

Abstract
Background and Aims
Hypoxia is a common feature of the tumor microenvironment (TME), which promotes tumor progression, metastasis, and therapeutic drug resistance through a myriad of cell activities in tumor and stroma cells. While targeting hypoxic TME is emerging as a promising strategy for treating solid tumors, preclinical development of this approach is lacking in the study of HCC.
Approach and Results
From a genome‐wide CRISPR/CRISPR‐associated 9 gene knockout screening, we identified aldolase A (ALDOA), a key enzyme in glycolysis and gluconeogenesis, as an essential driver for HCC cell growth under hypoxia. Knockdown of ALDOA in HCC cells leads to lactate depletion and consequently inhibits tumor growth. Supplementation with lactate partly rescues the inhibitory effects mediated by ALDOA knockdown. Upon hypoxia, ALDOA is induced by hypoxia‐inducible factor‐1α and fat mass and obesity–associated protein–mediated N6‐methyladenosine modification through transcriptional and posttranscriptional regulation, respectively. Analysis of The Cancer Genome Atlas shows that elevated levels of ALDOA are significantly correlated with poor prognosis of patients with HCC. In a screen of Food and Drug Administration–approved drugs based on structured hierarchical virtual platforms, we identified the sulfamonomethoxine derivative compound 5 (cpd‐5) as a potential inhibitor to target ALDOA, evidenced by the antitumor activity of cpd‐5 in preclinical patient‐derived xenograft models of HCC.
Conclusions
Our work identifies ALDOA as an essential driver for HCC cell growth under hypoxia, and we demonstrate that inhibition of ALDOA in the hypoxic TME is a promising therapeutic strategy for treating HCC.
Abbreviations
- ALDOA
aldolase A
- ALKBH5
alpha‐ketoglutarate‐dependent dioxygenase alkB homolog 5
- Cas9
CRISPR‐associated protein 9
- cpd‐5
compound 5
- DOX
doxycycline
- ECAR
extracellular acidification rate
- EdU
5‐ethynyl‐2′‐deoxyuridine
- FDA
Food and Drug Administration
- FTO
fat mass and obesity–associated protein
- G3P
glyceraldehyde‐3‐phosphate
- HIF‐1
hypoxia‐inducible factor
- HRE
hypoxia‐responsive element
- IC50
50% inhibition concentration
- IHC
immunohistochemistry
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- m6A
N6‐methyladenosine
- METTL3/14
methyltransferase‐like 3/14
- MFGE8
milk fat globule EGF factor 8
- OPLS‐DA
orthogonal PLS‐DA
- PCA
principal component analysis
- PDX
patient‐derived xenograft
- PLS‐DA
partial least squares discriminant analysis
- RNAi
RNA interference
- sg‐
single‐guide
- sh‐
short hairpin
- si‐
small interfering
- TCGA
The Cancer Genome Atlas
- TME
tumor microenvironment
- VEGFR
VEGF receptor
- YTHDF1/2
YTH N6‐methyladenosine RNA binding protein 1/2
HCC accounts for the majority of primary liver malignancies and is the fourth leading cause of cancer‐related death worldwide.( 1 ) The incidence of HCC is increasing due to the prevalence of HBV infection, HCV infection, alcohol abuse, and the increase of NAFLD.( 2 ) Surgical resection and transplantation are the main curative treatments for patients with HCC at the early‐onset stage with long‐term survival rates.( 3 ) Because most patients with HCC are diagnosed at the late stage, systemic therapies such as sorafenib treatment have been used as the frontline therapy for advanced HCC but still with limited clinical benefits for patients.( 4 ) Recently, emerging targeted therapies have produced potent and specific responses and minimized the toxicity in the treatment of advanced HCC. For example, lenvatinib, a multiple tyrosine kinase inhibitor against VEGF receptor 1 (VEGFR1), VEGFR2, and VEGFR3, received Food and Drug Administration (FDA) approval for the treatment of advanced HCC in 2018.( 5 ) Therefore, it is of great clinical importance and urgency to develop efficacious targeted intervention for advanced HCC.
Intratumoral hypoxia or oxygen deprivation is a common survival state of human advanced solid tumors, including HCC. The median O2 partial pressure in HCC is 6 mm Hg compared with 30 mm Hg in normal liver.( 6 ) The homeostatic response to the intratumoral hypoxic environment is mediated by hypoxia‐inducible factor (HIF‐1), consisting of an α‐subunit and a β‐subunit,( 7 ) which triggers a variety of cellular responses such as angiogenesis,( 8 ) erythropoiesis,( 9 ) apoptosis,( 10 ) metastasis,( 11 ) drug resistance,( 12 ) autophagy,( 13 ) and stemness.( 14 ) In a recent study, we reported that hypoxia induces sorafenib resistance in HCC in an N6‐methyladenosine (m6A)–dependent manner and that methyltransferase‐like 3 (METTL3) depletion enhances forkhead box O3–mediated autophagy under hypoxia.( 15 ) Targeting the hypoxic tumor microenvironment (TME) by specifically blocking glutamine metabolism enhances T‐cell oxidative phosphorylation and anticancer immune responses,( 16 ) highlighting the importance of developing effective therapies under hypoxic conditions. Loss‐of‐function genetic screening approaches have been widely used to study the molecular mechanisms and functional consequences of gene deletion in various tumor cells.( 17 , 18 ) These approaches include pooled RNA interference (RNAi) screens using a short hairpin RNA (shRNA) library for gene knockdown and pooled genetic screens using a CRISPR/CRISPR‐associated 9 (Cas9) system for gene knockout that simultaneously investigate the role of a group of genes on tumor cell growth, viability, metastasis, and drug resistance.( 19 , 20 ) However, these screens have not been applied to directly assess the role of the hypoxic TME in HCC. Examining the role of hypoxia in HCC can provide comprehensive insights into the molecular mechanisms of the occurrence and the hypoxia‐induced tumor growth in HCC and identify potential therapeutic targets specific for advanced HCC.
In this study, we performed a genome‐wide screen using a CRISPR/Cas9 genome editing system in HCC cells to identify genes that are required for cell growth and viability under hypoxic conditions and integrate with differentially expressed genes in patients with HCC from The Cancer Genome Atlas (TCGA) and a local cohort and in HCC cells responding to hypoxia. We identified aldolase A (ALDOA), a key enzyme in the glycolytic pathway by catalyzing the reversible conversion of fructose‐1,6‐bisphosphate to glyceraldehyde‐3‐phosphate (G3P) and dihydroxyacetone phosphate,( 21 ) as an essential gene for HCC under hypoxia. Up‐regulation of ALDOA promoted HCC cell proliferation and migration under hypoxia in vitro and in vivo. By metabolomics analysis, we found that the level of lactate was significantly decreased in ALDOA‐depleted HCC cells and that rescue of lactate accumulation in ALDOA‐depleted HCC cells alleviated inhibitory effects on tumor growth under hypoxia. Our findings suggest that targeting ALDOA is a promising strategy to challenge the hypoxic tumor microenvironment in patients with advanced HCC.
Materials and Methods
Plasmid Constructions, Cell Transfection, and Infection
The human CRISPR Knockout Pooled Library was obtained from Addgene (1000000049). The library was amplified and used according to instructions, as described.( 18 ) Model‐based analysis of genome‐wide CRISPR/Cas9 (MAGeCK) of the library sequencing data was performed by Novogene (Beijing, China). Stable knockdown of target genes was achieved by lentivirus‐based shRNA delivery. PLKO.1 vectors with antipuromycin or antihygromycin plasmid were constructed, while doxycycline (DOX)–induced stable knockdown of ALDOA was achieved by Tet‐pLKO‐puro (21915; Addgene). Stable knockout of ALDOA was accomplished by lentiCRISPRv2 (52961; Addgene). All primer sequences are listed in Supporting Table S1. For the ALDOA overexpressing system, ALDOA complementary DNA (KR709337) was cloned into pCDH‐puro lentiviral vector (CD510B‐1; System Biosciences). For shRNA knockdown and overexpression, pLKO.1, Tet‐pLKO‐puro, lentiCRISPRv2, and pCDH constructs together with packing and helper plasmids PAX2 and MD2G were cotransfected into HEK‐293T cells by the Calcium Phosphate Transfection Kit (CAPHOS‐1KT; Sigma). Viruses were collected, filtered, and titrated before infecting target cells with 8 mg/mL polybrene (TR‐1003, Sigma). The infected cells were screened by puromycin or hygromycin accordingly. Small interfering RNA (siRNA) duplexes were synthesized by TSINGKE (Guangzhou, China) and transfected into cells by Lipofectamine3000 (Thermo Fisher).
Seahorse
Liver cancer cells (shCtrl, shALDOA#1, and shALDOA#2) were seeded to a 96‐well Seahorse XF Cell Culture Microplate (8,000 cells per well). After 24 hours in hypoxia, the cell culture growth medium was changed to a prewarmed assay medium, and the cell culture microplate was placed into a 37°C non‐CO2 incubator for 45 minutes. Glycolysis (extracellular acidification rate [ECAR]) was measured in the XF Analyzer according to the manual of the Seahorse XF Cell Mito Stress Test Assay (Seahorse Bioscience of Agilent Technologies).
Liver cancer patient samples
The liver tumor and normal tissues (fresh tissues and paraffin‐embedded specimens) were obtained from the First Affiliated Hospital of Sun Yat‐Sen University which were approved by the institutional review board of the hospital. The study was compliant with all relevant ethical regulations regarding research involving human participants and the informed consents were collected.
