

Summary
Vitis vinifera is an economically important crop and a useful model in which to study chromatin dynamics. In contrast to the small and relatively simple genome of Arabidopsis thaliana, grapevine contains a complex genome of 487 Mb that exhibits extensive colonization by transposable elements. We used Hi‐C, ChIP‐seq and ATAC‐seq to measure how chromatin features correlate to the expression of 31 845 grapevine genes. ATAC‐seq revealed the presence of more than 16 000 open chromatin regions, of which we characterize nearly 5000 as possible distal enhancer candidates that occur in intergenic space > 2 kb from the nearest transcription start site (TSS). A motif search identified more than 480 transcription factor (TF) binding sites in these regions, with those for TCP family proteins in greatest abundance. These open chromatin regions are typically within 15 kb from their nearest promoter, and a gene ontology analysis indicated that their nearest genes are significantly enriched for TF activity. The presence of a candidate cis‐regulatory element (cCRE) > 2 kb upstream of the TSS, location in the active nuclear compartment as determined by Hi‐C, and the enrichment of H3K4me3, H3K4me1 and H3K27ac at the gene are correlated with gene expression. Taken together, these results suggest that regions of intergenic open chromatin identified by ATAC‐seq can be considered potential candidates for cis‐regulatory regions in V. vinifera. Our findings enhance the characterization of a valuable agricultural crop, and help to clarify the understanding of unique plant biology.
Keywords: chromatin, epigenetics, transcription factors, gene expression, plant biology, Vitis vinifera
Significance Statement
We use ATAC‐seq to identify open chromatin regions in Vitis vinifera, and show that they contain transcription factor binding sites and are correlated with gene expression, making them likely enhancer candidates. These findings add to the knowledge of biology of horticultural crops and provide possible targets for improved grape breeding strategies.
Introduction
Cis‐regulatory elements (CREs) are regions of DNA that influence expression of nearby genes through the binding of transcription factor (TF) proteins. Proper spatio‐temporal activation and silencing of genes is a critical part of development and, in multi‐cellular organisms, a specific gene regulatory program gives rise to each different cell type. A major part of this regulatory program includes the function of CREs, which play key roles in cellular processes such as cell differentiation and limb development, as well as tumor growth (Boulay et al., 2018; Mojica‐Vázquez et al., 2017; Xu et al., 2009). Distal, gene‐activating CREs are known as enhancers (Heintzman and Ren, 2009) and, while their precise mechanism has not been definitively established, enhancers have been shown to bind TFs and are believed to form a DNA loop with their target promoter to assist in either the initiation or prolongation of transcription, likely through the delivery of necessary transcription components (Calo and Wysocka, 2013; Pennacchio et al., 2013). Conversely, silencing elements disrupt transcription, either by blocking RNA polymerase or otherwise inhibiting transcription initiation (Rojo, 2001).
Although the functions of CREs are well‐documented in mammals and Drosophila, the complete set of regulatory elements in plant genomes remains largely unidentified and uncharacterized (Weber et al., 2016). Phylogenetic footprinting approaches that have been successful in identifying regulatory elements in vertebrates (Siepel, 2005) have not found widespread applications in plants, probably as a consequence of the difficulties in aligning their genomes arising from extreme structural variation due to very recent movement of transposable elements that is characteristic of most Angiosperm plant species (Morgante et al., 2007). Nevertheless, the limited studies performed have demonstrated the importance of certain CREs in plants, showing not only that these regions play a vital role in shaping useful traits in our current agricultural crops (Clark et al., 2006), but also that alterations in CREs represent a rich source of genetic diversity that can already be used to improve plant phenotypes (Rodríguez‐Leal et al., 2017). Furthermore, the engineering of entire gene regulatory networks is recognized as a potentially powerful tool to increase crop efficiency in the future (Nemhauser and Torii, 2016). To fully explore these strategies, cis‐regulatory regions must first be located within plant genomes, and their effects on gene expression must be established.
Early studies using enhancer‐trapping and QTL mapping revealed the general location of some enhancers in various plant species (Clark et al., 2006; Michael and McClung, 2003; Wu et al., 2003), but these techniques are slow and laborious, precluding extensive genome‐wide identification and characterization of CREs. More recently, advances in next‐generation sequencing technology have led to improved methods for identifying CREs, including DNAse‐seq and ATAC‐seq (Assay for Transposase‐Accessible Chromatin; Boyle et al., 2008; Buenrostro et al., 2013; Song and Crawford, 2010). These techniques are based on the property of TF‐binding regulatory regions to form relatively open chromatin (Crawford et al., 2004; Galas and Schmitz, 1978), making them sensitive to enzymatic cleavage, and allowing for CRE‐targeted sequencing and bioinformatics analysis. The advent of large‐scale single‐cell sequencing has further highlighted the correlation between ATAC‐seq identified regions and cell‐type‐specific gene regulation, confirming the utility of this technique in modern plant transcriptomics studies (Farmer et al., 2021; Xu et al., 2021).
Genome‐wide open chromatin searches have been performed in other plant species, including rice, maize and Arabidopsis (Oka et al., 2017; Rodgers‐Melnick et al., 2016; Sullivan et al., 2014; Zhang et al., 2012). A comparison of these studies shows the remarkable variety of plant genome sizes and structures, and highlights the unique insights that can be obtained from the exploration of different plant genomes. Grapevine (Vitis vinifera) provides an excellent crop in which to investigate CRE characteristics and function as its medium‐sized genome of 487 Mb (Jaillon et al., 2007) comprises approximately 50% repetitive elements in varying conformations across its 19 chromosomes; thus, the nucleus must maximize efficient gene transcription while minimizing deleterious effects of transposons. Grapevine is a highly heterozygous fruit tree (Salmaso et al., 2005; Thomas and Scott, 1993) whose long life span makes it commercially suitable for clonal propagation (Rühl et al., 2004), during which individuals can accumulate somatic mutations (Vondras et al., 2019). This widely‐grown crop includes hundreds of varieties adapted to specific climates worldwide, providing potential real‐world applications of new discoveries.
With this study, we use ATAC‐seq to search for regions of open chromatin in the grapevine leaf tissue, identifying 16 771 regions overall, with nearly 5000 of these occurring in intergenic space [> 2 kb away from the nearest transcription start site (TSS)], and therefore representing possible enhancers, or distal candidate CREs (cCREs). We perform sequence analysis of these open chromatin regions, and find that they contain more than 480 TF motifs and are enriched near TF genes. Additionally, we use ChIP‐seq to search for known epigenetic signatures of these cCREs and do not find evidence for strong enrichment of H3K27ac or H3K4me1 at these loci. We exploit the high levels of individual heterozygosity to perform an allele‐specific analysis of open chromatin regions and to relate this information to patterns of allele‐specific expression. Finally, we use Hi‐C to define nuclear organization patterns, and correlate these and other chromatin features with RNA‐seq data to illustrate the additive effects of global and local gene regulation. We find that the presence of open chromatin, H3K4me3, H3K4me1 and H3K27ac, and location in the active nuclear compartment all correlate with gene expression.
Results
Mapping ATAC‐seq peaks and histone modifications
To examine the local chromatin landscape of V. vinifera, we performed ATAC‐seq (Buenrostro et al., 2013) and ChIP‐seq with antibodies to three modified histones (H3K4me3, H3K27ac and H3K4me1) shown to fulfill important roles in eukaryotic organisms (for review, see Zentner and Henikoff, 2013) in young leaf tissue from Pinot Noir plants. Peaks were called with MACS v 2.1.0 (Zhang et al., 2008), and we identified 23 207 peaks for H3K4me3, 28 272 peaks for H3K27ac, 14 370 broad regions for H3K4me1 and 16 771 peaks for ATAC‐seq (the MACS2 narrow peak or broad peak output files for each peak set can be found in Data S1). We then divided the genome into eight compartments, including promoter (defined as the 2‐kb region upstream of the TSS), 5ʹ‐UTR, coding exons and introns, 3ʹ‐UTR, 3ʹ‐UTR‐adjacent (500 bp), genic transposable elements, intergenic transposable elements and non‐TE intergenic regions. We counted the number of peak summits (for H3K4me3, H3K27ac and ATAC‐seq) or broad region centers (for H3K4me1) found at each of these genomic locations to determine the correlation of chromatin features with specific genome fractions (Figure 1a). All three histone modifications are highly enriched in coding exons and introns, which contain 73.5% of H3K4me3 peak summits, 57.3% of H3K27ac peak summits and 79.8% of H3K4me1 broad region centers. The distributions of H3K4me3 and H3K27ac are remarkably similar, with 16 318 instances of peaks overlapping by at least 50% of each peak of the pair. The summits of ATAC‐seq peaks, representing regions of accessible chromatin, exist most frequently at the promoter (36.5%) and in other intergenic space (intergenic non‐TE: 5188 summits, 30.9%; intergenic TE: 139 summits, 0.83%). The distribution of TE families in which peak summits were found is shown in Figure S1. Meta‐profiles of either ChIP‐seq or ATAC‐seq enrichment at the TSS were produced from MACS2‐generated bedgraph files, and show the read pile‐up of these features at the TSS (n = 31 845) and 5 kb upstream or downstream. Areas of open chromatin (ATAC‐seq) are enriched at the TSS, downstream of which lie H3K4me3 and H3K27ac whose signals strongly overlap. H3K4me1 levels decrease just before the TSS before increasing again in concordance with a tapering of H3K4me3 signal. A slight increase and then depletion of all marks is seen at the transcription end site (TES; Figure S2).
Figure 1.
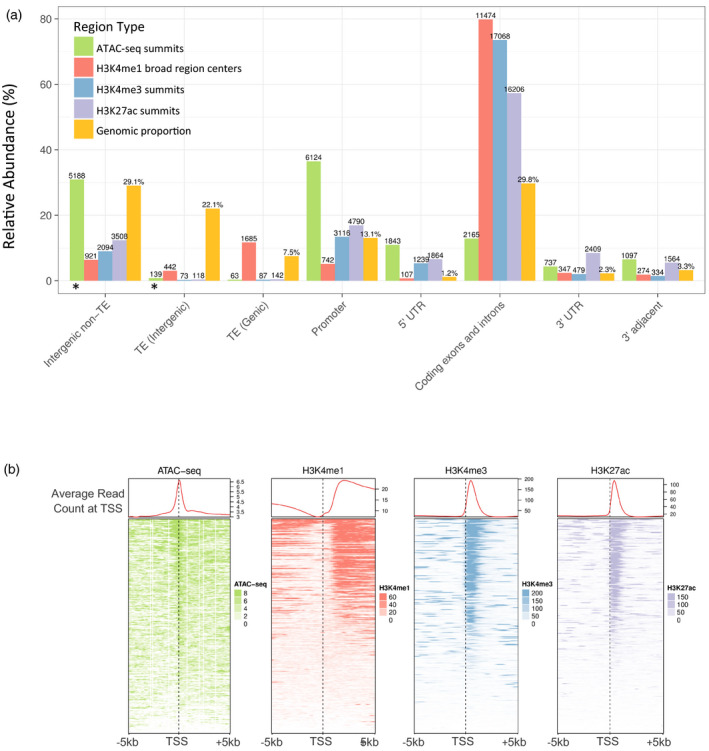
Location of epigenetic features in Vitis vinifera.
(a) Occurrence of histone modifications or accessible chromatin in different genomic regions compared with the overall proportion (yellow bars) of each region type in the genome. The y‐axis shows the percentage of the total of each feature type found at the genomic regions. Peak summits were used to classify the locations, except for H3K4me1, for which the center of broad enriched regions was used. The number of peaks (ATAC‐seq, H3K4me3, H3K27ac) or broad regions (H3K4me1) at each region is shown above each bar while, for the genomic proportions (yellow bars), the total base pair percent of each region type is shown. Percentages do not sum to 100% because some regions overlap in the genome. ‘3’ adjacent’ indicates the 500‐bp window directly downstream of the transcription end site (TES). ‘Promoter’ indicates the 2‐kb promoter region upstream of the TSS. Asterisks (*) indicate candidate cis‐regulatory element (CRE) categories.
