

Abstract
Permeabilization of the outer mitochondrial membrane by pore‐forming Bcl2 proteins is a crucial step for the induction of apoptosis. Despite a large set of data suggesting global conformational changes within pro‐apoptotic Bak during pore formation, high‐resolution structural details in a membrane environment remain sparse. Here, we used NMR and HDX‐MS (Hydrogen deuterium exchange mass spectrometry) in lipid nanodiscs to gain important high‐resolution structural insights into the conformational changes of Bak at the membrane that are dependent on a direct activation by BH3‐only proteins. Furthermore, we determined the first high‐resolution structure of the Bak transmembrane helix. Upon activation, α‐helix 1 in the soluble domain of Bak dissociates from the protein and adopts an unfolded and dynamic potentially membrane‐bound state. In line with this finding, comparative protein folding experiments with Bak and anti‐apoptotic BclxL suggest that α‐helix 1 in Bak is a metastable structural element contributing to its pro‐apoptotic features. Consequently, mutagenesis experiments aimed at stabilizing α‐helix 1 yielded Bak variants with delayed pore‐forming activity. These insights will contribute to a better mechanistic understanding of Bak‐mediated membrane permeabilization.
Keywords: apoptosis, HDX‐MS, lipid nanodiscs, membrane, NMR
Subject Categories: Autophagy & Cell Death, Membranes & Trafficking, Structural Biology
Outer mitochondrial membrane permeabilization by pore‐forming Bcl2 proteins, including Bak, is crucial for apoptosis. Here, a high‐resolution structural characterization reveals large conformational changes of Bak during activation at the membrane surface.

Introduction
Bak is a pro‐apoptotic member of the Bcl2 protein family, which regulate apoptosis by controlling the permeability of the outer mitochondrial membrane (OMM) (Adams & Cory, 2007; Dewson & Kluck, 2009). The Bcl2 family consists of three subgroups classified as pro‐ and anti‐apoptotic proteins. The pro‐apoptotic class includes both effectors and Bcl2‐homology (BH)3‐only proteins. Effectors, such as Bak and Bax, are able to form membrane pores consisting of protein oligomers, leading to downstream apoptosis (Chipuk & Green, 2008; Westphal et al, 2014b; Uren et al, 2017a). This process is prevented by the interaction with anti‐apoptotic Bcl2 proteins, such as Bcl2 and BclxL, which inhibit their oligomerization (Willis et al, 2007). The subgroup of BH3‐only proteins comprises of sensitizers, e.g., Bad and Noxa, which competitively bind anti‐apoptotic members, leading to the displacement of pro‐apoptotic Bax or Bak and activators, e.g., Bid, Bim and Puma, which additionally can directly activate the effectors (Letai et al, 2002; Moldoveanu et al, 2013; Hockings et al, 2015). Both effectors and anti‐apoptotic Bcl2 proteins are composed of four BH domains (Westphal et al, 2011), respectively, and have a highly conserved tertiary structure featuring a BH3 domain‐binding groove (Kvansakul & Hinds, 2015). Despite their structural homology, predominantly the effectors undergo distinct structural transformations upon the activation of pore formation by binding partners or a lipid bilayer membrane surface (O’neill et al, 2016; Bleicken et al, 2017). Previous studies have gained valuable structural information on this active state. High‐resolution structures of possible dimeric intermediate states of both, Bak and Bax, during pore formation induced by BH3‐only proteins in detergents, show that the core (α‐helices 2–5) and latch domain (α‐helices 6–8) separate during the activation process (Czabotar et al, 2013; Brouwer et al, 2014). By studying the distances and accessibility of specifically labeled residues on Bak and Bax in a membrane environment, the different helices could be localized with respect to each other and the membrane surface (Aluvila et al, 2014; Westphal et al, 2014a; Mandal et al, 2016). Additionally, antibody epitope binding assays revealed that helix 1 becomes solvent exposed after activation (Griffiths et al, 1999; Dewson et al, 2009). This process was determined as a key step for the downstream conformational transition (Alsop et al, 2015). Concerning the state of the pore, it has long been discussed that both Bak and Bax most likely form lipidic rather than proteinaceous pores (Basanez et al, 1999; Mandal et al, 2016; Salvador‐Gallego et al, 2016; Uren et al, 2017a; López et al, 2019; Flores‐Romero et al, 2020). While all of these studies reveal essential aspects, a higher‐resolution structural picture of the critical first step of Bak activation involving helix 1 dislodgement is still missing.
Here, we studied the structural and biophysical features of the membrane‐attached inactive and active conformations of Bak in lipid nanodiscs and determined the first high‐resolution structure of its C‐terminal transmembrane helix. For these studies, we first systematically evaluated the lipid‐ and concentration‐dependent Bak pore formations in liposomes and show that high protein doses as well as negatively charged lipids promote BH3‐independent autoactivation. Thus, in order to prevent premature activation of membrane‐attached Bak during structural studies at high protein concentrations, we utilized small lipid nanodiscs of a defined and stable size as a membrane mimetic. With this strategy, Bak could be stabilized in an inactive, membrane‐attached state and activated by the addition of BH3‐only proteins in a controlled manner for subsequent NMR and hydrogen‐deuterium exchange mass spectrometry (HDX‐MS) experiments. We could show that, before activation, Bak is in transient contact with the membrane surface via its latch domain and could demonstrate at high resolution that, upon activation, the first 66 amino acids of Bak adopt a highly dynamic, unfolded and solvent exposed conformation, confirming and expanding previous insights at lower resolution (Griffiths et al, 1999; Dewson et al, 2009; Alsop et al, 2015). Furthermore, protein folding studies reveal a molten globular intermediate state for Bak, but not for the anti‐apoptotic family member BclxL. NMR titration experiments with chemical denaturants suggest that helix 1 is the most affected structural element in Bak, consistent with the finding that this region is undergoing a complete unfolding transition upon activation in a membrane environment. Consequently, mutations in Bak aimed at enhancing helix 1 packing to the remaining helical bundle reduced its pore‐forming activity. In this study, we provide high‐resolution structural insights on Bak in various structural states and contribute to a better understanding of the molecular basis of Bak activation at the membrane surface. In particular, the interaction of the unfolded N‐terminus with the membrane surface might resemble a critical feature for stabilizing the detached structural state of helix 1 to promote the pore‐forming activity of Bak.
Results
Bak activation by binding to BH3‐only proteins or high protein concentrations
First, we validated the functionality of our protein preparations and determined optimized sample conditions by conducting established liposome assays that monitor the pore‐forming activity of Bcl2 proteins (Yethon et al, 2003; Kale et al, 2014). Since we used Bcl2 proteins lacking their C‐terminal transmembrane helix (BakΔTM or BclxLΔTM), we reestablished a stable membrane binding by replacing the transmembrane helix by a His6‐tag and using a lipid blend containing Ni2+‐caged lipids (Ni‐lipids) for our liposome preparations. This setup leads to an attachment of Bcl2 proteins in a native‐like orientation, as also described in an earlier study (Oh et al, 2010). In line with these previous results, no pore‐forming activity was observed for Bak if Ni‐lipids were omitted, even in the presence of the activator cBid (Fig EV1A). In contrast, when C‐terminal membrane binding was enabled by the presence of Ni‐lipids, BakΔTM selectively permeabilized the liposomes upon activation by cBid. High amounts of Ni‐lipids led to BakΔTM autoactivation, indicating that a high local density of Bak at the membrane surface facilitates its oligomerization that is required for pore formation. In order to mimic the native situation, where Bak does not show autoactivation properties under normal cellular conditions, the presence of 2% (w/w) Ni‐lipids was found to be optimal and was used in further assays unless indicated otherwise. In this setup, BakΔTM showed the expected activation profile, where pore formation was activated by cBid and inhibited by BclxLΔTM (Fig 1A). In agreement with the displacement model of Bcl2 proteins (Willis et al, 2007), the presence of cBid concentrations higher than BclxLΔTM reestablished pore formation (Fig 1B). Interestingly, as already seen in the Ni‐lipid titration, when BakΔTM was added at higher concentrations, it forms pores autonomously (Fig 1C and D). This behavior may be a relevant feature also in the cell (Dai et al, 2015) but turned out to be critical for our structural studies which require high protein concentrations. To obtain further insights on the impact of lipids on Bak pore formation in this setup, we used two different lipid blends, an E. coli polar lipid extract or a more commonly used synthetic lipid mixture that resembles the outer mitochondrial membrane (OMM lipids) (Kale et al, 2014). Compared to the OMM lipids, the E. coli extract is also rich in cardiolipin, a lipid widely discussed to be important for Bcl2 pore formation (Schafer et al, 2009; Landeta et al, 2011; Lai et al, 2019), and has a higher content of negatively charged lipids. A comparison of the concentration dependence of BakΔTM pore formation in both lipid systems shows that the E. coli lipid extract slightly favors autoactivation, presumably caused by the higher content in negative charges (Fig EV1B). At high protein concentrations, the addition of an activator BH3‐only protein, such as cBid, still accelerates the pore‐forming kinetics (Fig EV1C). The anti‐apoptotic protein BclxL inhibits Bak pore formation in both conditions, i.e., with or without the activator cBid, if used in excess (Fig EV1C and D). This further supports the model that BclxL directly binds to Bak to inhibit its pore‐forming activity. The activator cBid binds to BclxL with high affinity (Dai et al, 2011; Pogmore et al, 2016), thereby hindering the interaction between these two pro‐ and anti‐apoptotic players. Once the concentration of activator exceeds the concentration of BclxL, Bak is released from its complex with BclxL and at the same time is activated by the excess of cBid. This set of experiments clearly shows that our protein samples have the expected functional properties if attached to a membrane surface, which provides the basis for a detailed investigation of the associated structural transformations.
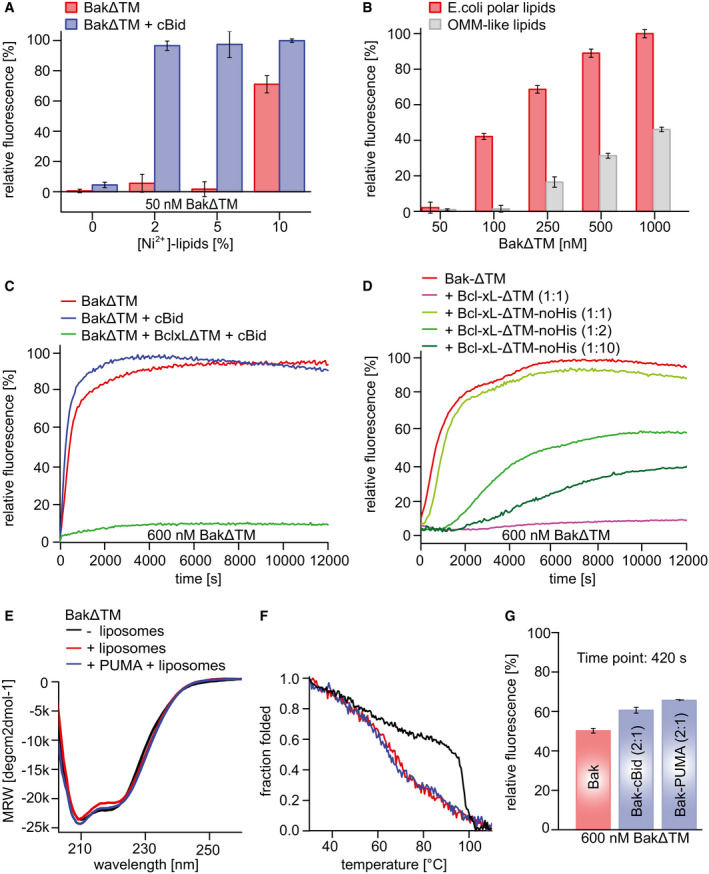
Figure EV1. Pore‐forming assay using BakΔTM and BclxLΔTM bound to Ni2+‐caged lipids in liposomes.
-
A, BThe data are averaged for the time frame of 9,000–10,000 s and the standard deviation calculated from three measurements, respectively. (A) Influence of Ni2+‐caged lipids on BakΔTM activity. Liposomes were formed with OMM‐like lipids and subjected to 50 nM BakΔTM ± 20 nM cBid. (B) Influence of the indicated lipid compositions on BakΔTM autoactivity.
-
CInfluence of cBid (300 nM) and BclxLΔTM (300 nM) on BakΔTM (600 nM), when autoactive.
-
DComparison of the effect of BclxLΔTM on 600 nM BakΔTM pore formation with and without a His‐tag. The inhibitory activity of BclxL is highly dependent on its stable membrane association.
-
EFar‐UV‐CD spectra of BakΔTM ± E. coli polar liposomes and the Puma‐BH3 peptide as indicated.
-
FCD‐detected thermal melting experiments of BakΔTM (black) with liposomes (red) and Puma‐BH3 (blue).
-
GPercentile dye release is shown after 420 s for 600 nM BakΔTM ± 300 nM of an activator. The data are averaged for the time frame of 9,000–10,000 s and the standard deviation calculated from three technical replicates, respectively. Both cBid and Puma‐BH3 lead to enhanced liposome permeabilization.
Data information: (C–G) Liposomes were formed using E. coli polar lipids.
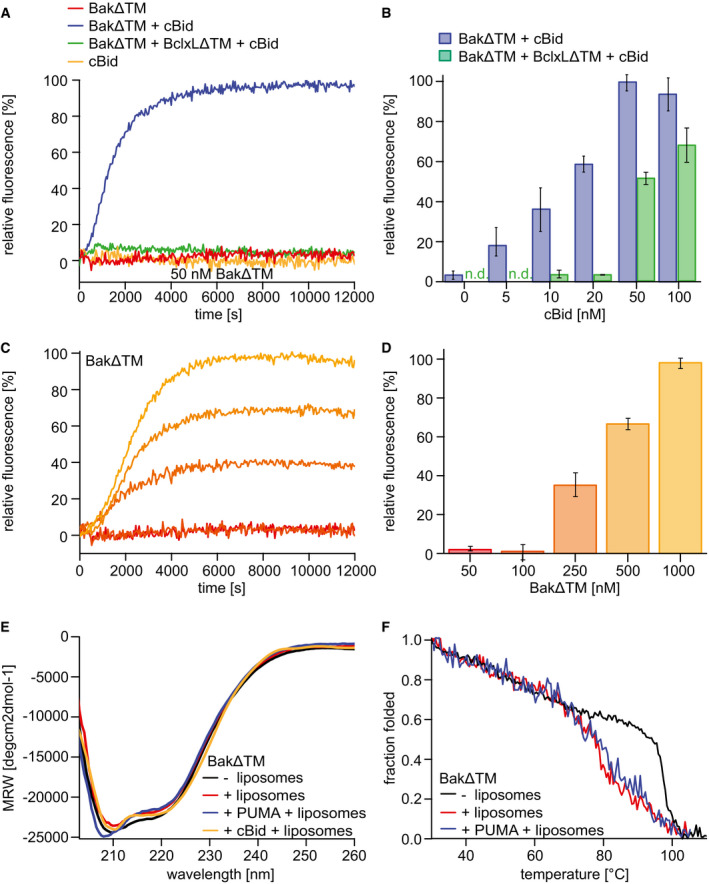
Figure 1. Functional and biophysical characterization of BakΔTM bound to liposomes.
