

ABSTRACT
Clostridium perfringens type F strains causing nonfoodborne human gastrointestinal diseases (NFD) typically produce NanI sialidase as their major secreted sialidase. Type F NFDs can persist for several weeks, indicating their pathogenesis involves intestinal colonization, including vegetative cell growth and adherence, with subsequent sporulation that fosters enterotoxin production and release. We previously reported that NanI contributes to type F NFD strain adherence and growth using Caco-2 cells. However, Caco-2 cells make minimal amounts of mucus, which is significant because the intestines are coated with adherent mucus. Therefore, it was important to assess if NanI contributes to the growth and adherence of type F NFD strains in the presence of adherent mucus. Consequently, the current study first demonstrated greater growth of nanI-carrying versus non-nanI-carrying type F strains in the presence of HT29-MTX-E12 cells, which produce an adherent mucus layer, versus their parental HT29 cells, which make minimal mucus. Demonstrating the specific importance of NanI for this effect, type F NFD strain F4969 or a complementing strain grew and adhered better than an isogenic nanI null mutant in the presence of HT29-MTX-E12 cells versus HT29 cells. Those effects involved mucus production by HT29-MTX-E12 cells since mucus reduction using N-acetyl cysteine reduced F4969 growth and adherence. Consistent with those in vitro results, NanI contributed to growth of F4969 in the mouse small intestine. By demonstrating a growth and adherence role for NanI in the presence of adherent mucus, these results further support NanI as a potential virulence factor during type F NFDs.
KEYWORDS: Clostridium perfringens, NanI sialidase, intestinal disease, bacterial attachment, bacterial growth, mucus
INTRODUCTION
Clostridium perfringens causes many human and animal diseases due to its ability to produce more than 20 different toxins (1–4). However, individual C. perfringens strains do not make all of those toxins, which allows this bacterium to be classified into 7 types (A to G) (5). Type F strains, which, by definition, must carry the genes encoding alpha-toxin and Clostridium perfringens enterotoxin (CPE) (5), are the 2nd most common cause of bacterial food poisoning (FP) in the United States, where ∼1 million cases occur annually (6, 7). These bacteria also cause about 5 to 10% of all cases of nonfoodborne human intestinal diseases (NFD), such as antibiotic-associated diarrhea (8, 9). Type F infections are typically a diarrheal disease but can also involve a lethal enterotoxemia (i.e., where CPE is produced in the intestines before it is absorbed into the circulation and damages internal organs like the liver) in patients with certain predisposing medical conditions (10, 11).
Both FP and NFD caused by type F strains involve initial multiplication of these bacteria in the intestine (4, 8, 12, 13). Those vegetative cells then commit to an in vivo sporulation which causes CPE production and, when the mother cell lyses at the completion of sporulation, release of CPE into the intestinal lumen (12–14). While type F FP is often an acute illness lasting only a day, type F NFD can last up to several weeks despite the presence of diarrhea (8, 9, 12). This extended disease duration requires type F NFD strains to colonize the intestines, which involves cycles of growth and adherence of their vegetative cells in the intestines, with subsequent sporulation and CPE production (8, 9). Considering the above, understanding type F NFDs requires knowledge of how vegetative cells of type F NFD strains grow and adhere in disease-relevant conditions.
Nearly all type F NFD strains, and a minority of type F FP strains, produce a secreted sialidase named NanI (15). For those strains, NanI is predominantly responsible for exosialidase activity present during vegetative growth, and this sialidase is also produced by sporulating cultures (where NanI is copresent with CPE) (15). NanI is emerging as an important potential virulence factor for NanI-positive type F strains, i.e., NanI sialidase can contribute to their in vitro growth, sporulation, and CPE production, as well as to their persistent intestinal colonization (16, 17). NanI can also enhance CPE binding to, and cytotoxicity for, Caco-2 cells (18). NanI can be proteolytically processed to a stable 60-kDa fragment when incubated in the presence of mouse small intestinal fluid, and compared to native NanI, this 60-kDa NanI fragment has more sialidase activity and greater ability to promote CPE binding/cytotoxicity for enterocyte-like Caco-2 cells (18).
Our previous studies showed that NanI can (i) enhance the adherence of C. perfringens type F NFD strain F4969 with intestinal-like Caco-2 cells (15), and (ii) generate sialic acids to support the growth of F4969 when this type F strain is cocultured with Caco-2 cells (16). While Caco-2 cells have cell surface sialylconjugates, they make minimal amounts of mucus (19). In addition, our previous study showed that NanI-positive type F strains can grow using a solution of solubilized, partially purified mucins, which are sialylated components of mucus (16). While those previous studies are informative, the intestines are covered with an adherent layer of mucus, which is a complex mixture of several constituents (20, 21). Therefore, it is physiologically relevant to evaluate NanI contributions to C. perfringens adherence and growth in the presence of cells containing substantial levels of adherent mucus.
Like Caco-2 cells, HT-29 cells (HT29) are a minimal mucus-producing human intestinal cell line that was isolated from a colon adenocarcinoma (22). However, the HT29-MTX-E12 cell line (MTX-E12), which was derived from HT29 cells treated with methotrexate (23), exhibits characteristics of mature goblet-like cells in the small and large intestine, e.g., these cells produce a substantial layer of adherent mucus and make tight junctions (23). Although MTX-E12 cells are not a perfect model since they do not produce significant quantities of Muc2 mucin, which is a component of intestinal mucus, these cells are commonly used in comparison with HT29 or Caco-2 cells to study the contributions of mucus to bacterial pathogenesis in the gastrointestinal (GI) tract (24, 25). Similarly, the current study first used MTX-E12 cells as a model cell line to evaluate if NanI is important for supporting C. perfringens growth and adherence in environments containing host cells rich in adherent mucus. NanI effects on these parameters were concurrently examined in the parental HT29 cells to allow comparisons between NanI effects in the presence of similar high- versus low-mucus-producing intestinal-like cells. Last, this study directly tested NanI contributions to type F strain growth in the mucus-rich intestine using a mouse ligated small intestinal loop model.
RESULTS
Western blot comparison of Muc5A production between the HT29 and MTX-E12 cell lines.
For exploring whether NanI contributes to type F strain growth and adherence in the presence of substantial levels of adherent mucus, the current study initially used the cell line named HT29-MTX-E12 (MTX-E12), which produces an adherent mucus layer and is a derivative of the mucus-poor HT29 human enterocyte-like cell line (23–25). Before starting those in vitro experiments, it was first important to confirm that, in our hands, substantially more mucus was produced by MTX-E12 cells than HT29 cells. For this purpose, production of the mucin Muc5Ac was compared between these two cell lines. Muc5Ac Western blot analysis (Fig. 1) showed that, consistent with stronger mucus production, much more Muc5Ac was made by MTX-E12 cells than HT29 cells. An actin Western blot analysis confirmed equal loading of all samples. The conclusion that, relative to HT29 cells, MTX-E12 cells were producing substantially more Muc5A, which was adherent, and this was also independently demonstrated by immunofluorescence microscopy (as described later in the Results).
FIG 1.

Muc5Ac Western blot analysis for HT29 and MTX-E12 cell lines. (Top) A 50-µg protein aliquot of each cell lysate supernatant (see Materials and Methods) was separated on an SDS-containing 8% polyacrylamide gel. After transfer to a PVDF membrane, a Muc5Ac Western blot analysis was performed. (Bottom) The same amount of each protein sample was loaded onto an SDS-containing 12% polyacrylamide gel and probed with anti-beta actin antibody for use as a loading control. Size of proteins in kilodaltons is shown at left. The results shown are representative of three repetitions.
Comparison of C. perfringens type F strain growth in the presence of HT29 or MTX-E12 cells.
Our previous study (16) used a Transwell 0.4-µm-pore-size membrane filter support system to evaluate if minimal mucus-producing Caco-2 cells can support the growth and survival of C. perfringens type F strain F4969. Therefore, the current study first employed this same cell culture model system to compare the growth of type F strains that do or do not carry the nanI gene when those bacteria are cultured in the presence of adherent mucus-producing MTX-E12 cells versus minimal mucus-producing HT29 cells, from which MTX-E12 cells are derived.
For these analyses, the lower chamber of Transwells contained phosphate-buffered saline (PBS) covering a monolayer of either HT29 or MTX-E12 cells. When the type F strains in PBS were added separately to the upper chamber of these Transwells for either 4 or 24 h (Fig. 2A), significantly higher optical density at 600 nm (OD600) values were observed in the presence of MTX-E12 versus HT29 cells for the two type F strains (F5603 and B40) carrying the nanI gene. In contrast, OD600 values for the two type F strains (SM101 and NCTC8239) that lack the nanI gene were similar in the presence of MTX-E12 or HT29 cells, with SM101 cultures actually showing lower OD600 values when grown for 24 h in the presence of MTX-E12 versus HT29 cells.
FIG 2.