Liver cancer patient samples The liver tumor and normal tissues (fresh tissues and paraffin‐embedded specimens) were obtained from the First Affiliated Hospital of Sun Yat‐Sen University which were approved by the institutional review board of the hospital. The study was compliant with all relevant ethical regulations regarding research involving human participants and the informed consents were collected. The detailed methodology can be found in the Supporting Materials and Methods.
Results
Genome‐Wide CRISPR/Cas9 Library Screening and Transcriptome Sequencing Identified ALDOA as an Essential Driver for HCC Cell Growth Under Hypoxia
Hypoxia is a hallmark of the solid TME with poor prognosis, which contributes to the plasticity and the heterogeneity of tumors, generates therapeutic resistance, and promotes malignant progression.( 22 ) Targeting the hypoxic TME is an unexplored strategy for cancer therapy. In this study, we performed transcriptome sequencing analysis of human HCC tumors and human HCC cell lines under hypoxia (Fig. 1A) and used a genome‐wide CRISPR/Cas9 knockout library to identify essential genes responding to hypoxia in HCC tumor cells (Fig. 1B). HCC cells were cultured in 20% O2 and 1% O2 for 3 days to mimic normoxia and hypoxia, respectively. We hypothesized that knockout of hypoxia‐responding essential genes would sensitize HCC cells to a hypoxic microenvironment and induce cell death or cell growth retardation. Under hypoxia, cells carrying single‐guide RNA (sgRNA) targeting hypoxia‐adaptation genes will be negatively selected in the knockout mutant cell pool and their corresponding sgRNA will be depleted in the library, determined by deep sequencing and MAGeCK analysis. From our CRISPR/Cas9 knockout library screening, we identified a subset of sgRNAs targeting 3,103 genes including protein‐coding genes; noncoding RNAs were significantly depleted (log fold change <−1.5) under hypoxia compared to those under normoxia (GSE168544), among which 215 genes were consistent with the ones with previous screening results for essential genes in K562 cells in hypoxia( 23 ) (Supporting Fig. S1A, GSE144527), and 227 genes harbored putative HIF1α binding elements in the promoters( 24 ) (Supporting Fig. S1B, GSE16347), respectively. From transcriptome analysis, we identified 613 genes with significant up‐regulation under hypoxia in the human cell line (GSE168544). Differentially expressed genes under hypoxia from transcriptome analysis and CRISPR/Cas9 knockout screening were then compared with the databases of our local pairs of HCC tumors (GSE143235) and of cancer/HCC‐related genes from TCGA. Based on the comparison, two genes were overlapped and associated with the hypoxic response in HCC (Fig. 1C‐E). We further validated the up‐regulation of these overlapped genes under hypoxia compared to normoxia by RT‐PCR (Supporting Fig. S1C). Knockdown of these two genes by shRNAs was performed to validate their essential role in hypoxia (Supporting Fig. S1D). Among the list of genes, fructose‐bisphosphate ALDOA, the key enzyme of glycolysis and gluconeogenesis, was identified as the most negatively selected gene under hypoxic conditions in HCC. Depletion of ALDOA dramatically inhibited growth of SMMC‐7721 cells under hypoxia (Fig. 1F). To assess the sensitivity of various ALDOA levels in HCC cells to hypoxia, we generated inducible ALDOA‐knockdown HCC cells and measured their response to hypoxia (Supporting Fig. S1E). Treatment with DOX at a high dose caused significant HCC cell death under hypoxia, and the inhibitory effects were shown in a dose‐dependent manner in hypoxia compare to those in normoxia (Fig. 1G,H). The results suggest that ALDOA may be an essential driver for HCC cell growth under hypoxia and indicate a potential therapeutic window by targeting ALDOA under hypoxia in HCC cells.
Fig. 1.

Genome‐wide CRISPR/Cas9 library screening and RNA sequencing identified ALDOA as an essential driver for HCC cell growth under hypoxia. (A) Strategy to screen targets for HCC under hypoxia in vivo and in vitro by RNA sequencing. (B) Schematic diagram of genome‐wide CRISPR/Cas9 knockout library for HCC cells under hypoxia. (C) Venn diagrams show overlapped essential driver genes for HCC cell growth in response to hypoxia. (D) Volcano plots reveal that ALDOA and milk fat globule EGF and factor V/VIII domain containing (MFGE8) targeting sgRNAs were negatively selected under hypoxia, indicating their essential role for HCC cells to survive in response to hypoxia. (E) ALDOA and MFGE8 were identified as the overlapped gene in the library screening as indicated by the black dot from volcano plots. (F) Cell viability of overlapped expressed genes in SMMC‐7721 cells under hypoxia. (G,H) Dox‐induced suppression of ALDOA inhibited the proliferation of HepG2 and SMMC‐7721 cells under hypoxia but not of cells under normoxia (G), and quantification analyses (H) are shown. ***P < 0.001. Abbreviations: KO, knockout; OD, optical density.
Knockdown of ALDOA Inhibited HCC Cell Proliferation and Migration Under Hypoxia In Vitro and In Vivo
To investigate the functional roles of ALDOA in HCC under hypoxia, we generated the stable ALDOA‐knockdown cell lines SMMC‐7721 and HepG2 by two distinct shRNA lentiviruses (shALDOA#1 and #2). Successful knockdown of ALDOA was verified at both the protein and RNA levels (Supporting Fig. S2A,B). Knockdown of ALDOA significantly inhibited growth of SMMC‐7721 and HepG2 cells under hypoxia (Fig. 2A,B) and induced cell cycle arrest (Fig. 2C). The 5‐ethynyl‐2′‐deoxyuridine (EdU) staining assay showed that knockdown of ALDOA suppressed cell proliferation under hypoxia (Fig. 2D). Furthermore, knockdown of ALDOA impaired HCC colony‐formation abilities and mammosphere‐formation abilities under hypoxia, as indicated by the reduced number and size (Fig. 2E,F). We further generated the ALDOA‐knockout HCC cell lines using the CRISPR/Cas9 system (Supporting Fig. S2C) and confirmed that knockout of ALDOA resulted in cell growth retardation and decrease of colony formation under hypoxia in HCC cells (Supporting Fig. S2D,E). However, overexpression of ALDOA in HCC cells failed to promote cell growth under hypoxia, which may be due to the high basal level of ALDOA under hypoxia (Supporting Fig. S2F,G).
Fig. 2.
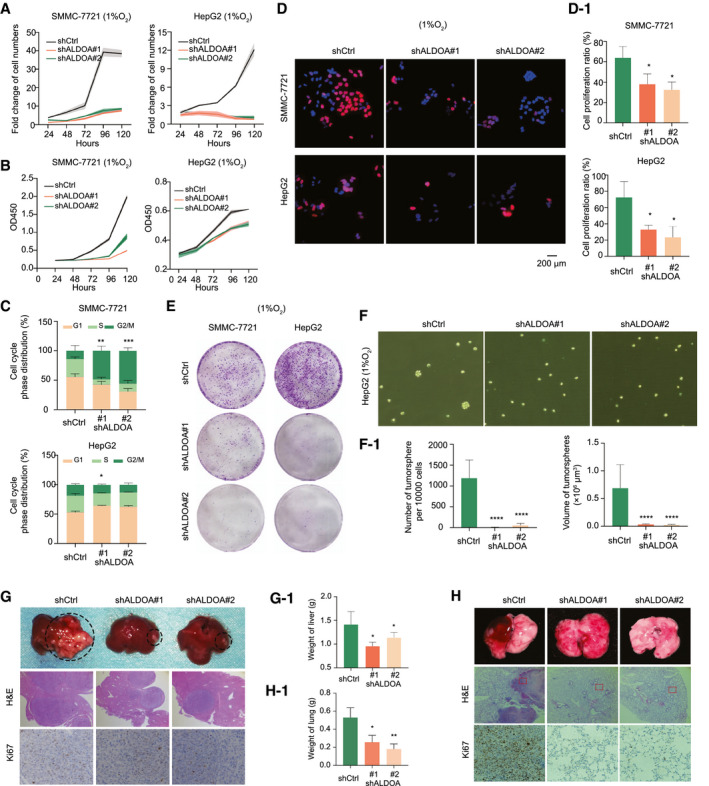
ALDOA promoted HCC cell proliferation and migration under hypoxia in vitro and in vivo. (A,B) Knockdown of ALDOA significantly reduced numbers (A) and growth (B) of SMMC‐7721 cells and HepG‐2 cells under hypoxia. (C) Knockdown of ALDOA induced HCC cell cycle arrest under hypoxia. (D) Knockdown of ALDOA significantly reduced HCC cell proliferation under hypoxia by EdU assay. Quantification of fold change is shown in D‐1. (E) Knockdown of ALDOA impaired colony formation in HCC cells under hypoxia. (F) Knockdown of ALDOA impaired mammosphere formation of HepG2 cells under hypoxia. Quantification of fold change is shown in F‐1. (G) Knockdown of ALDOA dramatically suppressed liver tumor growth in orthotopic xenograft mouse models. Stable ALDOA‐knockdown Hepa1‐6 cells (5 × 106), and control cells were injected into the liver of each female C57 mouse. Three weeks after injection, livers were separated for pathological analysis. Liver weights are shown in G‐1. (H) Knockdown of ALDOA abolished lung metastasis from HCC. Hepa1‐6 cells (5 × 106) were tail vein–injected into female C57 mice. Formation of metastatic foci in the lung was pictured after 2 weeks. Metastases were confirmed by pathological analysis. Lung weights are shown in H‐1. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Abbreviations: H&E, hematoxylin and eosin; OD, optical density.