(b) Read count profiles of modified histones or accessible chromatin at the TSS for 31 845 V. vinifera genes.
Intergenic regions of open chromatin are methylation‐depleted and contain TF binding sites
Of the 487 Mb of grapevine genome, 7.9 Mb (1.6%) can be classified as accessible chromatin in young leaf tissue as determined by ATAC‐seq (Figure 2a), with 5.4 Mb (11 332 regions, average size 476 bp) located either within genes or the upstream 2‐kb region and 2.5 Mb (5439 regions, average size 456 bp) occurring in intergenic regions. Distal intergenic regions of accessible chromatin, classified in this study as regions occurring neither in a gene nor in the 2‐kb promoter upstream of a TSS, are of particular interest as they are likely candidates for CREs, such as enhancers (Heintzman and Ren, 2009; Lu et al., 2017; Zhu et al., 2015). Because cytosine methylation has been shown to be incompatible with TF binding (O’Malley et al., 2016; Stadler et al., 2011; Yin et al., 2017) and, in accordance with this, active enhancers tend to be hypo‐methylated (For review, see Calo and Wysocka, 2013), we used previously‐generated bisulfite sequencing data from young leaf tissue (Magris et al., 2019) to analyze the CG, CHG and CHH methylation levels of intergenic ATAC‐seq peaks as well as those of 2173 randomly chosen, non‐TE intergenic regions of 500 bp each. For comparison, we included 5‐kb flanking regions upstream and downstream of the peaks (Figure 2b). We find that ATAC‐seq peak regions in grapevine have lower average methylation levels than control regions in all three contexts (CG: 16.8 versus 68.1%; CHG: 10.0 versus 45.0%; CHH: 1.4 versus 3.4%), providing support for a potential enhancer role for these regions. Enhancers typically feature specific TF binding sites of approximately 6–20 nucleotides contained within approximately 100–1000 bp of relatively accessible DNA (Hesselberth et al., 2009; Long et al., 2016; Mueller et al., 2017; Sung et al., 2014; Zhu et al., 2015). To determine if the intergenic open chromatin regions we found in grapevine might be associated with TFs, we used the MEME‐ChIP software from MEME Suite 5.1.1 (Bailey et al., 2009) to analyze a filtered subset of intergenic accessible regions and find any possible protein‐binding motifs, searching against the DAP Arabidopsis motifs database (O’Malley et al., 2016). Regions were filtered to exclude those adjacent to H3K4me3 enrichment and therefore suspected to be TSS‐adjacent peaks for unannotated genes, leaving 4902 regions to analyze. From our ATAC‐seq‐identified accessible regions, MEME‐ChIP discovered more than 400 centrally enriched motifs, with at least 20 of these belonging to the plant‐specific TCP protein family. Other common binding sites found include those for basic helix‐loop‐helix proteins (bHLH31, bHLH34, bHLH77, bHLH74), bZIP proteins (GBF3, GBF6, bZIP50, bZIP68), WRKY proteins, MYB proteins and others (examples of various TF families shown in Figure 2c). A full list of the centrally enriched motifs can be found in Data [Link], [Link]. The presence of numerous TF binding sites at intergenic accessible regions further supports a functional role for these loci in the regulation of gene expression, and suggests they could be cCREs.
Figure 2.
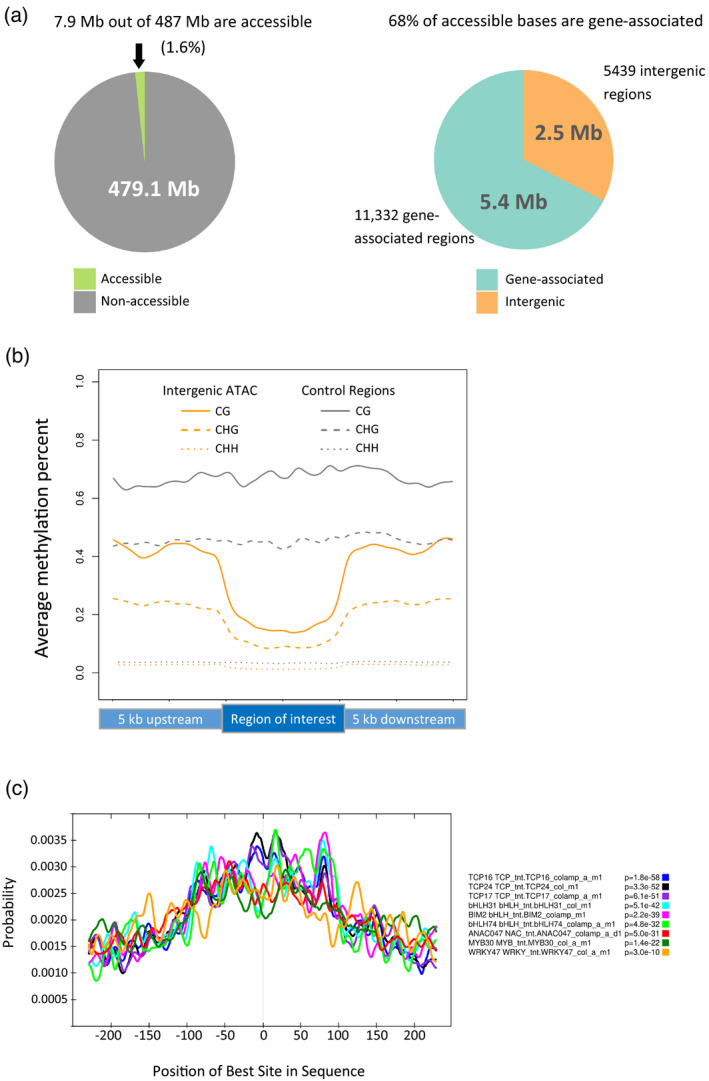
ATAC‐seq‐identified regions.
(a) Accessible regions comprise 7.9 Mb of the 487 Mb grapevine genome. Of these, 5.4 Mb is associated with genes and 2.5 Mb is intergenic.
(b) Comparison of methylation in CG (solid line), CHG (dashed line) or CHH (dotted line) contexts across intergenic ATAC‐seq peaks (orange) or 2173 500‐bp random intergenic, non‐TE regions (gray), all scaled to 100%.
(c) Centrimo output from MEME‐Suite showing the centrally located enrichment of nine example transcription factor (TF) binding sites found in intergenic accessible regions.
Hi‐C reveals enrichment of cCREs per expressed gene in inactive compartment
Next, we examined the global distribution of distal cCREs across nuclear compartments. In eukaryotic cells, chromosomes often 10s or 100s of megabases long must be efficiently folded so that an entire genome fits in the nucleus and is still transcribed as needed. Hi‐C experiments in mammals, yeast and Drosophila showed that the largest‐scale organization divides the nucleus into two general regions, the active and inactive compartments (Lieberman‐Aiden et al., 2009; Duan et al., 2010; Sexton et al., 2012), which correspond to loose, highly‐transcribed euchromatic zones and tightly‐packed heterochromatic zones, respectively. In the 135‐Mb genome of Arabidopsis, similar alternating compartments were described (Grob et al., 2014), revealing the boundaries between active and inactive euchromatin. Larger, more complex plant genomes feature diverse organizational patterns, with rice (Dong et al., 2018) and foxtail millet (Dong et al., 2017) chromosomes, for example, showing numerous alternating active/inactive regions, while barley chromosomes could only be resolved into three broad zones (Mascher et al., 2017). Because CREs interact with their target gene promoters to regulate expression, we reasoned they might be enriched in the active compartment, which is defined by longer‐range interactions and is characterized by higher gene expression. To test this hypothesis, we performed Hi‐C on young leaf tissue and used a principal component analysis (see Experimental Procedures) to assign 50‐kb genome windows to either the active or inactive compartment. We found that the grapevine genome in the nuclei of young leaves is nearly equally divided between the two compartments, which comprise approximately 230 Mb (47%, active compartment) and 248 Mb (51%, inactive compartment) of the genome, and do not adhere to a specific pattern across chromosomes (Figure 3a). A genome‐wide view of the compartments and density of genomic features, including genes, transposons, open chromatin (ATAC‐seq signal) and histone modifications, is shown in Figure S3. As observed in other plant species (Dong et al., 2017), chromosome composition in grapevine is reflected in its organization, with active compartment regions corresponding to regions of markedly higher gene density and lower TE density than inactive compartment regions (Figure S3). While there were overall more cCREs identified in the active compartment (2661 ATAC‐seq peaks) versus inactive compartment (1783 ATAC‐seq peaks), the ratio of identified cCREs to expressed genes was significantly greater in the inactive compartment (active: 2661 peaks/15 082 genes = 0.176; inactive: 1783 peaks/7630 genes = 0.234; P < 0.00001, two proportion z‐test). Additionally, both cCREs and promoter ATAC‐seq peaks in the inactive compartment were found to have slightly but significantly higher fold enrichment scores than peaks in the active compartment (Figure 3b). One technical explanation for this observation is that the computational algorithm used by MACS2 was more easily able to detect enrichment in the relatively closed background of the inactive compartment. Alternatively, the lower fold enrichment but higher gene expression in the active compartment (Figure 3c) could reflect genes that have a nearby cCRE and are highly expressed, but only in one or few cell types in the leaf. Regardless of the cause, this increase in observed local accessibility is not sufficient to fully counteract the repressive environment of the inactive compartment, as genes closest to intergenic ATAC‐seq peaks in this compartment still have lower average expression than non‐closest genes in the active compartment (Figure 3c).
Figure 3.
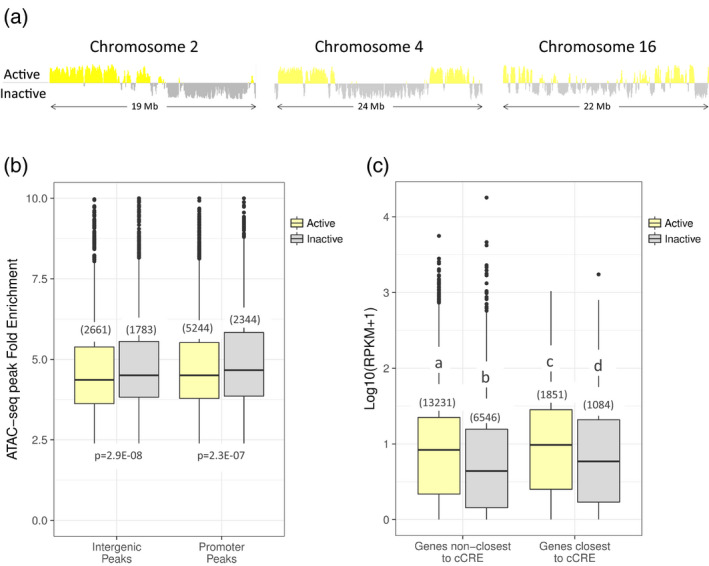
Active and inactive nuclear compartments in young grapevine leaves.
(a) Examples of active and inactive compartment organization patterns observed in grapevine leaf nuclei, as determined by a principal component analysis of Hi‐C interaction data. Positive yellow signal indicates active compartment regions, negative gray signal indicates inactive compartment regions.
(b) Fold enrichment of Tn5 transposase integration obtained from MACS2 at either intergenic or gene‐associated ATAC‐seq peaks in active (yellow) and inactive (gray) compartments, with the number of peaks in each boxplot shown in parentheses.
(c) Gene expression (log10[RPKM+1]) for expressed genes whose promoters are closest or not closest to a candidate cis‐regulatory element (cCRE) in active and inactive compartments, with the number of genes in each boxplot shown in parentheses. Significant differences (P < 0.05) in gene expression between subsets are indicated by unique letters (a–d). All P‐values shown were determined by an independent two‐sample or pairwise Wilcoxon test.
