA, Becht E, Irac SE, Khalilnezhad A, Narang V, Khalilnezhad S, et al. Single‐Cell analysis of human mononuclear phagocytes reveals subset‐defining markers and identifies circulating inflammatory dendritic cells. Immunity. 2019;51:573–89.e8. [DOI] [PubMed] [Google Scholar]
- 81. Bourdely P, Anselmi G, Vaivode K, Ramos RN, Missolo‐Koussou Y, Hidalgo S, et al. Transcriptional and functional analysis of CD1c(+) human dendritic cells identifies a CD163(+) subset priming CD8(+)CD103(+) T cells. Immunity. 2020;53: 335–52.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Blasius AL, Giurisato E, Cella M, Schreiber RD, Shaw AS, Colonna M. Bone marrow stromal cell antigen 2 is a specific marker of type I IFN‐producing cells in the naive mouse, but a promiscuous cell surface antigen following IFN stimulation. J Immunol. 2006;177:3260–5. [DOI] [PubMed] [Google Scholar]
- 83. Liu K, Victora GD, Schwickert TA, Guermonprez P, Meredith MM, Yao K, et al. In vivo analysis of dendritic cell development and homeostasis. Science. 2009;324:392–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Hornung V, Rothenfusser S, Britsch S, Krug A, Jahrsdorfer B, Giese T, et al. Quantitative expression of toll‐like receptor 1–10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J Immunol. 2002;168:4531–7. [DOI] [PubMed] [Google Scholar]
- 85. Leylek R, Alcantara‐Hernandez M, Lanzar Z, Ludtke A, Perez OA, Reizis B, et al. Integrated cross‐species analysis identifies a conserved transitional dendritic cell population. Cell Rep. 2019;29:3736–50.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Abbas A, Vu Manh TP, Valente M, Collinet N, Attaf N, Dong C, et al. The activation trajectory of plasmacytoid dendritic cells in vivo during a viral infection. Nat Immunol. 2020;21:983–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Coillard A, Segura E. In vivo differentiation of human monocytes. Front Immunol. 2019;10:1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Sprangers S, de Vries TJ, Everts V. Monocyte heterogeneity: consequences for monocyte‐derived immune cells. J Immunol Res. 2016;2016:1475435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Wang X, Cho B, Suzuki K, Xu Y, Green JA, An J, et al. Follicular dendritic cells help establish follicle identity and promote B cell retention in germinal centers. J Exp Med. 2011;208:2497–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Denzer K, van Eijk M, Kleijmeer MJ, Jakobson E, de Groot C, Geuze HJ. Follicular dendritic cells carry MHC class II‐expressing microvesicles at their surface. J Immunol. 2000;165:1259–65. [DOI] [PubMed] [Google Scholar]
- 91. McCloskey ML, Curotto de Lafaille MA, Carroll MC, Erlebacher A. Acquisition and presentation of follicular dendritic cell‐bound antigen by lymph node‐resident dendritic cells. J Exp Med. 2011;208:135–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. El Shikh ME, Pitzalis C. Follicular dendritic cells in health and disease. Front Immunol. 2012;3:292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Tew JG, Wu J, Fakher M, Szakal AK, Qin D. Follicular dendritic cells: beyond the necessity of T‐cell help. Trends Immunol. 2001;22:361–7. [DOI] [PubMed] [Google Scholar]
- 94. Mueller SN, Germain RN. Stromal cell contributions to the homeostasis and functionality of the immune system. Nat Rev Immunol. 2009;9:618–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Krautler NJ, Kana V, Kranich J, Tian Y, Perera D, Lemm D, et al. Follicular dendritic cells emerge from ubiquitous perivascular precursors. Cell. 2012;150:194–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Merad M, Ginhoux F, Collin M. Origin, homeostasis and function of Langerhans cells and other langerin‐expressing dendritic cells. Nat Rev Immunol. 2008;8:935–47. [DOI] [PubMed] [Google Scholar]