To determine whether the oncogenic role of ALDOA in HCC depends on hypoxia, we performed cell proliferation assays in normoxic conditions. We noticed a slightly reduced capacity for HCC cell growth in ALDOA‐knockdown or ALDOA‐knockout HCC cells under normoxia (Supporting Fig. S2H‐J), indicating that ALDOA is a hypoxia‐dependent oncogene in HCC. Moreover, because there is a total of three aldolase isozymes in humans,( 25 ) we further examined whether the other two aldolase enzymes, ALDOB and ALDOC, play similar roles as ALDOA does in HCC under hypoxia. ALDOB was undetectable in HCC cells, and knockdown of ALDOC failed to suppress cell growth in HCC under hypoxia (Supporting Fig. S2K,L).
Next, we investigated whether ALDOA plays a critical role in HCC migration under hypoxia. A wound‐healing assay and a transwell cell migration assay showed that depleting ALDOA resulted in suppression of HCC migration under hypoxia (Supporting Fig. S3A,B). By examining the epithelial–mesenchymal transition markers, we found that knockdown of ALDOA significantly induced expression of E‐cadherin at both the RNA and protein levels, while conversely it reduced expression of vimentin at both the RNA and protein levels (Supporting Fig. S3C,D). To further verify the oncogenic role of ALDOA in HCC in vivo, we performed an orthotopic xenograft mouse model and a tail vein injection metastasis model to test the effect of ALDOA‐knockdown on HCC tumorigenicity. Stable knockdown of ALDOA in Hepa1‐6 cells effectively suppressed tumor growth in the orthotopic liver microenvironment in C57 mice and attenuated lung metastasis as indicated by significant reductions of tumor size in liver and tissue weight when compared to the nonspecific shRNA control (Fig. 2G,H; Supporting Fig. S3E).
To determine the essentiality of ALDOA in normal cells, we generated two stable ALDOA‐knockout cell lines using the parental normal liver cell line LO2 in the CRISPR/Cas9 system (Supporting Fig. S4A). Knockout of ALDOA in LO2 cells had little effect on cell growth and colony formation under hypoxia (Supporting Fig. S4B,C). In vivo, we applied two mouse‐specific siRNAs to target ALDOA in normal mice to assess the effects caused by ALDOA depletion (Supporting Fig. S4D). Knockdown of ALDOA in vivo by siRNA showed no obvious effects on the visible pathological features of mouse livers and body weight (Supporting Fig. S4E,F). Targeting ALDOA by siRNAs in vivo had no impact on liver function according to liver function tests (Supporting Fig. S4G). Taken together, these data indicate that ALDOA plays a crucial role in promoting tumor growth and migration under hypoxia as a unique aldolase enzyme induced by hypoxia in HCC but has no effects in normal liver cells.
Transcriptome Sequencing and Metabolomics Analysis Identified Lactate as a Major Metabolic Player in HCC Mediated by ALDOA
To delineate the functional implications of ALDOA in HCC, transcriptome sequencing was performed to interrogate the differential gene expression in ALDOA‐knockdown HepG2 cells under hypoxia. We identified 313 genes with significant up‐regulation and 493 genes with significant down‐regulation (Fig. 3A, GSE168544). Gene enrichment analysis revealed gene sets involved in metabolic pathways, glycolysis/gluconeogenesis, biosynthesis of amino acids, and so on (Fig. 3B). Pathway relation networks of all enriched gene sets were analyzed by Cytoscape (Supporting Fig. S5), highlighting that the metabolic pathway functions as the core of the network mediated by ALDOA under hypoxia, which is listed in the heatmap (Fig. 3C). To examine the ultimate metabolite changes, we performed metabolomics analysis in the ALDOA‐knockdown HepG‐2 cells under hypoxia using gas chromatography‐mass spectrometry and carried out multivariate statistical analysis by principal component analysis (PCA), partial least squares discriminant analysis (PLS‐DA), and orthogonal PLS‐DA (OPLS‐DA) (Fig. 3D). In total, we identified 170 metabolites with significant changes in the ALDOA‐knockdown HepG2 cells under hypoxia. Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis of these 170 metabolites highlighted dysregulation of key metabolomes in the ALDOA‐knockdown HCC cells (Fig. 3E). By systematic integration of these metabolomic pathways, we observed the majority of variable pathways enriched in downstream pathways modulated by glycolysis (Fig. 3F), as indicated by the metabolites represented in this core pathway (Fig. 3G‐J). Among these, lactate, the terminal product of glycolysis, was identified as the major target in ALDOA‐mediating metabolism in HCC under hypoxia (Fig. 3F,J).
Fig. 3.
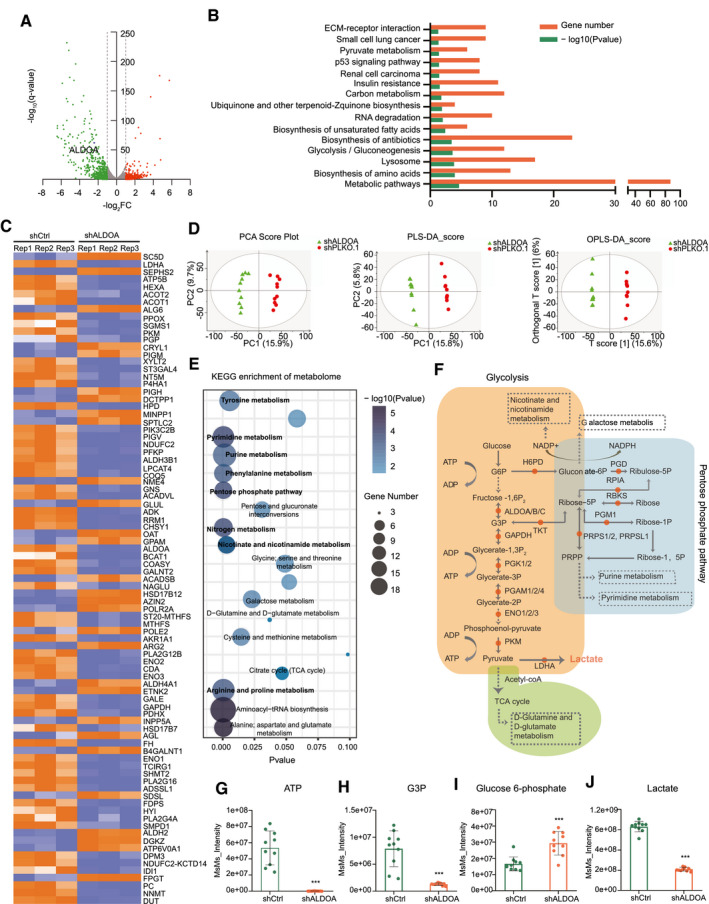
Transcriptome sequencing and metabonomic analysis identified lactate as a terminal metabolic player in HCC mediated by ALDOA. (A) Differentially expressed genes were identified in ALDOA knockdown HepG2 cells by RNA sequencing. (B) KEGG enrichment analysis of genes altered by ALDOA in HepG‐2 cells. (C) Heatmap of altered genes in metabolic pathway in ALDOA‐knockdown HepG2 cells under hypoxia. (D) Metabonomic analysis of ALDOA‐knockdown HepG2 cells under hypoxia by PCA, PLS‐DA, and OPLS‐DA. (E) KEGG enrichment analysis of metabolites altered by ALDOA in HepG2 cells under hypoxia. (F) Systematic integration of altered metabolites in ALDOA‐knockdown HepG2 cells under hypoxia. (G‐J) Levels of ATP (G), G3P (H), glucose 6‐phosphate (I), and lactate (J) in ALDOA‐knockdown HepG2 cells under hypoxia. Data were obtained from metabonomic analysis. ***P < 0.001. Abbreviations: ACOT1/2, acyl‐CoA thioesterase 1/2; ADP, adenosine diphosphate; ALDOA/B/C, fructose‐bisphosphate aldolase A/B/C; ALG6, alpha‐1,3‐glucosyltransferase; ATP, adenosine triphosphate; DUT, deoxyuridine triphosphatase; ECM, extracellular matrix; ENO1/2/3, enolase 1/2/3; FC, fold change; FH, fumarate hydratase; G3P, glyceraldehyde 3‐phosphate; G6P, glucose 6‐phosphate; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; H6PD, hexose‐‐phosphate dehydrogenase; HEXA, hexosaminidase subunit alpha; LDHA, lactate dehydrogenase A; OAT, ornithine aminotransferase; PC, principal component; PGAM1/2/4, phosphoglycerate mutase 1/2/4; PGD, phosphogluconate dehydrogenase; PGK1/2, phosphoglycerate kinase 1/2; PGM1, phosphoglucomutase 1; PGP, phosphoglycolate phosphatase; PKM, pyruvate kinase M1/2; PPOX, protoporphyrinogen oxidase; PRPS1/2, phosphoribosyl pyrophosphate synthetase 1/2; RBKS, ribokinase; RPIA, ribose 5‐phosphate isomerase A; SC5D, sterol‐C5‐desaturase; SEPHS2, selenophosphate synthetase 2; SGMS1, sphingomyelin synthase 1; TCA, tricarboxylic acid; TKT, transketolase.