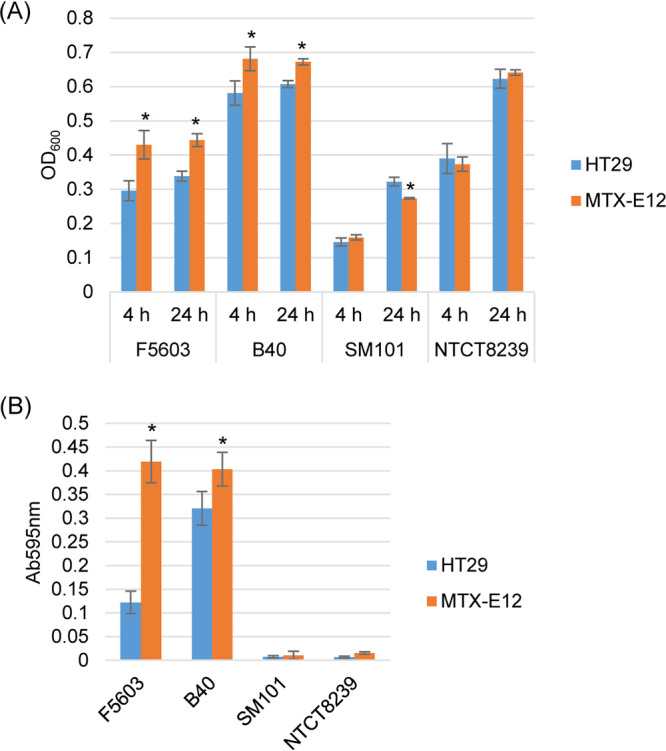
Comparison of C. perfringens type F strain growth in the presence of HT29 or MTX-E12 cells. (A) Postinoculation changes in the OD600 for type F strains B40, F5603, SM101, and NCTC8239 grown in PBS buffer and the presence of HT29 or MTX-E12 cells at 37°C for 4 h or 24 h. (B) Sialidase activity measured for 24-h cocultures using supernatants of samples from panel A. All panels show the mean values from three independent experiments. Error bars indicate the SD. *, P < 0.05 relative to culture with the HT29 cell line.
As expected from previous studies (15) showing that type F strains carrying the nanI gene usually make significantly more sialidase activity than type F strains that lack the nanI gene, F5603 and B40 culture supernatants contained significantly more sialidase activity than equivalent supernatants from SM101 and NCTC8239 (Fig. 2B). In addition, F5603 and B40 produced significantly more sialidase activity when cultured in the presence of MTX-E12 versus HT29 cells (Fig. 2B). These results suggested that NanI can help type F strains to grow better in the presence of mucus-producing host cells.
Analysis of NanI-producing type F strain F4969 growth and sialidase activity in the presence of mucus.
To confirm the Fig. 2 results and further characterize the growth of a NanI-producing type F in the presence of mucus-producing host cells, a time course growth study was performed with the NanI-producing type F NFD strain F4969 (15). Consistent with the Fig. 2 results, there was significantly more growth of wild-type F4969 when cocultured with MTX-E12 versus HT29 cells, with those growth differences beginning as soon as 2 h postinoculation (Fig. 3A). The significant difference detected between the two cell lines for their support of F4969 growth then continued at 4, 6, 8, 10 h, and even up to 24 h of coculture (Fig. 3A), i.e., at each of these time points, the OD600 values of F4969 were significantly higher when this type F strain was cultured in the presence of MTX-E12 versus HT29 cells, even though the F4969 inoculum was the same.
FIG 3.
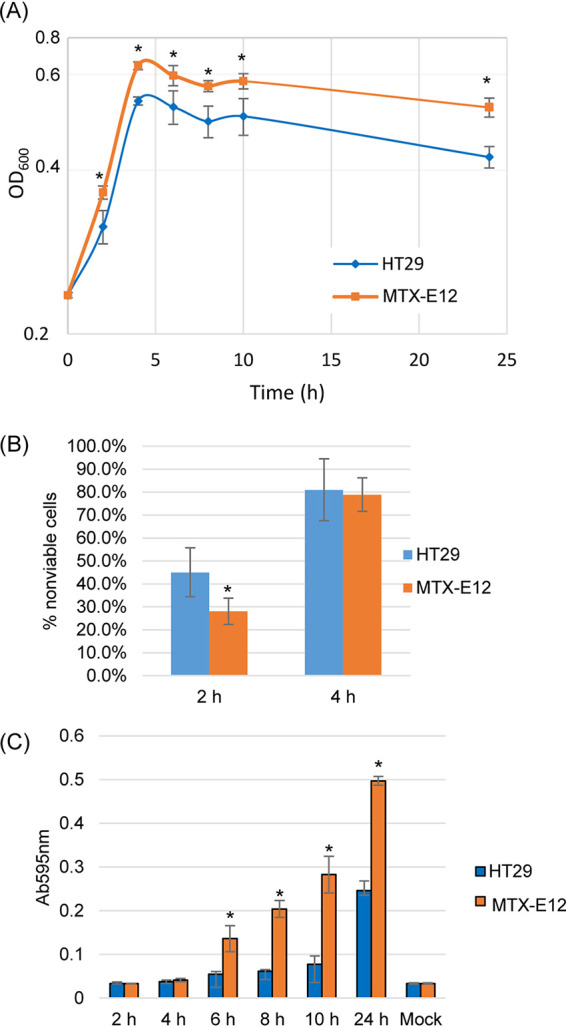
Comparison of growth, sialidase activity, and NanI activity levels when F4969 is cocultured in PBS buffer with the HT29 or MTX-E12 cell lines. (A) Postinoculation changes in the OD600 for cultures of wild-type F4969 growing in PBS buffer with HT29 or MTX-E12 cells at 37°C. Every 2 h up to 10 h of coculture, and then again at 24 h, a 0.5-ml aliquot of the coculture was removed, and the OD600 was determined. (B) HT29 and MTX-E12 cell cytotoxicity caused by coincubation with F4969 for 2-h and 4-h culture at 37°C. (C) F4969 sialidase activity measured every 2 h of coculture up to 10 h, and then at 24 h, using the supernatants of samples from panel A. All panels show the mean values from three independent experiments. Error bars indicate the SD. *, P < 0.05 relative to culture with HT29 cell line.
Experiments were then performed to begin determining why F4969 grows better in the presence of MTX-E12 cells. One possible explanation for the Fig. 3A growth differences might be that F4969 simply kills more MTX-E12 cells versus HT29 cells to obtain nutrients for growth. However, although there was already significantly more growth of F4969 when cocultured in the presence of MTX-E12 cells versus HT29 cells for 2 h (Fig. 3A), significantly less death of MTX-E12 cells versus HT29 cells was detected at that time point (Fig. 3B). By the 4-h peak of F4969 growth in Fig. 3A, there was equivalent death of MTX-E12 and HT29 cells (Fig. 3B).
An alternative explanation for Fig. 3A differences is that F4969 grows better in the presence of mucus-producing host cells because it uses NanI to liberate sialic acid, which C. perfringens can use for growth (26, 27). Consistent with that possibility, measurement of sialidase activity present in supernatants of the upper chambers of those F4969-infected Transwell cultures at 2-h intervals from 2 h to 10 h, and then again at 24 h (Fig. 3C), showed that supernatant sialidase activity increased over time when F4969 was cocultured with either MTX-E12 cells or HT29 cells. However, by 6 h and continuing thereafter, there was significantly more supernatant sialidase activity present when F4969 was cocultured with MTX-E12 cells versus HT29 cells. When the assay is incubated longer to increase sensitivity, higher sialidase activity was detected for F4969 cocultured, even for only 2 or 4 h with MTX-E12 cells versus HT29 cells (not shown).
Identification of the major sialidase responsible for supernatant sialidase activities when HT29 or MTX-E12 cultures were infected with F4969.
F4969 produces three sialidases, i.e., NanJ, NanI, and NanH (15), so it was important to discern which sialidase was the most important contributor to the increased supernatant sialidase activity that was detected (Fig. 3C) upon incubation of F4969 in the presence of MTX-E12 cells. For this purpose, a previously prepared F4969 nanI null mutant and nanI complementing strain (15) were used. Sialidase activity was determined at three time points (4, 8, and 24 h) when each of these C. perfringens strains was cocultured in the Transwell system with HT29 or MTX-E12 cells.
This experiment detected increased supernatant sialidase activity when wild-type F4969 was incubated in the presence of either HT29 or MTX-E12 cell cultures (Fig. 4A). However, consistent with Fig. 3C results, supernatant sialidase activity was significantly higher by 8 or 24 h postinfection when wild-type F4969 was incubated in the presence of MTX-E12 versus HT29 cultures.
FIG 4.