-
A–DLiposome permeabilization assays detecting the release of a fluorescence dye from liposomes. (A) Pore formation of 50 nM BakΔTM (red) is activated by 20 nM cBid (blue). 20 nM cBid alone (orange) shows no liposome permeabilization. Addition of 25 nM BclxLΔTM (green) inhibits pore formation. (B) Increasing amounts of cBid are titrated to 50 nM BakΔTM in the absence (blue) and presence (green) of 25 nM BclxLΔTM. (C,D) BakΔTM becomes autoactive at increasing concentrations. The concentrations are color‐coded as indicated in (D). (C) The averaged kinetics for increasing BakΔTM concentrations. Bar diagrams in (B) and (D) represent data from the kinetic experiments averaged between 9,000 and 10,000 s. The standard deviation was calculated from three measurements.
-
EFar‐UV‐CD spectra of BakΔTM ± liposomes and the activators cBid or Puma‐BH3 peptide as indicated.
-
FCD‐detected thermal melting experiments of BakΔTM (black) with liposomes (red) and Puma‐BH3 (blue).
Next, we compared the secondary structure content of BakΔTM in solution and when bound to liposomes (Figs 1E and EV1E). As the functional pore‐forming assay showed autoactivation at high concentrations, the µM‐concentrations of BakΔTM used for the CD measurements are expected to lead to activation even in the absence of an activator BH3 protein. Far‐UV CD spectra of these samples indicate that the α‐helical secondary structure content is comparable in all cases. Even though Bak activation most likely leads to a loss in secondary structure in certain areas, this is most likely compensated by a gain in secondary structure content in other parts of the protein, e.g., by the formation of a continuous instead of a partial α‐helical structure in the latch domain (Birkinshaw et al, 2021). CD‐detected thermal melting assays showed that Bak stability is markedly decreased if attached to a lipid bilayer surface (Fig 1F). The melting temperature of Bak in liposomes is not affected by the presence of a BH3 peptide, corroborating the notion that Bak is able to undergo a complete structural transformation even without an activator BH3 protein at the sample conditions used for the CD experiments. In E. coli lipids, bearing a higher content of negative charges, the melting temperature of the active pore state is decreased by ∼ 10°C (Fig EV1F), potentially reflecting its higher autoactivation potency. The melting temperatures for all conditions can be found in Appendix Table S1. Since different activator BH3 proteins and peptides caused a very similar effect in our pore‐forming assays (Fig EV1G), a short Puma‐BH3 peptide was used here instead of the cBid protein to minimize its contribution to the detected CD signal. Motivated by these initial experiments, we next pursued a more detailed structural investigation of the conformational transition of Bak.
Structural analysis of full‐length Bak in lipid nanodiscs using a segmental approach
In order to obtain a more detailed structural picture of full‐length Bak located at the membrane surface, we used NMR to analyze Bak in its membrane‐attached form. So far, NMR resonance assignments were available of N‐terminally truncated constructs of the Bak soluble domain alone (Moldoveanu et al, 2006; Ye et al, 2020), or in complex with a Bid‐BH3 peptide (Moldoveanu et al, 2013). A detailed comparison with our BakΔTM protein construct harboring an intact N‐terminus revealed markedly altered NMR backbone amide spectra. Therefore, we performed a sequence‐specific resonance assignment with a 1H,13C,15N‐labeled sample. This effort resulted in the assignment of 80% of all non‐proline residues in BakΔTM (Fig 2A). Despite the apparent NMR spectral changes, no pronounced structural changes in the protein backbone could be detected between our construct and a previously published crystal structure of N‐terminally truncated Bak (Moldoveanu et al, 2006), as probed by backbone amide residual dipolar couplings (RDCs) and chemical shift information (Fig EV2A–C).
Figure 2. Structural model of full‐length Bak derived from NMR.
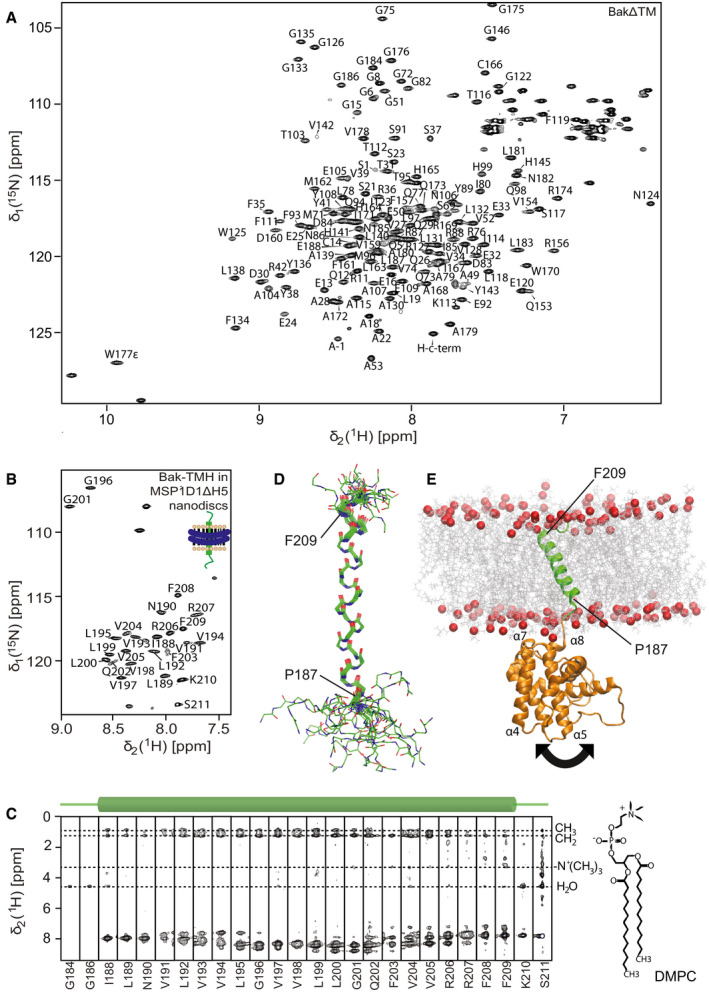
- NMR backbone resonance assignments labeled in a 2D‐[15N,1H]‐HSQC spectrum of BakΔTM.
- NMR backbone resonance assignment of the Bak‐TMH in MSP1D1ΔH5 nanodiscs.
- 3D‐15N‐edited‐[1H,1H]‐NOESY spectrum containing all sequential amide connectivities as well as the contacts to lipid tails and water exchange. The location of the transmembrane helix is indicated above.
- Overlay of the 20 lowest energy NMR structures of the Bak‐TMH in MSP1D1ΔH5 nanodiscs. A transmembrane helix is formed between Pro187 and Phe209 as indicated in the picture.
- Structural model of full‐length Bak obtained by an MD simulation and by utilizing our structural information. The arrow indicates the flexibility of the 4‐amino acid linker between the soluble domain and the TMH.
Figure EV2. NMR structural characterization of the Bak soluble domain and the TMH.
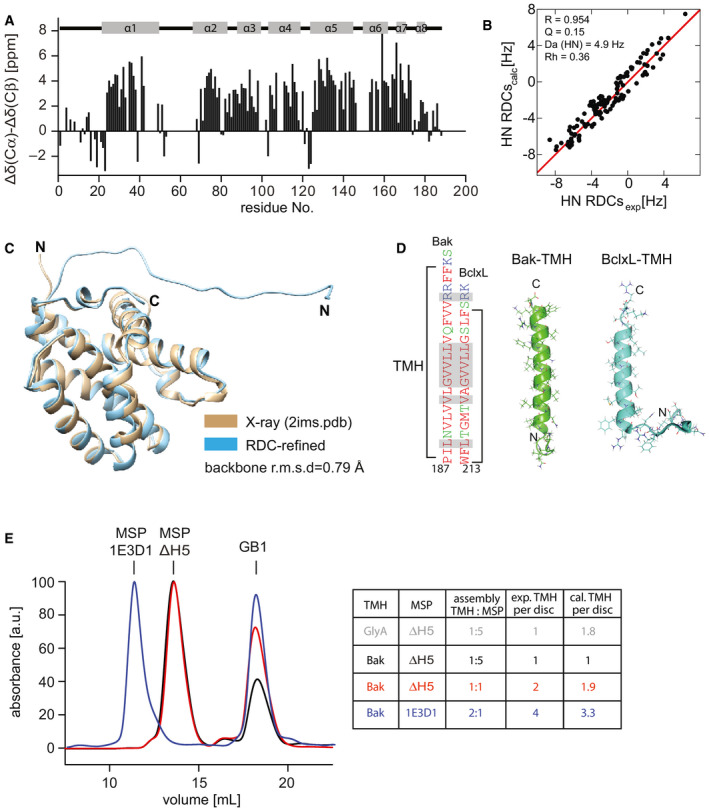
- NMR secondary chemical shifts of BakΔTM are in agreement with the α‐helical secondary structure content as present in a crystal structure (2IMS.pdb (Moldoveanu et al, 2006)) indicated at the top.
- Correlation between the experimental and back‐calculated RDCs for the NMR‐refined BakΔTM structure.
- Comparison of the refined BakΔTM structure containing the full N‐terminus with the published crystal structure (2IMS.pdb (Moldoveanu et al, 2006)), which was used as an initial structure for the refinement.
- Comparison of the sequence and structure of the Bak‐ and BclxL‐TMH in nanodiscs. The high similarity is shown by color coding the amino acids according to their polarity (red: apolar, green: polar, blue: basic). Identical amino acids are highlighted in the sequence in gray.
- SEC profiles of Bak‐TMH in nanodiscs after cleaving off the GB1 tag color‐coded according to the table on the right. The area under the curve was used to calculate the ratio of TMH/nanodisc. The table shows the applied assembly conditions as well as the expected and calculated amount of TMH/nanodisc. For a dimeric protein like the glycophorin A (GlyA) TMH, the expected and calculated ratios vary, which is not the case for the Bak‐TMH.
In order to gain structural information on the Bak transmembrane helix (TMH), we performed an in‐depth analysis by solution‐state NMR. The Bak‐TMH has previously been reported to adopt an α‐helical secondary structure in dimyristoyl‐glycero‐phosphocholine (DMPC) membranes (Martinez‐Senac et al, 2002; Torrecillas et al, 2005). However, a high‐resolution structure was not yet available. For high‐level production in E. coli, the Bak‐TMH was produced as an N‐terminal fusion protein with the expression helper and solubility tag GB1 (Zhou & Wagner, 2010), similar to what has previously been reported for the BclxL‐TMH and other systems (Raltchev et al, 2018; Steiner et al, 2020). Using multidimensional NMR experiments, we obtained backbone resonance assignments of 89% of all non‐proline residues in the Bak‐TMH protein construct both in dodecylphosphocholine (DPC) micelles (Appendix Fig S1A) and in DMPC/DMPG (3:1) MSP1D1ΔH5 nanodiscs (Hagn et al, 2013, 2018; Klöpfer & Hagn, 2019) (Fig 2B). Structures were calculated based on NOESY‐derived distance restraints, and hydrogen bonds were defined guided by initial structural models and the absence of a water exchange peak in the 15N‐edited 3D‐[1H,1H]‐NOESY spectrum (Fig 2C and Appendix Fig S1C). The statistics of the 20 lowest energy structures are shown in Appendix Table S1. The structure of the Bak‐TMH is characterized by a well‐defined continuous α‐helix, ranging from P187 to F209, and less defined dynamic flanking peptide stretches located outside the membrane, as probed by specific NOE contacts between backbone amides and the fatty acid tails of the lipids (Fig 2D and Appendix Fig S1B). Due to the high sequence similarity, the overall helix geometry is similar to the previously reported structure of the BclxL‐TMH (Raltchev et al, 2018) (Fig EV2D). However, the Bak‐TMH is more hydrophobic in its amino acid composition and longer, most likely resulting in a more tilted orientation within the membrane.
In order to establish a structural model of full‐length Bak attached to a membrane surface, we computationally fused the structures of the TMH and the RDC‐refined soluble domain, inserted it into a lipid bilayer membrane (DMPC:DMPG (3:1)) and subjected it to a molecular dynamic (MD) simulation to resolve structural clashes and equilibrate the overall geometry (Fig 2E). As expected from a C‐terminal membrane attachment mode, α‐helices 7 and 8 are facing the membrane surface in our MD‐based structural model, with some contribution of the loop region between α‐helices 4 and 5 and the N‐terminal tail. In agreement with its lipid binding properties, this surface region contains positively charged amino acids, which most likely interact with the negatively charged phosphate moieties of the lipid headgroups. To visualize the relative motions between the TMH and the soluble domain, we analyzed the distance between the centers of mass (COMs) of both domains for all frames of the 200‐ns simulation. The obtained quite large fluctuations (∼10 Å) in the calculated distances (Appendix Fig S2A and B) support the notion that both domains are quite flexible in their relative orientation, most likely due to the presence of a short flexible connecting linker. The structural model of the full‐length protein showed a tilted orientation of the TMH to match the hydrophobic thickness of the lipid bilayer membrane. Except for mediating stable membrane attachment, the functional role of the Bak‐TMH in the pore formation process is only poorly understood (Dai et al, 2011; Ferrer et al, 2012; Leshchiner et al, 2013). Therefore, we investigated whether the TMH alone has a tendency for oligomerization, which would suggest a potential active role in the oligomerization process of full‐length Bak in a lipid environment—a key step in activation (Bleicken et al, 2017; Cosentino & García‐Sáez, 2017; Moldoveanu & Czabotar, 2020). Oligomerization of the TMH in lipid nanodiscs was probed by a recently reported fusion protein assay (Häusler et al, 2020). Briefly, we assembled nanodiscs using different ratios of GB1‐fused Bak‐TMH and membrane scaffold protein (MSP) of different sizes. After isolating the GB1‐TMH‐containing nanodiscs, GB1 was cleaved off and the amount of TMH‐containing nanodiscs and GB1 was quantified by the peak integrals in size exclusion chromatography, taking into account the calculated extinction coefficients of the two species (Fig EV2E). If no oligomerization takes place, the ratio of Bak‐TMH to nanodisc is close to the theoretically expected value based on the chosen assembly conditions. For Bak, an assembly ratio of 1:5 (Bak‐TMH:MSP1D1ΔH5), favoring the incorporation of one TMH, resulted in the detection of one helix per nanodisc. This behavior is quite similar to what has been reported previously for the BclxL‐TMH (Raltchev et al, 2018) and is in contrast to the dimeric glycophorin A (GlyA) TMH for which the same assembly conditions gave the expected ratio of almost two helices per nanodisc. By varying the assembly conditions and the nanodisc sizes (using MSP1D1ΔH5 (8 nm) (Hagn et al, 2013) and MSP1E3D1 (12 nm) (Denisov et al, 2004)), we found that the number of Bak‐TMHs inserted in these nanodiscs is solely determined by the chosen assembly ratios. Thus, we conclude that it does not show any tendency to form oligomers in lipid bilayers. Taken together with the data showing that a stable membrane attachment is required for the full activity of both Bak and BclxL (Fig EV1A and D) (Oh et al, 2010), we conclude that the TMH of pro‐apoptotic Bak behaves very similar to the TMH of anti‐apoptotic BclxL and most likely does not play an active role in promoting the oligomerization of the full‐length protein.
Bak, but not anti‐apoptotic BclxL, can occupy a molten globule folding intermediate
Despite the high structural similarity of pro‐ and anti‐apoptotic Bcl2 proteins, their functional properties in a lipid bilayer membrane environment are very different (Kelekar & Thompson, 1998; Kvansakul & Hinds, 2015). Thus, we wondered whether the pore‐forming functionality of Bak can be monitored by protein folding experiments in comparison with the anti‐apoptotic family member BclxL.