Rescue of Lactate Alleviated ALDOA‐Dependent Liver Tumor Growth Under Hypoxia in HCC Cells
To validate that the effects of ALDOA‐dependent tumor inhibition are mainly mediated by lactate, we first measured glycolysis metabolism in ALDOA‐deficient HCC cells. Significant reduction of ECAR levels was detected in the ALDOA‐knockdown HCC cells under hypoxia, indicating the direct influence on glycolysis by ALDOA (Fig. 4A). Depletion of ALDOA significantly decreased both intracellular and extracellular lactate levels in HCC cells under hypoxia, which was consistent with our previous metabonomic analysis (Fig. 4B,C). Next, we rescued lactate levels in the ALDOA‐depleted HCC cells by addition of 4 mM lactate in cell culture medium. Supplementation of lactate partly rescued cell growth and cell proliferation in the ALDOA‐knockdown HCC cells (Fig. 4D,E), as well as restored partial mammosphere formation and colony formation (Fig. 4F,G) under hypoxia. To determine the effects of ALDOA‐dependent lactate in vivo, we performed subcutaneous implantation with stable ALDOA‐knockdown H22 cells into 4‐week‐old female Balb/c mice. Intratumoral injection of lactate (6 mmol/L) every 3 days in stable ALDOA‐knockdown tumors promoted HCC tumor growth in mice as indicated by the increase of tumor size and tumor weight, comparing to the stable ALDOA‐knockdown tumors with intratumoral injection of PBS (Fig. 4H‐J). Collectively, our results suggest that the lactate levels mediated by ALDOA affected HCC tumor growth under hypoxia and that restoration of the lactate levels in ALDOA‐knockdown HCC cells could alleviate the inhibitory effects mediated by depletion of ALDOA in vitro and in vivo.
Fig. 4.
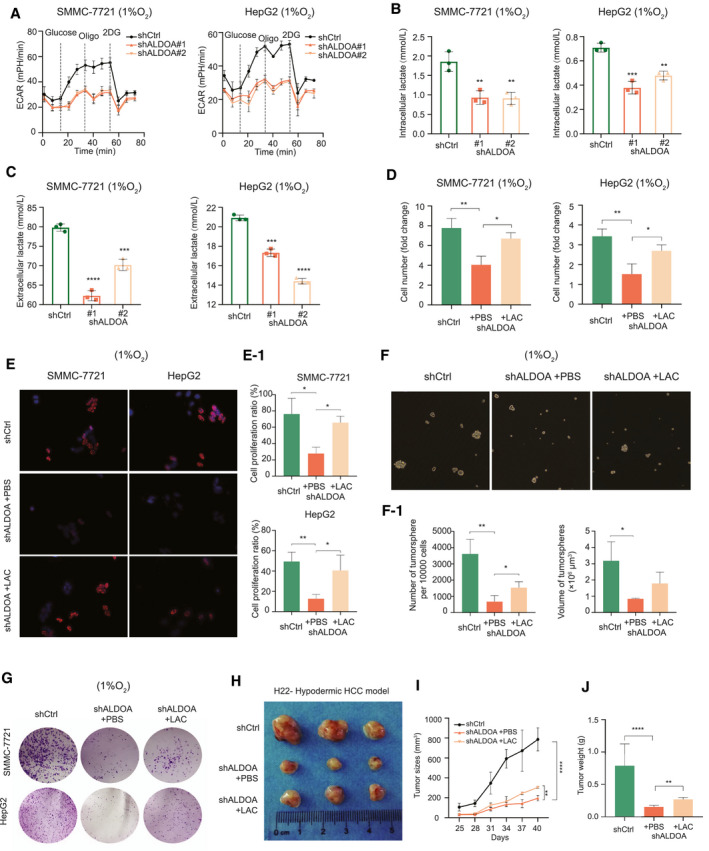
Rescue of lactate alleviated ALDOA‐dependent tumor growth under hypoxia in HCC cells. (A) Knockdown of ALDOA effectively decreased ECAR in HCC cells under hypoxia. (B) Knockdown of ALDOA decreased intracellular lactate levels in HCC cells under hypoxia. (C) Knockdown of ALDOA decreased extracellular lactate levels in HCC cells under hypoxia. (D) Supplementation of lactate in cell culture medium (4 mM) rescued cell proliferation in ALDOA‐knockdown HCC cells under hypoxia by Cell Counting Kit‐8 assays. (E) Supplementation of lactate in cell culture medium (4 mM) rescued cell proliferation in ALDOA‐knockdown HCC cells under hypoxia by EdU assay. Quantification of fold changes is shown in E‐1. (F) Supplementation of lactate in cell culture medium (4 mM) rescued mammosphere formation phenotype of ALDOA‐knockdown HepG2 cells under hypoxia. Quantification of fold changes is shown in F‐1. (G) Supplementation of lactate in cell culture medium (4 mM) rescued colony formation in ALDOA‐knockdown HCC cells under hypoxia. (H) Intratumoral injection of lactate (6 mM) effectively promoted ALDOA‐knockdown Hepa1‐6 cell growth in mice. (I,J) Tumor growth curve (I) and tumor weight (J) in ALDOA‐knockdown H22 cell–derived xenografts treated with lactate injection. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Abbreviations: 2DG, 2‐deoxyglucose; LAC, lactate.
ALDOA was Regulated by HIF‐1α and the m6A Modification Under Hypoxia
We next questioned the detailed molecular mechanisms by which ALDOA was up‐regulated under hypoxia. Previously, HIF‐1α, the transcriptional activator upon hypoxia stimulation, was found to be highly expressed in HCC and to activate a series of downstream targets that play important roles in clinical outcome in patients with HCC.( 26 , 27 ) Correlation analysis of the data set of HCC tumors revealed a significant positive correlation between ALDOA and HIF‐1α (Fig. 5A). Knockdown of HIF‐1α abrogated the induction of ALDOA in HCC cells under hypoxia by RT‐PCR assay (Fig. 5B; Supporting Fig. S6A). To further verify the correlation of ALDOA and HIF‐1α in vivo, we performed subcutaneous implantation of SMMC‐7721 in mice and measured the protein levels by immunofluorescence microscopy. A positive correlation of ALDOA and HIF‐1α was found at protein levels in vivo, and ALDOA was coexpressed with HIF‐1α in tumors, mostly in the nucleus (Fig. 5C). We further confirmed the coexpression of ALDOA and HIF‐1α in tumors of patients with HCC and HCC cell lines by an immunofluorescent localization assay (Fig. 5D; Supporting Fig. S6B). Interestingly, some of the HIF‐1α was located in the cytoplasm of various cells in the TME, indicating the heterogeneity in tumors of patients with HCC. By systematic analysis of the HIF‐1α chromatin immunoprecipitation (ChIP) sequencing profiles of HepG2 (GSE16347),( 28 ) we identified three putative hypoxia‐responsive elements (HREs) containing the HIF‐1α‐binding consensus sequence 5′‐A/GCGTG‐3′ in the promoter region of ALDOA (Fig. 5E). Subsequently, we generated three deleted mutants of the ALDOA promoter and cloned them into the pmirGLO vector with a dual luciferase reporter (Fig. 5E). Upon hypoxia treatment, we found a significant induction of luciferase signal in the wild‐type form of the ALDOA promoter compared with the normoxia treatment. However, deletion of sites 1 and 2 resulted in a dramatic decrease of luciferase signal, indicating that sites 1 and 2 were the functional elements recognized by HIF‐1α (Fig. 5F).
Fig. 5.
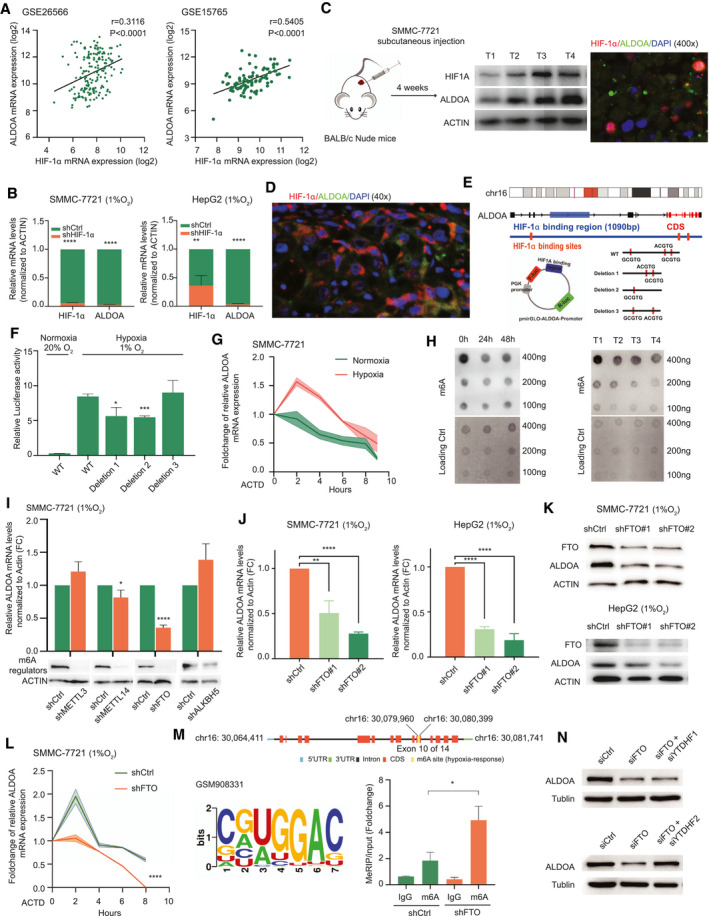
ALDOA was regulated by HIF‐1α and the m6A modification under hypoxia. (A) Correlation analysis of the RNA levels of HIF‐1α and ALDOA in HCC tumor samples. (B) Silencing of HIF‐1α decreased ALDOA expression at the mRNA level in HCC cells under hypoxia. (C) Correlation and colocalization analysis of ALDOA and HIF‐1α in a subcutaneous xenograft HCC mouse model. (D) Correlation and colocalization analysis of ALDOA and HIF‐1α in tumor tissues of patients with HCC by immunofluorescence histochemistry. (E) Wild‐type and mutants of HIF‐1α binding elements of ALDOA promoter were fused into a firefly luciferase reporter. Three potential HIF‐1α binding elements were found in the ALDOA promoter region, and mutant forms of ALDOA promoter were deleted as indicated. (F) Relative luciferase activity of the wild‐type and three mutant forms of ALDOA promoter in SMMC‐7721 cells under hypoxia. (G) Half‐life of ALDOA mRNA in SMMC‐7721 cells under normoxia or under hypoxia treated with actinomycin D. (H) Global mRNA m6A levels in SMMC‐7721 cells under hypoxia and in subcutaneous xenograft HCC tumors were determined by RNA m6A dot‐blotting assay. (I) Knockdown of key regulators of m6A modification affected the expression of ALDOA mRNA under hypoxia. (J) Knockdown of FTO significantly reduced the expression of ALDOA mRNA under hypoxia in HCC cells. (K) Knockdown of FTO significantly reduced the expression of ALDOA protein under hypoxia in HCC cells. (L) Half‐life of ALDOA mRNA in FTO‐knockdown SMMC‐7721 cells under hypoxia treated with actinomycin D. (M) Knockdown of FTO promoted m6A methylation in ALDOA mRNA by the m6A methylated RIP analysis in SMMC‐7721 cells. (N) Knockdown of YTHDF2, but not YTHDF1, rescued expression of ALDOA in FTO‐depleted SMMC‐7721 cells. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Abbreviations: ACTD, actinomycin D; CDS, coding sequence; FC, fold change; WT, wild type.