Sialidase activity, sialic acid release, and nanI gene expression of F4969, the F4969 nanI null mutant, and complementing strain when cocultured in PBS buffer with the HT29 or MTX-E12 cell lines. (A) Culture supernatant sialidase activity measured for 4, 8, or 24 h coculture of F4969 or its derivatives in PBS buffer and HT29 or MTX-E12 cells. Results shown are the average of three repetitions; error bars indicate the SD. Blue asterisks indicate a P value of <0.05 relative to nanI null mutant cultured with HT29 cell line. Orange asterisks indicate a P value of <0.05 relative to nanI null mutant cultured with MTX-E12 cell line. #, P < 0.05 relative to culture with HT29 cell line. (B) Sialic acid release by NanI when F4969 cocultures in PBS buffer with the HT29 or MTX-E12 cell lines at 6 h and 24 h. Sialic acid release due to NanI was calculated as the difference between the concentration of sialic acid released by coculture with F4969 versus the F4969 nanI null mutant strain. Shown is the mean of three repetitions. Error bars indicate SD. *, P < 0.05 relative to culture with HT29 cells. (C) Quantitative qRT-PCR analyses of nanI transcription was performed using 20 ng of F4969 RNA isolated from culture with HT29 or MTX-E12 cells for 4 h at 37°C. Average threshold cycle (CT) values were normalized to the value for the housekeeping 16S RNA gene, and the fold differences were calculated using the comparative CT (2−ΔΔCT) method. Shown is the mean of three repetitions. Error bars indicate SD. *, P < 0.05 relative to culture with HT29 cells.
In contrast, when Transwell cultures of either HT29 or MTX-E12 cells were infected with the F4969 nanI null mutant, there was little detectable supernatant sialidase activity, even after 24 h of incubation (Fig. 4A). The sharply reduced supernatant sialidase activity present in HT29 or MTX-E12 cultures infected with this nanI mutant was specifically due to nanI inactivation, rather than a secondary mutation, in that mutant since incubation of these cell cultures with a complementing strain resulted in strong supernatant sialidase activity. Together, these results indicated that NanI, rather than NanJ or NanH (or mammalian sialidases), was responsible for most of the exosialidase activity present in culture supernatants when F4969 is grown in the presence of host cells, including mucus-producing intestinal-like cells.
An experiment then examined whether the increased NanI activity observed when F4969 is cocultured in the presence of MTX-E12 cells impacts sialic acid levels released into the supernatants of those cultures, which could influence growth since free sialic acid can be used by C. perfringens (26, 27). In this experiment, increases in sialic acid supernatant levels due to NanI were calculated by subtracting supernatant sialic acid levels measured when MTX-E12 or HT29 cells were infected with wild-type F4969 versus the isogenic nanI null mutant. As shown in Fig. 4B, NanI caused nearly 2-fold higher sialic acid supernatant levels after coculture with MTX-E12 versus HT29 cells for either 6 or 24 h.
A final experiment evaluated whether the increased supernatant sialidase activity measured when F4969 encounters MTX-E12 versus HT29 cells is simply attributable to more NanI production due to the better growth of this strain in the presence of mucus or if it could also involve the presence of higher levels of nanI transcript levels in F4969 when that strain is cultured in the presence of mucus-producing cells. A nanI quantitative reverse transcription-PCR (qRT-PCR) assay was performed using F4969, which was cocultured with MTX-E12 cells or HT29 cells for 4 h at 37°C. Results of this experiment are shown in Fig. 4C and indicated that, compared to nanI transcript levels when F4969 was cocultured with HT29 cells, nanI transcript levels were significantly increased when F4969 was cocultured with MTX-E12 cells.
Assessing the specific contribution of NanI sialidase to F4969 growth in the presence of MTX-E12 versus HT29 cells.
Figure 4 showed that NanI is responsible for the strong supernatant sialidase activity induced when F4969 is cultured in the presence of HT29 cells or, particularly, MTX-E12 cells. Therefore, a similar experiment was performed to assess specifically whether NanI contributes to F4969 growth in the presence of those two cell lines. Results demonstrated that the nanI null mutant grew slower than wild-type F4969 or the complemented strain in the presence of either cell line. Within 4 h of culture in the presence of HT29 cells, the OD600 for the upper chamber of Transwells containing either F4969 or the complementing strain became significantly higher than those containing the nanI null mutant. In the presence of MTX-E12 cells, OD600 differences between the nanI mutant versus F4969 or the complementing strain reached statistical significance even faster, i.e., by 2 h of culture (Fig. 5A).
FIG 5.
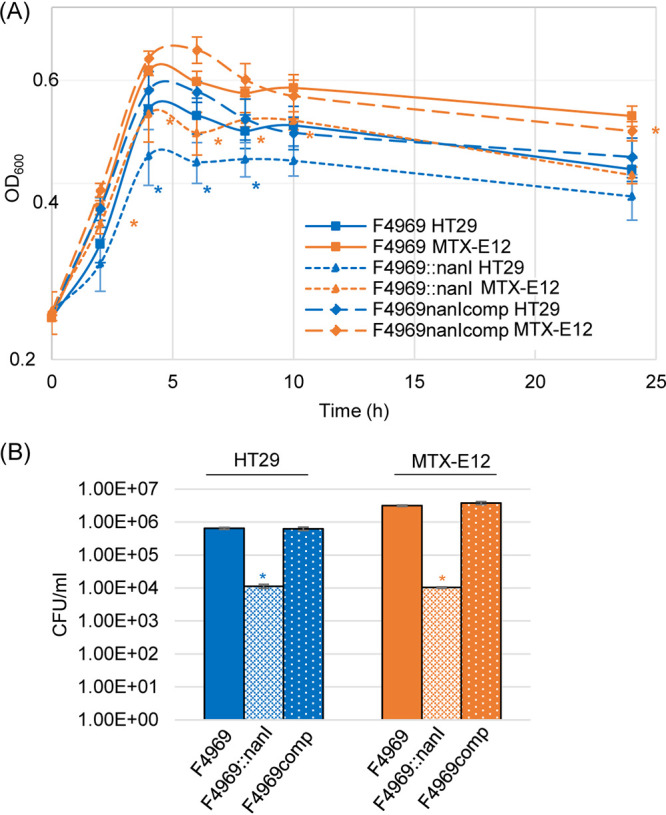
Comparison of growth and levels of vegetative cell survival by F4969, its isogenic nanI null mutant, and a complemented strain cocultured in PBS with HT29 or MTX-E12 cell lines. (A) F4969, the nanI null mutant, and a complemented strain were grown to 24 h at 37°C in PBS buffer with the presence of HT29 or MTX-E12 cells. At designated time points, the OD600 of each culture was determined. (B) Quantification of viable vegetative cells of F4969, its nanI null mutant, and a complemented strain when grown in PBS with HT29 or MTX-E12 cells. After the bacteria were cocultured with the host cells for 24 h at 37°C, those cultures were then plated onto BHI agar plates with 10-fold serial dilutions using PBS buffer. These BHI agar cultures were grown anaerobically overnight at 37°C for colony counting (log10 scale). Shown are the mean values from three independent experiments. Error bars indicate the SD. Blue asterisks indicate a P value of <0.05 relative to wild type in PBS cultured with the HT29 cell line. Orange asterisks represent a P value of <0.05 relative to wild type in PBS cultured with MTX-E12 cell line.
Another experiment was performed to compare the longer-term (24-h) viability of F4969 or its derivatives in the presence of HT29 versus MTX-E12 cells (Fig. 5B). When plate counting was performed for the 24-h cultures, considerably more viable vegetative cells of wild-type F4969 or the complementing strain were present when cocultured with MTX-E12 cells versus HT29 cells. Furthermore, compared to wild-type F4969 and complementing strains, significantly fewer viable vegetative cells of the nanI null mutant were present after 24 h culture in the presence of either host cell line. However, unlike wild-type F4969 or the complementing strain, there was no difference in viable vegetative cell numbers when the nanI null mutant was cultured for 24 h in the presence of the HT29 versus MTX-E12 cell lines. Collectively, Fig. 5 results further support the ability of NanI to contribute to C. perfringens growth and longer-term viability, particularly in a mucus-containing environment.
Evaluation of NanI effects on F4969 attachment to MTX-E12 versus HT29 cells.
We reported previously that NanI increases the attachment of F4969 to Caco-2 cells (15). The current experiment evaluated whether the presence of an adherent mucus layer impacts F4969 attachment to intestinal-like cells and if NanI impacts F4969 adherence to those mucus-producing host cells.
For these analyses, the adherence to HT29 cells versus MTX-E12 cells was compared between F4969 versus its isogenic nanI null mutant and the complemented strain. As shown in Fig. 6, the wild-type and complemented strains both attached to HT29 cells, with ∼4% to 5% of the total input bacteria being adherent. Quantitatively, this adhesion resembles the attachment of F4969 to Caco-2 cells, where ∼6% to 7% of the total input bacteria were adherent (15). However, the attachment of F4969 or the complemented strain to HT29 cells was significantly lower than their adhesion to MTX-E12 cells, where ∼20% to 30% of the total input bacteria attached. These results indicate that F4969 does attach well to host cells producing an adherent mucus layer and are consistent with the presence of that adherent mucus layer increasing the attachment of this type F strain to host cells.
FIG 6.