In agreement with previous reports (Vargas‐Uribe et al, 2013), BclxLΔTM has a thermal stability of ∼ 78°C as probed here by far‐UV‐circular dichroism (CD) spectroscopy and differential scanning calorimetry (DSC), both reporting a single unfolding transition at 78.1°C (Fig 3A). In contrast, BakΔTM shows a more complex unfolding profile in these experiments. As monitored by far‐UV CD melting experiments, a gradual decrease to ∼ 50% of the initial α‐helical secondary structure content could be detected up to a temperature of 90°C, followed by a final cooperative melting transition at 97.3°C (Fig 3A). Such a high melting point has also been reported for the other pore‐forming Bcl2 family member Bax (Yethon et al, 2003; Bleicken & Zeth, 2009). The observed high thermal stability correlated very well with the lack of amide proton exchange as observed in 2D‐[1H,15N]‐HSQC experiments with deuterated versus protonated BakΔTM (Fig EV3A). In the deuterated protein, several HSQC resonance signals in the protein core are absent, indicating that these residues are protected from the solvent. As indicated in Fig 3B, both, the center of helix 5 located in the protein core, and the periphery of helices 1, 2, 6, and 8, which are oriented toward helix 5, are affected. This suggests an exceptionally stable protein core that does not permit hydrogen back exchange. In contrast, for BclxLΔTM having a lower thermal stability, the NMR spectra are identical for protonated and deuterated samples (Fig EV3A).
Figure 3. Stability of the Bak and BclxL soluble domains.
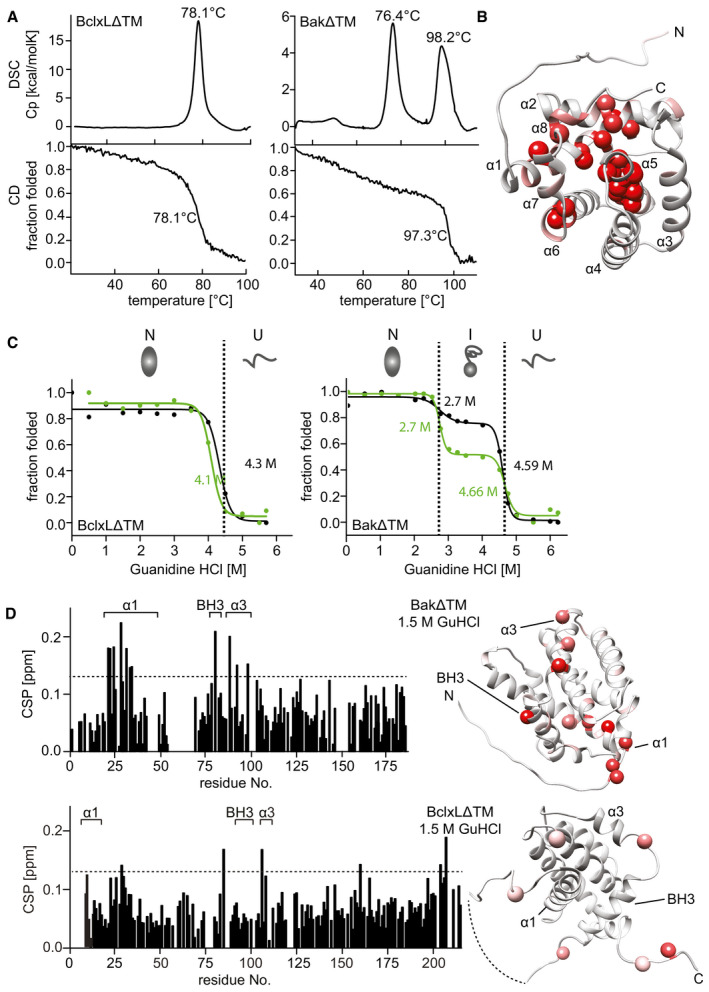
- Comparison of the thermal stability of BclxLΔTM (left) and BakΔTM (right) measured by DSC (top) and Far‐UV‐CD at 222 nm (bottom).
- RDC‐refined structure of BakΔTM. The NMR peak height ratio of the deuterated vs. protonated sample was used to identify solvent‐protected amino acid residues (red spheres).
- Comparison of the GuHCl‐induced chemical unfolding of the secondary structure (black), monitored by far‐UV‐CD at 222 nm, and tertiary structure (green), monitored by Trp‐fluorescence, of BclxLΔTM (left) and BakΔTM (right). The native (N), intermediate (I), and unfolded (U) states are indicated at the top.
- Chemical shift perturbation (CSP) plot derived from 2D‐[15N, 1H] HSQC spectra of BakΔTM (top) and BclxLΔTM (bottom) ± 1.5 M GuHCl. The most significant chemical shift perturbations (> mean value plus standard deviation, dotted line) are highlighted in red on the structures on the right.
Figure EV3. Biophysical characterization of soluble BakΔTM and BclxLΔTM.
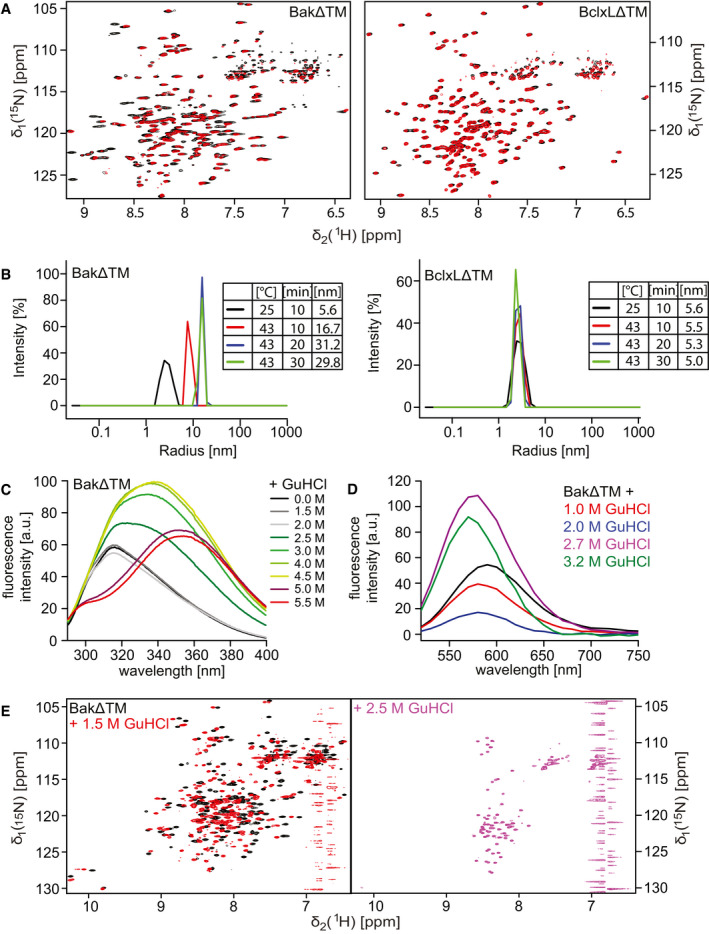
- 2D‐[15N, 1H]‐HSQC spectra of BakΔTM (left) and BclxLΔTM (right) using a protonated (black) or deuterated (red) sample.
- DLS experiments of BakΔTM (left) and BclxLΔTM (right) upon incubation at different temperatures and varying times. The tables on the right state the hydrodynamic diameter in [nm], which is calculated from the measured hydrodynamic radii.
- Tryptophan fluorescence emission spectra of BakΔTM at different GuHCl concentrations upon excitation at 280 nm.
- Fluorescence emission spectra of SYPRO Orange binding to BakΔTM at different GuHCl concentrations.
- 2D‐[15N, 1H]‐HSQC spectra of BakΔTM in the absence (black) and presence of 1.5 M GuHCl (red) and 2.5 M GuHCl (magenta).
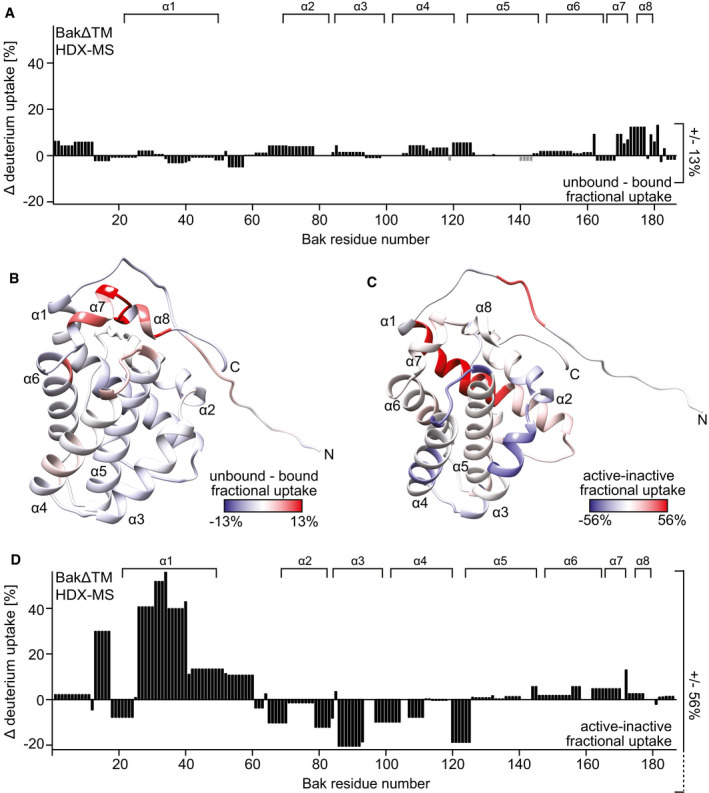
In order to obtain a more detailed characterization of the thermal unfolding behavior of Bak, we used differential scanning calorimetry (DSC). In these experiments, in addition to the high melting point of Bak (98.2°C), an earlier heat‐absorbing event at 76.4°C was detected (Fig 3A). As this event is only associated with a slight decrease in secondary structure content, as probed by CD spectroscopy, we assume that it represents the dissociation of the oligomer, which has been reported to form at a temperature of 43°C (Pagliari et al, 2005). To confirm this assumption, we conducted dynamic light scattering (DLS) measurements where BakΔTM forms larger assemblies when subjected to moderate heat (43°C), while BclxLΔTM stays in a monomeric state (Fig EV3B). Both proteins have a hydrodynamic diameter of ∼ 5.6 nm at 25°C, which is consistent with a monomeric state. This diameter is not altered for BclxLΔTM after prolonged incubation at 43°C. In contrast, for BakΔTM, a gradual increase in diameter up to 30 nm is detected, indicating the built‐up of larger oligomers.
In order to gain a better picture of the equilibrium folding landscape of these two Bcl2 proteins, we performed chemically induced protein unfolding experiments using the chaotropic agent guanidine hydrochloride (GuHCl). Unfolding of the secondary and the tertiary structure was monitored by far‐UV CD and fluorescence spectroscopy, respectively. For BclxLΔTM, both secondary and tertiary structures unfold at a GuHCl concentration of 4.2 M in a cooperative one‐step transition. Again, this profile is very different for BakΔTM (Fig 3C), where an equilibrium folding intermediate is populated, giving rise to a two‐step unfolding process with the first unfolding transition from the native to the intermediate state at a GuHCl concentration of 2.7 M and the final unfolding at 4.6 M GuHCl. The corresponding fluorescence spectra clearly indicate that the tryptophan side chains are present in three distinct environments during unfolding (Fig EV3C). Tryptophan residues that are buried inside the hydrophobic protein core typically show a blue‐shifted emission maximum between 309 and 335 nm, and the latter occurs in a partially buried hydrophobic environment. For a solvent exposed tryptophan, the emission maximum shifts to 355 nm (Royer, 2006). Considering the emission maximum at 315 nm, the tryptophan residues in BakΔTM can be assigned to be buried in the hydrophobic core up to a GuHCl concentration of 2 M. At 2.5 M GuHCl, the environment changes to a partially buried conformation with a maximum at 335 nm. At 4.5 M GuHCl concentration, the entire protein unfolds and the tryptophan fluorescence undergoes a redshift to an emission maximum at 355 nm. The detected change in tryptophan fluorescence indicates that these hydrophobic side chains are partially exposed to the solvent in the intermediate state. This was confirmed by the enhanced binding of the extrinsic hydrophobic fluorescence dye SYPRO Orange in the intermediate state (Fig EV3D). Due to the additional persistence of an almost native‐like secondary structure content, this intermediate state can be defined as a molten globule (Pedrote et al, 2018). A major loss in tertiary structure is also evident for the intermediate from 2D‐TROSY‐NMR experiments where a low chemical shift dispersion was detected (Fig EV3E). In contrast, for BclxLΔTM the native‐like folded state is still detectable up to 4.5 M GuHCl, suggesting that a 2‐state equilibrium between the folded and unfolded state occurs during unfolding (Appendix Fig S3). An analysis of chemical shift perturbations of BakΔTM induced by GuHCl concentrations below the first unfolding transition (up to 1.5 M) revealed that mainly helix 1 and to a lesser extent helix 3 and the BH3 domain are affected by the chaotropic agent (Fig 3D). For BclxLΔTM, the overall effect is smaller and not specific for helix 1. This suggests that a structural change within helix 1 at the N‐terminus might cause the appearance of the detected Bak molten globule folding intermediate. Since structural changes at the N‐terminus, including helix 1, have been suggested to be essential for the pore‐forming activity of Bak (Alsop et al, 2015), we conclude that the detected folding intermediate might also occur during the pore formation process in a lipid bilayer setting.
Structural characterization of partial Bak unfolding upon activation on a lipid bilayer surface
Next, we elucidated whether Bak unfolding can also be detected in a lipid bilayer environment using NMR spectroscopy. For this, we attached BakΔTM‐His6 to nanodiscs supplemented with Ni‐lipids to ensure proper membrane binding. To ensure that only nanodisc‐bound BakΔTM was present in our preparations, we subjected the nanodiscs to size exclusion chromatography (SEC) and retained only the fractions that contained the membrane scaffold protein (MSP) and BakΔTM (Fig 4A). Due to the stable attachment of soluble Bak to the lipid surface, the loaded nanodiscs eluted from the column earlier, consistent with a larger hydrodynamic radius. Lipid binding and the effect of this environment on BakΔTM stability was monitored by CD‐detected thermal melting experiments. While the empty MSP1D1ΔH5‐Ni2+‐nanodiscs unfolded at ∼ 80°C, an additional melting transition at ∼ 68°C appeared when BakΔTM is bound to the membrane surface (Fig EV4A), similar to the melting point of BakΔTM in liposomes (Fig EV1F).
Figure 4. NMR analysis of activated BakΔTM on lipid nanodiscs.