Combinations of cCREs and other local chromatin features are correlated with gene expression
Interestingly, most (81%) of the Vitis 31 845 genes are expressed in young leaves, but there is not a single epigenetic environment required for transcription. Instead, several distinct combinations of chromatin features can be found throughout the genome, suggesting that each gene is regulated in part through a customized environment. To measure correlations between these features and individual gene expression, we scored genes on seven epigenetic criteria associated with expression: overlap with (i) H3K4me1, (ii) H3K4me3, (iii) H3K27ac or (iv) a TSS ATAC‐seq peak, (v) the presence of a proximal ATAC‐seq peak from −200 bp to −2 kb upstream of the TSS, (vi) whether or not the promoter of the gene was the closest promoter to a distal cCRE > 2 kb upstream of the TSS (examples in Figure 4a and Figure S4 produced with IGV; Robinson et al., 2011), and (vii) location in the active versus inactive nuclear compartments. Expression levels, shown as log10‐transformed RPKM+1, were calculated for each group and shown in Figure 4(b), for genes with zero features (left boxplot, lightest blue) to seven features (right boxplot, darkest blue), with the number of genes measured for each group in parentheses above the box. A two‐sided pairwise Wilcoxon test was used to test for significance by measuring the P‐value of expression differences between groups. Genes associated with six or seven features (n = 457 and n = 27, respectively) showed the highest levels of expression, while those with zero features (n = 5272) had the lowest expression. Significant (P < 0.05) differences in gene expression were seen between all groups except for the seven‐feature group, as indicated by letters above the boxplots, showing that gene expression typically increases with each increment of an increasingly permissive locus. Genes were then grouped based on number of features, with the percentage of genes having each feature shown in Figure 4(c). To compare the effects of each individual feature on gene expression, we further divided these groups based on which features its members had and measured expression for each sub‐group (Figure S5). Single‐feature genes with only the ATAC‐seq features (TSS ATAC‐seq, cCRE, or Proximal Upstream ATAC‐seq) do not show significant expression differences from the zero‐feature gene sets (group ‘One’ boxplots, Figure S5), suggesting that, while in the inactive nuclear compartment and in the absence of other important chromatin features, local open chromatin is an insufficient catalyst for gene expression. In contrast, we see that in the active compartment, open chromatin in any position is correlated with greater expression than for genes in the active compartment with no other features (Figure S6). Taken together, these results argue against a uniform mechanism of gene regulation and suggest instead that a context‐specific, dynamic interplay of epigenetic features, including nuclear location, open chromatin and histone modifications, creates and maintains a uniquely appropriate environment for each gene. Finally, because the modified histones H3K27ac and H3K4me1 have been shown to fulfill additional roles at mammalian enhancers (Creyghton et al., 2010; Heintzman et al., 2009), we asked if these modifications might also be present at grapevine cCRE regions, but we found only low‐level variations in signal (Figure S7) that are not recognized as peaks by the MACS2 callpeak algorithm (Zhang et al., 2008). Our results agree with previously reported data in other plant species showing that plant enhancers are not strongly associated with H3K27ac or H3K4me1 (Oka et al., 2017; Yan et al., 2019; Zhu et al., 2015), as they are in mammals. These results emphasize the challenges of CRE identification in plants and highlight the efficacy of the ATAC‐seq approach.
Figure 4.
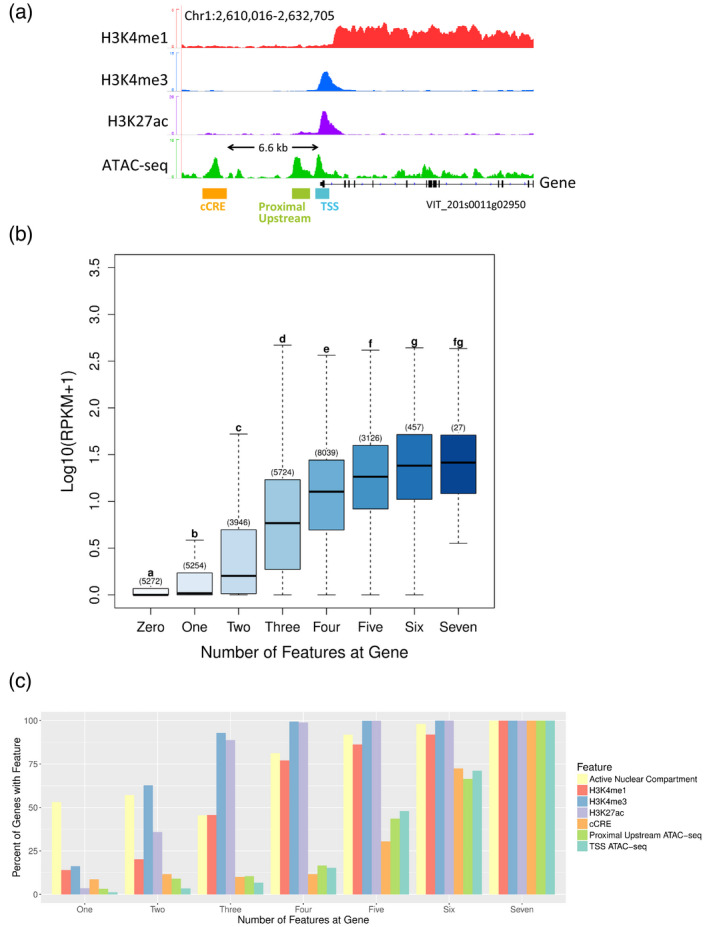
Correlation of local environment with gene expression.
(a) Examples of local features accounted for in (b) and (c). Not shown: nuclear compartment (active or inactive).
(b) Expression levels of genes calculated as log10 (RPKM + 1), stratified by total number of epigenetic features in gene environment. Number of genes included in each group is shown above each box, and significant differences (P < 0.05) between boxplots are indicated by letters (a–g). Each letter represents a statistically unique set of expression levels based on P‐values calculated by an independent pairwise Wilcoxon test.
(c) Percent of genes in each feature number group having the indicated epigenetic feature.
Upstream cCREs are correlated with higher expression in their closest promoter genes compared with downstream cCREs
After determining that distal cCREs were significantly associated with gene expression, we examined whether the position of the cCRE relative to the closest gene affected this relationship. Although mammalian CREs are frequently located 10s or 100s of kilobases away from their target promoter (Wang et al., 2019; Williamson et al., 2011; Yao et al., 2015), in more compact plant genomes CREs are often positioned within 10 kb of the gene they likely regulate (Lu et al., 2017; Maher et al., 2018). We found that approximately three‐quarters (76.3%, 3739 peaks) of cCRE regions in the grapevine genome are between 1 and 15 kb from the nearest promoter, and that about 66% of them (3232 peaks) are located upstream of their closest promoter (Table S1). Upstream peaks are significantly closer to their closest promoters than downstream peaks (dark blue boxplot versus dark red boxplot; Figure 5a), and the median intergenic spaces in which these peaks are found (light blue and light red boxplots) are significantly larger than the median length of all V. vinifera intergenic intervals (gray boxplot; Figure 5a). To compare the expression of genes associated with upstream or downstream peaks, we created three gene subsets of equal numbers (n = 1131 genes, the maximum number of unique genes closest to a downstream peak) that were also similar in size distribution (Figure S8a) to avoid the bias of shorter gene selection for closest downstream peak genes. Using these subsets, we compared the expression levels for genes with upstream peaks, downstream peaks, and also genes for which the promoter was not closest to any distal cCRE (Figure 5b). Notably, there is a significant difference (P = 0.002) in the expression levels of genes for which the cCRE is upstream versus downstream with respect to its closest promoter (Figure 5b). Because genes that are closest to a downstream cCRE show significantly more expression than genes that are not closest to any cCRE (gray boxplot, P = 2.0E‐07) but less expression than genes with an upstream cCRE, it is possible that cCRE position might influence its effect. Multiple possibilities could explain this phenomenon. Assuming downstream cCREs are nevertheless functional, some of these downstream peaks might regulate genes other than those whose promoters are closest, either the next‐closest gene or perhaps distal genes. For a fraction of these, it is also possible that their closest gene is unannotated in the reference genome. Furthermore, it is possible that the extra distance a downstream cCRE must cross to reach an upstream promoter could impede its function compared with upstream peaks. However, among all genes closest to a cCRE, we do not see a relationship between the distance between the cCRE and the promoter versus gene expression (Figure S8b), indicating that simple base pair distance has little or no effect on potential enhancer activity. Our observations support a model in which plant CREs are preferentially close to their target promoters, likely due to relatively short intergenic spaces compared with those of mammals (Murat et al., 2012), but that longer‐distance interactions are possible even without conserved mammalian‐like topologically associated domains (Dong et al., 2017; Wang et al., 2015).
Figure 5.
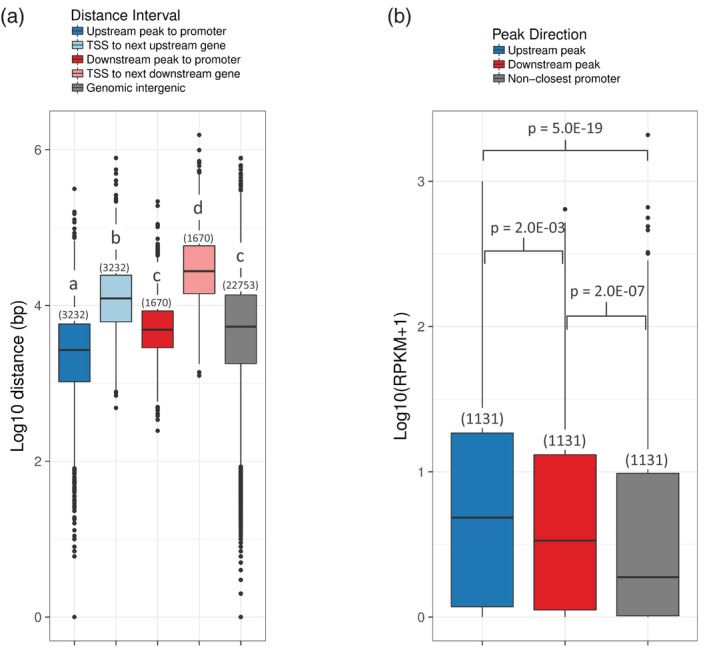
Distance of candidate cis‐regulatory elements (cCREs) to their closest promoter and expression levels of closest genes.
(a) Comparison of the distribution of candidate CRE distances from their nearest promoter for either upstream peaks (dark blue) versus the size of the intergenic interval (light blue) in which they are found or downstream peaks (dark red) versus the size of the intergenic interval in which they are found plus the length of the gene (light red). ‘Genomic Intergenic’ shows the average length of every intergenic interval remaining in the Vitis vinifera genome after subtracting the genes plus their 2‐kb upstream promoters. P‐value determined by an independent pairwise Wilcoxon test. Significant differences (P < 0.05) in promoter distance or intergenic interval length [log10(bp)] between subsets are indicated by unique letters (a–d).
(b) Expression levels of subsets of closest‐promoter genes for which the nearest cCRE is upstream (blue) or downstream (red), or of genes that are not closest to a cCRE (gray). Genes in each group were selected so that each subset had an equal number of genes with a statistically similar length (in bp) distribution. P‐value determined by an independent pairwise Wilcoxon test. Significant differences (P < 0.05) in expression were seen between each subset.