- 97. Sancho D, Joffre OP, Keller AM, Rogers NC, Martinez D, Hernanz‐Falcon P, et al. Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature. 2009;458:899–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Ahrens S, Zelenay S, Sancho D, Hanc P, Kjaer S, Feest C, et al. F‐actin is an evolutionarily conserved damage‐associated molecular pattern recognized by DNGR‐1, a receptor for dead cells. Immunity. 2012;36:635–45. [DOI] [PubMed] [Google Scholar]
- 99. van Dinther D, Veninga H, Iborra S, Borg EGF, Hoogterp L, Olesek K, et al. Functional CD169 on macrophages mediates interaction with dendritic cells for CD8(+) T cell cross‐priming. Cell Rep. 2018;22:1484–95. [DOI] [PubMed] [Google Scholar]
- 100. Das Mohapatra A, Tirrell I, Bénéchet AP, Pattnayak S, Khanna KM, Srivastava PK. Cross‐dressing of CD8α+ Dendritic Cells with Antigens from Live Mouse Tumor Cells Is a Major Mechanism of Cross‐priming. Cancer Immunology Research. 2020;8: 10:1287–1299. [DOI] [PubMed] [Google Scholar]
- 101. Granot T, Senda T, Carpenter DJ, Matsuoka N, Weiner J, Gordon CL, et al. Dendritic cells display subset and tissue‐specific maturation dynamics over human life. Immunity. 2017;46:504–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Calabro S, Liu D, Gallman A, Nascimento MS, Yu Z, Zhang TT, et al. Differential intrasplenic migration of dendritic cell subsets tailors adaptive immunity. Cell Rep. 2016;16:2472–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Nizzoli G, Larghi P, Paroni M, Crosti MC, Moro M, Neddermann P, et al. IL‐10 promotes homeostatic proliferation of human CD8(+) memory T cells and when produced by CD1c(+) DCs, shapes naive CD8(+) T‐cell priming. Eur J Immunol. 2016;46:1622–32. [DOI] [PubMed] [Google Scholar]
- 104. Sittig SP, Bakdash G, Weiden J, Skold AE, Tel J, Figdor CG, et al. A comparative study of the T cell stimulatory and polarizing capacity of human primary blood dendritic cell subsets. Mediators Inflamm. 2016;2016:3605643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Bosteels C, Neyt K, Vanheerswynghels M, van Helden MJ, Sichien D, Debeuf N, et al. Inflammatory Type 2 cDCs acquire features of cDC1s and macrophages to orchestrate immunity to respiratory virus infection. Immunity. 2020;52:1039–56.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Jegalian AG, Facchetti F, Jaffe ES. Plasmacytoid dendritic cells: physiologic roles and pathologic states. Adv Anat Pathol. 2009;16:392–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Brewitz A, Eickhoff S, Dahling S, Quast T, Bedoui S, Kroczek RA, et al. CD8(+) T cells orchestrate pDC‐XCR1(+) dendritic cell spatial and functional cooperativity to optimize priming. Immunity. 2017;46:205–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Fu C, Peng P, Loschko J, Feng L, Pham P, Cui W, et al. Plasmacytoid dendritic cells cross‐prime naive CD8 T cells by transferring antigen to conventional dendritic cells through exosomes. Proc Natl Acad Sci USA. 2020;117:23730–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Igyarto BZ, Jenison MC, Dudda JC, Roers A, Muller W, Koni PA, et al. Langerhans cells suppress contact hypersensitivity responses via cognate CD4 interaction and langerhans cell‐derived IL‐10. J Immunol. 2009;183:5085–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Kissenpfennig A, Henri S, Dubois B, Laplace‐Builhe C, Perrin P, Romani N, et al. Dynamics and function of Langerhans cells in vivo: dermal dendritic cells colonize lymph node areas distinct from slower migrating Langerhans cells. Immunity. 2005;22:643–54. [DOI] [PubMed] [Google Scholar]
- 111. Sere K, Baek JH, Ober‐Blobaum J, Muller‐Newen G, Tacke F, Yokota Y, et al. Two distinct types of Langerhans cells populate the skin during steady state and inflammation. Immunity. 2012;37:905–16. [DOI] [PubMed] [Google Scholar]