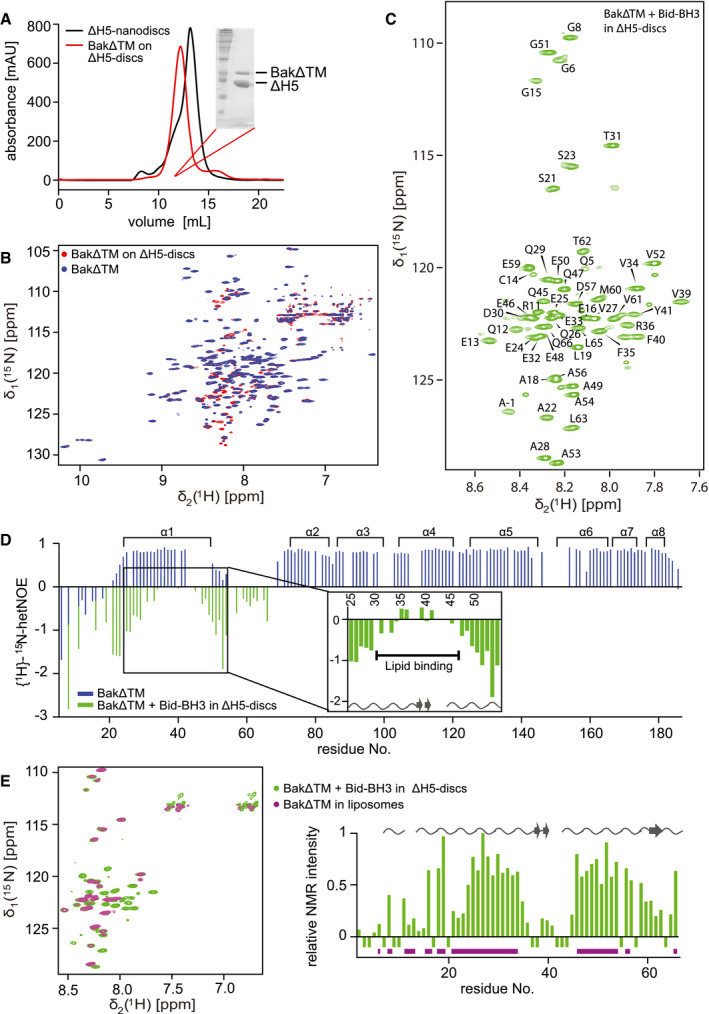
- Size exclusion chromatogram of nanodiscs (black) and BakΔTM bound to nanodiscs (red). Inset: SDS–PAGE confirms the presence of Bak‐loaded nanodiscs.
- Overlay of 2D‐[15N, 1H]‐TROSY spectra of BakΔTM with (red) and without (blue) nanodiscs.
- 2D‐[15N, 1H]‐TROSY spectrum of BakΔTM in nanodiscs after Bid‐BH3 activation with the assigned resonances indicated.
- {1H}–15N‐heteronuclear NOE of BakΔTM in the inactive state (blue) and the active state in nanodiscs after Bid‐BH3 activation (green). α‐Helices in Bak are shown at the top. Residues 25–54 of the activated state are magnified. Chemical shift‐derived secondary structure elements are indicated at the bottom.
- Left: Overlay of 2D‐[15N, 1H]‐TROSY spectra of BakΔTM in [Ni2+]‐caged liposomes (magenta) and Bid‐BH3‐activated BakΔTM in nanodiscs (green). Right: Plot of the relative NMR intensity of Bid‐BH3‐activated BakΔTM in nanodiscs. Peaks that are also visible in liposomes are indicated by horizontal lines in magenta at the bottom, while the chemical shift‐derived secondary structure elements are indicated at the top.
Figure EV4. Characterization of BakΔTM stability and pore‐forming activity in nanodiscs and liposomes.
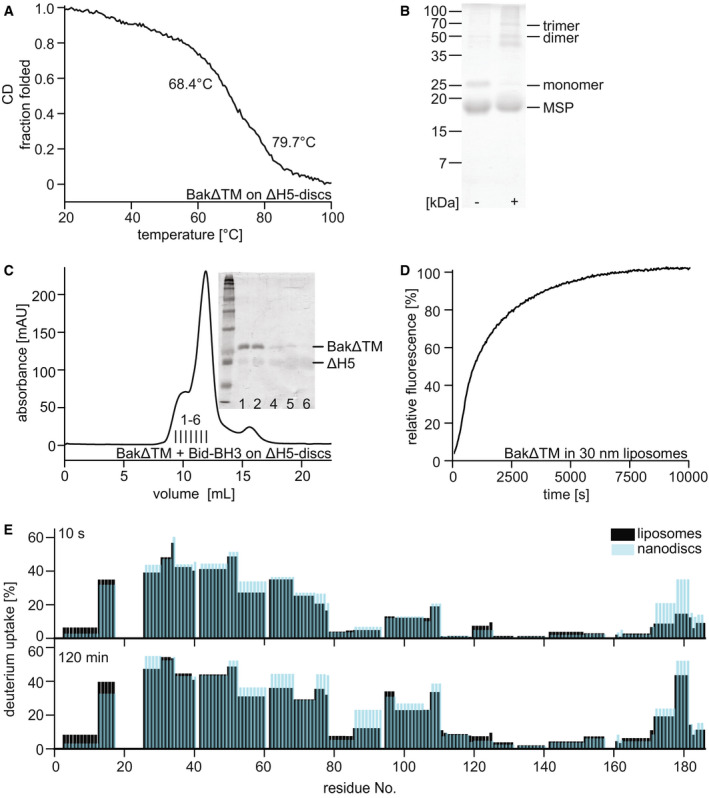
- Far‐UV‐CD thermal melting data of BakΔTM bound to lipid nanodiscs. The melting point at 68.4°C corresponds to BakΔTM, while the later unfolding event at 79.7°C shows the melting of the MSP forming the nanodisc.
- SDS–PAGE of Cu/phen crosslinking of BakΔTM bound to ΔH5 nanodiscs ± Bid‐BH3 peptide.
- Size exclusion chromatogram of BakΔTM bound to nanodiscs after activation by Bid‐BH3. Inset: SDS–PAGE shows that activated BakΔTM elutes before the main nanodisc peak. The MSP band is very faint due to increased destaining which is sometimes seen for MSP.
- Pore‐forming assay with 600 nM BakΔTM in liposomes of 30 nm size.
- Comparison of the deuterium uptake of the active Bak species in liposomes (black) and ΔH5 nanodiscs (blue). The HDX‐MS detected deuterium uptake is compared after 10 s at the top and 2 h at the bottom.
To obtain a structural fingerprint of Bak at the membrane surface, we recorded a 2D‐[15N, 1H]‐TROSY spectrum of 15N‐labeled BakΔTM bound to nanodiscs (Fig 4B). To our surprise, the obtained NMR spectrum is indicative of a well‐folded protein, similar to BakΔTM in solution. Notably, an additional set of weak NMR resonances appears, most likely originating from a minor population of activated Bak. Thus, the smaller and more constrained size of nanodiscs appeared to prevent premature spontaneous membrane insertion of the soluble domain of Bak at high protein concentrations, providing the opportunity for the selective stimulation of pore formation by BH3‐only proteins in this setup. The NMR‐detected compactly folded structural state of BakΔTM on a nanodisc surface seems to be in contrast to the CD data (Fig EV4A) where a lower melting temperature was detected for nanodisc‐attached Bak. However, despite the inhibiting effect of the constrained nanodisc environment, the high temperatures during the CD melting experiment most likely led to heat‐induced Bak activation, which was reported to take place previously (Pagliari et al, 2005). Thus, despite marked structural differences in these two membrane mimetics, the detected melting profiles were very similar. In our NMR experiments at 30°C, the addition of the inducer Bid‐BH3 peptide was required for activation in nanodiscs, as previously reported for the other pore‐forming family member Bax in nanodiscs (Xu et al, 2013). This activator‐dependent activation process resulted in a marked change in the NMR spectrum showing a very low chemical shift dispersion, which is characteristic for an unfolded protein. Interestingly, the position of these NMR signals is almost identical to the minor species that is already present prior to activation, suggesting that even in this trapped state, partial activation can take place (Fig 4C). Since Bak activation has to be accompanied by oligomerization to ultimately form a pore structure, we performed cysteine‐dependent crosslinking experiments with Bak bound to nanodiscs with and without the addition of Bid‐BH3. It could be shown that the inactive form is mainly monomeric, while dimers, trimers, and even higher oligomeric species can be detected for the activated form (Fig EV4B). The faint dimeric band detectable for the inactive state most likely originates from the minor population of active Bak. Furthermore, a SEC analysis shows that BakΔTM forms larger species upon activation (Fig EV4C), presumably leading to a disruption of the nanodisc structure. In the activated state, the number of NMR resonances is markedly reduced suggesting that only a specific part of BakΔTM is visible. To localize this part of Bak, we performed a sequence‐specific NMR backbone resonance assignment and could show that the visible resonances originate from the N‐terminal region up to amino acid Q66. This peptide stretch is composed of the unstructured N‐terminal tail (M1–E24), the first helix (α1, E25–E50), and the loop region before the second helix (G51‐S68). The observed sharp line width of the NMR resonances and the low chemical shift dispersion are indicative of a highly flexible structural state that is most likely located outside the membrane.
In order to probe the dynamic nature of Bak in solution and in its activated, membrane‐integrated form in the ns‐ps time scale, we conducted NMR {1H}–15N‐heteronuclear NOE (hetNOE) experiments (Fig 4D). High hetNOE values (∼ 0.8) represent rigid structures lacking fast internal motions, while flexible parts of the protein give rise to lower or negative values. As expected from the low chemical shift dispersion, the very N‐terminal region (up to aa 20) is highly dynamic in both states. In its inactive soluble form, BakΔTM is compactly folded and consequently less dynamic at this time scale. Flexibility can only be assigned to the loop regions between helices α1 and α2 as well as between α2 and α3. In contrast, the activated state was probed to be highly dynamic for all amino acids visible in the spectrum. A 13C secondary chemical shift‐derived secondary structure estimation confirms a mainly random‐coil conformation for all visible peaks. Interestingly, within this unfolded N‐terminal region of Bak, a less dynamic peptide stretch between E32 and E48 can be observed that has a tendency to adopt a more extended β‐sheet conformation, most likely indicating a transient interaction with the membrane surface. This is supported by the finding that a few amino acids in this region could not be assigned by NMR, possibly due to line‐broadening effects caused by association/dissociation dynamics in the intermediate exchange regime.
To exclude any unspecific effect caused by the presence of lipid nanodiscs, we verified our results by recording 2D‐[15N, 1H]‐TROSY spectra of BakΔTM bound to Ni‐lipid containing liposomes using the same lipid composition as for our nanodisc preparations.
The liposome pore‐forming assay data shown in Figs 1C and D and EV1B clearly show pore‐forming activity of BakΔTM without the need for further activation at protein concentrations of above 1 µM. In order to optimize this system for solution‐state NMR, we prepared liposomes of 30 nm in diameter that are still large enough to enable Bak pore‐forming activity (Fig EV4D). An NMR spectral overlay confirms that the conformation of the visible N‐terminal region in nanodiscs is identical to the pore‐forming state in liposomes (Fig 4E). All peaks visible in liposomes are congruent with assigned peaks in the spectrum of BakΔTM activated by Bid‐BH3 in nanodiscs. The peaks with a high intensity in nanodiscs are also visible in liposomes, while peaks that already showed pronounced line‐broadening in nanodiscs are not or only barely visible in liposomes. Additionally, a comparison of the HDX rates of autoactivated BakΔTM in liposomes and Bid‐BH3‐activated BakΔTM in nanodiscs clearly showed that the two states behave very similarly (Fig EV4E). With these experiments, we could show at a per‐residue resolution that the N‐terminus of Bak adopts a highly flexible and unfolded conformation and that a central part of helix 1 transiently interacts with the membrane.
In order to probe the structural state and solvent accessibility of the remaining parts of Bak that could not be detected by NMR, we used HDX‐MS. This method can be applied to monitor the existence of solvent‐protected regions that are present in secondary structure elements or otherwise shielded. The HDX peptide coverage maps show a high total coverage for the different BakΔTM samples, indicating sufficient data quality for further analysis (Fig EV5A). In the compactly folded inactive and soluble state, BakΔTM has a tightly packed protein core. Thus, only minor amide proton exchange after 10 s was detected for the core part of helix 5, as well as the surrounding helices, especially helices 1 and 4 (Fig EV5B and C, and Appendix Fig S4A). The surrounding helices show hydrogen exchange over time while helix 5 remains inaccessible, confirming the lack of hydrogen back exchange in deuterated Bak detected by NMR (Fig 3B). The hydrogen exchange properties of the inactive state are only slightly altered by the presence of a membrane (Figs 5A and EV5B and C). Merely on the surface of helices 7 and 8, the bound state displays reduced deuterium uptake, indicating that this area is more shielded in the membrane‐bound state as shown in red in Fig 5B. This correlates nicely with the membrane orientation of the soluble domain obtained in the MD simulations with our structural model of full‐length Bak (Fig 2E).
Figure EV5. HDX‐MS data of BakΔTM.
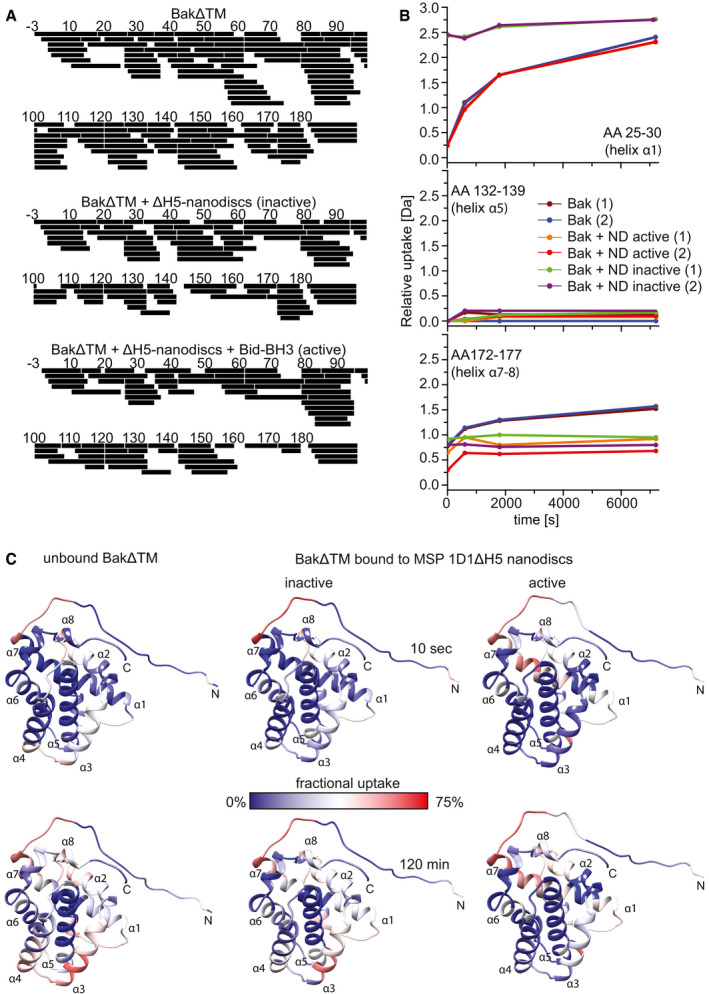
- HDX peptide coverage maps of the different BakΔTM samples. BakΔTM: Total coverage was 100% with 126 peptides and a redundancy of 6.96. BakΔTM + ∆H5 nanodiscs (inactive): Total coverage was 99% with 83 peptides and a redundancy of 4.47. BakΔTM + ∆H5 nanodiscs + Bid‐BH3 (active): Total coverage was 97% with 76 peptides and a redundancy of 4.25.