An imbalance in TSS and cCRE accessibility correlates with allele‐specific expression
Given the importance of the TSS environment for total gene expression, we then asked if an imbalance in expression between alleles could be explained in part by corresponding differences in chromatin accessibility and modified histone enrichment at each haplotype. We used allele‐specific expression estimates obtained from Pinot Noir leaf tissues, and then performed allele‐specific mapping of ATAC‐seq reads or ChIP‐seq reads. Where informative SNPs occurred in the peak regions we counted reads mapping to either allele (reference or alternative) and retained those for which coverage was equal or greater than 10 (5331 promoter ATAC‐seq peaks and 2867 intergenic ATAC‐seq peaks). For promoter ATAC‐seq peaks overlapping the TSS, intergenic peaks upstream of their closest promoter (distal cCREs), and TSS‐associated H3K4me3 and H3K27ac peaks, we calculated a d‐score (Xu et al., 2017), which reflects the ratio of sequencing reads mapping to one allele or the other and can range from −0.5 (all reads mapped to alternative allele) to 0.5 (all reads mapped to reference allele), with a value of 0.0 indicating peaks of equal magnitude at each allele. Similarly, we calculated d‐scores for allele‐specific expression, for which extreme values indicate monoallelic expression and 0.0 indicates equal expression of the two alleles, and we assessed the correlation between the gene expression d‐score and ATAC‐seq or ChIP‐seq peak d‐scores. We first filtered the genes to include only those for which the absolute value of the d‐score was greater than or equal to 0.3, indicating an expression bias toward one of the alleles. We then grouped the d‐scores of each chromatin feature according to the direction of allele bias of their associated gene and asked if the populations of feature d‐scores were significantly biased toward the same allele using a one‐sided Wilcoxon test. We found significant biases for TSS‐ATAC‐seq peaks (P = 0.0004, n = 57), intergenic ATAC‐seq peaks (distal cCREs; P = 0.026, n = 26) and H3K4me3 peaks (P < 2.2E‐16, n = 220; Figure 6a–c), suggesting that differences in chromatin accessibility, both at the TSS and at upstream cCREs, could be reflected in differences in allele expression. Surprisingly, neither TSS‐associated H3K27ac nor H3K4me1 d‐scores correlated with gene expression d‐scores (Figures 6d and S9), indicating that the roles of these modifications at genes might be separate from direct facilitation of transcription. Further investigation showed that these two modifications, and in particular H3K4me1, are directly correlated with CpG gene body methylation, suggesting an intriguing relationship between the coexistence of histone and DNA epigenetic marks (Figure [Link], [Link]) in the grapevine genome, as has been shown in Arabidopsis (Inagaki et al., 2017; Zhang et al., 2020). Together, these data confirm that large differences in expression between alleles are associated with equally large differences in the TSS environment deriving from the H3K4me3 histone modification and chromatin accessibility, while the weaker allele‐specific link between putative enhancers and genes implies a less‐direct relationship between upstream regulatory elements and their target genes.
Figure 6.
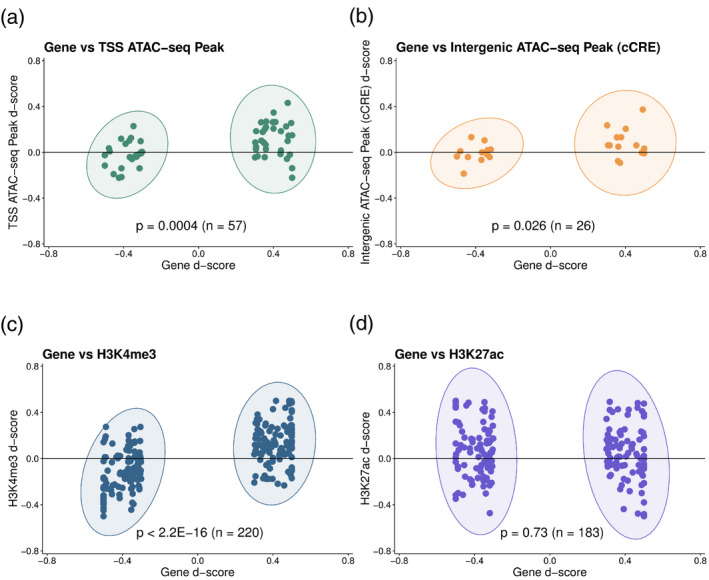
Correlation between d‐scores of allele‐specific expressed genes and (a) transcription start site (TSS)‐overlapping ATAC‐seq peaks, (b) upstream intergenic ATAC‐seq peaks [candidate cis‐regulatory elements (cCREs)], (c) TSS‐overlapping H3K4me3 peaks, and (d) TSS‐overlapping H3K27ac peaks. Panel (b) includes only allele‐specific expressed genes whose promoter was closest to an upstream intergenic ATAC‐seq peak. For each panel, P‐values were calculated between y‐axis values of groups contained in ellipses using a one‐tailed, independent two‐sample Wilcoxon test. The horizontal line in each plot marks a chromatin feature d‐score of zero.
Conserved cCREs are few but are located near highly conserved genes
Despite their universal function of facilitating transcription, enhancers themselves display a wide range of conservation, with some enhancer sequences strongly conserved and others undergoing accelerated evolution (Odom et al., 2007; Villar et al., 2015). To identify patterns of Vitis cCRE evolution, we used the VISTA‐Point tool for comparative genomics (Dubchak et al., 2000; Frazer et al., 2004), to find 3343 conserved, non‐coding, non‐TE sequences (CNSs) between V. vinifera and Prunus persica, a species with known orthology to grapevine (Verde et al., 2013). Most of these (3111 regions) do not overlap with the Vitis leaf cCREs identified in this study, and a manual BLAST search of several of these regions revealed a mixture of pseudogenes, mitochondrial genome fragments, and various other sequences. A MEME‐ChIP motif search of these non‐cCRE CNSs uncovered 33 significantly enriched motifs (Data [Link], [Link], [Link]), suggesting that some unknown conserved sequences might be regulatory elements, possibly in another tissue such as berry or root, although the maximum motif E‐value of 3.8E‐7 is well above that found for the Vitis leaf cCREs (2.6E‐124). The small fraction (188/3343; 5.6%) of CNSs that do overlap at least 50% with 169 grapevine cCREs suggest increased sequence constraint at a small subset of cCREs (169/4902; 3.4%), while the large majority of grapevine cCREs (4733/4902; 96.6%) do not represent regions of sequence conservation between the two species. For all cCREs for which nucleotide diversity data were available, we measured the diversity of these two groups (CNS‐overlapping or non‐overlapping cCREs) using data from 10 grapevine varieties (Magris et al., 2019), revealing two strikingly different patterns: the CNS‐overlapping cCREs appear to be under high sequence constraint, while the remaining cCREs are not (red boxplot and green boxplot, respectively; Figure 7a). Because this widespread lack of conservation seems to contradict recently published studies that showed a decrease in nucleotide diversity at the ATAC‐seq peak summit in maize and other plant species (Lu et al., 2019; Ricci et al., 2019), we considered the possibility that the majority of the regions we identified by ATAC‐seq were simply stochastic Tn5 insertions into the genome. However, the prevalence of common peaks between three separate transposition events argues against this possibility (Figure [Link], [Link]). Repeating the MEME‐ChIP analysis to compare the two categories of cCREs did not reveal notable differences in the TF binding motifs contained in each, so we then asked whether their conservation levels could be related instead to the genes they are closest to. As shown in Figure 7(b), genes closest to conserved cCREs indeed show a significantly lower ratio of non‐synonymous/synonymous (θN/θS) diversity, indicating that, like the nearest cCRE, these genes are under relatively high sequence constraint presumably due to strong purifying selection pressure. Notably, genes near the non‐conserved cCREs have higher θN/θS ratios than those near conserved cCREs, but significantly lower θN/θS ratios than genes not near any cCRE. Thus, we have identified three categories of genes differing in their sequence evolution patterns: (i) highly conserved genes that are near relatively conserved cCREs; (ii) moderately conserved genes that are near diversifying cCREs; (iii) less‐conserved genes that are not near cCREs.
Figure 7.
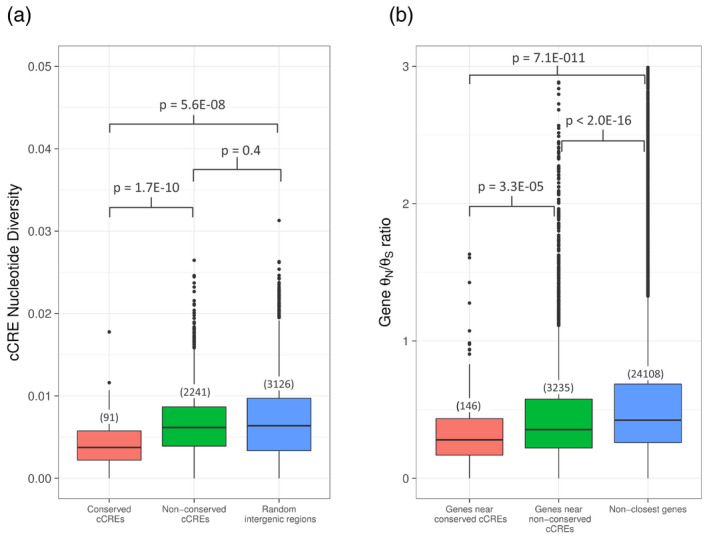
Conservation levels of candidate cis‐regulatory elements (cCREs) and their closest genes.
(a) Nucleotide diversity of cCREs sharing conservation (red boxplot) or not (green boxplot) with regions in Prunus persica as compared with that of non‐conserved, random intergenic regions (blue boxplot). The number of peaks or regions included in the analysis is listed above each boxplot, and P‐values were determined using an independent pairwise Wilcoxon test.
(b) Rates of non‐synonymous to synonymous (θN/θS) nucleotide substitutions for genes closest to conserved cCREs (red boxplot), non‐conserved cCREs (green boxplot), and genes not closest to a cCRE (blue boxplot). The number of genes included in the analysis is listed above each boxplot, and P‐values were determined using an independent pairwise Wilcoxon test.
Gene ontology reveals enrichment of cCREs near TF genes
Given the two different categories of cCREs we found (conserved or non‐conserved) and the different rates of evolution of their closest genes, we reasoned that these gene/cCRE pairs might fulfill different functions for the cell, and that a Gene Ontology search (Ashburner et al., 2000; The Gene Ontology Consortium et al., 2021) might reveal unique enrichments of genes in each group. Surprisingly, this was largely not the case. As shown in Table S2, both groups were enriched for molecular functions ‘DNA binding’ (GO:0003677) and ‘TF activity’ (GO:0003700), while significantly enriched biological processes showed greater variation, ranging from ‘cell differentiation’ (GO:0030154) to ‘biosynthetic process’ (GO:0009058). The most significant cellular compartment category for both groups was ‘nucleus’ (GO:0005634). These findings are consistent with previously reported data showing that the pool of genes targeted by MYB family TFs was enriched for other TF genes in Arabidopsis (Maher et al., 2018). Our results indicate that, regardless of conservation level, cCREs are preferentially located near TF genes, providing an intriguing clue to their function and potentially placing them in a position of importance within critical and broadly‐reaching regulatory cascades. However, more detailed studies are needed to understand why a select few of these genes are maintained under such strong sequence conservation.
Discussion
Appropriate gene expression is the critical function of a nucleus, and understanding how gene regulatory networks control transcription has become a major focus in a variety of fields, from medicine to plant breeding. Owing to the massive output of information from next‐generation sequencing, CREs have been identified as key players in gene transcription, and even small mutations in these regions have been shown to cause dramatic changes in phenotype (Cai et al., 2020; Rodríguez‐Leal et al., 2017). These regulatory elements represent an exciting new area of exploration in which levels of gene product can be controlled with high sensitivity, without affecting the coding sequence of the gene or needing to introduce RNA to a cell, as with RNAi, and are ideal sequences to be modified by CRISPR/Cas9 mediated genome editing techniques to achieve fine tuning of gene expression.