Next, we measured the half‐life of ALDOA mRNA under hypoxia by pretreating the cells with actinomycin D. In the presence of actinomycin D, significant decay of ALDOA mRNA was observed under hypoxia compared with the normoxia treatment, suggesting that ALDOA may be modulated in a posttranscriptional way under hypoxia as well (Fig. 5G). Previous reports have shown that the m6A modification plays important roles in regulating precursor mRNA maturation, translation, and degradation.( 29 , 30 , 31 , 32 ) In this regard, we tested whether the m6A modification was involved in posttranscriptional regulation of ALDOA under hypoxia. The m6A levels in HCC cell lines were decreased under hypoxia compared with those under normoxia, along with the decreased level of m6A in the HCC tumor tissues with a hypoxic microenvironment (Fig. 5H). We knocked down the key regulators of the m6A modification METTL3, METTL14, fat‐mass and obesity–associated protein (FTO), and alpha‐ketoglutarate‐dependent dioxygenase alkB homolog 5 (ALKBH5) in SMMC‐7721 cells and found that silencing FTO significantly reduced the mRNA expression level of ALDOA under hypoxia (Fig. 5I). As the m6A demethylase, FTO, but not ALKBH5, was induced in HCC cells under hypoxia (Supporting Fig. S6C), indicating the potential role of FTO in hypoxia. Knockdown of FTO caused elevated levels of m6A modification in HCC cells under hypoxia (Supporting Fig. S6D). We further validated the down‐regulation of ALDOA at both the RNA and protein levels in FTO‐knockdown HCC cells under hypoxia (Fig. 5J,K), indicating that ALDOA was posttranscriptionally regulated by FTO‐mediated m6A modification. Interestingly, silencing of FTO accelerated degradation of the mRNA of ALDOA under hypoxia compared with the control group (Fig. 5L).
To determine ALDOA as a bona fide target of FTO‐mediated m6A modification, we performed the m6A RNA immunoprecipitation (RIP) sequencing and identified the m6A site (GGAC) in the coding region of ALDOA mRNA (Fig. 5M, GSE143235). We performed the m6A‐RNA immunoprecipitation assay and analyzed with RT‐PCR. As expected, knockdown of FTO dramatically promoted the m6A level of ALDOA mRNA (Fig. 5M). One potential m6A site was identified in the coding regions of ALDOA and conserved among different species (Supporting Fig. S6E,F), and we generated the mutant form of ALDOA by replacing the adenosine base in the m6A consensus sequences with thymine to abolish the m6A modification (Supporting Fig. S6G). The m6A mutant ALDOA could partly rescue the reduction of mRNA expression level of ALDOA in FTO‐knockdown HCC cells upon hypoxia treatment compared with wild‐type ALDOA, indicating the functional role of the m6A modification in ALDOA under hypoxia (Supporting Fig. S6H). To delineate the detailed molecular mechanism by which ALDOA was regulated by the m6A modification, we silenced FTO and two m6A binding proteins, YTH N6‐methyladenosine RNA binding protein 1 (YTDHF1) and YTDHF2, in SMMC‐7721 cells by siRNAs (Supporting Fig. S6I‐K). Our results demonstrated that knockdown of YTHDF2, but not YTHDF1, rescued the expression level of ALDOA in FTO‐silenced HCC cells (Fig. 5N), suggesting that the posttranscriptional regulation of ALDOA under hypoxia was regulated by FTO‐mediated m6A modification in a YTHDF2‐dependent manner. Taken together, our results demonstrated that ALDOA was transcriptionally regulated by HIF‐1α and posttranscriptionally regulated by m6A modification under hypoxia.
Clinical Significance of ALDOA Inhibition in HCC
To determine the clinical significance of ALDOA in human cancers, we systematically analyzed the transcriptomic profiles of multiple tumors (GSE5364) and found that ALDOA was up‐regulated not only in HCC but also in breast cancer, lung cancer, and esophageal cancer (Fig. 6A). We further demonstrated the up‐regulation of ALDOA in TCGA and the HCC database (Fig. 6B; Supporting Fig. S7A), and the up‐regulated level of ALDOA was dependent on pathologic grades of HCC (Fig. 6C). By examining our local clinical cohort, we performed an immunohistochemistry (IHC) staining assay to detect the protein expression level of ALDOA in our clinical human HCC tissues and the corresponding nontumor adjunct liver tissues (Fig. 6D; Supporting Fig. S7B). Consistently, ALDOA protein was significantly overexpressed in HCC tissues compared to their adjunct normal tissues according to the quantification of IHC results (Fig. 6E). Moreover, as lactate was identified as the terminal metabolite mediated by ALDOA in HCC, we measured the level of lactate in our local patients with HCC. As indicated, a significantly higher level of lactate was found in patients with HCC compared to those in healthy people (Fig. 6F). We next examined the clinical prognostic impact of ALDOA in TCGA patients with HCC. Higher levels of ALDOA were associated with poorer overall survival in patients with HCC (Fig. 6G).
Fig. 6.
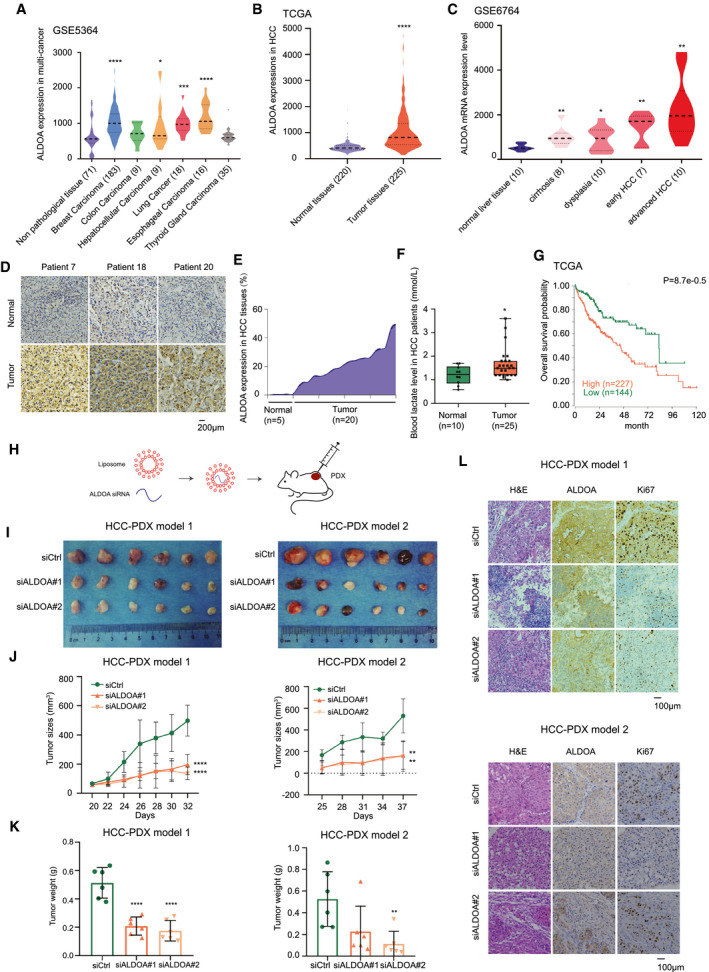
Clinical significance of ALDOA inhibition in HCC. (A) ALDOA expression in multiple tumors. (B) Up‐regulation of ALDOA in HCC tissues compared with normal liver tissues. (C) The expression level of ALDOA was correlated with pathological stages of HCC. (D) ALDOA expression level in clinical patients and their pericarcinomatous normal tissues by IHC assay. (E) Quantification of ALDOA expression in HCC tissues and normal liver tissues from IHC results. (F) Levels of blood lactate acid in patients with clinical HCC compared to healthy people. (G) Prognostic significance of ALDOA up‐regulation in HCC. (H) Schematic diagram of targeting ALDOA by siRNAs in vivo. (I) Inhibition of ALDOA by RNAi in two HCC‐PDXs. (J,K) Tumor growth curve (I) and tumor weight (J) of HCC‐PDXs treated with two in vivo–optimized siRNAs. (L) Hematoxylin and eosin staining and IHC images of ALDOA and Ki67 in randomly selected HCC‐PDX#1 and HCC‐PDX#2 tumors. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Abbreviation: H&E, hematoxylin and eosin.