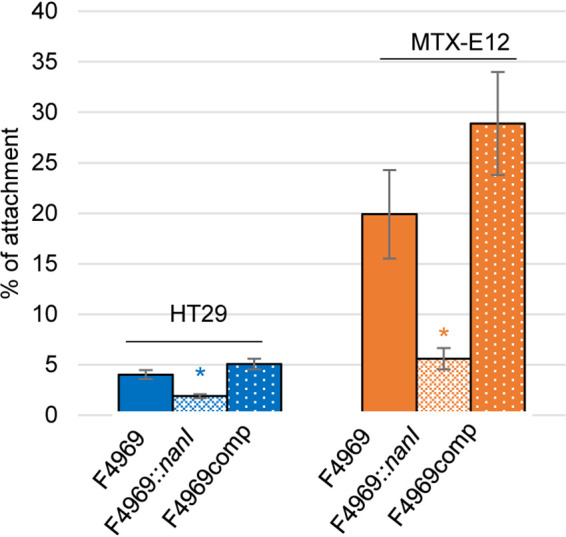
C. perfringens vegetative cell adherence to HT29 and MTX-E12 cell lines. HT29 or MTX-E12 cell monolayers were incubated for 2 h with washed vegetative cells of F4969, its nanI null mutant, or the complementing strain at 37°C under anaerobic conditions. Monolayers were then washed three times with PBS. After collection, the adherent bacteria were plated onto BHI agar plates for counting. Adherence was expressed as the percentage of attached bacteria relative to the total number of input bacteria. All data showed the mean values from three independent experiments. Error bars indicate the SD. Blue asterisks indicate a P value of <0.05 relative to wild-type adherence to the HT29 cell line. Orange asterisks represent a P value of <0.05 relative to wild-type adherence to the MTX-E12 cell line.
By comparison, the isogenic nanI null mutant strain exhibited only half as much adherence to HT29 cells as did wild-type F4969 or the complemented strain (Fig. 6). Compared to wild-type F4969 or the complemented strain, the nanI null mutant showed only ∼25% adherence to MTX-E12 cells. Note that, under the conditions used in Fig. 6, there was no growth of either F4969, the nanI null mutant, or the complemented strain during the 2-h duration of this experiment (data not shown), indicating that the measured adherence variations are not attributable to differences in the total number of bacterial cells present between F4969 and its derivatives. Collectively, these results indicated that NanI can be important for F4969 adherence to host cells, especially when an adherent mucus layer is present.
Immunofluorescence microscopy studies (Fig. 7) confirmed that wild-type F4969 and the complementing strain adhere better to the MTX-E12 cell line versus the HT29 cell line. This adherence involved NanI since, compared to F4969 or the complementing strain, fewer cells of the nanI mutant were visibly adherent to either MTX-E12 or HT29 cells in this experiment. Consistent with the plate count analyses for adherence, ImageJ fluorescence analysis of these photomicrographs indicated that F4969 adhered ∼2-fold better than the isogenic nanI mutant to HT29 cells and nearly 4-fold better than the nanI mutant to MTX-E12 cells.
FIG 7.

Microscopic comparison of mucin production by the HT29 and MTX-E12 cell lines, as well as detection of C. perfringens attachment to HT29 (A) and MTX-E12 (B) cells. Attachment of C. perfringens F4969, its nanI null mutant, and complementing strain to HT29 or MTX-E12 cell lines as detected by Olympus confocal laser scanning biological microscope (FluoView FV1000) with FV10-ASW (version 1.4) software. All pictures were taken at a magnification of ×400. Red, C. perfringens; green, Muc5Ac; blue, mammalian cell nuclei. Panels at right show enlarged view of attached wild-type F4969 and nanI null mutant in panels at left.
Muc5A immunostaining also supported Fig. 1 results indicating that MTX-E12 cells produce much more of an adherent mucus layer than HT29 cells.
Reduction of mucus in MTX-E12 cultures affects F4969 growth, sialidase production, and adherence.
To evaluate specifically whether F4969 grew better and was more adherent to MTX-E12 cells versus HT29 cells because those cells produce mucus, a pretreatment with N-acetyl cysteine (NAC) was used to reduce mucus levels in MTX-E12 cells, as previously reported (28). Consistent with the literature, NAC pretreatment of MTX-E12 cells reduced adherent Muc5A levels relative to untreated MTX-E12 cells, confirming a reduction in mucus levels (Fig. 8A). When those NAC-pretreated cells were tested for their ability to support growth of F4969, there was a significant decrease in F4969 growth relative to untreated MTX-E12 cells (Fig. 8B), supporting a role for mucus in the higher F4969 growth observed using MTX-E12 cells in Fig. 3. Consistent with the presence of mucus-inducing higher sialidase activity, NAC pretreatment of MTX-E12 cells significantly decreased sialidase activity in F4969-infected cultures (Fig. 8C). Last, NAC-induced reduction in mucus also significantly decreased F4969 adherence to MTX-E12 cells (Fig. 8D), supporting a role for mucus in the stronger adherence of F4969 to MTX-E12 cells versus HT29 cells.
FIG 8.
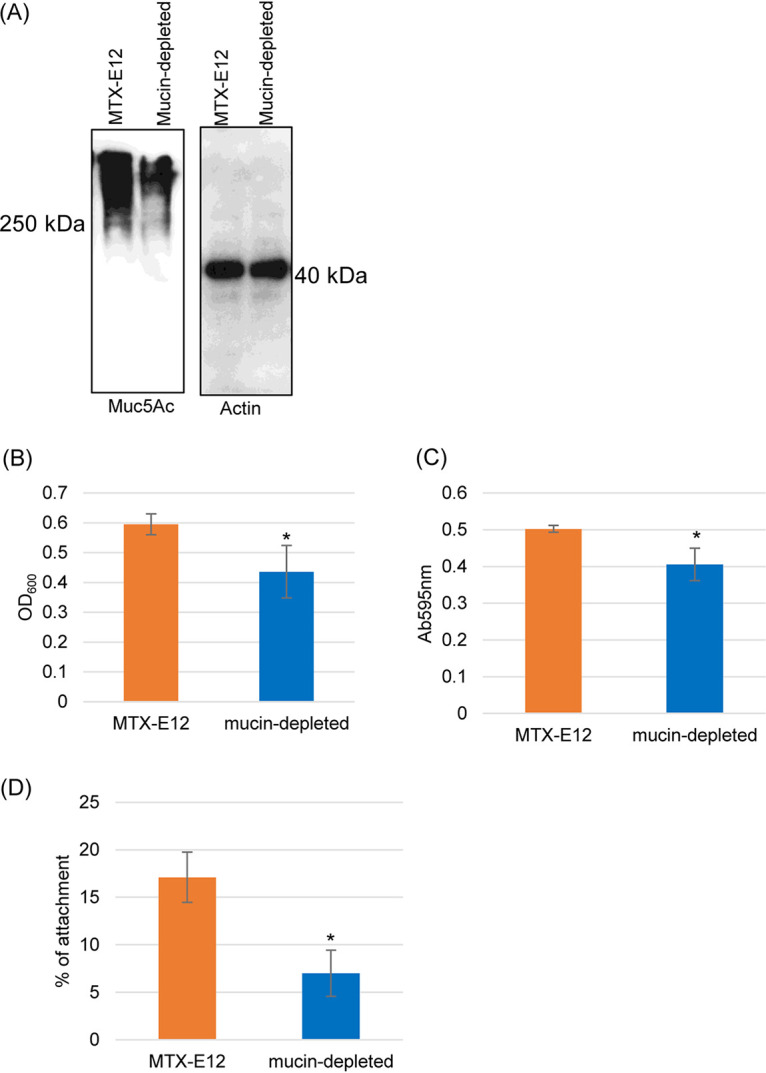
Reduction of mucus in MTX-E12 cultures using a NAC pretreatment to reduce adherent mucus affects F4969 growth, sialidase production, and adherence. (A) Muc5Ac Western blot analysis performed on an SDS-containing 8% polyacrylamide gel for MTX-E12 cells and MTX-E12 cells pretreated with NAC (see Materials and Methods). Size of proteins in kilodaltons is shown on the left. The same amount of each protein sample was loaded onto a SDS-containing 12% polyacrylamide gel or Western blotted with an anti-beta actin antibody for a loading control. Size of proteins in kilodaltons is shown at right. The gel results shown are representative of three repetitions. (B) Postinoculation changes in the OD600 for cultures of wild-type F4969 growing in PBS buffer with MTX-E12 or NAC-pretreated MTX-E12 cells at 37°C for 24 h. A 0.5-ml aliquot of each culture was removed, and the OD600 was determined. (C) Sialidase activity measured for F4969 growing in PBS buffer with MTX-E12 or NAC-pretreated MTX-E12 cells at 37°C for 24 h. Sialidase activity was measured in culture supernatants of samples from panel B. (D) Attachment of MTX-E12 or NAC-pretreated MTX-E12 cell monolayers incubated for 2 h with washed vegetative cells of F4969 at 37°C under anaerobic conditions. Monolayers were then washed three times with PBS. After collection, the adherent bacteria were plated onto BHI agar plates for counting. All panels show the mean values from three independent experiments. The error bars indicate the SD. *, P < 0.05 relative to coculture with MTX-E12 cell line.
NanI increases C. perfringens F4969 growth in the mouse small intestine.