Here, candidate CREs were identified in V. vinifera using ATAC‐seq. These regions of open chromatin were found to contain DNA binding motifs for several families of TFs, including, among others, bHLH proteins, bZIP proteins, MYB proteins and, most abundantly, TCP proteins, a large TF family exclusive to plants that has also been implicated in TAD‐like boundaries in rice (Liu et al., 2017). We find that such regions of open chromatin are correlated with increased expression of the gene whose promoter is closest to the ATAC‐seq peak, suggesting that these sites might be more likely to host activator proteins rather than repressors. We note that the level of enrichment for H3K27ac at these cCREs increases slightly, but not enough to be called a peak by MACS2. The absence of strong enrichment for histone modifications agrees with previously reported findings that H3K4me1 and H3K27ac cannot easily be used for de novo identification of plant enhancers, and raises the possibility that plants might have evolved unique epigenetic signatures at their enhancers as compared with their mammalian or Drosophila counterparts (Heintzman et al., 2009; Koenecke et al., 2016; Oka et al., 2017; Zhu et al., 2015), despite sharing the feature of low enhancer methylation (Calo and Wysocka, 2013). Combined with our results suggesting that upstream elements have stronger activating properties with respect to downstream elements, which might regulate long‐distance genes even in the likely absence of TADs (Dong et al., 2017; Wang et al., 2015), we provide further evidence that animal and plant regulatory elements have unique characteristics and that custom experimental approaches should be tailored to each.
A peculiar and seemingly contradictory feature of enhancers is their nucleotide diversity as a consequence of the selective forces acting on them. Assuming putative enhancers play an important role in gene expression, it is reasonable to expect that their sequence would be tightly constrained due to purifying selection, as previously shown for certain cases (Dickel et al., 2018; Lettice et al., 2017; Pennacchio et al., 2006). Indeed, two recent studies showed a decrease in nucleotide diversity at the summit of the open chromatin peak for several plant species (Lu et al., 2019; Ricci et al., 2019). In contrast, our analyses show that only a small fraction of candidate CREs in Vitis leaf tissue are under sequence constraint, while the majority permit an average nucleotide diversity similar to random intergenic regions. These results are consistent with the rapid enhancer evolution recently documented in another plant species, cotton (Gossypium hirsutum; Wang et al., 2017), and multiple studies that suggest mutations drive beneficial variation and evolution in enhancers (Odom et al., 2007; Villar et al., 2015) and that, while enhancer function is conserved, enhancer sequence conservation is not required (Blow et al., 2010; Hare et al., 2008; Plessy et al., 2005; Snetkova et al., 2021). This apparent paradox in CRE maintenance likely reflects the coexistence of multiple enhancer types as previously described (Li et al., 2019), whose characteristics are obscured with ensemble analyses. Our findings support a model in which different categories of enhancers fulfill separate functions and enhancer function can be maintained despite sequence diversification. We had previously reported (Magris et al., 2019) that genes under stronger purifying selection as estimated by the ratio of non‐synonymous to synonymous nucleotide diversity (θN/θS) are significantly less variable in their expression among grape genotypes. The observation that conserved cCREs are usually found near genes showing a low rate of sequence evolution may provide a mechanistic explanation for the correlation between expression variation and strength of purifying selection acting on the gene.
Of all the characteristics of open chromatin our study reveals, perhaps the most intriguing is the highly significant enrichment of TF genes located nearest to both conserved and not conserved cCREs, which is consistent with previous studies (Maher et al., 2018; Plessy et al., 2005). This result could simply reflect the requirement for high expression of these genes in young leaf tissue, but it could also hint at a mechanism of efficient enhancer‐activity propagation, in which a single TF gene can be upregulated by the cCRE, and the resulting high levels of TF protein are then available to move about the nucleus, facilitating gene expression in numerous other genes via binding at the TSS. This possibility is supported by grapevine leaf expression data (Figure 4b; Figures [Link], [Link] and [Link], [Link]), and would help to explain why a distal cCRE was found near only about 3700 out of approximately 32 000 grapevine genes, yet most of them are expressed nonetheless. Consequently, it is possible that disruption of a TF‐gene‐adjacent CRE could have a widespread and unexpected effect on distant genes.
Beyond cCREs, our study showed that the grapevine genome is broadly organized into active and inactive compartments, which alternate with no specific pattern along each chromosome. Genes in the active compartment are generally more expressed than those in the inactive compartment, but the presence of an upstream cCRE, while more frequent in the inactive compartment, promotes gene expression in either compartment (Figure 3c). We found that the seven chromatin features analyzed (active nuclear compartment, H3K27ac, H3K4me1, H3K4me3, distal, proximal and TSS open chromatin) have an additive effect on gene expression, with more features present at a gene generally correlated to greater expression (Figure 4b). Additionally, we show that allelic imbalances in grapevine gene expression are linked to corresponding allele‐biased levels of H3K4me3 and accessibility of the TSS and upstream cCRE (Figure 6a–c). Because TF occupancy promotes open chromatin, it is likely that differences in accessibility arise from altered TF binding, due either to the occurrence of a SNP within a conserved binding motif or to a local epigenetic change such as increased DNA methylation that could inhibit binding. Our analysis of allele‐specific accessible chromatin relies specifically on the presence of SNPs within the ATAC‐seq peak that allow for allele‐specific read assignment, and thus does not allow us to distinguish between possible alternative mechanisms. Still, these results provide valuable insights on the relationship between gene environment and expression levels, and can provide direction for both more efficient breeding efforts and improvement through genetic modification.
We note a conspicuous absence of transposon‐associated cCREs in our set of identified open chromatin regions. A growing body of evidence implicates transposable elements in shaping cis‐regulatory networks (Bourque et al., 2018; Long et al., 2016) and, in humans, Alu elements, comprising 10% of the genome, have even been shown to function as enhancers in in vitro assays (Su et al., 2014). In grapevine, TEs make up at least 41% of the genome (Jaillon et al., 2007), but less than 2% (n = 204) of our ATAC‐seq identified peaks are located within them, most of which occur in Gypsy or Copia LTR retrotransposons (Figure S1). While read mappability may partly account for this, it seems to point to a real deficiency of CREs within transposons in grapevine. This may be due to the already cited recent movement of TEs that may not have left enough time for their cooption into the regulatory networks of the host as predicted by Orgel and Crick when defining properties of selfish DNA (Orgel and Crick, 1980). It is still possible that in a species like grapevine where high levels of structural variation due to polymorphic TE insertion events are observed (G. Magris, M. Morgante, personal communication), TEs may play an indirect role in gene regulation by either changing the local epigenetic environment or by modifying the spacing of CREs.
Our study has two major limitations. First, due to high levels of chloroplast contamination, despite using different nuclear isolation methods, we were unable to obtain high sequencing coverage for two biological replicates. Although the low‐coverage replicate data confirmed that Tn5 insertions into the genome were not random, the lack of higher coverage also makes it likely that many potentially accessible genomic regions were not identified, which could explain why relatively few promoters (about 3700) were identified as having a nearby cCRE. Another limitation of this study is the lack of molecular data verifying a direct link between cCREs and their target genes. We have based our analyses and conclusions on the assumption that the closest promoter along the linear chromosome is the most likely target of enhancer activity. Although our results do indicate that the genes closest to regions of open chromatin are frequently upregulated, we also show that when the cCRE is downstream of its nearest promoter, the effect on expression is significantly less than if the cCRE is upstream (Figure 5b), showing one limitation of this assumption and raising additional questions about the three‐dimensional folding of the genome. To precisely analyze promoter‐enhancer interactions, a proximity ligation‐based technique that enriches for these loci prior to sequencing, such as ChIA‐PET (Tang et al., 2015) or Capture Hi‐C (Mifsud et al., 2015), could be used instead of whole‐genome Hi‐C to provide the higher resolution and signal‐to‐noise required. Another appealing method for a more direct assessment of the functionality of these regions would be targeted gene editing as was shown in tomato (Rodríguez‐Leal et al., 2017) that would mutate or destroy the TF binding motif of a particular region, followed by RNA‐seq or protein analysis to observe any changes in the level of gene product.
With this work we have identified candidate CREs in V. vinifera leaf tissue, showing that their presence is associated with gene expression and that their placement with respect to nearby genes has potentially important effects. We reveal two classes of cCREs, those conserved and non‐conserved, and show that the genes nearest to each class show corresponding levels of non‐synonymous to synonymous nucleotide substitutions. We also show that gene regulation in a complex plant genome is affected by both global and local chromatin features whose combinatorial effects are reflected in different levels of expression. Future studies focusing on different tissues and developmental stages in grapevine will help to clarify how regulatory dynamics change throughout the lifespan of this important crop, and will provide new targets for study and potential breeding efforts.
Experimental Procedures
ChIP‐seq
Young leaf material (1st–5th leaves) was collected from Pinot Noir plants (VCR18) grown in the field (Azienda Agraria A. Servedei, Udine, Italy and Vivai Cooperativo Rauscedo, Rauscedo, Italy) during late spring/early summer. Approximately 1 g of leaf material was formaldehyde‐crosslinked and used to isolate chromatin and perform the immunoprecipitation according to the manufacturer’s protocol of the Abcam Plant ChIP kit (ab117137), except for the final DNA purification which was performed with 1.6 × volumes of AmpureXP beads (Beckman Coulter) rather than spin columns. Chromatin was sheared with a Diagenode Bioruptor sonicator using 5 cycles of 30 sec on, 90 sec off on the ‘HI’ setting. For each immunoprecipitation, 3–5 µg of antibody was used of either H3K4me3 (Abcam ab8580), H3K4me1 (Abcam ab8895) or H3K27ac (Abcam ab4729). The resulting immunoprecipitated or input DNA was used to produce sequencing libraries using the Ovation Ultralow System V2 kit (Nugen 0344NB) with 15 cycles of polymerase chain reaction (PCR) amplification, and libraries were quantified and checked for quality using a Bioanalyzer (Agilent). H3K4me3 libraries were produced in duplicate, and H3K4me1 and H3K27ac libraries were performed in triplicate, and all libraries were sequenced (single‐end or paired‐end, 125 bp) on an Illumina Hi‐Seq 2500 sequencer. A total of 67 501 979 unique reads were mapped for H3K4me1, 41 048 912 unique reads for H3K4me3, 80 346 835 unique reads for H3K27ac, and 99 271 775 unique reads for input samples. Reads were trimmed and filtered for quality ERNE version 1.4 (Del Fabbro et al., 2013), and aligned to the V. vinifera 12Xv0 464 reference genome using Bowtie 2.0.2. Peaks or broad regions enriched in sample versus input DNA were identified using MACS2 (v 2.1.0; Zhang et al., 2008) using the default settings for narrow peaks for H3K4me3 and H3K27ac, and the –broad option for H3K4me1.
ATAC‐seq
Leaf material Pinot Noir (VCR18) was collected as for ChIP‐seq, and approximately 200 mg of tissue was frozen in liquid nitrogen and ground to a powder. Nuclei were isolated using a modified version of Protocol A from Lutz et al. (2011) that was scaled down for small volumes. Briefly, ground leaf tissue was thawed in Extraction Buffer 1 (0.4 m sucrose, 10 mm Tris‐HCl pH 8.0, 5 mm beta‐mercaptoethanol, 0.01 × volume protease inhibitor cocktail for plants; Sigma‐Aldrich, P9599). Nuclei were filtered through two layers of Miracloth and centrifuged for 20 min at 4°C at 1940 g . The pellet was resuspended in Extraction Buffer 2 (0.25 m sucrose, 10 mm Tris‐HCl pH 8.0, 10 mm MgCl2, 1% Triton X‐100, 5 mm beta‐mercaptoethanol, 0.01 × volume protease inhibitors) and was centrifuged for 10 min at 4°C at 12 000 g . The pellet was then resuspended in 100 uL of Extraction Buffer 3 (1.7 m sucrose, 10 mm Tris‐HCl pH 8.0, 0.15% Triton X‐100, 2 mm MgCl2, 5 mm beta‐mercaptotethanol, 0.01 × volume protease inhibitors) and layered over 300 uL of Extraction Buffer 3, and the tube was centrifuged for 45 min at 4°C at 14 000 g . The supernatant was removed and, to the isolated nuclei pellet, 1 × transposition mix containing 1 × TDE buffer and Tn5 enzyme (Illumina, FC‐121‐1031) was added, and the reaction was incubated at 37°C for 30 min. The entire reaction was used for PCR amplification in a 50‐µL reaction using Nextera primers (N7xx, N5xx) with 11 cycles of amplification. Libraries were cleaned using 0.7 × volume of AmpureXP beads (Beckman Coulter), and checked for quality and quantity using a Bioanalyzer (Agilent). Libraries were produced in duplicate with only one library sequenced to high coverage, and both libraries were sequenced with an Illumina Hi‐Seq 2500 (paired‐end, 125 bp) for a total of 16 511 418 uniquely mapping reads. Reads were trimmed and filtered for quality using ERNE version 1.4, and aligned to the V. vinifera 12Xv0 464 reference genome using Bowtie 2.0.2. Peaks were identified using MACS2 (v 2.1.0) using the default settings for narrow peaks, except for ‐‐shift −100 and ‐‐extsize 200.