To verify whether ALDOA acts as a potential therapeutic target in HCC, we performed two HCC patient‐derived xenograft (PDX) models in nude mice and used siRNAs to target ALDOA (Fig. 6H). Notably, depletion of ALDOA using two in vivo–optimized RNAis significantly reduced HCC‐PDX tumor growth in vivo, as indicated by decreased tumor size and tumor weight (Fig. 6I‐K). Pathologic analysis of ALDOA expression and cell proliferation (Ki67) by IHC assay was performed to confirm the effects of the RNAi treatment in HCC‐PDX models (Fig. 6L). Collectively, ALDOA was highly expressed in HCC and may act as a potential therapeutic target for HCC treatment.
Identification of Sulfamonomethoxine as a Potential Inhibitor to Target ALODA in HCC Mediating Metabolic Regulation of Lactate
To identify potential ALDOA inhibitors from FDA‐approved drugs, we applied a structure‐based hierarchical virtual screening strategy that was developed (Supporting Fig. S8A).( 33 ) We found sulfamonomethoxine to be a potential compound to inhibit ALDOA enzymatic activity at a 50% inhibitory concentration (IC50) of 349.6 ± 69.2 μM in HepG2 cells under hypoxia (Supporting Fig. S8B‐D). To improve the selectivity and potency of the ALDOA inhibitor, we performed a proof‐of‐concept structure–activity relationship test, the results of which accorded with the crystallographically determined binding mode of sulfamonomethoxine and its derivatives (Supporting Fig. S8E). Specifically, addition of a methoxy group on the pyrimidine ring along with an additional benzenesulfonylation on the aniline motif (compound 5 [cpd‐5]) was designed to improve sulfamonomethoxine’s interaction with ALDOA, as indicated by the predicted docking pose (Fig. 7A,B). Cpd‐5 increased the potency of the ALDOA IC50 to 106.7 ± 18.1 μM in HepG2 cells under hypoxia (Fig. 7C) but showed no potency in HepG2 cells with null ALDOA under hypoxia (Fig. 7D). To test the therapeutic effects of cpd‐5 in vivo, we treated the mice bearing HCC‐PDX tumors with intratumoral injection of cpd‐5 (10 mg/kg) 3 times per week. Notably, treatment with cpd‐5 significantly inhibited tumor growth in the HCC‐PDX model, as indicated by decreased tumor size, tumor weight, and lactate level (Fig. 7E‐H). However, treatment with cpd‐5 could also cause a reduction of the lactate level in non‐tumor‐bearing mice, suggesting its potential metabolic action in vivo (Fig. 7I). Collectively, our study identified a potential inhibitor (cpd‐5) to target ALDOA for therapeutic treatment of HCC, providing an alternative treatment strategy for patients with advanced HCC.
Fig. 7.
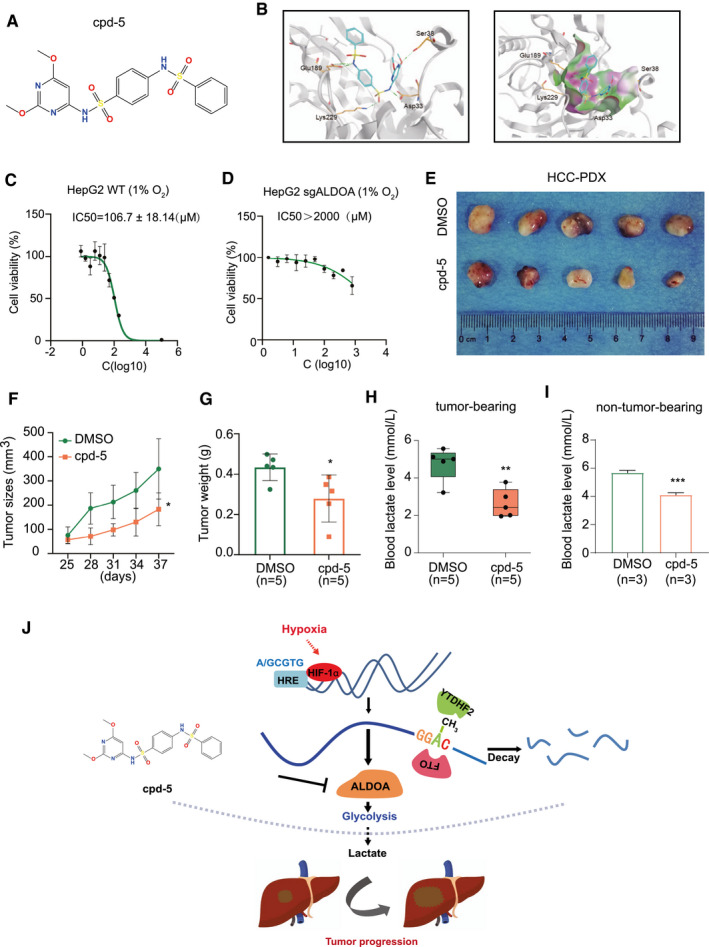
Identification of sulfamonomethoxine derivative cpd‐5 as a lead compound to inhibit ALDOA in HCC mediating metabolic regulation of lactate. (A) Structure of sulfamonomethoxine derivative cpd‐5. (B) Hydrogen interaction of ALDOA enzyme (Protein Data Bank code 5KY6) active site with cpd‐5 (left). Lipophilic potential surface (using a channel method) of ALDOA enzyme active site with cpd‐5 (right). Cpd‐5 and residues of ALDOA are represented by sticks and colored by atom type. (C) Viability of HepG2 cells treated with cpd‐5 under hypoxia. (D) Viability of ALDOA‐null HepG2 cells treated with cpd‐5 under hypoxia. (E) Inhibition of ALDOA by cpd‐5 suppressed tumor growth in the HCC‐PDX model. (F,G) Tumor growth curve (H) and tumor weight (I) of HCC‐PDX treated with cpd‐5. (H) Levels of blood lactate in HCC‐PDX mice treated with cpd‐5. (I) Levels of blood lactate in non‐tumor‐bearing mice treated with cpd‐5. (J) Working model. Under hypoxic conditions, ALDOA is up‐regulated by HIF‐1α and the m6A modification, leading to an increased level of glycolysis‐derived lactate, which promotes HCC tumor progression and migration. Cpd‐5 was developed as a potential ALDOA inhibitor for HCC therapy. *P < 0.05, **P < 0.01, ***P < 0.001. Abbreviation: WT, wild type.
Discussion
The TME is a rapidly evolving environment whose arrangement varies in the light of different anatomical regions of = tumor formation and the genetics and morphology of tumor cells.( 34 ) The proportion and composition of the TME shape tumor evolution and play an integral part in cancer biology, including tumorigenesis, tumor progression, and response to therapy.( 35 ) As the rapid growth of tumor cells causes a constraint in oxygen and nutrients that cannot be supplied by adjacent blood vessels, the hypoxic TME is subject to HIF‐driven responses in tumor cells to activate expression of specific genes involved in angiogenesis, metabolism, cell proliferation and migration, and immunosuppression.( 36 ) Emerging evidence has focused on targeting the TME as a rising strategy to impede tumor progression. In this scenario, we used an in vitro pooled loss‐of‐function genetic system to screen out the potential genes that were essential for HCC tumor cells under hypoxia, providing an approach for hypoxic TME target discovery. Deletion of ALDOA significantly retarded tumor growth and migration and sensitized the tumor cells to hypoxic treatment. ALDOA encodes a protein, fructose‐bisphosphate ALDOA, that regulates the key chemical reaction in glycolysis and gluconeogenesis.( 25 ) Previous studies have found that ALDOA was highly expressed in lung cancer, pancreatic cancer, and gastric cancer.( 37 , 38 , 39 ) Hu et al. showed that the activity of ALDOA was increased under stimulation of phosphoinositide 3‐kinase–dependent activation of Rac by releasing filamentous actin‐bound ALDOA, linking actin remodeling to glycolysis.( 40 ) Overexpression of ALDOA promoted formation of a cancer‐associated proton pump inhibitor with γ‐actin and accelerated lung cancer metastasis,( 37 ) suggesting its role as an oncogene in tumor progression. Grandjean et al. reported the critical role of ALDOA in connecting a feed‐forward mechanism of glycolysis–HIF‐1α signaling in hypoxic cancer cells and highlighted it as a potential therapeutic target to treat intractable hypoxic cancer cells.( 41 ) Consistently, our study identified ALDOA as a promising and essential target in HCC tumors under hypoxia, which was also recognized as an essential fitness defect of low oxygen in the leukemic cell line K562,( 23 ) indicating that ALDOA may not be a hepatocyte‐specific essential gene but highlighting the importance of mitochondrial metabolism in response to low oxygen. Although we observed no obvious damage in the liver with knockdown of ALDOA in normal mice, we could not exclude the possibility of potential toxicity toward other organs through indirect effects or other functions. As for liver cancer, knockdown of ALDOA inhibited cell growth under hypoxia and led to tumor growth retardation and suppression of migration in vivo and in vitro. ALODA was overexpressed in HCC tumors, and its level was highly associated with poor clinical outcome, indicating its potential role as a therapeutic target against HCC.