- 112. Hammad H, Lambrecht BN, Pochard P, Gosset P, Marquillies P, Tonnel AB, et al. Monocyte‐derived dendritic cells induce a house dust mite‐specific Th2 allergic inflammation in the lung of humanized SCID mice: involvement of CCR7. J Immunol. 2002;169:1524–34. [DOI] [PubMed] [Google Scholar]
- 113. Segura E, Touzot M, Bohineust A, Cappuccio A, Chiocchia G, Hosmalin A, et al. Human inflammatory dendritic cells induce Th17 cell differentiation. Immunity. 2013;38:336–48. [DOI] [PubMed] [Google Scholar]
- 114. van de Pavert SA, Olivier BJ, Goverse G, Vondenhoff MF, Greuter M, Beke P, et al. Chemokine CXCL13 is essential for lymph node initiation and is induced by retinoic acid and neuronal stimulation. Nat Immunol. 2009;10:1193–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Hase H, Kanno Y, Kojima M, Hasegawa K, Sakurai D, Kojima H, et al. BAFF/BLyS can potentiate B‐cell selection with the B‐cell coreceptor complex. Blood. 2004;103:2257–65. [DOI] [PubMed] [Google Scholar]
- 116. Das A, Heesters BA, Bialas A, O'Flynn J, Rifkin IR, Ochando J, et al. Follicular dendritic cell activation by TLR ligands promotes autoreactive B cell responses. Immunity. 2017;46:106–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Heesters BA, Chatterjee P, Kim YA, Gonzalez SF, Kuligowski MP, Kirchhausen T, et al. Endocytosis and recycling of immune complexes by follicular dendritic cells enhances B cell antigen binding and activation. Immunity. 2013;38:1164–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Tew JG, Mandel TE. Prolonged antigen half‐life in the lymphoid follicles of specifically immunized mice. Immunology. 1979;37:69–76. [PMC free article] [PubMed] [Google Scholar]
- 119. Jongbloed SL, Kassianos AJ, McDonald KJ, Clark GJ, Ju X, Angel CE, et al. Human CD141+ (BDCA‐3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross‐presents necrotic cell antigens. J Exp Med. 2010;207:1247–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Ferris ST, Durai V, Wu R, Theisen DJ, Ward JP, Bern MD, et al. cDC1 prime and are licensed by CD4(+) T cells to induce anti‐tumour immunity. Nature. 2020;584:624–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Krishnaswamy JK, Gowthaman U, Zhang B, Mattsson J, Szeponik L, Liu D, et al. Migratory CD11b+ conventional dendritic cells induce T follicular helper cell‐dependent antibody responses. Sci Immunol. 2017;2: 18:eaam9169. 10.1126/sciimmunol.aam9169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Melum GR, Farkas L, Scheel C, Van Dieren B, Gran E, Liu YJ, et al. A thymic stromal lymphopoietin‐responsive dendritic cell subset mediates allergic responses in the upper airway mucosa. J Allergy Clin Immunol. 2014;134:613–21.e7. [DOI] [PubMed] [Google Scholar]
- 123. Sanarico N, Ciaramella A, Sacchi A, Bernasconi D, Bossu P, Mariani F, et al. Human monocyte‐derived dendritic cells differentiated in the presence of IL‐2 produce proinflammatory cytokines and prime Th1 immune response. J Leukoc Biol. 2006;80:555–62. [DOI] [PubMed] [Google Scholar]
- 124. Qu C, Nguyen VA, Merad M, Randolph GJ. MHC class I/peptide transfer between dendritic cells overcomes poor cross‐presentation by monocyte‐derived APCs that engulf dying cells. J Immunol. 2009;182:3650–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Plantinga M, Guilliams M, Vanheerswynghels M, Deswarte K, Branco‐Madeira F, Toussaint W, et al. Conventional and monocyte‐derived CD11b(+) dendritic cells initiate and maintain T helper 2 cell‐mediated immunity to house dust mite allergen. Immunity. 2013;38:322–35. [DOI] [PubMed] [Google Scholar]