Allele‐specific ChIP‐seq and ATAC‐seq analysis
For allele‐specific mapping of ChIP‐seq or ATAC‐seq, filtered reads were aligned with Bowtie 2.0.2 (Langmead et al., 2009) to a reference V. vinifera genome for which SNPs were masked. Alignment files were then processed using markAllelicStatus.py of HiC‐Pro‐2.7.8 (Servant et al., 2015), and a VCF file containing phased Vitis SNPs to count the reads mapping to either haplotype at each heterozygous SNP occurring within peaks. For H3K4me1, we used only SNPs occurring in regions within 200 bp of the TSS. To account for mapping bias, this step was repeated using either input DNA (ChIP‐seq) or whole genome sequencing reads (ATAC‐seq and RNA‐seq) and, for each SNP, a correction factor (CF) was calculated [CF(ref) = 0.5*((reads(ref) + reads(alt))/reads(ref)], where reads(ref) is the number of reads mapping on the reference allele in the control experiment (input DNA or whole genome sequencing) and reads(alt) is the number of reads mapping on the alternative allele in the control experiment. CF(alt) was also calculated as 0.5*((reads(alt) + reads(ref))/reads(alt)). The number of experimental reads aligning to the reference and alternative allele were then multiplied by CF(ref) and CF(alt), respectively, to obtain their normalized value. From this new value, a corrected d‐score was calculated (d‐score = [normalized_reads(ref)/normalized_reads(ref) + normalized_reads(alt)] – 0.5) (Xu et al., 2017) such that for SNPs with an equal number of reads mapped to each haplotype, d‐score = 0, for SNPs with more reads mapped to the reference haplotype, d‐score > 0, and for SNPs with more reads mapped to the alternative haplotype, d‐score < 0.
Nucleotide diversity
Nucleotide diversity was estimated as previously described using a combined SNP dataset from 10 V. vinifera accessions (Magris et al., 2019). The average nucleotide diversity for each intergenic ATAC‐seq peak for which ND data was available was calculated and used in the resulting boxplot analysis. A random sampling of intergenic regions was included for comparison, and all regions were filtered to exclude those overlapping TEs, microsatellites and other regions for which no ND data was available.
DNA methylation
DNA methylation values were generated with bisulfite sequencing and estimated as previously reported (Magris et al., 2019), using young leaf tissue from Pinot Noir (VCR18). For the methylation metaprofile of ATAC‐seq peaks, full‐length intergenic ATAC‐seq peaks and flanking 5‐kb regions were each scaled to 100%, and for each percent the methylation averages for CG, CHG and CHH were calculated.
RNA‐seq
RNA‐seq data were produced from a prior experiment, using young leaves (1st–5th) collected in late spring to early summer. Leaf tissues for RNA extraction were sampled from mother stocks of ‘Pinot noir VCR18’ certified clone grown at the experimental station CASA40 of the Vivai Cooperativi Rauscedo (Rauscedo, Italy). Three biological replicates were sequenced for the transcriptome analysis. Each sample consisted of a mixture of the most distal leaves along the shoot, from the first leaf under the shoot apex to the fifth leaf from three vegetatively propagated plants along the raw in the vineyard, planted next to each other. Each biological replicate was separately processed during the steps of RNA extraction, library preparation, sequencing and data analysis. Total RNA was extracted with the Spectrum plant total RNA kit (Sigma, St Louis, MO, USA) from 200 mg of collected tissues ground in liquid nitrogen and stored at −80°C. Sequencing libraries were obtained using the TruSeq™ RNA Sample Preparation Kit v2 Set A (Illumina, RS‐122‐9001). Libraries were sequenced with the HiSeq2000 to obtain 50‐bp single‐end reads. Raw reads were processed for adapter removal, quality trimming and filtering for contaminants with package erne‐filter (from ERNE v2 (Del Fabbro et al., 2013). Filtered reads were aligned to the V. vinifera PN40024 reference genome (Jaillon et al., 2007) providing the V. vinifera V2.1 genes annotation through the aligner TopHat2 version 2.0.6 (Kim et al., 2013), default parameters. A total of 106 311 870 reads were mapped. Cufflinks version 2.2.0 (Trapnell et al., 2010, 2012) was used to estimate the transcript abundance in all varieties and tissues. Expression levels were reported as Reads Per Kilobase Million (RPKM) to normalize for the length of transcripts annotation and for the total number of reads aligned to the transcriptome.
Allele‐specific expression analysis
Allele‐specific gene expression was measured using using Allim (Pandey et al., 2013). By using haplotype‐specific reference genomes and RNA‐seq data, ALLIM quantified ASE for each replicate by determining the number of reads that can be unambiguously assigned to one of the haplotypes. For each replicate, statistical significance was assessed using a G‐test (the default test performed by ALLIM), and significance across replicates was assessed using Fisher’s meta‐analysis. The obtained P‐values were corrected for multiple testing according to the Benjamini–Hochberg procedure (Benjamini and Hochberg, 1995), and genes with a q‐value ≤ 0.05 were identified as showing evidence of allelic imbalance.
Hi‐C
In situ Hi‐C was performed on young grapevine leaves from Pinot Noir VCR18 using previously described methods (Louwers et al., 2009; Rao et al., 2014; Methods [Link], [Link], [Link], [Link]). Hi‐C libraries were sequenced with paired‐end, 125‐bp reads on an Illumina HiSeq 2500 sequencer by IGA Technology Services (Udine, Italy). Reads were filtered for contaminants and quality, and were processed with HOMER version 4.9 (Heinz et al., 2010) to produce genome‐wide contact maps. Principal component values were produced using the HOMER utility runHiCpca.pl, and A and B compartments were assigned to each 50‐kb window according to the sign of the first component (PC1) values.
Author Contributions
RS performed ATAC‐seq, ChIP‐seq and Hi‐C, integrated datasets and performed bioinformatics analyses, and drafted the manuscript; GM performed nucleotide diversity and gene nucleotide substitution analysis; M. Miculan performed RNA‐seq and whole‐genome transcriptome analysis; EP performed allele‐specific transcriptome analysis; MC and EDP performed bisulfite sequencing and methylation analysis; AT and FM performed Hi‐C computational analysis; AF and FM performed haplotype phasing used in allele‐specific analysis; M. Morgante designed the study, interpreted the results, and finalized the manuscript.
Conflict of Interest
All authors declare that they have no competing interests.
Supporting information
Figure S1. Distribution of TE‐located ATAC‐seq peak summits divided by TE family.
Figure S2. Read count profiles of modified histones or accessible chromatin at the TES.
Figure S3. IGV genome browser view of chromatin features across entire genome.
Figure S4. IGV genome browser views of chromatin features at specific sample genes.
Figure S5. Expression levels of genes stratified by number of local epigenetic features at each gene.
Figure S6. Expression levels of genes that are either in the Active Nuclear Compartment with no other chromatin features or in the Active Nuclear Compartment with a cCRE, Proximal Upstream ATAC‐seq peak or TSS ATAC‐seq peak.
Figure S7. Comparison of ChIP‐seq enrichment metaprofiles at cCREs, random intergenic regions matching the sizes of the cCREs, and TSSs.
Figure S8. Size distribution of the genes sets compared in Figure 4(a), and comparison of gene expression levels for nearby upstream and downstream cCREs.
Figure S9. Correlation between d‐scores of allele‐specific expressed genes and overlapping H3K4me1 enrichment.
Figure S10. Comparison of gene body methylation with respect to gene length and presence or absence of histone modifications.
Figure S11. Comparison of ATAC‐seq peaks from three separate Tn5 insertion reactions.
Table S1. Distribution of distances between cCREs and their closest promoter
Table S2. Gene ontology analysis results for genes near conserved cCREs versus genes near non‐conserved cCREs
Data S1. MACS2‐generated peak files for ATAC‐seq and ChIP‐seq.
Data S2. List of centrally enriched TF binding motifs found in intergenic ATAC‐seq peaks by MEME‐ChIP.
Data S3. List of centrally enriched TF binding motifs found in non‐cCRE CNSs by MEME‐ChIP.
Methods S1. Experimental procedures for Hi‐C in grapevine.
Acknowledgements
This work was funded by European Research Council grant agreement no. 294780 (project Novabreed), the Italian Ministry of Education and Research, National Research Council Epigenomics Flagship (project EPIGEN), and MIUR‐PRIN 2017 project 20178L3P38 ‘Regulation of gene expression in grapevine: analysis of genetic and epigenetic determinants’. The authors thank Dr Giusi Zaina, Nicoletta Felice and Dr Gabriele Di Gaspero for laboratory and field assistance, and IGA Technology Services (Udine, Italy) for DNA/RNA sequencing. Open Access Funding provided by Universita degli Studi di Udine within the CRUI‐CARE Agreement.
Data Availability Statement
Raw sequencing reads for ChIP‐seq, ATAC‐seq, RNA‐seq and Hi‐C are available at SRA accession PRJNA643441. Raw sequencing reads for BS‐seq are available under the SRA BioProject number SRP161872.