Previous studies have shown the influence of cellular metabolites on epigenetics and diseases including cancer.( 42 , 43 ) The metabolism of acyl‐CoA modulates the regulation of gene expression in various physiological processes by eight forms of histone acylation.( 44 ) By metabolomics analysis, we identified lactate as a terminal metabolic player driving tumor growth under hypoxia. Knockdown of ALDOA in HCC significantly affected glycolysis metabolism, leading to a dramatic reduction of lactate under hypoxia. Lactate was previously found to impact cancer‐related mortality, to function as a fuel for tumor metabolism, and to promote tumor progression and metastasis.( 45 , 46 ) A recent study reported that lactate connects to macrophage polarization in response to hypoxia and bacterial stimulation in the form of epigenetic modification. Lactate serves as a resource of lactylation of histone lysine residues and turns on gene expression to promote homeostasis of macrophage activity.( 47 ) Importantly, high levels of lactate in the TME derived from tumor cells are known to stimulate the recruitment of myeloid‐derived suppressor cells, dampening immunosurveillance and thus promoting immunosuppression.( 48 ) In our study, we demonstrated that rescue of lactate in ALDOA‐knockdown HCC cells partly restored tumor growth and migration in vitro and in vivo, suggesting the direct role of lactate, derived from ALDOA‐mediated glycolysis, in promoting HCC tumor growth and progression in a hypoxic TME.
We next delineated the molecular mechanisms by which ALDOA was up‐regulated under hypoxia in HCC cells. The main executors of the cellular response to hypoxia are HIF‐1 and HIF‐2, consisting of an O2‐regulated HIF‐1α or HIF‐2α subunit and a constitutively expressed HIF‐1β subunit.( 49 ) Upon hypoxia, HIF‐1α is stabilized by reducing its hydroxylation and translocated into the nucleus, where it forms the HIF‐1α/β dimer and binds with p300/cAMP response element–binding protein coactivator in the HRE to initiate a subset of downstream target gene activation in response to hypoxia.( 50 ) In analysis of an HIF‐1α ChIP‐chip database,( 24 ) we identified ALDOA as a potential direct target of HIF‐1α, suggesting its role in facilitating metabolic adaptation to hypoxia. As expected, we identified three HRE sites that are located in the promoter region of the ALDOA gene and validated the first two sites with functional effects on activation of ALDOA under hypoxia transcriptionally. Meanwhile, we found that ALDOA was regulated by the m6A modification in a posttranscriptional way. The m6A modification is the most pervasive internal modification of mRNA in human cells and mediated mRNA splicing, stability, degradation, and translation.( 29 , 31 , 32 ) Previously, ALKBH5, an alternative demethylase of m6A, was found to be induced upon hypoxia in breast cancer stem cells and mediated the m6A demethylation of NANOG mRNA, phonocopying the effect of hypoxia.( 51 ) In our study, we found that FTO instead of ALKBH5 was induced in HCC cells under hypoxia despite the presence of the functional redundancy between ALKBH5 and FTO in mediating m6A demethylation. Further, we showed that silencing FTO, a key demethylase of m6A, reduced ALDOA expression at both the mRNA and protein levels. The half‐life of ALDOA mRNA in FTO‐knockdown cells was significantly shorter than in control cells, indicating the negative regulation of stability of ALDOA mRNA by FTO‐mediated m6A modification. Previous reports have shown that YTHDF2 affects the localization and stability of targeted mRNAs.( 52 , 53 ) We identified YTHDF2 as a direct m6A reader for m6A‐methylated ALDOA mRNA, as indicated by the fact that silencing of YTHDF2 rescued ALDOA expression in FTO‐knockdown HCC cells. Our results suggested that the posttranscriptional regulation of ALDOA under hypoxia was regulated by FTO‐mediated m6A modification in a YTHDF2‐dependent manner.
Using a structure‐based hierarchical virtual screening approach, we identified sulfamonomethoxine as a potential inhibitor of ALDOA from FDA‐approved drugs. Sulfamonomethoxine is a common antibiotic widely used in human and veterinary medicine which inhibits dihydropteroate synthetase to block the synthesis of folic acid in bacteria.( 54 ) From the structural binding analysis, the metaposition methyl groups on the pyrimidine ring and aniline motif form hydrogen bonds to Asp33 and Glu189 at the active site of ALDOA, while a gap in the interaction remains on the surface. In this regard, we have designed a series of sulfamonomethoxine derivatives to improve the interaction. Cpd‐5 with introduction of a methoxy group on the pyrimidine ring along with an additional benzenesulfonylation on the aniline motif represented with higher binding score of interaction with ALDOA, where the metaposition methoxy group on the pyrimidine forms a hydrogen bound to Ser38 and the interaction gap was filled with benzenesulfonylation. We further validated the enhancement of cpd‐5 with lower IC50 and tested its therapeutic application in our HCC‐PDX mouse model and its impact on non‐tumor‐bearing mouse models. Reduction of lactate was further confirmed with the effects of cpd5 in vivo through targeting ALDOA‐mediated glycolysis, indicating that the pharmacological mechanism of action may have important implications in the development of a treatment strategy for HCC.
In summary, our study employed an unbiased genome‐wide CRISPR/Cas9 knockout screening pool to systematically identify ALODA as the essential driver gene for HCC cell growth under hypoxia. Knockdown of ALDOA led to depletion of lactate, a terminal metabolic player in HCC mediated by ALDOA, and thus suppressed HCC tumor growth and migration under hypoxia. Upon hypoxic conditions, ALDOA was up‐regulated by HIF‐1α‐mediated transcriptional activation and by FTO‐mediated m6A modification at the posttranscriptional level (Fig. 7J). Inhibition of ALDOA by the sulfamonomethoxine derivative cpd‐5 suppressed HCC tumor growth in vivo, highlighting a potential therapeutic strategy for HCC treatment.
Author Contributions
G.W. contributed to the study concept. G.W. and Y.N. contributed to the experimental designs. Y.N., Z.L., A.W., L.S., S.Y., H.L. and S.Z. contributed to the data generation. G.W., D.C., X.B., P.L., C.C., W.H. and X.L. contributed to the data analysis. G.W. and Y.N. drafted the manuscript. All authors provided critical review of the manuscript.
Supporting information
Supplementary Material
Supported in part by grants from the National Natural Science Foundation of China (82073869, 82122069, 21672266, 82022037, 81701834, 81871994); the Guangdong Basic and Applied Basic Research Foundation (2019A050510019, 2021B1515020004, 2019B151502063); the Guangdong Provincial Key Laboratory of Construction Foundation (2017B030314030, 2020B1212060034); the Guangzhou Science and Technology Planning Program (202002020051, 201902020018); the National Engineering Research Center for New Drug and Druggability Evaluation, Seed Program of Guangdong Province (2017B090903004); and the Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (2017BT01Y093).
Potential conflict of interest: Nothing to report.
References
Author names in bold designate shared co‐first authorship.