- 126. Kobayashi T, Glatz M, Horiuchi K, Kawasaki H, Akiyama H, Kaplan DH, et al. Dysbiosis and Staphylococcus aureus colonization drives inflammation in atopic dermatitis. Immunity. 2015;42:756–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Bertram KM, Botting RA, Baharlou H, Rhodes JW, Rana H, Graham JD, et al. Identification of HIV transmitting CD11c(+) human epidermal dendritic cells. Nat Commun. 2019;10:2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Yao C, Zurawski SM, Jarrett ES, Chicoine B, Crabtree J, Peterson EJ, et al. Skin dendritic cells induce follicular helper T cells and protective humoral immune responses. J Allergy Clin Immunol. 2015;136:1387–97.e1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Klechevsky E, Morita R, Liu M, Cao Y, Coquery S, Thompson‐Snipes L, et al. Functional specializations of human epidermal Langerhans cells and CD14+ dermal dendritic cells. Immunity. 2008;29:497–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Henri S, Poulin LF, Tamoutounour S, Ardouin L, Guilliams M, de Bovis B, et al. CD207+ CD103+ dermal dendritic cells cross‐present keratinocyte‐derived antigens irrespective of the presence of Langerhans cells. J Exp Med. 2010;207:189–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Liu K, Iyoda T, Saternus M, Kimura Y, Inaba K, Steinman RM. Immune tolerance after delivery of dying cells to dendritic cells in situ. J Exp Med. 2002;196:1091–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Lutz MB, Backer RA, Clausen BE. Revisiting current concepts on the tolerogenicity of steady‐state dendritic cell subsets and their maturation stages. J Immunol. 2021;206:1681–9. [DOI] [PubMed] [Google Scholar]
- 133. Guiducci C, Ott G, Chan JH, Damon E, Calacsan C, Matray T, et al. Properties regulating the nature of the plasmacytoid dendritic cell response to Toll‐like receptor 9 activation. J Exp Med. 2006;203:1999–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Ouabed A, Hubert FX, Chabannes D, Gautreau L, Heslan M, Josien R. Differential control of T regulatory cell proliferation and suppressive activity by mature plasmacytoid versus conventional spleen dendritic cells. J Immunol. 2008;180:5862–70. [DOI] [PubMed] [Google Scholar]
- 135. Flacher V, Tripp CH, Mairhofer DG, Steinman RM, Stoitzner P, Idoyaga J, et al. Murine Langerin+ dermal dendritic cells prime CD8+ T cells while Langerhans cells induce cross‐tolerance. EMBO Mol Med. 2014;6:1191–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Strandt H, Pinheiro DF, Kaplan DH, Wirth D, Gratz IK, Hammerl P, et al. Neoantigen expression in steady‐state langerhans cells induces CTL tolerance. J Immunol. 2017;199:1626–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Seneschal J, Clark RA, Gehad A, Baecher‐Allan CM, Kupper TS. Human epidermal Langerhans cells maintain immune homeostasis in skin by activating skin resident regulatory T cells. Immunity. 2012;36:873–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Trumpfheller C, Longhi MP, Caskey M, Idoyaga J, Bozzacco L, Keler T, et al. Dendritic cell‐targeted protein vaccines: a novel approach to induce T‐cell immunity. J Intern Med. 2012;271:183–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Dhodapkar MV, Sznol M, Zhao B, Wang D, Carvajal RD, Keohan ML, et al. Induction of antigen‐specific immunity with a vaccine targeting NY‐ESO‐1 to the dendritic cell receptor DEC‐205. Sci Transl Med. 2014;6:232ra51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Bhardwaj N, Friedlander PA, Pavlick AC, Ernstoff MS, Gastman BR, Hanks BA, et al. Flt3 ligand augments immune responses to anti‐DEC‐205‐NY‐ESO‐1 vaccine through expansion of dendritic cell subsets. Nature Cancer. 2020;1:1204–17. [DOI] [PubMed] [Google Scholar]