References
- Ashburner, M. , Ball, C.A. , Blake, J.A. , Botstein, D. , Butler, H. , Cherry, J.M. et al. (2000) Gene ontology: tool for the unification of biology. Nature Genetics, 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey, T.L. , Boden, M. , Buske, F.A. , Frith, M. , Grant, C.E. , Clementi, L. et al. (2009) MEME Suite: tools for motif discovery and searching. Nucleic Acids Research, 37, W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini, Y. & Hochberg, Y. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society Series B: Methodological, 57, 289–300. [Google Scholar]
- Blow, M.J. , McCulley, D.J. , Li, Z. , Zhang, T. , Akiyama, J.A. , Holt, A. et al. (2010) ChIP‐Seq identification of weakly conserved heart enhancers. Nature Genetics, 42, 806–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulay, G. , Volorio, A. , Iyer, S. , Broye, L.C. , Stamenkovic, I. , Riggi, N. et al. (2018) Epigenome editing of microsatellite repeats defines tumor‐specific enhancer functions and dependencies. Genes & Development, 32, 1008–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourque, G. , Burns, K.H. , Gehring, M. , Gorbunova, V. , Seluanov, A. & Hammell, M. et al. (2018) Ten things you should know about transposable elements. Genome Biology, 19(1), 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle, A.P. , Davis, S. , Shulha, H.P. , Meltzer, P. , Margulies, E.H. , Weng, Z. et al. (2008) High‐resolution mapping and characterization of open chromatin across the genome. Cell, 132, 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro, J.D. , Giresi, P.G. , Zaba, L.C. , Chang, H.Y. & Greenleaf, W.J. (2013) Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA‐binding proteins and nucleosome position. Nature Methods, 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai, Y.‐M. , Kallam, K. , Tidd, H. , Gendarini, G. , Salzman, A. & Patron, N.J. (2020) Rational design of minimal synthetic promoters for plants. Nucleic Acids Research, 48, 11845–11856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calo, E. & Wysocka, J. (2013) Modification of enhancer chromatin: what, how, and why? Molecular Cell, 49, 825–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark, R.M. , Wagler, T.N. , Quijada, P. & Doebley, J. (2006) A distant upstream enhancer at the maize domestication gene tb1 has pleiotropic effects on plant and inflorescent architecture. Nature Genetics, 38, 594–597. [DOI] [PubMed] [Google Scholar]
- Crawford, G.E. , Holt, I.E. , Mullikin, J.C. , Tai, D. , Blakesley, R. , Bouffard, G. et al. (2004) Identifying gene regulatory elements by genome‐wide recovery of DNase hypersensitive sites. Proceedings of the National Academy of Sciences, 101, 992–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton, M.P. , Cheng, A.W. , Welstead, G.G. , Kooistra, T. , Carey, B. , Steine, E. et al. (2010) Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proceedings of the National Academy of Sciences, 107, 21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Fabbro, C. , Scalabrin, S. , Morgante, M. & Giorgi, F.M. (2013) An extensive evaluation of read trimming effects on illumina NGS data analysis. PLoS ONE, 8(12), e85024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickel, D.E. , Ypsilanti, A.R. , Pla, R. , Zhu, Y. , Barozzi, I. , Mannion, B.J. et al. (2018) Ultraconserved enhancers are required for normal development. Cell, 172, 491–499.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong, P. , Tu, X. , Chu, P.‐Y. , Lü, P. , Zhu, N. , Grierson, D. et al. (2017) 3D chromatin architecture of large plant genomes determined by local A/B compartments. Mol. Plant, 10, 1497–1509. [DOI] [PubMed] [Google Scholar]
- Dong, Q. , Li, N. , Li, X. , Yuan, Z. , Xie, D. , Wang, X. et al. (2018) Genome‐wide Hi‐C analysis reveals extensive hierarchical chromatin interactions in rice. The Plant Journal, 94, 1141–1156. [DOI] [PubMed] [Google Scholar]
- Duan, Z. , Andronescu, M. , Schutz, K. , McIlwain, S. , Kim, Y.J. , Lee, C. et al. (2010) A three‐dimensional model of the yeast genome. Nature, 465, 363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubchak, I. , Brudno, M. , Loots, G.G. , Pachter, L. , Mayor, C. , Rubin, E.M. et al. (2000) Active conservation of noncoding sequences revealed by three‐way species comparisons. Genome Research, 10, 1304–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer, A. , Thibivilliers, S. , Ryu, K.H. , Schiefelbein, J. & Libault, M. (2021) Single‐nucleus RNA and ATAC sequencing reveals the impact of chromatin accessibility on gene expression in Arabidopsis roots at the single‐cell level. Molecular Plant, 14, 372–383. [DOI] [PubMed] [Google Scholar]
- Frazer, K.A. , Pachter, L. , Poliakov, A. , Rubin, E.M. & Dubchak, I. (2004) VISTA: computational tools for comparative genomics. Nucleic Acids Research, 32, W273–W279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galas, D.J. & Schmitz, A. (1978) DNAase footprinting a simple method for the detection of protein‐DNA binding specificity. Nucleic Acids Research, 5, 3157–3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grob, S. , Schmid, M.W. & Grossniklaus, U. (2014) Hi‐C analysis in Arabidopsis identifies the KNOT, a structure with similarities to the flamenco locus of Drosophila. Molecular Cell, 55(5), 678–693. 10.1016/j.molcel.2014.07.009 [DOI] [PubMed] [Google Scholar]
- Hare, E.E. , Peterson, B.K. , Iyer, V.N. , Meier, R. & Eisen, M.B. (2008) Sepsid even‐skipped enhancers are functionally conserved in drosophila despite lack of sequence conservation. PLoS Genetics, 4, e1000106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman, N.D. , Hon, G.C. , Hawkins, R.D. , Kheradpour, P. , Stark, A. , Harp, L.F. et al. (2009) Histone modifications at human enhancers reflect global cell‐type‐specific gene expression. Nature, 459, 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman, N.D. & Ren, B. (2009) Finding distal regulatory elements in the human genome. Current Opinion in Genetics & Development, 19, 541–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz, S. , Benner, C. , Spann, N. et al. (2010) Simple combinations of lineage‐determining transcription factors prime cis‐regulatory elements required for macrophage and B cell identities. Molecular Cell, 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesselberth, J.R. , Chen, X. , Zhang, Z. , Sabo, P.J. , Sandstrom, R. , Reynolds, A.P. et al. (2009) Global mapping of protein‐DNA interactions in vivo by digital genomic footprinting. Nature Methods, 6, 283–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki, S. , Takahashi, M. , Hosaka, A. , Ito, T. , Toyoda, A. , Fujiyama, A. et al. (2017) Gene‐body chromatin modification dynamics mediate epigenome differentiation in Arabidopsis . EMBO Journal, 36, 970–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaillon, O. , Aury, J.‐M. , Noel, B. , Policriti, A. , Clepet, C. & Casagrande, A. et al. (2007) The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature, 449, 463–467. [DOI] [PubMed] [Google Scholar]
- Kim, D. , Pertea, G. , Trapnell, C. , Pimentel, H. , Kelley, R. & Salzberg, S.L. (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biology, 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenecke, N. , Johnston, J. , Gaertner, B. , Natarajan, M. & Zeitlinger, J. (2016) Genome‐wide identification of Drosophila dorso‐ventral enhancers by differential histone acetylation analysis. Genome Biology, 17, 196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead, B. , Trapnell, C. , Pop, M. & Salzberg, S.L. (2009) Ultrafast and memory‐efficient alignment of short DNA sequences to the human genome. Genome Biology, 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lettice, L.A. , Devenney, P. , De Angelis, C. & Hill, R.E. (2017) The conserved sonic hedgehog limb enhancer consists of discrete functional elements that regulate precise spatial expression. Cell Reports, 20, 1396–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, S. , Kvon, E.Z. , Visel, A. , Pennacchio, L.A. & Ovcharenko, I. (2019) Stable enhancers are active in development, and fragile enhancers are associated with evolutionary adaptation. Genome Biology, 20(1), 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman‐Aiden, E. , van Berkum, N.l. , Williams, L. , Imakaev, M. , Ragoczy, T. , Telling, A. et al. (2009) Comprehensive mapping of long‐range interactions reveals folding principles of the human genome. Science, 326, 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, C. , Cheng, Y.‐J. , Wang, J.‐W. & Weigel, D. (2017) Prominent topologically associated domains differentiate global chromatin packing in rice from Arabidopsis. Nat. Plants, 3, 742–748. [DOI] [PubMed] [Google Scholar]
- Long, H.K. , Prescott, S.L. & Wysocka, J. (2016) Ever‐Changing landscapes: transcriptional enhancers in development and evolution. Cell, 167, 1170–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louwers, M. , Splinter, E. , van Driel, R. , de Laat, W. & Stam, M. (2009) Studying physical chromatin interactions in plants using chromosome conformation capture (3C). Nature Protocols, 4, 1216–1229. [DOI] [PubMed] [Google Scholar]
- Lu, Z. , Hofmeister, B.T. , Vollmers, C. , DuBois, R.M. & Schmitz, R.J. (2017) Combining ATAC‐seq with nuclei sorting for discovery of cis‐regulatory regions in plant genomes. Nucleic Acids Research, 45(6), e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, Z. , Marand, A.P. , Ricci, W.A. , Ethridge, C.L. , Zhang, X. & Schmitz, R.J. (2019) The prevalence, evolution and chromatin signatures of plant regulatory elements. Nat. Plants, 5, 1250–1259. [DOI] [PubMed] [Google Scholar]
- Lutz, K.A. , Wang, W. , Zdepski, A. & Michael, T.P. (2011) Isolation and analysis of high quality nuclear DNA with reduced organellar DNA for plant genome sequencing and resequencing. BMC Biotechnology, 11, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magris, G. , Di Gaspero, G. , Marroni, F. , Zenoni, S. , Tornielli, G.B. , Celii, M. et al. (2019) Genetic, epigenetic and genomic effects on variation of gene expression among grape varieties. The Plant Journal, 99(5), 895–909. [DOI] [PubMed] [Google Scholar]
- Maher, K.A. , Bajic, M. , Kajala, K. , Reynoso, M. , Pauluzzi, G. , West, D.A. et al. (2018) Profiling of accessible chromatin regions across multiple plant species and cell types reveals common gene regulatory principles and new control modules. The Plant Cell, 30, 15–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascher, M. , Gundlach, H. , Himmelbach, A. , Beier, S. , Twardziok, S.O. , Wicker, T. et al. (2017) A chromosome conformation capture ordered sequence of the barley genome. Nature, 544, 427–433. [DOI] [PubMed] [Google Scholar]
- Michael, T.P. & McClung, C.R. (2003) Enhancer trapping reveals widespread circadian clock transcriptional control in Arabidopsis. Plant Physiology, 132, 629–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mifsud, B. , Tavares‐Cadete, F. , Young, A.N. , Sugar, R. , Schoenfelder, S. , Ferreira, L. , et al. (2015) Mapping long‐range promoter contacts in human cells with high‐resolution capture Hi‐C. Nature Genetics, 47, 598–606. [DOI] [PubMed] [Google Scholar]
- Mojica‐Vázquez, L.H. , Benetah, M.H. , Baanannou, A. , Bernat‐Fabre, S. , Deplancke, B. , Cribbs, D.L. et al. (2017) Tissue‐specific enhancer repression through molecular integration of cell signaling inputs. PLoS Genetics, 13, e1006718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgante, M. , Depaoli, E. & Radovic, S. (2007) Transposable elements and the plant pan‐genomes. Current Opinion in Plant Biology, 10, 149–155. [DOI] [PubMed] [Google Scholar]
- Mueller, B. , Mieczkowski, J. , Kundu, S. , Wang, P. , Sadreyev, R. , Tolstorukov, M.Y. et al. (2017) Widespread changes in nucleosome accessibility without changes in nucleosome occupancy during a rapid transcriptional induction. Genes & Development, 31, 451–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murat, F. , de Peer, Y.V. & Salse, J. (2012) Decoding plant and animal genome plasticity from differential paleo‐evolutionary patterns and processes. Genome Biology Evolution, 4, 917–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemhauser, J.L. & Torii, K.U. (2016) Plant synthetic biology for molecular engineering of signalling and development. Nature Plants, 2, 16010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Malley, R.C. , Huang, S.C. , Song, L. , Lewsey, M.G. , Bartlett, A. , Nery, J.R. et al. (2016) Cistrome and epicistrome features shape the regulatory DNA landscape. Cell, 165, 1280–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odom, D.T. , Dowell, R.D. , Jacobsen, E.S. , Gordon, W. , Danford, T.W. , MacIsaac, K.D. et al. (2007) Tissue‐specific transcriptional regulation has diverged significantly between human and mouse. Nature Genetics, 39, 730–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka, R. , Zicola, J. , Weber, B. , Anderson, S.N. , Hodgman, C. , Gent, J.I. , et al. (2017) Genome‐wide mapping of transcriptional enhancer candidates using DNA and chromatin features in maize. Genome Biology, 18(1). 10.1186/s13059-017-1273-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orgel, L.E. & Crick, F.H.C. (1980) Selfish DNA: the ultimate parasite. Nature, 284, 604–607. [DOI] [PubMed] [Google Scholar]