- 1. Villanueva A. Hepatocellular carcinoma. N Engl J Med 2019;380:1450‐1462. [DOI] [PubMed] [Google Scholar]
- 2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin 2019;69:7‐34. [DOI] [PubMed] [Google Scholar]
- 3. Yang JD, Roberts LR. Hepatocellular carcinoma: a global view. Nat Rev Gastroenterol Hepatol 2010;7:448‐458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bruix J, Gores GJ, Mazzaferro V. Hepatocellular carcinoma: clinical frontiers and perspectives. Gut 2014;63:844‐855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kudo M, Finn RS, Qin S, Han K‐H, Ikeda K, Piscaglia F, et al. Lenvatinib versus sorafenib in first‐line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non‐inferiority trial. Lancet 2018;391:1163‐1173. [DOI] [PubMed] [Google Scholar]
- 6. Vaupel P, Hockel M, Mayer A. Detection and characterization of tumor hypoxia using pO2 histography. Antioxid Redox Signal 2007;9:1221‐1235. [DOI] [PubMed] [Google Scholar]
- 7. Semenza GL. Targeting HIF‐1 for cancer therapy. Nat Rev Cancer 2003;3:721‐732. [DOI] [PubMed] [Google Scholar]
- 8. Maxwell PH, Dachs GU, Gleadle JM, Nicholls LG, Harris AL, Stratford IJ, et al. Hypoxia‐inducible factor‐1 modulates gene expression in solid tumors and influences both angiogenesis and tumor growth. Proc Natl Acad Sci USA 1997;94:8104‐8109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Haase VH. Regulation of erythropoiesis by hypoxia‐inducible factors. Blood Rev 2013;27:41‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Carmeliet P, Dor Y, Herbert JM, Fukumura D, Brusselmans K, Dewerchin M, et al. Role of HIF‐1alpha in hypoxia‐mediated apoptosis, cell proliferation and tumour angiogenesis. Nature 1998;394:485‐490. [DOI] [PubMed] [Google Scholar]
- 11. Rankin EB, Giaccia AJ. Hypoxic control of metastasis. Science 2016;352:175‐180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Manoochehri Khoshinani H, Afshar S, Najafi R. Hypoxia: a double‐edged sword in cancer therapy. Cancer Invest 2016;34:536‐545. [DOI] [PubMed] [Google Scholar]
- 13. Daskalaki I, Gkikas I, Tavernarakis N. Hypoxia and selective autophagy in cancer development and therapy. Front Cell Dev Biol 2018;6:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cui CP, Wong CC, Kai AK, Ho DW, Lau EY, Tsui YM, et al. SENP1 promotes hypoxia‐induced cancer stemness by HIF‐1alpha deSUMOylation and SENP1/HIF‐1alpha positive feedback loop. Gut 2017;66:2149‐2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lin Z, Niu Y, Wan A, Chen D, Liang H, Chen X, et al. RNA m6A methylation regulates sorafenib resistance in liver cancer through FOXO3‐mediated autophagy. EMBO J 2020:e103181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Leone RD, Zhao L, Englert JM, Sun I‐M, Oh M‐H, Sun I‐H, et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 2019;366:1013‐1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Root DE, Hacohen N, Hahn WC, Lander ES, Sabatini DM. Genome‐scale loss‐of‐function screening with a lentiviral RNAi library. Nat Methods 2006;3:715‐719. [DOI] [PubMed] [Google Scholar]
- 18. Shalem O, Sanjana NE, Hartenian E, Shi XI, Scott DA, Mikkelsen TS, et al. Genome‐scale CRISPR‐Cas9 knockout screening in human cells. Science 2014;343:84‐87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Muller FL, Colla S, Aquilanti E, Manzo VE, Genovese G, Lee J, et al. Passenger deletions generate therapeutic vulnerabilities in cancer. Nature 2012;488:337‐342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Behan FM, Iorio F, Picco G, Gonçalves E, Beaver CM, Migliardi G, et al. Prioritization of cancer therapeutic targets using CRISPR‐Cas9 screens. Nature 2019;568:511‐516. [DOI] [PubMed] [Google Scholar]
- 21. Marin‐Hernandez A, Lopez‐Ramirez SY, Del Mazo‐Monsalvo I, Gallardo‐Perez JC, Rodriguez‐Enriquez S, Moreno‐Sanchez R, et al. Modeling cancer glycolysis under hypoglycemia, and the role played by the differential expression of glycolytic isoforms. FEBS J 2014;281:3325‐3345. [DOI] [PubMed] [Google Scholar]
- 22. Jing X, Yang F, Shao C, Wei K, Xie M, Shen H, et al. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol Cancer 2019;18:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jain IH, Calvo SE, Markhard AL, Skinner OS, To TL, Ast T, et al. Genetic screen for cell fitness in high or low oxygen highlights mitochondrial and lipid metabolism. Cell 2020;181:716‐727.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xia X, Lemieux ME, Li W, Carroll JS, Brown M, Liu XS, et al. Integrative analysis of HIF binding and transactivation reveals its role in maintaining histone methylation homeostasis. Proc Natl Acad Sci U S A 2009;106:4260‐4265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chang YC, Yang YC, Tien CP, Yang CJ, Hsiao M. Roles of aldolase family genes in human cancers and diseases. Trends Endocrinol Metab 2018;29:549‐559. [DOI] [PubMed] [Google Scholar]
- 26. Chiu D‐C, Xu I‐J, Lai R‐H, Tse A‐W, Wei LL, Koh H‐Y, et al. Hypoxia induces myeloid‐derived suppressor cell recruitment to hepatocellular carcinoma through chemokine (C‐C motif) ligand 26. Hepatology 2016;64:797‐813. [DOI] [PubMed] [Google Scholar]
- 27. Chiu D‐C, Tse A‐W, Xu I‐J, Di Cui J, Lai R‐H, Li LL, et al. Hypoxia inducible factor HIF‐1 promotes myeloid‐derived suppressor cells accumulation through ENTPD2/CD39L1 in hepatocellular carcinoma. Nat Commun 2017;8:517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xia X, Kung AL. Preferential binding of HIF‐1 to transcriptionally active loci determines cell‐type specific response to hypoxia. Genome Biol 2009;10:R113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dominissini D, Moshitch‐Moshkovitz S, Schwartz S, Salmon‐Divon M, Ungar L, Osenberg S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A‐seq. Nature 2012;485:201‐206. [DOI] [PubMed] [Google Scholar]
- 30. Wang X, Zhao B, Roundtree I, Lu Z, Han D, Ma H, et al. N6‐methyladenosine modulates messenger RNA translation efficiency. Cell 2015;161:1388‐1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yue Y, Liu J, He C. RNA N6‐methyladenosine methylation in post‐transcriptional gene expression regulation. Genes Dev 2015;29:1343‐1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Niu Y, Wan A, Lin Z, Lu X, Wan G. N6‐Methyladenosine modification: a novel pharmacological target for anti‐cancer drug development. Acta Pharm Sin B 2018;8:833‐843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lamb J. The Connectivity Map: a new tool for biomedical research. Nat Rev Cancer 2007;7:54‐60. [DOI] [PubMed] [Google Scholar]
- 34. Correia AL, Bissell MJ. The tumor microenvironment is a dominant force in multidrug resistance. Drug Resist Updat 2012;15:39‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. De Palma M, Biziato D, Petrova TV. Microenvironmental regulation of tumour angiogenesis. Nat Rev Cancer 2017;17:457‐474. [DOI] [PubMed] [Google Scholar]
- 36. Sormendi S, Wielockx B. Hypoxia pathway proteins as central mediators of metabolism in the tumor cells and their microenvironment. Front Immunol 2018;9:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chang Y‐C, Chiou J, Yang Y‐F, Su C‐Y, Lin Y‐F, Yang C‐N, et al. Therapeutic targeting of aldolase A interactions inhibits lung cancer metastasis and prolongs survival. Cancer Res 2019;79:4754‐4766. [DOI] [PubMed] [Google Scholar]
- 38. Ji S, Zhang BO, Liu J, Qin YI, Liang C, Shi SI, et al. ALDOA functions as an oncogene in the highly metastatic pancreatic cancer. Cancer Lett 2016;374:127‐135. [DOI] [PubMed] [Google Scholar]
- 39. Zhang C, Zhao LM, Wu H, Tian G, Dai SL, Zhao RY, et al. C/D‐Box Snord105b promotes tumorigenesis in gastric cancer via ALDOA/c‐myc pathway. Cell Physiol Biochem 2018;45:2471‐2482. [DOI] [PubMed] [Google Scholar]
- 40. Hu H, Juvekar A, Lyssiotis C, Lien E, Albeck J, Oh D, et al. Phosphoinositide 3‐kinase regulates glycolysis through mobilization of aldolase from the actin cytoskeleton. Cell 2016;164:433‐446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Grandjean G, de Jong PR, James B, Koh MY, Lemos R, Kingston J, et al. Definition of a novel feed‐forward mechanism for glycolysis–HIF1alpha signaling in hypoxic tumors highlights aldolase A as a therapeutic target. Cancer Res 2016;76:4259‐4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kaelin WG Jr, McKnight SL. Influence of metabolism on epigenetics and disease. Cell 2013;153:56‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sun L, Wan A, Zhou Z, Chen D, Liang H, Liu C, et al. RNA‐binding protein RALY reprogrammes mitochondrial metabolism via mediating miRNA processing in colorectal cancer. Gut 2020. Nov 20. 10.1136/gutjnl-2020-320652. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sabari BR, Zhang D, Allis CD, Zhao Y. Metabolic regulation of gene expression through histone acylations. Nat Rev Mol Cell Biol 2017;18:90‐101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Faubert B, Li KY, Cai L, Hensley CT, Kim J, Zacharias LG, et al. Lactate metabolism in human lung tumors. Cell 2017;171:358‐371.e59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tasdogan A, Faubert B, Ramesh V, Ubellacker JM, Shen BO, Solmonson A, et al. Metabolic heterogeneity confers differences in melanoma metastatic potential. Nature 2020;577:115‐120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang DI, Tang Z, Huang HE, Zhou G, Cui C, Weng Y, et al. Metabolic regulation of gene expression by histone lactylation. Nature 2019;574:575‐580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Husain Z, Huang Y, Seth P, Sukhatme VP. Tumor‐derived lactate modifies antitumor immune response: effect on myeloid‐derived suppressor cells and NK cells. J Immunol 2013;191:1486‐1495. [DOI] [PubMed] [Google Scholar]
- 49. Semenza GL. Hypoxia‐inducible factors in physiology and medicine. Cell 2012;148:399‐408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Majmundar AJ, Wong WJ, Simon MC. Hypoxia‐inducible factors and the response to hypoxic stress. Mol Cell 2010;40:294‐309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhang C, Samanta D, Lu H, Bullen JW, Zhang H, Chen I, et al. Hypoxia induces the breast cancer stem cell phenotype by HIF‐dependent and ALKBH5‐mediated m6A‐demethylation of NANOG mRNA. Proc Natl Acad Sci U S A 2016;113:E2047‐E2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Du H, Zhao YA, He J, Zhang Y, Xi H, Liu M, et al. YTHDF2 destabilizes m6A‐containing RNA through direct recruitment of the CCR4‐NOT deadenylase complex. Nat Commun 2016;7:12626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, et al. N6‐methyladenosine‐dependent regulation of messenger RNA stability. Nature 2014;505:117‐120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kuwano A. Inhibitory effect of sulfamonomethoxine on capsule formation of Bordetella bronchiseptica . Zentralbl Veterinarmed B 1991;38:685‐688. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material