- HDX graphs of three peptides, located in different helices as indicated, displaying the relative deuterium uptake during the reaction time. Two biological replicates were measured for each state. (ND = ∆H5 nanodiscs).
- HDX‐MS experiments of unbound BakΔTM and BakΔTM bound to Ni‐nanodiscs in the inactive state and after activation with the Bid‐derived BH3 peptide. HDX was mapped to the BakΔTM structure and colored according to the measured exchange rates after the incubation times indicated. Blue colors correspond to a lower (less accessible) and red colors to a higher (more accessible) fractional uptake of deuterium.
Figure 5. HDX‐MS analysis of active and inactive BakΔTM.
-
AThe HDX values after 120 min of BakΔTM bound to ΔH5 nanodiscs are subtracted from the values with soluble BakΔTM and plotted against the amino acid sequence. α‐Helical secondary structure is indicated at the top.
-
BThe HDX data displayed in (A) are color‐coded on the BakΔTM structure. Higher fractional uptake in the unbound state is colored in red, while less uptake is colored in blue.
-
C, DThe HDX of inactive BakΔTM subtracted from the HDX of Bid‐BH3‐activated BakΔTM, both bound to nanodiscs, is color‐coded on the BakΔTM structure in (C) and plotted against the amino acid sequence in (D). In this case, the HDX rates after 10 s were used for figure preparation. The position of the α‐helical secondary structure elements (α1–α8) is indicated at the top.
In contrast, in the active state, helix 1 shows rapid deuterium uptake while helices 4–7 display only low exchange even after 120 min (Fig EV5B and C and Appendix Fig S4B). This is in good agreement with the idea of an unfolded N‐terminus outside the membrane, as seen in NMR, while the rest of the protein is partially or fully protected from the solvent. A direct comparison of the two states shows 56% more deuterium uptake for helix 1 in the activated state (dark red) (Fig 5C and D). Helices 2, 3, and 4 become less accessible which fits perfectly to their transition from a peripheral location in the folded soluble Bak to a fully membrane‐inserted or otherwise protected location. No difference is seen for helix 5, which is already highly protected from the solvent in the inactive state and remains in a fully solvent inaccessible state after activation. The latch domain (helices 6–8) is slightly more solvent accessible in the activated state, possibly by adopting a membrane‐attached location as seen in a recent crystal structure (Birkinshaw et al, 2021).
Helix 1 inhibits Bak pore‐forming activity
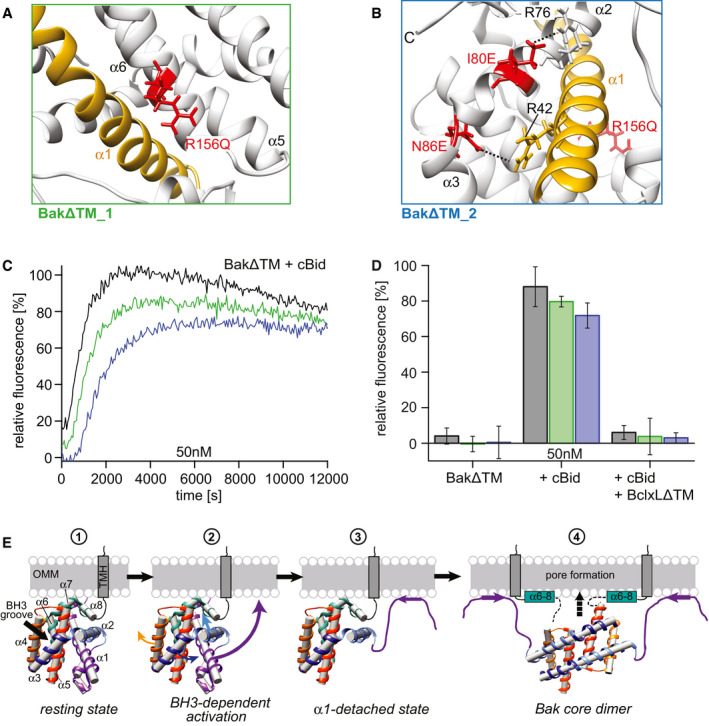
As shown above, the dissociation of the N‐terminal region, in particular helix 1, from the rest of the protein is an essential step for subsequent Bak activation, in agreement with previous literature (Griffiths et al, 1999; Dewson et al, 2009; Alsop et al, 2015). As probed by our protein folding studies (Figs 3D and EV3D), this region is sensitive to ionic chaotropic denaturants, suggesting that electrostatic interactions could play a role in this process. Thus, we analyzed the structures of BakΔTM (Moldoveanu et al, 2006) and BclxLΔTM (Muchmore et al, 1996) to identify amino acid positions which might be important for the interaction between helix 1 and the core helical bundle of Bak. In BclxL, the interactions between these structural elements appear to be highly optimized by stabilizing salt bridges. Thus, we hypothesized that introducing countercharges to unbalanced charges and reducing the overall number of charges in the hydrophobic protein core in Bak would stabilize the interaction of helix 1 and the helical bundle and consequently lead to a lower pore‐forming activity. To do so, we constructed two Bak variants in which distinct regions in Bak are modified. In variant 1 (BakΔTM_1), the interaction between helix 1 and the latch domain is stabilized by removing the unpaired charge at position 156 (R156Q) (Fig 6A). This mutation is aimed at stabilizing the hydrophobic patch in this region enabling the formation of a salt bridge between Glu32 and Arg36 in helix 1, similar to what can be found in BclxL. In variant 2 (BakΔTM_2), we aimed at additionally stabilizing the interaction between helix 1 and the BH3 domain at the C‐terminal end of helix 2 (Fig 6B). The mutations I80E and N86E in the BH3 domain were introduced as countercharges for R76 and R42, respectively.
Figure 6. The structural state of Bak helix 1 triggers pore formation.
-
ABakΔTM_1 (green frame): R156Q stabilizes the interaction between the latch domain (α6–α8) and helix 1 (gold).
-
BBakΔTM_2 (blue frame): Combines R156Q and I80E, N86E, which target the interaction between the BH3 region and helix 1.
-
C, DLiposome permeabilization assay comparing the variants in (A, green) and (B, blue) to wild‐type BakΔTM (black). (C) Comparison of 50 nM of wild‐type BakΔTM (black) to the two variants after activation by 25 nM cBid. (D) Bar diagram comparing the behavior of the BakΔTM variants alone and in the presence of 25 nM cBid and 25 nM BclxLΔTM. The data derived from the liposome permeabilization kinetics shown in panel (C) are averaged for the time interval of 9,000–10,000 s, and the standard deviations were derived from three technical replicates.
-
EModel of the structural changes of Bak during pore formation. Stage 1 depicts the resting state on the OMM surface. The linker between the TMH and soluble domain is most likely flexible. Stage 2 shows the effect induced by the binding of a BH3‐only protein to the Bak BH3 binding groove, where α3 and α4 are pushed apart, most likely weakening the interaction of α2 and α3 with α1, facilitating its detachment. Thereby, α1 unfolds and most likely binds to the membrane (stage 3). This enables the detachment of the core and latch domains, homodimerization (stage 4) and ultimately pore formation. Possibly, the unfolding of α1 and the detachment of the latch domain (α6–α8) might happen simultaneously. Since no structural information is available for the final Bak pore, only the dimeric form is depicted here. A membrane surface‐bound position of the latch domain is suggested by a recent crystal structure (Birkinshaw et al, 2021).
Protein production and stability were found to be similar to wild‐type BakΔTM for both constructs. However, in the pore‐forming assay both constructs displayed a delayed onset of pore formation after selective activation by cBid and could be inhibited by BclxL (Fig 6C and D). Mutation of R156 showed a smaller effect than the combined triple mutation with I80 and N86, in agreement with previous studies (Dewson et al, 2009; Weber et al, 2013; Westphal et al, 2014a; Li et al, 2017). In contrast, the removal of surface exposed positive charges in BakΔTM (R11Q, R169A, R174N) did not lead to reduced pore‐forming activity (Appendix Fig S5), suggesting a dominating contribution of hydrophobic interactions for this process. The mutations in Bak did not perturb its basic functional properties and did not alter the activation and inhibition pathway by cBid or BclxL, respectively, as probed in a pore‐forming assay. Furthermore, the secondary structure content (Appendix Fig S6A) and the chemical unfolding profile (Appendix Fig S6B) were unchanged in comparison with the wild‐type protein, suggesting that all protein variants are properly folded. These initial mutagenesis experiments are intended to provide additional hints on the relevance of helix 1 in initiating pore formation. However, a more systematic mutagenesis study would be required to obtain a better understanding of the relevant amino acid positions in Bak that govern its pro‐apoptotic functionality.
Discussion
In this study, we structurally investigated the pore‐forming Bcl2 protein Bak, which, in contrast to the second pore‐forming Bcl2 protein Bax (Suzuki et al, 2000), is permanently located at the mitochondrial outer membrane in healthy cells (Griffiths et al, 1999). This feature offers the advantage to selectively investigate the structural changes along the pore‐forming trajectory at the membrane. We first investigated the influence of various sample parameters, such as protein concentration, lipid composition, membrane location, and the interaction with pro‐survival BclxL, on the pore‐forming activity of Bak in liposomes (Figs 1 and EV1). In these experiments, the use of a Bak variant lacking its C‐terminal TMH (BakΔTM) and instead utilizing a C‐terminal hexahistidine tag and Ni‐NTA‐modified lipids (Oh et al, 2010) for stable membrane attachment proved essential for a rapid screening procedure and for obtaining a properly folded protein. Consistent with earlier studies (Willis et al, 2007; Chipuk & Green, 2008; Llambi et al, 2011; Westphal et al, 2014b), we found that at low protein concentrations (< 100 nM) Bak forms pores only after activation by an activator BH3 protein such as cBid or Puma‐BH3, and that Bak pore formation can be efficiently inhibited by membrane‐attached anti‐apoptotic BclxLΔTM. Therein, the inhibitory effect of BclxL could be neutralized if cBid was added but activation of Bak still required an excess of BH3‐only proteins over BclxL (Fig 1B). Furthermore, maximal BH3‐dependent activation of Bak pore formation was observed if cBid was present at least at an equimolar stoichiometry to Bak, suggesting that at low protein concentrations Bak activation is exclusively dependent on its interaction with activator BH3 proteins. These findings support existing models of Bcl2 protein pore formation that have been combined in a unified model (Llambi et al, 2011), which suggests that pro‐survival Bcl2 proteins are able to bind to both BH3‐only and effector Bcl2 proteins, and the relative protein levels determine the apoptotic outcome.
In contrast, when Bak is present at a higher concentration, a scenario that is encountered in most structural studies, it forms membrane pores in an autoactive manner, i.e., without the need for BH3‐mediated activation (Fig 1C and D) (Dai et al, 2015; O’neill et al, 2016). Such a concentration‐dependent activation pathway is consistent with earlier studies where knockout of BH3 activators in mice did not abrogate apoptosis (Willis et al, 2007) and where Bax/Bak expression in full Bcl2 protein knockout cells was sufficient to induce apoptosis (O’neill et al, 2016) even without BH3 activators, arguing for the existence of Bak/Bax autoactivation in vivo. In order to stabilize the pre‐pore state of BakΔTM that would otherwise readily insert into the membrane at the high protein concentrations required for structural investigations, we utilized lipid nanodiscs. Compared to liposomes, this membrane mimetic is smaller and constrained in size by the surrounding membrane scaffold protein (Hagn et al, 2018; Klöpfer & Hagn, 2019). In lipid nanodiscs, mainly monomeric Bak attached to the lipid surface was obtained (Fig EV4B), and concentration‐induced autoactivation was efficiently suppressed. Yet, upon BH3‐dependent activation BakΔTM was still able to form oligomers, as probed by cysteine‐mediated crosslinking and size exclusion chromatography (Fig EV4B and C). Most likely, this effect is caused by the high stability of the formed Bak oligomers, leading to nanodisc opening. Thus, we utilized this nanodisc setup to specifically trigger Bak membrane insertion by the addition of activator BH3 peptides and to investigate the stabilized associated structural states. This analysis revealed that the N‐terminal region of Bak (residues 1–66, encompassing helix 1 (α1) and the loop region to α2) adopts a flexible random‐coil conformation, confirming earlier studies at lower resolution obtained by epitope mapping and EPR spectroscopy (Griffiths et al, 1999; Dewson et al, 2008; Oh et al, 2010; Westphal et al, 2011; Alsop et al, 2015; Uren et al, 2017b). The excellent agreement with these previous studies obtained at lower protein concentrations indicates that the higher concentration used for NMR does not alter the activation pathway of Bak. Moreover, the NMR experiments provide novel insights on the interaction between a central part of α1 and the membrane surface (Fig 4D and E), which most likely stabilizes this dynamic partially unfolded conformation. The remaining parts of BakΔTM are most likely located inside the membrane and are therefore invisible in NMR due to line‐broadening effects resulting from the slower tumbling time of the membrane mimetic as well as possible sample inhomogeneities and intrinsic structural dynamics in the ms‐μs time scale. In order to obtain a more global structural picture of Bak, we used HDX‐MS to probe the structural state of the membrane incorporated parts of Bak and determined the high‐resolution NMR structure of its transmembrane helix (TMH) in lipid nanodiscs. This enabled the construction of a structural model of the full‐length, membrane‐anchored protein. Our data suggest that the TMH of Bak does not actively drive oligomerization but instead serves as a stable membrane anchor, similar to the TMH of anti‐apoptotic BclxL in a lipid environment (Raltchev et al, 2018). However, based on earlier studies showing contacts between the TMHs in the final pore (Iyer et al, 2015; Zhang et al, 2016), a stabilizing role of the TMH in the pore is possible.
The herein presented data contribute to a better structural and mechanistic understanding of the pore‐forming process of Bak, which is depicted in Fig 6E and is consistent with current structural models of pore formation by Bcl2 proteins (Mandal et al, 2016; Bleicken et al, 2018). Prior to membrane insertion (stage 1), Bak is present in a membrane‐attached state via its C‐terminal TMH. The latch helices α6–α8 are in contact with the membrane surface, as indicated by reduced HDX in this area if bound to lipid nanodiscs (Fig 5B). This resting state is compactly folded (Fig 4B) and does not insert into the membrane (Fig 1A). However, the proximity to the membrane destabilizes the soluble domain of Bak, as probed by the reduced thermal stability of BakΔTM in liposomes (Fig 1F). The main trigger to induce pore formation is the engagement of the Bak soluble domain with an activator BH3 protein, such as Bid or Puma (stage 2) (Letai et al, 2002; Moldoveanu et al, 2013; Hockings et al, 2015). Upon interaction with a BH3‐only protein, α3 and α4 in Bak form the canonical binding groove (Moldoveanu et al, 2013; Brouwer et al, 2017), which eventually leads to a shift in the location of α2 and α3 with respect to α1. The latter effect implies changes in the interaction between α2 and α1, possibly facilitating the dissociation of α1 from the helical bundle. As suggested by our NMR experiments, a central part of the unfolded α1 then most likely interacts with the surface of the membrane (Fig 4D and E), possibly contributing to the stabilization of its core‐detached conformation. Once α1 is detached (stage 3), the core helices α2–α5 become exposed and can form stable dimers (stage 4), a step that is considered to be essential for pore formation (Brouwer et al, 2014; Cowan et al, 2020). As evident from the crystal structures of Bak core dimers (Brouwer et al, 2014; Cowan et al, 2020), α2 of one Bak monomer needs to insert itself between α3 and α4 of the second monomer (Dewson et al, 2008). Thus, widening of the cleft between α3 and α4 by transient binding of activator BH3 proteins might contribute to an enhanced Bak homodimerization activity and ultimately pore formation. The participation of the Bak core helices in a final compact structural assembly, presumably a pore‐like structure, could be confirmed by the strong solvent protection in HDX‐MS experiments (Fig 5C and D), while solvent exchange rates of the latch helices (α6–α8) slightly increase, indicating a more exposed position, which is in excellent agreement with a recent crystal structure (Birkinshaw et al, 2021). Even though the HDX data can only provide ensemble‐averaged information, the tendencies seen in these data provide valuable insights on the global changes in solvent protection of BakΔTM upon activation in a lipid bilayer setting.