The ability of C. perfringens vegetative cells to grow in the intestine, which is coated with adherent mucus, during infections has long been assumed but, to our knowledge, never experimentally demonstrated in an animal model. Nor have the contributions, if any, of NanI to intestinal growth of NanI-positive C. perfringens strains, including type F NFD strains, yet been evaluated. Therefore, to compare the ability of F4969 versus its derivatives to grow in the intestine, ligated small intestinal loops were created in BALB/c mice. Similar numbers of each strain (∼106 CFU) were inoculated into those loops, which were then incubated for 2, 4, or 6 h. After each time point, samples of intestinal contents and mucosal scrapings were collected, combined, and resuspended in PBS for calculation of CFU per gram and to measure sialidase activity in the supernatants. There was no sporulation or CPE production under these experimental conditions (data not shown).
As shown in Fig. 9A, wild-type F4969 and its derivatives all grew in the small intestine of mice. However, compared to wild-type F4969 or the complemented strain, the isogenic nanI null mutant strain showed significantly less growth at 2, 4, and 6 h postinfection. In addition, sialidase activity in supernatants of the intestinal contents was significantly lower for the isogenic nanI null mutant versus wild-type F4969 or the complemented strain at all time points (Fig. 9B). The small amount of sialidase activity present in loops infected by the nanI null mutant likely involves production of other sialidases by F4969 and sialidase production by the relatively small numbers of normal microbiota present in the small intestines (29). These results documented that type F NFD strains can grow in the intestines and also indicated that NanI can be an important contributor to this growth in the small intestine.
FIG 9.
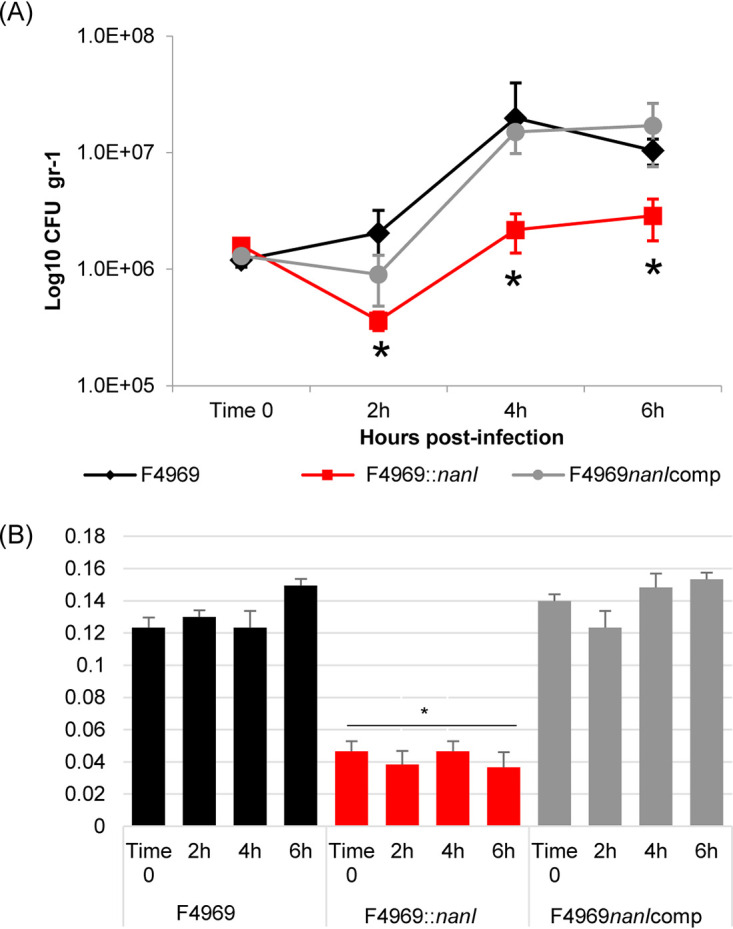
C. perfringens grows and produces sialidase in the intestines. (A) Shown are CFU per gram of intestinal tissue for F4969, its nanI null mutant, and the complementing strain as measured by plate counting at 2, 4, and 6 h postinfection in mouse small intestinal loops. (B) Sialidase activity present in supernatants of the intestinal contents of panel A loops at 2, 4, and 6 h postinfection. All data showed the mean values from 6 mice. Error bars indicate the SD. *, P < 0.05 relative to F4969 wild-type strain.
DISCUSSION
NanI-positive type F strains of Clostridium perfringens can cause NFDs that last up to several weeks (8, 9). This disease persistence indicates an important role for intestinal colonization by type F strains during type F NFD infections. Type F strain colonization during NFDs involves intestinal adherence by vegetative cells, as well as the growth and longer-term viability of type F strain cells in the intestines. This intestinal colonization allows for the cycles of vegetative cell growth and adherence, sporulation, and CPE production necessary to induce weeks-long intestinal pathology even in the presence of flushing effects from CPE-induced diarrhea. However, despite their pathogenic importance, the growth, survival, and adherence of type F strains in intestinal-like conditions have been the subject of limited study.
Previous studies reported that C. perfringens, including type F NFD strains, can use soluble sialic acid for their growth and survival because they possess a nan operon encoding various sialic acid transporters and enzymes involved in sialic acid metabolism (26, 27). However, in the intestine, sialic acid is often incorporated into macromolecules present either on the host surface or in adherent mucus that coats the intestinal epithelium (30). C. perfringens produces sialidases that could release sialic acid from those sialylated host macromolecules in the intestines (31). For most C. perfringens strains (including virtually all type F NFD strains) that produce three sialidases, NanI is their major sialidase (15, 32). Our previous study (16) showed that NanI can contribute to type F NFD strain growth and survival using human intestinal cell-like Caco-2 cells. Since Caco-2 cells produce minimal amounts of mucus, those observations support the ability of NanI to generate sialic acid from host cell surface molecules so that sialic acid can be used for growth and survival by NanI-positive type F strains.
However, compared to Caco-2 cells, the situation is more complicated in the intestine due to the presence of overlying adherent mucus. Therefore, the current study investigated whether the presence of adherent mucus impacts type F strain growth or longer-term viability and, if so, whether those effects involve NanI. To address these questions, the growth of several type F NFD strains were first compared using MTX-E12 cells, which produce a layer of adherent mucus, versus their parental HT29 cells, which make minimal mucus. Similar comparisons using these same cell lines have provided insights into the GI tract pathogenesis of other bacteria (24, 25, 33). Those analyses showed that all three tested type F NFD strains carrying the nanI gene grew significantly better using MTX-E12 cells versus HT29 cells, while this effect was not observed using two type F food poisoning strains lacking the nanI gene.
An isogenic nanI null mutant was then used to demonstrate that NanI is specifically involved in the ability of MTX-E12 cells to improve growth and longer-term viability of NanI-producing type F strains. Consistent with that observation, considerably more NanI sialidase activity and sialic acid release was measured when F4969 was cocultured with MTX-E12 cells versus parental HT29 cells. The increased sialic acid release caused by coinfection of MTX-E12 cells with F4969 likely contributes to the observed increase in growth and longer-term viability of F4969 in the presence of those mucus-producing host cells since C. perfringens can use sialic acid for growth and to maintain viability (26, 27). The stronger growth and sialidase production observed when F4969 was cocultured with MTX-E12 cells versus HT29 cells specifically involved mucus production by MTX-E12 cells since these effects were diminished when mucus levels in MTX-E12 cells were reduced using an NAC pretreatment.
The increased NanI activity detected in the presence of mucus likely involves, at least in part, the presence of MTX-E12 cells inducing higher nanI transcript levels in F4969. Based upon our previous study (26) showing that the presence of soluble sialic acid can increase nanI transcript levels, this observed increase in F4969 nanI transcript levels in the presence of MTX-E12 cells likely involves signaling induced by soluble sialic acid that was liberated from mucus and host cell sialylated molecules by NanI.
To help confirm the in vivo relevance of those results using MTX-E12 cells, the current study then assessed whether NanI contributes to the growth of C. perfringens type F NFD strain F4969 in the mucus-covered small intestine. While it has long been assumed that C. perfringens, including type F NFD strains, must initially multiply during intestinal infections, the intestinal growth of this bacterium has not yet, to our knowledge, been directly shown. Our previous study (17) on intestinal colonization did not address in vivo growth since the number of F4969 cells present in the challenged intestine were only assessed infrequently, i.e., at daily intervals. Furthermore, there was likely substantial loss of intestinal bacteria over the 3-day duration of the previous experiments since the intestine of these mice was not closed by ligation. Another previous study (34) from our group had challenged rabbit small intestinal loops with 108 cells of C. perfringens type C strain CN3685 for 6 h; the number of those type C bacteria recovered were equal to the input inoculum, i.e., suggesting that no intestinal growth had occurred.
In contrast, the current study was able to successfully demonstrate intestinal growth of F4969 by using a lower (and likely more reflective of early disease) challenge of 106 vegetative cells of this strain. Combining this new result with previous observation (34) suggests that intestinal challenge with very high numbers of C. perfringens may impair the ability of this bacterium to grow in the intestine, perhaps due to competition for nutrients or production of too much waste.