- Pandey, R.V. , Franssen, S.U. , Futschik, A. & Schlötterer, C. (2013) Allelic imbalance metre (Allim), a new tool for measuring allele‐specific gene expression with RNA‐seq data. Molecular Ecology Resources, 13, 740–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennacchio, L.A. , Ahituv, N. , Moses, A.M. , Prabhakar, S. , Nobrega, M.A. , Shoukry, M. et al. (2006) In vivo enhancer analysis of human conserved non‐coding sequences. Nature, 444, 499–502. [DOI] [PubMed] [Google Scholar]
- Pennacchio, L.A. , Bickmore, W. , Dean, A. , Nobrega, M.A. & Bejerano, G. (2013) Enhancers: five essential questions. Nature Reviews Genetics, 14, 288–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plessy, C. , Dickmeis, T. , Chalmel, F. & Strähle, U. (2005) Enhancer sequence conservation between vertebrates is favoured in developmental regulator genes. Trends in Genetics, 21, 207–210. [DOI] [PubMed] [Google Scholar]
- Rao, S. , Huntley, M. , Durand, N. , Stamenova, E. , Bochkov, I. , Robinson, J. et al. (2014) A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell, 159, 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci, W.A. , Lu, Z. , Ji, L. et al. (2019) Widespread long‐range cis‐regulatory elements in the maize genome. Nat. Plants, 5, 1237–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, J.T. , Thorvaldsdóttir, H. , Winckler, W. , Guttman, M. , Lander, E.S. , Getz, G. et al. (2011) Integrative genomics viewer. Nature Biotechnology, 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers‐Melnick, E. , Vera, D.L. , Bass, H.W. & Buckler, E.S. (2016) Open chromatin reveals the functional maize genome. Proceedings of the National Academy of Sciences, 113, E3177–E3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez‐Leal, D. , Lemmon, Z.H. , Man, J. , Bartlett, M.E. & Lippman, Z.B. (2017) Engineering Quantitative Trait Variation for Crop Improvement by Genome Editing. Cell, 171(2), 470–480.e8. [DOI] [PubMed] [Google Scholar]
- Rojo, F. (2001) Mechanisms of transcriptional repression. Current Opinion in Microbiology, 4, 145–151. [DOI] [PubMed] [Google Scholar]
- Rühl, E. , Konrad, H. , Lindner, B. & Bleser, E. (2004) Quality criteria and targets for clonal selection in grapevine. In Acta Horticulturae. International Society for Horticultural Science (ISHS), Leuven, pp. 29–33. [Google Scholar]
- Salmaso, M. , Faes, G. , Segala, C. , Stefanini, M. , Salakhutdinov, I. , Zyprian, E. et al. (2005) Genome diversity and gene haplotypes in the grapevine (Vitis vinifera L.), as revealed by single nucleotide polymorphisms. Molecular Breeding, 14, 385–395. [Google Scholar]
- Servant, N. , Varoquaux, N. , Lajoie, B.R. , Viara, E. , Chen, C.‐J. , Vert, J.‐P. et al. (2015) HiC‐Pro: an optimized and flexible pipeline for Hi‐C data processing. Genome Biology, 16, 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sexton, T. , Yaffe, E. , Kenigsberg, E. , Bantignies, F. , Leblanc, B. , Hoichman, M. et al. (2012) Three‐dimensional folding and functional organization principles of the drosophila genome. Cell, 148, 458–472. [DOI] [PubMed] [Google Scholar]
- Siepel, A. (2005) Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Research, 15, 1034–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snetkova, V. , Ypsilanti, A.R. , Akiyama, J.A. et al. (2021) Ultraconserved enhancer function does not require perfect sequence conservation. Nature Genetics, 53, 521–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, L. & Crawford, G.E. (2010) DNase‐seq: a high‐resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harbor Protocols, 2010(2), pdb.prot5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler, M.B. , Murr, R. , Burger, L. , Ivanek, R. , Lienert, F. , Schöler, A. et al. (2011) DNA‐binding factors shape the mouse methylome at distal regulatory regions. Nature, 480(7378), 490–495. 10.1038/nature10716 [DOI] [PubMed] [Google Scholar]
- Su, M. , Han, D. , Boyd‐Kirkup, J. , Yu, X. & Han, J‐D‐J. (2014) Evolution of Alu elements toward enhancers. Cell Reports, 7, 376–385. [DOI] [PubMed] [Google Scholar]
- Sullivan, A.M. , Arsovski, A.A. , Lempe, J. et al. (2014) Mapping and dynamics of regulatory DNA and transcription factor networks in A. thaliana. Cell Reports, 8, 2015–2030. [DOI] [PubMed] [Google Scholar]
- Sung, M.‐H. , Guertin, M.J. , Baek, S. & Hager, G.L. (2014) DNase footprint signatures are dictated by factor dynamics and dna sequence. Molecular Cell, 56, 275–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, Z. , Luo, O. , Li, X. , Zheng, M. , Zhu, J. , Szalaj, P. et al. (2015) CTCF‐mediated human 3D genome architecture reveals chromatin topology for transcription. Cell, 163, 1611–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Gene Ontology Consortium , Carbon, S. , Douglass, E. , Good, B.M. , Unni, D.R. , Harris, N.L. et al. (2021) The gene ontology resource: enriching a gold mine. Nucleic Acids Research, 49, D325–D334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, M.R. & Scott, N.S. (1993) Microsatellite repeats in grapevine reveal DNA polymorphisms when analysed as sequence‐tagged sites (STSs). Theoretical and Applied Genetics, 86, 985–990. [DOI] [PubMed] [Google Scholar]
- Trapnell, C. , Roberts, A. , Goff, L. et al. (2012) Differential gene and transcript expression analysis of RNA‐seq experiments with TopHat and Cufflinks. Nature Protocols, 7, 562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell, C. , Williams, B.A. , Pertea, G. , Mortazavi, A. , Kwan, G. , van Baren, M.J. et al. (2010) Transcript assembly and quantification by RNA‐Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotechnology, 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verde, I. , Abbott, A.G. , Scalabrin, S. et al. (2013) The high‐quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nature Genetics, 45, 487–494. [DOI] [PubMed] [Google Scholar]
- Villar, D. , Berthelot, C. , Aldridge, S. , Rayner, T. , Lukk, M. , Pignatelli, M. et al. (2015) Enhancer evolution across 20 mammalian species. Cell, 160, 554–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vondras, A.M. , Minio, A. , Blanco‐Ulate, B. , Figueroa‐Balderas, R. , Penn, M.A. , Zhou, Y. et al. (2019) The genomic diversification of grapevine clones. BMC Genomics, 20, 972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, C. , Liu, C. , Roqueiro, D. , Grimm, D. , Schwab, R. , Becker, C. et al. (2015) Genome‐wide analysis of local chromatin packing in Arabidopsis thaliana. Genome Research, 25, 246–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J. , Dai, X. , Berry, L.D. , Cogan, J.D. , Liu, Q. & Shyr, Y. (2019) HACER: an atlas of human active enhancers to interpret regulatory variants. Nucleic Acids Research, 47, D106–D112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, M. , Tu, L. , Lin, M. et al. (2017) Asymmetric subgenome selection and cis‐regulatory divergence during cotton domestication. Nature Genetics, 49, 579–587. [DOI] [PubMed] [Google Scholar]
- Weber, B. , Zicola, J. , Oka, R. & Stam, M. (2016) Plant enhancers: a call for discovery. Trends in Plant Science, 21, 974–987. [DOI] [PubMed] [Google Scholar]
- Williamson, I. , Hill, R.E. & Bickmore, W.A. (2011) Enhancers: from developmental genetics to the genetics of common human disease. Developmental Cell, 21, 17–19. [DOI] [PubMed] [Google Scholar]
- Wu, C. , Li, X. , Yuan, W. et al. (2003) Development of enhancer trap lines for functional analysis of the rice genome. The Plant Journal, 35, 418–427. [DOI] [PubMed] [Google Scholar]
- Xu, J. , Carter, A.C. , Gendrel, A.‐V. , Attia, M. , Loftus, J. , Greenleaf, W.J. et al. (2017) Landscape of monoallelic DNA accessibility in mouse embryonic stem cells and neural progenitor cells. Nature Genetics, 49, 377–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, J. , Watts, J.A. , Pope, S.D. , Gadue, P. , Kamps, M. , Plath, K. et al. (2009) Transcriptional competence and the active marking of tissue‐specific enhancers by defined transcription factors in embryonic and induced pluripotent stem cells. Genes & Development, 23, 2824–2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, X. , Crow, M. , Rice, B.R. , Li, F. , Harris, B. , Liu, L. et al. (2021) Single‐cell RNA sequencing of developing maize ears facilitates functional analysis and trait candidate gene discovery. Developmental Cell, 56, 557–568.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, W. , Chen, D. , Schumacher, J. , Durantini, D. , Engelhorn, J. , Chen, M. et al. (2019) Dynamic control of enhancer activity drives stage‐specific gene expression during flower morphogenesis. Nature Communications, 10, 1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao, L. , Berman, B.P. & Farnham, P.J. (2015) Demystifying the secret mission of enhancers: linking distal regulatory elements to target genes. Critical Reviews in Biochemistry and Molecular Biology, 50, 550–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin, Y. , Morgunova, E. , Jolma, A. , Kaasinen, E. , Sahu, B. , Khund‐Sayeed, S. et al. (2017) Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science, 356, eaaj2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zentner, G.E. & Henikoff, S. (2013) Regulation of nucleosome dynamics by histone modifications. Nature Structural & Molecular Biology, 20, 259. [DOI] [PubMed] [Google Scholar]
- Zhang, W. , Wu, Y. , Schnable, J.C. , Zeng, Z. , Freeling, M. , Crawford, G.E. et al. (2012) High‐resolution mapping of open chromatin in the rice genome. Genome Research, 22, 151–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Liu, T. , Meyer, C.A. , Eeckhoute, J. , Johnson, D.S. , Bernstein, B.E. et al. (2008) Model‐based analysis of ChIP‐Seq (MACS). Genome Biology, 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Wendte, J.M. , Ji, L. & Schmitz, R.J. (2020) Natural variation in DNA methylation homeostasis and the emergence of epialleles. Proceedings of the National Academy of Sciences, 117, 4874–4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, B. , Zhang, W. , Zhang, T. , Liu, B. & Jiang, J. (2015) Genome‐wide prediction and validation of intergenic enhancers in arabidopsis using open chromatin signatures. The Plant Cell, 27(9), 2415–2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Distribution of TE‐located ATAC‐seq peak summits divided by TE family.
Figure S2. Read count profiles of modified histones or accessible chromatin at the TES.
Figure S3. IGV genome browser view of chromatin features across entire genome.
Figure S4. IGV genome browser views of chromatin features at specific sample genes.
Figure S5. Expression levels of genes stratified by number of local epigenetic features at each gene.
Figure S6. Expression levels of genes that are either in the Active Nuclear Compartment with no other chromatin features or in the Active Nuclear Compartment with a cCRE, Proximal Upstream ATAC‐seq peak or TSS ATAC‐seq peak.
Figure S7. Comparison of ChIP‐seq enrichment metaprofiles at cCREs, random intergenic regions matching the sizes of the cCREs, and TSSs.
Figure S8. Size distribution of the genes sets compared in Figure 4(a), and comparison of gene expression levels for nearby upstream and downstream cCREs.
Figure S9. Correlation between d‐scores of allele‐specific expressed genes and overlapping H3K4me1 enrichment.
Figure S10. Comparison of gene body methylation with respect to gene length and presence or absence of histone modifications.
Figure S11. Comparison of ATAC‐seq peaks from three separate Tn5 insertion reactions.
Table S1. Distribution of distances between cCREs and their closest promoter
Table S2. Gene ontology analysis results for genes near conserved cCREs versus genes near non‐conserved cCREs
Data S1. MACS2‐generated peak files for ATAC‐seq and ChIP‐seq.
Data S2. List of centrally enriched TF binding motifs found in intergenic ATAC‐seq peaks by MEME‐ChIP.
Data S3. List of centrally enriched TF binding motifs found in non‐cCRE CNSs by MEME‐ChIP.
Methods S1. Experimental procedures for Hi‐C in grapevine.
Data Availability Statement
Raw sequencing reads for ChIP‐seq, ATAC‐seq, RNA‐seq and Hi‐C are available at SRA accession PRJNA643441. Raw sequencing reads for BS‐seq are available under the SRA BioProject number SRP161872.