At the same time, BakΔTM activation can be induced by higher BakΔTM concentrations on the membrane surface (Fig 1C and D) without the participation of activator BH3 proteins (Dai et al, 2015). Since Bak is stably attached to the membrane at any protein concentration, the only concentration‐dependent process during Bak activation is its homodimerization or oligomerization. Thus, we believe that Bak autoactivation on a membrane is only possible if there is a conformational equilibrium between the compactly folded state (stage 1) and the α1‐detached state (stage 2), which is strongly in favor of stage 1 when no activator is present. At high protein concentrations, the occurrence of spontaneously detached α1 is more likely, enabling its unfolding and membrane binding (stage 3). This process leads to the exposure of the Bak BH3 domain, which subsequently can activate further Bak molecules (Iyer et al, 2020) in an autocatalytic manner. Pore‐forming activity is further enhanced by the presence of negatively charged lipid headgroups (Fig EV1), which might strengthen the interaction of the positively charged residues in α1 and the membrane surface. In contrast, when no membrane is present, not even a BH3‐only activator can induce the large conformational changes leading to activation, as seen for the Bak soluble domain in complex with Bid‐BH3 (Moldoveanu et al, 2013). Thus, the stabilization of the detached conformation of α1 by a membrane surface might strongly facilitate the necessary global structural changes in Bak. In a cellular context, the exposed Bak BH3 domain is readily neutralized by anti‐apoptotic Bcl2 proteins, leading to low levels of complexes of activated Bak or Bax with pro‐survival Bcl2 proteins (Tan et al, 2006). The herein described activation pathways of Bak are in very good agreement with the unified model of Bcl2 proteins where both, direct activation of Bak/Bax and activation of Bak/Bax in complex with anti‐apoptotic Bcl2 proteins by BH3 proteins, are considered to take place in parallel (Llambi et al, 2011).
Interestingly, in contrast to our data, a previous study with full‐length Bak containing a mutated, amphipathic TMH, did not see autoactivation at higher concentrations (Leshchiner et al, 2013). Due to the amphipathic nature of the TMH, this mutant has reduced membrane binding properties (Ferrer et al, 2012; Westphal et al, 2014b) most likely caused by the interaction of the mutated TMH with its BH3 binding groove, as seen for the other pore‐forming member Bax (Suzuki et al, 2000). This explanation is in line with a previous study showing that mutations in the Bax TMH that reduce membrane residence also reduce its pore‐forming activity (Kuwana et al, 2020). Furthermore, that tendency is also evident from our Bak pore‐forming assays without Ni‐lipids, lacking stable membrane binding (Fig EV1A).
The relevance of α1 dissociation for pro‐apoptotic functionality is further corroborated by previous studies showing that deletion of the N‐terminal BH4 domain (containing α1) leads to a reversal of Bcl2 proteins from anti‐apoptotic to pro‐apoptotic functionality (Jonas et al, 2004) or increases the pore‐forming activity of pro‐apoptotic effector proteins (Xiao et al, 2016). This implicates that pore formation is intrinsically inhibited by the interaction between α1 and the remaining protein. In line with this notion, our mutagenesis experiments aiming at altering the α1 interaction within Bak led to a delayed onset of pore formation, consistent with an abolished pore‐forming activity of Bak if α1 is covalently tethered to the surrounding α‐helices (Alsop et al, 2015). A comparison of our mutation sites (I80, N86, R156) to a databank listing pathological phenotypes (https://www.oncomx.org/) (Türkmen et al, 2011; Kholoussi et al, 2014; Marcotte et al, 2017) reveals that two of the herein chosen positions in Bak, N86 and R156, occur in various cancers, presumably due to their lower apoptotic potency. In contrast, removal of surface exposed positive charges in Bak did not markedly alter the pore‐forming properties (Appendix Fig S5), suggesting a dominating role of hydrophobic interactions as a driving force for pore formation. Even though the measured effects are very likely caused by altered interactions involving α1, an impact on the downstream pore assembly process cannot be completely excluded from the herein presented data. Since no structure of a full Bak or Bax pore has been reported, future systematic rationally designed mutagenesis studies exploring the modulation of Bak functionality will be required to provide a more detailed understanding of the key residues that define pro‐ or anti‐apoptotic Bcl2 proteins. Nonetheless, in our protein folding studies, we observe clear differences in the folding pathway of anti‐ and pro‐apoptotic Bcl2 proteins (Fig 3), respectively, which suggest that, despite a high structural similarity, their distinct functional features are encoded in the stability and interaction pattern of individual secondary structure elements. The ability of effector Bcl2 proteins to undergo large conformational changes has been previously termed as “metastable” (Dewson & Kluck, 2009), further highlighting specific features in their intrinsic folding landscape. This study provides important insights into the conformational changes of Bak in the presence of membranes, while supporting the need for direct activation by BH3‐only proteins to efficiently induce these changes. These findings will contribute to a better mechanistic understanding of the initial steps of Bak activation that might be relevant for novel therapeutic approaches aiming at selective activation or inhibition of apoptosis.
Materials and Methods
Production of Bcl2 proteins in E. coli
All plasmids were transfected into E. coli BL21 (DE3), grown at 37°C, and protein production was induced at an OD600 of 0.6–0.8. The soluble domains of Bak (BakΔTM), consisting of residues 1–186, and BclxL (BclxLΔTM) (residues 1–212) were cloned into a pET21 vector with a LE‐HHHHHH motif at the C‐terminus. For BakΔTM, a MEAS‐header was added to increase expression levels as described (Moldoveanu et al, 2006). BakΔTM was induced with 0.1 mM IPTG and grown for 16–20 h at 20°C, while BclxLΔTM was induced with 1 mM IPTG and shaken for 4 h at 37°C. For the production of U‐[2H, 13C, 15N]‐BakΔTM, bacteria were grown in M9 medium supplemented with 1 g/l [98% 15N]‐NH4Cl and 2 g/l [98% 2H, 13C]‐glucose in 99% D2O (Eurisotope or Sigma‐Aldrich). The DNA encoding for Bak‐TMH (residues 183–211) was cloned into a pET15b vector N‐terminally fused to GB1 with a thrombin cleavage site and produced as inclusion bodies as described previously for BclxL‐TMH (Raltchev et al, 2018). Bid was cloned into a pET21a vector, and the caspase‐8 cleavage site was replaced by a thrombin cleavage site. It was produced in an identical manner as BclxLΔTM.
Purification of BakΔTM, BclxLΔTM, and cBid
Cell pellets were resuspended in Lysis Buffer (50 mM Tris pH 8, 200 mM NaCl, 1 mM EDTA) and incubated with lysozyme and a tablet of cOmplete™ EDTA‐free protease inhibitor cocktail (Roche). The final cell disruption was performed by sonicating for 10 min (1‐s pulse, 3‐s pause, 30% amplitude). After incubation with DNase I (Roche) and 5 mM MgSO4, the cell debris was spun down (50 000 g, 25 min, 4°C) and the supernatant applied to a Ni‐NTA gravity flow affinity column, equilibrated with Buffer A (20 mM Tris pH 8, 100 mM NaCl, 5 mM β‐mercaptoethanol (BME)). The beads were washed with 5 CV of Buffer A and 5 CV of Buffer A + 10 mM imidazole, followed by elution with 3 CV of Buffer A + 400 mM imidazole. 5% glycerol was added to Bak/BclxLΔTM before concentrating for size exclusion chromatography (SEC) on a ÄKTA™ Pure system equipped with a HiLoad 16/600 Superdex 75 pg column in 20 mM NaPi pH 7, 50 mM NaCl, 1 mM EDTA, 2 mM DTT. For binding experiments to Ni‐NTA lipids, EDTA and DTT were omitted in the buffer for the SEC and DTT was substituted for 5 mM BME. After the Ni‐affinity chromatography, Bid was dialyzed overnight in Buffer A. 20 U thrombin/ 1L cell pellet was added to mimic the cleavage to cBid. After cleavage was verified by SDS–PAGE, 5% glycerol was added, and the purification was continued as described above for Bak/BclxLΔTM.
Purification of Bak‐/BclxL‐TMH
The transmembrane domains were produced as GB1‐fusion proteins with a thrombin cleavage site and purified from inclusion bodies as reported before (Raltchev et al, 2018). The protein pellet was either dissolved in 5% sodium dodecyl sulfate (SDS) and purified using a HiPrep 16/60 Sephacryl S‐200 HR size exclusion column on a ÄKTA Start system at room temperature for subsequent nanodisc assemblies or dissolved in 4% dodecylphosphocholine (DPC) and purified using a HiLoad 16/600 Superdex 200 pg size exclusion column on a ÄKTA Pure system at 4°C for studies in micelles. The SEC was run in 20 mM Tris pH 7.5, 100 mM NaCl, 1 mM EDTA + 0.5% SDS, or 0.1% DPC. Protein prepared in DPC was used to study Bak‐TMH in micelles. Thrombin (25 U) was added for cleavage of the fusion protein, followed by a reverse Ni‐NTA gravity flow affinity chromatography step to separate the His‐tagged GB1 from the Bak‐TMH. Finally, the buffer was exchanged on a NAP‐25 column (GE Healthcare) using SEC buffer.
Preparation of liposomes
The respective lipids were mixed in chloroform and dried under a nitrogen gas flow (1 mg/ml for liposome permeabilization assays, 5 mg/ml for CD measurements). The lipids were resuspended in 1 ml of the respective buffer, sonicated in a sonication bath for 2 min, and subjected to ten freeze and thaw cycles before the suspension was subjected to eleven rounds of extrusion using a 100‐nm filter membrane unless indicated otherwise.
The following two lipid compositions were used in our studies, all presented as mass percentage. OMM lipids were prepared by mixing 46% l‐α‐phosphatidylcholine (PC), 25% l‐α‐phosphatidylethanolamine (PE), 11% l‐α‐phosphatidylinositol (PI), 10% 1,2‐dioleyl‐sn‐glycero‐3‐phospho‐L‐serine, and 7% 1,1’,2,2’‐tetra‐(9Z‐octadecenoyl)cardiolipin. According to the manufacturer, E. coli polar lipid extract consists of 67% phosphatidylethanolamine (PE), 23.2% phosphatidylglycerol (PG), and 9.8% cardiolipin (CL). 2–10% 18:1 DGS‐NTA (Ni2+) was added to the lipid mixtures as indicated in the corresponding method sections. All lipids were obtained from Avanti Polar Lipids.
Preparation of phospholipid nanodiscs
Phospholipid nanodiscs were prepared as previously described (Hagn et al, 2018). The nanodiscs for binding BakΔTM were prepared with E. coli polar lipids and four 18:1 DGS‐NTA (Ni2+) lipids per nanodisc. The nanodiscs assembled with Bak‐TMH were prepared using 75% dimyristoylphosphocholine (DMPC) and 25% dimyristoylglycerophosphoglycerol (DMPG).
Liposome permeabilization assay
The liposome permeabilization assay was performed at 30°C as described before (Yethon et al, 2003; Kale et al, 2014). 50 nM BakΔTM, 20 nM cBid/Puma‐BH3, and 25 nM BclxLΔTM were used for the measurements at low BakΔTM concentrations, while 600 nM BakΔTM and 300 nM cBid/Puma‐BH3/BclxLΔTM were used for measurements under autoactive BakΔTM conditions. The liposomes were formed as described above using either OMM‐like lipids with 2% (w/w) 18:1 DGS‐NTA(Ni) or E. coli polar lipids with 2% (w/w) 18:1 DGS‐NTA(Ni) with the addition of fluorescent dyes, as described (Yethon et al, 2003; Kale et al, 2014). For the measurements under autoactive conditions, E. coli polar lipids with 10% (w/w) 18:1 DGS‐NTA(Ni) were used. The data were averaged from three measurements and was normalized according to the equation (Ft−F0)/(F100−F0) with Ft being the measured fluorescence at time t, F0 being the initial fluorescence of liposomes without the addition of proteins, and F100 being the fluorescence measured after completely dissolving the liposomes and releasing all dye by adding 0.2% Triton X‐100. The data are presented as relative fluorescence with 100% representing the maximum liposome permeabilization seen for the respective conditions compared in the individual figures.
Cu/phen crosslinking
Cysteine‐mediated crosslinking was performed using the oxidizing complex CuII/phenanthroline (Cu/phen). Samples were prepared as described above, but the final size exclusion was performed without a reducing agent. A 240 mM Cu/phen stock solution in EtOH:H2O (1:4) was prepared freshly as described before (Teijido et al, 2012). After two steps of 1:10 dilution in H2O, 30 µM Cu/phen was added to the nanodisc samples (20 µM) and incubated for 2 h at 25°C while shaking. The results were analyzed with a non‐reducing SDS–PAGE.
Circular dichroism (CD) spectroscopy
CD spectroscopy was used for determining the secondary structure content of all protein samples as well as their stability. Measurements were conducted with a Jasco J‐715 spectropolarimeter with a 1‐mm path‐length quartz cuvette. Spectra were measured at 20°C with concentrations ranging from 10 to 20 µM. Liposomes were prepared using either OMM‐like lipids or E. coli polar lipids both with 5% 18:1 DGS‐NTA (Ni2+). For the spectrum of BakΔTM + cBid on liposomes, the spectrum of cBid on liposomes was subtracted. Thermal unfolding traces were measured by monitoring the ellipticity at 222 nm with a heating rate of 1°C/min. Chemical denaturant‐induced unfolding was detected by monitoring the ellipticity at 222 nm after incubating with varying guanidine hydrochloride (GuHCl) concentrations overnight at 4°C (each data point represents the average of a 30‐s recording with a data interval of 0.5 s). The averaged ellipticity was normalized, plotted against the GuHCl concentration, and fitted with a Boltzmann equation (Privalov, 1979).
Differential scanning calorimetry (DSC)
DSC measurements were performed on a Microcal PEAQ‐DSC instrument (Malvern) in 20 mM NaPi pH 7.0, 50 mM NaCl, 1 mM EDTA, 5 mM BME. All samples were dialyzed overnight in the buffer used as a reference. A temperature scan was performed from 25 to 100°C or 30 to 115°C, depending on the melting point of the sample, with a heating rate of 60°C/h. The sample concentration was 30 µM.
Dynamic light scattering (DLS)
DLS measurements were performed on a DynaPro NanoStar instrument. 60 µl of a 10–20 µM sample was centrifuged (20,000 x g, 20 min, 4°C) to sediment any oligomeric particles prior to the experiment. Measurements were done at 25°C and 43°C and at increasing incubation times.