The current study also showed that the intestinal growth of F4969 is impacted by the production of NanI. This in vivo growth promotion by NanI likely involves the liberation of sialic acid from sialylated macromolecules in intestinal mucus or on the surface of intestinal cells for use as a nutrient source (26, 27). However, it may also partially reflect NanI exposing underlying sugars and amino acids on those sialylated macromolecules, making those substrates accessible to liberation by other C. perfringens glycosidases and proteases.
This study also evaluated the effects of mucus on type F NFD strain adherence to host cells since this is another factor in intestinal colonization. Our previous in vivo study detected F4969 attachment to the mouse small intestinal mucosa and observed that many of those adherent bacteria appeared to be present in the adherent mucus layer (17). Since it is challenging to specifically evaluate the contribution of mucus on C. perfringens attachment in vivo, the current study instead compared F4969 attachment to MTX-E12 cells versus the parental HT29 cells. The results of that comparison supported the importance of adherent mucus as a proadhesion factor for type F NFD strains, i.e., F4969 adhered better to MTX-E12 cells versus HT29 cells, and this effect involves mucus production since NAC-induced reduction in mucus levels on MTX-E12 cells decreased F4969 adherence. Furthermore, using a nanI null mutant, it was determined that NanI production is important for attachment of F4969 to MTX-E12 cells, as well as HT29 cells. NanI could promote F4969 adherence to mucus-producing cells via at least two potential effects. First, by removing sialic acids from mucus and cell surface molecules, NanI could reduce charge repulsion effects. Second, NanI-induced removal of sialic acids from mucus or cell surface molecules could expose underlying F4969 adhesion sites that are otherwise masked.
Overall, the findings of the current study offer important new insights into understanding the potential contributions of NanI to type F NFD pathogenesis. However, many questions still remain unanswered regarding this topic. For example, while NanI increases CPE activity on, and binding to, Caco-2 cells (18), does this sialidase also exert similar effects in the presence of mucus? Also, while a number of studies have implicated NanI as a potential virulence factor, can this be proven in vivo for strains like F4969? The latter question is intriguing but challenging to address since it will require development and validation of a small animal model for studying the pathogenesis of type F NFD strain infections.
MATERIALS AND METHODS
C. perfringens strains, culture media, and chemicals.
C. perfringens strains used in this study included the type F food poisoning strains SM101 and NCTC8239, which do not carry the nanI gene and the type F human NFD strains F4969, F5603, and B40, which do carry the nanI gene (15). Also used were an F4969 nanI null mutant and the nanI-complemented mutant that had been prepared and characterized previously (15). Media used for preparing stock cultures and culturing C. perfringens included cooked meat medium (CMM; Difco Laboratories), FTG medium (fluid thioglycolate medium; Difco Laboratories), TH (Bacto Todd Hewitt Broth), TGY medium (3% tryptic soy broth [Becton, Dickinson], 2% glucose [Fisher Scientific], 1% yeast extract [Becton, Dickinson], and 0.1% sodium thioglycolate [Sigma-Aldrich]), and TSC medium (Difco SFP agar base [Becton, Dickinson] and 0.04% d-cycloserine [Sigma-Aldrich]). The media used to grow the complementing strain also has 30 mg/liter chloramphenicol (Cm; from Sigma-Aldrich). C. perfringens strains were cultured at 37°C under anaerobic conditions.
Culture of HT29 and MTX-E12 cells.
Authenticated HT29 and MTX-E12 cell lines were purchased from Sigma-Aldrich. HT29 cells were maintained in RPMI 1640 medium with l-glutamine and sodium bicarbonate (Sigma-Aldrich), supplemented with 10% (vol/vol) fetal bovine serum (FBS) and penicillin-streptomycin solution (100×; Corning). MTX-E12 cells were maintained in Dulbecco’s modification of Eagle’s medium (DMEM; with 4.5 g/liter glucose and l-glutamine without sodium pyruvate; Corning), supplemented with 10% (vol/vol) FBS, 1% nonessential amino acids, and penicillin-streptomycin solution (100×; Corning). For use in experiments, cells were grown in Costar 12-well or 6-well cluster plates. Medium was replaced every 2 days during the 3-week cultivation.
Muc5Ac Western blot analyses.
Cultured HT29 or MTX-E12 cells were resuspended in phosphate-buffered radioimmunoprecipitation assay (RIPA) lysis buffer (Alfa Aesar) containing protease inhibitor cocktail III (RPI Research Products International) before lysis for 10 s using a Qsonica sonicator. Those lysates were left to stand at 4°C for 15 min and then centrifuged at 20,000 × g for 30 min. Supernatants were stored at −80°C.
Those prepared cell lysate supernatants were used for Muc5Ac Western blot analyses. A 50-µg protein aliquot, as determined using the Pierce bicinchoninic acid (BCA) protein assay kit, of each supernatant was denatured in reducing sample buffer and then electrophoresed on an SDS-containing 8% polyacrylamide gel for 150 min at 20 mA. Thereafter, samples were transferred to a polyvinylidene difluoride (PVDF) membrane. The membrane was blocked with 1% Tween-5% bovine serum albumin (BSA)-PBS buffer and then incubated overnight with a 1:500 dilution of recombinant anti-mucin 5Ac antibody (clone EPR16904; Abcam) in 5% BSA-PBS buffer. The membranes were washed and incubated for 1 h with horseradish peroxidase-conjugated goat anti-rabbit IgG (1:10,000; Sigma-Aldrich) to detect the Muc5Ac antibody. The same amount of each protein sample was loaded onto an SDS containing 12% polyacrylamide gel and probed with anti-beta actin antibody (catalog no. ab8227; Abcam) to show that the loaded samples contained equal amounts of protein. The substrate used for detecting the bound goat anti-rabbit antibody was Pierce ECL Western blotting substrate.
Reduction of extracellular mucus on MTX-E12 cultures by pretreatment with NAC.
NAC is a mucolytic agent that can reduce adherent mucus levels in MTX-E12 cultures (28). To test the involvement of mucus in the enhanced growth and adherence of F4969 in the presence of MTX-E12 cells, 3-week-old cultures of MTX-E12 cells were pretreated with 10 mM NAC in PBS for 10 min followed by incubation in DMEM medium for 1 h. These cells were then pretreated again with 10 mM NAC in PBS for 10 min, followed by two washes with PBS prior to infection with F4969 to assess growth and viability as discussed above.
Measurement of C. perfringens growth, supernatant sialidase activity, and viable vegetative cell quantification.
To measure C. perfringens growth in the presence of HT29 or MTX-E12 cells, 12-well Corning Transwell plates with a polyester membrane containing 0.4-µm pores were used as employed previously to study C. perfringens growth using Caco-2 cells (16). In the current study, monolayers of HT29 or MTX-E12 cells were cultured in the lower Transwell chamber for 3 weeks to reach confluence. After these cultures were washed twice with PBS, a 1.5-ml aliquot of PBS buffer was added to these cells. A 0.6-ml aliquot of PBS buffer with 5% Oxyrase for broth (Oxyrase) was added into the upper Transwell insert chamber. These Transwell plates were then preincubated for 30 min at 37°C to create anaerobic conditions. A 40-µl aliquot of an overnight TGY (∼16-h) culture of the specified type F strain or F4969 derivative was added into the Transwell insert, the cultures were incubated at 37°C before growth, and sialidase activity measurements were performed, as described below. For these determinations, the cultures were removed from the top wells at specified time points (0, 2, 4, 6, 8, 10, or 24 h), and the optical density at 600 nm (OD600) was measured using a Bio-Rad SmartSpec reader.
The same culture lysate supernatants were also tested for their sialidase activity. For this analysis, a 60-µl aliquot of each supernatant was added to a 40-µl aliquot of the substrate (4 mM 5-bromo-4-chloro-3-indolyl-α-d-N-acetylneuraminic acid). The mixtures were then incubated at 37°C for 60 min. After that incubation, the absorbance at 595 nm was measured using a Bio-Rad microplate reader.
To quantify viable vegetative cells, overnight cultures of wild-type F4969, its isogenic nanI null mutant, or the complemented strain were serially diluted from 10−2 to 10−7 with sterile PBS buffer, and aliquots of those dilutions were plated onto brain heart infusion (BHI) agar. After anaerobic incubation overnight at 37°C, colonies appearing on BHI agar plates were counted, and those colony counts were used to calculate the numbers of viable vegetative cells per milliliter.
Measurement of HT29 and MTX-E12 cytotoxicity after infection with F4969.
To determine mammalian cell death after infection with F4969, 3-week cultures of HT29 or MTX-E12 cell lines grown in 12-well Transwells were incubated for 2 h or 4 h with F4969 as described above. Alternatively, some cultures of these cells were incubated with Hanks balanced salt solution (HBSS) with calcium and magnesium without phenol red (Corning) as a negative control or with 1% Triton-100 as positive cytotoxicity control. Following these treatments, the culture supernatant was removed for cytotoxicity detection using the Roche cytotoxicity detection kit (LDH).
Determination of culture supernatant sialic acid concentrations.