- 141. Sirvent S, Soria I, Cirauqui C, Cases B, Manzano AI, Diez‐Rivero CM, et al. Novel vaccines targeting dendritic cells by coupling allergoids to nonoxidized mannan enhance allergen uptake and induce functional regulatory T cells through programmed death ligand 1. J Allergy Clin Immunol. 2016;138:558–67.e11. [DOI] [PubMed] [Google Scholar]
- 142. Fossum E, Tesfaye DY, Bobic S, Gudjonsson A, Braathen R, Lahoud MH, et al. Targeting antigens to different receptors on conventional type 1 dendritic cells impacts the immune response. J Immunol. 2020;205:661–73. [DOI] [PubMed] [Google Scholar]
- 143. Oba T, Long MD, Keler T, Marsh HC, Minderman H, Abrams SI, et al. Overcoming primary and acquired resistance to anti‐PD‐L1 therapy by induction and activation of tumor‐residing cDC1s. Nat Commun. 2020;11:5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Joffre OP, Sancho D, Zelenay S, Keller AM, Reis e Sousa C. Efficient and versatile manipulation of the peripheral CD4+ T‐cell compartment by antigen targeting to DNGR‐1/CLEC9A. Eur J Immunol. 2010;40:1255–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Kato Y, Steiner TM, Park HY, Hitchcock RO, Zaid A, Hor JL, et al. Display of native antigen on cDC1 that have spatial access to both T and B cells underlies efficient humoral vaccination. J Immunol. 2020;205:1842–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Gudjonsson A, Lysen A, Balan S, Sundvold‐Gjerstad V, Arnold‐Schrauf C, Richter L, et al. Targeting influenza virus hemagglutinin to Xcr1(+) dendritic cells in the absence of receptor‐mediated endocytosis enhances protective antibody responses. J Immunol. 2017;198:2785–95. [DOI] [PubMed] [Google Scholar]
- 147. Gudjonsson A, Andersen TK, Sundvold‐Gjerstad V, Bogen B, Fossum E. Endocytosis deficient murine Xcl1‐fusion vaccine enhances protective antibody responses in mice. Front Immunol. 2019;10:1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Fossum E, Grodeland G, Terhorst D, Tveita AA, Vikse E, Mjaaland S, et al. Vaccine molecules targeting Xcr1 on cross‐presenting DCs induce protective CD8+ T‐cell responses against influenza virus. Eur J Immunol. 2015;45:624–35. [DOI] [PubMed] [Google Scholar]
- 149. Hartung E, Becker M, Bachem A, Reeg N, Jakel A, Hutloff A, et al. Induction of potent CD8 T cell cytotoxicity by specific targeting of antigen to cross‐presenting dendritic cells in vivo via murine or human XCR1. J Immunol. 2015;194:1069–79. [DOI] [PubMed] [Google Scholar]
- 150. Chappell CP, Giltiay NV, Draves KE, Chen C, Hayden‐Ledbetter MS, Shlomchik MJ, et al. Targeting antigens through blood dendritic cell antigen 2 on plasmacytoid dendritic cells promotes immunologic tolerance. J Immunol. 2014;192:5789–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Panda SK, Kolbeck R, Sanjuan MA. Plasmacytoid dendritic cells in autoimmunity. Curr Opin Immunol. 2017;44:20–5. [DOI] [PubMed] [Google Scholar]
- 152. Kranich J, Krautler NJ, Heinen E, Polymenidou M, Bridel C, Schildknecht A, et al. Follicular dendritic cells control engulfment of apoptotic bodies by secreting Mfge8. J Exp Med. 2008;205:1293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Rahman ZS, Shao WH, Khan TN, Zhen Y, Cohen PL. Impaired apoptotic cell clearance in the germinal center by Mer‐deficient tingible body macrophages leads to enhanced antibody‐forming cell and germinal center responses. J Immunol. 2010;185:5859–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Suurmond J, Diamond B. Autoantibodies in systemic autoimmune diseases: specificity and pathogenicity. J Clin Invest. 2015;125:2194–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Kim JH, Hu Y, Yongqing T, Kim J, Hughes VA, Le Nours J, et al. CD1a on Langerhans cells controls inflammatory skin disease. Nat Immunol. 2016;17:1159–66. [DOI] [PMC free article] [PubMed] [Google Scholar]