Fluorescence spectroscopy
Fluorescence spectroscopy was used for determining the global folding state of the protein samples. Tryptophan fluorescence was monitored from 290 nm to 400 nm after exciting at 280 nm using a Jasco FP‐8300 spectrofluorometer. All measurements were performed on 150–200 nM samples at 20°C while stirring at 300 rpm. Spectra were measured with a scanning speed of 100 nm/min, a bandwidth of 5 nm for both excitation and emission, and a response time of 0.5 s. Chemical unfolding was detected by monitoring the fluorescence after incubation with varying GuHCl concentrations overnight at 4°C. Reference spectra without protein were measured for all conditions and subtracted. For visualization, the wavelength of the fluorescence maxima was plotted against the GuHCl concentration.
Hydrogen deuterium exchange mass spectrometry (HDX‐MS)
For hydrogen/deuterium exchange (HDX) experiments, an ACQUITY UPLC M‐class system equipped with automated HDX technology (Waters, Milford, MA, USA) was used. HDX kinetics were determined at 20°C taking data points at 0, 10, 60, 600, 1,800, and 7,200 s in technical triplicates. At the respective data points of the kinetics, 3 µl of a solution of approximately 30 µM protein was diluted 1:20 into 99.9% D2O‐containing 20 mM sodium phosphate, pH 6.8 (titrated with HCl) or the respective H2O–containing reference buffer. Quenching of the reaction mixture was achieved by adding 1:1 200 mM KH2PO4, 200 mM Na2HPO4, pH 2.3 (titrated with HCl), containing 4 M guanidine hydrochloride, and 200 mM TCEP at 1°C. For on‐column peptic digest on a Waters Enzymate BEH pepsin column 2.1 × 30 mm at 20°C, 50 µl of the samples was applied. Peptides were separated by reversed‐phase chromatography at 0°C applying a gradient increasing the acetonitrile concentration stepwise from 5 to 35% in 6 min, from 35 to 40% in 1 min, and from 40 to 95% in 1 min. A Waters Acquity UPLC C18 1.7 µm Vangard 2.1 × 5 mm trapping‐column and a Waters Acquity UPLC BEH C18 1.7 µm 1 × 100 mm separation column were used, and the eluted peptides were analyzed using an in‐line Synapt G2‐S QTOF HDMS mass spectrometer (Waters, Milford, MA, USA). All experiments were performed in biological duplicate collecting MS data over an m/z range of 100–2,000. Mass accuracy was ensured by calibration with Glu‐fibrino peptide B (Waters, Milford, MA, USA). Peptides were identified by MSE ramping the collision energy automatically from 20 to 50 V. As the automated system handles all samples at identical conditions, deuterium levels were not corrected for back exchange and are therefore reported as relative deuterium levels (Wales & Engen, 2006). Data were analyzed using the PLGS 3.0.3 and DynamX 3.0 software packages (Waters, Milford, MA, USA).
Molecular dynamic simulations
Bak‐TMH was inserted into a hexagonal box of DMPC and DMPG lipid bilayer (3:1 ratio) using the CHARMM‐GUI web server (http://www.charmm‐gui.org) (Jo et al, 2008) in the presence of 0.15 M KCl. Equilibration of the system was done at 310 K in two phases with 3 cycles each. The force constants to fix the position of the protein and membrane were gradually reduced in each cycle. In the first phase, a time step of 1 fs and a simulation time of 50 ps in each cycle were used, whereas in the second phase, a time step of 2 fs and a simulation time of 200 ps for each cycle were used. The total equilibration time was 750 ps. The final MD simulation (129 ns duration) was carried out with the isothermal‐isobaric ensembles at 310 K using the program NAMD (Phillips et al, 2005). Long‐range electrostatic interactions were described using the particle‐mesh Ewald method (Darden et al, 1993). A smoothing function was applied to truncate short‐range electrostatic interactions. Analysis and visualization of the obtained trajectory was done with VMD (Humphrey et al, 1996).
NMR spectroscopy
NMR experiments were performed in 20 mM NaPi pH 6.8, 1 mM EDTA, 2 mM DTT on Bruker Avance III instruments operating at 600, 800, 900, and 950 MHz proton frequency, respectively, equipped with cryogenic probes. If Ni‐NTA lipids were used for nanodisc assembly, no EDTA was added and DTT was substituted for 5 mM BME. HSQC‐type triple resonance experiments (Sattler et al, 1999) were recorded for obtaining backbone resonance assignments of the soluble domain, while TROSY‐type 3D‐triple resonance experiments (Salzmann et al, 1998) were utilized for the nanodisc samples. The soluble domain was measured at 30°C both in solution (200–300 µM) and when bound to membrane mimetics (approximately 50–150 µM). Bak‐TMH (300 µM) was measured at 37°C in DPC micelles (∼ 300 mM DPC) and at 42°C in MSP1D1ΔH5 nanodiscs (Hagn et al, 2013, 2018; Klöpfer & Hagn, 2019). For the assignment of the Bak‐TMH, 3D‐TROSY‐HNCA and HNCO experiments with U‐[2H, 13C, 15N]‐labeled Bak‐TMH in DPC as well as 3D‐HNH‐ and 3D‐NNH‐NOESY experiments for U‐[2H, 15N]‐labeled Bak‐TMH in DPC and lipid nanodiscs were recorded. Spectral analysis was done with SPARKY4 (Goddard & Kneller, 2008) and NMRFAM‐SPARKY (Lee et al, 2015). For NOESY experiments, a mixing time of 200 ms was used. Structure calculations were done with XPlor‐NIH (Schwieters et al, 2003) using standard protocols. The 20 structures with the lowest restraint violation energies were used to obtain structural statistics of the ensemble. Ramachandran map analysis was performed with the RAMPAGE webserver (Lovell et al, 2003). {1H}–15N‐heteronuclear NOE experiments were recorded in a fully interleaved manner with 1,024 complex points in the direct dimension and 128 complex points in the indirect dimension. The proton saturation time was 2 s for activated BakΔTM in MSP1D1ΔH5 nanodiscs using a TROSY‐based experiment (Zhu et al, 2000) and 2.5 s for inactive BakΔTM using a HSQC‐based experiment. Amide residual dipolar couplings (RDCs) were measured with 2D‐TROSY and semi‐TROSY (Pervushin et al, 1997) experiments in the isotropic case and in the presence of 10 mg/ml Pf1 phage (Asla Biotech, Riga, Latvia). Fitting of the RDC alignment tensor was done with PALES (Zweckstetter & Bax, 2000). Refinement of the Bak crystal structure (PDB: 2IMS (Moldoveanu et al, 2006)) using NMR chemical shift and HN‐RDC restraints was done with XPlor‐NIH. NMR experiments on 400 µM BclxLΔTM were performed in 20 mM NaPi pH 7, 50 mM NaCl, 1 mM EDTA, 2 mM DTT at 30°C using either protonated or deuterated 15N‐labeled protein.
Author contributions
LES performed research on all aspects of this paper, analyzed data, and wrote the manuscript; FR performed HDX‐MS experiments under the supervision of MH; AS and LES performed DLS experiments. FH designed research, analyzed data, and wrote the manuscript. All authors commented on the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Acknowledgements
We gratefully acknowledge support from the DFG (CRC1035, project B13, to F.H.). The mass spectrometry facility used in this study was funded by the DFG (CRC1035, project Z1). FH was supported by the Helmholtz Society (VG‐NG‐1039). All NMR experiments were conducted at the Bavarian NMR Center (BNMRZ) at the Technical University of Munich supported by the Technical University of Munich and the Helmholtz Center Munich. Drs. Gerd Gemmecker and Sam Asami are gratefully acknowledged for general NMR support and Dr. Umut Günsel (TUM) for help with the preparation of the TOC figure. Open Access funding enabled and organized by Projekt DEAL.
The EMBO Journal (2021) 40: e107159.
See also: JJ Sandow et al (October 2021) and AM Ojoawo & T Moldoveanu (October 2021)
Data availability
The NMR backbone resonance chemical shift information and the structural coordinates of Bak‐TMH in DPC micelles and lipid nanodiscs have been deposited at the BMRB (accession codes 34621, 34622) and RCSB PDB (accession codes 7OFM, 7OFO) data banks, respectively. The NMR backbone resonance assignments of BakΔTM (aa 1–186) in its apostate have been deposited at the BMRB data bank (accession code 50942).
References
- Adams JM, Cory S (2007) Bcl‐2‐regulated apoptosis: mechanism and therapeutic potential. Curr Opin Immunol 19: 488–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsop AE, Fennell SC, Bartolo RC, Tan IK, Dewson G, Kluck RM (2015) Dissociation of Bak α1 helix from the core and latch domains is required for apoptosis. Nat Commun 6: 6841 [DOI] [PubMed] [Google Scholar]
- Aluvila S, Mandal T, Hustedt E, Fajer P, Choe JY, Oh KJ (2014) Organization of the mitochondrial apoptotic BAK pore: Oligomerization of the BAK homodimers. J Biol Chem 289: 2537–2551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basanez G, Nechushtan A, Drozhinin O, Chanturiya A, Choe E, Tutt S, Wood KA, Hsu Y‐T, Zimmerberg J, Youle RJ (1999) Bax, but not Bcl‐x L, decreases the lifetime of planar phospholipid bilayer membranes at subnanomolar concentrations. Proc Natl Acad Sci 96: 5492–5497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkinshaw RW, Iyer S, Lio D, Luo CS, Brouwer JM, Miller MS, Robin AY, Uren RT, Dewson G, Kluck RM et al (2021) Structure of detergent‐activated BAK dimers derived from the inert monomer. Mol Cell 81: 2123–2134.e5 [DOI] [PubMed] [Google Scholar]
- Bleicken S, Assafa TE, Stegmueller C, Wittig A, Garcia‐Saez AJ, Bordignon E (2018) Topology of active, membrane‐embedded Bax in the context of a toroidal pore. Cell Death Differ 25: 1717–1731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleicken S, Hantusch A, Das KK, Frickey T, Garcia‐Saez AJ (2017) Quantitative interactome of a membrane Bcl‐2 network identifies a hierarchy of complexes for apoptosis regulation. Nat Commun 8: 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleicken S, Zeth K (2009) Conformational changes and protein stability of the pro‐apoptotic protein Bax. J Bioenerg Biomembr 41: 29–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouwer JM, Lan P, Cowan AD, Bernardini JP, Birkinshaw RW, van Delft MF, Sleebs BE, Robin AY, Wardak A, Tan IK et al (2017) Conversion of Bim‐BH3 from activator to inhibitor of Bak through structure‐based design. Mol Cell 68: 659–672 [DOI] [PubMed] [Google Scholar]
- Brouwer JM, Westphal D, Dewson G, Robin AY, Uren RT, Bartolo R, Thompson GV, Colman PM, Kluck RM, Czabotar PE (2014) Bak core and latch domains separate during activation, and freed core domains form symmetric homodimers. Mol Cell 55: 938–946 [DOI] [PubMed] [Google Scholar]
- Chipuk JE, Green DR (2008) How do BCL‐2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol 18: 157–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosentino K, García‐Sáez AJ (2017) Bax and Bak pores: are we closing the circle? Trends Cell Biol 27: 266–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan AD, Smith NA, Sandow JJ, Kapp EA, Rustam YH, Murphy JM, Brouwer JM, Bernardini JP, Roy MJ, Wardak AZ et al (2020) BAK core dimers bind lipids and can be bridged by them. Nat Struct Mol Biol 27: 1024–1031 [DOI] [PubMed] [Google Scholar]
- Czabotar P, Westphal D, Dewson G, Ma S, Hockings C, Fairlie W, Lee E, Yao S, Robin A, Smith B et al (2013) Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis. Cell 152: 519–531 [DOI] [PubMed] [Google Scholar]
- Dai H, Ding H, Meng XW, Peterson KL, Schneider PA, Karp JE, Kaufmann SH (2015) Constitutive BAK activation as a determinant of drug sensitivity in malignant lymphohematopoietic cells. Genes Dev 29: 2140–2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai H, Smith A, Meng XW, Schneider PA, Pang YP, Kaufmann SH (2011) Transient binding of an activator BH3 domain to the Bak BH3‐binding groove initiates Bak oligomerization. J Cell Biol 194: 39–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darden T, York D, Pedersen L (1993) Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J Chem Phys 98: 10089–10092 [Google Scholar]
- Denisov IG, Grinkova YV, Lazarides AA, Sligar SG (2004) Directed self‐assembly of monodisperse phospholipid bilayer nanodiscs with controlled size. J Am Chem Soc 126: 3477–3487 [DOI] [PubMed] [Google Scholar]
- Dewson G, Kluck RM (2009) Mechanisms by which Bak and Bax permeabilise mitochondria during apoptosis. J Cell Sci 122: 2801–2808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewson G, Kratina T, Sim HW, Puthalakath H, Adams JM, Colman PM, Kluck RM (2008) To trigger apoptosis, Bak exposes its BH3 domain and homodimerizes via BH3:groove interactions. Mol Cell 30, 369–380 [DOI] [PubMed] [Google Scholar]
- Dewson G, Kratina T, Czabotar P, Day CL, Adams JM, Kluck RM (2009) Bak activation for apoptosis involves oligomerization of dimers via their α6 helices. Mol Cell 36: 696–703 [DOI] [PubMed] [Google Scholar]
- Ferrer PE, Frederick P, Gulbis JM, Dewson G, Kluck RM (2012) Translocation of a Bak C‐terminus mutant from cytosol to mitochondria to mediate cytochrome c release: Implications for Bak and Bax apoptotic function. PLoS One 7(3): e31510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores‐Romero H, Ros U, Garcia‐Saez AJ (2020) Pore formation in regulated cell death. EMBO J 39: e105753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard T, Kneller D (2008) Sparky 3. San Francisco, CA: University of California; [Google Scholar]
- Griffiths GJ, Dubrez L, Morgan CP, Jones NA, Whitehouse J, Corfe BM, Dive C, Hickman JA (1999) Cell damage‐induced conformational changes of the pro‐apoptotic protein Bak in vivo precede the onset of apoptosis. J Cell Biol 144: 903–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagn F, Etzkorn M, Raschle T, Wagner G (2013) Optimized phospholipid bilayer nanodiscs facilitate high‐resolution structure determination of membrane proteins. J Am Chem Soc 135: 1919–1925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagn F, Nasr ML, Wagner G (2018) Assembly of phospholipid nanodiscs of controlled size for structural studies of membrane proteins by NMR. Nat Protoc 13: 79–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Häusler E, Fredriksson K, Goba I, Peters C, Raltchev K, Sperl L, Steiner A, Weinkauf S, Hagn F (2020) Quantifying the insertion of membrane proteins into lipid bilayer nanodiscs using a fusion protein strategy. Biochim Biophys Acta Biomembr 1862: 183190 [DOI] [PubMed] [Google Scholar]
- Hockings C, Anwari K, Ninnis RL, Brouwer J, O'Hely M, Evangelista M, Hinds MG, Czabotar PE, Lee EF, Fairlie WD et al (2015) Bid chimeras indicate that most BH3‐only proteins can directly activate Bak and Bax, and show no preference for Bak versus Bax. Cell Death Dis 6: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten K (1996) VMD: Visual molecular dynamics. J Mol Graph 14: 33–38 [DOI] [PubMed] [Google Scholar]