Wild-type F4969 or the F4969 nanI null mutant were cocultured with HT29 and MTX-E12 cells, respectively, as described above, for 6 h or 24 h at 37°C. After this coculture, the sialic acid concentration in the culture supernatants was determined using the EnzyChrom neuraminidase assay (Bioassay Systems) kit. Briefly, a 20-µl aliquot of the upper well supernatant was added to 80 µl of working reagent (30 µl of double-distilled water [ddH2O], 50 µl of assay buffer, l µl of cofactors, l µl of enzyme, and 0.5 µl of dye reagent, all supplied in the kit). Reaction mixtures were incubated, with protection from light, for 20 min at 37°C, and A590 was then measured. A standard curve of sialic acid concentrations (supplied in the kit) was then used to determine the sialic acid concentration generated under each incubation condition. The difference in the concentration of sialic acid between F4969 and the F4969::nanI mutant represented the sialic acid generated by NanI.
C. perfringens RNA extraction and nanI qRT-PCR performance.
Saturated phenol (Fisher Scientific) was used to extract RNA from pelleted cells of F4969 cocultured with HT29 or MTX-E12 cells for 4 h at 37°C, as previously described (35). RNA samples were quantified by absorbance at 260 nm and then confirmed to be DNA free by PCR without reverse transcriptase (RT). The purified RNA was prepared to cDNA using a Maxima first-strand cDNA synthesis kit (Thermo Scientific) according to the manufacturer’s instructions. The nanI qRT-PCR primers used were as reported in a previous study (26). The nanI qRT-PCR was performed using the Applied Biosystems StepOnePlus qRT-PCR instrument. Each cDNA was diluted 5 times to 5 ng/µl. Power SYBR green PCR master mix (Thermo Fisher Scientific) was used to perform this qRT-PCR, as described in a previous paper (26). After qRT-PCR, the relative quantitation of mRNA expression was normalized to the level of constitutive expression of the housekeeping 16S RNA and calculated by the comparative threshold cycle (2−ΔΔCT) method.
C. perfringens attachment assays.
A C. perfringens attachment assay was performed as described previously (15). Briefly, a 1.5-ml aliquot of a TH overnight culture of wild-type F4969, its isogenic nanI null mutant, or the nanI-complemented strain was centrifuged, and the bacterial pellet was then washed three times with HBSS buffer. The washed bacteria were then resuspended in 1.0 ml of HBSS buffer.
Confluent monolayers of 3-week cultures of HT29 or MTX-E12 cells in 6-well plates were washed twice with HBSS buffer. A 1-ml aliquot of HBSS was added to each 6-well cluster plate with these cultured cells or to 6-well plates without any cultured cells (to calculate the number of input C. perfringens). A 10-µl aliquot of the washed bacteria was then added to each well. Under anaerobic conditions created using a Gas-Pak jar, these plates were incubated for 2 h at 37°C. The plates with the HT29 or MTX-E12 cells were washed three times with HBSS. A 1-ml aliquot of HBSS buffer was added to the washed cells, which were then scrapped and collected. These suspensions were then serially diluted from 10−2 to 10−7 with HBSS buffer and aliquots spread onto BHI agar plates. Following an overnight anaerobic incubation at 37°C, the colonies arising on these plates were counted to determine the number of CFU. The ratio of CFU added to the mammalian cell cultures to the total CFU was then calculated. In the presence of host cells, there was no growth of F4969 or its derivative strains for the 2-h duration of this experiment under the conditions used.
Immunofluorescence microscopy.
HT29 or MTX-E12 cells were grown for 3 weeks in an 8-well chamber slide (Fisher Scientific). At that time, a 2-µl aliquot of an overnight TH culture of wild-type F4969, its isogenic nanI null mutant, or the complemented strain was added to each chamber of the slide, which contained 100 µl of DMEM medium (control wells contained only 100 µl of DMEM medium without HT29 or MTX-E12 cells), and the slide was then incubated at 37°C for 2 h.
After this incubation, cells were washed three times with HBSS buffer. The slides were fixed with 100 µl of Carnoy’s solution (60% [vol/vol] ethanol, 30% [vol/vol] chloroform, and 10% [vol/vol] glacial acetic acid) for 2 h at 4°C. After 2 h, the Carnoy’s solution was removed, and the slide was air-dried for 20 min. The slide was blocked in 1% BSA in HBSS for 30 min at room temperature and then incubated with a 1:200 dilution of C. perfringens rabbit polyclonal antibody (Genway) and 1:250 dilution of an anti-mucin 5Ac (clone 45M1) mouse monoclonal IgG (catalog no. sc-21701; Santa Cruz Biotechnology) in HBSS with 1% BSA for 2 h at room temperature. It was then washed three times with HBSS buffer and incubated with 1:1,000 Alexa Fluor 596-conjugated anti-rabbit IgG (Thermo Fisher Scientific) and a 1:1,000 dilution of Alexa Fluor 488-conjugated anti-murine IgG (Thermo Fisher Scientific) in 1% BSA-HBSS for 1 h in the dark at room temperature. The slide was then treated with Hoechst 33342 stain (Invitrogen) at a 1:10,000 dilution for 10 min in the dark at room temperature. Following a final three washes with HBSS buffer, the chambers of the slide were removed, and a coverslip was mounted with Fluoro-Gel (Electron Microscopy Sciences). Imaging was performed by fluorescence microscopy using an Olympus confocal laser scanning biological microscope (FluoView FV1000) with FV10-ASW (version 1.4) software. All pictures were taken at a magnification of ×400.
C. perfringens attachment was quantified by ImageJ analysis (https://imagej.nih.gov) of 3 randomly chosen microscopy pictures for each treatment condition.
In vivo assessment of growth and sialidase activity.
All procedures involving animals were reviewed and approved by the University of California, Davis, Institutional Committee for Animal Care and Use (permit 21729). Similar numbers of male or female, 20- to 25-g BALB/c mice were kept in cages with sterile bedding and offered feed and water ad libitum. The day of the challenge, 0.1 ml of overnight fluid thioglycolate (FTG) broth culture of C. perfringens F4969 wild-type, an F4969 nanI null mutant, or F4969 nanI-complemented strains were inoculated into 10 ml of PBS containing 5% Oxyrase and incubated anaerobically for 30 min at 37°C. One milliliter of the PBS mix was then added to a microtube with screwed cap and incubated for 4 h at 37°C (CFU control), and a 20-µl aliquot of the supernatant was collected to measure sialidase activity (time zero), as previously described (18).
Another 1 ml of the PBS mix (∼106 CFU) for each strain was inoculated into a ligated small intestinal loop created in each of 18 mice per group of treatment. For this procedure, BALB/c mice were anesthetized using intraperitoneal administration of 0.2 ml/10 g of body weight with a mixture of xylazine (0.5 mg/ml) and ketamine (5 mg/ml). Immediately before surgery, the abdomen of each mouse was disinfected using iodine solution (betadine; Purdue Pharma LP). A midline laparotomy was then performed, and an ∼10- to 15-cm-long intestinal loop was prepared in the jejunum of each mouse by double ligation of the intestine, with the blood supply preserved. A new sterile 1-ml needle and syringe were used for each inoculation. Superglue (Henkel Corporation) was used to close, in one plane, the incision in the peritoneum, abdominal muscles, and skin. Animals remained deeply anesthetized until the end of the experiments at 2 h (n = 6), 4 h (n = 6), or 6 h (n = 6) of incubation, when they were humanely euthanized. At that time, ∼4-cm-long sections of the intestinal loops were removed aseptically to collect the intestinal contents and mucosal scrapings (using scalpel blades). These samples were weighed and resuspended in PBS. A 20-µl aliquot of the luminal supernatant was collected to measure sialidase activity, as previously described (18). Serial dilutions (101 to 108) in PBS were plated on selective tryptose sulfite cycloserine (TSC) agar plates for C. perfringens and anaerobically incubated at 37°C. Twenty-four hours later, black colonies, indicative of C. perfringens (36), were counted for calculation of the number of CFU per gram.
Statistical analyses.
All statistical analyses were performed using R (v3.3.1) (for the in vivo experiments) and GraphPad 8 (for the in vitro experiments). For comparison of sialidase activity in culture supernatant, one-way analysis of variance (ANOVA) was applied with post hoc analysis using Tukey’s multiple-comparison test. Bacterial counts were compared by negative binomial regression analysis. Differences were considered significant when the P value was less than 0.05.
ACKNOWLEDGMENTS
This study was generously supported by grant R21AI140010-02 (B.A.M. and F.A.U.) from the National Institute of Allergy and Infectious Diseases. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Contributor Information
Bruce A. McClane, Email: bamcc@pitt.edu.
Nancy E. Freitag, University of Illinois at Chicago
REFERENCES
- 1.Navarro MA, McClane BA, Uzal FA. 2018. Mechanisms of action and cell death associated with Clostridium perfringens toxins. Toxins (Basel) 10:212. 10.3390/toxins10050212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McDonel JL. 1986. Toxins of Clostridium perfringens types A, B, C, D, and E, p 477–517. In Dorner F, Drews H (ed), Pharmacology of bacterial toxins, Pergamon Press, Oxford, UK. [Google Scholar]
- 3.Uzal FA, Freedman JC, Shrestha A, Theoret JR, Garcia J, Awad MM, Adams V, Moore RJ, Rood JI, McClane BA. 2014. Towards an understanding of the role of Clostridium perfringens toxins in human and animal disease. Future Microbiol 9:361–377. 10.2217/fmb.13.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McClane BA, Uzal FA, Miyakawa MF, Lyerly D, Wilkins TD. 2006. The enterotoxic Clostridia, p 688–752. In Dworkin M, Falkow S, Rosenburg E, Schleifer H, Stackebrandt E (ed), The prokaryotes, 3rd ed, Springer Press, New York, NY. [Google Scholar]
- 5.Rood JI, Adams V, Lacey J, Lyras D, McClane BA, Melville SB, Moore RJ, Popoff MR, Sarker MR, Songer JG, Uzal FA, Van Immerseel F. 2018. Expansion of the Clostridium perfringens toxin-based typing scheme. Anaerobe 53:5–10. 10.1016/j.anaerobe.2018.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Centers for Disease Control and Prevention. 2011. CDC estimates of foodborne illness in the United States. https://www.cdc.gov/foodborneburden/pdfs/FACTSHEET_A_FINDINGS.pdf.
- 7.Scallan E, Hoekstra RM, Angulo FJ, Tauxe RV, Widdowson MA, Roy SL, Jones JL, Griffin PM. 2011. Foodborne illness acquired in the United States–major pathogens. Emerg Infect Dis 17:7–15. 10.3201/eid1701.p11101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carman RJ. 1997. Clostridium perfringens in spontaneous and antibiotic-associated diarrhoea of man and other animals. Rev Med Microbiol 8(Suppl 1):S43–S45. 10.1097/00013542-199712001-00024. [DOI] [Google Scholar]
- 9.Larcombe S, Hutton ML, Lyras D. 2016. Involvement of bacteria other than Clostridium difficile in antibiotic-associated diarrhoea. Trends Microbiol 24:463–476. 10.1016/j.tim.2016.02.001. [DOI] [PubMed] [Google Scholar]
- 10.Bos J, Smithee L, McClane B, Distefano RF, Uzal F, Songer JG, Mallonee S, Crutcher JM. 2005. Fatal necrotizing colitis following a foodborne outbreak of enterotoxigenic Clostridium perfringens type A infection. Clin Infect Dis 40:e78–e83. 10.1086/429829. [DOI] [PubMed] [Google Scholar]
- 11.Centers for Disease Control and Prevention. 2012. Fatal foodborne Clostridium perfringens illness at a state psychiatric hospital–Louisiana, 2010. MMWR Morb Mortal Wkly Rep 61:605–608. [PubMed] [Google Scholar]
- 12.McClane BA, Robertson SL, Li J. 2013. Clostridium perfringens, p 465–489. In Doyle MP, Buchanan RL (ed), Food microbiology: fundamentals and frontiers, 4th ed. ASM Press, Washington, DC. [Google Scholar]
- 13.Li J, Paredes-Sabja D, Sarker MR, McClane BA. 2016. Clostridium perfringens sporulation and sporulation-associated toxin production. Microbiol Spectr 4. 10.1128/microbiolspec.TBS-0022-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duncan CL. 1973. Time of enterotoxin formation and release during sporulation of Clostridium perfringens type A. J Bacteriol 113:932–936. 10.1128/jb.113.2.932-936.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li J, McClane BA. 2014. Contributions of NanI sialidase to Caco-2 cell adherence by Clostridium perfringens type A and C strains causing human intestinal disease. Infect Immun 82:4620–4630. 10.1128/IAI.02322-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li J, McClane BA. 2018. NanI sialidase can support the growth and survival of Clostridium perfringens strain F4969 in the presence of sialyated host macromolecules (Mucin) or Caco-2 cells. Infect Immun 86:e00547-17. 10.1128/IAI.00547-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Navarro MA, Li J, McClane BA, Morrell E, Beingesser J, Uzal FA. 2018. NanI sialidase is an important contributor to Clostridium perfringens type F strain F4969 intestinal colonization in mice. Infect Immun 86:e00462-18. 10.1128/IAI.00462-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Theoret JR, Li J, Navarro MA, Garcia JP, Uzal FA, McClane BA. 2018. Native or proteolytically activated NanI sialidase enhances the binding and cytotoxic activity of Clostridium perfringens enterotoxin and beta toxin. Infect Immun 86:e00730-17. 10.1128/IAI.00730-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nollevaux G, Deville C, El Moualij B, Zorzi W, Deloyer P, Schneider YJ, Peulen O, Dandrifosse G. 2006. Development of a serum-free co-culture of human intestinal epithelium cell-lines (Caco-2/HT29-5M21). BMC Cell Biol 7:20. 10.1186/1471-2121-7-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paone P, Cani PD. 2020. Mucus barrier, mucins and gut microbiota: the expected slimy partners? Gut 69:2232–2243. 10.1136/gutjnl-2020-322260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schroeder BO. 2019. Fight them or feed them: how the intestinal mucus layer manages the gut microbiota. Gastroenterol Rep (Oxf) 7:3–12. 10.1093/gastro/goy052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.von Kleist S, Chany E, Burtin P, King M, Fogh J. 1975. Immunohistology of the antigenic pattern of a continuous cell line from a human colon tumor. J Natl Cancer Inst 55:555–560. 10.1093/jnci/55.3.555. [DOI] [PubMed] [Google Scholar]
- 23.Behrens I, Stenberg P, Artursson P, Kissel T. 2001. Transport of lipophilic drug molecules in a new mucus-secreting cell culture model based on HT29-MTX cells. Pharm Res 18:1138–1145. 10.1023/A:1010974909998. [DOI] [PubMed] [Google Scholar]
- 24.Alemka A, Clyne M, Shanahan F, Tompkins T, Corcionivoschi N, Bourke B. 2010. Probiotic colonization of the adherent mucus layer of HT29MTXE12 cells attenuates Campylobacter jejuni virulence properties. Infect Immun 78:2812–2822. 10.1128/IAI.01249-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Naughton JA, Marino K, Dolan B, Reid C, Gough R, Gallagher ME, Kilcoyne M, Gerlach JQ, Joshi L, Rudd P, Carrington S, Bourke B, Clyne M. 2013. Divergent mechanisms of interaction of Helicobacter pylori and Campylobacter jejuni with mucus and mucins. Infect Immun 81:2838–2850. 10.1128/IAI.00415-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li J, Evans DR, Freedman JC, McClane BA. 2017. NanR regulates nanI sialidase expression by Clostridium perfringens F4969, a human enteropathogenic strain. Infect Immun 85:e00241-17. 10.1128/IAI.00241-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Therit B, Cheung JK, Rood JI, Melville SB. 2015. NanR, a transcriptional regulator that binds to the promoters of genes involved in sialic acid metabolism in the anaerobic pathogen Clostridium perfringens. PLoS One 10:e0133217. 10.1371/journal.pone.0133217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maares M, Keil C, Koza J, Straubing S, Schwerdtle T, Haase H. 2018. In vitro studies on zinc binding and buffering by intestinal mucins. Int J Mol Sci 19:2662. 10.3390/ijms19092662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sender R, Fuchs S, Milo R. 2016. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol 14:e1002533. 10.1371/journal.pbio.1002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haines-Menges BL, Whitaker WB, Lubin JB, Boyd EF. 2015. Host sialic acids: a delicacy for the pathogen with discerning taste. Microbiol Spectr 3. 10.1128/microbiolspec.MBP-0005-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li J, Uzal FA, McClane BA. 2016. Clostridium perfringens sialidases: potential contributors to intestinal pathogenesis and therapeutic targets. Toxins (Basel) 8:341. 10.3390/toxins8110341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li J, McClane BA. 2014. The sialidases of Clostridium perfringens type D strain CN3718 differ in their properties and sensitivities to inhibitors. Appl Environ Microbiol 80:1701–1709. 10.1128/AEM.03440-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dolan B, Naughton J, Tegtmeyer N, May FE, Clyne M. 2012. The interaction of Helicobacter pylori with the adherent mucus gel layer secreted by polarized HT29-MTX-E12 cells. PLoS One 7:e47300. 10.1371/journal.pone.0047300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sayeed S, Uzal FA, Fisher DJ, Saputo J, Vidal JE, Chen Y, Gupta P, Rood JI, McClane BA. 2008. Beta toxin is essential for the intestinal virulence of Clostridium perfringens type C disease isolate CN3685 in a rabbit ileal loop model. Mol Microbiol 67:15–30. 10.1111/j.1365-2958.2007.06007.x. [DOI] [PubMed] [Google Scholar]
- 35.Li J, Ma M, Sarker MR, McClane BA. 2013. CodY is a global regulator of virulence-associated properties for Clostridium perfringens type D strain CN3718. mBio 4:e00770-13. 10.1128/mBio.00770-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li J, Sayeed S, McClane BA. 2007. Prevalence of enterotoxigenic Clostridium perfringens isolates in Pittsburgh (Pennsylvania) area soils and home kitchens. Appl Environ Microbiol 73:7218–7224. 10.1128/AEM.01075-07. [DOI] [PMC free article] [PubMed] [Google Scholar]