- Iyer S, Bell F, Westphal D, Anwari K, Gulbis J, Smith BJ, Dewson G, Kluck RM (2015) Bak apoptotic pores involve a flexible C‐terminal region and juxtaposition of the C‐terminal transmembrane domains. Cell Death Differ 22: 1665–1675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer S, Uren RT, Dengler MA, Shi MX, Uno E, Adams JM, Dewson G, Kluck RM (2020) Robust autoactivation for apoptosis by BAK but not BAX highlights BAK as an important therapeutic target. Cell Death Dis 11: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S, Kim T, Iyer VG, Im W (2008) CHARMM‐GUI: a web‐based graphical user interface for CHARMM. J Comput Chem 29: 1859–1865 [DOI] [PubMed] [Google Scholar]
- Jonas EA, Hickman JA, Chachar M, Polster BM, Brandt TA, Fannjiang Y, Ivanovska I, Basanez G, Kinnally KW, Zimmerberg J et al (2004) Proapoptotic N‐truncated BCL‐xL protein activates endogenous mitochondrial channels in living synaptic terminals. Proc Natl Acad Sci U S A 101: 13590–13595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kale J, Chi X, Leber B, Andrews D (2014) Examining the molecular mechanism of bcl‐2 family proteins at membranes by fluorescence spectroscopy. Methods Enzymol 544 1–23 [DOI] [PubMed] [Google Scholar]
- Kelekar A, Thompson CB (1998) Bcl‐2‐family proteins: the role of the BH3 domain in apoptosis. Trends Cell Biol 8: 324–330 [DOI] [PubMed] [Google Scholar]
- Kholoussi NM, El‐Nabi SEH, Esmaiel NN, Abd El‐Bary NM, El‐Kased AF (2014) Evaluation of Bax and Bak gene mutations and expression in breast cancer. BioMed Res Int 2014: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klöpfer K, Hagn F (2019) Beyond detergent micelles: The advantages and applications of non‐micellar and lipid‐based membrane mimetics for solution‐state NMR. Prog Nucl Magn Reson Spectrosc 114–115: 271–283 [DOI] [PubMed] [Google Scholar]
- Kuwana T, King LE, Cosentino K, Suess J, Garcia‐Saez AJ, Gilmore AP, Newmeyer DD (2020) Mitochondrial residence of the apoptosis inducer BAX is more important than BAX oligomerization in promoting membrane permeabilization. J Biol Chem 295: 1623–1636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvansakul M, Hinds MG (2015) The Bcl‐2 family: structures, interactions and targets for drug discovery. Apoptosis 20: 136–150 [DOI] [PubMed] [Google Scholar]
- Lai YC, Li CC, Sung TC, Chang CW, Lan YJ, Chiang YW (2019) The role of cardiolipin in promoting the membrane pore‐forming activity of BAX oligomers. Biochim Biophys Acta Biomembr 1861: 268–280 [DOI] [PubMed] [Google Scholar]
- Landeta O, Landajuela A, Gil D, Taneva S, DiPrimo C, Sot B, Valle M, Frolov VA, Basañez G (2011) Reconstitution of proapoptotic BAK function in liposomes reveals a dual role for mitochondrial lipids in the BAK‐driven membrane permeabilization process. J Biol Chem 286: 8213–8230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W, Tonelli M, Markley JL (2015) NMRFAM‐SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics 31: 1325–1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leshchiner ES, Braun CR, Bird GH, Walensky LD (2013) Direct activation of full‐length proapoptotic BAK. Proc Natl Acad Sci U S A 110: E986–E995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ (2002) Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2: 183–192 [DOI] [PubMed] [Google Scholar]
- Li MX, Tan IKL, Ma SB, Hockings C, Kratina T, Dengler MA, Alsop AE, Kluck RM, Dewson G (2017) BAK α6 permits activation by BH3‐only proteins and homooligomerization via the canonical hydrophobic groove. Proc Natl Acad Sci USA 114: 7629–7634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llambi F, Moldoveanu T, Tait SWG, Bouchier‐Hayes L, Temirov J, McCormick LL, Dillon CP, Green DR (2011) A unified model of mammalian BCL‐2 protein family interactions at the mitochondria. Mol Cell 44: 517–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- López CA, Swift MF, Xu XP, Hanein D, Volkmann N, Gnanakaran S (2019) Biophysical characterization of a nanodisc with and without BAX: an integrative study using molecular dynamics simulations and Cryo‐EM. Structure 27: 988–999.e4 [DOI] [PubMed] [Google Scholar]
- Lovell SC, Davis IW, Arendall 3rd WB, de Bakker PIW, Word JM, Prisant MG, Richardson JS, Richardson DC (2003) Structure validation by Cα geometry: φ, ψ and Cβ deviation. Proteins 50: 437–450 [DOI] [PubMed] [Google Scholar]
- Mandal T, Shin S, Aluvila S, Chen HC, Grieve C, Choe JY, Cheng EH, Hustedt EJ, Oh KJ (2016) Assembly of Bak homodimers into higher order homooligomers in the mitochondrial apoptotic pore. Sci Rep 6: 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotte EL, Pankratz N, Amatruda JF, Frazier AL, Krailo M, Davies S, Starr JR, Lau CC, Roesler M, Langer E et al (2017) Variants in BAK1, SPRY4, and GAB2 are associated with pediatric germ cell tumors: a report from the children’s oncology group. Genes Chromosom Cancer 56: 548–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez‐Senac Mdel M, Senena C‐G, Gomez‐Fernandez JC (2002) The structure of the C‐terminal domain of the pro‐apoptotic protein Bak. Biophys J 81: 233–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moldoveanu T, Czabotar PE (2020) BAX, BAK, and BOK: a coming of age for the BCL‐2 family effector proteins. Cold Spring Harb Perspect Biol 12: 1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moldoveanu T, Grace CR, Llambi F, Nourse A, Fitzgerald P, Gehring K, Kriwacki RW, Green DR (2013) BID‐induced structural changes in BAK promote apoptosis. Nat Struct Mol Biol 20: 589–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moldoveanu T, Liu Q, Tocilj A, Watson M, Shore G, Gehring K (2006) The X‐ray structure of a BAK homodimer reveals an inhibitory zinc binding site. Mol Cell 24: 677–688 [DOI] [PubMed] [Google Scholar]
- Muchmore SW, Sattler M, Liang H, Meadows RP, Harlan JE, Yoon HS, Nettesheim D, Chang BS, Thompson CB, Wong S‐L et al (1996) X‐ray and NMR structure of human Bcl‐xL, an inhibitor of programmed cell death. Nature 381: 335–341 [DOI] [PubMed] [Google Scholar]
- Oh KJ, Singh P, Lee K, Foss K, Lee S, Park M, Lee S, Aluvila S, Park M, Singh P et al (2010) Conformational changes in BAK, a pore‐forming proapoptotic Bcl‐2 family member, upon membrane insertion and direct evidence for the existence of BH3‐BH3 contact interface in BAK homo‐oligomers. J Biol Chem 285: 28924–28937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill KL, Huang K, Zhang J, Chen YI, Luo XU (2016) Inactivation of prosurvival Bcl‐2 proteins activates Bax/Bak through the outer mitochondrial membrane. Genes Dev 30: 973–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliari LJ, Kuwana T, Bonzon C, Newmeyer DD, Tu S, Beere HM, Green DR (2005) The multidomain proapoptotic molecules Bax and Bak are directly activated by heat. Proc Natl Acad Sci U S A 102: 17975–17980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedrote MM, de Oliveira GAP, Felix AL, Mota MF, Marques MDA, Soares IN, Iqbal A, Norberto DR, Gomes AMO, Gratton E et al (2018) Aggregation‐primed molten globule conformers of the p53 core domain provide potential tools for studying p53C aggregation in cancer. J Biol Chem 293: 11374–11387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pervushin K, Riek R, Wider G, Wüthrich K (1997) Attenuated T2 relaxation by mutual cancellation of dipole‐dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc Natl Acad Sci U S A 94: 12366–12371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, Schulten K (2005) Scalable molecular dynamics with NAMD. J Comput Chem 26: 1781–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogmore J, Pemberton J, Chi X, Andrews D (2016) Using FRET to measure protein interactions between Bcl‐2 family proteins on mitochondrial membranes. Methods Mol Biol 1419: 197–212 [DOI] [PubMed] [Google Scholar]
- Privalov PL (1979) Stability of proteins: small globular proteins. Adv Protein Chem 33: 167–241 [DOI] [PubMed] [Google Scholar]
- Raltchev K, Pipercevic J, Hagn F (2018) Production and structural analysis of membrane‐anchored proteins in phospholipid nanodiscs. Chemistry 24: 5493–5499 [DOI] [PubMed] [Google Scholar]
- Royer CA (2006) Probing protein folding and conformational transitions with fluorescence. Chem Rev 106: 1769–1784 [DOI] [PubMed] [Google Scholar]
- Salvador‐Gallego R, Mund M, Cosentino K, Schneider J, Unsay J, Schraermeyer U, Engelhardt J, Ries J, García‐Sáez AJ (2016) Bax assembly into rings and arcs in apoptotic mitochondria is linked to membrane pores. EMBO J 35: 389–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzmann M, Pervushin K, Wider G, Senn H, Wüthrich K (1998) Trosy in triple‐resonance experiments: new perspectives for sequential NMR assignment of large proteins. Proc Natl Acad Sci U S A 95: 13585–13590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattler M, Schleucher J, Griesinger C (1999) Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Prog Nucl Magn Reson Spectrosc 34: 93–158 [Google Scholar]
- Schafer B, Quispe J, Choudhary V, Chipuk JE, Ajero TG, Du H, Schneiter R, Kuwana T (2009) Mitochondrial outer membrane proteins assist Bid in Bax‐mediated lipidic pore formation. Mol Biol Cell 20: 2276–2285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM (2003) The Xplor‐NIH NMR molecular structure determination package. J Magn Reson 160: 65–73 [DOI] [PubMed] [Google Scholar]
- Steiner A, Schlepckow K, Brunner B, Steiner H, Haass C, Hagn F (2020) γ‐Secretase cleavage of the Alzheimer risk factor TREM 2 is determined by its intrinsic structural dynamics. EMBO J 39: e104247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Youle RJ, Tjandra N (2000) Structure of bax: coregulation of dimer formation and intracellular localization. Cell 103: 645–654 [DOI] [PubMed] [Google Scholar]
- Tan Y, Wu C, De Veyra T, Greer PA (2006) Ubiquitous calpains promote both apoptosis and survival signals in response to different cell death stimuli. J Biol Chem 281: 17689–17698 [DOI] [PubMed] [Google Scholar]
- Teijido O, Ujwal R, Hillerdal CO, Kullman L, Rostovtseva TK, Abramson J (2012) Affixing N‐terminal α‐helix to the wall of the voltage‐dependent anion channel does not prevent its voltage gating. J Biol Chem 287: 11437–11445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrecillas A, Martínez‐Senac MM, Goormaghtigh E, De Godos A, Corbalán‐García S, Gómez‐Fernández JC (2005) Modulation of the membrane orientation and secondary structure of the C‐terminal domains of Bak and Bcl‐2 by lipids. Biochemistry 44: 10796–10809 [DOI] [PubMed] [Google Scholar]
- Türkmen S, Riehn M, Klopocki E, Molkentin M, Reinhardt R, Burmeister T (2011) A BACH2‐BCL2L1 fusion gene resulting from a t(6;20)(q15;q11.2) chromosomal translocation in the lymphoma cell line BLUE‐1. Genes Chromosomes Cancer 50: 389–396 [DOI] [PubMed] [Google Scholar]
- Uren RT, Iyer S, Kluck RM (2017a) Pore formation by dimeric Bak and Bax: an unusual pore? Philos Trans R Soc B Biol Sci 372: 20160218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uren RT, O’Hely M, Iyer S, Bartolo R, Shi MX, Brouwer JM, Alsop AE, Dewson G, Kluck RM (2017b) Disordered clusters of Bak dimers rupture mitochondria during apoptosis. Elife 6: e19944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas‐Uribe M, Rodnin MV, Ladokhin AS (2013) Comparison of membrane insertion pathways of the apoptotic regulator Bcl‐xL and the diphtheria toxin translocation domain. Biochemistry 52: 7901–7909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wales TE, Engen JR (2006) Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom Rev 25: 158–170 [DOI] [PubMed] [Google Scholar]
- Weber K, Harper N, Schwabe J, Cohen GM (2013) BIM‐Mediated Membrane Insertion of the BAK Pore Domain Is an Essential Requirement for Apoptosis. Cell Rep 5: 409–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphal D, Dewson G, Czabotar PE, Kluck RM (2011) Molecular biology of Bax and Bak activation and action. Biochim Biophys Acta 1813: 521–531 [DOI] [PubMed] [Google Scholar]
- Westphal D, Dewson G, Menard M, Frederick P, Iyer S, Bartolo R, Gibson L, Czabotar PE, Smith BJ, Adams JM et al (2014a) Apoptotic pore formation is associated with in‐plane insertion of Bak or Bax central helices into the mitochondrial outer membrane. Proc Natl Acad Sci 111: E4076–E4085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphal D, Kluck RM, Dewson G (2014b) Building blocks of the apoptotic pore: how Bax and Bak are activated and oligomerize during apoptosis. Cell Death Differ 21, 196–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis SN, Fletcher JI, Kaufmann T, van Delft MF, Chen L, Czabotar PE, Ierino H, Lee EF, Fairlie WD, Bouillet P et al (2007) Apoptosis initiated when BH3 ligands engage multiple Bcl‐2 homologs, not Bax or Bak. Science 315: 856–859 [DOI] [PubMed] [Google Scholar]
- Xiao K, Zhao W, Zhou L, Chang DC (2016) Alpha 5/6 helix domains together with N‐terminus determine the apoptotic potency of the Bcl‐2 family proteins. Apoptosis 21: 1214–1226 [DOI] [PubMed] [Google Scholar]
- Xu XP, Zhai D, Kim E, Swift M, Reed JC, Volkmann N, Hanein D (2013) Three‐dimensional structure of Bax‐mediated pores in membrane bilayers. Cell Death Dis 4: e683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye K, Meng WX, Sun H, Wu BO, Chen M, Pang Y‐P, Gao J, Wang H, Wang J, Kaufmann SH et al (2020) Characterization of an alternative BAK‐binding site for BH3 peptides. Nat Commun 11: 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yethon JA, Epand RF, Leber B, Epand RM, Andrews DW (2003) Interaction with a membrane surface triggers a reversible conformational change in Bax normally associated with induction of apoptosis. J Biol Chem 278: 48935–48941 [DOI] [PubMed] [Google Scholar]
- Zhang Z, Subramaniam S, Kale J, Liao C, Huang B, Brahmbhatt H, Condon SG, Lapolla SM, Hays FA, Ding J et al (2016) BH3‐in‐groove dimerization initiates and helix 9 dimerization expands Bax pore assembly in membranes. EMBO J 35: 208–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P, Wagner G (2010) Overcoming the solubility limit with solubility‐enhancement tags: successful applications in biomolecular NMR studies. J Biomol NMR 46, 23–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu G, Xia Y, Nicholson LK, Sze KH (2000) Protein dynamics measurements by TROSY‐based NMR experiments. J Magn Reson 143: 423–426 [DOI] [PubMed] [Google Scholar]
- Zweckstetter M, Bax A (2000) Prediction of sterically induced alignment in a dilute liquid crystalline phase: aid to protein structure determination by NMR [11]. J Am Chem Soc 122: 3791–3792 [Google Scholar]